hÄmostaseologie - haemophiliehannover.de · – hämophilie a (faktor viii), hämophilie b (faktor...
TRANSCRIPT

HÄMOSTASEOLOGIEHÄMOSTASEOLOGIEHÄMOSTASEOLOGIEHÄMOSTASEOLOGIEVorlesung zum Praktikumg
Andreas TiedeKlinik für Hämatologie, Hämostaseologie, Onkologie und Stammzelltransplantation
Tel. (0511) 600 604 30ti d d @ h h [email protected]

Grundlagen der Hämostase
EndothelverletzungEndothelverletzungEndothelverletzungEndothelverletzungEndothelverletzungEndothelverletzungEndothelverletzungEndothelverletzung
Subendothel (Kollagen)Subendothel (Kollagen) Gewebefaktor (TF)Gewebefaktor (TF)
Primäre Hämostase• Thrombozyten, VWFPrimäre Hämostase• Thrombozyten, VWF
Sekundäre Hämostase• Plasmatische GerinnungSekundäre Hämostase• Plasmatische Gerinnung

Primäre HämostasePrimäre Hämostase
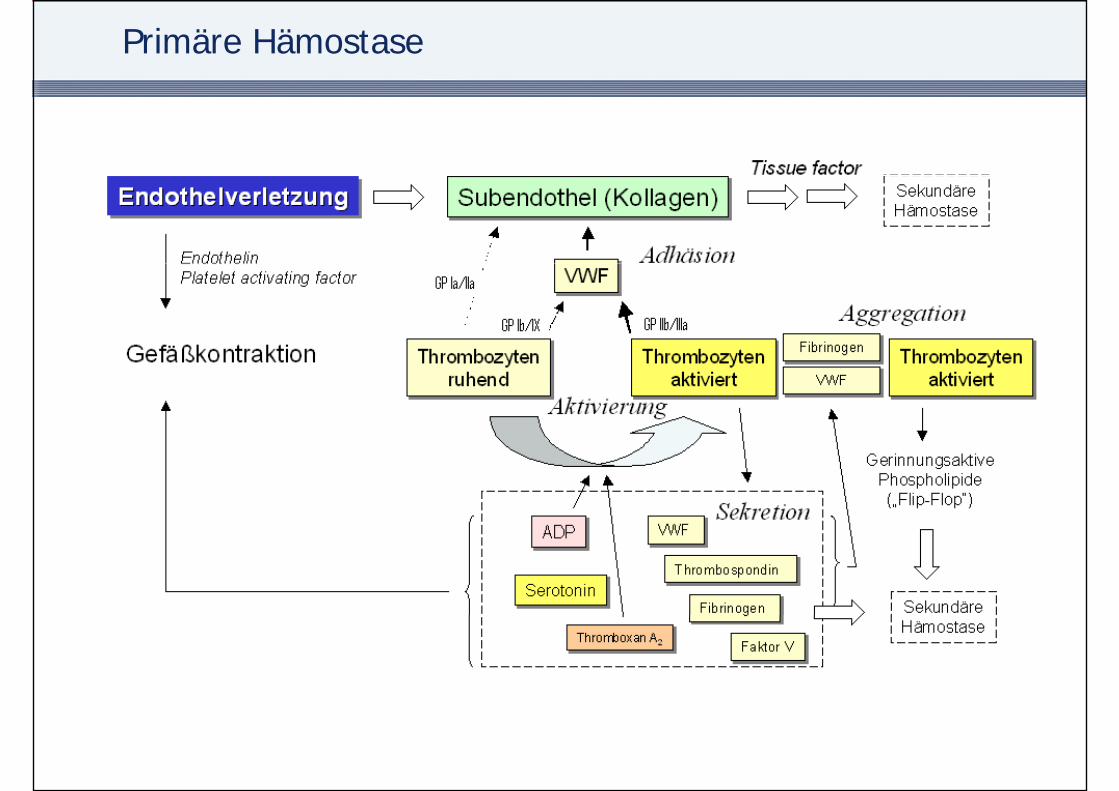
Primäre Hämostase

Beispiele für Thrombozytenrezeptoren
Name Ligand Funktion Therapeutischer Antagonist
GP I /II K ll Adhä iGP Ia/IIa(CD49/29)
Kollagen Adhäsion
GP Ib/IX/V(CD42)
VWF (GP Ib/IX)Thrombin (GP V)
AdhäsionAktivierung
GP IIb/IIIa Nach Aktivierung: Adhäsion Abciximab(CD41/61) Fibrinogen, VWF,
Vitronektin, Thrombospondin u. a.
Aggregation TirofibanEptifibatid
P2Y12ADP Aktivierung Ticlopidin,
Clopidogrel
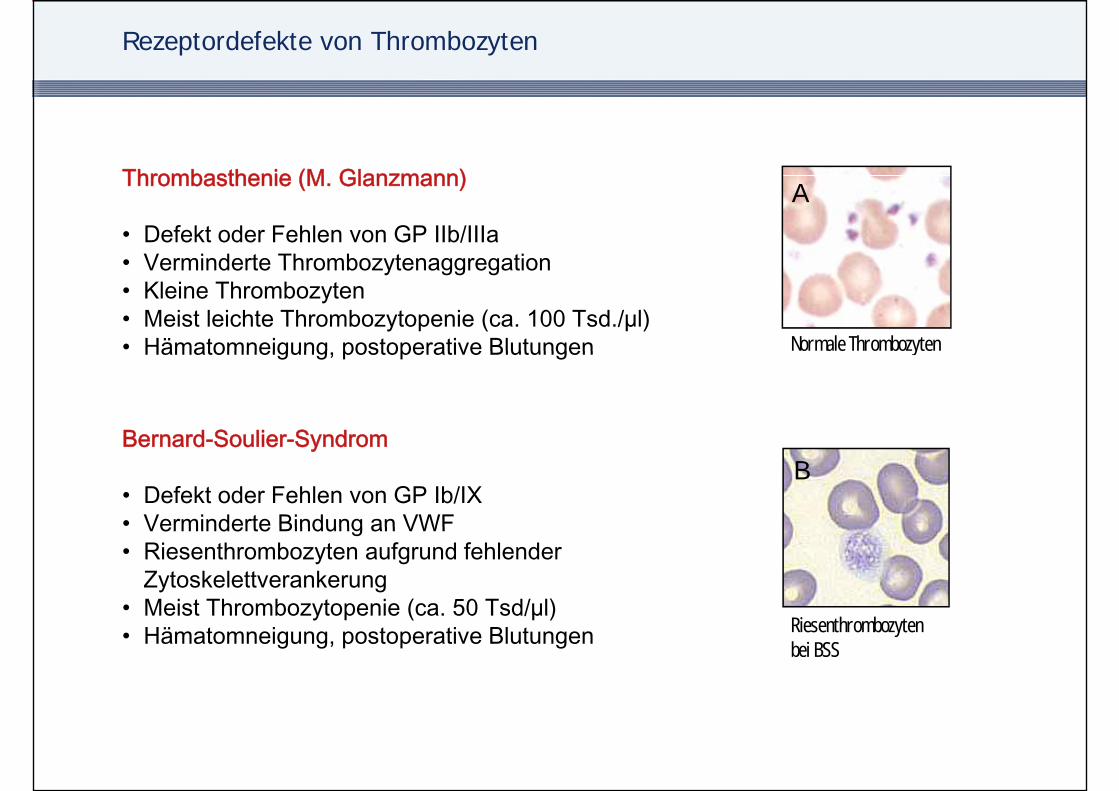
Rezeptordefekte von Thrombozyten
Thrombasthenie (M Glanzmann) AThrombasthenie (M. Glanzmann)
• Defekt oder Fehlen von GP IIb/IIIa• Verminderte Thrombozytenaggregation
Normale Thrombozyten
• Verminderte Thrombozytenaggregation• Kleine Thrombozyten• Meist leichte Thrombozytopenie (ca. 100 Tsd./µl)• Hämatomneigung postoperative Blutungen yHämatomneigung, postoperative Blutungen
Bernard Soulier SyndromB
Bernard-Soulier-Syndrom
• Defekt oder Fehlen von GP Ib/IX• Verminderte Bindung an VWFVerminderte Bindung an VWF• Riesenthrombozyten aufgrund fehlender
Zytoskelettverankerung• Meist Thrombozytopenie (ca. 50 Tsd/µl)
Riesenthrombozytenbei BSS
Meist Thrombozytopenie (ca. 50 Tsd/µl)• Hämatomneigung, postoperative Blutungen

Sekretionsdefekte von Thrombozyten
Storage Pool Disease
• Fehlen der Alpha-Granula (α-SDP) oderp ( )• Fehlen der Dense Bodies (δ-SPD) oder• Fehlen aller Granula (Grey Patelet
Syndrome)• Zytoskelettdefekte (Wiskot-Aldrich-
Syndrom) u.v.a.m.
α-Granula• Fibrinogen
F kt VDense bodies• ADP
Störungen metabolischer Wege
• z B Aspirin COX 2 Inhibitoren • Faktor V• VWF• Thrombospondin
• ADP• Serotonin• Kalzium
• z. B. Aspirin, COX-2-Inhibitoren
• P-Selektin• Zytokine u.a.
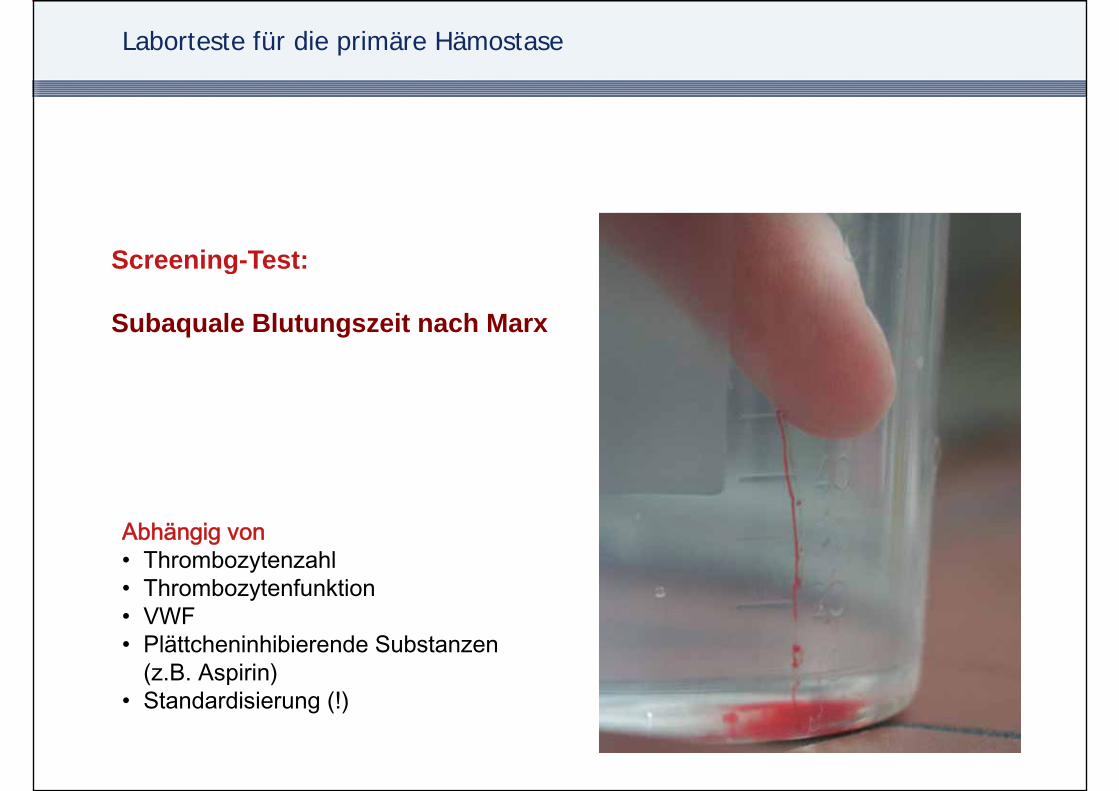
Laborteste für die primäre Hämostase
Screening-Test:Screening-Test:
Subaquale Blutungszeit nach Marx
Abhä iAbhängig von• Thrombozytenzahl• Thrombozytenfunktion• VWF• VWF• Plättcheninhibierende Substanzen
(z.B. Aspirin)• Standardisierung (!)• Standardisierung (!)

Laborteste für die primäre Hämostase
Screening-Test:Screening-Test:
Blutungszeit nach Ivy
Abhä iAbhängig von• Thrombozytenzahl• Thrombozytenfunktion• VWF• VWF• Plättcheninhibierende Substanzen
(z.B. Aspirin)• Standardisierung (!)• Standardisierung (!)

Laborteste für die primäre Hämostase
• VollbluttestScreening-Test: • Vollbluttest• Plättchenadhäsion und –aggregation• Hohe Scherkräfte
Screening-Test:PFA-100®
KollagenAd li
KollagenADPAdrenalin ADP
Verschlusszeit abhängig vonVerschlusszeit abhängig von• Thrombozytenzahl (nicht messbar <100.000 /µL)• Thrombozytopathie• VWF• VWF• Hämatokrit (<30 %)• Plättcheninhibierende Substanzent (z.B. Aspirin)

Thrombozytenfunktionsdiagnostik
Thrombozyten-Aggregation nach BORN
• Plättchenzahl 250.000/µL im Patientenplasma• Zugabe aktivierender/aggregierender Substanzen
Kollagen 1 2 µg/mL– Kollagen 1-2 µg/mL– Adrenalin (Epinephrin) 2,5-10 µmol/L– ADP 0,5-2 µmol/L– Ristocetin 0,5-1 mg/L
• Aggregation erhöht Lichtdurchlässigkeit in der Messküvette
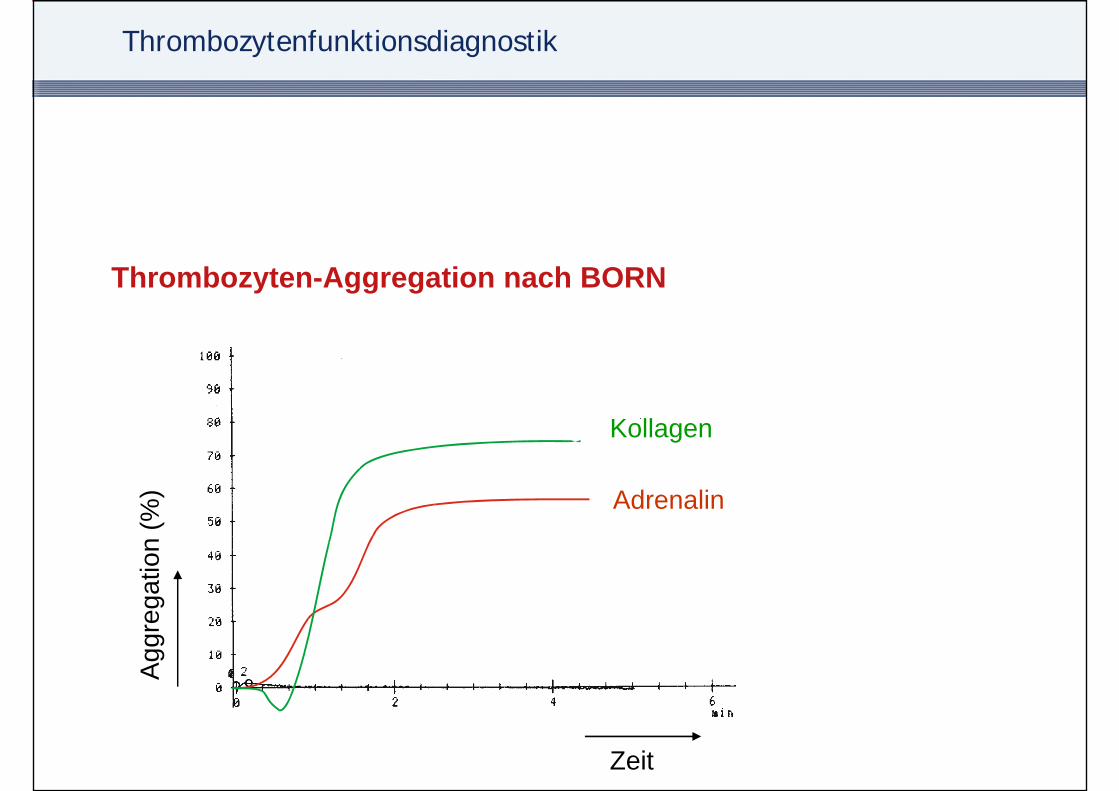
Thrombozytenfunktionsdiagnostik
Thrombozyten-Aggregation nach BORN
Kollagen
(%)
g
Adrenalin
egat
ion
(A
ggre
Zeit

Thrombozytopathien
MedikamenteC l i h (ASS )– Cyclooxigenasehemmer (ASS u.a.)
– Gp IIb/IIIa-Antagonisten (z.B. Tirofiban, ReoPro)– ADP-Rezeptorantagonisten (Clopidogrel)
RezeptordefekteRezeptordefekte– M. Glanzmann (gp IIb/IIIa)– Bernard-Soulier-Syndrom (gp Ib/V/IX)– Platelet-type VWS (gp Ib/V/IX)
D f kt d G l (St P l D f kt )Defekte der Granula (Storage-Pool Defekte)– Dense bodies (δ-SPD)– Alpha-Granula (α-SPD, Grey platelet syndrome)– Kombinierte Defekte (αδ-SPD)Kombinierte Defekte (αδ SPD)
Defekte im Arachidonsäuremetabolismus (Aspirin-like disease)Defekte in der SignaltransduktionDefekte der Zytoskelett-Regulation (z.B. Wiskott-Aldrich-Syndrom)y g ( y )Defekte in der Phospholipid-Exposition (Scott-Syndrom)
Beachte: Auch bei Thrombozytopathien häufig Thrombozytopenie

Thrombozytopenien
Verminderte ThrombozytenbildungErworbene Störungen der Megakaryopoese (Knochenmetastasen Aplastische Anämie Infekte– Erworbene Störungen der Megakaryopoese (Knochenmetastasen, Aplastische Anämie, Infekte, Medikamente, Strahlen)
– Angeborene Störungen der Megakaryopoese, z.B.• Amegakaryozytäre Thrombozytopenie (z.B. TAR-Syndrom)• Thrombozytopenien mit Riesenthrombozyten (z.B. May-Hegglin-Syndrom, Alport-Syndrom u.a.)
( S )• Thrombozytopenien mit Mikrothrombozyten (z.B. Wiskott-Aldrich-Syndrom)Erhöhter Thrombozytenverbrauch
– Immunthrombozytopenie (ITP)– Alloimmunthrombozytopenie (z.B. postpartal, nach Transfusion)
H l i– Hypersplenismus– Herzklappen– HIT, TTP, HUS, DIC, HELLP
SonstigesP d th b t i– Pseudothrombozytopenie
• Thrombozytenverklumpung in EDTA-Blut• Höhere Thrombozytenwerte in Citratblut

Von Willebrand Faktor
Genetik
• Chromosom 12
Synthese
• Chromosom 12
y
• Endothel
Funktion
• Primäre Hämostase:Thrombozytenadhäsion und –aggregation unter hohen Scherkräften
• Sekundäre Hämostase:Träger- und Schutzprotein für FVIII

Von Willebrand Faktor
Struktur:
C2C1BD4A3A2A1D3D‘D2D1 2813 aa
Signal-Peptid
Pro-VWF
2050 aa
FVIII gp IbHeparinKollagen
Kollagen gp IIb/IIIaBindungs-stellen:
g

Von Willebrand Faktor
MW (kD)
20.000
250250(Dimer)

VWF-Teste
Quantität• VWF-Antigen (ELISA)
Ristocetin-Cofaktor
t ge ( S )
Qualität (Funktion)Qualität (Funktion)• Ristocetin-Cofaktor• Kollagenbindungstest
FVIII Bi d t t• FVIII-Bindungstest
Qualität (Struktur)• Multimeranalyse
VWF in Pat -Plasma
Fixierte ThrombozytenRistocetin
VWF in Pat.-Plasma
Ristocetin

VWF-Teste
Quantität• VWF Antigen (ELISA)
Multimeranalyse
• VWF-Antigen (ELISA)
Qualität (Funktion)• Ristocetin-Cofaktor• Kollagenbindungstestg g• FVIII-Bindungstest
Qualität (Struktur)• Multimeranalyse
(SDS-Elektrophorese + Western Blot)(SDS Elektrophorese Western Blot)

Von Willebrand-Syndrom
KlassifikationKlassifikation Typ 1Typ 1 Typ 2Typ 2 Typ 3Typ 3
HäufigkeitHäufigkeit 70-80 % 10-20 % ca. 10 %
DefektDefektQuantitativ
(<50 %)
Qualitativ(MM-Struktur,
Funktion)
Quantitativ(<1 %)Funktion)
VererbungVererbung Autosomal- Variabel Autosomal-VererbungVererbung dominant Variabel rezessiv

Plasmatische GerinnungPlasmatische Gerinnung
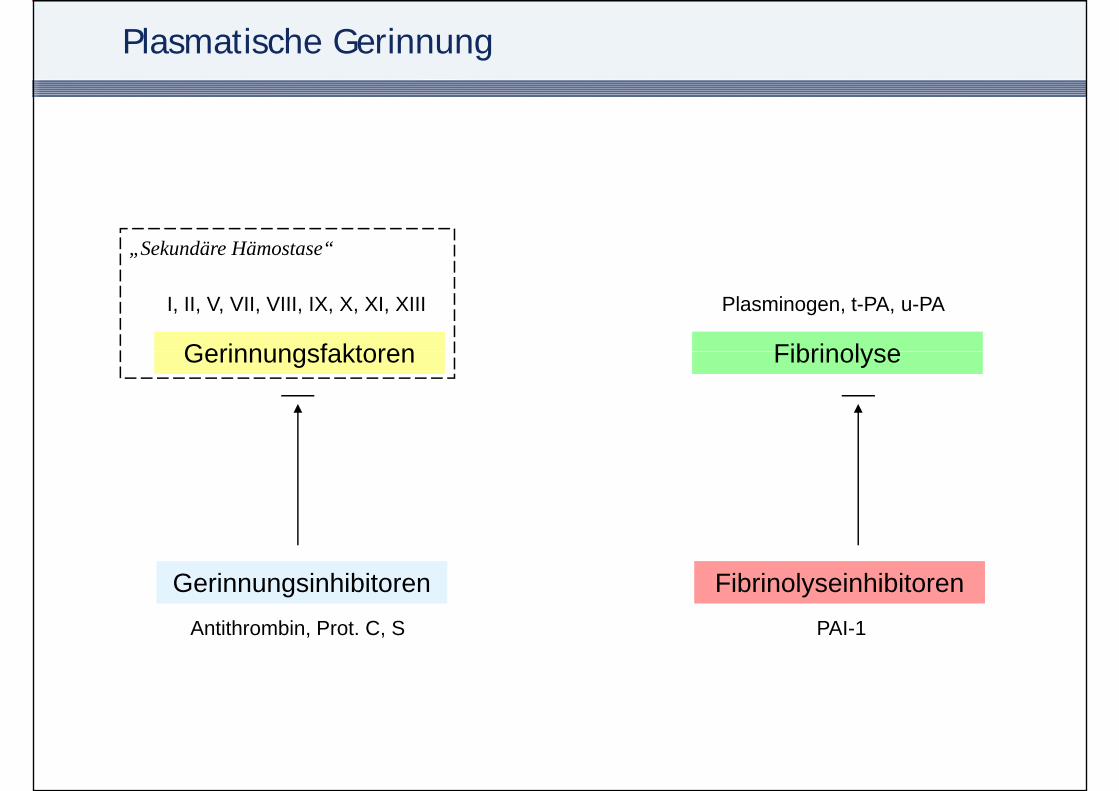
Plasmatische Gerinnung
„Sekundäre Hämostase“
Gerinnungsfaktoren Fibrinolyse
I, II, V, VII, VIII, IX, X, XI, XIII Plasminogen, t-PA, u-PA
Gerinnungsfaktoren Fibrinolyse
Gerinnungsinhibitoren FibrinolyseinhibitorenAntithrombin, Prot. C, S PAI-1
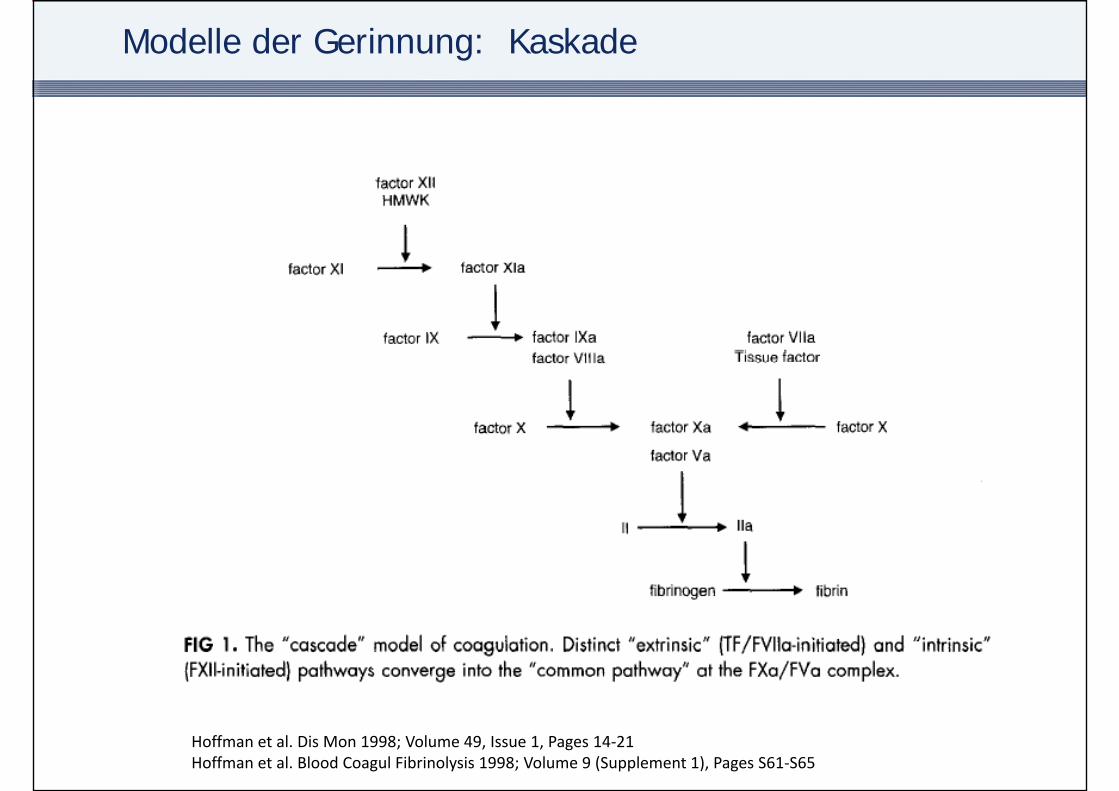
Modelle der Gerinnung: Kaskade
Hoffman et al. Dis Mon 1998; Volume 49, Issue 1, Pages 14‐21Hoffman et al. Blood Coagul Fibrinolysis 1998; Volume 9 (Supplement 1), Pages S61‐S65

Modelle der Gerinnung: Zellbasiertes Modell
Hoffman et al. Dis Mon 1998; Volume 49, Issue 1, Pages 14‐21Hoffman et al. Blood Coagul Fibrinolysis 1998; Volume 9 (Supplement 1), Pages S61‐S65
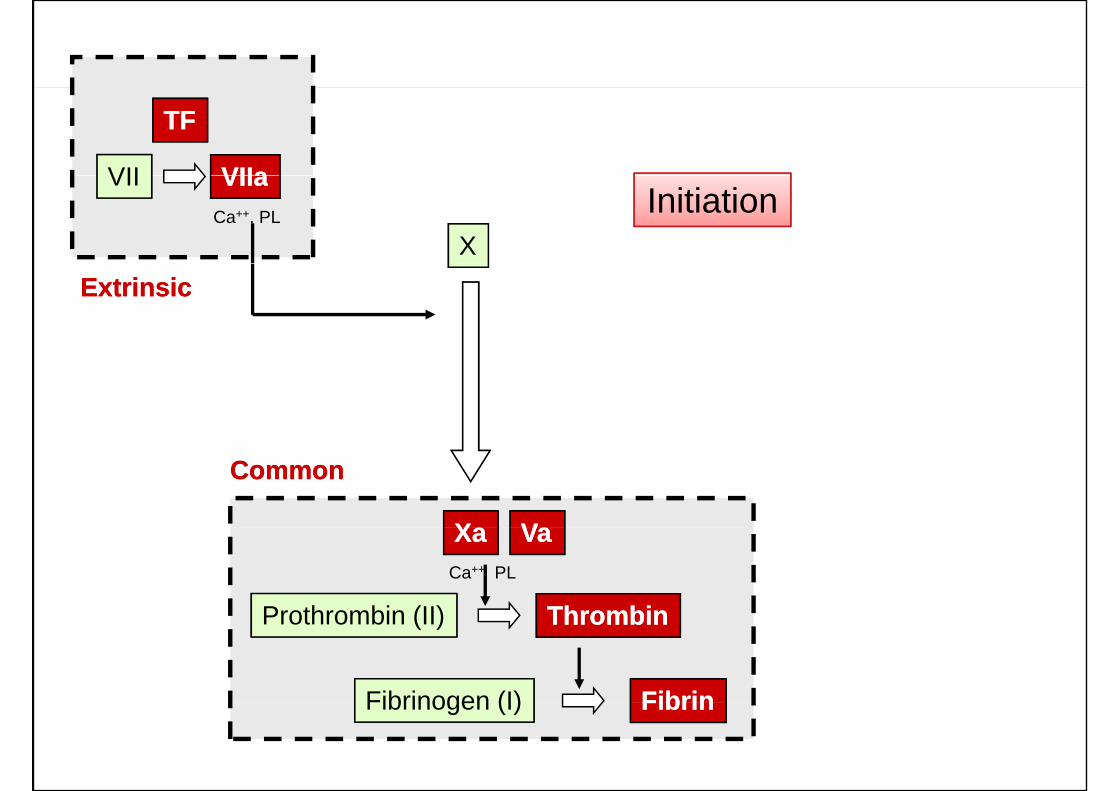
VII VIIaVIIa
TFTF
X
VII VIIaVIIaCa++, PL Initiation
ExtrinsicExtrinsic
XX VV
CommonCommon
Prothrombin (II) ThrombinThrombin
XaXa VaVaCa++ PL
Fibrinogen (I) FibrinFibrin
Prothrombin (II) ThrombinThrombin
Fibrinogen (I) FibrinFibrin
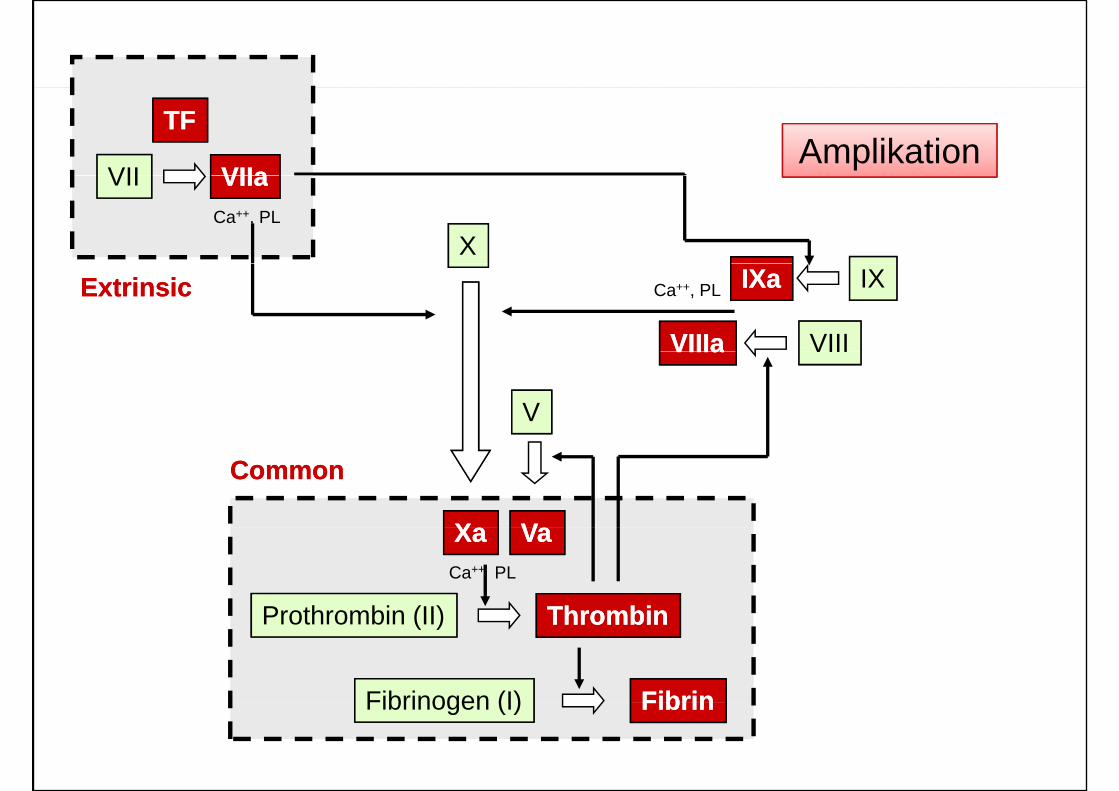
VII VIIaVIIa
TFTFAmplikation
X
VII VIIaVIIaCa++, PL
ExtrinsicExtrinsic
VIIIaVIIIa
IXaIXa
VIII
IXCa++, PL
VIIIaVIIIa VIII
V
XX VV
CommonCommon
Prothrombin (II) ThrombinThrombin
XaXa VaVaCa++ PL
Fibrinogen (I) FibrinFibrin
Prothrombin (II) ThrombinThrombin
Fibrinogen (I) FibrinFibrin

VII VIIaVIIa
TFTF Propagation
X
VII VIIaVIIaXIXIaXIaCa++, PL
ExtrinsicExtrinsic
VIIIaVIIIa
IXaIXa
VIII
IXCa++, PL
VIIIaVIIIa VIII
V
XX VV
CommonCommon
Prothrombin (II) ThrombinThrombin
XaXa VaVaCa++ PL
Fibrinogen (I) FibrinFibrin
Prothrombin (II) ThrombinThrombin
Fibrinogen (I) FibrinFibrin

Vorphase
TFTFXIIXIIa
HMWK KK
X
VII VIIaVIIaXIXIaXIaCa++, PL
XExtrinsicExtrinsic
VIIIVIII
IXaIXa
VIII
IXCa++, PL
VIIIaVIIIa VIII
IntrinsicIntrinsicV
CommonCommon
IntrinsicIntrinsic
XaXa VaVaCa++ PL Cave:
Ein Defekt der Vorphase‐Prothrombin (II) ThrombinThrombin
Ein Defekt der VorphaseFaktoren geht mit einer Verlängerung der APTT, aber mit keiner klinisch l lFibrinogen (I) FibrinFibrin relevanten Blutungsneigung
einher.

TFTFXIIXIIaXIIa
HMWK KK
X
VII VIIaVIIaXIXIaXIaCa++, PL
XExtrinsicExtrinsic
VIIIVIII
IXaIXa
VIII
IXCa++, PL
VIIIaVIIIa VIII
IntrinsicIntrinsicV
Cave:Ein Mangel an Faktor XIII geht in die Globalteste der
CommonCommon
IntrinsicIntrinsic
XIII
gGerinnung nicht ein.
XaXa VaVaCa++ PL
XIII
Prothrombin (II) ThrombinThrombin XIIIaXIIIa
Fibrinogen (I) FibrinFibrin Vernetztes FibrinVernetztes Fibrin
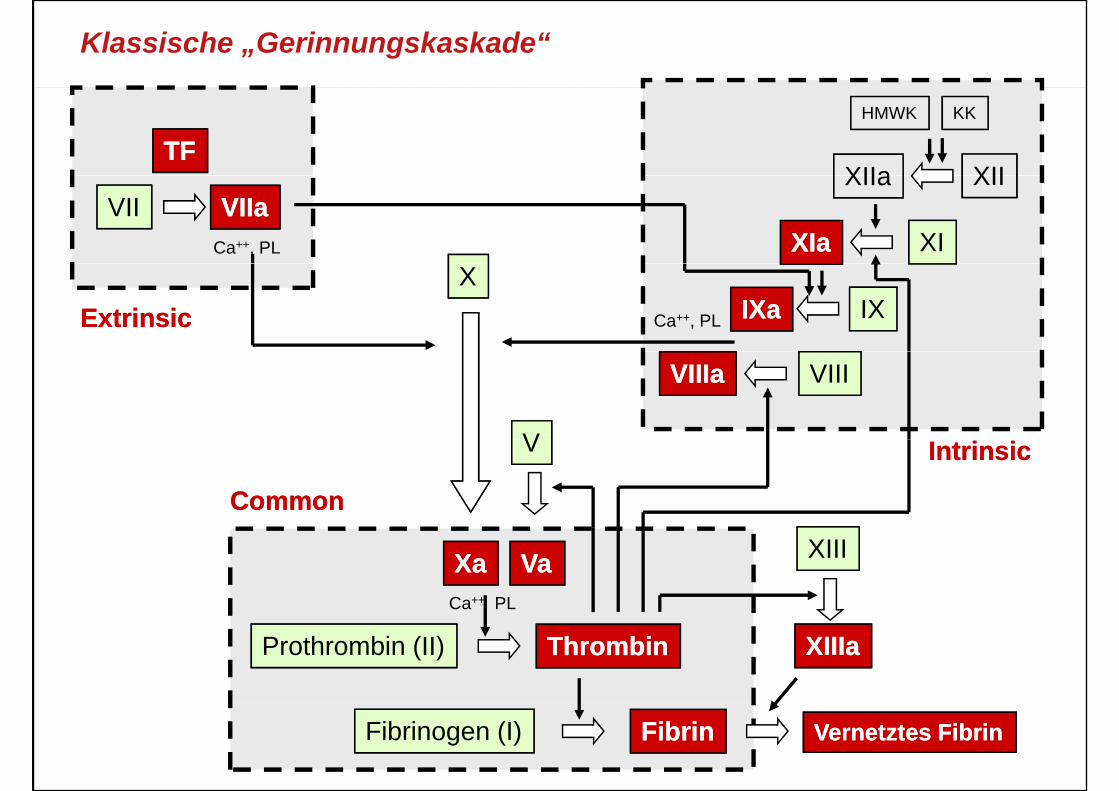
Klassische „Gerinnungskaskade“
TFTFXIIXIIa
HMWK KK
VII VIIaVIIaXIXIaXIa
XIIXIIa
Ca++, PL
XExtrinsicExtrinsic IXaIXa IXCa++, PL
VIIIaVIIIa VIII
V
CommonCommon
IntrinsicIntrinsicV
XaXa VaVaCa++ PL
XIII
Prothrombin (II) ThrombinThrombin XIIIaXIIIa
Fibrinogen (I) FibrinFibrin Vernetztes FibrinVernetztes Fibrin

Hämophilie: Faktor VIII und IX
XTFTF
Ca++, PLIXaIXa IX
VIIIaVIIIa VIII
VIIaVIIaCa++, PL
VIIIaVIIIa VIII
XX VV
Prothrombin (II) ThrombinThrombin
XaXa VaVaCa++ PL
Fibrinogen (I) FibrinFibrin
Prothrombin (II) ThrombinThrombin
Fibrinogen (I) FibrinFibrin

Hämorrhagische Diathese – Störungen der plasmatischen Gerinnung
• Angeborene Mangelzuständeg g– Hämophilie A (Faktor VIII), Hämophilie B (Faktor IX)– Afibrinogenämie, Hypofibrinogenämie– Seltene Mängel (Faktor V, VII, X, XI, XIII)
• Erworbene MangelzuständeL b k k– Lebererkrankungen
– (Auto-)Antikörper gegen Gerinnungsfaktoren– VerlustkoagulopathieVerlustkoagulopathie– Verbrauchskoagulopathie
• Sonstigesg– Hyperfibrinolyse– Hypothermie, Azidose

Physiologische Inhibitoren
TFXIIXIIa
HMWK KKTFPITFPI
tissue factor path a inhibitor
VII VIIaXIXIa
XIIXIIa
Ca++, PL
pathway inhibitor
XExtrinsicExtrinsic IXa IXCa++, PL
VIIIa VIII
VPCPC
CommonCommon
IntrinsicIntrinsicVPSPS
Xa VaCa++ PL
ANTITHROMBINANTITHROMBIN
Prothrombin (II) Thrombin
Fibrinogen (I) Fibrin

Antithrombin
• Irreversible Inaktivierung von IIa, Xa, XIa IXaXIa, IXa
• Wird durch Heparin beschleunigt und verstärkt
• Angeborener AT-Mangel bedingtAngeborener AT Mangel bedingt schwere Thrombophilie
AT+Heparin
ekt
AT allein
Ant
i-Xa-
Effe
A
Zeit(C) Jeffrey I. Weitz
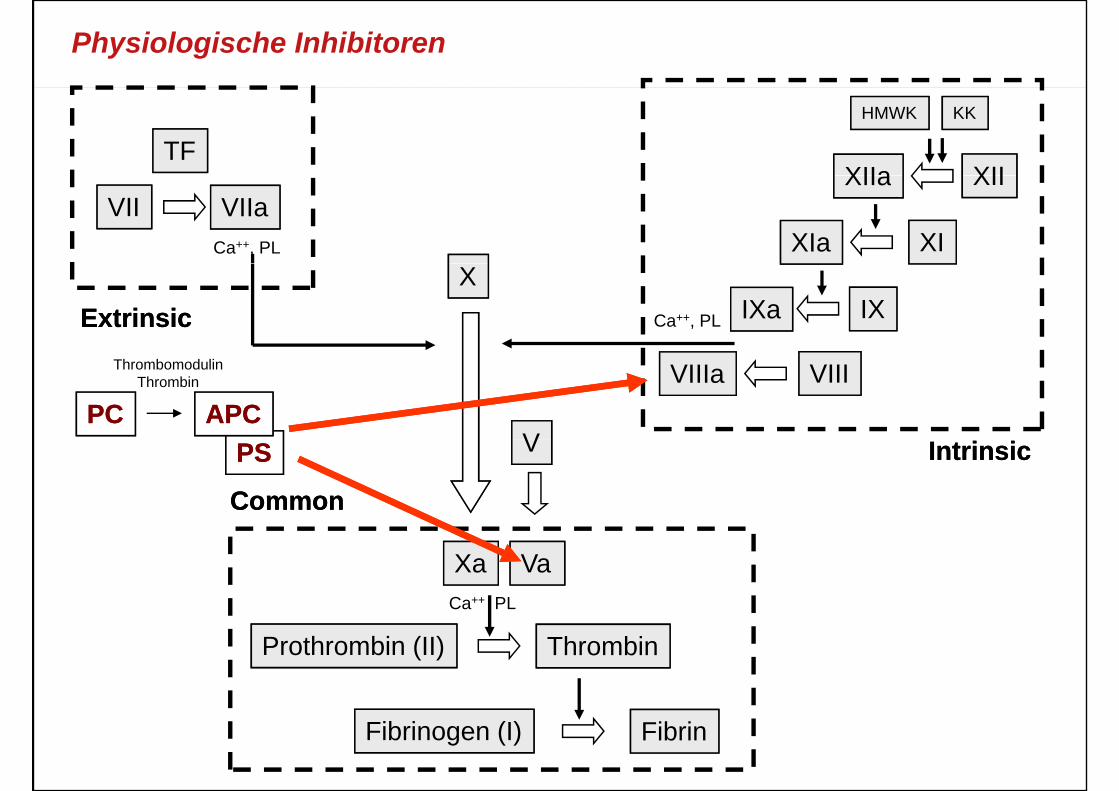
Physiologische Inhibitoren
TFXIIXIIa
HMWK KK
VII VIIaXIXIa
XIIXIIa
Ca++, PL
XExtrinsicExtrinsic IXa IXCa++, PL
VIIIa VIII
VAPCAPCPCPC
ThrombomodulinThrombin
CommonCommon
IntrinsicIntrinsicVPSPS
Xa VaCa++ PL
Prothrombin (II) Thrombin
Fibrinogen (I) Fibrin
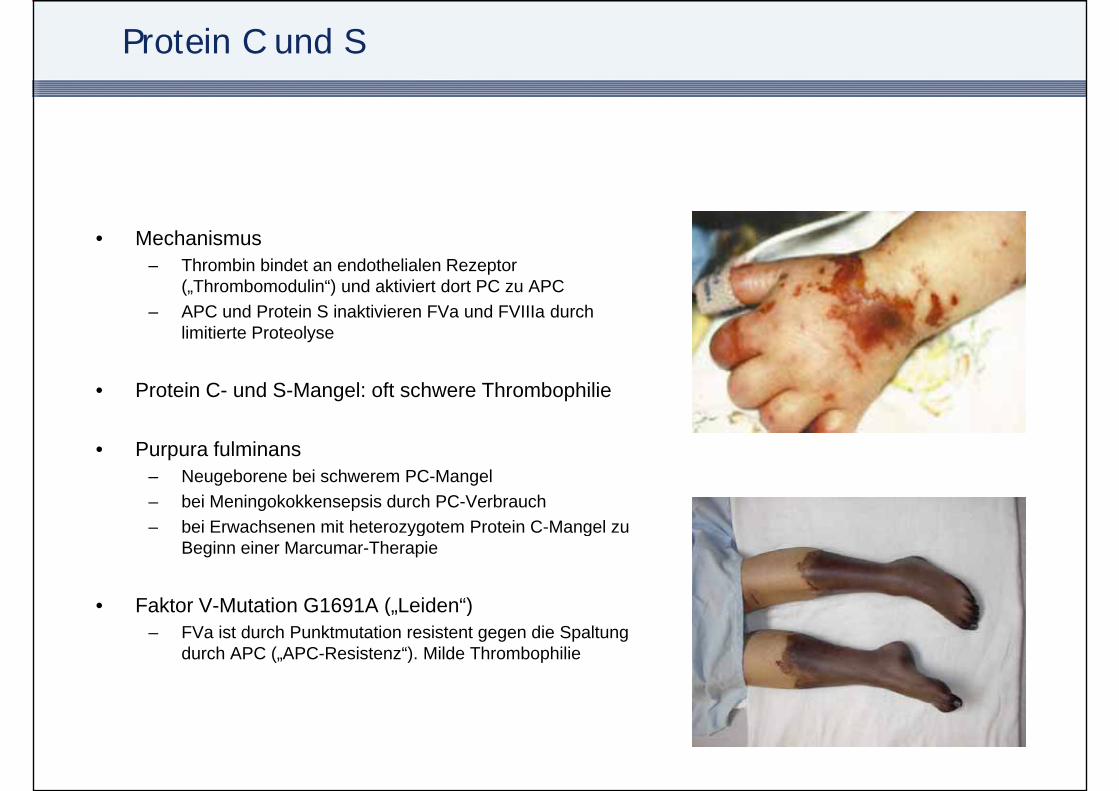
Protein C und S
• MechanismusThrombin bindet an endothelialen Rezeptor– Thrombin bindet an endothelialen Rezeptor („Thrombomodulin“) und aktiviert dort PC zu APC
– APC und Protein S inaktivieren FVa und FVIIIa durch limitierte Proteolyse
• Protein C- und S-Mangel: oft schwere Thrombophilie
• Purpura fulminans– Neugeborene bei schwerem PC-Mangel– bei Meningokokkensepsis durch PC-Verbrauch
bei Erwachsenen mit heterozygotem Protein C Mangel zu– bei Erwachsenen mit heterozygotem Protein C-Mangel zu Beginn einer Marcumar-Therapie
• Faktor V-Mutation G1691A („Leiden“)( )– FVa ist durch Punktmutation resistent gegen die Spaltung
durch APC („APC-Resistenz“). Milde Thrombophilie

Thrombophile Diathesen
• Mangel an Gerinnungsinhibitoreng g– Antithrombin– Protein C
Protein S– Protein S• Resistenz gegenüber der Wirkung von Gerinnungsinhibitoren
– FV-Variante R506Q („FV-Leiden“, APC-Resistenz)• Erhöhte Aktivität prokoagulatorischer Gerinnungsfaktoren
– Faktor II (z.B. bei Prothrombin-Genmutation G20210A)Faktor VIII– Faktor VIII
• Sonstiges– Lipoprotein (a)-Erhöhungp p ( ) g– Hyperhomozysteinämie– u.v.a.m.

GlobaltesteGlobalteste

TFXIIXIIa
HMWK KK
APTTAPTTPTPT(Q i k)(Q i k)
VII VIIaXIXIa
XIIXIIa
Ca++, PL
(Quick)(Quick)
XExtrinsicExtrinsic IXa IXCa++, PL
VIIIa VIII
VGlobalteste der Gerinnung
CommonCommon
IntrinsicIntrinsicV
Xa VaCa++ PL
Prothrombin (II) Thrombin ThrombinzeitThrombinzeitBatroxobinzeitBatroxobinzeitE i itE i it
Fibrinogen (I) FibrinEcarinzeitEcarinzeit

Thrombinzeit
• Fibrinogen-Mangel (Afibrinogenämie)g g ( g )• Fibrin-Bildungsstörungen („Dysfibrinogenämie“)• UF-Heparin (z. B. Monitoring bei F.XII-Mangel)p ( g g )• Thrombininhibitoren (Hirudin, für das Monitoring nicht geeignet,
da zu empfindlich)

APTT
• Faktoren des „Intrinsic Pathway“„ y• Faktoren des „Common Pathway“
• UF-Heparin• Hirudin

Quick-Test (Prothrombinzeit)
• Faktoren des „Extrinsic Pathway“„ y• Faktoren des „Common Pathway“
• Monitoring von Vitamin-K-Antagonisten (Coumarine)

Standardisierung des Quick-Test
Problem:Problem: Unterschiedliche Gewebsthromboplastine sind für Unterschiedliche Gewebsthromboplastine sind für F kF k M l hi dli h fi dli h!M l hi dli h fi dli h!FaktorenFaktoren--Mangel unterschiedlich empfindlich!Mangel unterschiedlich empfindlich!
Lösung:Lösung: ProthrombinratioProthrombinratioKorrekturfaktor: International Sensitivity index, ISIKorrekturfaktor: International Sensitivity index, ISI
Internationale normalisierte Ratio (INR), WHO 1983Internationale normalisierte Ratio (INR), WHO 1983( ),( ),
ISI
INR =PT [Patient]
PT [Normal]
ISI
Steuerung derAntikoagulanzien-PT [Normal] gTherapie (Marcumar etc.)

Standardisierung des Quick-Test
ISI: 1 1 1 0 1 32ISI: 1,1 1,0 1,32
Q ll H Ri d K i hQuelle: Human Rind Kaninchen
Q i k % 24 9 29Quick %: 24 9 29
INR 3 0 3 0 3 0INR: 3,0 3,0 3,0
Steuerung der Antikoagulanzientherapie (Marcumar etc.)

Eigenschaften der Globalteste
Keinen Einfluss auf die Globalteste haben:
• von Willebrand-Faktor• Faktor XIII
Antithrombin• Antithrombin• Protein C, Protein S• NM-Heparin (<1 E/ml)• Milde Hypofibrinogenämie• Milde Hypofibrinogenämie
– Referenzbereich Fibrinogen 2,0-3,5 g/l– Globalteste bei Fib. >0,6 g/l meist noch normal
• Fibrin(ogen)-Spaltprodukte in niedrigen Konzentrationen( g ) p p g– Referenzbereich D-Dimer <500 µg/l– Globalteste bei D-Dimer <50.000 µg/l i.d.R. normal

Fibrinbildung und FibrinolyseFibrinbildung und Fibrinolyse
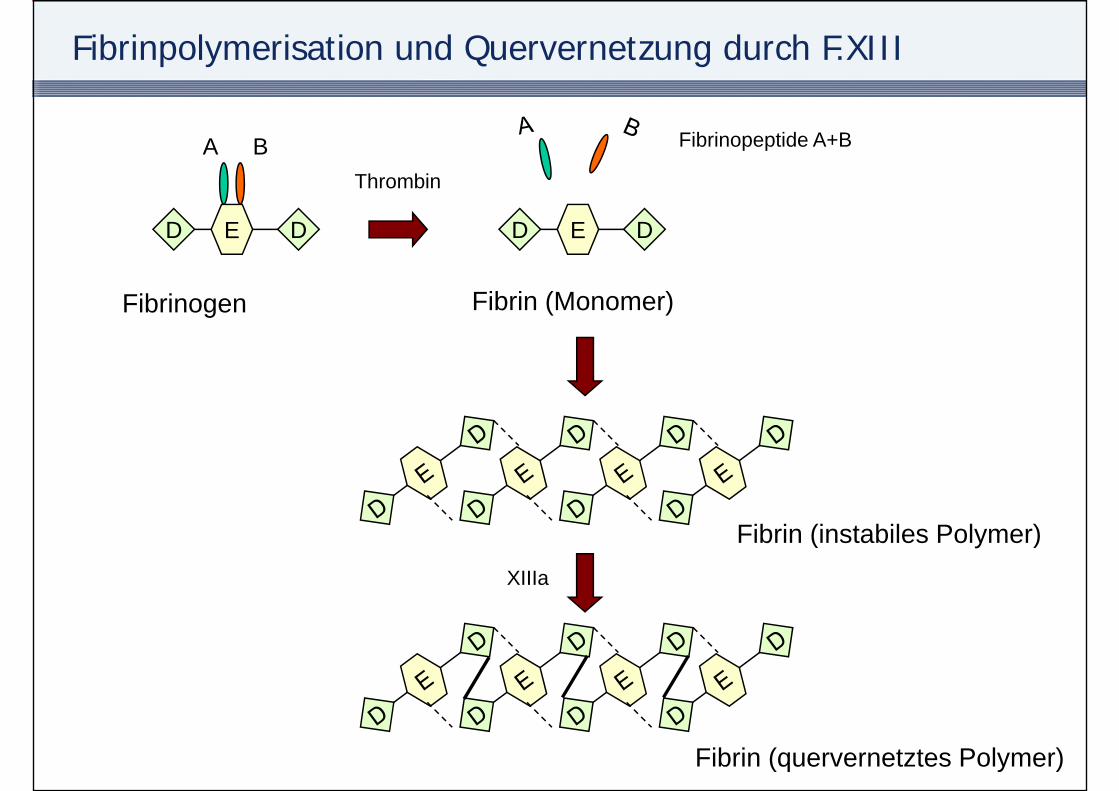
Fibrinpolymerisation und Quervernetzung durch F.XIII
A BTh bi
Fibrinopeptide A+B
ED D
Thrombin
ED D
Fibrinogen Fibrin (Monomer)
Fib i (i t bil P l )Fibrin (instabiles Polymer)XIIIa
Fibrin (quervernetztes Polymer)

Faktor XIII
• Synthese in der LeberSynthese in der Leber• Sehr lange HWZ (1 Woche)• Faktor XIII-Mangel
A b ( h lt )– Angeboren (sehr selten)– Erworben (häufig)
• bei schweren Blutungen• Leberfunktionsstörungen• Leberfunktionsstörungen
• Cave: Globalteste bei FXIII-Mangel normwertig!
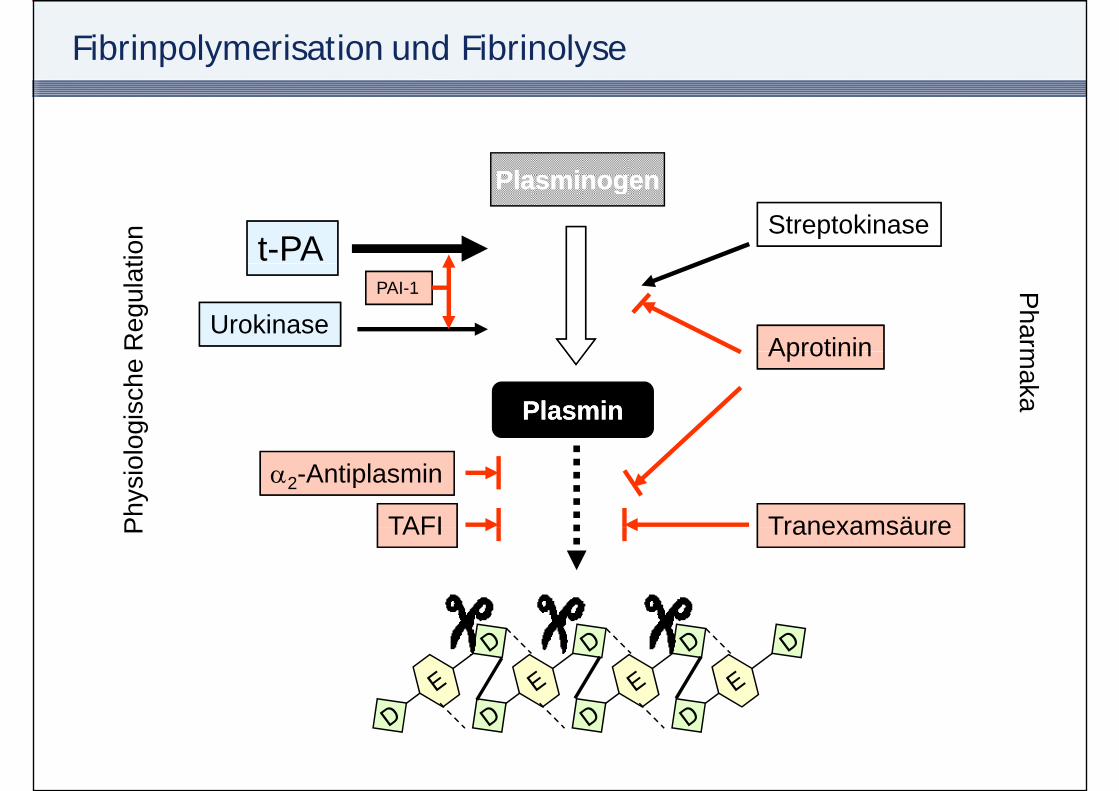
Fibrinpolymerisation und Fibrinolyse
PlasminogenPlasminogenPlasminogenPlasminogen
t-PAtion Streptokinase
UrokinasePAI-1
Reg
ulat
Aprotinin
Pharm
PlasminPlasmin
ogis
che Aprotinin m
aka
TAFI
α2-Antiplasmin
Phy
siol
o
TranexamsäureTAFIP Tranexamsäure

Fibrinpolymerisation und Fibrinolyse
PlasminogenPlasminogenPlasminogenPlasminogen
PlasminPlasmin
DD--DimerDimer
D-Dimer
• Erhöht bei– Thrombose und Lungenembolie– Hyperfibrinolyse– Postoperativ– Schwere Infekte
Malignome– Malignome– Schwangerschaft
• Negatives D-Dimer erlaubt den Ausschluss einer ThromboseNegatives D Dimer erlaubt den Ausschluss einer Thrombose oder Lungenembolie

AntikoagulanzienAntikoagulanzien

Physiologische Inhibitoren
TFXIIXIIa
HMWK KKTFPITFPI
tissue factor path a inhibitor
VII VIIaXIXIa
XIIXIIa
Ca++, PL
pathway inhibitor
XExtrinsicExtrinsic IXa IXCa++, PL
VIIIa VIII
VPCPC
CommonCommon
IntrinsicIntrinsicVPSPS
Xa VaCa++ PL
ANTITHROMBINANTITHROMBIN
Prothrombin (II) Thrombin
Fibrinogen (I) Fibrin
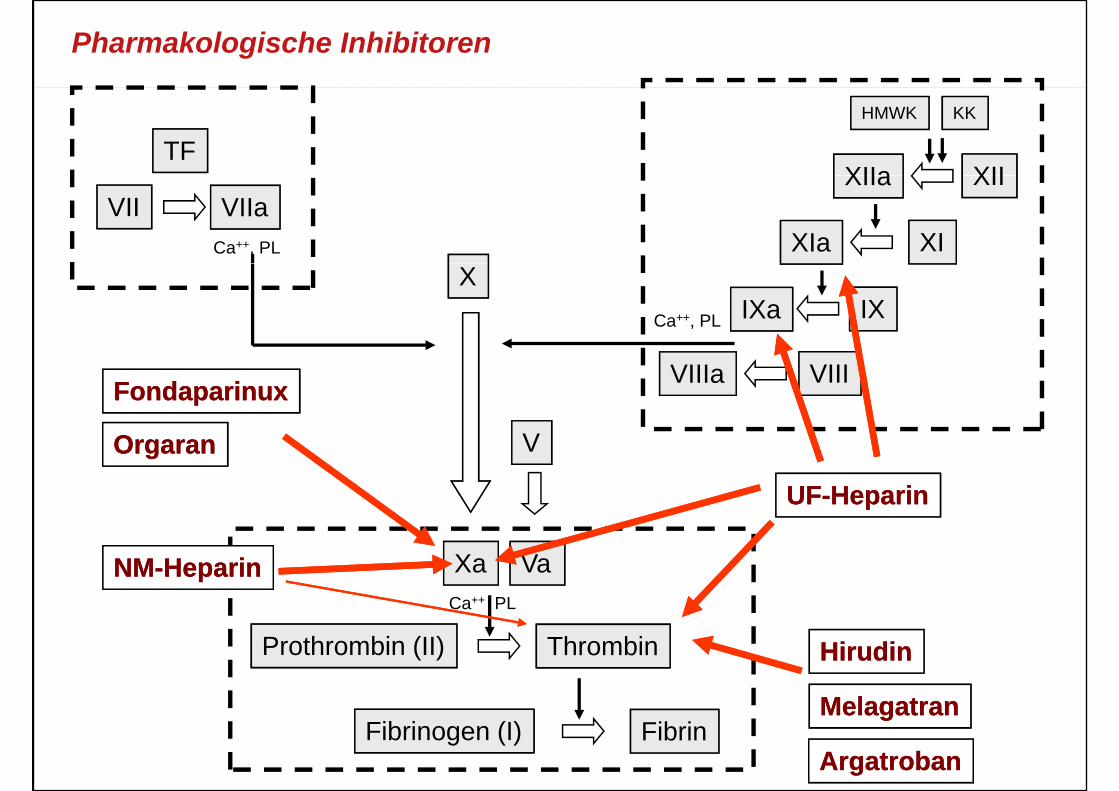
Pharmakologische Inhibitoren
TFXIIXIIa
HMWK KK
VII VIIaXIXIa
XIIXIIa
Ca++, PL
XIXa IXCa++, PL
VIIIa VIII
V
FondaparinuxFondaparinux
OO V
UFUF--HeparinHeparin
OrgaranOrgaran
Xa VaCa++ PL
NMNM--HeparinHeparin
Prothrombin (II) Thrombin HirudinHirudin
MelagatranMelagatranFibrinogen (I) Fibrin
MelagatranMelagatran
ArgatrobanArgatroban
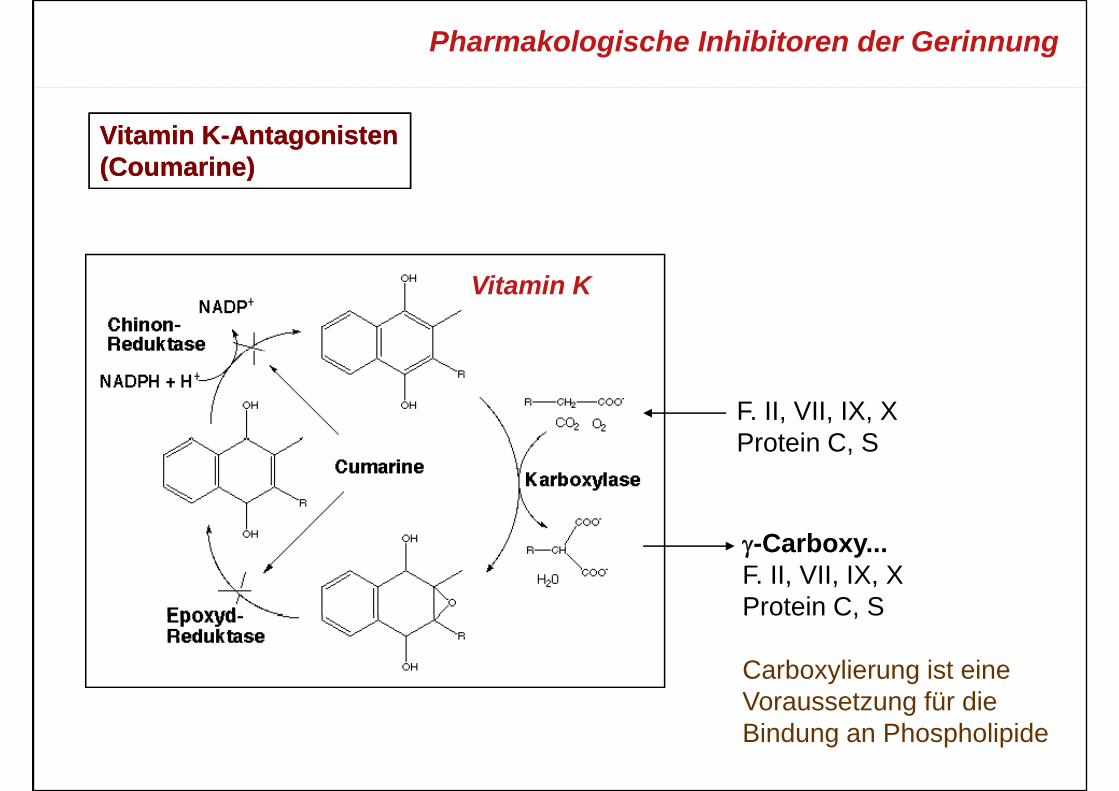
Pharmakologische Inhibitoren der Gerinnung
Vitamin KVitamin K--AntagonistenAntagonisten(Coumarine)(Coumarine)(Coumarine)(Coumarine)
Vitamin K
F. II, VII, IX, XProtein C SProtein C, S
γ-Carboxy...F. II, VII, IX, XProtein C, SProtein C, S
Carboxylierung ist eineVoraussetzung für dieVoraussetzung für dieBindung an Phospholipide

Vitamin K-abhängige Faktoren
TFXIIXIIa
HMWK KK
VII VIIaXIXIa
XIIXIIa
Ca++, PL
XExtrinsicExtrinsic IXa IXCa++, PL
VIIIa VIII
VVitamin KVitamin K--AntagonistenAntagonisten(Coumarine)(Coumarine)
CommonCommon
IntrinsicIntrinsicV(Coumarine)(Coumarine)
Xa VaCa++ PL
PSPC
Prothrombin (II) Thrombin
Fibrinogen (I) Fibrin

Heparin
Unfraktioniertes Heparin (UFH)Unfraktioniertes Heparin (UFH)
• Wirkung:• Bindung an Antithrombin, Affinität zu Gerinnungsfaktoren ↑g , g• Irreversible Bindung und Inaktivierung von F. IIa, Xa, IXa, XIa
• Monitoring: APTT
• Dosierung:• Prophylaxe
• 100-200 E/kg*d i.v. kontinuierlich• 5000-7500 E s.c. alle 8 bis 12 Std.
• Therapie akuter thromboembolischer Ereignisse („Vollheparinisierung“)80 E/k i l B l (5000 bi 10 000 E b l t)• 80 E/kg i.v. als Bolus (5000 bis 10.000 E absolut)
• 18 E/kg*h i.v. kontinuierlich• APTT-gesteuert (Ziel 2,5fache Verlängerung)
• Pharmakokinetik:• Schnelle Elimination unabhängig von Nierenfunktion• HWZ dosisabhängig ca 1 2 Std• HWZ dosisabhängig ca. 1-2 Std.

Heparin
Niedermolekulares Heparin (NMH)Niedermolekulares Heparin (NMH)
• Wirkung:• Bindung an Antithrombin Affinität zu Gerinnungsfaktoren ↑• Bindung an Antithrombin, Affinität zu Gerinnungsfaktoren ↑• Irreversible Bindung und Inaktivierung von F. Xa und (geringer) IIa
• Monitoring: Anti-XaMonitoring: Anti Xa• Monitoring indiziert bei KG <50 kg oder >100 kg, Niereninsuffizienz,
Schwangerschaft• Zielspiegel Prophylaxe: 0,2-0,4 E/mlZielspiegel Prophylaxe: 0,2 0,4 E/ml• Zielspiegel Therapie: 0,4-0,8 E/ml
• Dosierung:g• siehe Fachinfo (präparateabhängig)• z. B. therapeutische Dosierung von Enoxaparin: 100 E/kg 2x tgl. s.c.• z. B. therapeutische Dosierung von Tinzaparin: 175 E/kg 1x tgl. s.c.
• Pharmakokinetik:• Elimination unabhängig von Nierenfunktion• HWZ ca. 2-4 Std.

Anti-Xa
Anwendung Zielbereich Messzeitpunkt
• NM-Heparin• UF-Heparin• Danaparoid
• Prophylaxe: 0,2-0,4 E/ml• Therapie: 0,4-0,8 E/ml
• Kinetik des Medikaments!• Bei NMH 2-4 h nach
subkutaner Gabe• Fondaparinux
NMH Substrat
Patienten-plasma
1:40
XX
P-Nitroanilin
XaXa

Heparininduzierte Thrombozytopenie
Die wichtigste weil lebensbedrohliche Nebenwirkung der Heparintherapie!Die wichtigste, weil lebensbedrohliche Nebenwirkung der Heparintherapie!
Heparin
ThrPF4 Thr Thr
Neoantigen Antikörperbildungg p g
Thrombozyten Aktivierung
Th
Thrombozyten-AktivierungThrombozyten-Aggregation
THROMBOSETHROMBOSEThr Thrombozytopenie

Heparininduzierte Thrombozytopenie
Die wichtigste, weil lebensbedrohliche Nebenwirkung der Heparintherapie!
• Häufigkeit
Thr
Häufigkeit• 0,1 bis 3 % der behandelten Patienten• UFH >> LMWH• Chirurgische Pat. > internistische Pat.Chirurgische Pat. internistische Pat.
• Zeitfenster• Typischer Verlauf: 5-10 d nach Beginn der Heparintherapieyp g p p• früher („rapid onset“) oder später („delayed onset“) in 30 %
• Dran denken bei...• Jeder neuen oder progredienten Thrombose unter Heparin• Jedem Thrombozytenabfall >50 % unter Heparin
• Therapie• Heparin sofort absetzen• Thrombozytentransfusionen kontraindiziert
Alt ti A tik l ti ( B O A t b Hi di )• Alternative Antikoagulation (z. B. Orgaran, Argatroban, Hirudin)

Hirudin
Lepirudin, Desirudin, Bivalirudin („Hirudis medicinalis“ = medizinischer Blutegel)Lepirudin, Desirudin, Bivalirudin („Hirudis medicinalis medizinischer Blutegel)
• Wirkung:• Rekombinant hergestelltes Polypeptidg yp p• Direkter (d.h. antithrombinunabhängiger) Thrombininhibitor
• Monitoring:• APTT (cave: schlechte Dynamik im oberen Dosisbereich)• Ecarinzeit (Ecarin thrombinähnliches Enzym)
• Dosierung:• Therapeutische Antikoagulation (Lepirudin)
• 400 µg/kg als Bolus i.v., dann150 /k *h i l k ti i li h I f i• 150 µg/kg*h i.v. als kontinuierliche Infusion
• Prophylaxe (Desirudin)• 15 mg abs. s.c. alle 12 Std.
• Pharmakokinetik:• Elimination abhängig von Nierenfunktion• HWZ ca 1 Std (Lepirudin i v ) drastisch verlängert bei Niereninsuffizienz• HWZ ca. 1 Std. (Lepirudin i.v.), drastisch verlängert bei Niereninsuffizienz

Weitere Antikoagulanzien
Bezeichnung Route Target Monitoring Anwendung*
Danaparoid Parenteral F.Xa, IXa Anti-Xa Prophylaxe, HIT
Fondaparinux Parenteral F.Xa Anti-Xa Prophylaxe (Orthopädie)
Rivaroxaban Enteral F.Xa
Argatroban Parenteral F.IIa APTT HIT
rTFPI Parenteral FVIIa
NAPc2 Parenteral FVIIa
* Derzeit zugelassene Anwendung Derzeit zugelassene Anwendung