linfoma t paniculitis subcutánea, asociado a síndrome hemofagocitico
TRANSCRIPT

Linfoma T Paniculitis Subcutánea, Asociado a Síndrome Hemofagocitico
Dr. José Manuel LeonisMedicina Interna y Hematología
Servicio de Hematología
CHDr.AAM, 28 de septiembre de 2016
Caso Clínico

Motivo de Consulta
Ingresó el12/6/16 A CARGO DE MEDICINA INTERNA
BL, Femenina de 29 años
Historia de 3 meses de evolución, de fiebre N/C, sin predominiohorario, asociada a lesiones nodulares induradas eritematosas enextremidades (superior e inferior), tronco, cuadrante superior izq.del abdomen, rostro y edema facial.
Adenopatías no dolorosas cervicales, occipitales y axilares menoresde 2 centímetros.
No se describe hepatoesplenomegalia en el examen de ingreso.
Evaluada en 4 clínicas privadas, CSS Chorrera, HST, HNS, sin establecer diagnóstico.

Motivo de Consulta
Antecedentes:
APP: negados
AHF: HTA
Alergia: negada
Cirugía: Negada.
G1P1
Hospitalizaciones previas: negadas.
Etilismo: negado
Tabaquismo: negado
Drogas: negado.
TA: 110/80
Fc: 96
Sat: 98%
Peso:61.3 Kg

Inicio del cuadro


Historia Hematológica
Consideraciones diagnósticas por medicina
interna al ingreso:
Causa hematológica:
Linfoma
Causas inmunológicas:
LES, asociado a paniculitis
Sarcoidosis
Causas Infecciosas:
TBC ganglionar
Eritema nodoso.

Laboratorios
Antes del ingreso:
22-5-16
Hemoglobina 12.8 g/dL
Leucocitos 2,500 cel/ul
N 51%
L 36%
M 9%
E 4%
B 0
Plaquetas 186000 cel/ul
20-6-16
Hemoglobina 12.6 g/dL
Leucocitos 3,200 cel/ul
N 79%
L 15%
M 5%
E 0
B 0
Plaquetas 246000 cel/ul

Laboratorios
Antes del ingreso:
10-6-16
Glucosa 93 mg/dL
Creatinina 0.7 mg/dL
BUN 9 mg/dL
AST 42 U/L
ALT 41 U/L
Sodio 129 mmol/L
Potasio 4.5 mmol/L
CPK 64 U/l
24-5-16
Proteína C
Reactiva
35.2 mg/L
VES 26 mm/h
24-5-16
Urinalisis Normal

Laboratorios
Al ingreso:10-6-16
Glucosa 93 mg/dL
Creatinina 0.7 mg/dL
BUN 9 mg/dL
AST 42 U/L
ALT 41 U/L
Sodio 129 mmol/L
Potasio 4.5 mmol/L
CPK 64 U/l
LDH 442 U/L
Albumina 3.8 g/dL
Calcio 9.0 mg/dL
10-6-16
TP 11.8 seg
TPT 33 seg
Fib ----------
FSP: ovalocitos, dacriocitos, algunos
neutrófilos hiposegmentados

Interconsultas
• Reumatología: (23/6/16)
– Consideran como última posibilidad causas reumatológicas. (completan
panel inmunológico)
– Recomiendan evaluación por Hematología.
• Oftalmología: (27/6/16) (por uso de hidroxicloroquina)
– Sin lesiones a nivel ocular.
• Cirugía: (29/6/16)
– Programación de biopsia de adenopatía.
• Hematología: (4/7/16)
– Aspirado más biopsia de médula ósea.
• Dermatología: seguimiento de biopsia de piel.

Laboratorios
Perfil inmunológico (14/6/16)
ANA Negativo
C3 ; C4 Normales
ANCA PR-3 Negativo
ANCA MPO Negativo
ANA HEP-2 + mitocondrial. + moteado
ENAPLUS Negativo

Laboratorios
Serología Viral (13/6/16)
HIV Negativo
HBsAg No reactivo
Hepatitis A IgM No reactivo
Hepatitis C NHR
Toxoplasmosis IgG e IgM No reactivo
Treponema pallidum Negativo
EBV IgM Negativo
EBV IgG Positivo

Laboratorios
B2-MICROGLOBULINA 5.44 mg/L
Electroforesis de proteínas GAMMAPATIA POLICLONAL
COOMBS DIRECTO Negativo
Vitamina B12 NHR
CTFH 237.6 ug/dL
Ferritina
% SATURACIÓN DE HIERRO 18.08 %
Transferrina NHR
Hierrro 43 ug/dL

Patología
GANGLIO ZONA PERIAXILAR DERECHA (12-7-16)
Tejido subcutáneo con paniculitis
REVISION DE BIOPSIA DE PIEL (22-7-16)
Favorecen linfoma de células T de tipo paniculitis subcutánea.
Infiltrado es predominantemente lobulillar, hay necrosis grasa, prominente rimming
de linfocitos, prominente cariorrexis, “Bean Bag Histiocytes” y linfocitos
pleomórficos.
Inmunohistoquimica: CD8 predominio sobre CD4, CD56 Negativo
Principal diagnóstico diferencial es el linfoma de células T Gamma Delta; pero este
último se suele ulcerar y es CD56 positivo.

40
X
4
X
Infiltrado linfocítico atípico dentro del panículo
en un patrón lobular. Se identifican adipocitos
rodeados por linfocitos malignos y cariorrexis
(vista a alto poder 40X). No se identifica
afectación de la epidermis ni de la dermis (vista
a bajo poder 4X)
Dr. Jorge BonillaMR Patologí[email protected]
Dra. Mónica ChávezPatóloga

Vista a alto poder (100X). Infiltrado linfoide atípico, con células de pequeño a
mediano tamaño, núcleos hipercromáticos, nucleolos prominentes
ocasionales y escaso citoplasma, que rodean a los adipocitos. Hay necrosis,
cariorrexis y mitosis ocasionales.
100
X
Dr. Jorge BonillaMR Patologí[email protected]
Dra. Mónica ChávezPatóloga

Patología
MÉDULA ÓSEA (2-8-16)
Médula ósea normocelular
Sin evidencia de infiltración por linfoma.
INMUNOHISTOQUIMICA
Sin alteraciones fenotípicas.
(28-7-16)
Negativo para rearreglos del TCR CLONAL.

Gabinetes
• CAT C-T-A-P (15/6/16)
– Estructuras ganglionares a nivel de la región axilar
que llegan a medir hasta 1.8 cm.
– Esplenomegalia (13.6 cm)
– No hepatomegalia.
• USG tejidos blandos de la cara: (23/6/16)
– Lesiones nodulares superficiales en ambas regiones
malares.
• Eco: (FE 65%)
– Función sistólica y diastólica conservada.

Evolución ClínicaResumen
Feb - Marzo 2016 … junio 2016 … julio 2016 … Agosto 2016 … Septiembre
Inicio del cuadro
Hospitalización
Reporte de biopsia compatible con Linfoma T
paniculitis subcutánea.(22/7/16)
4/8/16 CHOP
25/8/16 CHOEP
14/7/16 CHOEP
Ferritina: 5,000 ng/ml
Fibrinógeno: 71.2 mg/dL
Citopenia: (Hb: 8.2 g/dL ;
Plaq: 82,700)
Fiebre
TG:452 mg/dL
Esplenomegalia
Paro cardiorespiratorio (TEP)

Caso clínicoEvolución
12 Junio 23 junio 5 julio
PDN 50 mg.
Hidroxicloroquina
(27/6 - 4/7/16)
LDH: 900 U/L
TG: 352 mg/d/L
TOA: 133 U/L
TGP: 76 U/L
LDH:
442 U/L

Caso clínicoEvolución
6 Julio 17 julio 29 julio
Solumedrol
250 mg iv
por 4 días
(23-26/7/17)
PDNPDN

Antes del ETOPOSIDO

Caso clínicoEvolución
1 agosto 22 agosto
CHOP (4/8/16)
38.3 C
4/8/16
CHOP
PDN
Neutropenia

Antes del ETOPOSIDO

Antes del ETOPOSIDO

Caso clínicoEvolución
23 agosto 3 septiembre
CHOEP (25/8/16)
LDH 735 U/L)

Síndrome hemofagocitico

Criterios cumplidos
• Fiebre.
• Hipofibrinogenemia (71.2 mg/dL)
• Aumento de la ferritina (5,029 ng/ml)
• Citopenias (Hb: 8.2 g/Dl ; Plaq: 82,700)
• Esplenomegalia
• Hipertrigliceridemia (452 mg/dL)

Aspirado de médula ósea:23/8/16)
Aparente aumento de la celularidad.
8-10 megacariocitos por campo de bajo poder.
Serie granulocitica con diferenciación terminal.
Serie eritroide residual con maduración
normocitica.
No se observan datos de hemofagocitosis.

Lo que no se observo en el aspirado de médula ósea

Primary hemophagocytic syndromes point to a direct link between
lymphocytecytotoxicity and homeostasis. Immunol Rev 2005 Feb;203:165-79.


Caso clínicoEvolución
4 septiembre 15 septiembre
CHOEP (14/9/16)
2 CHOEP6/9/16
TG 114 mg/dL
Fib 295 mg/dL
LDH: 251 U/L
LDH 423
U/L
Fib 120

Evolución
• Disminución de la agudeza visual
– Oftalmología:(15/9/16): – Sangrado macula, lesiones sospechosas de infiltración.
– Transfusión de crio-precipitado y PFC.
» TP:13.6 ; TPT: 44.8 ; Fib: 119 mg/dL
» Plaquetas: 80,000.
– Corticoide oftálmico
– Paro cardio-respiratorio ----- FV
(intubación)– Angiocat: TEP. (rama para el segmento lingular )
– En anticoagulación con clexane c/12h.

• USG HEPATOESPLENICO (13/9/16)
– Ascitis leve
– Hepatomegalia (21 cm)
– Esplenomegalia (13 cm)
– Colaterales esplenorenales.
– Efusión pleural D (pequeña)
– USG doppler supra hepática: no trombosis

• Ecocardiograma:(19/9/16)
– FE: 57%
– Función contráctil conservada.
– Función diastólica comprometida.
– No vegetación.
– Ligera efusión pericárdica.
– Reacción inflamatoria del pericardio.

Punción lumbar. (20/9/16)
– Cytospin: negativo
– Proteína LCR : normal
– Glucosa LCR: normal
– GB: 0
– GR: 0
– Hongos, BAAR, piógenos: negativo

Última evaluación (27/9/16)
• Afebril.
• Sin sangrado
• Sin distres respiratorio.
• No adenopatías
• Sin nuevas lesiones y las previas prácticamente
resueltas
• Plaquetas 46,000
• L: 550 N: 200
• TP 11.6 seg. TPT: 28.7 seg. Fib 145 mg/dL
• LDH: 155 UI
• Calcio: 8.5 mg/dL
• TG:115 mg/dL

Se plantea continuar tratamiento con:
Ciclosporina y prednisona.


Generalidad
• 1991: González et al. Reporta 8 pacientes con
patologías clínicas similares. Resulta en pobre pronóstico
complicado con SHF.
• 1994: SPTCL se consideraba un subtipo de linfoma
cutáneo.
• 2001: WHO clasifica este como una entidad.
• 2008: WHO separa en dos grupos.

• Edad promedio: 36 años
• 20% se presenta en pacientes <20 años
• Femenino.
• 20% se relaciona a enfermedad
autoinmune (LES, Sd. Sjögren, DM tipo1).

Presentación Clínica
• Típicamente se
presenta con 1
o más nódulos
paniculíticos
subcutáneos o
con placas
cuyos márgenes
son difíciles de
determinar.
Piernas (71%)
Brazos (62%)
Tronco (56%)
Cara (25%)



Los objetivos de este estudio fueron:
• Confirmar que SPTL-AB y SPTL-GD son realmente
entidades distintas.
• Establecer con mayor precisión el cuadro clínico-
patológico, inmunofenotipo, la respuesta al tratamiento,
factores pronósticos, y la supervivencia de estos 2 grupos
de SPTL.
• Averiguar qué casos deben ser tratados con terapias
agresivas y los que con terapias inmunosupresoras (no
agresivos).

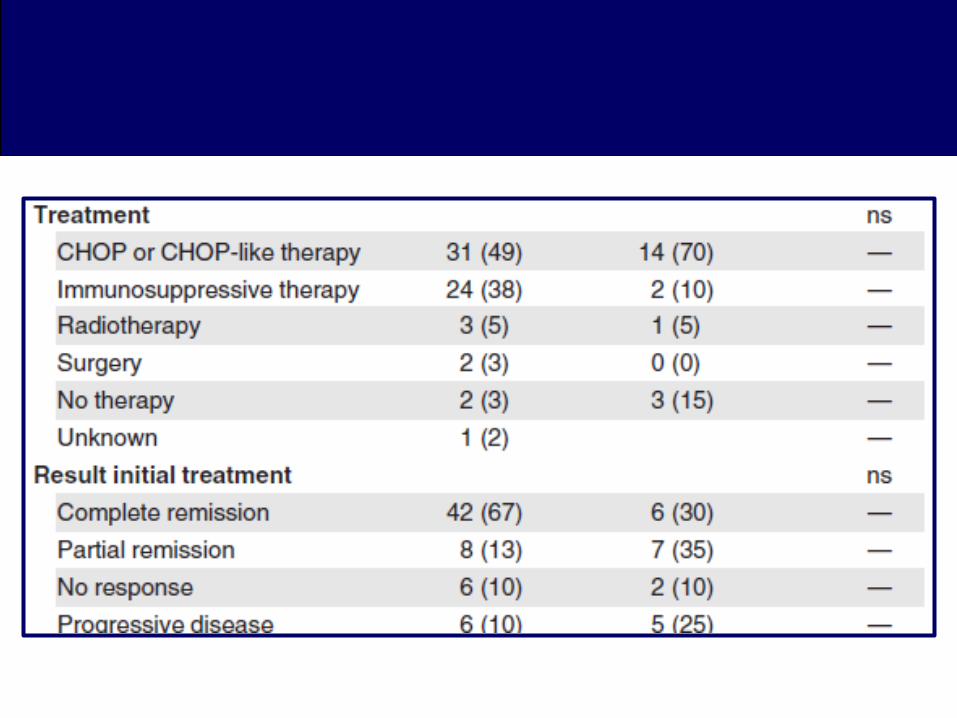

Treinta y un pacientes recibieron (CHOP) o cursos de tipo CHOP
como tratamiento inicial.
– En 4 de ellos en combinación con alemtuzumab (A-CHOP), y en
otros 3, CHOP seguido de TPH autólogo.
– Los resultados mostraron: RC 19 pacientes. RP en 3 pacientes.
4 pacientes sin respuesta, y la progresión de la enfermedad en 5
pacientes.
– De los 4 pacientes que recibieron A-CHOP (3 de 4 sin HPS) como
tratamiento inicial, sólo el 2 alcanzó una remisión completa, pero 1
de ellos desarrollaron CNS linfoma.
BLOOD, 15 JANUARY 2008 VOLUME 111, NUMBER 2

• Sólo 2 de los 19 pacientes con respuesta completa mostraron una
recaída.
• Tres de los 12 pacientes sin remisión completa después de la
terapia CHOP entró en remisión completa después del tratamiento
posterior, ya sea con una combinación de prednisona y metotrexato,
o después de un SCT alogénico (alo-SCT).
• En el momento del último seguimiento, 19 de los 31 pacientes
estaban en remisión completa, 8 tenían la enfermedad en curso,
mientras que 4 pacientes habían muerto, incluidos 3 de 6 pacientes
con HPS y uno de los efectos secundarios del tratamiento.
BLOOD, 15 JANUARY 2008 VOLUME 111, NUMBER 2

• Veinticuatro (24) pacientes habían sido tratados
inicialmente con terapias menos agresivas.
– Prednisona (19 casos), ciclosporina (5 casos), clorambucil (3
casos), metotrexato (2 casos), ciclofosfamida (1 caso), interferón-
alfa (1 caso), y gemcitabina (1 caso).
• Resultados.
– 16 RC ----- 9 recaídas ------ 5 lograron RC (PDN + Inmunosupresor)
– 5 RP
– 3 sin respuesta o progresión.
BLOOD, 15 JANUARY 2008 VOLUME 111, NUMBER 2

• Durante el seguimiento, 8 de estos 24 pacientes recibieron
CHOP, en 1 de ellos seguido de un auto-SCT.
– RC en 3 de 8 casos.
– En el momento del último seguimiento:
• 14 de estos 24 pacientes estaban en remisión
completa.
• 6 tenían la enfermedad en curso.
• 4 pacientes habían muerto, incluidos 3 de 4
pacientes con HPS asociado y 1 de la enfermedad
no relacionada.
BLOOD, 15 JANUARY 2008 VOLUME 111, NUMBER 2

BLOOD, 15 JANUARY 2008 VOLUME 111, NUMBER 2







Cancer Res
Treat.
2011;43(4):25
5-259

Curso agresivo clínica y el tratamiento de SPTCL con HPS no está bien
establecida.
La ciclofosfamida, doxorrubicina, vincristina, prednisolona terapia (CHOP) no
tiene éxito en la mayoría de los pacientes que sufren de SPTCL con HPS.
El papel de la quimioterapia de alta dosis seguida de hematopoyéticas trasplante de
células madre (HSCT) sigue siendo controvertido.
Hasta ahora, no ha habido tratamiento estándar establecido para SPTL debido
a su baja incidencia y la falta de ensayos clínicos.

TPH autólogo ------------- régimen de acondicionamiento (busulfán,
ciclofosfamida y etopósido)
CHOP ---- 6 CICLOS Desaparición de lesiones nodulares y fiebre.
Dos meses ------ Recaída
4 ciclos de ICE --------------- RC
4 meses después de TPH ------------- Recaída
2 ciclos de QT ----------------------------- Sin RESPUESTA
8 MESES DESPUES DE TPH ------- ciclosporina 4 mg / kg / día y prednisona.
DOS AÑOS SIN RECAÍDA.


Dosis 14 mg/m2 días1, 8, y 15. Ciclos de 28 días.
Romidepsin es bien tolerado.

• GRACIAS POR SU ATENCIÓN.