sibon igor chu bordeaux - collège des enseignants de … · 2016-11-25 · • pic de...
TRANSCRIPT

Les « stroke-like »
Sibon Igor
CHU Bordeaux

Stroke-like ?

Stroke-like ?

Stroke-like ?

Stroke-like ?

Stroke-like ?

Les déficits persistants non-vasculaires

Pathologies diverses
• Tumeurs
• Encéphalites
• Epilepsie avec déficit post-critique (paralysie de Todd)
• Affections du système nerveux périphérique– Syndrome de Guillain-Barré/Miller-Fisher– Vascularites du SNP– Atteinte de la jonction neuro-musculaire (myasthénie, botulisme)– Intoxications aux organo-phosphorés
• Troubles somatomorphes
• Dysfonctions neuronales…
Amort et al., Cerebrovasc Dis, 2011


Hypoglycémie:

Hypoglycémie:Réponses physiologiques
• 4.4 mmol/l: diminution sécrétion insuline
• 3.6 mmol/l: sécrétion adrénaline, glucagon, cortisol, GH
• 3.1 mmol/l: symptômes généraux: asthénie, faim, tremblement, anxiété, palpitations, sueurs
• 2.5 mmol/l: troubles cognitifs, confusion, diplopie, convulsions, coma…déficit neurologique focal.

Données d’imagerie
Œdème cytotoxique réversible
Nécrose laminaire

Resucrage et bilan étiologique
• Iatrogène (ADO, Insuline, antiarythmiques, AINS, ISRS, IEC, Bêtabloquants…)
• By-pass gastrique
• Insulinome
• Noninsulinoma pancreatogenous hypoglycemia syndrome (NIPHS)
• Hypoglycémie auto-immune (Syndrome d’Hirata)
• Hémopathies
• Encéphalite japonnaise
• Syndrome hyperinsulinisme-hyperamoniémie (hyperactivité de la glutamate dehydrogenase par mutation de GLUD1)

Dysfonction mitochondriale
• Une cellule = centaines de mitochondries
• ADNmt propre
• Usine énergétique

Généralités
• Siège de nombreux métabolismes intermédiaires:β-oxydation des acides gras, cycle de Krebs, phosphorylation oxydative, cycle de l’urée = production de l’énergie cellulaire
• 5 complexes majeursCI : NADH-CoQ réductase, CII: Succinyl -CoQ réductase, CIII: Ubiquinone-cyt. C réductase, CIV: Cytochrome C oxydase, CV: ATPase

Les mitochondriopathies
• Génétiques +++ ou Acquises
• Mutations de l’ADN mitochondrial ou nucléaire
• Peu de corrélation phénotype-génotype
• Manifestations cliniques dépendantes de la demande énergétique des tissus– SNP, SNC, endocrine, cœur, œil, oreilles, tube digestif, foie, reins,
moelle, peau

Spectre clinique

Les mitochondriopathies
• Génétiques +++ ou Acquises
• Mutations de l’ADN mitochondrial ou nucléaire
• Peu de corrélation phénotype-génotype
• Manifestations cliniques dépendantes de la demande énergétique des tissus– SNP, SNC, endocrine, cœur, œil, oreilles, tube digestif, foie, reins, moelle,
peau
• Manifestations aggravées par :– Fièvre, infections, stress et certains médicaments = demande énergétique
• Manifestations neurologiques variées– Directement liées à la mitochondriopathie– Secondaire aux dysfonctions systémiques

Expression neurologique des mitochondriopathies
Finsterer, Act Neurol Scand, 2006

AVC et mitochondriopathie
• Fréquent dans le MELAS…mais pas exclusif
• Localisation: lobes pariétaux et occipitaux ++
• Sujet jeune (troisième intention bilan AVC sujet jeune)
• Souvent précédé par un épisode migraineux
• Manifestations cliniques variées (déficit focal, migraines, épilepsie)
• Mécanisme méconnu: – hyperexcitabilité neuronale– variation taux ATP ?

Troisième intention (1),Rare mais impact thérapeutique
• Exploration du parenchyme :– Spectroscopie IRM : Si suspicion de mitochondriopathie ou d’autre maladie
métabolique.
• Exploration artérielle :– Angiographie par voie intra-artérielle (vascularite – dissection douteuse)– Scanner thoracique : recherche d’une malformation artério-veineuse pulmonaire.
• Cardiologie– Exploration rythmique : R-Test
• Biologie :– Hcy– Dosage des lactates et des pyruvates sanguins à jeun et en post prandial + LCR– Amoniémie sanguine + GDS +/- chromatographie des AA.– Activité enzymatique de l’alpha-galactosidase (Fabry) – Analyses génétiques par biologie moléculaire : suspicion clinique ou histoire
familiale évocatrice.– La mutation acquise (V617F) JAK2 sera recherchée en cas de polyglobulie ou
thrombocytémie.

• Neuro-psychiatriques– Céphalées 91 %– Troubles mnésiques 71%– Troubles de la sensibilité 48 %– Dépression 32 %
• Systémiques– Intolérance à l’effort 93 %– Surdité 77 %– Manifestations digestives 64 %– Petite taille 33 %– Diabète 33 %– Myocardiopathie 21 %
MELAS: Manifestations cliniques
Sproule et al; 2008

Apport de l’imagerie
• Aigue– Pas de systématisation artérielle– Prédominance des lésions corticales pariéto-occipitale– Migration des anomalies lésionnelles dans le temps– Œdème vasogénique puis cytotoxique– Hyperintensité T1 (nécrose laminaire)– Angio-IRM normale
• Chronique– Leucoencéphalopathie périventriculaire– Calcifications des noyaux gris centraux
Saneto et al; 2008

En image…
MELAS LeighSaneto et al; 2008

Le diagnostic positif
• Biologie:– Augmentation des taux de lactates mais attention: aciduries organiques,
aminoacidopathies, déficits en acide citrique, déficit en pyruvate deshydrogénaseet mauvais traitement du sérum ou du LCR
• Spectro-IRM:– Diminution du signal du NAA– Augmentation du pic de lactates
• Biopsie musculaire – Fibres vacuolisées– Inclusions basophiles (prolifération mitochondriale)– Fibres rouges déchiquetées marquées succinate DH– Modification expression COX
• Bio mol: m.3243A>G– Différents tissus possibles– Première intention si forte suspicion clinique syndromique
Filosto et al; 2008

Mitochondriopathies acquises
• Inhibiteurs de la polymérase γ (NRTI - VIH): diminue l’expression des protéines mitochondriales
– Myopathies et myocardiopathies (AZT)
• Chimiothérapies

Spectro-IRM

Variations génotypiques
Finsterer et al; 2004
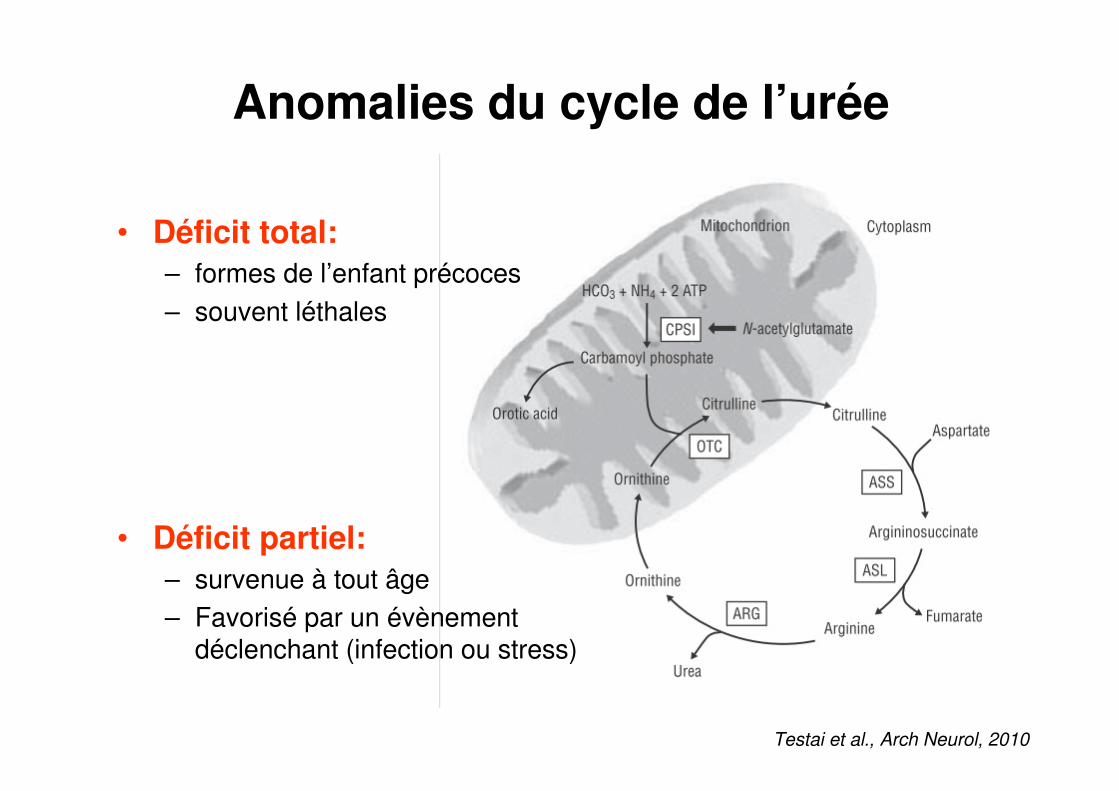
Anomalies du cycle de l’urée
• Déficit total:
– formes de l’enfant précoces – souvent léthales
• Déficit partiel:
– survenue à tout âge– Favorisé par un évènement
déclenchant (infection ou stress)
Testai et al., Arch Neurol, 2010

Physiopathologie
• Conséquences métaboliques– Hyperamoniémie– Augmentation des taux plasmatiques de glutamine
• Conséquences cellulaires– œdème astrocytaire et neuronal (+/- compression
microcirculation) – Altération du cycle glutamate-glutamine avec
phénomène d’excito-toxicité– Accumulation de glutamate intraneuronal avec mort
neuronale

• 20% des patients ont plus de 12 ans au moment de leur premier épisode d’hyperamoniémie
• AVC dans– Déficit en OTC– Déficit en CPS– Citrullinemie
• Stroke-like dans déficit en OTC ++
• Diagnostic biologique confirmé par:– analyses enzymatiques – biologie moléculaire
• Traitement– Réduction des apports en azote– Réduction des apports protéiques– Augmentation de l’excrétion du
azote (benzoate de sodium)– Place de la transplantation
hépatique non définie
Testai et al., Arch Neurol, 2010

Spectre clinico-radiologique
• Aspects cliniquesIntolérance aux protéines, troubles de croissance, insuffisance hépatique, troubles de la coagulation, stroke-like, épilepsie, confusion, coma
• Aspects IRM
• Mécanismes– Mutation OTC– Mutation ORNT1: Syndrome Hyperornithinemie-
hyperammonemie-homocitrullinurie– Mutation GLUD1: Syndrome hyperinsulinisme-
hyperamoniémie
Al-Hassnan et al; 2008

Aciduries organiques
Testai et al., Arch Neurol, 2010

Manifestations cliniques
• Début dans l’enfance (intervalle libre lié à l’activité résiduelle)
• Phase initiale: trouble de l’alimentation léthargie, vomissement, déshydratation
• « Sweaty feet » odor
• Pancytopénie et modifications glycémie
• Manifestations digestives: constipation, pancréatites
• Manifestations neurologiques: crises, retard intellectuel, mouvements anormaux (tremblement, chorée…), AVC:– Hémorragies cérebelleuses– Infarctus bilatéraux des ganglions de la base en l’absence de
décompensation métabolique
Testai et al., Arch Neurol, 2010

Diagnostic // Traitements
• Diagnostic– Spectroscopie IRM:
• Pic de lactate• Pic de glutamate/glutamine dans les ganglions de la base (acidurie
propionique)– Biologie
• Acyl carnitine• Acide aminés organiques sanguins et urinaires
– Evaluation activité enzymatique sur culture de fibroblastes ou leucocytes
– Biologie moléculaire (bonne relations génotype/phénotype)
• Traitement– Régime pauvre en protéines– L-carnitine– Contre-indication aspirine si acidurie isovalérique
Testai et al., Arch Neurol, 2010

Acidurie glutarique (GAT) (AR)
• GAT 1:– Déficit en glutaryl coenzyme A dehydrogenase– Altération métabolisme de la lysine, hydroxylysine et tryptophane.– Clinique: macrocéphalie progressive, crises comitiales (decompensation
métabolique ++°, crises encéphalopathiques (nécrose striatale) favorisée par épisodes infectieux
– AVC = stroke like (excitotoxicité striatale)– Diagnostic: dosage urinaire et mesure activité enzymatique– Traitement: régime sans lysine et pauvre en tryptophane; place de la
riboflavine (?; co-enzyme de GAT-1)
• GAT 2:– Déficit en flavoproteine ou flavoprotein oxyreductase. – Altération métabolisme mitochondrial des AA et β-oxydation des AG– Décompensations métaboliques déclenchées par le stress– Acidose, hypoglycemie sans cétose, hyperammonemie, and acidurie
organique.– AVC rares– Myopathie possible dans les formes tardives– Augmentation des CK

Stroke-like et maladies métaboliques
Keegan et al., Eur J Ped, 2003
Testai et al., Arch Neurol, 2010

Et les dysfonctions acquises…

Stroke-like migraine attacks after radiation therapy (SMART)
• Antécédent de radiothérapie encéphalique
• Absence d’arguments en faveur d’un résidu tumoral ou d’une récidive tumorale encéphalique
• Manifestations cliniques à début brutal, prolongées et réversibles évocatrices d’une atteinte corticale unilatérale de type– Déficit visuo-spatial, sensitif ou moteur unilatéral– Aphasie, Confusion, Crises comitiales
• Céphalées pendant les “attaques”
• Début une à plusieurs années après la radiothérapie
• Absence d’une autre pathologie potentiellement responsable
• Rehaussement cortical transitoire unilatéral dans le champ d’irradiation après injection de gadolinium
Black et al., Neurology 2006

Les déficits transitoires non-vasculaires

Pathologies diverses
• Aura migraineuse
• Epilepsie avec déficit post-critique (paralysie de Todd)
Amort et al., Cerebrovasc Dis, 2011

Paralysie de Todd
• 13.4 % de patients enregistrés en vidéo-EEG (44/513)
• Présentation clinique– Déficit moteur post-critique– Formes avec ataxie post-critique…ou aphasie…– Confusion fréquente
• Déficit unilatéral et controlatéral au foyer épileptogène
• Durée : 11s à 22 min en moyenne…parfois beaucoup plus…
• Parfois observé en l’absence de lésions IRM
• Favorisé par– Age– Epilepsie lésionnelle
• Apport diagnostic de l’EEG
Theodore., Epilepsy Behav , 2010

Pathologies diverses
• Aura migraineuse
• Epilepsie avec déficit post-critique (paralysie de Todd)
• HAnDL– déficit transitoire, céphalées, pléiocytose– Épisodes récurrents sur une période donnée– Hypoperfusion focale en imagerie
• Divers (VPPB, DDR…)
• Ictus amnésique
• Troubles somatomorphes
• Dysfonctions neuronales…
Amort et al., Cerebrovasc Dis, 2011

Canalopathies

Canaux ioniques
• Glycoprotéines transmembranaires
• Multiples sous-unités codées par des gènes différents
• Assemblées pour former un pore central
• Structure du pore définit la sélectivité de perméabilité d’un canal pour un ion
• Localisation SNC et SNP
• Impliqués dans l’excitabilité cellulaire– Canaux voltage dépendants
• Canaux potassiques: potentiel de repos et repolarisation• Canaux sodiques: potentiel d’action• Canaux calciques: contraction musculaire, neurotransmission, potentiel
d’action (cœur et CML), …• Canaux Chlore: hyperpolarisation
– Canaux ligands dépendants (Ach, GABA…)

Aspects cliniques
• Mutation d’une sous-unité d’un canal ionique => modification fonctionnelle– Gain de fonction: épilepsie, myokimie, myotonie…– Perte de fonction: paralysie
• Atteintes du Système nerveux central et périphérique

Atteintes Neurologiques
SNPSNC
Bernard et al; 2008

Aspects cliniques
• Mutation d’une sous-unité d’un canal ionique => modification fonctionnelle– Gain de fonction: épilepsie, myokimie, myotonie…– Perte de fonction: paralysie
• Atteintes du Système nerveux central et périphérique
• Hétérogénéité clinique et phénotypique– Une même manifestation clinique peut résulter de mutations
différentes– Une même mutation peut induire des phénotypes différents
Saneto et al; 2008

Migraine hémiplégique familiale
Rare: Sporadique = 0.002%Familliale = 0.003%
Ducros et al., Rev Neurol 2008

La clinique• Début au cours de la première ou seconde décade
• Le déficit moteur– Sévérité, localisation et durée variable (Uni ou bilatéral, alternant ou fixe)– Toujours associé à au moins un autre symptôme
• troubles sensitifs (98 %), troubles visuels (89 %), troubles du langage (79 %), «signes basilaires » (70 %).
• La céphalée– s’installe après, pendant ou avant l’aura.– 95 % des patients ont une céphalée lors de chaque crise de MH, 4 % ont parfois une céphalée et 1 %
n’en ont jamais – précédée de l’aura dans 75%
• Aura, visuelles, sensitives, phasiques, motrices ou vertébro-basilaires
• Faible nombre d’attaques annuelles
• Facteurs déclenchants: stress, traumatismes crâniens bénins, angiographie• Diminution de la fréquence des attaques avec le vieillissement• Divers
– 40 % des patients ont eu au moins une crise sévère avec aura prolongée (jusqu’à plusieurs semaines), confusion, hyperthermie jusqu’à 41 °C, signes méningés et troubles de la conscience allant de l’obnubilation au coma profond
– Signes cérébelleux permanents dans 4.5%– Associations possibles : tremblement essentiel, troubles cognitifs
Weiss et al, 2008
Russell et Ducros, 2011
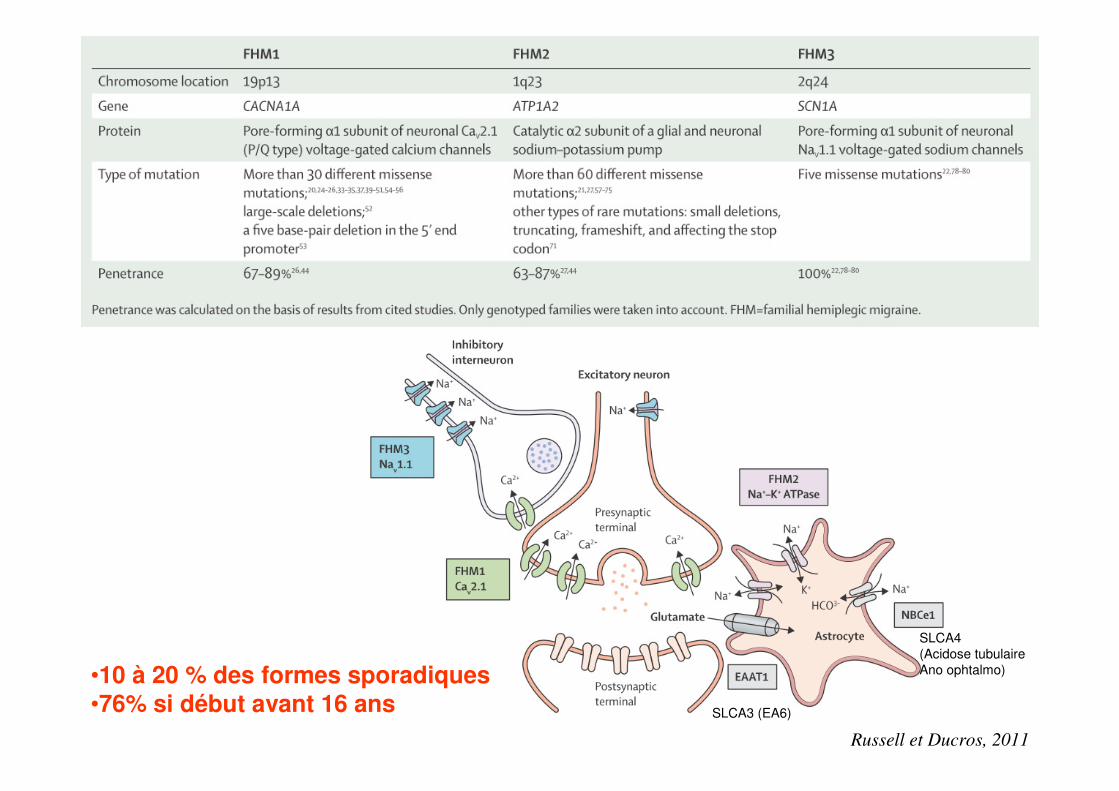
Russell et Ducros, 2011
SLCA3 (EA6)
SLCA4(Acidose tubulaireAno ophtalmo)•10 à 20 % des formes sporadiques
•76% si début avant 16 ans

Génétique MHF
• Influence mineure des gènes identifiés dans la survenue de formes sporadiques (10 à 20 %) sauf si survenue précoce (76% avant 16 ans)
• CACNA1A, – chromosome 19pl3 – sous-unité α1A des canaux calciques neuronaux Cav2.1 (type P/Q)– modulent la libération de neurotransmetteurs incluant les monoamines,
l’acétylcholine, le glutamate et le calcitonine gène related peptide (CGRP).
• ATP1A2,– bras long du chromosome 1 en q23 – sous-unité α2 d’une pompe Na+/K+ ATP dépendante– utilise l’hydrolyse de l’ATP pour faire rentrer du K+ dans la cellule en échange du
Na+ contre les gradients transmembranaires. – maintien d’un fort gradient transmembranaire de sodium est essentiel pour le
transport du glutamate et du calcium
• SCN1A,– chromosome 2 en q24 – canal sodique voltage-dépendant neuronal
Ducros et al., Rev Neurol 2008

Examens Complémentaires
Russell et Ducros, 2011

Prise en charge
• Diagnostic de certitude– Biologie moléculaire– Pas de diagnostic anténatal
• Thérapeutique– Crise
• antalgiques simples, de l’aspirine ou des anti-inflammatoires non stéroidiens
– Fond• Place des antiépileptiques stabilisateurs de membrane ?• Acetazolamide ?

Russell et Ducros, Lancet Neurol 2011


Ataxies épisodiques
Jen et al., Brain 2007
Late onset hereditary episodic ataxia, Damak et al; JNNP 2009
EA7: début avant 20 ans, déclenché par efforts et excitation, durée de quelques heures à plusieurs jours, associé à une faiblesse et vertige, gène localisé sur le chromosome 19q13
Kerber et al, Arch Neurol 2007

Prise en charge
• Diagnostic de certitude– Biologie moléculaire (EA1 et EA2)
• Prise en charge thérapeutique– EA1: Carabamazépine, acide valproique,
acetazolamide– EA2: Acétazolamide ++, 4-aminopyridine, 3-4-
aminopyridine

Paralysies Périodiques
• PP Hypokaliémique:– Déficit + hypokaliémie– +/- modifications ECG– Prédominance matinale– Déclenché par les facteurs abaissant les taux de potassium (sucre, sel, stress,
médicaments)– Mutations: CACNA1S – SCN4A (1/3 des patients)
• PP Hyperkaliémiques– Attaques brèves à partir adolescence– Potassium normal ou augmenté– Déclenché par repos après exercice, stress, fatigue, alimentation– Mutations : SCN4A– Formes acquises: Thyrotoxicose; Diarrhée; Alcool; Hyperadlostéronisme primaire ou
secondaire; Acidose tubulaire
• Syndrome d’Andersen-Tawil– Paralysie périodique– Arythmie cardiaque– Dysmorphie (hypertélorisme, micrognatie.).– Mutation KCNJ2
Finsterer, 2008
Hiraga et al., J of Neurol Sci, 2012

Cardiopathies héréditaires et canalopathies
Fréquence des Stroke et Stroke-Like ?
Johnson et al, 2008Makita, 2009

Angio-œdème héréditaireDéficit en C1-esterase inhibitor
• Autosomal dominant // Prévalence = 1/50000
• Manifestations cliniques– Œdèmes cutanés– Œdèmes des voies aériennes supérieures– Douleurs abdominales– Manifestations neurologiques
• Céphalées• Déficits transitoires répétés• Crises comitiales• Migraines prolongées
• Mécanisme moléculaire– Déficit en C1-INH => ↑ kallicréine => ↑ bradykinine => ↑ perméabilité vasculaire
• Traitement– Administration de C1-INH
Bonnaud et al., Neurology, 2012

Angiopathie Amyloïde
Augmentation du risque d’HIC
Attention aux AAP !!!
Charidimou et al., 2012

Risque de la thrombolyse ?
Taux de thrombolyse pour stroke-mimics
= 1,3 à 25% selon les séries
