metody instrumentalne w analizie chemicznej · w biologii, biochemii, medycynie do badania...
TRANSCRIPT

METODY INSTRUMENTALNEMETODY INSTRUMENTALNE
W ANALIZIE CHEMICZNEJW ANALIZIE CHEMICZNEJ
ZakZakłład Chemii Medycznej PUMad Chemii Medycznej PUM

Promieniowanie elektromagnetyczne:
fala
strumień fotonów
Fala to rozchodzące się w przestrzeni i w czasie,
spójne drganie pól elektrycznego
i magnetycznego.
2

Fotony są pozbawione masy spoczynkowej, niosą ze
sobą ściśle określoną energię, którą wyraża zależność
Plancka:
gdzie:
E energia promieniowania
h stała Plancka 6,625 • 10-34 Js
liczba falowa cm-1

Zajmuje się oddziaływaniem promieniowania elektromagnetycznego
z cząsteczkami.
W cząsteczce wieloatomowej można wyróżnić następujące rodzaje
energii:
translacji - swobodny ruch cząsteczki w przestrzeni
rotacji - związana z obrotem cząsteczki wokół osi środka masy
oscylacji - związana z drganiem atomów wokół położenia równowagi
elektronowa - energia kinetyczna (ruch) elektronów i potencjalna związana z przyciąganiem elektronów przez jądro i odpychaniem przez sąsiednie elektrony.
4

W wyniku absorpcji promieniowania elektromagnetycznego przez cząsteczki
następuje wzbudzenie odpowiednich poziomów energii , czego mierzalnym
efektem jest widmo.
pod wpływem promieniowania z zakresu mikrofal i dalekiej podczerwieni
(energia 4x10-2 – 4x10-3 KJ/mol) następuje wzbudzenie rotacyjnych poziomów
energii a efektem jest widmo rotacyjne.
pod wpływem promieniowania IR (podczerwień 4 – 240 KJ/mol)
wzbudzeniu ulegają oscylacyjne poziomy energii – efekt widmo oscylacyjne.
wzbudzenie elektronów jest wywołane promieniowaniem
elektromagnetycznym (400 KJ/mol) z zakresu UV – Vis (ultrafiolet i
widzialne) - efektem jest elektronowe widmo absorpcyjne.
5

Elektronowe widmo absorpcyjne jest widmem ciągłym
6

Podstawowe założenia do praw absorpcji:
monochromatyczność wiązki promieniowania elektromagnetycznego
jednorodność ośrodka absorbującego
7

8
Intensywność początkowa
promieniowania (Io)
Intensywność promieniowania
(I1) po przejściu przez próbkę
o grubości l
c- stężenie roztworu
α- współczynnik absorpcji 0,4343K

I prawo absorpcji (prawo Lamberta)
Wiązka promieniowania monochromatycznego po przejściu przez
jednorodny ośrodek absorpcyjny o grubości l ulega osłabieniu
wg równania:
I = Io e –Kl
A= log (I0 / I) = l
Absorbancja jest proporcjonalna do grubości warstwy absorbującej jeśli wiązka promieniowania elektromagnetycznego przechodzi przez jednorodny ośrodek absorbujący.
9
A- absorbancja, gęstość optyczna,
zdolność pochłaniania
promieniowania

II prawo absorpcji (prawo Lamberta - Beera)
Absorbancja wiązki promieniowania
monochromatycznego przechodzącej przez
jednorodny roztwór jest wprost proporcjonalna
do stężenia roztworu c i do grubości warstwy
absorbującej l.
A = l c
10

III Prawo absorpcji (addytywności absorpcji)
Absorbancja roztworu wieloskładnikowego równa się
sumie absorbancji poszczególnych składników.
A = A1 + A2 + A3 + ...... + An
11

Odchylenia od praw adsorpcji są wywołane przez:
1. Podstawowe ograniczenia praw
prawa absorpcji są spełniane dla roztworów rozcieńczonych
c 10 -2 mol/l
zakłada się, że jedynym oddziaływaniem promieniowania
elektromagnetycznego z substancją rozpuszczoną jest
absorpcja promieniowania (a nie np. fluorescencja)
12

2. Czynniki chemiczne
zmianie pH roztworu, także zmianie stężenia mogą towarzyszyć
reakcje takie jak: polimeryzacja, dysocjacja, asocjacja,
solwatacja, kompleksowanie - tym reakcjom towarzyszą zmiany
właściwości optycznych analizowanych roztworów.
3. Czynniki aparaturowe
brak monochromatyczności wiązki (główna przyczyna
odchylenia od praw)
promieniowanie rozproszone

Spektrometria w zakresie nadfioletu (ultrafiolet UV) i promieniowania
widzialnego (visible – Vis).
Absorpcja promieniowania w zakresie UV – Vis jest związana z
przejściami elektronów walencyjnych ( i π) oraz elektronów
wolnych par elektronowych (elektrony n).
14

Ilościowe oznaczanie metodą spektrofotometryczną UV – Vis należy
do metod porównawczych (porównanie z wzorcami, stosowanie
krzywych kalibracyjnych)
1. Metoda porównania z pojedynczym wzorcem
Ax – absorbancja roztworu badanego cx
Aw – absorbancja roztworu wzorcowego cw (pomiar w kuwetach o tej
samej grubości l i przy tej samej długości fali )
Ax = cx l Aw = cwl
15
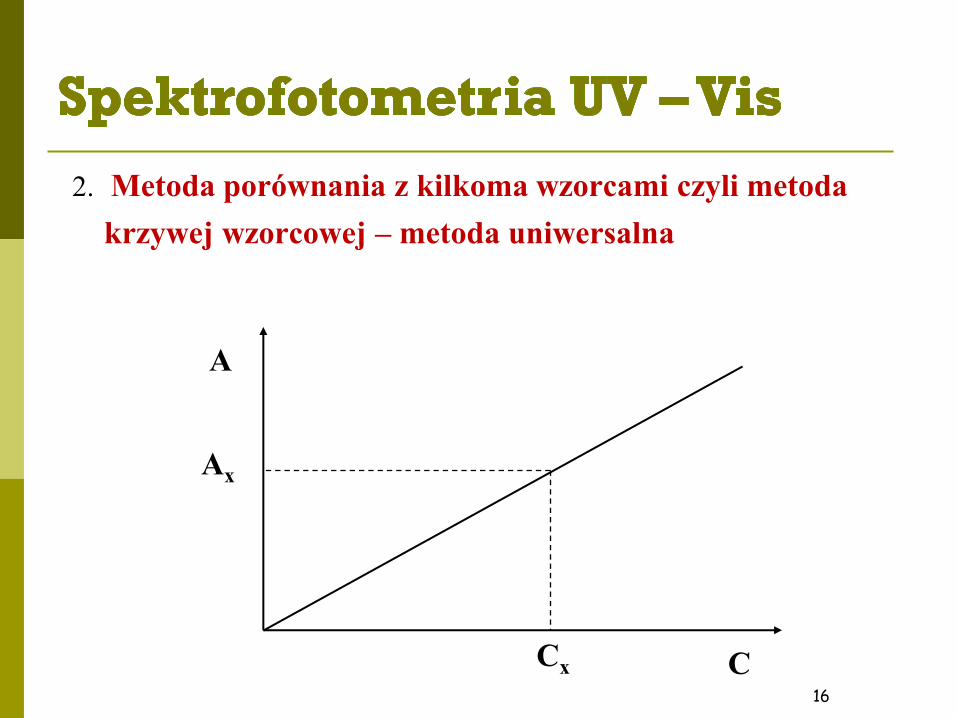
2. Metoda porównania z kilkoma wzorcami czyli metoda
krzywej wzorcowej – metoda uniwersalna
16
A
C
Ax
Cx

Analiza jakościowa
Elektronowe widmo absorpcyjne jest cechą charakterystyczną
związku chemicznego i określa jednoznacznie związek jednorodny.
W przypadku związku organicznego widma UV – Vis w zakresie
180 –800 nm są użyteczne do ustalania pewnych zależności
strukturalnych i identyfikacji grup funkcyjnych.
Użytecznym pojęciem jest wprowadzone przed wieloma laty określenia:
chromofor i auksochrom
17

Chromofory są to grupy absorbujące promieniowanie
elektromagnetyczne w zakresie 180 – 800 nm
grupy: karboksylowa, nitrozowa, alkenowa, azowa, tiolowa, benzenowa
Auksochromy są to grupy przesuwające absorpcję ( max i Emax) w
kierunku fal dłuższych (przesunięcie batochromowe)
grupy: aminowa, hydroksylowa, sulfhydrylowa
18

Cząstki w stanie wzbudzonym mogą emitować promieniowanie
przechodząc do stanu podstawowego - zjawisko to nazywamy
luminescencją - emisja światła przez ciała zimne.
W zależności od czynników które wywołały luminescencję rozróżniamy:
fotoluminescencja – substancja jest wzbudzona promieniowaniem elektromagnetycznym UV – Vis
chemiluminescencja – wzbudzenie jest wynikiem reakcji chemicznej
bioluminescencja - wzbudzenie jest wynikiem procesów biochemicznych
elektroluminescencja – wzbudzenie następuje w polu elektrycznym.
19

Proces fotoluminescencji cząstki można przedstawić
schematycznie: X + h X* X + ciepło + h
h ´- energia oddana na sposób
promienisty
W zależności od mechanizmu
przejść elektronowych:
fluorescencja
fosforescencja
20

Zastosowanie analityczne fluorescencyjnej analizy
ilościowej:
witaminy (tiamina)
hormony (adrenalina)
aminokwasy (tryptofan)
alkaloidy (chinina)
leki (antybiotyki, barbiturany)
środki spożywcze
substancje toksyczne w środowisku
tiamina
tiochrom

Absorpcja promieniowania IR wpływa na zmianę
energii oscylacyjnej cząsteczki
Zakresy długości fal odpowiadające podczerwieni:
0,8 – 2,5 m NIR bliska podczerwień
2,5 – 25 m MIR podstawowa
25 – 500 m FIR daleka podczerwień
22

Widmo IR jest właściwością charakterystyczną dla danego związku chemicznego.
Wykorzystuje się je do:
identyfikacji związków (ustala się grupy funkcyjne i strukturę badanego związku).
w analizie ilościowej (obowiązuje tu także prawo Lamberta-Beera).
w biologii, biochemii, medycynie do badania tkanek-mięśni, włókien i krwi.
23

Jądra atomowe niektórych izotopów umieszczone w jednorodnym polu
magnetycznym mogą absorbować promieniowanie elektromagnetyczne
o częstotliwości radiowej.
Z warunku rezonansu wynika, że można:
przy ustalonej częstotliwości radiowej zmieniać natężenie pola magnetycznego tak długo aż wystąpi rezonans
przy stałym natężeniu pola można zmieniać częstotliwość promieniowania radiowego aż do wystąpienia rezonansu
Promieniowanie o częstotliwości radiowej uzyskuje się z krystalicznego
oscylatora o dużej stabilności
Najczęściej bada się izotopy: 1H, 13C, 19F, 31P, 14N, 11Bi, 29Si
24

25
Tomografia MRI (magnetic resonance imaging)
Pierwszy raz wykorzystany w 1973r przez
Lauterbur’a,
Obecnie to istotna metoda stosowana w
diagnostyce klinicznej.
Magnes horyzontalny, do którego wsuwany
jest na leżąco pacjent utrzymuje stałe i
jednorodne pole magnetyczne.
Za pomocą dodatkowych cewek uzyskuje się
warunki rezonansowe jąder tylko w wybranej
warstwie, w przekroju.
Impuls pola zmiennego wzbudzającego rezonans trwa ułamek sekundy
Obraz żywego organizmu przedstawia mapę gęstości protonów wody w
tkankach danej części ciała.
Najczęściej obrazuje się nieruchome części ciała tj. mózg, rdzeń kręgowy,
kończyny.

Obraz mózgu otrzymany metodą tomografii zmienia się w zależności od tego ile świeżej utlenowanej krwi jest dostarczone do niego w trakcie myślenia, zapamiętywania czy reakcji na bodźce.
Uzyskuje się mapę funkcji mózgu
Do obrazowania narządów wewnętrznych konieczne jest dodatkowe sterowanie gradientem pola w rytm oddechów tzw. nawigator oddechowy
W badaniu serca sekwencje obrazujące są sterowane sygnałem EKG
26

PROTEin complement of the genOME
(składnik białkowy kodowany przez genom)
Proteomika zmierza do poznania wszystkich białek pojawiających się w danym organizmie w ciągu całego życia.
Wykorzystuje kilka technik służących do jednoczesnej analizy setek lub tysięcy białek.
Białka są trudnym obiektem badań ze względu na ich przestrzenną strukturę
i występowanie w wielkocząsteczkowych kompleksach.
Dynamiczny obraz proteomu zawiera:
zmiany w pojedynczej komórce związane z jej cyklem życiowym
rozmieszczenie białek w obrębie struktur komórki
wzajemne oddziaływanie komórek tworzących tkanki

Liczba białek proteomu jest znacznie
większa niż liczba kodujących ją genów.
Ludzki genom ma w przybliżeniu 30-50 tysięcy genów kodujących białka.
Do tej pory rozpoznano około 500 tysięcy białek, co oznacza, że na 1 gen przypada prawie 10 różnych wariantów białek
Na taki stan mają wpływ:
procesy molekularnego składania w trakcie translacji i powstawania informacyjnego kwasu rybonukleinowego
procesy potranslacyjne: fosforylacja czy glikozylacja, które mają wpływ na ostateczną strukturę białka
Pełna analiza białek- markerów białkowych zawartych w materiale biologicznym może być źródłem wielu informacji o zdrowiu pacjenta oraz wspomóc kliniczną diagnozę w monitorowaniu choroby czy prowadzeniu terapii.

Ludzka surowica zawiera wiele tysięcy białek/peptydów o stężeniu w zakresie 35-50 ∙ 10⁹ pg/ml do 0-5 pg/ml
Poznano 22 białka stanowiące 99% surowiczego proteomu, pozostały 1%
białek/peptydów nazwano peptydomem, zawierającym białka o masie cząsteczkowej
mniejszej niż 10 000 Da.
Małe białka są trudne do identyfikacji i wykonania analizy ilościowej z powodu ich małej
ilości w surowicy oraz tendencji przyłączania się do białek transportujących, takich jak
albumina.

W wykrywaniu markerów biologicznych stosuje się techniki pozwalające na
porównywanie złożonych mieszanin białkowych i ilościową ocenę ich
poszczególnych składników.

ETAP 1
Białka są rozdzielane w gradiencie pH w
zależności od punktu izoelektrycznego pI,
czyli wartości pH, przy której ładunek
wypadkowy białka jest równy zero
Stosuje się kapilary z żelem
poliakrylamidowym lub paski żeli z
immobilizowanym gradientem pH,
dającym wysoką rozdzielczość i
powtarzalność wyników Ogniskowanie izoelektryczne IEF
Analiza proteomiczna wykorzystuje głównie elektroforezę dwukierunkową 2D i
spektrometrię mas.

ETAP 2
Białka są rozdzielane zgodnie z ich masą
cząsteczkową metodą SDS-PAGE
Uzyskuje się rozdziały białek o
podobnych masach cząsteczkowych
Barwniki fluorescencyjne

Technika analityczna, której
podstawą jest pomiar stosunku
masy do ładunku elektrycznego
danego jonu.
Metoda oparta na jonizacji
cząsteczek lub atomów, a
następnie detekcji liczby jonów w
funkcji ich stosunku masy do
ładunku (m/z).
Wyniki działania spektrometru
mas są przedstawiane w postaci
tzw. widma masowego.

GC-MS - chromatografia gazowa w połączeniu ze spektrometrią mas;
metoda skojarzona, łącząca technikę separacji z detekcją MS; służy do ilościowego
oznaczania składników złożonych próbek.
Zastosowanie:
identyfikacja cukrów, amin, alkoholi, narkotyków, substancji dopingujących, pestycydów i dioksyn.
identyfikacja metabolizmu leków (farmakokinetyka)
badanie wpływu leków na funkcjonowanie organizmu (farmakologia)
odkrywanie nowych, bioaktywnych metabolitów
LC-MS - chromatografia cieczowa w połączeniu ze spektrometrią mas;
stosuje się chromatografię wysokosprawną (HPLC) i ultra wysokosprawną (UPLC).
Zalety:
jednoznaczna identyfikacja składników próbki
bardzo wysoka czułość układu analitycznego
możliwość identyfikacji wszystkich składników próbki
wysoka powtarzalność pomiarów
analiza ilościowa


Chromatografia (gr. chromatos = barwa + graphos = pisze)
technika służąca do rozdziału lub badania składu mieszanin
związków chemicznych.
Chromatografię stosuje się do :
wyodrębniania związków z mieszanin oczyszczania identyfikacji ilościowej i jakościowej preparacji związków
Wybór metody zależy od :
ilości mieszaniny identyfikacyjnej rodzaju związków stopnia skomplikowania podziału
36

Rozdział chromatograficzny polega na umieszczeniu badanej mieszaniny na ciekłej lub stałej fazie nieruchomej (stacjonarnej), a następnie na przepuszczeniu ciekłej bądź gazowej fazy ruchomej (mobilnej) przez nią lub też nad nią, czyli elucji mieszaniny z fazy nieruchomej.
Składniki mieszaniny o różnych stosunkach podziału są eluowane (migrują) z różnymi szybkościami. Te różnice w szybkości migracji prowadzą do rozdziału składników w czasie i przestrzeni.
37

Kryteria podziału metod chromatograficznych
38
I. Zastosowanie eluentu
1. chromatografia cieczowa
2. chromatografia gazowa
3. chromatografia fluidalna
IV. Stan skupienia faz
1. chromatografia gazowo-cieczowa (GLC)
2. chromatografia cieczowo-cieczowa (LLC)
3. chromatografia gazowo-stała (GSC)
4. chromatografia cieczowo-stała (LSC)
II. Natura sił fizyko-chemicznych procesu
1. chromatografia adsorpcyjna
2. chromatografia jonowymienna
3. chromatografia podziałowa
a. klasyczna
b. z odwróconymi fazami
c. par jonowych
4. chromatografia sitowo-żelowa (sączenie
molekularne)
5. chromatografia powinowactwa
6. elektroforeza kapilarna (CE)/ wysokosprawna
elektroforeza kapilarna (HPCE)
III. Geometria układu
1. chromatografia kolumnowa
2. chromatografia planarna
a. bibułowa
b. cienkowarstwowa (TLC)
V. Parametry procesu
1. wysokosprawna chromatografia cieczowa (HPLC)
2. szybka, białkowa chromatografia cieczowa (FPLC)

Klasyfikacja adsorbentów: 1. pod względem aktywności adsorpcyjnej
- słabe (skrobia, sacharoza)
- średnie (węglan wapnia)
- silne (aktywowany kwas krzemowy, tlenek glinu)
2. ze względu na polarność
- silnie polarne (tlenek glinu, tlenek krzemu)
- słabo polarne (węglan wapnia)
- niepolarne (węgiel aktywowany)
3. ze względu na właściwości chemiczne:
- kwaśne ( SiO2)
- zasadowe ( CaO)
- obojętne ( węgiel aktywowany)
- amfoteryczne ( Al2O3)
4. ze względu na naturę chemiczną:
- organiczne
- nieorganiczne
- mieszane
- specyficzne
39

Rozpuszczalniki (solwenty) - ułożone są w szereg eluotropowy, w zależności
od zdolności adsorpcyjnych w stosunku do substancji w nich rozpuszczonych
oraz elucyjnych.
Stosowane są do rozwinięcia chromatogramu jak i do elucji (wymywania
rozdzielonych substancji).
W chromatografii rozpuszczalniki uszeregowane są wraz ze wzrostem zdolności
wymywania zaadsorbowanych związków polarnych:
heksan ( najmniej polarny)
tetrachlorek węgla
benzen
eter dietylowy
aceton
chloroform
octan etylu
etanol
metanol
woda

Zdolności adsorpcyjne cząsteczek zależą od :
wielkości i budowy przestrzennej
liczby i położenia wiązań podwójnych
liczby podstawników polarnych i niepolarnych
liczby i rodzaju grup funkcyjnych
Stopień adsorpcji cząsteczek adsorbatu wzrasta wraz z:
ilością wiązań podwójnych
grup funkcyjnych
liczbą podstawników tego samego rodzaju

42

Rozpuszczone w fazie ruchomej składniki mieszaniny ulegają podziałowi między
dwie fazy. O rozdziale decyduje:
Prawo podziału solutu- prawo Nernsta
K - współczynnik w stanie równowagi zależy jedynie od temperatury i właściwości
substancji tworzących roztwory, a nie zależy od ilości substancji rozpuszczonej
C1 - stężenie solutu (substancji rozpuszczonej) w fazie stacjonarnej
C2 - stężenie solutu w fazie ruchomej
Ekstrakcja- metoda rozdziału składników mieszaniny lub oczyszczania
określonego składnika z towarzyszących zanieczyszczeń wykorzystująca
prawo podziału solutu.
43

Rozdział mieszaniny jest oparty na różnicy we współczynnikach podziału
składników mieszaniny (odmienną rozpuszczalnością) między dwie nie
mieszające się ze sobą fazy, z których jedna jest cieczą osadzoną na nośniku
(faza stacjonarna), a druga fazą ruchomą (ciecz, gaz). 44
Szybka metoda wstępnej orientacji w ilości i względnym
udziale składników mieszaniny związków organicznych.

Podstawę podziału stanowią reakcje wymiany jonowej między jonami z roztworu, a
jonami związanymi z fazą stacjonarną, którą stanowią jonity (kationity i anionity).
Rozdział składników mieszaniny wynika z różnic w ładunku elektrycznym jonów.
Jonity (wymieniacze jonowe) są to substancje, które mają właściwości pobierania
jonów z roztworów w zamian za oddawanie do roztworów równoważnych ilości innych
jonów tego samego znaku.
Żywica jonowymienna składa się ze szkieletu (matrycy) polimeru łańcuchów
węglowodorowych w postaci sieci przestrzennej. Z matrycą są trwale powiązane jony
(grupy) aktywne, które noszą nazwę jonów utrwalonych. Jony te mogą być wymieniane
na inne znajdujące się w roztworze.

Zastosowanie chromatografii jonowymiennej:
rozdział związków jonowych, kwasów karboksylowych, zasad (aminy),
aminokwasów, peptydów, białek, pochodnych kwasów nukleinowych,
cukrów
zmiękczanie wody – usuwanie jonów wapnia i magnezu
demineralizacja wody

Podstawę podziału stanowią różnice w rozmiarach cząsteczek – rozdział
zachodzi na skutek różnicy mas cząsteczkowych rozdzielanych związków.
Wypełnienie kolumn - żele o zdefiniowanych rozmiarach porów, zbliżonych
do rozmiarów cząsteczek analizowanych substancji;
sita molekularne
Faza nieruchoma - rozpuszczalnik zalegający wnętrze ziaren żelu.
Faza ruchoma - rozpuszczalnik przemieszczający się pomiędzy ziarnami żelu.
Substancje o dużych rozmiarach cząsteczek (większych od średnicy porów)
przesuwają się wzdłuż kolumny szybciej i przechodzą do wycieku. Cząsteczki
o określonych rozmiarach dyfundują w głąb ziaren żelu tylko na określoną
głębokość - im mniejsze rozmiary tym głębiej penetrują ziarna żelu – wolniej
migrują wzdłuż kolumny.
47

Przykładowe wypełnienia
1. Sephadex - na bazie dekstranu (wielkocząsteczkowy glikan)
2. Sepharose - granulowana forma agaru (glikan obojętny (D-galaktoza +
3,6-anhydro-L-galaktoza)
3. Sephacryl – całkowicie syntetyczne sito molekularne-kopolimer
allilodekstranu i N,N’- metylenobisakrylamidu
Zastosowanie chromatografii sitowej:
określanie rozkładu mas cząsteczkowych polimerów
rozdział substancji wielkocząsteczkowych takich jak: białka, kwasy
nukleinowe, peptydy, cukry, glikole, silikony.
Wada – nie nadaje się do rozdziału związków o zbliżonych masach
48

49

Szczególny typ chromatografii adsorpcyjnej, w której wykorzystuje się
wzajemne powinowactwo dwóch substancji:
chemicznie związany z nierozpuszczalnym nośnikiem – ligand (efektor)
substancja w fazie ruchomej specyficznie adsorbowana przez ligand
Substancja rozpuszczona w fazie ruchomej może oddziaływać z
unieruchomionym ligandem jak:
hormon z receptorem,
enzym z substratem,
enzym z inhibitorem,
przeciwciało z antygenem,
kwasy nukleinowe z białkami,
dopełniacz z przeciwciałami
50

Etapy chromatografii powinowactwa
1. Chemiczne związanie ligandu ze złożem (immobilizacja ligandu) –
złoża stosowane w filtracji żelowej
2. Specyficzne wiązanie substratu do unieruchomionego ligandu i
„odmycie” niespecyficznie zaadsorbowanych cząsteczek
białkowych
3. Elucja specyficznie związanego substratu – za pomocą eluentów:
specyficznych – np. inhibitorów enzymów
niespecyficznych – np. buforów o niskim/wysokim pH,
roztworów o dużej sile jonowej, związków rozrywających
wiązania wodorowe
51

52

Podstawą rozdziałów elektroforetycznych jest różnica w
prędkościach migracji cząstek obdarzonych ładunkiem w stałym
polu elektrycznym.
Kationy przyciągane są przez katodę (-), a aniony przez anodę (+).
Prędkość migracji w kierunku elektrod jest różna dla różnych
cząsteczek i zależy od:
natężenia pola elektrycznego
ładunku cząsteczki
rozmiaru cząsteczki
lepkości środowiska
Rozdział przeprowadza się w kapilarach ze stopionej krzemionki
(kwarcu) SiO2 wypełnionych roztworem elektrolitu nośnego;
kapilary o Фwewn. 25–75 µm i l ok. 50 cm.
53

Zastosowanie elektroforezy kapilarnej:
analiza biopolimerów (białka, polipeptydy, kwasy nukleinowe)
analiza związków biologicznie czynnych (aminokwasy, nukleozydy, nukleotydy)
analiza leków – kontrola jakości leków
analiza mieszanin racemicznych i ich rozdział na optycznie czynne enancjomery
54
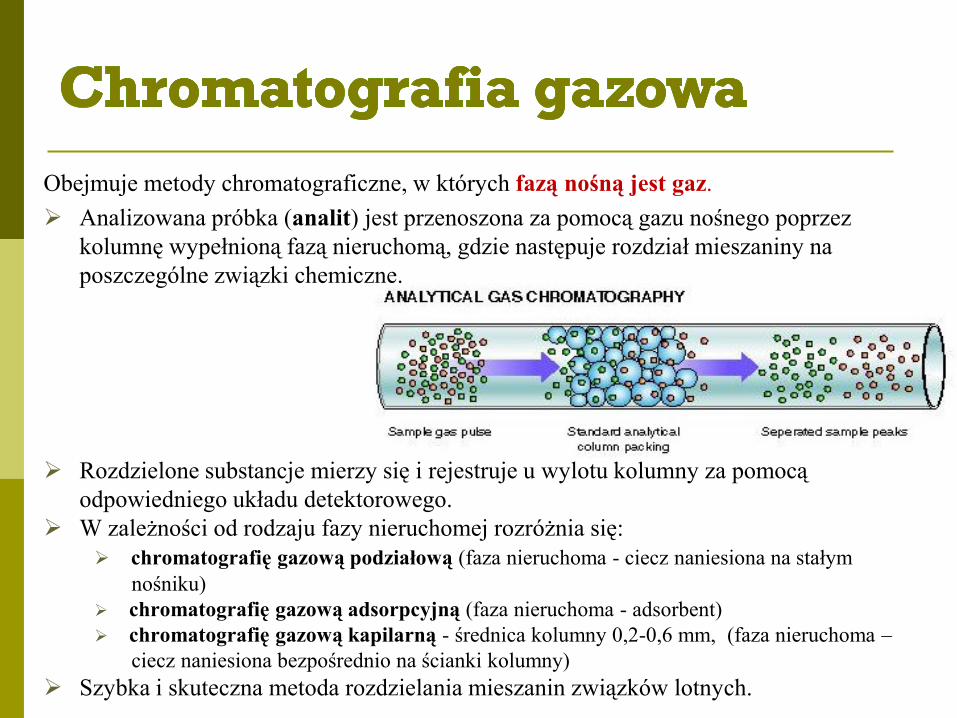
Obejmuje metody chromatograficzne, w których fazą nośną jest gaz.
Analizowana próbka (analit) jest przenoszona za pomocą gazu nośnego poprzez
kolumnę wypełnioną fazą nieruchomą, gdzie następuje rozdział mieszaniny na
poszczególne związki chemiczne.
Rozdzielone substancje mierzy się i rejestruje u wylotu kolumny za pomocą
odpowiedniego układu detektorowego.
W zależności od rodzaju fazy nieruchomej rozróżnia się:
chromatografię gazową podziałową (faza nieruchoma - ciecz naniesiona na stałym
nośniku)
chromatografię gazową adsorpcyjną (faza nieruchoma - adsorbent)
chromatografię gazową kapilarną - średnica kolumny 0,2-0,6 mm, (faza nieruchoma –
ciecz naniesiona bezpośrednio na ścianki kolumny)
Szybka i skuteczna metoda rozdzielania mieszanin związków lotnych.

Zastosowanie
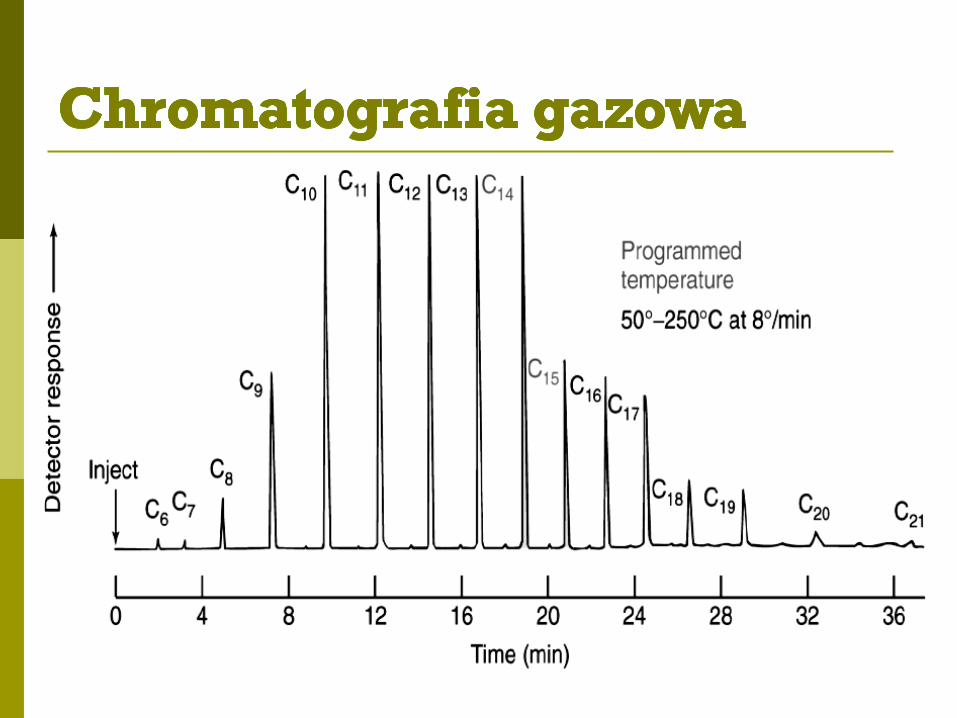
Analiza jakościowa:
identyfikacja związków na podstawie parametrów retencji - w tych samych
warunkach rozdziału takie same charakterystyki retencyjne
identyfikacja substancji poprzez porównanie ze wzorcem
Analiza ilościowa:
wysokość h i powierzchnia A piku są proporcjonalne do ilości oznaczanego
składnika: h = f(c) i A = f(c)
metoda krzywej kalibracyjnej
metoda wzorca wewnętrznego
metoda dodawania wzorca
56

58

Wysokociśnieniowa/wysokosprawna chromatografia cieczowa -
HPLC – High Pressure/Performance Liquid Chromatography
Faza ruchoma - ciecz wtłaczana pod wysokim ciśnieniem (20MPa-40MPa)
Faza stacjonarna - gęsto i jednorodnie upakowane złoże w kolumnie z metalu,
dobrze odpowietrzone
Wariant analityczny - węższe kolumny (4-8 mm); długość 5 – 25 cm
Wariant preparatywny - szersze kolumny (20 – 500 mm); długość 25 –100 cm
Zalety: szybki, zautomatyzowany i precyzyjny rozdział związków; łatwe
stosowanie gradientów; powtarzalność czasów retencji; ilościowe oznaczanie
związków (pole powierzchni pod pikiem)
Detekcja: spektroskopowa (UV-vis), elektrochemiczna, spektrofluorymetryczna
Wada: rozdział małych ilości związków
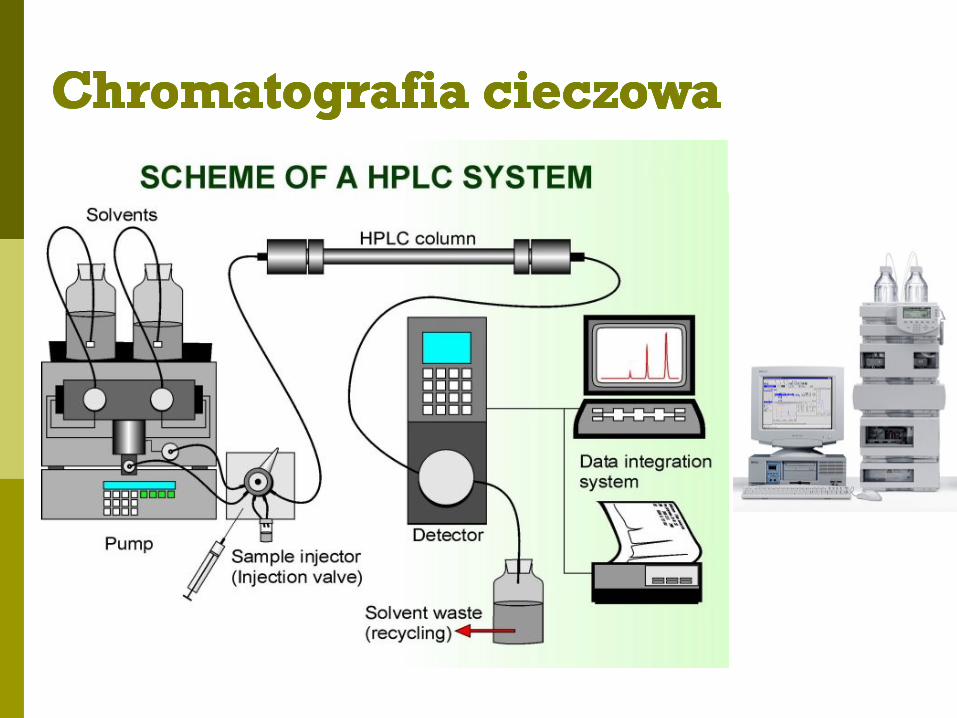
Schemat chromatografu cieczowego

Zastosowanie HPLC
Analiza związków:
biologicznie czynnych (białka, aminokwasy, witaminy,
kwasy nukleinowe, sterydy, polipeptydy, polisacharydy)
preparatów farmaceutycznych
środków ochrony roślin, pestycydów
węglowodorów policyklicznych (benzopiren w środowisku)
związków metaloorganicznych i kompleksowych
skomplikowanych mieszanin kationów np. rozdzielanie
pierwiastków ziem rzadkich 61

62 min 0 2 4 6 8 10
mAU
-2
0
2
4
6
8
10
VWD1 A, Wavelength=254 nm (US\16PUR051.D)
GTP GDP
UA
GMP
IMP
ATP
ADP
Hyp
AD
PR
Xan
AMP
Urd
NADP
NAD
In
o
Guo