genetic aspects of renal tubular transport: diversity and
TRANSCRIPT

Kidney International, Vol. 9 (1976) p. 149—1 71
Genetic aspects of renal tubular transport: Diversity andtopology of carriers
CHARLES R. SCRIVER, RUSSELL W. CHESNEY and RODERICK R. MCINNES
Medical Research Council Group in Genetics, deBelle Laboratory for Biochemical Genetics, McGill University-MontrealChildren's Hospital Research Institute, Montreal, Quebec, Canada
Mutations which cause the inborn errors of mem-brane transport can provide information about thenormal topology' of renal transepithelial transport.In recent years various reviews of tubular transport[1—6] have appeared which discussed the interrelationbetween disease and net tubular reabsorption of or-ganic solutes.2 Their emphasis was primarily on thefunctions which served solute transport and less onthe diseases associated with the disturbance of trans-port. In this review we have taken the opportunity todescribe, and to speculate on, the probable site in thetubular cell of the defect in transcellular movement ofthe solute in a number of inborn errors of tubulartransport. We hope that the speculations will stimu-late debate, formulation of hypotheses and furtherexperimental evaluation to advance our knowledge.The table included in this paper provides a catalogueof the currently accepted inborn errors of tubulartransport. It is these clinical "windows" which,through the expression of mutation, have revealedand helped to delineate an impressive array of spe-cific transport functions in tubular membrane.
Cellular uptake and transcellular transport
The epithelial cell of the renal tubule has luminaland basilar poles, with definite orientations to thefluids in contact with them; its luminal pole faces anultrafiltrate of plasma where solute is topologicallyoutside the body while the basilar pole is in contactwith peritubular interstitial fluid where solute is insidethe body. Cells of the proximal tubule, in both the
Topology—anatomical definition: the structure of a particularregion or part of the body (Webster).The term "solute" in this paper refers to organic solutes exceptwhen clearly indicated otherwise: for example, in the section onphosphate reabsorption.
© 1976, by the International Society of Nephrology.Published by Springer-Verlag New York
149
convoluted and pars recta segments where a majorfraction of electrolyte and organic solute transportoccurs, have a greatly enlarged membranous surfaceformed of closely packed microvilli: the latter in-crease the absorptive surface about forty-fold [7, 8].Microvilli are associated with an unstirred layer atthe external liquid-solid interface, which is the majorrate-limiting step in trans brush-border permeation atlow concentrations of many solutes [9]. The unstirredlayer also serves as a locale in which enzymes may actupon substrate, e.g., disaccharides, before transport[10]. Tubular epithelium is also a continuous sheet byvirtue of punctuate contacts, or tight junctions, be-tween cells at their luminal poles [11]. These junc-tions are probably impermeable to organic solutesunder normal conditions. Therefore, the solute mustenter epithelial cells to reach the peritubular spaceduring reclamative transport and to reach the lumenduring secretory transport. To achieve this vectorialprocess, an asymmetry of net solute flux is requiredduring reclamative and secretory transports. Howmutation informs us about the functional organ-ization of two sets of membranes (luminal or brushborder, and basolateral) and three pools (luminal,cellular and peritubular) ordered in series, and howthey achieve these net transtubular fluxes, is offundamental interest to us in this brief review.
Cellular uptake ofsolute. The current view of bio-logical membranes emphasizes a fluid-mosaic, struc-tural model, in which the proteins form a mosaic inthe bilaminar bed of lipid [12]. In the absence ofdiffusion channels, permeation of the plasma mem-brane by hydrophyllic solutes must be facilitated bycarriers [13]. Certain types of membrane protein ap-parently serve as carriers [14], thus providing a per-tinent focus for the effect of mutation on the trans-port process. Various chemical and kinetic probes[13] reveal the specificity of carriers and their abilityto recognize solutes. Since the transmembrane move-

150 Scriver el a!
ment of many solutes across the basolateral andbrush border membranes of tubular epithelium, bothin vivo and in vitro, can occur against a chemicalgradient in all parts of the nephron [5, 6], there mustalso be an investment of energy in transport in theform of a conjugate-driving force [15].
Assuming that the solute remains osmotically ac-tive upon entering the intracellular pool, and there isno evidence yet to the contrary [16], it follows thatthe plasma membrane plays a critical role in the formof a barrier to exodus after concentrative uptake. Amechanism for attainment of dynamic asymmetry inthe carrier as it functions on opposite sides of themembrane is required. A simple kinetic description ofthis event is possible [6] (Fig. I). The Michaelis equa-tion can be used to describe carrier-mediated andopposing fluxes2 of solute across the membranewhere entry (or influx) and exodus (or efflux) aredescribed by separate equations:
and
Vmax jflf [S]0lIlt
Km br + [S]0
Vmax elf [S]1Ueff
Km elf + [S]1
[S]0 and [S]1 are the concentrations of solute on theoutside and inside of the membrane, respectively.When uptake is concentrative, influx exceeds effluxuntil the steady state is achieved at which time influx
Fig. I. Diagram showing the apparent kinetics of steady-state entry(influx) and exodus (efflux) under conditions of active transport(accumulation against a chemical gradient) of L-proline across theplasma membrane of a cell. (See text for appropriate Michaelisequations and description of terms.) Where the rate of solute entryis not higher than the rate of its removal by metabolism, the lattercomes to influence transport in the transtubular orientation. Di-minution of metabolic "runout" may cause the internal soluteconcentration to rise (i.e., change from [S]. to [S]lb); when thischange occurs in relation to the luminal membrane, exodus (back-flux) will increase and egressed solute will be removed by theflowing column of urine. These considerations may be relevant tothe interpretation of solute loss in some hereditary disorders oftubular transport and metabolism and to the concept of maximalrates of tubular absorption (Tm). (From Scriver, Mclnnes andMohyuddin [97].)
equals efflux. In the latter state, [S]1 will be >> [SJ0; itfollows from equations 1 and 2 that Km err must thenbe >> Km ml The maintenance of this relationshipwhich is essential to concentrative uptake appears tobe a genetically determined property of the carrier.
Efflux across the luminal membrane occurs duringnet absorption of amino acids and glucose by theproximal tubule [17—19]; this backflux is quite inkeeping with the normal kinetics of efflux depicted inFig. 1. However, backflux is necessarily small if con-centrative uptake is an initial step during net trans-epithelial reclamation of solute, as is now known tobe the case for the transport of some amino acids andfor glucose [17—19].
The ability to generate asymmetry in transcellularmovement of solute, so that a vectorial flux occurs,has long been a subject of interest [20]. Transportacross epithelium has many features which are analo-gous to transport across the plasma membrane;but transepithelial transport also has unique char-
(I) acteristics which distinguish it from simple up-take into cells. In tubular epithelium the now well-recognized substrate specificity of the transport proc-ess, presumably invested in the transport proteins or
(2) reactive sites of the plasma membrane [13], and urn-ned by the appropriate kinetic, chemical and geneticprobes [1—6], is not necessarily identical at oppositepoles of the transporting cell. Uptake into the in-tracellular pool, as defined by equations I and 2, canpertain to both luminal and basilar and lateral mem-branes. However, if an identical net flux exists at bothsets of membranes, net transcellular flux, from urineto peritubular space, or the reverse would not occur.For example, unless the unidirectional flux of soluteoutward at the basilar pole of the cell is greater thanthe unidirectional flux outward at the luminal pole,there is nothing to prevent achievement of a trans-cellular steady state so that backflux becomes theequal of entry at the luminal site serving uptake fromglomerular ultrafiltrate, thus making net absorptivetransport impossible. During absorption, a mecha-nism must be found which maintains the interiminstantaneous uptake ratio below the "true" steady-state ratio across the luminal membrane; therefore,an explanation for "run out" from the intracellularspace in the direction of the peritubular space isrequired. Two mechanisms seem plausible. 1) Meta-bolism. If there is a large intracellular pool of thesolute, membrane transport will not be the rate-limit-ing step in its transcellular movement. The rate of itsintracellular utilization will determine its intracellularconcentration and, therefore, the number of mole-cules of the original solute species available for back-flux after uptake. It follows that a disturbance of
l/Ks 1/Ks. [S1b] [Sal [S0] 1/EPro]

\ Blood
o"%
Urine
1
Renal tubular transport 151
intracellular metabolism may perturb tubular ab-sorption through modulation of intracellular pooisize. Currently available techniques which measurenet reabsorption and transepithelial movement are,in essence, "black box" methods which largely fail toaccount for intracellular events acting on solute. 2)Dissimilar characteristics of luminal and basilar mem-branes. Luminal and basilar membranes are dis-similar both in their morphologic and functionalcharacteristics. Differences at the two poles have beendescribed, for example, in electropotential [21], andin the uptake of amino acids [18, 22—25] sugars [19,26, 27] and keto acids [28]. This functionalasymmetry clearly permits metabolically inert aminoacids, which have no "run out" into other metabolicpools, to be reclaimed from urine into blood against achemical gradient [29]. Accordingly, proximal tubulereclamation may be accomplished by a more per-missive efflux at the basilar membrane relative to theluminal pole. Relatively little work has been done toexamine the role of solute removal by peritubularblood flow in relation to urine flow, and the relationthis may have to transtubular transport kinetics.
We should recognize also that solutes such as,amino acids [6, 30, 31] and glucose [19] which aretaken up from peritubular fluid in many portions [16,26, 27] of the nephron evidently experience only min-imal net efflux across the luminal membrane and donot appear in bladder urine in significant amounts.Further evidence for diversification of transport inbrush border and basolateral membranes in variousparts of the nephron is seen in the distal tubule, whichtakes up amino acids, not at all from the lumen andonly slowly from peritubular fluid [6].
Functional classification ofinborn errors of tubular transport
Two membranes and three solute pools, arrangedin series, accommodate transepithelial transport. Inprinciple, the various inborn errors of reclamativetransport can then be assigned to specific disorders ofmembrane function according to Fig. 2. Mutationcan effect tubular reabsorption at the followingstages: JA) uptake activity (entry) at the luminalcarrier; JB) backflux (luminal exodus) permitted onthe carrier; 2) cellular utilization of absorbed solute(pool size) controlled by its metabolic disposal; 3)unidirectional flux from cell to peritubular fluid atbasilar and lateral membranes (peritubular exodus).
Disorders of secretory transport can also be servedby this model with due consideration for the pre-dominant direction of the fluxes.
Hormone-dependent inborn errors of tubular
Fig. 2. A model delineating possible sites for expression of mutantalleles causing hereditary disorders of tubular reabsorption. Appro-priate recognition of the vectorial flux in secretory transport allowsthe mode! to be adapted also to disorders of secretion. Defect IA,mutant site (carrier) blocks entry; defect !B, mutant carrier per-mits excessive exodus (backflux) from internal pool (); defect 2,blocked catabolic mutant state prevents metabolic "runout" toalternate form (0) leading to accumulation of original solute inthe internal pool (A) and exaggerated exodus (backflux) on normalcarrier; defect 3, defect in exodus at basilar (or lateral) membraneleads to intracellular accumulation as in defect 2. All lead toreduction of net transepithelial flux. Not shown are hormone-modulated defects involving hormone binding at specific sites onthe basilar membrane, and transduction of signal by internal mes-senger to influence the primary transport process (e.g., phosphatetransport modulation by parathyroid hormone or calcitonin, andwater transport modulation by vasopressin).
transport require consideration of two additionalsteps: 1) binding of hormone to the appropriateplasma membrane and 2) intracellular translation ofthe hormonal signal. We have selected examplesfrom Table 1 to describe defective mechanisms atdifferent stages of the transepithelial transport proc-ess.IA. Disorders of solute uptake of the luminal mem-brane. The casual student of biological transport islikely to assume that the hereditary disorders of tubu-lar transport express themselves at the luminal mem-brane. We know from studies of electrolyte transportthat other peritubular membranes play vital roles insolute migration across the tubule; no direct evidencefor a luminal membrane defect has been obtained inany disease listed in Table 1; only indirect evidence,and only in certain disorders of net reclamation, is athand. The examples we have chosen will highlight anumber of themes including the diversity of mem-brane sites used by even a single solute during tubulartransport, and the great specificity among sites ex-posed to a wide mixture of solutes. Consequently,mutation is likely to ablate only a fraction of anygiven transport function. Diversity has adaptive ad-vantage.
The Hartnup trait. Typical homozygotes have aselective impairment in the intestinal and tubular ab-sorption of a particular group of neutral a-amino
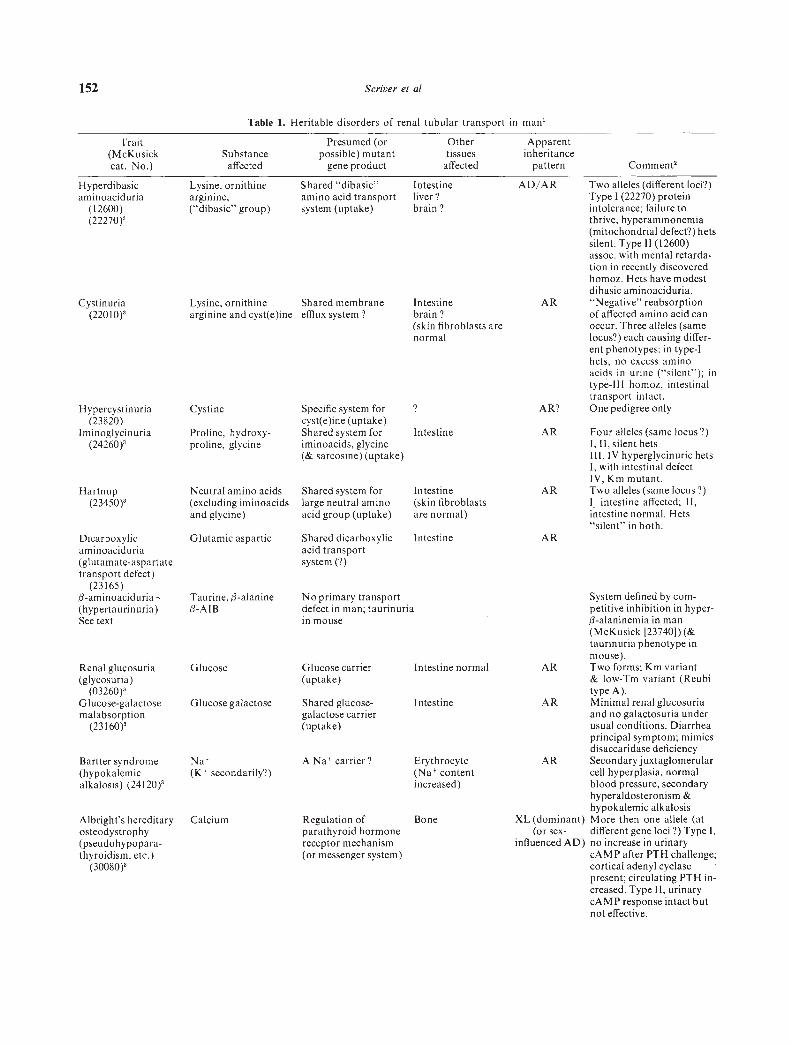
152 Scriver et al
Cystinuria(22010)'
Hypercystinuria(23820)
Iminoglycinuria(24260)'
Dicarboxylicaminoaciduria(glutamate-aspartatetransport defect)
(23165)-aminoaciduria -(hypertaurinuria)See text
Renal glucosuria(glycosuria)
(03260)'Glucose-galactosemalabsorption
(23160)'
Bartter syndrome(hypokalemicalkalosis) (24120)'
Albright's hereditaryosteodystrophy(pseudohypopara-thyroidism, etc.)
(30080)'
Specific system forcyst(e)ine (uptake)Shared system foriminoacids, glycine(& sarcosine) (uptake)
Intestinebrain?(skin fibroblasts arenormal
Shared dicarboxylic Intestineacid transportsystem (?)
Glucose carrier Intestine normal(uptake)
Shared glucose- Intestinegalactose carrier(uptake)
A Na carrier? Erythrocyte(Na contentincreased)
Regulation of Boneparathyroid hormonereceptor mechanism(or messenger system)
AD/AR Two alleles (different loci?)Type 1(22270) proteinintolerance; failure tothrive, hyperammonemia(mitochondrial defect?) betssilent. Type 11(12600)assoc. with mental retarda-tion in recently discoveredhomoz. Hets have modestdibasic aminoaciduria.
AR "Negative" reabsorptionof affected amino acid canoccur. Three alleles (samelocus?) each causing differ-ent phenotypes: in type-Ibets, no excess aminoacids in urine ("silent"); intype-Ill homoz, intestinaltransport intact.
AR? One pedigree only
AR Four alleles (same locus?)I, II, silent hetsIII, IV hyperglycinuric bets1, with intestinal defectIV, Km mutant.
AR Two alleles (same locus?)l intestine affected; II,intestine normal. Hets"silent" in both.
AR
System defined by com-petitive inhibition in hyper-fl-alaninemia in man(McKusick [23740]) (&taurinuria phenotype inmouse).
AR Two forms; Km variant& low-Tm variant (Reubitype A).
AR Minimal renal glucosuriaand no galactosuria underusual conditions. Diarrheaprincipal symptom; mimicsdisaccaridase deficiency
AR Secondary juxtaglomerularcell hyperplasia, normalblood pressure, secondaryhyperaldosteronism &hypokalemic alkalosis
XL (dominant) More then one allele (at(or sex- different gene loci ?) Type I,
influenced AD) no increase in urinarycAMP after PTI-I challenge;cortical adenyl cyclasepresent; circulating PTH in-creased. Type II, urinarycAMP response intact butnot effective.
Hyperdibasicaminoaciduria
(12600)(22270)'
Table 1. Heritable disorders of renal tubular transport in man'
Trait Presumed (or Other Apparent(McKusick Substance possible) mutant tissues inheritancecat. No.) affected gene product affected pattern Comment'
Lysine, ornithinearginine,("dibasic" group)
Shared "dibasic"amino acid transportsystem (uptake)
Intestineliver?brain?
Lysine, ornitbine Shared membrane
arginine and cyst(e)ine efflux system?
Cystine
Hartnup(23450)'
Intestine
Intestine
(skinfibroblasts
arenormal)
Shared system forlarge neutral aminoacid group (uptake)
Proline, hydroxy-proline, glycinc
Neutral amino acids(excluding iminoacidsand glycine)
Glutamic aspartic
Taurine, (3-alanine3-AIB
Glucose
Glucose galactose
Na(K secondarily?)
Calcium
No primary transportdefect in man; taurinuriain mouse
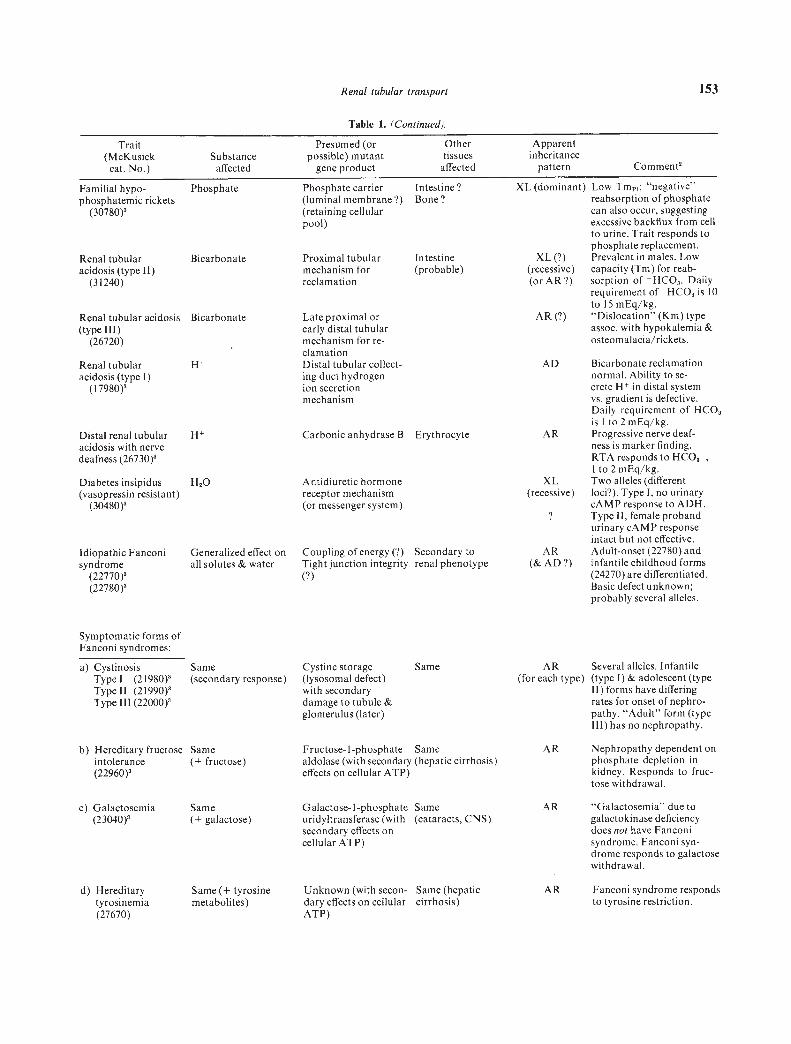
Renal tubular transport 153
Late proximal orearly distal tubularmechanism for re-clamationDistal tubular collect-ing duct hydrogenion secretionmechanism
Distal renal tubular H Carbonic anhydrase B Erythrocyteacidosis with nervedeafness (26730)2
Idiopathic Fanconisyndrome
(22770)(22780)
Symptomatic forms ofFanconi syndromes:
a) CystinosisType I (2l980)Type II (2l990)Type Ill (22000)
b) Hereditary fructoseintolerance(22960)
XL (dominant) Low Tm1; "negative"reabsorption of phosphatecan also occur, suggestingexcessive backflux from cellto urine. Trait responds tophosphate replacement.
XL (?) Prevalent in males. Low(recessive) capacity (Tm) for reab-(or AR?) sorption of HCO3. Daily
requirement of - HCO3 is 10to l5mEq/kg.
AR (?) "Dislocation" (Km) typeassoc. with hypokalemia &osteomalacia/rickets.
AD Bicarbonate reclamationnormal. Ability to se-crete H in distal systemvs. gradient is defective.Daily requirement of HCO3is Ito 2 mEq/kg.
AR Progressive nerve deaf-ness is marker finding.RTA responds to HCO3-,Ito 2 mEq/kg.
XL Two alleles (different(recessive) loci?). Type 1, no urinary
cAMP response to ADH.? Type II, female proband
urinary cAMP responseintact but not effective.
AR Adult-onset (22780) and(& AD?) infantile childhood forms
(24270) are differentiated.Basic defect unknown;probably several alleles.
AR Several alleles. Infantile(for each type) (type 1) & adolescent (type
11) forms have differingrates for onset of nephro-pathy. "Adult" form (typeIll) has no nephropathy.
AR Nephropathy dependent onphosphate depletion inkidney. Responds to fruc-tose withdrawal.
c) Galactosemia(23040)2
Same(+ galactose)
Galactose- I-phosphate Sameuridyltransferase (with (cataracts, CNS)secondary effects oncellular ATP)
AR "Galactosemia" due togalactokinase deficiencydoes not have Fanconisyndrome. Fanconi syn-drome responds to galactosewithdrawal.
d) Hereditarytyrosinemia(27670)
Same (+ tyrosinemetabolites)
Unknown (with secon- Same (hepaticdary effects on cellular cirrhosis)ATP)
AR Fanconi syndrome respondsto tyrosine restriction.
Table 1. (Continued).
Trait(McKusick Substance
Presumed (orpossible) mutant
Othertissues
Apparentinheritance
cat. No.) affected gene product affected pattern Comment2
Intestine?Bone?
Phosphate carrier(luminal membrane?)(retaining cellularpool)
Proximal tubularmechanism forreclamation
Familial hypo-phosphatemic rickets
(30780)
Renal tubular Bicarbonateacidosis (type II)
(31240)
Renal tubular acidosis Bicarbonate(type III)
(26720)
Renal tubularacidosis (type I)
(l7980)
Intestine(probable)
Diabetes insipidus(vasopressin resistant)
(30480)
[-120
Generalized effect onall solutes & water
Same(secondary response)
Same(+ fructose)
Antidiuretic hormonereceptor mechanism(or messenger system)
Coupling of energy (?) Secondary toTight junction integrity renal phenotype(?)
Cystine storage Same(lysosomal defect)with secondarydamage to tubule &glomerulus (later)
Fructose-I-phosphate Samealdolase (with secondary (hepatic cirrhosis)effects on cellular ATP)

154 Scriver et a!
Table 1. (Continued).
f) Lowe's oculocerebro Generalized disfunction Unknownrenal syndrome with defective urinary(30900) ammonia production
An oculocerebro-intestinal-renalsyndrome (involvingtissues with highy-glutamyl cycleactivity?)
XL Basic defect still(recessive) unknown. Treatment for
tubular reclamationdefects does not improvemental retardation or thecataracts & hydro-phthalmia.
Vitamin D dependency(pseudodeficiencyrickets) (26470)
Generalized defect. 25-hydroxyvitamin D-(Secondary response) lo-hydroxylase
(Vitamin D hormonesynthesis occurs inkidney mitochon-dna; deficiency af-fects intestinalabsorbtion of cal-cium & initiatesPTH response.)
AR Nephropathy dependent onPTH excess & hypocalcemia(phenocopy occurs in vita-min D deficiency.)
Miscellaneous
a) Glucoglycinuria(13810)
Glucose & glycine Unknown (the twosolutes do not share acommon carrier.
AD Asymptomatic. Normal-Tm(type-B) glucosuria. Possi-bility that this is aheterozygous manifestationof a Fanconi-like tubulopa-thy merits consideration.
b) Ludcr-Sheldonsyndrome (15250)
Generalized aminoacids glucose &phosphate
Unknown AD Same as for previous entry.Symptoms of Fanconisyndrome have occurredin probands.
c) Rowley-Rosenberg Generalizedsyndrome (26850) aminoaciduria
Unknown AR Associated components ofsyndrome; growth, retarda-tion, muscular hypoplasia,pulmonary involvement &right ventricular hyper-trophy.
A catalogue of 28 inherited disorders of tubular transport is provided herein. Each disease included in the table has a proven (see footnote3) or suspected pattern of inheritance and is to be found under its own five-digit catalogue number in the appropriate section (autosomaldominant, 10,000 series; autosomal recessive, 20,000 series; and X-linked, 30,000 series) of McKusick's Catalogue of Mendelian Inheritancein Man [161]; selected literature citations are given with each entry. Vignettes covering the major clinical features and the genetic aspects ofmany of these traits will also be found in the Compendium of Birth Defects, published by the National Foundation-March of Dimes [1621.Numerous probable inborn errors of tubular transport are not included in Table I because their tubular manifestations have yet to beclearly understood. These conditions (and their McKusick catalogue number, if available) include the following: Pyroglutamic aciduria(26613) due to a defect in glutathioñe synthesis and secondary overproduction of the pyroglutamic acid (5-oxo-proline) intermediate of they-glutamyl cycle; idiopathic hypercalcuria (hyperexcretory form) (23810); some patients with Leigh's necrotizing encephalopathy (25600);Familial nephrosis (25630) with a generalized tubulopathy; and Immerslund's syndrome (26110) with unexplained tubular proteinuria.2 hets = heterozygote, homoz = homozygote.
Proven pattern of inheritance.
acids [6, 32]; nonepithelial cells, such as cultured skinflbroblasts, do not have the defect [33]. Intestinaluptake of various dipeptides containing amino acidsaffected by the Hartnup trait is not impaired [34—36],because dipeptides are transported in the gut andkidney, at membrane sites which are independent ofthose used by their constituent free amino acids [37,38]. Following uptake, dipeptides are cleaved by in-
tracellular peptidases; the free amino acids then entermetabolic pools or leave the cell. The normal plasmaresponse curve in the Hartnup trait, following dipep-tide feeding, indicates that cleavage and absorption ofdipeptide-derived amino acids are normal, therefore,efflux of the released free amino acids across thebasilar plasma membrane must be intact, and thedefect in transepithelial absorption must be confined
Trait Presumed (or Other Apparent(McKusick Substance possible) mutant tissues inheritancecat. No.) affected gene product affected pattern Comment2
e) Wilson's disease Same (with proximal Unknown (seondary 1-lepatolenticular Fanconi syndrome responds(27790) and distal RTA) effects on cytochrome
oxidase system?)degeneration to depletion of copper
storage.
AR

Renal tubular transport 155
to a specific uptake carrier serving the large"Hartnup" group of amino acids on the luminalmembrane (viz. Fig. 26—21 in [6]). In the absence ofany comparable studies of dipeptide reabsorption bykidney, we reason by analogy that a similar locationof the defect in proximal tubule epithelium accountsfor the specific Hartnup hyperaminoaciduria.
Renal glucosuria and glucose-galactose ma!-absorption. Two autosomal recessive disorders ofhexose transport [39, 40] reveal the likelihood thatrenal tubular epithelium possesses two (or more) ge-netically distinct mechanisms for glucose transport;this may not be the case in the intestine. Thetransport defect in each trait almost certainly in-volves an uptake system on the luminal membrane.
The characteristics of D-hexose transport in kidneyare complex. Hexose transport mechanisms providesubstrates for the metabolic systems yielding energyfor basal renal work, and also for a component ofrenal transport work itself [41]. Hexoses enter prox-imal tubular cells, in vivo, from luminal and basilarpoles [26, 42]. However, luminal and basilar mem-branes clearly possess differing characteristics forhexose transport. By means of the sudden-injection,multiple-indicator dilution method [26], it has beenshown that there are D-glucose-preferring (G) sites(shared with D-galactose) and D-mannose-prefer-ring (M) sites in the luminal membrane [42].Confirmation of these data, and evidence for Na1-dependent D-glucose transport at the luminal mem-brane, has been obtained by a stop-flow micro-perfusion method [43] and by kinetic analysisof isolated preparations of brush border membranes[44]. It is likely also that G sites of luminal andbasilar membranes are not identical [26, 42]. Further-more, this delineation of hexose transport in the lu-minal membrane of kidney leads one to believe thatits properties are qualitatively different from thosepreviously defined for the luminal membrane of in-testinal epithelium [45].
By means of a technique using isolated, perfusedproximal tubule segments, the characterisitcs for truetranscellular transport of D-glucose have been re-vealed. D-glucose can be transported against a chem-ical gradient out of the tubule lumen. Active trans-port is therefore a property of the luminal membrane.Glucose reclamation occurs predominantly in theconvoluted portion of the proximal tubule but it alsotakes place in the pars recta. The unidirectional fluxesof D-glucose, from cell to lumen, and from cell toperitubular fluid, were dissected from the net trans-epithelial fluxes; outward flux at the basilar pole ex-ceeds exodus at the luminal border by a four-foldmargin. Basilar permeability to D-glucose is appar-
ently carried-mediated. When the maximum rate ofreabsorption (Tm() is reached, the limiting com-ponent is, accordingly, uptake at the luminal mem-brane (or intracellular metabolism), not permeabilityat the basilar membrane.
Tubular reabsorption of D-glucose has long beenknown to observe a Tm in mammalian kidney in vivo[46, 47] (Fig. 3). The observed "splay" in the titrationcurve relating filtered load to the threshold for gluco-suria (FminG), and the reabsorption rate, has evincedmuch argument. Some consider the observation to becompatible with ordinary Michaelis-Menten kineticsfor uptake by a single saturable system; others take itas evidence for anatomical heterogeneity among thenephrons performing the functions of filtration andreabsorption [1]. We believe a resolution of this clas-sical argument among renal physiologists is to befound in the hereditary disorders of glucose trans-port.
Reubi [48] observed two variations upon the nor-mal titration curve in familial renal glucosuria. Thetype-A variation is characterized by a low FminG andlow Tm(;; type-B glucosuria has a low FminG but anormal TmG (Fig. 3). A kinetic interpretation [3, 6,49] of glucose reabsorption ascribes the type-A vari-ant to a reduced number of transport sites, while thetype-B variant reflects reduced affinity of the hexosecarrier for glucose. With this in mind, should a hered-itary disorder of glucose transport involve the in-testine as well as the kidney, or should the type-A andtype-B phenotypes be found in the same pedigree,there would then be little room for the nephron heter-ogeneity hypothesis as an explanation for splay insolute absorption curves.
When in vivo data for hexose transport are com-pared with data obtained by the kidney cortex slicemethod, additional points of interest are found.The slice method, which exposes only the basilarmembranes of proximal tubular epithelium to theincubation medium (see following), reveals activetransport (uptake) of hexoses across these mem-branes [50. 51]; this finding is analogous to theevidence for active transport in vivo [19]. The slicedata also reveal more than one type of D-hexoseuptake [48]; this finding is also in keeping with the invivo observations [26, 42]. Kleinzeller [49] has delin-eated a number of homologies between D-hexoseuptake in vitro by kidney slices and during absorptionin vivo. However, Silverman, Aganon and Chinard[26, 42] have shown with different techniques thatbasilar membranes and luminal membranes are notidentical in their hexose carrier properties. Therefore,the genetic probes of glucose transport in man as-sume great importance since they may inform us

156 Scriver et a!
about the disposition of hexose carriers in the renaltubule in a manner not revealed by any previousstudy.
Glucose-galactose malabsorption is characterized bysevere impairment of hexose transport in the gut andminimal deficit in the renal tubule; familial renalglucosuria is a disorder of renal tubular reclamationof D-glucose without an intestinal defect.
In glucose-galactose malabsorption, an uptake sys-tem, which resembles the G-system in kidney brushborder [26, 42], is deleted in the luminal membrane ofintestinal epithelium of homozygotes [40, 52—54]. Onthe other hand, renal titration studies reveal little [40]or no [54, 55] deviation of TmG from normal inhomozygotes (Fig. 3). Endogenous glucose metabo-lism is normal in the disease [55, 56] and the hexosetransport defect is not expressed in the erythrocytes,as a representative of nonepithelial tissues [57].
In contrast to these findings in glucose-galactosemalabsorption, there is no aberration of glucosetransport in the intestine of homozygotes with fami-lial renal glucosuria [58]. However, the renal titrationdata reveal a very complex picture compared to therenal findings in glucose-galactose malabsorption.(Fig. 3), Studies in three unrelated pedigrees [39, 58]have revealed a "mild" form of homozygous type-Aglucosuria inherited from "silent" heterozygotes(COY pedigree, [58]); "severe" type-A glucosuria in-herited from "mild" type-A glucosuric heterozygotes(Hold pedigree, [39]) and type-B and "severe" type-Aglucosuria in sibs of a pedigree in which there arerelatives with "mild" type-A glucosuria (Hot ped-igree, [58]. A compelling argument for at least threemutant alleles, at a gene locus specifying a renalglucose transport system, is offered by these observa-tions. Of particular interest is the evidence that even
Fig. 3. Glucose titration curves showingmaximum transport (reabsorption) rate(TmG), and venous plasma threshold forglucosuria (Fmin0), in normal subjects andin the hereditary glucosurias. Theoreticaltype-A and type-B glucosuria titrationcurves according to Reubi (1954) areshown in left panel; note that the hypo-thetical curves do not conform to the ac-tual observations in vivo shown in thethree adjacent panels. Actual type-Aglucosuria data (2nd panel from left) wereredrawn from Elsas, Bossy and Rosenberg[391; type-B glucosuria data (3rd panelfrom left) were redrawn from Elsas andRosenberg [49]. The data for renal han-dling of D-glucose in glucose-galactosemalabsorption (right panel) are taken fromElsas et al (0) [40], Beauvais et al ()[54] and Abraham et al (A) [55].
"severe" type-A homozygotes retain about one-thirdthe normal Tm0 (Fig. 3); and that type-B probands(subject 11-4, Ho!, [58]) have a normal threshold forglucosuria (FminG) which is about one-third the nor-mal Tm(.
The combined evidence both from physiologic ob-servations in several mammalian species [19, 26, 42,47], and from the genetic studies in man suggests thefollowing synthesis and hypothesis. Efficient reclama-tion of hexose by the proximal tubule is controlled bythe luminal membrane. Three types of luminal hexosecarriers operate in parallel in this capacity. One is theM carrier [26, 42], a diffusional system of no furtherinterest to us here. The second, which we will call theG1 system, corresponds to the G-system of Silvermanet al [26, 42]; this carrier interacts with glucose andgalactose and is under control of a gene we will callthe "glucose-galatose" carrier (G1) locus. We pro-pose that the integrity of the G1 carrier is unmaskedin one condition—severe type-A glucosuria whichcauses deletion of a second (G2) glucose carrier.Evidence for the third carrier, which we call G2, isrevealed in homozygous glucose-galactose malab-sorption which causes deletion of the G1 carrier and"unmasks" the activity of the G2 carrier.
We estimate that the maximal capacity of the G1carrier is about one-third the total capacity for D-glucose transport in the normal nephron (Fig. 3).From observations in type-A glucosuria, we deducethat the affinity of the G1 system for D-glucOSe is lessthan that of the principle carrier (G2); this fact isrevealed by the displacen:mt of the titration curve forthe residual glucose transport in homozygous type-Aglucosuria (Fig. 3). The G2 carrier appears to be veryspecific for D-glucose and it has a high affinity andhigh capacity for D-glucose transport (Fig. 3).
Normal & variant Type—A Type—B Glucose—gulactost'gbucosuria glucosuria maiabsorption
Hots
)Tm6
P
0Fbomn Fmin6
0 250 500
E
ta Fmin6
Fm in6
I I I I
0 250 500 0 250 500Filtered glucose, nag ;ni'C /1. 73rn 2
0 250 500

Renal tubular transport 157
Mutation at the G1 locus causes glucose-galactosemalabsorption; mutation at the G2 locus causes fami-lial renal glucosuria. The G2 locus may not be ex-pressed in the intestine according to physiologic andgenetic evidence; or if there is an intestinal G2 carrier,it has not been affected by the mutations which per-mit homozygotes with renal glucosuria to survive andto be recognized [58]. More than one mutant allelehas already been described for the proposed G2 locus.One type impairs the capacity of the G2 carrier for D-glucose and causes "mild" or "severe" Reubi type-Aglucosuria; genetic and phenotypic evidence [40, 58]implies that there exist two different type-A alleles inthis respect. The other allele alters the affinity ofthe 02 carrier for substrate and causes type-Bglucosuria.
Hereditary taurinuria. We divert briefly to discussthe first of three animal models of hereditary loss oforganic solute. Such examples are rare [3] and, whenthey occur, the opportunity they provide for in vitroinvestigation is valuable.
We have used the kidney cortex slice method and invivo clearance studies, in parallel, to delineate thelocation of an inherited impairment of taurine rec-lamation [59, 60] which has been observed in themouse. Taurine is the prevalent 3-amino acid inmammalian body fluids and it serves as a marker fora well-documented, 3-amino-acid-preferring trans-port system in the kidney [38, 60—63].
Wedeen and Weiner have shown that kidney slicesdo not necessarily expose proximal tubule luminalmembranes to uptake from the incubation medium[64-66]. In response, some investigators have beenquick to reject the slice method as a useful techniqueto study solute transport in kidney. We have lessreactionary views; we believe the slice method pro-vides an opportunity to study pools-in-series (Fig. 4)which can highlight the topology of transepithelialtransport. Quick-freeze, soluble-label autoradiog-raphy has revealed the distribution of tritium-la-belIed, inert solutes after their uptake by cortex slices[64] (Fig. 4, top half). Inulin is confined to an extra-cellular space in contact witji basilar and lateralmembranes of proximal tubular epithelium; only inthe distal tubule does inulin penetrate the lumen, a-Aminoisobutyric acid (AIB) is concentrated withincells of the convoluted and straight portions of prox-imal tubule, little being found in the lumen; if AIBfluxes into the luminal pool, it is avidly reclaimed invitro, p-Aminohippuric acid (PAH) is accumulatedmaximally in the lumen of straight segment but alsoin the convoluted portion of proximal tubules. Integ-rity of the punctate contacts between epithelial cellsat their luminal pole is essential for these solute distri-
butions to occur in kidney cortex slices. Specific, netflux orientations across the isolated luminal mem-brane of the proximal tubule are also necessary toachieve the independent spatial distribution of AIBand PAH. The findings indicate that the peritubular,cytoplasmic and luminal pools of proximal tubules inslices exist in series, and that net reabsorptive andsecretory fluxes across the luminal membrane remainintact in slices.
The unique topology of slices was put to use in ourstudy of hereditary taurinuria in the mouse. Taurineis an inert metabolite in mouse kidney and is there-fore a useful probe of its transport functions. Weinvestigated three inbred strains of mice availablefrom the Jackson Laboratory, Bar Harbor, Maine:A/J is a normal taurine excretor (taut±) andC57BL/6J and PRO/Re are two homozygous hyper-taurinuric strains (taut-). Urine taurine is ten-foldgreater in taut_ animals, while plasma taurine is com-parable in the three strains. Net tubular reabsorptionof taurine is 96.7 1.3% (mean of the filteredload in A/J, and 83.9 + 0.8% and 78.7 5.0% inC57BL/6J and PRO/Re, respectively. Intracellulartaurine concentration in outer cortex in vivo is similarin the three strains. This important finding, wheninterpreted according to the kinetics described in Fig.1, indicates that backflux from an expanded in-tracellular pool of taurine is not the cause of hyper-taurinuria.
Other in vivo findings were of interest. fl-Alanine isa competitive inhibitor of taurine transport in kidney[38, 60, 63]. -Alanine inhibits taurine reabsorptionin vivo, in both taut+ and taut_ strains, indicating theretention of a residual taurine transport activity inthe nephrons of the latter.
Steady-state uptake of taurine at physiologic con-centrations (about 0.5 mM) by thin, outer-cortexslices is clearly greater in taut_ than in taut+ tissue.The higher uptake ratio by taut slices is not theresult of altered efflux at the basilar membrane slice;efflux from slices is the same in taut+ and tautstrains.
i-Alanine, which shares the taurine transport sys-tem in vitro [38] and in vivo [60—63], is also takenup more avidly by taut slices. /-Alanine which isvigorously oxidized by taut kidney cortex slices [38]is oxidized less by taut_ slices but normally by slicehomogenates with disrupted architecture.
These data suggest a block in concentrative uptakeof taurine at the luminal membrane of proximal tu-bule (Fig. 4). l-Taurine is not reclaimed efficientlyfrom the luminal "lacuna" of slices (the innermost ofthe three poo1s in series) once it has fluxed from cellinto lumen. Retention of solute in this pool leads to

HF
Inulin
Spaces
AIB
Lumen
PAH
158 Scriver et a!
Tau+ Tau
DR
Time Time
A/i C57BLPRO/Re

Renal tubular transport 159
Fig. 4. An interpretation of enhanced taurine uptake as observed inkidney cortex slices obtained from hyperlaurinuric mice (C57BL/6Jand PRO/Re strains designated taut in text), when compared withnormal mice (A/f, designated taut). Upper half. Figure showsdistribution of 3H-inulin, 3H- (or '4C-) a-amino-isobutyrate (AIB)and 3H-p-aminohippuric acid (PAl-I) after incubation of rat cor-tex slices. Inulin does not penetrate the lumen of the proximaltubule, and is confined to extracellular peritubular and glomerularspaces; AIB is concentrated by proximal tubule cells. PAH ismaximally concentrated in the lumen of proximal tubule. A con-cept of "pools-in-series" is implied by these findings (see text).(Rephotfigraphed from Wedeen and Weiner 163].) Lower half:Figure shows the enhanced distribution ratio (DR) during timecourse for taurine uptake by slices. The. interpretation for thisanomaly is shown in the two sketches at the bottom; it utilizes the"pools-in-series" hypothesis. The shaded area indicates the inulinspace; the adjacent clear area is the cytoplasmic pool; the luminalpool is drawn at the apex bounded by luminal membrane and tightjunctions (shown by solid bars). The luminal pool can be enteredfrom the extracellular space only through the cytoplasm. Taurinebecomes trapped in the luminal pool (stippled area) of taut_ slices.Four relevant permeation fluxes are shown: influx across basilarmembrane (J1); efflux into luminal pool (J2); reclamation flux fromluminal pool (J3); and efflux across basilar membrane (J4). J3exceeds J2 under normal conditions so that little amino acid isretained in the luminal pool. Assuming J3 = [Tau1J Xpermeability1,, where Ic is the lumen-to-cell movement, accumula-tion of taurine in luminal pool will occur if luminal membranepermeability1, is decreased, all other events being unchanged. Thechange in J3 in taut_ kidney is presumed to be the result of anhereditary impairment of transport at the urinary surface of thetaurine carrier in the luminal membrane (from Chesney RW, Scri-ver CR, Mohyuddin F, J. Cnn Invest, vol. 57, 1976, in press).
the higher uptake ratio observed in vitro. The in vivodata indicate "sequestration" of taurine in the urinepool, a finding also compatible with a luminal mem-brane transport defect. In vitro and in vivo data arethus concordant. We believe that in vivo and in vitrodata have been used in parallel, in this case, fortopological assignment of a hereditary transport de-fect to a specific membrane surface in the mamma-lian nephron. Appropriate studies—both with quick-freeze, soluble-label autoradiography to discernwhether labelled taurine accumulates in excess in thelumen of taut- slices, and with isolated brush bordermembranes to study taurine binding—will either af-firm or dispute these conclusions.lB. Defects in the integrity of the (luminal) plasmamembrane to efflux. No hereditary disorder of tubulartransport has been proven in this class. However,there is precedent for genetic control of exodus inprokaryocytes [67, 68]; and two disorders in man arereasonable candidates for this type of defect.
Wong, Kashket and Wilson [671 describe a geneticdefect of thiogalactoside transport in Escherichia coli,and Hectman and Scriver [68] found a mutant strainof Pseudomonasfluorescens defective in -alanine ac-cumulation. Both mutants are unable to concentratethe relevant free solute against a gradient in the in-tracellular pool, yet both retain the pertinent carrierin their plasma membranes. It was surmized in boththat the mutant carrier was unable to prevent soluteefflux following uptake into the intracellular pool.With these precedents in mind, it has been proposed[69, 70] that classical cystinuria and X-linked hypo-phosphaturia may be disorders in which the relevant,specific carriers in the luminal membrane are mutantso as to allow abnormal backflux, while still retainingtheir carrier functions for facilitated entry.
Cystinuria is an autosomal recessive disorder char-acterized by defective transport of the diamino dicar-boxylic amino acid cystine, and the diamino mon-ocarboxylic amino acids lysine, ornithine andarginine [71]. Two or more mutant alleles exist at thegene locus controlling the transport function in-volved in cystinuria [72]. Net tubular reabsorption ofthe four amino acids is greatly impaired in mutanthomozygotes and in "genetic compounds". It is par-tially impaired in heterozygotes for two of the threeproposed alleles [72].
While FminLYS and TmLYS may both be zero inhomozygous cystinuria, Lester and Cusworth [73]have shown that lysine infusion will still provokeenhanced excretion of ornithine, arginine and cystine.They, and others [74, 75], have also shown that theendogenous renal clearance of cystine and the otheraffected amino acids can exceed the clearance of in-

160 Scriver et a!
ulin, so that the "negative reabsorption"3 of aminoacids occurs in cystinuria. On the other hand,cyst(e)ine uptake is only slightly depressed, if at all, inslices prepared from human cystinuric kidney [72, 76,77, 78]; the representative dibasic amino acid lysineobserves a reduced rate of uptake without change inits apparent Km for uptake [72, 76, 78]. The carrierretains its ability to interact with the dibasic aminoacids. By contrast with these findings in kidney, theuptake of both cystine and the dibasic amino acids isclearly impaired in the intestine in homozygous cys-tinuria [721.
The original hypothesis of Dent and Rose [79], andof Robson and Rose [80], stated that cystinuria is adefect in a selective transport (uptake) system of thetubule shared by cystine and the dibasic amino acids.However, subsequent work in vitro has shown thatcystine and the dibasic amino acids do not share acommon system for uptake across the basilar plasmamembrane of the tubule as it is exposed in slices ofmammalian kidney [60, 81, 82]. On the other hand,cystine, cysteine and the three dibasic amino acids allinteract with each other at the luminal membrane, inthe normal proximal tubule, presumably on a sharedsite [83]. The discordance between the properties ofuptake sites on luminal and anti-luminal membranesis further ramified when efflux is considered.Schwartzman, Blair and Segal [84, 851 showed thatcysteine and the dibasic amino acids shared a mem-brane efflux site in kidney cortex slices. Theproperties of the efflux system appear not to be dupli-cated completely at the corresponding influx site; nordoes the efflux carrier experience the customary prop-erties of counterfiow. These properties might be ac-comodated by the behavior of carriers on the outersurfaces of luminal and basilar membranes when ex-posed in the slice model (see above, hereditary tauri-nuria discussion) except for the following facts.We know also that a mixed disulfide (cysteine-homocysteine) is prominent in cystinuric urine [86];that the renal arterial:venous extraction ratio is nor-mal for cysteine, and for cystine, in cystinuria [87];and that the intracellular cysteine:cystine ratio in nor-mal human and cystinuric renal cortex is normallyabout 10: 1, regardless of the form in which cyst(e)ine
The term "negative reabsorption" is sometimes used when it isfound that the excretion rate (UV) of a solute exceeds its load inglomerular filtrate (F), whereas the customary relationship is F >uv. The term "net secretion' could also be used in this instanceprovided it was not implied that UV > F was the result of aspecific energy-dependent secretion process. When used in thispaper, negative reabsorption implies an abnormal finding;secretion is reserved for normal functions in which UV > F is thecustomary relationship.
enters the cell [77, 88]. Each of these latterobservations suggests that intracellular cysteine is thesource of excess urinary disulfide in cystinuria.
These disparate observations at first defy coherentinterpretation. Moreover, the genetic evidence (TableI) tells us that mutation, in the form of isolatedhypercystinuria, and as hyperdibasicaminoaciduria,can impair tubular reabsorption of cystine and thedibasic amino acid quite independently, a findingwhich is explicable only if we assume that the luminalmembrane contains reactive sites which arecyst(e)ine-specific and dibasic amino acid-specific,each under the control of separate genes. However, toreiterate, the physiological evidence [83] tells us thata third species of uptake site in the luminalmembrane of the mammalian nephron is shared bythe five naturally occurring amino acids, but that thissite is apparently not present in the basilar membrane[61, 81, 82]. Loss of only the luminal membranesystem [83] (shared by cysteine, cystine, lysine, orni-thine and arginine) would indeed account for thecystinuria trait as we know it. But this simple inter-pretation would not explain the stimultaneous obser-vation of zero FminLYS, zero TmLYS, negative reab-sorption of amino acids and competitive interactionbetween the four amino acids during reabsorption.An alternative and seemingly unifying interpretationis to propose that cystinuria is a defect in a sharedeffiux system of the plasma membrane so that therelevant intracellular amino acids (cysteine, lysine,ornithine and arginine) experience exaggeratedexodus. This interpretation explains most of the invitro data and would account for backflux into urinefrom the intracellular pool to yield negative reabsorp-tion under certain conditions. The relevant aminoacids still interact on a shared luminal membraneuptake system which permits competitive inhibitionto occur under certain conditions.
The second candidate for defective luminal efflux isX-linked hypophosphatemia (familial hypophospha-temic rickets). In this "phosphopenic" form of rick-ets [70] tubular reabsorption of phosphate is selec-tively affected [891; a corresponding defect inintestinal absorption may also exist [901. The vener-able hypothesis of Albright ascribed the defect intubular reabsorption of phosphate in familial hypo-phosphatemia as a consequence of secondary hy-perparathyroidism initiated by a primary disorder ofvitamin D-dependent calcium absorption in the in-testine [911. However, this is implausible as there areno elevated serum concentrations of C-terminal im-munoreactive PTI-I in mutant hemizygotes unless cal-cium homeostasis has been altered [70]. Moreover,the anticipated generalized defect in tubular reab-

Renal tubular transport 161
sorption of solute which accompanies "calciopenic"secondary hyperparathyroidism [70] is not observedin the X-linked trait. Furthermore, normal urinarycyclic-AMP excretion under basal conditions [89, 92]is partial evidence against the defect being a selectivehyperresponsiveness of the tubule to normal levels ofcirculating PTH [93]. Mutant hemizygotes also mani-fest a low Tm1 and negative reabsorption of ortho-phosphate [89, 92, 94]; these two findings focusinterest on a primary transport defect involvingorthophosphate.
The fortunate discovery by Eicher and Southard,at the Jackson Laboratory, of an X-linked mutationwhich causes hypophosphatemic rickets in the mousehas allowed us to investigate, in preliminary fashion,the mechanism of hyperphosphaturia in this pre-sumed model of the human disease. Renal tubularreclamation of orthophosphate is equally diminishedin the mutant mouse and in man. Concentrative up-take of phosphate by cortical and medullary slicesappears to be normal in the mutant hemizygousmouse; and the intracellular concentration of totaland inorganic phosphate also appears to be normal[95].
A hypothesis is proposed to accomodate the find-ings in X-linked hypophosphatemia. We suggest thatthe mutation permits excessive luminal efflux (back-flux) of cytoplasmic phosphate ion to account fornegative reabsorption in the trait. We also suggestthat the equilibrium of phosphate between four poolsin series may determine its net transepithelial flux; theluminal, cytoplasmic, mitochondrial and peritubularspaces comprise the four phosphate pools. Partitionof phosphate in the mitochondrial pool may be foundto be important in the transepithelial movement ofphosphorus, modulating it in a manner analogous tothat proposed for transepithelial movement of cal-cium [96]. The hypothesis requires us to knowwhether concentration of phosphate in the cytoplasmavailable to backflux is dependent to any extent onthe amount in the mitochondrial pool. It may be ofsignificance that mitochondria, particularly of prox-imal tubule cells, are palisaded at the basilar mem-brane [7], forming an interface between peritubularand cytoplasmic pools. Events which diminish or in-crease phosphate activity in mitochondria may cometo influence phosphate activity in cytoplasm in aseries model of transport and may help to explain thevarious effects of calcium and PTH infusions re-ported in X-linked hypophosphatemia [89, 92, 93].
It is implied that a disorder of luminal membranebackflux could be expressed in any portion of neph-ron and could come to influence net reabsorption inboth the proximal and distal tubules. Any test of the
luminal membrane effiux hypothesis, in cystinuriaand X-linked hypophosphatemia for example, shouldconsider this possibility.2. Disorders of intracellular pool size. Interferencewith metabolic disposal ("run out") of solute couldlead to expansion of the intracellular pool in thepresence of continuing uptake (Fig. 2). Conse-quently, the effective concentration of solute whichinteracts with the luminal carrier at the intracel-lular interface could increase; under this condi-tion, backflux into the lumen and the moving columnof urine must increase, all other events being equal, asthe efflux component at the luminal membrane re-sponds to the elevated, internal solute concentration(Fig. 1). In this respect, intracellular metabolism ofsolute comes to influence its transtubular migration.One cannot avoid thinking of the possibility, thatunder this circumstance the Tm of a solute may beinfluenced by its renal metabolism [97, 98].
Renal tubular reabsorption of amino acids hasbeen carefully measured in phenylketonuria and sar-cosinemia, two blocked-catabolic mutant states inman, to determine whether impaired catabolism ofthe solute impedes its reabsorption [99—102]. Inphenylketonuria the hepatic conversion of phenylala-nine to tyrosine is almost completely blocked [103].Phenylalanine hydroxylase activity, with about one-fifth the specific activity of the hepatic enzyme, isfound in human renal cortex [104]. However, it is notknown whether the renal enzyme is an isozyme orwhether it is deficient in phenylketonuria. It is knownfrom measurement of phenylalanine reclamation thatthere is no abnormality of tubular reabsorption ofthis amino acid in phenylketonuria [99, 100, 101].
A similar observation has been made for sarcosinereabsorption in sarcosinemia [102]. It is likely thatless than 10% of total body sarcosine oxidation takesplace in mammalian kidney [102, 105]; a severe lossof sarcosine oxidation in one of our patients was notaccompanied by impaired renal reabsorption of thisamino acid [102], indicating that renal oxidation ofthis amino acid is unimportant in its reabsorption.Accumulation of sarcosine in the mutant state ac-tually enhances glycine reabsorption [102] eventhough sarcosine and glycine interact competitivelyon their shared uptake sites [6, 102]. Counterfiowbetween the raised intracellular sarcosine and urinaryglycine on the luminal carrier appears to be a satisfac-tory explanation for the behavior of glycine reabsorp-tion in sarcosinemia. The latter finding also suggestsa normal backflux exchange activity at the luminalmembrane in sarcosinuric kidney and no loss of car-rier integrity, It follows from the examples of phenyl-ketonuria and sarcosinemia that an initial increase in

162 Scriver et a!
the amount of an amino acid in its peritubular andluminal pools does not come to influence the capacityfor its net tubular reabsorption, beyond the normalkinetics of concentration-dependent uptake [13] atthe plasma membrane.
Another animal model (the third and last to bementioned in this review) has provided a valuableopportunity to examine directly the effect on nettubular absorption of an initial increase inintracellular pool size [97]. The mutant homozygousPRO/Re mouse has hyperprolinemia and less than 1%of the normal proline oxidase activity in kidney [106,107]. Under normal conditions, proline is reclaimedavidly from urine by the mammalian nephron [61,108, 109]; the kidney also takes up proline from pen-tubular plasma in vivo [110]. In the PRO/Re mouse,the endogenous proline concentration is eight timesnormal in plasma and four times normal in kidneycortex; by contrast it is fifty times normal in the urine[97]. The in vivo, steady-state uptake of proline acrossthe basilar plasma membrane is not impaired andefflux at this surface is normal; the integrity of thewell-documented proline transport systems in thesemembranes [Ill, 112] is also retained in PRO/Rekidney. Appropriate studies also reveal the luminalmembrane uptake of proline to be intact in thePRO/Re mouse [97].
Proline oxidation in normal mouse kidney is ofsuch large capacity that the intracellular proline poolis kept at a low level [97]; consequently, the normalmetabolic outflow comes to influence proline uptakerather dramatically. Normal slices do not observeany expansion of the soluble proline pool until theexternal substrate concentration is greatly elevated;and saturation of tubular reabsorption in vivo (Tmpro)is not observed in the normal mouse until the filteredproline load is augmented far beyond the limit whichis required to delineate the Tmpr in man.
The extraordinary hyperprolinuria which charac-terizes the PRO/Re mouse can be understood onlyby taking into account the elevated intracellular con-centration of free proline [97]. When we apply Mi-chaelis kinetics (Fig. 1), we see excessive prolinuria inthe PRO/Re phenotype as a simple consequence ofthe primary elevation in the concentration of in-tracellular proline in vivo (depicted as Sib in Fig. 1).Efflux of proline is thus enhanced on the same normalcarrier that moves it normally into the cell either fromurine, or from peritubular fluid. However, whenexodus occurs across the luminal membrane, prolinewill emerge into a moving column of fluid; since itsdistal tubular reclamation is insignificant [6], the"lost" proline will appear in bladder urine. Fractionalexcretion of proline is accordingly elevated in the
PRO/Re mutant. The in vivo topology of trans-epithelial transport permits this phenomenon to beobserved in PRO/Re kidney whereas the in vitrotopology does not.
With the hindsight afforded by the PRO/Remodel, it is of interest that the venous plasma thresh-old (FminPRO) for prolinunia in some human ho-mozygotes with autosomal recessive hyperprolinemiaappears to be slightly lower than the threshold ob-served in normal persons [108]. That finding suggeststhat renal proline oxidase activity may subtly in-fluence proline reabsorption even in man. It is alsoapparent from these observations that the classicalTm concept will require reevaluation since renalmetabolism of a solute does indeed influence its rateof tubular absorption.
3. Disorders of exodus at the antilurninal pole. Thistype of disorder would impede net transtubular mi-gration of solute (Fig. 3). The result would be en-hanced intracellular accumulation of the substrateleading to exaggerated backflux at the luminal sur-face. Thus far, only experimental models of thismechanism have been reported.
The rapid-injection, multiple-indicator dilutiontechnique has shown [10, 42, 113] that most soluteswhich interact competitively to augment their frac-tional excretion do so at the luminal membrane.However, an equivalent competition between solutesduring unidirectional exodus at the basilar plasmamembrane [84, 85] could also impede netreabsorption. The effect of artificially blocked exoduson fractional excretion has been studied in the ligatedureter preparation in vivo [31, 114]. L-Arginine en-hances renal clearance of L-lysine in the ligated dog[31] in part by causing the intracellular lysine concen-tration to increase through competitive interactionbetween lysine and arginine at the efflux site in thebasilar membranes. Corresponding experiments inthe rat [114] showed that L-lysine provokes cellularaccumulation of S-labelled products derived fromextracellular L-35 S cystine, and increases the renalclearance of L-cystine. These findings illustrate thegeneral theme of this review, that renal uptake andtranstubular migration are independent phenomena[115]; they also support the likelihood that blockedefflux of solute from the basilar pole of the epithelialcell can impede net reabsorption. It remains for in-vestigators to find a disease of tubular function inman which fits this interpretation.
Generalized disorders of tubular transport
The renal Fanconi syndrome [116] of various eti-ologies, and the X-linked, oculo-cerebro-renal syn-

I 50
1.00
0.50
Normal increase
100 200 300
Filtered L —proline. glinoles 113FF! 1/j. 2
+ 200
+ IOU
0
200 300
U
Renal tubular transport 163
drome of Lowe, Terry and MacLachlan [117] are ex-amples of generalized disruptions of tubulartransport activities.
The Fanconi syndrome can be defined as the in-tegrated clinical manifestations, of whatever cause,resulting from excessive urinary loss of three or moreclasses of solutes including the amino acids, mono-saccharides, electrolytes (phosphate, calcium, bi-carbonate, potassium, sodium), uric acid and protein(particularly /3-globulins); water loss also occurs.Substances not normally observed in any quantity inurine may also appear in excess, including lithium,magnesium, insulin and vitamin D and even ly-sozyme [118]. The increase in fractional excretion ofthese solutes is largely due to reduced net tubularreclamation. However, impairment of tubular secre-tion is also observed in the syndrome; for example,TmPAH is depressed [119].
Morphologic abnormalities may accompany theFanconi syndrome. The swan-neck lesion, which in-volves the initial portion of the proximal tubule, is ahallmark [120]. In all likelihood, this lesion is second-ary to the underlying cause of the syndrome. Theatrophy of the absorptive surface in the anatomiallesion further reduces the membrane activity avail-able for transport. Many species of transport sites arenonetheless still active, in the atrophied epithelium,since the normal competitive interactions betweenmolecules, which share reactive sites, are retained inthe Fanconi syndrome [79, 121] (Fig. 5).
A reduced Tm value characterizes the tubulartransport of various solutes in the Fanconi syndrome(Fig. 5). This finding alone could be attributed to asimple reduction in the activity or the number ofcarriers in the membrane (equationl). However,negative reabsorption has also been observed at highplasma solute concentrations in some patients withthe syndrome (Fig. 5) and this finding implies morethan the loss of carrier activity. The work ofBergeron, Vadeboncoeur and Laporte with the ma-leic acid model of the Fanconi syndrome [25, 122,123] is of particular interest in the latter context.Maleic acid is a noncompetitive inhibitor of soluteuptake by kidney in vitro [1251, and it causes theFanconi syndrome in vivo [1241. Within a few hoursof exposure to sodium maleate (400 mg/kg i.p.),there is a profound deterioration in the network ofperimitochondrial membranes [123]; this anatomicalabnormality and the Fanconi syndrome appear andthen abate in parallel after the maleate injection.Following peritubular capillary injection of leucine inthe maleate-treated rat, the amino acid appears intubular urine at a rate indicating direct transtubularflux [25]. There is also a reduction in the cellular
Fig. 5. Upper part: Renal titration curves relating proline reabsorp-tion to filtered load in normal subjects and three patients with tijeidiopathic Fanconi syndrome. The lower normal range for Tmpao1109] is shown by the shaded area. Reabsorption is depressed at allconcentrations of proline in ultrafiltrate in the Fanconi syndrome.In two subjects, excreted proline exceeded the filtered load whenthe plasma proline concentration was raised indicating "negative"reabsorption under these conditions. Lower part: Proline inhibitsglycine competitively on a shared, high-capacity system for uptake inthe normal subject /109, 111/. The upper normal range for frac-tional excretion of glycine in the presence of proline is shown bythe shaded area. Glycine excretion is abnormally increased, even atnormal endogenous levels of proline in the Fanconi syndrome,indicating excessive inhibiton of reclamation, or excessive back-flux, of glycine on its tubular transport systems [109, 111]. Theresponse to proline inhibition is unusual in two subjects and nega-tive reabsorption occurred. The latter finding indicates retention ofmembrane sites at which counter-flow inhibiton of uptake may beoccurring simultaneously. Negative reabsorption of other aminoacids was not observed indicating a selective interaction betweenproline and glycine at a site whose ability to retain solute has beencompromised in the Fanconi syndrome.
concentration of the amino acid while its fractionalexcretion is elevated [122]. Exaggeration of thesefindings to the point of "negative reabsorption" canbe imagined.
"Negative reabsorption" in the human Fanconisyndrome (Fig. 5) and transtubular backflux of solutewith reduced cellular accumulation in the maleic acidmodel in the rat suggest that two fundamental mech-anisms are potentially adrift in the syndrome. The

164 Scriver et al
defect in tubular transport may encompass enhancedexodus at the luminal surface after uptake of solute ateither pole (see' defect lA, Fig. 2). The mechanismmay be a loss of dynamic asymmetry in the carriersecondary to a disorder of energy metabolism and itscoupling to the carriers. The other defect concernsintegrity of punctate contacts between cells; integrityis maintained by a variety of factors including cellularmetabolism.
Hereditary fructose intolerance (HFI) [126] is adisease which illustrates how metabolism can be com-promised in the Fanconi syndrome. The metabolicabnormality induced by fructose in HF! is initiatedby cellular storage of fructose-i-phosphate (F1P) inliver, kidney and bowel [126]. The Fanconi syndromeaccompanies this metabolic event [127] waxing andwaning with exposure to and withdrawal of D-fruc-tose in the diet. Tissues normally assimilate fructoseand vigorously convert it to glucose and lactate.Fructokinase catalyzes the phosphorylation of fruc-tose to F1P; disposal of F1P requires fructose-l-phosphate aldolase which is present in splanchnictissues and muscle, The aldolase (B-isoenzyme) ofsplanchnic tissues has strong cleavage activity towardF1P and strong condensing activity towarddihydroxyacetone phosphate and D-glyceraldehydephosphate [129]. HF! is characterized by deficientaldolase-B activity.
The critical enzymes required for fructose metabo-lism in human kidney are found in the cortex but notin medulla [128]. This finding constitutes strong in-direct evidence that renal cortex metabolizes fructoseby the FIP route, and that storage of FTP wouldoccur in this region of kidney in HF!. The associationof a precise anatomical location of fructosemetabolism in kidney, and provocation of the Fan-coni syndrome by exposure to fructose in HFI, isintriguing. The normal activity of fructokinase per-mits large amount of FiPto accumulate in HF! uponexposure to fructose. This response is accompaniedby a fall in serum inorganic phosphorus while cellularadenosine triphosphate (ATP) is consumed to formthe F1P [129]. This relationship in HF! constitutes a"futile" hydrolysis of ATP, with depletion of high-energy phosphate and inorganic phosphate pools aridit is analogous to the situation in galactosemia inwhich the Fanconi syndrome also occurs [130].
Parathyroid hormone plays an important modu-lating role in the pathogenesis of the Fanconi syn-drome in HFI [131]. Administration of D-fructosemay not provoke the Fanconi syndrome in the ab-sence of the hormone [131]. Parathyroid hormonestimulates renal adenyl cyclase and ATP consump-tion. The "threshold" defect in ATP metabolism,
which appears to exist in HFI, highlights the energyrequirements of mechanisms serving transtubulartransport. From this perspective it is worth recall-ing that solutes of different species which do notshare common carriers in kidney membranes can stillimpede each others' reabsorption [132] or uptake[133] by mechanisms which do not involvecompetitive interaction [133]. Competition for avail-able energy may occur under such circumstances, amechanism which may explain the unusually intenseinhibition of amino acid reabsorption upon exposureto solute loads in the Fanconi syndrome (Fig. 5).
Disorders of net tubular secretion
There is no known primary hereditary disorderaffecting the active tubular secretion of organic so-lutes, with the possible exception of some forms ofhyperuricemia. However, one can speculate that or-ganic anions which escape excretion by nonionic dif-fusion [134], and which require conjugated transportsystems for their tubular excretion, are eligible sub-strates for inborn errors of tubular transport. Therecent finding that the proximal tubule contains li-gandin [135], an organic anion-binding protein whichis antigenically similar to the hepatic Y protein, yieldsa candidate for an organic anion carrier in kidney.An inherited deficiency of ligandin would provide asignificant test of its importance in tubular secretionof organic anions. In the meantime, we can questionwhether ontogeny of renal ligandin activity, similarto that of hepatic Y protein [136], is an explanationfor the abnormally low secretion of substances suchas PAH, chloramphenicol and penicillin in the hu-man newborn. An alteration in ligandin-dependentorganic acid excretion might even explain why a por-tion of gouty individuals with normal uric acid pro-duction have reduced net renal urate clearance withresultant hyperuricemia [137]. Finally, if it is an im-portant organic anion carrier, we could understandbetter why hyperuricemia occurs in type-I gly-cogenosis, diabetes mellitus, lactic acidosis, maplesyrup urine disease and fructose-i ,6-diphosphatasedeficiency when there is endogenous accumulation ofthe organic acids peculiar to each of these conditions[133].
Inherited defects of distal tubular acidification arelikely to represent true disorders of net tubular secre-tion. Classical renal tubular acidosis (type I or cRTA)occurs when the normal hydrogen ion gradient be-tween tubular urine and plasma is not maintained.Accordingly, there is an inability to lower the urinepH no matter how marked the systemic acidosis[139]. Three abnormalities of hydrogen ion secre-

Renal tubular transport 165
tion could occur: 1) H secretion may be normal butwith excessive back diffusion, so that the normal Hgradient (800: 1-1000:1) between urine and plasma isdissipated; 2) H secretion may be normal but onlyup to a limited capacity, above which no furtheracidification can occur; 3) H secretion may be de-creased at any urine pH. Recently findings suggestthat the third mechanism is an attractive explanationof cRTA [140]. Long ago, Pitts and Lotspeich [141]postulated that the elevated urinary Pco2 in alkalineurine, which is generally 35 mm Hg higher than inplasma, is the result of distal H ion secretion fol-lowed by delayed, noncatalyzed, dehydration ofHZCO3 to form CO2 and H2. The gradient betweenurine and plasma was assumed to be dependent onimpermeability of the collecting duct to C020. PakPoy and Wrong [142] were the first to observe thatthe normal Pco2 gradient was absent in patients withdistal RTA. This finding was extended by Halperin etal [140] who examined the urinary Pco2 gradientafter loading their patients with bicarbonate to max-imize H secretion. In their opinion [140] the mosttenable explanation for the negligible Pco2 gradientwhich they found is deficient H secretion, withoutproduction of carbonic acid for delayed dehydrationto CO2. Others [143] have provided counter-evidenceto this hypothesis, and suggest that the low urinaryPco2 in cRTA may represent excessive backflux ofH2C03 in alkalosis, and of H during acidosis.
Absence of erythrocyte carbonic anhydrase B hasbeen described in a family with distal RTA and deaf-ness [144]. This observation is intriguing, in view ofthe suggestion of Maren [145] that distal bicarbonatereclamation is entirely dependent on intracellularcarbonic anhydrase activity, which provides H ionsto combine with HC02. Distal tubular carbonic anhy-drase has not yet been shown to be abnormal in thesepatients.
Disorders accompanied by alteredtubule-hormone interaction
Hormones regulate several tubular transport func-tions. Such transport activities will be modified if thetubule is hyperresponsive to normal (or elevated)serum concentrations of the relevant hormone; or ifthe tubule receptor and translation system are unre-sponsive to the normal regulatory action of the hor-mone. Both phenomena can be illustrated by inher-ited disorders which involve parathyroid hormone(PTH). PTH-responsive, membrane-dependent activ-ities appear to be localized to the basilar surface ofouter cortical tubule segments [146].
Autosomal recessive vitamin D dependency
(ARVDD) [70] is a disorder in which the renal syn-thesis of la, 25-dihydroxycholecalciferol, a hormonalform of vitamin D, is apparently defective. A "calcio-penic" form [70] of postnatal rickets develops, ac-companied by secondary hyperparathyroidism; ageneralized defect in proximal tubular transportfollows. All abnormalities disappear when phar-macologic doses of vitamin D, or quasi-physio-logic doses of la-hydroxyvitamin D analogues, aregiven. The disorder of tubular transport in ARVDDis related, in some manner, to the constellation ofvitamin D hormone and calcium depletion and ofPTH excess. Depression of extracellular calcium ion[147] or elevation of cytoplasmic calcium [148] in-creases plasma membrane permeability. Calciotropichormones which can modify cytoplasmic calcium al-ter tubular transport of amino acids and other solutes[149]. Depression of cytoplasmic calcium also in-creases tight-junction permeability [150]. Depressionof cytoplasmic calcium is likely to occur in ARVDD,the result being a change in membrane andtranstubular permeability, so that reclamation of so-lute may be depressed or backflux enhanced. It is alsoapparent from recent work [96] that the excess ofPTH and the depletion of vitamin D hormone, whichcharacterized ARVDD, will combine to deplete mito-chondrial and cellular calcium and further alter cal-cium-dependent cellular permeability.
Pseudohypoparathyroidism of the classical type(PHP, type I) is an example of tubular unrespon-siveness to PTH. In this disease, there is a fail-ure of PTH infusion to augment urinary cyclic 3',5'-adenosine monophosphate (AMP) [151] and frac-tional excretion of phosphate is diminished. Endo-genous serum PTH levels are typically elevated whileserum calcium remains low and phosphorus is ele-vated. Recent studies [152] have shown that the PTHreceptor and adenyl cyclase in renal cortex are appar-ently intact in PHP-type 1; it is the mechanism ofcellular response to PTH and the effect on solutetransport which are defective. On the other hand,cyclic AMP-dependent mechanisms in the tubulesresponsive to hormones other than PTH seem to beintact although this facet of the problem has not beenrigorously examined to our knowledge. It is of inter-est that the renal adenyl cyclases responsive to PTH,calcitonin and vasopressin are preferentially locatedin outer cortex, inner cortex and medulla, respec-tively [146]. The basis for the insensitivity of adenylcyclase to PTH in the intact tubule in PTH-type Iremains obscure.
A variant of PUP known as type II PHP, has beenreported [153, 154]. In this trait, fractional excretionof phosphate remains insensitive to PTH infusion,
166 Scriver et a!
but a brisk increase in urinary cyclic AMP testifies toa responsive renal adenyl cyclase. PHP type II isfurther distinguished by a normal serum concen-tration of inorganic phosphate, a feature whichremains unexplained at present. Many facets of thetubular metabolism and handling of vitamin D, phos-phate and calcium, as well as of cyclic AMPmetabolism regulated by PTH, are pertinent to theinterpretation of PHP, but are beyond the scope ofthis review.
CommentaryIn this review we have used some of the inborn
errors of tubular transport listed in Table 1 to illus-trate, in a specific manner, the various components oftranstubular movement of solute. These experimentsof nature, as well as the steadily increasing body oflaboratory experimentation in man and animals, sup-
- -'
0.02
15 45 75 105 135 165 195Time, mm
7
6
a.54
3
Fig. 6. Renal clearance studies in the rat after bolus injection of 1'C-labelled x-amino-isobutyrate (AIB), an inert synthetic amino acid.Fractional excretion (left ordinate) rises above the 15-mm value(set at zero to accommodate interindividual variation between 0.03and 0.05 at 15 mm), to approach a steady state after 90 mm whilethe plasma concentration of A1B is falling (.—., (right ordinate).3H-inulin clearance remained stable during the experiment. Thereciprocal fall in fractional reabsorption of AIB with time couldreflect saturation of an intrarenal binding process; this explanationis unlikely since reinjection of AIB, or elevation of MB plasmaconcentration, did not alter the phenomenon. AIB equilibrationbetween peritubular fluid and epithelium followed by backflux intothe lumen is a more likely explanation. AIB concentration fell inthe cortex between 30 and 195 mm (from 1284 + 25 cpm/mg of wetwt of prepared slice, mean SD [N = 161 to 953 + 26cpm/mg[9N= 16], whereas the concentration remained unchanged in medulla(from 830 + 218 cpm/mg [N = 8] to 923 274 cpm/mg [N = 8]).When considered with the plasma AIB levels, these figures indicatethat although the distribution ratio of AIB is the same in Cortex andmedulla at 3'/2 hr, it is significantly less in the medulla than thecortex at 30 mm. These tissue changes were not an artefact ofchanging urine or plasma AIB concentration. The findings suggestthat a slowly accumulating tissue poo1 of AIB in the medullaryportion of the nephron is a potential source for enhanced luminalexodus (backflux) and, thus, of the observed increase in fractionalexcretion (Mclnnes RR, Scriber CR, unpublished data, [149]).
port what others have proposed recently [31, 97, 114,115]; that accumulation of solute by renal tubule cellsand its transtubular transport are each essentiallydifferent processes, the former being most evident inthe straight protion, the latter in the convoluted por-tion of proximal tubule [115]. The normal topologyof absorbing epithelium provides the framework forthese concepts which, being of fairly recent origin, arestill likely to be a source of controversy as well as astimulus for further investigation. On the other hand,if they are valid, it should not be surprising thattissues lacking the topological orientations of epithe-hum (e.g., erythrocytes, blood leukocytes and cul-tured skin fibroblasts) fail to show any abnormalityof solute transport wherever the investigation hasbeen performed, as in cystinuria [155], Hartnup dis-ease [33], iminoglycinuria [156], glucose-galactosemalabsorption [57] and X-linked hypophosphatemia[157], for example.
Various elegant experimental methods have pro-vided the important evidence for normal backfluxacross the luminal membrane, transtubular fluxes inboth directions and differential handling of soluteuptake and migration in different regions of the kid-ney (e.g., [19,, 25, 31, 83, 114, 115].) Even thetraditional "black box" clearance method appears tohonor this theme (Fig. 6). As a result of this newawareness, our interpretation of altered Tm values,fractional reabsorbtion rates relative to solute con-centration and other clinical indices of altered nettubular transport in the inborn errors of tubularfunction is likely to undergo refinement in the future.All of this will benefit diagnosis, counselling andtreatment of patients with hereditary disorders oftubular transport.
The expression of mutant alleles which affect thegene products controlling the various stages of trans-tubular transport is also complex [3, 61. The gene lociwhich determine the structure and quantity of therelevant proteins are likely to exhibit one or moremutant alleles per inborn error of transport. Geneticheterogeneity underlies a great many monogenic dis-eases [158] and the hereditary disorders of tubulartransport are no exception. Consequently, it s notsurprizing that, for example, three different mutantalleles, each apparently at the same "cystinuria" genelocus, occur among subjects with cystinuria [72]; andthat four mutant alleles, all apparently at the locusspecifying the shared transport system for iminoacids and glycine, are now required to explain thedifferent forms of familial renal iminoglycinuria [6,109]; and that different alleles at two independentgene loci controlling glucose transport are needed to
0.10
0.08 -
0.06
0.04 -//
//0-i
I I I I I

Renal tubular transport 167
explain glucose-galactose malabsorption and renalglucosuria [39,40J. The discovery of two differentphenotypes in the pseudohypoparathyroidism trait,one with blunted renal cyclic nucleotide formationand one without, again argues for mutant alleleswhich probably occur at different gene loci and whichcontrol different components of the relevant trans-port processes. It follows that recognition of geneticheterogeneity will also help to direct the physiciantoward more precise diagnosis, counselling and treat-ment.
Appropriate dissection of tubular function in manthrough genetic "probes" has revealed an importantgeneral underlying theme. Loss of a specific transportfunction rarely leaves the homozygous proband de-void of all function for the particular transport inquestion [160]. For example, homozygotes with renaliminoglycinuria retain a significant fraction of pro-line reclamation under endogenous conditions [109];and homozygotes with severe type-A glucosuria re-tain one-third of their maximum glucose reabsorptivecapacity in kidney and have lost none in the intestine[39, 58]. These manifestations of "residual" transportin mutants reflect functional redundancy in the mem-brane carriers and in the genes which specify trans-port options for solutes. The normal ontogeny oftubular function in the postnatal period also revealsdiversity among carriers, some being present at birth,others disappearing or appearing on schedule post-natally during renal maturation [112, 161]. Geneticsand ontogeny, in combination with kinetic and chem-ical probes [3, 6], have revealed a "horizontal" diver-sity of membrane sites which allows them to dis-tinguish the chemical identity of solutes; and a"vertical" diversity which allows them to recognize asolute differently when it is present at high or at lowconcentration.
This diversity among solute transport mechanismsthus provides an adaptive function which protects theorganism from the effect of transport mutations[1601; it also provides alternate systems which can beutilized in treatment. Replacement of solute hasbeen accomplished in several of the hereditarytubulopathies. Phosphate replacement is a valuablecomponent of treatment in X-linked hypophospha-ternia and the Fanconi syndrome; bicarbonate re-placement can be achieved in several forms of RTA.To know both the biological basis and thesymptomatic manifestations of the hereditary tubulo-pathies ought to provide present-day students ofMendel, Darwin and Garrod with the means to relaxselection effectively against these intriguing muta-tions in man.
Acknowledgments
This investigation was supported by the MedicalResearch Council of Canada. Dr. Chesney and Dr.Mclnnes held Fellowships from the MRC and theCystic Fibrosis Foundation of Canada, respectively,while at McGill University. Dr. Chesney is presentlywith the Department of Pediatrics, University ofWisconsin School of Medicine, Madison, Wisconsin.Dr. Mclnnes is now with the Genetics Unit, Massa-chusetts General Hospital, Boston, Massachusetts.We acknowledge the role our colleagues MichelBergeron, Andre Borle, FazI Mohyuddin, LeonRosenberg, Stanton Segal and Susan Tenenhousehave played in the development of the ideas ex-pressed in this paper; any errors are our sole respon-sibility, not theirs.
Reprint requests to Dr. Charles R. Scriver, Medical ResearchCouncil Group in Genetics, de Belle Laboratory for BiochemicalGenetics, McGill Unioersity-Montreal Children's Hospital ResearchInstitute, 2300 Tupper Street, Montreal, Quebec, Canada H3HI P3.
References
1. ROSENBERG LE: Hereditary diseases with membrane de-fects. Biological Membranes, edited by DOWBEN RM, Bos-ton, Little Brown and Co., 1969, PP. 255—295
2. YOUNG JA, FREEDMAN BS: Renal tubular transport of aminoacids. C/in (item 17:245—266, 1971
3. SCRIVER CR, HECHTMAN P: Human genetics of membranetransport with emphasis on amino acids. Adv Hum Genet1:21 1—274, 1970
4. MOREL F, DEROUFFIGNAC C: Kidney. Ann Rev Physiol35:17—54, 1973
5. SEGAL S, THIER SO: The renal handling of amino acid, inHandbook of Physiology, edited bYORLOFFJ, BERLINER RW,GELZER SR, Washington, D.C., American PhysiologicalSociety, 1973, pp. 653—676
6. SCRIVER CR, BERGERON M: Amino acid transport in kidney:The use of mutation to dissect membrane and transepithelialtransport, in Heritable Disorders of Amino Acid Metabolism,edited by NYHAN WL, New York, Wiley and Son, 1974, pp.5 15—592
7. SJflSTRAND FS, RHODIN J: The ultrastructure of the proximalconvoluted tubules of the mouse kidney as revealed by highresolution electron microscopy. Exp Cell Res 4:426—456, 1953
8. HEPTINSALL RM: Pathology of the Kidney. Boston, Little,Brown and Co. 1966, p. 75
9. DUGAS MC, RAMASWAMY K, CRANE RK: An analysis of theD-gluCose influx kinetics of in vitro hamster jejenum based onconsiderations of the mass-transfer coefficient. BiochemBiophys Ada 382:576—589, 1975
10. SILVERMAN M: Brush border disaccharidases in dog kidneyand their spatial relationship to glucose transport receptors. JC/in Invest 52:2486—2494, 1973
II. DIAMOND JM: Tight and leaky junctions of epithelia: A per-spective on kisses in the dark. Fed Proc 33:2220—2224, 1974
12. SINGER Si, NIcoLsoN GL: The fluid mosaic model of thestructure of cell membranes. Science 175:720—731, 1972

168 Scriver et a!
13. CHRISTENSEN HN, OXENDER DL, RONQUIST G: BiologicalTransport (2nd ed.) Reading, MA, W. A. Benjamin Inc.,1975
14. PARDEE AB: Membrane transport proteins. Science 162:632—637, 1968
15. SCHULTZ SG: Mechanisms of absorption, in Biological Mem-branes, edited by DOWBEN RM, Boston, Little, Brown andCo., 1969, pp. 59—108
16. UDENFRIEND 5, SALTZMAN-NIRENBERG P, GUROFF G: Astudy of cellular transport with fluorescent amino acidamino-naphthylalamine. Arch Biochem Biophys 116:261—270,1966
17. SILBERNAGL S, DEETJEN P: Glycine reabsorption in rat prox-imal tubules. Pflflgers Arch 323:323—350, 1971
18. CHAN YL, HUANG KC: Microperfusion studies on renaltubular transport of tryptophan derivatives in rats. Am JPhysiol 22 1:575—579, 1971
19. TUNE BM, BURG MB: Glucose transport by proximal renaltubules. Am J Physiol 221:580—585, 1971
20. OXENDER DL, CHRISTENSEN FIN: Transcellular concentra-tion as a consequence of intracellular accumulation. J BiolChem 234:2321—2324, 1959
21. BOULPAEP EL: Ion permeability of the peritubular and lu-minal membrane of the renal tubular cell, in Symposium uberTransport und Funk tion intracellulares Electrolyte, edited byKRUCK F, Munich, Urban und Schwarzenberg, 1967, p. 98
22. WINDHAGER FE, GIEBISCH G: Electrophysiology of the neph-ron. Physiol Rev 45:2 14, 1965
23. CIIINARD FP, DELEA AC: Luminal and anti-luminal trans-port characteristics of certain amino acids, in Proc IVlnter-national Congress of Nephrology, Stockholm, 1969, p. 294
24. BERGERON M, VADEIIONCOEUR M: Antiluminal transport ofL-arginine and L-leUCifle following microinjections in per-itubular capillaries of the rat. Nephron 8:355—366, 1971
25. BERGERON M, VADEBONCOFUR M: Microinjections of i,-leuc-inc into tubules and peritubular capillaries of the rat: 11. Themaleic acid model. Nephron 8:367—374, 1971
26. SILVERMAN M, AGANON MA, CHINARD FP: D-glucose inter-actions with renal tubule cell surfaces. Am J Physiol 218:735—742, 1970
27. KLEINZELLER A, KOLINSKA J, BENES I: Transport of glucoseand galactose in kidney cortex cells. BiochemJ 104:843, 1967
28. COHEN JJ: Specificity of substrate utilization by the dog kid-ney in vivo, in Renal Metabolism and Epidemiology of SomeRenal Diseases, edited by METCOFF J, New York, MaplePress, 1964, pp. 126—146
29. CHRISTENSEN HN, CLIFFORD IA: Excretion of 1-aminocyclo-pentane-carboxylic acid in man and the rat. Biochim BiophysActa 62:160—162, 1962
30. FOULKES EC: Effects of heavy metals on renal aspartatetransport and the nature of solute movement in kidney cortexslices. Biochem Biophys Ada 241:815—822, 1971
31. AUSIELLO DA, SEGAL 5, THIER SO: Cellular accumulation ofL-lysine in rat kidney cortex in vivo. Am J Physiol 222:1473—1478, 1972
32. SCRIVER CR: I-{artnup disease, a genetic modification of in-testinal and renal transport of certain neutral alpha aminoacids. NEnglJMed273:530—532, 1965
33. GROTH U, ROSENBERG L: Transport of dibasic amino acidscystine and tryptophan by cultured human fibroblasts: Ab-sence of a defect in cystinuria and Hartnup disease. J C/inInvest 51:2130—2142, 1972
34. ASATOOR AM, BANDOH JK, LANT AF, MILNE MD, NAVABF: Intestinal absorption of carnosine and its constitutentamino acids in man. Gut 11:250—254, 1970 (a)
35. ASATOOR AM, CHENG B, EDWARDS DG, LANT AF, MAT-THEWS DM, MILNE MD, N AVAil F, RICHARDS AJ: Intestinalabsorption of two dipeptides in Hartnup disease. Gui 11:380—387, 1970
36. NAVAB F, ASATOOR AM: Studies on intestinal absorption ofamino acids and a dipeptide n a case of Hartnup disease. Gut11:373—379, 1970
37. SILK DBA: Peptide absorption in man. Gui 15:494—501, 197438. NUTZENADEI, W, SCRIVER CR: Transport and metabolism of
fi-alanine and fl-alanyl-L-histidine (L-carnosine) by rat in-testinal enterocytes, kidney cortex and striated muscle invitro: Role in nutrition. Am J Physiol, in press.
39. ELSAS Li, BUSSE D, ROSENBERG LE: Autosomal recessiveinheritance of renal glycosuria. Metabolism 20:968—975, 1971
40. ELSAS Li, HILLMAN RE, PATTERSON JH, ROSENBERG LE:Renal and intestinal hexose transport in familial glucose-galactose malahsorption. J C/in Invest 49:576—585, 970
41. GARZA-QUINTERO R, COHEN ii, BRAND PH, YOUNG JK:Steady-state glucose oxidation by dog kidney in vivo: Rela-tion to Na reabsorption. Am J Physiol 228:549—555, 1975
42. SILVERMAN J, AGANON MA, CHINARD FP: Specificity of mon-osaccharide transport in dog kidney. Am J Physiol 2 18:743—750, 1970
43. ULLRICH KJ, RUMRICH G, KLOSS 5: Specificity of sodiumdependence of the active sugar transport in the proximalconvulution of the rat kidney. PflOgers Arch 351:35—48, 1974
44. SACKTOR B, CHESNEY RW, MITCHELL ME, ARONSON PS:The interactions of D-glucose with the renal brush border, inRecent Advances in Renal Physiology and Pharmacology,edited by FANELLI GM, WESSON LG, Baltimore, UniversityPark Press, 1974, pp. 151—166
45. CRANE RK: Hypothesis for mechanism of intestinal activetransport of sugars. Fed Proc 21:891—895, 1962
46. SHANNON IA, FISHER 5: The renal tubular reabsorption ofglucose in the normal dog. Am J Physiol 122:765—776, 1938
47. SMITH HW, GOLDRING W, CHASIS H, RANGES HA, BRADLEYSE: The application of saturation methods to the study ofglomerular and tubular function in the human kidney. J MtSinai Hosp 10:59—108, 1943
48. REUBI F: Physiopathologie et diagnostic du diabete renal.Rev Fr Etud Clin Biol 1:575—585, 1956
49. WOOLF LI, GOODWIN BL, PHELPS CE: Tm-limited renal tubu-lar reabsorption and the genetics of renal glucosuria. J TheorBiol 11:10—21, 1966
50. KLEINZELLER A: The specificity of the active sugar transportin kidney cortex cells. Biochim Biophys Ada 211:264—276,970
51. SEGAL 5, GENEL M, HOLTZAPPLE P REA C: Transport ofalpha-methyl-D-glucoside by human kidney cortex. Meiabo/-ism 22:67—75, 1973
52. MEEUWISSE GW, LINDQUIST B: Glucose-galactose nialab-sorption: A study with biopsy of the small intestinal mucosa.Acta Paediatr Scand 57:273—280, 1968
53. SCHNEIDER AJ, KINTER WB, STIRLING CE: Glucose-galactosemalabsorption: Report of a case with autoradiographic stud-ies of a mucosal biopsy. N Eng/ J Med 274:305—3 12, 1966
54. BEAUVAIS P, VAUDOUR G, DESJEUX JF, LE BALLE JC, GIROTiY, BRISSAUD HE, AYMARD P: La malabsorption con-genitale du glucose-galactose: Un nouveau cas avec étude invitro de l'absorption intestinale et étude du TmG. Arch FrPediatr 28:573—591, 1971
55. ABRAHAM iM, LEVIN B, OBERHOLZER VG, RUSSELL A: Glu-cose-galactose malabsorption. Arch Dis Child 42:592—597,1967
56. MEEUWISSE GW, LINDQUIST B: Glucose-galactose malab-

Renal tubular transport 169
sorption: Studies on the intermediate carbohydrate metabol-ism. Acta Paediatr Scand 59:74—79, 1970
57. MEvuwissE GW: Glucose-galactose malabsorption: A studyof the transfer of glucose across the red cell membrane. ScandiC/in Lab Invest 25:145—149, 1970
58. ELSAS U, ROSENBERG LE: Familial renal glycosuria: A gen-etic reappraisal of hexose transport by kidney and intestine. JC/in Invest 48:1845—1854, 1969
59. HARRIS H, SEARLE AG: Urinary amino acids in mice ofdifferent genotypes. Ann Eugenics (Lond) 17:165—167,1952—53
60. GILBERT JB, Ku Y, ROGERS LL, WILLIAMS RJ: The increasein urinary taurine after intraperitoneal administration ofamino acids to the mouse. J Biol Chem 235:1055—1060, 1960
61. WILsoN OH, SCRIVER CR:. Specificity of transport of neutraland basic amino acids in rat kidney. Am J Physiol 213:185—190, l967
62. GOLDMAN H, SCRIVER CR: A transport system in mammaliankidney with preference for fl-amino compounds. Pediatr Res1:212—213, 1967
63. SCRIVER CR, PUESCHEL S, DAVIES E: HYPER-fl-alaninemiaasociated with fl-aminoaciduria and y-aminobutyricaciduria,somnolence and seizures. N EngI J Med 274:636—643, 1966
64. WEDEEN RP, WEINER B: The distribution ofp-aminohippuricacid in rat kidney slices: 1. Tubular localization. Kidney Int3:205—213, 1973
65. WEDEEN RP, WEINER B: The distribution ofp-aminohippuricacid in rat kidney slices: 11. Depth of uptake. Kidney mt3:214—221, 1973
66. WEDEEN RP: Section freeze-dry autoradiography ofp-amino-hippuric acid and amino acid transport in the mammaliantubule, Chap. 8 in Radionuclides in Nephrology, edited byBLAUFOX MD, FUNCK-BRETANO J, New York, Grune andStratton, 1972, p. 209
67. WONG PTS, KASI-IKET ER, WILSON TH: Energy coupling inthe lactose transport system of Escherichia co/i. Proc Nat!Acad Sci USA 65:63—69, 1970
68. HECHTMAN P, SCRIVER CR: The isolation and properties of afl-alanine permeaseless mutant of Pseudomonas fluorescens.Biochim Biophys Acta 2 19:428—436, 1970
69. SCRIVER CR, WHELAN DT: Cystinuria: Concepts and newobservations, in Inherited Disorders of Sulphur Metabolism:Proc. 8th Symp S.S.1.E.M., edited by CARSON NAJ, RAINEDN, London, Livingstone, 1971, pp. 70—80
70. SCRIVER CR: Rickets and the pathogenesis of impaired tubu-lar transport of phosphate and other solutes. Am J Med57:43—49, 1974
71. THIER SO, SEGAL S: Cystinuria, in The Metabolic Basis ofInherited Disease (ed 3), edited by WYNGAARDEN JB, FRED-RICKSON DS, New York, McGraw-Hill Book Co. Inc., 1972,pp. 1504—1519
72. ROSENBERG LE, DOWNING S, DURANT JU, SEGAL S: Cys-tinuria: Biochemical evidence for three genetically distinctdiseases. J C/in Invest 45:365—371, 1966
73. LESTER FT, CUSWORTH DC: Lysine infusion in cystinuria:Theoretical renal thresholds for lysine. C/in Sci 44:99—111,1973
74. FRIMPTER GW, HORWITH M, FURTH E, FELLOWS RE,THOMPSON DO: Inulin and endogenous amino acid renalclearance in cystinuria: Evidence for tubular secretion. J C/inInvest 41:28 1—288, 1962
75. CRAWI-IALL JC, SCOWEN EF, THOMPSON CJ, WATTS RWE:The renal clearance of amino acids in cystinuria. J C/in Invest46:1l62—1I7l, 1967
76. Fox M, THIER S, ROSENBERG LE, KISER W, SEGAL S: Evi-
dence against a single renal transport defect in cystinuria. NEng/ J Med 270:556—561, 1964
77. SEGAL S, CRAWHALL JC: Transport of cysteine by humankidney cortex in vitro. Biochem Med 1:141—150, 1967
78. ROSENBERG LE, ALBRECHT 1, SEGAL S: Lysine transport inhuman kidney: Evidence for two systems. Science 155:1426—1428, 1967
79, DENT CE, ROSE GA: Amino acid metabolism in cystinuria. QJ Med20:205—219, 1951
80. ROBSON EB, ROSE GA: The effect of intravenous lysine onthe renal clearances of cystine, arginine and ornithine innormal subjects, in patients with cystinuria and Fanconisyndrome and in their relatives. C/in Sci 16:75—93, 1957
81. ROSENBERG LE, DOWNING SJ, SEGAL S: Competitive inhibi-tion of dibasic amino acid transport in rat kidney. J BiolChem 237:2265—2270, 1962
82. SEGAL S, SCHWARTZMAN L, BLAIR A, BERTOLI D: Dibasicamino acid transport in rat-kidney cortex slices. BiochimBiophysActa 135:127—135, 1967
83. SILBERNAGL 5, DEETJEN P: The tubular reabsorption of L-cystine and L-cysteine: A common transport system with L-arginine or not? PflOgers Arch 337:277—284, 1972
84. SCHWARTZMAN U, BLAIR A, SEGAL 5: A common renal trans-
port system for lysine, ornithine, arginine and cysteine. Bio-chem Biophys Res Commun 23:220—226, 1966
85. SCHWARTZMAN U, BLAIR A, SEGAL S: Exchange diffusion ofdibasic amino acids in rat-kidney cortex slices. BiochimBiophysActa 135:120—126, 1967
86. FRIMPTER GW: Cystinuria: Metabolism of the disulfide ofcysteine and homocysteine. iC/in Invest 42:1956—1964, 1963
87. ROSENBERG LE, DURANT JU, HOLLAND JM: Intestinal ab-sorption and renal extraction of cystine and cysteine in cys-tinuria. N Eng/J Med 273:1239—1243, 1965
88. CRAWHALL JC, SEGAL S: Intracellular ratio of cystine andcysteine in various tissues. Biochem J 105:891—898, 1967
89. GLORIEUX F, SCRIvER CR: X-linked hypophosphatemia:Loss of a PTH-sensitive component of phosphate transport.Science 175:997—1000, 1972
90. SHORT EM, BINDER HJ, ROSENBERG LE: Familial hypo-phosphatemic rickets: Defective transport of inorganic phos-phate by intestinal mucosa. Science 179:700—702, 1973
91. WILLIAMS TF, WINTERS RW: Familial (hereditary) vitamin-D-resistant rickets with hypophosphatemia, in The MetabolicBasis of Inherited Disease (3rd ed), edited by STANBURY JB,WYNGAARDEN JB, FREDRICKSON DS, 1972, pp. 1465—1485
92. GLORIEUX FH, SCRIVER CR: Transport metabolism andclinical use of inorganic phosphate in X-linked hypophos-phatemia, in The Clinical Aspects of Metabolic BoneDisease, edited by FRAME B, PARFITT AM, DUNCAN H,Amsterdam, Excerpta Medica, ICS 270, pp. 421—426
93. SHORT E, SEBASTIAN A, SPENCER M, MORRIS RC JR: Hyper-responsiveness of the phosphaturic effect of parathyroid hor-mone in X-linked hypophosphatemic vitamin D-resistantrickets (FUR) (abstract 282). J C/in Invest 53:75a, 1974
94. WILSON DR, YORK SE, JAWORSKI ZF, YENDT ER: Studies inhypophosphatemic vitamin D-refractory osteomalacia inadults: Oral phosphate supplements as an adjunct to therapy.Medicine 44:99—134, 1965
95. GLORIEUX FH, SCRIVER CR, EICHER EM, SOUTHARD JU,TRAVERS R: X-linked hypophosphatemia in Hyp/Y mouse(abstract). Pediatr Res 8:389, 1974
96. BORLE AB: Calcium metabolism at the cellular level. FedProc 32:1944—1950, 1973
97. SCRIVER CR, MCINNES RR, MOHYUDDIN F: Role of epithe-hal architecture and intracellular metabolism in prohine up-

170 Scriver et al
take and transtubular reclamation in PRO/Re mouse kidney.Proc Nat/AcadSci (ISA 72:1431-1435, 1975
98. COHEN JJ, CHESNEY RW, BRAND PH, NEVILLE HF, BLAN-CHARD CF: a-Ketoglutarate metabolism and potassium up-take by dog kidney slices. Am J Physiol 217:161—169, 1969
99. SCRIVER CR: The use of human genetic variation to studymembrane transport of amino acids in kidney. Am J DisChild 117:4—12, 1969
100. BRODEHL J, GELLISSEN K, KAAS WP:. The renal transport ofamino acids in untreated infants with phenylketonuria. AdaPaediatr Scand 59:24 1—248, 1970
101. SIJGITA M, SUGITA K, FURUKAWA T, ABE H: Studies on thetransport mechanism of amino acids in the renal tubules: I.Studies on the mechanism of aminoaciduria from the analyt-ical standpoint of titration curve. Jap CircJ 31:405—413, 1967
102. GLORIEUX FH, SCRIVER CR, DFI,VIN E, MOHYUDDIN F:Transport and metabolism of sarcosine in hypersarcosinemicand normal phenotypes. J C/in Invest 50:23 13—2333, 1971
103. FRIEDMAN PA, FISHER DII, KANG ES, KAUFMAN S: Detection
of hepatic phenylalanine 4-hydroxylase in classical phenyl-ketonuria. Proc Nail Acad Sd USA 70:552—556, 1973
104. AYLING JE, HELFAND GD, PIRs0N WD: Phenylalanine by-droxylase from human kidney. Enzyme 20:6—19, 1975
105. REHBERG ML, GERRITSEN T: Sarcosine metabolism in therat. Arch Biochem Biophys 127:661—665, 1968
106. BLAKE RL, RUSSELL ES: Hyperprolinemia and prolinuria ina new inbred strain of mice, PRO/Re. Science 176:809—8 Il,1972
107. BLAKE RL: Animal model for hyperprolinemia: Deficiency ofmouse proline oxidase activity. Biochem J 129:987—989, 1972
108. SCRIVER CR, EFRON ML, SCHAFER IA: Renal tubular trans-port of proline, hydroxy-proline and glycine in health and infamilial hyperprolinemia. J C/in Invest 43:374—385, 1964
109. SCRIVER CR: Renal tubular transport of proline, hydroxy-proline and glycine: III. Genetic basis for more than onemode of transport in human kidney. J C/in Invest47:823—835, 1968
110. FELIG P, OWEN OE, WAHREN J, CAHILL GF JR: Amino acidmetabolism during prolonged starvation. J C/in Invest48:584—594, 1969
Ill. MO1IYUDDIN F, SCRIVER CR: Amino acid transport in mam-malian kidney: Multiple systems for imino acids and glycinein rat kidney. Am J Physiol 219:1—8, 1970
112. BAERLOCHER K, SCRIVER CR, MOHYUDDIN F: Ontogeny ofiminoglycine transport in mammalian kidney. Proc Nat!AcadSci USA 65:1009—1016, 1970
113. FOULKES EC: Cellular localization of amino acid carriers inrenal tubules. Proc Soc Exp Biol Med 139:1032—1033, 1972
114. GRETH WE, THIER SO, SEGAL 5: Cellular accumulation ofL-cystine in rat kidney cortex in vivo. J C/in Invest 52:454—462,
1973
115. WEDEEN RP, THIER SO: Intrarenal distribution of non-metabolized amino acids in vivo. Am J Physiol 220:507—5 12,
1971
116. LEAF A: The syndrome of osteomalacia, renal glucosuria,amino-aciduria, and increased phosphorus clearance (theFanconi syndrome), in The Metabolic Basis of Inherited Dis-ease (2nd ed), edited by STANBURY JB, WYNGAARDEN JB,FREDRICKSON DS, New York, McGraw-Hill Book Co. Inc.,
1966, pp. 1205—1220
117. ABBASSI V, LOWE CU, CALCAGNO PL: Oculo-cerebro-renalsyndrome: A review. Am J Dis Child 115:145—168, 1968
118. BARRATT TM, CRAWFORD R: Lysozyme excretion as a meas-ure of renal tubular dysfunction in children. C/in Sci 39:457—465, 1970
119. HOUSTON IB, BoiCHis M, EDELMAN CM: Fanconi syndromewith renal dosium wasting and metabolic alkalosis. Am JMed 44:638—646, 1968
120. DARMADY EM, STRANECK F: Microdissection of the nephronin disease. Br Med Bull 13:21—25, 1957
121. SCRIVER CR, Got.DBI,00M RB, ROY CC: Hypophosphatemicrickets with renal hyperglycinuria, renal glucosuria andglycylprolinuria: A syndrome with evidence for renal tubularsecretion of phosphorus. Pediatrics 34:357—371, 1964
122. BERGERON M: Renal amino acid accumulation in maleate-treated rats. Rev Can Biol 30:267—272, 1971
123. LAPORTE P, BERGERON M: Effect membranaire du maleate auniveau du nephron proximal et distal. Rev Can Biol 32:275—279, 1973
124. HARRISON HE, HARRISON HC: Experimental production ofrenal glycosuria, phosphaturia and aminoaciduria by in-jection of maleic acid. Science 120:606—608, 1954
125. ROSENBERG LE, SEGAL 5: Maleic acid-induced inhibition ofamino acid transport in rat kidney. Biochem J 92:345—352,1964
126. FROESCH ER: Essential fructosuria and hereditary fructoseintolerance, in The Metabolic Basis of Inherited Disease(3rd ed), edited by STANBURY JB, WYNGAARDEN JB,FREDRICKSON DS, New York, McGraw-Hill Book Co. Inc.,1972, pp. 131—148
127. MORRIS RC JR: An experimental renal acidification defect inpatients with hereditary fructose intolerance: I. Its resem-blance to renal tubular acidosis. J Clin Invest 47:1389-1398,1968
128. KRANHOLD JF, LOH D, MORRIS RC: Renal fructose-metabo-lizing enzymes: Significance in hereditary fructose intoler-ance. Science 165:402—403, 1969
129. YU DT, BURCH H, PHILLIPS Mi: Pathogenesis of fructosehepatotoxicity. Lab Invest 30:85—92, 1974
130. SEGAL 5: Disorders of galactose metabolism, in The Meta-bolic Basis of Inherited Disease (3rd ed), edited by STANBURYJB, WYNGAARDEN JB, FREDRICKSON DS, New York,McGraw-Hill Book Co. Inc., 1972, pp. 174—195
131. MORRIS RC JR, MCSHERRY E, SEBASTIAN A: Modulation ofexperimental renal dysfunction of hereditary fructose intoler-ance by circulating parathyroid hormone. Proc NatlAcadSciUSA 68:132—135, 1971
132. DRUMMOND KN, MICHAEL AF: Specificity of the inhibitionof tubular phosphate reabsorption by certain amino acids.
Nature 201:1333, 1964
133. GENEL M, REA CF, SEGAI. 5: Transport interaction of sugars
and amino acids in mammalian kidney. Biochim BiophysActa 241:779—788, 1971
134. MILNE ME), SCRIBNER BH, CRAWFORD MA: Non-ionic diffu-
sion and the excretion of weak acids and bases. Am J Med24:709—729, 1958
135. KIRSCH R, FLEISHNER G, KAMISAKA K, ARIAS IM: Structuraland functional studies of ligandin, a major renal organicanion-binding protein. J C/in invest 55:1009—1019, 1975
136. LEVI AJ, GATMAITAN Z, ARIAS IM: Deficiency of hepaticorganic anion-binding protein, impaired organic anion up-take by liver and "physiologic" jaunice in newborn monkeys.N EngI J Med 283:1 136—1139, 1970
137. RIESELBACH RE, SORENSEN LB, SHELP WD, STEELE TH:Diminished renal orate secretion per ncphron as a basis forprimary gout. Ann Intern Med 73:359—366, 1970
138. CHESNEY RW: Comment on hyperuricemia with ketonuriaand ketosis. Pediatrics, in press
139. SELDIN DW, WILSON JD: Renal tubular acidosis, in Meta-bolic Basis of Inherited Disease, edited by STANBURY III,

Renal tubular transport 171
WYNGAARDEN JB, FREDRICKSON DS, New York, McGraw-Hill Book Co. Inc., 1972
140. HALPERIN ML, GOLDSTEIN MB, HAIG A, JOHNSON MD,STEINBAUGH BJ: Studies on the pathogenesis of type I (distal)renal tubular acidosis as revealed by the urinary pCO2 ten-sions. J C/in Invest 53:669—677, 1974
141. PirrS RF, LOTSPEICH WD: Bicarbonnate and the renal regu-lation of acid base balance. Am J Physiol 147:138, 1946
142. PAK Pov RK, WRONG 0: The urinary pCO2 in renal disease.Clin Sci 19:631—643, 1960
143. SEBASTIAN A, MCSHERRY E, MORRIS RC JR: On the mecha-nism of the inappropriately low urinary carbon dioxide ten-sion (1J-pCO.) in classic (type-I) renal tubular acidosis(cRTA). C/in Res 22:544A, 1974
144. SHAPIRA F, BEN-YOSEPI-I Y, EYAL FG, RUSSELL A: Enzymat-ically inactive red cell carbonic anhydrase B in a family withrenal tubular acidosis. J C/in Invest 53:59—63, 1974
145. MAREN TI-I: Chemistry of the renal reabsorption of bicarbo-nate. Can J Physiol Pharmaco/ 52:1041—1050, 1974
146, MARX SJ, FEDAK SA, AURBACI-I GD: Preparation of charac-terization of a hormone-responsive renal plasma membranefraction. J Biol Chem 247:6913—6918, 1972
147. BAKER PK: Sodium-calcium exchange across the nerve cellmembrane, in Calcium and Ce/lu/ar Function, edited byCUTHBERT AW, London, MacMillan and Co., 1970, pp.96— 107
148. BAKER PF: Transport and metabolism and calcium ions innerve, in Progress in Biophysics and Mo/ecu/ar Biology, editedby BUTLER JAy, NOBLE V, New York, Pergamon Press,1972, p. 179
149. MCINNES RR, SCRIVER CR: Mechanism of the amino-aciduria produced by calciotropic hormones and dibutyrylcAMP (abstract). C/in Res 538A, 1974
ISO. ROSE B, LOEWENSTEIN WR: Permeability of cell junctiondepends on local calcium activity. Nature 254:250—252, 1975
151. CHASE LR, MELSON GL, AURBACH GD: Pseudohypopa-
rathyroidism: Defective excretion of 3', 5-AMP in responseto parathyroid hormone. J C/in Invest 48:1832—1844, 1969
152. MARCUS R, WILBER JF, AURBACH GD: Parathyroid hor-mone-sensitive adenyl cyclase from the renal cortex of apatient with pseudohypoparathyroidism. J C/in Endocrinol33:537—54!, 1971
153. DREZNER M, NEELON FA, LEB0vITz HE: Pseudohypo-parathyroidism, type II: A possible defect in the receptionof the cyclic AMP signal. N Eng/ J Med 289:1056—1060,1973
154. RODRIGUEZ HJ, VILLARREAL H, KLAHR 5, SLATAPOLSKY E:Pseudohypoparathyroidism, type II: Restoration of normalrenal responsiveness to parathyroid hormone by calcium ad-ministration. J C/in Endocrino/ Metab 39:693—701, 1974
155. ROSENBERG LE, DOWNING S: Transport of neutral anddibasic amino acids by human leukocytes: Absence of defectin cystinuria. J C/in Invest 44:1382—1393, 1965
156. TADA K, MORIKAWA T, ARAKAWA T: Prolinuria: Transportof proline by leukocytes. Tokoku J Exp Med 90:189—193,1966
157. TENENHOUSE HS, SCRIVER CR: Orthophosphate transport inthe erythrocyte of normal Subjects and of patients with X-linked hypophosphatemia. J C/in Invest 55:644—654, 1975
158. CHILDS B, DER KALOUSTIAN M: Genetic heterogeneity. NEnglJ Med 279:1205—1212, 1267—1274, 1968
159. SCRIVER CR: The human biochemical genetics of amino acidtransport. Pediatrics 44:348—357, 1969
160. CHRISTENSEN HN: On the development of amino acid trans-port systems. Fed Proc 32:19—28, 1973
161. MCKUSICK VA: Mende/ian inheritance in Man: Catalogs ofA utosomal Dominant A ulosoma/ Recessive and X-Linked Phe-notypes (4th ed). Baltimore, Johns Hopkins Press, 1975
162. BERGSMA D: Birth Defects: At/as and Compendium. NationalFound-March of Dimes, Baltimore, Williams & WilkinsCo., 1972