journal of colloid and interface science - fyzhao.ciac.jl.cn · changchun institute of applied...
TRANSCRIPT

Journal of Colloid and Interface Science 463 (2016) 75–82
Contents lists available at ScienceDirect
Journal of Colloid and Interface Science
journal homepage: www.elsevier .com/locate / jc is
Highly selective Pt/ordered mesoporous TiO2–SiO2 catalystsfor hydrogenation of cinnamaldehyde: The promoting role of Ti2+
http://dx.doi.org/10.1016/j.jcis.2015.10.0260021-9797/� 2015 Elsevier Inc. All rights reserved.
⇑ Corresponding authors at: State Key Laboratory of Electroanalytical Chemistry,Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Chang-chun 130022, PR China.
E-mail addresses: [email protected] (C. Zhang), [email protected] (F. Zhao).
Qifan Wu a,b,c, Chao Zhang a,b,⇑, Bin Zhang a,b,c, Xiaoru Li a,b,c, Zhong Ying a,b,c, Tong Liu a,b,c, Weiwei Lin a,b,Yancun Yu a,b, Haiyang Cheng a,b, Fengyu Zhao a,b,⇑a State Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, PR Chinab Laboratory of Green Chemistry and Process, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, PR ChinacUniversity of Chinese Academy of Sciences, Beijing 100049, PR China
g r a p h i c a l a b s t r a c t
a r t i c l e i n f o
Article history:Received 18 August 2015Revised 9 October 2015Accepted 9 October 2015Available online 22 October 2015
Keywords:Mesoporous TiO2–SiO2
Ti2+
Selective hydrogenationCinnamaldehydeSMSI
a b s t r a c t
A highly selective and stable catalyst based on Pt nanoparticles confined in Mesoporous TiO2–SiO2 frame-works were prepared and employed for selective hydrogenation of cinnamaldehyde to cinnamyl alcohol.The as-prepared Pt/MesoTiO2–SiO2-M catalyst displayed excellent selectivity to cinnamyl alcohol(around 91%) at nearly complete conversion. Ti2+ and stronger metal-support interaction (SMSI) playedkey roles on the adsorption behavior of cinnamaldehyde and activation of C@O bonds. The existence ofamorphous SiO2 and mixed TiO2 phases (anatase and rutile) was helpful for the formation of Ti2+ sitesand SMSI. The electron-enriched Pt surfaces and the formed Pt-TiOx system benefited the enhanced activ-ity and selectivity.
� 2015 Elsevier Inc. All rights reserved.
1. Introduction
Rational design and selection of support is extremely importantin heterogeneous catalysis in view of the strong metal-supportinteractions (SMSI) [1–3]. The merits of mesoporous frameworkas support have been widely accepted owing to the facts that mass

76 Q. Wu et al. / Journal of Colloid and Interface Science 463 (2016) 75–82
transfer and the accessibility to active sites for bulky compoundsare greatly improved; the stability and sintering resistance of cat-alysts are enhanced [4,5]. Besides, the catalyst’s confinement effectin mesoporous frameworks is also useful for improving the selec-tivity and activity. The metal nanoparticles (NPs) confined in thesupports always lead to unique characters, such as special surfaceelectronic states, favorable chemoselectivity and enantioselectivity[6–9]. For instances, the Rh NPs confined inside carbon nanotubesdisplayed a striking enhancement of catalytic activity for ethanolproduction [10]. In addition to the mesoporous frameworks-induced confinement effect, the compositions of support have alsotestified to be very useful for modulating the catalytic perfor-mances [11,12].
Selective hydrogenation of organic substrates, such as a,b-unsaturated aldehydes is crucial in the manufacture of flavors,pharmaceuticals [13–27]. Besides, selective cinnamaldehyde(CAL) hydrogenation is also an important model reaction for inves-tigating the hydrogenation behaviors of C@C and C@O bonds, andthe catalytic performances of the catalysts [25,26,28]. Consideringthe hydrogenation of C@C bond is much more thermodynamicallyfavorable, most efforts have been cast toward improving the selec-tivity of the desired product of cinnamyl alcohol (COL). Severalstrategies have been developed for enhancing the yields of COL,such as using bimetallic catalysts [14,28,29], controlling the sizesof metal NPs [30–32], modifying the surface via capping agents[15,21,25], introducing promoters [33,34], using supercritical liq-uid medium [35–38], tuning the composition of support [39],and so on. In hydrogenation of CAL, the adsorption energies andbehaviors of C@C and C@O bonds were highly related to the sizesand exposed facets of metal NPs, components of support, SMSI,etc., and the adsorption mode of CAL on metal surface was con-firmed to determine the hydrogenation selectivity. However, somequestions have not been clarified yet, such as how does the phasecomponent and surface defect site on the support affect the cat-alytic properties, which is still puzzling the researchers in this field.Therefore, the study on the influences of phase component of sup-port and defect sites on the selective hydrogenation of C@C andC@O bonds is highly important and desired.
The hydrogenation catalysts with TiO2 or SiO2 as support havebeen widely studied, but none of them gave satisfied results forthe selective hydrogenation of CAL to cinnamyl alcohol (COL). Inview of the rational mesoporous structures, enhanced thermal sta-bility [40], mesoporous TiO2–SiO2 composite is expected to be asuitable support for CAL hydrogenation once the surface electroncharacters is optimized. In this work, a series of Pt/ordered meso-porous TiO2–SiO2 catalysts were prepared and studied to discoverthe influences of surface defects and SMSI on the selective hydro-genation of CAL. The adsorption behavior of reactant CAL could bealtered dramatically, and it caused a large increase of reaction rateand selectivity of COL. SMSI and Ti2+ were demonstrated to beresponsible for the enhanced catalytic performances. This workreveals the promoting role of Ti2+ in selective hydrogenation ofCAL over the Pt/ordered mesoporous TiO2–SiO2. It provides newinsights into the relationship between selective hydrogenation ofCAL and SMSI. It is expected that these findings would be usefulfor designing the catalysts for selective hydrogenation of other a,b-unsaturated aldehydes.
2. Experimental section
2.1. Synthesis of ordered mesoporous TiO2–SiO2 composite
In a typical synthesis procedure, 0.50 g of tri-block copolymersEO106PO70EO106 (Pluronic F127), 0.60 g of 1,3,5-trimethylbenzene(TMB) and 1.25 g of KCl were dissolved in 50 ml of 1 M HCl at room
temperature. After 1 h stirring, given amount of tetraethyl orthosil-icate (TEOS) and tetrabutyl titanate (TBT) were added to this solu-tion (The Ti/Si mole ratio was 4:6). The synthesis compositionF127/KCl/TEOS + TBT/TMB/HCl/H2O (in molars) was 0.00256/1.083/0.387/0.258/0.323/3.87 /100. A given amount of H2SO4 wasadded into the above solution to adjust the phases of TiO2. Afterstirring for 24 h at room temperature, the mixture was transferredinto an autoclave and heated at 140 �C for 24 h. The as-made pro-duct was then washed by ethanol and water for three times, col-lected by centrifuge and dried at 50 �C overnight. Finally, theproduct was calcined at 550 �C for 6 h.
2.2. Synthesis of catalysts
Catalysts with 2 wt.% Pt-loading supported on the aboveordered mesoporous TiO2–SiO2 supports, mesoporous SiO2, andTiO2(P25), were prepared by the incipient wetness impregnationmethod. Aqueous solution of H2PtCl6 was added dropwisely toform support slurry. Then the slurry was dried in oven overnight.Prior to usage, the catalysts were reduced in pure H2 at 150 �Cfor 2 h with a heating rate of 2.5 �C/min.
2.3. Catalysts characterization
X-ray powder diffraction (XRD) measurements were carried ona Bruker D8 ADVANCE diffractmeter, using Cu Ka radiation(k = 0.154 nm). Nitrogen porosimetry measurement was per-formed on a Micromeritics ASAP 2020 M instrument. X-ray photo-electron spectroscopy (XPS) measurements were carried on the VGMicrotech 3000 Multilab. Transmission electron microscopy (TEM)images were performed on a JEOL JEM-2010 instrument operatingat an accelerating voltage of 200 kV. Energy dispersive X-ray spec-troscopy (EDS) measurements were carried on a scanning electronmicroscope (SEM Hitachi S-4800).
2.4. Catalytic testing
The selective hydrogenation of cinnamaldehyde was performedin an 80-mL stainless steel reactor with an inner Teflon coating.The autoclave was charged with 0.2 g catalysts, 10 mL isopropanoland 4 mmol cinnamaldehyde, then flushed with H2 for more thanthree times and pressurized to 4.0 MPa. Then the reactor was putinto the water bath, and preheated at 80 �C for 15 min. The reac-tion was recorded with starting the stirrer at a speed of1300 rpm. After reaction, the vessel was cooled to room tempera-ture naturally, and the liquid phase was analyzed using a gas chro-matograph (Shimadzu, 2010) equipped with a capillary column(Restek-50 30 m � 0.25 mm � 0.25 lm,) and a flame ionizationdetector (FID).
3. Results and discussion
3.1. Catalysts characterization
Ordered mesoporous TiO2–SiO2 supports (The Ti/Si ratio is 4:6)were successfully prepared through F127 as templates [4]. Thesemesoporous supports were denoted as MesoSiO2, MesoTiO2–SiO2-A or R or M, where the ‘‘A”, ‘‘R”, and ‘‘M” indicated that thephase of TiO2 was anatase, rutile and their mixtures, respectively.It is well known that the phase of TiO2 depends on the additivesto the precursor solution and calcination temperature. In presentwork, the phases of the TiO2 were adjusted by simply controllingthe proportion of sulfate and chloride ions [41,42]. Pure TiO2 sup-port with similar porous structure failed to be obtained in presentwork. With F127 as template agent, titanium can be uniformly

Fig. 2. N2 adsorption and desorption isotherms for (a) Meso SiO2, (b) MesoTiO2–SiO2-M, (c) MesoTiO2–SiO2-A (d) MesoTiO2–SiO2-R.
Q. Wu et al. / Journal of Colloid and Interface Science 463 (2016) 75–82 77
dispersed in the mesoporous frameworks via the assistance ofTAOASi bonds. It is proposed that the formation of the compositescould undergo the hydrolysis, condensation and polymerization ofthe TEOS and TBT. It is well known that the hydrolysis and conden-sation rates of TEOS can be adjusted by acidity. Therefore, the syn-chronicity of hydrolysis and condensation of both TEOS and TBTachieved with the assistance of HCl during the hydrothermal pro-cedures. The composites were composed of amorphous silica andsmall crystallized titanium oxides based on the XRD patterns asshown in Fig. 1. The wide diffraction peaks indicated the smallcrystalline size of TiO2 in the supports. The weak peaks centeredat 39.7� in Fig. 1(d–f) can be indexed to the Pt NPs in the reducedcatalysts. Contrast to MesoTiO2–SiO2, a drastically decline of TiO2
crystallinity in the Pt/MesoTiO2–SiO2 catalysts took place duringthe impregnation and following reduction processes, as impliedby the broadened peaks and decreased intensities.
Fig. 2 shows the N2 sorption isotherms of the MesoSiO2,MesoTiO2–SiO2-M, MesoTiO2–SiO2-R, and MesoTiO2–SiO2-A. Allthese supports had similar ordered mesoporous structures, regard-less of their composition and phases. The N2 sorption isothermsshowed typical IV-type isotherm with an associated H2 type hys-teresis loop indicating the presence of significant mesopores. Thespecific surface area for MesoSiO2, MesoTiO2–SiO2-M, MesoTiO2–SiO2-R, and MesoTiO2–SiO2-A, was 212, 394, 388, and 412 m2/g,respectively. The introduction of Ti caused an increase of specificsurface area, and a slight decrease of pore diameter, which agreedwith the previous reports [11]. The average pore diameters in thesesupports were at a range of 5–8 nm as evaluated by BJHmethod. Asmentioned above, the phases of TiO2 were modulated by adjustingthe contents of HCl and H2SO4 additives. The two anions couldaffect the behaviors of F127 (template agent) in the precursor solu-tion, thus resulted in small deviation among the specific surfacearea of the MesoTiO2–SiO2-R, MesoTiO2–SiO2-A, and MesoTiO2–SiO2-M supports.
TEM images display a perfect replication of hexagonal orderedpore structure and the uniform pore diameter for MesoTiO2–SiO2-M support (Fig. 3a). The pore size was around 8 nm, whichwas a little larger than the value calculated from the N2 adsorptionmeasurement. As described in previous literatures, the TiO2 com-ponent can be finely dispersed in the mesoporous frameworks ofTiO2–SiO2 composites through the assistance of TiAOASi bonds[43]. The thoroughly removal of residue precursors by Soxhletextraction facilitated the following impregnation, and thenallowed the Pt NPs to be dispersed uniformly into the channelsof the supports. Fig. 3(b–f) present representative TME images of
Fig. 1. XRD patterns of MesoTiO2–SiO2 and Pt/MesoTiO2–SiO2 with differenttitanium phases. (a and d) anatase, (b and e) rutile, and (c and f) mixed phases.
MesoTiO2–SiO2-M support, Pt/MesoTiO2–SiO2-R, Pt/MesoTiO2–SiO2-M, Pt/MesoTiO2–SiO2-A, and Pt/TiO2 (P25), respectively. Theordered porous structures of the catalysts were observed clearlyin the TEM images of Fig. 3(a, b, e) taken along the channels.Fig. 3(c and d) display the TEM images of Pt/MesoTiO2–SiO2-Mtaken along a normal direction. Pt NPs were confined in the meos-porous frameworks with a fine dispersion, as observed from all thecatalysts with TiO2–SiO2 supports in Fig. 3(b–e). The average Ptsizes in the Pt/MesoTiO2–SiO2-M, Pt/MesoTiO2–SiO2-A, Pt/MesoTiO2–SiO2-R was about 5.9, 5.2 and 6.2 nm, respectively.However, the Pt size in Pt/TiO2 (P25) was much smaller; its averagediameter was around 2.5 nm, as seen in Fig. 3f. It seems that theseSi-containing mesoporous supports caused rapid growth of Ptnuclei during the reduction of catalysts and produced larger PtNPs, although they had bigger surface area than TiO2 (P25). Theexact Ti/Si ratios of the catalysts were identified by EDS (Fig. S1–3), and the values for the Pt/MesoTiO2–SiO2-A, Pt/MesoTiO2–SiO2-R, Pt/MesoTiO2–SiO2-M were all around 3:7, which wasslightly lower than the precursor ratios because of incompletehydrolysis of Ti precursors.
3.2. Catalytic performance and SMSI
Generally, the selective hydrogenation of CAL may produce cin-namyl alcohol (COL) via C@O hydrogenation; hydrocinnamalde-hyde (HCAL) via C@C hydrogenation; and hydrocinnamyl alcohol(HCOL) via both C@C and C@O hydrogenation as shown inScheme 1. The influence of reaction conditions (including reactiontime, temperature, pressure of H2) solvents, sizes of metal catalystson the product distribution had been extensively studied, and sev-eral useful hypothesis to describe the reaction pathway were pro-posed for the CAL hydrogenation. However, influences of SMSI anddefect sites of support on catalytic performance are not very clearyet.
The catalytic performances of the as-prepared Pt/MesoTiO2–SiO2-R, Pt/MesoTiO2–SiO2-M, Pt/MesoTiO2–SiO2-A catalysts withdifferent TiO2 phases were examined and compared with boththe Pt/TiO2 (P25) and Pt/MesoSiO2 catalysts in term of activityand selectivity, the results are listed in Table 1. Clearly, the cata-lysts with MesoTiO2–SiO2 supports showed superior catalytic per-formances to those with a single component support. It is knownthat the selectivity to COL deceases slightly along the increasingof conversion rate, because further hydrogenation of COL to HCOLwould take place simultaneously. Therefore a high selectivity to

Fig. 3. TEM images of MesoTiO2–SiO2-M (a), Pt/MesoTiO2–SiO2-R (b), Pt/MesoTiO2–SiO2-M at different magnifications (c) and (d), Pt/MesoTiO2–SiO2-A (e), and Pt/TiO2 (P25)(f).
Scheme 1. Reaction pathway for hydrogenation of cinnamaldehyde.
78 Q. Wu et al. / Journal of Colloid and Interface Science 463 (2016) 75–82
COL at near complete conversion of CAL is very meaningful. It isnoteworthy that the Pt/MesoTiO2–SiO2-M catalyst shows a highselectivity of 90.9% to COL at conversion of 98.8%, which is compa-
rable to the excellent catalysts in the literature [17,25]. In contrast,the selectivity to COL over Pt/TiO2 (P25) at similar conversion or Pt/MesoSiO2 catalysts (at lower conversion of 31%) was only around

Table 1The catalytic performances of several supported Pt catalysts.
Entry Catalyst Time (h) Conversion (%) COL Selectivity HCAL (%) HCOL Average reaction rate (mol g�1 h�1)
1 Pt/MesoTiO2–SiO2-A 1.5 85.3 84.0 3.3 6.5 1.1 ⁄ 10�2
2 2 90.2 81.6 2.3 5.5 9.0 ⁄ 10�3
3 5 99.0 63.8 1.2 22.5 3.9 ⁄ 10�3
4 Pt/MesoTiO2–SiO2-R 1.5 97.7 71.7 1.7 14.5 1.3 ⁄ 10�2
5 Pt/MesoTiO2–SiO2-M 0.5 98.8 90.9 0.4 7.8 3.9 ⁄ 10�2
6 2.5 99.8 85.6 0.5 8.5 8.0 ⁄ 10�3
7 Pt/TiO2 (P25) 5.5 98.4 43.7 2.6 23.6 3.6 ⁄ 10�3
8 Pt/MesoSiO2 7 30.9 39.1 40.4 5.4 8.9 ⁄ 10�4
Reaction conditions: 0.2 g catalyst, the loading of Pt was 2 wt.% for all the catalysts, 4 mmol CAL, 10 mL isopropanol, 80 �C, 4 MPa H2.Average reaction rate was calculated by the moles of CAL converted over per gram catalyst per hour.
Q. Wu et al. / Journal of Colloid and Interface Science 463 (2016) 75–82 79
40%, it was much lower than the value over the Pt/MesoTiO2–SiO2
catalysts. Besides, the activity of the catalysts was also affectedgreatly by the support. At the similar conversion of CAL (around98%), the average reaction rate over Pt/MesoTiO2–SiO2-A, Pt/MesoTiO2–SiO2-R, and Pt/MesoTiO2–SiO2-M (3.9 ⁄ 10�3,1.3 ⁄ 10�2, 3.9 ⁄ 10�2 mol g�1 h�1) were higher than that(3.6 ⁄ 10�3 mol g�1 h�1) of Pt/TiO2 (P25). While for Pt/MesoSiO2,the reaction rate was quite slow, only 31% conversion was achievedat reaction time of 7 h. In particular, the reaction rate of Pt/MesoTiO2–SiO2-M was almost 10 times that of Pt/TiO2 (P25),although the later had much smaller Pt particles. Therefore, itwas speculated that the metal particle size was a less influentialfactor compared with the composition and structure of the sup-ports for the present reaction, and the metal-support interactionmight play an important role in accelerating the reaction rateand promoting the product selectivity.
Consequently, the metal-support interaction was studied usingXPS characterization, and the chemical states of Pt and Ti in the Pt/MesoTiO2–SiO2 catalysts were investigated in detail. Contrast to Pt/TiO2 (P25), the Ti 2p spectra shifted obviously to higher bindingenergy direction for all the Pt/MesoTiO2–SiO2 catalysts, and theshift of Pt/MesoTiO2–SiO2-M was most prominent (Fig. 4). Accord-ingly, the Pt 4f spectra shifted to lower binding energy value for allthe three Pt/MesoTiO2–SiO2 catalysts by contrast with the Pt/TiO2
(P25). It was clearly that stronger interactions existed betweenMesoTiO2–SiO2 and Pt metal particles (SMSI) [42,44,45], in whichthe electron transferred from the surface of TiO2–SiO2 to the Pt par-ticles. Evidently, the presence of silica facilitated the formation ofSMSI, and it became much stronger with the existence of themix-phase TiO2. It is speculated that the SMSI in this work could
Fig. 4. XPS spectra of Ti 2p region (a) and Pt 4f region (b) for Pt/P25, Pt/Mes
include both electron transfer and the formation of Pt-TiOx. Com-monly, electrons can transfer from the reducible support to themetal, result in enrichment of electrons on the metal surfaces. Suchcharge transfer is quite complicated, which might be ascribed tothe ensemble of Ti defects, oxygen vacancies and intermetallicbonds, etc. [2,45–47].
Generally, the local increase of electron density on Pt in similarsystemwas considered as the source of SMSI’s enhancement of cat-alytic activity. The electron enrichments on the Pt surface mayactivate the electronphilic C@O group by enhancing its adsorption,resulting in the higher activity and selectivity to the unsaturatedalcohols. It is also assumed that Pt-TiOx system was firstly formedin the interfacial area and then followed by the partial migration ofTi and Pt species, where the encapsulation of Ti species by Pt NPs orthe diffusion of Pt into the TiO2 lattice could take place during thereduction process. Noteworthy is the fact that the peaks of Ti2+
were present in all the Pt/MesoTiO2–SiO2 catalysts. Indeed, theTi3+ and Ti2+ can be produced under reduction atmosphere, espe-cially the TiO2 size is small [48–50]. In addition, the presence of sil-ica in the form of TiAOASi and SiAOASi bonds might facilitate theTi2+ defects [44]. As displayed in the fitted XPS spectra in Fig. 5, theexamined four catalysts showed quite different Ti2+ content, andthe maximum Ti2+ content achieved in the Pt/MesoTiO2–SiO2-M.Oppositely, Ti2+ species were not detected for the Pt/TiO2 (P25),indicating silica contributed to the Ti2+ formation. Clearly, the for-mation of Pt-TiOx (in which the dominated Ti species was Ti2+) wasprimarily caused by the introduction of silica, and it was furtherpromoted by the mix-phased TiO2. The migration of Pt-TiOx systemhence provided special active sites on the Pt surfaces or near theinterfacial area, which can activate the C@O bonds via the Ti2+
oTiO2–SiO2-A, Pt/MesoTiO2–SiO2-R, and Pt/MesoTiO2–SiO2-M catalysts.
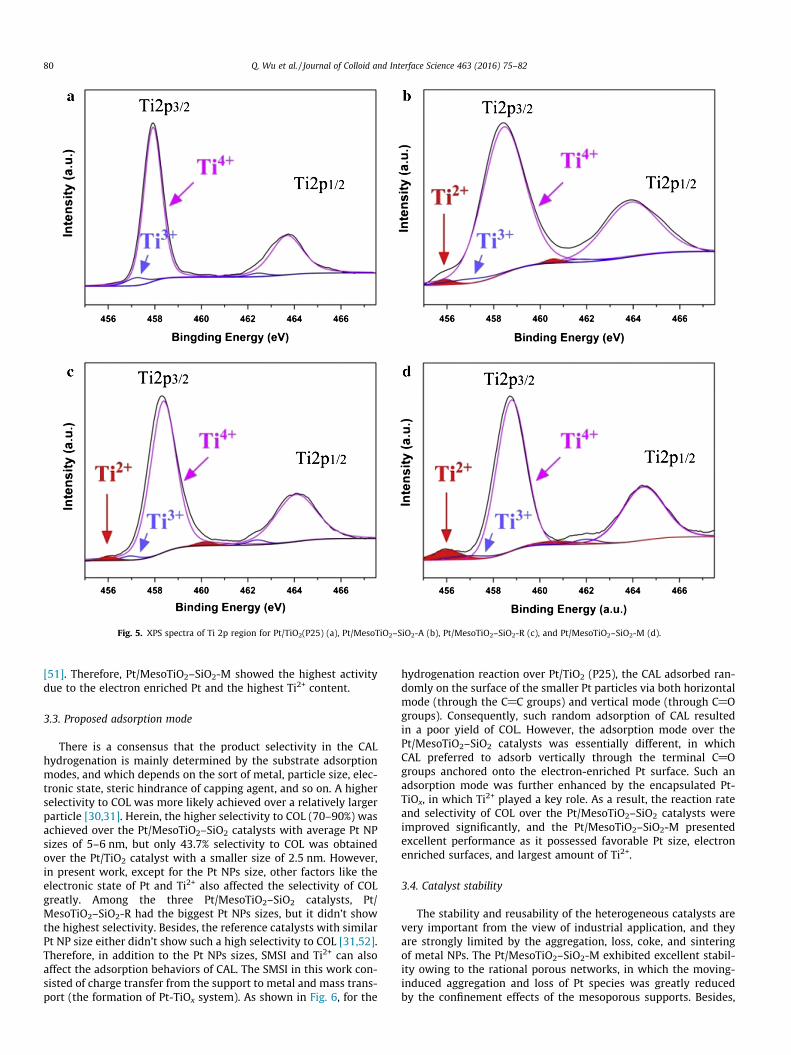
Fig. 5. XPS spectra of Ti 2p region for Pt/TiO2(P25) (a), Pt/MesoTiO2–SiO2-A (b), Pt/MesoTiO2–SiO2-R (c), and Pt/MesoTiO2–SiO2-M (d).
80 Q. Wu et al. / Journal of Colloid and Interface Science 463 (2016) 75–82
[51]. Therefore, Pt/MesoTiO2–SiO2-M showed the highest activitydue to the electron enriched Pt and the highest Ti2+ content.
3.3. Proposed adsorption mode
There is a consensus that the product selectivity in the CALhydrogenation is mainly determined by the substrate adsorptionmodes, and which depends on the sort of metal, particle size, elec-tronic state, steric hindrance of capping agent, and so on. A higherselectivity to COL was more likely achieved over a relatively largerparticle [30,31]. Herein, the higher selectivity to COL (70–90%) wasachieved over the Pt/MesoTiO2–SiO2 catalysts with average Pt NPsizes of 5–6 nm, but only 43.7% selectivity to COL was obtainedover the Pt/TiO2 catalyst with a smaller size of 2.5 nm. However,in present work, except for the Pt NPs size, other factors like theelectronic state of Pt and Ti2+ also affected the selectivity of COLgreatly. Among the three Pt/MesoTiO2–SiO2 catalysts, Pt/MesoTiO2–SiO2-R had the biggest Pt NPs sizes, but it didn’t showthe highest selectivity. Besides, the reference catalysts with similarPt NP size either didn’t show such a high selectivity to COL [31,52].Therefore, in addition to the Pt NPs sizes, SMSI and Ti2+ can alsoaffect the adsorption behaviors of CAL. The SMSI in this work con-sisted of charge transfer from the support to metal and mass trans-port (the formation of Pt-TiOx system). As shown in Fig. 6, for the
hydrogenation reaction over Pt/TiO2 (P25), the CAL adsorbed ran-domly on the surface of the smaller Pt particles via both horizontalmode (through the C@C groups) and vertical mode (through C@Ogroups). Consequently, such random adsorption of CAL resultedin a poor yield of COL. However, the adsorption mode over thePt/MesoTiO2–SiO2 catalysts was essentially different, in whichCAL preferred to adsorb vertically through the terminal C@Ogroups anchored onto the electron-enriched Pt surface. Such anadsorption mode was further enhanced by the encapsulated Pt-TiOx, in which Ti2+ played a key role. As a result, the reaction rateand selectivity of COL over the Pt/MesoTiO2–SiO2 catalysts wereimproved significantly, and the Pt/MesoTiO2–SiO2-M presentedexcellent performance as it possessed favorable Pt size, electronenriched surfaces, and largest amount of Ti2+.
3.4. Catalyst stability
The stability and reusability of the heterogeneous catalysts arevery important from the view of industrial application, and theyare strongly limited by the aggregation, loss, coke, and sinteringof metal NPs. The Pt/MesoTiO2–SiO2-M exhibited excellent stabil-ity owing to the rational porous networks, in which the moving-induced aggregation and loss of Pt species was greatly reducedby the confinement effects of the mesoporous supports. Besides,

Fig. 6. Proposed adsorption, activation and hydrogenation model for CAL over Pt/TiO2 (P25) and Pt/MesoTiO2–SiO2-M.
Fig. 7. Reusability of the Pt/MesoTiO2–SiO2-M catalyst (reaction condition: 0.2 gcatalyst, 4 mmol CAL, 10 mL isopropanol, 80 �C, 4 MPa H2, 0.5 h).
Q. Wu et al. / Journal of Colloid and Interface Science 463 (2016) 75–82 81
the rational ordered porous frameworks facilitates the rapid des-orption of the organic components from the active sites, and thensuppress the coke formation. As expected, Pt/MesoTiO2–SiO2-Mcan keep the higher selectivity and activity during the five recycles(Fig. 7). In brief, the Pt/MesoTiO2–SiO2-M displayed excellent cat-alytic performance toward selective hydrogenation of CAL to COL,and it is expected to be found potential applications for the hydro-genation of other a,b-unsaturated aldehydes.
4. Conclusions
In summary, the Pt/MesoTiO2–SiO2 catalysts were prepared andthey presented excellent catalytic activity and selectivity for theselective hydrogenation of CAL to COL. The roles of SMSI and Ti2+
on the catalytic performances in the hydrogenation of CAL wererevealed. Ti2+ species was formed in the catalysts with assistanceof silica, and it played key roles on the formation of Pt-TiOx, SMSI,and the adsorption modes of CAL. The charge transfer inducedelectron-enriched Pt NPs, and the Pt-TiOx systems were favorablefor the vertical adsorption of CAL (via C@O groups). Moreover,the Pt/MesoTiO2–SiO2-M catalyst showed an excellent stability,high activity and selectivity in five cycles. The confinement effectof the mesoporous structures in the Pt/MesoTiO2–SiO2-M catalystsuccessfully suppressed the aggregation and loss of active species,and coke formation as reported in the ordinary heterogeneous cat-alysts, which could also enhance the transfer and desorption of theproduct, as well as assist the fine dispersion of metal NPs on thesupports. We believe this work can provide useful knowledge for
the a,b-unsaturated aldehydes hydrogenation, and for understand-ing the Ti2+ and Pt-TiOx system, as well as for rational design ofstable heterogeneous catalysts.
Acknowledgements
We gratefully acknowledge the financial support provided bythe National Natural Science Foundation of China (21273222),(21473179) and Youth Innovation Promotion Association, CAS,China.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.jcis.2015.10.026.
References
[1] U. Diebold, Surf. Sci. Rep. 48 (2003) 53–229.[2] Q. Fu, T. Wagner, Surf. Sci. Rep. 62 (2007) 431–498.[3] M. Englisch, A. Jentys, J.A. Lercher, J. Catal. 166 (1997) 25–35.[4] G. Ma, X. Yan, Y. Li, L. Xiao, Z. Huang, Y. Lu, J. Fan, J. Am. Chem. Soc. 132 (2010)
9596–9597.[5] S. Wang, Q. Zhao, H. Wei, J.-Q. Wang, M. Cho, H.S. Cho, O. Terasaki, Y. Wan, J.
Am. Chem. Soc. 135 (2013) 11849–11860.[6] Y.S. Choi, E.G. Moschetta, J.T. Miller, M. Fasulo, M.J. McMurdo, R.M. Rioux, T.D.
Tilley, ACS Catal. 1 (2011) 1166–1177.[7] H. Yue, Y. Zhao, S. Zhao, B. Wang, X. Ma, J. Gong, Nat. Commun. 4 (2013) 2339.[8] C. Li, Catal. Rev. -Sci. Eng. 46 (2004) 419–492.[9] S.H. Cho, B.Q. Ma, S.T. Nguyen, J.T. Hupp, T.E. Albrecht-Schmitt, Chem.
Commun. (2006) 2563–2565.[10] X. Pan, Z. Fan, W. Chen, Y. Ding, H. Luo, X. Bao, Nat. Mater. 6 (2007) 507–511.[11] M. Bidaoui, C. Especel, N. Bouchenafa-Saib, D. Duprez, O. Mohammedi, S.
Royer, Appl. Catal. A-Gen. 445 (2012) 14–25.[12] T. Ekou, A. Flura, L. Ekou, C. Especel, S. Royer, J. Mol. Catal. A-Chem. 353 (2012)
148–155.[13] H.G. Manyar, B. Yang, H. Daly, H. Moor, S. McMonagle, Y. Tao, G.D. Yadav, A.
Goguet, P. Hu, C. Hardacre, ChemCatChem 5 (2013) 506–512.[14] R.Y. Zheng, M.D. Porosoff, J.L. Weiner, S.L. Lu, Y.X. Zhu, J.G.G. Chen, Appl. Catal.
A-Gen. 419 (2012) 126–132.[15] B.H. Wu, H.Q. Huang, J. Yang, N.F. Zheng, G. Fu, Angew. Chem. Int. Ed. 51 (2012)
3440–3443.[16] B.F. Machado, S. Morales-Torres, A.F. Perez-Cadenas, F.J. Maldonado-Hodar, F.
Carrasco-Marin, A.M.T. Silva, J.L. Figueiredo, J.L. Faria, Appl. Catal. A-Gen. 425(2012) 161–169.
[17] S. Bhogeswararao, D. Srinivas, J. Catal. 285 (2012) 31–40.[18] J. Teddy, A. Falqui, A. Corrias, D. Carta, P. Lecante, I. Gerber, P. Serp, J. Catal. 278
(2011) 59–70.[19] Y. Tang, S. Miao, H.N. Pham, A. Datye, X. Zheng, B.H. Shanks, Appl. Catal. A-Gen.
406 (2011) 81–88.[20] C.Y. Hsu, T.C. Chiu, M.H. Shih, W.J. Tsai, W.Y. Cheu, C.H. Liu, J. Phys. Chem. C
114 (2010) 4502–4510.[21] C. Kim, H. Lee, Catal. Commun. 10 (2009) 1305–1309.[22] S.C. Tsang, N. Cailuo, W. Oduro, A.T.S. Kong, L. Clifton, K.M.K. Yu, B. Thiebaut, J.
Cookson, P. Bishop, ACS Nano 2 (2008) 2547–2553.[23] A.M. Ruppert, T. Paryjczak, Appl. Catal. A-Gen. 320 (2007) 80–90.[24] J.P. Breen, R. Burch, J. Gomez-Lopez, K. Griffin, M. Hayes, Appl. Catal. A-Gen.
268 (2004) 267–274.[25] K.R. Kahsar, D.K. Schwartz, J.W. Medlin, J. Am. Chem. Soc. 136 (2014) 520–526.[26] M.S. Ide, B. Hao, M. Neurock, R.J. Davis, ACS Catal. 2 (2012) 671–683.

82 Q. Wu et al. / Journal of Colloid and Interface Science 463 (2016) 75–82
[27] H. Daly, H.G. Manyar, R. Morgan, J.M. Thompson, J.J. Delgado, R. Burch, C.Hardacre, ACS Catal. 4 (2014) 2470–2478.
[28] K.-Q. Sun, Y.-C. Hong, G.-R. Zhang, B.-Q. Xu, ACS Catal. 1 (2011) 1336–1346.[29] H. Vu, F. Goncalves, R. Philippe, E. Lamouroux, M. Corrias, Y. Kihn, D. Plee, P.
Kalck, P. Serp, J. Catal. 240 (2006) 18–22.[30] A.J. Plomp, H. Vuori, A.O.I. Krause, K.P. de Jong, J.H. Bitter, Appl. Catal. A-Gen.
351 (2008) 9–15.[31] A.K. Prashar, S. Mayadevi, R. Nandini Devi, Catal. Commun. 28 (2012) 42–46.[32] Y. Zhu, F. Zaera, Catal. Sci. Technol. 4 (2014) 955–962.[33] E.V. Ramos-Fernandez, J. Ruiz-Martinez, J.C. Serrano-Ruiz, J. Silvestre-Albero,
A. Sepulveda-Escribano, F. Rodriguez-Reinoso, Appl. Catal. A-Gen. 402 (2011)50–58.
[34] W. Lin, H. Cheng, L. He, Y. Yu, F. Zhao, J. Catal. 303 (2013) 110–116.[35] B. Zhao, J. Chen, X. Liu, Z. Liu, Z. Hao, J. Xiao, Z. Liu, Ind. Eng. Chem. Res. 51
(2012) 11112–11121.[36] C.M. Piqueras, V. Gutierrez, D.A. Veg, M.A. Volpe, Appl. Catal. A-Gen. 467
(2013) 253–260.[37] F.Y. Zhao, S. Fujita, S. Akihara, M. Arai, J. Phys. Chem. A 109 (2005) 4419–4424.[38] F.Y. Zhao, S. Fujita, J.M. Sun, Y. Ikushima, M. Arai, Chem. Commun. (2004)
2326–2327.[39] A. Ungureanu, B. Dragoi, A. Chirieac, C. Ciotonea, S. Royer, D. Duprez, A.S.
Mamede, E. Dumitriu, ACS Appl. Mater. Interf. 5 (2013) 3010–3025.[40] G. Calleja, D.P. Serrano, R. Sanz, P. Pizarro, Micro. Meso. Mater. 111 (2008)
429–440.
[41] M. Yan, F. Chen, J. Zhang, M. Anpo, J. Phys. Chem. B 109 (2005) 8673–8678.[42] J. Ohyama, A. Yamamoto, K. Teramura, T. Shishido, T. Tanaka, ACS Catal. 1
(2011) 187–192.[43] W. Dong, Y. Sun, C.W. Lee, W. Hua, X. Lu, Y. Shi, S. Zhang, J. Chen, D. Zhao, J. Am.
Chem. Soc. 129 (2007) 13894–13904.[44] A. Lewera, L. Timperman, A. Roguska, N. Alonso-Vante, J. Phys. Chem. C 115
(2011) 20153–20159.[45] D.W. Goodman, Catal. Lett. 99 (2005) 1–4.[46] X.Y. Liu, A. Wang, T. Zhang, C.-Y. Mou, Nano Today 8 (2013) 403–416.[47] M.S. Kim, S.H. Chung, C.J. Yoo, M.S. Lee, I.H. Cho, D.W. Lee, K.Y. Lee, Appl. Catal.
B-Environ. 142 (2013) 354–361.[48] G.C. Vásquez, M.A. Peche-Herrero, D. Maestre, A. Cremades, J. Ramírez-
Castellanos, J.M. González-Calbet, J. Piqueras, J. Phys. Chem. C 117 (2013)1941–1947.
[49] K.D. Schierbaum, S. Fischer, M.C. Torquemada, J.L. deSegovia, E. Roman, J.A.MartinGago, Surf. Sci. 345 (1996) 261–273.
[50] F. Sedona, G.A. Rizzi, S. Agnoli, F.X. Llabrés i Xamena, A. Papageorgiou, D.Ostermann, M. Sambi, P. Finetti, K. Schierbaum, G. Granozzi, J. Phys. Chem. B109 (2005) 24411–24426.
[51] A. Dandekar, M.A. Vannice, J. Catal. 183 (1999) 344–354.[52] W.O. Oduro, N. Cailuo, K.M.K. Yu, H.W. Yang, S.C. Tsang, Phys. Chem. Chem.
Phys. 13 (2011) 2590–2602.