in copyright - non-commercial use permitted rights ...32699/... · prom.nr. 3165...
TRANSCRIPT

Research Collection
Doctoral Thesis
Untersuchungen über die Turboextraktion von Chinarinde
Author(s): Walter, Werner
Publication Date: 1961
Permanent Link: https://doi.org/10.3929/ethz-a-000090201
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 3165
Untersuchungen über die
Turboextraktion von Chinarinde
Von der
EIDGENÖSSISCHEN TECHNISCHEN
HOCHSCHULE IN ZÜRICH
zur Erlangung
der Würde eines Doktors der Naturwissenschaften
genehmigte
PROMOTIONSARBEIT
vorgelegt von
WERNER WALTER
dipl. Apotheker
von Winterthur (Kt. Zürich)
Referent: Herr Prof. Dr. K. Steiger
Korreferent: Herr Prof. Dr. H. Flück
Juris-Verlag Zürich
1961
Leer - Vide - Empty

Meinen lieben Eltern
Leer - Vide - Empty

Die vorliegende Arbeit entstand unter der Leitung von Herrn Prof. Dr. K.
Steiger-Trippi an der galenischen Abteilung des pharmazeutischen Institutes
der Eidgenössischen Technischen Hochschule.
Für die zahlreichen wertvollen Anregungen und das stets rege Interesse an
meiner Arbeit möchte ich meinem Lehrer an dieser Stelle nochmals herzlich danken.
Die Mithilfe bei der statistischen Sicherung unserer Resultate sei hier auch Herrn
Prof. Dr. A. Linder nochmals gebührend verdankt.

Leer - Vide - Empty

- 7 -
INHALTSVERZEICHNIS
1 Theoretisches über die Extraktion 13
11 Der Extraktionsvorgang 13
12 Die Einflüsse auf den Extraktionsvorgang 14
121 Einflüsse allgemeiner Art 14
122 Aeussere Einflüsse 14
2 Die Extraktionsmethoden 15
21 Die bisher gebräuchlichen, mit Losungsmitteln arbeitenden Extraktions¬
methoden 15
211 Diskontinuierliche, einstufige Verfahren 15
211.1 Die Mazeration 16
211.2 Die Digestion 16
211.3 Die Schuttelmazeration 16
211.4 Die Extraktion mittels Vibromixer 17
211.5 Anwendung von Schall 17
211.6 Industrielle Verfahren 17
212 Diskontinuierliche, mehrstufige Verfahren 17
212.1 Die Abkochung (Decoctum) und der Aufguss (Infusio) 18
212.2 Alle übrigen Verfahren 18
213 Kontinuierliche Verfahren 19
213.1 Die Perkolation 19
213.2 Die Diakolation 19
213.3 Die Evakolation 20
213.4 Die Soxhletextraktion 20
213.5 Industrielle Verfahren 21
22 Neuere, mit Losungsmitteln arbeitende Extraktionsmethoden 21
221 Diskontinuierliche, einstufige Verfahren 21
221.1 Die Ultraschall-Extraktion 21
221.2 Die Turbo-oder Wirbelextraktion 21
221.3 Das Zentnfugieren 22
222 Diskontinuierliche, mehrstufige Verfahren 22
222.1 Die Returbo- oder Diturboextraktion 22
23 Zusammenstellung der Extraktionsverfahren 23
3 Extraktionsapparate 24
31 Die bisher gebräuchlichen, mit Losungsmitteln arbeitenden Extraktions¬
apparate 24
311 Apparate fur diskontinuierliche Extraktion 24
311.1 Die Mazeration 24
311.2 Die Digestion, Decoction und Infusion 24
311.3 Die Schuttelmazeration 24
311.4 Die Extraktion mittels Vibromixer 24
311.5 Die Anwendung von Schall 24
311.6 Die Apparate der Industrie 25
312 Apparate fur kontinuierliche Extraktion 25
312.1 Die Perkolation 25

- 8 -
312.2 Die Diakolation 25
312.3 Die Evakolation 25
312.4 Die Soxhletextraktion 25
312.5 Apparate der Industrie 26
32 Neuere Extraktionsapparate 26
321 Die Ultraschall-Extraktion 26
322 Die Turboextraktion 27
323 Die Zentrifugierung 27
4 Arbeiten über die Drogenextraktion mit besonderer Berücksichtigungder Chinarinde 29
41 Untersuchungen betreffend das Menstruum 29
42 Grundlagenforschung betreffend einzelne Extraktionsmethoden 31
421 Diskontinuierliche Verfahren 31
421.1 Die Mazeration 31
421.2 Die Ultraschall-Extraktion 32
421.3 Die Turboextraktion 33
422 Diskontinuierliche, mehrstufige Verfahren 33
422.1 Die mehrstufige Mazeration 33
423 Kontinuierliche Verfahren 34
423.1 Die Perkolation 34
423.2 Die Diakolation 36
423.3 Die Evakolation 36
423.4 Die Heissperkolation 37
423.5 Die fraktionierte Perkolation oder Re-Perkolation 37
43 Vergleichende Arbeiten 37
431 Bisher gebräuchliche Methoden 37
431.1 Vergleich diskontinuierlicher, einstufiger Verfahren 37
Allgemeine Vorschriften für:
Die Mazeration in einigen Arzneibüchern 38
Die Perkolation in einigen Arzneibüchern 39
Die Herstellung von:
Chinarinden-Trockenextrakt in einigen Arzneibüchern 40
Chinarindentinktur in einigen Arzneibüchern 41
Perkolationsvorschriften verschiedener Autoren für Chinarinde 41
431.2 Vergleich diskontinuierlicher, einstufiger mit diskontinuierlichen,mehrstufigen Verfahren 42
431.3 Vergleich diskontinuierlicher mit kontinuierlichen Verfahren 42
431.4 Vergleich kontinuierlich arbeitender Verfahren 43
432 Vergleiche mit neuen Methoden 44
432.1 Die Ultraschall-Extraktion 44
432.2 Die Turboextraktion 45
Tabelle der im Text erwähnten Literatur 46
5 Zielsetzung der eigenen Untersuchungen 47
51 Einleitung 47
52 Bietet die Turboextraktion Vorteile vor den gebräuchlichen Extraktions¬
methoden der Chinarinde? 47

- 9 -
6 Auswahl der Droge und des Menstruums 48
61 Auswahl und Charakterisierung der Droge 48
62 Auswahl des Menstruums 50
7 Beschreibung der verwendeten Apparate 51
71 Ueberprüfung des Rührgerätes 51
711 Das für unsere Versuche verwendete Gerät POLYTRON-45 ST 51
712 Die minimale Charge 51
713 Die Tourenzahl in Luft, Wasser und im Extraktionsgemisch in
Abhängigkeit von Stromstärke und Spannung am Rührmotor 53
714 Der durch das Rühren ohne Kühlung bedingte Temperaturanstiegim Extraktionsgemisch 54
72 Die Kühleinrichtung 55
721 Die Kühlspirale um das Extraktionsgefäss 56
722 Der Thermostat 58
723 Die Kühlung des Thermostaten 59
724 Steuerung der Kühleinrichtung 59
73 Heizung des Extraktionsgefässes 59
74 Zentrifuge zur Abtrennung der Droge von der Extraktlösung 59
8 Die Herstellung der Extraktlösungen, die Abtrennung vom Drogen¬material und die Bestimmung der Inhaltsstofie 61
81 Die Herstellung der Extraktlösungen 61
811 Die Mazeration 61
812 Die Perkolation 61
813 Die Turboextraktion 62
814 Die Diturboextraktion 63
82 Die Abtrennung der Extraktlösungen von der teilweise extrahierten
Droge 63
821 Abtrennung der Extraktlösungen von der mazerierten Droge 63
822 Abtrennung der Extraktlösungen nach der Turboextraktion 64
822.1 Filtration 64
822.2 Zentrifugieren 65
822.3 Filtration des in zwei Hälften aufgeteilten Extraktionsgemisches 66
83 Messgrössen 66831 in allen Extrakten 67
832 in allen Trockenrückständen 67
84 Bestimmungsmethoden 67
841 Gesamtalkaloide 67
841.1 Alkaloidbestimmung in Chinin-HCl-Lösung mittels Ionen¬
austauschern 68
841.2 Bestimmung des Gesamtalkaloidgehaltes in Chinarinde 70
841.21 Mittels Ionenaustauschern 70
841.22 Nach der Methode der Ph.Helv. V.Suppl.I 73
841.3 Bestimmung der Gesamtalkaloide in den Extraktlösungen 73
842 Die Bestimmung der Gerbstoffe 74
842.1 In der Droge (Uebersicht über die Methoden) 74

- 10 -
842.11 Nach dem Vorschlag für die Ph. Helv. VI 77
842.111 Vorversuche mit Kastanienholzextrakt 77
842.112 Hauptversuche mit Succiruba-Rinde 81
842.12 Die kolorimetrische Hautpulvermethode 83
(Kolorimeter, Cuvetten, Reagens)842.121 Vorversuche mit Kastanienholzextrakt 85
842.121.1 Die Extinktionskurve 85
842.121.2 Die Haltbarkeit der gefärbten Lösungen 87
842.121.3 Der für die Messungen geeignete Konzentrationsbereich 88
842.121.4 Die notwendige Menge Reagens pro 100 ml Messlösung 88
842.121.5 Der Einfluss der Konzentration der Sodalösung 89
842.121.6 Aufnahme der Eichkurve mit Kastanienholzextrakt 89
842.122 Hauptversuche mit Chinarinde 93
842.13 Zusammenfassung der bisherigen Resultate der Gerbstoff¬
bestimmung 94
842.2 Die Bestimmung der Gerbstoffe in der Extraktlösung 94
842.21 Einfluss von Menstruum auf die kolorimetrische Messung 94
842.22 Einfluss der Eigenfärbung der verdünnten Gerbstofflösung 95
842.23 Prüfung des Hautpulvers auf Abgabe menstruumlöslicher,mit Phosphorwolframsäure eine Färbung erzeugender Stoffe 95
842.24 Einfluss des Alkoholgehaltes der zu entgerbenden Lösung 95
842.25 Endgültige Vorschrift der Gerbstoffbestimmung 99
842.26 Kontrolle von deren Streuung an einer Extraktlösung 100
843 Die Bestimmung der Totalextraktivstoffe 101
843.1 der Droge 101
843.2 in der Extraktlösung 101
844 Die Bestimmung des spezifischen Gewichtes und der Viskosität 102
844.1 Die Bestimmung des spezifischen Gewichtes 102
844.11 der Droge 102
844.12 der Extraktlösung 104
844.2 Die Bestimmung der Viskosität der Extraktlösungen 104
845 Die Bestimmung der Korngrössenverteilung 104
845.1 in der nicht extrahierten Droge 104
845.2 in der extrahierten Droge 105
846 Die Bestimmung der Asche und ihrer in Salzsäure unlöslichen Anteile 106
847 Die Bestimmung des Wassergehaltes 106
848 Die Bestimmung des Alkoholgehaltes 106
848.1 Die Alkoholbestimmung im Menstruum nach Ph. Helv. V 107
848.2 Die Alkoholbestimmung in den Extraktlösungen 107
849 Die Bestimmung der Tourenzahl 107
85 Die Berechnung der Ausbeuten 108
9 Versuchsergebnisse 110
91 Der Wirkstoffgehalt der Droge 110
92 Die Mazeration 110
93 Die Perkolation 111

- 11 -
94 Die Turboextraktion 113
941 Die Nachextraktion wahrend der Trennung von extrahierter
Droge und Extraktlosung (Versuchsreihe 1) 113
942 Einfluss der Tourenzahl auf den Wirkstoffgehalt der Extrakt¬
losung (Versuchsreihe 2) 116
943 Einfluss der Extraktionszeit (Versuchsreihe 3) 118
943.1 Resultate 118
943.2 Die statistische Sicherung der Resultate der Versuchs¬
reihen 2 und 3 120
944 Einfluss der Temperatur auf den Wirkstoffgehalt der Extrakt¬
losung 124
945 Einfluss der zweistufigen Turboextraktion auf den Wirkstoffgehaltder Extraktlosung 125
946 Wirkung des Ruhrgerates auf den Zerkleinerungsgrad der Droge 129
946.1 Das spezifische Gewicht der Chinarinde in den einzelnen
Siebfraktionen 129
946.2 Die Korngrossenverteilung des extrahierten Drogenmatenalsals Funktion der Versuchsbedingungen 130
946.21 Der Einfluss der Trennverfahren auf die Korngrossenver¬teilung 132
946.22 Der Einfluss von Tourenzahl, Ruhrzeit und Temperatur auf
die Korngrossenverteilung 134
946.3 Die Oberflachenzunahme des extrahierten Drogenmatenalsdurch Turboextraktion 140
10 Diskussion und Schlussfolgerungen 145
101 Bestimmungsmethoden 145
1011 Alkaloide 145
1012 Gerbstoffe 145
102 Extraktionsmethoden 146
1021 Mazeration 146
1022 Perkolation 146
1023 Turboextraktion 146
1023.1 Zeitaufwand 146
1023.2 Ausbeuten an Alkaloiden, Gerbstoffen und Totalextraktiv¬
stoffen 147
1023.3 Extraktquahtat in Bezug auf Konzentration und Ballaststoffe 147
1023.4 Energieaufwand 149
1023.5 Anpassungsfähigkeit dieser neuen Extraktionsmethode an die
Erfordernisse des Laboratoriums 149
1023.6 Weitere Untersuchungsresultate 150
1024 Diturboextraktion 150
103 Verfahrensvorschlag 150
Literaturverzeichnis 153

Leer - Vide - Empty

- 13 -
1 THEORETISCHES UEBER DIE EXTRAKTION
Unter Extraktion wird in dieser Arbeit das Ausziehen fester Phasen mitHilfe
von Lösungsmitteln verstanden. Auf Ausnahmen, wie die Extraktion aus flüssigen
Phasen, wird speziell hingewiesen.
11 Der Extraktionsvorgang
Die Drogenextraktion, ohne äussere Beeinflussung, setzt sich aus drei gröss¬
tenteils parallel verlaufenden Prozessen zusammen:
1. der Quellung der Zellwand. Diese ist nach Feinstein eine unbedingteVoraussetzung für die Extraktion.
2. dem Lösungsvorgang. Dabei werden einerseits die schon bei der Drogen¬zerkleinerung aufgerissenen Zellen ausgewaschen; anderseits werden in
unverletzten Zellen Wirkstoffe durch hineindiffundiertes Menstruum gelöst.
3. dem langsameren Diffusionsvorgang der Wirkstofflösung.
Mit dem Aufbau der Zellwand befasste sich eingehend Frey-Wyssling .
Die Zellwand von nicht verholzten Parenchymzellen, die im Wesentlichen als Trä¬
ger von hydrophilen Wirkstoffen in Frage kommen, baut sich auf aus fadenförmigen
Zellulosemolekülen, die streckenweise parallel verlaufen (Micell, ungestörter Kri¬
stallbereich) und streckenweise höchstens angenähert parallel verlaufen. Viele sol¬
che Assoziationen werden durch Nebenvalenzkräfte zu grösseren Einheiten, den
Fibrillen, zusammengehalten, die die kleinsten, elektronenmikroskopisch sichtba¬
ren Einheiten der Zellwand darstellen. Die Fibrillen bilden z.T. durch Verflech¬
tungen und zum Teil wieder durch Nebenvalenzbindungen die pflanzliche Zellwand.
In der Zellwand finden sich neben den Fibrillen andere, gelartige polymère
Kohlehydrate wie Pektin und Hemicellulosen, die als Matrix bezeichnet werden und
keine fibrilläre Anordnung aufweisen. Sie machen bei der dünnen Primärmembran
ca. 40%, bei der dickeren Sekundärmembran nur ca. 5% aus.
Diese Zellwände werden durch die aus Pektin bestehenden Mittellamellen zum
pflanzlichen Gewebe verkittet. In den Membranen finden sich zwischen den Fibrilleno
die ca. 100 bis über 1000 A grossen Interfibrillarräume. Innerhalb der Fibrillen
finden sich kleinere Intermicellarräume. In gequollenem Zustande enthalten diese
Räume Wasser und die gequollenen Matrixsubstanzen.
Die interfibrillären Räume stellen den wesentlichen Weg dar, auf dem die Al-
kaloide nach aussen diffundieren können. Die Permeabilität für molekulardisperse
Lösungen, eingeschlossen die grossen Chininmoleküle, ist nachgewiesen. Feinstein
unterscheidet:
a) die Zelldiffusion, d.h. den Durchtritt des mit Extraktivstoffen angereicher¬ten Menstruums durch die Zellwand.
b) die Kapillardiffusion, d.h. die Diffusion dieser Lösung durch die mit Men¬
struum angefüllten Kapillargänge zwischen den Zellen und einzelnen Drogen¬
partikeln.

- 14 -
12 Die Einflüsse auf den Extraktionsvorgang
121 Einflüsse allgemeiner Art
Als solche kommen in Frage:
Die Natur und Löslichkeit der auszuziehenden Wirkstoffe.
Die Beschaffenheit des Zellmaterials der Droge.Die Zusammensetzung des Menstruums.
122 Aeussere Einflüsse
Die Temperatur.
Der Ersatz des mit Extraktivstoffen beladenen Menstruums, der Extraktlösung,durch frisches Menstruum. Dieser kann durch Schütteln oder Rühren (z.B.Ma¬zeration), durch Schwerkraft (z.B. Perkolation) oder durch Erzeugung eines
künstlichen Schwerefeldes (z. B. Zentrifugierung) erfolgen.
Die Zerkleinerung der Droge, welche eine Vergrösserung der inneren Ober¬
fläche des Drogenmaterials zur Folge hat. Die Partikelgrösse kann vor oder
während der Extraktion reduziert werden.
Bei gegebener Droge ist die Wahl des Menstruums von entscheidender Bedeu¬
tung. Es beeinflusst alle Phasen des Extraktionsvorganges, insbesondere die Quel¬
lung des Drogenmaterials und die Lösung der Inhaltsstoffe. Erstere steigt im allge-
meinen mit zunehmendem Wassergehalt des Extraktionsmittels ' '. Grundlegende4)516)
Arbeiten über die Quellung stammen von Hu sa und Mitarbeitern ' ' '. Die Autoren
fanden z. B. für Chinarinde rascheste und stärkste Quellung in Wasser mit 24 Vol-%4)
Alkohol.Bei Kastanienholz und Belladonnawurzel stellten sie eine Zunahme der
Quellung mit zunehmender Pulverfeinheit ' und eine Abnahme der Quellung'von
Wasser über Glycerin nach Alkohol fest.
Das Herauslösen der Wirkstoffe erfolgt, je nach deren Natur, meist mit Alko¬
hol-Wasser-Mischungen mit eventuellen Zusätzen von Säuren, Laugen oder Glycerin.
Freie Gerbstoffe sind meist gut wasserlöslich. Glycoside erfordern Menstrua mit
maximal ca. 30 % Alkohol. Alkaloide lösen sich mit steigendem Alkoholgehalt bes¬
ser, wobei jedoch die Quellung abnimmt. Das Optimum für die Extraktion von Alka-
loiddrogen liegt deshalb bei 50 - 70 % Alkohol. Weitere diesbezügliche Literaturan¬
gaben werden unter "Arbeiten über die Drogenextraktion mit besonderer Berücksich¬
tigung der Chinarinde" zusammengestellt (s. pg. 29).
Der Ersatz des mit Extraktivstoffen beladenen Menstruums durch frisches Lö¬
sungsmittel reguliert den Diffusionsprozess.
Die weitgehende Drogenzerkleinerung vergrössert die Austauschoberfläche und
beschleunigt damit die Diffusion des Lösungsmittels in die Zelle hinein.
Da beim Zerkleinern der Droge viele Zellen aufgerissen werden, erfolgt in
diesen Fällen eine direkte Lösung des Zellinhaltes und der Düfusionsvorgang wird
übersprungen. Die Extraktionsgeschwindigkeit steigt daher mit abnehmender Korn-
grösse der Drogenpartikel.
Bei einem Vergleich mehrerer Extraktionsmethoden, wie es in der vorliegen¬
den Arbeit der Fall ist, sind Droge, Menstruum und meist auch die Temperatur als
konstant gegeben. Die Ausbeute kann daher nur noch durch den ausreichenden Nach¬
schub frischen Menstruums und die Drogenzerkleinerung beeinflusst werden.

- 15 -
2 DIE EXTRAKTIONSMETHODEN
21 Die bisher gebräuchlichen, mit Lösungsmitteln
arbeitenden Extraktionsmethoden
Spezielle, ohne Lösungsmittel arbeitende Extraktionsverfahren wie Sublima¬
tion, Destillation, Vakuumdestillation, Ausschmelzen und Abpressen werden hier
nicht berücksichtigt. Diese sind wichtig für die Gewinnung von leichtflüchtigen In¬
haltsstoffen sowie von Oelen und Fetten.
Es wird daher hier, bei gegebenem Menstruum und Extraktionsgut von der be¬
nötigten Korngrösse, ein Extraktionsverfahren wie folgt definiert:
Ein Extraktionsverfahren umschliesst alles, von den vorbereitenden Arbeiten bis
zum Ablauf des letzten Tropfens klarer, nicht defäkierter, wenn nötig sofort filtrier¬
ter Extraktlösung. Das Abpressen der Droge gehört nicht dazu.
Die Abtrennung von Droge und Extraktlösung richtet sich nach dem Zweck der
Extraktion. Sie erfolgt z.B. durch Abpressen, wenn maximale Ausbeute erwünscht,
oder durch Filtration oder Zentrifugieren, wenn rascheste Abtrennung und Analyse
der Extrakte wichtig ist.
Die Extraktionsmethoden können in drei Hauptgruppen eingeteilt werden:
211 Diskontinuierliche, einstufige Verfahren
212 Diskontinuierliche, mehrstufige Verfahren
213 Kontinuierliche Verfahren,
wobei die Art und Weise des Ersatzes von Extraktlösung durch frisches Menstruum
massgebend ist. Dieses Einteilungsprinzip ist andern vorzuziehen, weil vor allem
der Nachschub von frischem Menstruum einer der wesentlichen Punkte bei jedem
Extraktionsvorgang ist. Beinahe die gleiche Einteilung findet sich auch in Uli -
manns Encyclopädie der technischen Chemie ' für die Extraktionsapparate der
Industrie.
211 Diskontinuierliche, einstufige Verfahren
Diskontinuierliche, einstufige Extraktionsverfahren sind alle jene, bei denen
die gesamten Mengen von Extraktionsgut und Lösungsmittel, künftig nur Menstruum
genannt, schon bei Beginn des Extraktionsvorganges gemischt weraen.
Vorteile dieser Verfahren:
Der Arbeitsaufwand ist gering

- 16 -
Wenig Kontrolle ist notwendigDie Toleranz bezüglich Korngrösse des Extraktionsgutes ist gross.
Nachteile dieser Verfahren:
Die Einstellung eines Gleichgewichts-Zustandes zwischen extrahiertem Stoff
und Extraktlösung bedingt beschränkte Extraktkonzentration.
Für eine befriedigende Ausbeute ist viel Menstruum notwendig.
Ein erheblicher Teil der Extraktlösung wird im extrahierten Stoff zurückge¬halten.
Nach der Extraktion ist meist eine Trennung von Rückstand und Auszug not¬
wendig.
211.1 Die Mazeration
Die Mazeration ist eine bei gewöhnlicher Temperatur vorgenommene, einma¬
lige Extraktion fester Stoffe. Die zu Beginn der Extraktion miteinander vermisch¬
ten, gesamten Mengen von Extraktionsgut und Menstruum werden während mehreren
Tagen häufig umgeschüttelt. Die Extraktflüssigkeit wird sodann von der Droge abge-
trennt. Diese Definition weicht von derjenigen der Ph.Helv. V'in folgenden Punkten
ab: Durch die Ausdrücke "wiederholte Extraktion" und "unter häufigem Umschütteln
während einer bestimmten Zeit" schliesst die Ph.Helv. V unter Mazeration auch die
mehrstufige- und eventuell die Schüttelmazeration ein. Die Trennung von Droge und
Extraktlösung erfolgt durch Kolieren und Abpressen. Erst nach achttägiger Defalca¬
tion wird weiter verarbeitet. Der Vorteil der Einfachheit und des geringen apparati¬
ven Aufwandes wird bei der Mazeration durch lange Dauer und mittelmässige Aus-9}
beute erkauft. Trotzdem schlug U. Bogs'dieses Verfahren im Jahre 1958 als
neue Arzneibuchmethode vor.
211.2 Die Digestion
Die Digestion ist eine Mazeration bei erhöhter Temperatur, wobei nach Bedarf
das Gefäss mit einem Rückflusskühler versehen wird. Jermstad u. Oestby'
sowie Bari'digerierten z.B. Drogen 3 Stunden am Rückflusskühler auf dem
Wasserbad. Die Definition der Ph.Helv. V'schreibt jedoch eine Temperatur von
40° - 50 C und häufiges Umschütteln in einem geschlossenen Gefäss vor.
211.3 Die Schüttelmazeration
Die Schüttelmazeration ist eine bei gewöhnlicher Temperatur vorgenommene,
einmalige Extraktion fester Stoffe, bei der das Extraktionsgemisch während der ge¬
samten Dauer des Extraktionsvorganges ununterbrochen geschüttelt wird.

- 17 -
Hier wird zum ersten Mal durch Zufuhr grösserer Mengen mechanischer Ener¬
gie eine gute Durchmischung und dadurch ein rascher Ausgleich des Konzentrations¬
gefälles von der intra- zur extrazellulären Flüssigkeit erhalten. Der Zeitaufwand
9)ist je nach Droge ein Bruchteil desjenigen für die gewöhnliche Mazeration .
211.4 Die Extraktion mittels Vibromixer
Die Extraktion mittels Vibromixer ist eine einmalige Extraktion pulverförmi-
ger oder flüssiger Stoffe mit Hilfe von Lösungsmitteln. Ein durch Wechselstromfre¬
quenz erregter Schwingungsüberträger im Extraktionsgemisch sorgt für dessen in¬
tensive Durchwirbelung während der ganzen Dauer des Ausziehens. Prinzipiell un¬
terscheidet sich dieses Verfahren nur durch die höhere Frequenz von der Schüttel-
mazeration, von der es sich auch bezüglich Zeitaufwand kaum unterscheiden dürfte.
211.5 Die Anwendung von Schall
Die Schall-Extraktion ist eine einmalige Extraktion pulverförmiger oder flüs¬
siger Stoffe mit Hilfe von Lösungsmitteln. Ein durch Schallenergie erregter Schwin¬
gungsüberträger im Extraktionsgemisch sorgt für dessen intensive Durchwirbelung12)
während der ganzen Dauer des Ausziehens '.
Dies ist kein grundsätzlich neues Extraktionsverfahren, denn es handelt sich
auch dabei nur um eine weitere Steigerung der Frequenz gegenüber der Vibromixer-
Methode. Zudem wurde Schall-Energie bisher oft in Kombination mit einem der her¬
kömmlichen Extraktionsverfahren verwendet.
211.6 Industrielle Verfahren
Die diskontinuierlichen Verfahren der Industrie unterscheiden sich von den
bisher genannten Methoden nur durch höheren apparativen Aufwand, bedingt durch
die grösseren Chargen.
Infuse und Dekokte, als stets frisch zu bereitende Auszüge, kommen für die
Industrie kaum in Betracht.
212 Diskontinuierliche, mehrstufige Verfahren
Diskontinuierliche, mehrstufige Verfahren sind alle jene, bei welchen die ge¬
samte Menge des Extraktionsgutes in mehreren Stufen mit je einer Komponente des1Q\
Menstruums ' oder je einem Teil der gesamten Menstruum-Menge extrahiert wird.

- 18 -
Die Abtrennung von Droge und Extraktflüssigkeit kann zwischen den einzelnen Stufen
erfolgen.131
M.Herzog' erzielte durch Mazeration in drei Stufen, wobei erst mit Was¬
ser, dann zweimal mit durch Zumischen von Alkohol erhöhter Alkoholkonzentration
extrahiert wurde, z. T. höhere Wirkstoffgehalte in den Extrakten als durch Mazera¬
tion mit dem zuvor gemischten Menstruum.
Gegenüber den diskontinuierlichen, einstufigen Verfahren entsteht folgender
Vorteil:
Der Rückstand im extrahierten Stoff ist bedeutend geringer, die Extraktiv-14)
Stoffausbeute daher bei gleichem Menstruumverbrauch grösser. Risch 'schlägt
die Dimazeration für das DAB-7 vor.
Ein Nachteil ist der etwas grössere Arbeitsaufwand. Im übrigen gelten die
Vor- und Nachteile der diskontinuierlichen, einstufigen Verfahren.
212.1 Die Abkochung (Decoctum) und der Aufguss (Infusio)
Diese beiden Verfahren benützen, wie die Digestion, Wärmeenergie zur Ver¬
besserung der Ausbeute. Die Methoden sind hier der Vollständigkeit halber erwähnt.
Nach Definition der Ph. Helv. V sind Abkochungen und Aufgüsse wässerige
Auszüge, welche bei Bedarf stets frisch herzustellen sind. Die Ph. Helv. V gibt hier
mit Recht eine Rezepturvorschrift im Gegensatz zu allen andern Herstellungsverfah¬
ren von Extrakten.
Nach Ph.Helv. V wird dabei die Alkaloiddroge erst gründlich durchfeuchtet,
dann 15 Minuten mit der Hälfte der vorgeschriebenen Wassermenge unter häufigem
Umrühren mazeriert und anschliessend durch Watte filtriert. Der Drogenrest wird
dann mit der zweiten Hälfte des Wassers:
für Abkochungen 15 Minuten in bedecktem Gefäss auf dem Wasserbade erhitzt
oder
für Aufgüsse siedend heiss Übergossen und 15 Minuten in bedecktem Gefäss
stehen gelassen.
Dann wird jeweils durch dieselbe Watte zum ersten Auszug filtriert. Nur die Vor¬
schrift betreffend die Frisch-Herstellung unterscheidet diese Methoden von einer
Kombination von Mazeration und Digestion.
212.2 Alle übrigen Verfahren
Alle diskontinuierlichen Extraktionsmethoden können nach dem Beispiel der
Mazeration mehrstufig ausgeführt werden, so dass sich eine Beschreibung erübrigt.

- 19 -
213 Kontinuierliche Verfahren
Kontinuierlich arbeitende Extraktionsverfahren sind alle jene, bei welchen
während des ganzen Extraktionsvorganges frisches Menstruum zufliesst und mit
Extraktivstoffen beladene Extraktflüssigkeit abgeführt wird.
Vorteile gegenüber den diskontinuierlichen, mehrstufigen Verfahren:
Das Arbeiten ist sehr rationell dank dem optimalen Konzentrationsgefälle zwi¬
schen der intra- und der extrazellulären Flüssigkeit.
Der Menstruumverbrauch ist daher geringer und die Extraktlösung konzentrier¬
ter.
Der Aufwand an Energie und Kontrollen ist nicht grösser als bei allen diskon¬
tinuierlichen Methoden.
Extraktionsrückstand und Extraktlösung werden im kontinuierlichen Verfahren
meist automatisch getrennt.
Nachteile gegenüber den diskontinuierlichen, mehrstufigen Verfahren:
Extraktionsverlauf und Ausbeute sind stark von der Korngrösse des Extrak¬
tionsgutes abhängig.
Die vorbereitenden Arbeiten sind etwas grösser.
213.1 Die Perkolation
Die Perkolation ist eine kontinuierliche Extraktion bei gewöhnlicher Tempera¬
tur. Nach angemessener Mazerationszeit im Perkolator durchmesst das Lösungs¬
mittel langsam, in einmaligem Durchgang das Extraktionsgut.17)
Die Ph.Helv.V gibt keine eigentliche Definition, sondern eine Beschreibung
des Verfahrens.
Die Perkolationsmethode zeichnet sich aus durch:
Kontinuität
Sehr geringen Energie- und bescheidenen Arbeitsaufwand
Konzentrierte Vorläufe bei langsamem Abfluss
Geringe notwendige UeberwachungKeine Erwärmung des Drogenmaterials
Nachteilig sind hingegen:
Der noch bedeutende Zeitaufwand
Die Beschränkung auf bestimmte Zerkleinerungsgrade des Extraktionsgutes.
213.2 Die Diakolation
1 o\
Die Diakolation ist eine Abwandlung der Perkolation, bei welcher die Dro¬
gensäule je nach Extraktionsgut bis auf mehrere Meter verlängert wird. Das Men¬
struum treibt man mittels Ueberdruck (bis 1,5 atü) durch die in Röhren abgefüllte
Droge. Die in der Drogensäule zurückgehaltene Extraktlösung wird durch Wasser
verdrängt, so dass der Menstruumverbrauch nur 100 - 150 % der Drogenmenge be¬
trägt.

- 20 -
Vorteile gegenüber der Perkolation sind:
Geringer Menstruum-Verbrauch
Wegfall des Abpressens der Droge dank Verdrängung der in ihr zurückgehal¬tenen Extraktlösung mit Wasser
Konzentrierte Extrakte, die nicht oder nur wenig eingeengt werden müssen
Ersatz der Mazeration nach dem Füllen des Diakolators durch entsprechendlangsameren Zufluss
Nachteilig wirken sich aus:
sehr langsamer Menstruumdurchfluss (z. B. ca. 1, 5 Tropfen pro Minute für
Chinarinde)Grosse Gefahr der Verstopfung des Diakolators
Bedeutende Mehrarbeit bei der Montage der Apparatur
Ausser den hier erwähnten Punkten gilt dieselbe Arbeitsvorschrift wie für die
Perkolation.
213.3 Die Evakolation
19)Auch die Evakolation nach Kessler 'ist eine Abwandlung der Perkolation.
Droge und Auffanggefäss werden dabei vorerst evakuiert. Daraufhin treibt man die
vorgeschriebene Menge Menstruum mittels Atmosphärendurck langsam durch das
Extraktionsgut. Die in der Drogensäule zurückgehaltene Extraktlösung wird durch
Wasser verdrängt.
Die vorbereitenden Arbeiten entsprechen denjenigen einer Perkolation. Die
Vorzüge sind dieselben wie bei der Diakolation.
Nachteile sind:
Der sehr langsame Menstruumdurchfluss (5 Tropfen/Minute)Eine gewisse Mehrarbeit bei der Montage der Apparatur.
20)Ein Vorgänger der Evakolation war die Mulkolation ', welche sich nur durch
die Verwendung eines Diakolator-Röhrensystems von der Evakolation unterscheidet.
213.4 Die Soxhlet-Extraktion
Die Soxhlet-Extraktion ist eine kontinuierliche Extraktion fester Stoffe, bei
der das Lösungsmittel kontinuierlich aus dem Menstruum- und zugleich Extraktsam-
melgefäss destilliert, über der Droge kondensiert und erneut zu deren Extraktion
verwendet wird. Das mit Extraktivstoffen beladene Menstruum fliesst zurück ins
Sammelgefäss.
Das Verfahren hat folgende Vorteile:
Sehr geringer LösungsmittelbedarfHohe Extraktkonzentrationen sind möglichEin optimales Konzentrationsgefälle von der intra- zur extrazellulären Flüs¬
sigkeit im DrogenmaterialUeberwachung ist kaum notwendig

- 21 -
aber auch Nachteile wie:
Erwärmung des Extraktionsgutes bis zum Siedepunkt des Losungsmittels.Ausschluss von Losungsmittelgemischen mit stark verschiedenen Siedepunk¬ten der Komponenten.
213.5 Industrielle Verfahren
Dank der schon zu Beginn dieses Abschnittes erwähnten, grossen Vorteile der
kontinuierlichen Extraktionsmethoden sind diese Verfahren heute auch in der Indu¬
strie weit verbreitet. Es sind vor allem die Perkolation und, sowohl fur festes wie
fur flussiges Extraktionsgut verwendete Verfahren, die nach dem Gegenstromprin¬
zip arbeiten.
Einige wichtige Apparate sind unter 312. 5 (s. pg. 26) beschrieben.
22 Neuere, mit Losungsmitteln arbeitende Extraktionsmethoden
221 Diskontinuierliche, einstufige Verfahren
221.1 Ultraschall-Extraktion
Die Ultraschall-Extraktion ist eine einmalige Extraktion pulverformiger oder
flussiger Stoffe mit Hilfe von Losungsmitteln, bei der ein durch Ultraschall erreg¬
ter Schwingungsubertrager im Extraktionsgemisch fur intensive Durchwirbelung21)
wahrend der ganzen Dauer des Ausziehens sorgt .
Im übrigen gilt das schon unter 211.5, Anwendung von Schall, Gesagte.
221.2 Die Turbo- oder Wirbel-Extraktion
Die Turbo- oder Wirbel-Extraktion ist die Extraktion von Inhaltsstoffen aus
einem flussigen oder festen Extraktionsgut, z. B. einer Droge, mit Hilfe eines ge-
eigneten Losungsmittels unter intensiver Durchwirbelung mittels Ruhrgeraten.
Vorteile der Turbo-Extraktion:
Dank der intensiven Durchwirbelung des Extraktionsgutes wird fur einen ra¬
schen Ausgleich des Konzentrationsgefalles zwischen der intra- und der extra¬
zellularen Flüssigkeit gesorgt.
Durch die hohe mechanische Beanspruchung werden viele Zellen aufgerissen,so dass deren Inhaltsstoffe direkt, d.h. ohne Behinderung durch die Zellwand,ausgewaschen werden können.

- 22 -
Die Drogenteilchen erfahren, besonders bei höheren Tourenzahlen, plötzlichhohe Beschleunigungen. Es ist also bei Unterschieden im spezifischen Gewicht
zwischen Zellwand und flüssigem Zellinhalt durchaus denkbar, dass Flüssig¬keit entgegen den osmotischen Druckverhältnissen in Zellen hinein oder aus
Zellen herausgepresst wird.
Zudem wird durch die wiederholte Beschleunigung der Teilchen mit anschlies¬
sendem Aufprall auf den Statorzahnkranz ein intermittierender Druck auf die
Teilchen ausgeübt, was nach S te ig er 23) ebenfalls die Extraktion günstigbeeinflusst.
Das Extraktionsgemisch erwärmt sich bei der Turboextraktion. Die höhere
Temperatur kann zu höheren Ausbeuten führen.
Die vier erwähnten Möglichkeiten versprechen daher eine Beschleunigung des?2)
Extraktionsvorganges und eine höhere Wirkstoffausbeute als bei der Mazeration '.
Nachteile der Turboextraktion sind:
Der starke Anstieg der Temperatur im Extraktionsgemisch kann, wenn ohne
Kühlung gearbeitet wird24)( zum teilweisen Verlust von flüchtigen oder ther-
molabilen Wirkstoffen und von Menstruum Anlass geben.
Die weitgehende Zerkleinerung des extrahierten Stoffes erschwert dessen
Trennung von der Extraktlösung9).
221.3 Das Zentrifugieren
Das Zentrifugieren ist vorwiegend eine Trennungsmethode spezifisch ungleich
schwerer Stoffe durch Schleudern des Gemisches auf einer Kreisbahn. Unterschiede
in den spezifischen Gewichten von intra- und extrazellulärer Flüssigkeit und Zell¬
wand in Extraktionsmischungen können daher im starken künstlichen Schwerefeld zu
einer beschleunigten Diffusion und somit besserer Extraktausbeute führen.
222 Diskontinuierliche, mehrstufige Verfahren
222.1 Die Returboextraktion oder Diturboextraktion
Die Diturboextraktion ist eine mehrstufige Wirbelextraktion. In jeder Stufe
wird die gesamte Menge des Extraktionsgutes mit einem Teil der gesamten Men-
struum-Menge gewirbelt, anschliessend die Extraktlösung vom Rückstand getrennt
und zur Extraktausbeute der ersten Stufe gegeben.
Vorteil der Diturboextraktion:
Die im extrahierten Stoff der ersten Stufe zurückgehaltene Extraktlösung kann
noch grösstenteils gewonnen werden.
Der Arbeits- und Zeitaufwand ist, wenn auch ca. doppelt so gross wie bei ge¬
wöhnlicher Wirbelextraktion, immer noch sehr bescheiden im Vergleich zu
andern Extraktionsmethoden.
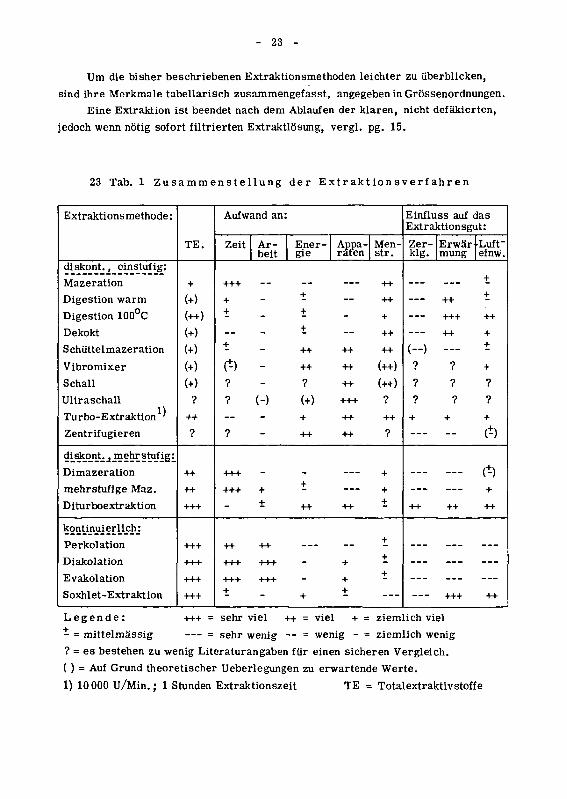
- 23 -
Um die bisher beschriebenen Extraktionsmethoden leichter zu überblicken,
sind ihre Merkmale tabellarisch zusammengefasst, angegeben in Grössenordnungen.
Eine Extraktion ist beendet nach dem Ablaufen der klaren, nicht defäkierten,
jedoch wenn nötig sofort filtrierten Extraktlösung, vergl. pg. 15.
23 Tab. 1 Zusammenstellung der Extraktionsverfahren
Extraktionsméthode :
TE.
Aufwand an: Einfluss auf das
Extraktionsgut:
Zeit Ar¬beit
Ener¬
gieAppa¬raten
Men-str.
Zer-
klg.Erwär
mung
Luft"einw.
diskont., einstufig:
Mazeration + +++ — — — ++ — —
+
Digestion warm (+) + -
+— ++ — ++
+
Digestion 100°C (++)+
-
+- + — +++ ++
Dekokt (+) — -
+~ ++ ... ++ +
Schüttelmazeration (+)+
- ++ ++ ++ (--) —
+
Vibromixer (+) (+-) - ++ ++ (++) ? ? +
Schall (+) ? - ? ++ (++) ? ? ?
Ultraschall ? ? (-) (+) +++ ? ? ? ?
Turbo-Extraktion '++ — - + ++ ++ + + +
Zentrifugieren ? ? - ++ ++ ? — — (*)
diskont.j mehrstufig:
Dimazeration ++ +++ - - — + ... — (+-)
mehrstufige Maz. ++ +++ ++
— + ... ... +
Diturboextraktion +++ -±
++ +++
++ ++ ++
kontinuierlich:
Perkolation +++ ++ ++ — —
+... ... —
Diakolation +++ +++ +++ - ++— ... —
Evakolation +++ +++ +++ - ++
... ... —
Soxhlet-Extraktion ++++
- ++
... +++ ++
Legende: +++ = sehr viel ++ = viel + = ziemlich viel
- = mittelmässig = sehr wenig -- = wenig - = ziemlich wenig
? = es bestehen zu wenig Literaturan gaben für einen sicheren Vergleich.
( ) = Auf Grund theoretischer Ueberlegungen zu erwartende Werte.
1) 10 000 U/Min.; 1 Stunden Extraktionszeit TE = Totalextraktivstoffe

- 24 -
3 EXTRAKTIONSAPPARATE
31 Die bisher gebräuchlichen, mit Lösungsmitteln
arbeitenden Extraktionsapparate
311 Apparate für diskontinuierliche Extraktion
311.1 Die Mazeration
Die Mazeration ist wohl die einfachste und anspruchsloseste aller Extraktions¬
methoden, indem sie in jedem gut verschliessbaren Gefäss aus inertem Material
(z. B. Glas, Porzellan, Steingut oder Spezialmetalle) ausgeführt werden kann.
Darum wird wohl diesem Verfahren, trotz seiner Nachteile, besonders im
9)Kleinbetrieb noch heute oft der Vorzug gegeben.
311.2 Die Digestion, Dekoktion und Infusion
Diese drei Verfahren benötigen zusätzlich eine Heizung in Form eines Wärme¬
bades (z. B. die altbekannte Infundierbüchse) und, bei hohen Temperaturen oder leicht
flüchtigen Lösungsmitteln, einen Rückflusskühler auf dem Extraktionsgefäss.
311. 3 Die Schüttelmazeration
Eine automatische Schütteleinrichtung ist hier fast unumgänglich. Im übrigen
ist das Verfahren genau so anspruchslos wie die Mazeration.
311.4 Die Extraktion mittels Vibromixer
Der Vibromixer ist ein Mischgerät, das mit Hilfe eines mit Wechselstromfre-
quenz gespiesenen Elektromagneten einen vertikalen Stab zu Schwingungen in seiner
Längsachse anregt. Der eigentliche Rührkopf ist meist eine gelochte Platte, die
senkrecht zum vertikalen Stab montiert ist. Der Stab mit Rührkopf ist auswechsel¬
bar.
311.5 Die Anwendung von Schall
Für die Beschallung während einer Extraktion dient am einfachsten ein elektri-14)
sches Horn. So verwendeten Schultz u. Klotz ' ein Bosch-Horn. Andere Schall¬
quellen von befriedigender Leistung, z. B. ein Tongenerator, erfüllen den gleichen
Zweck.

- 25 -
311.6 Die Apparate der Industrie
Für diskontinuierliche Extraktion im Grossbetrieb wird wohl am häufigsten
mazeriert oder digeriert. Es dienen dazu entweder feststehende, bei Bedarf heiz-
oder kühlbare Tanks mit Rührwerk oder Tanks, die sich um eine oft schräg durch
diesen verlaufende Achse drehen und deren Innenwand zur intensiven Vermengung25)
des Extraktionsgemisches mit Lamellen versehen ist '.
312 Apparate für kontinuierliche Extraktion
312.1 Die Perkolation
Der Perkolator ist ein zylindrisches oder sich nach unten leicht verengendes
Glasrohr aus inertem Material, das unten mit einer Einrichtung zur Regulierung
des Abflusses versehen werden kann. Der Perkolator muss nach Vorschrift der Ph.
Helv. V aus Glas, Porzellan, Steingut oder versilbertem Metall bestehen.
Eine Abwandlung ist der Dünnschichtperkolator nach Köhler,eine verti¬
kal stehende Metallplatte mit den Zu- und Abflussleitungen, die, nach dazwischen
legen einer Dichtung, mit einer dicken Glasplatte verschraubt wird. Zwischen die
beiden Platten füllt man das Extraktionsgut.
312.2 Die Diakolation
Der Diakolator nach Br eddin'besteht aus mehreren, senkrecht in einem
Gestell montierten Röhren, welche in Serie geschaltet sind. Ihr innerer Durchmes¬
ser ist 1, 7 cm, ihre Länge je 80 cm. Am Ende des Systems ist ein offenes Abfluss-
röhrchen befestigt. Der Menstruumzufluss wird reguliert. Er erfolgt unter einem
Druck bis 1, 5 atü.
312.3 Die Evakolation
19}
Beim Evakolator nach Kessler ' dient zur Drogenaufnahme ein langes,
ziemlich enges Glasrohr, welches oben luftdicht und verschliessbar ans Menstruum-
gefäss angeschlossen und unten gleicherweise mit dem Auffanggefäss verbunden
wird.
312.4 Die Soxhlet-Extraktion
Der Soxhletapparat besteht aus einem heizbaren Extraktauffang- und Menstruum-

- 26 -
gefäss, welches luftdicht an ein Extraktionsgefäss angeschlossen wird. Ueber die¬
sem Gefäss befindet sich ein Rückflusskühler. Die Extraktlösung fliesst durch eine
enge Leitung nach dem Heberprinzip vom Extraktionsgut wieder zum Menstruum
hinunter. Das Menstruum wird dort verdampft. Der Dampf steigt zum Kühler empor,
kondensiert dort und fällt wieder auf das Extraktionsgut.
312.5 Apparate der Industrie
Der Perkolator:
Der Perkolator mit entsprechend grossem Fassungsvermögen wird auch in
der Industrie oft verwendet.
Der Hildebrandt-Extraktor ':
Einer der wichtigsten Extraktionsapparate der Industrie ist der Hildebrandt-
Extraktor. Er besteht aus einem U-Rohr mit Förderschnecken, die das Extraktions¬
gut durch das Röhrensystem transportieren, welches vom Menstruum im Gegen¬
strom durchflössen wird.
Der Boll mann-Extraktor ':
Der Boll mann-Extraktor ist ein langsam laufendes, vertikal stehendes Be¬
cherwerk. Die aufsteigenden Drogen-Behälter mit vorextrahierter Droge werden im
Gegenstrom mit frischem Lösungsmittel extrahiert. Der unten gesammelte Auszug
dient zur Vorextraktion der Frischdroge in den absteigenden Behältern nach Entlee¬
rung und Neufüllung der Behälter im oberen Umkehrpunkt. Die Vorextraktion der
Droge geschieht also im Gleichstrom und liefert die Extraktlösung zur weiteren Auf¬
arbeitung.
32 Neuere Extraktionsapparate
Apparate für diskontinuierliche Extraktion
321 Die Ultraschall-Extraktion
Die Erzeugung der Hochfrequenzspannungen erfolgt heute fast ausschliesslich
mittels Elektronenröhren, meist in einer einzigen Röhre in Selbsterregerschaltung '.
Die Umwandlung der Hochfrequenzspannung, die von einigen 100 V bis zu einigen
10 000 V betragen kann, in mechanische Energie erfolgt entweder magnetostriktiv

- 27 -
21)oder piezoelektrisch; vergl. Baud: "Die Anwendung des Ultraschalles"
Im ersten Falle schwingt ein Nickelstab, dessen Länge auf die Frequenz abge¬
stimmt wurde, als Spulenkern im Hochfrequenz-Feld. Dieses System findet für Fre¬
quenzen bis ca. 200 000 Hz Anwendung.
Bei der piezoelektrischen Umwandlung wird die Hochfrequenzspannung an eine
Quarzplatte von einer auf die Frequenz abgestimmten Dicke angelegt. Das Medium,
in welchem der Quarz schwingt, z.B. Luft oder irgend eine nicht leitende Flüssig¬
keit wie Paraffinöl, ist von ausschlaggebender Bedeutung. Auf piezoelektrischem
Wege können Frequenzen von ca. 200 000 Hz bis 100 MHz erzeugt werden. Die opti¬
male Ultraschall-Frequenz muss von Fall zu Fall ausprobiert werden.
322 Die Turbo-Extraktion
Für die Turbo-Extraktion kommt ein Gerät in Betracht, dessen Tourenzahl
mindestens ca. 3000 Umdrehungen pro Minute beträgt und dessen Rührkopf so kon¬
struiert ist, dass im zu bearbeitenden Material neben dem Mischeffekt eine möglichst
weitgehende Teilchenzerkleinerung stattfindet.
Die handelsüblichen Geräte findet man hauptsächlich in zwei Ausführungen:
a) Der Motor ist durch eine Welle mit dem Rührkopf verbunden, der in das zu
bearbeitende Gemisch eingetaucht wird; so die Geräte: POLYTRON.%,HO-
MOREX2>, HOMOMIXERgv, KOTTHOFF MISCHSIRENE^.i;
b) Das Gefäss mit dem zu bearbeitenden Gemisch enthält unten den Rührkopfoder die Messer und wird direkt auf dem Sockel des Gerätes, der den Mo¬
tor enthält, befestigt. Schon im Jahre 1937 wurde ein solches Gerät mit 3
Liter Fassungsvermögen und einer Motorleistung von 0,5 PS beschrieben,-^").Neuere Geräte dieser Bauart sind: TURMIX».
,CUISTO„> etc. '
1) Hersteller: Max Wullimann, Maschinen- und Apparatebau, Selzach,SO/Vertretung: Mobil Aarau AG., Hallwylstrasse 11, Aarau.
2) Hersteller & Vertrieb: Alfred Brogle & Co., Elisabethenstrasse 44, Basel 10.3) Hersteller; Eppenbach Inc., Long Island City, N.Y. (USA); Vertretung wie 2).4) Hersteller: Hans Kotthoff, Weisser Strasse 74, Köln-Rodenkirchen.5) Hersteller: Kühnle, Kopp & Rausch AG., Frankenthal, Pfalz.6) Hersteller: Techag AG., Zürich.
7) Hersteller: Ed. AerneS.A. (300 W), Zürich.

- 28 -
323 Die Zentrifugen
Alle diskontinuierlich arbeitenden Zentrifugen und evtl. auch Durchlaufzentri¬
fugen kommen für diese Extraktions- -und Trennungsmethode in Frage. Dieses Ver¬
fahren soll vor allem für die Extraktion der Antibiotica benützt werden. Dass eine
Beschleunigung der Extraktion eintritt, geht später aus unseren Versuchen hervor.

- 29 -
4 ARBEITEN UEBER DIE DROGENEXTRAKTION MIT BESONDERER
BERUECKSICHTIGUNG DER CHINARINDE
41 Untersuchungen betreffend das Menstruum
Da diese Untersuchungen für alle in der Folge aufgeführten Extraktionsmetho¬
den Bedeutung besitzen, werden sie hier vorweg genommen. Es wird zuerst die Quel¬
lung und der Einfluss von Alkoholkonzentration und Säurezusatz sowie anschliessend
der Effekt anderer Zusätze zum Menstruum auf die Extraktion besprochen. Abschlies¬
send folgen noch einige neuere Arbeiten mit andern Lösungsmitteln.
Die Quellung der Zellwand ist eine unbedingte Voraussetzung für eine
gute Extraktion der Inhaltsstoffe aus intakten Zellen. Dies betonte schon Feinstein4-6)
im Jahre 1936, wobei er sich auf Vorarbeiten von Hu sa und Mitarbeitern ' stüt¬
zen konnte. Die Autoren überprüften die Quellung von Kastanienholz und Belladonna¬
wurzel in Alkohol, Glycerin und Wasser '. Sie konnten in Alkohol-Wasser-Mischun¬
gen eine Abnahme der Quellung von Belladonnawurzel mit steigender Alkoholkonzentra-
5) 4)tion feststellen '. Husa und Jones beobachteten im Jahre 1937 bei Chinarinde
rascheste und stärkste Quellung in Wasser mit 24 Vol. % Alkohol. Versuche von
2)
Lang 'im Jahre 1950 zeigten, dass das Quellungsvermögen der Droge mit steigen-4)
der Alkoholkonzentration abnimmt, was schon Husa und Jones ' beobachteten.
Anderseits nahm das Lösungsvermögen des Menstruums für adsorptiv gebundene
Stoffe mit steigender Alkoholkonzentration zu. Unter anderem wurden Modellversuche2)
mit Alkaloid-Tanniden ausgeführt. Lang' schliesst daraus auf beste Extraktions¬
ausbeuten im Schnittpunkt dieser beiden Kurven, d.h. bei ca. 50 Vol.-% Alkohol.
301Auch Thomson weist auf gute Alkaloidausbeuten mit 44 bis 45-proz. Al-
31)
kohol hin. Thörn ' erzielte beste Ausbeuten aus Ephedrablatt mit Spir. 70 Vol.-%.
Für die Extraktion der Solanaceen-Alkaloide Hyoscyamin und Atropin bewährte sich
nach Märki Alkohol von 70 Vol.-% mit Zusatz von 1 % Ameisen- oder Apfelsäu¬
re am besten. Die Ph.Helv. V schreibt für die Herstellung von Chinarinden-Trocken¬
extrakt eine Alkoholkonzentration von ca. 43 Gew. -% vor (46 Gew. -% des
33)Menstruums Alkohol à 92,5 Gew. -%). Eine Untersuchung von Ch. Béguin ergab,
dass diese Alkoholkonzentration sehr nahe dem Optimum liegt. Mit 60 - 70 Gew. -%
Alkohol wurden mittels Perkolation alkaloidreiche Vorläufe, dafür alkaloidärmere
Nachläufe gewonnen. Ameisensäurezusatz erhöhte die Alkaloidausbeute, wo-
34)bei ein Zusatz von 1,0 - 1,25 % optimal war. Nach Schrader wirkt sich
42 Gew. -%-iger Alkohol günstig auf die Extraktion aus, da dieser die höchste Vis-35)
kosität besitzt. Belcot und Rapeanu'schlugen für Chinarinden-Trockenex¬
trakt ebenfalls Menstruum und Methode der Ph.Helv. V vor, für Chinarinden-Fluid-
extrakt ein ebenfalls saures, jedoch glycerinhaltiges Menstruum höherer Alkoholkon¬
zentration nach USP X vor. Fuch s' hat 1941 den Einfluss der Zusammensetzung

- 30 -
des Menstruums auf die Extraktion der Chinarinde überprüft. Sie fand für angesäu¬
erte Alkohol-Wasser-Mischungen beste Alkaloid- und Totalextraktivstoff-Ausbeuten
bei 60 Gew. -% Alkohol mit Zusatz von 2 Mol Milchsäure. Ein grösserer Säurezusatz
erhöhte die Alkaloidausbeuten nur unwesentlich. Das Ph.Helv.V-Menstruum extra¬
hierte die Alkaloide schlechter, jedoch mit hohem Gerbstoffanteil im Extrakt. Die
mit diesen beiden Menstrua nach Ph.Helv.V hergestellten Trockenextrakte zeigten
aber durchaus vergleichbare Wirkstoffgehalte, da bei der Ballaststoff-Beseitigung,
zur Erzielung eines in möglichst verdünntem Alkohol löslichen Trockenextraktes,
die ganze Alkaloid-Mehrausbeute mit dem erstgenannten Menstruum wieder verlo-37)
ren geht. Schill '
berichtet, dass das mit 42 Vol. -% Alkohol bei Zusatz von 1 %
Ameisensäure erreichte pH nicht tief genug sei, um die Alkaloid-Gerbstoff-Verbin-
dungen der Chinarinde zu löslichen Verbindungen umzusetzen, was nach Fuchs '
nur bedingt richtig ist. Herzog' erzielte bei Zusatz von 2 % Milchsäure die be¬
sten Alkaloidausbeuten mit 60 Vol. -% Alkohol, die besten Totalextraktivstoff-Aus¬
beuten mit 45 Vol. -% Alkohol in 5 - 6 Monate gelagerten Tinkturen. Der Verfasser
schlägt daher für die Herstellung von Chinarindentinktur DAB-7 60 Vol. -% Alkohol39)
vor '.
Andere Zusätze zum Menstruum zwecks Verbesserung der Extraktions¬
ausbeute sind verschiedentlich ausprobiert worden. Husa und Magid 'fanden
bei Zugabe von Glycerin zum Menstruum eine Verzögerung der Alkaloidextraktion
aus Belladonnawurzel-Pulver proportional zur Glycerinkonzentration. Fuchs '
erhielt mit ammoniakalischem Menstruum sowohl geringere Alkaloid- als auch tie¬
fere Totalextraktivstoff-Ausbeuten verglichen mit den sauren Menstrua. Meyer'
ist ein Verfahren patentiert worden, wonach durch Zugabe biochemischer Salze
oder von deren Mischungen zum Menstruum eine Wirkungserhöhung und geschmack-42)
liehe Verbesserung von Kolazubereitungen erreicht wird. Butler u. Wiese '
konnten mit 20 mg-% Sorbitan-Monolaurat bezogen auf das fertige Fluidextrakt die
Wirkstoffausbeute bei Chinarinde um 1,4 - 1,7 % steigern. Bro chmann-Han s -
43)sen erreichte bei wässriger Extraktion von Chinarinde mit 0,5 % kationenakti¬
ven Netzmitteln bessere Alkaloidausbeuten als ohne Netzmittel. Alkoholische Men¬
strua mit nichtionogenen Netzmitteln ergaben keine günstigeren Resultate.
Abschliessend seien nun noch einige Extrakt ionsver suche mit an-
44)dern Menstrua erwähnt. Märki verwendete u.a. für Chinarinde Isopropa¬
nol und erhielt bei gleicher Ausbeute einen etwas weniger hygroskopischen Trocken¬
extrakt als mit Aethanol. Der Unterschied ist jedoch zu gering, als dass Isopropanol
praktische oder ökonomische Vorteile gegenüber Aethanol bieten würde. Du com -
45)mun gelang die völlige Erschöpfung von Chinarinde mit 4 Teilperkolaten einer
Mischung von (Isopropanol 30 T + Aceton 30 T + Wasser 36 T + Ameisensäure 4 T)

- 31 -
gegenüber 6 Teilperkolaten des Ph.Helv.V-Menstruums. Applezweig und Mitar-4g\
beiter ' beschreiben Apparaturen zur Perkolation von Chinarinde mit 0, ln-Schwe-
felsäure, in welchen der Auszug anschliessend einen sulfonierten Kohle-Kationen-47)
austauscher "ZEOCARB" passiert. Pinxteren ' weist darauf hin, dass neutrale
wässerige Menstrua vorwiegend rechtsdrehende, salzsaure Menstrua vorwiegend48)
linksdrehende Chinaalkaloide erfassen. Campo u. Gramling perkolierten
Chinarinde erfolgreich mit Tetrahydrofuran bei 10 - 15 % Wasserzusatz. Ein azeo-
tropes Gemisch Tetrahydrofuran-Wasser (5, 3 % Wasser) löst Chinin und Cinchonin
selektiv.
42 Grundl agenf or schung betreffend einzelne Extraktionsmethoden
Es wird hier nur noch auf die am häufigsten verwendeten Methoden eingegangen.
421 Diskontinuierliche Verfahren
421.1 Die Mazeration
Der Zerkleinerungsgrad der Droge:49)
Bohrisch u. Kürschner ' fanden an 12 Drogen, dass sich sowohl ge¬
schnittenes als auch gepulvertes Material für die Mazeration gleich gut eignet, aller¬
dings bei einer Mazerationszeit von 8 Tagen. Chinarinde ist nicht untersucht worden.
Vakuum:
Der Einfluss eines Vakuums auf die Extraktionsausbeute ist von Hu sa und
50)Jones untersucht worden. Es gelang aber bei Calisaya-Rinde und bei Belladon¬
nawurzel nicht, die Ausbeute mit dieser Methode entscheidend zu verbessern.
Temperatur:
Den Temperatureinfluss auf die Wirkstoffextraktion aus Chinarinde hat Gr aet-
zer untersucht. Er fand bei einer Temperaturhöhung von 20 C auf 60°C in den
Mazeraten statt 77,1 % 95, 7 % des Totalalkaloidgehaltes der Droge; die Extraktiv¬
stoffausbeute stieg von 86,1 % auf 104, 8 %.
Zeit:eil
Marschak mazerierte 8 Drogen bis zum erreichten Gleichgewichtszu¬
stand im Extraktionsgemisch. Es wurde 1 T Chinarinde (Sieb: 0,75 mm Maschen-

- 32 -
weite) mit 5 T Alkohol 68 - 69 Vol. -% angesetzt. Er fand folgende Mazerationszeiten:
6-7 Tage für Chinatinktur
3 bis max. 4-5 Tage für alle andern untersuchten Tinkturen.
Jermstad u. Oestby mazerierten 5 Drogen vom Feinheitsgrad Sieb V nach
Ph.Helv. V während 3, 8 und 10 Tagen und fanden 8-10 Tage als beste Mazerations¬
zeit. Angesetzt wurden 100 T Droge mit 1000 T Alkohol 70 Vol. -% mit Zusatz von
0,5 oder 1,9% Salzsäure. Der Sprung von nur 3 auf 8 Tage ist so gross, dass aus
dieser Untersuchung die kürzeste, mögliche Mazerationszeit nicht hervorgeht. To -
53)
mi c u. Kosak 'schlagen für die zweite Ausgabe der jugoslavischen Pharmaco-
9)poe eine Mazerationszeit von 6 Tagen vor. Nach Bogs genügt erfahrungsgemäss
eine Mazerationszeit von 5 bis höchstens 8 Tagen.
Eine mathematische, formelmässige Beschreibung des Mazerationsvorganges
gaben 1953 Schultz u. Klotz54\
421.2 Die Ultraschall-Extraktion
Mit der starken Entwicklung der Technik in den letzten Jahren entstanden auch
neue Apparate, und es fehlte nicht an Versuchen, diese für Extraktionszwecke nutz¬
bar zu machen.
Meyer' beobachtete in je 2 ml verschiedener Flüssigkeiten bei einer Ultra¬
schallbehandlung von konstanter Schallintensität während 10 Sekunden folgende Tem¬
peraturanstiege:
Gelatine-Gel 1 °C Wasser 2 °CGelatine-Lösung 1 °C Alkohol 3,5 °CStearinsäure 36 C Glycerin 10 C
Paraffinöl 10 °C Wachs 44 °C
Ferner wurden in den Zellen der Wasserpest bei Ultraschall-Behandlung eine inten¬
sive Wirbelbildung und zuletzt eine Ablösung des Protoplasmas bzw. des Chloro-
plasts von den Zellwänden und eine Zusammenballung an andern Stellen festgestellt.
Längere Pflanzenzellen und Algen wurden zerrissen.
Durch Ultraschall-Behandlung von Hopfen in wässerigem Milieu bei 50 C an
Stelle einer Extraktion mit siedendem Wasser ohne Beschallung verbesserte
Specht 'die Bitterstoffausbeute aus Hopfen bei gleichzeitig geringerer Gerbstoff -
ausbeute. Die Ausbeute ist eine Funktion der Beschallungsdauer und des Verhältnis¬
ses Hopfen zu Flüssigkeitsmenge.
Weitere Literaturangaben befinden sich sowohl unter 2 Extraktionsmethoden,
als auch im Abschnitt 432, "Vergleiche mit neuen Methoden".

- 33 -
421.3 Die Turbo-Extraktion
57)Schon im Jahre 1948 extrahierten Bay u. Gisvold frische Digitalisblat-
ter mit einem hochtourigen Mischgerat innert 10 Minuten und erhielten Extrakte, die
mit denen der USP vergleichbar waren .
59)
D ean und Mitarbeiter 'stellten aus Tollkraut- und Stechapfelblatt Sieb 40
mit Hilfe einer Kolloidmuhle bei 3600 U/Minute die entsprechenden USP- und NF-
Tinkturen her.22)
1953 veröffentlichte Melichar 'eine erste grundlegende Arbeit über die
Turbo-Extraktion. Der Einfluss von Extraktionsdauer, Diffusionsoberflache und
Temperatur auf Trockenruckstand, Alkaloidgehalt, Dichte und Brechungsindex von
Enzian-, Kalmus-, Tollkraut- und Chinarinden-Tinktur wurde beschrieben. Eine
24)neuere Arbeit vom gleichen Autor behandelt die Warmecharakteristik der Wirbel-
extraküon in Theorie und Praxis. Er verwendet einen TURMK-Mischer mit 8000
bis 13000 U/Minute. Bei 7-10 Minuten Extraktionszeit stieg die Temperatur in
500 g des reinen Menstruums (Alkohol, Wasser oder Mischungen davon):
bis zu einem Grenzwert von 50 C bei Stufe 1/2bis zu einem Grenzwert von 65 C bei Stufe 1/1und bei Zugabe von 50 - 75 g Wermut:
bis zu einem Grenzwert von 77°C bei Stufe 1/1
Melichar beschreibt schliesslich Vorrichtungen fur isotherme Wirbeiextraktion.
Er benutzt einen TURMIX-Mischer fur Temperaturen von +10 C bis +30 C, bei wel¬
chem ein Kuhlrohrsystem von oben in das Extraktionsgemisch eingetaucht wird. Die
Temperaturkonstanz soll - 0,2 C sein.
Fur anspruchsvollere Arbeiten wird eine speziell konstruierte Apparatur mit
Doppelmantel-Extraktionsgefass und thermostatisch regulierter Kuhlmantel-Tempe¬
ratur beschrieben. Die Temperaturen des Extraktionsgemisches schwanken von 0 C
bis zum Siedepunkt des Extraktionsmittels. Die Reguliergenauigkeit ist - 0,05 C bis
±0,1°C.
422 Diskontinuierliche, mehrstufige Verfahren
422.1 Die mehrstufige Mazeration
Die belgische Pharmacopoe' versteht unter Mazeration eine Dimazeration,
wobei die Droge erst 24 Stunden mazeriert, dann ausgepresst und anschliessend
nochmals 12 Stunden gleich behandelt wird.
Auch nach der russischen Pharmacopoe 1958 'wird die Droge erst 6-12
Stunden mazeriert und nach dem Abpressen nochmals 4-6 Stunden mazeriert.

- 34 -
Das danische Arzneibuch'schreibt fur die Tinkturen-Herstellung eine Ma¬
zeration wahrend 5 Tagen vor. Andere Präparate, z.B. Extrakte, werden durch Di-
mazeration oder Perkolation gewonnen.64)
1958 benutzte Melichar ' die isotherme Wirbeiextraktion zur Ueberprufung
der zwei- und mehrstufigen Mazeration. Die fur beste Ausbeute notwendige Auftei¬
lung des Menstruums auf die verschiedenen Stufen wurde berechnet. Der experimen¬
telle Nachweis ergab, dass die Ausbeuten der Dimazeration nur mit einer statisti¬
schen Sicherheit von 50 - 80 % vom Aufteilungsverhaltnis des Extraktionsmittels
abhangig sind.
Es wurde gezeigt, dass die einstufige Mazeration mit 99 - 99, 9 % statistischer
Sicherheit niedrigere Ausbeuten als die zweistufige Mazeration ergibt.
423 Kontinuierliche Verfahren
423.1 Die Perkolation
Die folgende Zusammenstellung umfasst Arbeiten betreffend den Zerkleinerungs-
grad der Droge oder einzelne Phasen der Perkolation. Die umfassenderen Arbeiten
sind am Schluss dieses Abschnittes m einer Tabelle zusammengestellt.
Zerkleinerungsgrad:65)
Nach Graetzer eignen sich Chinarindenpulver von der Korngrosse Sieb
IV, IVa oder V nach Ph. Helv. V am besten fur die Perkolation. Auch Herzog'
schlagt Chinarinde grob gepulvert nach DAB 6 vor.
Quellung:
Die Quellung und Druckerzeugung im Perkolator als Funktion der Flussigkeits-
menge untersuchten Husa u. Magid an verschiedenen Drogen. Mitzunehmen¬
der Flussigkeitsmenge nimmt die Quellung zu und die Druckerzeugung im Perkolator
ab, auch wenn dieser sofort nach der Befeuchtung des Drogenmaterials gefüllt wird.
Chinarinde erzeugte so den höchsten Druck bei den untersuchten Drogen.
Mazeration der Droge vor und nach dem Einfüllen in der Perkolator:
Dieses Problem wurde durch Husa und Huyck 'untersucht. Sie fanden
bei Cahsaya-Rinde, dass die Mazeration der Droge vor dem Einfüllen in den Perko¬
lator ohne Einfluss auf die Alkaloidausbeute war, eine Mazeration nach dem Einfül¬
len in der Perkolator diese jedoch verbesserte.
Muhlemann fand hingegen bei der Perkolation von Thymianblatt höhere

- 35 -
Ausbeuten an ätherischem Oel, wenn die Droge ohne Befeuchtung, also auch ohne
vorherige Mazeration in den Perkolator gefüllt wurde. Einfüllen der feuchten Droge
in den Perkolator verschlechterte die Ausbeute.
Perkolatorform :
69)
Mit der Perkolatorform befasst sich eingehend Feinstein '. Er fand nur
einen sehr geringen Einfluss der üblichen Perkolatorformen auf die Wirkstoffaus¬
beute. Mit zunehmender Drogensaulenhohe stieg die Wirkstoffkonzentration im er-
70)sten Teilperkolat und sank in den Nachlaufen, was Feinstein auf das langsa-
71) 72)
mereEinstromen des Menstruums zurückfuhrt. Auch Koch und Breddin'
betonten die intensivere Extraktion bei zunehmender Drogensaulenlange, jedoch glei-73)
eher Durchstromungs-Geschwindigkeit. Husa u. Huyck empfehlen fur Bella¬
donna-Wurzel den amerikanischen Perkolator und betonen, dass dieser ebenso ge¬
eignet sei wie ein Glasrohr von gleicher Hohe und gleichem Volumen.
Füllen des Perkolators:
Das Füllen des Perkolators mit feuchtem Drogenmatenal soll nach Husa u.
Huyck74', Feinstein75', G raetzer76'
und S chill77' in einem Male mit
möglichst wenig Druck erfolgen. Fur die Perkolation mehrerer Kilogramm Droge78)
hat Munzel vorgeschlagen, erst Menstruum in den Perkolator zu geben, dann
dann Droge sedimentieren zu lassen, so dass letztere immer von Menstruum be¬
deckt ist. Dies geschieht stufenweise, bis der Perkolator voll ist. Nach 12 Stunden
Mazeration wird abfliessen gelassen.
Oekonomische Perkolation:75)
Als ökonomische Perkolation schlagt Feinstein 'das Auffangen von 4 Teil-
79)
perkolaten vor. Buchi ' untersuchte die Alkaloidausbeute als Funktion der Anzahl
der aufgefangenen Teilperkolate und fand bei Chinarinde:
87 % der Alkaloide in den ersten 4 Teilperkolaten94 % der Alkaloide in den ersten 6 Teilperkolaten99 % der Alkaloide in den ersten 14 Teilperkolaten
Temperatureinflusse:
Den Temperatureinfluss bei der Perkolation untersuchte Graetzer '. Er
musste feststellen, dass bei höherer Temperatur der grosste Teil der Mehrausbeu¬
te durch stärkere Niederschlagsbildung bei der Defakation, bis auf eine kleine Zu¬
nahme der Totalextraktivstoffe, wieder verloren ging.
Eine mathematische Abhandlung über den gesamten Diffusions- und Auswasch-
prozess bei der Perkolation veröffentlichten S chu ltz und Klotz .

- 36 -
423.2 Die Diakolation
Die Arbeiten von Br eddin hat Feinstein eingehend referiert, so dass
diese hier weggelassen werden können. Feinstein musste seine Diakolationsver-
suche infolge des ausserordentlich langsamen Menstruumdurchflusses vorzeitig ab¬
brechen.
Graetzer '
perkolierte Chinarinde in einem schwach konischen Perkolator
bei 0 bis 3 atü und erreichte dadurch eine raschere Extraktion der Alkaloide und To¬
talextraktivstoffe. 3 atü ergaben aber in 5 Teilperkolaten und der Pressflüssigkeit
einzig eine um 4 % höhere Alkaloidausbeute gegenüber der Normaldruckperkolation.
Gramberg versuchte die Ausdrängung des Extraktes mit Vakuumhilfe zu
84)
beschleunigen, Keller ' diakolierte zweimal, einmal durch Vorextraktion stark
quellender Drogen mit dem Alkoholanteil des Menstruums und dann mit dem abge¬
sparten Wasser. Hu s a u. Mitarbeiter ' diakolierten Calisaya-Rinde mit 1 bis 5
Glasrohren in Serie, die durch ebenfalls mit Droge gefüllte U-Rohre gleichen Innen-
Durchmessers miteinander verbunden wurden. Die Wirksamkeit der Extraktion stieg
mit zunehmender Drogensäulenlänge. Mit 5 Röhren in Serie und soviel Menstruum,
dass 1 ml Perkolat einem Gramm Droge entsprach, wurden 58 % der Totalalkaloide
extrahiert.
Dietmann empfiehlt Abpressen der Droge an Stelle des Nachdrängens mit
Wasser. G rote 'empfiehlt Anfärbung des zur Ausdrängung verwendeten Wassers
mit zur Drogenfarbe gegensätzlichen Speisefarben.
Gstirner'bringt in seinem Buch eine Aufzählung der für Diakolation geeig¬
neten Drogen, unter anderem auch Chinarinde. Das Verfahren hat auch Eingang ins
89)National Formulary X gefunden, wobei vermerkt wird, dass die Methode jeder
Droge erst anzupassen sei.
423.3 Die Evakolation
90)
Auch für dieses Verfahren hat F ei n st e i n'die Literatur bis und mit dem
Jahre 1935 zusammengetragen. Er selbst konnte auch mit dieser Methode keine bes-
91)seren Ausbeuten erhalten als mittels gewöhnlicher Perkolation
.
Graetzer 'führte, z.T. gestützt auf die Arbeiten von H usa und Jones ,'
Vakuumperkolationen sowohl im gewöhnlichen Perkolator als auch im Evakolator
durch. Die Evakolation führte zu einer geringen, praktisch unbedeutenden Verbesse¬
rung der Extraktion.

- 37 -
423.4 Die Heissperkolation
Diese Methode wird in der F.Ital.VI93\ dem NF X89) und der Brit.Ph. 195894^
beschrieben. Das Menstruum ist dabei siedendes Wasser und der Alkohol wird erst
nach dem Einengen der Perkolate zugefügt.
423.5 Die fraktionierte Perkolation oder Reperkolation
93)Dieses Verfahren ist sowohl in der italienischen Pharmacopoe als auch im
QQ\ 95}
National Formulary X'und in der dänischen Pharmacopoe 1948 ' beschrieben.
Die Droge wird dabei in mehrere (in den erwähnten Vorschriften sind es drei) Teile
aufgeteilt, von denen jeder mit dem Nachlauf der vorhergehenden Perkolation befeuch¬
tet und perkoliert wird. Der gesamte Abfluss der letzten Perkolation ergibt nach Zu¬
mischung des Vorlaufes jeder vorausgehenden Teilperkolation die Extraktlösung.81)
Nach Feinstein ist dieses Verfahren für Chinarinde ungünstiger als Perkola¬
tion.
43 Vergleichende Arbeiten
431 Bisher gebräuchliche Methoden
431.1 Vergleich diskontinuierlicher, einstufiger Verfahren
Bari'verglich schon 1926 Chinatinkturen, welche nach 20 verschiedenen
Verfahren hergestellt wurden. Die durch 6 Tage lange Digestion bei 37 C mit
70-proz. Alkohol hergestellten Tinkturen waren sowohl frisch wie nach 12-monati-
ger Lagerung gehaltreicher bei besserer Ausnützung der Droge als solche, die durch
8-tägige Mazeration mit 50-proz. Alkohol gewonnen wurden. Auch in einer spä¬
teren Arbeit extrahierte der Autor durch 3-stündige Digestion unter Erwärmung
im Wasserbad am Rückflusskühler mehr Cinchona-Wirkstoffe als durch Mazeration.
24 Tinkturen wurden je auf 6 verschiedene Arten hergestellt und untereinander ver¬
glichen. Droge, Menstruum und Ansatzverhältnis blieben konstant. Die gleiche Be¬
obachtung machten Jermstad u. O est by an Chinarinde und Bogs an an¬
dern Drogen. Letzterer weist jedoch auf die stärkere Bodensatzbildung in warm
hergestellten Tinkturen hin.

- 38 -
Tab. 2 Mazeration, allgemeine Vorschriften in einigen Arzneibüchern
Arzneibuch: Seite Jahr Dr. Zg. Me. Zeit P Def.
Bd. T. mm Teile Tage Tage
USPXV 820 1955 - 40*) 3/4 3 -*) -
Brit. Ph. 682 1958 - D D 7 --
Norske F. 12 1939 - D D 8 + 2-3
Svenska F. XI
S.639: Tinct.
S.223: Extr.
639
223
1946
1946 1
D
D
D
5
2
5
2
2
+
+
+
eini¬
ge1
Ph.Dan.IX
incl. Add. 1954I, 426
I, 56
1948
1948 1
D
D
D
5
2
5
2
1
+
+
+
~
Nederl. Ph. VI 577 1958 - D D 5 + 2
Ph. Belg. IV 284 1930 1 D 4-8
2-4
1
0,5+
+
DAB 6
(f.Extr. sicc.)690 1926 - D D 10
+
Ph.Helv.V 5 1933 - 3 D - + 8
F.Espan.DC I, 664
I, 664
1954
1954
1
1
D
D
9
6
4
10
4
4
+
+
+
F.Ital.VI
für Tinct.
455 1940 - D 1/21/2
5
3
+
+
Ph.Int.I II, 232 1955 1 grobpulv.
10
3/45
1/42
Dr. = Droge Zg. = Zerkleinerungsgrad Bd. = Band
T. = Teile P. = Abpressen der DrogeMe. = Menstruum Def. = Defäkation
D = je nach dem einzelnen Drogenartikel
*) Alle Teilchen müssen ein Sieb von 0,42 mm Maschenweite passieren. Davon
dürfen höchsten 40 % durch ein Sieb von 0,177 mm Maschenweite fallen.
Mit 3/4 des Menstruums wird extrahiert, 1/4 davon wird für das Auswaschen
der abgetrennten, extrahierten Droge verwendet.
**) Gleiches Vorgehen wie nach USP XV.
Eventuelle Addenda zu den angegebenen Arzneibüchern sind bis und mit dem
Jahr 1959 berücksichtigt.

- 39 -
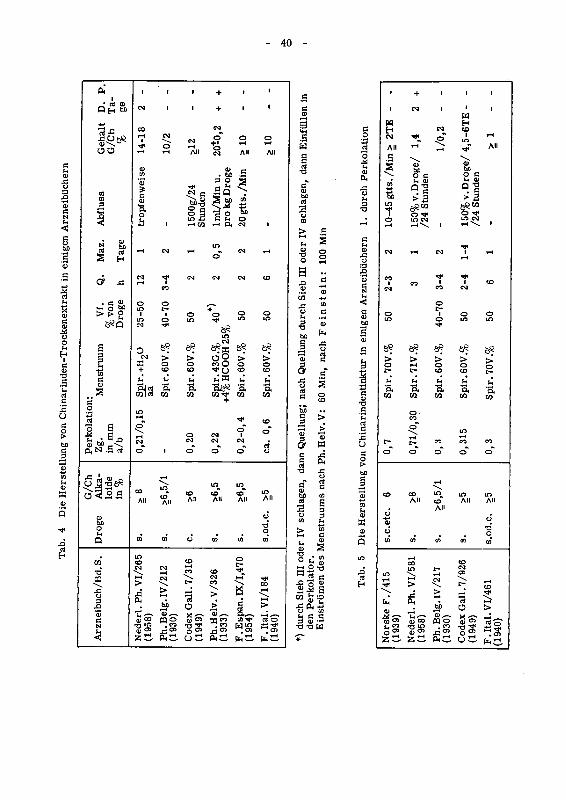
Tab. 3 Perkolation, allgemeine Vorschriften in einigen Arzneibuchern
Arzneibuch/Bd. S. JahrPerkolationsvorgang:
zg.mm
Vorf.
%Qg.h
Maz.
Tage
Abfluss P.
USP XV/820 1955 40*) 60-80
(NFX)
1/4 1 1-3-5 ml/Min -
Bnt.Ph./682 1958 D - 4 1 - +
Norske F./13 1939 D 50 2-3 2 10-45 gtts. /Min -
SvenskaF.Xl/237 1946 D 40-50 +**> 2 10-100 gtts. /Min -
Ph.Dan.IX/1, 56,57 1948 D 50 N. 2 lOgtts-(Min.Kg)"1 -
Nederl.Ph. VI/261 1958 D 25-50 12 1 tropfenweise -
Tinkturen: 577
(Extrakte: 261)1958 D 25-50 3 1 150% v. Droge/24
Stunden
+
Ph. Belg.IV/285(Fluidextrakte)
1930 D 40-70 3-4 2 - -
Codex Gall. 7/453 1949 0,3151,25
50 2-4 1-4 150% v.Droge/24 Stunden
-
DAB 6/237(Fluidextrakte)
1926 D D 12 2 10-70 gtts /Min-.1-10 Kg Droge
-
Ph.Helv.V/6 1933 D D 2 0,5 1 ml/Min +
F.Espan.DC/lI, 812 1954 D 50 2 2 20 gtts./Min +
F.Ital.Vl/177-178 1940 D 50 6 1 - -
Ph. Int. 1/11,232 1955 gem. D 4 1 1 ml/Min -
Ph.Hung. V/1,178 1954 D D 3 1 2% v.Drogenvo¬lumen/Stunde
+
Bd. = Band Zg. = ZerkleinerungsgradS. = Seite Vorf. = Vorfeuchtung
Qg. = Quellung N. = wahrend der Nacht
Maz. = Mazeration gem. = gemahlenP. = Abpressen D. = je nach den einzelnen Drogenprapa
raten
*) Alle Teilchen müssen ein Sieb von 0,42 mm Maschenweite passieren. Davon dür¬
fen höchstens 40 % durch ein Sieb von 0,177 mm Maschenweite fallen.
**) einige Zeit.
Eventuelle Addenda zu den angegebenen Arzneibuchern sind bis und mit dem
Jahr 1959 berücksichtigt.
--
1^
16
50
Spir.70V.%
0,3
>5
s.od.c.
(1940)
VI/461
Ital.
F.
Stunden
/24
-150%v.Droge/4,5-6TE-
1-4
2-4
50
Spir.60V.%
0,315
>5
s.
(1949)
7/926
Gall.
Codex
--
1/0,
22
3-4
40-70
Spir.60V.%
0,3
>6,5/l
s.
(1930)Belg.IV/217
Ph.
Stunden
/24
+2
1,4
v.Droge/
150%
13
Spir.71V.%
0,71
/0,3
0>8
s.
(1958)
VI/581
Ph.
Nederl.
--
2TE
>gtts./Min
10-45
22-3
50
70V.%
Spir.
0,7
6.
s.c.etc
(193
9)F./415
Norske
Perkolation
durch
1.
Arzneibuchern
einigen
in
Chinanndentinktur
von
Herstellung
Die
5Tab.
Mm
100
stein:
ein
Fnach
Min,
60
Ph.Helv.V:
nach
Menstruums
des
Einströmen
Perkolator.
den
in
Einfüllen
dann
schlagen,
IV
oder
HI
Sieb
durch
Quellung
nach
Quellung;
dann
schl
agen
,IV
oder
HI
Sieb
durch
*)
--
10
>-
16
50
Spir.60V.%
0,6
ca.
--
10
>20gtts./Min
22
50
Spir.60V.%
0,2-0,4
Droge
kg
pro
+4%HCOOH25%
++
20^0,2
lml/Minu.
0,5
240
*^Spir.43G.%
0,22
Stunden
--
>12
1500g/24
12
50
Spir.60V.%
0,20
--
10/2
-2
3-4
40-70
Spir.60V.%
aa
-2
14-18
tropfenweise
112
25-50
Sj>i
r.+H
200,
21/0
,15
>5
s.od.c.
>6,5
s.
>6,5
s.
>6
c.
>6,5
/ls.
8>
S.
(1940)
VI/184
Ital.
F.
(195
4)F.Espan.K/1,470
(193
3)Ph.Helv.V/326
(194
9)7/316
Gall.
Codex
(193
0)IV/212
Belg.
Ph.
(195
8)VI/265
Ph.
Nederl.
ge
%Tage
hDroge
a/b
Ta-
G/Ch
von
%mm
in
P.
D.
Gehalt
Abfluss
Maz.
Q.
Vf.
Menstruum
Zg.
Perkolation:
in%
loide
Alka-
Droge
G/Ch
S.
Arzneibuch/Bd.
Arzneibuchern
einigen
in
Chinarinden-Trockenextrakt
von
Herstellung
Die
4Tab.
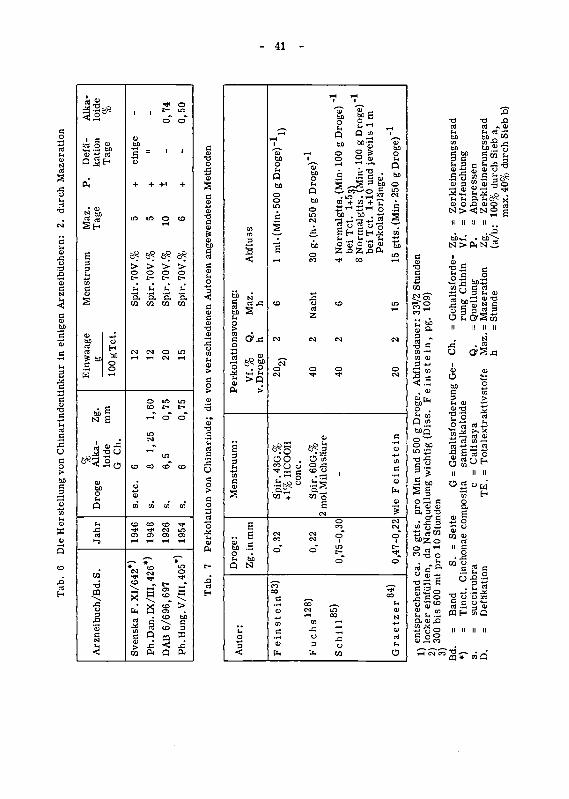
b)Sieb
durch
40%
max.
a,
Sieb
durch
100%
(a/b
:Stunde
=h
Zerkleinerungsgrad
=Zg.
Mazeration
=Maz.
Totalextraktivstoffe
=TE.
Defakation
=D.
Abpressen
=P.
Quellung
=Q.
Calisaya
=c
succirubra
=s.
Vorfeuchtung
=Vf.
Chinin
rung
samtalkaloide
composita
Cinchonae
Tinct.
=*)
Zerkleinerungsgrad
=Zg.
Gehaltsforde-
=Ch.
Ge-
Gehaltsforderung
=G
Seite
=S.
Band
=Bd.
Stunden
10
pro
ml
600
bis
300
109)
pg.
Feinstein,
(Diss.
wichtig
Nachquellung
da
einf
ülle
n,locker
Stunden
33'/2
Abflussdauer:
Droge.
g500
und
Min
pro
gtts.
30
ca.
entsprechend
Droge)"1
g(Min-250
gtts.
15
15
220
Feinstein
wie
0,47-0,22
Graetzer84)
Perkolatorlange.
m1
jeweils
und
1+10
Tct.
bei
Drog
e)"1
g(Min-100
Normalgtts.
8
.I+
53)
Tct.
bei
"
Droge)
g(Min-100
Normalgtts.
46
240
0,75-0,30
Schill
85'
Droge)"1
gg-(h-250
30
Nacht
240
Milchsäure
mol
2Spir.60G.%
0,22
Fuch
s128
)
"^
Droge)
gml-(Min-500
16
220
2)conc.
HCOOH
+1%
43G.%
Spir.
0,32
ein83)
st
ein
F
Abfluss
hh
v.Droge
Maz.
Q.
Vi.%
Perkolationsvorgang:
Menstruum:
mm
in
Zg.
Droge:
Autor:
Methoden
angewendeten
Autoren
verschiedenen
von
die
Chinarinde;
von
Perkolation
7Tab.
0,50
-+
670V.%
Spir
.15
0,75
6s.
1954
405*
'V/m,
Ph.Hung.
0,74
-
+10
70V.%
Spir.
20
0,75
6,5
s.
1926
6/69
6,69
7DAB
-n
+5
70V.%
Spir
.12
1,60
1,25
8s.
1948
Ph.D
an.I
X/ni
,426
*>
-
einige
+5
Spir.70V.%
12
6s.etc.
1946
F.XI
/642
*)Svenska
lOOgTct.
%loide
Alka-
Tage
kation
Defa¬
P.
Tage
Maz.
Menstruum
g
Einwaage
mm
Zg.
Ch.
Gloide
Alka-
%
Droge
Jahr
Arzneibuch/Bd.S.
Mazeration
durch
2.
Arzneibuchern:
einigen
in
Chinanndentinktur
von
Herstellung
Die
6Tab.

- 42 -
97)Thörn verglich die Ausbeuten der Extraktion von Ephedra-Blatt mittels
Infusion (I), Dekoktion (II), Mazeration (III) und Perkolation (IV). Ohne Salzsäurezu-
98)satz zum Menstruum nahmen die Ausbeuten von (I - IV) zu. Eichenberger
fand mittels Dekoktion bessere Ausbeuten als mittels Mazeration.
431.2 Vergleich diskontinuierlicher, einstufiger mit diskontinuierlichen, mehrstu¬
figen Verfahren.
99)Rosen thaler beobachtete in Chinatinkturen, die bei gleichen Ansätzen
durch Mazeration (I), Dimazeration (II) oder Perkolation (III) hergestellt wurden,
von I - III eine Zunahme des Wirkstoffgehaltes. Auch Bari gelang durch Di¬
mazeration eine Erhöhung der Wirkstoffkonzentration der Chinatinkturen. Vor allem
aus seiner zweiten Arbeit, in welcher Droge, Menstruum und Ansatzverhältnis kon¬
stant gehalten wurden, geht dies deutlich hervor. Die Digestionsausbeuten waren,
je nach der Ausführung der Digestion, grösser oder etwas kleiner als die Ausbeuten
der Dimazeration. Eschenbrenner und Gärtner ' beschreiben eine Mehr -
ausbeute bei Dimazeration von 48 % gegenüber einfacher Mazeration bei Chinarin¬
den-Tinktur; Weber 'und G stirner ' beweisen die Ueberlegenheit des
103)13)erstgenannten Verfahrens an andern Drogen. Herzog konnte durch stufen¬
weise Erhöhung der Alkoholkonzentration die Mazerationsausbeute bei verschiedenen
Drogen verbessern.
431.3 Vergleich diskontinuierlicher mit kontinuierlichen Verfahren.
Vergleichende Arbeiten über diese Verfahren, vor allem die Gegenüberstellung
von Mazeration und Perkolation, sind recht zahlreich. Bei allen Autoren er¬
wiesen sich die kontinuierlichen Verfahren den diskontinuierlichen, ein- oder mehr¬
stufigen Verfahren überlegen in Bezug auf Extraktion der Inhaltsstoffe.
Der Vergleich diskontinuierlicher Methoden mit der Perkolation von Chinarinde
beschäftigte folgende Autoren:
Rosenthaler,Bari ', welcher durch sechstägige Digestion bei 37 C,
also ein diskontinuierliches Verfahren, in gelagerten Tinkturen Ausbeuten wie durch
das Perkolationsverfahren erzielte. Infolge gleichzeitiger Veränderung mehrerer
Faktoren in dieser Arbeit sind die gewonnenen Resultate jedoch nur bedingt vergleich¬
bar.
Jermstad und Oestby'fanden nach dreistündigen Digestionen auf dem
Wasserbad am Rückflusskühler gleiche Resultate wie nach einer achttägigen Maze¬
ration.

PerkolationsmethodediesicherwiesDrogenandernbeiAuchExtraktionsleistung.
derSteigerungeinePerkolationEvakolation,Diakolation,Reihenfolgederinfand
'Frommwerden.abgebrochenDurchflussesdesVerlangsamungstarkerinfolge
mussteDiakolationdieundResultate,schlechterebedeutendReperkolationdieten,
Totalextraktivstoffausbeu-undAlkaloid-gleichepraktischergabenVerfahrenbeiden
letztenDieEvakolation.undPerkolationDiakolation,mitReperkolationdieverglich
Feinsteinausgeführt:ChinarindeanwurdenUntersuchungenfolgendenDie91)
Modifikationen.derenund
PerkolationderzwischenVergleicheausschliesslichfastsindArbeitenDiese
Verfahren.arbeitenderkontinuierlichVergleich431.4
mitarbeiteteBogsBrechnuss.ausAlkaloidegleichvielejedochextraktivstoffe, 9)
Baldriantinktur.undTollkraut-
gleichvijedochextraktivstoffe,
TotalmehrPerkolationStufenmazeration,Mazeration,Reihenfolgederinhierte
'extra¬HerzogExtraktausbeuten.bessereEphedrablattmitPerkolatDekokt,13)
salzsauresMazerat,Infus,ReihenfolgederinbeobachtetenEichenberger98)
undThörnStunden.36nachMazeratangesetztesMenstruumTeilen9undge97)
Drogenmen-gleichendermiteinwieHarzgleichvielenthieltTeilperkolatersteDas
'.Jalapenknollenund'PodophyllumHarzdrogendiebenützten''beiter
Mitar¬undsaHuMazeration.dervonundDimazerationdervongefolgtMethode,
bestealsPerkolationdiebezeichnetundTinkturen8überprüfte'
Weber
überlegen.Methodendiskontinuierlichenden
kontinuierlichendieausnahmslossichzeigenhierAuchzusammengefasst.beiten
Ar¬durchgeführteDrogenandernmiteinigenochsindAbschnittfolgendenIm
ist.möglichMazerationmittelsdiesalsher,Wirkstoffkonzentrationhöherermit
TinkturenEvakolationsprinzip,demnachalsoVakuumhilfe,mitRöhrenperkolation
durchstellte'MosigMazeration.durchalsChinatinkturengehaltreichere
'HÖH'undGstirner',BarigewannenDiakolationMittels
wurden.
hergestelltArzneibüchernandernundPh.Helv.VderDAB-6,demnachdierinde,
China¬ausPräparateandereundChinarinden-Tinkturverglich'Thomson
30)
Perkolation.
deranhandTurboextraktiondieüberprüftenMitarbeiterundMelichar22)
mazeration).
Di-einealsbesserdabeiextrahiertenWasserbadimDigestionStunden(3Tinkturen
24anExtraktionsmethoden6jeArbeitumfassendeneinerinprüft'Bari
-43-

- 44 -
65)überlegen. Graetzer gelang durch Evakolation nur eine sehr geringe Erhöhung
der Alkaloidausbeute, analoge Vakuumanwendung im schwach konischen Perkolator
war erfolglos. Ueberdruck beschleunigte die Extraktion. Bei Totalextraktion stieg
jedoch nur die Alkaloidausbeute um etwa 4 %, während die Totalextraktivstoff-Aus¬
beute sank. Olszewski '
gibt der Evakolation nach K e s s 1 e r' den Vorzug,
welche bei erhöhter Ausbeute die Extraktionsdauer von Chinarinde und zwei anderen
Drogen verkürzt haben soll.
Mit andern Drogen wurden folgende Arbeiten ausgeführt: Hu s a und Mitarbei¬
ter extrahierten mit einer modifizierten Diakolation schon im ersten Teilperkolat
99 % des Totalharzgeahltes von Podophyllum und Jalapenknollen gegenüber
85 - 90 % im ersten Teilperkolat bei Perkolation. Mühle mann 'verglich die
Evakolation mit der Perkolation. Er gewann viel höhere Ausbeuten an ätherischem
Oel durch Evakolation aus Fol.Thymi. Dies bestätigten auch Gstirner und Ber-
niker,fanden aber zugleich für andere Drogen eine Ueberlegenheit der Perko¬
lation.
112)Hu s a und Mitarbeiter empfehlen für Belladonnawurzel die Reperkolation,
welche bei gleicher Alkaloidausbeute weniger Ballaststoffe extrahierte als die Per-
113) 31)kolation '. Nach Thor n
' wird dagegen Ephedrablatt durch Perkolation besser
114)ausgezogen. Soll ne r betont mit Recht: "Die günstigste Extraktionsmethode
muss unter Berücksichtigung der Löslichkeitsverhältnisse der Wirkstoffe einer Dro¬
ge in jedem Einzelfall gesondert ausgearbeitet werden".
432 Vergleiche mit neuen Methoden
Da die kontinuierlichen Extraktionsmethoden schon eine Erschöpfung der Droge
mit angemessenen Menstruum-Mengen erlauben, tendieren die neueren Arbeiten vor
allem nach einer Verkürzung der Extraktionszeit.
432.1 Die Ultraschall-Extraktion
12)Schultz und Klotz konnten die Extraktion von Chinarinde mit verdünn¬
ter Ameisensäure durch Schallwellen begünstigen. Ultraschall von 2,4 MHz bei 20 W
Leistung verbesserte nur die Extraktion der ungequollenen Droge. Head und Mit-
115)arbeiter beobachteten eine raschere Extraktion der Chinarinde durch Ultraschall-
Mazeration, verglichen mit der Soxhlet-Extraktion. Bei mehrstündiger Extraktion
erfolgt jedoch ein Ausgleich und schon nach 7 Stunden war die Soxhlet-Extraktion in
Bezug auf die Wirkstoffausbeute überlegen. Diese Unterschiede sind m.E. vor allem
auf den Vergleich eines diskontinuierlichen mit einem kontinuierlichen Verfahren zu¬
rückzuführen. Zwei parallel ausgeführte Perkolationen mit und ohne Ultraschall wür¬
den dessen Einfluss vermutlich besser zeigen.

- 45 -
1 1 OS
Thompson extrahierte Erdnüsse mit n-Hexan. Die Oelausbeute nach 6
Minuten, ausgedruckt in Prozent der Soxhletextraktion, stieg in der Reihenfolge:
Mazeration unter Ruhren bei 675 U/Mm; Ultraschall (400 kHz: Leistung: 6,5 W/cm );
Soxhletextraktion; Ultraschall (400 kHz; Leistung 63,3 W/cm ); Mazeration unter
Ruhren mit 1350 U/Min.
432.2 Die Turbo-Extraktion
22)Melichar veröffentlichte 1953 eine Arbeit, in welcher auf den bedeuten¬
den Zeitgewinn bei der Turboextraktion hingewiesen wird. Er fand bei 8000 - 13000
U/Min nach 7-10 Minuten in den Extrakten, unter anderem aus Chinarinde, Trocken¬
ruckstande von der Grossenordnung der maximalen Ausbeuten bei der Mazeration.
Der Alkaloidgehalt soll sogar denjenigen einer Perkolation ubertroffen haben. Die
Präparate waren, selbst nach einjähriger Lagerung, Mazeraten und Perkolaten eben¬
bürtig.
Bogs erhielt mit andern Drogen sowohl fur Alkaloide sowie fur Totalextrak-
tivstoffe Ausbeuten, die geringer als bei der Perkolation, jedoch hoher als bei der
117)Mazeration waren. Melichar benutzte die Wirbeiextraktion auch mit Erfolg
als Ersatz der Infusion und Dekoktion, wobei vor allem Zeitgewinn und Wegfall stär¬
kerer Erwärmung von Vorteil sind.
Alle aufgeführten, vergleichenden Arbeiten sind abschliessend in einer Tabelle
zusammengefasst.

- 46 -
Tab. 8 Vergleichende Untersuchungen betreffend die Extraktionsmethoden mit
besonderer Berücksichtigung der Chinarinde
Methode : Mazera- Turboex- US.
tion traktion
Dimaze¬
ration
Dk. Perkolation
Vergleich mit:
Mazeration
Mazeration und
Ultraschall
Digestion
9,22 116
12
£,10, 11., 96 11,96
105 13,96,98,103
11 10,11,96
Decoction
Infusion
Dimazeration
Stufen-Mazerat.
97 117
97 117
11,96,99Ï507IOT71O2
13,103
98
98
11 11,96,99,101
13,103
Perkolation
Diakolation (D.)
modifiz. D.
Evakolation
fraktionierte
Perkolation
Repetitions-Diakolation
Soxhletextrakt.
Ultraschall und
Soxhletextrakt.
9,10,11,22 9,22307Ü779"97~
—
IUI, 1077108
11,104
107,108
115
116
115
11,109
107,108
109 109,95,110SÏÏ71TT
31,112,113
118 *)
*) Nur im Literaturverzeichnis erwähnt, jedoch nicht im Text.
Zahl = Arbeiten mit Chinarinde
ZjLhl = Allgemeine Aussagen auf Grund von Erfahrungen mit mehreren Drogen
Die Zahlen in dieser Tabelle entsprechen den Nummern im Literaturverzeichnis
dieser Arbeit.
Dk. = Diakolation modifiz.D. = modifizierte Diakolation
US. = Ultraschall

- 47 -
5 ZIELSETZUNG DER EIGENEN UNTERSUCHUNGEN
51 E ml eitung
Aus den vergleichenden Untersuchungen geht eindeutig hervor, dass die Lei¬
stungsfähigkeit der verschiedenen Extraktionsmethoden in der Reihenfolge: 1. dis¬
kontinuierliche, einstufige, 2. diskontinuierliche, mehrstufige und 3. kontinuierli¬
che Verfahren zunimmt. Die Perkolation, als eine der besten und bisher gebräuch¬
lichsten Methoden, erfordert aber noch mehrere Tage.
Wenn es mit Hilfe eines hochtourigen Ruhrgerates gelange, innert kurzer Zeit
Extrakte angemessener Konzentration herzustellen, so ware damit eine Methode ge¬
schaffen, um kleine Chargen von Extrakten im Apotheken-Laboratorium "ex tempore"
herzustellen. Da die Zeit wirtschaftlich der entscheidende Faktor bei solchen Prozes¬
sen darstellt, schien es interessant, ein Verfahren zu prüfen, das in kürzerer Zeit ge¬
brauchsfertige Extrakte liefert, als die bisherigen Methoden. Trotzdem es sich hier
um ein diskontinuierliches Verfahren handelt, sind die Resultate von Melichar und
Mitarbeitern ' recht ermutigend (s. pg. 21/22, Ziffer 221.2).
52 Bietet die Turbo-Extraktion Vorteile vor den gebrauchlichen
Extraktionsmethoden der Chinarinde9
Mit den nachfolgenden Untersuchungen soll versucht werden, die Turbo-Ex¬
traktion von Chinarinde mit den gebrauchlichsten Extraktionsmethoden zu verglei¬
chen. Dabei sollen vor allem verglichen werden:
Der Zeitaufwand
Die Ausbeuten an Alkaloiden und Gerbstoffen
Die Extraktqualltat in Bezug auf Konzentration und Ballaststoffe
Der EnergieaufwandDie Anpassungsfähigkeit dieser neuen Extraktionsmethode an die Erforder¬
nisse des Laboratoriums.

- 48 -
6 AUSWAHL DER DROGE UND DES MENSTRUUMS
61 Auswahl und Charakterisierung der Droge
Das Ziel dieser Arbeit ist der Vergleich einer neuen Extraktionsmethode mit
bisher gebräuchlichen Verfahren. Daher schien es sehr zweckmässig, eine Droge
zu wählen, die schon mehreren Autoren als Testobjekt diente, so dass dadurch Ver¬
gleichsmöglichkeiten entstehen. Dies trifft zu bei Chinarinde; zudem ist diese Droge
etwas schwer extrahierbar, was für die exakte Kontrolle des Einflusses der Rührzeit
auf die Wirbelextraktion wichtig ist. Bei hoher Tourenzahl kann eine zu rasche Ein¬
stellung des Extraktionsgleichgewichtes die Versuche stark erschweren.
Der Gesamtalkaloid- oder Hauptwirkstoffgehalt der Chinarinde lässt sich über-
119')dies hinreichend genau bestimmen, was schon Feinstein 'betont.
Obwohl den Amerikanern Woodward und Doering'im Jahre 1944 die
Totalsynthese des Chinins gelang, haben die Drogenpräparate noch ihre volle Bedeu¬
tung, da Reinalkaloide und deren Mischungen keinen vollwertigen Ersatz für den von
der Pflanze gelieferten Wirkstoffkomplex darstellen.
Die Eigenschaften der für diese Arbeit verwendeten Droge seien hier festgehal¬
ten. Die Verfahren werden unter 84, den Bestimmungsmethoden, beschrieben (s.
Pg. 67).

- 49 -
Normierung der verwendeten Chinarinde
Alkaloidgehalt nach Ph.Helv. V (= AD %) 11,65 %
Gerbstoffgehalt (= GD %)
a) nach vorgesehener Methode für Ph.Helv. VI 10,48 %
b) nach eigener kolorimetrischer Methode 7,34 %
(dieser Gehalt, 7, 34 %, diente als Grundlage für die
weiteren Berechnungen. )
Totalextraktivstoffgehalt (TED %) nach der Methode von Fuchs 51,02 %(Diss. ETH 1941)
Wassergehalt nach Ph. Helv. V 8,89%
Aschegehalt nach Ph. Helv. V und 2, 96 %
deren in HCl unlöslicher Anteil nach Ph.Helv. V 0,86 %
Dichte (bestimmt im Teerpyknometer nach Evakuieren, 1,405 g/cmmit Benzin als Suspensionsmittel)
Siebanalysen der Frischdroge, Deklaration Sieb IV:
Anzahl Analysen:
Drogeneinwaage je:
durch
Sieb
auf
Sieb
m IV
IV IVa
IVa V
V VI
VI vn
VH Din 60
Din 60 Din 80
Din 80 Din 100
* Din 100
V. = auf Grundbrett der Maschine gesammelt
Z. = in die Luft zerstäubt (feine Anteile)
Total:
17
25 g
42,6
23,0
19,1
4,0
2,1
6,2
0,2
2,9
100,1 «
7
25 g
43,1
23,1
18,6
4,1
2,5
1,3
1,3
1,1
0,7
0,1
_A°
99, 9 %
Die römischen Ziffern bedeuten die Siebnummern der Ph.Helv. V.

- 50 -
62 Auswahl des Menstruums
Schon im allgemeinen Teil wurde darauf hingewiesen, dass das von der Ph.
Helv. V für die Herstellung von Chinarinden-Trockenextrakt vorgeschriebene Men-
121) 76)struum für diesen Zweck gut geeignet sei. Auch Feinstein 'und Graetzer '
verwendeten für ihre Arbeiten dieses Menstruum; darum wurde es auch für alle
nachstehend beschriebenen Extraktionsversuche gewählt, nicht zuletzt auch im Hin¬
blick auf die daraus entstehenden Vergleichsmöglichkeiten.
Das Menstruum besteht aus:
Acidum formicicum Ph.Helv. V (24 - 25 %) 4 Teile
Spiritus Ph.Helv. V (92,1 - 92,8 Gew.%) 46 Teile
Aqua destillata 50 Teile
Durch die Säurezugabe sollen schwerlösliche Alkaloid-Gerbstoff-Komplexe
möglichst in leichtlösliche Alkaloidsalze und freie Gerbstoffe aufgespalten werden.
121)Feinstein weist darauf hin, dass ca. ein Drittel der zugesetzten Amei¬
sensäure mit Alkohol zu Aethylformiat verestert wird. Das Reaktionsgleichgewicht
ist bei Zimmertemperatur nach 8-12 Tagen erreicht. Für die vorliegenden Unter¬
suchungen wurde daher ausschliesslich gealtertes Menstruum verwendet. Dieses
zeigte folgende Eigenschaften:
Dichte 0, 935 g/cm? (15°C)0, 931 g/cm (20°C)
Viskosität in Centipoise (cP) 2,595 cP
Gehalt an freier Ameisensäure 0, 65 %
Alkoholgehalt 42,95 Gew.% (15°C)
50,48 Vol. %(15°C)
Vergleiche das Kapitel Bestimmungsmethoden, pg. 67.

- 51 -
7. BESCHREIBUNG DER VERWENDETEN APPARATE
71 Ueberprufung des Ruhrgerates
711 Das fur unsere Versuche verwendete Gerat
POLYTRON-45 ST
Alle Wirbeiextraktionen wurden mit dem "POLYTRON-45 ST".* durchgeführt,
das uns von der Firma Mobil Aarau AG. in verdankenswerter Weise zur Verfugung
gestellt wurde.
Das Gerat besteht aus Motor, Gehäuse, Achse und Ruhrkopf. Der oben ange¬
ordnete Motor ist durch einen langen Schaft mit dem Ruhrkopf verbunden. Der Sta¬
tor des Ruhrkopfes ist mit einem Rohr, in welchem die Rotorachse lauft, an dem
Motorgehäuse befestigt. Im Ruhrkopf ist die Rotorwelle mittels einer Teflondich¬
tung im Stator gelagert.
Fur das "POLYTRON-45 ST".* macht die Firma die folgenden Angaben:
1-10 Liter
5 cm
28,5 cm
34'000 Hz
1 V4A-Stahl
1 V4A-Stahl
70 P ( = 7000 cP)1 cm
250 W (neue Gerate 350 W)max. 16'000 U/Min (stufenlos regulier¬
bar)Motortype : Kollektor (Fabrikat: Epple)Gewicht des Gerätes : ca. 12, 7 kg
Um eine zweckentsprechende Planung unserer Versuche zu ermöglichen, wa¬
ren noch die folgenden Punkte abzuklären:
712 Die minimale Charge
Auf Grund unserer Bestimmungsmethoden (s. pg. 67) genügen 350 g Extrakt¬
losung auch in den verdünnten Extrakten zur Ausfuhrung aller Analysen. Es wurden
Chargen (Kapazität)minimale Halsweite des Behalters
Lange des Ruhrers (Schaftlange)maximale WirkfrequenzAnzahl Zahnkranze Stator
Rotor
maximale Viskosität (Poise)maximale KorngrösseMotorleistungDrehzahl pro Minute
1) Hersteller: Max Wulhmann; Maschinen- und Apparatebau; Selzach (Solothurn).
Vertrieb durch: Mobil Aarau AG., Hallwylstrasse 11, Aarau.

- 52 -
darum jeweils 500 g Menstruum mit 50 g Chinarinde eingesetzt, so dass ca. 500 g
Extraktlösung gewonnen werden konnten (Verlust durch Extraktflüssigkeit in der ex¬
trahierten Droge).
Vorversuche mit 550 g Wasser in einem 1 L-Becherglas zeigten, dass schon
bei ca. 3300 u/Min (= Position 2 am Regulierwiderstand des Gerätes) beim Rührkopf
Luft ins Wasser gewirbelt wird. Bei ca. 10 500 U/Min (= Position 9 am Regulierwi¬
derstand) erfolgt periodisches Leerdrehen des Rührkopfes unter Anstieg der Touren¬
zahl und Herausspritzen des Becherglasinhaltes.
Um in unseren Versuchen eine konstante Tourenzahl zu garantieren und die
Lösungsmittelverluste möglichst tief zu halten, wurden ein Plexiglasdeckel auf dem
Extraktionsgefäss und über dem Rührkopf eine vertikal verstellbare Abdeckplatte
aus Plexiglas montiert. Der Rührkopf wurde tief in das Extraktionsgemisch getaucht.
Die Anordnung war für alle Versuche einheitlich folgendermassen:
BG1-» 75 mm
Aussen-0 108,5 mmInnen - 0 102 mm
Fig. 1 Masse des Rühkopfes mit Abdeckplatte und des Extraktionsgefässes
AP: Abdeckplatte EG: ExtraktionsgemischBG1: Becherglas RK: Rührkopf
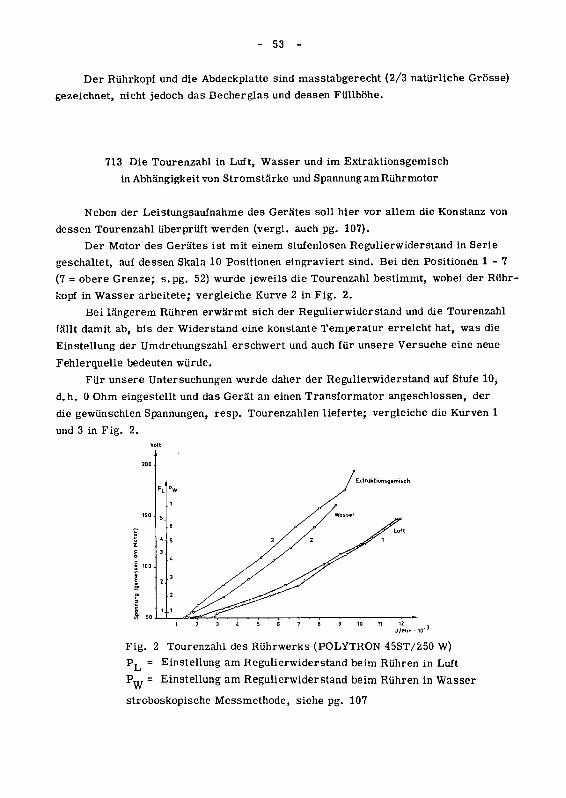
- 53 -
Der Rührkopf und die Abdeckplatte sind masstabgerecht (2/3 natürliche Grösse)
gezeichnet, nicht jedoch das Becher glas und dessen Füllhöhe.
713 Die Tourenzahl in Luft, Wasser und im Extraktionsgemisch
in Abhängigkeit von Stromstärke und Spannung am Rührmotor
Neben der Leistungsaufnahme des Gerätes soll hier vor allem die Konstanz von
dessen Tourenzahl überprüft werden (vergl. auch pg. 107).
Der Motor des Gerätes ist mit einem stufenlosen Regulierwiderstand in Serie
geschaltet, auf dessen Skala 10 Positionen eingraviert sind. Bei den Positionen 1-7
(7 = obere Grenze; s.pg. 52) wurde jeweils die Tourenzahl bestimmt, wobei der Rühr-
kopf in Wasser arbeitete; vergleiche Kurve 2 in Fig. 2.
Bei längerem Rühren erwärmt sich der Regulierwider stand und die Tourenzahl
fällt damit ab, bis der Widerstand eine konstante Temperatur erreicht hat, was die
Einstellung der Umdrehungszahl erschwert und auch für unsere Versuche eine neue
Fehlerquelle bedeuten würde.
Für unsere Untersuchungen wurde daher der Regulierwiderstand auf Stufe 10,
d.h. 0 Ohm eingestellt und das Gerät an einen Transformator angeschlossen, der
die gewünschten Spannungen, resp. Tourenzahlen lieferte; vergleiche die Kurven 1
und 3 in Fig. 2.
PL Pw
/ Extrahtionsgemisch
5
.7
.6
/Woss
^^LuftL
3
5
L
3 ,/ / 2^T
2
1
3 y/
1 J^^^gZ^^9 10 11 12
U/Mm 10"'
Fig. 2 Tourenzahl des Rührwerks (POLYTRON 45ST/250 W)
PL = Einstellung am Regulierwiderstand beim Rühren in Luft
Pw = Einstellung am Regulierwiderstand beim Rühren in Wasser
stroboskopische Messmethode, siehe pg. 107

- 54 -
Sei 3000 und mehr Umdrehungen pro Minute wurden maximale Tourenzahl¬
schwankungen von - 250 U/Min festgestellt. Unterhalb 3000 U/Min wird die Motor¬
drehzahl mit abnehmender Tourenzahl labiler.
Ein spezieller Einfluss des Temperaturanstieges im ungekühlten Extraktions¬
gemisch auf die Tourenzahl konnte nicht festgestellt werden.
Die folgende Tabelle 9 zeigt die Leistungsaufnahme des Rührmotors.
Tab. 9 Die Leistungsaufnahme des Rührmotors
Volt Ampere Watt U/Min Medium:
50,5 0,44 22,2 2700 Luft
71,5* 0,44 31,4 5000 Luft
112,0 0,49 54,9 9300 Luft
122,0* 0,50 61,0 10000 Luft
50,5 0,42 21,2 1000 Chinarinde + Menstruum
109,5 0,79 86,5 5000 „50 g „ „
500 g „
194,0 1,16 225,0 10000 M ii m h
*) Dies sind nur Vergleichswerte, welche die vermehrte Leistungsaufnahme in Ab¬
hängigkeit des Mediums direkt abzulesen gestatten.
714 Der durch das Rühren ohne Kühlung bedingte Temperaturanstieg
im Extraktionsgemisch
Da in der Apotheke nicht immer eine leistungsfähige Kühleinrichtung zur Ver¬
fügung stehen wird, wurde der Temperaturanstieg als Funktion der Rührzeit für die
in dieser Arbeit zu verwendenden Tourenzahlen bestimmt. Dies ist später wichtig
für die Beurteilung der Methode.
50 g Chinarinde wurden mit 500 g Menstruum für Chinarinden-Trockenextrakt
nach Ph. Helv.V im bedeckten Gefäss während einer Stunde gerührt und der Tempe¬
raturanstieg beobachtet. Die Versuche erfolgten sowohl in einem Becherglas von 1
Liter Inhalt (das Extraktionsgefäss in unseren Versuchen), als auch in einem Dewar-
gefäss von 2,5 Liter Inhalt (12 cm Innendurchmesser) bei 1000, 5000 und 10 000U/Min.
Die Resultate zeigen die folgenden Kurven in Fig. 3.
Es ist daraus ersichtlich, dass eine Erhöhung der Tourenzahl von 5000 auf
10 000 U/Min zu einem viermal rascheren Temperaturanstieg in den ersten 15 - 20

- 55 -
TURBOEXTRAKTION
Temperatur des ungekühlten Extraktionsgemisches
10 000 U/Min
5 10 30 60 65
Minuten
Fig. 3 Temperaturanstieg als Funktion der Zeit bei Turboextraktion mit verschie¬
denen Tourenzahlen
Minuten führt. Nach 25 - 30 Minuten bleibt die Temperatur konstant. Diese Steige¬
rung der Temperatur ist nur gerechtfertigt, wenn auch die Extraktion in dieser Zeit
viermal schneller erfolgt.
Der etwas grössere Wärmeverlust im Becherglas ist bemerkbar bei 1000 und
5000 U/Min, nicht mehr jedoch bei der grossen Selbsterwärmung bei 10 000 u/Min.
72 Die Kühleinrichtung
Um in einer Versuchsreihe nur einen veränderlichen Faktor zu haben, musste
die gewünschte Temperatur, meist 20 C, während der ganzen Extraktionsdauer kon¬
stant gehalten werden können. Dazu wurde folgende Apparatur aufgebaut:

- 56 -
721 Die Kühlspirale um das Extraktionsgefäss
Das Extraktionsgefäss (1 L-Becherglas) wurde mit einer dicht anliegenden
Kühlspirale aus geglühtem Kupferrohr umgeben (Rohr: Länge 4 m/Aussen-0 6,5 mm),
das sich, im Gegensatz zu Glas, durch gute Wärmeleitung auszeichnet.
Das Ganze wurde zur besseren Wärmeableitung in ein Becherglas von 3 Litern
Inhalt gestellt, welches bis zum obern Rand mit Wasser gefüllt war. Extraktionsge¬
fäss und Kühlmantel enthielten je ein Thermometer.
5 9 9
Fig. 4 Extraktionsapparatur
1 Kontaktthermometer
2 Rührwerk (POLYTRON)3 Stockthermometer
4 Thermometer für Kühlmantel
5 Heizung6 Befestigung der Abdeckplatte7 Kühlspiralen-Anschluss
8 Abdeckplatte9 Rührkopf (0 = 4,5 cm)
10 Extraktionsgefäss (1 Liter)11 Kühlmantel-Gefäss (3 Liter)12 Heizplatten13 Wasser im Kühlmantel
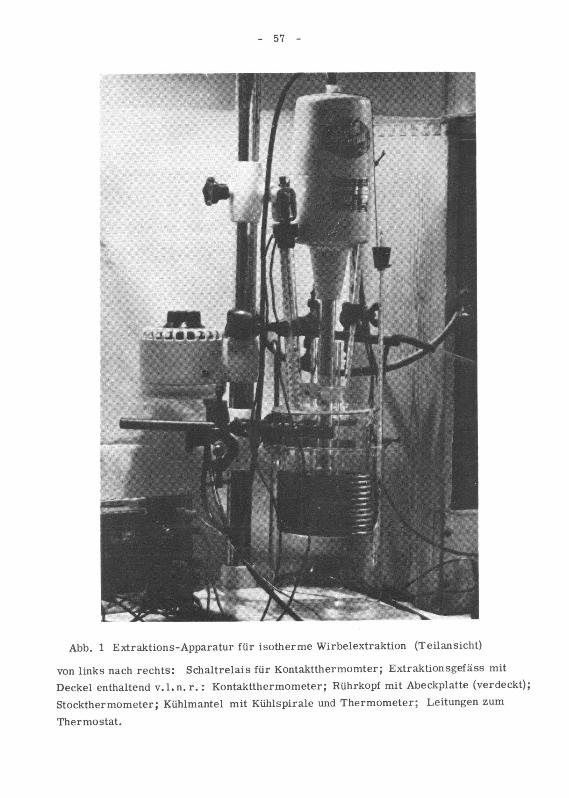
- 57 -
*!
Ï 1fc?M*=
Abb. 1 Extraktions-Apparatur fur isotherme Wirbeiextraktion (Teilansicht)
von links nach rechts: Schaltrelais fur Kontaktthermomter; Extraktionsgefass mit
Deckel enthaltend v.l.n. r. : Kontaktthermometer; Ruhrkopf mit Abeckplatte (verdeckt);
Stockthermometer; Kuhlmantel mit Kuhlspirale und Thermometer; Leitungen zum
Thermostat.

- 58 -
722 Der Thermostat
Der zum Hopple r-Viskosimeter gehörende Thermostat diente bei allen Wir¬
belextraktionen nur zur Kühlung. Er wurde als Wärmeaustauscher und Umwälzpum¬
pe verwendet.
Dieser Thermostat benützt zur Wärmeübertragung destilliertes Wasser, wel¬
ches durch das mit diesem verbundene, geschlossene System gepumpt wird, in die¬
sem Fall durch die das Extraktionsgefäss umgebende Kühlspirale. Im Kessel des
Thermostaten sind ferner: die Umwälzpumpe, eine Kühl spirale, eine elektrische
Heizung und ein Kontaktthermometer; die beiden letzteren wurden hier ausgeschaltet,
Fig. 5 Ultrathermostat (Höppler)
1 Kontaktthermometer 6 Anschluss für Kühlung2 Motor zu Umwälzpumpe 7 Umwälzpumpe3 Relais zu Kontaktthermometer 8 Destilliertes Wasser
4 Umwälzpumpen-Anschlüsse 9 Gefässmantel
5 Einfüllöffnung 10 Heizspirale

- 59 -
723 Die Kühlung des Thermostaten
Zur Kühlung des Thermostaten wurde aus einem hoch montierten Gefäss Eis¬
wasser durch dessen Kühlspirale geleitet. Schemazeichnung des Thermostaten s.
Fig. 5 pg. 58.
724 Steuerung der Kühleinrichtung
Ein in das Extraktionsgemisch tauchendes Kontaktthermometer schaltete beim
Erreichen der eingestellten Temperatur über ein Quecksilberrelais die Umwälzpum¬
pe des Thermostaten ein. Dadurch wurde, solange notwendig, Wasser von ca. 4 C
durch die Kühlspirale um das Extraktionsgefäss gepumpt.
73 Heizung des E xtr aktions gef ässes
Die Heizung wurde nur für die Versuche bei 40 C zur Erwärmung des Kühlman¬
tels und des Menstruums im Extraktionsgefäss benötigt.
Sie bestand aus zwei horizontalen, parallelen Aluminiumplatten von 9,3 cm
Durchmesser, die unter dem Extraktionsgefäss im Kühlmantel angeordnet waren,
und an die 220 V Spannung angelegt wurde. Der Plattenabstand wurde so eingestellt,
dass im Stromkreis ca. 1, 5 A Heizstrom flössen.
Für die bei 40 C durchgeführten Extraktionsversuche mit 1000 U/Min und 5 Mi¬
nuten Extraktionszeit wurde ausnahmsweise der Thermostat auf 40 C eingestellt.
74 Zentrifuge zur Abrennung der Droge von der Extraktlösung
Es wurde die hängende Laboratoriumszentrifuge ECCO, Type E/5/IV verwen¬
det. Der Einsatz erlaubte gleichzeitiges Zentrifugieren mit 4 Gläsern à ca. 75 ml
Inhalt mit einer Tourenzahl von maximal 3000 U/Min.

- 60 -
«ïAfflS^Sv
Abb. 2 Extraktions-Apparatur für isotherme Wirbelextraktion (Gesamtansicht)
hinten von links nach rechts: Verteilerwürfel; Schaltrelais für Kontaktthermome¬
ter; Extraktions-Apparatur; Ultrathermostat mit Eiswasserkühlung;
vorn von links nach rechts: Amperemeter für Heizungskontrolle; Messinstrumente
für Stromstärke und Spannung am Rührwerk;
am Boden: Transformator.

- 61 -
8 DIE HERSTELLUNG DER EXTRAKTLOESUNGEN, DIE ABTRENNUNG VOM
DROGENMATERIAL UND DIE BESTIMMUNG DER INHALTSSTOFFE
Alle Versuche wurden im Doppel ausgeführt, dies gilt auch für die Perkolation.
81 Die Herstellung der Extraktlösungen
(eingesetzte Mengen an Droge und Menstruum)
Bei allen Extraktionsmethoden, mit Ausnahme der Perkolation, wurden 50 g
Droge mit 500 g Menstruum Ph.Helv. V eingesetzt.
811 Die Mazeration
Es wurden 6 Mazerationen gleichzeitig in 1 L-Erlenmeyerkolben mit Korkstop¬
fen durchgeführt. Je zwei Proben wurden 4, 8 und 16 Tage mazeriert, wobei jede
dreimal täglich eine halbe Minute umgeschüttelt wurde.
812 Die Perkolation
500 g Droge wurden in einem schwach konischen, sog. amerikanischen Perko¬
lator von 3, 5 L Inhalt genau nach der in der Ph. Helv. V beschriebenen Methode aus¬
gezogen. Es wurde soviel Menstruum verwendet, dass 6 Teilperkolate (= TP) von
500 g aufgefangen werden konnten. Der Aufsog beim Einströmen in der Perkolator
betrug 1180 g Menstruum. Ueber der Droge lagen aus technischen Gründen (grosser
Perkolator) noch 1310 g Menstruum. Mit Vorfeuchtung und 6 TP war der Menstruum-
verbrauch 5690 g.

- 62 -
813 Die Turboextraktion
Die Herstellung einer Extraktlösung erfolgte einheitlich folgendermassen:
1) Vorkühlung des Ultrathermostaten mittels Eiswasser auf ca. 4 C bei arbeitender
Umwälzpumpe. Die Kühlspirale am Extraktionsgefäss wurde während der Vorküh¬
lung ausgeschaltet. Ausnahme: Für die bei 40°C und 1000 U/Min mit je 5 Min Ex¬
traktionszeit hergestellten Auszüge wurde der Thermostat auf 40°C eingestellt.2) Einfüllen von 500 g Menstruum ins Extraktionsgefäss.3) Fertigmontage der Apparatur (Rührgerätj Anschluss der Kühlspirale und des
Schaltrelais).4) Einstellen der gewünschten Temperatur am Kontaktthermometer. Umrühren bei
niedriger Tourenzahl, bis die gewünschte Temperatur erreicht ist. Für Versuche
bei höherer Temperatur gleichzeitiges Heizen.
5) Zugabe von 50 g Chinarinde und Einschalten der gewünschten Tourenzahl.
6) Genau 45 Min nach Schluss des Wirbeins wurde die erste Hälfte des Extraktions¬
gemisches 35 Min bei 2500 U/Min zentrifugiert, genau 2 Stunden nach Schluss
ebenso die zweite Hälfte.
Diese Aufteilung war durch die geringe Kapazität der verfügbaren Zentrifuge
bedingt. Die relativ lange Zeit war notwendig zur Demontierung der Apparatur und
um die Verdunstungsverluste während der Extraktion und während des Zentrifugie-
rens (Extraktions-, resp. Zentrifugierverluste) festzustellen. Diese Daten sind für
die Berechnung der Ausbeuten unbedingt notwendig.
In der folgenden Tabelle 10 sind die ausgeführten Versuche zusammengefasst:
Tab. 10 Durchgeführte Versuche
Versuch Versuch sbedii
No. in die¬
ser Arbeit
Touren¬
zahl
U/Min
Zeit
Min
Tu 1
Tu 2
Tu 3
Tu 4
0
10 000
0
0
0
5
0
0
Tu 5
Tu 6
Tu 7
Tu 8
Tu 9
Tu 10
1000
5 000
10 000
1000
5 000
10 000
60
60
60
5
5
5
Tu 11
Tu 12
Tu 13
0
1000
10 000
0
5
60
*) Trennung von extrahierter Droge und Extraktlösung durch Filtration, jedoch unter
Aufteilung des Extraktionsgemisches in zwei Hälften und analogen Bedingungen wie
beim Zentrifugieren. Die Laufzeit der Zentrifuge wurde dabei durch die Filtrations¬
zeit ersetzt.
Jeder Versuch (No. Tul - Tul3) in dieser Tabelle umfasst 2 Parallelversuche.
tingen Trennverfahren
Temp.
°C
20 Filtration
20 FiltrationVersuchsreihe 1
20
20ZentrifugierenFiltration +
Mazeration *)
(Einfluss der Trenn¬
verfahren)
20 Zentrifugieren Versuchsreihe 2
20 h (Einfluss der Tourenzahl)20 M
20 M ' Versuchsreihe 3
20 II (Einfluss der Extraktions¬
20 11 zeit)
40 II Versuchsreihe 4
40 II (Einfluss der Temperatur)40 II

- 63 -
814 Die Diturboextraktion
Bei der Diturboextraktion wurde genau gleich vorgegangen wie bei der Turbo¬
extraktion. Nach der 1. Extraktion wurde zentrifugiert und die über der Droge ste¬
hende Flüssigkeit abgegossen. Dann wurde der Drogenrest der ersten Stufe mit der
darin zurückgehaltenen Extraktflüssigkeit mit soviel Menstruum Ph.Helv.V versetzt,
dass wiederum 500 g Flüssigkeit die Droge umgaben. Darauf setzte man erneut eine
2. Turboextraktion an.
Es wurden als 5. Versuchsreihe zwei Diturboextraktionen bei 20 C als Paral¬
lelversuche ausgeführt (Versuch Tu 14; I = erste Stufe; II = zweite Stufe). Für alle
Stufen wurden gewählt:
Die Tourenzahl : 5000 U/MinDie Extraktionszeit : 5 Min
82 Die Abtrennung der Extraktlösungen von der teilweise
extrahierten Droge
Um nach beendeter Extraktion eine nachträgliche Mazeration der Droge durch
die Extraktlösung zu verhindern, musste eine sofortige Trennung erfolgen. Dies ist
um so wichtiger, je grösser das Konzentrationsgefälle von der intra- zur extrazel¬
lulären Flüssigkeit im Extraktionsgemisch nach beendeter Extraktion ist. Ferner
musste eine Verdunstung von Lösungsmitteln vermieden werden.
821 Abtrennung der Extraktlösung von der mazerierten Droge
Nach der gewünschten Mazerationszeit wurde ein doppelt gelochter Gummi¬
stopfen auf das Extraktionsgefäss gesetzt, durch welchen ein umgekehrtes, in der
Höhe verstellbares U-Rohr bis in die überstehende Flüssigkeit reichte. An der zwei¬
ten Oeffnung wurde eine Gummiballon-Druckpumpe angesetzt und so der grösste Teil
der überstehenden, weitgehend klaren Extraktlösung dekantiert.
Diese Lösung wurde, wie im folgenden Text unter b) beschrieben, filtriert.
Der Rückstand mit der extrahierten Droge wurde wie folgt filtriert:
a) zuerst durch einen zweiteiligen Aluminiumtrichter nur mit Gaze-Einlage(filtrierende Fläche 20 cm^, Durchmesser 5,1 cm, Gaze: Schuss: 10,6 Fä-

- 64 -
den pro cm, Kette (Zettel): 13,1 Fäden pro cm (= 23, 7 Fäden pro cm ).Die Filtereinrichtung besitzt keine weitere, den Durchfluss hemmende Schi¬
kanen wie z. B. eine Siebplatte), bis das Filtrat höchstens noch stark trüb
war.
b) anschliessend durch ein Chromatographierohr von 3, 2 cm Innen-Durchmes-
ser und 39 cm Länge. Auf die Siebplatte wurde ein zugeschnittenes Rundfil¬
ter, LS-14 Schleicher und Schüll, gelegt. Zur Beschleunigung der Filtration
erzeugte man mit einer Gummiballon-Druckpumpe einen schwachen Ueber-
druck im gefüllten Chromarohr.
822 Abtrennung der Extraktlösung nach der Turboextraktion
Das Trennungsverfahren sollte möglichst wenig von der Korngrössenverteilung
der extrahierten Droge abhängig sein und bei allen Turboextrakten etwa gleichviel
und möglichst kurze Zeit benötigen.
822.1 Filtration
Mit einem bei 10 000 U/Min während einer Stunde gewirbelten Extraktionsge¬
misch wurden Filtrationsversuche durchgeführt. Nur eine Methode, welche auch
diese Suspension von z.T. feinsten Partikeln noch zu klären vermag, ist für die fol¬
genden Versuche geeignet. Eine Abtrennung der groben Drogenteilchen (Grobfiltra-
tion) mit nachfolgender Klärung der Lösung (Feinfiltration) scheint unumgänglich.
Alle Filtrationsversuche mit Watte, Glaswatte oder Gaze scheiterten, danach eini¬
gen Minuten auch diese groben Filter verstopften.
Auch eine Nutsche von 10 cm Durchmesser mit eingelegtem Rundfilter LS-14
Schleicher & Schüll filtrierte nach 10 Min nur noch tropfenweise, selbst bei Anwen¬
dung von Vakuum, das jedoch schon wegen der Menstruumverdunstung nicht in Fra¬
ge kommt.
Am besten bewährte sich bei den wenig gewirbelten Extraktlösungen (10 000 U/
Min während 5 Min) unter den Trennungsmethoden durch Filtration die im Abschnitt
821 beschriebene Aufteilung in die Grob- und Feinfiltration (s. pg. 63). Der Zeit¬
aufwand ist jedoch hier wesentlich grösser als in den nicht gewirbelten Extrakten.
Dieses Filtrationsverfahren wurde daher nur für die entsprechenden Vergleichsver¬
suche verwendet.
Bei der Trennung von extrahierter Droge und Extraktlösung in zwei aufeinan¬
derfolgenden Prozessen a) und b) wurden folgende Filtrationszeiten festgestellt:

- 65 -
Tab. 11 Filtrationszeiten fur die Grobfiltration (a) und die Feinfiltration (b)
Extraktionsgemisch: *) nach a) nach b)
40 g mit 5000 U/Mm 1 Stunde extrahierte 30 Mm 70 Min
Chinarinde mit 500 g Menstruum (Vorversuch)Ausbeute: 346 g Extraktlosung
175 g feuchter Drogenrest
50 g nicht extrahierte Chinarinde mit 500 g 70 Mm 100 Mm
Menstruum
Ausbeute: 380 g Extraktlosung158 g feuchter Drogenrest
50 g Chinarinde mit 500 g Menstruum mit 10 000 240 Min 260 Min
U/Min wahrend 5 Minuten gewirbeltAusbeute: 371 g Extraktlosung
156 g feuchter Drogenrest
*) Diese Mischungen wurden z. T. aus Versuchsruckstanden nur fur diese Filtrations-
versuche hergestellt.
a) = Grobfiltration b) = Feinfiltration (s.pg. 63)
Diese Filtration erwies sich somit als zu stark von der Korngrosse der Droge
abhangig, was der letzte Versuch deutlich zeigt. Es wurde daher nach einem allge¬
mein brauchbaren Trennungsverfahren gesucht.
822.2 Zentrifugleren
Fur das mit 10 000 U/Min wahrend 5 Minuten gewirbelte Extraktionsgemisch
genügte eine minimale Zentrifugierzeit von 20 Min bei 2500 U/Min zur Klarung. Da
die Losungsmittelverdunstung aus den gefüllten, austarierten Zentrifugenglasern in¬
folge verschiedener Fullhohen unterschiedlich ist, wurde aus Sicherheitsgründen die
Maximaldrehzahl von ca. 3000 U/Min nicht voll ausgenutzt.
Um auch feinste Suspensionen sicher zu klaren, werden alle Extraktionsgemi-
sche nach erfolgter Wirbelextraktion 35 Mm bei 2500 U/Min zentrifugiert (siehe auch
unter 813 Herstellung der Extraktlosungen, Turboextraktion, pg. 62).
Die Filtration dient nur zu Vergleichszwecken in Spezialfällen.
Bei 2500 U/Min ergibt sich im Schwerpunkt der Flüssigkeit im Zentrifugenglas
eine Zentrifugalbeschleunigung von 698- g, wobei g in diesem Falle die Erdbeschleuni¬
gung bedeutet.
Berechnung:
P = auf ein Teilchen wirkende Schwerkraft
P' = Zentrifugalkraft (Zentralkraft)m = Masse eines Teilchens g' = Zentrifugalbeschleunigung

- 66 -
v = Geschwindigkeit eines Teilchens im Schwerpunkt der Flüssigkeits¬säule im Zentrifugenglas auf seiner Kreisbahn
r = Bahnradius dieses Teilchens mit der Geschwindigkeit v. Angabe in m.
n = Drehzahl der Zentrifuge, angegeben in U/Min.g = Erdbeschleunigung (9, 81 m • sec-2)
Auf Grund des Newton'schen Kraftwirkungsgesetztes ist P = nvg
oder n„ 2 2
r>t iv
„
,n-2irr>2 1 n -ir . rP
'= m • g1 = m • — = m • (———) • - = ——— m&
r 60 r 900
2 2
g. =
250° •*. r = 68 500 sec"2 • r = 68 500 • 0,1 m • sec"2
900 sec2
6850
9,81= 698 und somit ist P' = 698 P
Die auf die Teilchen wirkende Zentrifugalkraft ist siebenhundertmal grösser
als die entsprechende Gravitationskraft der Erde.
822.3 Filtration des in zwei Hälften aufgeteilten Extraktionsge¬
misches unter analogen zeitlichen Bedingungen, wie sie
beim Z entr if ugier en in zwei Chargen auf treten (s. 813,pg. 62)
Jede Hälfte wird dabei grobfiltriert nach a) (s.pg. 63/64), statt zentrifugiert,
und anschliessend feinfiltriert nach b) (s.pg.64).
Diese Methode ist nur anwendbar bei Drogenmaterial mit wenig pulverförmi-
gen Anteilen. Sie wird in dieser Arbeit nur für die nicht extrahierte Droge verwen¬
det, welche sofort nach Vermischung mit dem Menstruum wieder von diesem getrennt
wird. Ein Vergleich mit der Zentrifugierung als Trennmethode gibt Hinweise auf die
Nachextraktion der Droge durch das reine Zentrifugieren (s.pg. 114 ).
83 Messgrössen
Alle Bestimmungen, mit Ausnahme der Siebanalyse, wurden im Doppel ausge¬
führt. Für eine zweite Siebanalyse war jeweils zu wenig Droge vorhanden.

- 67 -
831 in allen Extrakten
wurden bestimmt:
a) Alkaloide (Gehalt in Prozent der Extraktlösung)b) Gerbstoffe ( " " " " " )c) Totalextraktivstoffe ( " " " " " )d) Dichte in g/cm3e) Viskosität in cP (= Centipoise)f ) Daneben wurden alle Lösungsmittelverluste sowie die Gewichte von dekan¬
tierter Extraktlösung und abzentrifugierter Droge vor und nach der Trock¬
nung festgehalten. Nur der Menstruumverlust während der Extraktion wurde
ergänzt. Mit Hilfe dieser Gewichtsangaben kann auch die in der Droge zu¬
rückgehaltene Extraktlösung berechnet werden.
832 in allen Drogenrückständen
wurde bestimmt:
Die Korngrössenverteilung, d.h. der Anteil der einzelnen Siebfraktionen in
Prozent des Gewichtes der extrahierten Droge.
84 Bestimmungsmethoden
841 Gesamtalkaloide
Die Gesamtalkaloide in Chinarinde und deren Präparaten werden meist nach
der klassischen Methode wie folgt quantitativ bestimmt:
a) Aufschluss der Alkaloidkomplexe mit angesäuertem Wasser.
b) Ausschütteln des alkalisierten Aufschlusses mit Aether-Chloroform-Mi-
schung.c) Destillation des Lösungsmittels und anschliessend Titration der in Alkohol
und Wasser gelösten Alkaloide.
Diese Methode benützt auch die Ph.Helv. V, Suppl.I ', gestützt auf Arbeiten
von Wojahn 'und Vogel und Eubel '. Nach ihr arbeiteten auch Fein¬
stein125', Graetzer76'und Fuchs126'. Mit der Titration der China-Alkaloide
127)befasste sich Poethke .
Er empfiehlt unter anderem Methylrot, evtl. gemischt
mit Methylenblau. Zudem sei eine Alkoholkonzentration der Alkaloidlösung von 30 -
128)40 % am günstigsten. Saunders u. Srivastava empfehlen eine potentiome-

- 68 -
1291
trische Titration der Alkaloide. Lebrun und Mitarbeiter ' beschreiben schliess¬
lich die "Brüsseler Methode vom Mai 1949". Dabei werden die Alkaloide alkalisch
aufgeschlossen, dann mit Benzol extrahiert, die Hydrochloride gebildet und die Ge-
samtalkaloide titriert. Chinin und Cinchonidin werden in die entsprechenden Tartra¬
te übergeführt. Im Tartratgemisch wird der Chininsalzanteil bestimmt. Mittels Ta¬
belle kann aus der Linksdrehung der Tartrate der Gehalt an wasserfreiem Chinin
und Cinchonidin errechnet werden.
Mit der raschen Entwicklung der Kunstharzindustrie wurden auch speziell grob¬
porige Ionenaustauscher in den Handel gebracht, die evtl. auch für den Austausch der
grossen Chininmoleküle geeignet sind.
Jindra u. Pohorsky'
geben eine Vorschrift zur Bestimmung von Alka-
loiden, unter anderem auch für Chinarinde und -Extrakt. Sie arbeiteten mit dem
schwach basischen Anionen-Austauscher AMBERLITE IR-4B (Alkaloide aufschlies-
sen - mit Aether bei NHq-Ueberschuss ausschütteln - Verdampfungsrückstand des
aliquoten Teils in HgSO^ lösen und über die Säule giessen - Eluat potentiometrisch
titrieren). Furr er131' bestimmte die Alkaloide in Chinarinde und Chinatrocken¬
extrakt mittels Säulen- und Kontaktverfahren mit Hilfe des stark sauren Kationen-
Austauschers DUOLITE-C-10. Fischer'arbeitete mit dem stark basischen
Anionen-Austauscher DOWEX-2 im Schüttelverfahren bei 40 - 45°C. Die Methode
soll allgemein für Alkaloide anwendbar sein. Eine später veröffentlichte Untersu-
133)chung vom gleichen Autor gibt Beispiele von Morphin- und Chininbestimmungen.
1341
Kamp' arbeitet mit dem Kationenaustauscher IMAC-C-22 nach der Methode von
131)Fur rer
Die folgenden Autoren arbeiteten schliesslich mit Al90„-Säulen. Tunmann1351
u. H üb mann ' studierten die Alkaloidbasen-Chromatographie auf saurem, Me-1361
thylrot enthaltendem AlgO,. Pfandl und Klotz ' benützten basisches AUO,
und eluierten mit Alkohol von 86 Gew.%.
Im Folgenden soll untersucht werden, ob sich die von Furr er vorgeschlage¬
ne Methode unter Verwendung z.T. neuerer Austauscherharze für die Alkaloidbe-
stimmung in Chinapräparaten eigne. Die Ionenaustauscher-Methode und diejenige
der Ph.Helv. V, Suppl. I, sollen bezüglich Genauigkeit und Zeitaufwand miteinander
verglichen werden.
841.1 Alkaloidbe Stimmung in Chinin-HC 1-Lösung mittels Ionen¬
austauschern .
Diese Vorversuche sollen einen Hinweis geben, ob die vorgesehenen Kationen¬
austauscher für die grossen Chininmoleküle brauchbar sind.

- 69 -
Die verwendeten, stark sauren Kationenaustauscher sind:
Harz: Vernet- Gerüst: aktive Gruppen:
zung:
NALCITEjx HCR-1 Zu- Polystyrol + Divinylbenzol Aryl-SOgNanaii— >t H n mHCR-2
HCR-4
me: 1-4
DUOLITE2v C-10 gross- Phenolharz -CHgSOgNa" C-25 porig Kohlenwasserstoff Aryl-SOnNa
131)Gearbeitet wurde nach den Vorschlägen von Fur rer
'mit Säulen. Die in
der Na+-Form in feuchtem Zustand gelieferten Harze (DUOLITE-C-10 trocken gelie¬
fert) wurden erst mit HCl-2n in die H -Form und anschliessend mitNH, ca. 10-proz.
in die NH.+-Form übergeführt.
Die grobkörnigen DUOLITE-Harze wurden zerkleinert und so gemischt, dass
im feuchten Zustand die gleiche Korngrössenverteilung wiebeidenNALCITE-Harzen
resultierte.
Für die Vorversuche diente eine Stammlösung von Chinin-HCl mit einem durch
alkalische Ausschüttelung bestimmten Gehalt an Chininbase von 1,238 % (= 97, 26 %
der Einwaage an Base). Chinin-Einwaage (als Chinin-HCl mit 5,12 % H,0) entspre¬
chend 1,273 % Base. Dies ist die Grundlage für alle weiteren Berechnungen.
Bei den Bestimmungen wurde wie folgt vorgegangen:
Harzsäule: 8 x 200 mm (Harzvolumen 10 ml/NH4+-Form; 90 % des Harzes von
Korngrösse 0,17 - 0,47 mm (ideal nach Fur r er : 0,17 -0,22 mm)).Rohrlänge 300 mm. Durchflussgeschwindigkeit allgemein 1 ml pro Min.
1) Aufguss von genau 10 ml Chinin-HCl-Stammlösung auf die Säule.
2) Mit Wasser die Säule spühlen bis Abfluss Cl"-frei.
3) Elution des Chinins mit NH, 5 % in Spiritus.
4) Austauscher NH?" -frei waschen.
Die Ausbeuten, ausgedrückt in Prozent der Einwaage, sind in der folgenden
Tabelle 13 zusammengefasst.
Die Durchflussgeschwindigkeit durch die Kolonne war 1 ml pro Minute. Der
Prozess bis und mit Elution dauerte 100 Minuten. Das anschliessende Rückspülen
dauerte meist länger als 290 Minuten.
1) Hersteller: National Aluminate Corporation, Chicago 38, 6216 West, 66th Place.
Vertretung: Hydrochemie, Dreikönigstrasse 21, Claridenhof, Zürich.
2) Hersteller: Chemical Process Company, Redwood City, California, P.O. Box829.
Vertretung: Hydrochemie, Zürich.

- 70 -
Tab. 13 Gefundene Chininbase in Prozent der Einwaage mit verschiedenen Ionen¬
austauschermaterialien
Nalcite-HCR-1 Nalcite-HCR-2 Nalcite-HCR-4
No Ausbeute Zeit
% Min
AI 98,23 400
A4 96,24 468
No Ausbeute Zeit
% Min
A2 100,22 390
A5 92,96 535
A7 102,31 573
No Ausbeute Zeit
% Min
A3 95,89 430
A6 94,40 966
A8 102,23 1460
A9 98,80 1400
Duolite-C-10 Duolite-C-25
No Ausbeute Zeit
% Min
A10 97,09 495
A12 97,22 712
A14 97,46 1079
No Ausbeute Zeit
% Min
All 95,62 409
A13 95,01 500
A15 96,11 675
Die Einwaage war jeweils
0,1273 g Base.
Die Duolite-Harze ergeben geringere Streuung der Resultate als die Nalcite-
Austauscher, letztere dagegen höhere Ausbeuten.131)
Da schon Fur r er' mit Duolite-C-10-Harz arbeitete, wurden in diesem
Falle die Nalcite-Harze zu Versuchen mit Chinarinde herangezogen.
841.2 Die Bestimmung des Ge samtalkaloidgehalte s in Chinarinde
841.21 Die Bestimmung mittels Ionenaustauschern
Die Austauschersäule wird wie oben (vergleiche a)) vorbereitet. Der Versuch
wird jedoch wie folgt durchgeführt:
Zeit:
Min
1) Aufschluss der Alkaloide nach der Ph.Helv. V, Suppl.I-Methode 30
2) Heissen Aufschluss durch ein Papierfilter auf die Kolonne giessen 15
3) Rückstand viermal mit heisser, 2-proz. Ameisensäure auswaschen und
durch das Filter auf die Kolonne giessen 40
4) Kolonne dreimal mit je 10 ml destilliertem Wasser nachspülen 30
5) Entfärbung der Säule mit 50 ml ammoniakalischer Ammoniumchlorid-
Lösung (= Entfärber) 30
6) Kolonne dreimal mit 10 ml destilliertem Wasser auswaschen 30
7) Es wurden in unseren Versuchen alle Filtrate aufgefangen und auf Alka¬
loide untersucht

- 71 -
Zeit:
Min
8) Elution mit alkoholischer, 5-proz. Ammoniaklösung 55
9) Rückspülen der Kolonne Minimum: 190
10) Lösungsmittel verdampfen 15
11) Titration nach Ph.Helv.V 5_Gesamter Zeitaufwand ohne Aufschluss der Alkaloide: 210
Gesamter Zeitaufwnad mit Aufschluss der Alkaloide: 240
Bei diesen Versuchen zeigten sich folgende Schwierigkeiten:
1) Schon bei der Elution der Farbstoffe aus den Kolonnen konnte ein geringer Alka¬
loid-Durchbruch festgestellt werden. Alle abfliessenden Lösungen, mit Ausnahme
des Haupteluates, das nach Vorschrift aufgearbeitet wurde, wurden daher vereinigt,
eingeengt und darin nach Ph.Helv.V, Suppl.I, wie in Chinarindenauszügen der Ge-
samtalkaloidgehalt bestimmt.
2) Die Elution erfolge meist ziemlich langsam, so dass oft mehrere 100 ml Eluier-
mittel benötigt wurden.
3) Trotz der vorgängigen Entfärbung waren die Eluate noch etwas rotbraun gefärbt,
so dass der Mischindikator (Methylrot 0,1-proz. 1 T + Methylenblau 0,05-proz.137)
1 T) , sogenannter Tashiro-Indikator, verwendet werden musste. Auch127)
Poethke und Horn'
empfehlen diese Indikatormischung mit Umschlag grün-
rotviolett (sauer).
4) Trotz Berücksichtigung der Alkaloidgehalte in allen abfliessenden Lösungen sind
die Ausbeuten sehr unterschiedlich.
Die Resultate sind in der folgenden Tabelle 14 zusammengefasst:

- 72 -
Tab. 14 Resultate und Zeitaufwand der Gesamtalkaloidbestimmung in Chinarinde
mittels verschiedener Kationenaustauscherharze
nach Ph.V.x totale Ausbeute:
Ver¬
such
Harz Eluier-
mittel
Eluat Ent-
färber2)%
von Droge
% der
Alkaloide
Zeit
Min
A16 HCR-1 E - 0,685,88
9,7083,30
420
AI 7 HCR-2 E - 0,484,15
9,8984,91 390
AI8 HCR-4 E+Er - 0,191,61
8,7174,75 490
AI 9 HCR-1 Er+E' -
_
7,8667,53 490
A20 HCR-2 E' - 0,191,61
7,9768,47 700
A21 HCR-4 E1 - 0,191,61
6,4255,09 460
A22 HCR-1 E' 9,2979,73
1,3711,76
10,6691,50 630
A23 HCR-2 E* 8,2670,92
0,726,15
10,1186,83 750
A24 HCR-4 E' 9,3380,10
0,615,21
12,58108,04 670
A25 HCR-1 E'+(E) 9,7583,71
1,6414,04
11,5499,09 540
A26 HCR-4 E' 9,2579,43
0,474,01
11,2896,82 810
1) Nach Ph. V. bedeutet: diese Fraktion wurde eingeengt und anschliessend darin der
Alkaloidgehalt nach Ph.Helv. V wie in unseren Extraktlösungen bestimmt.
2) inklusive Auswaschflüssigkeit.
In der Tabelle 14 bedeutet die obere Zahlenreihe in jedem Versuch jeweils den Al¬
kaloidgehalt in % der Droge, die untere Zahlenreihe denjenigen in % des Alkaloid-
gehaltes der Droge.
Die in den Kolonnen "totale Ausbeute" angegebenen Werte sind Summen von Teil¬
ausbeuten und zwar in den
Versuchen A16-A21: Eluat + evtl. Nacheluate + nach Ph.Helv.V aufgearbeiteteEntfärberlösung.
Versuchen A22-A26: nach Ph.Helv. V aufgearbeitetes Eluat und ebenso behandel¬
te Entfärberlösung. Dazu die Nacheluate.
Eluiermittel:
E = NH3-23,4% 21, 35 g + Spiritus 95 Vol.% ad 100 g
Er = E redestilliert 87, 38 g + 12,62 g NH,- 23,4 % (entsprechend dem NH,-Ver¬
lust)J J
E' = 5 % NH3-Gas in Spir. 96 Vol.%
E und Er haben gleichen NH,-Gehalt. Der Alkoholgehalt nimmt von Er über E
nach E' etwas zu.

- 73 -
Versuch
No.
Alkaloide
%v. DrogeVersuch
No.
Alkaloide
%v. Droge
A27 11,65 A30 11,68
A28 11,65 A31 11,65
A29 11,63 A32 11,62
MA27-•A32 U>6466 ...= 11, 647%
Für alle Harze wurde die Durchflussgeschwindigkeit auf 1 ml/Min reguliert.
Eine Bestimmung dauert daher 4 Stunden ohne Berücksichtigung des Rückspulens der
Kolonnen.
841.22 Bestimmung nach der Methode der Ph.Helv.V, Suppl.I'
Die Ph.Helv. V-Methode gab folgende Resultate:
Tab. 15 Die Genauigkeit der Alkaloidbestimmungsmethode der Ph.Helv. V, Suppl.I,
überprüft an Chinarinde
Die maximalen Abweichungen der
Resultate in Prozent vom arithme¬
tischen Mittel (M) sind:
+ 0,29% - 0,23%
Die Streuung s ist 0,000 43
Die mittlere quadratische Abweichung s ist 0,0207
Eine Bestimmung dauert 120 - 150 Min.
Der Vergleich der beiden Verfahren zeigt eindeutig die Ueberlegenheit der kon¬
ventionellen Arzneibuch-Methode. Diese arbeitet bedeutend rascher, als es mit Ionen¬
austauschern möglich ist und ergibt bessere Alkaloidausbeuten bei geringerer Streu¬
ung der Resultate.
Die Gesamtalkaloide werden daher in unseren Arbeiten nach Ph.Helv.V, Suppl.
I, bestimmt, wobei sich für die Extraktlösungen die nachstehend beschriebene An¬
passung der Bestimmungsmethode als notwendig erwies.
841.3 Die Bestimmung der Gesamtalkaloide in den Extraktlö¬
sungen
50 ml Extraktlösung werden auf dem Wasserbad in einer tarierten Kristalli¬
sierschale von ca. 8 cm Durchmesser zur Trockene verdampft (Dauer ca. 3 Stun¬
den), im Exsiccator 30 Min erkalten gelassen und gewogen.
Dieser Roh-Trockenrückstand wird ausgedrückt in Prozent der in der Droge be¬
stimmten Totalextraktivstoffe und dient zur Schätzung der Gesamtalkaloide.
Von der Extraktlösung werden ungefähr 15 g, entsprechend ca. 0,1 - 0,13 g
geschätztem Alkaloidgehalt, in einen 150 ml Normalschlifferlenmeyer gewogen.

- 74 -
Versuch
No.
Alkaloide
% der Lsg.
A3 3 0,830
A34 0,854
A3 5 0,817
Nach Zugabe von 1,5 g Ameisensäure Ph.Helv. V und, wenn nötig Wasser ad 20 g,
wird die gut durchgemischte Probe so lange in ein siedendes Wasserbad gestellt,
bis der Kolbeninhalt noch maximal 10 g wiegt. Nach dem Abkühlen wird die Probe
auf 10 g aufgefüllt und nach der Vorschrift für Chinarinde nach Ph.Helv. V, Suppl.I,
aufgearbeitet.125}
Dieselbe Methode mit geringen Variationen benützten auch Feinstein,
Graetzer 'und Fuchs '. Feinstein gibt vor dem Einengen der Extrakt¬
lösung (Entfernung des Alkohols) 5 ml HCl-2n zu, Fuchs dagegen 2 ml HCOOH
nach Ph.Helv.V, Suppl.I (Verfahren von Wojahn und Erdelmeier ').Die oben beschriebene Methode ergab folgende Resultate:
Tab. 16 Die Genauigkeit der Alkaloidbestimmungsmethode in den Chinarinde-Extrakt¬
lösungen
Versuch Alkaloide M = arithmetisches Mittel
No. % der Lsg.
A36 0 850Die maximalen Abweichungen in
AT7 0 8fiSProzent von M sind:
A38 0^873 "3'63% +2>97%
MA33-A38 = °'848 % (°>8478 %) LsS- = Lösung
Die Streuung s2 beträgt : 0,000438
Die mittlere quadratische Abweichung ist : 0,020 94
Als Stammlösung für diese Versuche diente eine durch Schüttelmazeration
während 5 Stunden 40 Minuten hergestellte Extraktlösung.
50 g Chinarinde wurden mit 500 g Menstruum Ph.Helv. V angesetzt.
842 Die Bestimmung der Gerbstoffe
842.1 Die Bestimmung der Gerbstoffe in der Droge
Obwohl der therapeutische Wert der Gerbstoffe in Chinarinde umstritten ist,
wird deren Bestimmung einen besseren Einblick in die Extraktion dieser Droge er¬
möglichen. Dies ist schon deshalb wichtig, weil die Chinaalkaloide z.T. gebunden
an Gerbstoffe vorliegen.
Uebersicht über die bisher beschriebenen Gerbstoffbestimmungs-Methoden:
Eine reichhaltige Literatur-Zusammenstellung betreffend die quantitative
Gerbstoffanalyse geben G n a m m undFlück.Danach kommen folgende
Methoden in Betracht:

- 75 -
1) Ausnutzung der Aktivität der Gerbstoffe gegen biologisches Material (diese Metho¬
den beruhen auf Eigenschaften, die auch bei der therapeutischen Anwendung von
Gerbstoffen wichtig sind).
a) gravi metrische Hautpulvermethode (z.B. internationale Hautpulvermethode und
141)Vorschlag fur die Ph.Helv. VI ). Man unterscheidet je nach Arbeitsweise:
<x) Schuttelmethode142)143)
ß) Filtermethode144)145)
Herr mann betont, dass die sichersten analytischen Werte mit der interna¬
tionalen Hautpulvermethode gewonnen werden, wenn alle Punkte genau befolgt wer¬
den. In einer spateren Arbeit gibt der Verfasser einen Ueberblick über quali-147)
tative und quantitative Gerbstoffbestimmungsmethoden. Nick empfiehlt, die
zeitraubende Hautpulvermethode durch eine Mikro-Bestimmungsmethode mit Haut¬
pulver zu ersetzen. Das homöopathische Arzneibuch bestimmt die Gerbstoffe
mittels Hautpulvermethode mit Veraschung oder kolorimetrisch nach Fallung mit
149)Zn-Acetat. In emer neueren Arbeit erhielt Her rmann mittels gravimetri-
scher Hautpulvermethode, photometrischer Phosphorwolframat-Methode und ei¬
nem massanalytischen Verfahren mit Na-Hypojodit gut vergleichbare Werte. Er
weist darauf hin, dass auch Phlobaphene, Polyphenolcarbonsauren und Catechine
an Hautpulver adsorbiert werden.
b) kolonmetrische Hautpulvermethode
Prinzip: vor und nach der Entgerbung der Losung mittels Hautpulver werden
die mit einem geeigneten Reagens, z.B. Phosphorwolframsaure nach Fol in,
erzeugten Färbungen kolonmetnert. Die Intensitatsdifferenz dieser Färbungen
kann mittels Eichkurve in Gerbstoffe umgerechnet werden. Dieses Verfahren deu-
149)tet Herr mann ' schon an. Bei sehr stark gefärbten Extrakten, die durch die
Entgerbung stark aufgehellt werden, muss evtl. die Eigenfarbung berücksichtigt
werden (Blindmessung mit Wasser statt Folin-Reagens und Sodalosung).
Der Nachteil der direkten kolonmetrischen Methoden, siehe pg. 76, dass auch
Nichtgerbstoffe wie Flavonole die gleiche Färbung ergeben, fallt bei dieser Methode
dahin, da dieser Fehler bei der Differenzbildung der Färbungen eliminiert wird.151)
c) Agglutinationsmethode'
Prinzip: Diejenige Gerbstoffmenge, die gerade noch totale Agglutination einer
bestimmten Menge roter Blutkörperchen in eiweissfreier (= gewaschener) Suspen¬
sion hervorruft, dient zur Berechnung des Gerbstoffgehaltes. Die totale Aggluti¬
nation wird nach 12 Stunden festgestellt:
mikroskopisch, oder
durch Bestimmung der Blaufärbung mit Eisenchloridlosung.
Nachteil: grosse Streuung bei hohem Gerbstoffgehalt; Blutbeschaffung.
d) kolonmetrische Agglutinationsmethode ':
Das Prinzip ist genau dasselbe wie bei der kolonmetrischen Hautpulvermethode,
nur dass die Entgerbung durch rote Blutkörperchen (von Hammel, Rind oder Zie-

- 76 -
ge) erfolgt. Die Resultate sind stets niedriger als nach der Hautpulvermethode, die
auch Phlobaphene, Farbstoffe und Polyoxycarbonsäuren als Gerbstoffe bestimmt. In
152)seiner neuesten Arbeit verwendet Gstirner Milcheiweiss zur Entgerbung der
Lösung. Vor und nach der Entgerbung wird mit Phosphorwolframsäure in alkalischer
Lösung der Gerbstoffgehalt bestimmt.
2) Fällungsmethoden mitSalzen von Cu, Zn, Sn, Pb etc. :
Prinzip: Die Gerbstoffe werden mit Metallsalzen gefällt und nach einem der fol¬
genden Verfahren bestimmt:
Wägung des Niederschlages
Bestimmung des nicht verbrauchten Metallsalzes und Errechnung des Gerb¬
stoffgehaltes
Waschen des Niederschlages und Ueberführung mittels geeigneter Reagentienin eine kolorimetrisch zu bestimmende Lösung.
153)Fuchs verwendet eine gravimetrische Fällungsmethode, wobei jedoch erst die
Alkaloide durch Aufschluss und alkalische Ausschüttelung mit Aether und Chloroform
entfernt wurden.145)
Herr mann 'ist der Ansicht, dass Methoden, die nicht auf der Gerbwirkung der
Gerbstoffe beruhen, sondern auf deren Fällung oder Löslichkeit, ungeeignet seien;
Metallsalzfällungen seien angängig ', wenn neben viel Gerbstoff wenig Flavone,
Phenolcarbonsäuren und durch Schwermetalle fällbare Stoffe vorhanden seien. H err-
149)mann erhielt bei 10 Stoffen mit der gravimetrischen Cu-Acetat-Methode zu tie¬
fe Werte, trotzdem auch NichtgerbstoffewieKaffesäure als Gerbstoff erfasst wurden.154)
Fuchs und Mitarbeiter 'empfehlen eine Grenzwert-Fällungsmethode, wobei mit
einer für vollkommene Gerbstoffausfällung ungenügenden Menge Schwermetallsalz¬
lösung gefällt, dann filtriert wird. Entsteht bei weiterer Zugabe von Reagens im Fil¬
trat noch ein Gerbstoffniederschlag, so enthält die Droge genügend Gerbstoff.
155)Gnamm erwähnt schon die kolorimetrische Gerbstoffbestimmungsmethode von
Menaul mit Na-Wolframat (Vorschlag 1923; Fällung mit Bleiacetat, Lösung in
Schwefelsäure, Färbung mit Na-Wolframat, Vergleich mit Tanninlösung von bekann¬
tem Gerbstoffgehalt). Diemair und Mitarbeiter'fällten die Gerbstoffe aus Wein
mit Gelatinelösung und Bleiacetat. Der abzentrifugierte Niederschlag wird aufgear¬
beitet und kolorimetrisch mit Na-Wolframat Reagens ausgewertet. Auch die Arbeiten157)
von Friedrich ' basieren auf den Untersuchungen von Menaul und Diemair
(siehe unter: 842.121.1, Aufnahme der Extinktionskurve). Nick ' machte darauf
aufmerksam, dass einmal ausgefällter Gerbstoff nur schwer und unvollständig wieder
in Lösung zu bringen ist. Richter ' fällte die Gerbstoffe mit Zinkacetat. Der
zentrifugierte Niederschlag wird aufgearbeitet und mit Phosphorwolfram säure in

- 77 -
soda-alkalischer Lösung kolorimetriert. Die Werte sollen nahe bei den nach der
internationalen Hautpulvermethode gewonnenen Resultaten liegen. Auch nach
F lück'liefern diese Methoden meist höhere, gelegentlich auch niedrigere
Werte als die internationale Hautpulvermethode. Die Differenzen betragen bis
über 20 %. Der Gerbstoff wird nicht quantitativ gefällt, dafür werden auch ande¬
re Stoffe aus dem Drogenauszug mitgerissen.
3) Verfahren, die die Löslichkeitsverhältnisse der Gerbstoff benützen, wie z. B. die159)
S chultemethode '. Diese Verfahren geben praktisch mit jedem Pflanzen¬
material "Gerbstoffgehalte" und werden daher heute als allgemeine Methoden ab¬
gelehnt.
4) Oxydimetrische Methoden: Der Oxydationswert von Auszügen vor und nach dem
Ausschütteln mit
Carbo adsorbens, oder
Gelatine-NaCl-Lösung
wird bestimmt. So arbeitet z.B. die massanalytische Methode nach Löwen¬
thal.Die Methode ergibt meistens zu hohe Werte, da auch Nichtgerbstoffe
145)adsorbiert werden. Dies bestätigt auch Herrmann
,der die Methode in ei¬
ner späteren Arbeit als einfach und für sehr verdünnte Lösungen geeignet be-lfil)
zeichnet. Bolotnikow u. Schreiber 'schlugen diese Methode für das
neue russische Arzneibuch vor.
842.11 Die Bestimmung der Gerbstoffe nach dem Vorschlag für die Ph.Helv. VI
In Anlehnung an die bewährte internationale Hautpulvermethode nach dem
Schüttelverfahren wurde ein etwas vereinfachter Vorschlag einer Ph.Helv. VI-Vor-
schrift - in Zukunft kurz Ph. Helv. VI-Methode benannt - ausgearbeitet (Vorschlag141)
vom 4.1.1956 ')• Die Methode der Ph.Helv. VI soll nun auf ihre Eignung für un¬
sere Gerbstoffbestimmungen geprüft werden. Rutschmann ' fand mit ihr für
Chinarinde im Mittel 5, 5 % Gerbstoffe. Die maximalen Abweichungen vom arithme¬
tischen Mittel waren + 18,0 % - 20,0 %.
842.111 Vorversuche mit Kastanienholzextrakt von bekannter Zusammensetzung
Es war leider unmöglich, Chinarindenpulver zu beschaffen, dessen Gerbstoff¬
gehalt schon nach dem Schüttelverfahren der international angewendeten Hautpulver¬
methode bestimmt worden war. Es wurde daher zur Abklärung der unter 842.11 be¬
schriebenen Frage unter anderem als Testsubstanz Kastanienholzextrakt benützt,
der auch der "Eidgenössischen Materialprüfungs- und Versuchsanstalt für Industrie,

- 78 -
Bauwesen und Gewerbe" (EMPA), Abteilung III, in St. Gallen zur Ueberprüfung ihrer
Methode dient und uns in verdankenswerter Weise zur Verfügung gestellt wurde. Die
Zusammensetzung dieses Musters wurde uns bekanntgegeben, wobei die Werte einzig
ab und zu nach dem neu zu bestimmenden Wassergehalt zu korrigieren waren.
Methodik der Gerbstoffbestimmung mittels Hautpulver:
Prinzip:
Hautpulver, als tierisches Protein, wird durch eine gerbstoffhaltige Lösung de¬
naturiert und die Lösung dadurch entgerbt. Dazu wird die Lösung entweder mit Haut¬
pulver geschüttelt (Schüttelverfahren), oder durch Hautpulver filtriert (Filterverfahren).
Vor und nach dieser Entgerbung wird der Trockenrückstand der Lösung bestimmt
und die Differenz als Gerbstoffgehalt berechnet.141)
Die für die Ph.Helv. VI vorgesehene Methode lautet zur Zeit folgendermassen ':
"Die im Einzelartikel angegebene Menge Droge von vorgeschriebenem Zerklei¬
nerungsgrad wird in einem Erlenmeyerkolben von 750 ml Inhalt mit 250 ml Wasser
Übergossen, hierauf am Rückflusskühler unter gelegentlichem Umschwenken so zum
Sieden erhitzt, dass bis zum Eintritt des Siedens 10 Min nötig sind, und anschliessend
während 30 Min gekocht. Hierauf wird unter dem Wasserstrahl unter Umschwenken
auf Zimmertemperatur abgekühlt und durch ein Faltenfilter von 10 cm Durchmesser
klarfiltriert (= Filtrat I), wobei die ersten 10 ml Filtrat zu verwerfen sind.
50 ml des Filtrates werden in einem tarierten Veraschungsgefäss (Porzellan¬tiegel, Nickelschale etc.) eingedampft, der Rückstand bei 103 - 105°C bis zur Ge¬
wichtskonstanz getrocknet und gewogen (= T<). Der Trockenrückstand wird verascht
und gewogen (= A^).Weitere 70 ml des Filtrates werden mit 3 g Hautpulver-R während 30 Min stän¬
dig geschüttelt. Hierauf wird durch ein Leinen- oder Baumwolltuch gegossen und der
Rückstand leicht von Hand ausgepresst, und die abgetrennte Flüssigkeit durch ein
Filter von 10 cm Durchmesser filtriert. Vom klaren Filtrat (= Filtrat II) werden50 ml wie oben angegeben zur Trockene verdampft, der Rückstand bei 103° - 105°C
getrocknet und gewogen (= T2). Der Trockenrückstand wird verascht und gewogen(= A2).
Vom Hautpulver wird in einem Blindversuch unter den gleichen Mengenverhält¬nissen der Anteil an wasserlöslichen Bestandteilen durch Ermittlung des Trocken¬
rückstandes bestimmt (= H).Die Berechnung der Gerbstoffmenge erfolgt nach folgendem Schema:
O = lösliche organische Stoffe
M = Nichtgerbstoffe plus Lösliches aus Hautpulver-R
N = Nichtgerbstoffe
Gerbstoff in V5 der eingewogenen Droge."
Diese Vorschrift wurde so weit als möglich eingehalten.
Die verwendeten Apparate und Untensilien:
Um jeglichen störenden Effekt von Kork oder Gummi auszuschalten, wird die
Extraktion der Gerbstoffe in einer Normalschliff-Apparatur ausgeführt, und zwar in
einem Rundkolben von 500 ml. Die Filtration der gerb Stoffhaltigen, sowie der ent-
Tl -Al
T2 " A2M - H

- 79 -
gerbten, vom Hautpulver befreiten Extraktlosungen erfolgt erst grob durch Watte,
dann durch gewohnliche, gefaltete Rundfilter von 11 cm Durchmesser, Typ LS-14
(Schleicher & Schull). Durch einmalige Filtration wird völlige Klarung erreicht. Als
Veraschungsgefasse dienen offene Porzellanschalen von 8-9 cm Durchmesser. Ver¬
ascht wird auf einem Dreibein mit gelochtem Asbestcarton, wobei der Lochdurchmes¬
ser jeweils ca. 3/4 des Schalendurchmessers betragt.
Das Schuttein der Gerbstofflosung mit Hautpulver erfolgt in 150 ml Normal-
schhff-Erlenmeyerkolben auf einer elektrischen Schuttelmaschine (Horizontal-Am¬
plitude: 16 cm; Frequenz: 156 Hin- und Herbewegungen/Min) wahrend 30 Minuten.
Da ein Baumwolltuch zuviel Flüssigkeit aufsaugt ,wird die entgerbte Ex¬
traktlosung erst durch ein Leinentuch gegossen, das zur Entfernung der Appretur1421
mit destilliertem Wasser ausgekocht wurde '. Die Trocknung samtlicher Ruck¬
stande bei 103 - 105 C erfolgt im elektrischen Trockenschrank.
Eingesetzte Mengen an Gerbstoffdroge:
Die internationale Hautpulvermethode der Gerbereifachleute schreibt vor, dass
sich in der zu entgerbenden Losung pro Liter max. 4 - 0, 25 g an Hautpulver zu ad-
142)143)sorbierende Stoffe oder max. 10 g Trockenruckstand befinden dürfen
Auf Grund der neu bestimmten Zusammensetzung unseres Testmusters müs¬
sen, um 1 g gerbende Stoffe in 250 ml Extraktlosung zu erhalten, 1,4144 g Extrakt
eingewogen werden, vergl. Tabelle 17.
Tab. 17 Zusammensetzung unserer Testsubstanz Kastamenholzextrakt, nach dem
Schuttelverfahren der internationalen Hautpulvermethode bestimmt:
I II III IV
Datum 13.11.57 16.1.58 14.2.58
gerbende Stoffe 71,9% 70,70 % 70,57 % 70,46 %
lösliche Nichtgerbstoffe 18,1% 17,80% 17,77% 17,74 %
Unlösliches 0,0% 0,0 % 0,0 % 0,0 %
Wasser 10,0% 11,50% 11,66% 11,80%
Total 100,0% 100,0 % 100,0 % 100,0 %
notige Einwaage :
(fur 1 g Gerbstoff)1,4144 g 1,4170 g 1,4192 g
Kolonne I
Kolonnen II, in, IVAngaben der E. M. P. A. (s.pg. 77/78).errechnete Zusammensetzung nach erneuter
Bestimmung des Wassergehaltes.

- 80 -
Eingesetzte Hautpulvermenge:
Da die Resultate der Gerbstoffbestimmungsmethoden stark von der Qualität des
Hautpulvers abhängen, verschafften wir uns solches von der Gerbereischule Freiberg
in Sachsen, wie es auch von der E.M. P. A. in St. Gallen verwendet und verkauft wird.
Zur Entgerbung mittels Hautpulver wurden 75 ml statt der vorschriftsgemässen
70 ml filtrierter Extraktlösung abpipettiert, um sicher 50 ml entgerbte Extraktlösung
zurückgewinnen zu können.
Die für die Ph.Helv. VI vorgeschlagene Hautpulvermethode macht keine Anga¬
ben über den zulässigen Wassergehalt des Hautpulvers, der aber, für konstante Ein¬
waagen mit verschiedenen Hautpulvermustern (in Apotheken, Industrie etc.), bekannt
sein muss. Bei der internationalen Methode wird das Hautpulver vor dem Schütteln
mit der Gerbstofflösung auf einen normierten Feuchtigkeitsgehalt gebracht.
In unserem Falle wurde daher der Feuchtigkeitsgehalt nach den allgemeinen
Vorschriften der Ph.Helv.V bestimmt und die genau 3 g des getrockneten Materials
entsprechende Menge eingewogen. Der Wassergehalt des Hautpulvers (Partie 153/
Freiberg) beträgt 11,19 %. Demnach ist die einzuwägende Menge 3, 3780 g.
Die Resultate sind in der folgenden Tabelle zusammengefasst.
Tab. 18 Gerbstoffgehalt von lufttrockenem Kastanienholzextrakt (70, 70 % Gerbstoffe),
bestimmt nach der für die Ph.Helv. VI vorgesehenen Methode:
Versuch
No.
Gerbstoffgehalt (% der
Einwaage)Maximale Abweichungen in % des
arithmetischen Mittels
Ph.Helv. VI dito,-A Ph.Helv.VI dito, -A
Gl 70,58 70,02 -0,85
G2 71,40 71,05 + 1,17
G3 71,03 69,51 -1,02
G4 70,66 69,70
G5 72,31 70,86 +1,57
MG1-G5 71,196 70,228
Ph.Helv. VI dito, -A
Die Streuung s beträgt : 0,494 0,478
Die mittlere quadratische Ab¬
weichung s ist : 0,703 0,691
dito, -A bedeutet: Gerbstoffbestimmung nach Ph.Helv. VI, jedoch ohne Berück-
si chtigung der Aschen der gerbstoffhaltigen (Aj) und der ent¬
gerbten Lösungen (A2) (s.pg. 78).

- 81 -
Schlussfolgerungen aus den bisherigen Resultaten:
Wie die Versuche zeigen, liefert die für die Ph.Helv. VI vorgesehene Hautpul¬
ver-Konventionsmethode, zum Mindesten bei Drogen mit hohem Gerbstoffgehalt,
Resultate, die mit denen des Schüttelverfahrens der internationalen Hautpulverme¬
thode gut übereinstimmen. Die Resultate nach Ph.Helv. VI streuen um den nach der
internationalen Hautpulvermethode gefundenen Gerbstoffgehalt.
Die Abweichungen der Resultate vom arithmetischen Mittel liegen zwar mit ma¬
ximal+1, 57/-0,85 % bedeutend höher als nach der an der E.M. P.A. in St. Gallen
angewandten Methode, deren Ergebnisse erfahrungsgemäss nicht mehr als 0, 5 %
absolut auseinander liegen .
Wenn trotz genau gleichen Vorgehens bei allen Versuchen grössere Unterschie¬
de zwischen den Versuchsergebnissen entstanden, so liegt dies vermutlich an der
weniger schonenden und weniger genau reproduzierbaren Methode der Extraktion
und Filtration der Gerbstoffe bei der Ph.Helv. VI-Methode; dies aus folgenden Grün¬
den:
1) Wenn die Gerbstoffextraktion durch Erwärmen auf einem Asbestdrahtnetz
über offener Flamme vorgenommen wird, besteht, besonders am Anfang,
die Gefahr örtlicher Ueberhitzung oder sogar Verkohlung von Drogenparti¬
keln, wenn die Droge nicht vollkommen benetzt wurde. Die Droge sollte,
sofern diese schwerer als Wasser ist, auf die Flüssigkeit gegeben werden.
2) Das Erhitzen zum Sieden innert 10 Minuten kann nur auf ca. 10 % genau
eingestellt werden.
3) Nach der Extraktion sollte, wie bei der internationalen Vorschrift, auf em
bestimmtes, konstantes Volumen ergänzt werden.
4) Die Filtration dauerte minimal 3 Stunden. Unterschiedliche Filtrationszei¬
ten können, infolge Verdunstens von Lösungsmittel, zu verschiedenen Kon¬
zentrationszunahmen der entgerbten Lösungen und dadurch zu Fehlern füh¬
ren.
Aus den Versuchsergebnissen geht ferner hervor, dass 3 g trockenes Hautpulver auf
75 ml Extraktlösung auch bei hohem Gerbstoffgehalt genügen.
842.112 Hauptversuche mit Succirubra-Rinde
Bei dieser Versuchsreihe wurde, wenn immer möglich, genau gleich vorgegan¬
gen, wie es schon unter 842.111 in der Ph.Helv. VI-Methode (s.pg. 78) bei den Vor¬
versuchen mit Kastanienholzextrakt beschrieben wurde.
Die folgenden Punkte mussten abgeändert werden:
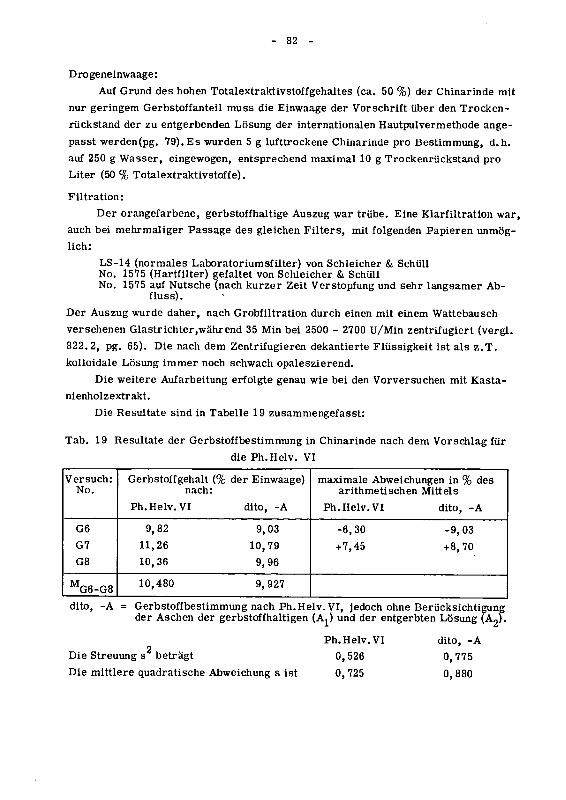
- 82 -
Drogeneinwaage :
Auf Grund des hohen Totalextraktivstoffgehaltes (ca. 50 %) der Chinarinde mit
nur geringem Gerbstoffanteil muss die Einwaage der Vorschrift über den Trocken¬
rückstand der zu entgerbenden Lösung der internationalen Hautpulvermethode ange-
passt werden(pg. 79).Es wurden 5 g lufttrockene Chinarinde pro Bestimmung, d.h.
auf 250 g Wasser, eingewogen, entsprechend maximal 10 g Trockenrückstand pro
Liter (50 % Totalextraktivstoffe).
Filtration:
Der orangefarbene, gerbstoffhaltige Auszug war trübe. Eine Klarfiltration war,
auch bei mehrmaliger Passage des gleichen Filters, mit folgenden Papieren unmög¬
lich:
LS-14 (normales Laboratoriumsfilter) von Schleicher & Schüll
No. 1575 (Hartfilter) gefaltet von Schleicher & Schüll
No. 1575 auf Nutsche (nach kurzer Zeit Verstopfung und sehr langsamer Ab-
fluss).
Der Auszug wurde daher, nach Grobfiltration durch einen mit einem Wattebausch
versehenen Glastrichter,während 35 Min bei 2500 - 2700 U/Min zentrifugiert (vergl.
822.2, pg. 65). Die nach dem Zentrifugieren dekantierte Flüssigkeit ist als z.T.
kolloidale Lösung immer noch schwach opaleszierend.
Die weitere Aufarbeitung erfolgte genau wie bei den Vorversuchen mit Kasta-
nienholzextrakt.
Die Resultate sind in Tabelle 19 zusammengefasst:
Tab. 19 Resultate der Gerbstoffbestimmung in Chinarinde nach dem Vorschlag für
die Ph.Helv. VI
Versuch:
No.
Gerbstoff gehalt (% der Einwaage)nach:
Ph.Helv.VI dito, -A
maximale Abweichungen in % des
arithmetischen Mittels
Ph.Helv.VI dito, -A
G6
G7
G8
9,82 9,03
11,26 10,79
10,36 9,96
-6,30 -9,03
+7,45 +8,70
MG6-G8 10,480 9,927
dito, -A = Gerbstoffbestimmung nach Ph.Helv. VI, jedoch ohne Berücksichtigungder Aschen der gerbstoffhaltigen (A..) und der entgerbten Lösung (A?).
Ph.Helv.VI dito, -A
Die Streuung s beträgt 0,526 0,775
Die mittlere quadratische Abweichung s ist 0,725 0,880

- 83 -
Schlussfolgerung aus den Gerbstoffbestimmungen der Chinarinde:
Trotzdem auch hier, wie beim Kastanienholzextrakt, die drei Versuche paral¬
lel, unter gleichen Bedingungen und gleichzeitig durchgeführt wurden, liegen die Ein¬
zelwerte bedeutend weiter auseinander. Dies kann z.T. auf den viel geringeren Gerb¬
stoffgehalt von Chinarinde, z. T. auf die Differenz bei der Herstellung des Extraktes
aus Chinarinde zurückgeführt werden. Als Vergleich diene die Arbeit von Rut s ch -
mann . Die Autorin fand unter 6 gerbstoffhaltigen Drogen am schlechtesten re¬
produzierbare Werte mit Chinarinde.
Diese Methode arbeitet zu ungenau, besonders unter Berücksichtigung des gros¬
sen Zeitaufwandes für die Wägungen bis zur Gewichtskonstanz, sowohl der leeren
Schalen als auch der eingedampften Auszüge. Es sollte daher eine geeignetere Me¬
thode der Gerbstoffbestimmung gesucht werden.
842.12 Die unseren Bedürfnissen und Möglichkeiten angepasste, kolorimetrische
Hautpulvermethode:
Die Wahl fiel auf die kolorimetrische Bestimmung der Gerbstoffe vor und nach
der Entgerbung mit Hautpulver (s. unter 842.1, pg. 75). Da die Entgerbung mit Haut¬
pulver weitgehend den biochemischen Vorgängen bei der Therapie entspricht, kann
auf Grund dieser Bestimmungen direkt auf die therapeutische Wirksamkeit geschlos¬
sen werden.
Der Ersatz der Gravimetrie durch die Kolorimetrie verspricht eine starke
Vereinfachung bei bedeutender Zeitersparnis. Es ist auch zu erwarten, dass die ko¬
lorimetrische Methode dank ihrer Empfindlichkeit zu besser reproduzierbaren Wer¬
ten führt und somit die hauptsächlichen Fehlerquellen bei der gravimetrischen Haut¬
pulvermethode eliminiert.
Das Kolorimeter:
Für die folgenden Arbeiten wird das Spektralphotometer und Kolorimeter
"Spectronic-20" *) verwendet. Das Instrument erlaubt Messungen in den Bereichen:
375 - 650 mu mit Photozelle RCA - 5581 /5-52 / USA
650 - 950 mu mit Photozelle RCA - IP 40 / 4 - 29A / USA und Rotfiltereinsatz.
Die effektive Bandbreite ist über den ganzen Wellenlängenbereich konstant und be¬
trägt 20 mu.
*) Hersteller: Bausch & Lomb Optical Co., Rochester, N.Y., USA;
Vertretung für die Schweiz: Bachofen & Co., Streulistr. 19, Zürich 7/32.

- 84 -
Der genaueste Ablesebereich des Instrumentes liegt bei 25 - 65 % Transmissi¬
on. Es wurden zylindrische Cuvetten von 1 Zoll (= 2,48 cm) Durchmesser verwendet,
um auch schwächste Färbungen noch erfassen zu können.
Das Kolorimeter erlaubt Extinctions- und Transmissionsgrad-Ablesung. Bei
allen Messungen wurden, dank der linearen Skala, Transmissionsgrade (\^) abgele¬
sen und anhand der Formel £ = 2 - log&j, die Extinktion (£.) berechnet.
Da sich bei verschiedener Stellung der Cuvetten unterschiedliche Resultate
zeigten, wurde die günstigste Cuvettenlage bestimmt. Die Cuvetten wurden zu diesem
Zwecke numeriert, mit Wasser gefüllt und im Cuvettenhalter unter Lichtabschluss
um 360 gedreht unter gleichzeitiger Beobachtung der Transmissionsgrad-Schwan¬
kungen. In der folgenden Zeichnung ist für jede Cuvette der günstigste Bereich an
deren Halter markiert:
1
+ 3%Cuvette:
Schwankungen von :
(bei Drehung um 360° bei 675 mp) ^ ^
Cuvettenstellung: -|—/ \
M : Marke auf Cuvette j I
C : Cuvettenhalter -^A M I
Fig. 6 Die günstigste Stellung der Cuvetten
Der Bereich mit den geringsten Schwankungen bei Drehung der Cuvette wurde
festgestellt, dessen Mitte als günstigste Cuvettenstellung markiert und diese Stel¬
lungen bei allen zukünftigen Messungen eingehalten.
Die Transmissionsgrade der mit Wasser gefüllten Cuvetten:
Bei der vorgesehenen Differenzmessung der Farbintensität fallen bei Verwen¬
dung der gleichen 2 Cuvetten alle Blindwerte weg. Diese Daten wurden daher nur für
den Fall des Verlustes einer Cuvette hier festgehalten. Es wurden unabhängig von
der Wellenlänge (Grauwert) die folgenden Resultate gefunden:
Tab. 20 Transmissionsgrade der Cuvetten
Cuvette: Transmissionsgrad: Extinktion:
1 99,04% 0,00419
2 99,22% 0,003 40
3 100,00% 0,000 00
Für alle Messungen wurde Cuvette 3 mit Wasser gefüllt, Cuvette 2 mit der zu kolo-

- 85 -
rimetrierenden Lösung. Cuvette 1 diente als Reserve.
Herstellung des Phosphorwolframsäure-Reagens.
Es wurde das schon von Gstirner und Mitarbeitern sowie von Rich¬
ter'verwendete Phosphorwolframsäure-Reagens nach Folin benützt.
Herstellung: 100 g Na-Wolframat + 80 ml 85-proz. Phosphorsäure werden mit
750 ml destilliertem Wasser 3 Stunden am Rückflusskühler gekocht.Nach dem Erkalten wird mit Wasser auf 1000 ml ergänzt.
Dieses Reagens ergibt mit Gerbstoffen und gewissen Polyphenolen in alkalischer Lö-
152)sung eine intensive Blaufärbung. G stirner hat die Spezifität dieser Reaktion an
über 20 Substanzen geprüft. Er fand, dass einwertige Phenole nicht reagieren, dage¬
gen zweiwertige o- und p-ständige Phenole. Dreiwerte Phenole mit vicinaler Stellung
der Hydroxylgruppen reagieren stark. Ascorbinsäure reagiert ebenfalls positiv.
842.121 Vorversuche mit Kastanienholzextrakt
Einwaagen:
Unser Kastanienholzextrakt enthält 70, 70 % Gerbstoffe. Auf Grund der interna¬
tionalen Hautpulvermethode muss die zu entgerbende Lösung 4 - 0, 25 g Gerbstoffe
oder 5,658 - 0,35 g Kastanienholzextrakt pro Liter enthalten.
Um schon von Anfang an analog zu diesen Versuchsbedingungen zu arbeiten,
wurde für alle Versuche von einer Stammlösung mit 0,5658 g Kastanienholzextrakt
in 100 ml ausgegangen und diese für die Messungen je nach Bedarf verdünnt.
Herstellung der Messlösungen:
Z.T. gestützt auf die Arbeiten von Gstirner 'und Richter 'wurden
alle Messlösungen wie folgt hergestellt:
10 ml entsprechend verdünnte Gerbstofflösung werden in einen 100 ml Mess¬
kolben abpipettiert, 3 ml Folin-Reagens zugegeben, umgeschwenkt, mit einer Lö¬
sung von 15 % Na2CO, sicc. in Wasser ad 100 ml aufgefüllt und gut gemischt. Ge¬
nau nach 5 Min wird bei Wellenlänge 720 mju kolorimetriert. Als Vergleichslösung
dient Wasser, dessen Transmissionsgrad gleich 100 % angenommen wird.
Die einzelnen Punkte dieser Vorschrift werden noch genauer untersucht wer¬
den.
157)842.121.1 Die Extinktionskurve: Friedrich hat in seiner Arbeit
die Extinktionskurve für eine Lösung von 100 mg Tannin (Merck) pro Liter bei 1 cm
Schichtdicke aufgenommen. 1 ml der Tanninlösung +0,5 ml Folin -Reagens +
8, 5 ml einer Lösung von 500 g Na„C03 crist. pro Liter wurden gemischt und je¬
weils nach 2 Min kolorimetriert. Die Werte sind in der folgenden Tabelle 21 an¬
gegeben. Die Beständigkeit der Färbung nahm mit zunehmender Konzentration der

- 86 -
Gerbstofflösung ab. Diese Erscheinung zeigt sich sowohl in einer frischen, als auch
in einer im Verlaufe einer Bestimmungsmethode mit Pb-Acetat oder Zn-Acetat dena¬
turierten und anschliessend aufgearbeiteten Gerbstofflösung.
Tab. 21 Werte der Extinktionskurve für Tannin (-Merck) nach Friedrich
Wellenlänge Extinktion Wellenlänge Extinktion
nui mju
750 0,725 530 0,348
720 0,785 500 0,255
660 0,750 470 0,202
610 0,625 420 0,170
570 0,475
Die Intensität der Blaufärbung ist nach Friedrich von der Art der Gerbstoffe ab¬
hängig. Das Adsorptionsmaximum der blauen Komplexverbindung liegt jedoch, wie
mehrere Autoren annehmen, unabhängig vom Gerbstoff, bei 720 mu.
Die zu messenden Lösungen sind klar und haben keine Eigenfärbung neben der
blauen Phosphorwolframat-Komplexfarbe.
Die Wellenlänge 720 mp wurde auch für diese Arbeit gewählt.
Ueberprüfung der Extinktionskurve:
Da die blaue Farbe der Lösung nach einigen Stunden in Gelb übergeht, müsste
für jeden Punkt der Extinktionskurve eine frische Lösung hergestellt und nach genau
gleicher Zeit, z. B. 5 Min, kolorimetriert werden.
Für die Ueberprüfung des Extinktionsmaximums wurde eine durch den Wolfram-
Gerbstoff-Komplex blau gefärbte Lösung mit 0,4 mg Kastanienholzextraktgerbstoffen
pro 100 ml (= Messlösung) kolorimetriert.
5 Minuten werden für die Bereitung aller Messlösungen genügen.
Nach genau 5, sowie nach jeder weiteren Minute, wurde die Messlösung gegen
Wasser als Blindwert kolorimetriert. Für jede Messung wurde die Wellenlänge ge¬
ändert.
Aus zwei zeitlich 4 Minuten auseinanderliegenden Messungen bei gleicher
Wellenlänge wurde die Transmissionsgradzunahme pro Minute als 0, 75 % absolut
berechnet. Alle Transmissionsgrade wurden sodann auf den Zeitpunkt der ersten
Messung nach 5 Minuten umgerechnet, wobei sich eindeutig ein Extinktionsmaxi¬
mum (Transmissionsgradminimum) bei 720 mu ergab.
Bei der kurzen Versuchsdauer von nur 7 Minuten spielt die Verschiebung der
Extinktionsmaxima der blauen Messlösung durch deren Verfärbung nach gelb noch
keine Rolle ; die Resultate sind in Tabelle 22 (pg. 87) zusammengefasst.

Tab. 22 Ueberprufung des Extinktionsmaximums
berechnete Werte:Ablesungen:
Zeit Wellen- Trg.Min lange (H20 = 100 %)
5 650 mp 22, 7 %
6 700 mu 21,0%
7 720 mu 21,5 %
8 750 mu 24,0 %
9 800 mu 32, 0 %
10 710 mp 24,0 %
11 720 mp 24,5 %
12 730 mu 25,5%
Trg. - Trg. corr
Zunahme (5 Min)(0,75%/Min)
0 % 22,7 %
0, 75 % 20,25 %
1,50 % 20,00 %
2, 25 % 21,75 %
3, 00 % 29,00 %
3, 75 % 20,25 %
4,50 % 20,00 %
5, 25 % 20,25 %
Trg. = abgelesener Transmissionsgrad als Prozente desjenigen von Wasser
Trg. corr. = abgelesener Transmissionsgrad, vermindert um dessen Zunahme
durch Entfärbung der Messlosung seit der ersten Messung nach 5 Mi¬
nuten (Ausschaltung des veränderlichen Faktors Zeit).
Ein analoger Versuch mit üblicher Photozelle (RCA/5581/5-52) und ohne Rot¬
filter zeigte standige Abnahme des Transmissionsgrades von 436 - 650 mju.
842.121.2 Die Haltbarkeit der gefärbten Losungen: 10 ml einer Ka-
stanienholzextraktlosung von 5,656 mg/100 ml wurden, wie auf pg. 85 beschrieben,
kolonmetnert und die Vergleichsmessungen in verschiedenen Zeitabstanden wieder¬
holt.
Tab. 23 Transmissionsgradzunahme der blau gefärbten Messlosung als Funktion
der Zeit
jede Messung erfolgte gegen
Wasser, dessen Transmis-
sionsgrad gleich 100 % ge¬setzt wurde.
Die Transmissionsgradzunahme betragt durchschnittlich 0, 9 Skalenteile (%)
pro Minute und ist linear proportional zur Zeit.
Nach einigen Stunden ist die ursprünglich blaue Losung gelb gefärbt.
Zeit in Transmissions-
Minuten: grad in % :
3 18,7
5 19,9
31 44,0

- 88 -
842.121.3 Der für die Messung geeignete Gerbstoffkonzentra-
tionsbereich (siehe pg. 84): 0, 2829 g Kastanienholzextrakt wurden im Mess¬
kolben auf 1000 ml ergänzt als Stammlösung (Stl.I) und Stufe I für die folgende Ver¬
dünnungsreihe, welche kolorimetriert wurde.
Tab. 24 Ermittlung der höchsten, noch genau messbaren Gerbstoffkonzentration
Stufe II
Stl.I
ml
10
20
30
Stl.
+ H20ad ml
100
100
100
Stufe ni *) Messresultat:
Drogen-konz.:
mg/100 ml
Drogen-konz.:
mg/100 ml
Gerbstoff -
konz. :
mg/100 ml
Trans¬
mission:
£% v.H20
Extink¬
tion:
2,829 0,2829 0,2 49,5 0,30 539
5,658 0,5658 0,4 21,0 0,67 778
8,487 0, 8487 0,6 9,5 1,02 228
Stammlösung
100 ml von Stammlösung I werden im Messkolben auf 1000 ml ergänzt (Stl. H;
Drogenkonzentration 2,829 mg/100 ml) als Stufe I der nachfolgenden Untersuchun¬
gen:
Tab. 25 Ermittlung der niedersten, noch genau messbaren Gerbstoffkonzentration
Stufe II
sti.n
ml
100
75
50
35
25
10
+ H20ad ml
100
100
100
100
100
100
Stufe III *) Messresultat:
Drogen-konz. :
mg/1001
Drogen-konz.:
ml mg/100 ml
Gerbstoff-
konz.:
mg/100 ml
Trans¬
mission
&% v.H20
Extink¬
tion:
2,829 0,2829 0,20 49,50 0,30 54
2,122 0,2122 0,15 61,00 0,2147
1,414 0,1414 0,10 74,00 0,1308
0,990 0,0990 0,07 81,50 0,0888
0,707 0,0707 0,05 86,00 0,0655
0,283 0,0283 0,02 93,75 0,0280
Stl. = Stammlösung
*) = Messlösung; siehe unter "Herstellung der Messlösungen" (pg. 85).
In den günstigten Messbereich des Instrumentes fallen Gerbstoffkonzentrationen
von 0,07 mg/100 ml bis 0,40 mg/100 ml.
842.121.4 Die notwendige Menge Folin-Reagen s pro 100 ml Mess¬
lösung: 10 ml einer Kastanienholzextrakt-Stammlösung mit 5,656 mg/100 ml
wurden unter Zusatz verschiedener Mengen F o 1 in-Reagens kolorimetriert. Es
wurden 3 solche Versuchsreihen durchgeführt.

- 89 -
Die Gerbstoffkonzentration der Messlösung ist 0,4 mg/100 ml (entsprechend
0,5656 mg Extrakt/100 ml) und bleibt konstant. Es ergaben sich die Extinktionswerte
der folgenden Tabelle 26:
Tab. 26 Einfluss der Reagenszugabe auf die Intensität der Blaufärbung
Reagenszugabe pro 100 ml Messlösung:
2 ml 3 ml 4 ml
Extinktionen:
1.
2.
Reihe
Reihe
0,6716
0,6882
0,6716
0,6778
0,6656
0,6576
M,« = Dure
schnittswert
3. Reihe 0,6666 0,6676 0,6676
M1-3
0,6755 0,6723 0,6636
Die Versuche zeigen, dass 3 ml Fol in-Reagens genügen, um auch in den
konzentriertesten, für unsere Messungen in Frage kommenden Gerbstofflösungen
eine vollkommene Blaufärbung hervorzurufen.
842.121.5 Der Einfluss der Konzentration der Sodalösung: 10ml
Stammlösung mit 5,658 mg Droge/100 ml wurden nach Zugabe des F ol in -Reagens
mit 10-, 15- oder 20-proz. Sodalösung (Na2COn sicc. ) ad 100 ml verdünnt und ko-
lorimetriert. Dabei wurden folgende Extinktionswerte erhalten:
Tab. 27 Einfluss der Konzentration der Sodalösung auf die Intensität der Blaufärbung
Sodalösung:
10 % 15% 20%
Extinktionen:
1. Reihe 0,6925 0, 6990 0,6778
2. Reihe 0,7055 0,6925 0,6861
Durchschnittswert 0,6990 0,69575 0,6820
Wie die Versuche zeigen, nimmt die Intensität der Färbung mit zunehmender
Konzentration der Sodalösung etwas ab.
Unterschiedliche Extinktionswerte trotz Messlösungen gleicher Herstellung
und gleicher Gerbstoffkonzentration liegen im Bereiche des Versuchsfehlers.
842.121.6 Aufnahme der Eichkurve mit Kastanienholzextr ak t :
Dies geschah nur in Ermangelung einer Standard-Chinarinde.

- 90 -
Die Eichkurve soll:
a) zeigen, ob die Blaufärbung mit Phosphorwolframsäure dem Lambert-Beer-
schen Gesetz folgt, d.h. ob die Extinktion eine lineare Funktion der Konzentra¬
tion der Gerbstofflösung sei.
b) ermöglichen, aus der Extinktion einer Lösung die zugehörige Gerbstoffkonzentra¬
tion rasch abzulesen.
Infolge Angleichung der kolorimetrischen an die internationale Hautpulverme¬
thode wird ein unterschiedlicher Gerbstoffgehalt der zu analysierenden Lösung schon
vor der Entgerbung durch entsprechende Vorverdünnung ausgeglichen. Die gerbstoff-
haltige und die entgerbte Lösung eines Versuches wurden nach gleicher Verdünnung
kolorimetriert. Die Messlösungen aller zukünftigen Bestimmungen haben daher un¬
gefähr den gleichen Gerbstoffgehalt von ca. 0,32 mg/100 ml (Messung £„). Ihr Nicht-
gerbstoffgehalt£2 ist viel geringer, ca. 0,075 mg/100 ml (Messung £,), variiert aber
zwischen den einzelnen Bestimmungen trotzdem nur wenig. Die Extinktionen der an
Hautpulver adsorbierten Stoffe (£. -£„ = £„) der Lösungen ändern wenig und machen
daher nur ein kurzes Stück der Extinktionskurve aus.
Die Eichkurve wurde nur zur Kontrolle von Punkt a) (s. oben) über einen mög¬
lichst weiten Bereich ausgedehnt.
Die gefundenen Werte sind Vergleichswerte, die aussagen, wieviel Kastanien-
holzextraktgerbstoffen der Gerbstoffgehalt einer Droge entspricht.
Für die Ausmessung von 3 Eichkurven wurden 3 Stammlösungen folgender Kon¬
zentration hergestellt:
Stammlösung A mit 5,656 mg/100 ml für 5 Punkte der Eichkurve, dann daraus
Stammlösung B mit 2,828 mg/100 ml für die niederen Gerbstoffkonzentrationen.
Stammlösung C mit 565,6 mg/100 ml wurde entgerbt und diente nach 100- sowie
200-facher Verdünnung (Tab. 28, No. 15/16 NG.) als Ausgangslösung für die Kolo-
rimetrierung der Nichtgerbstoffe. Die Resultate finden sich in Tabelle 28, pg. 91
Prüfung des Hautpulvers auf Abgabe wasserlöslicher, mit F o 1 in-Reagens eine
Färbung erzeugender Stoffe:
In Zukunft werden nur noch 50 statt 75 ml der zu analysierenden Lösung mit
ca. 2 g lufttrockenem Hautpulver (2,0 statt 3,0 g getrocknetes Material) entgerbt,
da nur 10 ml des daraus gewonnenen Filtrates für eine kolorimetrische Bestimmung
benötigt werden.
2, 2381 g Hautpulver wurden mit 50 ml Wasser 30 Min geschüttelt, dann klar¬
filtriert. 10 ml Filtrat, im Messkolben auf 1 L ergänzt, davon 10 ml aufgearbeitet
und kolorimetriert, ergaben einen Transmissionsgrad von 98,25 % gegenüber 99,22 %
der gleichen, nur mit Wasser gefüllten Cuvette. Da dieser Fehler von ca. 1 Prozent
bei weitem innerhalb der Streuung der Resultate der Gerbstoffbestimmung liegt,
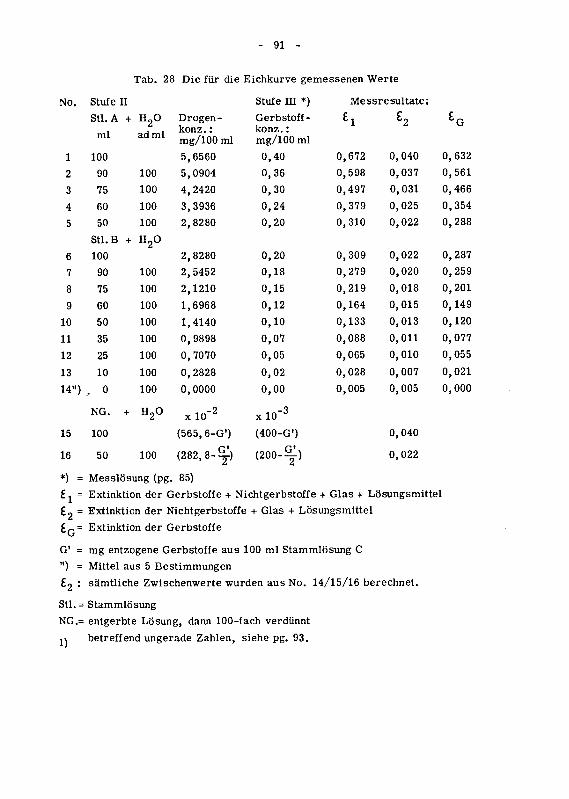
- 91 -
Tab. 28 Die für die Eichkurve gemessenen Werte
No. Stufe II Stufe in *) Messresultate;
Stl.A +
ml
H20ad ml
Drogen -
konz. :
mg/100 ml
Gerbstoff-
konz.:
mg/100 ml
£1 h £G
1 100 5,6560 0,40 0,672 0,040 0,632
2 90 100 5,0904 0,36 0,598 0,037 0,561
3 75 100 4,2420 0,30 0,497 0,031 0,466
4 60 100 3,3936 0,24 0,379 0,025 0,354
5 50
Stl.B +
100
H20
2,8280 0,20 0,310 0,022 0,288
6 100 2,8280 0,20 0,309 0,022 0,287
7 90 100 2,5452 0,18 0,279 0,020 0,259
8 75 100 2,1210 0,15 0,219 0,018 0,201
9 60 100 1,6968 0,12 0,164 0,015 0,149
10 50 100 1,4140 0,10 0,133 0,013 0,120
11 35 100 0,9898 0,07 0,088 0,011 0,077
12 25 100 0,7070 0,05 0,065 0,010 0,055
13 10 100 0,2828 0,02 0,028 0,007 0,021
14").
0 100 0,0000 0,00 0,005 0,005 0,000
NG. + H20 xlO"2 xlO"3
15 100 (565, 6-G') (400-G') 0,040
16 50 100 (282,8-^) (200-Ç) 0,022
*) = Messlösung (pg. 85)
£1 = Extinktion der Gerbstoffe + Nichtgerbstoffe + Glas + Lösungsmittel
£2 = Extinktion der Nichtgerbstoffe + Glas + Lösungsmittel
£G= Extinktion der Gerbstoffe
G' = mg entzogene Gerbstoffe aus 100 ml Stammlösung C
") = Mittel aus 5 Bestimmungen
£2 : sämtliche Zwischenwerte wurden aus No. 14/15/16 berechnet.
Stl. = Stammlösung
NG.= entgerbte Lösung, dann 100-fach verdünnt
y,betreffend ungerade Zahlen, siehe pg. 93.
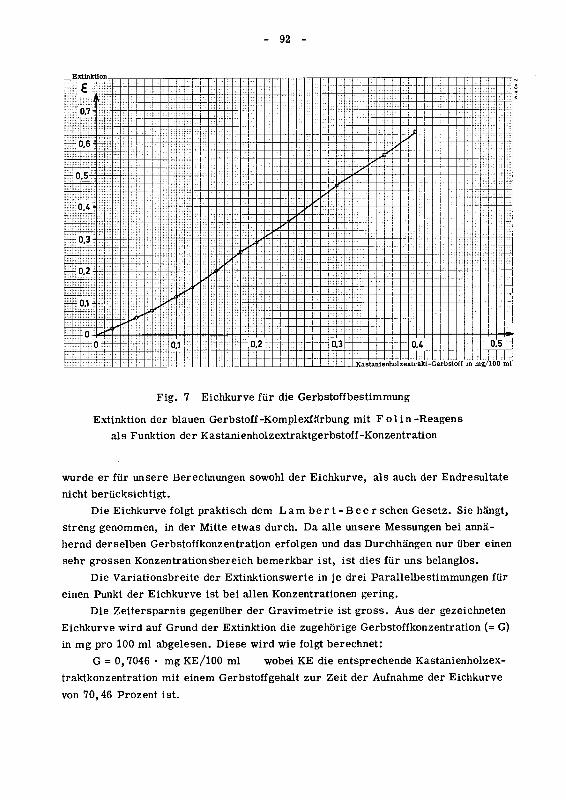
- 92 -
Extinktion.
Fi i1
!-
i i-
1
1 | ! i
\-
-i
!-
-0.7-
1
,i 1
1
i
' ;
y-0.6-
't I ! i
~\ ,
| !- - -/
y1I 1 i 1
*
I-
ZL0.5
1 I ,
--
i
- ._ - —
Ii
'
!
-0,4-. 1
"*"
"- j --
1>
-i-013-1
_.
i\
,I
- 1 _ 1
W-- -
1 i l
! i -
'
"
1-0.1 :
'! i
i \-
i
, |!
~H1
-t" p-
1 T
'
01 0,2-1
- -|
0 i U,J | 0.4
1
0 3
1 i'
4,0 ml
,
1 I 1 i Kastanienholzextrakt-Gerbstoff in mk/io
Fig. 7 Eichkurve für die Gerbstoffbestimmung
Extinktion der blauen Gerbstoff-Komplexfärbung mit F o 1 i n-Reagens
als Funktion der Kastanienholzextraktgerbstoff-Konzentration
wurde er für unsere Berechnungen sowohl der Eichkurve, als auch der Endresultate
nicht berücksichtigt.
Die Eichkurve folgt praktisch dem Lam bert-Beer sehen Gesetz. Sie hängt,
streng genommen, in der Mitte etwas durch. Da alle unsere Messungen bei annä¬
hernd derselben Gerbstoffkonzentration erfolgen und das Durchhängen nur über einen
sehr grossen Konzentrationsbereich bemerkbar ist, ist dies für uns belanglos.
Die Variationsbreite der Extinktionswerte in je drei Parallelbestimmungen für
einen Punkt der Eichkurve ist bei allen Konzentrationen gering.
Die Zeitersparnis gegenüber der Gravimetrie ist gross. Aus der gezeichneten
Eichkurve wird auf Grund der Extinktion die zugehörige Gerbstoffkonzentration (= G)
in mg pro 100 ml abgelesen. Diese wird wie folgt berechnet:
G = 0,7046 • mg KE/100 ml wobei KE die entsprechende Kastanienholzex-
traktkonzentration mit einem Gerbstoffgehalt zur Zeit der Aufnahme der Eichkurve
von 70,46 Prozent ist.
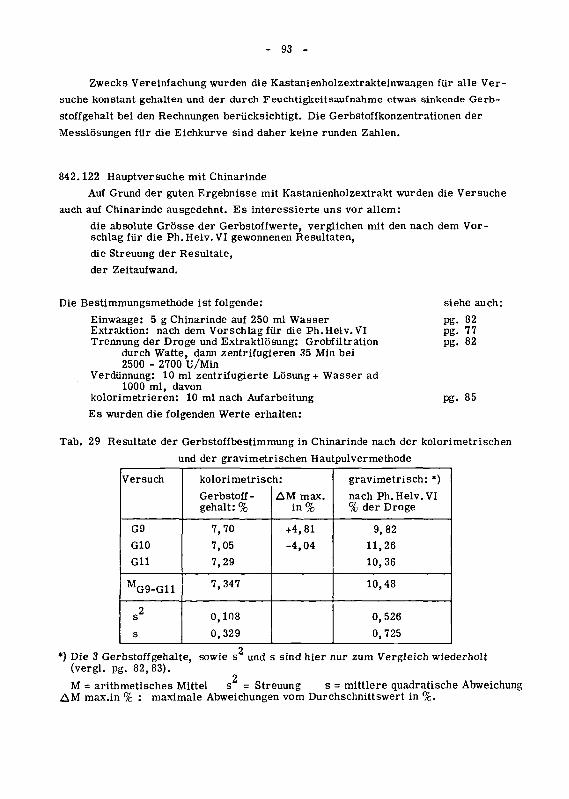
- 93 -
Zwecks Vereinfachung wurden die Kastanienholzextrakteinwaagen für alle Ver¬
suche konstant gehalten und der durch Feuchtigkeitsaufnahme etwas sinkende Gerb-
stoffgehalt bei den Rechnungen berücksichtigt. Die Gerbstoffkonzentrationen der
Messlösungen für die Eichkurve sind daher keine runden Zahlen.
842.122 Hauptversuche mit Chinarinde
Auf Grund der guten Ergebnisse mit Kastanienholzextrakt wurden die Versuche
auch auf Chinarinde ausgedehnt. Es interessierte uns vor allem:
die absolute Grösse der Gerbstoffwerte, verglichen mit den nach dem Vor¬
schlag für die Ph.Helv. VI gewonnenen Resultaten,
die Streuung der Resultate,
der Zeitaufwand.
Die Bestimmungsmethode ist folgende:
Einwaage: 5 g Chinarinde auf 250 ml Wasser
Extraktion: nach dem Vorschlag für die Ph.Helv. VI
Trennung der Droge und Extraktlösung: Grobfiltration
durch Watte, dann zentrifugieren 35 Min bei
2500 - 2700 U/MinVerdünnung: 10 ml zentrifugierte Lösung + Wasser ad
1000 ml, davon
kolorimetrieren: 10 ml nach Aufarbeitung
Es wurden die folgenden Werte erhalten:
siehe auch:
pg. 82
Pg. 77
pg. 82
pg. 85
Tab. 29 Resultate der Gerbstoffbestimmung in Chinarinde nach der kolorimetrischen
und der gravimetrischen Hautpulvermethode
Versuch kolorimetrisc
Gerbstoff-
gehalt: %
h:
AM max.
in%
gravimetrisch: *)
nach Ph.Helv. VI
% der Droge
G9
G10
Gll
7,70
7,05
7,29
+4,81
-4,04
9,82
11,26
10,36
MG9-G11 7,347 10,48
s2
s
0,108
0,329
0,526
0,725
*) Die 3 Gerbstoffgehalte, sowie s und s sind hier nur zum Vergleich wiederholt
(vergl. pg. 82,83).2
M = arithmetisches Mittel s = Streuung s = mittlere quadratische AbweichungAM max.in % : maximale Abweichungen vom Durchschnittswert in %.
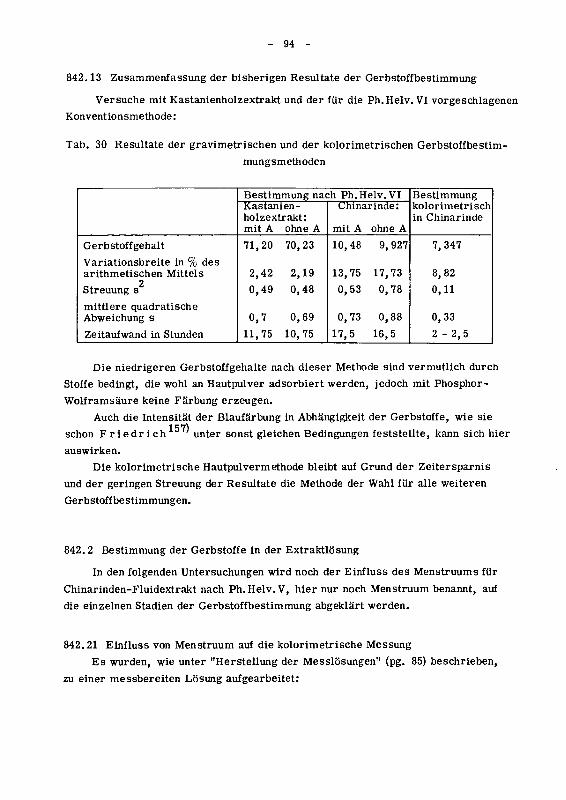
- 94 -
842.13 Zusammenfassung der bisherigen Resultate der Gerbstoffbestimmung
Versuche mit Kastanienholzextrakt und der für die Ph.Helv. VI vorgeschlagenen
Konventionsmethode:
Tab. 30 Resultate der gravimetrischen und der kolorimetrischen Gerbstoffbestim¬
mungsmethoden
Bestimmung nach Ph.Helv. VI Bestimmungkolorimetrisch
in Chinarinde
Kastanien¬
holzextrakt:
mit A ohne A
Chinarinde:
mit A ohne A
Gerbstoffgehalt
Variationsbreite in % des
arithmetischen Mittels
Streuung s
mittlere quadratischeAbweichung s
Zeitaufwand in Stunden
71,20 70,23
2,42 2,19
0,49 0,48
0,7 0,69
11,75 10,75
10,48 9,927
13,75 17,73
0,53 0,78
0,73 0,88
17,5 16,5
7,347
8,82
0,11
0,33
2 - 2,5
Die niedrigeren Gerb Stoffgehalte nach dieser Methode sind vermutlich durch
Stoffe bedingt, die wohl an Hautpulver adsorbiert werden, jedoch mit Phosphor-
Wolframsäure keine Färbung erzeugen.
Auch die Intensität der Blaufärbung in Abhängigkeit der Gerbstoffe, wie sie
157)schon Friedrich unter sonst gleichen Bedingungen feststellte, kann sich hier
auswirken.
Die kolorimetrische Hautpulvermethode bleibt auf Grund der Zeitersparnis
und der geringen Streuung der Resultate die Methode der Wahl für alle weiteren
Gerbstoffbestimmungen.
842.2 Bestimmung der Gerbstoffe in der Extraktlösung
In den folgenden Untersuchungen wird noch der Einfluss des Menstruums für
Chinarinden-Fluidextrakt nach Ph.Helv.V, hier nur noch Menstruum benannt, auf
die einzelnen Stadien der Gerbstoffbestimmung abgeklärt werden.
842.21 Einfluss von Menstruum auf die kolorimetrische Messung
Es wurden, wie unter "Herstellung der Messlösungen" (pg. 85) beschrieben,
zu einer messbereiten Lösung aufgearbeitet:

- 95 -
a) 10 ml Menstruum. Dabei entsteht nur eine feine weisse Fällung, bedingt durch
den Alkoholgehalt.
b) 10 ml des wie vorher in den Extrakten 1+99 verdünnten Menstruums. Der Trans¬
missionsgrad dieser Lösung ist 98,75 %, derjenige der mit Wasser gefüllten Cu¬
vette 99,22%.
Das Menstruum hat also auf die kolorimetrische Messung selbst keinen Einfluss von
Bedeutung.
842.22 Einfluss der Eigenfärbung der verdünnten Gerbstofflösung
Nach 1000- bis 4000-facher Verdünnung der Extraktlösungen wie unter den
Versuchsbedingungen, jedoch ohne Zugabe von Reagens und Sodalösung, ist keine mit
dem "Spectronic-20"-Kolorimeter messbare Färbung mehr festzustellen.
842.23 Prüfung des Hautpulvers auf Abgabe menstruumlöslicher, mit Phosphor¬
wolframsäure eine Färbung erzeugender Stoffe
2,2386 g Hautpulver wurden mit 50 ml Menstruum 30 Min geschüttelt, dann
klarfiltriert; 10 ml Filtrat im Messkolben auf 1 L ergänzt, davon 10 ml aufgearbei¬
tet und kolorimetriert. Der Transmissionsgrad der Lösung war 98 % (Wasser: 99,22%).
Diese Korrektur wird bei allen Messungen weggelassen (siehe auch pg. 90, 92, "Abgabe
wasserlöslicher Stoffe durch das Hautpulver").
842.24 Einfluss des Alkoholgehaltes der zu entgerbenden Lösung
Mit steigendem Alkoholgehalt der entgerbten Lösung nimmt deren Verdunstung
bei der Filtration zu und die Nichtgerbstofflösung wird dabei konzentrierter. Dies
täuscht einen zu niedrigen Gerbstoffgehalt vor.
Anderseits kann durch den Alkohol auch der Entgerbungsprozess selbst beein-
flusst werden.
Um das Ausmass dieser Fehler zu bestimmen, sind 20 ml eines ersten Teil-
perkolates mit 23 % Totalextraktivstoffen mit Menstruum Ph.Helv. V (Me.) ad 50 ml
ergänzt worden. Diese Lösung diente als Stammlösung (Stl.). In Verdünnungen aus
je 10 ml Stammlösung mit verschiedenen Mengen Menstruum Ph.Helv. V und Wasser
sind die Gerbstoffe mittels kolorimetrischer Hautpulvermethode bestimmt worden.
Die Resultate sind in der folgenden Tabelle 31 zusammengefasst:

- 96 -
Tab. 31 Einfluss des Alkoholgehaltes der zu entgerbenden Lösung auf die gefundenen
Gerbstoffgehalte
No. Zusam
SU.
ml
mensetzung der Prüflösungen+ Me. + H20
ml ad ml
Alkohol
Gew.%
Dichte
g/cm
G% Faktor
F
G12 10 0 100 0,00 1,0011 5,98 1,013
G13 10 0 100 3,70 0,9969 6,26 0,968
G14 10
10
1120
100
100
9,82 0,9866 6,06 1,000
1,022
G15 10
10
10
10
30
35
40
45
100
100
100
100
16,06 0,9762 5,66 1,070
1,084
1,098
1,112
G16 10 50 100 24,60 0, 9624 5,37 1,127
Stl. = Stammlösung G % = bestimmter Gerbstoffgehalt in Prozenten
Me. = Menstruum der ExtraktlösungF = Korrekturfaktoren für Bestimmungen mit andern Alkoholgehalten der zu
entgerbenden Lösung als 9,82 Gew.%.
Der Trockenrückstand unserer Extraktlösung ist im allgemeinen 4 g/100 ml,
darf jedoch nach der internationalen Hautpulvermethode nur 1 g/100 ml der zu ent¬
gerbenden Lösung erreichen. 1 T unserer Extraktlösung wird daher mit 3 T Wasser
verdünnt, wobei sich im allgemeinen ein Alkoholgehalt von 9,82 Gew.% ergibt.
Extraktlösungen mit andern Alkoholgehalten müssen auf 9,82 Gew.% Alkohol
eingestellt, oder die gefundenen Gerbstoffwerte mittels der angegebenen Korrektur -
faktoren umgerechnet werden.
Versuche ergaben, dass 9 - 11 g Extraktlösung an das Hautpulver und an alle
Filter adsorbiert werden. Auf Grund dieser Tatsache wurden in denselben 5 Lösun¬
gen die Verdunstungsverluste während der Filtration der entgerbten Lösungen be¬
stimmt.
Die dadurch bedingten Konzentrationszunahmen und damit auch Extinktionszu¬
nahmen der entgerbten Lösungen wurden berechnet. Daraus konnte schliesslich die
Extinktion der Gerbstoffe und deren Konzentration in der Extraktlösung unter Aus¬
schaltung der Verdunstung von entgerbter Extraktlösung berechnet werden.
Die Werte sind in Tabelle 32 zusammengefasst und anschliessend graphisch
dargestellt.
Wenn trotz dieser Korrektur, je nach Alkoholkonzentration, unterschiedliche
Gerbstoffgehalte gefunden werden, so ist dies auf den Einfluss der Alkoholkonzen¬
tration auf den Entgerbungsprozess selbst zurückzuführen.
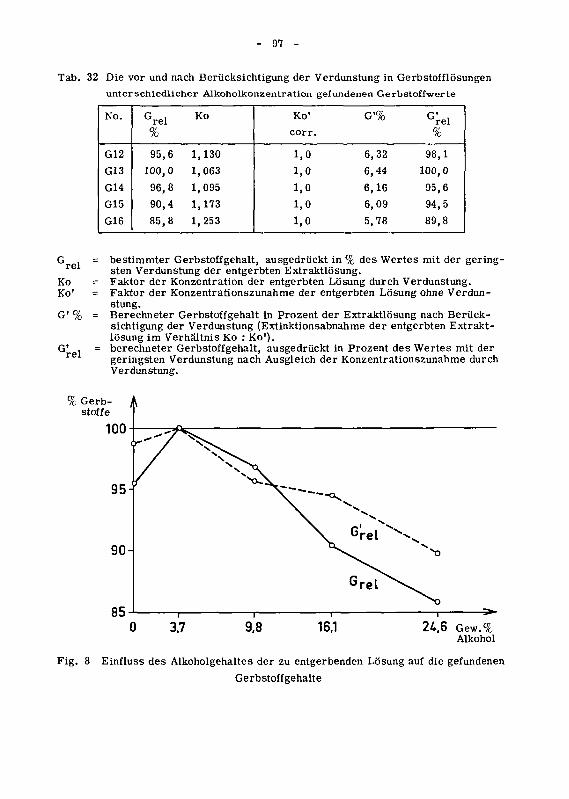
- 97 -
Tab. 32 Die vor und nach Berücksichtigung der Verdunstung in Gerbstofflosungen
unterschiedlicher Alkoholkonzentration gefundenen Gerbstoffwerte
No. Grel%
Ko Ko'
corr.
G'% Grel%
G12 95,6 1,130 1,0 6,32 98,1
G13 100,0 1,063 1,0 6,44 100,0
G14 96,8 1,095 1,0 6,16 95,6
G15 90,4 1,173 1,0 6,09 94,5
G16 85,8 1,253 1,0 5,78 89,8
G,
= bestimmter Gerbstoffgehalt, ausgedruckt in % des Wertes mit der gering-r
sten Verdunstung der entgerbten Extraktlosung.Ko = Faktor der Konzentration der entgerbten Losung durch Verdunstung.Ko' = Faktor der Konzentrationszunahme der entgerbten Losung ohne Verdun¬
stung.G' % = Berechneter Gerbstoffgehalt in Prozent der Extraktlosung nach Berück¬
sichtigung der Verdunstung (Extinktionsabnahme der entgerbten Extrakt¬
losung im Verhältnis Ko : Ko').G' ,
= berechneter Gerbstoffgehalt, ausgedruckt in Prozent des Wertes mit derr
geringsten Verdunstung nach Ausgleich der Konzentrationszunahme durch
Verdunstung.
% Gerb- A
85 J1 1 1 1 >•
0 3.7 9.8 16.1 24,6 Gew.%Alkohol
Fig. 8 Einfluss des Alkoholgehaltes der zu entgerbenden Losung auf die gefundenen
Gerbstoffgehalte
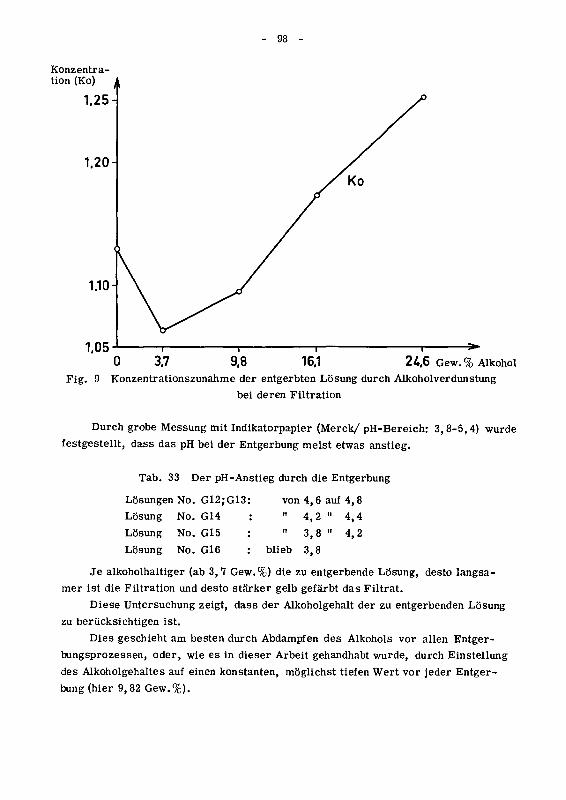
98 -
Konzentra-
1.05
Fig. 9
0 3.7 9,8 16,1 24.6 Gew.% Alkohol
Konzentrationszunahme der entgerbten Lösung durch Alkoholverdunstung
bei deren Filtration
Durch grobe Messung mit Indikatorpapier (Merck/ pH-Bereich: 3,8-5,4) wurde
festgestellt, dass das pH bei der Entgerbung meist etwas anstieg.
Tab. 33 Der pH-Anstieg durch die Entgerbung
Lösungen No. G12;G13:
Lösung No. G14
Lösung No. G15
Lösung No. G16
von 4,6 auf 4,8"
4,2" 4,4
"
3,8"
4,2
blieb 3,8
Je alkoholhaltiger (ab 3,7 Gew.%) die zu entgerbende Lösung, desto langsa¬
mer ist die Filtration und desto stärker gelb gefärbt das Filtrat.
Diese Untersuchung zeigt, dass der Alkoholgehalt der zu entgerbenden Lösung
zu berücksichtigen ist.
Dies geschieht am besten durch Abdampfen des Alkohols vor allen Entger-
bungsprozessen, oder, wie es in dieser Arbeit gehandhabt wurde, durch Einstellung
des Alkoholgehaltes auf einen konstanten, möglichst tiefen Wert vor jeder Entger¬
bung (hier 9,82 Gew.%).

- 99 -
Es können aber auch die Resultate mit den auf pg. 96 (Tab. 31) angegebenen
Umrechnungsfaktoren je nach Alkoholgehalt (oder Menstruumgehalt) der zu entger¬
benden Lösung umgerechnet werden.
Auf Grund all dieser Untersuchungen ergab sich die
842.25 endgültige Vorschrift der Gerbstoffbestimmung
1) Bestimmung des approximativen Trockenrückstandes der Extraktlösung
50 ml Extraktlösung werden auf dem Wasserbad in einer tarierten Kristalli¬
sierschale von ca. 8 cm Durchmesser zur Trockene eingedampft (ca. 3 Stunden).Nach dem Abtrocknen der Kristallisierschalen lässt man im Exsiccator 30 Minuten
erkalten, wägt und berechnet den approximativen Trockenrückstand, welcher wegen
ungenügender Trocknung noch ca. 10 Prozent Wasser enthält.
2) Herstellung der Stammlösung aus der Extraktlösung
Von der Extraktlösung wird nun soviel abpipettiert, dass beim Ergänzen mit
Wasser auf 100 ml eine Stammlösung von maximal 1 g Trockenrückstand oder
0,4 ± 0,025 g an Hautpulver adsorbierbare Stoffe pro 100 ml entsteht (= Anforderun¬
gen der internationalen Hautpulverméthode).
Wässerige Auszüge werden direkt in einen 100 ml Messkolben pipettiert, eben¬
so 25 ml unserer Extraktlösungen, die beim Auffüllen auf 100 ml einen gleichen,schwachen Alkoholgehalt von ca. 9, 8 Gew. % ergeben.
Wenn wegen zu geringem Trockenrückstand grössere Mengen als 25 ml Ex¬
traktlösung für die Herstellung der Stammlösung benötigt werden, so muss der Al¬
kohol erst im Vakuum abdestilliert werden (in einem mit doppelt gelochtem Gummi¬
stopfen, Kapillare und Absaugrohr versehenen 300 ml Langhals-Rundkolben); dann
wird der Rückstand mit 25 ml Menstruum und anschliessend mit Wasser quantitativin einen 100 ml Messkolben gespült, mit Wasser bis zur Marke aufgefüllt und gut
durchgemischt (= Stammlösung).
3) Herstellung der Kolorimetrierlösung
10 ml der gut gemischten Stammlösung werden in einen 1 1-Messkolben abpi¬
pettiert und mit destilliertem Wasser auf 1 1 ergänzt. Nach gutem Umschütteln wer¬
den 10 ml dieser Lösung in einen 100 ml-Messkolben abpipettiert, 3 ml Phosphor¬
wolframsäurereagens nach F olin zugegeben und mit einer Lösung von 15% NagCOosicc. in Wasser ad 100 ml aufgefüllt. Im Moment der Zugabe der Sodalösung wird
die Zeit genommen, und genau 5 Minuten später wird bei Wellenlänge 720 mu in Cu-
vetten von 1 Zoll 0 (Schichtdicke 2,48 cm) kolorimetriert (vergl. auch pg. 84/85).
4) Herstellung der entgerbten Lösung
50 ml der Stammlösung werden mit soviel Hautpulver von bekanntem Wasser¬
gehalt versetzt, dass die Einwaage 2 g wasserfreiem Hautpulver entspricht. Dann
wird während 30 Minuten in der Schüttelmaschine geschüttelt (156 Hin- und Herbewe¬
gungen pro Minute, horizontale Verschiebung: 2x8 cm, also 16 cm).Das Gemisch wird durch ein Leinentuch von ca. 15 cm x 15 cm, das zur Entfernung
der Appretur mit destilliertem Wasser ausgekocht wurde, in einen mit etwas Watte
versehenen Glastrichter von ca. 10 cm Durchmesser koliert und durch die Watte fil¬
triert. Dieses zweite Filtrat wird noch einmal durch ein Faltenfilter von ca. 10 cm
Durchmesser - aus einem Rundfilter von Schleicher & Schüll, No. LS-14, herge¬stellt - klarfiltriert.
10 ml dieser gut durchmischten Lösung, welche nun keine Gerbstoffe mehr ent¬
hält, werden genau gleich behandelt wie die nicht entgerbte Lösung unter 3.

- 100 -
5) Berechnung des Gerbstoffgehaltes
Die Differenz der Extinktionen der beiden Bestimmungen ergibt mittels der Eich¬
kurve den Gerbstoffgehalt.Der gefundene Vergleichswert sagt aus, wieviel Kastanienholzextrakt-Gerbstof-
fen der Gerbstoffgehalt unserer Chinarinde entspricht.Da die Hautpulvermethode die Aktivität gegenüber biologischem Material aus¬
nützt, darf man wohl annehmen, dass die beiden Gerbstoff-Vergleichswerte, zum
mindesten in therapeutischer Hinsicht, gleichwertig sind.
842.26 Kontrolle der Streuung der kolorimetrischen Hautpulvermethode an einer
Extraktslösung
Zur Feststellung der Streuung der beschriebenen Bestimmungsmethode wurde
eine Extraktlösung mit einem Roh-Trockenrückstand von 3,46 g/100 ml verwendet.
Pro Bestimmung wurden 25 ml Extraktlösung mit Wasser erst auf 100 ml verdünnt
und, wie oben beschrieben, verarbeitet. Es wurden folgende Gerbstoffgehalte ge¬
funden:
Tab. 34 Streuung der kolorimetrischen Hautpulvermethode, bestimmt an einer
Extraktlösung
s = Streuungs = mittlere quadratische
Abweichung
s2 = 0,000 010 *)
Datum:
1959
No. Gerbstoffe 2
% v. Lösungs
3.2.
M(3.2.)
4.3.
M(4.3.)
5.3.
M(5.3.)
Gl 7
G18
G19
G20
G21
G22
G23
G24
î;So6 0,000008
1,004
1,028
{'°23 0,000022
l'020
1,022
{'q37 0,000000 5
1,0365
0,0032
M = arithmetisches Mittel
*) Da unsere 8 Versuche nicht,
wie später die 2 Parallelbe¬
stimmungen in den Extrakten,
alle am gleichen Tag ausge¬
führt werden konnten, der Ge¬
halt der an Hautpulver adsor¬
bierbaren Stoffe jedoch während der Alterung der Extraktlösungen zunimmt, wurde2
s als arithmetisches Mittel der Streuungen an den drei Tagen bestimmt. Auf dieseo
Weise wird der Einfluss der Alterung auf s und s eliminiert.
Auf Grund der geringen Streuung der Resultate auch in den Extraktlösungen
wird die Methode in der beschriebenen Form für alle folgenden Gerbstoffbestim¬
mungen verwendet.

- 101 -
843 Die Bestimmung der Totalextraktivstoffe
843.1 Die Bestimmung der Total extraktiv Stoff e der Droge
Die Wahl fiel auf das schon von Fuchs126)
verwendete Verfahren, da dieses
125)vermutlich die vollständigste Drogenextraktion bewirkt. Feinstein und
Graetzer extrahierten weniger lange und nur mit Menstruum Ph.Helv. V und
Wasser.
Unsere Vorschrift lautet wie folgt:
5,00 g Droge werden im Soxhlet-Apparat je 3 Stunden mit je 100 g der folgendenLösungsmittel extrahiert: 1. Alkohol 95 Vol.%; 2. Wasser + 0,6 % HCl + 0,6 %HCOOH; nach Neutralisation des Drogenrückstandes mit NH3. 3. Wasser + 1,2 %NH3.
Die vereinigten Auszüge werden auf dem Wasserbad in einer tarierten Kristal¬
lisierschale von ca. 8 cm Durchmesser zur Trockene eingedampft, anschliessend
bei 103° - 105° C bis zur Gewichtskonstanz getrocknet und gewogen.
Nach einheitlicher Trocknungszeit von 61 h 50 Min (= Zeit bis zur maximalen
Gewichtsabnahme von 0, 5 mg/Stunde für 2,5 g Trockenrückstand) wurden die folgen¬
den Werte erhalten:
Tab. 35 Die Streuung der von uns verwendeten Bestimmungsmethode der Total¬
extraktivstoffe in Chinarinde
Versuch
No.
Totalextraktiv¬
stoffe % v. Droge
Versuch
No.
Totalextraktiv¬
stoffe % v. Droge
TRI
TR2
TR3
51,14
51,42
51,15
TR4
TR5
TR6
49,99
51,09
51,35
MTRl-6 51,023
M = arithmetisches Mittel
Die Streuung s2 beträgt: 0, 27Die mittlere quadratische Abweichung s ist: 0,52Die maximalen Abweichungen der Resultate in % des arithme¬
tischen Mittels sind: + 0,78% -2,02%
843.2 Bestimmung der Totalextraktivstoffe in der Extraktlösung
Von den Extraktflüssigkeiten wurden 50 ml oder soviel, dass ca. 2 g Trocken¬
rückstand zu erwarten waren, genau wie die vereinigten Soxhletauszüge aus dem
Drogenmaterial (843.1) aufgearbeitet.

- 102 -
844 Die Bestimmung des spezifischen Gewichtes und der Viskosität
844.1 Die Bestimmung des spezifischen Gewichtes
844.11 Dessen Bestimmung in der Droge
In Ermangelung eines besondern Gerätes erfolgten die Messungen in einem
Wägegläschen von 27 mm Durchmesser und 76 mm Höhe mit eingeschliffenem, un¬
ten flachem Glasstopfen mit Kerbe von ca. 2mm Breite, wie es die Ph.Helv. V für
Balsame und Teere vorschlägt .
Als Medium zur Bestimmung des effektiven Drogenvolumens kam nur eine
Flüssigkeit mit folgenden Eigenschaften in Frage:
hoher Siedepunkt, damit keine Fehler durch Verdunstung entstehen,niederes spezifisches Gewicht, damit die Droge rasch sedimentiert,geringer Ausdehnungskoeffizient (geringer Temperatureinfluss),geringes Lösungsvermögen für Drogeninhaltsstoffe.
Bei Vorversuchen mit Aceton, Benzin, Benzol, Essigester, Petroläther und Wasser
erwies sich Benzin mit einem Siedpunkt von 60 - 120 C als am besten geeignet.
Um Temperaturfehler möglichst auszuschalten, erfolgten alle Messungen bei
20 C, oder wurden entsprechend korrigiert, weshalb auch die Dichte des Benzins
bei dieser Temperatur von Bedeutung ist. Für eine Dichtebestimmung wurde wie
folgt vorgegangen:
In das Pyknometer, dessen Leergewicht, Fassungsvermögen für Wasser so¬
wie Benzin von je 20 C bestimmt wurden, wird eine gewisse Menge Drogenpulver
eingewogen. Dann wird Benzin von bekannter Dichte bei 20 C zugegeben, gut umge¬
schwenkt, das Ganze zur Entfernung der Luft aus den Drogenteilchen im Exsiccator
zweimal während bestimmter Zeit gründlich evakuiert.
Anschliessend wird die Temperatur des Benzins im Pyknometer auf - 0,1 C
genau gemessen, der Deckel aufgesetzt, wobei das überschüssige Benzin ausge¬
drängt wird, und nach genau zwei Minuten das Gewicht des gefüllten Pyknometers
bestimmt. (Die Gewichtsabnahme durch Verdunstung beträgt 2,6 - 4, 2 mg pro
Minute.)
Die Dichtezunahme des Benzins beim Evakuieren währena der festgesetzten
Zeit, bedingt durch stärkeres Verdunsten von dessen tiefer siedenden Anteilen, ist
berücksichtigt.
Die Volumenänderung des Benzins wurde bei allen Wägungen, die nicht bei
20 C erfolgten, errechnet und das Gewicht korrigiert.
Die Berechnung geschieht folgendermassen:

- 103 -
C+B
Pyknometer leer
Pyknometer + Benzin
Pyknometer + Benzin +
Chinarinde m
V
Pyknometer + Chinarinde
Temperatur, bei der gewogen
wurde
Masse
VolumenDichte von Benzin in
g/ml bei 20° C
Dichte von Chinarinde in g/ml bei 20 C
Gewicht des über der Droge stehenden Benzins bei 20 C nach dem Eva¬
kuieren
Gewicht des über der Droge stehenden Benzins bei 20 C vor dem Eva¬
kuieren
Gewichtszunahme des nach dem Evakuieren verschlossenen, mit Benzin
gefüllten Pyknometers durch Verdunstung niedrig siedender, leichter An¬
teile des Benzins beim Evakuieren} ausgedrückt in mg/g (0,240 mg/g)
durch Ausdehnung des Benzins bedingte Gewichtsabnahme des mit Benzin
gefüllten Pyknometers, ausgedrückt in mg/g • °C (1,08 mg»g-1'0C-l)
y = (Prj.D " Pc'20° nacn Evakuation
(PC+B " PcV y - by(t' - 20°) = y(l - b(t' - 20°))
(P,C+B pcV
1 - b(t* - 20°)
2 x = y- ay = y(l-a) = (pc+B - pc^20° vor Evakuation
<U =S
(PC " PL>C V
BPB-PL>20° " (Pc+B-PC^O^M"
dB20° • (PC " PL>
^B-^O0 -
^'Vf • (1 ' a)
1 -b -(f -20°)
dC =
dn,no(PP-PT) • (l-Kf- 20°))JB20" KrC rh
(PB-PL)20° -d-b(f-20^) - (Pc+B-Pc)t.-d-a)
Die Dichten der einzelnen Siebfraktionen folgen später bei der Besprechung der
Wirkung des Rührgerätes auf den Zerkleinerungsgrad der Droge.

- 104 -
Die Dichte von Chinarinde nahm mit steigender Pulverfeinheit etwas zu. Als
mittlere Korngrösse kann die auf Sieb IVa gesammelte Fraktion betrachtet werden,
d.h. es sind gewichtsmässig im Pulver der nicht extrahierten Droge gleichviele fei¬
nere wie gröbere Anteile vorhanden. Diese Fraktion hat ein spezifisches Gewicht von
1,4056 g/cm3.
844.12 Die Bestimmung der Dichte der Extraktlösungen
Diese erfolgte in einem Pyknometer von 10 ml Inhalt, dessen Glasstopfen, um
die Füllung zu erleichtern, mit einer Kapillare versehen war ', bei 20 C.
844.2 Die Bestimmung des Viskosität der Extraktlösungen
Als weiteres Kriterium wurde die Viskosität aller hergestellten Extraktlösun-1 RR
gen mit dem Ostwald Viskosimeter bestimmt, s.Ph.Helv.V, Suppl.in/pg. 47 )
Die Berechnung ist im Kommentar zur Ph. Helv. V ' beschrieben.
845 Die Bestimmung der Korngrössenverteilung
Anhand dieser Daten soll versucht werden, die durch das Wirbeln erfolgte
Teilchenzerkleinerung und damit die Vergrösserung der inneren Oberfläche zu er¬
fassen.
845.1 Die Bestimmung der Korngrössenverteilung in der nicht
extrahierten Droge
Es wurde die Prüfsiebmaschine "LAVIB", Grösse II *), mit Schütteleinrich¬
tung verwendet und von oben nach unten die folgenden Siebe mit Innendurchmesser
20 cm eingesetzt:
*) N.v. Technisch Materieel 's-Gravenhage (Holland)/"TEMA"; Ooievaarlaan 6.

- 105 -
Tab. 36 Die für unsere Siebanalysen verwendeten Siebsätze
1. Siebanalyse (a)verwendet:
entsprechendnach Ph.Helv.V
2. Siebanalyse (b)verwendet:
Sieb: Maschen¬
weite:
Sieb: Maschen¬
weite:
Sieb: Maschen¬
weite:
Din 14 0,43 mm
Din 20 0, 30 mm
D 90 0,21 mm
D 120 0,17 mm
Din 40 0,15 mm
IV 0, 47 mm
IVa 0, 32 mm
V 0, 22 mm
VI 0,17 mm
VH 0,15 mm
D 120 0,170 mm
Din 40 0,150 mm
Din 60 0,102 mm
Din 80 0,075 mm
Din 100 0,060 mm
Die Analyse a erfolgte mit 25 g Drogenpulver, welche erst von Hand durch
Sieb Din 6 geschüttelt wurden.
Teilchen grösser als Sieb Din 6 (1,02 mm Maschenweite; entsprechend nach
Ph.Helv.V ca. Sieb III mit sogar 1,5 mm Maschenweite) konnten nie festgestellt
werden.
Die Analyse b erfolgte mit der Fraktion kleiner Sieb VH von Analyse a.
Die Siebe D 120 und Din 40 mussten auch bei Siebanalyse b mitverwendet
werden, da nur genau 5 Siebe in die Maschine eingespannt werden können.
Für jede Analyse wurde das Drogenpulver und 5 mit Gummi überzogene Me¬
tallkugeln auf das oberste Sieb gegeben und die Maschine während genau 5 Minuten
in Betrieb gesetzt.
845.2 Die Bestimmung der Korngrös senverteilung in der extra¬
hierten Droge
In zwei Vorversuchen zeigte sich, dass die feinen Anteile einer stark extra¬
hierten und von der Extraktlösung durch Zentrifugieren abgetrennten Droge beim
Trocknen stark zusammenkleben. Diese Agglomerate mussten durch leichten Druck
im Mörser zerteilt werden. Die mikroskopische Kontrolle der einzelnen Siebfraktio¬
nen, sowohl in Paraffin, perliquid., als auch in Glycerin 50 % sowie Chloralhydrat,
zeigte, dass die Teilchen grösser als Sieb m ausschliesslich, diejenigen auf Sieb
IV und IVa teilweise, aus feinen, zusammenklebenden Anteilen bestanden.
Die extrahierte Droge wurde daher noch viermal mit je der Hälfte ihres Ge¬
wichtes mit Menstruum Ph.Helv.V vermischt und sofort zentrifugiert. Anschlies¬
send wird analog mit Spir. 96 Vol.% gewaschen. Dieses Vorgehen verhindert ein
Zusammenkleben der Drogenteilchen nach dem Trocknen. Dann wird die Droge auf

- 106 -
einem Tonteller bei Zimmertemperatur zum Trocknen ausgebreitet.
Nach einigen Tagen werden nach gutem Durchmischen 25 g davon den Siebanaly¬
sen, wie unter 845.1 beschrieben, unterworfen.
Alle Siebfraktionen bis und mit Sieb Din 60 wurden anschliessend mikroskopisch
in Chloralhydrat eingelegt kontrolliert.
Weitere, gelegentlich verwendete Bestimmungsmethoden:
846 Die Bestimmung der Asche und ihrer in Salzsäure unlöslichen Anteile
Die Bestimmung wurde nur zur Drogennormierung durchgeführt und erfolgte mit
2 g Chinarinde nach Vorschrift der Ph.Helv. V '. Der Seesand wurde jedoch wegge¬
lassen, um eine Verunreinigung des Tiegels von aussen, sowie Spritzer in den Tiegel
zu vermeiden. Der Tiegel wurde zudem schräg auf die Oeffnung im Asbestcarton ge¬
setzt mit angelehntem Deckel, so dass Dämpfe entweichen können. Resultate: siehe
unter Drogennormierung, pg. 49.
847 Die Bestimmung des Wassergehaltes
Die Bestimmung des Wassergehaltes erfolgte jeweils mit 2 g Chinarinde nach
Vorschrift der Ph.Helv. V169\
848 Die Bestimmung des Alkoholgehaltes der Extrakte
Dabei handelt es sich nur um einige Einzeluntersuchungen. In der Literatur fin¬
det man zahlreiche Vorschläge. Die Ph.Helv. V ' bestimmt den Alkoholgehalt durch
Destillation. Jermstad und Oestby' bezeichnen diese Methode als sehr ein-
171)fach und zuverlässig. Baschilowa und Figurowskij
'
schlagen für Tinkturen
die Bestimmung der Alkoholkonzentration durch Bestimmung des Siedepunktes vor.
Fischer und Mitarbeiter ' schlagen nach einer Vorfällung (Reinigung) das Aus¬
schütteln des Alkohols mit Benzol vor mit anschliessender Bestimmung des Brechungs-173)
indexes. Fischer und Auer ' benützen für die Bestimmung des Alkoholgehaltes
die kritische Mischungstemperatur mit Testlösungen auf dem Kof le r-Heiztisch.174)
Krutzsch ' konstruierte ein weiterentwickeltes Alkrumeter. In einem Pyknome-

- 107 -
ter, das mit Thermometer und Steigrohr versehen ist, wird der für eine bestimmte
Volumenzunahme nötige Temperaturanstieg festgestellt. Mittels Eichkurve kann der
entsprechende Alkoholgehalt abgelesen werden. Bei hohen Extraktivstoffgehalten
kommt noch eine Dichtemessung mittels Pyknometer dazu.
Unsere Bestimmungen wurden nach der Methode der Ph.Helv.V ' durchge-125)
führt, wie sie auch schon Feinstein ; mit Erfolg benützte.
848.1 Die Alkoholbestimmung im Menstruum nach Ph.Helv.V
Im Menstruum wurde die Ameisensäure mit 5 ml 2n-NaOH vollkommen neutra¬
lisiert. Dann wurde weiter genau nach der Vorschrift der Ph.Helv.V ' verfahren.
Der Alkoholgehalt unseres Menstruums beträgt 42,95 Gew.% oder 50,484 Vol.%
bei 15° C.
848.2 Die Alkoholbestimmung in den Extraktlösungen
Die Gerbstoffe in den Extraktlösungen wurden mit 10 ml Bleiacetatlösung Ph.
175)Helv.V ' gefällt, und die Ameisensäure wiederum mit 5 ml 2n-NaOH neutralisiert.
Im weitern wurde genau nach Ph.Helv.V ' verfahren.
849 Die Bestimmung der Tourenzahl
Die Tourenzahl des Rührgerätes wurde mit Hilfe des "Strobotac" Typ 631-BS8 *)
bestimmt. Der Apparat hat zwei Messbereiche: Low: 600 - 3600 U/Min und High:
2400 - 14 400 U/Min.
Da der Rührkopf in das Extraktionsgemisch taucht, ist die einzige für eine Dreh¬
zahlmessung in Frage kommende Markierungsstelle der unten am Rotor im Motorge¬
häuse befestigte Kühlventilator. Dieser besitzt 6 Flügel, die von unten durch die Kühl¬
luftschlitze zugänglich sind.
Die Flügel wurden nun in abwechselnder Reihenfolge mit weissen und schwarzen
Selbstklebestreifen markiert. Einer der drei weissen Streifen wurde zudem mit rotem
Fettstift gefärbt.
Die richtige Tourenzahl ist dann gefunden, wenn:
*) Hersteller: General Radio Company, Cambridge 39, Massachusetts, New York,
Chicago, Los Angeles.

- 108
a) Bei einer bestimmten Drehzahl, welche der Stroboskopfrequenz entspricht, ein
stehendes Bild erhalten wird, d.h. zwei weisse, drei schwarze und ein roter Flü¬
gel sichtbar sind.
b) Bei der doppelten Stroboskopfrequenz 4 graue und zwei graurote Flügel zu sehen
sind (Belichtung nach je 180° Drehung).
c) Bei der halben Stroboskopfrequenz dasselbe, aber schwächer ausgeleuchtete Bild
wie unter a) sichtbar ist.
85 Die Berechnung der Ausbeuten
In dieser Arbeit werden die Wirkstoffgehalte (WG.) in Zukunft folgendermassen
bezeichnet:
AD%
GD%
TED%
A%
G%
TE%
A%D
G%D
TE%D
WG
Gesamtalkaloidgehalt der Droge, ausgedrückt in Prozent (Droge =100%)
Gerbstoffgehalt der Droge, ausgedrückt in Prozent (Droge = 100%)
Totalextraktivstoffgehalt der Droge, ausgedrückt in Prozent (Droge = 100%)
Gesamtalkaloidgehalt
Gerbstoffgehalt
Totalextraktivstoffgehalt_,
in Prozent der fertigen Extrakt¬
lösung, wobei nur der Verlust wäh¬
rend der Extraktion (= Extraktions¬
verlust = e) ergänzt wurde.
Gesamtalkaloidgehalt
Gerbstoffgehalt )ausgedrückt in % [ AD% Gesamtalkaloid-G.
StSÄJTX GD^ Gerbstoff-Gehaltes
100% angenommen [ TED% Totalextraktiv¬
stoff-Gehaltes
Wirkstoffgehalt (= allgemeiner Ausdruck an Stelle von A, G oder TE,z.B. WGD%)
Die Werte AD%, GD% und TED% sind im Kapitel Auswahl der Droge (pg.49) zu
finden.
Alle Wirkstoffgehalte wurden vorerst als Prozente der gewonnenen Extraktlösun¬
gen berechnet, als A%, G%, TE%, allgemein WG%.
Der Wirkstoffgehalt in 500 g Extraktlösung (entsprechend 50 g Droge), ausge¬
drückt in Gramm ist demnach: 5 • WG% g
Der in 50 g Droge bestimmte Wirkstoffgehalt in Gramm (= - g) sei 100 %.
Daraus ergibt sich die folgende Proportion für die Berechnung des gesuchten
Wirkstoffgehaltes, ausgedrückt in Prozenten des Wirkstoffgehaltes der Droge:

- 109 -
WGD%
2: 100% = 5-WG% g : WG%D
oderWG%D =±°ll°0%_^G%g
WGD%g
In dieser Formel muss, je nach Bedarf, WG durch A, G oder TE ersetzt werden.
Wenn allein der Extraktionsverlust berücksichtigt wird, können auf sehr einfache
Weise die folgenden, aufschlussreichen Daten berechnet werden, die auch zur Kontrolle
der Versuche nützlich sind:
Es werden die folgenden Abkürzungen benützt (alle Angaben ausser n in g):
x : Gesamtmenge an Extraktlösung im Extraktionsgemisch nach der Extraktion
T : totale Einwaage von Droge und Menstruum = 550 g = konstant
Z : Verlust während dem Zentrifugieren (= Zentrifugierverlust)
V : an Extraktionsgefäss und eventuell andern Hilfsgefässen hängen gebliebene Ex¬
traktlösung
V : ausser Z noch verdunstetes Lösungsmittel nach Ergänzen des Verlustes wäh¬
rend der Extraktion (= e)
n : Totalextraktivstoffgehalt in Prozent in der Extraktlösung
e : Verlust während der Extraktion (= Extraktionsverlust)
y' : Gewicht der extrahierten Droge
Dk : dekantierte Extraktlösung nach dem Zentrifugieren (e vor dem Zentrifugierenergänzt)
x' : nach der Extraktion in der Droge zurückgehaltene Extraktlösung
V. : V + V (totaler Menstruumverlust)
Aus diesen Angaben können x, x' und y' berechnet werden wie folgt:
3) x -
WH T - (Z + Vt)dennx = 10T.(z+v)+x. n
1 - n/100 u l 1ÜU
4) x' = x - Dk
5) y" =
rjyT - x- ^2- = T-(Z+x)
Das Gewicht der extrahierten Droge lag, bedingt durch das Auswaschen vor der
Trocknung und Verluste beim Manipulieren, meist ca. 10 % (2 - 3 g) unter dem be¬
rechneten Wert.

- 110 -
9 VERSUCHSERGEBNISSE
91 Der Wirkstoffgehalt der Droge
Die für alle folgenden Versuche verwendete Chinarinde enthält:
11,65 % Alkaloide, nach Ph.Helv.V bestimmt (= AD%) = 100 %
7,34 % Gerbstoffe, nach der kolorimetrischen Hautpulvermethodebestimmt (= GD%) = 100 %
Für die Drogennormierung wurden die Gerbstoffe mit siedendem Wasser am Rück¬
flusskühler während 30 Min ausgezogen. Alle übrigen Extrakte wurden jedoch mit al¬
koholhaltigem Menstruum hergestellt, wobei scheinbar mehr an Hautpulver adsorbier¬
bare und mit Phosphorwolframsäure eine Färbung erzeugende Stoffe in den Auszug
gelangen. Daher kommen die Ausbeuten in den Extrakten von über 100 %, verglichen
mit der Drogennormierung.
51,02 % Totalextraktivstoffe, nach der Methode von Fuchs '
bestimmt (= TED%) = 100 %
Weitere Daten über die Droge sind im Abschnitt Drogennormierung (pg.49) und unter
den Bestimmungsmethoden zusammengestellt.
92 Die Mazeration
Die Mazerate zeigten in Abhängigkeit von der Mazerationszeit in 500 g Extrakt¬
lösung die folgenden Ergebnisse. Alle Werte sind Mittel aus zwei Parallelversuchen
mit je zwei Bestimmungen.
Tab. 37 Dichte, Viskosität und Wirkstoffgehalte der Mazerate
Versuch
No.
Zeit
TageTemp.°C
Dichte
g/mlViskosität
in cP
Alkaloide
A%D*)Gerbstoffe
G%D*)Totalex¬
traktiv¬
stoffe
TE%D*)
Mazl
Maz2
Maz3
4
8
16
20
20
20
0,945
0,945
0,945
2,98
2,98
2,97
72,1
73,5
73,6
147,8
147,8
143,0
75,4
75,2
75,2
*) Methode der Ausrechnung und Abkürzungen, s.pg. 108-109.
- Ill -
In den Mazeraten wurden zudem folgende mittleren Teilresultate gefunden:
Tab. 38 Die Gewichte der gesamten, der in der Droge zurückgehaltenen und der ab¬
getrennten Extraktlösung sowie der extrahierten Droge nach der Mazeration
Versuch
No.
x*)
g %vonx
x' *)
g % von x
Dk. *)
g %vonx
y'*)
theor. gewogen
Mazl
Maz2
Maz3
508 100
509 100
508 100
136 26,8
130 25,5
132 26,0
372 73,2
379 74,5
376 74,0
30,4 g 27,2 g
30.4 g 28,1g
30.5 g 27,4 g
MMazl-3 508 100 133 26,2 375 73,8 30,4 g 27,6 g
*) Methode der Ausrechnung und Abkürzungen, s.pg. 108-109.
Aus dieser Tabelle geht klar hervor, dass zwar die Alkaloidextraktion als einzi¬
ge nach 4 Tagen noch nicht beendet ist. Die Mehrausbeute in den nächsten 4 Tagen ist
jedoch sehr klein und den Zeitaufwand nicht wert.
Man kann daher sagen, dass unter den gegebenen Bedingungen eine Mazerations¬
zeit von 4 Tagen für Chinarinde genügt.
Ohne Auspressen der Droge konnten 73,8 % der Extraktlösung gewonnen werden.
26, 2 % blieben nach dem Filtrieren in der Droge. Durch Zentrifugieren an Stelle der
Filtration wären die Mazerationsausbeuten evtl. noch um wenige Prozente höher aus¬
gefallen.
93 Die Perkolation
Die mit dem gleichen Drogenmaterial unter möglichst analogen Bedingungen
hergestellten Perkolate und Mazerate sind wichtige Vergleichswerte für die spätere
Beurteilung der Turboextraktion.
Die Perkolationen zeigten den folgenden Verlauf:
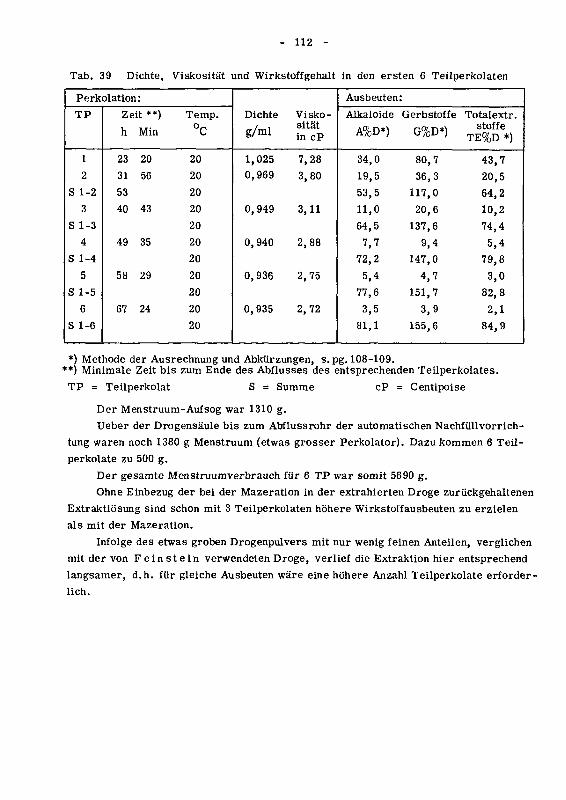
- 112 -
Tab. 39 Dichte, Viskosität und Wirkstoffgehalt in den ersten 6 Teilperkolaten
Perkolation :
Dichte Visko-
, . sität&ml in cP
Ausbeuten:
TP Zeit **) Temp,
h Min °C
Alkaloide Gerbstoffe Totalextr.
A%D*) G%D*) TES^}1
2
S 1-2
3
S 1-3
4
S 1-4
5
S 1-5
6
S 1-6
23 20 20
31 56 20
53 20
40 43 20
20
49 35 20
20
58 29 20
20
67 24 20
20
1,025 7,28
0,969 3,80
0,949 3,11
0,940 2,88
0,936 2,75
0,935 2,72
34,0 80,7 43,7
19,5 36,3 20,5
53,5 117,0 64,2
11.0 20,6 10,2
64.5 137,6 74,4
7,7 9,4 5,4
72,2 147,0 79,8
5.4 4,7 3,0
77.6 151,7 82,8
3.5 3,9 2,1
81.1 155,6 84,9
*) Methode der Ausrechnung und Abkürzungen, s.pg. 108-109.
**) Minimale Zeit bis zum Ende des Abflusses des entsprechenden Teilperkolates.
TP = Teilperkolat S = Summe cP = Centipoise
Der Menstruum-Aufsog war 1310 g.
Ueber der Drogensäule bis zum Abflussrohr der automatischen Nachfüllvorrich¬
tung waren noch 1380 g Menstruum (etwas grosser Perkolator). Dazu kommen 6 Teil¬
perkolate zu 500 g.
Der gesamte Menstruumverbrauch für 6 TP war somit 5690 g.
Ohne Einbezug der bei der Mazeration in der extrahierten Droge zurückgehaltenen
Extraktlösung sind schon mit 3 Teilperkolaten höhere Wirkstoifausbeuten zu erzielen
als mit der Mazeration.
Infolge des etwas groben Drogenpulvers mit nur wenig feinen Anteilen, verglichen
mit der von Feinstein verwendeten Droge, verlief die Extraktion hier entsprechend
langsamer, d.h. für gleiche Ausbeuten wäre eine höhere Anzahl Teilperkolate erforder¬
lich.

- 113 -
94 Die Turboextraktion
941 Die Nachextraktion während der Trennung von extrahierter Droge
und Extraktlösung (= Versuchsreihe 1)
Aus unseren Vorversuchen mussten wir schliessen, dass während des Zentrifu-
gierens noch beträchtliche Mengen Wirkstoffe aus der Droge extrahiert werden.
Die nachträgliche Extraktion bei der Abtrennung der extrahierten Droge von der
Extraktlösung ist umso stärker, je gehaltreicher die Droge ist. Um deren stärkste
Auswirkung zu studieren, wurden die Trennungsverfahren (Filtration und Zentrifugie-
ren) an der nicht extrahierten Droge, sowie an einem bei 10 000 U/Min während 5 Min
gewirbelten Extraktionsgemisch, ausprobiert.
Die Resultate sind in der folgenden Tabelle 40 zusammengefasst.
Die Nachextraktion der Droge durch das Filtrieren oder Zentrifugieren während der
Trennung von extrahierter Droge und Extraktlösung:
Diese Untersuchungen erfolgten vor allem an der nicht extrahierten Droge, wel¬
che dank ihrem hohen Wirkstoffgehalt durch das Trennverfahren am stärksten extra¬
hiert wird. Die gefundenen Wirkstoffgehalte sind daher obere Grenzwerte, welche bei
einer durch Turboextraktion vorextrahierten Droge nie erreicht werden.
Tab. 40 Einfluss verschiedener Trennverfahren von extrahierter Droge und Extrakt¬
lösung auf den Wirkstoffgehalt der Extraktlösung
Turboextraktion :
Trv. Versuch
No.
Drehzahl
U/MinZeit
Min
Temp.°C
Dichte
g/mlVisko¬
sität
in cP
Alka- Gerb- Totalex-
loide Stoffe tr. Stoffe
A%D*) G%D*) TE%D*)
F Tul 0 0 20 0,938 2,78 41,8 78,8 40,3
F Tu2 10000 5 20 0,945 2,93 66,4 136,3 67,8
ZM Tu3 0 0 20 0,947 2,94 73,3 141,4 73,1
ZM TulO 10000 5 20 0,950 2,99 79,9 146,9 73,5
FM Tu4 0 0 20 0,943 2,90 61,4 122,8 61,9
F = Filtration; Methodik siehe unter 821 a und b, pg. 63, 64.
cP = CentipoiseTrv. = Trennverfahren
ZM = Zentrifugieren unter Aufteilung des Extraktionsgemisches in zwei Hälften,die nacheinander zentrifugiert werden (vergl. 813, pg.62)
FM = Filtration des in zwei Hälfte aufgeteilten Extraktionsgemisches unter ana¬
logen zeitlichen Bedingungen, wie sie beim Zentrifugieren bestehen (vergl.822.3, pg.66)
*) Methode der Ausrechnung und Abkürzungen, s.pg. 108-109.
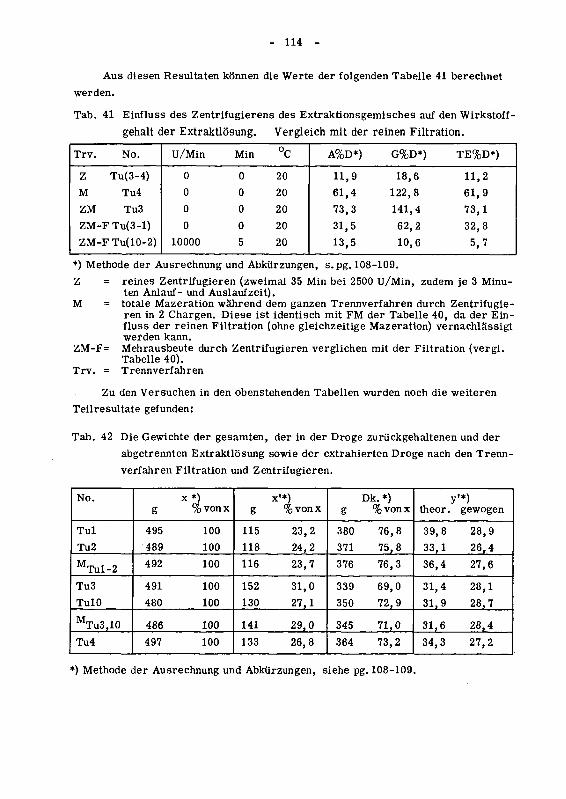
- 114 -
Aus diesen Resultaten können die Werte der folgenden Tabelle 41 berechnet
werden.
Tab. 41 Einfluss des Zentrifugierens des Extraktionsgemisches auf den Wirkstoff¬
gehalt der Extraktlösung. Vergleich mit der reinen Filtration.
Trv. No. U/Min Min °C A%D*) G%D*) TE%D*)
Z Tu(3-4) 0 0 20 11,9 18,6 11,2
M Tu4 0 0 20 61,4 122,8 61,9
ZM Tu3 0 0 20 73,3 141,4 73,1
ZM-FTu(3-l) 0 0 20 31,5 62,2 32,8
ZM-FTu(10-2) 10000 5 20 13,5 10,6 5,7
*) Methode der Ausrechnung und Abkürzungen, s.pg. 108-109.
Z = reines Zentrifugieren (zweimal 35 Min bei 2500 U/Min, zudem je 3 Minu¬
ten Anlauf- und Auslaufzeit).M = totale Mazeration während dem ganzen Trennverfahren durch Zentrifugie¬
ren in 2 Chargen. Diese ist identisch mit FM der Tabelle 40, da der Ein¬
fluss der reinen Filtration (ohne gleichzeitige Mazeration) vernachlässigtwerden kann.
ZM-F= Mehrausbeute durch Zentrifugieren verglichen mit der Filtration (vergl.Tabelle 40).
Trv. = Trennverfahren
Zu den Versuchen in den obenstehenden Tabellen wurden noch die weiteren
Teilresultate gefunden:
Tab. 42 Die Gewichte der gesamten, der in der Droge zurückgehaltenen und der
abgetrennten Extraktlösung sowie der extrahierten Droge nach den Trenn¬
verfahren Filtration und Zentrifugieren.
No. x*)
g %vonxX'*)
g %vonxDk.*)
g %vonxy'*)
theor. gewogen
Tul
Tu2
495 100
489 100
115 23,2
118 24,2
380 76,8
371 75,8
39,8 28,9
33,1 26,4
MTul-2 492 100 116 23,7 376 76,3 36,4 27,6
Tu3
TulO
491 100
480 100
152 31,0
130 27,1
339 69,0
350 72,9
31,4 28,1
31,9 28,7
MTu3,10 486 100 141 29,0 345 71,0 31,6 28,4
Tu4 497 100 133 26,8 364 73,2 34,3 27,2
*) Methode der Ausrechnung und Abkürzungen, siehe pg. 108-109.
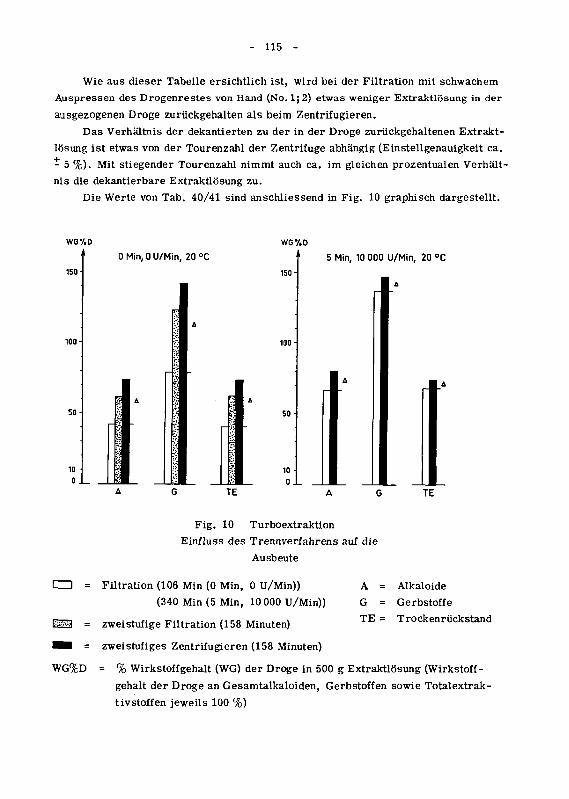
- 115 -
Wie aus dieser Tabelle ersichtlich ist, wird bei der Filtration mit schwachem
Auspressen des Drogenrestes von Hand (No. 1; 2) etwas weniger Extraktlosung in der
ausgezogenen Droge zurückgehalten als beim Zentrifugleren.
Das Verhältnis der dekantierten zu der in der Droge zurückgehaltenen Extrakt¬
losung ist etwas von der Tourenzahl der Zentrifuge abhangig (Einstellgenauigkeit ca.
- 5 %). Mit stiegender Tourenzahl nimmt auch ca. im gleichen prozentualen Verhält¬
nis die dekantierbare Extraktlosung zu.
Die Werte von Tab. 40/41 sind anschliessend in Fig. 10 graphisch dargestellt.
WG%D
OMin,OU/Min, 20 °C
TE
WGV.D
150
10
0 J_
5 Min, 10 000 U/Min, 20 °C
1TE
Fig. 10 Turboextraktion
Einfluss des Trennverfahrens auf die
Ausbeute
A
G
TE
Alkaloide
Gerbstoffe
Trockenruckstand
CH = Filtration (106 Min (0 Min, 0 U/Min))
(340 Min (5 Min, 10 000 U/Mm))
ESH3 = zweistufige Filtration (158 Minuten)
^H = zweistufiges Zentrifugieren (158 Minuten)
WG%D = % Wirkstoffgehalt (WG) der Droge in 500 g Extraktlosung (Wirkstoff -
gehalt der Droge an Gesamtalkaloiden, Gerbstoffen sowie Totalextrak¬
tivstoffen jeweils 100 %)
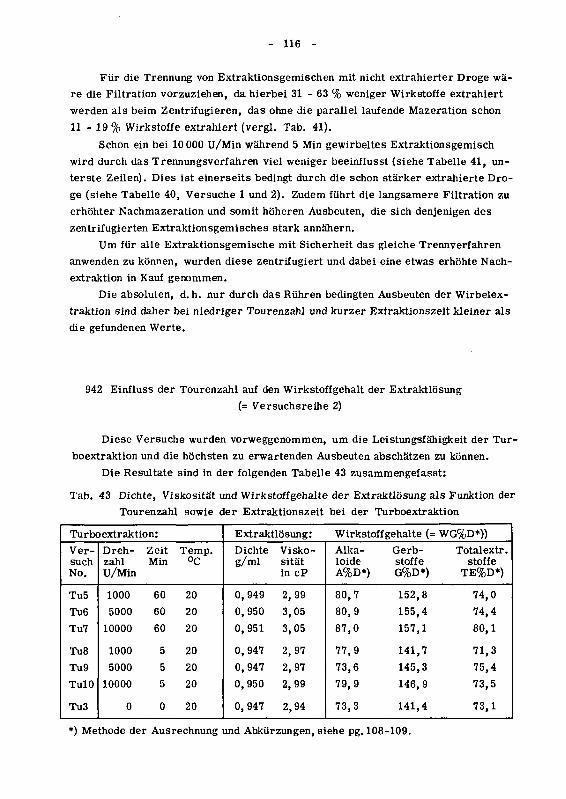
- 116 -
Für die Trennung von Extraktionsgemischen mit nicht extrahierter Droge wä¬
re die Filtration vorzuziehen, da hierbei 31 - 63 % weniger Wirkstoffe extrahiert
werden als beim Zentrifugieren, das ohne die parallel laufende Mazeration schon
11 - 19 % Wirkstoffe extrahiert (vergl. Tab. 41).
Schon ein bei 10 000 U/Min während 5 Min gewirbeltes Extraktionsgemisch
wird durch das Trennungsverfahren viel weniger beeinflusst (siehe Tabelle 41, un¬
terste Zeilen). Dies ist einerseits bedingt durch die schon stärker extrahierte Dro¬
ge (siehe Tabelle 40, Versuche 1 und 2). Zudem führt die langsamere Filtration zu
erhöhter Nachmazeration und somit höheren Ausbeuten, die sich denjenigen des
zentrifugierten Extraktionsgemisches stark annähern.
Um für alle Extraktionsgemische mit Sicherheit das gleiche Trennverfahren
anwenden zu können, wurden diese zentrifugiert und dabei eine etwas erhöhte Nach¬
extraktion in Kauf genommen.
Die absoluten, d.h. nur durch das Rühren bedingten Ausbeuten der Wirbelex¬
traktion sind daher bei niedriger Tourenzahl und kurzer Extraktionszeit kleiner als
die gefundenen Werte.
942 Einfluss der Tourenzahl auf den Wirkstoffgehalt der Extraktlösung
(= Versuchsreihe 2)
Diese Versuche wurden vorweggenommen, um die Leistungsfähigkeit der Tur¬
boextraktion und die höchsten zu erwartenden Ausbeuten abschätzen zu können.
Die Resultate sind in der folgenden Tabelle 43 zusammengefasst:
Tab. 43 Dichte, Viskosität und Wirkstoffgehalte der Extraktlösung als Funktion der
Tourenzahl sowie der Extraktionszeit bei der Turboextraktion
Turboextraktion: Extraktlösung: Wirkstoffgehalte (= WG%D*))
Ver¬
such
No.
Dreh- Zeit Temp,zahl Min °C
U/Min
Dichte Visko-
g/ml sität
in cP
Alka- Gerb- Totalextr.
loide stoffe Stoffe
A%D*) G%D*) TE%D*)
Tu5
Tu6
Tu7
Tu8
Tu9
TulO
Tu3
1000 60 20
5000 60 20
10000 60 20
1000 5 20
5000 5 20
10000 5 20
0 0 20
0, 949 2, 99
0,950 3,05
0,951 3,05
0, 947 2, 97
0, 947 2, 97
0, 950 2, 99
0,947 2,94
80,7 152,8 74,0
80,9 155,4 74,4
87,0 157,1 80,1
77,9 141,7 71,3
73,6 145,3 75,4
79,9 146,9 73,5
73,3 141,4 73,1
*) Methode der Ausrechnung und Abkürzungen, siehe pg. 108-109.

- 117 -
Dazu kommen noch die folgenden Teilresultate:
Tab. 44 Die Gewichte der gesamten, der in der Droge zurückgehaltenen und der
abgetrennten Extraktlösung sowie der extrahierten Droge als Funktion der
Tourenzahl sowie der Extraktionszeit bei der Turboextraktion.
No.x*) x' *) Dk. *) y' *)
g % vonx g % vonx g %vonx theor. gewogen
Tu5 482 100 145 30,1 337 69,9 31,7 28,8
Tu6 486 100 121 24,9 365 75,1 31,4 28,2
Tu7 485 100 118 24,3 367 75,7 29,9 26,2
Tu8 490 100 144 29,4 346 70,6 32,2 27,7
Tu9 488 100 147 30,1 341 69,9 31,2 27,2
TulO 480 100 130 27,1 350 72,9 31,9 28,7
MTu5-l 3485 100 134 27,6 351 72,4 31,4 27,8
*) Methode der Ausrechnung und Abkürzungen siehe pg. 108-109.
Wie aus der Tabelle hervorgeht, wird desto weniger Extraktlösung in der aus¬
gezogenen Droge zurückgehalten, je mehr die letztere bei der Extraktion zerkleinert
wurde.
Die Werte der Versuchsreihe 2 sind nachstehend noch graphisch dargestellt.
(Vergl. Fig. 11, pg.118.)
Die Wirkstoffgehalte der 16-tägigen Mazeration in 500 g Extraktlösung sowie
die Ausbeuten der Perkolation mit 4 und mit 6 Teilperkolaten sind als Vergleichs¬
masstab für unsere Versuche als horizontale Linien eingetragen.
Bei einer Wirbelzeit von einer Stunde erreicht man schon bei 5000 U/Min in
500 g Extraktlösung Wirkstoffgehalte, die in Bezug auf Alkaloide und Gerbstoffe den¬
jenigen in 6 Teilperkolaten entsprechen. Der Totalextraktivstoffgehalt liegt tiefer
als in 6 Teilperkolaten.
Bei 10 000 U/Min werden in einer Stunde 87 % der Drogenalkaloide extrahiert.
Auch der Gerbstoffgehalt ist höher als in 6 Teilperkolaten, nicht jedoch der Total¬
extraktivstoffgehalt, was auch Melichar'feststellte. Die Alkaloide und Gerb¬
stoffe sind offenbar leichter löslich im Menstruum als alle übrigen Extraktivstoffe.
Auch Hagelstein stellt fest, dass keine Beziehung zwischen dem Totalex¬
traktivstoff- und dem Alkaloidgehalt bei Chinarindentinktur besteht, so dass für die
Wertbestimmung unbedingt beide Daten bestimmt werden müssen.
Die Dichte und die Viskosität zeigten im Verlaufe all dieser Versuche nur ver¬
hältnismässig geringe Schwankungen.

- 118 -
WG. ALKALOIDE WG. GERBSTOFFE WG TROCKEN-55 t RUCKSTAND
5 10
• 1000 R/Min -1000 R/Min -1000R/Min
Fig. 11 Ausbeute der Turboextraktion als Funktion der Tourenzahl
%WG. = % Wirkstoffgehalt (WG.) der Droge in 500 g Extraktlösung (Wirk¬
stoffgehalt der Droge an Gesamtalkaloiden, Gerbstoffen sowie Totalextrak¬
tivstoffen jeweils = 100 %). Die horizontalen Linien bedeuten % Wirk¬
stoffgehalt bei 16-tägiger Mazeration (M) in 500 g Extraktlösung oder
nach Perkolation in 4 - 6 Teilperkolaten (TP).
I l = 0 Minuten Extraktionszeit (Nullwert)
E7TT3 5 " "
= 60
943 Einfluss der Extraktionszeit auf den Wirkstoffgehalt der Extraktlösung
(= Versuchsreihe 3)
943.1 Resultate:
Das Zahlenmaterial dazu ergibt sich aus der Gegenüberstellung der Auszüge
der Versuchsreihe 2 (pg. 116-117), die sich nur in der Extraktionszeit (0, 5, 60 Mi¬
nuten) voneinander unterscheiden. Die entsprechende graphische Darstellung sieht
wie folgt aus:

- 119 -
WG ALKALOIDE WG. GERBSTOFFE
165-
160-
TPIbS- 1-6
150-
-
TP
U5- 11-4
M
un- MÜ. 1 ;
TROCKEN-
RÜCKSTAND
fl IFig. 12 Ausbeute der Turboextraktion als Funktion der Zeit
%WG. = % Wirkstoffgehalt (WG.) der Droge in 500 g Extraktlösung (Wirk¬
stoffgehalt der Droge an Gesamtalkaloiden, Gerbstoffen sowie Totalextrak¬
tivstoffen jeweils = 100 %). Die horizontalen Linien bedeuten % Wirkstoff¬
gehalt bei 16-tägiger Mazeration (M) in 500 g Extraktlösung oder nach Per-
kolation in 4 - 6 Teilperkolaten (TP).
= 0 Touren pro Minute (= R/Min)
= 1000 " " "
F^TT-?! = 5000
=10000
Sowohl eine Erhöhung der Tourenzahl bei konstanter Extraktionszeit als auch
eine Verlängerung der Extraktionszeit bei konstanter Tourenzahl steigert den Wirk¬
stoffgehalt der Extraktlösungen. Dies gilt vor allem für die Alkaloid- und Gerbstoff¬
ausbeuten.
Bei Tourenzahlen unter 5000 U/Min bestimmt vorwiegend die Extraktionszeit
den Wirkstoffgehalt der Extraktlösungen.
Bei 10 000 U/Min werden in 5 Minuten 8, 5 % mehr Alkaloide extrahiert als bei
5000 U/Min.

- 120 -
943.2 Die statistische Sicherung der Resultate der Versuchs¬
reihen 2 und 3
Der Einfluss von Tourenzahl und Extraktionszeit auf die Wirkstoffgehalte in
500 g Extraktlösung wurde statistisch überprüft.
Es wurde mit Hilfe der einzelnen Bestimmungen je eine Streuungszerlegung
für den Alkaloid-, den Gerbstoff- sowie den Totalextraktivstoffgehalt durchgerech¬
net. In der Arbeit sind jeweils nur die arithmetischen Mittel aus 4 Bestimmungen
(2 Parallelbestimmungen in 2 Parallelversuchen) angegeben.
Die Einzelresultate wurden zur Vereinfachung der Rechnung umgeformt (s. pg.
122, Tab. 45). In der folgenden Tabelle 45 sind die Summen von je vier solchen um¬
geformten Einzelresultaten (je 2 Parallelbestimmungen in 2 Parallelversuchen) auf¬
geführt. (Würden diese Summen durch 4000 dividiert und der bei der Umformung
subtrahierte Betrag anschliessend addiert, so ergäben sich die zu den entsprechen¬
den Versuchsnummern gehörenden, in dieser Arbeit angegebenen Resultate.) Die
Umformung hat keinen Einfluss auf die statistische Prüfung.
Die Signifikanz der Differenzen zwischen den in Tab. 45 angegebenen Versuchs¬
ergebnissen wurde nun mittels des t-Testes statistisch überprüft.177)
Linder '
gibt dafür folgende drei Formeln:
1 Nl N2(1) s2 = fs (x!-x')2 + S (x»-x")2l
Nl + N2 zi=l i=l
(2)x'-x» \ N1'N2
sd V N1+N2
(3) n = Nx + N2 - 2
Es bedeuten:
s ,= Streuung (für das besondere Problem "Unterschied zweier Durchschnitte")
N1 = Anzahl Messungen oder Zählungen der ersten Versuchsreihe
N2 = Anzahl Messungen oder Zählungen der zweiten Versuchsreihe
x! = Einzelwert der ersten Versuchsreihe
x'.' = Einzelwert der zweiten Versuchsreihe_ix' = Durchschnitt der ersten Versuchsreihe
x" = Durchschnitt der zweiten Versuchsreihe
S = Summenzeichen (= summiere von i=l bis i=Nj resp. N,)
n = Zahl der Freiheitsgrade (=Nj+N2-2), da wir zum berechnen von x' und

- 121 -
x" zwei lineare Beziehungen benötigen.
t = statistische Masszahl, d.h. der für den Vergleich mit der "Standard-
Tabelle" umgerechnete Unterschied der_beiden Durchschnitte (vor der
Umrechnung ist dieser Unterschied d = x' - x").
Für unser berechnetes n sind im Buche von Linder die Sicherheits-
schwellen t angegeben, und zwar für eine Wahrscheinlichkeit von 1:1000, 1:100, so¬
wie 5:100, dass eine zufällige Abweichung fälschlicherweise als wesentlich beurteilt
wird.
t für P=0,01 wird in Zukunft als tQ Q<bezeichnet.
P = Wahrscheinlichkeit
Es bedeuten:
t > tn oioder *-0 001
* Resultate sind gesichert,
'o Oli<'<'o Ol: Hinweis auf einen wesentlichen Unterschied,
t„fii-
>t : der Unterschied ist bloss zufällig, die Resultate sind nicht gesichert.
Da alle zu vergleichenden Werte aus 4 Einzelresultaten entstanden, N also
gleich 4 und konstant ist, und da die mittlere quadratische Abweichung für ein- und
denselben Wirkstoff ebenfalls konstant ist, kann die Formel (2) nach (x'-x") aufge¬
löst, d.h. die kleinste gesicherte Differenz für die zu vergleichenden Durchschnitts¬
werte oder, nach Multiplikation mit 4 für die zu vergleichenden Summen für tn „-,
tQ Q,und t„
00«berechnet werden.
N == 4 = konstant
(4) x' - X"*** l-sd
\ /N1'N2 \[TV N1+N2
(5) 4 -(x> • x") =
P d= dp
somit ist
0,001, 0,01 oder 0,05
Wird in Formel 5 einer der angegebenen Werte für P eingesetzt, so ergibt
sich der entsprechende Wert d (dQ „„.,, d0 _-,d„ Q1-).
Die Differenz zweier Summen in Tabelle 45 sei d.
Die Resultate sind dann wie folgt zu beurteilen:
d > 1 ». oder d-n01
: Resultate sind gesichert,
dQ nc<d <d0 0.: Hinweis auf einen wesentlichen Unterschied,
(L nr>d : der Unterschied ist bloss zufällig, die Resultate sind nicht gesichert.
Die Grössenordnung der so erhaltenen statistischen Sicherung der Resultate
ist in Tabelle 46 zusammengefasst.
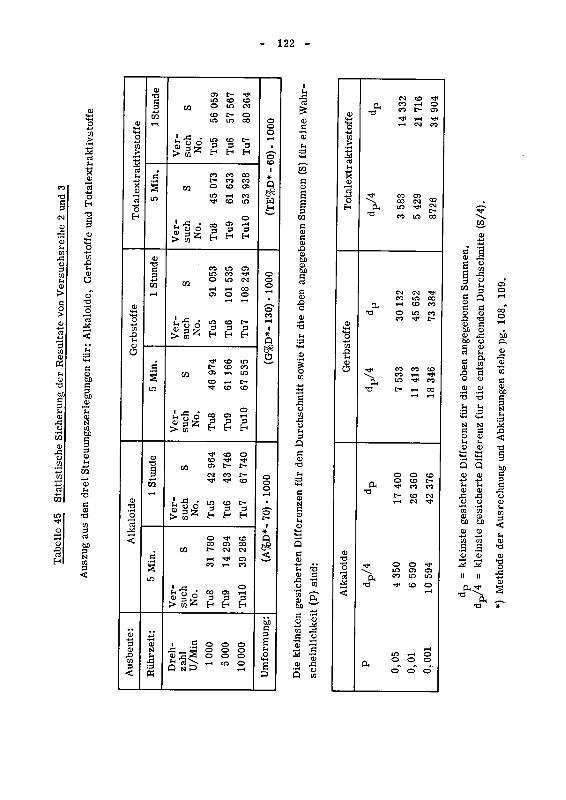
109.
108,
pg.
siehe
Abkürzungen
und
Ausrechnung
der
Methode
*)
(S/4).
Durchschnitte
entsprechenden
die
fur
Differenz
gesicherte
kleinste
=dp/4
Summen.
angegebenen
oben
die
für
Differenz
gesicherte
kleinste
=dp
904
34
8726
716
21
429
5
332
14
583
3
dpdp
/4
384
73
346
18
652
45
413
11
132
30
533
7
dpdp
/4
376
42
594
10
0,001
360
26
590
60,01
400
17
350
40,05
dp
dp/4
P
Totalextraktivstoffe
Gerbstoffe
Alkaloide
sind:
(P)
scheinlichkeit
,Wahr-
eine
für
(S)
Summen
angegebenen
oben
die
für
sowie
Durchschnitt
den
für
Differenzen
gesicherten
kleinsten
Die
CO
to
1000
(TE%D*-60).
1000
(G%D*-130)-
1000
•70)
(A%D*-
Umformung:
264
80
Tu7
567
57
Tu6
059
56
Tu5
No.
Ssuch
Ver¬
938
53
TulO
633
61
Tu9
073
45
Tu8
No.
Ssuch
Ver¬
249
108
Tu7
535
101
Tu6
053
91
Tu5
No.
Ssuch
Ver¬
535
67
TulO
166
61
Tu9
974
46
Tu8
No.
Ssuch
Ver¬
740
67
Tu7
746
43
Tu6
964
42
Tu5
No.
Ssuch
Ver¬
286
39
TulO
294
14
Tu9
780
31
Tu8
No.
Ssuch
Ver¬
000
10
000
51000
U/Min
zahl
Dreh¬
Stunde
1Min.
5Stunde
1Min.
5Stunde
1Min.
5Rührzeit:
Totalextraktivstoffe
Gerbstoffe
Alkaloide
Ausbeute:
Totalextraktivstoffe
und
Gerbstoffe
Alka
loid
e,für:
Streuungszerlegungen
drei
den
aus
Auszug
3und
2Versuchsreihe
von
Resultate
der
Sicherung
Statistische
45
Tabelle

- 123 -
Tabelle 46 Grössenordnung der statistischen Sicherung der Resultate der Versuchsreihen 2 und 3
Extraktionszeit:
5 Minuten 1 Stunde
Dreh¬ Zeiteinfluss:
zahl
U/Min
1 000
5 Min——1 Stunde
% d
A:«95 (11184)
G: <99 (44 079)1 1
1 TE:<95 (10 986) 1%
A: - ?
d
(-17 486)
%A: «95
d
( 782)
G:«95 (14 192) G: «95 (10 482)
TE: >95 (16 560) TE: «95 ( 1 508)
% d
A: « 95 (7 506)% d
A:>99 (29 452)
% d
A: < 99 (24 776)
| G: «95 (20 561)
TE: «95 ( 8 865)
G: < 99 (40 369)
TE: - ? (-4 066)
G: «95 (17 196)
1 TE: > 99 (24 205)
%A: <99
d
(24 992)
%A: < 99
d
(23 994)
G: « 95 ( 6 369) G: «95 ( 6 714)
TE: - ? (-7 695)
1 A: > 99 (28 454)
TE: > 99 (22 697)
10 0001
G: < 99 (40 714)1
TE: > 99 (26 326)
< = etwas kleiner als ...
> = etwas grosser als ...
- ? = Abnahme statt Zunahme der Ausbeute
<?: = viel kleiner als .
»= viel grösser als .
A = Alkalolde
G = Gerbstoffe
TE = Totalextraktivstoffe
In Klammern sind die Differenzen (d) der Werte S der vorhergehenden Tabelle 45 angegeben. (Die Werte d wurden mit den
entsprechenden Werten dp verglichen, um die Prozente zu erhalten).

- 124 -
In Worten ausgedrückt heisst das für eine bestimmte Menge Extraktlösung:
1. stark gesichert sind: die Zunahmen der Wirkstoffgehalte bei Verlängerungder Extraktionszeit von 5 Min auf 1 Stunde. Ausnahmen bilden der Alkaloid-
gehalt bei 1000 U/Min und der Totalextraktivstoffgehalt bei 5000 U/Min.Die Zunahmen des Alkaloidgehaltes und des Totalextraktivstoffgehal-
tes bei Erhöhung der Tourenzahl von 1000 auf 10 000 U/Min bei 1 Stunde
Extraktionszeit.
2. schwach gesichert sind: die Zunahmen des Wirkstoffgehaltes bei Erhöhungder Tourenzahl von 1000 auf 10 000 U/Min bei 5 Minuten Extraktionszeit.
Die Zunahme des Gerbstoffgehaltes bei Erhöhung der Tourenzahl von
1000 auf 10 000 U/Min bei 1 Stunde Extraktionszeit.
Die Zunahme des Alkaloidgehaltes bei 1000 U/Min und Verlängerungder Extraktionszeit von 5 Min auf 1 Stunde.
3. nicht gesichert sind: die Zunahme des Alkaloidgehaltes bei 5 Min Extrak¬
tionszeit und Erhöhung der Tourenzahl von 1000 auf 5000 U/Min.Die Zunahme des Totalextraktivstoffgehaltes bei 5 Min Extraktions¬
zeit und Erhöhung der Tourenzahl von 5000 auf 10 000 U/Min.Diese Differenzen sind zufällig, d.h. nicht signifikant. Sie sind auf
den Einfluss der Nachextraktion beim Zentrifugieren zurückzuführen.
944 Einfluss der Temperatur auf den Wirkstoffgehalt der Extraktlösung
Die Schwankungen der Ausbeuten als Funktion von Rührzeit und Tourenzahl
waren nicht so gross wie erwartet. Dies ist hauptsächlich auf die unvermeidbare,
bei nur schwach ausgezogenen Drogen starke Nachextraktion während der Aufarbei¬
tung zurückzuführen. Es wurden daher bei 40 C nur die in der folgenden Tabelle 47
zusammengefassten Versuche ausgeführt (= Versuchsreihe 4).
Tab. 47 Dichte, Viskosität und Wirkstoffgehalte der Extraktlösungen als Funktion
der Temperatur bei der Turboextraktion
Turboextraktion: Extraktlösung: Ausbeuten (WG%D*))
Ver¬
such
No.
Dreh- Zeit Temp,zahl
U/Min Min C
Dichte Visko¬
sität
g/ml in cP
Alkaloide Gerbstoffe Totalextr.
Stoffe
A%D *) G%D *) TE%D *)
Tüll
Tu3
Tul2
Tu8
Tul3
Tu7
0 0 40
0 0 20
1000 5 40
1000 5 20
10000 60 40
10000 60 20
0, 945 3,010,947 2,940,949 2,980,947 2,970,958 2,970,951 3,05
73,9 152,5 79,273,3 141,4 73,1**)78.0 154,8 80,077,9 141,7 71,3**)83.1 163,6 82,587,0 157,1 80,1**)
*) Methode der Ausrechnung und Abkürzungen, siehe pg. 108-109.
**) Diese Werte sind nur zum Vergleich aufgeführt.
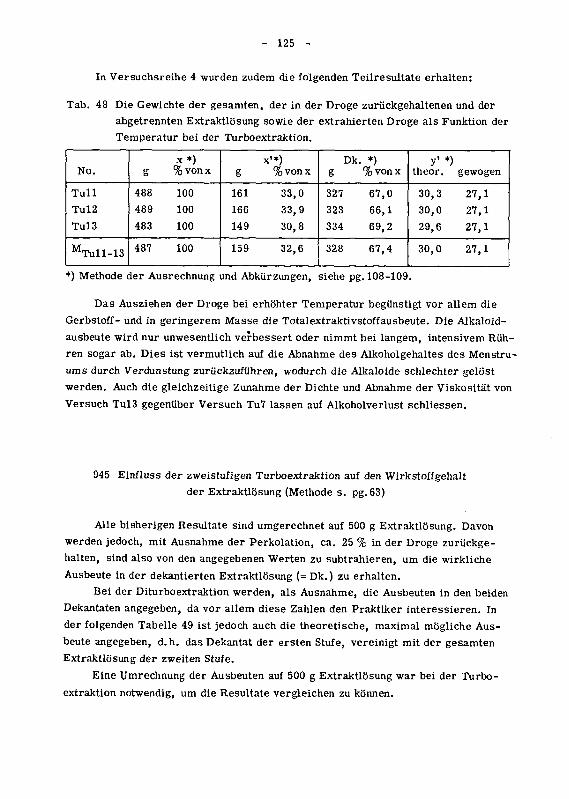
- 125 -
In Versuchsreihe 4 wurden zudem die folgenden Teilresultate erhalten:
Tab. 48 Die Gewichte der gesamten, der in der Droge zurückgehaltenen und der
abgetrennten Extraktlösung sowie der extrahierten Droge als Funktion der
Temperatur bei der Turboextraktion.
No.
x*)
g %vonxx'*)
g %vonxDk. *)
g %vonxy. *)
theor. gewogen
Tüll
Tul2
Tul3
488 100
489 100
483 100
161 33,0
166 33,9
149 30,8
327 67,0
323 66,1
334 69,2
30,3 27,1
30,0 27,1
29,6 27,1
MTull-13 487 100 159 32,6 328 67,4 30,0 27,1
*) Methode der Ausrechnung und Abkürzungen, siehe pg. 108-109.
Das Ausziehen der Droge bei erhöhter Temperatur begünstigt vor allem die
Gerbstoff- und in geringerem Masse die Totalextraktivstoffausbeute. Die Alkaloid-
ausbeute wird nur unwesentlich verbessert oder nimmt bei langem, intensivem Rüh¬
ren sogar ab. Dies ist vermutlich auf die Abnahme des Alkoholgehaltes des Menstru-
ums durch Verdunstung zurückzuführen, wodurch die Alkaloide schlechter gelöst
werden. Auch die gleichzeitige Zunahme der Dichte und Abnahme der Viskosität von
Versuch Tul3 gegenüber Versuch Tu7 lassen auf Alkoholverlust schliessen.
945 Einfluss der zweistufigen Turboextraktion auf den Wirkstoffgehalt
der Extraktlösung (Methode s. pg. 63)
Alle bisherigen Resultate sind umgerechnet auf 500 g Extraktlösung. Davon
werden jedoch, mit Ausnahme der Perkolation, ca. 25 % in der Droge zurückge¬
halten, sind also von den angegebenen Werten zu subtrahieren, um die wirkliche
Ausbeute in der dekantierten Extraktlösung (= Dk. ) zu erhalten.
Bei der Diturboextraktion werden, als Ausnahme, die Ausbeuten in den beiden
Dekantaten angegeben, da vor allem diese Zahlen den Praktiker interessieren. In
der folgenden Tabelle 49 ist jedoch auch die theoretische, maximal mögliche Aus¬
beute angegeben, d.h. das Dekantat der ersten Stufe, vereinigt mit der gesamten
Extraktlösung der zweiten Stufe.
Eine Umrechnung der Ausbeuten auf 500 g Extraktlösung war bei der Turbo¬
extraktion notwendig, um die Resultate vergleichen zu können.
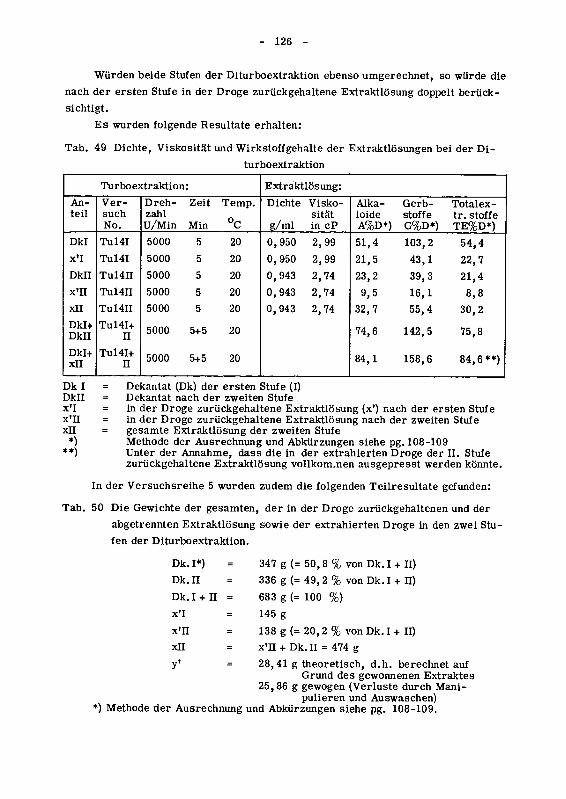
- 126 -
Würden beide Stufen der Diturboextraktion ebenso umgerechnet, so würde die
nach der ersten Stufe in der Droge zurückgehaltene Extraktlösung doppelt berück¬
sichtigt.
Es wurden folgende Resultate erhalten:
Tab. 49 Dichte, Viskosität und Wirkstoffgehalte der Extraktlösungen bei der Di¬
turboextraktion
Turboextraktion: Extraktlösung:
An¬
teil
Ver¬
such
No.
Dreh¬
zahl
U/Min
Zeit
Min
Temp.
°C
Dichte Visko¬
sität
g/ml in cP
Alka-
loide
A%D*)
Gerb¬
stoffe
G%D*)
Totalex-
tr. Stoffe
TE%D*)
Dkl Tul4I 5000 5 20 0,950 2,99 51,4 103,2 54,4
x*I Tul4I 5000 5 20 0, 950 2, 99 21,5 43,1 22,7
Dkn Tul4H 5000 5 20 0,943 2,74 23,2 39,3 21,4
x'n Tul4II 5000 5 20 0,943 2,74 9,5 16,1 8,8
xll TuHII 5000 5 20 0,943 2,74 32,7 55,4 30,2
Dkl+
Dkll
Tul4I+
n5000 5+5 20 74,6 142,5 75,8
Dkl+
xll
Tul4I+
n5000 5+5 20 84,1 158,6 84,6**)
Dk I = Dekantat (Dk) der ersten Stufe (I)Dkll = Dekantat nach der zweiten Stufe
x'I = in der Droge zurückgehaltene Extraktlösung (x') nach der ersten Stufe
x'H = in der Droge zurückgehaltene Extraktlösung nach der zweiten Stufe
xll = gesamte Extraktlösung der zweiten Stufe
*) Methode der Ausrechnung und Abkürzungen siehe pg. 108-109
**) Unter der Annahme, dass die in der extrahierten Droge der II. Stufe
zurückgehaltene Extraktlösung vollkom.nen ausgepresst werden könnte.
In der Versuchsreihe 5 wurden zudem die folgenden Teilresultate gefunden:
Tab. 50 Die Gewichte der gesamten, der in der Droge zurückgehaltenen und der
abgetrennten Extraktlösung sowie der extrahierten Droge in den zwei Stu¬
fen der Diturboextraktion.
Dk. I*) = 347 g (= 50,8 % von Dk. I + II)
Dk. II = 336 g (= 49,2 % von Dk. I + n)
Dk.I + II = 683 g(= 100 %)
x'I = 145 g
x'H = 138 g (= 20,2 % von Dk. I + II)
xll = x'H + Dk. II = 474 g
y' = 28,41 g theoretisch, d.h. berechnet auf
Grund des gewonnenen Extraktes
25,86 g gewogen (Verluste durch Mani¬
pulieren und Auswaschen)*) Methode der Ausrechnung und Abkürzungen siehe pg. 108-109.
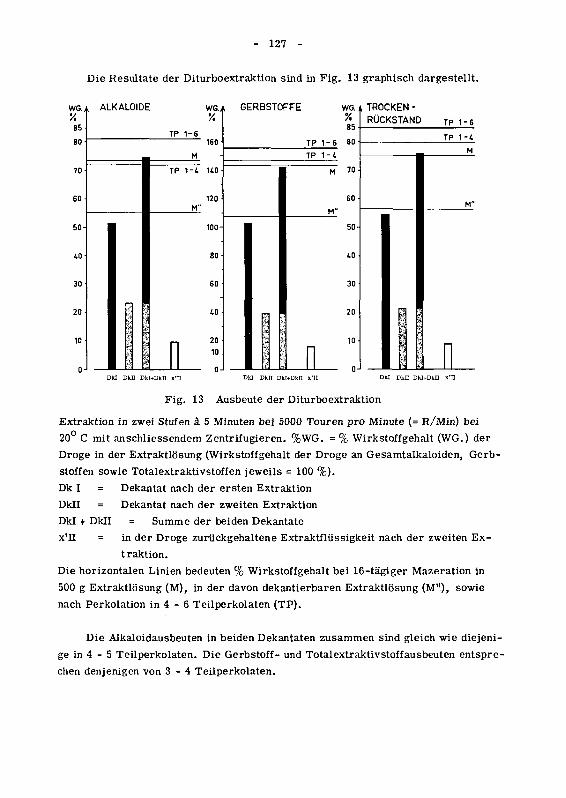
- 127 -
Die Resultate der Diturboextraktion sind in Fig. 13 graphisch dargestellt.
WG
•/.
85
80
ALKALOIDE
TP i-s
WG k GERBSTOFFE
%
160
M
TP 1-A U0
120
M"
100
80
60
20
10
%85
TP 1-6 80
WG iTROCKEN -
TP 1-4
M 70
60
M"
50-
Li
20
10
RUCKSTAND TP 1-6
Dkl Dkn Dkl+Dkn x'n Dkl DkD Dkl+Dkn x'II Dkl Dkll Dkl+DkU x'n
Fig. 13 Ausbeute der Diturboextraktion
Extraktion in zwei Stufen à 5 Minuten bei 5000 Touren pro Minute (= R/Min) bei
20° C mit anschliessendem Zentrifugieren. %WG. = % Wirkstoffgehalt (WG.) der
Droge in der Extraktlösung (Wirkstoffgehalt der Droge an Gesamtalkaloiden, Gerb¬
stoffen sowie Totalextraktivstoffen jeweils = 100 %).
Dk I = Dekantat nach der ersten Extraktion
Dkll = Dekantat nach der zweiten Extraktion
Dkl + Dkll = Summe der beiden Dekantate
x'U = in der Droge zurückgehaltene Extraktflüssigkeit nach der zweiten Ex¬
traktion.
Die horizontalen Linien bedeuten % Wirkstoffgehalt bei 16-tägiger Mazeration in
500 g Extraktlösung (M), in der davon dekantierbaren Extraktlösung (M"), sowie
nach Perkolation in 4 - 6 Teilperkolaten (TP).
Die Alkaloidausbeuten in beiden Dekantaten zusammen sind gleich wie diejeni¬
ge in 4 - 5 Teilperkolaten. Die Gerbstoff- und Totalextraktivstoffausbeuten entspre¬
chen denjenigen von 3-4 Teilperkolaten.

- 128 -
Die Ausbeuten sind auch höher als in den Dekantaten der 16-tägigen Mazera¬
tion.
Durch Abpressen könnten noch ca. 5 Gew.% mehr Dekantat und damit Inhalts¬
stoffe gewonnen werden.
Die Ausbeuten in den Dekantaten der Diturboextraktion (14I+14II) sowie der
einstufigen Turboextraktion mit 10000 U/Min während einer Stunde (No. Tu7) sind
in der folgenden Tabelle 51 verglichen:
Tab. 51 Dichte, Viskosität und Ausbeuten in den dekantierten Extraktlösungen nach
Diturboextraktion oder Turboextraktion durch Wirbeln während 1 Stunde
bei 10 000 U/Min bei 20° C
Versuch
No.
Dreh- Zeit
zahl
U/Min Min
Dichte Visko¬
sität
g/ml in cP
Alka- Gerb- Totalex-
loide Stoffe tr. Stoffe
% % %
Tul4I+14II
Tu7
5000 5+5
10000 60 0,951 3,05
74,6 142,5 75,8
63,9 115,3 58,8
Me
Me%
10,7 27,2 17,0
16,74 23,6 28,9
Me = absolute Mehrausbeute der Diturboextraktion (Versuch Tul4I+14n) ge¬
genüber der einstufigen Turboextraktion (Versuch Tu7).
Me % = Me, ausgedrückt in Prozent der einstufigen Turboextraktion (VersuchTu7).
Aus diesen Zahlen geht deutlich hervor, dass die mehrstufige Turboextraktion
die Wirkstoffausbeute mehr steigert als eine hohe Tourenzahl bei langer Extraktions¬
zeit. Dies kommt daher, weil nach jeder Stufe ca. 25 - 30 % der Extraktflüssigkeit
in der Droge zurückgehalten und in der folgenden Stufe grösstenteils ausgewaschen
werden.
Zudem wird durch die erneute Menstruumzugabe das für die Extraktion uner-
lässliche Konzentrationsgefälle von der intra- zur extrazellulären Flüssigkeit wie¬
der hergestellt.
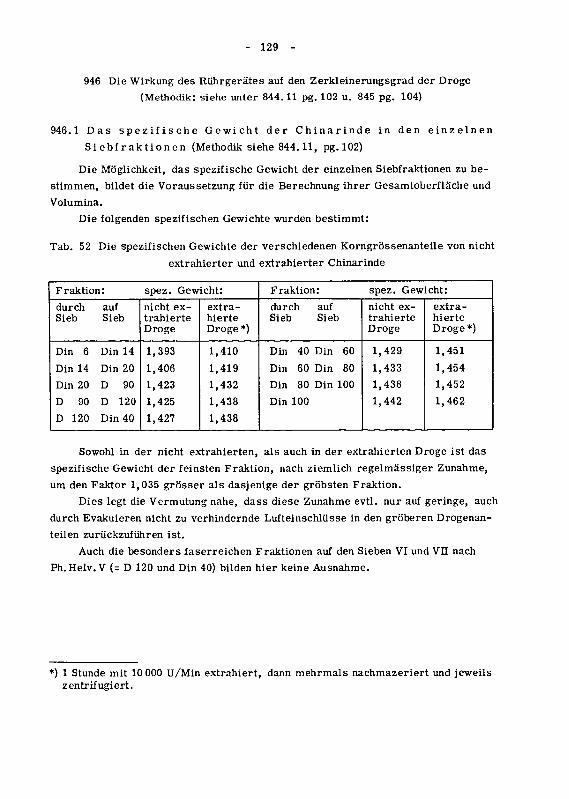
- 129 -
946 Die Wirkung des Rührgerätes auf den Zerkleinerungsgrad der Droge
(Methodik: siehe unter 844.11 pg. 102 u. 845 pg. 104)
946.1 Das spezifische Gewicht der Chinarinde in den einzelnen
Siebfraktionen (Methodik siehe 844.11, pg. 102)
Die Möglichkeit, das spezifische Gewicht der einzelnen Siebfraktionen zu be¬
stimmen, bildet die Voraussetzung für die Berechnung ihrer Gesamtoberfläche und
Volumina.
Die folgenden spezifischen Gewichte wurden bestimmt:
Tab. 52 Die spezifischen Gewichte der verschiedenen Korngrössenanteile von nicht
extrahierter und extrahierter Chinarinde
Fraktion: spez. Gewicht: Fraktion: spez. Gewicht:
durch auf
Sieb Sieb
nicht ex¬
trahierte
Droge
extra¬
hierte
Droge *)
durch auf
Sieb Sieb
nicht ex¬
trahierte
Droge
extra¬
hierte
Droge*)
Din 6 Din 14
Din 14 Din 20
Din 20 D 90
D 90 D 120
D 120 Din 40
1,393
1,406
1,423
1,425
1,427
1,410
1,419
1,432
1,438
1,438
Din 40 Din 60
Din 60 Din 80
Din 80 Din 100
Din 100
1,429
1,433
1,438
1,442
1,451
1,454
1,452
1,462
Sowohl in der nicht extrahierten, als auch in der extrahierten Droge ist das
spezifische Gewicht der feinsten Fraktion, nach ziemlich regelmässiger Zunahme,
um den Faktor 1,035 grösser als dasjenige der gröbsten Fraktion.
Dies legt die Vermutung nahe, dass diese Zunahme evtl. nur auf geringe, auch
durch Evakuieren nicht zu verhindernde Lufteinschlüsse in den gröberen Drogenan¬
teilen zurückzuführen ist.
Auch die besonders faserreichen Fraktionen auf den Sieben VI und VII nach
Ph. Helv. V (= D 120 und Din 40) bilden hier keine Ausnahme.
*) 1 Stunde mit 10 000 U/Min extrahiert, dann mehrmals nachmazeriert und jeweils
zentrifugiert.

- 130 -
946.2 Die Korngrössenverteilung des extrahierten Drogenmate¬
rials als Funktion der Versuchsbedingungen
Um die Siebanalysen auf möglichst einfache Weise graphisch darstellen zu kön¬
nen, wurde die Summenhäufigkeit in % ( H %) als Funktion des Logarithmus der Ma¬
schenweite im Wahrscheinlichkeitsnetz eingezeichnet.
Auf der Ordinatenachse ist für jede Siebnummer das Gewicht aller Teilchen
einer Analyse in Prozent angegeben, die dieses Sieb passieren. Das Ausgangsge¬
wicht der für die Siebanalyse eingesetzten Drogenmenge wird als 100 % angenommen
(a oder a+b ist 100 %; siehe unter 845, pg. 104).
Auf der Abszissenachse sind die Logarithmen der in Hundertstelsmillimetern
ausgedrückten Maschenweiten der verwendeten Siebe aufgetragen.
Die Maschenweiten und die Logarithmen der aus praktischen Gründen in Hun¬
dertstelsmillimetern angegeben Maschenweiten (positive Logarithmen) sind für die
verwendeten Siebe in Tabelle 53 angegeben.
Tab. 53 Maschenweite (Mw), Logarithmus von Mw und Logarithmus von 100 Mw
für die verwendeten Siebe
Sieb: Maschenweite: log Mw: log 100 • Mw:
Din 100 0,060 mm 0,778 -2 0,778
Din 80 0,075 mm 0,875 -2 0,875
Din 60 0,102 mm 0,009 -1 1,009
Din 40 0,150 mm 0,176 -1 1,176
D 120 0,170 mm 0,230 -1 1,230
D 90 0,210 mm 0,322 -1 1,322
Din 20 0,300 mm 0,477 -1 1,477
Din 14 0,430 mm 0,633 -1 1,633
Din 6 1,020 mm 0,009 2,009
Auf diese Weise kann eine Normalverteilung ganz und eine asymmetrische
Verteilung weitgehend in eine Gerade transformiert werden. In unserem Falle liegt
keine normale Verteilung vor.
Je steiler die Gerade verläuft, desto geringer ist die Streuung der zugehörigen
Verteilung.
Der gesamte Verlust (V +Z ) der Siebanalyse a wurde von 100 % subtrahiert,
so dass die
Summe aller Siebfraktionen von a = 100 % - (V +Z )%3. 3.

- 131 -
V oder V, : grobe Anteile auf dem Tisch im Umkreis der Maschineab
Z oder Z, : feine, in die Luft zerstäubte Anteilea b
'
Der Index bezeichnet die Serie der Siebanalyse, a: weitmaschige Siebe,
b: feinmaschige Siebe.
Die Fraktion 5 Sieb VII der Analyse a diente für die Siebanalyse b. Zfa wurde
zur Fraktion ^ Sieb Din 100 addiert. VY wurde subtrahiert, d.h. bildet die Ergän¬
zung auf das Total von Analyse b.
(Eine Addition von Z zur Fraktion kleiner oder gleich wie Sieb VE der Sieb¬
analyse a und gleichzeitige Aufteilung von Z auf die Analyse b entsprechend den An¬
teilen der verschiedenen ï raktionen der Analyse b hebt alle Kurven etwas an, hat je¬
doch sonst keinen Einfluss auf die graphische Darstellung (Fig. 14)).
S
9
!
3
3
r
„ ¥i
- -
-
1
- -
. 4a.p . -X-
_!_ \SH
1
-
I ! /ff. J^ /tx
-- Vf^ r '" -, ,_r--
"-1/ ! |
3 "1 1 ;!'! ;
t
Fig. 14 Turboextraktion: Einfluss der Trennverfahren auf die Korn-
grössenverteilung in der extrahierten Droge
Die Siebanalysen sind im Wahrscheinlichkeitsnetz eingetragen
n. e.Dr.
P
Z
Mw
nicht extrahierte DrogeDroge nach Perkolation
Droge nicht gewirbelt, jedoch beim Abtrennen
vom Menstruum und Auswaschen mehrmals zen-
trifugiert (Durchschnittswert der Versuche
Tu(l,4,3,11)).Maschenweite in mm.

- 132 -
946.21 Der Einfluss der Trennverfahren auf die Korngrössenverteilung
Diese Untersuchungen sollen vor allem darüber Auskunft geben, ob die Zellen
durch die Extraktion "geschwächt" werden, wodurch diese bei der Siebanalyse stär¬
ker zerkleinert würden.
Ueberdies soll beobachtet werden, ob durch das Zentrifugieren allein schon
eventuell ein Zerreissen von Zellen stattfindet.
Die folgende Tabelle 54 zeigt die Anteile der verschiedenen Korngrössen in
Prozent der gesiebten Pulvermenge, sowie die Summenhäufigkeit (= S%). Letztere
gibt für jedes Sieb den prozentualen Anteil der Gesamtpulvermenge an, der dieses
Sieb passiert.
Tab. 54 Anteile der verschiedenen Korngrössen und Summenhäufigkeit in Prozent
der gesiebten Pulvermenge der nicht turboextrahierten Drogenpulver
durch
Sieb:
Drogenmaterial:
nicht extrahierte
Droge:% s%
nach
Perkolation:
nach M«az.(l-3)Mazeration: zentrif\igiert:
Din 100
Din 80
Din 60
Din 40
D 120
V.
1,79 1,79
1,13 2,92
1.32 4,24
1.33 5,57
0,27 5,84
0,03 5,87
0,61 0,61
0,51 1,12
1,87 2,99
2,27 5,26
0,41 5,67
0,16 5,83
2,27 2,27
0,90 3,17
1,99 5,16
2,11 7,27
0,46 7,73
0,09 7,82
3,22 3,22
1,44 4,66
2,95 7,61
2,81 10,42
0,39 10,81
0,08 10,89
tot.b 5,87 5,83 7,82 10,89
Din 40
D 120
D 90
Din 20
Din 14
Din 6
V.+Z.
5,87 5,87
2,18 8,05
4,10 12,15
18,57 30,72
23,09 53,81
43,15 96,96
3,04 100,00
5,83 5,83
2,54 8,37
6,44 14,81
25,07 39,88
23,74 63,62
34,81 98,43
1,57100,00
7,82 7,82
2,43 10,25
5,59 15,84
21,20 37,04
23,07 60,11
36,46 96,57
3,43 100,00
10,89 10,89
2,63 13,52
6,67 20,19
22,75 42,94
22,35 65,29
29,58 94,87
5,13 100,00
tot. a 100,0 100,0 100,0 100,0
Die Werte von Tab. 54 sind in Figur 14/15 graphisch dargestellt.
*) Die beiden Kurven der arithmetischen Mittel der Versuche Tul und Tu4 sowie
der Versuche Tu3 und Tüll überdecken sich fast, so dass hier der Durchschnitts¬
wert der Versuche Tul,Tu4,Tu3 und Tüll berechnet und aufgetragen ist (vergl.auch pg. 62, Begründung siehe pg. 139).

- 133 -
Gewichtsprozente des
extrahiercen Drogen
pulvers
Fig. 15 Einfluss des Trennverfahrens auf die Korngrossenverteilung in der extra¬
hierten Droge
n. e.Dr. = nicht extrahierte DrogeP = Droge nach PerkolationM = Droge nach Mazeration (nicht ausgewaschen und zentrifugiert)Z = Droge nicht gewirbelt, jedoch beim Abtrennen vom Menstruum
und Auswaschen mehrmals zentrifugiert (Durchschnittswertder Versuche Tu(l,4,3,11))
Die Abtrennung der Droge von der Extraktlosung erfolgte:
bei der Mazeration und bei den Versuchen Tul und Tu4 durch Filtration, bei den
Versuchen Tu3 und Tüll durch Zentrifugleren.
Die mazerierte oder perkolierte Droge wurde direkt getrocknet. In allen an¬
dern Versuchen wurde die Droge jedoch beim Auswaschen noch fünfmal abzentrifu-
giert, dann zur Trocknung auf einem Tonteller ausgebreitet.
Die Siebanalysen zeigen eindeutig, dass das Zellmatenal auch nach erschöp¬
fender Extraktion, einer Perkolation mit 10 Teilperkolaten, in getrocknetem Zustand
mechanisch noch so stabil ist, dass keine merkbare, stärkere Zerkleinerung wah¬
rend der Siebanalyse eintritt.
Sogar wenn bei der Siebanalyse der perkoherten Droge die m die Luft zerstaub¬
ten Anteile der ersten Hälfte der Siebanalyse (Z = 1, 31 %) statt zum Verlust zu der
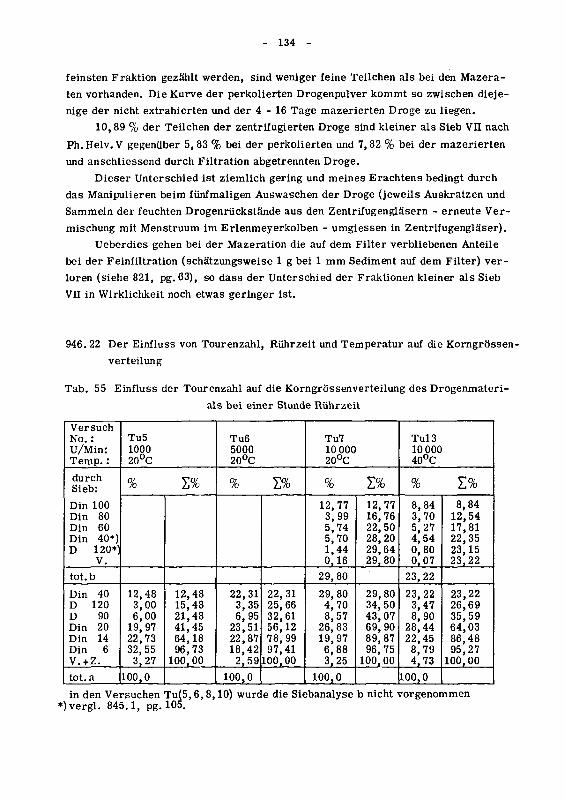
- 134 -
feinsten Fraktion gezählt werden, sind weniger feine Teilchen als bei den Mazera-
ten vorhanden. Die Kurve der perkolierten Drogenpulver kommt so zwischen dieje¬
nige der nicht extrahierten und der 4-16 Tage mazerierten Droge zu liegen.
10,89 % der Teilchen der zentrifugierten Droge sind kleiner als Sieb VII nach
Ph.Helv. V gegenüber 5,83 % bei der perkolierten und 7,82 % bei der mazerierten
und anschliessend durch Filtration abgetrennten Droge.
Dieser Unterschied ist ziemlich gering und meines Erachtens bedingt durch
das Manipulieren beim fünfmaligen Auswaschen der Droge (jeweils Auskratzen und
Sammeln der feuchten Drogenrückstände aus den Zentrifugengläsern - erneute Ver¬
mischung mit Menstruum im Erlenmeyerkolben - umgiessen in Zentrifugengläser).
Ueberdies gehen bei der Mazeration die auf dem Filter verbliebenen Anteile
bei der Feinfiltration (schätzungsweise 1 g bei 1 mm Sediment auf dem Filter) ver¬
loren (siehe 821, pg. 63), so dass der Unterschied der Fraktionen kleiner als Sieb
VII in Wirklichkeit noch etwas geringer ist.
946. 22 Der Einfluss von Tourenzahl, Rührzeit und Temperatur auf die Korngrössen-
verteilung
Tab. 55 Einfluss der Tourenzahl auf die Korngrössenverteilung des Drogenmateri¬
als bei einer Stunde Rührzeit
Versuch
No.:
U/Min:Temp. :
Tu5
1000
20°C
Tu6
5000
20°C
Tu7
10 000
20°C
Tul3
10 000
40°C
durch
Sieb:
Din 100
Din 80
Din 60
Din 40*)D 120*)
V.
% 2% % 1% % 1% % E%
12,773,995,745,701,440,16
12,7716,7622,5028,2029,6429,80
8,843,705,274,540,800,07
8,8412,5417,8122,3523,1523,22
tot.b 29,80 23,22
Din 40
D 120
D 90
Din 20
Din 14
Din 6
V.+Z.
12,483,006,0019,9722,7332,553,27
12,4815,4821,4841,4564,1896,73
100,00
22,313,356,9523,5122,8718,422,59
22,3125,6632,6156,1278,9997,41
100,00
29,804,708,57
26,8319,976,883,25
29,8034,5043,0769,9089,8796,75
100,00
23,223,478,90
28,4422,458,794,73
23,2226,6935,5964,0386,4895,27
100,00
tot. a 100,0 100,0 100,0 J00,0
in den Versuchen Tu(5,6,8,10) wurde die Siebanalyse b nicht vorgenommen
*)vergl. 845.1, pg. 105.
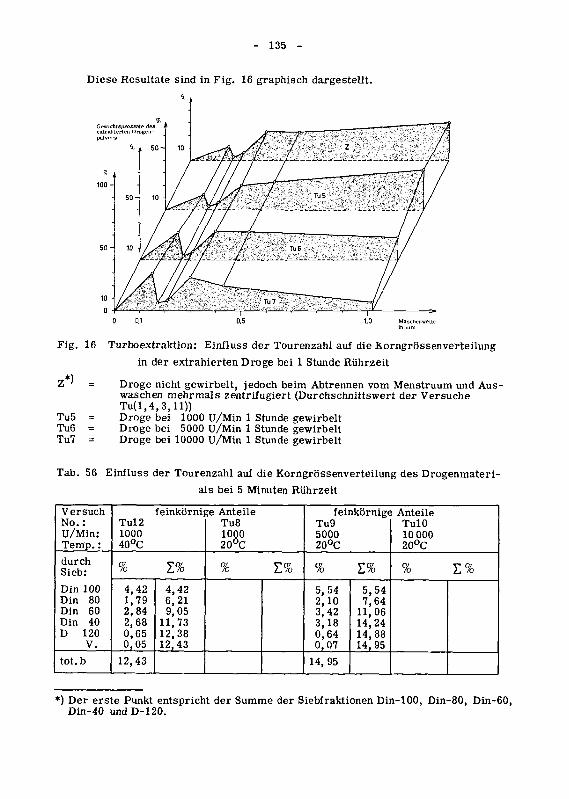
- 135 -
Diese Resultate sind in Fig. 16 graphisch dargestellt.
0 01 0 5 10 Maschenweue
Fig. 16 Turboextraktion; Einfluss der Tourenzahl auf die Rorngrössenverteilung
in der extrahierten Droge bei 1 Stunde Rührzeit
Z '= Droge nicht gewirbelt, jedoch beim Abtrennen vom Menstruum und Aus¬
waschen mehrmals zentrifugiert (Durchschnittswert der Versuche
Tu(l,4,3,ll))Tu5 = Droge bei 1000 U/Min 1 Stunde gewirbeltTu6 = Droge bei 5000 U/Min 1 Stunde gewirbeltTu7 = Droge bei 10000 U/Min 1 Stunde gewirbelt
Tab. 56 Einfluss der Tourenzahl auf die Korngrössenverteilung des Drogenmateri¬
als bei 5 Minuten Rührzeit
Versuch
No.:
U/Min:Temp. :
feinkörni
Tul2
1000
40°C
le Anteile
Tu8
1000
20°C
feinkörnigeTu9
5000
20°C
Anteile
TulO
10 000
20°C
durch
Sieb;
Din 100
Din 80
Din 60
Din 40
D 120
V.
% 1% % z% % T.% % E%
4,421,792,842,680,650,05
4,426,219,05
11,7312,3812,43
5,542,103,423,180,640,07
5,547,6411,0614,2414,8814,95
tot.b 12,43 14,95
*) Der erste Punkt entspricht der Summe der Siebfraktionen Din-100, Din-80, Din-60,Din-40 undD-120.
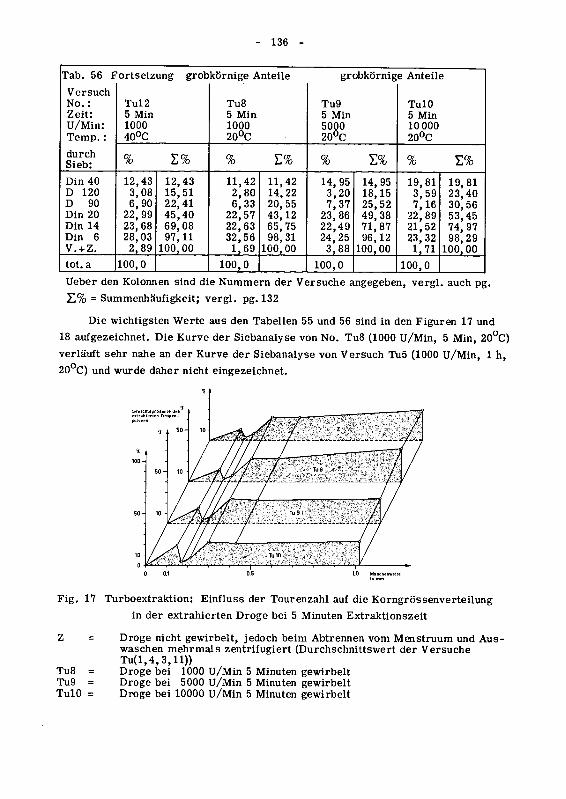
- 136 -
Tab. 56 F
Versuch
No.:
Zeit:
U/Min:Temp. :
durch
Sieb:
ortsetzung grobkörnige Anteile
Tul2 Tu8
5 Min 5 Min
1000 1000
40°C 20°C
grobkörnige Anteile
Tu9 TulO
5 Min 5 Min
5000 10 000
20°C 20°C
% s% % E% % S% % s%
Dm 40
D 120
D 90
Din 20
Din 14
Din 6
V. + Z.
12,433,086,90
22,9923,6828,032,89
12,4315,5122,4145,4069,0897,11
100,00
11,422,806,33
22,5722,6332,561,69
11,4214,2220,5543,1265,7598,31
100,00
14,953,207,37
23,8622,4924,253,88
14,9518,1525,5249,3871,8796,12
100,00
19,813,597,16
22,8921,5223,321,71
19,8123,4030,5653,4574,9798,29
100,00
tot. a 100,0 100,0 100,0 100,0
Ueber den Kolonnen sind die Nummern der Versuche angegeben, vergl. auch pg.
Z% = Summenhäufigkeit; vergl. pg. 132
Die wichtigsten Werte aus den Tabellen 55 und 56 sind in den Figuren 17 und
18 aufgezeichnet. Die Kurve der Siebanalyse von No. Tu8 (1000 U/Min, 5 Min, 20°C)verläuft sehr nahe an der Kurve der Siebanalyse von Versuch Tu5 (1000 U/Min, 1 h,
20 C) und wurde daher nicht eingezeichnet.
Fig. 17 Turboextraktion: Einfluss der Tourenzahl auf die Korngrössenverteilung
in der extrahierten Droge bei 5 Minuten Extraktionszeit
Z = Droge nicht gewirbelt, jedoch beim Abtrennen vom Menstruum und Aus¬
waschen mehrmals zentnfugiert (Durchschnittswert der Versuche
Tu(l,4,3,ll))Tu8 = Droge bei 1000 U/Min 5 Minuten gewirbeltTu9 = Droge bei 5000 U/Min 5 Minuten gewirbeltTulO = Droge bei 10000 U/Min 5 Minuten gewirbelt
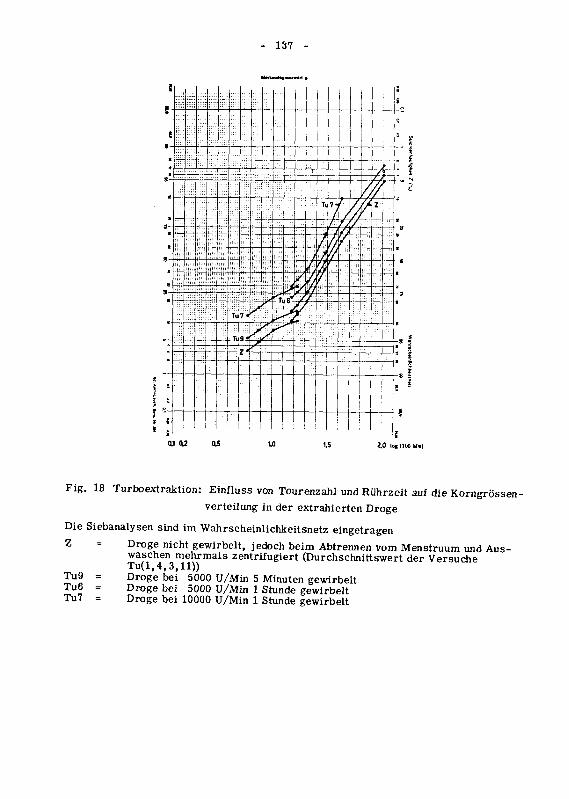
- 137 -
{„ T
S
I'
I2
t — „_ _
a ^f...• 1 "—-
« _____
—=^--
t -ifM1.
3 ___ ^3
; z# :- ^-^5*
-JÂ" -Z8-~
~p>&m zm :
^'V"^^^&Ä- ———— z* — — — S
—— — —
I*
1 e1 1
Ï".1 ,
! |
1s
ai <u as 2.0 log (100 H«)
Fig. 18 Turboextraktion: Einfluss von Tourenzahl und Rührzeit auf die Korngrössen-
verteilung in der extrahierten Droge
Die Siebanalysen sind im Wahrscheinlichkeitsnetz eingetragenZ = Droge nicht gewirbelt, jedoch beim Abtrennen vom Menstruum und Aus¬
waschen mehrmals zentrifugiert (Durchschnittswert der VersucheTu(l,4,3,ll))
Tu9 = Droge bei 5000 U/Min 5 Minuten gewirbeltTu6 = Droge bei 5000 U/Min 1 Stunde gewirbeltTu7 = Droge bei 10000 U/Min 1 Stunde gewirbelt

- 138 -
Tab. 57 Einfluss von Intensität und Dauer der Turboextraktion auf den prozentualen
Anteil der Teilchen kleiner als Sieb Din 40 (Drogenmenge pro Siebanalyse =
100 %)
Versuch No. :
U/Min:Minuten:
Tu(l,4,3,ll)0*)0
Tu8
1000
5
Tu5
1000
60
Tu9
5000
5
Tu6
5000
60
TulO10000
5
Tu7
10 000
60
Prozente: 10,89 11,42 12,48 14,95 22,31 19,81 29,80
') Droge nur mit Menstruum gemischt und anschliessend durch Zentrifugierenwieder von der Extraktlösung getrennt.
Einfluss der Tourenzahl:
Aus Tabelle 57 ist ersichtlich, dass bei Drehzahlen bis zu 1000 U/Min keine
merkliche Zunahme der feinen Drogenanteile stattfindet.
Der prozentuale Anteil der Teilchen kleiner als Sieb Din 40 ist eine lineare
Funktion der Tourenzahl für 5 Min Extraktionszeit, also eine Gerade.
Bei 1 Stunde Extraktionszeit nimmt diese Siebfraktion bei Erhöhung der Tou¬
renzahl von 1000 auf 5000 U/Min von 12,48 auf 22,31 Prozent zu (Faktor 1,79), bei
Steigerung der Tourenzahl von 5000 auf 10 000 U/Min von 22,31 auf 29,8 Prozent
(Faktor 1,34).
Beim Vergleich der nur zentrifugierten mit der turboextrahierten Droge ergibt
sich folgendes Bild:
Eine Extraktion mit 5000 U/Min während 5 Minuten vermehrt den Anteil der
Teilchen kleiner als Sieb Din 40 um ca. 50 Prozent, den Anteil der Teilchen kleiner
als Sieb Din 100 um ca. 72 Prozent.
Bei einer Extraktion mit 10 000 U/Min während einer Stunde wird der Anteil
der Teilchen kleiner als Sieb Din 40 um ca. 174 Prozent, der Anteil der Teilchen
kleiner als Sieb Din 100 um ca. 300 Prozent vermehrt (siehe Tabellen 54/56, pg.
132, 135, 136).
Einfluss der Rührzeit:
Eine Verlängerung der Rührzeit von 5 Min auf 60 Min vermehrt die Teilchen
kleiner als Sieb Din 40 bei:
1000 U/Min um den Faktor 1,09, d.h. von 11,42 auf 12,48 %
5000 U/Min um den Faktor 1,49, d.h. von 14, 95 auf 22,31 %
10000 U/Min um den Faktor 1,50, d.h. von 19,81 auf 29,80 %
Die Faktoren sind aus den Werten in den Tabellen 55 und 56 berechnet.
Die Drogenzerkleinerung steigt also mit der Rührzeit und der Tourenzahl.
Bei 5000 U/Min ist eine deutliche Drogenzerkleinerung feststellbar, und die zuge¬
führte Energie ist bezüglich Drogenzerkleinerung besser ausgenützt als bei 1000U/Min.

- 139 -
Einfluss der Temperatur:
Die Siebanalysen der bei erhöhter Temperatur gewirbelten Drogen sind der
Vollständigkeit halber in den Tabellen aufgeführt. In den graphischen Darstellungen
wurden sie weggelassen, um die Uebersicht zu verbessern.
Die mit dem Menstruum von 40 C nur vermischte und anschliessend zentrifu-
gierte Droge (Versuch Tüll) zeigt bei der Siebanalyse 9,56 % Anteile kleiner oder
gleich Sieb Din 40 gegenüber 11, 56 % der bei 20°C ebenso behandelten Droge (Ver¬
such Tu3).
Nach einer Ruhrzeit von 5 Min mit 1000 U/Min zeigt jedoch die bei 40 C ex¬
trahierte Droge 8, 8 % mehr Anteile kleiner als Sieb Din 40, überhaupt mehr feine
Anteile als der 20°C-Versuch.
Da diese Schwankungen offensichtlich innerhalb der Versuchsfehler liegen,
wurde in Tab. 54, pg. 132, Versuch Tüll ebenfalls, trotz erhöhter Auszugstempe¬
ratur von 40°C, in den Mittelwert der nur zentrifugierten Drogenruckstande einbe¬
zogen.
Die bei 40 C mit 10 000 U/Min wahrend einer Stunde extrahierte Droge (Ver¬
such Tul3) hat 23, 22 % Anteile S Sieb Din 40 gegenüber 29,80 % der bei 20°C eben¬
so behandelten Droge (Tabelle 55, Versuch Tu7, Tul3).
Scheinbar werden unter diesen Versuchsbedingungen zusätzlich harzartige
Stoffe aus der Droge extrahiert. Feine Drogenanteile konnten dadurch wahrend der
Trocknung und nachfolgenden Siebanalyse an andern Partikeln haften bleiben. Bei
der mikroskopischen Prüfung dieser Pulver konnten in keiner Fraktion typische
Agglomerate gefunden werden. Einzig bei Vorversuchen mit Drogenpulvern, welche
nach der Extraktion nicht ausgewaschen wurden, konnten verklebte Einzelteilchen
beobachtet werden, die wahrscheinlich erst beim Trocknen zu grosseren Agglomera¬
ten verwachsen waren. Die Einzelpartikel dieser Agglomerate gehorten ausnahms¬
los in die Fraktionen der feinsten Pulver. Diese Klumpchen zerfallen meist schon
beim Einlegen in Chloralhydrat, sicher aber beim Anpressen des Deckglases mit
einer Prapariernadel.
Em Anhaften von feinen Teilchen an den grossen Zellverbanden, welche die
Siebe m, IV und IVa passieren, ist jedoch auch anzunehmen. Mikroskopisch ist
dies, auch bei Anpressen des Deckglases mit einer Prapariernadel, nur äusserst
schwer feststellbar; diese Teilchen werden daher nicht zu den Agglomeraten gezahlt.

- 140 -
946.3 Die Oberflächenzunahme des extrahierten Drogenmateri¬
als durch Turboextraktion
Da die Oberfläche der Teilchen, besonders bei groben Drogenpartikeln, nicht
genau erfasst werden kann, muss für die Berechnung ein Idealfall angenommen wer¬
den.
Die Berechnung der Oberfläche erfolgte daher unter der Annahme, dass jede
Siebfraktion aus Kugeln besteht, deren Durchmesser gerade der Maschenweite des
feinsten, von ihr passierten Siebes entspreche.
Die Oberfläche der groben Teilchen dürfte in Wirklichkeit infolge von aufgeris¬
senen Zellen grösser sein, während diejenigen der feinsten Teilchen sich der Kugel¬
form eher annähern.
Auch die Grössenverteilung der Partikel innerhalb einer Siebfraktion wurde
für unsere Betrachtung nicht berücksichtigt. Da wir mit dem grösstmöglichen Durch¬
messer rechnen, sind die Ergebnisse der Oberflächenberechnung sicher alle zu
klein, aber unter sich durchaus vergleichbar.
Die Berechnung geschieht wie folgt für jede Siebfraktion:
V
G
d
F
V,.
Volumen der Siebfraktion
Gewicht der Siebfraktion
spezifisches Gewicht der Siebfraktion
Oberfläche der gesamten Siebfraktion
Volumen eines Teilchens (Kügelchens) dieser Siebfraktion
Oberfläche eines Teilchens (Kügelchens) dieser Siebfraktion
Radius eines Teilchens (Kügelchens) dieser Siebfraktion
1) VG
d2) V„ =
2iL
41t 3— r
33) F,, = 4wr'
4) FG -3 -4t vù
d -411^
G
d
Die Gewichte der verschiedenen Siebfraktionen sind in den Tabellen 54 - 56
(pg. 132,135,136) und ihre spezifischen Gewichte in Tabelle 52 (pg.129) zu finden.
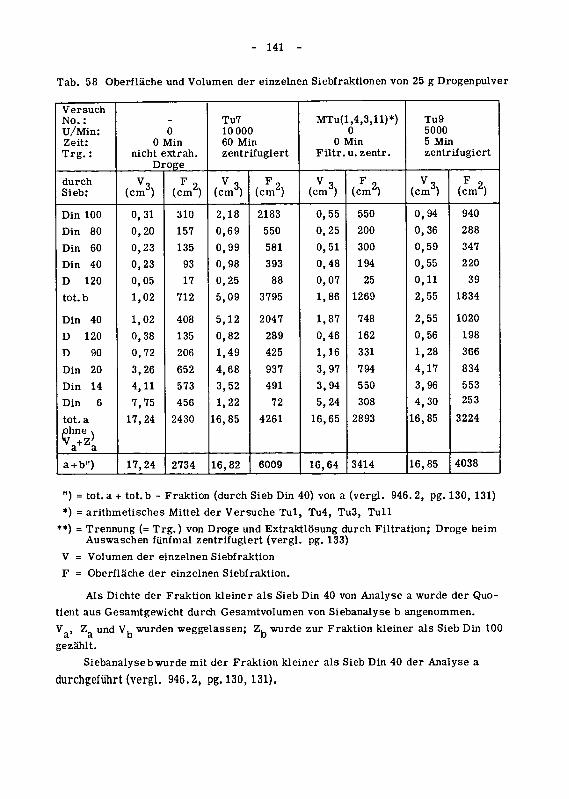
- 141 -
Tab. 58 Oberfläche und Volumen der einzelnen Siebfraktionen von 25 g Drogenpulver
Versuch
No.:
U/Min:Zeit:
Trg.:
0
0 Min
nicht extrah.
Droge
Tu7
10 000
60 Min
zentrifugiert
MTu(l,4,3,ll)*)0
0 Min
Filtr.u. zentr.
Tu9
5000
5 Min
zentrifugiert
durch
Sieb:V3
(cmJ)F2
(cm2)V3
(cmJ)F2
(cm2) (cm ) (cm2)V3
(cm3)
F2
(cm2)
Din 100
Din 80
Din 60
Din 40
D 120
tot.b
Din 40
D 120
D 90
Din 20
Din 14
Din 6
tot. a
ohne i
a a
0,31
0,20
0,23
0,23
0,05
1,02
1,02
0,38
0,72
3,26
4,11
7,75
17,24
310
157
135
93
17
712
408
135
206
652
573
456
2430
2,18
0,69
0,99
0,98
0,25
5,09
5,12
0,82
1,49
4,68
3,52
1,22
16,85
2183
550
581
393
88
3795
2047
289
425
937
491
72
4261
0,55
0,25
0,51
0,48
0,07
1,86
1,87
0,46
1,16
3,97
3,94
5,24
16,65
550
200
300
194
25
1269
748
162
331
794
550
308
2893
0,94
0,36
0,59
0,55
0,11
2,55
2,55
0,56
1,28
4,17
3,96
4,30
16,85
940
288
347
220
39
1834
1020
198
366
834
553
253
3224
a+b") 17,24 2734 16,82 6009 16,64 3414 16,85 4038
") = tot. a + tot.b - Fraktion (durch Sieb Din 40) von a (vergl. 946. 2, pg. 130, 131)
*) = arithmetisches Mittel der Versuche Tul, Tu4, Tu3, Tüll
**) = Trennung (= Trg.) von Droge und Extraktlösung durch Filtration; Droge beim
Auswaschen fünfmal zentrifugiert (vergl. pg. 133)
V = Volumen der einzelnen Siebfraktion
F = Oberfläche der einzelnen Siebfraktion.
Als Dichte der Fraktion kleiner als Sieb Din 40 von Analyse a wurde der Quo¬
tient aus Gesamtgewicht durch Gesamtvolumen von Siebanalyse b angenommen.
V,Z und V. wurden weggelassen; Z. wurde zur Fraktion kleiner als Sieb Din 100
gezählt.
Siebanalyseb wurde mit der Fraktion kleiner als Sieb Din 40 der Analyse a
durchgeführt (vergl. 946.2, pg. 130, 131).

- 142 -
Tab. 59 Die Oberfläche und das Volumen der einzelnen Siebfraktionen von Tab. 58
2in Prozent der Gesamtoberfläche (2734 cm = 100 %) und des Gesamtvolu-
o
mens (17,24 cm = 100 %) aller Fraktionen der Siebanalyse der nicht ex¬
trahierten Droge
Versuch
No.:
U/Min:Zeit:
Trg.:
0
0 Min
n. extr. Droge
Tu7
10 000
60 Min
zentrifugiert
MTu(l,4,3,ll)*)0
0 Min
Filtr.u. zentr.
Tu9
5000
5 Min
zentrifugiert
durch
Sieb:
Din 100
Din 80
Din 60
Din 40
D 120
V
%
F
%V
%F
%V
%F
%V
%F
%
1,8
1,2
1,3
1,3
0,3
11,3
5,8
4,9
3,4
0,6
12,6
4,0
5,7
5,7
1,5
79,8
20,1
21,2
14,4
3,2
3,2
1,4
3,0
2,8
0,4
20,1
7,3
11,0
7,1
0,9
5,5
2,1
3,4
3,2
0,6
34,4
10,5
12,7
8,1
1,4
tot.b
Din 40
D 120
D 90
Din 20
Din 14
Din 6
5,9
5,9
2,2
4,2
18,9
23,8
45,0
26,0
14,9
4,9
7,5
23,9
21,0
16,7
29,5
29,7
4,7
8,6
27,2
20,4
7,1
138,7
74,9
10,6
15,5
34,3
18,0
2,6
10,8
10,8
2,7
6,7
23,0
22,9
30,4
46,4
27,4
5,9
12,1
29,0
20,1
11,3
14,8
14,8
3,2
7,4
24,2
23,0
25,0
67,1
37,3
7,2
13,4
30,5
20,2
9,3
tot. a 100,0 88,9 97,7 155,9 96,5 105,8 97,6 117,9
a + b ") 100,0 100,0 97,7 219,7 96,5 124,8 97,6 147,7
") = tot. a + tot. b minus Fraktion kleiner als Sieb Din 40 von Analyse a
F = Oberfläche der einzelnen Siebfraktionen
V = Volumen der einzelnen Siebfraktionen
No. = Versuchsnummer, vergl. auch pg. 62 (gleiche Nummern wie in Tab. 10/58)
Tabelle 59 zeigt uns, dass die Oberfläche des Drogenmaterials während der
Turboextraktion wie folgt vergrössert wird:
bei 5000 U/Min während 5 Minuten 1, 5 mal
bei 10000 U/Min während 60 Minuten 2,2 mal
beim blossen Abtrennen der Droge von der Extraktlösung durch Zentrifu-
gieren: 1, 25 mal
Die Werte von Tabelle 59 sind in den Figuren 19 und 20 graphisch dargestellt.
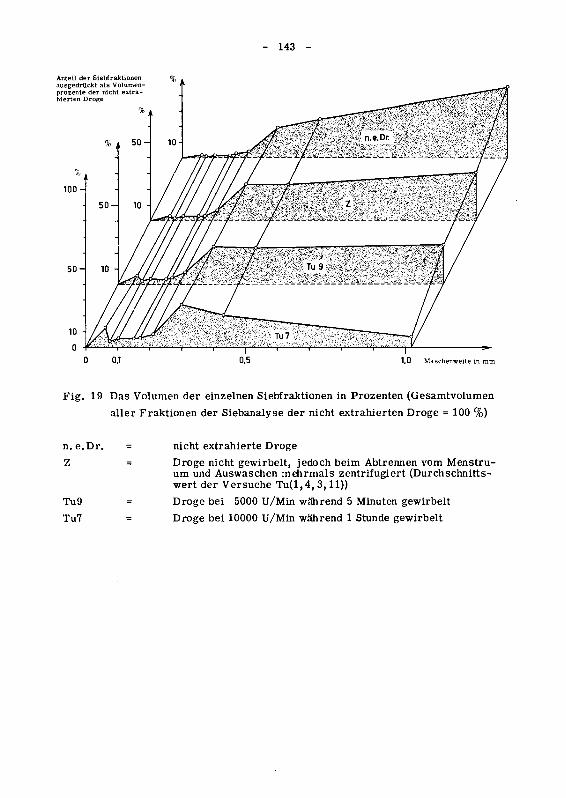
- 143 -
0 0,1 0,5 1,0 Maschenweite in mm
Fig. 19 Das Volumen der einzelnen Siebfraktionen in Prozenten (Gesamtvolumen
aller Fraktionen der Siebanalyse der nicht extrahierten Droge = 100 %)
n. e.Dr. = nicht extrahierte Droge
Z = Droge nicht gewirbelt, jedoch beim Abtrennen vom Menstru¬
um und Auswaschen mehrmals zentrifugiert (Durchschnitts¬wert der Versuche Tu(l,4, 3,11))
Tu9 = Droge bei 5000 U/Min während 5 Minuten gewirbelt
Tu7 = Droge bei 10000 U/Min während 1 Stunde gewirbelt

- 144 -
Anteil der Siebfraktionen
ausgedrückt in Prozenten
der Oberfläche der nicht
extrahierten Droge
0 0,1 0,5 1,0 Maschenweite in mm
Fig. 20 Die Oberfläche der einzelnen Siebfraktionen in Prozenten (Gesamtoberfläche
aller Fraktionen der Siebanalyse der nicht extrahierten Droge = 100 %)
n. e.Dr. = nicht extrahierte Droge
Z = Droge nicht gewirbelt, jedoch beim Abtrennen vom Menstru¬
um und Auswaschen mehrmals zentrifugiert (Durchschnitts¬wert der Versuche Tu(l,4,3,ll))
Tu9 = Droge bei 5000 U/Min während 5 Minuten gewirbelt
Tu7 = Droge bei 10000 U/Min während 1 Stunde gewirbelt

- 145 -
10 DISKUSSION UND SCHLUSSFOLGERUNGEN
101 Bestimmungsmethoden
1011 Alkaloide
Die Alkaloidbestimmungsmethode der Ph.Helv.V bewährte sich gut. Der Zeit¬
aufwand ist gering. Die maximalen Abweichungen in Prozent vom arithmetischen
Mittel sind:
in der Droge
in der Extraktlösung
in der Extraktlösung meist
+ 0,29% - 0,23%
+ 2,97% - 3,63%
< + 1,00 %<- 1,00%
Pg.73
74
Extraktionsversuche
Vorversuche
1012 Gerbstoffe
Die kolorimetrische Hautpulvermethode ist bezüglich Zeitaufwand der ent¬
sprechenden gravimetri sehenBestimmung überlegen und daher für serienmässige
Bestimmungen zu empfehlen.
Die maximalen Abweichungen der Resultate vom arithmetischen Mittel sind in
Prozent:
Vorversuche: % % pagina:
Zeitaufwand
in Minuten:
in der Droge gravimetrisch + 7,45 - 6,30 82 1050
in der Droge kolorimetrisch + 4,80 - 4,00 93 150
in der Extraktlösung kolori¬
metrisch
+ 0,59*) - 0,49 100 150
Extraktionsversuche:
In der Extraktlösung kolori¬
metrisch. .
meist
+ 1,90 - 1,90 **)
< + 1,00 < - 1,00
*) Abweichungen von M (vergl. Tab. 34, pg. 100, Werte vom 4.3.1959)
**) Nur in 4 Parallelbestimmungen vorgekommen.

- 146 -
102 Extraktionsmethoden
1021 Mazeration
Für die maximal mögliche Extraktion von Chinarinde von der Korngrösse Sieb IV
nach Ph.Helv.V genügt eine Mazerationszeit von 4 Tagen, wenn das Extraktionsge¬
misch dreimal täglich je l/2 Minute ungeschüttelt wird.
Die Wirkstoffausbeute entspricht derjenigen von 2 Teilperkolaten der ausge¬
führten Perkolationen und blieb auch niedriger als in einigen Turboextrakten.
1022 Perkolation
Die Perkolation liefert die konzentriertesten Extraktlösungen und in 6 Teil¬
perkolaten auch die besten Ausbeuten aller untersuchten Verfahren. Die Methode
erfordert jedoch mehrere Tage.
1023 Turboextraktion
Die fünf in der Zielsetzung dieser Untersuchungen aufgeführten Fragen (vergl.
pg. 47) lassen sich nun wie folgt beantworten:
1023.1 Der Zeitaufwand
Es ist innert ca. 1,5-2 Stunden möglich, mittels Turboextraktion eine klare,
zentrifugierte Extraktlösung herzustellen, deren Wirkstoffausbeute (Dekantat) bei
gleichem Ansatzverhältnis höher ist als diejenige nach einer viertägigen Mazeration,
siehe Tabelle 60, pg. 148.
Nach intensiver Turboextraktion mit hoher Tourenzahl müssen die Extrakte
zentrifugiert werden, oder man muss 1 -2 Stunden sedimentieren lassen. Eine Fil¬
tration ist infolge der vielen feinen Anteile sehr erschwert.

- 147 -
1023.2 Die Ausbeuten an Alkaloiden, Gerbstoffen und Totalex¬
traktivstoffen
1 Teil Chinarinde Sieb IV nach Ph.Helv. V mit 10 Teilen Menstruum nach Ph.
Helv.V fur Chinarinden-Trockenextrakt wahrend 1 Stunde bei 5000 U/Min turboex¬
trahiert, ergeben in der Extraktlosung einen Alkaloidgehalt, der demjenigen eines
perkoherten Extraktes mit 6 Teilperkolaten entspricht. Der Gerbstoffgehalt hegt
etwas tiefer, der Totalextraktivstoffgehalt wesentlich tiefer als in 6 Teilperkolaten.
Die Wirkstoffausbeute steigt mit zunehmender Wirbeizeit. Die statistische Si¬
cherung dieser Zunahme des Wirkstoffgehaltes in je 500 g der Extraktlosungen
(WG%D) betragt bei 10 000 U/Min und bei einer Verlängerung der Extraktionszeit
von 5 Minuten auf 60 Minuten ca. 99,0 %.
Ferner sind mit 95 - 99 % statistisch gesichert:
Die Zunahme der Alkaloidgehalte (A%D) in der Extraktlosung bei einer Erhöhung
der Tourenzahl von 5000 auf 10 000 U/Min, sowohl bei 5 als auch bei 60 Min Extrak¬
tion szeit.
Die Zunahme der Gerbstoff gehalte (G%D) in 500 g Extraktlosung bei einer Verlänge¬
rung der Extraktionszeit von 5 auf 60 Minuten bei 1000, 5000 und 10 000 U/Min. Im
übrigen sei auf die Tabelle 60 verwiesen.
Eine Turboextraktion mit 10 000 U/Min wahrend einer Stunde bei 20 C lieferte
in 500 g Extraktlosung die höchsten Wirkstoffgehalte aller untersuchten Verfahren.
Davon können jedoch nur 367 g dekantiert werden (der Rest wird in der extrahierten
Droge zurückgehalten (= 118 g), wenn der Drogenruckstand nach dem Zentrifugieren
nicht abgepresst wird, vergl. Tab. 44, pg. 117, No.Tu7). Die Ausbeute entspricht
somit ca. derjenigen einer Perkolation (Pulver Sieb IV) mit 3 Teilperkolaten.
Eine Temperaturerhöhung auf 40 C hat nur geringen oder negativen Einfluss
auf die Alkaloidextraktion. Die Gerbstoff- und die Totalextraktivstoffausbeute neh¬
men jedoch zu.
1023.3 Die Extraktqualitat in Bezug auf Konzentration und Bal¬
laststoffe
Bei allen bei 20 C ausgeführten Turboextraktionen konnte beobachtet werden,
dass dabei, verglichen mit den Mazeraten und den Perkolaten, die Alkaloidausbeute
im besondern und die Gerbstoffausbeuten bedeutend mehr zunehmen als die Total¬
extraktivstoffe; siehe auch 92,pg. 110; 93,pg.ll2; 942,pg.ll6.
Die so gewonnenen Extrakte sind also armer an Ballaststoffen als Perkolate
oder Mazerate, haben jedoch einen hohen Wirkstoffgehalt.
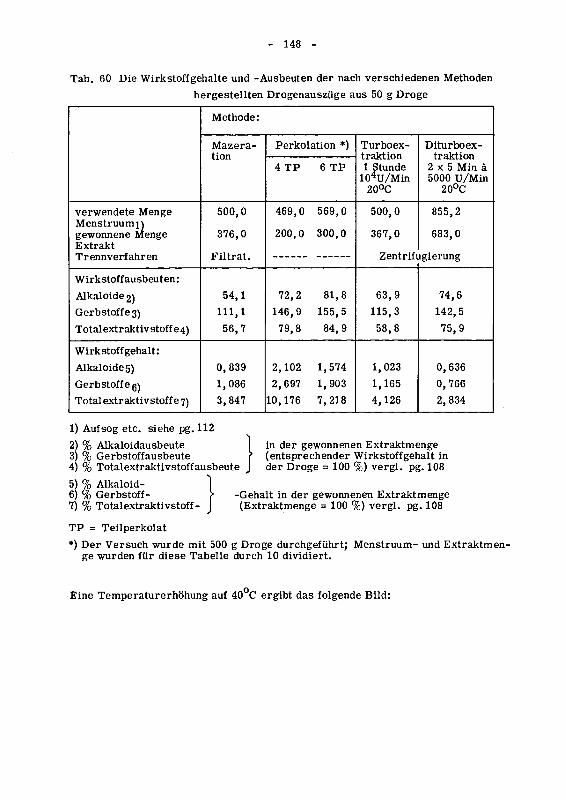
- 148 -
Tab. 60 Die Wirkstoffgehalte und -Ausbeuten der nach verschiedenen Methoden
hergestellten Drogenauszüge aus 50 g Droge
Methode:
Mazera¬
tion
Perkolation *) Turboex¬
traktion
1 Stunde
104U/Min20°C
Diturboex-
traktion
2x5 Min à
5000 U/Min20°C
4 TP 6 TP
verwendete Menge
Menstruumi)gewonnene MengeExtrakt
Trennverfahren
500,0
376,0
Filtrat.
469,0 569,0
200,0 300,0
500,0 855,2
367,0 683,0
Zentrifugierung
Wirkstoffausbeuten :
Alkaloide 2)
Gerbstoffe 3)
Totalextraktivstoff e4)
54,1
111,1
56,7
72,2 81,8
146,9 155,5
79,8 84,9
63,9
115,3
58,8
74,6
142,5
75,9
Wirkstoffgehalt:
Alkaloide 5)
GerbStoffe g)
Totalextraktivstoffe 7)
0,839
1,086
3,847
2,102 1,574
2,697 1,903
10,176 7,218
1,023
1,165
4,126
0,636
0,766
2,834
1) Aufsog etc. siehe pg. 112
2) % Alkaloidausbeute I in der gewonnenen Extraktmenge3) % Gerbstoffausbeute f (entsprechender WirkStoffgehalt in
4) % Totalextraktivstoffausbeute J der Droge = 100 %) vergl. pg. 108
5) % Alkaloid- )6) % Gerbstoff- ? -Gehalt in der gewonnenen Extraktmenge7) % Totalextraktivstoff- J (Extraktmenge = 100 %) vergl. pg. 108
TP = Teilperkolat
*) Der Versuch wurde mit 500 g Droge durchgeführt; Menstruum- und Extraktmen¬
ge wurden für diese Tabelle durch 10 dividiert.
Eine Temperaturerhöhung auf 40 C ergibt das folgende Bild:
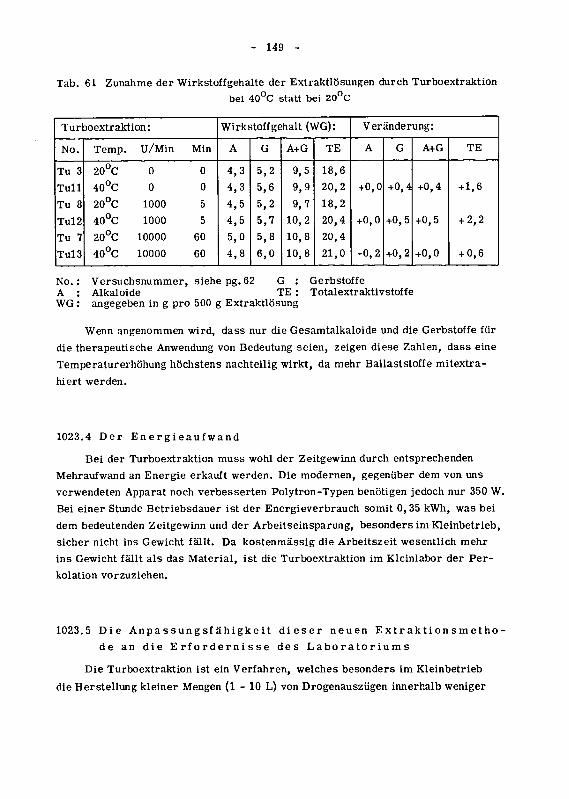
- 149 -
Tab. 61 Zunahme der Wirkstoffgehalte der Extraktlösungen durch Turboextraktion
bei 40°C statt bei 20°C
Turboextraktion: Wirkstoffgehalt (WG): Veränderung:
No. Temp. U/Min Min A G A+G TE A G A+G TE
Tu 3
Tüll
Tu 8
Tul2
Tu 7
Tul3
20°C 0 0
40°C 0 0
20°C 1000 5
40°C 1000 5
20°C 10000 60
40°C 10000 60
4,3
4,3
4,5
4,5
5,0
4,8
5,2
5,6
5,2
5,7
5,8
6,0
9,5
9,9
9,7
10,2
10,8
10,8
18,6
20,2
18,2
20,4
20,4
21,0
+0,0
+0,0
-0,2
+0,4
+0,5
+0,2
+0,4
+0,5
+0,0
+ 1,6
+ 2,2
+ 0,6
No. : Versuchsnummer, siehe pg. 62 G : Gerbstoffe
A : Alkaloide TE : Totalextraktivstoffe
WG : angegeben in g pro 500 g Extraktlösung
Wenn angenommen wird, dass nur die Gesamtalkaloide und die Gerbstoffe für
die therapeutische Anwendung von Bedeutung seien, zeigen diese Zahlen, dass eine
Temperaturerhöhung höchstens nachteilig wirkt, da mehr Ballaststoffe mitextra¬
hiert werden.
1023.4 Der Energieaufwand
Bei der Turboextraktion muss wohl der Zeitgewinn durch entsprechenden
Mehraufwand an Energie erkauft werden. Die modernen, gegenüber dem von uns
verwendeten Apparat noch verbesserten Polytron-Typen benötigen jedoch nur 350 W.
Bei einer Stunde Betriebsdauer ist der Energieverbrauch somit 0,35 kWh, was bei
dem bedeutenden Zeitgewinn und der Arbeitseinsparung, besonders im Kleinbetrieb,
sicher nicht ins Gewicht fällt. Da kostenmässig die Arbeitszeit wesentlich mehr
ins Gewicht fällt als das Material, ist die Turboextraktion im Kleinlabor der Per-
kolation vorzuziehen.
1023.5 Die Anpassungsfähigkeit dieser neuen Extraktionsmetho¬
de an die Erfordernisse des Laboratoriums
Die Turboextraktion ist ein Verfahren, welches besonders im Kleinbetrieb
die Herstellung kleiner Mengen (1 - 10 L) von Drogenauszügen innerhalb weniger

- 150 -
Stunden ermöglicht. Eine Trennung der Extraktlösung von der extrahierten Droge
ist ohne Schwierigkeiten nur durch Sedimentation oder Zentrifugieren möglich.
Letzteres bewirkt zudem eine stärkere Nachextraktion der Droge, sofern noch ein
Konzentrationsgefälle von der intra- zur extrazellulären Extraktflüssigkeit besteht.
1023.6 Weitere Untersuchungsresultate
Es wird gezeigt, dass während der Turboextraktion die innere Oberfläche von
25 g Droge bei einer Rührzeit von einer Stunde mit 10 000 U/Min z.B. ca. von
2 22750 cm auf 6000 cm vergrössert wird (vergl. Tab. 58, pg. 141),
1024 Diturboextraktion
Mittels mehrstufiger Turboextraktion ist es möglich, trotz kürzerer Gesamt¬
wirbelzeit bedeutend höhere Wirkstoffausbeuten zu erzielen als durch einstufige
Wirbelextraktion.
Ca. 30 Prozent der Extraktlösung bleiben nach jeder Stufe in der zentrifugier-
ten Droge zurück.
Ziemlich sicher wäre es auch möglich, durch Erhöhung des Drogenanteils im
Ansatzverhältnis die Diturboextraktion noch wirtschaftlicher zu gestalten. In dieser
Arbeit wurde jedoch absichtlich das Ansatzverhältnis immer konstant gehalten.
103 Verfahrensvorschlag
Auf Grund aller bisherigen Beobachtungen kann eine Droge bei Anwendung der
Turboextraction am wirtschaftlichsten wie folgt ausgezogen werden:
durch Wirbelung in 2 - 3 Stufen während je 5 - 10 Minuten bei 5000 U/Min, mit jeV2
bis l/3 der totalen Menstruum-Menge. Zwischen den einzelnen Stufen wird die Ex¬
traktflüssigkeit abzentrifugiert.

- 151 -
Literaturverzeichnis
1) K. Feinstein, "Theoretische und praktische Untersuchungen über das Perkola-
tionsverfahren nebst einem Ueberblick über dessen Entwicklung,Diss. ETH 1936, pg. 104-106.
A. Frey-Wyssling: "Die pflanzliche Zellwand", Springer-Verlag, Berlin 1959.
2) W. Lang, Aren.Pharm. 283, Vol. j)5 (1950), pg. 115-121.
3) William J.Husa und Paul Fehder, J.amer.pharm.Ass.sei.Ed. 24,616-619 (1935).
—
4) William J.Husa und George R.Jones, J. amer, pharm. Ass. sei. Ed. 26,20-23 (1937).
—
5) William J.Husa und C . L. Huy ck,
J. amer, pharm. Ass. sei. Ed. 24,446-450 (1935).
~"
6) William J.Husa und Louis Magid, J. amer, pharm. Ass. sei.Ed. 23,891 (1934).
—
7) Ulimanns Encyclopädie der technischen Chemie, 1. Band, chemischer Apparate¬bau und Verfahrenstechnik, Urban und Schwarzenberg, Mün¬
chen-Berlin 1951 (= Ullmann).
8) Ph. Helv. V, allgemeiner Teil, pg. 5.
9)U.Bogs, Dtsch. Apoth. Ztg. j)8, 917-921(1958).
10) A. Jermstad und O. Oe st by ,Pharm. Acta Helv. £, 129-140(1934).
11) S.V.Bari, Pharm. Ztg. 80, 854, 880, 1265(1935).
12) O. E.Schultz und J.Klotz, Arzneim. Forsch. 4, 325(1954), ref. J.amer.
pharm. Ass. sei. Ed. 45, 239 (19567.
13) M.Herzog, Dtsch. Apoth. Ztg. 98, 200(1958), ref.F.Gstirner/Arch. Pharm.292, 64, 18-25,~~3"7-44 (1959).
14) C. Risch, Süddtsch. Apoth. Ztg. J59, 84 (1949).
15) Ph. Helv. V, spezieller Teil, pg. 295, Decocta.
16) Ph. Helv. V, spezieller Teil, pg. 507, Infusa.
17) Ph. Helv. V, allgemeiner Teil, pg. 6.
18) H .B r edd in
,"Das Diakolationsverfahren", Kirchhain N. L. 1935, dito: Pharm.
Ztg. ^76, 400, 802 (1931), ref.Diss. Feinstein, pg. 60, 61, 164, 165.
19) E.Kessler
,Pharm. Ztg. 80, 1080(1935), ref.Pharm. Zhalle 77, 120(1936),
Süddtsch. Apoth. Ztg. 75, 437(1935), Pharm. Zïg". jtt, 1308(1936).
20) E. Kessler, Pharm. Ztg. 2£, 635, 823 (1934), ref. Diss. Feinstein, pg. 61.
21)R.V.Baud, Schweiz. Apoth. Ztg. 87, 873-877(1949), 89, 365-372(1951), 90,413-419 (1952).
— — —
22) M. Melichar, V.Rusek und J.Solich, Cescosl. Farmac. 2, 338(1953).ref. F. Gstirner: "Prüfung und Verarbeitung von"Arzneidrogen"Band II, Springer Verlag, Berlin 1955, pg. 43, Pharm. Zhalle 94,63 (1955), Chem.Zbl. 127, 1091 (1956).
—
23) K.Steiger, Diss. Bern 1936.
24) M. Melichar,
Pharmazie 13, 325-329(1958).

- 152 -
25) Ulimann, pg. 682 (vergl. ').
26) H. Köhler, Dtsch. Apoth. Ztg. 98, 53-55 (1958), ref. Pharm. Zhalle £7, 339(1958).
27) Ullmann, pg. 684 Hildebrandt-Extraktor: DRP 547*040 (vergl. 7))Bollmann -Extraktor: DRP 303'846
28) F. O.W. Meyer, Pharmazie 2, 510 (1947), ref. Pharm. Zhalle j87, 173 (1948).
29) Pharm. Zhalle W, 56 (1937).
30) W. Thomson, Pharm. Zhalle 95, 318-325 (1956).
31)N.Thörn, Farm.Rev. 9.11.1940, pg. 681-686.
32) W. Märki, "Vergleichende Untersuchungen an Extr, Belladonnae unter Berück¬
sichtigung des Gehaltes an Hyoszyamin, Atropin, Scopolamin",Diss. ETH 1945, pg. 60, 91.
33) Ch. Béguin, Pharm. Acta Helv. U, 362-369, 370-374, 375-377(1938).
34) Schrader, Pharm. Ztg. J78, 1159 (1933), ref. F. Gstirner: "Prüfung und Ver¬
arbeitung von Arzneidrogen", II. Band, pg. 32.
35)E.Belcot und H.Rapeanu, Curierul farmac. 10, No. 1, 1-10, No. 2, 3-14
(1940), ref.Chem.Zbl. Ill, I, 2815(1940), ref. Pharm. Zhalle
81, 548 (1940).
36) D .Fuchs
, "Untersuchungen zur Herstellung von China-Trockenextrakt",Diss. ETH 1941.
37) W.Schill, Pharmazie Z, 218(1948), ref. Pharm. Zhalle 88, 81(1949).
38) M. Herzog, Pharmazie ]_, 331, 332 (1952), ref. Pharm. Zhalle 92, 21 (1953).
39) M. Herzog, Pharmazie _7, 464-472 (1952), ref. Pharm. Zhalle j)2, 21 (1953).
40) William J. Husa und Louis Magid, J.amer.pharm.Ass. sei. Ed. 24,839, 840 (1935).
—
41) F. O.W. Meyer, Pharm.Wschr. 167(1942), ref. Pharm. Zhalle 86, 46-52
(1947).—
42) Wanda J.Butler und Gail A.Wiese, J. amer, pharm. Ass. sei. Ed. 42,382 (1953), ref. F. Gstirner, Band II, pg. 16 (vergl. 22)).
43) E.Br ochm ann-Hanssen, J.amer.pharm. Ass.sei.Ed. ^3_, 27(1954).
44) W. Märki, Pharm.Acta Helv. t3. 210(1938).
45) F.Ducommun, Pharm.Acta Helv. ^3_, 185(1938).
46) N. Applezweig und S .E
. Ronzone , Ind.engng.Chem.ind.Ed. 38, 576(1946),ref.Chem.Zbl. 118, I, 352 (1947), Pharm. Zhalle§&, 238 (1947).
47) Pinxteren, Pharm.Weekbl. 66, 929 (1929), ref.Arch.Pharm. ^85, 50 (1952).
48)J.M.Campo und L . G. Gr a mling, J. amer, pharm. Ass. sei. Ed. ^5, 242(1956).
49) P. Bohrisch und F.Kür s chner
,Pharm. Zhalle 54, 1-13 (1913).
50) William J.Husa und G.R.Jones, J. amer, pharm. Ass. sei. Ed. 2]_, 852-859,862-865 (1938).
51) J. Graetzer, "Untersuchungen über die Extraktion durch Perkolation", Diss.
ETH 1941, pg. 71.
52) J. A. Marschak, Pharm. Zhalle 74, 145-148(1933).

- 153 -
53)D.Tomlc und R.Kosak, Farmaceutski Glasnik (Kroat. Pharm. Anzeiger),jj, 115(1949), ref. Pharm. Zhalle £9, 282 (1950).
54) O. E. Schultz und J.Klotz, Arzneim. Forsch. 3, 471, 525(1953).
55) F.O.W.Meyer, Pharmazie 2, 116-123(1947), ref. Pharm. Zhalle 86, 336
(1947).~
56) W.Specht, Z.Lebensmittelunters, u. Forsch. 94, 157(1952), ref.Pharm.
Zhalle 92, 106 (1953).
W.Specht, Brauwelt B 6200(1951), ref. Pharm. Zhalle j)£, 146(1953).
57) G. Bay und O. Gis void, J. amer, pharm. Ass. sei. Ed. J37, 314-316(1948).
58)R.E.King und O.Gisvold, J.amer.pharm.Ass.sei.Ed. 39, 109-112 (1950).
59)S.J.Dean, D.C.Brodie, E. Br ochmann-Hanssen und S
. Riegel¬mann, J.amer.pharm.Ass.sei.Ed. 42_, 88-90(1953).
60) M. Melichar,
Pharmazie ^3, 330-333(1958).
61) Ph. Belg. IV, pg. 284.
62) Ph. Russica (State Pharmacopoeia of the Union of Soviet Socialist Republics)VIII (1958), pg. 98.
63) Ph. Dan. IX, Band I, pg. 56 : Dimazeration
Band in, pg. 426 : Mazeration von Tinkturen.
64) M . Melichar ,Acta pharm.Brunensis et Bratislavensis I (1958) dito: Arch.
Pharm. 292, 64, 177-193 (1959).51)
65) Diss. Graetzer, pg. 103 (vergl. ')•
66) William J.Husa und Louis Magid, J.amer.pharm.Ass.sei.Ed. 25,11-15 (1936).
67) William J.Husa und C .L . Huy ck, J. amer, pharm. Ass. sei. Ed. 25,
311-313 (1936).
68) H. Mühlemann, Pharm.Acta Helv. 16, 121-140(1941), ref.Pharm.Zhalle 84,140 (1943).
69) Diss. Feinstein, pg. 92 (vergl. ').
70) Diss. Feinstein, pg. 99 (vergl. ')
71) C. Koch, Pharm. Ztg. £0, 552 (1935), ref. in Diss. Feinstem.
72) H.Breddin, Pharm. Ztg. 81, 112-114(1936).
73) William J.Husa und C .L
. Huy ck, J.amer.pharm.Ass.sei. Ed. 27,205-207 (1938).
74) William J.Husa und C.L.Huyck, J. amer, pharm. Ass. sei. Ed. 25,110-112 (1936).
—
75) Diss. Feinstein, pg. 158-160 (vergl. ').
76) Diss. Graetzer, pg. 21 (vergl. 51)).77) W.Schill, Pharm. Ztg. 86, 413-419(1950).
78) K. Münzel, Festschrift Paul Casparis 167-173 (1949), ref.Scientia pharm.
Jj), 137(1951).

- 154 -
79)J.Büchi, Arch.Pharm. 285, 57, 40-51(1952), siehe auch Pharm. Acta Helv.
V2, 326 (r9"3"7)7~
80) Diss. Graetzer, pg. 77 (vergl. 51)).
81) Diss. Feinstein, pg. 60, 61, 164, 165 (vergl. *)).82) Diss. Graetzer, pg. 95, 96 (vergl. 51)).
83) Gramberg, die dtsch.Apoth., No. 43 (1934), ref. F. Gstirner: "Prüfung und
Verarbeitung von Arzneidrogen", Band II, pg. 40.
84) F. Keller, die dtsch.Apoth. No. 41 (1934), ref. Gstirner, II. Band, pg. 40.
85) William J.Husa und C.L.Huyck, J.amer.pharm.Ass.sei.Ed. 27,290-295 (1938); Apparate-Beschreibung
W. J.Husa und Clifford D.Pacenta, J. Amer, pharm. Ass. sei. Ed. 30,635-636 (1941).
~
86) H.Dietmann, Pharm.Ind. U, 277-283 (1950), ref. Pharm. Zhalle 90, 341 (1951).
87) B. Grote, Dtsch.Apoth. Ztg. 93, 656 (1953), ref. F. Gstirner, II. Band, pg. 40.
88) F. Gstirner: "Prüfung und Verarbeitung von Arzneidrogen", Band II, pg. 40.
89) NF X (1955), pg. 676, 677.
90) Diss. Feinstein, pg. 61, 62, 166, 167 (vergl. 1').91) Diss. Feinstein, pg. 168 (vergl. ').
92) Diss. Graetzer, pg. 87, 88 (vergl. 51^).93) F.Ital. VI, pg. 178.
94) Brit.Ph. 1958, pg. 962.
95) Ph.Dan. IX/l. Band, 1948, pg. 57.
96) S.B ar i, Pharm. Ztg. 71, 622-624 (1926).
97)N.Thörn, Farm.Rev. No. 46, 698-702(16.11.1940).
98) K. Eichenberger, "Ueber Herstellung und Eigenschaften einiger Ephedra-Zu-bereitungen", Diss. ETH 1944.
99) L. Rosenthaler, Schweiz. Apoth. Ztg. 61, No. 3, 25-29 (1923).
100) H. Eschenbrenner und R . Gär tner,
Pharm. Ztg. J8, 160 (1933).
101) Weber, Ber. ung. pharm. Ges. 9, 434(1933), ref. Pharm. Zhalle 75, 136(1934).
102) F. Gstirner, Mitt.dtsch.pharm.Ges. 2£, No. 2; 21 (1959).
103) M. Herzog, Dtsch. Apoth. Ztg. £6^ No. 10; 200-202 (1956).
104) F. Gstirner, Pharm. Ztg. 2?_, 310-312 (1934).
105) K. Höll, Pharm. Ztg. 80, 1185 (1935).
106) A. Mos ig, Dtsch. Apoth. Ztg. 54, 368-371 (1939).
107) William J.Husa und D . W.Lee, J. amer, pharm. Ass. sei. Ed. 28,593-597 (1939).
~~
108) William J.Husa und Thomas J.Macek, J. amer, pharm. Ass. sei. Ed.
29, 455-458 (1940).
109) H. R. Fromm, Diss. Halle 1939, ref. Dtsch. Apoth. Ztg. £5, 275 (1940).
110) Z.Olszews ki und M
.Der kaezowa, Acta Polon.pharmac. 13, 147-153
(1956), ref. Pharm. Zhalle 96, 530(1957).

- 155 -
111) F. Gstirner und B.Berniker, Pharm. Ztg. 94, 420-423, 480-483(1958),réf. Pharm. Zhalle 97, 344 (1958).~~
112)William J.Husa und C.L.Huyck, J.amer.pharm.Ass.sei.Ed. 25,392-393 (1936).
—
113)William J.Husa und C.L.Huyck, J.amer.pharm.Ass.sei.Ed. 27,106-113 (1938).
114)Söllner, Pharm. Ztg. 101, 20(1956).
115)W.F.Head Jr., H.M.Beal und W.M.Lauter, J. amer, pharm. Ass.sei. Ed. -45, 239 (1956).
116) D. Thompson und D.G.Sutherland, Ind.engng.Chem. 47, 1167 u.f.
(1955).~
117) J. Solich, V.Rusek und E.Benesovä, Cescosl. farmac. 4, 512-514
(1955), ref. Pharm. Zhalle 95, 451 (1956), Chem.Zbl. 128,2277 (1957).
~~
118) William J.Husa und C.L.Huyck, J. amer, pharm. Ass. sei. Ed. 27,211-217 (1938).
—
119) Diss. Feinstein, pg. 73 (vergl. 1^).120) R. B. Woodward und W. E
. Doering, J.amer.ehem.Soc. 67, 860 (1945),C. H. Brieskorn, Süddtsch. Apoth. Ztg. 88, "Bl (1948), ref.
Pharm. Zhalle £8, 20(1949).
121) Diss. Feinstein, pg. 75 (vergl. 1)).
122) Ph.Helv. V, Suppl. I, pg. 109.
123) H. Wojahn, Dtsch. Apoth. Ztg. 54, 1224(1939), ref. Pharm. Zhalle 82, 164(1941).
124)E.Vogel und J.Eubel, Pharm. Zhalle 81, 397-401 (1940).
125) Diss. Feinstein, pg. 81-83, 84 (vergl. !)).
126) Diss. Fuchs, pg. 19-21 (vergl. 36*).127) W. Poethke und D.Horn, Pharm. Zhalle 93, 414-422 (1954).
128) L. Saunders und R .S
.S ri vas tava
,J. Pharm. Pharmacol, j}, 78-86(1951),
ref. Pharm. Zhalle 91, 16(1952).
129) I. Lebrun,M.Warlet und G. Schichar e vitch
,Ind. chim. Belge 15,
328-338(1950), ref.Chem. Zbl. 122, 3536-37 1(1951), PKirm.
Zhalle_91, 128 (1952).
130)A.Jindra und J.Pohorsky, J. Pharm.Pharmacol. 3, 344-350(1951), ref.
Pharm. Zhalle 90, 405 (1951).
131) F.F u r r e r
,"Die Verwendung neuer Austauschadsorbentien auf Harzbasis zur
Bestimmung und Gewinnung von Alkaloiden", Diss. ETH,Zürich 1954.
132) A. Fischer,
Pharm.Ind. 1_7, 418-420(1955), ref. Pharm. Zhalle j)6, 165(1957).
133) A. Fischer,
Dtsch. Apoth. Ztg. 97, 23-25(1957), ref. Pharm. Zhalle 96, 165
358 (1957).
134) W. Kamp, "Het bepalen en scheiden van alkaloiden in enige plantaardigegeneesmiddelen en nun preparaten met behulp van ionenuit-
wisselaars op harsbasis", Diss. Amsterdam 1956, ref. Pharm.
Weekbl. 92, 1-24 (1957), Pharm. Zhalle 96, 282 (1957).

- 156 -
135) P. Tunmann und W. Hubmann, Arch.Pharm. 287, 59, 281-290(1954), ref.
Pharm.Zhalle 95, 69 (1956).
136)K.Pfandl und J.Klotz, Dtsch.Apoth.Ztg. 98, 27, 28(1958), ref.Pharm.
Zhalle 97, 325 (1958).—
137) "The extra Pharmacopoeia" (Martindale), London 1955, pg. 472.
138)H.Wojahn und K. E r de Im e ie r,
Dtsch. Apoth. Ztg. j>4, 226 (1939).
139) H. Gnamm, "Die Gerbstoffe und Gerbmittel", wissenschaftliche Verlags-GmbH,Stuttgart 1949, pg. 140-143.
140) Eidgenössische Pharmakopoe-Kommission, 37. Sitzung, Bemerkungen zum Ent¬
wurf betreffend Bestimmung des Gerbstoffgehaltes von Drogen,4.1.1956, H. Flück.
141) Eidgenössische Pharmakopoe-Kommission, 37. Sitzung, allgemeine Bestim¬
mungen Ph.Helv. VI, Bestimmung des Gerbstoffgehaltes von
Drogen, Entwurf 4.1.1956, H. Flück.
142) R.Weber, "Arbeitsvorschrift der Gerbstoffbestimmung" der EMPA HI,St. Gallen.
143) Gnamm, pg. 111 u. folgende (siehe auch ').
144) Gnamm , pg. 125 u. folgende (siehe auch "9)j_
145) K. Herrmann, Pharmazie 5, 488-490 (1950), ref.Pharm. Zhalle 90, 406
(1951).~~ ~~
146) K. Herrmann, Pharmazie 2, 320-325 (1952).
147) E. Nick, Pharm.Ind. 1_5, 382-384 (1953), ref. Pharm. Zhalle £3, 384 (1954).
148) G. Kuhlmann, Dtsch.Apoth. Ztg. 97, 552-554(1957), ref. Pharm. Zhalle 96,549 (1957).
—
149) K. Herrmann, 5. Arbeitstagung der deutschen Gesellschaft für Arzneipflan¬zenforschung und Therapie 10. -13.10.57, ref. Pharm. Zhalle
97, 301 (1958).
150) F. Gstirner , A.Bopp und H. Hopmann, "Ueber eine kolorimetrische
Agglutinationsmethode zur Bestimmung von Gerbstoffen", Arch.
Pharm. 289, 61, 188-196 (1956).
151) R. Robert, Ber.dtsch.pharm.Ges. 24, 470 (1914), Collegium 321 (1915), 164
213 (1916).A.Boettcher, Virchows Arch. 36, 342(1866)R.Wasicky, Pharm.Post 92, 7Ö5~(1917) 1c;n>
W.Brandt und F. SchlunT, pharm. Ztg. 598 (1924) ref. in lou\
152) F. Gstirner und G.GoGho, Arch.Pharm. 292, 64, 621-632(1959).
153) Diss. Fuchs, pg. 19-24 (vergl. 36V154)L.Fuchs, M.Wichtl und H
. Haring, Scientia pharm. 20, 209-216 (1952),ref. Pharm. Zhalle 93, 70(1954).
155) Gnamm, pg. 138, 139 (vergl. 139^156) W. Diemair
,H. Janecke und G. Kr ieger ,
Z. anal. Chem. 133, 346-352
(1951), ref. Pharm. Zhalle 91, 166(1952).
157) H. Friedrich, Pharmazie 9, 138-155(1954).

- 157 -
158) J. Richter, "Arbeitsvorschriften für das Pulfrich-Photometer", Sammlung II,"Photometrische Bestimmungen in der Pharmazie, Lebensmit¬
telchemie, Toxicologie und Arbeitsmedizin", Berlin 1956,Gustav Fischer Verlag, Jena, VEB Carl Zeiss, Gerbstoffe,Tannin, pg. 61. Daselbst ref.
W.Lang, Pharmazie j>, 137 (1951)H.Friedrich, Pharmazie £, 138(1954).
159) K. Herrmann, Pharmazie £, 490(1950)Schulte, Pharm. Wbl. 59, 412(1922), ref. Pharm. Jahresber.
5i7, 149 (1922).~
160) K. Steiger, persönliche Mitteilung, "Methode zur quantitativen Tanninbestim¬
mung", massanalytische Bestimmung von Löwenthal, St. Gallen,26.3.1944.
161) S. M. Bolotnikow und M.S.Schreiber, Apothekenwesen 3, 10 (1954),ref. Pharm. Zhalle 94, 61 (1955).
162) Th. Rutschmann, "Dosage des tanins dans quelques drogues au moyen de la
méthode de la poudre de peau", Galenica-Wettbewerb 1956/57.
163) R. Weber,
mündliche Mitteilung; EMPA, St. Gallen (3.12.1958).
164) Ph.Helv. V, allgemeiner Teil, pg. 25.
165) Kommentar zur Ph. Helv. V, pg. 67.
166) Ph.Helv. V, Suppl. IH, pg. 47.
167) Kommentar zur Ph.Helv. V, pg. 71-74.
168) Ph.Helv. V, allgemeiner Teil, pg. 28, 29, siehe auch Kommentar zu Ph.Helv.
V, pg. 82, 83.
169) Ph.Helv. V, allgemeiner Teil, pg. 26, 27.
170) A . Jermstad und O.Oestby, Norskfarm. Tidsskrift 4 (1935), ref.Pharm.
Zhalle 77, 606 (1936).
171) V. M. Baschilowa und N. A . Figurowskij/Aptetschnoje Djelo (Apo¬thekenwesen) 3. Heft 6, 41 (1954), ref. Pharm. Zhalle 95,199-200 (1956J".
—
172) R. Fischer und F . Kolmay r,
Pharm. Zhalle 93, 54-61, 87-92 (1954).R.Fischer und W. Mühlber ger, Scientia pharm. 7A, 17-24 (1956),
ref. Pharm. Zhalle 96, 115(1957).
173) R. Fischer undH.Auer, Pharm. Zhalle 9£, 497-503 (1957).
174) J. Krutzsch, Dtsch. Apoth. Ztg. 98, 609-610 (1958), ref. Pharm. Zhalle 97,493 (1958).
~
175) Ph. Helv. V, pg. 1039.
176) F. Hagelstein, Pharm.Zhalle 77, 103-106(1936).
177) A.Linder
,"Statistische Methoden für Naturwissenschafter, Mediziner und
Ingenieure", Birkhäuser Verlag Basel 1945, pg. 53.
178) A. Linder, statist. Methoden, pg. 140 (siehe 177)).

Lebenslauf
Geboren wurde ich am 20. Dezember 1930 in Basel als Sohn von Dr. Ing. -Chem.
Max Walter und Mathilde, geb. Kern von Zurich. Primarschule und Gymnasium be¬
suchte ich in Basel und erwarb im Frühjahr 1950 das Maturitätszeugnis Typus C am
mathematisch-naturwissenschaftlichen Gymnasium.
Nachbestandener eidgenossischer Erganzungsprufung in der lateinischen Sprache
und beendetem naturwissenschaftlichem Vorstudium an der Universität Basel absol¬
vierte ich das Praktikum in der Burgfelder Apotheke, Frl. Dr. A. Senglet und bei
Herrn E. Hausmann, Barfusser Apotheke in Basel, anschliessend das Assistenten¬
jahr in den Apotheken Pharmacie M. Droz, Neuchâtel und Adler-Apotheke, L. Friedli
Biel. Das eidgenossische Apothekerdiplom erhielt ich im Herbst 1956 nach Abschluss
des Fachstudiums an der Universität Basel.
Nach halbjähriger Tätigkeit als Apotheken-Verwalter begann ich im Frühjahr 1957
die vorliegende Arbeit an der Eidgenossischen Technischen Hochschule, deren prak¬
tischer Teil im Frühjahr 1959 beendet war.
Seither arbeite ich in den wissenschaftlichen Laboratorien der Firma Siegfried
A.G. in Zofingen.