mitochondrial translocation of cofilin is an early step in apoptosis induction
TRANSCRIPT

L E T T E R S
NATURE CELL BIOLOGY VOLUME 5 | NUMBER 12 | DECEMBER 2003 1083
Mitochondrial translocation of cofilin is an early step inapoptosis inductionBoon Tin Chua1, Christiane Volbracht1, Kuan Onn Tan2, Rong Li3, Victor C. Yu2,4 and Peng Li1,5,6
Increasing evidence suggests that movement of key proteins inor out of mitochondria during apoptosis is essential for theregulation of apoptosis. Here, we report identification of theactin-binding protein cofilin by a proteomic approach, as sucha factor translocated from cytosol into mitochondria afterinduction of apoptosis. We found that after induction ofapoptosis, cofilin was translocated to mitochondria beforerelease of cytochrome c. Reduction of cofilin protein levelswith small-interfering RNA (siRNA) resulted in inhibition ofboth cytochrome c release and apoptosis. Onlydephosphorylated cofilin was translocated to mitochondria,and the cofilin S3D mutant, which mimicks thephosphorylated form, suppressed mitochondrial translocationand apoptosis. Translocation was achieved through exposure ofan amino-terminal mitochondrial targeting signal incombination with carboxy-terminal sequences. When correctlytargeted to mitochondria, cofilin induced massive apoptosis.The apoptosis-inducing ability of cofilin, but not itsmitochondrial localization, was dependent on the functionalactin-binding domain. Thus, domains involved inmitochondrial targeting and actin binding are indispensable forits pro-apoptotic function. Our data suggest that cofilin has animportant function during the initiation phase of apoptosis.
Mitochondria are the major organelles involved in the signal trans-duction and biochemical execution of apoptosis1. Many types ofapoptotic signals converge at the level of mitochondria and induce therelease of apoptogenic mitochondrial proteins that directly promoteapoptosis. Cytochrome c is released into the cytoplasm2 and in turnactivates caspase-9 by forming a complex with Apaf-1 (ref. 3), activat-ing the execution caspase cascade. Bcl-2 family members are known toregulate the release of cytochrome c by controlling mitochondrialmembrane integrity4. Translocation of pro-apoptotic Bcl-2 familyproteins to mitochondria has been suggested to be important for initi-ating apoptotic signalling from the organelle4. Activation of these pro-apoptotic proteins can be achieved by post-transcriptionalmodifications such as dephosphorylation of BAD5. Direct signalling
to the mitochondria through translocation is not restricted to Bcl-2family members, as several other proteins (including the transcriptionfactor TR3 (ref. 6) and the Peutz-Jegher gene product LKB1 (ref. 7))have also been shown to translocate into the mitochondria, therebyinducing apoptosis. Although the role of mitochondria in controllingdownstream apoptotic events such as caspase activation is relativelywell characterized, mechanisms by which upstream apoptotic signalsare transduced into mitochondria remain largely elusive.
Intrigued by the observation that protein movement in or out of themitochondria seems to be essential for regulation of apoptosis, we iso-lated mitochondria from staurosporine (STS)-treated and untreatedHL60 cells and investigated their mitochondrial protein compositionsusing two-dimensional gel electrophoresis and silver staining. STS wasused as it is a general kinase inhibitor that rapidly induces caspasedependant apoptosis. We observed a significant increase in distinctprotein spots in mitochondrial fractions from STS-treated apoptoticcells, compared with untreated controls. Using mass spectrometricanalysis, we identified one of the spots as cofilin (Fig. 1a, arrows).Cofilin is a member of the cofilin/actin depolymerizing factor (ADF)family, which regulates actin dynamics by increasing the rate of actindepolymerization and facilitating actin filament turnover8. Westernblot analysis confirmed that cofilin was found in mitochondrial frac-tions only in the presence of STS (Fig. 1b, top), and this was concomi-tant with the disappearance of cofilin from cytosolic fractions (Fig. 1b,bottom). Translocation of cofilin was detected as early as 5 min afterSTS treatment (see Supplementary Information, Fig. S1a) and was notblocked by the broad-range caspase inhibitor zVAD-fmk (Fig. 1b, top)or by Bcl-xL in the presence of apoptotic stimuli (see SupplementaryInformation, Fig. S1c). Translocation of cofilin was further confirmedin SH-SY5Y cells by indirect immunofluorescence microscopy (seeSupplementary Information, Fig. S1b). Mitochondrial translocationof cofilin did not occur through increased association of actin withmitochondria, as similar levels of actin were detected in mitochondrialfractions from control and apoptotic cells (Fig. 1b, top). The gradient-purified mitochondrial fraction was free of cytosolic protein contami-nation, as no cytoplasmic Grb2b protein was detected (Fig. 1b, top).An early accumulation of cofilin in mitochondrial fractions was
1Laboratories of Apoptosis Regulation, 2Mechanisms of Apoptosis in Mammalian Cells and 3Proteomics, Institute of Molecular and Cell Biology, National Institute ofSingapore, 30 Medical Drive, 117609, Singapore. 4Adjunct staff of the Department of Pharmacology, National University of Singapore, 18 Medical Drive, Singapore,119260. 5Department of Biology, Hong Kong University of Science and Technology, Clear water bay, Kowloon, Hong Kong. 6Correspondence should be addressed to P.L. (e-mail: [email protected]).
Published online: 23 November 2003; DOI: 10.1038/ncb1070
print ncb1070 13/11/03 1:59 pm Page 1083
© 2003 NaturePublishing Group
© 2003 Nature Publishing Group

L E T T E R S
1084 NATURE CELL BIOLOGY VOLUME 5 | NUMBER 12 | DECEMBER 2003
observed in several different cell types (SH-SY5Y, COS-7 and HeLacells; data not shown). In addition, stimulation of apoptosis withetoposide also resulted in early accumulation of cofilin in mitochondr-ial fractions, indicating that mitochondrial translocation of cofilin is ageneral phenomenon that occurs before caspase activation. The pre-cise location of cofilin at mitochondria was further determined usingproteinase K-mediated digestion of isolated mitochondria. Cofilin wasfound to be associated with the outer mitochondrial membrane (seeSupplementary Information, Fig. S1d).
To further corroborate that mitochondrial translocation of cofilin iscrucial for the initiation of apoptosis, we used siRNA to knock downcofilin protein levels in SH-SY5Y cells. Endogenous cofilin protein lev-els were efficiently and specifically reduced in the presence of a cofilin-specific double-stranded RNA (dsRNA) oligomer (Fig. 2a). As acontrol, we used an siRNA containing a two-nucleotide mutation ofthe cofilin sequence. This control siRNA did not affect cofilin proteinlevels (Fig. 2a). Using immunofluorescence micrscopy, we determinedthat cofilin expression was reduced to undetectable levels in 40% ofcells (data not shown). SH-SY5Y cells lacking cofilin were healthy andhad a normal morphology (Fig. 2b). In the presence of STS, cells with
normal levels of cofilin underwent apoptosis, displaying typical mor-phological changes such as diffuse cytochrome c staining in the cyto-plasm, and nuclear condensation and fragmentation (Fig. 2b,arrowhead). In cofilin knock-down cells, however, the punctuatecytochrome c staining pattern was maintained and cells did not displayany apoptotic nuclear morphology (Fig. 2b, arrow). When cofilin pro-tein was reduced, cells were protected from the effects of STS andetoposide treatment at 8 and 24 h, respectively (Fig. 2c, d). Cofilinknock-down cells displayed long-term resistance to STS- (seeSupplementary Information, Fig. S2a) or etoposide-induced apoptosis(data not shown), and were subsequently able to proliferate and formcolonies (see Supplementary Information, Fig. S2b). Thus, our dataindicate that cofilin is crucial for the initiation of apoptosis induced bySTS and etoposide.
As cofilin activity is negatively regulated by LIM kinases, whichphosphorylate cofilin on Ser 3 and thereby inactivate its depolymer-ization activity9,10, we separated the two forms of cofilin from cytosolicand mitochondrial fractions on 2D gels and detected both forms withan anti-cofilin antibody. Both phosphorylated and dephosphorylatedcofilin were identified in the cytosolic fraction of control cells (Fig. 3a;also see Supplementary Information, Fig. S3a). After induction ofapoptosis with STS for 1 h, only dephosphorylated cofilin was presentin both the cytosolic and mitochondrial fractions (Fig. 3a). When cellswere treated with etoposide, both forms of cofilin were found in thecytosolic fraction. However, only the dephosphorylated form of cofilinappeared in the gradient-purified mitochondrial fraction (Fig. 3a). Asdephosphorylation of cofilin occurs in response to diverse signals thatstimulate motility and shape changes without triggering apoptosis, wetested whether dephosphorylated cofilin is localized at mitochondriaduring these processes. We activated the differentiated neutrophil-likeHL60 cells11 with the protein kinase C activator 12-O-tetrade-canoylphorbol-13-acetate (TPA), which results in dephosphorylationof cofilin12. As expected, both phosphorylated and dephosphorylatedcofilin were found in whole-cell lysates from neutrophil-like HL60cells, whereas only dephosphorylated cofilin was present after activa-tion with TPA (see Supplementary Information, Fig. S3b). No cofilinproteins were detected in gradient-purified mitochondrial fractionsfrom TPA-stimulated cells (Fig. 3b), suggesting that dephosphorylatedcofilin only localizes to mitochondria during apoptotic induction.
We then investigated whether the phosphorylation status of cofilincan influence its ability to translocate to mitochondria and induceapoptosis. Two cofilin mutants were generated that mimick either thedephosphorylated or phosphorylated forms by changing Ser 3 to ala-nine (S3A) or aspartate (S3D), respectively13. Wild-type (wt) ormutant forms of cofilin (S3A and S3D) were expressed at comparablelevels (data not shown). COS-7 cells overexpressing haemagglutinin(HA)-tagged wild type (wt) or mutant forms of cofilin (S3A and S3D)had normal morphology, with no detectable mitochondrial localiza-tion of exogenous cofilin (data not shown) and no increase in basalapoptosis when compared with vector control 24 h after transfection(9.5 ± 3.7%, 14.5 ± 5.8%, 13.8 ± 3.7% and 4.8 ± 0.8% for vector con-trol, wild-type, S3A and S3D cofilin, respectively). Interestingly, wild-type and S3A cofilin accumulated in gradient-purified mitochondrialfractions from STS-treated cells, whereas no cofilin S3D was detected(Fig. 3c, top). Importantly, overexpression of cofilin S3D reducedmitochondrial accumulation of endogenous cofilin in STS-treatedcells (Fig. 3c, middle). Therefore, mutant cofilin S3D not only failed totranslocate to mitochondria, but functioned in a dominant-negativemanner to suppress translocation of endogenous cofilin to mitochon-dria. Accordingly, overexpression of cofilin S3D reduced STS-inducedapoptosis by 30%, whereas the level of apoptosis for wild-type- and
0 h 4 h2 h1 h 2 h + zVAD-fmkMitochondria
STS:
Cytosol
0 h 4 h2 h1 h 0 h 2 h
zVAD-fmk
STS:
Control STS
Mitochondria
19K pI 8−8.5Cofilin
H+ H+ OH−OH−
Who
le-ce
ll lys
ate
Cofilin
Bcl-2
Bad
Actin
Grb2
Cyt c
Cofilin
a
b
Figure 1 Translocation of cofilin at an early stage of apoptosis. (a) 2D PAGEanalysis identifies cofilin in mitochondrial fractions from STS-treated HL60cells. Analysis of silver-stained gradient-purified mitochondrial proteinsfrom cells incubated for 1 h in the presence or absence of 1 µM STS. (b) Mitochondrial accumulation of cofilin before cytochrome c release.Western blot analysis of mitochondrial (top) and cytosolic (bottom) fractionstreated with 1 µM STS and 50 µM zVAD-fmk.
print ncb1070 13/11/03 1:59 pm Page 1084
© 2003 NaturePublishing Group
© 2003 Nature Publishing Group

L E T T E R S
NATURE CELL BIOLOGY VOLUME 5 | NUMBER 12 | DECEMBER 2003 1085
S3A-expressing cells did not differ significantly when compared withthat of vector control (Fig. 3d). Thus, our data indicate that the mito-chondrial translocation of cofilin and its ability to induce apoptosis areboth diminished by phosphorylation at Ser 3.
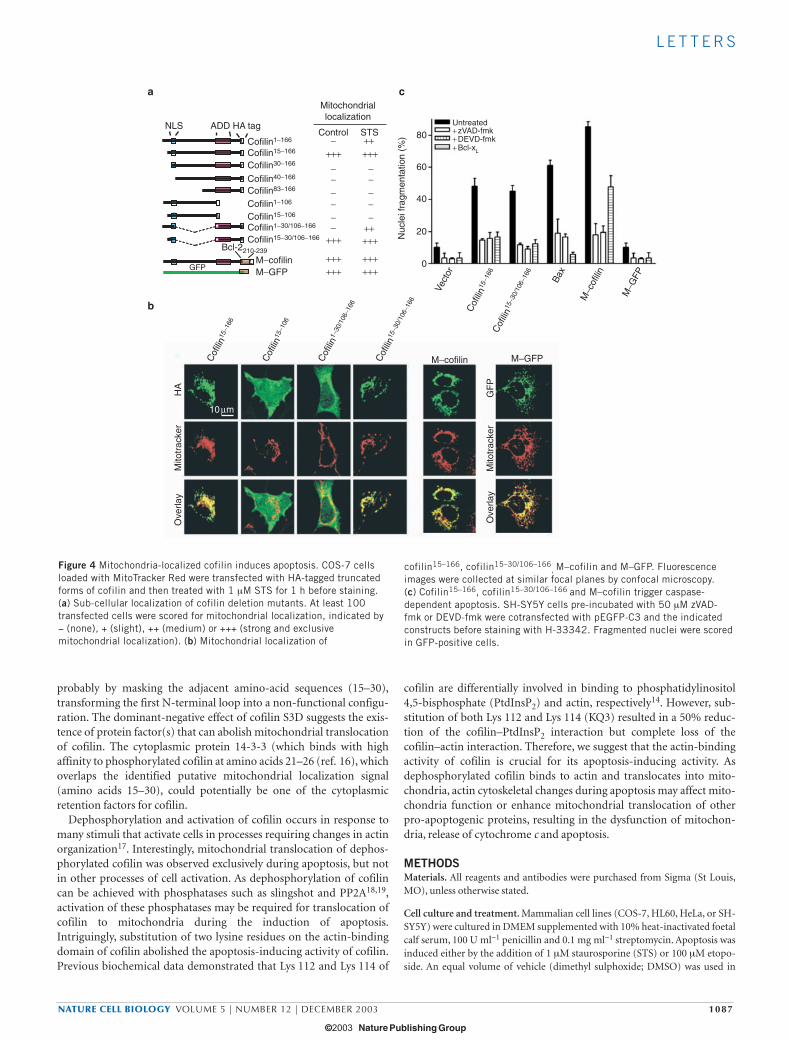
To identify region(s) responsible for mitochondrial localization ofcofilin, we generated C-terminal HA-tagged truncated forms of cofilinand investigated their sub-cellular localization by immunofluores-cence microscopy with anti-HA antibodies. Surprisingly, althoughother deletion mutants displayed no mitochondrial localization in theabsence of STS (Fig. 4a), a mutant in which the first 15 N-terminalamino acids were deleted (cofilin15–166) had a predominantly mito-chondrial localization (Fig. 4b). Deletion of an additional 15 aminoacids at the N-terminus (cofilin30–166) abolished its mitochondriallocalization, suggesting that amino acids 15–30 are required for target-ing of cofilin into mitochondria. However, deletion of C-terminalresidues 106–166 (cofilin15–106) also abolished mitochondrial localiza-tion (Fig. 4a, b), indicating that additional regions in the C terminusare also required for mitochondrial localization. Indeed, green fluores-cent protein (GFP) fusions containing residues 15–30 of cofilin werenot localized to mitochondria (data not shown), suggesting that this
region is not sufficient to target cofilin to the mitochondria. Mostimportantly, when C-terminal residues 106–166 were fused to the pro-posed mitochondrial-targeting region (cofilin15–30/106–166), mitochon-drial localization was restored (Fig. 4a, b). As expected, fusion ofamino acids 106–166 to the N-terminal 1–30 amino acids resulted inmitochondrial localization after STS treatment, but not in the absenceof apoptotic stimuli (Fig. 4a, b). These data indicate that two regions ofcofilin are required for its mitochondrial localization: the N-terminaldomain consisting of amino acids 15–30 and the C-terminal region(amino acids 106–166).
Next, we evaluated whether mitochondrial-localized cofilin mutantsare sufficient to induce apoptosis. Cells overexpressing exogenouscofilin15–166 or cofilin15–30/106–166 displayed typical apoptotic pheno-types, such as the clustering of mitochondria around the nucleus (Fig.4b) and triggered apoptosis in 50% of the cells within 16 h of transfec-tion, a potency comparable with that induced by overexpression of Bax(Fig. 4c). Apoptosis triggered by mitochondrial-localized cofilinmutants was blocked by the caspase inhibitors zVAD-fmk and DEVD-fmk, and also through co-expression of Bcl-xL (Fig. 4c). In an alterna-tive approach, we targeted full-length cofilin expression to the outer
a bCofilinNuclei
Cyt cNuclei
Con
trol
ST
S (
4 h)Cofilin
Tubulin
Apo
ptos
is (
%)
Apo
ptos
is (
%)
c d
20 µm
STS treatment (h) Etoposide treatment (h)
100
75
50
25
00 2 4 8 0 2 4 8 24
80
60
40
20
0
UntransfectedControl siRNACofilin siRNA
UntransfectedControl siRNACofilin siRNA
+ C
ontro
l siR
NA
+ C
ofilin
siR
NA
Figure 2 Silencing of cofilin expression blocks cytochrome c release andinhibits apoptosis. SH-SY5Y cells were transfected with control or cofilinsiRNA. (a) Reduced cofilin expression in the presence of cofilin siRNA.Western blot analysis of total cell lysates. (b–d) Lack of cytochrome c releaseand apoptosis in cofilin knock-down cells. Cells treated with 1 µM STS were
fixed and stained for cofilin and cytochrome c (b) or for cofilin only (c-d).Nuclei were stained with H-33342. In b, arrows indicate cofilin knock-downcells and arrowheads indicate cofilin-expressing cells. In c and d, apoptosiswas scored according to nuclei morphology in 300 cofilin knock-down cellsafter treatment with 1 µM STS (c) or 100 µM etoposide (d).
print ncb1070 13/11/03 1:59 pm Page 1085
© 2003 NaturePublishing Group
© 2003 Nature Publishing Group

L E T T E R S
1086 NATURE CELL BIOLOGY VOLUME 5 | NUMBER 12 | DECEMBER 2003
mitochondrial membrane by fusing HA-tagged cofilin with the mito-chondria-targeting sequence of Bcl-2 (M–cofilin). As expected,M–cofilin colocalized with the mitochondrial marker Mitotracker Red(Fig. 4b). Overexpression of M–cofilin triggered massive apoptosis (Fig.4c) and induced release of cytochrome c into the cytoplasm (seeSupplementary Information, Fig. S4a). M–cofilin-triggered apoptosiswas also completely blocked by caspase inhibitors and partially pre-vented by co-expression with Bcl-xL (Fig. 4c). As a control, overexpres-sion of GFP targeted to mitochondria (M–GFP) neither resulted inmorphological changes (Fig. 4b) nor in apoptosis (Fig. 4c). These dataindicate that cofilin becomes a potent inducer of caspase-dependentapoptosis once it is targeted to mitochondria. The pro-apoptotic mutantof cofilin (cofilin15–166), however, was ineffective in triggering the effluxof cytochrome c from isolated mitochondria (see SupplementaryInformation, Fig. S4b), suggesting that additional modification eventsand/or cofactors activated during apoptosis are required.
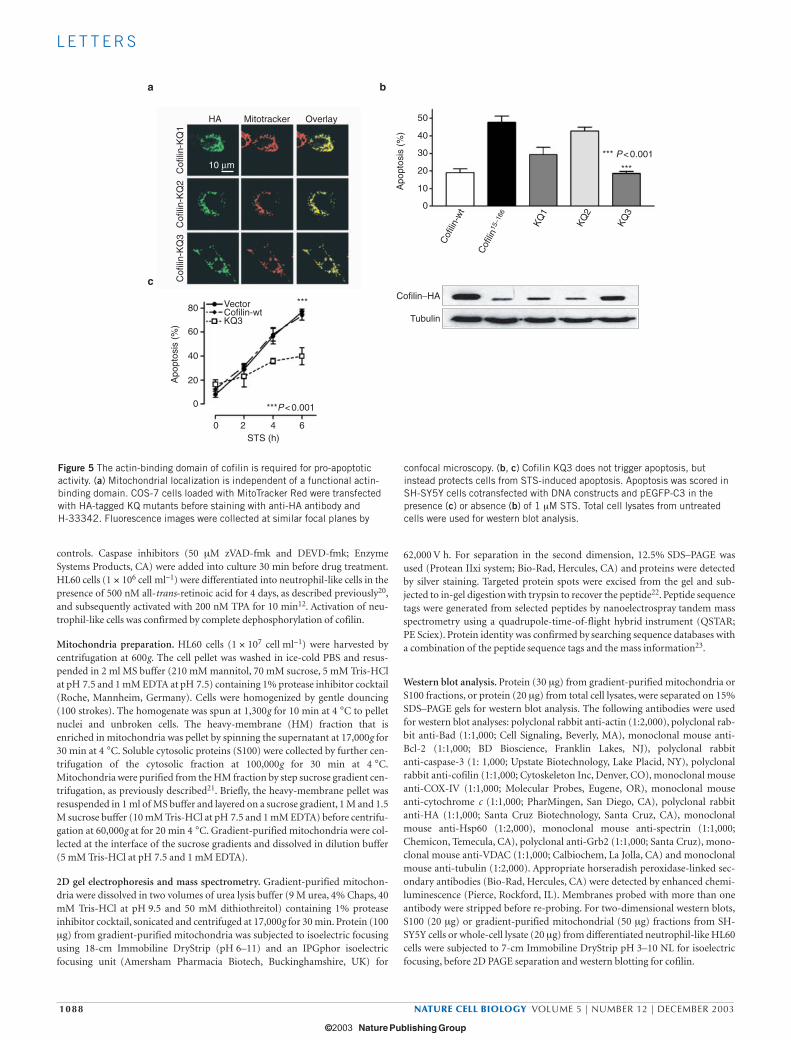
To access the role of the actin-binding domain at the C-terminalregion of cofilin on mitochondrial localization and apoptosis, we gen-erated mutants of mitochondrial-localized cofilin15–166 in which one orboth lysine residues (Lys 112 and Lys 114) were mutated to glutamine(KQ1 (K112Q); KQ2 (K114Q); KQ3 (K112Q, K114Q). Substitution ofboth lysines with glutamine has been shown abolish the F-actin-bind-ing and -depolymerizing activity of cofilin, whereas single amino-acidsubstitution (K112Q or K114Q) partially reduces these activities14.Similarly to cofilin15–166
, the cofilin KQ mutants with either single ordouble substitutions displayed mitochondrial localization (Fig. 5a),
indicating that a functional actin-binding domain is not required fortranslocation of cofilin to mitochondria. Surprisingly, although thecofilin KQ1 mutant had reduced apoptosis-inducing activity, cofilinKQ3 lost all apoptosis-inducing activity (Fig. 5b). Interestingly, theoverexpressed KQ3 mutant functioned in a dominant-negative man-ner to inhibit STS-induced apoptosis (Fig. 5c; 78 ± 3.7% and38 ± 7.1% for wild type and KQ3, respectively). These data suggest thata functional actin-binding domain of cofilin is not required for mito-chondrial localization, but is crucial for its apoptosis-inducing activity.
Mitochondrial targeting seems to be an essential step for engagingthe pro-apoptotic effect of cofilin. Mitochondrial translocation ofcofilin was achieved through three primary determinants: first, expo-sure of N-terminal sequences (15–30 aa); second, the C-terminalregion; third, the dephosphorylation of cofilin at Ser 3. According tothe sequence similarity between destrin and cofilin, and the nuclearmagnetic resonance structure of destrin15, the cofilin N-terminalregion may form a flexible loop that can mask the adjacent sequences(amino acids 15–30) in the absence of apoptotic stimuli. After induc-tion of apoptosis or deletion of this inhibitory region in cofilin15–166,for example, this mitochondrial localization signal maybe unmaskedand cofilin is then translocated to mitochondria to induce apoptosis.Although we demonstrated that the C-terminal regions are importantfor mitochondrial localization of cofilin, a functional actin-bindingdomain is not required. Only dephosphorylated cofilin accumulated inmitochondria after induction of apoptosis. In contrast, phosphoryla-tion of cofilin at Ser 3 suppressed mitochondrial translocation most
Con
trol
ST
S
Eto
posi
de
Cytosol Mitochondria
a b
c d
− +−+TPA:
Cell lysate Mitochondria
− +−+STS:
Cofilin-wt Cofilin-S3A
+−
Cofilin-S3D
Mitochondria
20.6K
H+ H+OH− OH−
Cofilinp-Cofilin
Apo
ptos
is (
%)
Cofilin
Bcl-2
Cofilin−HA
Cofilin
Hsp60
VectorCofilin-wtCofilin-S3ACofilin-S3D
80
60
40
20
0
0 2 4 6
STS treatment (h)
*** P < 0.001
Figure 3 Phosphorylation status of cofilin. (a) Only dephosphorylated cofilinis localized at mitochondria after apoptotic stimulation. Subcellularfractions of SH-SY5Y cells treated with 1 µM STS or 100 µM etoposide for 1h were subjected to 2D western blotting for cofilin. (b) Dephosphorylatedcofilin does not accumulate in mitochondria from activated neutrophil-like
cells. Western blot analysis of sub-cellular fractions is shown. (c) CofilinS3D blocks translocation of endogenous cofilin. Gradient-purifiedmitochondria were used for western blot analysis. (d) Cofilin S3D inhibitsSTS-induced apoptosis. pEGFP-C3 was cotransfected and apoptosis scoredin GFP-positive cells. *** represents p<0.001, ** represents p<0.01.
print ncb1070 13/11/03 1:59 pm Page 1086
© 2003 NaturePublishing Group
© 2003 Nature Publishing Group
L E T T E R S
NATURE CELL BIOLOGY VOLUME 5 | NUMBER 12 | DECEMBER 2003 1087
probably by masking the adjacent amino-acid sequences (15–30),transforming the first N-terminal loop into a non-functional configu-ration. The dominant-negative effect of cofilin S3D suggests the exis-tence of protein factor(s) that can abolish mitochondrial translocationof cofilin. The cytoplasmic protein 14-3-3 (which binds with highaffinity to phosphorylated cofilin at amino acids 21–26 (ref. 16), whichoverlaps the identified putative mitochondrial localization signal(amino acids 15–30), could potentially be one of the cytoplasmicretention factors for cofilin.
Dephosphorylation and activation of cofilin occurs in response tomany stimuli that activate cells in processes requiring changes in actinorganization17. Interestingly, mitochondrial translocation of dephos-phorylated cofilin was observed exclusively during apoptosis, but notin other processes of cell activation. As dephosphorylation of cofilincan be achieved with phosphatases such as slingshot and PP2A18,19,activation of these phosphatases may be required for translocation ofcofilin to mitochondria during the induction of apoptosis.Intriguingly, substitution of two lysine residues on the actin-bindingdomain of cofilin abolished the apoptosis-inducing activity of cofilin.Previous biochemical data demonstrated that Lys 112 and Lys 114 of
cofilin are differentially involved in binding to phosphatidylinositol4,5-bisphosphate (PtdInsP2) and actin, respectively14. However, sub-stitution of both Lys 112 and Lys 114 (KQ3) resulted in a 50% reduc-tion of the cofilin–PtdInsP2 interaction but complete loss of thecofilin–actin interaction. Therefore, we suggest that the actin-bindingactivity of cofilin is crucial for its apoptosis-inducing activity. Asdephosphorylated cofilin binds to actin and translocates into mito-chondria, actin cytoskeletal changes during apoptosis may affect mito-chondria function or enhance mitochondrial translocation of otherpro-apoptogenic proteins, resulting in the dysfunction of mitochon-dria, release of cytochrome c and apoptosis.
METHODSMaterials. All reagents and antibodies were purchased from Sigma (St Louis,MO), unless otherwise stated.
Cell culture and treatment. Mammalian cell lines (COS-7, HL60, HeLa, or SH-SY5Y) were cultured in DMEM supplemented with 10% heat-inactivated foetalcalf serum, 100 U ml−1 penicillin and 0.1 mg ml−1 streptomycin. Apoptosis wasinduced either by the addition of 1 µM staurosporine (STS) or 100 µM etopo-side. An equal volume of vehicle (dimethyl sulphoxide; DMSO) was used in
a c
STSControlCofilin1−166
Cofilin15−166
Cofilin30−166
Cofilin40−166
Cofilin83−166
Cofilin1−106
Cofilin15−106
Cofilin1−30/106−166
Cofilin15−30/106−166
M−cofilin
− +++++ +++
− −− −− −− −
−−++−
+++ +++
+++ ++++++ +++
NLS
GFP
Nuc
lei f
ragm
enta
tion
(%)
Vect
or
Bax
M−c
ofilin
M−G
FP
Cof
ilin15
−166
Cof
ilin15
−30/
106−
166
b
Cof
ilin15
−166
Cof
ilin15
−106
Cof
ilin1−
30/1
06−1
66
HA
Mito
trac
ker
Ove
rlay
GF
PM
itotr
acke
rO
verla
y
Cof
ilin15
−30/
106−
166
10 µm
ADD HA tag
Bcl-2210-239
M−GFP
Mitochondriallocalization
Untreated+ zVAD-fmk+ DEVD-fmk+ Bcl-xL
80
60
40
20
0
M−cofilin M−GFP
Figure 4 Mitochondria-localized cofilin induces apoptosis. COS-7 cellsloaded with MitoTracker Red were transfected with HA-tagged truncatedforms of cofilin and then treated with 1 µM STS for 1 h before staining.(a) Sub-cellular localization of cofilin deletion mutants. At least 100transfected cells were scored for mitochondrial localization, indicated by− (none), + (slight), ++ (medium) or +++ (strong and exclusivemitochondrial localization). (b) Mitochondrial localization of
cofilin15–166, cofilin15–30/106–166, M–cofilin and M–GFP. Fluorescence
images were collected at similar focal planes by confocal microscopy.(c) Cofilin15–166, cofilin15–30/106–166 and M–cofilin trigger caspase-dependent apoptosis. SH-SY5Y cells pre-incubated with 50 µM zVAD-fmk or DEVD-fmk were cotransfected with pEGFP-C3 and the indicatedconstructs before staining with H-33342. Fragmented nuclei were scoredin GFP-positive cells.
print ncb1070 13/11/03 2:00 pm Page 1087
© 2003 NaturePublishing Group
© 2003 Nature Publishing Group
L E T T E R S
1088 NATURE CELL BIOLOGY VOLUME 5 | NUMBER 12 | DECEMBER 2003
controls. Caspase inhibitors (50 µM zVAD-fmk and DEVD-fmk; EnzymeSystems Products, CA) were added into culture 30 min before drug treatment.HL60 cells (1 × 106 cell ml−1) were differentiated into neutrophil-like cells in thepresence of 500 nM all-trans-retinoic acid for 4 days, as described previously20,and subsequently activated with 200 nM TPA for 10 min12. Activation of neu-trophil-like cells was confirmed by complete dephosphorylation of cofilin.
Mitochondria preparation. HL60 cells (1 × 107 cell ml−1) were harvested bycentrifugation at 600g. The cell pellet was washed in ice-cold PBS and resus-pended in 2 ml MS buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris-HClat pH 7.5 and 1 mM EDTA at pH 7.5) containing 1% protease inhibitor cocktail(Roche, Mannheim, Germany). Cells were homogenized by gentle douncing(100 strokes). The homogenate was spun at 1,300g for 10 min at 4 °C to pelletnuclei and unbroken cells. The heavy-membrane (HM) fraction that isenriched in mitochondria was pellet by spinning the supernatant at 17,000g for30 min at 4 °C. Soluble cytosolic proteins (S100) were collected by further cen-trifugation of the cytosolic fraction at 100,000g for 30 min at 4 °C.Mitochondria were purified from the HM fraction by step sucrose gradient cen-trifugation, as previously described21. Briefly, the heavy-membrane pellet wasresuspended in 1 ml of MS buffer and layered on a sucrose gradient, 1 M and 1.5M sucrose buffer (10 mM Tris-HCl at pH 7.5 and 1 mM EDTA) before centrifu-gation at 60,000g at for 20 min 4 °C. Gradient-purified mitochondria were col-lected at the interface of the sucrose gradients and dissolved in dilution buffer(5 mM Tris-HCl at pH 7.5 and 1 mM EDTA).
2D gel electrophoresis and mass spectrometry. Gradient-purified mitochon-dria were dissolved in two volumes of urea lysis buffer (9 M urea, 4% Chaps, 40mM Tris-HCl at pH 9.5 and 50 mM dithiothreitol) containing 1% proteaseinhibitor cocktail, sonicated and centrifuged at 17,000g for 30 min. Protein (100µg) from gradient-purified mitochondria was subjected to isoelectric focusingusing 18-cm Immobiline DryStrip (pH 6–11) and an IPGphor isoelectricfocusing unit (Amersham Pharmacia Biotech, Buckinghamshire, UK) for
62,000 V h. For separation in the second dimension, 12.5% SDS–PAGE wasused (Protean IIxi system; Bio-Rad, Hercules, CA) and proteins were detectedby silver staining. Targeted protein spots were excised from the gel and sub-jected to in-gel digestion with trypsin to recover the peptide22. Peptide sequencetags were generated from selected peptides by nanoelectrospray tandem massspectrometry using a quadrupole-time-of-flight hybrid instrument (QSTAR;PE Sciex). Protein identity was confirmed by searching sequence databases witha combination of the peptide sequence tags and the mass information23.
Western blot analysis. Protein (30 µg) from gradient-purified mitochondria orS100 fractions, or protein (20 µg) from total cell lysates, were separated on 15%SDS–PAGE gels for western blot analysis. The following antibodies were usedfor western blot analyses: polyclonal rabbit anti-actin (1:2,000), polyclonal rab-bit anti-Bad (1:1,000; Cell Signaling, Beverly, MA), monoclonal mouse anti-Bcl-2 (1:1,000; BD Bioscience, Franklin Lakes, NJ), polyclonal rabbitanti-caspase-3 (1: 1,000; Upstate Biotechnology, Lake Placid, NY), polyclonalrabbit anti-cofilin (1:1,000; Cytoskeleton Inc, Denver, CO), monoclonal mouseanti-COX-IV (1:1,000; Molecular Probes, Eugene, OR), monoclonal mouseanti-cytochrome c (1:1,000; PharMingen, San Diego, CA), polyclonal rabbitanti-HA (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA), monoclonalmouse anti-Hsp60 (1:2,000), monoclonal mouse anti-spectrin (1:1,000;Chemicon, Temecula, CA), polyclonal anti-Grb2 (1:1,000; Santa Cruz), mono-clonal mouse anti-VDAC (1:1,000; Calbiochem, La Jolla, CA) and monoclonalmouse anti-tubulin (1:2,000). Appropriate horseradish peroxidase-linked sec-ondary antibodies (Bio-Rad, Hercules, CA) were detected by enhanced chemi-luminescence (Pierce, Rockford, IL). Membranes probed with more than oneantibody were stripped before re-probing. For two-dimensional western blots,S100 (20 µg) or gradient-purified mitochondrial (50 µg) fractions from SH-SY5Y cells or whole-cell lysate (20 µg) from differentiated neutrophil-like HL60cells were subjected to 7-cm Immobiline DryStrip pH 3–10 NL for isoelectricfocusing, before 2D PAGE separation and western blotting for cofilin.
c
a b
Apo
ptos
is (
%)
10 µmA
popt
osis
(%
)
STS (h)
HA Mitotracker Overlay
Cof
ilin-
KQ
3C
ofili
n-K
Q2
Cof
ilin-
KQ
1
50
40
30
20
10
0
Cof
ilin-w
t
Cof
ilin15
−166
KQ1
KQ2
KQ3
*** P < 0.001
***P < 0.001
***
Cofilin−HA
Tubulin80
60
40
20
0
0 2 4 6
VectorCofilin-wtKQ3
***
Figure 5 The actin-binding domain of cofilin is required for pro-apoptoticactivity. (a) Mitochondrial localization is independent of a functional actin-binding domain. COS-7 cells loaded with MitoTracker Red were transfectedwith HA-tagged KQ mutants before staining with anti-HA antibody and H-33342. Fluorescence images were collected at similar focal planes by
confocal microscopy. (b, c) Cofilin KQ3 does not trigger apoptosis, butinstead protects cells from STS-induced apoptosis. Apoptosis was scored inSH-SY5Y cells cotransfected with DNA constructs and pEGFP-C3 in thepresence (c) or absence (b) of 1 µM STS. Total cell lysates from untreatedcells were used for western blot analysis.
print ncb1070 13/11/03 2:00 pm Page 1088
© 2003 NaturePublishing Group
© 2003 Nature Publishing Group

L E T T E R S
NATURE CELL BIOLOGY VOLUME 5 | NUMBER 12 | DECEMBER 2003 1089
Immunofluorescence microscopy. COS-7 or SH-SY5Y cells seeded on 12-mmcoverslips were loaded with 50 nM MitoTracker Red CMXRos (MolecularProbes, Eugene, OR) for 1 h and washed twice with DMEM before treatment.After the desired period of treatment, cells were washed once in PBS and fixedin 4% paraformaldehyde before permeabilization in 0.1% Triton X-100.Immunostaining was performed using the following antibodies: polyclonalrabbit anti-cofilin (1:200; Cytoskeleton, Denver, CO), monoclonal mouse anti-cytochrome c (1:400; PharMingen, San Diego, CA), monclonal mouse anti-mycc (1:250; Santa Cruz Biotechnology, Santa Cruz, CA) and polyclonal rabbit anti-HA (1:250; Santa Cruz) followed by appropriate secondary antibodies conju-gated with either Alexa Fluor 488 or Alexa Fluor 568 (Molecular Probes). Nucleiwere counterstained with 250 ng ml−1 H-33342 (Molecular Probes).Fluorescence images were collected and analysed with either a MRC-1024 MPlaser-scanning confocal microscope (Bio-Rad) or a microscope equipped withan AxioCam CCD camera (Carl Zeiss, Oberkochen, Germany).
Molecular cloning of cofilin, point and deletion mutants, and mitochondrial-targeted fusion proteins. Wild-type cofilin was generated by PCR from SH-SY5YcDNA library with the following pair of primers: 5′-gCC CAT ATg ATg gCC TCCggT gTg gCT gT-3′ and 5′-CgC ggA TCC gCC AAA ggC TTg CCC TCC Ag-3′, inwhich NdeI and BamH1 restriction sites were added to the forward and reverseprimers, respectively. The 500-bp fragment was cloned in-frame into a mam-malian expression vector pXJ40 with 3′-HA tag. TheS3A and S3D cofilin mutantswere generated by site-directed mutagenesis . A series of N- or C-terminal dele-tion mutants of cofilin were generated by PCR using primers containing a 5′EcoRI site and a 3′ HindIII site using a pXJ-cofilin-wt construct as template. Themitochondrial-targeted cofilin (M–cofilin) and green fluorescent protein(M–GFP) were constructed by amplification of the C-terminal Bcl-2 mitochon-dria-targeting signal peptide (Phe 210–Lys 239) using a pair of primers corre-sponding to this region. The 100-bp fragments were cloned in-frame to the Cterminus of the pXJ-cofilin-wt construct or pEGFP-C3 vector (M–GFP). CofilinKQ mutants were generated by site-directed mutagenesis of the pXJ-cofilin15–166
construct, substituting Lys 112 and/or Lys 114 with glutamine.
Transient transfections. Transient transfections were performed withLipofectAMINE2000 (Invitrogen, San Diego, CA) in accordance with the man-ufacturer’s instructions. Briefly, 6 × 104 SH-SY5Y or COS-7 cells per well wereseeded on 24-well plates overnight before transfection with 0.4 µg cofilin, Baxor Bcl-xL constructs. DNA was mixed with the liposome reagent at a ration of1:1 before addition to cells. For cotransfections, an additional 0.2 µg pEGFP-C3and/or 0.8 µg pXJ-BclxL-myc were included.
siRNA. The siRNA sequence targeting cofilin was designed from codons 64–84,relative to the start codon (Dharmacon Research, Lafayette, CO). As a control,two single-nucleotide mutations (C71G and A73U) were introduced into thesiRNA sequence used for cofilin silencing. 1 × 104 SH-SY5Y cells per well wereseeded in 24-well plates overnight. Cells were transfected with 0.8 µg siRNAs for72 h using Oligofectamine (Invitrogen, San Diego, CA), according to manufac-turer’s instructions. Through immunofluorescence microscopy analysis, wedetermined that the efficiency of cofilin knock-down is approximately 40% inSH-SY5Y cells.
Apoptosis assay. Apoptosis was assessed by examination of nuclear morphol-ogy. Cells were loaded with 1 µg ml−1 H-33342 (cell-permeable, blue fluores-cent chromatin stain) for 10 min. Apoptosis was characterized by scoringcondensed and fragmented highly fluorescent nuclei. Each set of experimentswas repeated at least three times, with at least 300 cells counted in each instance.
Accession number. The accession number for cofilin is NM_005507.
Note: Supplementary Information is available on the Nature Cell Biology website.
ACKNOWLEDGEMENTSWe are grateful to Y. Samstag (Institute for Immunology, Heidelberg, Germany)and H. Abe (Chiba University, Chiba, Japan) for anti-cofilin antibodies. We are alsograteful to B. Luen Tang, E. Manser, A. Porter, S.-L. Chan and N. Fu for criticalcomments and valuable discussion about the manuscript. This work wassupported by Agency for Science, Technology and Research (A*STAR) inSingapore.
COMPETING FINANCIAL INTERESTSThe authors declare that they have no competing financial interests.
Received 17 July 2003; accepted 20 October 2003Published online at http://www.nature.com/naturecellbiology.
1. Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 15,2922–2933 (2001).
2. Liu, X., Kim, C. N., Yang, J., Jemmerson, R. & Wang, X. Induction of apoptotic pro-gram in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157(1996).
3. Li, P. et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 com-plex initiates an apoptotic protease cascade. Cell 91, 479–489 (1997).
4. Cory, S. & Adams, J. M. The Bcl2 family: regulators of the cellular life-or-deathswitch. Nature Rev. Cancer 2, 647–656 (2002).
5. Zha, J., Harada, H., Yang, E., Jockel, J. & Korsmeyer, S. J. Serine phosphorylation ofdeath agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87, 619–628 (1996).
6. Li, H. et al. Cytochrome c release and apoptosis induced by mitochondrial targeting ofnuclear orphan receptor TR3. Science 289, 1159–1164 (2000).
7. Karuman, P. et al. The Peutz-Jegher gene product LKB1 is a mediator of p53-depend-ent cell death. Mol. Cell 7, 1307–1319 (2001).
8. Chen, H., Bernstein, B. W. & Bamburg, J. R. Regulating actin-filament dynamics invivo. Trends Biochem. Sci. 25, 19–23 (2000).
9. Arber, S. et al. Regulation of actin dynamics through phosphorylation of cofilin byLIM- kinase. Nature 393, 805–809 (1998).
10. Yang, N. et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediatedactin reorganization. Nature 393, 809–812 (1998).
11. Breitman, T. R., Selonick, S. E. & Collins, S. J. Induction of differentiation of thehuman promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl Acad.Sci. USA 77, 2936–2940 (1980).
12. Zhan, Q., Bamburg, J. R. & Badwey, J. A. Products of phosphoinositide specific phos-pholipase c can trigger dephosphorylation of cofilin in chemoattractant stimulatedneutrophils. Cell Motil. Cytoskeleton 54, 1–15 (2003).
13. Nagaoka, R., Abe, H. & Obinata, T. Site-directed mutagenesis of the phosphorylationsite of cofilin: its role in cofilin–actin interaction and cytoplasmic localization. CellMotil. Cytoskeleton 35, 200–209 (1996).
14. Moriyama, K., Yonezawa, N., Sakai, H., Yahara, I. & Nishida, E. Mutational analysis ofan actin-binding site of cofilin and characterization of chimeric proteins betweencofilin and destrin. J. Biol. Chem. 267, 7240–7244 (1992).
15. Hatanaka, H. et al. Tertiary structure of destrin and structural similarity between twoactin-regulating protein families. Cell 85, 1047–1055 (1996).
16. Gohla, A. & Bokoch, G. M. 14-3-3 regulates actin dynamics by stabilizing phosphory-lated cofilin. Curr. Biol. 12, 1704–1710 (2002).
17. Bamburg, J. R. Proteins of the ADF/cofilin family: essential regulators of actin dynam-ics. Annu. Rev. Cell Dev. Biol. 15, 185–230 (1999).
18. Niwa, R., Nagata-Ohashi, K., Takeichi, M., Mizuno, K. & Uemura, T. Control of actinreorganization by Slingshot, a family of phosphatases that dephosphorylateADF/cofilin. Cell 108, 233–246 (2002).
19. Ambach, A. et al. The serine phosphatases PP1 and PP2A associate with and activatethe actin-binding protein cofilin in human T lymphocytes. Eur. J. Immunol. 30,3422–3431 (2000).
20. Drayson, M. T., Michell, R. H., Durham, J. & Brown, G. Cell proliferation and CD11bexpression are controlled independently during HL60 cell differentiation initiated by1,25 α-dihydroxyvitamin D(3) or all-trans-retinoic acid. Exp. Cell Res. 266, 126–134(2001).
21. Spector, D. L., Goldman, R. D. & Leinwand, L. A. in Cells: A Laboratory Manual41.1–41.7 (Cold Spring Habor Laboratory, 1997).
22. Shevchenko, A., Wilm, M., Vorm, O. & Mann, M. Mass spectrometric sequencing ofproteins silver-stained polyacrylamide gels. Anal Chem. 68, 850–858 (1996).
23. Wilm, M. et al. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 379, 466–469 (1996).
print ncb1070 13/11/03 2:00 pm Page 1089
© 2003 NaturePublishing Group
© 2003 Nature Publishing Group

S U P P L E M E N TA RY I N F O R M AT I O N
WWW.NATURE.COM/NATURECELLBIOLOGY 1
MATERIAL AND METHODS-SUPPLEMENTARYProteinase K digestion and alkaline phosphatase assayGradient purified mitochondria (50 µg) were re-suspended in MS buffer (with-out protease inhibitors) and incubated with 10 ng/ml proteinase K on ice for 10minutes. The reaction mixture was spun at 17,000g for 15 minutes and pelletwas collected and dissolved in SDS lysis buffer (2% SDS) for western blotting.S100 proteins (20 ?g from untreated SH-SY5Y cells) were incubated with 20Units calf intestinal alkaline phosphatase (Promega, Manheim, Germany) at37oC for 30 minutes to dephosphorylate cytosolic proteins.
Production of recombinant cofilin wt, cofilin15-166 and t-BidPXJ-cofilin wt, -cofilin15-166, -cofilin15-30/106-166, and -M-cofilin were sub-cloned into pET 21b vector and mouse t-Bid was cloned into pET-H in framewith His tag. The constructs were transformed into E.coli strain BL21 (DE3) forexpression of recombinant proteins that were purified. Aliquots of fractionseluted from Ni2+ column (Qiagen, Germany) were separated by SDS-PAGE andvisualized by Coomassie blue staining to determine positive fractions. Fractionswere dialyzed and protein concentrations were determined using Bradfordassay (Bio-Rad).
Isolation of mitochondria and the in vitro assay of cytochrome creleaseTo isolate mitochondria for the cytochrome c release assay, SH-SY5Y cells weresuspended in isolation buffer (320 mM sucrose, 1 mM EDTA, 50 mM HEPESpH 7.5 and 1 mM dithiothreitol) and disrupted by 10 expulsions through a 27-gauge needle. Disrupted cells were centrifuged two times at 1,000g for 5 min-utes to remove cell debris and nuclei. The supernatants were centrifuged at
5,000g to pellet the mitochondria. Mitochondria (15 µg) were re-suspended inassay buffer (250 mM sucrose, 2 mM KH2PO4, 5 mM sodium succinate, 2 mMEGTA and 10 mM HEPES pH 7.5) at 0.5 mg/ml and treated at 300C with theindicated proteins for 30 minutes followed by centrifugation at 10,000g for 10minutes. Proteins from mitochondrial pellets and supernatant fractions wereseparated by 13.5% SDS–PAGE and the cytochrome c content was analyzed byWestern blot.
Viability assayViability of cells was accessed using the cell proliferation reagent WST-1 (RocheDiagnostics, Germany) according to the manufacturer’s instruction. Briefly,SH-SY5Y cells transfected with cofilin or control siRNA were treated with 1 µMSTS for 6 hours or 100 µM etoposide for 10 hours, respectively. The percentageof viable cells was quantified by their WST-1-reducing capacity after incubationwith WST-1 (1:10 dilution) for 30 minutes. The viability of untreated cells wasset to 100%, and the viability of treated cells was expressed as percentage of for-mazan absorbance (A460 nm-A690 nm) compared with that of control cells.
Clonogenicity assaySH-SY5Y cells transfected with cofilin and control siRNA were treated with 1µM STS for 6 hours. Cells were trypsinised and plated onto 96 well round bot-tom plate with a density of 1 cell/well by serial dilution method. Cell colonieswere counted 8 days after plating and the percentage of clonogenic cells werecalculated by the ratio of the theoretical Poisson density of the number of nega-tive wells observed, against the initial plating density using the equation: %clonogenicity = (Ln ? [96/negative wells])/(plate density) ? 100, where nega-tive wells is the number of wells that have failed to grow colonies and plate den-sity is the original cell plating density per well.
© 2003 Nature Publishing Group

S U P P L E M E N TA RY I N F O R M AT I O N
2 WWW.NATURE.COM/NATURECELLBIOLOGY
Con
trol
STS
Cofilin Myc-Bcl-xL Overlay
a
b
c
dCofilin
Bcl-2
Bad
0min 45min15minSTS: 5min
0h 1h 1h + PKSTS:
Cofilin
Bcl-2
Cyt c
COX-IV
12.5 mm
11.5 mm
Fig S1. (a) Mitochondrial accumulation of cofilin at early phase of apoptosis.Gradient purified mitochondria from HL60 cells treated with 1 µM STS and50 µM zVAD-fmk were subjected to Western blot analysis. (b) Sub-cellularlocalization of cofilin. SH-SY5Y cells were loaded with MitoTracker Red andtreated with 1 µM STS and 50 µM zVAD-fmk. After 0.5 hour, cells werefixed and stained for cofilin. (c) Mitochondrial translocation of cofilin isblocked by Bcl-xL over-expression. SH-SY5Y transfected with a myc (c)-
tagged Bcl-xL DNA construct were treated with 1 µM STS for 1 hour, fixedand stained for cofilin and myc. (b and c) Specimens were imaged byconfocal microscopy using Normaski optics and/or fluorescence at similarfocal planes. (d) Cofilin is localized to the outer mitochondria membrane.Gradient purified mitochondria from HL60 cells were digested by proteinaseK, before subjected to Western blot analysis.
© 2003 Nature Publishing Group

S U P P L E M E N TA RY I N F O R M AT I O N
WWW.NATURE.COM/NATURECELLBIOLOGY 3
cofilin siRNA
controlsiRNA
controlsiRNA
cofilin siRNA
Via
bilit
y (%
S
D)
Clo
noge
nici
ty (%
S
D)
a b
Fig S2. Silencing cofilin (promotes) protects cell (viability) from STS-mediated apoptosis. Cells transfected with either cofilin or control siRNAwere treated with 1 µM STS for 6 hours. Cell cultures transfected withcofilin siRNA contained approximately 40% cells displaying cofilin knock-down phenotype (determined by cofilin immunostaining). Viability in themixed cultures (containing cofilin knock-down cells and cofilin positivecells) treated with STS was assessed by measuring WST-1 reductioncapacity (a) and clonogenicity (b). Data are means ± SD from threeindependent experiments.
a bCytosol Whole cell lysate
Con
trol
AP
Con
trol
PM
A
20.6kDa
H+ H+OH- OH-
pCofilin Cofilin
Fig S3. (a) Separation of phosphorylated and dephosphorylated form ofcytosolic cofilin. S100 fractions from untreated SH-SY5Y cells wereincubated with calf intestinal alkaline phosphatase (AP) to generatedephosphorylated cofilin. (b) Activation of neutrophil-like cells leads tocomplete dephosphorylation of cofilin. Differentiated neutrophil-like HL60cells were activated with the phorbol ester PMA (phorbol 12-myristate 13-acetate). Whole cell lysate were subjected to 2-D PAGE and Western blottingfor cofilin.
© 2003 Nature Publishing Group

S U P P L E M E N TA RY I N F O R M AT I O N
4 WWW.NATURE.COM/NATURECELLBIOLOGY
a bG
FP
Cyt
c
Nuc
leus
HA
Cyt
c
Nuc
leus
M-GFP M-cofilin
8.0 mm
Mit
o pe
llet
Supe
rnat
ant T
rito
n-X
t-B
id
cofi
lin15
-166
(3.5
µg)
cofi
lin15
-166
(2.5
µg)
cofi
lin15
-166
(1.5
µg)
cofi
linw
t (1
.5µg
)
GST
PB
S
Cyt c
VDAC
Cyt c
t-B
id
cofi
lin15
-166
cofi
linw
t
GST
(1.5
µg)
Coomassie stain
Fig S4. (a) M-cofilin-induced cytochrome c release precedes apoptoticnuclear changes. SH-SY5Y cells transfected with the different plasmidconstructs were stained for cytochrome c and/or HA simultaneously. Nucleiwere stained with H-33342. (b) Recombinant cofilin15-166 does not inducecytochrome c release from isolated mitochondria. Mitochondria wereincubated with different concentrations of the recombinant proteins GST, t-Bid, cofilin wt and cofilin15-166 or with control buffer (PBS). Western blot
analysis of proteins from supernatant (upper panel) and mitochondrialpellets (middle panel). Recombinant t-Bid was used as positive and GST asnegative control. Equal loading of mitochondrial pellets was controlled byVDAC. Recombinant protein fractions (lower panel) of GST, t-Bid, cofilin wtand cofilin15-166 were separated by SDS-PAGE. The Coomassie blue bandscorresponding to the recombinant proteins used in the experiment werealigned.
© 2003 Nature Publishing Group