sequencealignmentinbioinformatics 100204112518-phpapp02
TRANSCRIPT

PILLAI ASWATHY VISWANATHBOTANY

• An alignment is an arrangement of two or more sequence (DNA, RNA or protein) which shows whether the two sequence aligned are similar or different
• Helps in inferring functional , Structural or evolutionary relationship between the sequence
• Sequence alignment methods are used to find the best- matching sequences

The sequence alignment is made between a known sequence and unknown sequence or between two unknown sequences.
The known sequence is called reference sequence,the unknown sequence is called query sequence

Sequences that are very much alike may have similar secondary and 3D structure, similar function and likely a common ancestral sequence.
Such sequence are termed as homologous and shares a common ancestors

In sequence alignment,the sequence to be compared are written one above the other.
A T C G………..1 -- T C A………...2 -2 +2 +2 -1 = 1 there are match and mismatch characters To reduce mismatch a “gap’’ is added A T C G ………..1 T C A --………...2 -1 -1 -1 -2 = -5
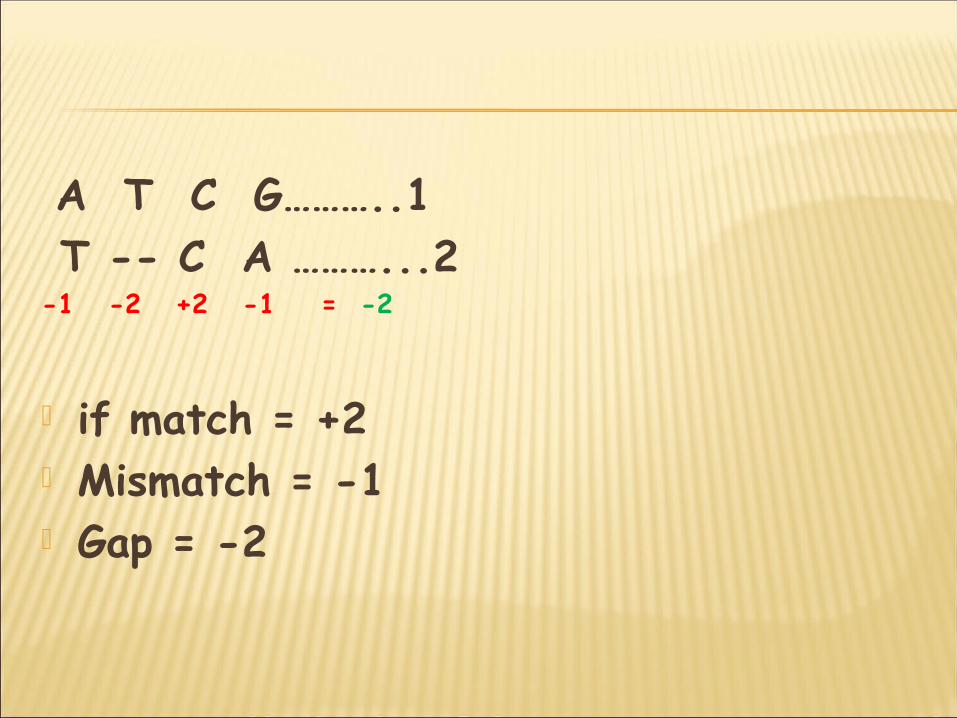
A T C G………..1 T -- C A ………...2-1 -2 +2 -1 = -2
if match = +2 Mismatch = -1 Gap = -2

Very short or very similar sequences can be aligned by hand.
However, most interesting problems require the alignment of lengthy, highly variable or extremely numerous sequences that cannot be aligned solely by human effort.
Computational approaches to sequence alignment

Different computational methods,called dynamic programming algorithems
They are required for finding the best alignment of the sequence
There Are Mainly Two Types Of Sequence Alignment
Global AlignmentLocal Alignment

A general global alignment technique is the Needleman–Wunsch algorithm, which is based on dynamic programming.
The Smith–Waterman algorithm is a general local alignment method also based on dynamic programming.

In global alignment ,an attempt is made to align the entire sequence ( end to end alignment )
It two sequences have approximately the same length and are quite similar,they are suitable for global alingment
Suitable for aligning two closely related sequences

Global Alignment are usually done for comparing homologous genes
Like comparing two genes with same function or comparing two proteins with similar function

Finds local regions with the highest level of similarity between the two sequence
Any two sequences can be locally aligned as local alignment finds stretches of sequence with high level of matches without considering the alingnment of rest of the sequence regions
Suitable for aligning more divergent sequence or distantly related sequence

Sequences which are suspected to have similarity or even dissimilar sequences can be compared with local alignment method. It finds local regions with high level of similarity.

These two algorithms make all possible pair wise comparisons to all of the data base sequence and find the the best alignment of sequence
But the process is often too slow for searching large database.some times it takes hours for a search
So faster algorithem,such as BLAST and FASTA have been developed

Blast and Fasta are two software that are used to compare biological sequences of DNA, amino acids, proteins and nucleotides of different species and look for the similarities.
These algorithms were written keeping speed in mind because as the data bank of the sequences swelled once DNA was isolated in the laboratory by the scientists in mid 1980s there raised a need to compare and find identical genes for further research at high speed.
Blast is an acronym for Basic Local Alignment Search Tool and uses localized approach in comparing the two sequences.
Fasta is a software known as Fast A where A stands for All because it works with the alphabet like Fast A for DNA sequencing and Fast P for protein.
Both Blast and Fasta are very fast in comparing any genome database and are therefore very viable monetarily as well as in saving time.

One of the most widely used bioinformatics software Blast was developed in 1990 and since then have been available to everyone at NCBI site.
This software can be accessed by any one and can be modified according to ones need.
Blast is the software in which input data of a sequence to be compared is in Fasta format and output data can be obtained in plain text, HTML or XML.
Blast works on the principle of searching for localized similarities between the two sequences and after short listing the similar sequences it searches for neighborhood similarities.

The software searches for high number of similar local regions and gives the result after a threshold value is reached.
This process differs from earlier software in which entire sequence was searched and compared which took a lot of time.
Blast is used for many purposes like DNA mapping, comparing two identical genes in different species, creating phylogenetic tree.

For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene;
BLAST will identify sequences in the human genome that resemble the mouse gene based on similarity of sequence.
The BLAST algorithm and program were designed by Stephen Altschul, Warren Gish, Webb Miller, Eugene Myers, and David J. Lipman at the National Institutes of Health and was published in the Journal of Molecular Biology in 1990

Fasta program was written in 1985 for comparing protein sequences only but was later modified to conduct searches on DNA also.
Fasta software uses the principle of finding the similarity between the two sequences statistically.
This software matches one sequence of DNA or protein with the other by local sequence alignment method.
It searches for local region for similarity and not the best match between two sequences.

Since this software compares localized similarities at times it can come up with a mismatch.
In a sequence Fasta takes a small part known as k-tuples where tuple can be from 1 to 6 and matches with k-tuples of other sequence and once a threshold value of matching is reached it comes up with the result.
It is a program that is used to shortlist prospects of matching sequence from a large number for full comparison as it is very fast.

Blast is much faster than Fasta. Blast is much more accurate than Fasta. For closely matched sequences Blast is very accurate
and for dissimilar sequence Fasta is better software. Blast can be modified according to the need but
Fasta cannot be modified. Blast has to use Fasta input format to get the output
data. Blast is much more versatile and widely used than
Fasta.

Global and local sequence alignments can be of two types:
pair wise alignemnt multiple sequence alignemnt

This is primarily a method for comparing two sequence to find the best matching in local and global alignments
The purpose of pair wise alignment is to find related gene or gene product in a database of known sequence
It is used for the identification of sequence of unknown structure of function
Another important use is the study of molecular evolution.

Multiple alignments is an alingnment that compares more than two sequences
Here an unknown sequence is matched with several known sequence to reveal the relatedness of sequences ,with out making pair wise alignment first
A multiple alignment contains a distribution of closely and distantly related sequences
It provides information about the most similar regions in the set

Thus it is more informative about evolutionary relationship
This is used to build phylogenetic trees. It begins with the most closely related sequence and ends the most distant
The most commonly used multiple alignment software is the CLUSTAL.

Similar sequence are aligned in pairs first and distanly related sequence are added later
The aligned scores thus obtained are used to cluster the sequences to generate the final multiple alignment