funktionalisiertes graphen als multifunktionaler
TRANSCRIPT

Funktionalisiertes Graphen
als multifunktionaler Nanofüllstoff für
Keramik-Nanokomposite
INAUGURALDISSERTATION
zur Erlangung des Doktorgrades
der Fakultät für Chemie und Pharmazie
der Albert-Ludwigs-Universität Freiburg im Breisgau
vorgelegt von
Wenli Zhang
aus Shandong, VR China
2018

I

II

III

IV
Funktionalisiertes Graphen
als multifunktionaler Nanofüllstoff für
Keramik-Nanokomposite
INAUGURALDISSERTATION
zur Erlangung des Doktorgrades
der Fakultät für Chemie und Pharmazie
der Albert-Ludwigs-Universität Freiburg im Breisgau
vorgelegt von
Wenli Zhang
aus Shandong, VR China
2018


Vorsitzender des Promotionsausschusses: Prof. Dr. Stefan Weber
Dekan: Prof. Dr. Manfred Jung
Referent: Prof. Dr. Rolf Mülhaupt
Korreferent: Prof. Dr. Dr. Christian Friedrich
Datum der mündlichen Prüfung: 27.06.2018

I
Nicht weil es schwer ist,
wagen wir es nicht,
Sondern weil wir es nicht wagen,
ist es schwer.
Seneca


Veröffentlichungen
I
Die nachfolgende Arbeit wurde von Dezember 2014 bis November 2017 am Institut für
Makromolekulare Chemie und am Freiburger Materialforschungszentrum (FMF) der
Albert-Ludwigs-Universität Freiburg im Arbeitskreis von Herrn Prof. Dr. Rolf
Mülhaupt durchgeführt. An dieser Stelle möchte ich mich bei allen Kollegen für die
Unterstützung und Zusammenarbeit bei der Durchführung dieser Arbeit herzlich
bedanken.
Herrn Prof. Dr. Rolf Mülhaupt danke ich herzlich für die Aufnahme in seinen
Arbeitskreis sowie die Bereitstellung des interessanten und herausfordernden Themas,
sein stetes Interesse an meinen Ergebnissen, die zahlreichen Hilfestellungen und die
gewährleisteten Freiheiten bei meiner Arbeit.
Herrn Prof. Dr. Dr. Christian Friedrich danke ich für die freundliche Übernahme des
Korreferats sowie für die wissenschaftlichen Fragestellungen.
Herzlicher Dank gilt Herrn Dr. Andreas Kailer, Herrn Dr. Christian Schröder und Frau
Dr. Bernadette Schlüter vom Fraunhofer-Institut für Werkstoffmechanik in Freiburg,
Herrn Martin Knoch und Herrn Dr. Ulrich Degenhardt von der Firma FCT
Ingenieurkeramik GmbH in Sonneberg, Herrn Jörg Thelke von der Firma
EagleBurgmann Germany GmbH & Co. KG in Wolfratshausen, Herrn Dr. Csaba
Balázsi und Frau Dr. Katalin Balázsi vom Institute for Technical Physics and Materials
Science, Centre for Energy Research in Budapest, Herrn Prof. Jan Dusza und Herrn
Richard Sedlák von The Institute of Materials Research of SAS in Košice sowie Herrn
Prof. Dr. Michael Moseler und Herrn Dr. Andreas Klemenz vom Freiburger
Materialforschungszentrum für die fruchtbare Kooperation innerhalb des BMBF-
Projekts CERAPHENE. Ferne danke ich PD Dr. Detlef Klimm vom Institut für
Kristallzüchtung für die erfolgreiche Zusammenarbeit.
Für das gute Arbeitsklima und die Unterstützung bei der Forschung und Entwicklung
bedanke ich mich bei meinen Arbeitskollegen, insbesondere Fabian, Folke, Ion, Höffi,
Kris, Laura, Mark, Benny und Hannes, am Institut für Makromolekulare Chemie und
am Freiburger Materialforschungszentrum.
Dr. Yi Thomann und Vincent Ahmadi möchte ich für die Aufnahme von AFM-
Aufnahmen und hilfreiche Hinweise zu den Ergebnissen danken. Dr. Ralf Thomann,

Veröffentlichungen
II
Dr. Katalin Balázsi, Kris Bielefeld und Vladimir Girman danke ich für die TEM- und
SEM-Bilder sowie EDX-Messungen, Andreas Warmbold und PD Dr. Detlef Klimm für
die TGA- und BET-Messungen, und Angelika Siegel für die Elementaranalysen.
Weiterhin bedanke ich mich bei den anderen Kollegen, die mich in neue Geräte und
Techniken eingewiesen haben. Sie ermöglichten mir die Verwendung aller für meine
Untersuchungen notwendigen Geräte und Materialien.
Meinen Bürokollegen Steffen, Michael, J. D., Rukia und Fritz bedanke ich mich für die
angenehme Atmosphäre. Kris Bielefeld, Steffen Wiedmann, Florian Mönkemeyer,
Vitalij Schimpf, Burkhardt Pössel, Tobias Trötschler und Carla Vlad danke ich für die
kritische Durchsicht sowie sprachliche und wissenschaftliche Korrektur dieser Arbeit.
Frau Aritina Illmann, Frau Carmen Hermann, Frau Katrin Kiefer, Frau Beate Gloderer
und Frau Nicola Weis danke ich für die administrative Unterstützung. Ein besonderes
Dankeschön richte ich an die Mitarbeiter der Werkstatt und des Magazins,
stellvertretend Alexander, Eric, Sascha, Harald, Svetlana und Mona, für die
Unterstützung bei technischen Problemen.
Meinen Prktikanten Yuan Yang, Ye Tang und Zhibin Dong danke ich für ihr
herausragendes Engagement und ausgezeichnete Mitarbeit im Rahmen ihrer
Mitarbeiterpraktika.
Weiter möchte ich mich bei meinen Freunden und Kollegen Zucong Zhang, Yi Wei,
Kaitong Wu, Liang Xu, Yuqing Guan und Luyao Zhang für die schöne und
unvergessliche Zeit in Freiburg bedanken.
Mein allergrößter Dank gilt definitiv meinen Eltern Yanjie Zhang und Yanzhen Liu,
meinem Bruder Wenbo Zhang, meiner Schwägerin Cuiping Liu sowie allen anderen
Familienmitgliedern, die mich nicht nur stets in jeder Hinsicht während des gesamten
Studiums unterstützt haben, sondern immer auch ein sehr großes Interesse an mir und
meiner Arbeit zeigen.
Abschließend danke ich besonders meiner Frau Jingting Luan, die immer an meiner
Seite steht, mir eine große Stütze ist, immer an mich glaubt, für die schöne Zeit, die ich
mit ihr erleben darf.

Veröffentlichungen
III
Veröffentlichungen
1. F. Beckert, S. Bodendorfer, W. Zhang, R. Thomann, R. Mülhaupt:
Mechanochemical Route to Graphene-Supported Iron Catalysts for Olefin
Polymerization and in Situ Formation of Carbon/Polyolefin Nanocomposites,
Macromolecules 2014, 47, 7036-7042.
2. W. Zhang, C. Schröder, B. Schlüter, J. Dusza, R. Sedlák, R. Mülhaupt, A. Kailer:
Effect of Mechanochemically Functionalized Graphene on the Tribological
Properties of Silicon Carbide/Graphene Nanocomposites in Aqueous
Environment, Manuskript liegt vor.
Patentanmeldung
1. W. Zhang, R. Mülhaupt, M. Knoch, J. Thelke: Verfahren zur Herstellung eines
mit einem graphenhaltigen Material ummantelten partikulären Trägermaterials
und eines keramischen Bauteils, sowie keramisches Bauteil,
DE 10 2017 211 663, 2017.
Berichte
1. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) 1. Zwischenbericht, 01.08.2015.
2. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) 2. Zwischenbericht, 01.02.2016.
3. W. Zhang, R. Mülhaupt, A. Kailer, B. Schlüter, C, Schröder, M. Knoch, J.
Thelke, M. Moseler, A. Klemenz: Reib- und verschleißarme Graphen-Keramik-
Komposite für tribologische Anwendungen in wässrigen Medien,
CERAPHENE (BMBF) Meilensteinbericht, 30.04.2016.

Veröffentlichungen
IV
4. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) 3. Zwischenbericht, 22.07.2016.
5. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) 4. Zwischenbericht, 22.01.2017.
6. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) 5. Zwischenbericht, 26.07.2017.
7. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) Abschlussbericht, 02.02.2018.
Vorträge
1. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) Projekttreffen, Budapest, 06.05.2015.
2. W. Zhang, R. Mülhaupt: Reib- und verschleißarme Keramik-Graphen-
Nanokomposite für tribologische Anwendungen in wässrigen Medien, Seminar
des Arbeitskreis Mülhaupt, Freiburg, 10.06.2015.
3. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) Projekttreffen, Kosice, 18.11.2015.
4. W. Zhang, R. Mülhaupt: Reib- und verschleißarme Keramik-Graphen-
Nanokomposite für tribologische Anwendungen in wässrigen Medien, Seminar
des Arbeitskreis Mülhaupt, Freiburg, 09.12.2015.
5. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) Projekttreffen, Sonneberg, 17.05.2016.
6. W. Zhang, R. Mülhaupt: Reib- und verschleißarme Keramik-Graphen-
Nanokomposite für tribologische Anwendungen in wässrigen Medien, Seminar
des Arbeitskreis Mülhaupt, Freiburg, 29.06.2016.
7. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) Projekttreffen, Wolfratshausen,
16.11.2016.

Veröffentlichungen
V
8. A. Kailer, B. Schlüter, C, Schröder, W. Zhang: Tribology of SiC and
SiC/graphene ceramic composites in aqueous environment, Euro Friction Wear
and Wear Protection 2017, Ettlingen, 22.02.2017.
9. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) Projekttreffen, Kosice, 17.05.2017.
10. W. Zhang, R. Mülhaupt: Reib- und verschleißarme Keramik-Graphen-
Nanokomposite für tribologische Anwendungen in wässrigen Medien, Seminar
des Arbeitskreis Mülhaupt, Freiburg, 05.07.2017.
11. A. Kailer, C. Schröder, B. Schlüter, W. Zhang, R. Mülhaupt, A. Klemenz, M.
Moseler, C. Balazsi, K. Balazsi, J. Dusza, A. Kovalcikova, R. Sedlak, M. Knoch,
J. Thelke, J. Otschick: Low friction and wear resistant graphene-ceramics
composites for tribological application in aqueous environments, 15th
Conference & Exhibition of the European Ceramic Society (ECerS2017),
Budapest, 11.07.2017.
12. W. Zhang, R. Mülhaupt: Herstellung funktionalisierter Graphene für Keramik-
Nanokomposite, CERAPHENE (BMBF) Projekttreffen, Freiburg, 16.10.2017.
13. C. Schröder, B. Schlüter, W. Zhang, A. Kailer, R. Mülhaupt, J. Thelke, U.
Degenhardt: Investigation of graphenecontaining silicon carbides under
tribological loading and waterlubricated conditions, 93. DKG-Jahrestagung und
Symposium Hochleistungskeramik 2018, München, 11.04.2018.
Posterpräsentationen
1. W. Zhang, R. Mülhaupt: Functionalized Graphene for Ceramic Nanocomposites,
15th Conference & Exhibition of the European Ceramic Society (ECerS2017),
Budapest, 09. – 13.07.2017.
2. W. Zhang, C. Schröder, M. Knoch, A. Kailer, R. Mülhaupt: Effect of
Functionalized Graphene on Electrical and Tribological Properties of Silicon
Carbide Nanocomposites, 2017 International Graphene Innovation Conference
(GRAPCHINA 2017), Nanjing, 24. – 26.09.2017.


Inhaltsverzeichnis
I
Inhaltsverzeichnis
Abkürzungsverzeichnis ............................................................................................... I
1 Einleitung .......................................................................................................... - 1 -
1.1 Graphen.................................................................................................................. - 2 -
1.1.1 Herstellung von Graphen ............................................................................................ - 4 -
1.2 Keramik ................................................................................................................ - 13 -
1.2.1 Siliciumcarbid .......................................................................................................... - 14 -
1.2.2 Siliciumnitrid ............................................................................................................ - 15 -
1.3 Tribologie ............................................................................................................. - 16 -
1.3.1 Reibung .................................................................................................................... - 17 -
1.3.2 Verschleiß ................................................................................................................. - 19 -
1.4 Keramik-Graphen-Nanokomposite ...................................................................... - 19 -
2 Aufgabenstellung ........................................................................................... - 23 -
2.1 Graphensynthese und Charakterisierung ............................................................. - 25 -
2.2 Graphen-Nanokomposite auf Basis von Siliciumcarbid ...................................... - 26 -
2.3 Graphen-Nanokomposite auf Basis von Siliciumnitrid ........................................ - 28 -
3 Graphensynthese und Charakterisierung ................................................... - 29 -
3.1 Einleitung ............................................................................................................. - 29 -
3.2 Graphitoxid und thermisch reduziertes Graphitoxid ........................................... - 31 -
3.2.1 Dispersionsstabilität ................................................................................................. - 37 -
3.3 Mechanochemisch funktionalisiertes Multilagen-Graphen ................................. - 40 -
3.3.1 Dispersionsstabilität ................................................................................................. - 48 -
3.4 Graphenbeschichtetes Siliciumcarbid .................................................................. - 52 -
3.4.1 Synthese von GSiC aus Furfurylalkohol .................................................................. - 53 -
3.4.2 Synthese von GSiC aus Glucose .............................................................................. - 55 -
3.4.3 Synthese von GSiC aus Dopamin ............................................................................. - 56 -
3.4.4 Charakterisierung der GSiCs .................................................................................... - 59 -
3.5 Benchmark Kohlenstofffüllstoffe .......................................................................... - 64 -
3.6 Thermische Stabilität ........................................................................................... - 69 -
3.7 Zusammenfassung ................................................................................................ - 72 -

Inhaltsverzeichnis
II
4 Graphen-Nanokomposite auf Basis von Siliciumcarbid ............................ - 75 -
4.1 Einleitung ............................................................................................................. - 75 -
4.2 Herstellung ........................................................................................................... - 76 -
4.3 Dichte & Porosität ............................................................................................... - 80 -
4.4 Morphologie ......................................................................................................... - 85 -
4.5 Mechanische Eigenschaften ................................................................................. - 89 -
4.6 Elektrische Eigenschaften .................................................................................... - 99 -
4.7 Tribologische Eigenschaften .............................................................................. - 104 -
4.7.1 Stift-Scheibe-Tribometer ........................................................................................ - 104 -
4.7.2 Scheibe-Scheibe-Tribometer .................................................................................. - 116 -
4.7.3 Simulation: Atomistische Modellierung ................................................................. - 121 -
4.8 Sintern mittels SPS-Verfahren ............................................................................ - 125 -
4.9 Zusammenfassung .............................................................................................. - 133 -
5 Graphen-Nanokomposite auf Basis von Siliciumnitrid ............................ - 137 -
5.1 Einleitung ........................................................................................................... - 137 -
5.2 Herstellung ......................................................................................................... - 138 -
5.3 Dichte & Porosität ............................................................................................. - 140 -
5.4 Morphologie ....................................................................................................... - 141 -
5.5 Mechanische Eigenschaften ............................................................................... - 144 -
5.6 Tribologische Eigenschaften .............................................................................. - 147 -
5.7 Zusammenfassung .............................................................................................. - 149 -
6 Zusammenfassende Diskussion .................................................................. - 153 -
6.1 Graphensynthese und Charakterisierung ........................................................... - 156 -
6.2 Graphen-Nanokomposite auf Basis von Siliciumcarbid .................................... - 162 -
6.3 Graphen-Nanokomposite auf Basis von Siliciumnitrid ...................................... - 172 -
6.4 Fazit und Ausblick .............................................................................................. - 177 -

Inhaltsverzeichnis
III
7 Experimenteller Teil .................................................................................... - 180 -
7.1 Verwendete Chemikalien .................................................................................... - 180 -
7.1.1 Chemikalien und Lösungsmittel ............................................................................. - 180 -
7.1.2 Keramik-Rohstoffe & Benchmark Kohlenstofffüllstoffe ....................................... - 181 -
7.2 Herstellung von funktionalisiertem Graphen ..................................................... - 181 -
7.2.1 Herstellung von Graphitoxid und thermisch reduziertem Graphitoxid .................. - 181 -
7.2.2 Herstellung von mechanochemisch funktionalisiertem Multilagen-Graphen ......... - 183 -
7.2.3 Herstellung von graphenbeschichtetem Siliciumcarbid und Siliciumnitrid............ - 183 -
7.3 Herstellung von SiC-Graphen-Nanokompositen ................................................ - 186 -
7.3.1 Konventioneller Sinter-Ofen .................................................................................. - 186 -
7.3.2 Spark Plasma Sintern/Feld-Aktiviertes Sintern ...................................................... - 187 -
7.4 Herstellung von Si3N4-Graphen-Nanokompositen ............................................. - 188 -
7.5 Charakterisierungsmethoden und verwendete Geräte ....................................... - 189 -
7.5.1 Atomkraftmikroskopie (AFM) ............................................................................... - 189 -
7.5.2 Biegefestigkeit ........................................................................................................ - 189 -
7.5.3 Bruchzähigkeit ........................................................................................................ - 190 -
7.5.4 Brunauer-Emmett-Teller Analyse (BET) ................................................................ - 191 -
7.5.5 Drehrohrofen .......................................................................................................... - 191 -
7.5.6 Elementaranalyse (EA) ........................................................................................... - 191 -
7.5.7 Energiedispersive Röntgenspektroskopie (EDX) ................................................... - 192 -
7.5.8 FT-IR-Spektroskopie .............................................................................................. - 192 -
7.5.9 Härte ....................................................................................................................... - 192 -
7.5.10 Hochdruckhomogenisator ....................................................................................... - 193 -
7.5.11 Kryomühle .............................................................................................................. - 193 -
7.5.12 Leitfähigkeitsmessung ............................................................................................ - 193 -
7.5.13 LUMiSizer .............................................................................................................. - 194 -
7.5.14 Rasterelektronenmikroskopie (SEM) ..................................................................... - 195 -
7.5.15 Thermogravimetrische Analyse (TGA) .................................................................. - 196 -
7.5.16 Transmissionselektronenmikroskopie (TEM) ........................................................ - 196 -
7.5.17 Tribologie ............................................................................................................... - 197 -
7.5.18 Ultraschalllanze ...................................................................................................... - 198 -
8 Kurzzusammenfassung ............................................................................... - 199 -
9 Lebenslauf..................................................................................................... - 201 -
10 Literaturverzeichnis .................................................................................... - 202 -

Inhaltsverzeichnis
IV

Abkürzungsverzeichnis
I
Abkürzungsverzeichnis
2D Zweidimensional
°C Grad Celsius
Å Ångström
ABET Spezifische Oberfläche bestimmt aus BET-Messungen
a.u. Willkürliche Einheite
AFM Atomkraftmikroskopie
At.-% Atomprozent
BET Brunauer-Emmett-Teller
ca. circa
CNT Carbon Nanotubes
CRGO Chemisch reduziertes Graphitoxid
CVD Chemical Vapor Deposition
d Tag
DICY Dicyandiamid
DIN Deutsches Institut für Normung
EA Elementaranalyse
EDX Energiedispersive Röntgenspektroskopie
FT-IR Fourier-Transformations-Infrarot-Spektroskopie
Gew.-% Gewichtsprozent
GO Graphitoxid
h Stunde
HH Hochdruckhomogenisator
K Kelvin
K1C Kritischer Spannungsintensitätsfaktor
L Liter
LM Lösungsmittel

Abkürzungsverzeichnis
II
m Meter
MG Multilagen-Graphen
Milliarde Mrd.
min Minute
N Newton
n. b. Nicht bestimmt
Pa Pascal
RT Raumtemperatur
Rspez Spezifischer Widerstand
S Siemens
s Sekunde
SEM Rasterelektronenmikroskopie
SiC Siliciumcarbid
Si3N4 Siliciumnitrid
TEM Transmissionenelektronenmikroskopie
TGA Thermogravimetrische Analyse
TRGO Thermisch reduziertes Graphitoxid
U/min Umdrehungen pro Minute
XPS Röntgenphotoelektronenspektroskopie
Ω Ohm
µ Reibungskoeffizient
σB Biegefestigkeit
σS Spezifische Leitfähigkeit

1 Einleitung
- 1 -
1 Einleitung
Keramische Werkstoffe spielen weltweit eine immer wichtigere Rolle, da sie sich in einer
Vielzahl von Anwendungen aufgrund ihrer technischen Eigenschaften bewährt haben.
Besonders hervorzuheben sind hierbei die sehr hohe Härte, Steifigkeit, Druck- und
Verschleißfestigkeit sowie sehr gute Korrosions-, Temperatur- und Wärmeformbeständigkeit.
Weitere Vorteile vieler Keramiken sind die leichte Zugänglichkeit der Rohstoffmaterialien und
die Vielseitigkeit ihrer Anwendung.[1–4]
Unter Keramik versteht man diejenigen Werkstoffe, welche aus nichtmetallischen
anorganischen Massen geformt und durch Einwirkung hoher Temperaturen verfestigt werden.[1]
Keramik war die erste durch Menschen künstlich hergestellte Werkstoffklasse, welche in der
Altsteinzeit vor etwa 30 000 Jahren erfunden wurde. Das berühmteste Beispiel eines
altsteinzeitlichen Keramikobjektes ist die mehr als 24 000 Jahre alte Venus von Dolní Věstonice,
welche 1925 während archäologischer Ausgrabungen in Dolní Věstonice gefunden wurde.
Aufgrund der gut formbaren „Erden“ bzw. Rohstoffe und ihres einfachen
Herstellungsverfahrens durch Brennen wurden Keramiken über die Jahrtausende stets
weiterentwickelt und die Verwendung hat sich weit verbreitet.[2,3] Heutzutage sind keramische
Werkstoffe aus vielen Technologiebereichen wie der Elektronik (Isolatoren), der
Hochtemperaturtechnik (Industrieöfen) und dem Maschinen- und Anlagenbau (Gleit- und
Regelelemente) nicht mehr wegzudenken.[3] Im Jahr 2014 wurden über 110 Mio. Tonnen
keramische Werkstoffe produziert und mit einem Umsatz von 157 Mrd. $ weltweit verkauft.[5]
Die Eigenschaften von keramischen Werkstoffen können entweder durch Variation der
verwendeten Sinterhilfsmittel oder durch die Inkorporierung funktionaler Füllstoffe
substanziell verbessert werden. Für keramische Werkstoffe gehören Kohlenstofffüllstoffe zu
den wenigen sinnvollen Möglichkeiten um eine Verstärkung oder Funktionalisierung
hervorzurufen. Seit 2003 wurden kohlenstoffbasierte Nanomaterialien wie Carbon Nanotubes
(CNTs) und Graphene in der Keramikkompositherstellung als Füllstoffe eingesetzt, die deren

1 Einleitung
- 2 -
thermischen, mechanischen, elektrischen und tribologischen Eigenschaften verbessern.[6] Im
Vergleich zu den füllstofffreien Varianten zeigen die neuartigen Keramik-Kohlenstoff-
Nanokomposite unkonventionelle Eigenschaftskombinationen wie z. B. deutlich verbesserte
mechanische Festigkeiten, höhere Verschleißbeständigkeit und auch eine stark erhöhte
elektrische Leitfähigkeit.
1.1 Graphen
Graphen bezeichnet eine Nanomodifikation des Kohlenstoffs, bei der die Kohlenstoffatome
durch die Ausbildung eines konjugierten Bindungssystems sechsgliedriger Ringe eine
2-dimensionale Schicht ausbilden.[7,8] Stapelt man solche Graphenschichten übereinander,
erhält man einen 3-dimensionalen Kristall, den Graphit. Graphen besteht folglich aus einer
einzelnen Schicht des Kristallgitters von Graphit. Im Gegensatz dazu kann man sich die 0- bzw.
1-dimensionalen Fullerene bzw. Carbon Nanotubes (CNT) als aufgerolltes kugelförmiges bzw.
zylinderförmiges Graphen vorstellen. Im Vergleich zu den 0-, 1- und 3-dimensionalen
Nanomodifikationen sind Graphene ultradünne großflächige 2D-Makromoleküle aus
Kohlenstoff mit ultrahohem Aspektverhältnis (siehe Abbildung 1).[9,10]
Abbildung 1: Kohlenstoffbasierte-Nanomodifikationen in verschiedenen Dimensionen.[9]

1 Einleitung
- 3 -
Graphen weist einzigartige und faszinierende Eigenschaften auf, die es sowohl für die
Grundlagenforschung als auch für Anwendungen interessant macht. Beispielsweise sind
Graphene außerordentlich steif und fest. Die Steifigkeit von Graphen beträgt 1 TPa und ist
damit fast so groß wie die des Diamanten. Gleichzeitig besitzt es eine Zugfestigkeit von
130 GPa, was dem 125-fachen von Stahl entspricht.[11] Zusätzlich verfügt Graphen über eine
einzigartige elektronische Struktur, da es im Gegensatz zu den Halbleitern keine Bandlücke
aufweist. Die elektrische Leitfähigkeit innerhalb der Schichten entspricht 2.6 · 104 S∙cm-1.[12]
Außerdem zeichnet sich das Monolagengraphen durch eine große spezifische Oberfläche von
2630 m2∙g-1[13] und eine hohe Wärmeleitfähigkeit von 5000 W m-1 K-1[14] aus. Graphen hat,
sofern es als Monolage vorliegt, eine sehr gute Lichtdurchlässigkeit, da eine einzelne
Graphenschicht das Licht nur um ca. 2.3 % abschwächt.[15,16] Mit diesen hervorragenden
Eigenschaften stellt Graphen einen vielversprechenden Füllstoff für die Werkstoffentwicklung
dar. Graphene bieten völlig neue Möglichkeiten mit hohem Anwendungspotential, die mit
anderen Kohlenstoff-Nanopartikeln wie CNT, Fulleren und leitfähigem Carbon Black nicht zu
realisieren sind.[17–19] Damit stellt Graphen ein wissenschaftlich interessantes Material dar.
Im Jahr 1948 wurden die ersten TEM-Aufnahmen von Graphen mit geringer Lagenzahl von
RUESS et al. veröffentlicht.[20] Zu den Pionieren der Graphenherstellung gehört BOEHM, der
bereits 1962 über einlagige Kohlenstofffolien berichtete, die durch alkalische Behandlung von
Graphitoxid-Dispersionen und anschließende Reduktion mit Hydrazinhydrat hergestellt
wurden.[21] Die erste Definition des Begriffs Graphen aus dem Jahr 1994 geht auch auf BOEHM
zurück.[22,23] NOVOSELOV et al. beschrieben 2004 die erste Darstellung von idealem Graphen
durch Abblätterung von hochgeordnetem pyrolytischem Graphit. Auf diese Weise lassen sich
lokal dünne Graphenfilme herstellen, welche dünner als 50 nm und somit optisch fast
vollständig transparent sind. Ab diesem Zeitpunkt kam es zu einem sprunghaften Anstieg des
Interesses an diesem Thema und damit auch zu zahlreichen Publikationen.[24] Bei der von
NOVOSELOV et al. verwendeten Methode handelt es sich um ein sehr zeitaufwendiges Verfahren,
bei dem man zwar hochwertige Monolagengraphene erhält, jedoch nur in sehr geringer Menge,
welche daher ausschließlich für grundlagenanalytische Zwecke benutzt werden können. Für die
Produktion größerer Graphenmengen ist die mechanische Exfolierung nach diesem Vorbild

1 Einleitung
- 4 -
nicht geeignet. Das rasant gestiegene Interesse hat daher die Entwicklung einer ganzen Reihe
unterschiedlicher Herstellungsmethoden von Graphenen vorangetrieben. Dabei wird die
Bezeichnung Graphen nicht nur für das ideale Monolagenmaterial, sondern für mono- bis
multilagige Graphenderivate verwendet, welche graphenartigen Charakter aufweisen und sich
je nach Herstellungsverfahren in ihren Eigenschaften unterscheiden.
1.1.1 Herstellung von Graphen
Die Herstellungsmethoden von idealem Graphen und Graphenderivaten lassen sich in Bottom-
up- und Top-down-Prozesse unterteilen (siehe Abbildung 2). Dabei werden Graphene bei Top-
down-Prozessen über mechanische, thermische oder chemische Exfolierung ausgehend von
Graphit erhalten, während die Bottom-up-Methoden auf dem Schichtaufbau aus molekularen
Kohlenstoffquellen beruhen.[25]
Abbildung 2: Übersicht der Top-down- und Bottom-up-Verfahren zur Herstellung von Graphenen.

1 Einleitung
- 5 -
Bei Bottom-up-Methoden werden Graphene aus niedermolekularen organischen Molekülen
mittels organisch-chemischer Synthese schrittweise aufgebaut. Im Bereich klassischer Bottom-
up-Methoden stellt das CVD-Verfahren (Chemical Vapour Deposition) einen wichtigen
Prozess zur Herstellung von idealem Graphen dar. Dabei wurde eine gasförmige
Kohlenstoffquelle (z. B. Methan, Ethen oder Acetylen) über eine katalytische Metalloberfläche
(z. B. Ni, Co\MgO, Au oder Cu) geleitet. Dort wurden großflächige Monolagen-
Graphenschichten durch starkes Erhitzen auf über 600 °C abgeschieden.[26–34]
Eine weitere Möglichkeit der Graphendarstellung stellt das epitaktische Wachstum einzelner
Schichten auf Siliciumcarbid (SiC) dar. Bei diesem Verfahren wurde ein Teil des Siliciums aus
SiC durch Sublimation unter starkem Erhitzen (1300 – 1800 °C) entfernt. Dabei bildeten sich
aus dem zurückbleibenden Kohlenstoff hochwertige, großflächige Mono- und
Weniglagengraphene auf der Oberfläche von SiC.[35–38]
Des Weiteren konnte Graphen aus aromatischen Kohlenwasserstoffen über
Suzukikupplung[39–41] oder thermische Rekombination[42] hergestellt werden. Typische Mengen,
die bei den oben genannten Bottom-up-Methoden erhalten werden, liegen im Bereich von
wenigen Milligramm und damit weit unter jenen, welche für die Herstellung von Keramik-
Kompositen benötigt werden.
Im Gegensatz zu den bisher beschriebenen Verfahren entwickelte X. LI[43] eine einfach
durchführbare, kostengünstige Bottom-up-Methode zur Herstellung von Graphen mit relativ
hohem Durchsatz. Hier wurde das funktionalisierte Graphen unter Verwendung von Glucose
als Kohlenstoffquelle über eine templatvermittelte Reaktion hergestellt. Auf ähnliche Weise
wurde diese Methode von M. BECKERT[44,45] und R. LI
[46] erweitert, indem sie statt Glucose
Furfurylalkohol bzw. Dopamin als Kohlenstoffquelle benutzten (siehe Kapitel 1.1.1.4).
Top-down-Prozesse zur Herstellung von Graphenen beruhen üblicherweise auf der
mechanischen, thermischen oder chemischen Exfolierung von Graphit oder auf dessen
oxidierter Form Graphitoxid (GO). Um den Zeit- und Kostenaufwand gering zu halten und eine
Scale-up-Fähigkeit zu gewährleisten werden Top-down-Prozesse in vielen Fällen gegenüber
Bottom-up-Methoden bevorzugt. Top-down-Verfahren basieren auf der Überwindung der
relativ schwachen Van-der-Waals-Wechselwirkungen zwischen den Schichten des Graphits.

1 Einleitung
- 6 -
Neben der von A. GEIM verwendeten Scotch-Tape-Methode[24,47] konnte Graphen über
Graphitinterkalate hergestellt werden. Beispielsweise wurde Graphit mit einer Säure wie
Salpetersäure oder Schwefelsäure interkaliert, thermisch oder mittels Mikrowellenstrahlung
exfoliert und durch weiteres Vermahlen in Graphen überführt.[48] Die in dieser Arbeit
eingesetzten Benchmark-Graphene Grade C und Grade M in Kapitel 3.4 wurden laut
Herstellerangabe nach dieser Methode hergestellt.[49,50]
In jüngster Zeit wurden größere Mengen an Graphenen durch Nass- oder Trockenvermahlung
von Graphit in einer Kugelmühle hergestellt. Als Medium konnte flüssiges H2O,[51] Ethanol,[52]
DMF[53] und THF,[53,54] fester Schwefel,[55] Melamin[53,56] und Trockeneis[57] sowie gasförmiges
CO2,[54,55] N2
[54,55,58] und Ar[54,55] verwendet werden (siehe Kapitel 1.1.1.3). Eine weitere CO2-
basierte Methode stellt die Vermahlung von Graphit in überkritischem CO2 dar. Bei dieser
Methode interkalierte das überkritische CO2 zwischen die Graphitschichten. Durch die
anschließende, plötzliche Druckentlastung wurden die Schichten exfoliert.[59]
Des Weiteren kann Graphen aus Graphitoxid durch thermische oder chemische Reduktion
hergestellt werden. Diese Methode zählt aufgrund preiswerter Edukte, etablierter Verfahren und
hoher Produktionsmengen zu den wichtigsten Herstellungsverfahren (siehe Kapitel 1.1.1.1 und
1.1.1.2).
1.1.1.1 Graphitoxid (GO)
Im Vergleich zu Graphit wird der Kohlenstoffanteil von Graphitoxid (GO) durch die
Einführung sauerstoffhaltiger funktionellen Gruppen deutlich reduziert. In seiner maximal
oxidierten Form besitzt GO ein Verhältnis von Kohlenstoff zu Sauerstoff von 1.5:1 bis 2.5:1.[60]
GO verfügt ebenfalls über eine Schichtstruktur, wobei die Abstände zwischen den
Molekülebenen größer und unregelmäßiger als im Graphit sind.[61] Seit Kurzem ist GO als am
weitesten verbreitete Vorstufe für die Herstellung von funktionalisiertem Graphen in großem
Maßstab interessant geworden.[62,63] Aus GO wird thermisch reduziertes Graphitoxid (TRGO)
durch thermische bzw. chemisch reduziertes Graphitoxid (CRGO) durch chemische Reduktion
gewonnen. GO kann durch unterschiedliche Verfahren aus Graphit unter Einwirkung starker
Oxidationsmittel in Anwesenheit starker Säuren gewonnen werden.[62,64]

1 Einleitung
- 7 -
Die erste Darstellung von GO gelang dem britischen Chemiker BRODIE bereits 1859.[65] Er
behandelte Graphit mit einer Mischung aus Kaliumchlorat (KClO3) und rauchender
Salpetersäure (HNO3). Nach der Oxidation erhielt er ein weißes, kaum verunreinigtes
kristallines Material. Während der Reaktion wurde gasförmiges und explosives Chlordioxid
(ClO2) freigesetzt, welches hier als eigentliches Oxidationsmittel wirkt.
Eine schnellere, ungefährlichere und besser handhabbare Methode mit höherer Ausbeute stellt
die Oxidation nach HUMMERS und OFFEMAN von 1957 dar.[64] Sie verwendeten eine Mischung
aus Schwefelsäure (H2SO4), Natriumnitrat (NaNO3) und Kaliumpermanganat (KMnO4). Das
eigentliche Oxidationsmittel ist im Vergleich zu den älteren Methoden, die gefährliches
Chlordioxid (ClO2) verwenden, Dimanganheptaoxid (Mn2O7), welches aus KMnO4 in
Anwesenheit von H2SO4 entsteht. Aufgrund der beschriebenen Vorteile ist diese Methode bis
heute im Einsatz und wurde zur Herstellung des in dieser Arbeit verwendeten GOs
angewandt.[61] Struktur und Eigenschaften des GOs werden durch die verwendete
Synthesemethode und den Oxidationsgrad bestimmt. Die Schichtstruktur des eingesetzten
Graphits bleibt typischerweise erhalten, aber die Lagen sind unregelmäßig hinsichtlich ihrer
Planarität und weisen einen bis zu doppelt so großen Abstand von ungefähr 0.7 nm auf. Neben
Epoxidgruppen wurden experimentell auch Carbonyl-, Hydroxyl- sowie Phenol-Gruppen
nachgewiesen.[23,60,66–69]
1.1.1.2 Thermisch reduziertes Graphitoxid (TRGO)
Thermisch reduziertes Graphitoxid (TRGO) stellt dank seiner Scale-up-Fähigkeit bis jetzt die
beste Möglichkeit dar, um große Mengen Graphen zu erhalten. Durch die Reduktion von GO
wird der Kohlenstoffanteil wieder auf ca. 80 Gew.-% erhöht, wobei funktionelle Gruppen
abgespalten werden und sich das konjugierte π-System regeneriert.
TRGO wird durch rasches Erhitzen von GO unter Schutzgasatmosphäre erzeugt.[62,70–73] Dabei
zersetzen sich hauptsächlich die sauerstoffhaltigen funktionellen Gruppen, insbesondere die
Epoxid- und Hydroxyl-Gruppen. Durch das entstehende CO und CO2 wirkt ein großer

1 Einleitung
- 8 -
Gasdruck zwischen den Schichten, der die Van-der-Waals-Kräfte übersteigt und somit die
Schichten auseinanderreißt. Theoretisch wird ein Druck von 40 MPa bei 300 °C und 130 MPa
bei 1000 °C erreicht. Nach Berechnungen mit HAMAKER-Konstanten ist ein Druck von 2.5 MPa
notwendig, um zwei gestapelte Graphen-Schichten zu trennen. Das so entstehende TRGO
verfügt über eine sehr geringe Schüttdichte und hohe Porosität und besteht laut Literatur zu
80 % aus Monolagen.[62,74,75]
Die Eigenschaften von TRGO lassen sich über die Reduktionstemperatur steuern.[76] Je höher
diese ist, desto mehr funktionelle Gruppen lösen sich ab. Es bilden sich mehr sp2-Zentren und
die Leitfähigkeit, die spezifische Oberfläche und der E-Modul steigen. Die sauerstoffhaltigen
funktionellen Gruppen werden durch das Erhitzen jedoch nicht vollständig entfernt. Die
verbleibenden sp3-Zentren verursachen die gewellte Struktur der TRGO-Plättchen. Zudem
verschlechtern diese Defektstellen die mechanischen und physikalischen Eigenschaften im
Vergleich zum idealen Graphen. So beobachtet man eine geringere spezifische Oberfläche von
600 – 950 m2∙g-1[75] (ideal: 2630 m2∙g-1[13]), eine Abnahme des E-Moduls auf 0.25 TPa[8] (ideal:
1.03 TPa[11]) sowie eine reduzierte Leitfähigkeit von maximal 60 S∙cm-1[75] (ideal:
2.6 ∙ 104 S∙cm-1[12]).
1.1.1.3 Mechanochemisch funktionalisiertes Multilagen-Graphen (MG)
Der Zugang zu funktionalisiertem und idealem Graphen wurde bereits auf verschiedene Arten
beschrieben. Ideales Graphen kann bisher nur im Milligrammbereich hergestellt werden und
liegt damit weit unter den Mengen, welche für die Herstellung von Keramikkompositen
benötigt werden. TRGO und CRGO sind zwar in größerem Maßstab verfügbar, die
Herstellungsprozesse sind jedoch sehr kosten- und zeitaufwendig. Zusätzlich beinhalten TRGO
und CRGO den Nachteil, dass die Flächenstruktur des Graphens durch Fehlstellen gestört ist.
Deswegen liegt ein aktueller Forschungsfokus auf einer neuen, umweltfreundlichen, einfachen
und billigen Methode zur Graphenherstellung. Eine Möglichkeit zur Herstellung von
mechanochemisch funktionalisiertem Multilagen-Graphen (MG) ist die direkte mechanische
Exfolierung unter Verwendung von verschiedenen Additiven.[52,77–80]

1 Einleitung
- 9 -
Bereits seit den 1950er Jahren ist die Verwendung der mechanischen Vermahlung von Graphit
bekannt. Zur damaligen Zeit wurde sie ausschließlich verwendet um eine rhomboedrische
Modifikation von Graphit zu erhalten.[81] Die während der Vermahlung entstehenden
Scherkräfte dienen zur Komprimierung, Abblätterung und Zerkleinerung der graphitischen
Schichtstruktur und ermöglichen die Darstellung großer Mengen an Graphenen mit
vergleichsweise geringen Kosten und hoher Ausbeute.[82] Im Labormaßstab wird die
Vermahlung vor allem in Planetenkugelmühlen[57,83,84] oder Attritormühlen[52] durchgeführt. Je
nach Reaktionsbedingungen (Gasatmosphäre, Kugel- und Kammermaterial, beigemischte
Edukte, Mahldauer) konnten funktionalisierte[57,83,84] und unfunktionalisierte[52] MG mit
unterschiedlicher Größe[54], hauptsächlich breiter Partikelgrößenverteilung, Geometrie[85–87]
und Funktionalität[54] hergestellt werden.
Abbildung 3: Mechanismusvorschlag von I. JEON für die mechanochemische Funktionalisierung und
Exfolierung von Graphit zur Herstellung von MG.[84]
JEON et al. stellten verschiedene funktionalisierte MG durch trockene Vermahlung von Graphit
unter H2, CO2, SO2 oder CO2/SO2 Atmosphäre her (siehe Abbildung 3). Die mechanische
Einwirkung während des Vermahlungsprozesses führt zur Spaltung von C–C-Bindungen und
zur Bildung hochenergetischer Kohlenstoffzentren. Dabei entstehen durch heterolytische
Spaltung Carbanionen und Carbokationen und durch homolytische Spaltung Radikale.
Anschließend können diese entweder mit umgebenden Gasen, beigemischten Edukten oder
nach Öffnung der Kammer mit Luftsauerstoff und Kohlendioxid reagieren. Über die Wahl
geeigneter Gase und beigemischter Edukte ermöglicht sich die Herstellung von MG mit
verschiedenen funktionellen Gruppen (siehe Tabelle 1).

1 Einleitung
- 10 -
Tabelle 1: Übersicht über die literaturbekannten MG mit unterschiedlichen funktionellen Gruppen.
Gasatmosphäre Beigemischte Edukte Funktionelle Gruppe Ref.
Ar - -OH [55]
CO2 (Trockeneis oder Gas) - -CO2H [55,57]
H2 - -H [57,84]
N2 - -N=N- (Pyrazol/Pyridazin) [55,83]
NH3 - -NH2 [57,84]
SO3, CO2/SO3 - -SO3H [57,84]
Cl2, Br2, I2 - -Cl, -Br, -I [88]
Ar Maleinsäureanhydrid -OH, COOH [89]
Ar Maleimid -COOH, -OH, Imid, Anhydrid [89]
Ar S8 -SH, -SO3H [90,91]
Vakuum Prot -P(=O)(OH)2 [92]
n. b. Asparaginsäure, KMnO4 -O-, -COOH, =O, -OH [93]
Luft Melamin/Ethanol keine [52,56]
Tabelle 1 gibt einen Überblick über die literaturbekannten Verfahren zur Herstellung von
verschiedenen MG. Durch Vermahlung mit Ar und den anschließenden Kontakt mit Luft
konnten Hydroxylgruppen eingeführt werden (MG-Ar). Diese funktionellen Gruppen konnten
mittels FT-IR-Spektroskopie nachgewiesen werden.[54] Die Vermahlung unter CO2 führte zu
MG-CO2 mit kantenständigen Carboxylgruppen. Das hergestellte MG-CO2 besteht vorwiegend
aus weniger als fünf Lagen. Dabei beobachteten die Autoren eine Erhöhung der Oberfläche auf
390 m2∙g-1 (Graphit: 2.8 m2∙g-1), des Weiteren beobachteten sie eine elektrische Leitfähigkeit
von 1.1 × 10-2 S∙cm-1. Durch Erhitzen auf 900 °C unter Argon-Atmosphäre erhält man
reduziertes MG-CO2, welches eine doppelt so große Oberfläche von 630 m2∙g-1 und eine
enorme Steigerung der Leitfähigkeit auf 1214 S∙cm-1 zeigte.[57] Die Vermahlung mit N2 liefert
MG-N2 mit kovalent gebundenem Stickstoff. Dieser liegt in Form von aromatischem Pyrazol
und Pridazin vor und konnte mittels XPS-Analytik nachgewiesen werden.[83] In ähnlicher Weise
wurde MG mit Wasserstoff-, Amino- oder Sulfonsäure-Gruppen durch Vermahlung unter H2,
NH3 oder SO3 erhalten. Einen weiteren Weg zur Herstellung von MG mit Sulfonsäure-Gruppen
stellt die Zugabe von festem Schwefel dar. Außerdem konnten durch Vermahlung mit Phosphor
Phosphonsäure-Gruppen in MG generiert werden, was eine Verwendung als
Flammschutzmittel denkbar macht.[92,94] Des Weiteren führte die Vermahlung mit Halogenen

1 Einleitung
- 11 -
zu halogenierten MG.[88] Darüber hinaus konnte eine Cycloaddition von Maleimid an Graphit
durch Vermahlung realisiert werden, welche mit Raman-Spektren nachgewiesen werden
konnte.[89] Statt HUMMERS-Methode konnte das Graphitoxid auch lösungsmittelfrei durch
Vermahlung von Graphit in Anwesenheit von Asparaginsäure und KMnO4 hergestellt werden.
Das erhaltene GO zeigte eine dünne Schichtstruktur mit hohem Aspektverhältnis.[93] Im
Vergleich dazu konnte unfunktionalisiertes MG auch durch Vermahlung von Graphit in
Ethanol[52] oder mit Melamin[56] hergestellt werden.
Der Herstellungsprozess von MG in einer Kugelmühle ist sehr einfach durchführbar und
gleichzeitig kostengünstig. Durch ein einfaches Scale-up bildet es einen Zugang zu einem
neuen Füllstoffmaterial.
1.1.1.4 Funktionalisiertes Graphen aus Glucose, Dopamin und Furfurylalkohol
Auf alternative Route konnte Graphen nachhaltig aus Glucose mit relativ hoher Ausbeute über
eine einfach durchführbare, kostengünstige Bottom-up-Methode hergestellt werden, welche
keine organischen Lösungsmittel, Katalysatoren, umweltbelasteten Chemikalien oder
Nachreinigungen erfordert.[43,95–97] In der Forschungsgruppe von X. LI[43]
wurde
funktionalisiertes Graphen aus Glucose in Anwesenheit einer graphitischen Kohlenstoffnitrit-
Vorlage über eine templatvermittelte Reaktion synthetisiert (siehe Abbildung 32 in Kapitel
3.4.2). In einem thermischen Prozess (600 °C) unter Schutzatmosphäre wurden die Edukte
Glucose und Dicyandiamid mittels Polykondensation in Graphen und Kohlenstoffnitrit
umgewandelt, wobei sich das entstehende Graphen durch Donor-Akzeptor-Wechselwirkungen
auf der Oberfläche des Kohlenstoffnitrit-Templates schichtartig niederschlug. Bei
anschließender thermischer Behandlung (ab 750 °C) wurde das Kohlenstoffnitrit-Template
vollständig zersetzt und das Graphen blieb zurück. Es resultierte ein mit Stickstoff dotiertes
funktionalisiertes Graphen, welches über eine sehr große spezifische Oberfläche (820 m2∙g-1)
und eine hohe spezifische Leitfähigkeit (8 S∙cm-1) sowie eine dünne gefaltete Schichtstruktur
(Länge: 10 – 100 µm; Dicke: 0.3 – 15 nm) verfügte.

1 Einleitung
- 12 -
Neben der Glucose lässt sich das funktionalisierte Graphen auch aus biobasiertem Dopamin auf
ähnliche Weise gewinnen.[46,98–101] Bei den Forschungsarbeiten von R. LI[46] wurde SiO2/Si-
oder Kupferfolie als Vorlage für die templatvermittelte Synthese von funktionalisiertem
Graphen verwendet (siehe Abbildung 33 in Kapitel 3.4.3). In diesem Verfahren wurde das
Template in eine wässrige Dopamin-Lösung (pH 8.5) eingetaucht, wobei das Dopamin bei
Raumtemperatur auto-polymerisierte[102] und eine dünne Beschichtung aus Polydopamin auf
der Oberfläche des Templates bildete.[103] Durch anschließendes Tempern bei 800 – 1000 °C
wurde der so erhaltene Polydopaminfilm pyrolysiert und in funktionalisiertes Graphen
umgesetzt, welches neben Kohlenstoff auch Wasserstoff, Stickstoff und Sauerstoff in Form von
C-OH-, C=N-, C-O-C- und C-N-Gruppen aufweist. Die Eigenschaften des funktionalisierten
Graphens konnten durch Variation der Reaktionstemperatur eingestellt werden. Dabei führte
die Erhöhung der Pyrolyse-Temperatur von 800 auf 1000 °C zu einer starken Zunahme der
elektrischen Leitfähigkeit von ca. 150 auf 1200 S∙cm-1. Laut XPS-Analyse wurde mit
zunehmender Temperatur ein abnehmendes N/C- und O/C-Verhältnis und ein zunehmender sp2-
Kohlenstoff-Anteil beobachtet, was auf den Abbau von funktionellen Gruppen und den Aufbau
von graphitischer Struktur hinweist. Zudem ist das so hergestellte Graphen hoch
lichtdurchlässig (bis zu 93 %), was eine Anwendung als transparente Elektrode ermöglicht.
Des Weiteren wurde eine Graphensynthese aus Furfurylalkohol als nachwachsende
Kohlenstoffquelle in verschiedenen Forschungsgruppen untersucht.[44,45,104–108] Furfurylalkohol
wird industriell durch katalytische Reduktion aus Furfural gewonnen, welches aus Cellulose
durch Zweifachdestillation gewonnen wird.[109] Die Herstellung von funktionalisiertem
Graphen bei M. BECKERT et al.[44,45] erfolgte ebenfalls über eine templatvermittelte Reaktion.
Dabei wurde ein graphitisches Kohlenstoffnitrid-Template in situ aus Harnstoff und
Dicyandiamid für die templatvermittelte Synthese von funktionalisiertem Graphen hergestellt
(siehe Abbildung 30 in Kapitel 3.4.1). Auf der Oberfläche des Templates wurde
Polyfurfurylalkohol durch eine säurekatalytische Polymerisation aus Furfurylalkohol
synthetisiert, welcher während einer anschließenden thermischen Behandlung (< 750 °C) zu
funktionalisiertem Graphen pyrolysierte. Nach vollständiger Umwandlung zu Graphen wurde
das Gemisch auf 800 °C aufgeheizt, wobei das Kohlenstoffnitrit-Template sich vollständig

1 Einleitung
- 13 -
zersetzte und das funktionalisierte Graphen zurückblieb. Je nach Reaktionsbedingung enthielt
das erhaltene Graphen 8 – 30 Gew.-% Stickstoff, was eine erfolgreiche Funktionalisierung des
Graphens bestätigt. Zusätzlich war eine typisch gefaltete Schichtenstruktur (Dicke: 2 nm) und
eine hohe spezifische Oberfläche von bis zu 470 m2∙g-1 zu beobachten.
Alle vorgestellten Herstellungsmethoden aus organischen Kohlenstoffquellen sind zwar
umweltfreundlich und einfach durchführbar, benötigen aber ein Template, welches
anschließend aufwändig mechanisch (Übertragung/Transfer), chemisch (Ätzen/Lösen) oder
thermisch (Pyrolyse) entfernt werden muss. Dieser große Nachteil beschränkt die Produktion
von funktionalisiertem Graphen in industriellem Maßstab über eine templatvermittelte
Reaktion.
1.2 Keramik
Keramiken sind anorganische, nichtmetallische und in Wasser schwer lösliche Werkstoffe,
welche bei Raumtemperatur aus einer Rohmasse geformt werden und ihre typischen
Werkstoffeigenschaften erst durch den Sinterprozess bei Temperaturen oberhalb 800 °C
erhalten.[110] Je nach Verwendungszweck werden Keramiken in technische Keramik,
Gefäßkeramik, Zierkeramik, Sanitärkeramik, Baukeramik und Ofenkeramik eingeteilt. Zur
technischen Keramiken gehören Hochleistungskeramik, Konstruktionskeramik,
Funktionskeramik, Elektrokeramik, Schneidkeramik und Biokeramik. Alternativ werden
technische Keramiken nach der chemischen Zusammensetzung in Silikatkeramik, Oxidkeramik
und Nicht-Oxid-Keramik eingeteilt.[1–3]
Silikatkeramiken als älteste Gruppe aller Keramiken umfassen generell alle Rohstoffe, die
[SiO4]4−-Tetraeder in der Kristallstruktur eingebaut haben, wie z. B. Tonminerale und Kaoline.
Sie finden in der Wärmetechnik, der Mess- und Regeltechnik, der Verfahrens- und
Umwelttechnik sowie der Hoch- und Niederspannungstechnik Anwendung (z. B. als Isolatoren,
Sicherungspatronen, Katalysatoren oder Gehäuse).

1 Einleitung
- 14 -
Oxidkeramiken bestehen im Wesentlichen aus einphasigen Metalloxiden, wie z. B. Al2O3, ZrO2
und BeO. Häufig werden sie in der Elektrotechnik und der Elektronik z. B. als
Strukturwerkstoff verwendet.
Nicht-Oxid-Keramiken werden aus oxidfreien Rohstoffen hergestellt, wobei es sich dabei meist
um Carbide oder Nitride handelt, wie z. B. SiC, B4C, Si3N4 und BN. Aufgrund des hohen
Anteils kovalenter Bindungen der carbidischen und nitridischen Kristallstrukturen in Nicht-
Oxid-Keramik verfügen diese über hervorragende chemische (Korrosions- und
Temperaturbeständigkeit), mechanische (E-Modul, Härte, Festigkeit) und tribologische
(Verschleißbeständigkeit) Eigenschaften.
Die Rohstoffe für Silikatkeramiken sind meist natürlichen Ursprungs, während Oxid- und
Nicht-Oxid-Keramiken im Wesentlichen aus synthetischen Rohstoffen hergestellt wurden.
Dafür verfügen die Oxid- und die Nicht-Oxid-Keramiken über höhere Reinheiten und spielen
eine größere Rolle in technischen Anwendungen im Vergleich zu Silikatkeramiken. Die am
häufigsten eingesetzten Keramiken sind Werkstoffe aus den Gruppen SiC, Si3N4, Al2O3 und
ZrO2.[1–3]
1.2.1 Siliciumcarbid
Keramische Werkstoffe auf Basis von Siliciumcarbid (SiC) sind in der Technik sicherlich mit
Abstand die bedeutendsten oxidfreien Keramiken. Aufgrund seiner herausragenden
mechanischen, thermischen und chemischen Eigenschaften hat sich SiC in weiten Bereichen
der Industrie und Energietechnik etabliert. Hierbei zeichnet es sich generell durch eine extrem
hohe Härte von 9.6 Mohs bzw. 2600 Vickers/Knoop und eine daraus resultierende große
Abriebfestigkeit, exzellente chemische Beständigkeit (vom stark sauren bis in den stark
basischen Bereich) sowie die Zulassung im Lebensmittel-Bereich aus. Zusätzlich verfügt es
über eine hohe Biegefestigkeit von 120 – 600 MPa und ein E-Modul von bis zu 450 GPa (auch
bei hohen Temperaturen bis 1500 °C) sowie eine sehr gute Temperaturbeständigkeit (bis
1600 °C) bei geringem Wärmeausdehnungskoeffizienten (4.8 ∙ 10-6 K-1), aber hoher

1 Einleitung
- 15 -
Wärmeleitfähigkeit (100 – 350 W m-1 K-1). Im Vergleich zu anderen Keramiken wie z. B.
Wolframcarbid (15.63 g/cm3) und Zirconiumdioxid (5.68 g/cm3) verfügte SiC über eine geringe
Dichte von 3.21 g/cm3. Im Vergleich zu Metall und Kunststoff eignet sich SiC insbesondere bei
der Anwendung im Maschinenbau als hoch verschleißfestes und korrosionsbeständiges
Material unter extremen Bedingungen, z. B. als Gleitringdichtungen in Chemiepumpen,
Gleitlager für sehr hohe Anwendungstemperaturen, für Konstruktionsbauteile von
Hochtemperatur- und Chemie-Anlagen sowie für Kupplungsscheiben und Bremsscheiben
hochwertiger Sportwagen.[111–115] Die wichtigste Herstellungsmethode von SiC erfolgt nach
dem ACHESON-Verfahren aus Quarzsand (SiO2) und Koks (C) bei 2000 °C. Je nach
Herstellungstechnik können Siliciumcarbidkeramiken in drucklos gesintert (SSiC),
rekristallisiert (RSiC), nitridgebunden (NSiC), siliciuminfiltriert (SiSiC),
flüssigphasengesintert (LPSiC), heiß gepresst (HPSiC) und heißisostatisch gepresst (HIPSiC)
eingeteilt werden. Die genauen Werte der oben genannten, typischen Eigenschaften variieren je
nach Herstellungsprozess unterschiedlich stark.[1–3]
1.2.2 Siliciumnitrid
Unter den Nitridkeramiken spielen keramische Werkstoffe auf Basis von Siliciumnitrid (Si3N4)
derzeit eine klar dominierende Rolle in der Technik. Durch die Kombination von
hervorragenden Werkstoffeigenschaften, wie z. B. hoher Biegefestigkeit von 330 – 1000 MPa,
Bruchzähigkeit von 4.0 – 8.5 MPa·m0.5 und hohem E-Modul von 180 – 800 GPa sowie kleinen
Defektgrößen, niedrigem Wärmeausdehnungskoeffizienten von 3.0 – 3.5 ∙ 10-6 K-1 mit
ausgezeichneter Thermoschock-Beständigkeit und hervorragender Verschleißbeständigkeit
sowie guter chemischer und thermischer Beständigkeit bis 1300 °C, eignet sich Si3N4-Keramik
insbesondere für Maschinenbauteile und Lagertechnik mit sehr hohen thermischen,
mechanischen und tribologischen Beanspruchungen und Zuverlässigkeitsanforderungen.[1–3]
Si3N4 wird im Wesentlichen durch direkte Nitridierung von reinem Silicium mit Stickstoff bei
1000 bis 1400 °C synthetisiert. Je nach Herstellungstechnik können Siliciumnitridkeramiken in
heißisostatisch gepresst (HIPSN), gesintert (SSN), reaktionsgebunden (RBSN) und heiß
gepresst (HPSN) eingeteilt werden.[1,2]

1 Einleitung
- 16 -
1.3 Tribologie
Der Begriff Tribologie wurde 1966 von P. JOST eingeführt und von ihm definiert als „die
Wissenschaft und die Technologie der aufeinander einwirkenden, in Relativbewegung
befindlichen Oberflächen und der damit zusammenhängenden praktischen Vorgänge“[116]. Sie
umfasst die Gesamtgebiete von Reibung, Verschleiß sowie Schmierung und dient zur
Optimierung von Bewegungsvorgängen bzw. zur Minderung von Verschleiß und
Reibungskoeffizient. Damit können die Lebensdauer von Maschinen und Anlagen verlängert,
die Energie- und Rohstoffressourcen geschont, der Materialeinsatz reduziert, die
Umweltschäden durch Emissionen gemindert sowie der Arbeitsschutz und die Arbeitssicherheit
verbessert werden.[117] Jährlich entsteht ein Verlust von etwa 5 % des Bruttosozialproduktes in
Industrieländern nur durch Reibung und Verschleiß. Das bedeutet allein für Deutschland ca.
35 Milliarden Euro pro Jahr.[118] Aus diesem Grund ist das wissenschaftliche und technische
Interesse an Tribologie in den letzten Jahren deutlich gestiegen.[119] Damit verbreitet sich die
Anwendung der Tribologie auf alle Bereiche in der Technik mit mechanischen
Bewegungssystemen, wie z. B. Konstruktion, Schmierstoff, Maschinenbau, Fertigungstechnik,
Antriebs- und Fördertechnik, Fahrzeugtechnik, Energieversorgung und Medizintechnik.
Abbildung 4: Darstellung eines tribologischen Systems.[2]

1 Einleitung
- 17 -
Wie in Abbildung 4 dargestellt, besteht ein tribologisches System (Tribosystem) aus Grund-
und Gegenkörper (z. B. Metall oder Keramik), Zwischenstoff (z. B. Schmierstoff) sowie
Umgebungsmedium (z. B. Wasser, Öl oder Gas). Da Reibung und Verschleiß keine reinen
Werkstoffkennwerte (beispielsweise Härte oder E-Modul), sondern Systemeigenschaften sind,
werden sie im Allgemeinen als Verlustgrößen des betreffenden tribologischen Systems
angesehen. Diese Eigenschaften hängen nicht nur von den Materialeigenschaften ab, sondern
auch von den auftretenden vielfältigen Wechselwirkungen und den Größen des
Beanspruchungskollektives, welches aus Eingangsgrößen wie z. B. Normalkraft FN,
Geschwindigkeit v, Temperatur T und Beanspruchungsdauer t sowie den Bewegungsformen
und Störgrößen wie z. B. Vibrationen gebildet wird. Neben Verlustgrößen werden Nutzgrößen
wie die übertragene Kraft erhalten.[2]
1.3.1 Reibung
Reibung ist ein Bewegungswiderstand und äußert sich als die Widerstandskraft, die der
Relativbewegung von zwei sich berührenden Körpern entgegenwirkt. Zur Klassifikation wird
Reibung in äußere und innere Reibung eingeteilt. Zur äußeren Reibung zählen die statische
Ruhereibung und dynamische Bewegungsreibung, während die innere Reibung vor allem durch
die Viskosität beschrieben wird.[117] Grundlage der Tribologie sind die zwei amontonsschen
Gesetze, welche das Verhältnis der Reibungskraft und Normalkraft darstellen. Die
Reibungskraft FR ist von der Ausdehnung der Reibfläche unabhängig und zu der
Normalkraft FN direkt proportional. Der dimensionslose Proportionalitätsfaktor ist der
Reibungskoeffizient µ, welcher von der Materialpaarung und dem Zwischenstoff abhängig ist
und zu den bekanntesten Rechengrößen innerhalb der Tribologie zählt.[120] Im Falle
dynamischer Reibung kann das Verhältnis zwischen Reibkraft bzw. Reibungskoeffizient und
Reibgeschwindigkeit mit Hilfe einer Stribeck-Kurve (Abbildung 5) beschrieben werden.

1 Einleitung
- 18 -
Abbildung 5: Schematisches Diagramm und Prinzip von Stribeck-Kurve.[121]
Zu Beginn eines geschmierten tribologischen Systems trennen nur einige Moleküle des
Schmierstoffs teilweise den Grundkörper vom Gegenkörper. In diesem Grenzreibungsbereich
sind der Verschleiß und der Reibungskoeffizient am höchsten, da die beiden Kontaktkörper fast
ungeschmiert aneinander reiben. Mit zunehmender Gleitgeschwindigkeit wird mehr
Schmierstoff in den Bereich zwischen den Kontaktkörpern befördert, somit nimmt die
Schichtdicke des Schmierfilms zu und der Reibungskoeffizient bis zu einem Minimum ab.[122]
Es herrscht ein Mischreibungsbereich, bei dem nur noch die Rauheitshügel von Grund- und
Gegenkörper ungeschmiert sind. Aufgrund des restlichen Kontakts zwischen Grund- und
Gegenkörper nimmt der Verschleiß zwar im Vergleich zum Grenzreibungsbereich deutlich ab,
aber ist immer noch sehr groß. Bei weiter ansteigender Gleitgeschwindigkeit bzw.
Schmierfilmdicke sind Grund- und Gegenkörper vollständig durch den Schmierstoff getrennt.
In diesem Bereich liegt hydrodynamische Reibung vor, wobei der Verschleiß am niedrigsten ist.
Mit zunehmender Geschwindigkeit erhöht sich der Reibungskoeffizient jedoch wieder, da die
innere Reibung des Schmierstoffs steigt.

1 Einleitung
- 19 -
1.3.2 Verschleiß
Nach der 1997 zurückgezogenen DIN 50320 war Verschleiß als „der fortschreitende
Materialverlust aus der Oberfläche eines festen Grundkörpers, hervorgerufen durch
mechanische Ursachen, d. h. Kontakt- und Relativbewegung eines festen, flüssigen oder
gasförmigen Gegenkörpers“ definiert. In der Praxis wird Verschleiß je nach Beanspruchung in
viele verschiedene Verschleißarten wie z. B. Wälz-, Gleit- und Rollverschleiß aufgegliedert und
hauptsächlich durch die folgenden vier Verschleißmechanismen bestimmt: adhäsiver und
abrasiver Verschleiß, Oberflächenzerrüttung sowie Tribooxidation. Verschleiß ist meist
unerwünscht und tritt oft an Maschinen und Anlagen mit daraus resultierender
Schadenentstehung auf, z. B. an Lagern, Gleitringdichtungen, Kupplungen, Getrieben,
Wasserhähnen und Bremsen. Daher ist die Verringerung oder evtl. Vermeidung von Verschleiß
von hoher wirtschaftlicher Bedeutung, da von ihm die Lebensdauer von Maschinen und
Geräten sowie die resultierten Kosten und benötigten Rohstoffressourcen abhängen.[2] In solch
hoch beanspruchten Tribosystemen werden keramische Werkstoffe wegen ihrer hervorragenden
Verschleißfestigkeit oft eingesetzt. Im Vergleich zu Stahl zeichnen sich die keramischen
Werkstoffe zusätzlich durch ihre geringe Dichte, gute chemische und thermische Beständigkeit
sowie hohe Härte und Druckfestigkeit aus.
1.4 Keramik-Graphen-Nanokomposite
Kohlenstoffbasierte Nanofüllstoffe stellen eine bekannte Möglichkeit zur Verbesserung der
Eigenschaften von keramischen Werkstoffen dar.[123,124] Deswegen wurde in den vergangenen
Jahren verstärkt versucht, durch die Zugabe von Kohlenstoffadditiven, z. B. Carbon Black,
CNTs und Graphenen, die mechanischen,[125] elektrischen,[126] thermischen[127] und
tribologischen[128] Eigenschaften von Keramiken zu verbessern.[123] Dabei wurde besonders
Graphen aufgrund seiner herausragenden Eigenschaften (siehe Kapitel 1.1) in Keramik-
Nanokompositen verwendet, was sich auch in einem rasanten Zuwachs wissenschaftlicher
Publikationen zu diesem Thema widerspiegelt. In Abbildung 6 ist zu sehen, dass sich das

1 Einleitung
- 20 -
Interesse sowohl an Graphen und als auch an Graphen-Keramik-Nanokompositen in den letzten
Jahren drastisch gesteigert hat.
2000 2002 2004 2006 2008 2010 2012 2014 2016
0
5000
10000
15000
20000
25000
30000
35000
Graphene
Graphene + Ceramic
Jahr
An
za
hl P
ub
lika
tio
nen
"G
rap
he
ne
"
0
50
100
150
200
250
300
350
An
za
hl P
ub
lika
tio
nen
"G
rap
he
ne
+ C
era
mic
"
Abbildung 6: Weltweite Veröffentlichungen zu „Graphene“ bzw. „Graphene + Ceramic“ in den Jahren
2000 bis 2016.[129]
Mit Graphen können die mechanischen Eigenschaften von keramischen Werkstoffen wesentlich
verbessert werden. Die erste Veröffentlichung zu graphenverstärkter Si3N4-Keramik aus dem
Jahr 2011 zeigte bereits, dass die Risszähigkeit erheblich gesteigert werden konnte.[130] Das mit
Spark-Plasma-Sintern (SPS) hergestellte Si3N4-Nanokomposit erreichte mit 1.5 Vol.-% TRGO
eine Risszähigkeit von 6.6 MPa·m0.5, was einer Erhöhung um ca. 140 % im Vergleich zur
Referenzprobe (2.8 MPa·m0.5) entspricht. Bei den durch heißisostatisches Pressen (HIP)
gesinterten Si3N4-Nanokompositen konnte die Risszähigkeit durch Einlagerung von
1.0 Gew.-% Multilagengraphen von 7 auf bis zu 10 MPa·m0.5 erhöht werden. Diese für Si3N4-
Keramiken sehr hohen Werte können erreicht werden, weil Risse durch das Graphen abgelenkt,
verzweigt oder überbrückt werden.[131,132] MIRANZO et al. berichteten 2016 von SiC-TRGO-
Nanokompositen, die durch in situ Reduktion von Graphitoxid (GO) hergestellt wurden. Mit
5 Vol.-% TRGO wurde die Risszähigkeit um 162 % im Vergleich zur Referenzprobe
(3.2 MPa·m0.5) erhöht, während eine Steigerung der Biegefestigkeit von 370 auf 620 MPa

1 Einleitung
- 21 -
beobachtet wurde.[133] Entsprechende Ergebnisse gibt es auch bei oxidkeramischen
Nanokompositen. Die Biegefestigkeit von Al2O3 stieg bei Zugabe von 0.2 Gew.-% TRGO von
ca. 350 auf 620 MPa. Zudem wurde eine wesentliche Erhöhung der Risszähigkeit von 3.4 auf
5.1 MPa∙m0.5 beobachtet.[134] Bei Al2O3/ZrO2-Mischkeramiken wurde von LIU et al. durch
Zugabe von 0.8 Vol.-% Graphen bereits eine Erhöhung der Risszähigkeit um 40 % im Vergleich
zum Referenzmaterial berichtet.[135]
Neben der mechanischen Eigenschaftsverbesserung kann die elektrische Leitfähigkeit der
keramischen Werkstoffe durch Zugabe von Graphen drastisch verbessert werden.
MIRANZO et al. beobachteten bei SPS-gesintertem SiC mit 4 Vol.-% epitaktischem Graphen
eine Erhöhung der elektrischen Leitfähigkeit von ~ 10-9 auf bis zu 1 S∙cm-1.[136] Dieselbe
Forschungsgruppe erreichte mit 20 Vol.-% Füllstoffanteil eine Zunahme der elektrischen
Leitfähigkeit des SiC von 2 × 10-8 (reine Matrix) auf 44 S∙cm-1 (gefüllte Probe).[137] Eine
ähnliche Verbesserung der elektrischen Leitfähigkeit von Al2O3 wurde von SUVACI et al.
berichtet. Durch Beimischung von 9 Vol.-% Graphen erhöhte sich die elektrische Leitfähigkeit
um 9 Größenordnungen auf ca. 0.01 S∙cm-1.[138] OSENDI et al. stellten Si3N4-Nanokomposite
mit 25 Vol.-% kommerziellem Graphen her und erreichten eine Zunahme der elektrischen
Leitfähigkeit von 10-13 auf 104 S∙cm-1.[139] Grund dafür ist, dass Graphen ein durchgehendes
Netzwerk in der Keramikmatrix bildet, welches den elektrischen Strom leiten kann.
Wie aus den bisherigen Ausführungen zu erkennen ist Graphen sehr interessant für
tribologische Systeme und könnte in diesem Bereich vielseitig verwendet werden. So können
Graphene als Beschichtung oder als Additiv in Beschichtungen,[140–142] in Schmiermedien[143,144]
und als Füllstoff in Kompositwerkstoffen[123] zu einer tribologischen Verbesserung beitragen.
Die Verwendung von Graphen als Nanofüllstoff in verschiedenen Kompositwerkstoffen wie
z. B. Metallen,[145,146] Polymeren oder Keramiken[123] ist von großer Bedeutung in
tribologischen Systemen. So gibt es zahlreiche Veröffentlichungen, die verbessertes Reib- und
Verschleißverhalten der mit Graphitoxiden modifizierten oder mit funktionalisierten Graphenen
gefüllten Polymere wie z. B. UHMWPE,[147] Epoxidharzen,[148] PA,[142,149] PEEK[150] und
Polyimiden[151] sowie Keramiken wie z. B. SiC,[152] Si3N4,[132,153,154] Al2O3,
[155,156] SiO2,[157]

1 Einleitung
- 22 -
Bioglas,[158] B4C,[159] ZrO2[160] und CaSiO3
[161] erwähnen. Für Keramiken können ihre
tribologischen Eigenschaften bereits mit sehr niedrigen Graphengehalten (1 – 10 Gew.-%)
deutlich verbessert werden.
Im Jahr 2013 gab es die ersten Publikationen über die tribologischen Eigenschaften von
graphenverstärkten Si3N4-Keramiken. HVIZDOS et al.[132] und BELMONTE et al.[162] untersuchten
Si3N4-Graphen-Nanokomposite unter trockenen oder Isooctan-geschmierten Bedingungen.
Beide Forschungsgruppen zeigten, dass die Verschleißbeständigkeit des Nanokomposits mit
3.0 Gew.-% Füllstoffanteil trotz unverminderter Reibung unabhängig der Testbedingungen um
ca. 60 % verbessert werden konnte.
SEDLAK et al. beschäftigten sich mit der Herstellung von B4C-Graphen-Nanokompositen. Mit
6.8 Vol.-% Füllstoffanteil konnte der Reibungskoeffizient von 0.58 auf 0.35 reduziert werden,
zudem wurde die Verschleißbeständigkeit um 77 % verbessert.[159]
ZHANG et al. berichteten 2016 von Al2O3-Graphen-Nanokompositen, die nach dem SPS-
Verfahren hergestellt wurden. Mit 0.5 Gew.-% Graphen wurde die Verschleißbeständigkeit um
65 % erhöht, während eine Reduzierung des Reibungskoeffizienten um 18 % beobachtet
wurde.[156]
Diese tribologischen Leistungen durch Zugabe von Graphen begründen sich vor allem durch
seinen selbst-schmierenden Effekt. Zusätzlich kann Graphen während der tribologischen
Untersuchungen einen haftenden und schützenden Tribofilm aus Kohlenstoff auf der
Oberfläche der Nanokomposite bilden, was die Reibungskoeffizienten und die
Verschleißvolumina stark reduziert.[152,154,156,158,163]

2 Aufgabenstellung
- 23 -
2 Aufgabenstellung
Zahlreiche Untersuchungen haben insbesondere die mechanische Verstärkung von Polymeren
durch Zugabe von Graphen gezeigt.[164–171] Für keramische Werkstoffe gehört die Verwendung
von Graphen auch zu den seltenen sinnvollen Möglichkeiten, um eine Verstärkung oder
Funktionalisierung zu erzielen. Deswegen wurden Graphene seit sechs Jahren[123] in der
Keramikkomposit-Herstellung als Füllstoffe eingesetzt, um deren thermische,[172,173]
mechanische,[130,133] elektrische[137,139] und tribologische[132,152] Eigenschaften zu verbessern.[123]
Damit ein Nanofüllstoff die gewünschten Effekte erzielt, ist ein hohes Aspektverhältnis
(Längen-/Dicke-Verhältnis), eine gute Dispergierung sowie eine gute Kompatibilisierung
zwischen Keramikmatrix und Füllstoff wichtige Voraussetzung. Keramische Werkstoffe auf
Basis von Siliciumcarbid (SiC) und Siliciumnitrid (Si3N4) zählen zu den bedeutendsten Nicht-
Oxid-Keramiken und werden heute aufgrund ihrer exzellenten chemischen und thermischen
Beständigkeit und sehr guten mechanischen und tribologischen Eigenschaften sowie der
Gewichtseinsparung in vielen Bereichen der Industrie und Energietechnik eingesetzt.[1,2]
Deswegen wurden SiC und Si3N4 in dieser Dissertation als vielversprechende Matrix-Systeme
eingesetzt werden. Um die Effizienz, Lebensdauer, Zuverlässigkeit und Leistungsfähigkeit
keramischer Komponenten in den Anwendungen unter Wasser- bzw. Mangelschmierung, aber
auch zur Förderung anderer Medien gezielt zu verbessern, sollen leistungsfähigere Werkstoffe
durch die Kombination von Graphen und den genannten Keramiksystemen synthetisiert werden.
Das bisher größte Problem bei der Anwendung von Graphen als Füllstoff in Keramiken ist die
Entstehung von Poren, was zur Verschlechterung der mechanischen Eigenschaften führt. Daher
liegt ein kombinatorischer Anstieg der mechanischen und tribologischen Eigenschaften mit
vollständiger Verdichtung durch Graphen in Si3N4- und SiC-Nanokompositen bisher nicht vor.
Zentrales Ziel der vorliegenden Dissertation war die Herstellung und Charakterisierung von
neuartigen Keramik-Nanokompositen auf Basis von Siliciumcarbid (SiC) und Siliciumnitrid
(Si3N4) mit bislang unbekannten Materialeigenschaftskombinationen. Die Nanokomposite
sollen entweder aus dem Gemisch von reinem Keramikrohstoff und Graphen oder aus

2 Aufgabenstellung
- 24 -
graphenbeschichtetem Keramikrohstoff durch Sintern hergestellt werden. Um eine homogene
Durchmischung von Graphen und Keramik zu gewährleisten bzw. die Porenvolumina von
hergestellten Keramik-Graphen-Nanokompositen zu minimieren, sollen Graphene mittels
Präkursor-Verfahren direkt auf die Oberfläche der Keramikrohstoffe aufgebracht werden,
wobei graphenbeschichtete SiC (GSiC) und Si3N4 (GSi3N4) erhalten werden. Im Vergleich dazu
kommen als reine Graphene thermisch reduziertes Graphitoxid (TRGO) und mechanochemisch
funktionalisiertes Multilagen-Graphen (MG) aus trockener Vermahlung von Graphit zum
Einsatz, dessen Eigenschaften (Funktionalität und Morphologie) durch die einfache Variation
der Prozessbedingungen (Reduktionstemperatur bzw. Mahldauer und Gasatmosphäre)
gesteuert werden können. Außerdem werden kommerzielle plättchenförmige
Kohlenstofffüllstoffe Graphit, Nanographit, Grade M (Multilagen-Graphen) und MG-Ethanol
(unfunktionalisiertes Multilagen-Graphen), sowie die sphärischen Kohlenstofffüllstoffe Carbon
Black und Grade C als Referenz untersucht, um diese als Füllstoff in Keramik-Nanokompositen
mit selbst hergestellten Graphenen zu vergleichen. Der Fokus liegt dabei neben der
Eigenschaftsverbesserung auch auf der Verfügbarkeit und Kosteneffizienz der eingesetzten
Kohlenstofffüllstoffe. Die Herstellung und Analytik der Keramik-Graphen-Nanokomposite
fand aufgrund des hohen Material- und Zeitaufwandes in enger Zusammenarbeit mit anderen
Forschungsinstituten und Industrien innerhalb des CERAPHENE-Projekts statt. Durch die
Kooperation insbesondere mit Industriepartnern aus dem Bereich Keramikherstellung und der
Anwendungen im Bereich Gleitlager und Gleitringdichtungssysteme wird sichergestellt, dass
die gewonnenen Erkenntnisse nicht nur wissenschaftlich, sondern auch wirtschaftlich verwertet
werden.
Durch die Einarbeitung von Graphen in Keramik wird erwartet, dass es als Füllstoff in
tribologisch beanspruchten Keramikkomponenten durch mechanische Verstärkung und
chemische Passivierung sowohl die Reibung als auch den Verschleiß erheblich senkt. Damit
wird der Energieverlust beim Betrieb der Keramikkomponenten beispielsweise für den
Anlagenbau (Wälz- & Gleitlager), insbesondere in Pumpen, Mischern und Rührwerken,
vermindert und die Lebensdauer erhöht. Außerdem sollen die erwarteten Nanokomposite
verbesserte mechanische und elektrische Eigenschaften aufweisen. Mit erhöhten mechanischen

2 Aufgabenstellung
- 25 -
Eigenschaften wird die Belastbarkeit sowie die Zuverlässigkeit der Werkstoffe erhöht. Darüber
hinaus könnten durch erhöhte elektrische Leitfähigkeiten der Keramiken einerseits
Schutzvorrichtungen zur weiteren Verminderung von Reibung, Verschleiß und Korrosion
entwickelt werden.[174] Andererseits würde ein geringer elektrischer Widerstand ermöglichen,
den Keramikwerkstoff mittels Elektroerosionsverfahren zu bearbeiten, was eine Erweiterung
der geometrisch darstellbaren Formen und Bauteilgeometrien bedeuten würde.[124,175–177]
2.1 Graphensynthese und Charakterisierung
Teilziel der Arbeit ist die Entwicklung eines Syntheseprozesses zur Herstellung von
funktionalisierten Graphenen in großem Maßstab (10 – 200 g) durch angepasste chemische und
thermische Verfahrensschritte für die Herstellung von Keramik-Graphen-Nanokompositen.
Gemäß dem Top-down-Ansatz soll einerseits das thermisch reduzierte Graphitoxid (TRGO)
durch thermische Reduktion und Expansion des Graphitoxids (GO), welches nach HUMMERS-
Verfahren aus Graphit synthetisiert wurde, hergestellt werden. Der Einfluss der
Reduktionstemperatur auf den Funktionalisierungsgrad, die Temperaturbeständigkeit, die
spezifische Oberfläche und den Exfolierungsgrad des TRGOs soll untersucht werden.
Andererseits soll das mechanochemisch funktionalisierte Multilagen-Graphen (MG) durch
einen einstufigen Trockenvermahlungsprozess in einer Planetenkugelmühle unter Ar-, CO2-
oder N2-Atmosphäre direkt aus Graphit hergestellt werden. Dabei soll der Einfluss der
Prozessparameter (Mahldauer und Gas-Atmosphäre) auf die Funktionalisierung und
Partikelstruktur des MGs analysiert werden. Hierbei ist die Anzahl funktioneller Gruppen in
TRGO und MG entscheidend, um die Dispergierungsfähigkeit in Lösemitteln und letztendlich
die Grenzflächeneigenschaften zwischen Graphen und Keramik gezielt einzustellen. Darüber
hinaus sollen stabile tensidfreie MG- und TRGO-Dispersionen durch den Einsatz eines
Hochdruckhomogenisators (HH) hergestellt werden. Dabei soll Wasser statt flüssigen
organischen Stoffen als Lösungsmittel verwendet werden, was einerseits umweltfreundlich ist
und andererseits die weitere Verarbeitung bei der Herstellung von Nanokompositen leichter
macht.

2 Aufgabenstellung
- 26 -
Als Alternative zum Top-down-Ansatz soll das Graphen mittels Bottom-up-Methode aus drei
verschiedenen Graphen-Präkursoren, nämlich Furfurylalkohol, Glucose und Dopamin, über
eine templatvermittelte Reaktion direkt auf die Oberfläche von SiC (GSiC) und Si3N4 (GSi3N4)
synthetisiert werden. Dies bildet ein essentielles Ziel dieser Arbeit. Dabei wird der Einfluss der
des Edukts sowie der Eduktmenge auf die Materialeingenschaften wie Morphologie,
Graphengehalt und spezifische Oberfläche untersucht. Im Vergleich zu TRGO und MG ist
hierbei weder ein Einsatz von fossilen Kohlenstoffquellen (z. B. Graphit) noch eine
nachträgliche Behandlung (z. B. Reinigung und Homogenisierung) erforderlich. Zusätzlich ist
bei den Keramik-Nanokompositen mit GSiC oder GSi3N4 im Vergleich zu den konventionellen
Graphenen höhere Verdichtung und als Folge bessere Werkstoffeigenschaften zu erwarten.
Plättchenförmige Kohlenstofffüllstoffe Graphit und Nanographit (von AMG Mining), Grade M
(Multilagen-Graphen von XG Sciences) und MG-Ethanol (unfunktionalisiertes Multilagen-
Graphen von Projektpartner Dr. C. BALÁZSI aus Institute for Technical Physics and Materials
Science, Centre for Energy Research) sowie die sphärischen Kohlenstofffüllstoffe Carbon
Black (von Orion Engineered Carbons) und Grade C (Graphen von XG Sciences) werden als
Referenzen verwendet, um den verstärkenden Füllstoffcharakter der selbst hergestellten
Graphene beurteilen zu können. Dazu sollen die Unterschiede der Morphologie, der
Funktionalität und des Aspektverhältnisses charakterisiert werden. Darüber hinaus soll die
thermische Stabilität der eingesetzten Graphen-Füllstoffe bis zu 2000 °C per TGA-
Untersuchung evaluiert werden, um sicherzustellen, dass die Füllstoffe während des
Sintervorgangs nicht abbauen.
2.2 Graphen-Nanokomposite auf Basis von Siliciumcarbid
Im Vergleich zu den anderen technischen Keramiken waren SiC-Graphen-Nanokomposite zu
Beginn dieser Arbeit kaum erforscht. Im Zeitraum dieser Dissertation wurden nur einige wenige
Publikationen über graphengefülltes SiC veröffentlicht. Die Einarbeitung von GO oder
kommerziellem Graphen in SiC erzielte zwar verschleißarme und elektrisch leitfähige[137]

2 Aufgabenstellung
- 27 -
Systeme aber auf Kosten der mechanischen Eigenschaften und der Reibung.[152] Bisher ist kein
SiC-Graphen-Nanokomposit, welches über reduzierte Reibung und reduzierten Verschleiß bei
gleichbleibenden mechanischen Eigenschaften verfügt, literaturbekannt. Der zweite
Schwerpunkt dieser Arbeit liegt auf der Herstellung tribologisch verstärkter SiC-Graphen-
Nanokomposite mit verbesserten elektrischen und mechanischen Eigenschaften mittels
drucklosen Sinterverfahren. Zu diesem Zweck sollen die selbst synthetisierten TRGO, MG und
GSiC, sowie die kommerziellen erhältlichen Kohlenstofffüllstoffe als funktionelle Füllstoffe
verwendet werden. Die erhaltenen SiC-Nanokomposite werden schließlich morphologisch,
mechanisch, elektrisch und tribologisch charakterisiert. Außerdem soll der Einfluss des
Funktionalitätsgrads von TRGO und MG, welcher durch die Variation der
Reduktionstemperatur bzw. Mahldauer und Gasatmosphäre eingestellt wurde, auf die Porosität,
die mechanischen, elektrischen und tribologischen Eigenschaften von entsprechenden
Nanokompositen ermittelt werden. Zudem soll der technische Nutzen von SiC-Graphen-
Nanokompositen, insbesondere im Hinblick auf die mögliche Anwendung in Pumpen, bewertet
werden. Dabei sollen Gleitringe aus den SiC-Graphen-Nanokompositen hergestellt und dessen
tribologische Eigenschaften im Vergleich zu den kommerziellen Gleitringprodukten der Firma
3M unter anwendungsnahen Bedingungen untersucht werden.
Des Weiteren soll die Möglichkeit zur Herstellung von SiC-Graphen-Nanokompositen mittels
Spark-Plasma-Sinterverfahren, welches schnellere Sinterzyklen mit verbesserten
Werkstoffeigenschaften ermöglicht, statt dem konventionellen drucklosen Sinterverfahren
untersucht werden. Wegen seines extrem hohen spezifischen Durchgangswiderstandes ist die
Verwendung des SPS-Verfahrens bei reinem SiC unmöglich. Die Beimischung von Graphen
könnte eine elektrische Leitfähigkeit dieser Pulvermischungen bewirken, wodurch das SPS-
Verfahren zur Herstellung der SiC-Nanokomposite eingesetzt werden könnte. Weiterhin sollen
die mittels SPS-Verfahren gewonnenen Nanokomposite charakterisiert und anschließend mit
den aus konventionellen Sinterverfahren hergestellten Nanokompositen verglichen werden.
Das Interesse gilt, neben der tribologischen Charakterisierung, der Porosität und der
Biegefestigkeit sowie der elektrischen Leitfähigkeit der Produkte.

2 Aufgabenstellung
- 28 -
2.3 Graphen-Nanokomposite auf Basis von Siliciumnitrid
Im Vergleich zu SiC-Werkstoffen wurden die Si3N4-Graphen-Nanokomposite intensiver
erforscht. Die Herstellung der Si3N4-Nanokomposite mit reduzierter Porosität konnte durch die
Zugabe von Graphen verwirklicht werden.[154] Darüber hinaus führte die Inkorporierung von
Graphen zu verbesserten mechanischen Eigenschaften.[130,178–181] Außerdem konnten die
tribologischen Eigenschaften durch die Einarbeitung von Graphen optimiert
werden.[131,132,154,162,178,182] Allerdings liegt ein kombinatorischer Anstieg der mechanischen und
tribologischen Eigenschaften mit vollständiger Verdichtung durch Graphen in Si3N4-
Nanokompositen bisher nicht vor. Daher stellt die Erreichung dieser Eigenschaftenkombination
ein angestrebtes Ziel dieser Dissertation dar. Des Weiteren wurde der Einfluss weder von
mechanochemisch funktionalisiertem Multilagen-Graphen (MG) noch von GSi3N4 auf die
Eigenschaften von Si3N4-Werkstoffen bislang untersucht. Die Herstellung von Si3N4-Graphen-
Nanokompositen soll sowohl mit den selbst hergestellten Graphenen als auch mit den
kommerziellen Benchmark-Graphenen verwirklicht werden. Die Charakterisierung der
Porosität, der Morphologie, der mechanischen und tribologischen Eigenschaften soll als
Funktion des Füllstoffs erfolgen.

3 Graphensynthese und Charakterisierung
- 29 -
3 Graphensynthese und Charakterisierung
3.1 Einleitung
In jüngster Zeit wurden kohlenstoffbasierte Nanomaterialien wie Carbon Black (CB), Carbon
Nanotubes (CNTs), und (funktionalisiertes) Graphen in der Keramikkompositherstellung als
Füllstoffe eingesetzt, um deren thermische, mechanische, elektrische und tribologische
Eigenschaften zu verbessern. Damit ein Nanofüllstoff die gewünschten Effekte erzielt, sind ein
hohes Aspektverhältnis (Länge-/Dicke-Verhältnis), eine homogene Verteilung sowie eine gute
Kompatibilisierung zwischen Keramikmatrix und Füllstoff wichtige Voraussetzungen.
Das übergeordnete Ziel dieser Arbeiten war die Herstellung und Charakterisierung von
neuartigen Keramik-Graphen-Nanokompositen mit bislang unbekannten
Materialeigenschaftskombinationen. Zu diesem Zweck wurden verschiedene funktionalisierte
Graphene durch angepasste chemische und thermische Verfahrensschritte hergestellt. Dabei
erfolgte die Herstellung entweder mittels Top-down-Ansatz aus Graphit oder als Bottom-up-
Ansatz aus einer Kohlenstoffquelle (Abbildung 7).
Abbildung 7: Übersicht der eingesetzten Graphene zur Herstellung von Keramik-Graphen-
Nanokompositen.

3 Graphensynthese und Charakterisierung
- 30 -
Wie in Abbildung 7 dargestellt, wurde das Graphen gemäß des Top-down-Ansatzes einerseits
über die thermische Reduktion von Graphitoxid (TRGO), andererseits über mechanochemische
Funktionalisierung von Graphit (MG) hergestellt. Für die spätere Herstellung von Keramik-
Graphen-Nanokompositen werden TRGO und MG in Form als stabile Dispersionen benutzt,
welche mittels Hochdruckhomogenisator hergestellt wurden. Um eine homogene
Durchmischung von Graphen und Keramik zu gewährleisten bzw. die Porenvolumina von
hergestellten Keramik-Graphen-Nanokompositen zu minimieren wurde auch versucht,
Keramikrohstoffe vor dem Sintern gemäß eines Bottom-up-Ansatzes mit Graphen zu
beschichten. Als Referenzen wurden kommerzielle Benchmark-Kohlenstofffüllstoffe
untersucht, um diese mit den selbst hergestellten Graphenen zu vergleichen. Der Fokus liegt
dabei neben der Eigenschaftsverbesserung auch auf der Verfügbarkeit und Kosteneffizienz der
eingesetzten Füllstoffe. In diesem Kapitel werden die verschiedenen eingesetzten
Füllstoffsysteme hinsichtlich ihrer unterschiedlichen Herstellungsweisen sowie der hieraus
resultierenden Eigenschaften vorgestellt.
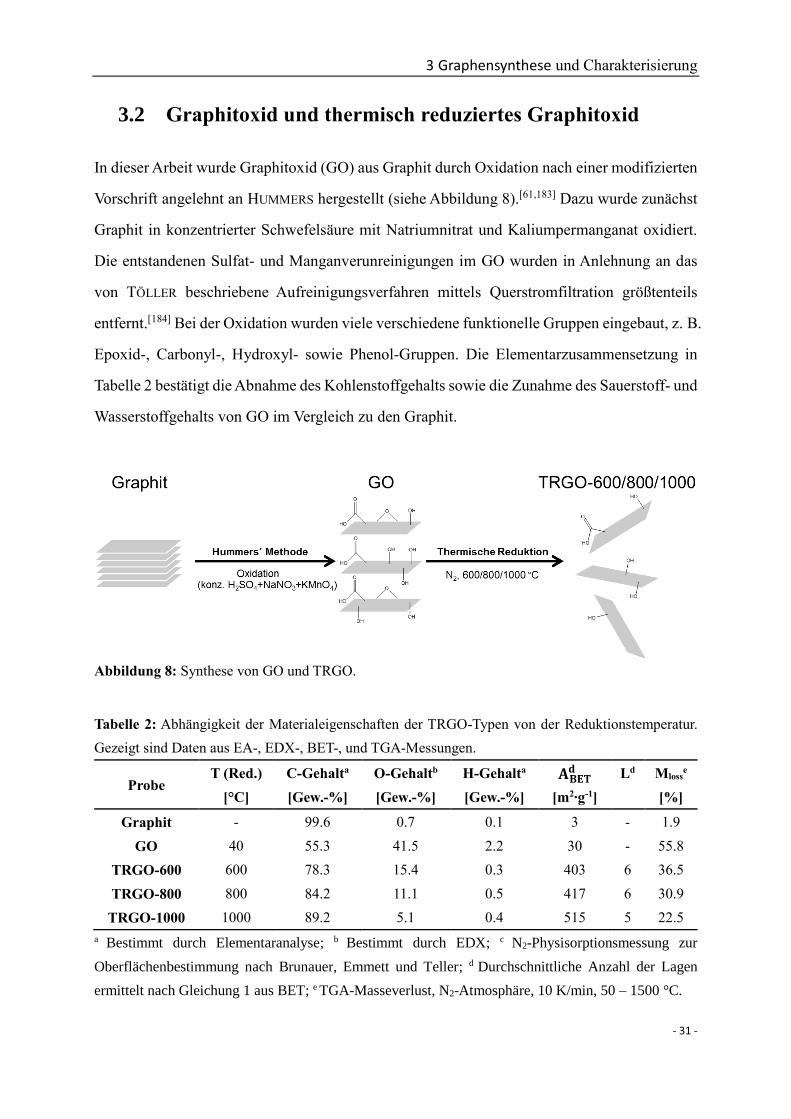
3 Graphensynthese und Charakterisierung
- 31 -
3.2 Graphitoxid und thermisch reduziertes Graphitoxid
In dieser Arbeit wurde Graphitoxid (GO) aus Graphit durch Oxidation nach einer modifizierten
Vorschrift angelehnt an HUMMERS hergestellt (siehe Abbildung 8).[61,183] Dazu wurde zunächst
Graphit in konzentrierter Schwefelsäure mit Natriumnitrat und Kaliumpermanganat oxidiert.
Die entstandenen Sulfat- und Manganverunreinigungen im GO wurden in Anlehnung an das
von TÖLLER beschriebene Aufreinigungsverfahren mittels Querstromfiltration größtenteils
entfernt.[184] Bei der Oxidation wurden viele verschiedene funktionelle Gruppen eingebaut, z. B.
Epoxid-, Carbonyl-, Hydroxyl- sowie Phenol-Gruppen. Die Elementarzusammensetzung in
Tabelle 2 bestätigt die Abnahme des Kohlenstoffgehalts sowie die Zunahme des Sauerstoff- und
Wasserstoffgehalts von GO im Vergleich zu den Graphit.
Abbildung 8: Synthese von GO und TRGO.
Tabelle 2: Abhängigkeit der Materialeigenschaften der TRGO-Typen von der Reduktionstemperatur.
Gezeigt sind Daten aus EA-, EDX-, BET-, und TGA-Messungen.
Probe T (Red.)
[°C]
C-Gehalta
[Gew.-%]
O-Gehaltb
[Gew.-%]
H-Gehalta
[Gew.-%]
𝐀𝐁𝐄𝐓𝐝
[m2∙g-1]
Ld Mlosse
[%]
Graphit - 99.6 0.7 0.1 3 - 1.9
GO 40 55.3 41.5 2.2 30 - 55.8
TRGO-600 600 78.3 15.4 0.3 403 6 36.5
TRGO-800 800 84.2 11.1 0.5 417 6 30.9
TRGO-1000 1000 89.2 5.1 0.4 515 5 22.5
a Bestimmt durch Elementaranalyse; b Bestimmt durch EDX; c N2-Physisorptionsmessung zur
Oberflächenbestimmung nach Brunauer, Emmett und Teller; d Durchschnittliche Anzahl der Lagen
ermittelt nach Gleichung 1 aus BET; e TGA-Masseverlust, N2-Atmosphäre, 10 K/min, 50 – 1500 °C.

3 Graphensynthese und Charakterisierung
- 32 -
Die Morphologie des Graphits und von GO wurde mittels SEM- und TEM-Aufnahmen
analysiert. Der kommerzielle Graphit zeichnete sich typischerweise durch zwiebelartig
aufeinander gelagerte Plättchen aus. Zudem verfügte er über eine enge Partikelgrößenverteilung
mit einer Kantenlänge von ca. 400 µm und einer einheitlichen Dicke von 1.7 µm. Graphitoxid
verfügte ebenfalls über eine gut erkennbare, aber viel dünnere Schichtstruktur, wobei die
Abstände zwischen den einzelnen Kohlenstoffschichten aufgrund der eingebauten
funktionellen Gruppen größer und unregelmäßiger als im Graphit sind (siehe Abbildung 9).[185]
Abbildung 9: SEM-Aufnahmen von Graphit (a) und TEM-Aufnahmen von GO (b) bei variierenden
Vergrößerungen.
Thermisch reduziertes Graphitoxid (TRGO) wurde aus getrocknetem und vermahlenem
Graphitoxid (GO) durch starkes Erhitzen bei verschiedenen Reduktionstemperaturen (600 °C,
800 °C, 1000 °C) im Drehrohrofen hergestellt. Für die TRGO-Proben wurde der vereinfachte
Probencode eingesetzt, welcher aus dem TRGO sowie der Reduktionstemperatur in °C besteht.
Die thermische Reduktion zu TRGO erfolgte unter Stickstoffatmosphäre, damit eine Reaktion
des GO bzw. TRGO mit Sauerstoff ausgeschlossen werden konnte. Bei schnellem Erhitzen von
Graphitoxid werden die funktionellen Gruppen wie Carboxyl-, Epoxid-, und Hydroxylgruppen
sowie die adsorbierte Luftfeuchtigkeit abgespalten und als Kohlenstoffmonoxid, -dioxid und
Wasserdampf freigesetzt. Der dabei entstehende Druck zwischen den Kohlenstofflagen reißt
die durch π-Wechselwirkungen zusammengehaltenen Schichten auseinander. Durch die
Expansion der Schichten wird das Volumen drastisch vergrößert und die Schüttdichte nimmt
deutlich ab. Man erhaltet bei 600 °C Reduktionstemperatur eine spezifische Oberfläche von
rund 400 m2∙g-1, was mit der Exfolierung korreliert. Die Beziehung zwischen

3 Graphensynthese und Charakterisierung
- 33 -
Reduktionstemperatur und Morphologie, Größe der Oberfläche, Funktionalität sowie
thermische Stabilität wurden untersucht. Charakteristisch für den Reduktionsgrad des TRGO
ist sein Sauerstoffgehalt. Dieser nahm proportional mit höherer Reduktionstemperatur ab (siehe
O-Gehalt in Tabelle 2), was auf die Abspaltung der Sauerstofffunktionalitäten und die
Rückgewinnung der sp2-Zentren zurückzuführen ist. Das konjugierte π-System vergrößert sich,
wodurch die Leitfähigkeit, spezifische Oberfläche und der E-Modul anstiegen.[23,75] Nach der
Reduktion verbleiben noch 5 – 15 Gew.-% Sauerstoff im TRGO (siehe Tabelle 2), welcher
hauptsächlich in Form von kantenständigen Hydroxylgruppen (wegen besserer
Temperaturbeständigkeit) vorliegt, die die hohe Polarität von TRGO hervorrufen und eine gute
Dispergierbarkeit in Wasser gewährleisten sowie eine verbesserte Kompatibilität mit der
Keramik-Matrix im Vergleich zum unfunktionalisierten Graphen ermöglichen.[186–189] Die
Morphologie des TRGO wurde mittels SEM (Abbildung 10), TEM (Abbildung 11) und AFM
(Abbildung 12) untersucht.
Abbildung 10: SEM-Aufnahmen von (links) TRGO-600, (Mitte) TRGO-800 und (rechts) TRGO-1000.

3 Graphensynthese und Charakterisierung
- 34 -
Abbildung 11: TEM-Aufnahmen von TRGO-600 (a), TRGO-800 (b) und TRGO-1000 (c).
Abbildung 12: TEM- (a) und AFM-Aufnahmen (b) von TRGO.
Wie in den SEM- und TEM-Aufnahmen in Abbildung 10 und Abbildung 11 zu sehen, zeigen
alle TRGOs eine dünne gewellte und gefaltete Schichtstruktur, die durch das Entweichen der
Gase beim Reduktionsprozess entstehen. Die Reduktionstemperatur beeinflusst die
Morphologie nur gering. Die TEM- und AFM-Aufnahmen (Abbildung 12) zeigen eine
uneinheitliche Dicke der TRGO-Flocken. Die gemessene Schichtdicke von 0.8 nm bestätigt,
dass zumindest teilweise exfolierte Monolagen vorliegen. Im Gegensatz dazu erscheint die
mittlere Stelle deutlich dicker, was auf die Anlagerung weiterer Schichten zurückzuführen ist.
Die durchschnittliche Anzahl an Graphenlagen ist über Gleichung 1 bestimmbar. Die
Schichtzahl (L) von TRGO entspricht dem Wert der spezifischen Oberfläche des idealen
Graphens (Sideal: 2630 m2∙g-1) dividiert durch die gemessene spezifische Oberfläche des TRGOs
(STRGO).[190] Die berechnete Schichtzahl wurde mit zunehmender Reduktionstemperatur
niedriger und liegt insgesamt im Bereich zwischen 5 und 6 Lagen (siehe Tabelle 2). Diese Werte

3 Graphensynthese und Charakterisierung
- 35 -
stimmen mit den Ergebnissen der morphologischen Untersuchungen überein. Die TEM-
Aufnahme unter hoher Auflösung in Abbildung 12 zeigt, dass eine TRGO-Flocke aus sieben
Graphenlagen besteht, was in etwa der berechneten durchschnittlichen Schichtzahl entspricht.
TRGOTRGO
ideal
SS
SL
2630 (1)
Tabelle 2 zeigt charakteristische Messwerte verschiedener TRGO-Typen aus einer
Versuchsreihe mit verschiedenen Reduktionstemperaturen. Die thermische Behandlung
beeinflusst die Materialeigenschaften deutlich. In Abbildung 13 sind die Zunahme des
Kohlenstoffgehalts sowie die Abnahme des Sauerstoffgehalts in TRGO mit höherer
Reduktionstemperatur aufgetragen. Dieser Effekt beruht auf der Abspaltung der
Sauerstofffunktionalitäten und der Bildung von sp²-Zentren. Dabei vergrößert sich das
konjugierte π-System und die spezifische Oberfläche steigt. Man erhaltet bei 600 °C
Reduktionstemperatur eine spezifische Oberfläche von rund 400 m²/g und bei 1000 °C ca.
520 m²/g. Nach der Reduktion verbleibt immer noch Sauerstoff im TRGO, welcher
hauptsächlich in Form von kantenständigen Hydroxylgruppen vorliegt, die zu einer hohen
Polarität des Materials führen und eine gute Dispergierbarkeit in Wasser gewährleisten. Diese
funktionellen Gruppen wurden teilweise auch bei der TGA-Messung abgebaut. Bei den TRGO-
Proben wird ein kontinuierlicher Masseverlust detektiert, der mit abnehmendem
Sauerstoffgehalt der Probe niedriger ausfällt.
600 700 800 900 1000
10
20
80
90
C
O
Geh
alt
[G
ew
.-%
]
T (Red.) [°C]
Abbildung 13: Auswirkung der Reduktionstemperatur auf die elementare Zusammensetzung von
TRGO.

3 Graphensynthese und Charakterisierung
- 36 -
Die funktionellen Gruppen in GO und TRGO wurden mittels FT-IR-Spektroskopie
charakterisiert. In Abbildung 14 sind die FT-IR-Spektren von Graphit, GO und TRGO
dargestellt.
4000 3500 3000 2500 2000 1500 1000 500
c) TRGO
b) GO
Inte
nsitä
t [a
.u.]
Wellenzahl [cm-1]
a) Graphit
Abbildung 14: FT-IR-Spektrum von a) Graphit, b) GO und c) TRGO.
Das breite Signal der OH-Streckschwingung der Hydroxylgruppen bei 3444 cm-1 ist in allen
Spektren zu erkennen und kann dem angelagerten Wasser zugeordnet werden. Im Gegensatz zu
dem Graphit zeigten GO und TRGO zwei neue Banden bei 2923 und 2852 cm-1, welche von
symmetrischen CH2-Valenzschwingungen und asymmetrischen CH3-Streckschwingungen
stammten und damit das Vorliegen von sp3-Zentren bestätigten. Das Signal der C=O-
Schwingung von CO2 lag bei 2362 cm-1 und stammte nicht von den Proben, sondern wurde
durch CO2 aus der Luft verursacht. Die Intensität des Signals ergab sich aus der hohen CO2-
Konzentration in der Messkammer während der Aufnahme des Hintergrundspektrums. Bei
1740 cm-1 ist die C=O-Streckschwingung von Carbonyl- und Carboxylgruppen zu sehen, deren
Intensität durch die Reduktion stark verringert wurde. Das weitere Signal der OH-
Streckschwingung bei 1633 cm-1 ist vermutlich auf eingelagertes Wasser oder
Hydroxylgruppen zurückzuführen.[68] Ähnlich wie das Signal bei 3444 cm-1 ist dieses Signal
bei GO viel größer als bei TRGO und Graphit, was auf die hohe Menge an hydrophilen
funktionellen Gruppen zurückzuführen ist. Die Bande bei 1543 cm-1 ließ sich einer C=C-

3 Graphensynthese und Charakterisierung
- 37 -
Steckschwingung innerhalb eines aromatischen Kohlenstoffgerüst zuordnen.[191] Die Signale
der phenolischen OH-Gruppen[192] bei 1380 und 1256 cm-1 sowie der epoxischen C-O-
Streckschwingung[193] bei 1048 cm-1 von GO nahmen bei TRGO stark ab, was auf den Abbau
der sauerstoffhaltigen funktionellen Gruppen bei thermischer Behandlung hinwies.
3.2.1 Dispersionsstabilität
Zur Herstellung von Nanokompositen mit effizienter Matrixverstärkung ist neben einem großen
Aspektverhältnis die Dispergierung des Füllstoffs in der Matrix maßgeblich.[167] Die
Homogenität der Füllstoffverteilung bestimmt im Wesentlichen die zu erwartenden
Materialeigenschaften. Je besser die Dispergierung des Füllstoffs erfolgt, desto höher ist der
Grenzflächenkontakt zwischen Füllstoff und Matrixmaterial und umso höher ist die
Materialverstärkung.[194]
Eines der größten Probleme für die industrielle Anwendung von Graphen ist seine schlechte
Dispergierbarkeit. Um die Wechselwirkung zwischen dem Matrixmaterial und dem Füllstoff zu
verbessern bzw. die Agglomeratbildung zu vermeiden wird das pulverförmige TRGO als
Füllstoff für die spätere Herstellung von Keramik-TRGO-Nanokompositen in Form als stabile
Dispersionen benutzt. Um die stabile Graphen-Dispersion zu bekommen stehen die drei
folgenden Methoden zur Verfügung: chemische, physikalische und mechanische Ansatz.
Als chemische Methoden werden die Zugabe von Tensiden oder die organische Modifizierung
der Füllstoffoberfläche bezeichnet, welche die Graphenpartikel stabilisiert und deren
Ausfällung verhindert.[195–197] Nachteilig beim Einsatz von Tensiden sind die Verringerung der
elektrischen Leitfähigkeit, der nötige Aufwand zur Entfernung der Tenside, da diese
unerwünschte Verunreinigung in Nanokompositen sind, sowie die damit verbundenen höheren
Kosten.[198]
Von der Vielzahl zur Verfügung stehender physikalischer Dispergierungsmethoden soll hier
eine Methode, die im Rahmen dieser Arbeit Anwendung gefunden hat, vorgestellt werden.

3 Graphensynthese und Charakterisierung
- 38 -
Hierbei handelt es sich um die Stabilisierung durch elektrostatische Abstoßung gleichartiger
Ladungen. Mit zunehmender Annäherung der Teilchen werden die repulsiven
Wechselwirkungen größer, wodurch eine weitere Annäherung verhindert wird.[199–204] Damit
wird bei mit polaren Gruppen funktionalisiertem Graphen eine bessere Dispergierbarkeit in
Wasser als bei unfunktionalisiertem Graphen erwartet, welche eine elektrostatische
Stabilisierung durch deprotonierte funktionelle Gruppen ermöglicht.
Die dritte Möglichkeit zur Graphendispergierung bedient sich der mechanischen Behandlung
durch hohe Scherkräfte oder mittels Ultraschallbehandlung. Dabei werden verbliebene
Schichtpakete durch hochenergetische Scherung oder Vibrationen exfoliert.[205,206] Frequenz,
Leistung und Einwirkungszeit sind wobei die wichtigsten Variablen.[198,207]
Auf Grundlage früherer Erkenntnisse wurden in der vorliegenden Arbeit stabile tensidfreie
TRGO-Dispersionen mit Hilfe eines Hochdruckhomogenisators (HH) hergestellt (siehe
Abbildung 15).[184]
Rührer
Geschlossener Rotor
Sägezahnklinge
Rotor-Stator
Kolloidmühle
Homogenisator
0
200000
400000
600000
800000
Sch
erg
esch
win
dig
keit
[1/s
]
Abbildung 15: Funktionsweise des Hochdruckhomogenisators[208] (links) und erzeugte Scherkräfte des
Homogenisators im Vergleich zu den anderen Prozessen[209] (rechts).
Hierbei wurde die Dispersion unter hohem Druck von 1000 bar durch einen dünnen
Homogenisierungsspalt gefördert. Nach dem Verlassen des Homogenisierungsspalts stieß die
Dispersion auf den Prallring und verließ anschließend das Ventil über einen Auslass. Die
auftretende große Geschwindigkeitsänderung durch spontane Entspannung der Flüssigkeit von

3 Graphensynthese und Charakterisierung
- 39 -
1000 bar auf Normaldruck führte zu hohen Scherkräften, welche zur effektiven
Deagglomerierung und Homogenisierung der TRGO-Partikel führten.[198]
Die Dispersionsstabilität wurde qualitativ mittels eines einfachen optischen Tests untersucht.
Abbildung 16 zeigt den Einfluss des HH-Prozesses auf die Stabilität der TRGO-Dispersionen
in Wasser nach 3 min (frisch), 5 und 50 Tagen.
Abbildung 16: Auswirkung des HH-Prozesses auf die Stabilität der TRGO-Dispersionen in Wasser
(pH 10, 1 g/L).
Die durch den HH-Prozess erhaltenen TRGO-Dispersionen erschienen homogen schwarz und
blieben über mindestens 50 Tage stabil. Dies zeigt, dass die Mehrheit der Partikel in exfolierter
Form und kaum noch Agglomerate vorliegen. Im Gegensatz dazu zeigten die TRGO-
Dispersionen nach Standard-Ultraschallbehandlung eine wesentlich geringere
Dispersionsstabilität, die schließlich nach 5 Tagen zur Agglomeration und folglich zu
Sedimentierung und Phasentrennung führte.
Zusammenfassend konnten homogene TRGO-Dispersionen mittels HH hergestellt werden,
welche über langen Zeitraum stabil bleiben. Dies ermöglicht die Verwendung von TRGO in
Form als stabile Dispersion für die Herstellung von Keramik-Graphen-Nanokompositen.

3 Graphensynthese und Charakterisierung
- 40 -
3.3 Mechanochemisch funktionalisiertes Multilagen-Graphen
In Anlehnung an die Arbeiten von I. -Y. JEON[57,83,84] und F. BECKERT
[94] wurde das
mechanochemisch funktionalisierte Multilagen-Graphen (MG) durch mechanochemische
Exfolierung und Funktionalisierung von Graphit über kosten- und zeiteffiziente Mahlprozesse
in einer Planetenkugelmühle hergestellt. Die Variation der Prozessparameter ermöglicht eine
individuelle Anpassung der Partikelgeometrie, Größe und Funktionalisierung. Durch die
Vermahlung von Graphit in einer Kugelmühle unter verschiedenen Gasatmosphären wurde
hierbei die Einführung weiterer funktioneller Gruppen realisiert. Anders als bei der Herstellung
von TRGO wurden hier keine umwelt- und gesundheitsgefährdenden Chemikalien und
Lösungsmittel eingesetzt. In der Planetenkugelmühle wurde der Graphit durch
hochenergetische Schläge der Mahlkugeln und die entstehenden hohen Scherkräfte zerkleinert.
Um Metallabrieb-Verunreinigungen auszuschließen, kamen in dieser Arbeit eine abriebfeste
Keramik-Mahlkammer und 96 Stücke Keramikkugeln zum Einsatz (siehe Abbildung 17).
Abbildung 17: Keramikmahlkammer (links) und Kugeln (rechts) für die Herstellung von MG.
Teile der in diesem Kapitel vorgestellten Ergebnisse wurden in Zusammenarbeit mit F. BECKERT
durchgeführt und bereits bei Macromolecules unter dem Titel „Mechanochemical Route to
Graphene-Supported Iron Catalysts for Olefin Polymerization and in Situ Formation of
Carbon/Polyolefin Nanocomposites“ veröffentlicht.[55]
In dieser Arbeit wurde der getrocknete Graphit (KFL 99.5, AMG Mining) unter verschiedenen
Atmosphären, wie CO2, N2 und Argon, vermahlen. Die Proben wurden bei einer Drehzahl von
250 U/min für eine Dauer von 24 bis 120 h behandelt, wobei sich der Prozess unter N2 und

3 Graphensynthese und Charakterisierung
- 41 -
Argon wegen des hohen Abriebes (siehe Zr-Gehalt in Tabelle 3) auf 72 h beschränkt.
Abbildung 18 zeigt die vereinfachte Herstellung des MG-Materials sowie den Aufbau der
Probencodes. Es wurde untersucht, welche Auswirkungen die Mahldauer und Gasatmosphäre
auf die Partikelgeometrie, -größe, -größenverteilung, Dispersionsstabilität sowie den
Funktionalisierungsgrad hatte. Die typischen Eigenschaften der erhaltenen MG-Materialien
wurden in Tabelle 3 aufgeführt.
Abbildung 18: Herstellung von MG unter verschiedenen Gasatmosphären (X) sowie Mahldauern (Y).
Tabelle 3: Abhängigkeit der Materialeigenschaften der MG-Typen von der Gasatmosphäre und
Mahldauer. Gezeigt sind Daten aus EA-, EDX-, BET.-, und TGA-Messungen.
a Bestimmt durch Elementaranalyse; b Bestimmt durch EDX; c N2-Physisorptionsmessung zur
Oberflächenbestimmung nach Brunauer, Emmett und Teller; d N2-Atmospäre, 10 K/min, 50 – 1500 °C.
Probe
C-
Gehalta
H-
Gehalta
N-
Gehalta
O-
Gehaltb
Zr-
Gehaltb
𝐀𝐁𝐄𝐓𝐜 TGAd-
Masseverlust
[Gew.-%] [m²/g] [Gew.-%]
Graphit 99.6 0.2 0.2 0.7 - 3 2.5
MG-Ar-24h 89.1 0.6 0.6 6.9 0.6 444 9.6
MG-Ar-48h 85.7 0.7 0.5 8.4 2.2 351 13.3
MG-Ar-72h 81.7 0.6 0.6 9.3 3.8 275 17.3
MG-CO2-24h 91.5 0.7 - 8.0 - 283 6.4
MG-CO2-48h 88.2 0.8 - 10.4 - 416 10.9
MG-CO2-72h 86.4 0.9 - 11.7 - 539 13.7
MG-CO2-96h 87.9 0.5 - 10.9 - 561 20.7
MG-CO2-120h 86.4 0.6 - 11.1 0.5 575 28.0
MG-N2-24h 82.2 1.0 4.0 8.4 - 449 20.0
MG-N2-48h 80.2 0.8 6.1 9.0 1.9 269 24.5
MG-N2-72h 75.2 1.1 7.2 7.6 4.5 219 29.3

3 Graphensynthese und Charakterisierung
- 42 -
Die Abhängigkeit der Morphologie der verschiedenen MG von der Gasatmosphäre wurden
mittels SEM- (Abbildung 19) und TEM-Aufnahmen (Abbildung 20) untersucht.
Abbildung 19: SEM-Aufnahmen der MG in Abhängigkeit der Atmosphäre und Mahldauer: a)
Graphit, b) MG-Ar-48h, c) MG-N2-48h, d) MG-CO2-24h, e) MG-CO2-48h, f) MG-CO2-72h.
Anhand der SEM-Aufnahmen in Abbildung 19 ist zu erkennen, dass durch die Vermahlung eine
drastische Veränderung der ursprünglichen Plättchenstruktur erfolgte. Der kommerzielle
Graphit zeichnet sich durch eine typischerweise zwiebelschalenartig aufeinander gelagerte
Plättchenstruktur aus. Der Graphit verfügt über eine enge Partikelgrößenverteilung mit einer
Kantenlänge von ca. 400 µm und eine einheitliche Dicke von 1.7 µm sowie eine glatte
Oberfläche. Die Übersichtsaufnahmen in Abbildung 19 zeigen für alle MG-Materialien eine
drastische Abnahme der Partikelgröße und des Aspektverhältnisses mit einer sehr breiten
Partikelgrößenverteilung unabhängig von der Gasatmosphäre. Die längere Vermahlung unter
CO2 führte zu einer weiteren Abnahme der Partikelgröße, welche bei MG-Ar und MG-N2 nicht
beobachtet wurde. Nach 24 h Vermahlung sind noch deutlich große Plättchen mit einer
Kantenlänge von 200 µm zu erkennen. Trotzdem zeigt diese Probe bereits eine breite
Partikelgrößenverteilung. Durch die weitere Vermahlung reduziert sich der Anteil an großen

3 Graphensynthese und Charakterisierung
- 43 -
Partikeln immer weiter, bis schließlich nach 72 h statt großer Plättchen nur noch sphärische
Partikel mit einer breiten Partikelgrößenverteilung zu finden sind.
Abbildung 20: TEM-Aufnahmen der MG in Abhängigkeit der Atmosphäre und Mahldauer:
a) MG-CO2-24h, b) MG-CO2-48h, c) MG-CO2-72h, d) MG-CO2-120h e) MG-Ar-24h, f) MG-Ar-48h,
g) MG-N2-24h, c) MG-N2-72h.
Mit vergrößerten TEM-Aufnahmen (Abbildung 20) wird deutlich, dass die Vermahlung unter
CO2 als einzige Methode die Plättchenstruktur der Partikel erhaltet. Bei MG-Ar und MG-N2
sind keinerlei große Plättchen, sondern nur noch sphärische Partikel zu erkennen. Auffallend
ist die Partikelgröße bei MG-Ar und MG-N2, welche sich als fast unabhängig von der
Mahldauer erweist. Im Vergleich dazu zeigt das MG-CO2 neben den größtenteils kleinen
sphärischen Partikeln auch erhaltene Plättchen mit dünnen Schichtstrukturen und Abmessungen
von wenigen Hundert Nanometern evtl. bei einer Mahldauer von bis zu 120 h.
Dieser große Unterschied der Morphologie in Abhängigkeit der Gasatmosphäre lässt sich mit
dem von F. BECKERT[94] vorgeschlagenen Reaktionsmechanismus in Abbildung 21 erklären.
Während der Vermahlung von Graphit erfolgt eine homolytische Spaltung der C-C-Bindungen
im Graphit wegen der hohen Scherkräfte. Bei der Vermahlung unter CO2-Druck kommt es zur
Addition der CO2-Moleküle und der Bildung von Carboxylradikalen. Anschließend erfolgt die
Absättigung der Carboxylradikale durch eine interne Redox-Reaktion. Unter N2-Atmosphäre

3 Graphensynthese und Charakterisierung
- 44 -
erfolgt eine Anbindung von Stickstoff. Zusätzlich erfolgt eine erhebliche Funktionalisierung
mit Hydroxylgruppen durch die heftige Reaktion verbliebener Kohlenstoffradikale mit
Sauerstoff und Luftfeuchtigkeit beim Öffnen der Kammer. Dieselbe Reaktion erfolgt auch bei
der Vermahlung unter Argon. Diese heftige Reaktion von reaktiven Kohlenstoffradikalen mit
Sauerstoff und Luftfeuchtigkeit führte zur vollständigen Zerstörung der Schichtstruktur von
Graphit. Aus diesem Grund wurden nur kleine sphärische Partikel bei MG-Ar und MG-N2
beobachtet. Diese Hypothese wurde auch durch die unterschiedliche Reaktion des Graphens
beim Öffnen des Mahlbechers gestützt. Es wurde beobachtet, dass beim Öffnen des
Mahlbechers nach Vermahlung unter Ar oder N2 eine heftige Reaktion stattfand, sich eine
Temperaturerhöhung des Mahlbechers und Mahlguts einstellte sowie kleine Glutherde
entwickelten. Die heftige Reaktion konnte nur bei MG-Ar und MG-N2 beobachtet werden,
während bei MG-CO2 keine Temperaturerhöhung festgestellt werden konnte.
Abbildung 21: Mechanismusvorschlag von F. BECKERT für die mechanochemische Funktionalisierung
von Graphit in einer Planetenkugelmühle.[94]

3 Graphensynthese und Charakterisierung
- 45 -
Die funktionellen Gruppen in verschiedenen MG konnten mittels FT-IR-Spektroskopie
bestimmt werden. In Abbildung 22 sind die FT-IR-Spektren von Graphit, MG-Ar-48h,
MG-CO2-48h und MG-N2-48h dargestellt.
4000 3500 3000 2500 2000 1500 1000
d) MG-N2-48h
c) MG-CO2-48h
b) MG-Ar-48h
Inte
nsität [a
.u.]
Wellenzahl [cm-1]
a) Graphit
Abbildung 22: FT-IR-Spektren von a) Graphit, b) MG-Ar-48h, c) MG-CO2-48h, d) MG-N2-48h.
Das breite Signal der OH-Streckschwingung der Hydroxylgruppen bei 3450 cm-1 und das
Signal der OH-Vibrationsschwingung bei 1635 cm-1 sind bei allen Spektren zu erkennen und
konnten auf die Bildung von Hydroxylgruppen hinweisen oder dem angelagerten Wasser
zugeordnet werden. Im Gegensatz zum Graphit und MG-CO2 zeigten MG-Ar und MG-N2 zwei
neue Banden bei 2923 und 2852 cm-1, welche von symmetrischen CH2-Valenzschwingungen
und asymmetrischen CH3-Streckschwingungen stammten und das Vorliegen von sp3-Zentren
sowie die Reaktion der reaktiven Kohlenstoffradikale mit Luftfeuchtigkeit bestätigten. Das
Signal der C=O-Schwingung von CO2 liegt bei 2362 cm-1 und stammte nicht von den Proben,
sondern wird durch CO2 aus der Luft verursacht. Die Intensität des Signals ergab sich aus der
hohen CO2-Konzentration in der Messkammer während der Aufnahme des
Hintergrundspektrums. Durch die Vermahlung von Graphit unter CO2-Atmosphäre wurden
Carboxylgruppen an das MG angebunden, welche mit der C=O-Streckschwingung bei
1733 cm-1, der asymmetrischen Streckschwingung bei 1578 cm-1 und der C-O-
Vibrationsschwingung bei 1384 cm-1 nachgewiesen werden konnten. Die Vermahlung von

3 Graphensynthese und Charakterisierung
- 46 -
Graphit unter N2-Atmosphäre führte zu mechanochemischer Stickstofffixierung. Die
Absorptionsbande bei 1630 cm-1 könnte auf C-N- oder NH-Vibrationsschwingung
zurückzuführen sein. Die Auswertung der FT-IR-Spektren stützt sowohl den
Mechanismusvorschlag in Abbildung 22 als auch die Ergebnisse der XPS-Spektroskopie[94] von
F. BECKERT.
Die Mahldauer und Gasatmosphäre wirkten sich nicht nur auf die Morphologie und
Funktionalisierung, sondern auch auf den Funktionalisierungsgrad und die spezifische
Oberfläche aus. Die hergestellten MG-Produkte wurden mittels EDX auf ihren
Funktionalisierungsgrad sowie mittels BET auf ihre spezifische Oberfläche hin untersucht.
0 24 48 72 96 1200
2
4
6
8
10
12
MG-N2
MG-Ar
O-G
eh
alt
[G
ew
.-%
]
Mahldauer [h]
MG-CO2
Abbildung 23: Funktionalisierungsgrad (O-Gehalt) in Abhängigkeit der Mahldauer und Gasatmosphäre.
Wie aus Abbildung 23 hervorgeht, nahm der Sauerstoffgehalt mit steigender Mahldauer
unabhängig von der Gasatmosphäre stetig zu, was auf eine zunehmende Funktionalisierung
hindeutete. Die leichte Abnahme des Sauerstoffgehalts von MG-N2 nach 48 h könnte mit dem
Fehler des Messgerätes erklärt werden. Die funktionellen Gruppen wie die Hydroxyl- oder
Carboxyl-Funktionen können die Wechselwirkung und Kompatibilität zwischen Graphen und
Matrixmaterial verbessern und für eine effektivere Dispergierung sorgen. Dabei erhöhte sich
der Funktionalisierungsgrad bei allen MG innerhalb der ersten 24 h stark auf ca. 8 Gew.-%,
während die weitere Vermahlung bis zu 72 bzw. 120 h nur noch zu einer moderaten Zunahme
führte. Bei MG-Ar und MG-N2 erreichte der Funktionalisierungsgrad nur bis zu 9 Gew.-%,
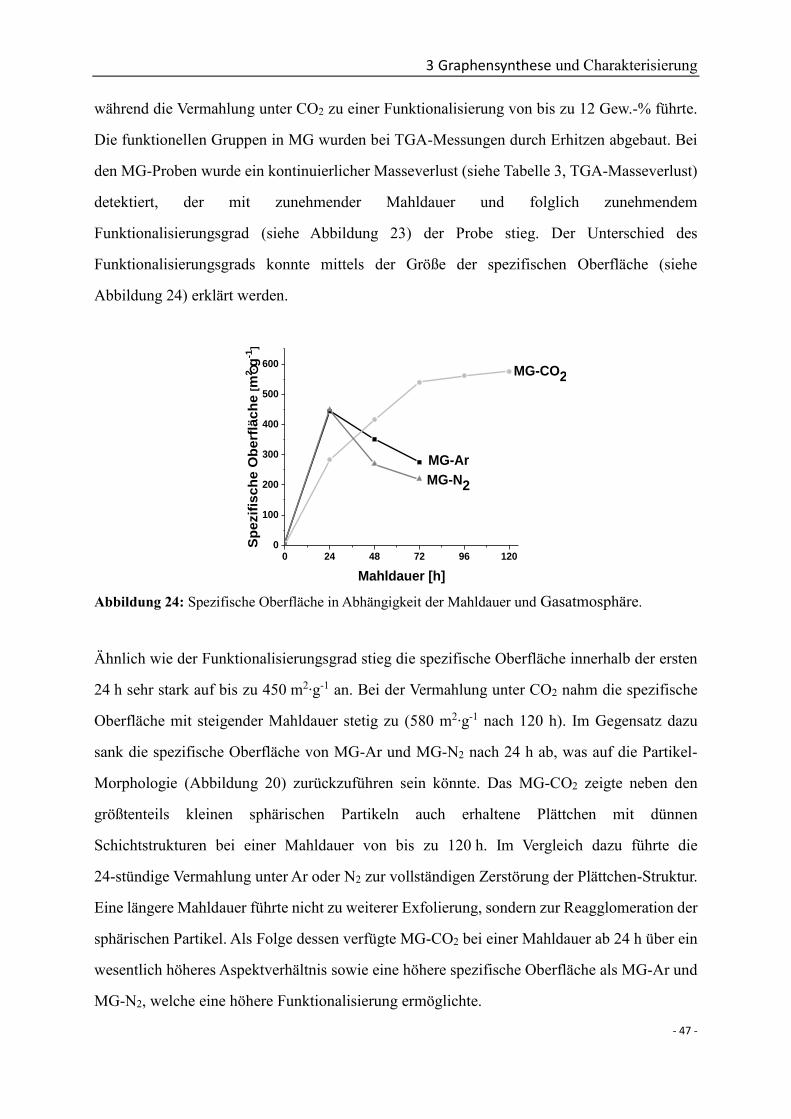
3 Graphensynthese und Charakterisierung
- 47 -
während die Vermahlung unter CO2 zu einer Funktionalisierung von bis zu 12 Gew.-% führte.
Die funktionellen Gruppen in MG wurden bei TGA-Messungen durch Erhitzen abgebaut. Bei
den MG-Proben wurde ein kontinuierlicher Masseverlust (siehe Tabelle 3, TGA-Masseverlust)
detektiert, der mit zunehmender Mahldauer und folglich zunehmendem
Funktionalisierungsgrad (siehe Abbildung 23) der Probe stieg. Der Unterschied des
Funktionalisierungsgrads konnte mittels der Größe der spezifischen Oberfläche (siehe
Abbildung 24) erklärt werden.
0 24 48 72 96 1200
100
200
300
400
500
600
MG-Ar
MG-N2
Sp
ezif
isc
he
Ob
erf
läc
he
[m
2g
-1]
Mahldauer [h]
MG-CO2
Abbildung 24: Spezifische Oberfläche in Abhängigkeit der Mahldauer und Gasatmosphäre.
Ähnlich wie der Funktionalisierungsgrad stieg die spezifische Oberfläche innerhalb der ersten
24 h sehr stark auf bis zu 450 m2∙g-1 an. Bei der Vermahlung unter CO2 nahm die spezifische
Oberfläche mit steigender Mahldauer stetig zu (580 m2∙g-1 nach 120 h). Im Gegensatz dazu
sank die spezifische Oberfläche von MG-Ar und MG-N2 nach 24 h ab, was auf die Partikel-
Morphologie (Abbildung 20) zurückzuführen sein könnte. Das MG-CO2 zeigte neben den
größtenteils kleinen sphärischen Partikeln auch erhaltene Plättchen mit dünnen
Schichtstrukturen bei einer Mahldauer von bis zu 120 h. Im Vergleich dazu führte die
24-stündige Vermahlung unter Ar oder N2 zur vollständigen Zerstörung der Plättchen-Struktur.
Eine längere Mahldauer führte nicht zu weiterer Exfolierung, sondern zur Reagglomeration der
sphärischen Partikel. Als Folge dessen verfügte MG-CO2 bei einer Mahldauer ab 24 h über ein
wesentlich höheres Aspektverhältnis sowie eine höhere spezifische Oberfläche als MG-Ar und
MG-N2, welche eine höhere Funktionalisierung ermöglichte.

3 Graphensynthese und Charakterisierung
- 48 -
3.3.1 Dispersionsstabilität
Die Verfügbarkeit von stabiler Graphen-Dispersion in verschiedenen Medien, insbesondere in
umweltfreundlichen Lösungsmitteln, wie z. B. Wasser, spielt eine wichtige Rolle für die
möglichen Verarbeitungstechniken sowie folglich für die industrielle Anwendung. Um die
Wechselwirkung zwischen dem Matrixmaterial und dem Füllstoff zu verbessern bzw. die
Agglomeratbildung zu vermeiden wird das pulverförmige MG als Füllstoff für die spätere
Herstellung von Keramik-MG-Nanokompositen in Form als stabile Dispersionen benutzt.
Dafür wurden stabile tensidfreie MG-Dispersionen in Wasser in der vorliegenden Arbeit auf
Grundlage der Erkenntnisse in Kapitel 3.1.1 mit Hilfe des Hochdruckhomogenisators (HH)
hergestellt.
Analog zu der Herstellung von TRGO-Dispersionen wurden die MG-Dispersionen unter hohem
Druck von 1000 bar durch einen dünnen Homogenisierungsspalt gefördert. Nach dem Verlassen
des Homogenisierungsspalts stießen die Dispersionen auf den Prallring und verließen
anschließend das Ventil über einen Auslass. Die auftretende große Geschwindigkeitsänderung
durch spontane Entspannung der Flüssigkeit von 1000 bar auf Normaldruck führte zu hohen
Scherkräften, welche zur effektiven Deagglomerierung und Homogenisierung der MG-Partikel
führten (siehe Abbildung 15 in Kapitel 3.1.1).[198]
Die direkte beschleunigte Stabilitätsanalyse sowie die Partikelgrößenverteilung der MG-
Dispersionen erfolgten mit der analytischen Photozentrifuge LUMiSizer der Firma LUM GmbH.
Der LUMiSizer zeichnete ortsaufgelöste Transmissionsprofile im Verlauf eines
Sedimentationsvorgangs auf. Die Messung erfolgte bei einer Drehzahl von 1000 U/min bei
einer Temperatur von 25 °C. Ein Vergleich mit Graphit konnte nicht erfolgen, da Graphit
keinerlei Dispergierbarkeit zeigte.
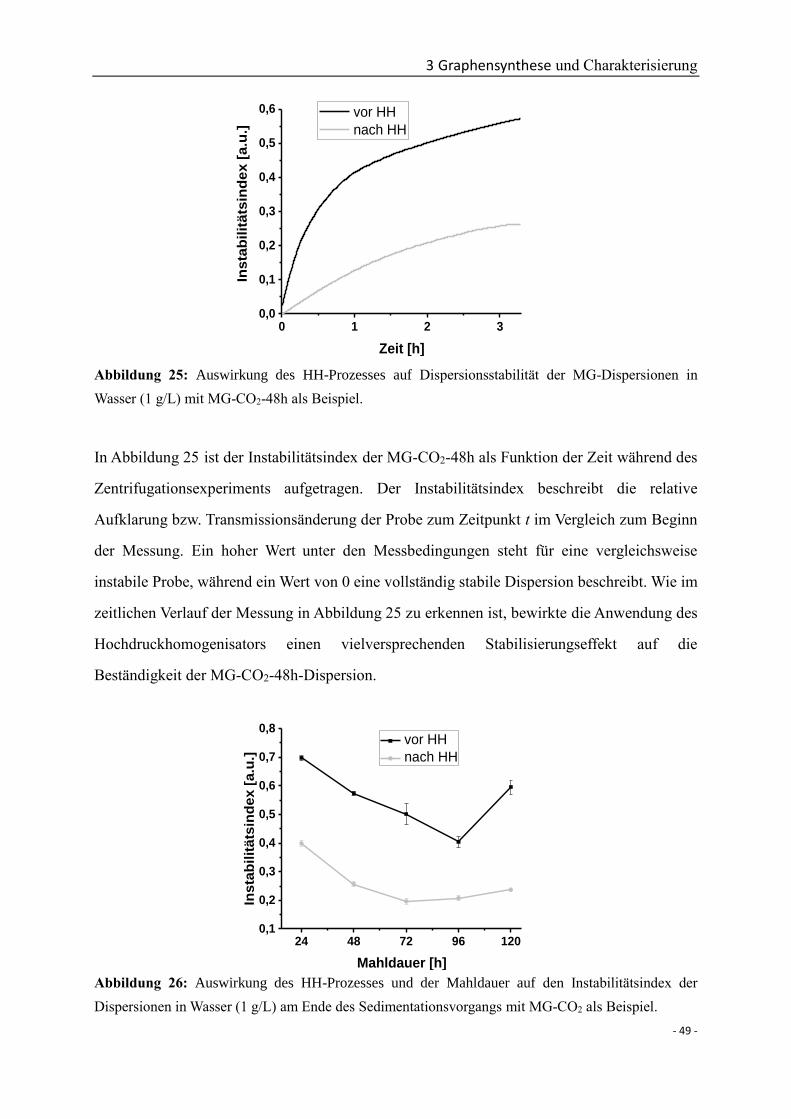
3 Graphensynthese und Charakterisierung
- 49 -
0 1 2 30,0
0,1
0,2
0,3
0,4
0,5
0,6
Insta
bilit
äts
ind
ex [
a.u
.]
Zeit [h]
vor HH
nach HH
Abbildung 25: Auswirkung des HH-Prozesses auf Dispersionsstabilität der MG-Dispersionen in
Wasser (1 g/L) mit MG-CO2-48h als Beispiel.
In Abbildung 25 ist der Instabilitätsindex der MG-CO2-48h als Funktion der Zeit während des
Zentrifugationsexperiments aufgetragen. Der Instabilitätsindex beschreibt die relative
Aufklarung bzw. Transmissionsänderung der Probe zum Zeitpunkt t im Vergleich zum Beginn
der Messung. Ein hoher Wert unter den Messbedingungen steht für eine vergleichsweise
instabile Probe, während ein Wert von 0 eine vollständig stabile Dispersion beschreibt. Wie im
zeitlichen Verlauf der Messung in Abbildung 25 zu erkennen ist, bewirkte die Anwendung des
Hochdruckhomogenisators einen vielversprechenden Stabilisierungseffekt auf die
Beständigkeit der MG-CO2-48h-Dispersion.
24 48 72 96 1200,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Ins
tab
ilit
äts
ind
ex
[a
.u.]
Mahldauer [h]
vor HH
nach HH
Abbildung 26: Auswirkung des HH-Prozesses und der Mahldauer auf den Instabilitätsindex der
Dispersionen in Wasser (1 g/L) am Ende des Sedimentationsvorgangs mit MG-CO2 als Beispiel.

3 Graphensynthese und Charakterisierung
- 50 -
Der Einfluss der Mahldauer auf die Stabilität der Dispersionen wurde ebenfalls untersucht. Die
Instabilitätsindizes am Ende des Sedimentationsvorgangs in Abhängigkeit der Mahldauer sind
in Abbildung 26 aufgetragen. Es wurde eine zunehmende Stabilität mit erhöhter Mahldauer
festgestellt. Eine längere Mahldauer führte zu einer Verbesserung der Exfolierung und damit
zu einer zunehmenden Funktionalisierung mit hydrophilen funktionellen Gruppen, welche mit
dem Dispersionsmedium Wasser attraktiv wechselwirken können, was in einer Stabilisierung
der Dispersionen resultiert. Im Vergleich zur Mahldauer spielte der HH-Prozess sogar noch
eine größere Rolle bei der Stabilisierung von MG-Dispersionen. Nach der HH-Behandlung
zeigten die MG-Dispersionen einen niedrigen Instabilitätsindex und damit eine höhere
Stabilität als zuvor. Zusammenfassend stellt der HH-Prozess eine gute Methode dar,
plättchenartige und sphärische MG-Partikel in Wasser zu dispergieren.
Der HH-Prozess zeichnete sich nicht nur durch die Stabilisierung der Dispersion, sondern auch
durch Zerkleinerung der MG-Agglomerate in Dispersion, aus. Die Auswirkung des HH-
Prozesses auf den Partikeldurchmesser bzw. die Partikelgrößenverteilung wurde am Beispiel
der Probe MG-CO2-48h untersucht (siehe Abbildung 27).
10 100 1000 10000
Partikelgröße [nm]
vor HH
nach HH
Re
lati
ve H
äu
fig
ke
it [
a.u
.]
Abbildung 27: Auswirkung des HH-Prozesses auf die Partikelgrößenverteilung.
Anhand der Auftragungen in Abbildung 27 konnte festgestellt werden, dass sich nach dem HH-
Prozess die mittlere Partikelgröße reduzierte und eine einheitlichere Partikelgrößenverteilung
erreicht wurde, was auf den Deagglomerationseffekt im HH-Prozess zurückzuführen ist.

3 Graphensynthese und Charakterisierung
- 51 -
Die Stabilität der Dispersion wurde neben der Auflösung großer Agglomerate durch die hohen
Scherkräfte in dem Hochdruckhomogenisator auch durch eine elektrostatische Abstoßung der
Flocken hervorgerufen. Dieser Effekt ließ sich durch Messung des Zetapotentials analysieren.
Das Zetapotential ist das elektrokinetische Potential in einer Dispersion zwischen dem
Dispersionsmedium und der stationären Schicht um die Partikel und ist ein Hauptindikator für
die Stabilität einer kolloidalen Dispersion. Die Höhe zeigt den Grad an elektrostatischer
Abstoßung zwischen benachbarten Partikeln in einer Dispersion an. Je höher das Potential,
desto stabiler ist die Dispersion und desto unwahrscheinlicher ist eine Agglomeration. Bei
geringem Potential können die anziehenden Kräfte überwiegen und es kommt zur Ausflockung.
Ein Zetapotential mit einem Betrag größer als 30 mV bedeutet, dass eine Dispersion
elektrostatisch ausreichend stabilisiert ist. Der Grund für die Stabilisierung sind die zwischen
gleich geladenen Teilchen vorliegenden repulsiven Wechselwirkungen. Mit zunehmender
Annäherung der Teilchen werden die repulsiven Wechselwirkungen größer, wodurch eine
weitere Annäherung verhindert wird.[199–204] Die Zetapotentiale der TRGO und der
verschiedenen MG-Dispersionen wurden in Abbildung 28 dargestellt.
TRGO
MG-CO2
MG-N2
MG-Ar
-60
-50
-40
-30
-20
-10
0
Zeta
po
ten
tial [m
V]
Abbildung 28: Zetapotential von TRGO und MG-Dispersionen in Wasser (0.005 mg/mL).
Anhand der Auftragungen in Abbildung 28 ist zu sehen, dass all die gemessenen Zetapotentiale
zwischen -40 – -60 mV lagen, welche auf stabile Dispersionen hinwiesen. Die elektrostatische
Stabilisierung erfolgte durch deprotonierte funktionelle Gruppen in TRGO oder MG, z. B. OH-
oder COOH-Gruppen.

3 Graphensynthese und Charakterisierung
- 52 -
3.4 Graphenbeschichtetes Siliciumcarbid
In jüngster Vergangenheit wurden (funktionalisierte) Graphene in den Keramiken als Füllstoffe
eingesetzt um deren thermische (Wärmeleitfähigkeit), mechanische (Risszähigkeit,
Biegefestigkeit), elektrische und tribologische (Verschleißbeständigkeit) Eigenschaften zu
verbessern. Das bisher größte Problem bei der Anwendung von Graphen als Füllstoff in
Keramiken ist die Entstehung von Poren. Die bislang veröffentlichten Artikel über SiC-
Graphen-Nanokomposite zeigten im Vergleich zum reinen SiC niedrige E-Moduln und
Härten.[152] Die Gründe hierfür sind vor allem die Porosität und die geringere Dichte der
SiC-Graphen-Nanokomposite. Des Weiteren sind schwache Partikel-Matrix-Anbindungen und
Fehlstellen im Gefüge mitverantwortlich für die verschlechterten mechanischen Eigenschaften.
In dieser Arbeit wurde, um die Entstehung von Poren während der Herstellung der SiC-
Graphen-Nanokomposite zu vermeiden, der SiC-Rohstoff vor dem Sintern mit Graphen
beschichtet. Abbildung 29 zeigt die allgemeine Synthese von graphenbeschichtetem SiC
(GSiC). Hier wurde das funktionalisierte Graphen mit Hilfe der Kohlenstoffquelle über eine
templatvermittelte Reaktion hergestellt, wobei SiC als Template diente.
Abbildung 29: Synthese von graphenbeschichtetem SiC.
Wie in Abbildung 7 gezeigt, wurde das graphenbeschichtete SiC aus drei verschiedenen
Graphen-Präkursoren hergestellt. In Anlehnung an die Arbeiten von M. BECKERT, X. LI und
R. LI wird respektive Furfurylalkohol[44,45], Glucose[43] und Dopamin[46] als Kohlenstoffquelle

3 Graphensynthese und Charakterisierung
- 53 -
verwendet, um GSiC herzustellen. Aus Gründen des Umweltschutzes wird das funktionalisierte
Graphen hier, im Gegensatz zu den konventionellen Methoden der Graphenherstellung aus
fossilen Kohlenstoffquellen wie Graphit, aus nachhaltigen, bio-basierten Rohstoffen in situ auf
der Oberfläche von SiC hergestellt.
Teile der in diesem Kapitel vorgestellten Ergebnisse wurden in Zusammenarbeit mit M. KNOCH
von der Firma FCT Ingenieurkeramik GmbH in Sonneberg und J. THELKE von der Firma
EagleBurgmann Germany GmbH & Co. KG in Wolfratshausen durchgeführt und bereits am
07.07.2017 unter dem Titel „Verfahren zur Herstellung eines mit graphenhaltigen Material
ummantelten partikulären Trägermaterials und eines keramischen Bauteils, sowie keramisches
Bauteil“ unter dem Aktenzeichen „DE 10 2017 211 663“ als Patentanmeldung eingereicht.[210]
3.4.1 Synthese von GSiC aus Furfurylalkohol
Die Herstellung von graphenbeschichtetem SiC aus Furfurylalkohol (GSiC-F) erfolgte in
Anlehnung an die Arbeiten von M. BECKERT. Bei ihren Arbeiten wurde ein graphitisches
Kohlenstoffnitrid-Template (g-C3N4) in situ aus Harnstoff und Dicyandiamid für die
templatvermittelte Synthese von funktionalisiertem Graphen hergestellt (siehe
Abbildung 30).[44,45] Im Vergleich dazu fiel in dieser Arbeit die Zwischenstufe des g-C3N4-
Templates weg, wobei das SiC selbst als Template diente, was eine homogene Durchmischung
von Graphen und SiC erwarten ließ.
Abbildung 30: Mechanismusvorschlag von M. BECKERT für die in situ templatvermittelte Synthese von
funktionalisiertem Graphen durch die Karbonisierung von Polyfurfurylalkohol in Anwesenheit von
Harnstoff und Dicyandiamid.[44,45]

3 Graphensynthese und Charakterisierung
- 54 -
Zuerst wurde das SiC (90 Gew.-%) in Wasser dispergiert. Nach Zugabe und Dispergierung von
Furfurylalkohol (10 Gew.-%) wurde die kationische Polymerisation des Furfurylalkohols durch
die Zugabe einer wässerigen p-Toluolsulfonsäurelösung (p-TsOH, 1 mol% bezogen auf
Furfurylalkohol) initiiert. Nach einstündiger Polymerisation bei 70 °C unter Rühren wurde die
Temperatur auf 100 °C erhöht, um das Reaktionsgemisch zu trocknen. Nach vollständiger
Trocknung und Zerkleinerung wurde das entstandene Polyfurfurylalkohol-beschichtete SiC zur
Karbonisierung in einem Rohrofen unter Stickstoff-Atmosphäre erhitzt. Zunächst wurde die
Probe bei 150 °C für 21 h erhitzt, um Lösungsmittelreste zu entfernen und den unreagierten
Furfurylalkohol zu polymerisieren. Danach erfolgte die Karbonisierung bei 800 °C für 6 h, um
eine vollständige Graphenisierung zu gewährleisten. Durch die Beschichtung mit Graphen
veränderte sich die Farbe des SiC stark (siehe Abbildung 31), was vermutlich auf das Entstehen
von Mehrlagen-Graphen zurückzuführen ist.
Abbildung 31: Makroskopische Optik des SiC im Vergleich zum GSiC-F.
Die Charakterisierung der hergestellten GSiC-F erfolgte durch EDX- und BET-Messungen,
TGA-Untersuchungen sowie TEM-Aufnahmen in Kapitel 3.4.4.
Die Herstellung von graphenbeschichtetem Si3N4 (GSi3N4-F) mit Furfurylalkohol als
Kohlenstoffquelle erfolgte mit der gleichen Methode und wird hier nicht näher erläutert.

3 Graphensynthese und Charakterisierung
- 55 -
3.4.2 Synthese von GSiC aus Glucose
Die Herstellung von graphenbeschichtetem SiC aus Glucose (GSiC-G) erfolgte in Anlehnung
an die Arbeiten von X. LI. Bei seinen Arbeiten wurde eine graphitische Kohlenstoffnitrid-
Schablone (g-C3N4) in situ aus Dicyandiamid (DCDA) für die templatvermittelte Synthese von
funktionalisiertem Graphen hergestellt (siehe Abbildung 32).[43] Ähnlich wie bei der
Herstellung von GSiC-F fiel in dieser Arbeit die Zwischenstufe der g-C3N4-Schablone auch
weg, wobei das SiC selbst als Template diente.
Abbildung 32: Mechanismusvorschlag von X. LI für die in situ templatvermittelte Synthese von
Graphen durch die Karbonisierung von Glucose in Anwesenheit von DCDA.[43]
Zuerst wurde das SiC (90.9 Gew.-%) in Wasser in einem Ultraschallbad dispergiert. Nach
Zugabe und Dispergierung von Glucose (9.1 Gew.-%) wurde das Reaktionsgemisch mittels
Ultraschalllanze weiter homogenisiert. Anschließend erfolgte die Trocknung mittels
Gefriertrocknung, um ein pulverförmiges Produkt zu bekommen, was die nachfolgende
Verarbeitung vereinfacht. Nach Zerkleinerung wurde das entstandene glucosebeschichtete SiC
zur Karbonisierung in einem Rohrofen unter Stickstoff-Atmosphäre erhitzt. Zunächst wurde
die Probe bei 150 °C für 21 h erhitzt, um Lösungsmittelreste und das angebundene
Kristallwasser in Glucose zu entfernen. Danach erfolgte die Karbonisierung bei 800 °C für 6 h,
um eine vollständige Graphenisierung zu gewährleisten. Ähnlich wie bei der Herstellung von
GSiC-F veränderte sich die Farbe des SiC stark durch die Beschichtung mit Graphen, was
vermutlich auch hier auf das Entstehen von Mehrlagen-Graphen zurückzuführen ist. Die
Charakterisierung der hergestellten GSiC-G erfolgte durch BET-Messungen, TGA-
Untersuchungen und TEM-Aufnahmen in Kapitel 3.4.4.

3 Graphensynthese und Charakterisierung
- 56 -
3.4.3 Synthese von GSiC aus Dopamin
Die Herstellung von graphenbeschichtetem SiC aus Dopamin (GSiC-D) erfolgte in Anlehnung
an die Arbeiten von R. LI.[211] Bei seinen Arbeiten wurde SiO2/Si oder Kupfer als Vorlage für
die templatvermittelte Synthese von funktionalisiertem Graphen verwendet (Abbildung 33). Im
Vergleich dazu diente in dieser Arbeit das Matrixmaterial-SiC selbst als Template, was zu einer
homogenen Durchmischung von Graphen und SiC führen soll.
Abbildung 33: Mechanismusvorschlag von R. LI für die in situ templatvermittelte Synthese von
funktionalisiertem Graphen durch die Pyrolyse von Polydopamin.[211]
Zuerst wurde das SiC (90 Gew.-%) in einer Tris(hydroxymethyl)aminomethan-Lösung (TRIS)
mittels Ultraschall dispergiert. Nach Zugabe und Dispergierung von Dopamin (10 Gew.-%)
initiierte die Polymerisation des Dopamins von selbst. Nach mind. 72 h Polymerisation bei
Raumtemperatur unter Rühren wurde das Reaktionsgemisch mittels Gefriertrocknung vom
Lösungsmittel befreit. Nach Zerkleinerung wurde das entstandene Polydopamin-beschichtete
SiC zur Karbonisierung in einem Rohrofen unter Stickstoff-Atmosphäre erhitzt. Zunächst
wurde die Probe bei 150 °C für 21 h erhitzt, um Lösungsmittelreste zu entfernen und das
unreagierte Dopamin zu polymerisieren. Danach erfolgte die Karbonisierung bei 1000 °C für
5 h, um eine vollständige Graphenisierung zu gewährleisten. Ähnlich wie bei der Herstellung
von GSiC-F und GSiC-D veränderte sich die Farbe des SiC stark durch die Beschichtung mit
Graphen, was vermutlich auch auf die erfolgreiche Herstellung von Mehrlagen-Graphen
zurückzuführen ist.
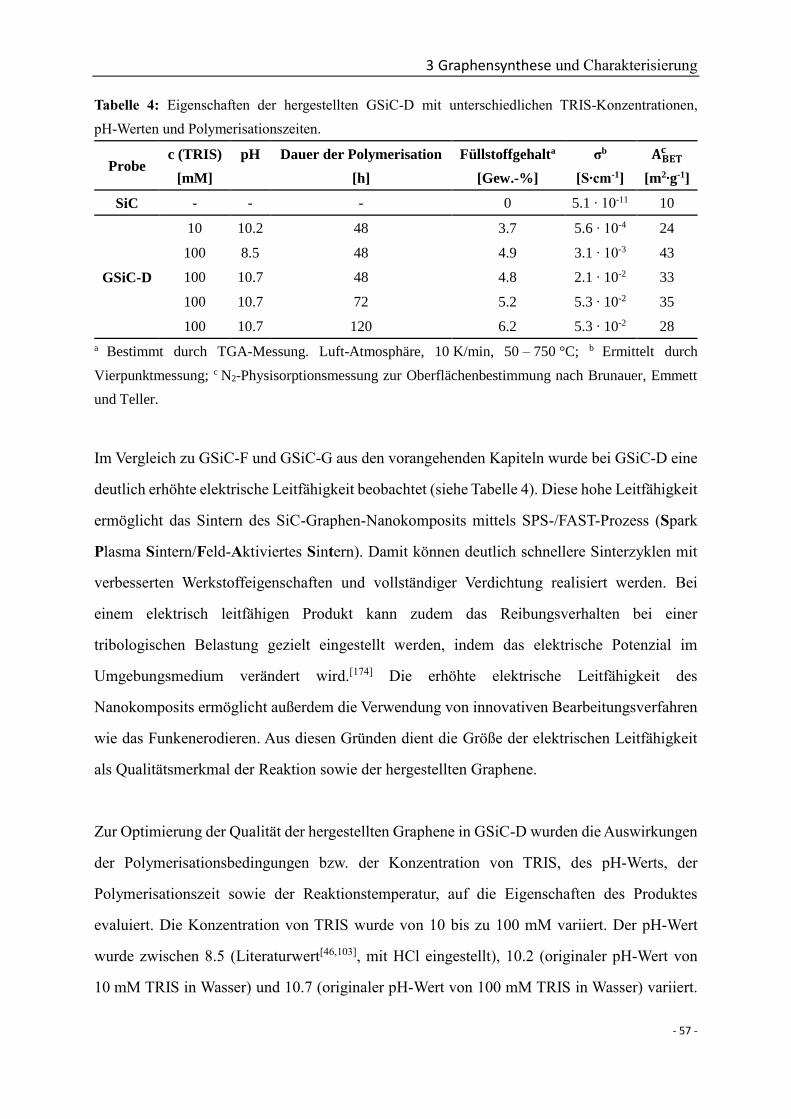
3 Graphensynthese und Charakterisierung
- 57 -
Tabelle 4: Eigenschaften der hergestellten GSiC-D mit unterschiedlichen TRIS-Konzentrationen,
pH-Werten und Polymerisationszeiten.
Probe c (TRIS)
[mM]
pH Dauer der Polymerisation
[h]
Füllstoffgehalta
[Gew.-%]
σb
[S∙cm-1]
𝐀𝐁𝐄𝐓𝐜
[m2∙g-1]
SiC - - - 0 5.1 ∙ 10-11 10
GSiC-D
10 10.2 48 3.7 5.6 ∙ 10-4 24
100 8.5 48 4.9 3.1 ∙ 10-3 43
100 10.7 48 4.8 2.1 ∙ 10-2 33
100 10.7 72 5.2 5.3 ∙ 10-2 35
100 10.7 120 6.2 5.3 ∙ 10-2 28
a Bestimmt durch TGA-Messung. Luft-Atmosphäre, 10 K/min, 50 – 750 °C; b Ermittelt durch
Vierpunktmessung; c N2-Physisorptionsmessung zur Oberflächenbestimmung nach Brunauer, Emmett
und Teller.
Im Vergleich zu GSiC-F und GSiC-G aus den vorangehenden Kapiteln wurde bei GSiC-D eine
deutlich erhöhte elektrische Leitfähigkeit beobachtet (siehe Tabelle 4). Diese hohe Leitfähigkeit
ermöglicht das Sintern des SiC-Graphen-Nanokomposits mittels SPS-/FAST-Prozess (Spark
Plasma Sintern/Feld-Aktiviertes Sintern). Damit können deutlich schnellere Sinterzyklen mit
verbesserten Werkstoffeigenschaften und vollständiger Verdichtung realisiert werden. Bei
einem elektrisch leitfähigen Produkt kann zudem das Reibungsverhalten bei einer
tribologischen Belastung gezielt eingestellt werden, indem das elektrische Potenzial im
Umgebungsmedium verändert wird.[174] Die erhöhte elektrische Leitfähigkeit des
Nanokomposits ermöglicht außerdem die Verwendung von innovativen Bearbeitungsverfahren
wie das Funkenerodieren. Aus diesen Gründen dient die Größe der elektrischen Leitfähigkeit
als Qualitätsmerkmal der Reaktion sowie der hergestellten Graphene.
Zur Optimierung der Qualität der hergestellten Graphene in GSiC-D wurden die Auswirkungen
der Polymerisationsbedingungen bzw. der Konzentration von TRIS, des pH-Werts, der
Polymerisationszeit sowie der Reaktionstemperatur, auf die Eigenschaften des Produktes
evaluiert. Die Konzentration von TRIS wurde von 10 bis zu 100 mM variiert. Der pH-Wert
wurde zwischen 8.5 (Literaturwert[46,103], mit HCl eingestellt), 10.2 (originaler pH-Wert von
10 mM TRIS in Wasser) und 10.7 (originaler pH-Wert von 100 mM TRIS in Wasser) variiert.

3 Graphensynthese und Charakterisierung
- 58 -
Des Weiteren wurde die Polymerisationszeit zwischen 24 und 120 h variiert, wobei die
Polymerisation entweder bei Raumtemperatur oder 60 °C stattfand.
Zunächst wurden der Einfluss der Konzentration und des pH-Werts der TRIS-Lösung auf die
elektrische Leitfähigkeit des GSiC-D untersucht. Dabei wurden die Reaktionen für 48 h bei
Raumtemperatur durchgeführt. Durch die Beschichtung mit Graphen veränderte sich die
spezifische Leitfähigkeit des SiC stark. Höhere Leitfähigkeit lässt sich auf die vollständige
Polymerisation und Graphenisierung zurückführen. Tabelle 4 zeigt den Einfluss der
Konzentration und des pH-Werts der TRIS-Lösung auf die Eigenschaften von GSiC-D.
Wie Tabelle 4 zu entnehmen ist, wies die Probe, welche mit hoher TRIS-Konzentration und bei
einem hohen pH-Wert hergestellt wurde, die höchste elektrische Leitfähigkeit auf. Bei diesen
Reaktionsbedingungen wurde das Dopamin vollständiger polymerisiert, was zu einem erhöhten
Füllstoffgehalt führte. Im Vergleich zum Referenz-SiC steigt die spezifische Oberfläche bei
allen Systemen, was mit den Ergebnissen bei GSiC-F und GSiC-G in den vorangegangenen
Kapiteln übereinstimmt. Unter diesen Reaktionsbedingungen wurde die Reaktionszeit auf bis
zu 120 h verlängert. Mit Hinblick auf den Füllstoffgehalt in GSiC-D, stieg dieser mit
zunehmender Reaktionszeit kontinuierlich. Im Vergleich dazu stieg die elektrische
Leitfähigkeit zunächst auf 0.05 S∙cm-1 nach 72 h Reaktionszeit, danach blieb dieser Wert
konstant. Aus wirtschaftlichen Gründen ist es wünschenswert mit möglichst wenig Graphen
eine hohe elektrische Leitfähigkeit zu erzeugen. Für weitere Versuche wurden die
Reaktionsbedingungen mit 100 mM TRIS als Lösungsmedium, einem pH-Wert von 10.7 und
einer Reaktionszeit von 72 h gewählt, da diese die höchste elektrische Leitfähigkeit und eine
hohe spezifische Oberfläche ergaben.
Die Charakterisierung der hergestellten GSiC-D erfolgte durch BET-Messungen, TGA-
Untersuchungen und TEM-Aufnahmen in Kapitel 3.4.4.

3 Graphensynthese und Charakterisierung
- 59 -
3.4.4 Charakterisierung der GSiCs
Die Strukturen der synthetisierten funktionalisierten Graphene bzw. der Beschichtungen auf der
SiC-Matrix wurden mittels TEM-Aufnahmen in Abbildung 34 untersucht.
Abbildung 34: TEM-Aufnahmen von a) SiC, b) GSiC-F, c) GSiC-G und d) GSiC-D.
Die Aufnahmen (Abbildung 34 a) entsprechen dabei der SiC-Referenz, welche sich durch
kleine körnige Partikel mit breiter Partikelgrößenverteilung (mittlerer Korndurchmesser der
Verteilung bei 50 % Durchgang d50 = 0.6 µm) auszeichnete. Die dünnen Graphenschichten sind
unabhängig von der Kohlenstoffquelle am Rand der SiC-Partikel leicht zu erkennen
(Aufnahmen b - d). Das funktionalisierte Graphen wurde direkt auf der Oberfläche von SiC
hergestellt, wodurch eine homogene Verteilung von Graphen in der SiC-Matrix erfolgte. Das

3 Graphensynthese und Charakterisierung
- 60 -
pulverförmige SiC-Trägermaterial und das hergestellte Graphen sind vermutlich durch
physikalische und/oder chemische Wechselwirkungen zwischen dem SiC-Trägermaterial und
dem Graphen geprägt. Dadurch wurde eine dauerhaft gute Beschichtung mit Graphen auf der
Oberfläche des SiC-Trägermaterials erhalten. Die Dicke der Graphenbeschichtung auf der SiC-
Oberfläche wurde mittels hochauflösende Transmissionselektronenmikroskopie (HRTEM)
charakterisiert (siehe Abbildung 35).
Abbildung 35: HRTEM-Aufnahmen von GSiC. Das SiC wurde bei a) GSiC-F, b) GSiC-G und
c) GSiC-D mit ca. 10, 10 – 20 und 5 Graphen-Lagen beschichtet.
Mit den HRTEM-Aufnahmen (Abbildung 35 a) konnte eine Graphen-Schichtdicke von ca.
5 nm bei GSiC-F gemessen und die Anzahl der Graphenlagen auf ca. 10 bestimmt werden. Bei
GSiC-G in Abbildung 35 (b) wurden mehrere SiC-Partikel aufgenommen, welche durch 10 bis
20 Graphenlagen mit einer Dicke von 5 – 10 nm komplett beschichtet wurden. Im Vergleich
dazu wurde GSiC-D in Abbildung 35 (c) teilweise mit Nanographen beschichtet, welches über
eine Dicke von ca. 10 nm bzw. 5 Graphenlagen und eine Länge von ca. 30 nm verfügte.
Die erfolgreiche Herstellung der Graphenbeschichtung auf der SiC-Oberfläche konnte
zusätzlich mittels EDX am Beispiel von GSiC-F nachgewiesen werden (siehe Abbildung 36
und Tabelle 5).
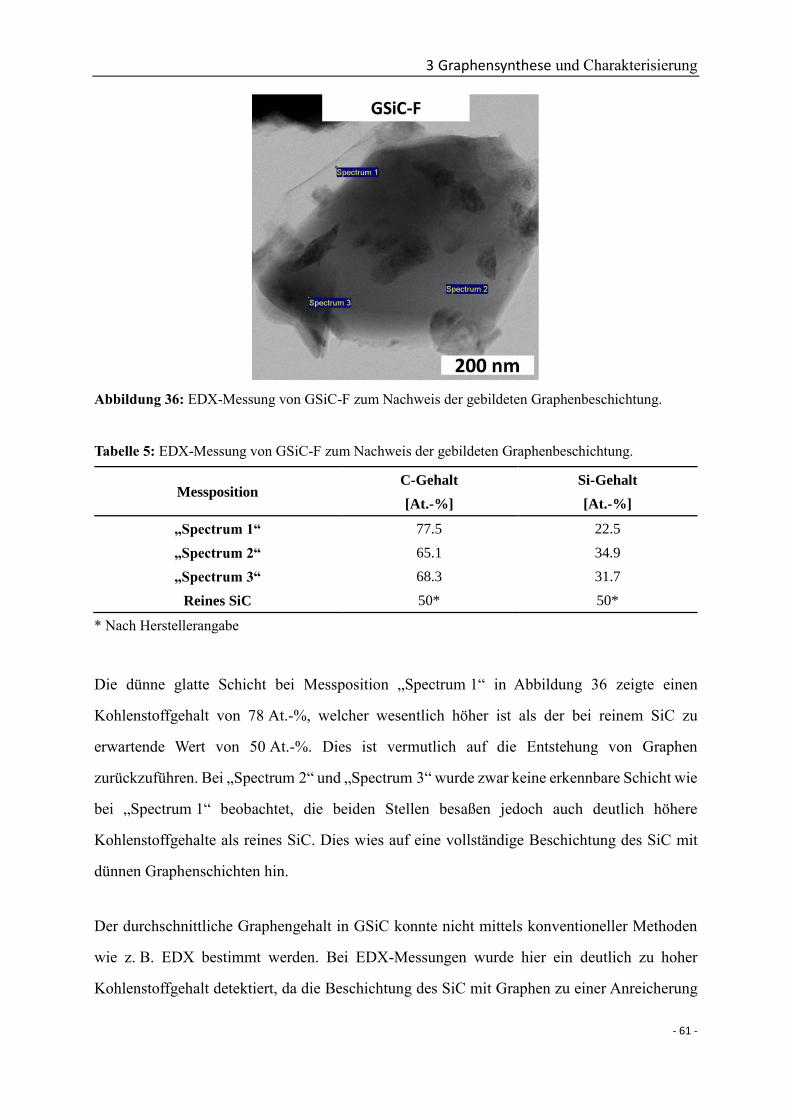
3 Graphensynthese und Charakterisierung
- 61 -
Abbildung 36: EDX-Messung von GSiC-F zum Nachweis der gebildeten Graphenbeschichtung.
Tabelle 5: EDX-Messung von GSiC-F zum Nachweis der gebildeten Graphenbeschichtung.
Messposition C-Gehalt
[At.-%]
Si-Gehalt
[At.-%]
„Spectrum 1“ 77.5 22.5
„Spectrum 2“ 65.1 34.9
„Spectrum 3“ 68.3 31.7
Reines SiC 50* 50*
* Nach Herstellerangabe
Die dünne glatte Schicht bei Messposition „Spectrum 1“ in Abbildung 36 zeigte einen
Kohlenstoffgehalt von 78 At.-%, welcher wesentlich höher ist als der bei reinem SiC zu
erwartende Wert von 50 At.-%. Dies ist vermutlich auf die Entstehung von Graphen
zurückzuführen. Bei „Spectrum 2“ und „Spectrum 3“ wurde zwar keine erkennbare Schicht wie
bei „Spectrum 1“ beobachtet, die beiden Stellen besaßen jedoch auch deutlich höhere
Kohlenstoffgehalte als reines SiC. Dies wies auf eine vollständige Beschichtung des SiC mit
dünnen Graphenschichten hin.
Der durchschnittliche Graphengehalt in GSiC konnte nicht mittels konventioneller Methoden
wie z. B. EDX bestimmt werden. Bei EDX-Messungen wurde hier ein deutlich zu hoher
Kohlenstoffgehalt detektiert, da die Beschichtung des SiC mit Graphen zu einer Anreicherung

3 Graphensynthese und Charakterisierung
- 62 -
des Kohlenstoffs an der Oberfläche führte (siehe Tabelle 5). In dieser Arbeit wurde der
Füllstoffgehalt in GSiC per TGA gemessen (siehe Abbildung 37).
100 200 300 400 500 600 700
94
95
96
97
98
99
100
GSiC-DGSiC-F
GSiC-G
Gew
icht [%
]
Temperatur [°C]
SiC
Abbildung 37: Bestimmung des Füllstoffgehalts in GSiC. Messung unter Luft-Atmosphäre bei einer
Heizrate von 10 K/min im Temperaturbereich von 50 bis 750 °C.
Die Messung wurde im Temperaturbereich 50 – 750 °C durchgeführt und erfolgte mit einer
Aufheizrate von 10 °C/min unter Luft-Atmosphäre. In diesem Temperaturbereich ist das SiC
gegenüber Sauerstoff beständig. Im Gegensatz dazu beginnt das Graphen bei ca. 400 °C
abzubauen bzw. zu verbrennen. Bei der TGA-Messung wurde das Graphen in den GSiC-Proben
durch Erhitzen abgebaut. Der Masseverlust im Vergleich zum reinen SiC wurde als
Füllstoffgehalt ausgewiesen. Bei allen GSiC-Proben wurde ein starker Masseverlust zwischen
400 und 600 °C detektiert, was vermutlich auf den Abbau des Graphens in GSiC
zurückzuführen ist (siehe Abbildung 37). Die Differenz des Masseverlusts zwischen SiC und
GSiC-F ist 4.4 Gew.-%, was einem Füllstoffgehalt von 4.4 Gew.-% in GSiC-F entspricht.
Ähnlich wurden die Graphengehalte in GSiC-G und GSiC-D auf 2.0 bzw. 5.1 Gew.-% bestimmt.
Weiterhin wurde die Kontrollierbarkeit des Füllstoffgehalts durch Variation des Gewichtsanteils
der Kohlenstoffquelle im Reaktionsgemisch mit SiC untersucht. Das Gewichtsverhältnis von
Kohlenstoffquelle (Furfurylalkohol, Glucose und Dopamin) zu SiC wurde von 9:91 bis zu
40:60 variiert. Abbildung 38 zeigt den Einfluss des Gewichtsanteils der Kohlenstoffquelle auf
den Füllstoffgehalt in allen GSiCs.

3 Graphensynthese und Charakterisierung
- 63 -
0 5 10 15 20 25 30 35 400
2
4
6
8
10
12
14
16
GSiC-GGSiC-D
Gra
ph
en
ge
ha
lt [
Ge
w.-
%]
Anteil der Kohlenstoffquelle [Gew.-%]
GSiC-F
Abbildung 38: Anteil an Graphen in GSiC in Abhängigkeit des eingesetzten Anteil der
Kohlenstoffquelle.
Der Füllstoffgehalt stieg bei allen GSiC-Systemen nahezu linear mit dem
Kohlenstoffquellengehalt an, was den gut kontrollierbaren Charakter der Reaktion zur
Herstellung von GSiC bestätigte. Dabei besetzen GSiC-F und GSiC-D eine gleiche Steigung,
welche höher als bei GSiC-G ist, was auf eine kleinere relative Ausbeute bei GSiC-G als von
GSiC-F und GSiC-D hindeutete. Folglich ist diese Methode hervorragend geeignet um GSiC
mit definiertem Füllstoffgehalt herzustellen, da sich dieser gezielt über die Menge der
eingesetzten Kohlenstoffquelle steuern lässt.
0 2 4 6 8 10 12 14 160
20
40
60
80
100
120
GSiC-D
GSiC-G
Sp
ezif
isch
e O
berf
läch
e [
m2g
-1]
Graphengehalt [Gew.-%]
GSiC-F
Abbildung 39: Spezifische Oberfläche von GSiC in Abhängigkeit des Füllstoffgehalts.

3 Graphensynthese und Charakterisierung
- 64 -
Zusätzlich wurde das Verhältnis zwischen Füllstoffgehalt und spezifischer Oberfläche
untersucht. Im Vergleich zum SiC, zeichnet sich das hergestellte Graphen durch viel dünnere
Schichten mit höherem Aspektverhältnis aus (siehe Abbildung 34). Dadurch ist die spezifische
Oberfläche unabhängig von der Kohlenstoffquelle linear vom Füllstoffgehalt abhängig (siehe
Abbildung 39). Des Weiteren ermöglicht diese hohe spezifische Oberfläche eine homogene
Verteilung bei einer späteren Verwendung als Füllstoff in Polymer- und Keramik-Matrizes.
3.5 Benchmark Kohlenstofffüllstoffe
In dieser Arbeit wurden verschiedene Benchmark-Kohlenstofffüllstoffe als
Vergleichsmaterialien untersucht. Davon sind Graphit (KFL 99.5) und Nanographit (V-HF 98)
von AMG Mining, Grade M und Grade C kommerziell von XG Sciences und sphärischer Leitruß
Carbon Black von Orion Engineered Carbons erhältlich. Zusätzlich wurde eine Multilagen-
Graphen-Variante „MG-Ethanol“ in Kooperation mit Projektpartner Dr. C. BALÁZSI vom
Institute for Technical Physics and Materials Science, Centre for Energy Research in Budapest
hergestellt. Diese Arbeit dient dabei auch der Evaluierung von Benchmark-
Kohlenstofffüllstoffen in Keramik-Graphen-Nanokompositen gegenüber den selbst
hergestellten Graphenen. Der Fokus liegt dabei neben der Eigenschaftsverbesserung auch auf
der Verfügbarkeit und Kosteneffizienz der eingesetzten Graphene.
Der Nanographit wurde durch mechanische Exfolierung aus Graphit hergestellt. Details der
Synthese sind aus Geheimhaltungsgründen nicht bekannt. Der Graphit kann auf Grund seiner
Schmiermitteleigenschaften selbst als Festschmierstoff benutzt werden, da die einzelnen Lagen
nur von van-der-Waals-Kräfte zusammengehalten werden. Darüber hinaus findet Graphit
bereits Anwendung als Füllstoff in den SiC-Werkstoffen. Dabei zeigt das kommerzielle SiC
Typ G der Firma 3M, welches mit Graphit gefüllt ist, bessere tribologische und elektrische
Eigenschaften als das ungefüllte Typ F (vgl. Tabelle 8 in Kapitel 4.2). Deswegen wurden im
Rahmen dieser Arbeit Graphit und Nanographit auch als Benchmark-Kohlenstofffüllstoffe
verwendet.

3 Graphensynthese und Charakterisierung
- 65 -
Abbildung 40: Allgemeine Herstellungsmethode von Grade M und Grade C laut
Herstellerangaben.[212,213].
Die kommerziell erhältlichen Graphentypen Grade M und Grade C werden laut
Herstellerangaben durch einfache mechanische Interkalation (siehe Abbildung 40 a),
Exfolierung (b) und Reduzierung der Größe (c) hergestellt, ähnlich wie in der Literatur
beschrieben.[24,49,50] Durch Interkalation mit den Molekülen/Ionen zwischen den
Graphitschichten wurde der Schichtabstand vergrößert. Als typische Interkalate werden in der
Literatur Alkalimetalle, Metallhalogenide, Säuren oder eine Kombinationen von Alkalimetallen
und organischen Verbindungen (z. B. THF, Benzol, Ethylen, Styrol oder Butadien)
verwendet.[212] Durch Exfolierung des Graphits mit den Interkalaten können Multilagen-
Graphene hergestellt werden. Die relativ großen Partikel werden dann in nachfolgenden
Mahlprozessen entsprechend zerkleinert, um zuerst Grade M und anschließend Grade C zu
erhalten. Ob die Exfolierung des Graphits mit den Interkalaten direkt während des
Mahlprozesses oder durch thermische Behandlung erfolgt ist nicht bekannt. Zur Verarbeitung
zeichnet sich Graphen von XG Sciences durch eine gute Dispergierbarkeit in Wasser oder in
organischen Lösungsmitteln aus.[212]
Das MG-Ethanol wurde durch Nassvermahlen von Graphit (10 g, Aldrich) in einer
Attritormühle mit Ethanol als Dispersionsmedium bei einer Mahldauer von 10 h und einer
Drehzahl von 4000 U/min hergestellt. Die Vermahlung und Exfolierung des Graphits erfolgte
dabei durch mechanische Scherkräfte in der Mahlkammer aus Siliciumnitrid (750 mL), welche
mit Keramikperlen aus Zirconia (d = 1 mm) befüllt war.[52] Im Vergleich zu den
funktionalisierten MGs in Kapitel 3.3 verfügt MG-Ethanol über keine funktionelle Gruppen.
Die Verwendung von MG-Ethanol als Referenz dient zur Evaluierung des Einflusses von
funktionellen Gruppen auf die Eigenschaften der hergestellten Keramik-Nanokomposite.

3 Graphensynthese und Charakterisierung
- 66 -
Gegenüber der kurzen Historie von Graphen werden andere Kohlenstofffüllstoffe insbesondere
Carbon Black (Industrieruß) und Graphit bereits deutlich länger erforscht und großindustriell
verwendet. Darüber hinaus wird Carbon Black schon bei der Herstellung von SiC als Standard-
Sinterhilfsmittel genutzt. Daher wurde Carbon Black auch als Referenz zur Evaluierung der
Füllstoffqualität von Graphen herangezogen.
In diesem Abschnitt werden die kommerziell erhältlichen Kohlenstoff-Nanofüllstoffe in Bezug
auf ihre Eigenschaften untersucht. Im Einzelnen wird die Oberfläche (BET), die elementare
Zusammensetzung (EA & EDX), das thermische Abbauverhalten (TGA) und die Morphologie
(Aspektverhältnis und Partikelgeometrie) mittels SEM- und TEM-Aufnahmen untersucht.
Tabelle 6: Eigenschaften der verwendeten kommerziellen Kohlenstofffüllstoffe. Gezeigt sind Daten aus
EA-, BET-, und TGA-Messungen.
Probe C-Gehalta
[Gew.-%]
O-Gehaltb
[Gew.-%]
H-Gehalta
[Gew.-%]
𝐀𝐁𝐄𝐓𝐜
[m2∙g-1]
Mlossd
[%]
Geometrie
Graphit 99.6 0.7 0.2 3 1.9 Plättchen
Nanographit 96.9 3.8 0.2 34 2.5 Plättchen
MG-Ethanol 98.3 1.7 0.2 94 13.3 Plättchen
Carbon Black 90.1 8.1 1.8 44 21.4 Sphärisch
Grade C 88.0 10.7 1.1 794 21.4 Sphärisch
Grade M 90.9 5.9 0.9 88 10.1 Plättchen
a Bestimmt durch Elementaranalyse; b Bestimmt durch EDX; c N2-Physisorptionsmessung zur
Oberflächenbestimmung nach Brunauer, Emmett und Teller; d TGA-Masseverlust, N2-Atmosphäre,
10 K/min, 50 – 1500 °C.
Eine Übersicht über die Eigenschaften der verwendeten Nanofüllstoffe gibt Tabelle 6.
Charakteristisch für die verschiedenen Graphentypen war der Sauerstoffgehalt. Dieser ist
proportional zur Funktionalisierung mit –OH oder –COOH Gruppen. Grade C zeigt den
höchsten Sauerstoffgehalt bzw. die höchste Dichte funktioneller Gruppen, welche Polarität
erzeugen. Daher sollte Grade C auch die beste Dispergierung in Wasser und damit auch die
beste Verteilung in der Keramik-Matrix ergeben. Die Detektion des Sauerstoffgehalts bei
Carbon Black lässt sich vermutlich auf Oberflächenfunktionalisierung zurückführen. Im
Vergleich dazu weisen der Nanographit und das MG-Ethanol einen deutlich niedrigen

3 Graphensynthese und Charakterisierung
- 67 -
Sauerstoffgehalt auf, was auf eine geringe Funktionalisierung beim Exfolierungsprozess
zurückzuführen ist. Diese funktionellen Gruppen wie –COOH und –OH wurden teilweise auch
bei der TGA-Messung durch Erhitzen abgebaut und als Kohlenstoffmonooxid und
Kohlenstoffdioxid freigesetzt. Dafür zeigten die Benchmark-Kohlenstoffe einen
kontinuierlichen Masseverlust, welcher mit zunehmendem Funktionalisierungsgrad korreliert.
Die Bestimmung der spezifischen Oberfläche durch BET-Messung ergab für Graphit 3 m2∙g-1,
für Carbon Black und Nanographit ca. 40 m2∙g-1, für Grade M und MG-Ethanol ca. 90 m2∙g-1
und für Grade C 800 m2∙g-1 (siehe Tabelle 6). Die hohe Oberfläche von Grade C lässt sich auf
die geringe Partikelgröße zurückführen.
Abbildung 41: SEM- Aufnahmen von a) Graphit, b) Nanographit, c) MG-Ethanol, d) Carbon Black,
e) Grade C und f) Grade M.
Abbildung 41 zeigt die SEM-Aufnahmen der Partikelmorphologie von Graphit, Nanographit,
MG-Ethanol, Carbon Black, Grade C und Grade M im Vergleich. Der Graphit (a),
Nanographit (b), MG-Ethanol (c) und das Grade M (f) zeigen deutlich Plättchenstrukturen,
während das Carbon Black (d) und Grade C (e) sphärische Morphologie aufweisen. Der
schichtartige Graphit (a) und Grade M (f) verfügen über eine Lateralabmessung von über
100 µm, während die Partikeldimension des Nanographits (b) und MG-Ethanols (c) in einer
Größenordnung von maximal 3 µm liegt. Dagegen bestehen das sphärische Carbon Black (d)

3 Graphensynthese und Charakterisierung
- 68 -
und Grade C (e) aus kumulierten Partikelclustern mit einer maximalen Größe von 10 µm.
Darüber hinaus wurden die vergrößerten Partikel von MG-Ethanol, Carbon Black, Grade C und
Grade M mittels TEM-Aufnahmen bei hohen Auflösungen untersucht (siehe Abbildung 42).
Abbildung 42: TEM- Aufnahmen von a) MG-Ethanol, b) Carbon Black, c) Grade C und d) Grade M.
Anhand der TEM-Aufnahmen in Abbildung 42 zeichnet sich MG-Ethanol (a) und Grade M (d)
wie TRGO (vgl. Abbildung 11 in Kapitel 3.1) durch eine ähnliche Plättchen-Morphologie mit
einer Lateralabmessung von bis zu 3 µm aus. MG-Ethanol und Grade M bilden eine glatte
Plättchen Struktur mit scharfen Kanten aus. Es treten Agglomerate, aber auch exfolierte
Multilagen-Strukturen auf, was die Ergebnisse der BET-Messungen erklärt. Die
durchschnittliche Anzahl an Graphenlagen L ist über Gleichung 1 (siehe Kapitel 3.1)
bestimmbar. Die Schichtzahl von MG-Ethanol bzw. Grade M entspricht dem dividierten Wert
der spezifischen Oberfläche von idealem Graphen (Sideal: 2630 m2∙g-1) durch die gemessene
spezifische Oberfläche des MG-Ethanol bzw. Grade M.[190] Die berechnete Anzahl liegt jeweils
bei ca. 30 Lagen. Carbon Black setzte sich aus nahezu uniformen, sphärischen Primärpartikeln
mit einheitlichem Durchmesser von ca. 50 nm zusammen. Im Vergleich dazu besteht Grade C
aus annähernd sphärischen Partikeln mit uneinheitlichen Durchmessern von maximal 500 nm.
Es war eine große morphologische Ähnlichkeit zu den unter Ar- oder N2-Atmosphäre
vermahlenen Nanopartikeln (siehe Abbildung 20 in Kapitel 3.2) zu beobachten. Die Kanten der
Partikel sind sehr uneinheitlich, was auf eine hohe mechanische Beanspruchung bei der
Herstellung hinweist. Aufgrund des geringen Aspektverhältnisses waren für die mit Grade C
und Carbon Black gefüllten Nanokomposite deutlich geringere Steigerungen der Risszähigkeit
und der Leitfähigkeit zu erwarten, als mit Grade M oder MG-Ethanol.
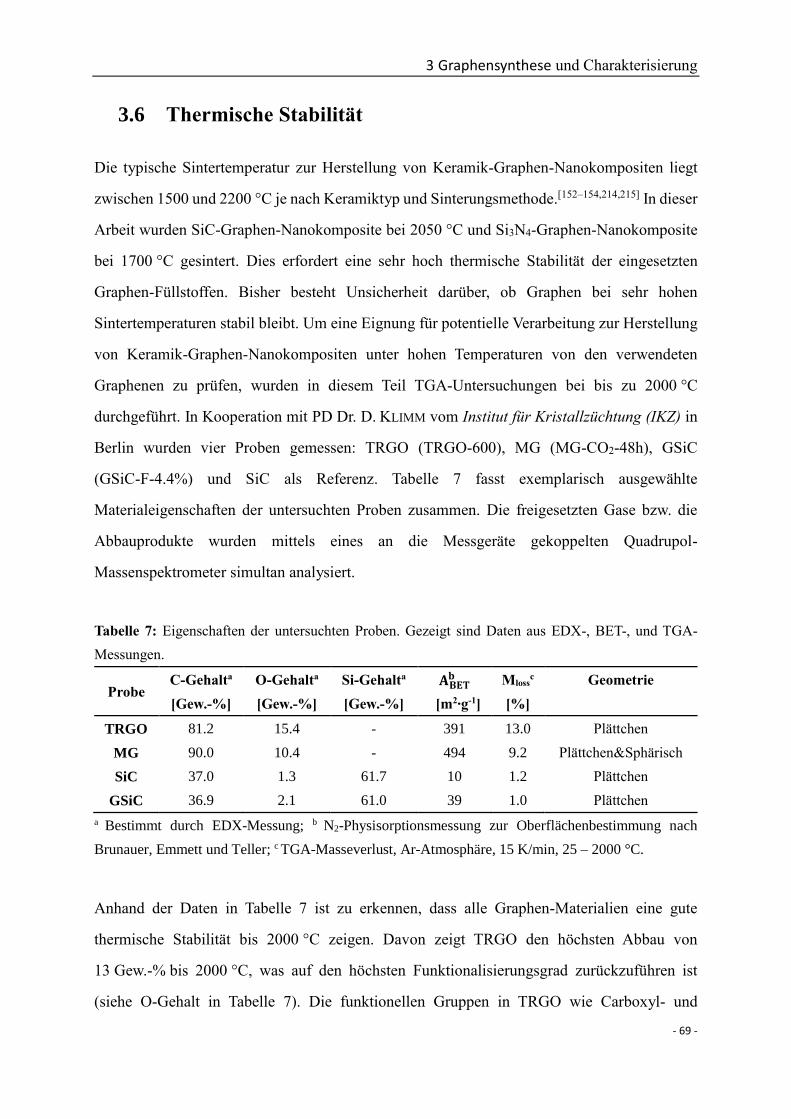
3 Graphensynthese und Charakterisierung
- 69 -
3.6 Thermische Stabilität
Die typische Sintertemperatur zur Herstellung von Keramik-Graphen-Nanokompositen liegt
zwischen 1500 und 2200 °C je nach Keramiktyp und Sinterungsmethode.[152–154,214,215] In dieser
Arbeit wurden SiC-Graphen-Nanokomposite bei 2050 °C und Si3N4-Graphen-Nanokomposite
bei 1700 °C gesintert. Dies erfordert eine sehr hoch thermische Stabilität der eingesetzten
Graphen-Füllstoffen. Bisher besteht Unsicherheit darüber, ob Graphen bei sehr hohen
Sintertemperaturen stabil bleibt. Um eine Eignung für potentielle Verarbeitung zur Herstellung
von Keramik-Graphen-Nanokompositen unter hohen Temperaturen von den verwendeten
Graphenen zu prüfen, wurden in diesem Teil TGA-Untersuchungen bei bis zu 2000 °C
durchgeführt. In Kooperation mit PD Dr. D. KLIMM vom Institut für Kristallzüchtung (IKZ) in
Berlin wurden vier Proben gemessen: TRGO (TRGO-600), MG (MG-CO2-48h), GSiC
(GSiC-F-4.4%) und SiC als Referenz. Tabelle 7 fasst exemplarisch ausgewählte
Materialeigenschaften der untersuchten Proben zusammen. Die freigesetzten Gase bzw. die
Abbauprodukte wurden mittels eines an die Messgeräte gekoppelten Quadrupol-
Massenspektrometer simultan analysiert.
Tabelle 7: Eigenschaften der untersuchten Proben. Gezeigt sind Daten aus EDX-, BET-, und TGA-
Messungen.
Probe C-Gehalta
[Gew.-%]
O-Gehalta
[Gew.-%]
Si-Gehalta
[Gew.-%]
𝐀𝐁𝐄𝐓𝐛
[m2∙g-1]
Mlossc
[%]
Geometrie
TRGO 81.2 15.4 - 391 13.0 Plättchen
MG 90.0 10.4 - 494 9.2 Plättchen&Sphärisch
SiC 37.0 1.3 61.7 10 1.2 Plättchen
GSiC 36.9 2.1 61.0 39 1.0 Plättchen
a Bestimmt durch EDX-Messung; b N2-Physisorptionsmessung zur Oberflächenbestimmung nach
Brunauer, Emmett und Teller; c TGA-Masseverlust, Ar-Atmosphäre, 15 K/min, 25 – 2000 °C.
Anhand der Daten in Tabelle 7 ist zu erkennen, dass alle Graphen-Materialien eine gute
thermische Stabilität bis 2000 °C zeigen. Davon zeigt TRGO den höchsten Abbau von
13 Gew.-% bis 2000 °C, was auf den höchsten Funktionalisierungsgrad zurückzuführen ist
(siehe O-Gehalt in Tabelle 7). Die funktionellen Gruppen in TRGO wie Carboxyl- und

3 Graphensynthese und Charakterisierung
- 70 -
Epoxidgruppen wurden während der thermischen Behandlung abgespalten und als
Kohlenstoffmonooxid freigesetzt, welches mittels Massenspektrometer nachgewiesen wurde.
In Abbildung 43 wurden die ermittelte TGA-Kurve von MG und die Massenspektroskopie von
freigesetzten Gasen als Beispiel dargestellt und ausgewertet.
500 1000 1500 200080
85
90
95
100
Gewicht
m/z-Wert 18 (H2O)
m/z-Wert 28 (CO)
m/z-Wert 44 (CO2)
Re
lati
ve
s G
ew
ich
t [%
]
Temperatur [°C]
9.2 %
In
ten
sit
ät
[a.u
.]
Abbildung 43: TGA von MG und Massenspektroskopie von freigesetzten Gasen. Messung unter Ar-
Atmosphäre bei einer Heizrate von 15 K/min im Temperaturbereich von 25 bis 2000 °C.
Wegen der hohen Sauerstofffunktionalisierung wurde bei MG bis 2000 °C auch ein großer
Masseverlust von 9 Gew.-% detektiert. Dabei wies MG in Abbildung 43 bis 1000 °C einen
kontinuierlichen Masseverlust auf. Im Bereich von 100 bis 250 °C wurde das freigesetzte Gas
mit einem Masse-zu-Ladung-Verhältnis (m/z-Werte) von 18 detektiert, was Wasser
zugeschrieben wurde, das im hydrophilen MG vorhanden ist. Zwischen 90 und 600 °C trat das
freigesetzte Gas bei einem m/z-Wert von 44 auf, was auf das Entweichen von nicht gebundenen
CO2 zwischen den Graphenschichten sowie thermische Zersetzung der
Sauerstofffunktionalitäten zu CO2 zurückzuführen ist. Weiterhin wurde das freigesetzte Gas aus
MG zwischen 150 und 1000 °C mit einem m/z-Wert von 28 beobachtet, welches auf den Abbau
der funktionellen Carboxyl- und Hydroxylgruppen hinwies und dem damit verbundenen
Entweichen von CO zugeordnet werden konnte.

3 Graphensynthese und Charakterisierung
- 71 -
Im Vergleich dazu wiesen SiC und GSiC sehr ähnliche thermische Eigenschaften mit sehr
kleinem Masseverlust von ca. 1 Gew.-% auf, was auf den geringen Graphengehalt (4.4 Gew.-%)
in GSiC und die gute thermische Stabilität der funktionalisierten Graphene in GSiC
zurückzuführen ist. Erst über 1200 °C zeigten SiC und GSiC einen geringfügigen Masseverlust
von 1 Gew.-% bis 1600 °C. Im gleichen Bereich wurde im Massenspektrometer ein Signal mit
einem m/z-Wert von 28 detektiert, welches Siliciumionen zugeordnet werden könnte. Dies
entspricht dem literaturbekannten Verhalten, dass hohe Temperaturen von 1300 – 1800 °C die
Sublimation von Silicium von der SiC-Oberfläche ermöglichen.[35–38] Dabei haften die
zurückgebliebenen Kohlenstoffatome auf der Oberfläche des Siliciumcarbid-Substrats und
konnten dort durch anschließende Graphenisierung zu einer Graphenschicht anordnen, was die
weitere Sublimation von Silicium verhindert. Dies führte dazu, dass hier nur ein geringer
Masseverlust detektiert wurde.[35–38]
Zusammenfassend zeigten alle Graphen-Materialien zeigten eine gute thermische Stabilität bis
2000 °C mit einem maximalen Masseverlust von 13 Gew.-%. Damit eigenen sich alle getesteten
Materialien als Füllstoffe zur Herstellung von SiC- und Si3N4-Graphen-Nanokomposite durch
Sinterung unter hohen Temperaturen (1700 – 2050 °C). Des Weiteren werden bei den GSiC-
Nanokompositen in späteren Kapiteln aufgrund ihrer hervorragenden thermischen Stabilität
vollständigere Verdichtung als die TRGO- und MG-CO2-Varianten erwartet.

3 Graphensynthese und Charakterisierung
- 72 -
3.7 Zusammenfassung
Im Rahmen dieser Arbeit wurden verschiedene Graphen-Materialien entweder mittels Top-
down-Ansatz aus Graphit oder Bottom-up-Methode aus verschiedenen Kohlenstoffquellen für
die Erzeugung multifunktionaler Keramik-Graphen-Nanokomposite hergestellt. Davon erfolgte
die Herstellung von TRGO mittels Top-Down-Ansatz durch thermische Reduktion und
Expansion des GO, welches nach HUMMERS-Verfahren aus Graphit synthetisiert wurde. Mit
Änderung der Reduktionstemperatur konnten die Materialeigenschaften des TRGO gezielt
eingestellt werden. Damit ging eine Steigerung der Temperaturbeständigkeit, der spezifischen
Oberfläche und des Exfolierungsgrades. Außerdem wurde die Funktionalität reduziert, was
mittels EDX- und IR-Analyse nachgewiesen werden konnte. Im Vergleich zu TRGO-600 und
TRGO-800 zeigte TRGO-1000 die höchste Oberfläche von 515 m2∙g-1 und den höchsten
Exfolierungsgrad mit einer durchschnittlichen Anzahl an Graphenlagen von fünf. Anhand der
SEM-, TEM- und AFM-Aufnahmen zeigten alle TRGO eine dünne, gewellte und gefaltete
Mono- bis Weniglagen-Struktur.
Als Alternative zu TRGO konnte das mechanochemisch funktionalisierte Multilagen-Graphen
(MG) mittels Top-down-Ansatz durch einen einstufigen Trockenvermahlungsprozess direkt aus
Graphit hergestellt werden. Die Synthese erfolgte in einer Planetenkugelmühle unter
verschiedenen Gas-Atmosphären (Ar, CO2 und N2). Die Variation der Prozessparameter
(Mahldauer und Gas-Atmosphäre) ermöglichte eine individuelle Anpassung der
Partikelgeometrie, Größe und Funktionalisierung. So konnte z. B. die Plättchenstruktur nach
der Vermahlung unter CO2 erhalten werden, während bei MG-Ar und MG-N2 nur noch
sphärische Partikel zu erkennen waren. Somit wird eine Verbesserung der elektrischen
Leitfähigkeit der mit MG-CO2 (insbesondere bei geringerer Mahldauer) gefüllten
Nanokomposite im Gegensatz zu den MG-Ar/N2-Vaterianten erwartet. Des Weiteren führte die
Vermahlung unter CO2 zu funktionalisiertem Graphen mit Carboxylgruppen, während die
Vermahlung unter N2 oder Ar zu MG mit Hydroxylgruppen führte. Darüber hinaus konnten
stabile tensidfreie MG- und TRGO-Dispersionen in Wasser mit Hilfe des
Hochdruckhomogenisators (HH) hergestellt werden. Aufgrund der hohen Scherkräfte blieben

3 Graphensynthese und Charakterisierung
- 73 -
die Dispersionen über einen langen Zeitraum (z. B. TRGO über 50 Tage) stabil. Zusätzlich
wurde die Stabilität der Dispersionen durch eine elektrostatische Abstoßung der Graphen-
Flocken hervorgerufen, was durch die Messung des Zetapotentials nachgewiesen wurde.
Als Bottom-up-Methode wurde das Graphen aus drei verschiedenen Graphen-Präkursoren,
respektive Furfurylalkohol, Glucose und Dopamin, über eine templatvermittelte Reaktion
direkt auf die Oberfläche von SiC hergestellt. Im Vergleich zu TRGO und MG war hier weder
ein Einsatz von fossilen Kohlenstoffquellen (z. B. Graphit) noch nachträgliche Behandlung
(z. B. Reinigung und Homogenisierung) erforderlich. Zusätzlich ist bei den Nanokompositen
mit GSiC im Vergleich zu den konventionellen Graphenen weniger Porosität bzw.
vollständigere Verdichtung zu erwarten. Anhand der TEM-Aufnahmen und EDX-Analysen
konnte eine erfolgreiche Herstellung der GSiCs nachgewiesen werden. Außerdem konnte die
Dicke der Beschichtung mittels TEM-Aufnahmen analysiert werden. Je nach Kohlenstoffquelle
variierte die Anzahl der Graphenlagen zwischen 5 und 20 mit einer Dicke von 5 – 10 nm. Des
Weiteren konnte der Graphengehalt und die spezifische Oberfläche von GSiC durch Variation
des Gewichtsanteils der Kohlenstoffquelle im Reaktionsgemisch mit SiC kontrolliert werden.
Neben den selbst hergestellten Graphenen wurden die plättchenförmigen Kohlenstofffüllstoffe
Graphite, Nanographit, Grade M und MG-Ethanol sowie die sphärischen Kohlenstofffüllstoffe
Carbon Black und Grade C als Referenzen verwendet. Dabei verfügten Grade M und
MG-Ethanol über ein höheres Aspektverhältnis und eine dünnere Schichtstruktur als Graphit
und Nanographit. Im Hinblick auf die Funktionalität nahm der O-Gehalt von Grade C
(10.7 Gew.-%) über Carbon Black und Grade M (6 – 8 Gew.-%) bis hin zu Nanographit
(3.8 Gew.-%) ab. Dagegen beinhalteten Graphit und MG-Ethanol kaum funktionelle Gruppen,
was eine schlechtere Dispergierung in Wasser und als Folge eine schlechtere Verteilung in den
Keramik-Nanokompositen sowie eine geringere Verstärkungswirkung erwarten lässt.
In den späteren Kapiteln werden SiC-Graphen-Nanokomposite bei 2050 °C und Si3N4-
Graphen-Nanokomposite bei 1700 °C hergestellt, was eine sehr hohe thermische Stabilität der
eingesetzten Graphen-Füllstoffe erfordert. Dafür wurde die Eignung für potentielle

3 Graphensynthese und Charakterisierung
- 74 -
Verarbeitung unter hohen Temperaturen von den verwendeten Graphenen mittels TGA-
Untersuchungen bis zu 2000 °C ausgewertet. Dabei zeigten alle Graphen-Materialien eine gute
thermische Stabilität bis 2000 °C mit einem maximalen Masseverlust von 13 Gew.-%
(TRGO-600) gefolgt von 9 Gew.-% (MG-CO2-48h). Zusätzlich ist zu sehen, dass SiC und
GSiC-F sehr ähnliche thermische Eigenschaften mit geringfügigem Masseverlust (ca. 1 Gew.-%)
zeigten, was auf den geringen Graphengehalt (4.4 Gew.-%) in GSiC-F und die gute thermische
Stabilität der funktionalisierten Graphene in GSiC-F zurückzuführen ist. Dafür werden bei den
GSiC-Nanokompositen in späteren Kapiteln aufgrund ihrer hervorragenden thermischen
Stabilität die vollständigeren Verdichtungen und als Folge die besseren Werkstoffeigenschaften
im Vergleich zu den TRGO- und MG-CO2-Varianten erwartet.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 75 -
4 Graphen-Nanokomposite auf Basis von
Siliciumcarbid
4.1 Einleitung
Keramische Werkstoffe auf Basis von Siliciumcarbid (SiC) sind in der Technik sicherlich die
bedeutendste Nicht-Oxid-Keramik.[2] Aufgrund seiner herausragenden mechanischen,
thermischen und chemischen Eigenschaften findet SiC eine breite Anwendung in vielen
Bereichen der Industrie und Energietechnik.[1] Hierfür zeichnet es sich generell durch eine
extrem hohe Härte und Abriebfestigkeit, Korrosions- und Oxidationsbeständigkeit, hohe
Festigkeit auch bei hohen Temperaturen, sowie gute Temperaturwechselbeständigkeit mit
geringem Wärmeausdehnungskoeffizienten, aber hoher Wärmeleitfähigkeit aus.[1] Im Vergleich
zu Metall und Kunststoff eignet sich das SiC insbesondere bei der Anwendung im
Maschinenbau in besonders verschleißfesten und korrosionsbeständigen Bauteilen unter
extremen Bedingungen, z. B. Gleitringdichtungen in Chemiepumpen sowie Gleitlagern für sehr
hohe Anwendungstemperaturen (bis zu 1500 °C).[111–113] Um die Effizienz, Lebensdauer,
Zuverlässigkeit und Leistungsfähigkeit keramischer Komponenten in solchen Anwendungen
unter Wasser- bzw. Mangelschmierung (d.h. ohne Schmierstoffe), aber auch zur Förderung
anderer Medien zu verbessern, werden jedoch leistungsfähigere Werkstoffe gebraucht. Eine
Senkung der Reibung in solchen Lagerungen würde zu erheblichen Energieeinsparungen führen.
Durch Erhöhung der Lebensdauer und der Zuverlässigkeit dieser sicherheitsrelevanten
Komponenten können zudem die Anlagenverfügbarkeit und damit die Ressourceneffizienz
erhöht und die Gefahr für Menschen und Umwelt vermindert werden. Ferner schränkt die
elektrische Isolation einerseits die Verwendung von innovativen Bearbeitungsverfahren wie
dem Funkenerodieren, andererseits die Anwendung in der Sensorik (z. B. Abgassensorik) und
in Brennstoffzellen ein,[1,124,175–177] sodass in den vergangenen Jahren verstärkt versucht wurde,
durch die Zugabe von Kohlenstoffadditiven, z. B. Carbon Black und CNTs, die
mechanischen[125], elektrischen[126] und tribologischen[128] Eigenschaften von SiC zu verbessern.
Trotzdem ist die Anwendung von CNTs als Füllstoff in Keramik aufgrund der

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 76 -
Nanotoxizitätsproblematiken und hohen Materialkosten sowie der schlechten Dispergierbarkeit
eingeschränkt, da CNTs synthesebedingt zu hochaggregierten Clustern verschlaufen.[216,217]
Angesichts der steigenden Verfügbarkeit, unproblematischen Dispergierbarkeit sowie dem
geringen Syntheseaufwand wurde in jüngster Zeit funktionalisiertes Graphen als funktionaler
Füllstoff in Keramik zur Verstärkung oder Funktionalisierung eingesetzt.[198] Von dieser neuen
Kohlenstoffmodifikation wird erwartet, dass sie als Füllstoff in tribologisch beanspruchten SiC-
Komponenten durch mechanische Verstärkung und chemische Passivierung sowohl die
Reibung als auch den Verschleiß erheblich senkt.[152] Im Vergleich zu den anderen technischen
Keramiken wurden SiC-Graphen-Nanokomposite bisher kaum erforscht. Im Zeitraum dieser
Arbeit wurden nur die folgenden Publikationen über graphengefülltes SiC veröffentlicht. Die
Gruppe von BELMONTE stellten SiC-Graphen via SPS-Sinterverfahren her. Sie erreichten mit
20 Vol.-% Füllstoffanteil eine Zunahme der elektrischen Leitfähigkeit von 2 × 10-8 (reine
Matrix) auf 44 S∙cm-1 (gefüllten Probe).[137] Des Weiteren beobachteten sie mit 20 Vol.-%
Füllstoffanteil eine Verbesserung der Verschleißbeständigkeit um 70 % bei tribologischen
Messungen unter trockenen Bedingungen.[152] MIRANZO et al. berichteten 2016 von SiC-
TRGO-Nanokompositen, die nach dem in situ Reduktion von Graphitoxid (GO) hergestellt
wurden. Mit 5 Vol.-% TRGO wurde die Risszähigkeit um 162 % erhöht, wobei eine Steigerung
der Biegefestigkeit von 370 auf 620 MPa beobachtet wurde.[133]
4.2 Herstellung
In dieser Arbeit wurden SiC-Graphen-Nanokomposite in Zusammenarbeit mit M. KNOCH von
der Firma FCT Ingenieurkeramik GmbH in Sonneberg hergestellt. Die im Kapitel 3
dargestellten Graphene wurden hier als funktionelle Füllstoffe eingesetzt. Der Einfluss der
Füllstoffarten im Hinblick auf die Funktionalität, mechanische und tribologische Eigenschaften
sowie die Leitvermögen in einem kommerziell erhältlichen SiC (Ref. SiC) der Firma FCT
Ingenieurkeramik GmbH wurde evaluiert. Die erwarteten Nanokomposit-Produkte sollen eine
geringere Reibung, höhere Verschleißbeständigkeit, verbesserte Risszähigkeit sowie erhöhte

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 77 -
elektrische Leitfähigkeit besitzen. Damit wird die Schadenstoleranz und Energieeffizienz
verbessert, die Lebensdauer und die Belastbarkeit und damit einhergehend die Zuverlässigkeit
erhöht. Somit verspricht die Entwicklung von technisch nutzbaren Werkstoffen und
Komponenten auf Basis von SiC-Graphen-Nanokompositen einen großen technischen und
wirtschaftlichen Nutzen auch weit über den angesprochenen Anwendungsbereich hinaus.
Weitere vorteilhafte Anwendungen für diese neuen Keramiken sind zum Beispiel Umform- und
Zerspanungswerkzeuge, die einen geringeren Verschleiß und Kühlschmierstoffbedarf haben,
keramische Wälzlager mit erhöhter Bruchzähigkeit, Dickschichtsysteme als Beschichtungen in
Wärmekraftmaschinen, die zu höheren Betriebstemperaturen und Wirkungsgraden führen und
letztlich alle Maschinenelemente, die hohen tribologischen Beanspruchungen ausgesetzt sind.
Als Referenzwerkstoffe wurden das ungefüllte SiC (Ref. SiC) sowie zwei kommerzielle SiC-
Produkte Typ F und Typ G der Firma 3M untersucht. Laut Herstellerangaben ist Typ F ein
feinkörniger Konstruktionswerkstoff mit hoher Härte und guter Korrosionsbeständigkeit mit
einer mittleren Korngröße kleiner als 5 μm. Im Vergleich dazu verfügt Typ G über eine größere
Korngröße zwischen 10 und 1000 µm sowie homogen im Gefüge verteilten Graphite mit einer
Größe von 50 – 120 μm. Tabelle 8 fasst exemplarisch ausgewählte Materialeigenschaften der
ungefüllten Matrix sowie die kommerziellen Produkte von 3M zusammen.
Tabelle 8: Materialeigenschaften der eingesetzten Ref. SiC und kommerziellen Produkten Typ F und
Typ G von 3M nach den technischen Datenblättern.
Eigenschaften Einheit Ref. SiCa Typ Fb Typ Gb
Dichte g/cm3 3.08 – 3.17 > 3.15 > 3.10
Porosität % < 3.0 < 2.0 < 2.0
Mittlere Korngröße µm 1 – 5 < 5 10 – 1000
Phasenzusammensetzung α - SiC + C/B4C α - SiC α - SiC + Graphit
Vickers-Härte GPa 26.0 24.5 24.5
E-Modul GPa 420 430 390
Biegefestigkeit MPa 460 400 250
Bruchzähigkeit MPa·m0.5 3.5 4.0 3.5
Spez. Leitfähigkeit S∙cm-1 3 × 10-8 < 10-8 10-5 – 10-4
a technisches Datenblatt FCT SiC Standardwerkstoffe SC-S; b technisches Datenblatt 3MTM
Siliciumcarbid Portfolio.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 78 -
Abbildung 44: Flussdiagramm zur Herstellung von SiC-Graphen-Nanokompositen.
Wie in Abbildung 44 dargestellt, erfolgte die Herstellung der SiC-Graphen-Nanokomposite
zunächst durch Mischung und Homogenisierung des Graphens und der Sinterhilfsmittel in der
SiC-Matrix. Dies erfolgte durch einen Turbula Shaker Mixer innerhalb von 4 h. Hierfür wurde
das Graphen in Form einer stabilen Dispersion verwendet, welche durch den in Kapitel 3.1.1
beschriebenen Hochdruckhomogenisierungsprozess in Wasser hergestellt wurde. Nach Erhalt
stabiler Dispersionen erfolgte die Rücktrocknung und Granulierung der Probe über eine
Gefriertrocknung. Dabei wurde das Wasser bei Unterdruck durch Sublimation entfernt,
wodurch die Ausbildung von harten Granulaten und Agglomeraten vermieden wurde.
Anschließend wurden aus dem so gewonnenen Granulat Probeplatten durch kalt isostatisches
Pressen bei 2000 bar Druck verdichtet und zuletzt im Ofen drucklos bei 2050 °C unter Argon
für 4 h gesintert. Die diese Weise erhaltenen Nanokomposite wurden daraufhin mittels
Hartbearbeitung (Fräsen und Abschleifen) zu Prüfkörpern geformt, um die Dichte, Porosität,
Biegefestigkeit, Bruchzähigkeit und die elektrischen und tribologischen Eigenschaften sowie
zu Gleitring-Prüfkörpern für die anwendungsnahen Prüfstandteste bestimmen zu können.
Zusätzlich erfolgte die Charakterisierung der Verteilung der Füllstoffe in der SiC-Matrix mittels
SEM-Aufnahmen.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 79 -
Die meisten Nanokomposite wurden mit einem Füllstoffgehalt von 2.0 Gew.-% hergestellt. Bei
Graphenanteilen über 2.0 Gew.-% ist aufgrund vermehrter Partikel-Partikel-Wechselwirkungen
damit zu rechnen, dass sich die Nanokomposite bei der Standardsinterung schlechter verdichten
lassen und damit die Restporosität im Bauteil zu hoch wird. Ein höherer Anteil an Graphen
könnte die erreichbare Sinterdichte deutlich reduzieren, was zu den schlechten mechanischen
Eigenschaften führt.[132,133,152,154,159] Unter Berücksichtigung der Analyse der mechanischen
Eigenschaften wurde der Füllstoffgehalt bei den vielversprechenden GSiC-Systemen auf bis zu
8.0 Gew.-% erhöht.
Abbildung 45: Mit GO gefüllte SiC-Nanokomposite a) vor und b) nach Sintertest.
Wie bereits ausgeführt, wurden die aus den Granulaten hergestellten Proben gesintert.
Wiederum auffallend bei den Sinterversuchen war die Probe mit GO. Diese konnte nicht
gesintert werden, da sie unter den Sinterbedingungen zerfiel, wie in Abbildung 45 dargestellt.
Ähnlich wie bei der Herstellung von TRGO, beim Erhitzen der mit GO gefüllten Probe während
des Sinterprozesses, wurden die hoch sauerstoffhaltigen funktionellen Gruppen im GO wie
Carboxyl-, Epoxid- und Hydroxylgruppen sowie die adsorbierte Luftfeuchtigkeit abgespalten
und als Kohlenstoffmonoxid, Kohlenstoffdioxid und Wasserdampf freigesetzt. Der
resultierende Gasdruck riss die gepresste Probenplatte auseinander. Alle weiteren Proben ließen
sich fehlerfrei ausheizen und sintern.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 80 -
4.3 Dichte & Porosität
Die Porosität ist eine dimensionslose Messgröße und definiert als 1 minus dem Quotienten aus
Rohdichte und Reindichte. Sie ist eine der wichtigsten Eigenschaften bei der Entwicklung von
keramischen Werkstoffen und erlaubt Aussagen über den Zustand von hergestellten
Komponenten nach der Sinterung.[2] Bei unzureichender Verdichtung bilden sich Poren, welche
einen signifikanten Einfluss auf die Eigenschaften keramischer Werkstoffe haben. So sinkt
beispielsweise der E-Modul mit zunehmender Porosität.[214] Der Zusammenhang zwischen
Porosität und E-Modul (E) wird durch die Gleichung 2 beschrieben, wobei E0 dem E-Modul
von porenfreier Keramik, P der Porosität und b dem empirischen Materialkonstant
entspricht.[218,219]
bPeEE 0 (2)
Ebenso wird die Korrosionsbeständigkeit der keramischen Werkstoffe mit zunehmender
Porosität stark reduziert. Durch offene Poren an der Oberflächenschicht der keramischen
Werkstoffe wird die Reaktionsfläche wesentlich erhöht. Dies begünstigt die Korrosion
erheblich, wobei Poren mit einem großen effektiven Porenradius besonders schädlich sind.
Darüber hinaus beeinflusst die Porosität die Thermoschockbeständigkeit, die Festigkeit, die
Härte,[153,154,159,214] die Wärmeleitfähigkeit, sowie die magnetischen Eigenschaften der
keramischen Werkstoffe wesentlich.[3] Deshalb kann es z. B. für mechanische Anwendungen
wichtig sein, eine Dichte nahe der jeweiligen theoretischen Dichte ohne Dichtegradienten zu
erreichen, um möglichst gute mechanische Eigenschaften zu erzielen.
Zur Beurteilung der hergestellten Nanokomposite wurden diese Kennwerte mit Hilfe der
archimedischen Dichtemessung in Zusammenarbeit mit M. KNOCH von der Firma FCT
Ingenieurkeramik GmbH in Sonneberg bestimmt. Die Ergebnisse dieser Messungen sind in
Tabelle 9 zusammengefasst. Zur besseren Vergleichbarkeit wurden die Dichtewerte auf die
theoretisch erreichbare Dichte bezogen. Der Einfluss von Füllstofftyp auf die Dichte sowie die
resultierende Porosität der SiC-Nanokomposite wurde im Folgenden graphisch ausgewertet.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 81 -
Tabelle 9: Übersicht der Dichtewerte und Porosität von SiC-Nanokompositen in Abhängigkeit von
Füllstofftypen und -gehalten.
Probe
Füllstoffgehalt
[Gew.-%]
Dichte
[g/cm3]
Reindichte
[g/cm3]
Relative Dichte
[%]
Porosität
[%]
SiC
Referenz 0 3.170 3.174 99.9 0.1
TR
GO
TRGO-600 2 3.101 3.149 98.5 1.5
TRGO-800 2 3.046 3.149 96.7 3.3
TRGO-1000 2 3.035 3.149 96.4 3.6
MG
MG-Ethanol 2 2.940 3.149 93.3 6.7
MG-Ar-24h 2 3.137 3.149 99.6 0.4
MG-Ar-48h 2 3.147 3.149 99.9 0.1
MG-Ar-72h 2 3.148 3.149 100.0 0.0
MG-CO2-24h 2 3.076 3.149 97.7 2.3
MG-CO2-48h 2 3.106 3.149 98.6 1.4
MG-CO2-72h 2 3.114 3.149 98.9 1.1
MG-CO2-96h 2 3.106 3.149 98.6 1.4
MG-CO2-120h 2 3.132 3.149 99.4 0.6
MG-N2-24h 2 3.142 3.149 99.8 0.2
MG-N2-48h 2 3.142 3.149 99.8 0.2
MG-N2-72h 2 3.143 3.149 99.8 0.2
GS
iC
GSiC-F-2% 2 3.147 3.149 99.9 0.1
GSiC-F-4% 4 3.077 3.126 98.4 1.6
GSiC-F-8% 8 2.920 3.082 94.7 5.3
GSiC-G-2% 2 3.121 3.149 99.1 0.9
GSiC-G-4% 4 3.067 3.126 98.1 1.9
GSiC-D-2% 2 3.142 3.149 99.8 0.2
GSiC-D-4% 4 3.093 3.126 99.0 1.0
Ben
chm
ark
-
Syst
em
Grade C 2 3.094 3.149 98.2 1.8
Grade M 2 2.929 3.149 93.0 7.0
Graphit 2 3.070 3.149 97.5 2.5
Nanographit 2 3.097 3.149 98.3 1.7
Carbon Black 2 3.149 3.149 100.0 0.0
* Nach dem technischen Datenblatt FCT SiC Standardwerkstoffe SC-S wurde das Kriterium für die
Beurteilung der Dichtewerte aufgestellt: Eine Dichte höher als 3.08 bzw. eine relative Dichte höher als
97 % gilt als erfolgreiche Sinterung.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 82 -
TRGO-600
TRGO-800
TRGO-1000
96,0
96,5
97,0
97,5
98,0
98,5
99,0 Relative Dichte
O-Gehalt
Re
lati
ve
Dic
hte
[%
]
0
2
4
6
8
10
12
14
16
Sa
ue
rsto
ffg
eh
alt
[G
ew
.-%
]
MG-CO2-24h
MG-CO2-48h
MG-CO2-72h
MG-CO2-96h
MG-CO2-120h97,0
97,5
98,0
98,5
99,0
99,5
100,0
Relative Dichte
O-Gehalt
Re
lati
ve
Dic
hte
[%
]
7,0
7,5
8,0
8,5
9,0
9,5
10,0
10,5
11,0
11,5
12,0
Sa
ue
rsto
ffg
eh
alt
[G
ew
.-%
]
Abbildung 46: Relative Dichte der gesinterten SiC-Nanokomposite in Abhängigkeit des
Funktionalitätsgrads bei je 2.0 Gew.-% TRGO (links) und MG-CO2 (rechts), korreliert mit
Sauerstoffgehalt.
Abbildung 46 (links) stellte den Einfluss der TRGO-Typen, welche bei unterschiedlicher
Reduktionstemperatur (Tred.) von GO hergestellt wurden, auf den erhaltenen Dichten dar. Ein
Vergleich der Komposite mit je 2.0 Gew.-% TRGO zeigte, dass unabhängig vom
Funktionalisierungsgrad des TRGO-Materials eine Abnahme der relativen Dichte um bzw. eine
Porosität von 1.5 bis 3.6 % in Bezug auf die ungefüllte Referenz induziert wurde. Diese
reduzierte Dichte war jedoch in allen Fällen mit einem abnehmenden Funktionalitätsgrad des
eingesetzten TRGO verbunden. So lässt sich tendenziell eine Abnahme der relativen Dichte bei
sinkendem Funktionalitätsgrad, also höherer Tred., erkennen. Diese Tendenz konnte auf die
bessere Verteilung der höher funktionalisierten Füllstoffe (TRGO-600) in der SiC-Matrix
zurückführen. Somit war die TRGO-600-Probe zwar weniger gut exfoliert und weniger
thermisch stabil, bildete aber aufgrund ihrer homogenen Verteilung dennoch wenige
Agglomerate in der Matrix und gewährleistete eine hohe Verdichtung. Der Zusammenhang
zwischen dem Funktionalitätsgrad bzw. Sauerstoff-Gehalt des Füllstoffes und der relativen
Dichte der SiC-Nanokomposite wurde auch bei den Werkstoffen mit den hier verwendeten MG-
CO2-Füllstoffen bestätigt (siehe Abbildung 46 rechts). Längere Mahldauer führte zu vermehrten
hydrophilen funktionellen Gruppen (Sauerstoff-Gehalt), welche die Bildung von Agglomeraten
verhinderten und zu einer homogenen Verteilung von MG-CO2 in der Keramik-Matrix führten.
Deswegen wurde bei MG-CO2 auch eine zunehmende Dichte mit steigender Mahldauer
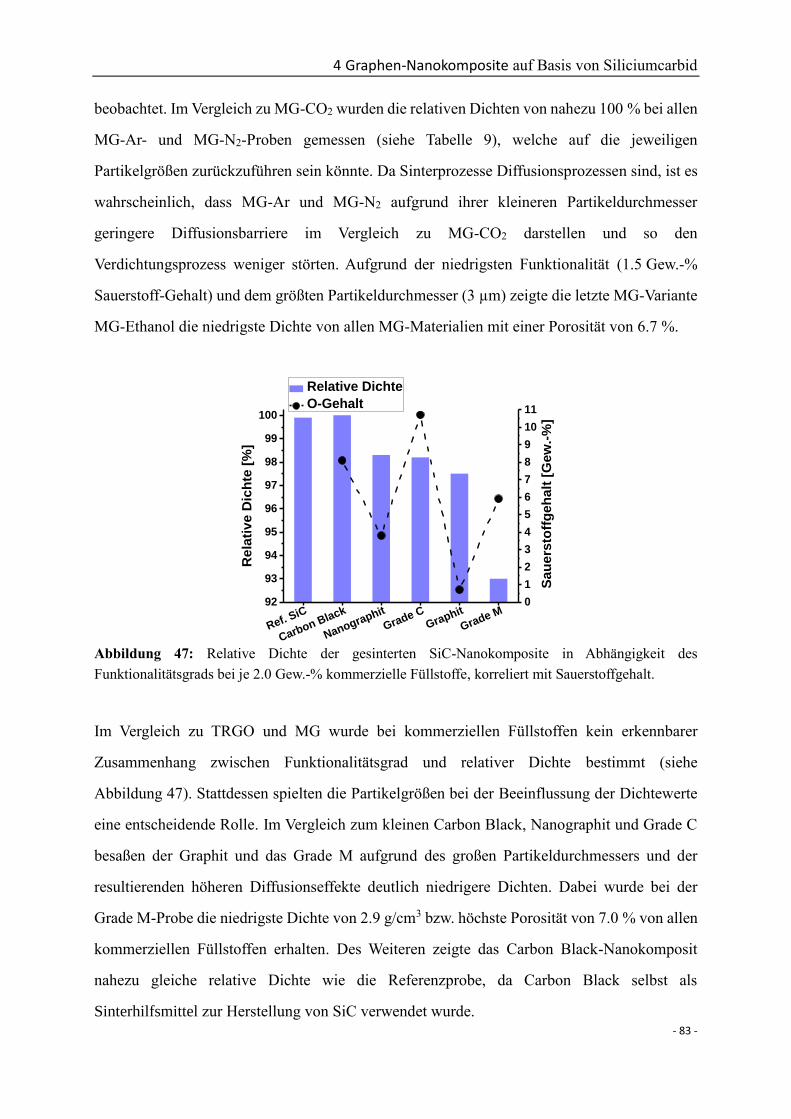
4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 83 -
beobachtet. Im Vergleich zu MG-CO2 wurden die relativen Dichten von nahezu 100 % bei allen
MG-Ar- und MG-N2-Proben gemessen (siehe Tabelle 9), welche auf die jeweiligen
Partikelgrößen zurückzuführen sein könnte. Da Sinterprozesse Diffusionsprozessen sind, ist es
wahrscheinlich, dass MG-Ar und MG-N2 aufgrund ihrer kleineren Partikeldurchmesser
geringere Diffusionsbarriere im Vergleich zu MG-CO2 darstellen und so den
Verdichtungsprozess weniger störten. Aufgrund der niedrigsten Funktionalität (1.5 Gew.-%
Sauerstoff-Gehalt) und dem größten Partikeldurchmesser (3 µm) zeigte die letzte MG-Variante
MG-Ethanol die niedrigste Dichte von allen MG-Materialien mit einer Porosität von 6.7 %.
Ref. SiC
Carbon Black
Nanographit
Grade CGraphit
Grade M92
93
94
95
96
97
98
99
100
Relative Dichte
O-Gehalt
Re
lati
ve
Dic
hte
[%
]
0
1
2
3
4
5
6
7
8
9
10
11
Sa
ue
rsto
ffg
eh
alt
[G
ew
.-%
]
Abbildung 47: Relative Dichte der gesinterten SiC-Nanokomposite in Abhängigkeit des
Funktionalitätsgrads bei je 2.0 Gew.-% kommerzielle Füllstoffe, korreliert mit Sauerstoffgehalt.
Im Vergleich zu TRGO und MG wurde bei kommerziellen Füllstoffen kein erkennbarer
Zusammenhang zwischen Funktionalitätsgrad und relativer Dichte bestimmt (siehe
Abbildung 47). Stattdessen spielten die Partikelgrößen bei der Beeinflussung der Dichtewerte
eine entscheidende Rolle. Im Vergleich zum kleinen Carbon Black, Nanographit und Grade C
besaßen der Graphit und das Grade M aufgrund des großen Partikeldurchmessers und der
resultierenden höheren Diffusionseffekte deutlich niedrigere Dichten. Dabei wurde bei der
Grade M-Probe die niedrigste Dichte von 2.9 g/cm3 bzw. höchste Porosität von 7.0 % von allen
kommerziellen Füllstoffen erhalten. Des Weiteren zeigte das Carbon Black-Nanokomposit
nahezu gleiche relative Dichte wie die Referenzprobe, da Carbon Black selbst als
Sinterhilfsmittel zur Herstellung von SiC verwendet wurde.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 84 -
Aufgrund der vorteilhaften Herstellungsverfahren erreichten die allen GSiC-Proben mit
2 Gew.-% Graphengehalt eine vollständige Verdichtung mit einer relativen Dichte nahe der
theoretischen Enddichte (siehe Tabelle 9). Dafür wurde der Trend bei steigenden Füllgraden bis
zu 8 Gew.-% untersucht (siehe Abbildung 48).
0 2 4 6 894
95
96
97
98
99
100
Rela
tive D
ich
te [
%]
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
6
5
4
3
2
1
0
Po
rosit
ät
[%]
Abbildung 48: Relative Dichte und Porosität der gesinterten SiC-Nanokomposite mit GSiC-F (aus
Furfurylalkohol), GSiC-G (aus Glucose) und GSiC-D (aus Dopamin) bei variierender Graphen-
Konzentration.
Wie in Abbildung 48 dargestellt, wurden die Dichtewerte vom Füllstoffgrad stark beeinflusst.
Alle GSiC-Proben, welche mit 2 Gew.-% gefüllt waren, zeigten eine vollständige Verdichtung.
Im Vergleich dazu nahm die Dichte aller GSiC-Proben mit steigendem Füllstoffgehalt ständig
ab, was mit der Tendenz in der Literatur übereinstimmt.[159,214] Eine ausreichend hohe
Verdichtung mit einer relativen Dichte über 98 % kann bis zu einem Füllstoffgehalt von
4 Gew.-% Graphen in GSiC-Nanokomposite erreicht werden. Bei höheren
Füllstoffkonzentrationen traten starke Partikel-Partikel-Wechselwirkungen auf, die zur
Agglomeratbildung führten. Der mit 8 Gew.-% Graphen gefüllte GSiC-F-Werkstoff besaß den
niedrigsten Dichtewert von 2.9 g/cm3, was einer Porosität von 5.3 % entspricht. Daher wurde
der Füllstoffgehalt bei GSiC-G und GSiC-D auf maximal 4 Gew.-% beschränkt.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 85 -
4.4 Morphologie
Zur Herstellung von Nanokompositen mit effizienter Matrixverstärkung ist neben einem großen
Aspektverhältnis die Dispergierung des Füllstoffs in der Matrix maßgeblich.[167] Die
Homogenität der Füllstoffverteilung bestimmt im Wesentlichen die zu erwartenden
Materialeigenschaften. Je besser die Dispergierung des Füllstoffs erfolgt, desto höher ist der
Grenzflächenkontakt zwischen dem Füllstoff und dem Matrixmaterial und desto höher ist die
Materialeigenschaftsverstärkung.[194]
Die Morphologie der erhaltenen Nanokomposite wurde in Zusammenarbeit mit Prof. J. DUSZA
von The Institute of Materials Research of SAS (IMR) in Košice mittels SEM-Aufnahmen der
polierten Oberfläche untersucht, um einen Einblick in die Füllstoffdispersion sowie
Kompatibilisierung der Füllstoffe in der SiC-Matrix zu erhalten. Die Geometrie und
Funktionalität der Füllstoffe wurde auf ihre Verteilung in der SiC-Matrix analysiert.
Diesbezüglich wurden vertretend SiC mit TRGO, MG und kommerziellem Grade M, Grade C
sowie Carbon Black bei einem Füllstoffgehalt von 2 Gew.-% und GSiC-F bei 2, 4 und 8 Gew.-%
untersucht. Die Ergebnisse der morphologischen Charakterisierung der hergestellten
Nanokomposite sind in folgenden dargestellt.
Abbildung 49: SEM-Aufnahmen des gesinterten a) reinen SiC und b) SiC-Nanokomposits mit Carbon
Black (2 Gew.-%).
Die ungefüllte SiC-Probe (Abbildung 49 a) zeigte deutlich weniger schwarze Bereiche als alle
Kompositmaterialien. Hier konnte es sich um das Sinterhilfsmittel Carbon Black handeln, da
es durch Phasenanalyse mit Raman-Spektroskopie als Kohlenstoff identifiziert wurde. Dadurch

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 86 -
erschien die Carbon Black-Probe (Abbildung 49 b) eine ähnliche homogene Füllstoffverteilung,
aber mit deutlich höherer Konzentration.
Abbildung 50: SEM-Aufnahmen des gesinterten SiC-Nanokomposite mit TRGO unterschiedlichen
Funktionalitätsgrades (je 2 Gew.-%).
Aus diesen Aufnahmen von TRGO-Nanokompositen in Abbildung 50 geht klar hervor, dass
alle Werkstoffe eine ziemlich gleichmäßige Verteilung der Füllstoffpartikel aufwiesen. Hierbei
wurde die Dispergierung durch die große spezifische Oberfläche und die hohe Funktionalität
von TRGO begünstigt. Darüber hinaus in diesen Kompositmaterialien ließen sich zwar neben
gut exfolierten Strukturen auch kleine verbliebenen Schichtagglomerate von ca. 10 µm Länge
erkennen, diese bestehen aber meistens aus wenigen Lagen. Die hohen Scherkräfte beim
Hochdruckhomogenisierungsprozess reichten offenbar nicht aus, um alle Schichtpakete
vollständig aufzubrechen. Des Weiteren war eine klare Abstufung zwischen den
Nanokompositen mit TRGO-600 bis TRGO-1000 anhand der SEM-Aufnahmen aufgrund der
geringen Unterschiede nicht möglich, was mit den nicht unterscheidbaren morphologischen
Ergebnissen von TRGO in Kapitel 3.1 korreliert.
Abbildung 51: SEM-Aufnahmen des gesinterten SiC-Nanokomposite mit a) MG-CO2-24h,
b) MG-CO2-120h und c) MG-Ethanol (je 2 Gew.-%).

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 87 -
Im Gegensatz zu TRGO spielten die Herstellungsbedingungen von MG-CO2 eine wesentliche
Rolle auf ihre Füllstoffverteilung in der SiC-Matrix. Dabei nahm die Agglomeratgröße mit
steigender Mahldauer von MG-CO2 drastisch ab und korrelierte nicht linear mit der
zunehmenden spezifischen Oberfläche und Funktionalität (vgl. Tabelle 3 in Kapitel 3.2). Die
größere Anzahl funktioneller Gruppen in den MG-CO2-Materialien längerer Mahldauer
konnten eine bessere Kompatibilität und Verteilung in den Matrixmaterialien bewirken. Des
Weiteren wurde der Sintervorgang durch die Verringerung der Partikelgröße, welche mit
längerer Mahldauer beobachtet wurde, deutlich begünstigt. So waren im Nanokomposit mit
MG-CO2-24h noch Partikel über 20 µm zu sehen, während die Probe mit MG-Material längerer
Mahldauer einheitlichere kleine Partikel (< 5 µm) zeigten. Dafür besaß MG-CO2-120h-Probe
eine wesentlich einheitlichere Füllstoffverteilung sowie geringere Porosität als MG-CO2-24h-
Probe. Im Vergleich zu den Kompositen mit funktionalisierten MG-CO2 waren in SEM-
Aufnahmen der unfunktionalisierte MG-Ethanol gefüllten SiC-Nanokomposite deutlich
größere Agglomerate und offene Poren über 30 µm erkennbar. Diese ungleichmäßige
Füllstoffverteilung war auf die fehlenden funktionellen Gruppen im MG-Ethanol und große
Partikelgröße zurückzuführen und korreliert direkt mit der kleinsten Dichte (2.9 g/cm3) und
größten Porosität (6.7 %) von allen Werkstoffvarianten.
Abbildung 52: SEM-Aufnahmen des gesinterten GSiC-F-Nanokomposite mit unterschiedlichen
Graphengehalten.
Im Vergleich der SEM-Aufnahmen aller Nanokomposite mit 2 Gew.-% Graphengehalt zeigte
die GSiC-F-Probe (Abbildung 52 a) die beste Füllstoffverteilung mit einer ähnlichen
porenfreien Morphologie wie die Referenzprobe (siehe Abbildung 49 a), was auf das

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 88 -
vorteilhafte Herstellungsverfahren zurückzuführen war. Trotzdem stieg die Menge an
Agglomeraten mit zunehmenden Füllstoffgehalt bei den GSiC-F-Proben wegen vermehrter
Partikel-Partikel-Wechselwirkungen stetig an, was mit der abnehmender Dichte und
zunehmenden Porosität in Kapitel 4.3 einhergehend.
Abbildung 53: SEM-Aufnahmen des gesinterten SiC-Nanokomposite mit kommerziellen
Referenzfüllstoffen a) Grade C und b) Grade M (je 2 Gew.-%).
Die morphologische Betrachtung der Komposite mit kommerziellen Füllstoffen ergab für die
Grade C gefüllte Werkstoffvariante eine nahe homogene Verteilung mit ziemlich kleinen
Agglomeraten, was vergleichbar mit dem von MG-CO2 gefüllten SiC war. Im Gegensatz hierzu
wurde bei dem System mit Grade M eine hauptsächlich inhomogene Füllstoffverteilung mit
massiv erscheinender Schichtenagglomerate mit einer Größe über 30 µm in der Matrix neben
füllstofffreien Bereichen identifiziert, welche auf Ausfällung des Graphens zurückzuführen ist.
Im Vergleich zu Grade C lässt sich die wesentliche schlechtere Dispergierung von Grade M
wiederum durch seine Partikelgröße erklären. Da der Sinterprozess ein Diffusionsprozess ist,
störten die größeren Partikel von Grade M die Sinterprozesse. Zusätzlich war die kleine
spezifische Oberfläche von Grade M für die schlechte Dispergierung verantwortlich, da sie zur
geringen SiC/Füllstoffgrenzflächen führte. Damit wurde hier eine große Porosität (7.0 %) und
inhomogene Füllstoffverteilung erhalten.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 89 -
4.5 Mechanische Eigenschaften
Im Hinblick auf die verschiedenen Einsatzgebiete der SiC-Werkstoffe sind die mechanischen
Eigenschaften wichtige Parameter. In diesem Abschnitt wird der Einfluss des Graphentyps und
des Füllgrads auf die mechanischen Eigenschaften der SiC-Nanokomposite untersucht. Wie in
der Einleitung beschrieben, spielt die Füllstoff-Dispergierung eine wichtige Rolle bei der
Beeinflussung der mechanischen Eigenschaften. Homogene Dispergierung führt zu stärkerer
Wechselwirkung zwischen Füllstoff und Keramikmatrix und somit zu einer positiven
Beeinflussung unter sonst gleichen Bedingungen, während Risse und Defekte durch
Inhomogenität im Werkstoff entstehen. Im Bereich solcher Fehlstellen kommt es zu einer
Spannungsüberhöhung im Werkstoff, welche um ein Vielfaches höher als die Nennspannung
liegt und zu Sprödbruchanfälligkeit der Keramik führt.[3]
Die mechanischen Eigenschaften wurden durch Analyse der Biegefestigkeit und
Bruchzähigkeit in Zusammenarbeit mit Prof. J. DUSZA von The Institute of Materials Research
of SAS (IMR) in Košice untersucht. Die Biegefestigkeit ist als die maximale nominale
Randfaserspannung im Moment des Versagens definiert. Bei der 4-Punkt-Biegeprüfung wurde
die Biegesteifigkeit bei Raumtemperatur an SiC-Graphen-Nanokompositen ermittelt. Die
Bruchzähigkeit wurde mittels der „Single-Edge-V-Notched Beam (SEVNB)” Methode
bestimmt. Die Ergebnisse der Biegefestigkeit (σB) und der Bruchzähigkeit (SEVNB) sind in
Abhängigkeit des Füllstofftyps und der Konzentration in Tabelle 10 aufgeführt. Diese enthält
ebenfalls die Ergebnisse der elektrischen Eigenschaften (σS), welche in folgendem Kapitel 4.6
ausführlich diskutiert werden sollen. Der Einfluss von Füllstofftyp auf die Biegefestigkeit und
die Bruchzähigkeit wurde im Folgenden graphisch ausgewertet.
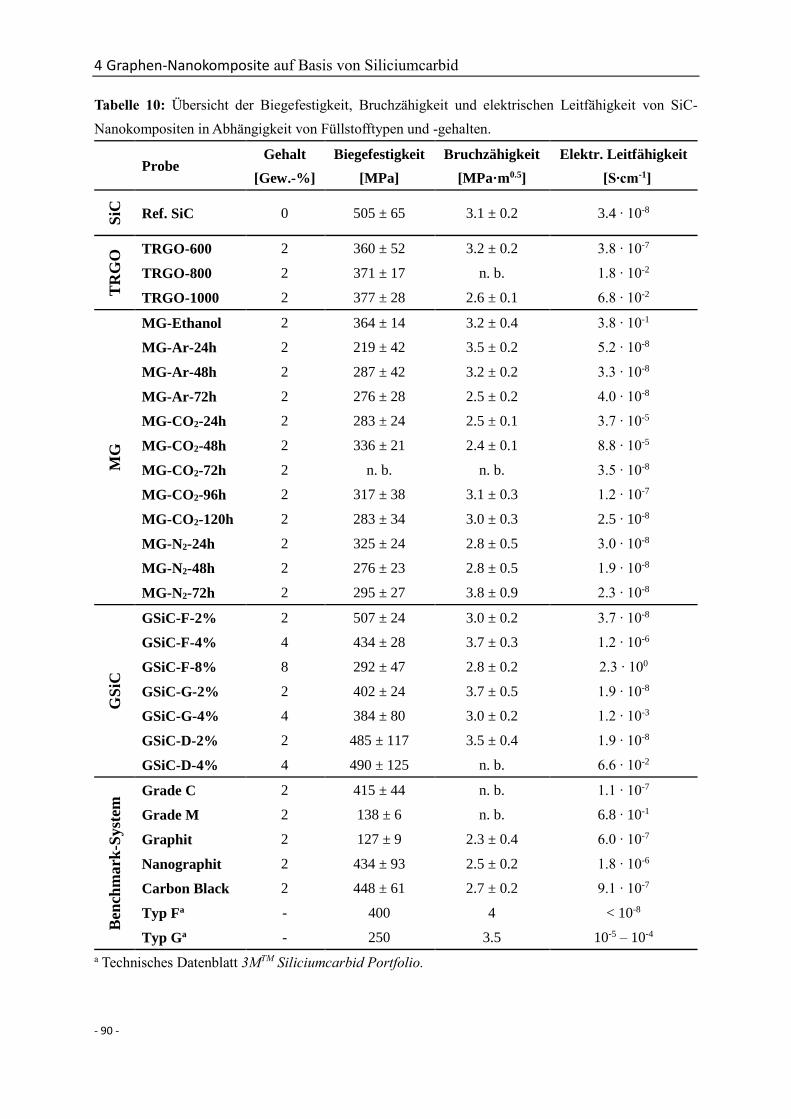
4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 90 -
Tabelle 10: Übersicht der Biegefestigkeit, Bruchzähigkeit und elektrischen Leitfähigkeit von SiC-
Nanokompositen in Abhängigkeit von Füllstofftypen und -gehalten.
Probe
Gehalt
[Gew.-%]
Biegefestigkeit
[MPa]
Bruchzähigkeit
[MPa·m0.5]
Elektr. Leitfähigkeit
[S∙cm-1]
SiC
Ref. SiC 0 505 ± 65 3.1 ± 0.2 3.4 ∙ 10-8
TR
GO
TRGO-600 2 360 ± 52 3.2 ± 0.2 3.8 ∙ 10-7
TRGO-800 2 371 ± 17 n. b. 1.8 ∙ 10-2
TRGO-1000 2 377 ± 28 2.6 ± 0.1 6.8 ∙ 10-2
MG
MG-Ethanol 2 364 ± 14 3.2 ± 0.4 3.8 ∙ 10-1
MG-Ar-24h 2 219 ± 42 3.5 ± 0.2 5.2 ∙ 10-8
MG-Ar-48h 2 287 ± 42 3.2 ± 0.2 3.3 ∙ 10-8
MG-Ar-72h 2 276 ± 28 2.5 ± 0.2 4.0 ∙ 10-8
MG-CO2-24h 2 283 ± 24 2.5 ± 0.1 3.7 ∙ 10-5
MG-CO2-48h 2 336 ± 21 2.4 ± 0.1 8.8 ∙ 10-5
MG-CO2-72h 2 n. b. n. b. 3.5 ∙ 10-8
MG-CO2-96h 2 317 ± 38 3.1 ± 0.3 1.2 ∙ 10-7
MG-CO2-120h 2 283 ± 34 3.0 ± 0.3 2.5 ∙ 10-8
MG-N2-24h 2 325 ± 24 2.8 ± 0.5 3.0 ∙ 10-8
MG-N2-48h 2 276 ± 23 2.8 ± 0.5 1.9 ∙ 10-8
MG-N2-72h 2 295 ± 27 3.8 ± 0.9 2.3 ∙ 10-8
GS
iC
GSiC-F-2% 2 507 ± 24 3.0 ± 0.2 3.7 ∙ 10-8
GSiC-F-4% 4 434 ± 28 3.7 ± 0.3 1.2 ∙ 10-6
GSiC-F-8% 8 292 ± 47 2.8 ± 0.2 2.3 ∙ 100
GSiC-G-2% 2 402 ± 24 3.7 ± 0.5 1.9 ∙ 10-8
GSiC-G-4% 4 384 ± 80 3.0 ± 0.2 1.2 ∙ 10-3
GSiC-D-2% 2 485 ± 117 3.5 ± 0.4 1.9 ∙ 10-8
GSiC-D-4% 4 490 ± 125 n. b. 6.6 ∙ 10-2
Ben
chm
ark
-Syst
em Grade C 2 415 ± 44 n. b. 1.1 ∙ 10-7
Grade M 2 138 ± 6 n. b. 6.8 ∙ 10-1
Graphit 2 127 ± 9 2.3 ± 0.4 6.0 ∙ 10-7
Nanographit 2 434 ± 93 2.5 ± 0.2 1.8 ∙ 10-6
Carbon Black 2 448 ± 61 2.7 ± 0.2 9.1 ∙ 10-7
Typ Fa - 400 4 < 10-8
Typ Ga - 250 3.5 10-5 – 10-4
a Technisches Datenblatt 3MTM Siliciumcarbid Portfolio.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 91 -
Die in Tabelle 10 dargestellten SiC-Graphen-Nanokomposite wiesen meistens niedrigere
Biegefestigkeiten (von 250 bis 400 MPa) gegenüber reinem SiC (500 MPa) auf. Ähnlich wie
in der Literatur beschrieben, führte die Einarbeitung von Graphen zu einer leichten Abnahme
der mechanischen Eigenschaften von keramischen Werkstoffen.[152,154,220] Die Gründe dafür
sind vor allem die unzureichende Homogenität sowie die Porosität und die geringere Dichte der
SiC-Graphen-Komposite. Des Weiteren sind schwache Partikel-Matrix-Anbindungen und
Fehlstellen im Gefüge mitverantwortlich für die verschlechterten mechanischen Eigenschaften.
Aufgrund der drastischen Abnahme der Biegefestigkeit wurde der maximale Füllstoffgehalt bei
diesen Nanokompositen auf 2 Gew.-% beschränkt.
Ref. SiC
Carbon Black
Nanographit
Grade CGraphit
Grade M
100
200
300
400
500
600 Biegefestigkeit
Relative Dichte
Bie
gefe
sti
gkeit
[M
Pa]
92
93
94
95
96
97
98
99
100
Rela
tive D
ich
te [
%]
MG-Ar-24h
MG-Ar-48h
MG-Ar-72h
150
200
250
300
350 Biegefestigkeit
Relative Dichte
Bie
gefe
sti
gkeit
[M
Pa]
99,2
99,4
99,6
99,8
100,0
Rela
tive D
ich
te [
%]
Abbildung 54: 4-Punkt-Biegefestigkeit der gesinterten SiC-Nanokomposite in Abhängigkeit der
relativen Dichte bei je 2 Gew.-% kommerzielle Füllstoffe (links) und MG-Ar (rechts), korreliert mit
relativer Dichte.
Untersuchung der mechanischen Eigenschaften der mit kommerziellen Füllstoffen gefüllten
SiC-Komposite zeigten eine beträchtliche Abhängigkeit der Biegefestigkeit von der relativen
Dichte der Nanokomposite, welche durch Partikelgrößen der Füllstoffe beeinflusst wird (siehe
Abbildung 54 links). Ein Vergleich der Komposite mit je 2.0 Gew.-% Füllstoff zeigte, dass
unabhängig von Füllstofftypen eine Abnahme der Biegefestigkeit zwischen 11 und 75 % in
Bezug auf die ungefüllte Referenzprobe induziert wurde. Die Reduktion steht in allen Fällen
mit der relativen Dichte der Nanokomposite im Zusammenhang und korreliert mit den
morphologischen Untersuchungen in Kapitel 4.4. Auf diese Weise lässt sich eine tendenzielle
Abnahme der Biegefestigkeit bei sinkender relativer Dichte, also höherer Porosität, erkennen.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 92 -
Dabei führen bei 2 Gew.-% Füllstoffgehalt die unfunktionalisierten Füllstoffe Grade M und
Graphit aufgrund der hohen Porosität in den Nanokompositen zu einer maximalen Abnahme
der Biegefestigkeit um 70 % auf 130 MPa. Ein ähnlicher Einfluss von relativer Dichte auf die
Festigkeit konnte auch bei MG-Ar beobachtet werden (siehe Abbildung 54 rechts). Jedoch lässt
sich diese Tendenz im Rahmen des Fehlers bei MG-Ar verringern.
TRGO-600
TRGO-800
TRGO-1000
200
250
300
350
400
450 Biegefestigkeit
Relative Dichte
Bie
gefe
sti
gkeit
[M
Pa]
96,0
96,5
97,0
97,5
98,0
98,5
99,0
Rela
tive D
ich
te [
%]
MG-CO2-24h
MG-CO2-48h
MG-CO2-96h
MG-CO2-120h
MG-N2-24h
MG-N2-48h
MG-N2-72h
200
250
300
350
400 Biegefestigkeit
Relative Dichte
Bie
gefe
sti
gkeit
[M
Pa]
97,0
97,5
98,0
98,5
99,0
99,5
100,0
Rela
tive D
ich
te [
%]
Abbildung 55: 4-Punkt-Biegefestigkeit der gesinterten SiC-Nanokomposite in Abhängigkeit der
relativen Dichte bei je 2 Gew.-% TRGO (links) sowie MG-CO2 und MG-N2 (rechts), korreliert mit
relativer Dichte.
Im Gegensatz dazu wurde bei TRGO (Abbildung 55 links) sowie MG-CO2 und MG-N2
(Abbildung 55 rechts) kein erkennbarer Zusammenhang zwischen Biegefestigkeit und relativer
Dichte gefunden. Bei den Systemen mit TRGO verläuft die Biegefestigkeit unabhängig von der
TRGO-Typen im Rahmen der Fehlergrenzen konstant (ca. 370 MPa), was vergleichbar mit der
von kommerziellem SiC Typ F (400 MPa) der Firma 3M ist. Der Einfluss des
Funktionalitätsgrades des eingesetzten TRGOs, welche durch die unterschiedliche
Reduktionstemperatur (Tred) eingestellt werden kann, ist hierbei sehr gering, obwohl gerade in
diesen Proben eine abnehmende Dichte mit höherer Tred beobachtet wurde. Eine mögliche
Ursache für diese Unterschiede könnte in den Exfolierungsgrad der Füllstoffe liegen. Somit war
in der TRGO-1000-Probe zwar eine höhere Porosität vorhanden, das TRGO konnte aber
aufgrund der besseren Exfolierung demzufolge höheren Oberfläche einen besseren
Grenzflächenkontakt mit der Matrix bilden. So kann eine bessere Kraftübertragung zwischen
SiC und dem Füllstoff gewährleistet werden.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 93 -
Ähnlich wie bei dem TRGO-System bewirkte der Einsatz von MG-CO2 und MG-N2 eine
reduzierte Biegefestigkeit. Der in Abbildung 55 (rechts) abgebildete Werteverlauf verdeutlicht,
dass die Biegefestigkeiten von MG-CO2 und MG-N2 unter Berücksichtigung der Fehlertoleranz
keine signifikante Abhängigkeit von der relativer Dichte, der Mahldauer bzw. dem
Funktionalitätsgrad aufwiesen. Dies korreliert mit den ähnlichen morphologischen Ergebnissen
von MG-CO2 und MG-N2 aus Kapitel 3.2. Dabei lagen die Biegefestigkeiten aller
MG-Materialien im Bereich zwischen 280 und 340 MPa.
Dementgegen bildeten die Proben GSiC-F (507 MPa) und GSiC-D (485 MPa) eine Ausnahme.
Sie wiesen eine ähnliche Biegefestigkeit wie das Referenzmaterial (505 MPa) und somit höhere
Werte als die kommerziellen Produkte Typ-F (400 MPa) und Typ-G (250 MPa) der Firma 3M
auf. Der Unterschied lässt sich wiederum mit dem vorteilhaften Herstellungsverfahren erklären.
Wie bereits aus den SEM-Aufnahmen in Kapitel 4.4 sowie den Dichtemessungen (Tabelle 9)
hervorging, wurde unter Verwendung von GSiC, bei dem das Graphen über eine
templatvermittelte Reaktion direkt auf die Oberfläche von SiC aufgebracht wurde, eine deutlich
homogenere Verteilung von Graphen in der SiC-Matrix, eine stärkere Anbindung mit dem SiC
sowie eine vollständige Verdichtung der Nanokomposite erzielte als unter Zugabe von anderen
Graphenen. Diese Erklärung konnte auch mit den Prüfkörpern von GSiC (GSiC-G), TRGO
(TRGO-600) und MG (MG-Ar-24h) bei der 4-Punkte-Biegeprüfung in Abbildung 56
nachgewiesen werden.
Abbildung 56: Prüfkörper von GSiC, TRGO und MG für 4-Punkt-Biegeprüfung.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 94 -
Durch die einheitliche Verteilung der Graphenpartikel in der SiC-Matrix wurden die Kräfte im
4-Punkt-Biegeversuch gleichmäßiger verteilt, weshalb der Werkstoff resistenter gegen
mechanische Beanspruchung war. Deshalb wurden die GSiC-Proben deutlich einheitlicher
gebrochen (siehe Abbildung 56) als die TRGO- und MG-Proben, die eine unzureichende
Homogenität und Fehlstellen aufwiesen. Diese These wurde mittels SEM-Aufnahmen der
Bruchflächen der 4-Punkt-Biegefestigkeitsprüfkörper im Folgenden bekräftigt.
Abbildung 57: SEM-Aufnahmen an Bruchflächen des gesinterten a) reinen SiC und b) SiC-
Nanokomposits mit Carbon Black (2 Gew.-%) nach den 4-Punkt-Biegeprüfungen.
Anhand der SEM-Aufnahmen in Abbildung 57 ist ersichtlich, dass die Bruchfläche von mit
Carbon Black gefülltem Nanokomposit analog zu der ungefüllten Referenzprobe eine ähnliche
Rauigkeit aufweist. Dies ist darauf zurückzuführen, dass Carbon Black selbst als
Sinterhilfsmittel zur Herstellung von SiC verwendet wurde. Diese Ergebnisse korrelieren mit
den morphologischen Ergebnissen der Oberflächenanalyse aus Kapitel 4.4.
Aus der Aufnahme in Abbildung 58 b) geht hervor, dass das unfunktionalisierte MG-Ethanol
keine starke Wechselwirkung mit der SiC-Matrix einging und demzufolge die schlechteste
Verteilung im Vergleich zu den anderen mit Graphen gefüllten Kompositen aufwies, da hier die
größten Poren und Fehlstellen (über 40 µm) zu sehen sind. Dies korreliert ebenfalls gut mit den
Ergebnissen aus Kapitel 4.4. Bei den anderen funktionalisierten Graphenen, z. B. TRGO
(Abbildung 58 a), MG-Ar (Abbildung 58 c) und MG-CO2 (Abbildung 58 d), wurden ebenfalls
inhomogen verteilte Agglomerate über 20 µm Länge gefunden. In diesem Fall korrelierten die
Schlussfolgerungen aus den SEM-Aufnahmen der Bruchflächen nicht mit den Ergebnissen der

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 95 -
SEM-Aufnahmen der Oberflächen aus Kapitel 4.4, denn diese eine nahe homogene Verteilung
der funktionalisierten Graphenen zeigten. Damit konnte aber die Abnahme der Biegefestigkeit
(von 505 auf bis zu 287 MPa) besser erklärt werden. Bei den Nanokompositen mit TRGO
(Abbildung 58 a) und MG-CO2-48h (Abbildung 58 d) ließen sich Schichtagglomerate erkennen,
während bei MG-Ar-48h mit sphärischen aggregierten Partikeln gefüllte Fehlstellen gefunden
wurden. Des Weiteren wurde bei MG-Ar-48h (Abbildung 58 c) ein Riss mit einer Länge über
50 µm identifiziert, welcher auf eine ungleichmäßige Füllstoffverteilung und auf die
Agglomeratbildung zurückzuführen ist. Trotz der hohen Scherkräfte im
Hochdruckhomogenisatorprozess ist es nicht möglich, alle Graphen-Agglomerate
aufzubrechen. Diese Fehlstellen führten zur drastischen Reduktion der maximalen Belastungen
der Werkstoffe bzw. der Biegefestigkeit.
Abbildung 58: SEM-Aufnahmen an Bruchflächen des gesinterten SiC-Nanokomposite mit
a) TRGO-600, b) MG-Ethanol, c) MG-Ar-48h und d) MG-CO2-48h (je 2 Gew.-%) nach den 4-Punkt-
Biegeprüfungen.
Dem entgegen wies das GSiC-F-Nanokomposit deutlich homogenere Füllstoffverteilung mit
einer porenfreien Struktur auf (siehe Abbildung 59). Dabei erschien die glatte Bruchfläche
analog dem ungefüllten SiC. Dies unterstützt die These, dass sich dieses

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 96 -
Beschichtungsverfahren zur Herstellung von GSiC im Vergleich zu den anderen Graphenen
offensichtlich vorteilhaft auswirkt, da die Bildung von Agglomeraten vermieden und eine
homogenere Verteilung der Füllstoffe verwirklicht werden kann. Damit wurde die Bruchenergie
weitreichender im Material verteilt. Somit erreichten die GSiC-Proben eine sehr hohe
Biegefestigkeit.
Abbildung 59: SEM-Aufnahmen an Bruchflächen des gesinterten GSiC-F-Nanokomposite mit
2 Gew.-% Graphengehalt nach den 4-Punkt-Biegeprüfungen bei unterschiedlichen Auflösungen.
Aufgrund dieser positiven Resultate der Biegefestigkeit von GSiC-Proben wurde der Trend bei
steigenden Füllgraden bis zu 8 Gew.-% untersucht. Der Einfluss des Füllstoffgehalts auf die
Biegefestigkeit wird in Abbildung 60 dargestellt.
0 2 4 6 8100
200
300
400
500
600
4-P
un
kt-
Bie
ge
fes
tig
ke
it [
MP
a]
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
Typ F
Typ G
Abbildung 60: 4-Punkt-Biegefestigkeit der gesinterten SiC-Nanokomposite mit GSiC-F (aus
Furfurylalkohol), GSiC-G (aus Glucose) und GSiC-D (aus Dopamin) bei variierender Graphen-
Konzentration.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 97 -
Bei GSiC-F nimmt die Biegefestigkeit trotz dem vorteilhaften Herstellungsverfahren mit
steigendem Füllstoffgehalt ständig ab, während die Werte bei GSiC-G und GSiC-D nahezu
konstant blieben. Dieser Unterschied lässt sich auf die unterschiedlichen Kohlenstoffquellen
bzw. Molekülstruktur und Trocknungsverfahren zurückführen. Das Lösungsmittel Ethanol bei
den GSiC-F-Proben wurde mittels Rotationsverdampfung entfernt, während das Lösungsmittel
Wasser bei den GSiC-G- und GSiC-D-Proben durch Gefriertrocknung entfernt wurde. Im
Unterschied zu dem Rotationsverdampfer wurde das Lösungsmittel bei der Gefriertrocknung
aus festen Zustand durch Sublimation bei Unterdruck entfernt, wodurch eine Ausbildung von
harten Granulaten und Agglomeraten vermieden wurde. Die Menge der bei der Herstellung von
GSiC-F entstandenen Agglomerate ist proportional zum Füllstoffgehalt, wegen vermehrter
Partikel-Partikel Wechselwirkungen. Deswegen wurde hierbei eine stetige Abnahme der Dichte,
Zunahme der Porosität und Reduzierung der Biegefestigkeit mit steigendem Füllstoffgehalt
gemessen, was mit den Tendenzen in der Literatur übereinstimmt.[159,214] Dennoch wiesen alle
GSiC-Proben mit 4 Gew.-% Füllstoffgehalt aufgrund der vorteilhaften Herstellungsverfahren
immer eine vergleichbare hohe Biegefestigkeit wie der kommerzielle Typ F (400 MPa). Die mit
8 Gew.-% GSiC-F gefüllte Keramik besitzt eine erhebliche Abnahme der Biegefestigkeit um
40 % auf 290 MPa, welche stets höher als das andere kommerzielle SiC Typ G (250 MPa) der
Firma 3M.
Neben der Biegefestigkeit wurde der Einfluss der Füllstofftypen auf die Bruchzähigkeit der
gesinterten SiC-Nanokomposite untersucht. Hinsichtlich der Bruchzähigkeit zeigten die
meisten Werkstoffvarianten keine Erhöhung (siehe Tabelle 10). So fiel die Bruchzähigkeit z. B.
für die Systeme mit Graphit auf 2.3 MPa∙m0.5 (-26 %) ab. Die Mikrostrukturanalysen in
Kapitel 4.4 hatten gezeigt, dass bei den meisten Nanokompositen die Verteilung der Graphen-
Füllstoffe ungleichmäßig und die Verdichtung der Werkstoffe nach dem Sintern nicht
ausreichend war. Somit ist es nicht verwunderlich, dass eine Mehrzahl der Nanokomposite
zunächst keine Verbesserung der Eigenschaften zeigte. Bei einigen Werkstoffvarianten wurde
allerdings eine höhere Bruchzähigkeit beobachtet. In der Reihe der gleichgefüllten Materialien
zeigten MG-N2-72h und GSiC-G die höchsten Werte der Bruchzähigkeit, welches einer
Erhöhung der Bruchzähigkeit um 20 % auf ca. 3.8 MPa∙m0.5 im Vergleich zum ungefüllten

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 98 -
System entspricht. Auch einige weitere Varianten, wie z. B. TRGO-600, MG-Ethanol,
MG-Ar-24h und GSiC-D, zeigten geringfügig höhere Bruchzähigkeiten als der
Referenzwerkstoff.
Da sich in den GSiC-F-Proben bis zu 4 Gew.-% keine Abnahme der Bruchzähigkeiten zeigte,
wurden sie auch bei höheren Füllgraden (bis zu 8 Gew.-%) untersucht. Der Einfluss des
Füllstoffgehalts auf die Bruchzähigkeit wird in Abbildung 61 dargestellt.
0 2 4 6 82,0
2,5
3,0
3,5
4,0
4,5
Bru
ch
zä
hig
ke
it [
MP
a. m
0,5
]
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
Abbildung 61: Bruchzähigkeit der gesinterten SiC-Nanokomposite mit GSiC-F (aus Furfurylalkohol),
GSiC-G (aus Glucose) und GSiC-D (aus Dopamin) bei variierender Graphen-Konzentration.
Bei GSiC-F und GSiC-G nahm die Bruchzähigkeit mit steigendem Füllstoffgehalt zunächst zu
und anschließend nach Erreichen der höchsten Werte ab, was mit der Literatur
übereinstimmt.[152,214,220] Dies lässt sich unter Umständen durch die abnehmende Dichte und
Biegefestigkeit mit steigendem Füllstoffgehalt erklären. Bei 4 Gew.-% GSiC-F und 2 Gew.-%
GSiC-G wurden die höchste Werte der Bruchzähigkeit gemessen, welche einer Erhöhung der
Bruchzähigkeit um 20 % im Vergleich zum ungefüllten System entsprechen. Einen Vergleich
zu den kommerziellen SiC Typ F und Typ G von 3M könnte nicht durchgeführt werden, da die
Bruchzähigkeit von Typ F und Typ G im technischen Datenblatt 3MTM Siliciumcarbid Portfolio
nach einer anderen Methode bestimmt wurden.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 99 -
4.6 Elektrische Eigenschaften
Ein Werkstoff zeigt erst eine elektrische Leitfähigkeit, wenn Ladungsträger vorhanden und über
große Distanzen beweglich sind. Reines Siliciumcarbid besitzt Halbleiter-Eigenschaften mit
einer Bandlücke zwischen 2.4 und 3.4 eV je nach Kristallstrukturen.[221–223] Abhängig von den
Sintermethoden, Sinterhilfsmitteln und Kristallstrukturen (α oder β-SiC) liegt die elektrische
Leitfähigkeit von reinem SiC im Bereich 10-2 und 10-15 S∙cm-1.[224–226] Einerseits sind die
Halbleiter-Eigenschaften von SiC in vielen Anwendungen, z. B. im Elektrobereich als
Varistoren, Leuchtdioden und Schaltungen, von großer Bedeutung, andererseits muss das SiC
wegen seiner extrem hohen Härte und hohen spezifischen Durchgangswiderstand mittels
Diamant bearbeitet werden, was die Kosten stark erhöht und seine Anwendung erheblich
beschränkt. Deshalb ist eine gewisse elektrische Leitfähigkeit an SiC-Oberflächen
wünschenswert, um die Verwendung von dem innovativen und billigen Bearbeitungsverfahren
wie das Funkenerodieren zu ermöglichen.[227] Ein Weg um zu leitfähigen Keramiken zu
gelangen, stellt der Einsatz elektrisch leitfähiger Füllstoffe auf Basis von Kohlenstoff
dar.[139,228–230] Die elektrischen Eigenschaften von Kompositen mit kohlenstoffbasierten
Füllstoffen sind von vielen Faktoren abhängig, unter anderen Füllstoffgehalt,
Aspektverhältnisse, Probendicke und der angewendeten Testmethode.[75,231] Maßgeblich für
eine elektrische Leitfähigkeit in Keramiken ist die Ausbildung eines geschlossenen Netzwerk
durchgesehener leitfähigen Partikelstränge, eines sogenannten Perkolationsnetzwerkes. Ist
genügend Füllstoff eingebracht, so wird die Perkolationsschwelle überschritten und der
Leitwert der Nanokomposite steigt exponentiell an.[232] Ein solcher keramische Werkstoff mit
elektrischer Leitfähigkeit von mehr als 0.01 S∙cm-1 kann als Superionenleiter bezeichnet
werden.[1,177,233] Eine elektrische Leitfähigkeit in diese Klasse ermöglicht das
Funkenerodieren[234] und wird durch Zugabe von Graphen erwünscht.
Bessere Dispergierung und Exfolierung des Füllstoffs führt zur effektiveren Ausbildung eines
Perkolationsnetzwerks und damit zu einer höheren elektrischen Leitfähigkeit. Die Messungen
der elektrischen Leitfähigkeiten der gesinterten SiC-Nanokomposite erfolgten in
Zusammenarbeit mit M. KNOCH von der Firma FCT Ingenieurkeramik GmbH in Sonneberg an
ausgefrästen Prüfkörpern, die an den Kanten mit Leitsilber überzogen wurden, um einen

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 100 -
besseren Kontakt zwischen dem Probenkörper und der Klemme zu gewährleisten. Der
spezifische Durchgangswiderstand Rspez wurde über Gleichung 3 bestimmt, mit der Länge d
zwischen den Leitsilberkontakten, der Querschnittsfläche A des Probenkörpers sowie den
gemessenen Widerstand Rgem. Die spezifische Leitfähigkeit σs ergibt sich nach Gleichung 4 aus
dem Kehrwert des spezifischen Widerstandes. Die erhaltenen Ergebnisse wurden bereits in
Tabelle 10 aufgelistet und sind in folgenden graphisch dargestellt.
d
ARR gemspez (3)
spez
sR
1 (4)
Die hierbei erhaltenen Ergebnisse wurden bereits in Tabelle 10 aufgeführt. Anhand der
aufgetragenen Daten ist zu erkennen, dass bereits die Einarbeitung von 2 Gew.-% jedes
Graphentyps eine deutliche Erhöhung in Bezug auf die spezifischen elektrischen
Leitfähigkeiten induzierte. Die gesinterten SiC-Nanokomposite mit TRGO, MG-Ethanol und
Grade M erzielten mit 2 Gew.-% Füllstoffgehalt eine vollständige leitfähige Netzwerkstruktur.
Hierbei wurde die elektrische Leitfähigkeit um maximal sieben Größenordnungen im Vergleich
zum reinen SiC auf bis 0.7 S∙cm-1 erhöht. Das ist ein großer Vorteil sowohl gegenüber der
Referenzprobe (3 ∙ 10-8 S∙cm-1) als auch gegenüber den kommerziellen SiC Typen F
(< 10-8 S∙cm-1) und G (10-4 – 10-5 S∙cm-1) der Firma 3M. Diese hohe Leitfähigkeit ermöglicht
einerseits innovative Bearbeitungsverfahren wie das Funkenerodieren und andererseits das
Sinterverfahren des SiC-Graphen-Nanokomposits mittels SPS-/FAST-Prozess. Damit können
deutlich schnellere Sinterzyklen mit verbesserten Werkstoffeigenschaften und vollständiger
Verdichtung realisiert werden. Bei einem elektrisch leitfähigen Produkt kann zudem das
Reibungsverhalten bei einer tribologischen Belastung gezielt eingestellt werden, indem das
elektrische Potenzial im Umgebungsmedium verändert wird.[174] Die erhöhte elektrische
Leitfähigkeit des Nanokomposits ermöglicht außerdem die Anwendung in der Sensorik (z. B.
Abgassensorik) und Brennstoffzellen.[1,177] In Hinsicht auf SiC-Nanokomposite mit
verschiedenen TRGO- und MG-CO2-Proben wurde eine drastische Auswirkung der
Reduktionstemperatur bzw. der Mahldauer auf die elektrische Leitfähigkeit des Nanokomposits
festgestellt (Abbildung 62).

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 101 -
TRGO-600
TRGO-800
TRGO-1000
10-8
10-7
10-6
10-5
10-4
10-3
10-2
10-1 Elektrische Leitfäigkeit
C-Gehalt
Ele
ktr
isch
e L
eit
fäh
igkeit
[S
. cm
-1]
76
78
80
82
84
86
88
90
Ko
hle
nsto
ffg
eh
alt
[G
ew
.-%
]
MG-CO2-24h
MG-CO2-48h
MG-CO2-72h
MG-CO2-96h
MG-CO2-120h10-8
10-7
10-6
10-5
10-4
Elektrische Leitfähigkeit
C-Gehalt
Ele
ktr
isc
he
Le
itfä
hig
ke
it [
S. c
m-1
]
86
87
88
89
90
91
92
Ko
hle
ns
toff
ge
ha
lt [
Ge
w.-
%]
Abbildung 62: Elektrische Leitfähigkeit der gesinterten SiC-Nanokomposite in Abhängigkeit des
Funktionalitätsgrads bei je 2.0 Gew.-% TRGO (links) und MG-CO2 (rechts), korreliert mit
Kohlenstoffgehalt.
Wie in Abbildung 62 (links) zu sehen ist, lagen die Leitfähigkeiten der TRGO-Proben zwischen
10-7 und 10-1 S∙cm-1. Diese erhöhte elektrische Leitfähigkeit im resultierenden
SiC-Nanokomposit war jedoch in allen Fällen mit dem Funktionalisierungsgrad bzw. dem
Kohlenstoffgehalt des Füllstoffes, welcher durch Änderung der Reduktionstemperatur (Tred)
eingestellt werden konnte, verbunden. So führte der durch Erhöhung der Tred hervorgerufene
steigende Kohlenstoffgehalt bzw. der sinkende Funktionalisierungsgrad der TRGO-Proben zu
einer höheren Leitfähigkeit der entsprechenden Nanokomposite. Dies lässt sich auf die
Wiederherstellung einer großflächigen konjugierten sp2-Struktur in TRGO[235] bzw. die
Ausheilung von Defekten im TRGO durch thermische Regraphitisierung[236] zurückführen, was
durch hohe Reduktionstemperatur stark begünstigt wurde.[184] Die Variation der Tred bzw. des
Funktionalitätsgrades des TRGO bietet somit die Möglichkeit zur gezielten Einstellung der
elektrischen Leitfähigkeit des Kompositmaterials. Diese Erklärung gilt auch bei den
Nanokompositen mit MG-CO2 (siehe Abbildung 62 rechts). Längere Mahldauer führte zu
vermehrten hydrophilen funktionellen Gruppen (Sauerstoff-Gehalt), welche Defekte in der
Graphitstruktur darstellten und den elektrischen Transport störten. Somit korrelierte die
elektrische Leitfähigkeit der Nanokomposite mit der Mahldauer bzw. dem Kohlenstoff- und
Sauerstoffgehalt sowie die jeweiligen Partikelgrößen von MG-CO2. Im Gegensatz dazu wurde
keiner ähnliche Einfluss von der Mahldauer auf die resultierenden elektrischen Leitfähigkeiten
bei den Werkstoffen mit MG-Ar und MG-N2 identifiziert (siehe Tabelle 10), was auf die

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 102 -
jeweiligen Partikelgrößen zurückzuführen sein könnte. Während bei MG-CO2 Plättchen-
Partikel nach einer Vermahlung von 120 h immer vorhanden sind, besaßen MG-Ar und MG-N2
bereits nach 24 h nur noch sphärische amorphe Partikel, welche eine leitfähige
Netzwerkstruktur bzw. einen elektrischen Transport mit identischer Wirkung erschwerten.
Dafür erlangen die mit MG-Ar und MG-N2 gefüllten Nanokomposite nahezu unveränderte
elektrische Leitfähigkeiten. Im Einklang zu diesen Ergebnissen wurde der höchste
Leitfähigkeitswert (0.4 S∙cm-1) aller MG-Füllstoffe für die letzte MG-Variante MG-Ethanol
gefunden, welches mittels Nassvermahlung von Graphit erzeugt wurde. Dies lässt sich auf seine
niedrigste Funktionalität (1.5 Gew.-% Sauerstoff-Gehalt und 98.3 Gew.-% Kohlenstoff-Gehalt)
und seinen größten Partikeldurchmesser (3 µm) zurückführen.
Ref. SiC
Graphit
Nanographit
Grade M
Carbon Black
Grade C10-8
10-7
10-6
10-5
10-4
10-3
10-2
10-1
100
Elektrische Leitfäigkeit
C-Gehalt
Ele
ktr
isc
he
Le
itfä
hig
ke
it [
S. c
m-1
]
86
88
90
92
94
96
98
100
Ko
hle
ns
toff
ge
ha
lt [
Ge
w.-
%]
Abbildung 63: Elektrische Leitfähigkeit der gesinterten SiC-Nanokomposite in Abhängigkeit des
Funktionalitätsgrads bei je 2.0 Gew.-% kommerzielle Füllstoffe, korreliert mit Kohlenstoffgehalt.
Der Zusammenhang zwischen dem Funktionalitätsgrad bzw. Sauerstoff-Gehalt des Füllstoffes
und der elektrischen Leitfähigkeiten der SiC-Nanokomposite wurde auch bei den Werkstoffen
mit den kommerziellen Füllstoffen Grade M, Carbon Black und Grade C bestätigt (siehe
Abbildung 63). Aufgrund seinem hohen Aspektverhältnis und seiner vollständigen
graphitischen Struktur mit ausgedehntem konjugierten π-System sowie seinem hohen
Kohlenstoffgehalt erzielte die Probe mit Grade M eine Erhöhung der elektrischen Leitfähigkeit
um mehr als sieben Größenordnungen auf knapp 1 S∙cm-1.[237] Im Vergleich dazu besaßen die
Graphite und Nanographite zwar sehr hohe C-Gehalt, aber konnten aufgrund ihrer inhärent
ausgeprägten Mikrofüllstoffcharaktere bzw. ihrer kleinen Aspektverhältnisse keine effektive
leitfähige Netzwerkstruktur bilden und somit bewirkten geringfügige verbesserte Leitvermögen.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 103 -
Da die GSiC-Proben sehr gute mechanischen Eigenschaften und vollständige Verdichtung
(siehe Kapitel 4.5 bzw. 4.3) erzielten, wurde die elektrische Leitfähigkeit bei steigenden
Füllgraden bis zu 8 Gew.-% untersucht (siehe Abbildung 64).
0 2 4 6 810-9
10-8
10-7
10-6
10-5
10-4
10-3
10-2
10-1
100
101
Ele
ktr
isc
he
Le
itfä
hig
ke
it [
S. c
m-1
]
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
Typ G
Typ F
Abbildung 64: Elektrische Leitfähigkeit der gesinterten SiC-Nanokomposite mit GSiC-F (aus
Furfurylalkohol), GSiC-G (aus Glucose) und GSiC-D (aus Dopamin) bei variierender Graphen-
Konzentration.
Im Vergleich zu Grade M bewirkten alle GSiC mit 2.0 Gew.-% Füllstoffgehalt aufgrund ihrer
kleinen Aspektverhältnisse kein verbessertes Leitvermögen. Die weitere Zugabe von Graphen
führte erst zu einer starken Erhöhung der Leitfähigkeit. Die vielen Füllstoffkontakte
untereinander verstärkten das Perkolationsnetzwerk und führten schließlich zur Verbesserung
der elektrischen Leitfähigkeit. Analog zur Arbeit von MIRANZO[238] konnten durch die
Beimischung von Graphen elektrisch leitfähige SiC-Nanokomposite hergestellt werden.
Die Leitfähigkeiten stiegen kontinuierlich mit zunehmendem Graphengehalt und erreichten ein
Maximum bei 3 S∙cm-1 mit 8 Gew.-% GSiC-F. Aufgrund der starken Abnahme der
mechanischen Eigenschaften mit steigendem Füllstoffgehalt wurde GSiC-G und GSiC-D nur
bis 4 Gew.-% hergestellt. Für Füllgrade um 4 Gew.-% zeigte die GSiC-D den höchsten Leitwert
von 0.07 S∙cm-1, was einer Erhöhung um sechs Größenordnungen im Vergleich zum reinen SiC
sowie um sieben Größenordnungen zum Typ F und drei Größenordnungen zum Typ G der
Firma 3M entspricht. Dies korrelierte mit der elektrischen Leitfähigkeit der Pulvermischung
von GSiC vor dem Sintern, bei der nur GSiC-D eine Erhöhung der Leitfähigkeit detektiert
wurde (siehe Tabelle 4 in Kapitel 3.3.3).

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 104 -
4.7 Tribologische Eigenschaften
4.7.1 Stift-Scheibe-Tribometer
Keramische Werkstoffe werden häufig aufgrund ihrer herausragenden tribologischen
Eigenschaften bzw. Verschleißbeständigkeit eingesetzt.[1–3] Die Messungen der tribologischen
Eigenschaften der im Rahmen dieser Arbeit gesinterten SiC-Nanokomposite mittels Stift-
Scheibe-Tribometer erfolgten in Zusammenarbeit mit Dr. C. SCHRÖDER, Dr. B. SCHLÜTER und
Dr. A. KAILER vom Fraunhofer-Institut für Werkstoffmechanik in Freiburg an ausgefrästen
Prüfkörpern (siehe Abbildung 65).
Abbildung 65: Schematische Zeichnung der SiC-Stift Geometrie (links) für die Stift-Scheibe
tribologische Untersuchung (rechts).
Wie die Abbildung 65 dargestellt, wurden die Stift-Prüfkörper aus gesinterten SiC-Graphen-
Nanokompositen mit den Abmessungen 4 × 4 × 10 mm ausgefräst, an jeweils einer Stirnseite
wurde eine Kugelkalotte mit Radius 5 mm gefertigt. Der Prüfkörper wurde gegen einen
Gleitring aus drucklos gesintertem Siliciumcarbid (SSiC) der Firma EagleBurgmann Germany
GmbH & Co. KG gepresst. Der Radius der durch die Gleitreibung zwischen Stift und Gleitring
gebildeten Reibspur war 26.5 mm. Die aufgebrachte Normalkraft betrug 50 N, die
Gleitgeschwindigkeit 0.1 m/s. Die Versuche erfolgten im wässrigen Medium bei
Raumtemperatur für eine Belastungsdauer von 4 h. Aus der tribologischen Messung wird der
Reibungskoeffizient (µ) der Nanokomposite erhalten, was dem Verhältnis aus der gemessenen
Reibungskraft zur Normalkraft entspricht. In Abbildung 66 ist der zeitliche Verlauf des
gemessenen Reibungsverhaltens der verschiedenen Nanokomposite zu sehen. Bei jeder Probe
wurde die Messung mindestens zweimal wiederholt und die Ergebnisse anschließend gemittelt.
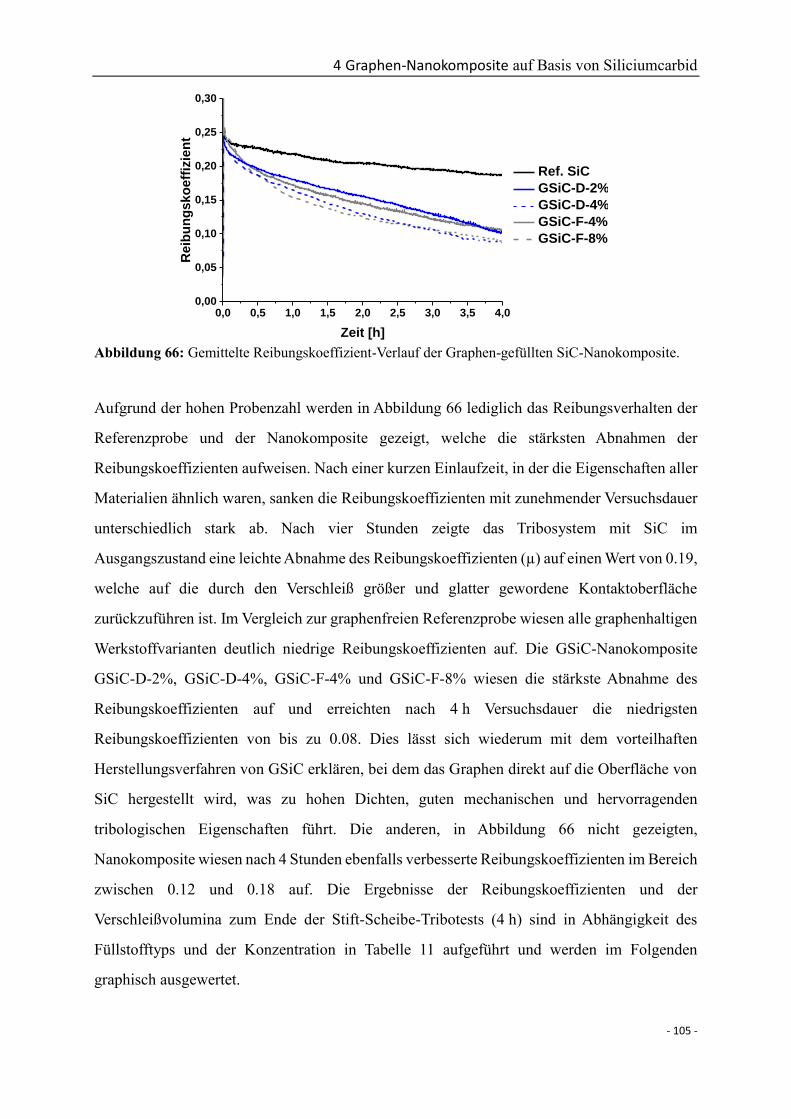
4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 105 -
0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,00,00
0,05
0,10
0,15
0,20
0,25
0,30
Ref. SiC
GSiC-D-2%
GSiC-D-4%
GSiC-F-4%
GSiC-F-8%
Re
ibu
ng
sk
oe
ffiz
ien
t
Zeit [h]
Abbildung 66: Gemittelte Reibungskoeffizient-Verlauf der Graphen-gefüllten SiC-Nanokomposite.
Aufgrund der hohen Probenzahl werden in Abbildung 66 lediglich das Reibungsverhalten der
Referenzprobe und der Nanokomposite gezeigt, welche die stärksten Abnahmen der
Reibungskoeffizienten aufweisen. Nach einer kurzen Einlaufzeit, in der die Eigenschaften aller
Materialien ähnlich waren, sanken die Reibungskoeffizienten mit zunehmender Versuchsdauer
unterschiedlich stark ab. Nach vier Stunden zeigte das Tribosystem mit SiC im
Ausgangszustand eine leichte Abnahme des Reibungskoeffizienten (µ) auf einen Wert von 0.19,
welche auf die durch den Verschleiß größer und glatter gewordene Kontaktoberfläche
zurückzuführen ist. Im Vergleich zur graphenfreien Referenzprobe wiesen alle graphenhaltigen
Werkstoffvarianten deutlich niedrige Reibungskoeffizienten auf. Die GSiC-Nanokomposite
GSiC-D-2%, GSiC-D-4%, GSiC-F-4% und GSiC-F-8% wiesen die stärkste Abnahme des
Reibungskoeffizienten auf und erreichten nach 4 h Versuchsdauer die niedrigsten
Reibungskoeffizienten von bis zu 0.08. Dies lässt sich wiederum mit dem vorteilhaften
Herstellungsverfahren von GSiC erklären, bei dem das Graphen direkt auf die Oberfläche von
SiC hergestellt wird, was zu hohen Dichten, guten mechanischen und hervorragenden
tribologischen Eigenschaften führt. Die anderen, in Abbildung 66 nicht gezeigten,
Nanokomposite wiesen nach 4 Stunden ebenfalls verbesserte Reibungskoeffizienten im Bereich
zwischen 0.12 und 0.18 auf. Die Ergebnisse der Reibungskoeffizienten und der
Verschleißvolumina zum Ende der Stift-Scheibe-Tribotests (4 h) sind in Abhängigkeit des
Füllstofftyps und der Konzentration in Tabelle 11 aufgeführt und werden im Folgenden
graphisch ausgewertet.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 106 -
Tabelle 11: Übersicht des Reibungskoeffizienten nach 4 h und des generierten Verschleißvolumens von
SiC-Nanokompositen in Abhängigkeit von Füllstofftypen und -gehalten.
Probe Gehalt
[Gew.-%]
Reibungskoeffizienta
Verschleißvolumenb
[mm3]
SiC
Ref. SiC 0 0.19 ± 0.01 0.22 ± 0.02
TR
GO
TRGO-600 2 0.15 ± 0.01 0.21 ± 0.01
TRGO-800 2 0.14 ± 0.01 0.20 ± 0.02
TRGO-1000 2 0.15 ± 0.02 0.22 ± 0.02
MG
MG-Ethanol 2 0.17± 0.02 0.26 ± 0.01
MG-Ar-24h 2 0.13 ± 0.02 0.18 ± 0.03
MG-Ar-48h 2 0.16 ± 0.04 0.21 ± 0.06
MG-Ar-72h 2 0.17 ± 0.01 0.20 ± 0.03
MG-CO2-24h 2 0.16 ± 0.01 0.23 ± 0.01
MG-CO2-48h 2 0.16 ± 0.01 0.20 ± 0.02
MG-CO2-72h 2 0.14 ± 0.01 0.20 ± 0.02
MG-CO2-96h 2 0.12 ± 0.03 0.19 ± 0.03
MG-CO2-120h 2 0.13 ± 0.01 0.17 ± 0.02
MG-N2-24h 2 0.18 ± 0.02 0.21 ± 0.02
MG-N2-48h 2 0.13 ± 0.04 0.18 ± 0.05
MG-N2-72h 2 0.14 ± 0.01 0.16 ± 0.02
GS
iC
GSiC-F-2% 2 0.13 ± 0.01 0.15 ± 0.02
GSiC-F-4% 4 0.11 ± 0.05 0.14 ± 0.02
GSiC-F-8% 8 0.09 ± 0.04 0.13 ± 0.02
GSiC-G-2% 2 0.16 ± 0.05 0.17 ± 0.01
GSiC-G-4% 4 0.15 ± 0.03 0.15 ± 0.03
GSiC-D-2% 2 0.10 ± 0.05 0.19 ± 0.04
GSiC-D-4% 4 0.09 ± 0.05 0.17 ± 0.04
Ben
chm
ark
-
Syst
em
Grade C 2 0.12 ± 0.03 0.18 ± 0.05
Grade M 2 0.14 ± 0.01 0.21 ± 0.01
Graphit 2 0.17 ± 0.02 0.25 ± 0.02
Nanographit 2 n. b. n. b.
Carbon Black 2 0.15 ± 0.02 0.20 ± 0.02
a Reibungskoeffizient zum Ende des Stift-Scheibe-Tribotests (4 h); b Generierte Verschleißvolumen nach
Stift-Scheibe-Tribotest (4 h).

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 107 -
Wie der Reibungskoeffizient der verschiedenen TRGO-Nanokomposite in Tabelle 11 zeigt, hat
die Reduktionstemperatur bei der Herstellung von TRGO keinen Einfluss auf die
Reibungskoeffizienten. Nach der vierstündigen Untersuchung lagen die Reibungskoeffizienten
im Bereich von ca. 0.14, was einer Verbesserung um 26 % entspricht. Bei den
Benchmarksystemen mit MG-Ethanol, Grade C, Grade M, Carbon Black und Graphit liegt der
Reibungskoeffizient nach der tribologischen Untersuchung zwischen 0.12 und 0.17. Dabei
lagen die Reibungskoeffizienten der Graphit-, MG-Ethanol- und Carbon Black-gefüllten
Proben unterhalb der Referenzprobe, jedoch höher als die beiden kommerziell erhältlichen
Benchmarkgraphene (Grade C und Grade M). Hervorzuheben ist hierbei das Grade C-System
mit einem finalen Reibungskoeffizienten nach 4 h von 0.12.
24 48 72 96 120
0,05
0,10
0,15
0,20
Reib
un
gsko
eff
izie
nt
Mahldauer [h]
Ref.
MG-Ar
MG-CO2
MG-N2
Abbildung 67: Reibungskoeffizient zum Ende des Stift-Scheibe-Tribotests der gesinterten SiC-
Nanokomposite mit je 2 Gew.-% MG-Ar, MG-N2 und MG-CO2 Füllgehalt.
Die Reibungskoeffizienten der MG-Nanokomposite wurden nach Herstellungsbedingungen
bzw. Mahlspezifikation in Abbildung 67 dargestellt. Bei den SiC-Nanokompositen, in denen
MG-CO2 bzw. MG-N2 eingearbeitet war, sanken die Reibungskoeffizienten mit steigender
Mahldauer stetig ab. Diese Tendenz ist primär von der Partikelgröße, der spezifischen
Oberfläche und der Funktionalität abhängig (siehe Kapitel 3.2). Eine längere Mahldauer führte
zu einer Abnahme der Partikelgröße, steigenden spezifischen Oberfläche und Zunahme der

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 108 -
Funktionalität, wodurch eine homogene Verteilung in der SiC-Matrix gewährleistet und bessere
mechanische und tribologische Eigenschaften erreicht wurden. Im Gegensatz zu den MG-CO2-
und MG-N2-Proben nimmt der Reibungskoeffizient nach vierstündiger Untersuchung mit
steigender Mahldauer zu. Dieses Verhalten könnte an der Partikelmorphologie liegen. Im
Vergleich zum plättchenartigen MG-CO2 führte die 24 h Vermahlung unter Argon zur
vollständigen Zerstörung der Plättchen-Strukturen. Eine längere Mahldauer führt zur keinen
Abblätterungen, sondern Reagglomeration der sphärischen Partikel. Als Folge verfügen MG-
Ar bei einer Mahldauer ab 24 h über eine abnehmende spezifische Oberfläche bei steigender
Mahldauer, was zu einer schlechten Verteilung in SiC-Matrix und schlechten tribologischen
Eigenschaften führt.
0 2 4 6 8
0,05
0,10
0,15
0,20
Re
ibu
ng
sk
oe
ffiz
ien
t
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
Abbildung 68: Reibungskoeffizient zum Ende des Stift-Scheibe-Tribotests der gesinterten SiC-
Nanokomposite mit GSiC-F (aus Furfurylalkohol), GSiC-G (aus Glucose) und GSiC-D (aus Dopamin)
bei variierender Graphen-Konzentration.
Abbildung 68 zeigt den Einfluss der Kohlenstoffquelle auf dem Reibungskoeffizienten der
Nanokomposite mit graphenbeschichteten SiC (GSiC) bei verschiedenen Füllgraden (bis zu
8 Gew.-%). Der niedrigste Reibungskoeffizient aller Nanokomposite bei 2.0 Gew.-%
Füllstoffanteil wurde bei GSiC-D (0.10) gemessen (vgl. GSiC-F 0.13 und GSiC-G 0.16). Die
Erhöhung des Graphenanteils in GSiC führte zu einer kontinuierlichen Abnahme des
Reibungskoeffizienten. Die unterschiedlichen Kurvenverläufe waren hauptsächlich auf die
unterschiedlichen Präkursoren zurückzuführen. Dies führte wahrscheinlich zu

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 109 -
unterschiedlichen Qualitäten der Graphene, wie z.B. Partikelgröße, Funktionalität und
Partikelgrößenverteilung, die bei der Weiterverbreitung in der Keramik zur Beeinflussung der
mechanischen und tribologischen Eigenschaften führen können. Des Weiteren wurden die
niedrigsten Reibungskoeffizienten (µ = 0.09) aller mit Graphen gefüllten Proben bei
GSiC-D-4% und GSiC-F-8% beobachtet, was einer Reduzierung des Reibungskoeffizienten
um 54 % gegenüber der zur Referenzprobe entsprach.
Zusammenfassend wiesen alle Nanokomposite niedrigere Reibungskoeffizienten als die
ungefüllte Referenzprobe auf. Die mit 4 Gew.-% GSiC-D und 8 Gew.-% GSiC-F gefüllten SiC
besaßen zum Ende der tribologischen Messung die niedrigsten Reibungskoeffizienten, gefolgt
von 2 Gew.-% GSiC-D und 4 Gew.-% GSiC-F. Bei diesen Proben wurde der
Reibungskoeffizient bis zu 54 % reduziert. Gegenüber der Referenzprobe zeigten die Proben
mit je 2 Gew.-% GSiC-F, MG-CO2-96h und MG-CO2-120h sowie Grade C eine Reduzierung
des Reibungskoeffizienten um ca. 35 %.
Nach den tribologischen Untersuchungen wurde der Einfluss von Füllstofftypen auf die
generierten Verschleißvolumina bzw. die Verschleißbeständigkeit der SiC-Nanokomposite
untersucht. Die Ergebnisse der generierten Verschleißvolumina wurden bereits in Tabelle 11
aufgeführt. Hinsichtlich der Verschleißbeständigkeit zeigten die meisten Tribosysteme mit
Ausnahme des Systems mit MG-Ethanol, MG-CO2-24h und Graphit verbesserte
Verschleißbeständigkeit im Vergleich zum SiC-System. Die Verschleißwerte zeigten analog zu
den Reibungskoeffizienten erkennbare Tendenzen. Diese lassen sich auf die
Verschleißmechanismen zurückführen. Je höher die Reibung ist, desto größer sind die
Wechselwirkungen der Elemente und desto größer sind die tribologischen Beanspruchungen,
die zu Verschleiß führen. Die Benchmarksysteme Grade C, Grade M sowie Carbon Black
zeigten leicht niedrige Verschleißwerte im Vergleich zum Referenzmaterial. Bei den TRGO-
Nanokompositen hat die Reduktionstemperatur keinen Einfluss auf die Verschleißwerte, was
mit der Tendenz der Reibungskoeffizientswerte übereinstimmt. Exemplarisch werden
Verschleißvolumina und Reibungskoeffizienten der tribologischen Untersuchung abhängig von
der Mahldauer von MG-CO2 und MG-N2 in Abbildung 69 dargestellt.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 110 -
MG-CO2-24h
MG-CO2-48h
MG-CO2-72h
MG-CO2-96h
MG-CO2-120h
0,15
0,17
0,19
0,21
0,23
0,25 Verschleißvolumen
Reibungskoeffizient
Vers
ch
leiß
vo
lum
en
[m
m3]
Ref.
0,11
0,12
0,13
0,14
0,15
0,16
Reib
un
gsko
eff
izie
nt
MG-N2-24h
MG-N2-48h
MG-N2-72h
0,13
0,15
0,17
0,19
0,21
0,23
Verschleißvolumen
Reibungskoeffizient
Vers
ch
leiß
vo
lum
en
[m
m3]
Ref.
0,10
0,12
0,14
0,16
0,18
0,20
Reib
un
gsko
eff
izie
nt
Abbildung 69: Verschleißvolumina der gesinterten SiC-Nanokomposite in Abhängigkeit des
Funktionalitätsgrads bei je 2.0 Gew.-% MG-CO2 (links) und MG-N2 (rechts), korreliert mit dem
Reibungskoeffizienten zum Ende beim 4 h-Tribotest.
Aus Abbildung 69 ist ersichtlich, dass die Zugabe von MG-CO2-24h eine geringfügige
Reduktion der Verschleißbeständigkeit im Vergleich zu reinem SiC (V = 0.22 mm3) induzierte.
Dies lässt sich auf seine hohe Porosität, schlechten mechanischen Eigenschaften und seinen
hohen Reibungskoeffizienten zurückführen. Im Vergleich der MG-CO2-Systeme bei
verschiedener Mahldauer zeigen hohe Mahldauer eine starke Verbesserung der
Verschleißbeständigkeit, was mit abnehmenden Reibungskoeffizienten korreliert. So fiel das
Verschleißvolumen für das Werkstoffvariant mit MG-CO2-120h auf 0.17 mm3 (-23 %) ab. Diese
Tendenz gilt ebenfalls bei MG-N2-Systemen. Dabei erzielte das Nanokomposit mit MG-N2-72h
eine Reduzierung des Verschleißvolumens um 27 %.
Des Weiteren ist in Tabelle 11 zu erkennen, dass die GSiC-Nanokomposite unter allen
Nanokompositen mit 2 Gew.-% Füllstoffgehalt den geringsten Verschleiß (V ~ 0.15 mm3)
aufwiesen. Dies entspricht einer Verbesserung der Verschleißbeständigkeit bis zu 31 %. Dieses
hervorragende Resultat lässt sich wiederum auf das vorteilhafte Herstellungsverfahren
zurückführen, welches eine vollständige Verdichtung gewährleistet und somit sehr gut
mechanische und tribologische Eigenschaften zur Folge hat. Aufgrund dieser Eigenschaften
wurde der Trend bei steigenden Füllgraden bis zu 8 Gew.-% untersucht. Der Einfluss von
Füllstoffgehalt auf die Verschleißbeständigkeit wird in Abbildung 70 dargestellt.
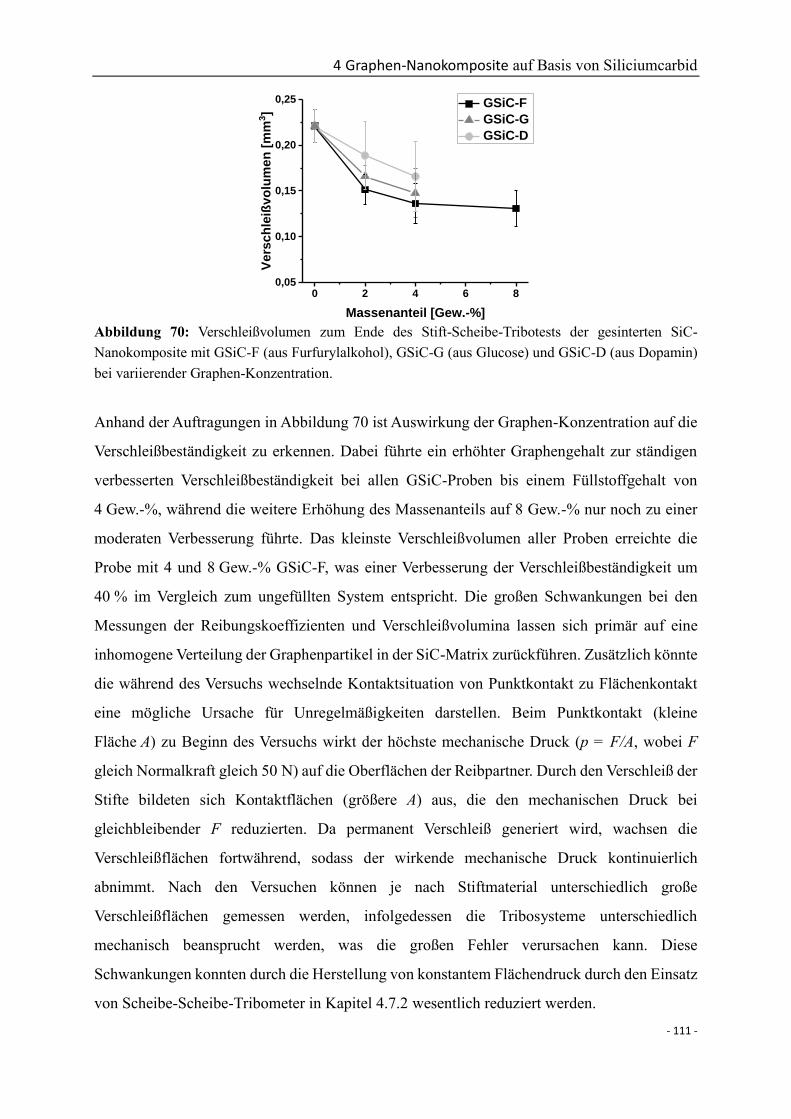
4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 111 -
0 2 4 6 80,05
0,10
0,15
0,20
0,25
Ve
rsc
hle
ißv
olu
me
n [
mm
3]
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
Abbildung 70: Verschleißvolumen zum Ende des Stift-Scheibe-Tribotests der gesinterten SiC-
Nanokomposite mit GSiC-F (aus Furfurylalkohol), GSiC-G (aus Glucose) und GSiC-D (aus Dopamin)
bei variierender Graphen-Konzentration.
Anhand der Auftragungen in Abbildung 70 ist Auswirkung der Graphen-Konzentration auf die
Verschleißbeständigkeit zu erkennen. Dabei führte ein erhöhter Graphengehalt zur ständigen
verbesserten Verschleißbeständigkeit bei allen GSiC-Proben bis einem Füllstoffgehalt von
4 Gew.-%, während die weitere Erhöhung des Massenanteils auf 8 Gew.-% nur noch zu einer
moderaten Verbesserung führte. Das kleinste Verschleißvolumen aller Proben erreichte die
Probe mit 4 und 8 Gew.-% GSiC-F, was einer Verbesserung der Verschleißbeständigkeit um
40 % im Vergleich zum ungefüllten System entspricht. Die großen Schwankungen bei den
Messungen der Reibungskoeffizienten und Verschleißvolumina lassen sich primär auf eine
inhomogene Verteilung der Graphenpartikel in der SiC-Matrix zurückführen. Zusätzlich könnte
die während des Versuchs wechselnde Kontaktsituation von Punktkontakt zu Flächenkontakt
eine mögliche Ursache für Unregelmäßigkeiten darstellen. Beim Punktkontakt (kleine
Fläche A) zu Beginn des Versuchs wirkt der höchste mechanische Druck (p = F/A, wobei F
gleich Normalkraft gleich 50 N) auf die Oberflächen der Reibpartner. Durch den Verschleiß der
Stifte bildeten sich Kontaktflächen (größere A) aus, die den mechanischen Druck bei
gleichbleibender F reduzierten. Da permanent Verschleiß generiert wird, wachsen die
Verschleißflächen fortwährend, sodass der wirkende mechanische Druck kontinuierlich
abnimmt. Nach den Versuchen können je nach Stiftmaterial unterschiedlich große
Verschleißflächen gemessen werden, infolgedessen die Tribosysteme unterschiedlich
mechanisch beansprucht werden, was die großen Fehler verursachen kann. Diese
Schwankungen konnten durch die Herstellung von konstantem Flächendruck durch den Einsatz
von Scheibe-Scheibe-Tribometer in Kapitel 4.7.2 wesentlich reduziert werden.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 112 -
Um die tribologischen Reibmechanismen zu erklären, wurden die Nanokomposite, SiC +
2 Gew.-% Grade C, SiC + 4 Gew.-% GSiC-F und SiC + 8 Gew.-% GSiC-F sowie die
Referenzprobe nach den Stift-Scheibe-Untersuchungen (4 h) für die AFM-Analyse ausgewählt,
da sie sehr niedrige Reibungskoeffizienten und Verschleißvolumina zeigten.
Abbildung 71: AFM-Aufnahmen der Referenzprobe in der Kontaktstelle vor (links) und nach (Mitte
und rechts) tribologischer Messung.
Abbildung 72: AFM-Aufnahmen der ausgewählten SiC-Graphen-Nanokomposite in der Kontaktstelle
nach tribologischen Messungen bei unterschiedlichen Auflösungen.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 113 -
Die entsprechenden AFM-Aufnahmen werden in den Abbildungen 71 und 72 dargestellt. Nach
den tribologischen Untersuchungen zeigten alle Oberflächen eine unterschiedliche
Morphologie. Im Gegensatz zu der vollständig glatten Oberfläche der Referenzprobe
(Abbildung 71) weisen die Nanokomposite (Abbildung 72) neben der glatt geriebenen
Oberfläche auch hervorstehende Kohlenstoff-Erhebungen auf, die bei den Materialvarianten
unterschiedlich ausgeprägt sind. Die Kohlenstoff-Erhebungen könnten Graphen-Flocken sein.
Während der tribologischen Untersuchungen könnte das Matrixmaterial SiC durch
tribologische Beanspruchung abgerieben werden, während das Graphen auf der Oberfläche
anhaftet und somit anreichert. Dadurch könnte ein schützender Tribofilm bzw. Graphenfilm
gebildet werden, was den Reibungskoeffizienten und das Verschleißvolumen stark reduziert.
Ähnliche Graphen-Tribofilme wurden bei der Arbeit von BELMONTE et al. mittels Raman-
Spektroskopie nachgewiesen.[152] Sie berichteten über einen schützenden Tribofilm, der durch
Anreicherung der Graphene auf der Oberfläche des Nanokomposits durch die tribologische
Untersuchung entsteht. In dieser Arbeit wurde die tribologische Untersuchung unter trockenen
Bedingungen vorgenommen und eine beachtliche Reduzierung der Verschleißvolumina bei
unveränderten Reibungskoeffizienten erreicht.
Zusammenfassend liefern die GSiC-Nanokomposite die vielversprechendsten tribologischen
Eigenschaften mit reduzierter Reibung und gleichzeitig reduziertem Verschleiß. Dabei zeigen
die GSiC-Varianten GSiC-D-4% und GSiC-F-8% mit Abstand die niedrigsten
Reibungskoeffizienten und Verschleißvolumina. Die individuellen Unterschiede können
primär auf den verwendeten Präkursoren oder auf deren Verarbeitungsverfahren zurückgeführt
werden. Beispielsweise wurden die GSiC-G- und GSiC-D-Proben gefriergetrocknet, während
die GSiC-F-Proben mittels Rotationsverdampfer vom Lösungsmittel befreit wurden. Die
resultierenden unterschiedlichen Partikelgrößen und Funktionalitäten beeinflussen die
Eigenschaften der Keramikkomposite. Die MG, welche für 96 h bzw. 120 h unter CO2
gemahlen wurden, liefern ebenfalls deutlich verbesserte tribologische Eigenschaften. Weniger
erfolgsversprechend ist die Einarbeitung von MG-Ethanol. Dies lässt sich auf ungleichmäßige
Verteilung des Graphens zurückführen.
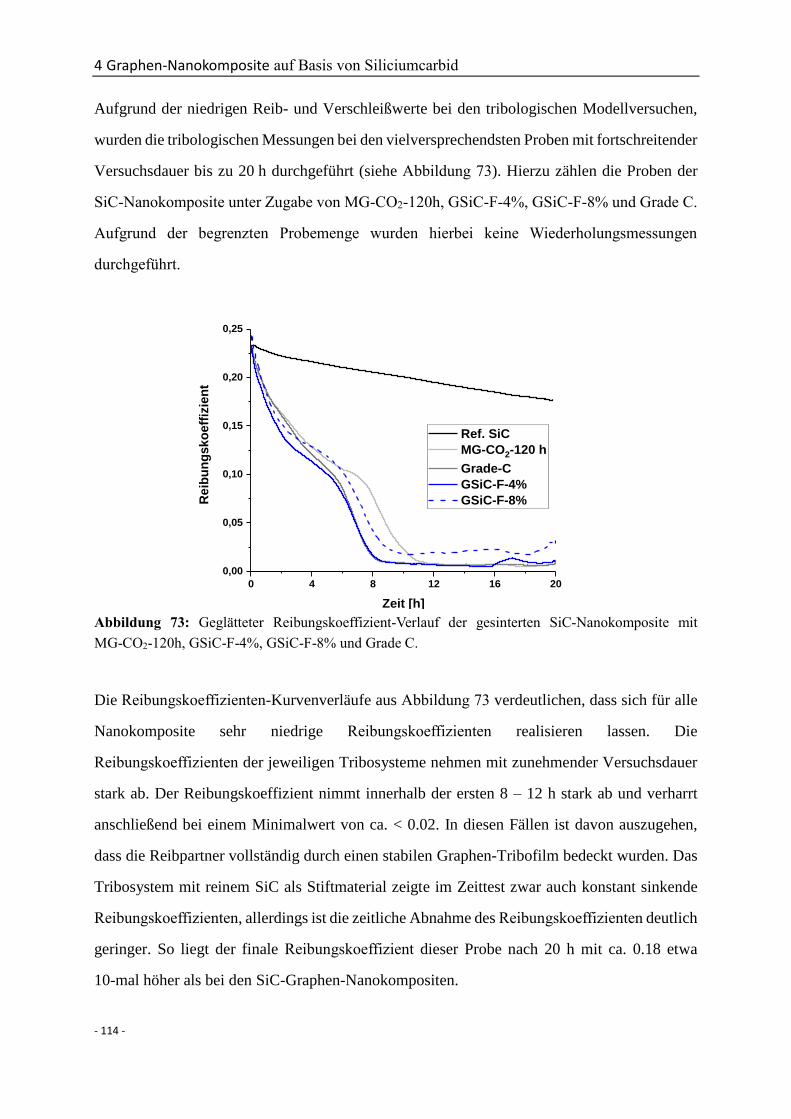
4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 114 -
Aufgrund der niedrigen Reib- und Verschleißwerte bei den tribologischen Modellversuchen,
wurden die tribologischen Messungen bei den vielversprechendsten Proben mit fortschreitender
Versuchsdauer bis zu 20 h durchgeführt (siehe Abbildung 73). Hierzu zählen die Proben der
SiC-Nanokomposite unter Zugabe von MG-CO2-120h, GSiC-F-4%, GSiC-F-8% und Grade C.
Aufgrund der begrenzten Probemenge wurden hierbei keine Wiederholungsmessungen
durchgeführt.
0 4 8 12 16 20
0,00
0,05
0,10
0,15
0,20
0,25
Reib
un
gsko
eff
izie
nt
Zeit [h]
Ref. SiC
MG-CO2-120 h
Grade-C
GSiC-F-4%
GSiC-F-8%
Abbildung 73: Geglätteter Reibungskoeffizient-Verlauf der gesinterten SiC-Nanokomposite mit
MG-CO2-120h, GSiC-F-4%, GSiC-F-8% und Grade C.
Die Reibungskoeffizienten-Kurvenverläufe aus Abbildung 73 verdeutlichen, dass sich für alle
Nanokomposite sehr niedrige Reibungskoeffizienten realisieren lassen. Die
Reibungskoeffizienten der jeweiligen Tribosysteme nehmen mit zunehmender Versuchsdauer
stark ab. Der Reibungskoeffizient nimmt innerhalb der ersten 8 – 12 h stark ab und verharrt
anschließend bei einem Minimalwert von ca. < 0.02. In diesen Fällen ist davon auszugehen,
dass die Reibpartner vollständig durch einen stabilen Graphen-Tribofilm bedeckt wurden. Das
Tribosystem mit reinem SiC als Stiftmaterial zeigte im Zeittest zwar auch konstant sinkende
Reibungskoeffizienten, allerdings ist die zeitliche Abnahme des Reibungskoeffizienten deutlich
geringer. So liegt der finale Reibungskoeffizient dieser Probe nach 20 h mit ca. 0.18 etwa
10-mal höher als bei den SiC-Graphen-Nanokompositen.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 115 -
Ref. SiC
MG-CO2-120h
GSiC-F-4%
GSiC-F-8%
Grade C
0,0
0,2
0,4
0,6
0,8
1,0
1,2
Vers
ch
leiß
vo
lum
en
[m
m3]
Abbildung 74: Verschleißvolumen der gesinterten SiC-Nanokomposite mit MG-CO2-120h,
GSiC-F-4%, GSiC-F-8% und Grade C nach 20 h Stift-Scheibe-Tribotest.
Die generierten Verschleißvolumina der Stifte wurden nach materieller Zusammensetzung in
Abbildung 74 eingetragen. Es ist zu erkennen, dass der Verschleiß der graphenhaltigen
Varianten wesentlich niedriger ist. Der niedrigste Verschleiß wurde mit dem GSiC-F-4%
erreicht, was einer Verbesserung der Verschleißbeständigkeit um 80 % entspricht.
Zusammenfassend lässt sich feststellen, dass mit zunehmender Versuchsdauer (bis 8 – 12 h)
die SiC-Graphen-Nanokomposite stetig bessere Reibungskoeffizienten und
Verschleißbeständigkeit im Vergleich zum Referenzsystem zeigen. Außerdem nimmt die
Überlegenheit der Nanokomposite gegenüber der Referenzprobe innerhalb dieser
Versuchsdauer stetig zu.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 116 -
4.7.2 Scheibe-Scheibe-Tribometer
Neben dem Stift-Scheibe-Tribotest wurden die tribologischen Eigenschaften der gesinterten
SiC-Nanokomposite auch mit einem oszillierenden Scheibe-Scheibe-Tribometer in
Zusammenarbeit mit J. THELKE von der Firma EagleBurgmann Germany GmbH & Co. KG in
Wolfratshausen an Gleitring-Prüfkörpern untersucht (siehe Abbildung 75). Anders als bei dem
Stift-Scheibe-Tribometer (Punktkontakt) wird bei dem Scheibe-Scheibe-Tribometer ein
Flächenkontakt gebildet. Die Gleitflächen wurden von EagleBurgmann Germany GmbH & Co.
KG durch Politur mit speziellem Diamantkorn auf eine Oberfläche mit Rauheit von 0.15 µm
gebracht.
Abbildung 75: Schematische Zeichnung der Geometrie der Keramik-Graphen-Nanokomposite für die
Scheibe-Scheibe tribologische Untersuchung.
Die Untersuchungen erfolgten an Werkstoffpaaren (Gleitring und Gegenring), welche aus den
ausgewählten Nanokompositen Carbon Black, TRGO-600, MG-Ethanol, MG-CO2-48h,
MG-CO2-72h, MG-CO2-120h und GSiC-G zu je 2 Gew.-%, sowie aus GSiC-F (4 und 8 Gew.-%)
und GSiC-D (4 Gew.-%) gefräst wurden. Die Prüfkörper verfügen über eine Kontaktfläche von
450 mm2 mit einem Außen- und Innendurchmesser von 51.8 bzw. 44.6 mm. Als
Referenzwerkstoffe wurden das ungefüllte SiC (Ref. SiC) sowie zwei kommerzielle SiC-
Produkte Typ F und Typ G der Firma 3M ebenfalls untersucht. Während in Kapitel 4.7.1 die
gemessenen statischen Reibungskoeffizienten wiedergegeben werden, werden in diesem
Kapitel die geschwindigkeits- und kraftabhängigen Reibungsverhalten der Tribosysteme unter
naher Betriebsbedingungen im wässrigen Medium bei Raumtemperatur für eine
Belastungsdauer von 320 min ausgewertet (siehe Abbildung 76). Es wurden zur Auswertung
pro Werkstoffpaarung mindestens drei Testläufe durchgeführt und der Mittelwert gebildet.
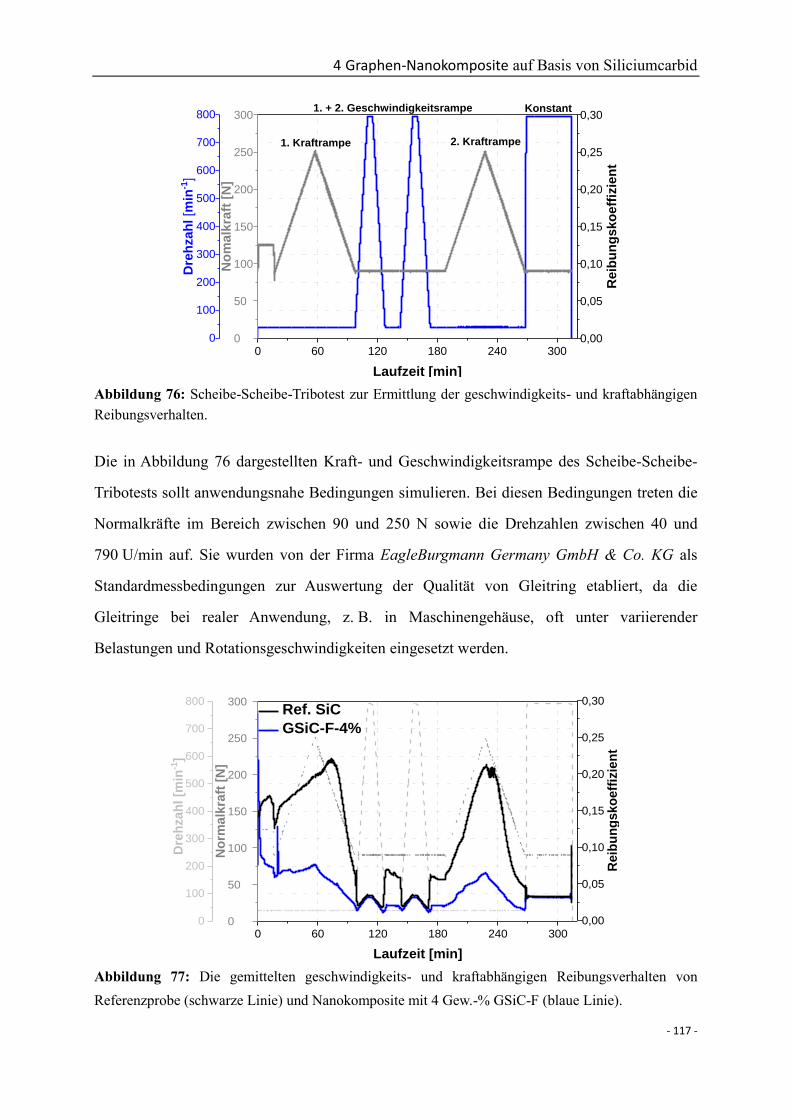
4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 117 -
0 60 120 180 240 3000
100
200
300
400
500
600
700
800Konstant
2. Kraftrampe
1. + 2. Geschwindigkeitsrampe
Dre
hza
hl
[min
-1]
Laufzeit [min]
1. Kraftrampe
0
50
100
150
200
250
300
No
ma
lkra
ft [
N]
0,00
0,05
0,10
0,15
0,20
0,25
0,30
Reib
un
gs
ko
eff
izie
nt
Abbildung 76: Scheibe-Scheibe-Tribotest zur Ermittlung der geschwindigkeits- und kraftabhängigen
Reibungsverhalten.
Die in Abbildung 76 dargestellten Kraft- und Geschwindigkeitsrampe des Scheibe-Scheibe-
Tribotests sollt anwendungsnahe Bedingungen simulieren. Bei diesen Bedingungen treten die
Normalkräfte im Bereich zwischen 90 und 250 N sowie die Drehzahlen zwischen 40 und
790 U/min auf. Sie wurden von der Firma EagleBurgmann Germany GmbH & Co. KG als
Standardmessbedingungen zur Auswertung der Qualität von Gleitring etabliert, da die
Gleitringe bei realer Anwendung, z. B. in Maschinengehäuse, oft unter variierender
Belastungen und Rotationsgeschwindigkeiten eingesetzt werden.
0 60 120 180 240 3000
100
200
300
400
500
600
700
800
Dre
hzah
l [m
in-1
]
Laufzeit [min]
0
50
100
150
200
250
300
No
rma
lkra
ft [
N]
0,00
0,05
0,10
0,15
0,20
0,25
0,30 Ref. SiC
GSiC-F-4%
Re
ibu
ng
sk
oe
ffiz
ien
t
Abbildung 77: Die gemittelten geschwindigkeits- und kraftabhängigen Reibungsverhalten von
Referenzprobe (schwarze Linie) und Nanokomposite mit 4 Gew.-% GSiC-F (blaue Linie).

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 118 -
Die Abbildung 77 (schwarze Linie) zeigte den gemittelten Reibungskoeffizientenverlauf der
Referenzprobe unter verschiedenen Belastungsbedingungen in Kraft- und
Geschwindigkeitsrampen. Der Reibungskoeffizient nach der Einlaufphase bei der zweiten
Kraftrampe unter maximaler Belastung (250 N) ist ein Kernelement der Auswertung und lag
bei Ref. SiC um 0.21. Im Vergleich hierzu wies die GSiC-F Probe mit 4 Gew.-% Graphengehalt
in allen Bereichen deutlich verbesserte Reibeigenschaften mit reduziertem
Reibungskoeffizienten auf (siehe Abbildung 77 blaue Linie). Beispielsweise ist der
Reibungskoeffizient bei der zweiten Kraftrampe etwa 0.065, was einer Reduktion um 69 %
gegenüber der Referenzprobe entspricht.
Zur besseren Vergleichbarkeit wurden die Reibungskoeffizienten aller Graphen-gefüllten SiC-
Nanokomposite nach Einlaufphase bei zweiter Kraftrampe unter maximal Belastung (250 N)
in Abbildung 78 aufgetragen.
Ref
. SiC
Typ
F
Typ
GTR
GO
-600
MG
-Eth
anol
MG
-CO
2-48
hM
G-C
O2-
72h
MG
-CO
2-12
0hG
SiC
-F-4
%G
SiC
-F-8
%G
SiC
-G-2
%G
SiC
-D-4
%Car
bon
Bla
ck
0,00
0,05
0,10
0,15
0,20
0,25 Referenz
TRGO
MG
GSiC
Carbon Black
Re
ibun
gsko
eff
izie
nt
Abbildung 78: Gemittelte Reibungskoeffizienten aller Graphen-gefüllten SiC-Nanokomposite nach
Einlaufphase bei zweiter Kraftrampe unter 250 N.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 119 -
Durch den Vergleich der Referenzprobe (Ref. SiC) mit kommerziellen Referenzen (Typ F und
Typ G) der Firma 3M zeigte die kommerzielle Referenzprobe Typ G den niedrigsten
Reibungskoeffizienten (0.20) und gefolgte von Ref. SiC (0.21) und Typ F (0.23) auf, was sich
auf die im Typ G enthaltenen Graphite zurückführen lässt. Des Weiteren konnte durch
Beimischung von Graphen eine deutliche Verminderung des Reibungskoeffizienten bei
Festkörperreibung gegenüber den graphenfreien Siliciumcarbidvarianten, bzw. der
Referenzprobe und den kommerziellen Typ F und Typ G, erzielt werden. Ähnlich wie bei dem
Stift-Scheibe-Tribotest in Kapitel 4.7.1 wurde der höchste Reibungskoeffizient für das
Nanokomposit mit unfunktionalisiertem MG-Ethanol (µ = 0.18) erhalten. Für die TRGO-600-
Probe sank der Reibungskoeffizient auf etwa 0.12 ab. Bei den SiC-Nanokompositen mit
MG-CO2 sanken die Reibungskoeffizienten mit steigender Mahldauer stetig ab, was mit den
Ergebnissen des Stift-Scheibe-Tribotests übereinstimmt. Diese Tendenz hängt wiederum vor
allem mit der Partikelgröße, der spezifischen Oberfläche und der Funktionalität von MG-CO2
(vgl. Kapitel 3.2) sowie den resultierenden Dichte der Nanokomposite zusammen (vgl.
Abbildung 46 in Kapitel 4.3). Längere Mahldauer führte zur Abnahme der Partikelgröße, der
steigenden spezifischen Oberfläche und der Zunahme der Funktionalität, wodurch eine
homogene Verteilung in der SiC-Matrix gewährleistete und vollständigere Verdichtung sowie
bessere mechanische und tribologische Eigenschaften hervorrief. Darüber hinaus wiesen alle
GSiC-Proben ähnlich wie bei dem Stift-Scheibe-Tribotest in Kapitel 4.7.1 die niedrigsten
Reibungskoeffizienten mit einer maximalen Reduzierung bis zu 78 % auf, was wiederum auf
das vorteilhafte Herstellungsverfahren zurückzuführen ist. Hier wurde das Graphen mittels
Präkursor-Verfahren direkt auf die SiC-Oberfläche aufgebracht und war damit sehr gut in der
Matrix verteilt. Dies führte nicht nur zu einer sehr hohen Sinterdichte, guten mechanischen
Eigenschaften, sondern auch zu herausragenden Reibungs- und Verschleißverhalten.
Darüber hinaus wurden die Belastungsgrenze der Nanokomposite untersucht. Hierbei wurde
die tribologische Messung (Scheibe-Scheibe) mit einer maximalen Belastung von 700 statt
250 N durchgeführt. Diese zeitliche Belastung wird in Abbildung 79 (rechts) aufgetragen und
mit Standard-Messbedingung (siehe Abbildung 79 links) verglichen.
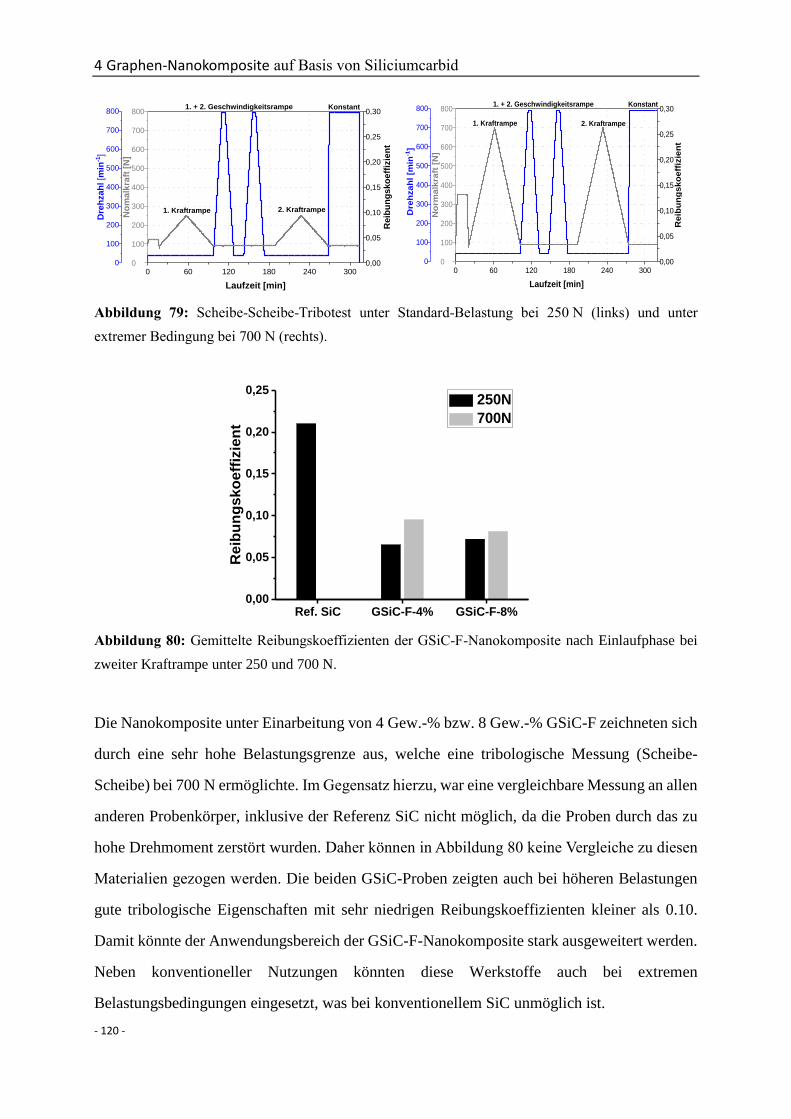
4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 120 -
0 60 120 180 240 3000
100
200
300
400
500
600
700
800Konstant
2. Kraftrampe
1. + 2. GeschwindigkeitsrampeD
reh
za
hl
[min
-1]
Laufzeit [min]
1. Kraftrampe
0
100
200
300
400
500
600
700
800
No
ma
lkra
ft [
N]
0,00
0,05
0,10
0,15
0,20
0,25
0,30
Reib
un
gs
ko
eff
izie
nt
0 60 120 180 240 3000
100
200
300
400
500
600
700
800
Dre
hzah
l [m
in-1
]
Laufzeit [min]
0
100
200
300
400
500
600
700
800
No
rmalk
raft
[N
]
0,00
0,05
0,10
0,15
0,20
0,25
0,30Konstant1. + 2. Geschwindigkeitsrampe
2. Kraftrampe1. Kraftrampe
Reib
un
gsko
eff
izie
nt
Abbildung 79: Scheibe-Scheibe-Tribotest unter Standard-Belastung bei 250 N (links) und unter
extremer Bedingung bei 700 N (rechts).
Ref. SiC GSiC-F-4% GSiC-F-8%0,00
0,05
0,10
0,15
0,20
0,25
Reib
un
gsko
eff
izie
nt
250N
700N
Abbildung 80: Gemittelte Reibungskoeffizienten der GSiC-F-Nanokomposite nach Einlaufphase bei
zweiter Kraftrampe unter 250 und 700 N.
Die Nanokomposite unter Einarbeitung von 4 Gew.-% bzw. 8 Gew.-% GSiC-F zeichneten sich
durch eine sehr hohe Belastungsgrenze aus, welche eine tribologische Messung (Scheibe-
Scheibe) bei 700 N ermöglichte. Im Gegensatz hierzu, war eine vergleichbare Messung an allen
anderen Probenkörper, inklusive der Referenz SiC nicht möglich, da die Proben durch das zu
hohe Drehmoment zerstört wurden. Daher können in Abbildung 80 keine Vergleiche zu diesen
Materialien gezogen werden. Die beiden GSiC-Proben zeigten auch bei höheren Belastungen
gute tribologische Eigenschaften mit sehr niedrigen Reibungskoeffizienten kleiner als 0.10.
Damit könnte der Anwendungsbereich der GSiC-F-Nanokomposite stark ausgeweitert werden.
Neben konventioneller Nutzungen könnten diese Werkstoffe auch bei extremen
Belastungsbedingungen eingesetzt, was bei konventionellem SiC unmöglich ist.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 121 -
4.7.3 Simulation: Atomistische Modellierung
Zur Aufklärung der grundlegenden Mechanismen von Reibung und Verschleiß wurden,
begleitend zu den experimentellen Untersuchungen, molekularstatische Simulationen in
Zusammenarbeit mit Dr. A. KLEMENZ und Prof. Dr. M. MOSELER vom Freiburger
Materialforschungszentrum (FMF) modelliert.
Als Modellsysteme dienten hierbei SiC-Einkristalle verschiedener Größe, mit und ohne
Bedeckung der Oberflächen mit Graphen. Analog zur Arbeit von KLEMENZ et al. wurden
kristalline SiC Quader mit (100), (110) und (111) Oberflächen als Substrate verwendet, deren
Seitenlängen so gewählt wurden, dass sich möglichst geringe Missverhältnisse zwischen den
Substraten und den aufgebrachten Graphenlagen ergaben.[239] Als Nanoindenter wurden starre,
inerte Kugeln mit Radien von 15 und 30 Å und glatter Oberfläche verwendet. Die Nanoindenter
wurden mit flächenzentrierten Platten mit (111) Oberflächen und einem Nächste-Nachbar
Abstand von 1.5 Å modelliert, deren z-Koordinate so modifiziert wurde, dass die Atome eine
Kugeloberfläche beschrieben. Wechselwirkungen zwischen den Substrat- und den
Graphenatomen wurden mit einem gescreenten Bond-Order-Potential[240,241] modelliert,
Wechselwirkungen zwischen den Nanoindenter- und den Graphen-Substratatomen mit einem
rein repulsiven Lennard-Jones Potential.
Bei den Simulationen der tribologischen Eigenschaften wurden die Nanoindenter mit
konstanter Höhe parallel zu den Substratoberflächen bewegt. Die resultierende Reibkräfte
wurden gegen die Normalkräfte im Fall der Referenzprobe (Ref. SiC) und mit Graphen
bedeckte SiC (GSiC) in Abbildung 81 aufgetragen.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 122 -
0 100 200 300 400 500 600 700
0
20
40
60
80
100
120
Reib
kra
ft [
nN
]
Normalkraft [nN]
Ref. SiC
GSiC
Abbildung 81: Reibkräfte aufgetragen gegen die Normalkräfte beim Kratzen eines Nanoindenters über
eine unbedeckte (Ref. SiC) und eine mit Graphen bedeckte (GSiC) SiC-Oberfläche.
Dabei zeigte sich, dass eine Bedeckung der Oberflächen mit Graphen in der Lage ist, die
Reibkräfte über einen weiten Bereich hinweg deutlich zu verringern, was den Ergebnissen der
experimentellen Untersuchungen entspricht. Abhängig von der gewählten Normalkraft
reduzierte die Bedeckung die Reibkräfte teilweise auf weniger als 50 % des Ausgangswertes.
Im Falle unbedeckter Substratoberflächen ließen sich drei verschiedene Bereiche unterscheiden
(siehe Abbildung 82).
Abbildung 82: Reibkräfte und Modellbilder der Simulationen mit unbedeckten SiC-Oberflächen
(Ref. SiC). Links: Reibkräfte, aufgetragen gegen die Normalkräfte und ein Modellbild aus dem
elastischen Bereich (I), Phasenumwandlung (II) und Amorphisierung im Substrat (III).

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 123 -
Bei geringen Eindringtiefen (Bereich I) verformten sich unbedeckte Substrate rein elastisch und
es traten nur sehr geringe Reibkräfte auf. Bei höheren Eindringtiefen (Bereich II) stellte sich in
der Reibspur lokal ein Phasenübergang von der ursprünglichen kubischen Kristallstruktur zu
einer hexagonalen Struktur ein. Eine genauere Klassifizierung der entstehenden Phase war nicht
möglich, da diese lediglich eine Dicke von etwa zwei Atomlagen aufwies. Beim Übergang vom
Bereich rein elastischer Deformation zum Bereich des Phasenübergangs (I → II) erhöhten sich
die Reibkräfte sprunghaft und stiegen mit wachsender Eindringtiefe deutlich an. Mit
weiterwachsender Eindringtiefe stellte sich schließlich die plastische Deformation des Substrats
ein (Bereich III). In diesem Bereich war nochmals ein deutlicher Anstieg der Reibkräfte mit
wachsender Eindringtiefe zu beobachten. Im Gegensatz dazu ließen sich bei der Untersuchung
graphenbedeckter Oberflächen lediglich zwei verschiedene Bereiche voneinander
unterscheiden (siehe Abbildung 83).
Abbildung 83: Reibkräfte und Modellbilder der Simulationen mit Graphen bedeckten SiC-Oberflächen
(GSiC). Oben: Reibkräfte, aufgetragen gegen die Normalkräfte und ein Modellbild aus dem elastischen
Bereich (I), unten: Amorphisierung im Substrat und lokale Rissbildung im Graphen (II).
Bei geringen Eindringtiefen (Bereich I) war eine rein elastische Deformation der Substrate zu
beobachten. Die auftretenden Reibkräfte waren bei sehr geringen Eindringtiefen geringfügig
höher, als bei unbedeckten Substraten (siehe Abbildung 82). In einem weiteren, deutlich davon
abgesetzten Bereich (Bereich II) kam es zu Amorphisierungen im Substrat, zur Bildung lokal

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 124 -
begrenzter Defekte im Graphen und zur Ausbildung kovalenter Bindungen zwischen den
Kohlenstoffatomen des Graphens und den Substratatomen. In diesem Bereich war ein deutlich
stärkerer Anstieg der Reibkräfte mit den Normalkräften zu beobachten, die Reibkräfte blieben
jedoch auch nach Einsetzen der Rissbildung im Graphen zunächst unterhalb derer, die bei
unbedeckten Oberflächen beobachtet wurden (siehe Abbildung 83 II). Mit zunehmender
Eindringtiefe nach der Bildung der ersten Risse näherten sich die Reibkräfte denen an, die bei
unbedeckten Oberflächen beobachtet wurden. Eine Umwandlung der Kristallstruktur, wie im
Falle unbedeckter Substrate, war hier nicht zu beobachten. Die beobachteten Effekte können
mit der Steifigkeit des Graphens und mit der Bildung kovalenter Bindungen zwischen Graphen
und Substrat erklärt werden. Diese unterdrücken einerseits die Phasenumwandlung in der
Reibspur und führen andererseits dazu, dass das Graphen nach der Bildung erster Risse nicht
sofort vollständig zerreißt und einen Teil seiner Tragfähigkeit behält.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 125 -
4.8 Sintern mittels SPS-Verfahren
Das SPS-/FAST-Verfahren (Spark Plasma Sintern/Feld-Aktiviertes Sintern) ist eine neue und
innovative Sintertechnologie, die bei der Verarbeitung zahlreicher Materialien zunehmend an
Bedeutung gewinnt.[242,243] Das SPS-Verfahren basiert auf einer modifizierten Heißpresstechnik
und erfolgt im Wesentlichen durch Gleichstrom, der mit hoher Stromstärke und niedriger
Spannung durch das Presswerkzeug und durch den Sinterkörper geleitet wird. Durch den
ohmschen Widerstand des Sinterkörpers wird die elektrische Leistung des Stroms in
Wärmeleistung umgesetzt, wodurch der Sinterkörper aufgeheizt wird.[242,244] Somit wird die
Probe von innen heraus erwärmt und nicht wie beim konventionellen Sinterverfahren von außen.
Das SPS-Verfahren ermöglicht eine vollständige Verdichtung der Werkstoff-Komposite unter
Einstellung spezieller, mittels anderer Sinterverfahren nicht realisierbarer Werkstoff-
Eigenschaften. Durch das direkte Aufheizen von Werkstoffen beim SPS können deutlich
schnellere Sinterzyklen, mit verbesserten Werkstoffeigenschaften realisiert werden als bei
konventionellen Sinterverfahren mit indirektem Beheizen der Proben. Voraussetzung hierfür
ist, dass der Pressling bereits einen ausreichend niedrigen Widerstand (R) besitzt, um eine hohe
elektrische Leistung (P = U2/R) bei gleicher elektrischer Spannung (U) zu gewährleisten und
somit ein effizientes Aufheizen zu ermöglichen.[245,246]
Siliciumcarbid besitzt Halbleiter-Eigenschaften. Je nach Kristallstrukturen liegt die Bandlücke
zwischen 2.4 und 3.4 eV.[221–223] Abhängig von der Sintermethode, Sinterhilfsmittel und
Kristallstrukturen liegt die elektrische Leitfähigkeit von reinem SiC im Bereich von 10-2 bis
10-15 S∙cm-1.[224–226] Wegen seinen extrem hohen spezifischen Durchgangswiderstand ist die
Verwendung des SPS-Verfahrens bei reinem SiC unmöglich. Bei den wenigen
literaturbekannten „SPS-Verfahren“ zur Herstellung von SiC-Graphen-Nanokompositen
handelt es sich strenggenommen um „schnelle Heißpressungen“, da der Strom in diesen Fällen
nicht durch die hochwiederständige Pulververschüttung, sondern durch die leitfähige Matrize
oder Graphitfolie fließt.[133,136,152,173,238] Ein Strom durch die hochohmige Pulververschüttung
könnte zwar durch Anlegung einer starken Spannung realisiert werden, dies ist aber nicht
anzustreben, da davon auszugehen ist, dass dabei die Durchschlagspannung des Materials

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 126 -
überschritten wird und es dem zufolge zu Spannungsdurchschlag kommt, was sowohl die Probe
als auch die Apparatur beschädigen kann.[247,248] Aus diesem Grund wird die innovative SPS-
/FAST-Technik für die Herstellung von SiC-Werkstoffen nicht im industriellen Maßstab
angewendet. Gelingt es dagegen, mittels des Graphenzusatzes bereits bei Raumtemperatur eine
ausreichende elektrische Leitfähigkeit in der SiC-Pulverschüttung zu erzeugen, so sollte das
Verfahren vorteilhaft auch im industriellen Maßstab zur Herstellung von vollständig dichten
SiC-Bauteilen mit exzellenten mechanischen Eigenschaften einsetzbar sein.
Wie bereits in dem Kapitel 4.6 beschreiben wird durch Beimischung von Graphen wurde die
elektrische Leitfähigkeit von SiC stark erhöht. Deshalb wurde die Möglichkeit zur Herstellung
von SiC-Graphen-Nanokomposite mittel SPS-Verfahren in diesem Kapitel untersucht. Die
Eignung für den SPS-Prozess wurde mittels der in Abbildung 84 dargestellten Versuchsanlage
in Zusammenarbeit mit M. KNOCH von der Firma FCT Ingenieurkeramik GmbH in Sonneberg
untersucht.
Abbildung 84: Versuchsaufbau für SPS-Sinterung.
Hierbei wurde das Pulver zwischen zwei gut leitfähigen Graphitstempeln (Ø 60 mm) mit einem
Druck von 60 MPa in einer Si3N4-Matrize verdichtet. An den Stempeln wurde ein Strom mit
einer elektrischen Spannung von maximal 10 V angelegt. Wenn der Pressling eine ausreichend

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 127 -
hohe Leitfähigkeit bzw. einen niedrigen Widerstand aufweist, kommt es zur Erwärmung des
Materials. Aus Vorversuchen war bekannt, dass der spezifische Widerstand maximal 20 Ω cm
betragen darf, damit überhaupt eine Erwärmung erkennbar wird. Die Tabelle 12 zeigt hierzu
die Versuchsergebnisse des Erwärmungsvermögens der SiC-Graphen-Pulvermischungen mit
TRGO-600, MG-Ethanol, MG-CO2-48h, GSiC-F am Beispiel unterschiedlicher
Füllstoffgehalten für SPS-Untersuchungen.
Tabelle 12: Erwärmungsvermögen der SiC-Graphen-Pulvermischungen für SPS-Untersuchungen. Die
Temperatur 2050 °C wurde kenngezeichnet, da sie für eine erfolgreiche Sinterung erforderlich ist.
Probe
Graphengehalt [Gew.-%]
2 3 4 5 8
Rsp
[Ω cm]
Tmax
[°C]
Rsp
[Ω cm]
Tmax
[°C]
Rsp
[Ω cm]
Tmax
[°C]
Rsp
[Ω cm]
Tmax
[°C]
Rsp
[Ω cm]
Tmax
[°C]
TRGO-600 20 82 5 200 - 2050 - - - -
MG-Ethanol >20 RT - - - - >20 RT - -
MG-CO2-48h >20 RT >20 RT - - - - - -
GSiC-F >20 RT - - >20 RT - - 10 169
Anhand der aufgetragenen Daten in Tabelle 12 ist zu erkennen, dass bei den SPS-Versuchen
lediglich die TRGO-600-haltigen SiC-Nanokomposite eine Temperaturerhöhung induziert
wurde und somit die beste Eignung für den SPS-Prozess zeigten. Bei Zugabe von 2 Gew.-%
Graphen wurde nur die Probe TRGO-600 bis 80 °C aufgeheizt. Mit 3 Gew.-% desselben
Füllstoffs stieg die Temperatur bis 200 °C. Bei einem Füllgrad von 4 Gew.-% TRGO-600
erwärmte sich die Probe auf bis zu 2050 °C, sodass der Sinterprozess von SiC ermöglicht
werden kann. Bei den anderen Proben wurde wegen dem hohen elektrischen Widerstand
(> 20 Ω cm) keine Temperaturänderung beobachtet. Erst ab 8 Gew.-% Füllstoffgehalt wurde
bei der Probe GSiC-F eine detektierbare Temperaturerhöhung auf 170 °C gefunden, welche
allerdings noch immer nicht ausreicht um einen erfolgreichen SPS-Prozess gewährleisten zu
können. Der deutlich niedrigere elektrische Widerstand von der TRGO-gefüllten Probe lässt
sich auf ihr hohes Aspektverhältnis zurückführen, welches die Erzeugung leitfähiger
Netzwerkstrukturen mit geringerem Füllstoffbedarf ermöglicht.[237]

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 128 -
Durch das direkte Aufheizen des SiC-TRGO-Nanokomposits durch SPS wurden deutlich
schnellere Sinterzyklen als bei konventionellen Sinterverfahren mit indirekter Beheizung der
Proben realisiert. Dabei wurden die Zeitaufwände der Aufheizung und Sinterung von 25 h bzw.
4 h auf 35 min bzw. 5 min stark reduziert, was die Herstellungskosten deutlich reduziert und
gleichzeitig die Anwendung der SiC-Werkstoffe erweitert.
Die auf diese Weise erhaltenen SiC-TRGO-600-Nanokomposite wurden daraufhin auf ihre
Dichte bzw. Porosität, mechanischen (Biegefestigkeit), elektrischen und tribologischen
Eigenschaften untersucht. Des Weiteren wurden sie mit der Referenzprobe SiC und den
TRGO-600-haltigen SiC-Nanokomposite, welche nach Standard Sinterverfahren (SSiC)
hergestellten wurden, verglichen. Ein Vergleich mit der SPS-gesinterten Referenzprobe konnte
nicht durchgeführt werden, da reines SiC keine Eignung für den SPS-Prozess zeigte. Die
erhaltene Ergebnisse sind in Abhängigkeit des Sinterverfahrens in Tabelle 13 aufgeführt, und
sind im Folgenden graphisch ausgewertet.
Tabelle 13: Eigenschaften der SiC-TRGO-Nanokomposite nach dem Standard(SSiC)- und SPS-
Sinterprozess.
Probe Sintermethode
SSiC/SPS
Gehalt
[Gew.-%]
Relative Dichte
[%]
σsp
[S∙cm-1]
Biegefestigkeita
[MPa] µb
Ref. SiC SSiC 0 99.9 3.4 ∙ 10-8 505 ± 65 0.19
TRGO-600 SSiC 2 98.5 3.8 ∙ 10-7 360 ± 52 0.15
TRGO-600 SSiC 4 89.9 n. b. 201 ± 17 n. b.
TRGO-600 SPS 4 99.1 6.0 358 ± 38 0.10
a 4-Punkt-Biegefestigkeit; b Reibungskoeffizient nach 4 h Stift-Scheibe-Tribotest.
Wie bereits erklärt ist die Dichte eine der wichtigsten Eigenschaften bei der Entwicklung von
keramischen Werkstoffen, da sie Aussagen über den Zustand von hergestellten Komponenten
nach der Sinterung erlaubt.[2] Bei unzureichender Verdichtung entsteht Porosität, welche einen
signifikanten Einfluss auf die Eigenschaften keramischer Werkstoffen hat.[214] Die Dichte sowie
die resultierende Porosität der TRGO-Nanokomposite nach dem SPS-Prozess werden in
Abbildung 85 dargestellt und mit den nach Standard-Sinterprozess (SSiC) hergestellten
Nanokompositen verglichen.
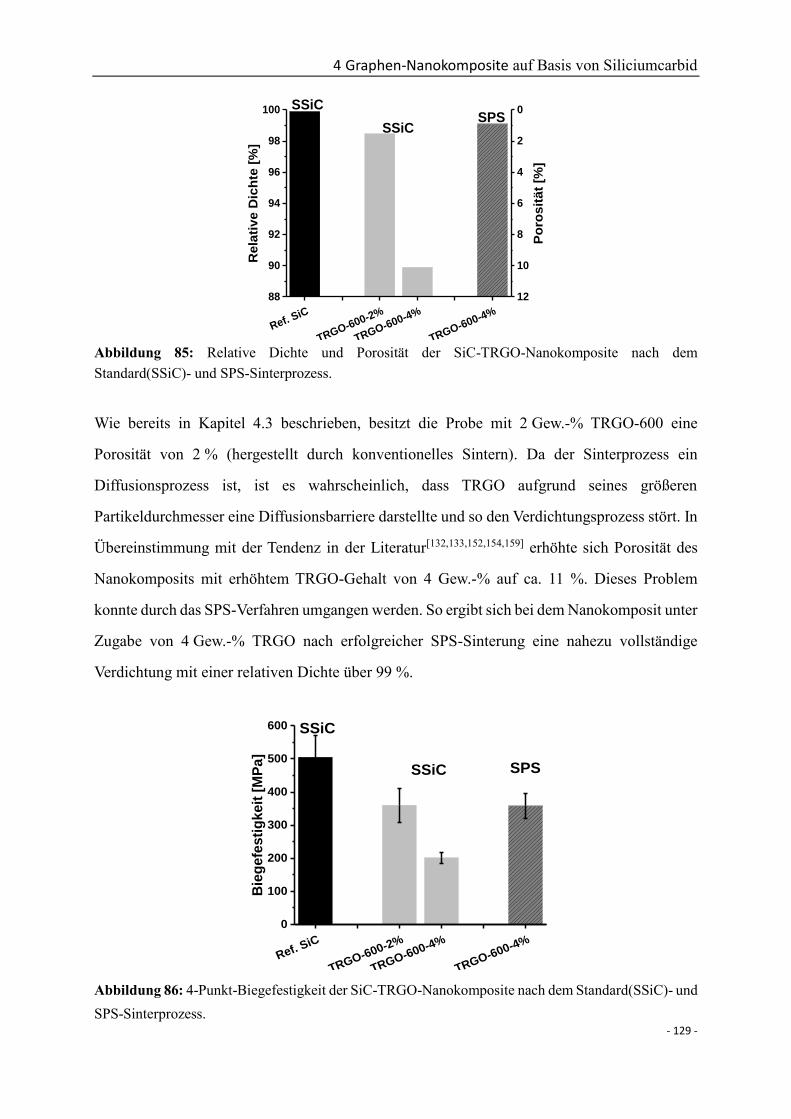
4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 129 -
Ref. SiC
TRGO-600-2%
TRGO-600-4%
TRGO-600-4%
88
90
92
94
96
98
100
SSiC
Re
lati
ve
Dic
hte
[%
]
SPSSSiC
12
10
8
6
4
2
0
Po
ros
itä
t [%
]
Abbildung 85: Relative Dichte und Porosität der SiC-TRGO-Nanokomposite nach dem
Standard(SSiC)- und SPS-Sinterprozess.
Wie bereits in Kapitel 4.3 beschrieben, besitzt die Probe mit 2 Gew.-% TRGO-600 eine
Porosität von 2 % (hergestellt durch konventionelles Sintern). Da der Sinterprozess ein
Diffusionsprozess ist, ist es wahrscheinlich, dass TRGO aufgrund seines größeren
Partikeldurchmesser eine Diffusionsbarriere darstellte und so den Verdichtungsprozess stört. In
Übereinstimmung mit der Tendenz in der Literatur[132,133,152,154,159] erhöhte sich Porosität des
Nanokomposits mit erhöhtem TRGO-Gehalt von 4 Gew.-% auf ca. 11 %. Dieses Problem
konnte durch das SPS-Verfahren umgangen werden. So ergibt sich bei dem Nanokomposit unter
Zugabe von 4 Gew.-% TRGO nach erfolgreicher SPS-Sinterung eine nahezu vollständige
Verdichtung mit einer relativen Dichte über 99 %.
Ref. SiC
TRGO-600-2%
TRGO-600-4%
TRGO-600-4%
0
100
200
300
400
500
600
SSiC
Bie
gefe
sti
gkeit
[M
Pa]
SPS
SSiC
Abbildung 86: 4-Punkt-Biegefestigkeit der SiC-TRGO-Nanokomposite nach dem Standard(SSiC)- und
SPS-Sinterprozess.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 130 -
Je nach Einsatzgebiet der SiC-Werkstoffe sind die mechanischen Eigenschaften wichtige
Parameter. Eine weitere Möglichkeit der Untersuchung dieser Materialien bietet die 4-Punkt-
Biegefestigkeit. Die hierbei gewonnenen Ergebnisse der TRGO-Nanokomposite, die über den
SPS-Prozess dargestellt wurden, werden in Abbildung 86 mit SSiC, welches mit dem
Standardprozess dargestellt wurde, verglichen.
Wie bereits in Kapitel 4.5 beschrieben, wurden durch den Einsatz von TRGO Fehlstellen und
Agglomerate induziert, welche die mechanischen Eigenschaften stark negativ beeinflusst. Das
SiC-Nanokomposit mit 2 Gew.-% TRGO-600 zeigte nach Standardsinterung eine Reduzierung
der Biegefestigkeit um 30 % auf 360 MPa. Durch Erhöhung des Graphenanteils auf 4 Gew.-%
wurde die Biegefestigkeit weiter auf 201 MPa reduziert. Das Absenken der Biegefestigkeit
konnte durch das SPS-Verfahren reduziert werden. Das Nanokomposit mit 4 Gew.-%
TRGO-600 zeigte nach erfolgreicher SPS-Sinterung eine Biegefestigkeit von 360 MPa, was
einer Erhöhung der Biegefestigkeit um etwa 80 % im Vergleich zu dem Nanokomposit nach
Standardsinterung (SSiC) entspricht.
Ref. SiC
TRGO-600-2%
TRGO-600-4%
10-8
10-7
10-6
10-5
10-4
10-3
10-2
10-1
100
101
SSiC
SPS
SSiC
Ele
ktr
isch
e L
eit
fäh
igkeit
[S
. cm
-1]
Abbildung 87: Elektrische Leitfähigkeit der SiC-TRGO-Nanokomposite nach dem Standard(SSiC)-
und SPS-Sinterprozess.
Je nach den Sintermethoden, Sinterhilfsmitteln und Kristallstrukturen liegt die elektrische
Leitfähigkeit von reinem SiC im Bereich 10-2 und 10-15 S∙cm-1.[224–226] Der hohe elektrische
Widerstand von SiC schränkt einerseits die Verwendung von innovativem
Bearbeitungsverfahren wie das Funkenerodieren, andererseits die Anwendung in der Sensorik

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 131 -
(z. B. Abgassensorik) und Brennstoffzellen ein.[1,124,175–177] Wie bereits in Kapitel 4.6
beschrieben, konnte durch Zugabe von Graphen die elektrische Leitfähigkeit stark erhöht
werden. Die elektrischen Leitfähigkeiten der TRGO-Nanokomposite, die mittels SPS-Prozess
dargestellt wurden, werden in Abbildung 87 dargestellt und mit der nach Standard-
Sinterprozess hergestellten Probe (SSiC) verglichen.
Wie erwartet zeigte die Referenzprobe nach Standardsinterung eine sehr geringe spezifische
Leitfähigkeit von 3 ∙ 10-8 S∙cm-1, welche durch Zugabe von 2 Gew.-% TRGO-600 auf
4 ∙ 10-7 S∙cm-1 leicht erhöht werden konnte. Die elektrische Eigenschaft konnte durch das SPS-
Verfahren noch wesentlich verbessert werden. So zeigte das Nanokomposit mit 4 Gew.-%
TRGO-600 nach erfolgreicher SPS-Sinterung eine Erhöhung der spezifischen Leitfähigkeit um
acht Größenordnungen auf 6 S∙cm-1. Ein Vergleich mit dem Nanokomposit mit 4 Gew.-%
TRGO-600 (SSiC), wurde wegen seiner zu hohen Porosität und seinen zu schlechten
mechanischen Eigenschaften nicht durchgeführt.
Die tribologischen Eigenschaften des hergestellten SiC-Nanokomposits wurden mittels Stift-
Scheibe-Tribometer analysiert. Das erhaltene Reibungsverhalten und Verschleißvolumen des
TRGO-Nanokomposits nach SPS-Prozess werden in Abbildung 88 dargestellt und mit den
Daten der nach Standard-Sinterprozess erhaltenen Probe (SSiC) verglichen.
0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,00,00
0,05
0,10
0,15
0,20
0,25
0,30
Re
ibu
ng
sk
oe
ffiz
ien
t
Zeit [h]
Ref. SiC (SSiC)
TRGO-600-2% (SSiC)
TRGO-600-4% (SPS)
Ref. SiC
TRGO-600-2%
TRGO-600-4%
0,00
0,05
0,10
0,15
0,20
0,25 SSiC
Ve
rsc
hle
ißv
olu
me
n [
mm
3]
SPSSSiC
Abbildung 88: Reibungskoeffizient-Verlauf (links) und Verschleißvolumen (rechts) nach 4 h Stift-
Scheibe-Tribotest von SiC-TRGO-Nanokomposite nach dem Standard(SSiC)- und SPS-Sinterprozess.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 132 -
Aus Abbildung 88 (links), welche den Reibungskoeffizienten (µ) in Abhängigkeit von der
Belastungsdauer zeigt, sind die gemessenen Reibverhalten der Nanokomposite TRGO-600-2%
(SSiC) und TRGO-600-4% (SPS) ersichtlich. Wie bereits in Kapitel 4.7.1 beschrieben, konnten
die Reibungskoeffizienten der SiC-Werkstoffe durch Beimischung von Graphen reduziert
werden. Nach 4 h Untersuchung zeigte die Referenzprobe (SSiC) einen Reibungskoeffizienten
von 0.19, welche durch Einarbeitung von 2 Gew.-% TRGO-600 auf 0.15 reduziert werden
konnte. Die tribologischen Eigenschaften konnten durch das SPS-Verfahren weiter verbessert
werden. Im Vergleich dazu zeigte das Nanokomposit mit 4 Gew.-% TRGO-600 nach
erfolgreicher SPS-Sinterung eine Reduzierung des Reibungskoeffizienten um 50 % auf 0.10.
Nach den tribologischen Untersuchungen wurden die generierten Verschleißvolumina der Stifte
ermittelt und in Abbildung 88 (rechts) dargestellt. Im Hinsicht auf die Verschleißbeständigkeit
zeigte der Einsatz von SPS-Verfahren eine deutliche Verbesserung im Vergleich zur
Standardsinterung. Die Referenzprobe und das mit 2 Gew.-% TRGO-600 gefüllte
Nanokomposit zeigen nach Standardsinterung ähnliche Verschleißvolumina, während das
Verschleißvolumen von dem mit 4 Gew.-% TRGO-600 gefüllten Nanokomposit um etwa 30 %
auf 0.16 mm3 reduziert wird. Aufgrund der hohen Porosität und der daraus folgenden schlechten
mechanischen Eigenschaften des Nanokomposits mit 4 Gew.-% TRGO-600, welches durch den
Standardsinterprozess dargestellt wurde, war eine vergleichende tribologische Untersuchung
derselben nicht möglich.
Zusammenfassend wurde SiC-Nanokomposit durch Zugabe von TRGO-600 (4 Gew.-%)
erstmals mittels SPS-Verfahren hergestellt, ohne dabei auf leitfähige Matrizen oder
Graphitfolien zurückzugreifen. Hierbei wurde die Verarbeitungszeit stark verkürzt. Das
erhaltene Keramiknanokomposit erreichte eine nahezu vollständige Verdichtung, gute
mechanische Eigenschaften und deutlich höhere Leitfähigkeit sowie einen reduzierten
Reibungskoeffizienten mit verbesserter Verschleißbeständigkeit als die konventionell
gesinterte Variante. Somit besteht durch Zugabe von TRGO die Chance, SiC-Nanokomposite
zu entwickeln, die im SPS-Prozess schneller und effizienter gesintert werden können.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 133 -
4.9 Zusammenfassung
In diesem Kapitel wurden SiC-Graphen-Nanokomposite mit verschiedenen Graphenen, welche
in Kapitel 3 diskutiert wurden, hergestellt. Der Einfluss der Füllstoffe auf die elektrischen,
mechanischen und tribologischen Eigenschaften der SiC-Werkstoffe wurde untersucht.
Da die Beimischung von Graphen hohe Porositäten (bis zu 7 %) verursachen kann, wurden die
meisten Werkstoffvarianten mit einem Füllstoffgehalt von 2 Gew.-% hergestellt. Die typischen
Eigenschaften der ausgewählten Nanokomposite mit GSiC-F, TRGO-1000, dem
funktionalisierten MG-CO2-120h und dem unfunktionalisierten MG-Ethanol wurden in
Tabelle 14 aufgelistet und in Abbildung 89 graphisch dargestellt.
94
98
96
100
300
400
500
2,6
2,8
3,0
3,2
10-7
10-5
10-3
10-1
0,18
0,16
0,14
0,12
0,24
0,21
0,18
0,15
Verschleiß
[mm3]
Reibungskoeffizient
Elektrische Leitfäigkeit [S.cm-1]
Bruchzähigkeit
[MPa.m0.5]
Biegefestigkeit
[MPa]
Relative Dichte [%]
GSiC-F
MG-CO2-120h
MG-Ethanol
TRGO-1000
Ref. SiC
Abbildung 89: Eigenschaftsprofil der gesinterten SiC-Nanokomposite mit jeweils 2 Gew.-% GSiC-F,
MG-CO2-120h, MG-Ethanol und TRGO-1000.
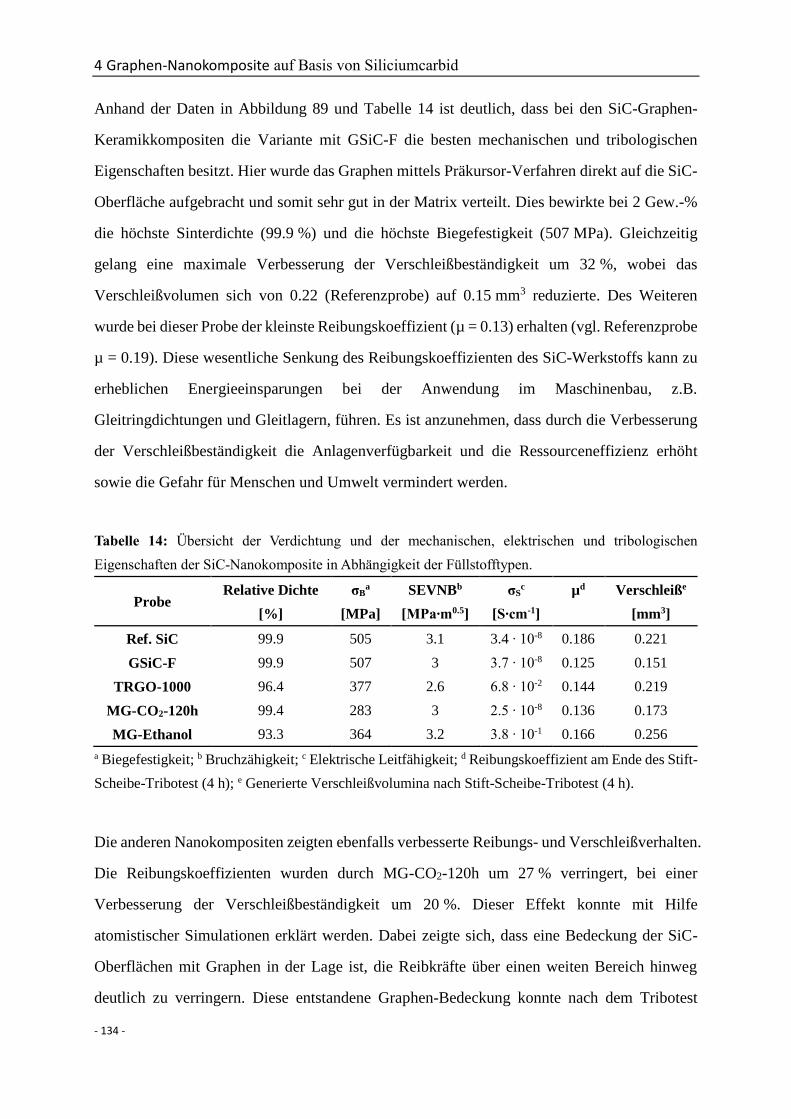
4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 134 -
Anhand der Daten in Abbildung 89 und Tabelle 14 ist deutlich, dass bei den SiC-Graphen-
Keramikkompositen die Variante mit GSiC-F die besten mechanischen und tribologischen
Eigenschaften besitzt. Hier wurde das Graphen mittels Präkursor-Verfahren direkt auf die SiC-
Oberfläche aufgebracht und somit sehr gut in der Matrix verteilt. Dies bewirkte bei 2 Gew.-%
die höchste Sinterdichte (99.9 %) und die höchste Biegefestigkeit (507 MPa). Gleichzeitig
gelang eine maximale Verbesserung der Verschleißbeständigkeit um 32 %, wobei das
Verschleißvolumen sich von 0.22 (Referenzprobe) auf 0.15 mm3 reduzierte. Des Weiteren
wurde bei dieser Probe der kleinste Reibungskoeffizient (µ = 0.13) erhalten (vgl. Referenzprobe
µ = 0.19). Diese wesentliche Senkung des Reibungskoeffizienten des SiC-Werkstoffs kann zu
erheblichen Energieeinsparungen bei der Anwendung im Maschinenbau, z.B.
Gleitringdichtungen und Gleitlagern, führen. Es ist anzunehmen, dass durch die Verbesserung
der Verschleißbeständigkeit die Anlagenverfügbarkeit und die Ressourceneffizienz erhöht
sowie die Gefahr für Menschen und Umwelt vermindert werden.
Tabelle 14: Übersicht der Verdichtung und der mechanischen, elektrischen und tribologischen
Eigenschaften der SiC-Nanokomposite in Abhängigkeit der Füllstofftypen.
Probe Relative Dichte
[%]
σBa
[MPa]
SEVNBb
[MPa∙m0.5]
σSc
[S∙cm-1]
µd
Verschleiße
[mm3]
Ref. SiC 99.9 505 3.1 3.4 ∙ 10-8 0.186 0.221
GSiC-F 99.9 507 3 3.7 ∙ 10-8 0.125 0.151
TRGO-1000 96.4 377 2.6 6.8 ∙ 10-2 0.144 0.219
MG-CO2-120h 99.4 283 3 2.5 ∙ 10-8 0.136 0.173
MG-Ethanol 93.3 364 3.2 3.8 ∙ 10-1 0.166 0.256
a Biegefestigkeit; b Bruchzähigkeit; c Elektrische Leitfähigkeit; d Reibungskoeffizient am Ende des Stift-
Scheibe-Tribotest (4 h); e Generierte Verschleißvolumina nach Stift-Scheibe-Tribotest (4 h).
Die anderen Nanokompositen zeigten ebenfalls verbesserte Reibungs- und Verschleißverhalten.
Die Reibungskoeffizienten wurden durch MG-CO2-120h um 27 % verringert, bei einer
Verbesserung der Verschleißbeständigkeit um 20 %. Dieser Effekt konnte mit Hilfe
atomistischer Simulationen erklärt werden. Dabei zeigte sich, dass eine Bedeckung der SiC-
Oberflächen mit Graphen in der Lage ist, die Reibkräfte über einen weiten Bereich hinweg
deutlich zu verringern. Diese entstandene Graphen-Bedeckung konnte nach dem Tribotest

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 135 -
anhand von AFM-Aufnahmen nachgewiesen werden. Weiterhin wurde für die Nanokomposite
mit TRGO-1000 und MG-Ethanol eine wesentliche Zunahme der elektrischen Leitfähigkeit von
3 ∙ 10-8 auf 0.07 S∙cm-1 (+6 Größenordnungen) bzw. 0.38 S∙cm-1 (+7 Größenordnungen)
beobachtet, welche aufgrund ihrer großen Plättchenstruktur leitfähige Netzwerkstruktur leichter
bilden konnten. Trotz der tribologischen und elektrischen Eigenschaften sind diese Füllstoffe
aufgrund der schlechteren Verteilung der Graphen-Partikel in der SiC-Matrix und der daraus
folgenden geringeren Matrix-Anbindung weniger für die Herstellung von SiC-Graphen-
Nanokompositen geeignet. Die Einarbeitung dieser Graphene führte zu unvollständiger
Verdichtung und in der Folge zu schlechten mechanischen Eigenschaften. Dabei sank die
Biegefestigkeit durch die Zugabe von MG-CO2-120h von 505 auf 283 MPa und gefolgt von
MG-Ethanol (364 MPa) und TRGO-1000 (377 MPa).
Aufgrund der hervorragenden Eigenschaften der GSiC-Proben wurde der Graphengehalt auf
bis zu 8 Gew.-% erhöht. Trotz der vorteilhaften Herstellungsmethode sanken die Dichtewerte
der GSiC-Nanokomposite mit steigendem Füllstoffgehalt um bis zu 5 % und die mechanischen
Eigenschaften um bis zu 40 %.
Des Weiteren wurden aus den SiC-Graphen-Nanokompositen Gleitringe hergestellt und unter
anwendungsnahen Bedingungen untersucht. Durch die Einarbeitung von Graphen konnte eine
deutliche Verminderung des Reibungskoeffizienten gegenüber der graphenfreien
Referenzprobe (0.21) und den kommerziellen Typ F (0.23) und Typ G (0.20) der Firma 3M,
erzielt werden. Unter allen Werkstoffvarianten wiesen die GSiC-Proben die niedrigsten
Reibungskoeffizienten mit einer maximalen Reduzierung um bis zu 78 % auf 0.047 auf.
Darüber hinaus zeichneten sich die Nanokomposite mit 4 Gew.-% und 8 Gew.-% GSiC-F durch
eine sehr hohe Belastungsgrenze aus, welche eine tribologische Messung bei 700 N statt 250 N
ermöglichte. Damit könnte der Anwendungsbereich von SiC-Werkstoffen ausgeweitet werden,
wie z.B. in mechanischen Hochlastpumpen zur Treibstoffförderung in
Flüssigkeitsraketentriebwerken.

4 Graphen-Nanokomposite auf Basis von Siliciumcarbid
- 136 -
Durch das direkte Aufheizen von Werkstoffen beim SPS-Sinterverfahren können deutlich
schnellere Sinterzyklen mit verbesserten Werkstoffeigenschaften realisiert werden als bei
konventionellen Sinterverfahren mit indirektem Beheizen der Proben. Wegen seinen extrem
hohen spezifischen Durchgangswiderstand ist die Verwendung des SPS-Verfahrens bei reinem
SiC unmöglich. Durch Beimischung von Graphen konnte die elektrische Leitfähigkeit von SiC
stark erhöht werden und die Anwendung von SPS-Verfahren ermöglicht werden. Dabei zeigte
TRGO in SPS-Versuchen die beste Eignung. Deshalb wurde eine mit 4 Gew.-% TRGO-600
gefüllte Keramikprobe hergestellt, die nach erfolgreicher SPS-Verdichtung eine deutlich
niedrigere Porosität (0.9 %), als die konventionell gesinterte Variante (10.1 %) aufwies. Als
Folge erzielte das TRGO-Nanokomposit nach SPS-Sinterung eine höhere Biegefestigkeit
(358 MPa, +78 %) und niedrigeren Reibungskoeffizienten (0.10, -33 %) als die nach Standard-
Sinterung erhaltene Variante. Weiterhin wurde die elektrische Leitfähigkeit um
8 Größenordnung auf 6 S∙cm-1gegenüber reinem SiC (3 ∙ 10-8 S∙cm-1) erhöht. Ferner ermöglicht
das hohe elektrische Leitvermögen einerseits die Verwendung von innovativem
Bearbeitungsverfahren wie das Funkenerodieren, was eine Erweiterung der geometrisch
darstellbaren Formen und Bauteilgeometrien bedeuten wird, andererseits die Anwendung des
SiC-Werkstoffs in der Sensorik (z. B. Abgassensorik) und Brennstoffzellen.
Zusammenfassend zeigt die GSiC-Probe bei den SiC-Graphen-Nanokompositen mit Standard-
Sinterprozess die vielversprechendsten Eigenschaften. Die vorteilhafte Herstellungsmethode
von Graphen in GSiC führte zu einer sehr hohen Sinterdichte, guten mechanischen
Eigenschaften und damit auch zu herausragenden Reibungs- und Verschleißverhalten. Für das
SPS-Sinterverfahren stellte sich der Füllstoff TRGO-600 in SiC als vielversprechend heraus.
Das erhaltene Keramik-Nanokomposit erreichte wesentlich verbesserte
Werkstoffeigenschaften als die konventionell gesinterte Variante. Somit besteht durch Zugabe
von Graphen die Chance, Pulver zu entwickeln, die im SPS-Prozess schnell und effizient
gesintert werden können.

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 137 -
5 Graphen-Nanokomposite auf Basis von
Siliciumnitrid
5.1 Einleitung
Der Einsatz von keramischen Werkstoffen auf Basis von Siliciumnitrid (Si3N4) weist hohe
Produktionsmenge insbesondere im Bereich Gleit- und Wälzlager und als
Konstruktionswerkstoff im Maschinen-, Motoren- und Turbinenbau auf. Dies beruht auf seiner
hohen Steifigkeit (4.0 – 8.5 MPa·m0.5) und seinem geringen Wärmeausdehnungskoeffizienten
(3.0 – 3.5 ∙ 10-6 K-1) sowie seinem geringen spezifischen Gewicht (3.2 – 3.6 g/cm3) und der
Möglichkeit des Einsatzes unter Mangelschmierung oder Trockenlaufbedingungen. Des
Weiteren zeichnet es sich durch eine extrem hohe Härte, Korrosions- und
Thermoschockbeständigkeit sowie hohe Festigkeit (330 – 1000 MPa) auch bei hohen
Temperaturen (bis 1300 °C) aus.[1,2]
Um die Effizienz, Lebensdauer, Zuverlässigkeit und Leistungsfähigkeit von Si3N4-
Komponenten in den Anwendungen unter Wasser- bzw. Mangelschmierung zu verbessern,
werden jedoch leistungsfähigere Werkstoffe benötigt. Diese Materialien sollten eine Senkung
der Reibung ermöglichen, was zu erheblichen Energieeinsparungen führen kann. Durch
Erhöhung der Verschleißbeständigkeit und Bruchzähigkeit könnten die Lebensdauer und die
Zuverlässigkeit erhöht werden. Als Folge daraus ergibt sich eine bessere Ressourceneffizienz.
So wurde in den vergangenen Jahren verstärkt versucht, durch die Zugabe von
Kohlenstoffadditiven, z. B. Carbon Black, CNTs und Graphene, die mechanischen und
tribologischen Eigenschaften von Si3N4 zu verbessern.[131,132,179,180,249]
Mit Graphen konnten die mechanischen Eigenschaften von Si3N4-Werkstoffen wesentlich
verbessert werden. Die erste Publikation zu graphenverstärkter Si3N4-Keramik in 2011 zeigte
bereits, dass die Risszähigkeit erheblich gesteigert werden konnte.[130] Das mit Spark-Plasma-
Sintern (SPS) hergestellte Si3N4-Nanokomposit erreichte mit 1.5 Vol.-% TRGO eine
Risszähigkeit von 6.6 MPa·m0.5, welche einer Erhöhung von ca. 140 % im Vergleich zur

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 138 -
Referenzprobe (2.8 MPa·m0.5) entspricht. Bei den nach heißisostatischem Pressen (HIP)
gesinterten Si3N4-Nanokompositen konnte die Risszähigkeit durch Einlagerung von
1.0 Gew.-% Mehrlagengraphen von 7 auf bis zu 10 MPa·m0.5 verbessert werden. Laut der
Literatur wurden die Risse durch das Graphen abgelenkt, verzweigt oder überbrückt. Zudem
wurde auch eine wesentliche Verminderung des Verschleißes erreicht. Mit 3.0 Gew.-%
Mehrlagengraphen wurde der Verschleiß um ca. 60 % reduziert.[131,132] Die Si3N4-Graphen-
Nanokomposite wurden zwar intensiv erforscht liegt ein kombinatorischer Anstieg der
mechanischen und tribologischen Eigenschaften mit vollständiger Verdichtung durch Graphen
in Si3N4-Nanokompositen bisher nicht vor. Daher stellte die Erreichung dieser
Eigenschaftenkombinationen ein angestrebtes Ziel dieser Kapitel dar.
5.2 Herstellung
In dieser Arbeit wurden Si3N4-Graphen-Nanokomposite in Zusammenarbeit mit Dr. C. BALÁZSI
und Dr. K. BALÁZSI vom Institute for Technical Physics and Materials Science, Centre for
Energy Research in Budapest und Dr. C. SCHRÖDER, Dr. B. SCHLÜTER und Dr. A. KAILER vom
Fraunhofer-Institut für Werkstoffmechanik in Freiburg hergestellt und charakterisiert. Neben
GSi3N4-F (graphenbeschichtetes Si3N4 aus Furfurylalkohol), TRGO-600 (thermisch reduziertes
Graphitoxid) und MG-CO2-48h (mechanochemisch funktionalisiertes Multilagen-Graphen)
wurden MG-Ethanol (unfunktionalisiertes Multilagen-Graphen) sowie die kommerziell
erhältlichen Füllstoffe Grade C und Grade M als funktionelle Füllstoffe eingesetzt. Der Einfluss
der Füllstoffarten wurde im Hinblick auf die Funktionalität sowie die mechanischen und
tribologischen Eigenschaften von Si3N4-Graphen-Nanokompositen evaluiert. Das angestrebte
Ziel der Zugabe von Graphen ist die Verbesserung der mechanischen (Bruchzähigkeit und Härte)
und tribologischen (Reibung und Verschleiß) Eigenschaften der Si3N4-Werkstoffe im Vergleich
zur reinen Matrix. Damit soll die Schadenstoleranz und Energieeffizienz verbessert sowie die
Lebensdauer, Belastbarkeit und die Zuverlässigkeit erhöht werden.
Die Herstellung der Si3N4-Graphen-Nanokomposite erfolgte zunächst durch Mischung und
Homogenisierung des Graphens, der Sinterhilfsmittel und der Si3N4-Matrix in Ethanol per

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 139 -
Attritormühle. Nach Erhalt stabiler Dispersionen erfolgte die Rücktrocknung und Granulierung
der Probe. Anschließend wurden aus dem gewonnenen Granulat Probeplatten durch
heißisostatisches Pressen (HIP) bei 20 MPa Druck verdichtet und im Ofen bei 20 MPa Druck
und 1700 °C unter Stickstoff-Atmosphäre für 3 h gesintert (siehe Abbildung 90).
Abbildung 90: Flussdiagramm zur Herstellung von Si3N4-Graphen-Nanokomposite.
Zur besseren Vergleichbarkeit mit der Literatur[153,162,250–253] wurden die meisten
Nanokomposite mit einem Füllstoffanteil von 3.0 Gew.-% hergestellt. Bei Graphenanteilen über
3.0 Gew.-% ist aufgrund vermehrter Partikel-Partikel-Wechselwirkungen damit zu rechnen,
dass sich die Nanokomposite bei der Standardsinterung schlechter verdichten lassen und damit
die Restporosität im Bauteil zu hoch wird. In der Literatur führt dies zu schlechteren
mechanischen Eigenschaften.[132,133,152,154,159] Die Herstellung von graphenbeschichtetem Si3N4
(GSi3N4-F), bei der das Graphen über eine templatvermittelte Synthese aus Furfurylalkohol
ähnlich wie GSiC-F direkt auf der Oberfläche von Si3N4 hergestellt wurde (vgl. Kapitel 3.4.1),
war eine Bulk-Reaktion. Daher konnte der erhaltene Graphengehalt in GSi3N4-F nicht weiter
eingestellt werden. Aus diesem Grund wurde das GSi3N4-F-Nanomoposit mit einem
Füllstoffgehalt von 0.5 Gew.-% hergestellt.
Die auf diese Weise erhaltenen Si3N4-Graphen-Nanokomposite wurden daraufhin auf ihre
Dichte, Porosität, mechanischen und tribologischen Eigenschaften in den folgenden Kapiteln
untersucht. Zusätzlich erfolgte die Charakterisierung der Verteilung der Füllstoffe in der Si3N4-
Matrix mittels TEM-Aufnahmen. Anschließend wurden die Eigenschaften der hergestellten
Si3N4-Graphen-Nanokomposite mit der Literatur verglichen.

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 140 -
5.3 Dichte & Porosität
Dichte ist eine der wichtigsten Eigenschaften bei der Entwicklung von keramischen
Werkstoffen. Sie erlaubt Aussagen über den Zustand von hergestellten Komponenten nach der
Sinterung.[2] Bei unzureichender Verdichtung kommt es zu Porosität, welche einen
signifikanten Einfluss auf die Eigenschaften keramischer Werkstoffen hat.[214] Zur Beurteilung
der hergestellten Nanokomposite wurden diese Kennwerte mit Hilfe der archimedischen
Dichtemessung bestimmt. Die Dichte sowie resultierende Porosität der Si3N4-Nanokomposite
nach dem Sinterprozess wurden in Tabelle 15 aufgeführt und mit der Referenzprobe verglichen.
Zur besseren Vergleichbarkeit wurden die Dichtewerte auf die theoretisch erreichbare Dichte
bezogen.
Tabelle 15: Füllstoffabhängigkeit der Dichtewerte und Porosität von Si3N4-Nanokompositen, geordnet
nach sinkender Dichte.
Probe Füllstoffgehalt
[Gew.-%]
Dichte
[g/cm3]
Reindichtea
[g/cm3]
Relative Dichte
[%]
Porosität
[%]
Ref. Si3N4 0 3.225 3.295 97.9 2.1
GSi3N4-F 0.5b 3.201 3.287 97.4 2.6
MG-CO2-48h 3.0 3.029 3.251 93.2 6.8
Grade M 3.0 2.675 3.251 82.3 17.7
MG-Ethanol 3.0 2.589 3.251 79.6 20.4
TRGO-600 3.0 2.516 3.251 77.4 22.6
Grade C 3.0 2.412 3.251 74.2 25.8
a berechnet nach Gewichtsanteil und theoretischen Dichten von Si3N4, Sinteradditiven und Graphen; b bestimmt durch TGA-Messung. Luft-Atmosphäre, 10 K/min, 50 – 750 °C.
Die in Tabelle 15 dargestellten Si3N4-Graphen-Nanokomposite wiesen meistens niedrigere
relative Dichten im Bereich von 74 bis 93 % gegenüber reinem Si3N4 (97.9 %) auf, was den
Ergebnissen in der Literatur entspricht.[153,154] Die Gründe dafür sind vor allem die
unzureichende Homogenität sowie die schwachen Partikel-Matrix-Anbindungen und
Fehlstellen im Gefüge. Daher werden bei diesen Nanokompositen schlechtere mechanische
Eigenschaften als bei der Referenzprobe erwartet. Weiterhin ist die Einwirkung der
Funktionalisierung von Graphen auf die Eigenschaften der hergestellten Keramik-

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 141 -
Nanokomposite zu erkennen. Im Vergleich zu unfunktionalisiertem MG-Ethanol verfügt
MG-CO2-48h über hydrophile funktionelle Gruppen, welche mit dem Dispersionsmedium
Ethanol attraktiv wechselwirken konnten, was in einer Stabilisierung der Dispersionen
resultierte. Damit erzielte das Nanokomposit mit MG-CO2-48h eine bessere Verdichtung mit
viel geringerer Porosität (6.8 %) als die mit MG-Ethanol gefüllte Probe (20.4 %).
Dementgegen bildet die Probe GSi3N4-F die einzige Ausnahme. Sie weist eine ähnliche relative
Dichte (97.4 %) als das Referenzmaterial (97.9 %) auf. Der Unterschied deckte sich mit den
Dichtewerten der GSiC-F-Nanokomposite in Kapitel 4.3 und lässt sich vor allem mit dem
vorteilhaften Herstellungsverfahren von GSi3N4-F und zusätzlich mit der geringeren
Füllstoffmenge erklären. Die Verwendung von GSi3N4-F, bei dem Graphen über eine
templatvermittelte Reaktion direkt auf die Oberfläche von Si3N4 aufgebracht wurde,
gewährleistete eine deutlich homogenere Verteilung von Graphen in der Si3N4-Matrix. Dies
führte zu einer stärkeren Anbindung an das Si3N4 und hat zur Folge eine vollständigere
Verdichtung des Nanokomposits im Vergleich zu anderen Graphenen. Daher werden beim
GSi3N4-F-Nanokomposit bessere mechanische und tribologische Eigenschaften als bei den
restlichen Proben erwartet.
5.4 Morphologie
Die Homogenität der Füllstoffverteilung bestimmt im Wesentlichen die effiziente
Matrixverstärkung.[167] Bessere Dispergierung führt zu höherem Grenzflächenkontakt zwischen
dem Füllstoff und dem Matrixmaterial, was bessere Materialeigenschaften liefert.[194] Um einen
Einblick in die Füllstoffdispergierung sowie Kompatibilisierung der Füllstoffe in der Si3N4-
Matrix zu erhalten, wurde die Untersuchung der Morphologie mittels TEM-Aufnahmen
durchgeführt. Die Ergebnisse der morphologischen Charakterisierung der hergestellten
Nanokomposite sind im Folgenden dargestellt.

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 142 -
Abbildung 91: TEM-Aufnahmen des gesinterten a) reinen Si3N4 und b) GSi3N4-F-Nanokomposits.
Abbildung 91 zeigt TEM-Aufnahmen von der ungefüllten Referenzprobe und dem GSi3N4-F-
Nanokompoist. Beide weist eine ähnliche porenfreie Morphologie auf. Dies wurde durch die
vorteilhaften Herstellungsbedingungen von Graphen in GSi3N4-F und zusätzlich durch die
geringere Füllstoffmenge massiv begünstigt wurde. Dies wird anhand der höchsten relativen
Dichte (97.4 %) aller Nanokomposite in Kapitel 5.3 bestätigt. Dies deckte sich mit der
Morphologie der GSiC-F-Nanokomposite in Kapitel 4.4 und unterstützte die These, dass sich
das mit dem Beschichtungsverfahren hergestellte GSi3N4 im Vergleich zu den anderen
Graphenen offensichtlich vorteilig auswirkte, um die Bildung von Agglomeraten zu vermeiden
und eine homogenere Verteilung der Füllstoffe zu verwirklichen.
Im Gegensatz sind große Poren (helle Bereiche) bei allen anderen Nanokompositen im µm-
Bereich zu erkennen (siehe Abbildung 92). Die Graphene (mit Pfeilen kenngezeichnet) gingen
keine starke Wechselwirkung mit der Si3N4-Matrix ein und wiesen hauptsächlich eine
inhomogene Morphologie auf, welche auf einen unvollständigen Sinterprozess durch Zugabe
von Graphen hinweist. Dabei wurden bei den Nanokompositen mit Grade C, TRGO-600, MG-
Ethanol und Grade M ungefüllte Löcher identifiziert. Diese Ergebnisse stimmten mit den
Dichtewerten in Kapitel 5.3 überein, da diese Nanokomposite eine sehr hohe Porosität von über
17 % besaßen.

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 143 -
Abbildung 92: TEM-Aufnahmen der gesinterten Si3N4-Nanokomposite mit a) Grade C, b) TRGO-600,
c) unfunktionalisiertem MG-Ethanol, d) Grade M und e) funktionalisiertem MG-CO2-48h, geordnet
nach sinkender Porosität. Die Pfeile deuten auf Graphen-Agglomerate, während die hellen Bereiche
offene Poren darstellen.
Weiterhin geht aus den TEM-Aufnahmen der Nanokomposite mit MG-Ethanol (Abbildung 92 c)
und MG-CO2-48h (Abbildung 92 e) klar hervor, dass die Herstellungsbedingungen bzw. die
Funktionalisierungen von MG eine wesentliche Rolle in ihrer Füllstoffverteilung in der Si3N4-
Matrix spielten. Die größere Anzahl funktioneller Gruppen in den MG-CO2-Materialien
konnten eine bessere Kompatibilität und Verteilung in den Matrixmaterialien bewirken. So
waren im Nanokomposit mit MG-CO2-48h zwar auch Poren über 2 µm zu sehen, sie wurden
aber meist mit kleinen Graphen-Partikeln befüllt. Im Vergleich dazu waren in TEM-Aufnahmen
der mit unfunktionalisiertem MG-Ethanol gefüllten Si3N4-Nanokomposite offene Poren
erkennbar, welche nicht mit Graphen-Partikeln befüllt wurden. Dies bestätigte die Ergebnisse
aus den Dichte-Untersuchungen in Kapitel 5.3, dass das MG-CO2-48h-Nanokomposit deutlich
geringere Porosität (6.8 %) als die Variante mit MG-Ethanol (20.4 %) aufweist.
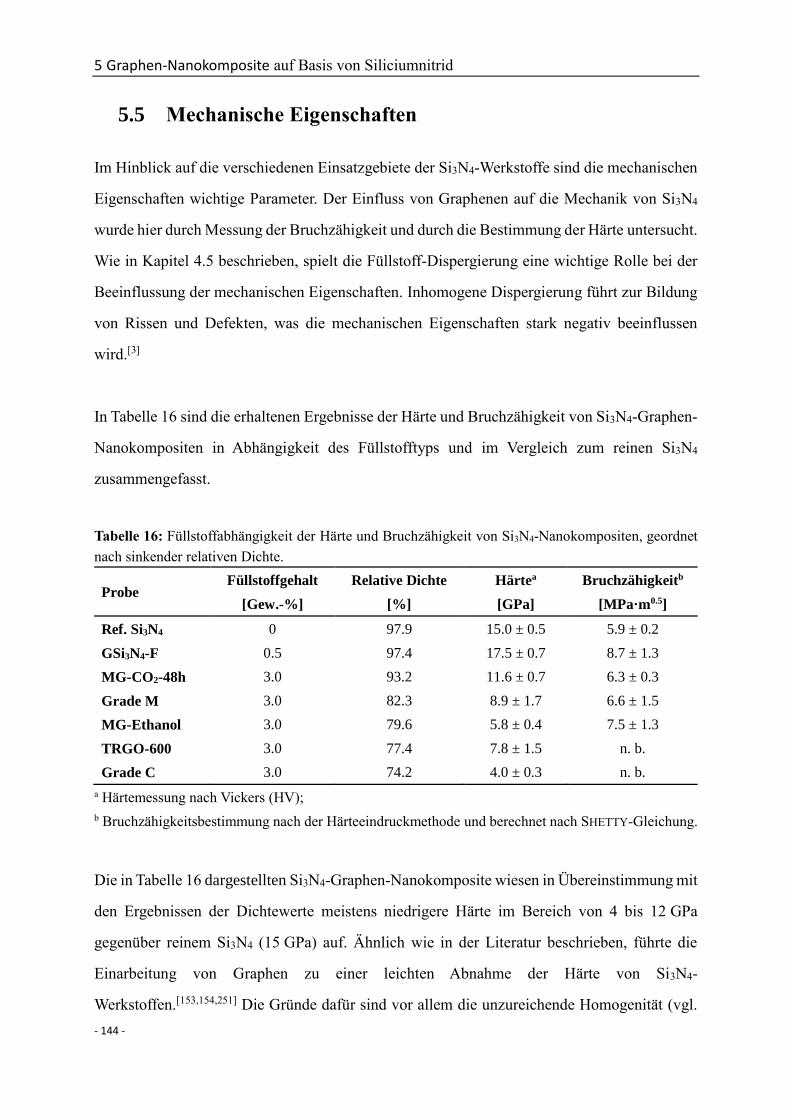
5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 144 -
5.5 Mechanische Eigenschaften
Im Hinblick auf die verschiedenen Einsatzgebiete der Si3N4-Werkstoffe sind die mechanischen
Eigenschaften wichtige Parameter. Der Einfluss von Graphenen auf die Mechanik von Si3N4
wurde hier durch Messung der Bruchzähigkeit und durch die Bestimmung der Härte untersucht.
Wie in Kapitel 4.5 beschrieben, spielt die Füllstoff-Dispergierung eine wichtige Rolle bei der
Beeinflussung der mechanischen Eigenschaften. Inhomogene Dispergierung führt zur Bildung
von Rissen und Defekten, was die mechanischen Eigenschaften stark negativ beeinflussen
wird.[3]
In Tabelle 16 sind die erhaltenen Ergebnisse der Härte und Bruchzähigkeit von Si3N4-Graphen-
Nanokompositen in Abhängigkeit des Füllstofftyps und im Vergleich zum reinen Si3N4
zusammengefasst.
Tabelle 16: Füllstoffabhängigkeit der Härte und Bruchzähigkeit von Si3N4-Nanokompositen, geordnet
nach sinkender relativen Dichte.
Probe Füllstoffgehalt
[Gew.-%]
Relative Dichte
[%]
Härtea
[GPa]
Bruchzähigkeitb
[MPa·m0.5]
Ref. Si3N4 0 97.9 15.0 ± 0.5 5.9 ± 0.2
GSi3N4-F 0.5 97.4 17.5 ± 0.7 8.7 ± 1.3
MG-CO2-48h 3.0 93.2 11.6 ± 0.7 6.3 ± 0.3
Grade M 3.0 82.3 8.9 ± 1.7 6.6 ± 1.5
MG-Ethanol 3.0 79.6 5.8 ± 0.4 7.5 ± 1.3
TRGO-600 3.0 77.4 7.8 ± 1.5 n. b.
Grade C 3.0 74.2 4.0 ± 0.3 n. b.
a Härtemessung nach Vickers (HV);
b Bruchzähigkeitsbestimmung nach der Härteeindruckmethode und berechnet nach SHETTY-Gleichung.
Die in Tabelle 16 dargestellten Si3N4-Graphen-Nanokomposite wiesen in Übereinstimmung mit
den Ergebnissen der Dichtewerte meistens niedrigere Härte im Bereich von 4 bis 12 GPa
gegenüber reinem Si3N4 (15 GPa) auf. Ähnlich wie in der Literatur beschrieben, führte die
Einarbeitung von Graphen zu einer leichten Abnahme der Härte von Si3N4-
Werkstoffen.[153,154,251] Die Gründe dafür sind vor allem die unzureichende Homogenität (vgl.

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 145 -
TEM-Aufnahmen in Kapitel 5.4) sowie die entstandene Porosität und die reduzierte Dichte
(siehe Tabelle 15 in Kapitel 5.3) der Si3N4-Graphen-Nanokomposite.
Im Gegensatz dazu bildete die Probe GSi3N4-F die einzige Ausnahme. Dabei wurde die Härte
um 17 % auf 17.5 GPa im Vergleich zur Referenzprobe erhöht. Dies lässt sich auf die homogene
Verteilung der Graphene in GSi3N4-F und die resultierende vollständige Verdichtung
zurückführen. Des Weiteren hängt die Härte beträchtlich von der relativen Dichte des
Si3N4-Nanokomposits ab (siehe Abbildung 93).
Ref. Si3N4
GSi3N4-F
MG-CO2-48h
Grade M
TRGO-600
MG-Ethanol
Grade C
4
6
8
10
12
14
16
18 Härte
Relative Dichte
Hä
rte
[G
Pa
]
75
80
85
90
95
100
Re
lati
ve
Dic
hte
[%
]
Abbildung 93: Härte der gesinterten Si3N4-Nanokomposite in Abhängigkeit von Graphentypen,
korreliert mit relativer Dichte.
Ein Vergleich aller Si3N4-Nanokomposite zeigte, dass die Härtewerte unabhängig von
Füllstofftypen in allen Fällen mit der relativen Dichte der Nanokomposite korrelieren, was mit
den morphologischen Untersuchungen in Kapitel 5.4 korreliert. So ließ sich tendenziell eine
Abnahme der Härte bei sinkender relativer Dichte, also höherer Porosität, erkennen. Dabei
führten das unfunktionalisierte MG-Ethanol und das Grade C zur maximalen Abnahme der
Härte um mehr als 60 % auf bis zu 4 GPa bei 3 Gew.-% Füllstoffgehalt, was mit ihrer großen
Porosität korrelierte.

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 146 -
Ergänzend zu den Härte-Experimenten wurde der Einfluss von Graphenen auf die
Bruchzähigkeit des Si3N4 untersucht. Anhand der Ergebnisse in Tabelle 16 wird deutlich, dass
anders als bei den Härtewerten die Einarbeitung aller Graphenderivate zur Erhöhung der
Bruchzähigkeit führte, was mit den Ergebnissen in der Literatur übereinstimmte.[130–132] Bei
dem GSi3N4-F-Nanokomposit nahm die Bruchzähigkeit im Vergleich zum ungefüllten Si3N4
(5.9 MPa·m0.5) um 48 % auf 8.7 MPa·m0.5 zu, während MG-Ethanol (+27 %, 7.5 MPa·m0.5),
Grade M (+12%, 6.6 MPa·m0.5) und MG-CO2-48h (+7 %, 6.3 MPa·m0.5) geringeren Zuwachs
bewirkten. Analog zur Literatur kann davon ausgegangen werden, dass die Graphene ein
durchgehendes Netzwerk entlang der Korngrenzen bilden könnten.[130–132] Des Weiteren könnte
die Zugabe von Graphenen zu Verzahnungen der Rissflanken hinter der Rissspitze führen,
welche die Rissspitze teilweise abschirmen. Dadurch könnten die Risse abgelenkt, verzweigt
oder überbrückt werden und die Bruchzähigkeiten somit erhöht werden.
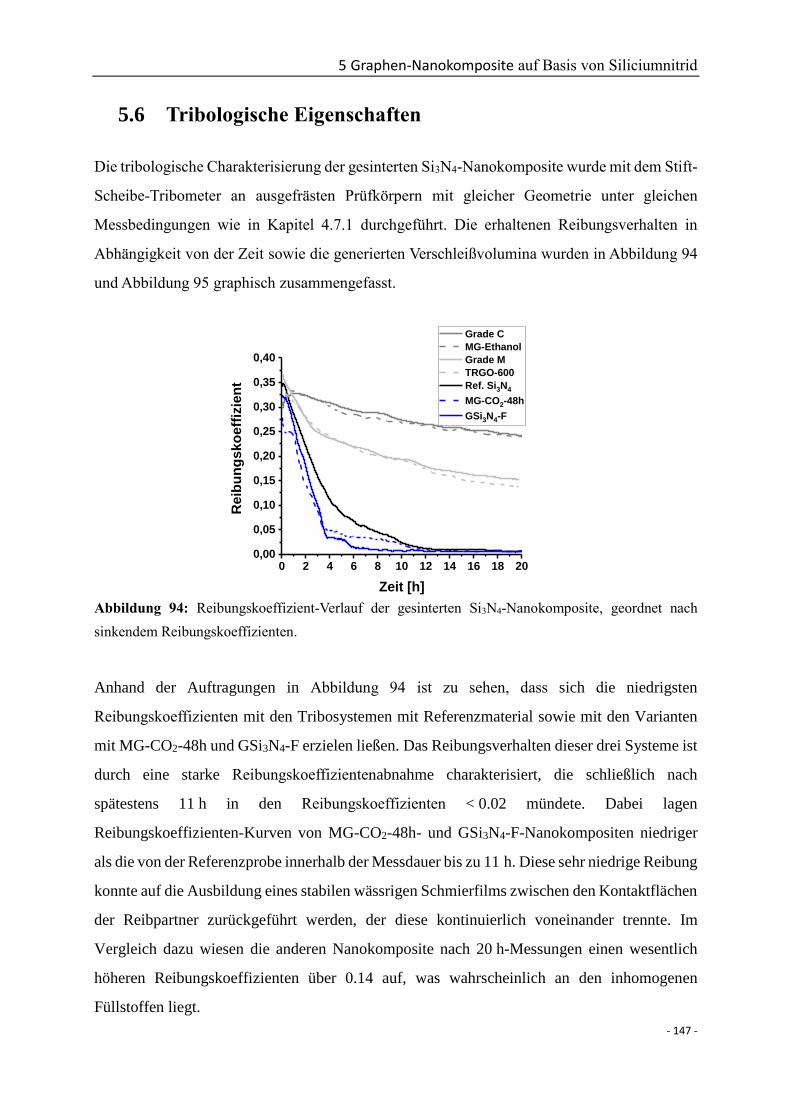
5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 147 -
5.6 Tribologische Eigenschaften
Die tribologische Charakterisierung der gesinterten Si3N4-Nanokomposite wurde mit dem Stift-
Scheibe-Tribometer an ausgefrästen Prüfkörpern mit gleicher Geometrie unter gleichen
Messbedingungen wie in Kapitel 4.7.1 durchgeführt. Die erhaltenen Reibungsverhalten in
Abhängigkeit von der Zeit sowie die generierten Verschleißvolumina wurden in Abbildung 94
und Abbildung 95 graphisch zusammengefasst.
0 2 4 6 8 10 12 14 16 18 200,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
Re
ibu
ng
sk
oe
ffiz
ien
t
Zeit [h]
Grade C
MG-Ethanol
Grade M
TRGO-600
Ref. Si3N4
MG-CO2-48h
GSi3N4-F
Abbildung 94: Reibungskoeffizient-Verlauf der gesinterten Si3N4-Nanokomposite, geordnet nach
sinkendem Reibungskoeffizienten.
Anhand der Auftragungen in Abbildung 94 ist zu sehen, dass sich die niedrigsten
Reibungskoeffizienten mit den Tribosystemen mit Referenzmaterial sowie mit den Varianten
mit MG-CO2-48h und GSi3N4-F erzielen ließen. Das Reibungsverhalten dieser drei Systeme ist
durch eine starke Reibungskoeffizientenabnahme charakterisiert, die schließlich nach
spätestens 11 h in den Reibungskoeffizienten < 0.02 mündete. Dabei lagen
Reibungskoeffizienten-Kurven von MG-CO2-48h- und GSi3N4-F-Nanokompositen niedriger
als die von der Referenzprobe innerhalb der Messdauer bis zu 11 h. Diese sehr niedrige Reibung
konnte auf die Ausbildung eines stabilen wässrigen Schmierfilms zwischen den Kontaktflächen
der Reibpartner zurückgeführt werden, der diese kontinuierlich voneinander trennte. Im
Vergleich dazu wiesen die anderen Nanokomposite nach 20 h-Messungen einen wesentlich
höheren Reibungskoeffizienten über 0.14 auf, was wahrscheinlich an den inhomogenen
Füllstoffen liegt.

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 148 -
MG-CO2-48h
GSi3N4-F
Ref. Si3N4
Grade M
TRGO-600
Grade C
MG-Ethanol
0,1
0,2
0,3
0,4
0,5 Verschleißvolumen
Porosität
Vers
ch
leiß
vo
lum
en
[m
m3]
0
5
10
15
20
25
30
Po
rosit
ät
[%]
Abbildung 95: Verschleißvolumina der gesinterten Si3N4-Nanokomposite nach Stift-Scheibe-Tribotest
in Abhängigkeit des Füllstofftyps, korreliert mit der Porosität.
Nach den tribologischen Untersuchungen wurde der Einfluss von Füllstofftypen auf die
generierten Verschleißvolumina bzw. die Verschleißbeständigkeit der Si3N4-Nanokomposite
untersucht (siehe Abbildung 95). Hinsichtlich der Verschleißbeständigkeit zeigten die
Tribosysteme mit GSi3N4-F und MG-CO2-48h verbesserte Verhältnisse im Vergleich zum
System mit Si3N4 im Ausgangszustand. Die Verschleißwerte zeigten analog zu den
Reibungskoeffizienten steigende Tendenzen und wurden erwartungsgemäß durch die relative
Dichte bzw. die Porosität wesentlich beeinflusst. Damit konnte festgestellt werden, dass die
Porosität nicht nur die mechanischen Eigenschaften, sondern auch die Verschleißbeständigkeit
von keramischen Werkstoffen erheblich beeinflusst. Dabei nahm das generierte
Verschleißvolumen der Probe mit MG-CO2-48h um 42 % auf 0.19 mm3 und gefolgt von
GSi3N4-F-Probe (28 % bzw. 0.24 mm3) gegenüber der Referenzprobe (0.34 mm3) ab. Im
Vergleich dazu wiesen die Werkstoffvarianten mit Grade M, TRGO-600, Grade C und
MG-Ethanol, welche über eine große Porosität von über mind. 17 % verfügten, höhere
Verschleißvolumina (bis 0.49 mm3) als reines Si3N4 auf, was eine schlechtere
Verschleißbeständigkeit bedeutet. Dieser Unterschied lässt sich auf die mechanischen
Schwächungen (reduzierte Härte) und die erhöhten Reibungskoeffizienten zurückführen.

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 149 -
5.7 Zusammenfassung
In diesem Kapitel wurden Si3N4-Graphen-Nanokomposite mit verschiedenen Graphenen,
welche in Kapitel 3 erwähnt wurden, durch heißisostatisches Pressen (HIP) hergestellt. Der
Einfluss des Füllstoffs wurde auf die mechanischen und tribologischen Eigenschaften der
Si3N4-Werkstoffe hin untersucht. Die erhaltenen Eigenschaften aller Si3N4-Graphen-
Nanokomposite wurden in Tabelle 17 zusammengefasst und in Abbildung 96 graphisch
dargestellt.
80
90
100
8 12 16
6
7
8
9
0,40,30,2Verschleiß [mm3]
Bruchzähigkeit [MPa.m0.5]
Härte [GPa]
Relative Dichte [%] Ref. Si3N4
GSi3N4-F
MG-CO2-48h
Grade M
MG-Ethanol
TRGO-600
Grade C
Abbildung 96: Eigenschaftsprofil der gesinterten Si3N4-Nanokomposite, Anordnung der Legende nach
sinkender relativen Dichte.
Anhand der Auftragung des Eigenschaftsprofils in Abbildung 96 und der Daten in Tabelle 17
ist zu erkennen, dass die GSi3N4-F-Probe die beste Verstärkungswirkung in der Si3N4-Matrix
induzierte. Die Einarbeitung des GSi3N4-F führte zur vollständigen Verdichtung mit höchster
Härte (17.5 GPa, +17 %), höchster Bruchzähigkeit (8.7 MPa∙m0.5, +48 %) und verbesserter

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 150 -
Verschleißbeständigkeit bzw. reduziertem Verschleißvolumen (0.24 mm3, -28 %), was sich
tendenziell mit den Ergebnissen der SiC-Graphen-Nanokomposite deckt. Dies lässt sich mit
dem vorteilhaften Herstellungsverfahren sowie geringem Füllstoffgehalt erklären, bei dem das
Graphen direkt auf der Oberfläche von Si3N4 erzeugt wurde, was eine homogene Verteilung
von Graphen gewährleistet und in eine porenfreie Struktur und gute mechanische und
tribologische Eigenschaften resultiert. Durch diese wesentlich verstärkten mechanischen und
tribologischen Eigenschaften könnte die Lebensdauer, Zuverlässigkeit und Leistungsfähigkeit
der Si3N4-Komponenten im Betrieb beispielsweise im Anlagenbau (Wälz- & Gleitlager),
insbesondere in Pumpen, Mischern und Rührwerken verbessert werden.
Tabelle 17: Übersicht der Verdichtung und der mechanischen und tribologischen Eigenschaften der
Si3N4-Nanokomposite in Abhängigkeit von Füllstofftypen und -gehalten.
Probe Füllstoffgehalt
[Gew.-%]
Relative Dichte
[%]
Härtea
[GPa]
Bruchzähigkeitb
[MPa·m0.5]
Verschleißc
[mm3]
Ref. Si3N4 0 97.9 15.0 ± 0.5 5.9 ± 0.2 0.335
GSi3N4-F 0.5 97.4 17.5 ± 0.7 8.7 ± 1.3 0.240
MG-CO2-48h 3.0 93.2 11.6 ± 0.7 6.3 ± 0.3 0.194
Grade M 3.0 82.3 8.9 ± 1.7 6.6 ± 1.5 0.388
MG-Ethanol 3.0 79.6 5.8 ± 0.4 7.5 ± 1.3 0.490
TRGO-600 3.0 77.4 7.8 ± 1.5 n. b. 0.397
Grade C 3.0 74.2 4.0 ± 0.3 n. b. 0.472
a Vickers-Härte (HV); b bestimmt nach der Härteeindruckmethode; c Generierte Verschleißvolumina
nach Stift-Scheibe-Tribotest (4 h).
Die Inkorporierung von MG-CO2-48h erreichte zwar die beste Verschleißbeständigkeit mit
Abnahme des Verschleißes um 42 %, aber führte gleichzeitig zur Abnahme der Härte (11.6 GPa,
-23 %) und der relativen Dichte (-4.7 %). Zudem gelang es, den knapp reibfreien Zustand
(Reibungskoeffizienten < 0.02) der Werkstoffe durch Zusatz von GSiC-F (nach 6 h) und
MG-CO2-48h (nach 10 h) schneller als die Referenzprobe (nach 12 h) zu erreichen. Weiterhin
wurde die Bruchzähigkeit bei allen Nanokompositen verbessert. Abhängig von Füllstofftypen
wurde die Bruchzähigkeit auf 6.3 bis 8.7 MPa∙m0.5 erhöht, was einer Verstärkung zwischen 7
und 48 % entspricht. Die Risse in den Werkstoffen könnten durch die Graphene abgelenkt,
verzweigt oder überbrückt werden.[130–132]

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 151 -
Im Vergleich dazu wiesen die Nanokomposite mit MG-Ethanol, TRGO-600 sowie den
kommerziellen Grade C und Grade M unvollständige Verdichtung mit wesentlich geringerer
Härte und Verschleißbeständigkeit gegenüber reinem Si3N4 auf. Die Gründe sind vor allem die
ungleichmäßige Verteilung und Agglomeratbildung, was die Fehlstellen und die hohe Porosität
(siehe TEM-Aufnahmen in Kapitel 5.4) verursacht sowie zur Härteabnahme (bis -73 %) und zu
höheren Reibungskoeffizienten (bis zu Reibungskoeffizienten von 0.24) und reduzierter
Verschleißbeständigkeit (bis -46 %) führt.
Tabelle 18: Vergleich der Eigenschaften von literaturbekannten Si3N4-Graphen-Nanokompositen mit
GSi3N4.
Sinterart Graphen Dichte Mechanik Tribologie Lit.
Typ Gehalt Härte KICa µb Verschleiß
HIP GSi3N4-F 0.5 Gew.-% = + + + +
HIP MLG 1 Gew.-% - + + = = [131,132,249]
HIP MLG 3 Gew.-% - - n. b. - + [131,132]
HIP MLG 1/3 Gew.-% - - + - + [178]
HIP MLG 0.5/1.0 Gew.-% = = = n. b. n. b. [254]
HIP MLG 2 – 10 Gew.-% - - - n. b. n. b. [254]
HIP MLG 1/3 Gew.-% +/- - -/+ - - [154]
HIP MLG 3 Gew.-% n. b. n. b. n. b. + - [182]
HIP/GPS MLG 1 Gew.-% = - + n. b. n. b. [179]
HIP/SPS MLG 3 Gew.-% n. b. - - n. b. n. b. [250,251]
SPS TRGO 0.02 – 1.5 Vol.-% - - + n. b. n. b. [130]
SPS MLG/GO 1 – 8 Vol.-% - - + n. b. n. b. [180]
SPS MLG 3 Gew.-% - - - + + [162]
SPS MLG 1/3 Gew.-% +/- - - + + [154]
SPS MLG 1 – 5 Gew.-% - +/- +/- n. b. n. b. [181]
+: verbessert; -: verschlechtert; =: nicht geändert; +/-: abhängig von Graphengehalt;
a Bruchzähigkeit; b Reibungskoeffizient.
Trotz der Tatsache, dass die Si3N4-Graphen-Nanokomposite zwar intensiv erforscht wurden,
gibt es kein literaturbekanntes Beispiel, was eine vollständige Verdichtung, verstärkte
mechanischen Eigenschaften und gleichzeitig verbessertes tribologisches Verhalten beschreibt
(siehe Tabelle 18). Dementgegen wurde das GSi3N4-F-Nanokomposit mit nur sehr geringem

5 Graphen-Nanokomposite auf Basis von Siliciumnitrid
- 152 -
Füllstoffgehalt von 0.5 Gew.-% in dieser Arbeit erfolgreich hergestellt, welches die
unkonventionellen Eigenschaftskombinationen mit vollständiger Verdichtung, steigenden
mechanischen und tribologischen Eigenschaften zeigte. Eine Übersicht der Eigenschaften von
literaturbekannten Si3N4-Graphen-Nanokompositen und GSi3N4-F-Probe wurde in Tabelle 18
aufgelistet und verglichen. Beispielsweise wurde bei der Arbeit von RUTKOWSKI et al. ebenfalls
einen Graphengehalt von 0.5 Gew.-% verwendet, um Si3N4-Graphen-Nanokomposit
herzustellen. Das erhaltene Nanokomposit zeigte unveränderte Bruchzähigkeit von 8 GPa und
Härte von 13 GPa wie die Referenzprobe. Die tribologischen Eigenschaften des
Nanokomposits wurden zwar gemessen, liegt der Vergleich mit der Referenzprobe nicht vor.[254]
Zusammenfassend konnten die mechanischen und tribologischen Eigenschaften von Si3N4-
Werkstoffen durch die Beimischung von Graphen wesentlich verbessert werden. Die besten
Eigenschaften wurden bei dem GSi3N4-F-Nanokomposit erhalten, welches eine Erhöhung der
Härte um 17 %, der Bruchzähigkeit um 48 % und der Verschleißbeständigkeit um 28 % im
Vergleich zu der Referenzprobe zeigte.

6 Zusammenfassende Diskussion
- 153 -
6 Zusammenfassende Diskussion
In jüngster Zeit hat die Verwendung der (funktionalisierten) Graphene aufgrund ihrer
hervorragenden und vielseitigen Materialeigenschaften zunehmend an Bedeutung gewonnen.
Für keramische Werkstoffe gehören insbesondere (funktionalisierte) Graphene zu den wenigen
sinnvollen und vielversprechenden Nanofüllstoffen, um Materialeigenschaften gezielt
maßzuschneidern. Das bisher größte Problem bei der Anwendung von Graphen als Füllstoff in
Keramiken ist die Entstehung von Poren, was zu unvollständiger Verdichtung und daraus
resultierenden Verschlechterung der mechanischen Eigenschaften führt. Daher liegt ein
kombinatorischer Anstieg der mechanischen und tribologischen Eigenschaften mit
vollständiger Verdichtung durch Graphen in Si3N4- und SiC-Nanokompositen bisher nicht vor.
Um eine homogene Durchmischung von Graphen und Keramik zu gewährleisten bzw. die
Porenvolumina von hergestellten Keramik-Graphen-Nanokompositen zu minimieren, wurden
Graphene mittels Präkursor-Verfahren direkt auf die Oberfläche der Keramikrohstoffe
aufgebracht, wobei graphenbeschichtete SiC (GSiC) und Si3N4 (GSi3N4) erhalten wurden. Das
übergeordnete Ziel dieser Arbeiten war die Herstellung und Charakterisierung von neuartigen
Keramik-Graphen-Nanokompositen mit bislang unbekannten Materialeigenschafts-
kombinationen aus vollständiger Verdichtung, verbesserter mechanischen Eigenschaften,
erhöhter elektrischen Eigenschaften und tribologischer Verstärkung. Damit werden die
Effizienz, Lebensdauer, Zuverlässigkeit und Leistungsfähigkeit keramischer Komponenten in
den Anwendungen unter Wasser- bzw. Mangelschmierung, aber auch zur Förderung anderer
Medien verbessert.
Um das Ziel dieser Arbeit zu realisieren wurden zunächst verschiedene Graphenderivate
entweder aus Graphit mittels Top-down-Methode oder aus einer Kohlenstoffquelle anhand
Bottom-up-Ansatzes hergestellt und dessen Eigenschaften in Relation zu Benchmark-
Kohlenstofffüllstoffen evaluiert (Kapitel 6.1). Anschließend wurden Keramik-Graphen-
Nanokomposite auf Basis von SiC (Kapitel 6.2) und Si3N4 (Kapitel 6.3) aus Graphen,
Keramikrohrstoff und Sinteradditive durch Sintern in enger Zusammenarbeit mit anderen

6 Zusammenfassende Diskussion
- 154 -
Forschungsinstituten und Industrien innerhalb des CERAPHENE-Projekts hergestellt und
charakterisiert. Durch Zugabe von Graphenen können thermische, elektrische und tribologische
Eigenschaften von Keramiken verbessert werden. Trotzdem besteht die Beschränkung der
Anwendung von Graphen in Keramiken meistens auf der Entstehung von Poren, welche durch
Graphenagglomerate verursacht werden und zu den verschlechterten mechanischen
Eigenschaften führen. Daher ist bisher kein SiC-Graphen-Nanokomposit, welches über
reduzierte Reibung und gleichzeitig reduzierten Verschleiß ohne Verschlechterung
mechanischen Eigenschaften verfügt, literaturbekannt. Im Vergleich zu SiC-Werkstoffen
wurden die Si3N4-Graphen-Nanokomposite zwar intensiver erforscht, jedoch liegt ein
kombinatorischer Anstieg der mechanischen und tribologischen Eigenschaften mit
vollständiger Verdichtung durch Graphen in Si3N4-Nanokompositen bisher nicht vor. Diese
Probleme sollen in dieser Arbeit durch Beschichtungsverfahren, bei dem das Graphen gemäß
eines Bottom-up-Ansatzes direkt auf der Oberfläche von Keramikrohstoffen hergestellt werden,
umgangen werden. Dabei sollen Furfurylalkohol, Glucose bzw. Dopamin als bio-basierte
Kohlenstoffquelle verwendet werden, um graphenbeschichtetes SiC (GSiC) und Si3N4 (GSi3N4)
herzustellen. Im Vergleich dazu kommen als reine Graphene thermisch reduziertes Graphitoxid
(TRGO) und mechanochemisch funktionalisiertes Multilagen-Graphen (MG) aus trockener
Vermahlung von Graphit unter CO2 (MG-CO2), Ar (MG-Ar) oder N2 (MG-N2) zum Einsatz.
Außerdem werden kommerzielle plättchenförmige Kohlenstofffüllstoffe Graphit, Nanographit,
Grade M (Multilagen-Graphen) und MG-Ethanol (unfunktionalisiertes Multilagen-Graphen),
sowie die sphärischen Kohlenstofffüllstoffe Carbon Black und Grade C als Referenz untersucht,
um diese als Füllstoff in Keramik-Nanokompositen mit selbst hergestellten Graphenen zu
vergleichen. Die erwarteten Nanokomposite sollten vor allem über eine vollständige
Verdichtung, verbesserte mechanische Belastbarkeit und erhöhte elektrische Leitfähigkeit
sowie optimierte Reibungs- und Verschleißeigenschaften verfügen. Die Übersicht der
Vorgehensweise ist in Abbildung 97 gezeigt.

6 Zusammenfassende Diskussion
- 155 -
Abbildung 97: Übersicht der Vorgehensweise zur Herstellung und Charakterisierung der Keramik-
Graphen-Nanokomposite.

6 Zusammenfassende Diskussion
- 156 -
6.1 Graphensynthese und Charakterisierung
Die in dieser Arbeit verwendeten Graphenderivate wurden durch angepasste chemische und
thermische Verfahrensschritte in großem Maßstab (10 – 200 g) hergestellt. Die typischen
Eigenschaften der hergestellten Graphenvarianten werden in Tabelle 19 aufgelistet. Gemäß dem
Top-down-Ansatz wurde einerseits das thermisch reduzierte Graphitoxid (TRGO) durch
thermische Reduktion und Expansion des GO, welches nach dem HUMMERS-Verfahren aus
Graphit synthetisiert wurde, hergestellt. Durch Variierung der Reduktionstemperatur konnten
die Materialeigenschaften des TRGO gezielt eingestellt werden. Mit erhöhter
Reduktionstemperatur ging eine Steigerung der Temperaturbeständigkeit, der spezifischen
Oberfläche und des Exfolierungsgrades einher. Außerdem nahm der Sauerstoffgehalt, welcher
charakteristisch für den Funktionalisierungsgrad ist, mit höherer Reduktionstemperatur von
TRGO-600 über TRGO-800 bis hin zu TRGO-1000 von 15.4 über 11.1 auf 5.1 Gew.-% ab.
Während der thermischen Behandlung vergrößert sich das konjugierte π-System, wodurch die
Leitfähigkeit, spezifische Oberfläche und der E-Modul anstiegen.[23,75] Im Vergleich zu TRGO-
600 und TRGO-800 zeigte TRGO-1000 die höchste Oberfläche von 515 m2∙g-1 und den
höchsten Exfolierungsgrad mit einer durchschnittlichen Anzahl an Graphenlagen von fünf.
Anhand der SEM-, TEM- und AFM-Aufnahmen zeigten alle TRGO eine dünne, gewellte und
gefaltete Mono- bis Weniglagen-Struktur, die durch das Entweichen der Gase beim
Reduktionsprozess entstehen (siehe Abbildung 98).
Abbildung 98: SEM- (a), TEM- (b) und AFM-Aufnahmen (c) von TRGO.

6 Zusammenfassende Diskussion
- 157 -
Tabelle 19: Übersicht der verwendeten Kohlenstofffüllstoffe und ihrer Eigenschaften.
Probe C-Gehalta
[Gew.-%]
O-Gehaltb
[Gew.-%]
H-Gehalta
[Gew.-%]
N-Gehalta
[Gew.-%]
𝐀𝐁𝐄𝐓𝐜
[m2∙g-1]
Mlossd
[%] Geometrie
GO 55.3 41.5 2.2 - 30 55.8 Plättchen
TRGO-600 78.3 15.4 0.3 0.3 403 36.5 Plättchen
TRGO-800 84.2 11.1 0.5 - 417 30.9 Plättchen
TRGO-1000 89.2 5.1 0.4 0.4 515 22.5 Plättchen
MG-Ar-24h 89.1 6.9 0.6 0.6 444 9.6 Sphärisch
MG-Ar-48h 85.7 8.4 0.7 0.5 351 13.3 Sphärisch
MG-Ar-72h 81.7 9.3 0.6 0.6 275 17.3 Sphärisch
MG-CO2-24h 91.5 8.0 0.7 - 283 6.4 P & Se
MG-CO2-48h 88.2 10.4 0.8 - 416 10.9 P & Se
MG-CO2-72h 86.4 11.7 0.9 - 539 13.7 P & Se
MG-CO2-96h 87.9 10.9 0.5 - 561 20.7 P & Se
MG-CO2-120h 86.4 11.1 0.6 - 575 28.0 P & Se
MG-N2-24h 82.2 8.4 1.0 4.0 449 20.0 Sphärisch
MG-N2-48h 80.2 9.0 0.8 6.1 269 24.5 Sphärisch
MG-N2-72h 75.2 7.6 1.1 7.2 219 29.3 Sphärisch
Graphit 99.6 0.7 0.2 0.2 3 1.9 Plättchen
Nanographit 96.9 3.8 0.2 - 34 2.5 Plättchen
MG-Ethanol 98.3 1.7 0.2 - 94 13.3 Plättchen
Carbon Black 90.1 8.1 1.8 - 44 21.4 Sphärisch
Grade C 88.0 10.7 1.1 0.5 794 21.4 Sphärisch
Grade M 90.9 5.9 0.9 - 88 10.1 Plättchen
a Bestimmt durch Elementaranalyse; b Bestimmt durch EDX; c N2-Physisorptionsmessung zur
Oberflächenbestimmung nach Brunauer, Emmett und Teller; d TGA-Masseverlust, N2-Atmosphäre,
10 K/min, 50 – 1500 °C; e Plättchen und Sphärisch.
Weiterhin wurde das mechanochemisch funktionalisierte Multilagen-Graphen (MG) durch
einen einstufigen Trockenvermahlungsprozess in einer Planetenkugelmühle unter Ar-, CO2-
oder N2-Atmosphäre direkt aus Graphit hergestellt. Die Variation der Prozessparameter
(Mahldauer und Gas-Atmosphäre) ermöglichte eine individuelle Einstellung der
Partikelmorphologie und chemischen Struktur der resultierenden Materialien. So konnte z. B.
die Plättchenstruktur nach der Vermahlung unter CO2 erhalten werden, während bei MG-Ar
und MG-N2 nur noch sphärische Partikel zu erkennen waren. Des Weiteren führte die
Vermahlung unter CO2 zu funktionalisiertem Graphen mit Carboxylgruppen, während die

6 Zusammenfassende Diskussion
- 158 -
Vermahlung unter N2 oder Ar zu MG mit Hydroxylgruppen führte. Darüber hinaus nahm der
Sauerstoffgehalt mit steigender Mahldauer unabhängig von der Gasatmosphäre zu, was auf eine
zunehmende Funktionalisierung hindeutete. Dabei erhöhte sich der Funktionalisierungsgrad bei
allen MG innerhalb der ersten 24 h stark auf ca. 8 Gew.-%, während die weitere Vermahlung
bis zu 72 bzw. 120 h nur noch zu einer moderaten Zunahme führte. Bei MG-Ar und MG-N2
erreichte der Funktionalisierungsgrad nur bis zu 9 Gew.-%, während die Vermahlung unter CO2
zu einer Funktionalisierung von bis zu 12 Gew.-% erreichte. Ähnlich wie der
Funktionalisierungsgrad stieg die spezifische Oberfläche innerhalb der ersten 24 h sehr stark
auf bis zu 450 m2∙g-1 an. Bei der Vermahlung unter CO2 nahm die spezifische Oberfläche mit
steigender Mahldauer stetig zu (580 m2∙g-1 nach 120 h). Im Gegensatz dazu sank die spezifische
Oberfläche von MG-Ar und MG-N2 nach 24 h ab, was auf die Reagglomeration der sphärischen
Partikel zurückzuführen sein könnte.
Da eine homogene Füllstoffverteilung bzw. -dispergierung die Voraussetzung für eine optimale
Verstärkungswirkung ist, wurden stabile tensidfreie MG- und TRGO-Dispersionen in Wasser
durch den Einsatz eines Hochdruckhomogenisators (HH) unter hohem Druck von 1000 bar
hergestellt. Aufgrund der hohen Scherkräfte in dem HH-Prozess wurden die großen Graphen-
Agglomerate aufgelöst und blieben die Dispersionen über einen langen Zeitraum (z. B. TRGO
über 50 Tage) stabil. Zusätzlich wurde die Stabilität der Dispersionen durch eine
elektrostatische Abstoßung der deprotonierten funktionellen Gruppen in Graphen-Flocken wie
die Hydroxyl- oder Carboxyl-Funktionen hervorgerufen, was durch die Messung des
Zetapotentials nachgewiesen wurde. All die gemessenen Zetapotentiale von MG- und TRGO-
Dispersionen lagen zwischen -40 – -60 mV, welche auf stabile Dispersionen hinwiesen.
Als kommerzielle Referenzen wurden die plättchenförmigen Kohlenstofffüllstoffe Graphit
(KFL 99.5, AMG Mining), Nanographit (mikronisiertes Graphit, V-HF 98, AMG Mining),
Grade M (Multilagen-Graphen, XG Sciences) und MG-Ethanol (unfunktionalisiertes
Multilagen-Graphen, Institute for Technical Physics and Materials Science, Centre for Energy
Research) sowie die sphärischen Kohlenstofffüllstoffe Carbon Black (Orion Engineered
Carbons) und Grade C (XG Sciences) untersucht, um diese mit den selbst hergestellten
Graphenen zu vergleichen. Ähnlich wie MG-Ar, MG-CO2 und MG-N2 beruht die Herstellung
von MG-Ethanol, Grade M und Grade C sowie Nanographit, laut Herstellerangaben, ebenso

6 Zusammenfassende Diskussion
- 159 -
auf der mechanischen Exfolierung von Graphit. Der Fokus liegt dabei neben der
Eigenschaftsverbesserung auch auf der Verfügbarkeit und Kosteneffizienz der eingesetzten
Füllstoffe. Dabei verfügten Grade M und MG-Ethanol ähnlich wie TRGO über ein höheres
Aspektverhältnis und eine dünnere Schichtstruktur als Graphit und Nanographit, während
Carbon Black und Grade C ähnliche sphärische Struktur wie MG-Ar und MG-N2 zeigten. Im
Hinblick auf die Funktionalität nahm der O-Gehalt von Grade C (10.7 Gew.-%) über Carbon
Black und Grade M (6 – 8 Gew.-%) bis hin zu Nanographit (3.8 Gew.-%) ab. Dagegen
beinhalteten Graphit und MG-Ethanol kaum funktionelle Gruppen.
Abbildung 99: Synthese von graphenbeschichtetem Keramikrohstoff.
Das bisher größte Problem bei der Anwendung von Graphen als Füllstoff in Keramiken ist die
Entstehung von Poren, was zu verschlechterten mechanischen Eigenschaften führt. Um eine
homogene Durchmischung von Graphen und Keramik zu gewährleisten bzw. die
Porenvolumina von hergestellten Keramik-Graphen-Nanokompositen zu minimieren, wurden
Keramikrohstoffe (SiC/Si3N4) vor dem Sintern gemäß eines Bottom-up-Ansatzes mit Graphen
beschichtet (siehe Abbildung 99). In Anlehnung an die Arbeiten von M. BECKERT, X. LI und
R. LI wurden Furfurylalkohol[44,45], Glucose[43] bzw. Dopamin[46] als bio-basierte
Kohlenstoffquelle verwendet, um graphenbeschichtetes SiC (GSiC) und Si3N4 (GSi3N4)
herzustellen. Im Vergleich zu TRGO und MG war hier weder ein Einsatz von fossilen
Kohlenstoffquellen (z. B. Graphit) noch nachträgliche Behandlung (z. B. Reinigung und
Homogenisierung) erforderlich. Anhand der TEM-Aufnahmen und EDX-Analysen konnte eine

6 Zusammenfassende Diskussion
- 160 -
erfolgreiche Herstellung der GSiCs nachgewiesen werden. Unabhängig von der
Kohlenstoffquelle sind die dünnen Graphenschichten am Rand der SiC-Partikel erkennbar
(siehe Abbildung 100 b). Außerdem konnte die Dicke der Beschichtung mittels TEM-
Aufnahmen analysiert werden. Je nach Kohlenstoffquelle variierte die Anzahl der
Graphenlagen zwischen 5 und 20 mit einer Dicke von 5 – 10 nm (siehe Abbildung 100 c).
Abbildung 100: TEM-Aufnahmen von SiC vor (a) und nach (b und c) Beschichtung mit Graphen.
Des Weiteren konnte der Graphengehalt (Abbildung 101 links) und die spezifische Oberfläche
(Abbildung 101 rechts) von GSiC in dieser Arbeit durch Variation des Gewichtsanteils der
Kohlenstoffquelle im Reaktionsgemisch mit SiC leicht kontrolliert werden. Das
Gewichtsverhältnis von Kohlenstoffquelle (Furfurylalkohol, Glucose und Dopamin) zu SiC
wurde von 9:91 bis zu 40:60 variiert. Der Füllstoffgehalt stieg bei allen GSiC-Systemen nahezu
linear mit dem Kohlenstoffquellengehalt an, was den gut kontrollierbaren Charakter der
Reaktion zur Herstellung von GSiC bestätigte. Folglich ist diese Methode hervorragend
geeignet um GSiC mit definiertem Füllstoffgehalt herzustellen, da sich dieser gezielt über die
Menge der eingesetzten Kohlenstoffquelle steuern lässt. Im Vergleich dazu stellten Miranzo
et al. zwar Graphene von 4 Vol.-% durch das epitaktische Wachstum in SiC-Nanokomposite
in situ während des SPS-Sinterprozesses, jedoch ist der Graphengehalt nicht wie in dieser
Arbeit leicht kontrollierbar.[136] Außerdem ist die spezifische Oberfläche unabhängig von der
Kohlenstoffquelle linear vom Füllstoffgehalt abhängig und ermöglicht diese hohe spezifische
Oberfläche eine homogene Verteilung bei einer späteren Verwendung als Füllstoff in Polymer-
und Keramik-Matrizes.

6 Zusammenfassende Diskussion
- 161 -
0 5 10 15 20 25 30 35 400
2
4
6
8
10
12
14
16
GSiC-GGSiC-D
Gra
ph
en
geh
alt
[G
ew
.-%
]
Anteil der Kohlenstoffquelle [Gew.-%]
GSiC-F
0 2 4 6 8 10 12 14 160
20
40
60
80
100
120
GSiC-D
GSiC-G
Sp
ezif
isc
he
Ob
erf
läc
he [
m2g
-1]
Graphengehalt [Gew.-%]
GSiC-F
Abbildung 101: Graphengehalt (links) und spezifische Oberfläche (rechts) von GSiC-F (aus
Furfurylalkohol), GSiC-G (aus Glucose) und GSiC-D (aus Dopamin) in Abhängigkeit des eingesetzten
Anteil der Kohlenstoffquelle.
Alle Graphen-Materialien zeigten eine gute thermische Stabilität bis 2000 °C mit einem
maximalen Masseverlust von 13 Gew.-% (TRGO-600) gefolgt von 9 Gew.-% (MG-CO2-48h).
Damit eigenen sich alle getesteten Materialien als Füllstoffe zur Herstellung von SiC- und
Si3N4-Graphen-Nanokomposite durch Sinterung unter hohen Temperaturen (1700 – 2050 °C).
Zusätzlich ist zu sehen, dass SiC und GSiC-F sehr ähnliche thermische Eigenschaften mit
geringfügigem Masseverlust (ca. 1 Gew.-%) zeigten, was auf den geringen Graphengehalt
(4.4 Gew.-%) in GSiC-F und die gute thermische Stabilität der funktionalisierten Graphene in
GSiC-F zurückzuführen ist. Des Weiteren wurden bei den GSiC-Nanokompositen aufgrund
ihrer hervorragenden thermischen Stabilität weniger Poren und vollständigere Verdichtung als
die TRGO- und MG-CO2-Varianten gezeigt (siehe Kapitel 6.2).
Zusammenfassend wurden verschiedene Graphen-Füllstoffe nach unterschiedlichen Methoden
für die Kompositbildung mit SiC und Si3N4 hergestellt und charakterisiert. Diese große Vielfalt
Kohlenstofffüllstoffe erlaubte die systematische Evaluierung des Einflusses der Partikelgrößen,
Geometrien, Exfolierungsgrads und Funktionalitäten der Füllstoffe auf die Verdichtung, die
morphologischen, mechanischen, elektrischen und tribologischen Eigenschaften von Keramik-
Nanokompositen. Dadurch könnten die eingesetzten Nanofüllstoffe hinsichtlich ihrer
Eigenschaftsverbesserung sowie ihrer Verfügbarkeit und Kosteneffizienz eingestuft werden.

6 Zusammenfassende Diskussion
- 162 -
6.2 Graphen-Nanokomposite auf Basis von Siliciumcarbid
Im Vergleich zu den anderen technischen Keramiken waren die SiC-Graphen-Nanokompositen
im Labor- und Industriemaßstab zu Beginn dieser Arbeit kaum erforscht. Seitdem wurden
einige wenige Publikationen über graphenhaltiges SiC veröffentlicht.[127,133,136,152,173,214,238] Die
Einarbeitung von GO oder kommerziellem Multilagen-Graphen in SiC erzielte dabei zwar
verschleißarme und elektrisch leitfähige[137] sowie thermische leitfähige[127,173] Systeme jedoch
auf Kosten der Verdichtung,[127,214] der mechanischen Eigenschaften (Biegefestigkeit[255],
Härte[152,214] oder E-Modul[152]) und der Reibung.[152] Bisher ist kein SiC-Graphen-
Nanokomposit, welches über reduzierte Reibung und reduzierten Verschleiß ohne
Verschlechterung der mechanischen Eigenschaften verfügt, literaturbekannt. Ebenfalls ist die
Verwendung von MG und GSiC als Füllstoffe zur Herstellung SiC-Graphen-Nanokomposit
bisher nicht bekannt. Die Einarbeitung von Graphen zielte auf tribologisch verstärkte SiC-
Nanokomposite mit verbesserter elektrischen und mechanischen Eigenschaften. Als Füllstoffe
wurden sowohl die selbst synthetisierten TRGO, MG und GSiC als auch die kommerziellen
erhältlichen Kohlenstofffüllstoffe verwendet. Die meisten Nanokomposite wurden mit einem
Füllstoffgehalt von 2.0 Gew.-% hergestellt, um eine hohe Sinterdichte und demzufolge gute
mechanischen Eigenschaften zu gewährleisten.[132,133,152,154,159] Unter Berücksichtigung der
Analyse der mechanischen Eigenschaften wurde der Füllstoffgehalt bei den vielversprechenden
GSiC-Systemen auf bis zu 8.0 Gew.-% erhöht. Die nach dem Standard-Sinterverfahren bzw.
via druckloser Sinterung (SSiC) erhaltenen SiC-Nanokomposite wurden auf charakteristische
Materialeigenschaften untersucht. Im Vergleich zu literaturbekannten drucklosgesinterten SiC-
Nanokompositen mit unfunktionalisierten Multilagengraphen[127,214,255] wurden in dieser Arbeit
erstmals drucklosgesinterte SiC-Nanokomposite aus TRGO, MG und GSiC hergestellt. Dabei
wurden die Proben nach Homogenisierung und Trocknung durch kalt isostatisches Pressen bei
2000 bar Druck verdichtet und im Ofen drucklos bei 2050 °C unter Argon für 4 h gesintert.
Anschließend wurde der Einfluss des Funktionalitätsgrads von TRGO und MG, welcher durch
die Variation der Reduktionstemperatur bzw. Mahldauer und Gasatmosphäre eingestellt wurde,
auf die relative Dichte, mechanischen, elektrischen und tribologischen Eigenschaften von
entsprechenden Nanokompositen ermittelt und in Abbildung 102 graphisch dargestellt.

6 Zusammenfassende Diskussion
- 163 -
TRGO-600
TRGO-800
TRGO-1000
96,0
96,5
97,0
97,5
98,0
98,5
99,0 Relative Dichte
O-Gehalt
Re
lati
ve
Dic
hte
[%
]
0
2
4
6
8
10
12
14
16
Sa
ue
rsto
ffg
eh
alt
[G
ew
.-%
]
MG-CO2-24h
MG-CO2-48h
MG-CO2-72h
MG-CO2-96h
MG-CO2-120h97,0
97,5
98,0
98,5
99,0
99,5
Relative Dichte
O-Gehalt
Rela
tive D
ich
te [
%]
7,0
7,5
8,0
8,5
9,0
9,5
10,0
10,5
11,0
11,5
12,0
Sau
ers
toff
geh
alt
[G
ew
.-%
]
TRGO-600
TRGO-800
TRGO-1000
10-8
10-7
10-6
10-5
10-4
10-3
10-2
10-1 Elektrische Leitfäigkeit
C-Gehalt
Ele
ktr
isch
e L
eit
fäig
keit
[S
. cm
-1]
76
78
80
82
84
86
88
90K
oh
len
sto
ffg
eh
alt
[G
ew
.-%
]
MG-CO2-24h
MG-CO2-48h
MG-CO2-72h
MG-CO2-96h
MG-CO2-120h10-8
10-7
10-6
10-5
10-4
Elektrische Leitfähigkeit
C-Gehalt
Ele
ktr
isch
e L
eit
fäh
igkeit
[S
. cm
-1]
86
87
88
89
90
91
92
Ko
hle
nsto
ffg
eh
alt
[G
ew
.-%
]
TRGO-600
TRGO-800
TRGO-1000
0,15
0,17
0,19
0,21
0,23
0,25 Verschleißvolumen
Reibungskoeffizient
Ve
rsc
hle
ißv
olu
me
n [
mm
3]
0,11
0,12
0,13
0,14
0,15
0,16
Re
ibu
ng
sk
oe
ffiz
ien
t
MG-CO2-24h
MG-CO2-48h
MG-CO2-72h
MG-CO2-96h
MG-CO2-120h
0,15
0,17
0,19
0,21
0,23
0,25 Verschleißvolumen
Reibungskoeffizient
Ve
rsc
hle
ißv
olu
me
n [
mm
3]
0,11
0,12
0,13
0,14
0,15
0,16
Re
ibu
ng
sk
oe
ffiz
ien
t
Abbildung 102: Relative Dichte (oben), elektrische Leitfähigkeit (Mitte) und tribologische
Eigenschaften (unten) der gesinterten SiC-Nanokomposite in Abhängigkeit des Funktionalitätsgrads bei
je 2.0 Gew.-% TRGO (links) und MG-CO2 (rechts).
Die TRGO- und MG-CO2-Nanokomposite in Abbildung 102 (oben) zeigten eine
kontinuierliche Abnahme der relativen Dichte bei sinkendem Funktionalitätsgrad
(Sauerstoffgehalt), was auf die schlechtere Verteilung und Kompatibilisierung der weniger

6 Zusammenfassende Diskussion
- 164 -
funktionalisierten Füllstoffe in der SiC-Matrix zurückführen ist. Dies korreliert auch mit der
morphologischen Untersuchung, da die Agglomeratgröße mit steigender Mahldauer von
MG-CO2 drastisch abnahm. Im Vergleich zu MG-CO2 wurden die relativen Dichten von nahezu
100 % bei allen MG-Ar- und MG-N2-Proben gemessen, welche auf die kleineren
Partikeldurchmesser und demzufolge geringeren Diffusionsbarriere zurückzuführen sein
könnte. Bei kommerziellen Füllstoffen besaßen Graphit, das Grade M und das MG-Ethanol
aufgrund des großen Partikeldurchmessers und der resultierenden höheren Diffusionsbarriere
deutlich niedrigere Dichten im Vergleich zur kleinen Carbon Black, Nanographit und Grade C.
Die mit TRGO, MG und kommerziellen Kohlenstofffüllstoffen dargestellten SiC-Graphen-
Nanokomposite wiesen niedrigere Biegefestigkeiten (von 250 bis 400 MPa) gegenüber reinem
SiC (500 MPa) auf. Die Gründe dafür sind vor allem die unzureichende Homogenität sowie die
Porosität und die geringere Dichte der SiC-Graphen-Komposite, welche mit den
Bruchverhalten sowie den morphologischen Untersuchungen der Bruchfläche mittels SEM von
Prüfkörpern bei der 4-Punkte-Biegeprüfung nachgewiesen werden konnten. Im Gegensatz dazu
zeigten einige Nanokomposite verbesserte (nicht in Abbildung 102 enthaltene) Bruchzähigkeit
bis zu 3.8 MPa∙m0.5, welches einer Erhöhung um 20 % im Vergleich zum ungefüllten System
(3.1 MPa∙m0.5) entspricht. Eine ähnliche Erhöhung der Bruchzähigkeit wurde bei den Arbeiten
von ZHANG[255] und FINO
[214] mit 2 Gew.-% bzw. mit 2 Vol.-% Graphen erreicht.
Des Weiteren führte die Einarbeitung von 2 Gew.-% jedes Graphentyps zu einer deutlichen
Erhöhung spezifischen elektrischen Leitfähigkeiten. Die gesinterten SiC-Nanokomposite mit
TRGO, MG-Ethanol und Grade M erzielten aufgrund der großen Plättchenstruktur mit
2 Gew.-% Füllstoffgehalt eine leitfähige Netzwerkstruktur mit einer elektrischen Leitfähigkeit
von bis zu 0.7 S∙cm-1. Das ist ein großer Vorteil im Gegensatz zur Referenzprobe (3 ∙ 10-8 S∙cm-1)
und auch zum kommerziellen SiC Typ F (<10-8 S∙cm-1) und Typ G (10-4 – 10-5 S∙cm-1) der Firma
3M. Des Weiteren erzielten die erhaltenen Nanokomposite höhere Leitfähigkeit beim gleichen
Füllstoffgehalt gegenüber den Arbeiten von MIRANZO[238] und AI
[127]. Hinsichtlich der TRGO-
Proben führte der durch Erhöhung der Tred hervorgerufene steigende Kohlenstoffgehalt bzw.
der sinkende Funktionalisierungsgrad zu einer höheren Leitfähigkeit der entsprechenden
Nanokomposite (siehe Abbildung 102 Mitte links). Dies lässt sich auf die Wiederherstellung
einer großflächigen konjugierten sp2-Struktur in TRGO[235] bzw. die Ausheilung von Defekten

6 Zusammenfassende Diskussion
- 165 -
im TRGO durch thermische Regraphitisierung[236] zurückführen. Ähnlicher Zusammenhang
zwischen Kohlenstoffgehalt und Leitfähigkeit wurde ebenfalls bei MG-CO2-Nanokompositen
beobachtet (siehe Abbildung 102 Mitte rechts).
Die Zugabe von Graphen verwirklichte unabhängig von Graphentypen ebenfalls tribologische
Verstärkung, welche auf der Erhöhung der Verschleißbeständigkeit und Reduzierung des
Reibungskoeffizienten beruht. Nach der 4 h-Stift-Scheibe-Tribotest wurden die
Reibungskoeffizienten (µ) und Verschleißvolumina der mit Graphen gefüllten Tribosysteme
von 0.19 auf bis zu 0.07 bzw. von 0.22 auf bis zu 0.10 mm3 reduziert. Die Funktionalität des
TRGOs hatte keinen Einfluss auf die Reibungskoeffizienten und Verschleißbeständigkeit. Nach
der 4 h-Untersuchung lagen die Reibungskoeffizienten und Verschleißvolumen im Bereich von
ca. 0.14 bzw. 0.21 mm3 (siehe Abbildung 102 unten links). Im Gegensatz dazu sanken die
Reibungskoeffizienten und Verschleißvolumina bei den SiC-Nanokompositen mit MG-CO2
(siehe Abbildung 102 unten rechts) und MG-N2 mit steigender Mahldauer bzw. steigender
Funktionalität stetig auf bis zu 0.12 bzw. 0.16 mm3 ab. Im Vergleich zu der einzigen Arbeit mit
tribologischer Untersuchung der SiC-Graphen-Nanokomposite von BELMONTE[152] erzielt diese
Arbeit nicht nur eine Verbesserung der Verschleißbeständigkeit, sondern gleichzeitig auch eine
starke Reduzierung der Reibung. Weiterhin wurden erstmals die tribologischen Eigenschaften
der SiC-Graphen-Nanokomposite in wässerigen Medien untersucht, da die SiC-Komponenten
oft in solchen Anwendungen unter Wasserschmierung bzw. zur Förderung Wasser in
Wasserkraftwerk/Pumpe eingesetzt werden. Bei den vielversprechenden GSiC-
Nanokompositen wurde der Einfluss von Graphengehalt und Füllstofftyp auf die
Materialeigenschaften untersucht und in Abbildung 103 zusammengefasst.
Wie in Abbildung 103 (oben links) dargestellt, zeigten alle GSiC-Proben mit 2 Gew.-% eine
nahezu vollständige Verdichtung mit einer relativen Dichte über 99 %. Dank des vorteilhaften
Herstellungsverfahrens von Graphen in GSiC, bei dem das Graphen direkt auf der Oberfläche
von SiC erzeugt wurde, war es möglich, eine hohe relative Dichte bis zu 99 % mit 4 Gew.-%
Füllstoffgehalt zu gewährleisten. Der mit 8 Gew.-% Graphen gefüllte GSiC-F-Werkstoff besaß
die größte Porosität von 5.3 %, welche aber immer noch geringer als das Nanokomposit in der
Arbeit von FINO mit gleichem Füllstoffgrad ausfiel[214]. Daher wurde der Füllstoffgehalt bei
GSiC-G und GSiC-D auf maximal 4 Gew.-% beschränkt.

6 Zusammenfassende Diskussion
- 166 -
0 2 4 6 894
95
96
97
98
99
100
Re
lati
ve
Dic
hte
[%
]
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
6
5
4
3
2
1
0
Po
ros
itä
t [%
]
0 2 4 6 810-9
10-8
10-7
10-6
10-5
10-4
10-3
10-2
10-1
100
101
Ele
ktr
isc
he
Le
itfä
igk
eit
[S
. cm
-1]
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
Typ G
Typ F
0 2 4 6 8100
200
300
400
500
600
4-P
un
kt-
Bie
gefe
sti
gkeit
[M
Pa]
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
Typ F
Typ G
0 2 4 6 82,0
2,5
3,0
3,5
4,0
4,5
Bru
ch
zä
hig
ke
it [
MP
a. m
0,5
]
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
0 2 4 6 8
0,05
0,10
0,15
0,20
Reib
un
gsko
eff
izie
nt
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
0 2 4 6 80,05
0,10
0,15
0,20
0,25
Vers
ch
leiß
vo
lum
en
[m
m3]
Massenanteil [Gew.-%]
GSiC-F
GSiC-G
GSiC-D
Abbildung 103: Relative Dichte (oben links), elektrische Leitfähigkeit (oben rechts), mechanische
(Mitte) und tribologische Eigenschaften (unten) der gesinterten SiC-Nanokomposite mit GSiC-F (aus
Furfurylalkohol), GSiC-G (aus Glucose) und GSiC-D (aus Dopamin) bei variierender Graphen-
Konzentration.
Die Leitfähigkeiten stiegen kontinuierlich mit zunehmendem Graphengehalt und erreichten ein
Maximum bei 3 S∙cm-1 mit 8 Gew.-% GSiC-F, was einer Erhöhung um acht Größenordnungen
im Vergleich zum reinen SiC sowie um neun Größenordnungen zum Typ F und fünf
Größenordnungen zum Typ G der Firma 3M entspricht. Für Füllgrade um 4 Gew.-% zeigte die

6 Zusammenfassende Diskussion
- 167 -
GSiC-D den höchsten Leitwert von 0.07 S∙cm-1 (siehe Abbildung 103 oben rechts). Das hohe
elektrische Leitvermögen ermöglicht einerseits innovative Bearbeitungsverfahren wie das
Funkenerodieren und andererseits die Anwendung des SiC-Werkstoffs in der Sensorik (z. B.
Abgassensorik) und Brennstoffzellen.[1,177] Im Gegensatz zur Abnahme der Biegefestigkeit bei
den Nanokompositen mit allen anderen Graphenvarianten wiesen lediglich die Nanokomposite
mit GSiC-F-2% (507 MPa) sowie GSiC-D-2% (485 MPa) und GSiC-D-4% (490 MPa) eine
vergleichbare Biegefestigkeit wie das Referenzmaterial (505 MPa) und somit höhere Werte als
die kommerziellen Produkte Typ-F (400 MPa) und Typ-G (250 MPa) der Firma 3M auf. Des
Weiteren wurde die Bruchzähigkeit von 3.1 auf bis zu 3.7 MPa·m0.5 erhöht (siehe Abbildung
103 Mitte). Mit je 2 Gew.-% Füllstoffgehalt wurde der niedrigste Reibungskoeffizient bei
GSiC-D (0.10) erreicht, gefolgt von GSiC-F (0.13) und GSiC-G (0.16). Die Erhöhung des
Graphenanteils in GSiC führte zu einer kontinuierlichen Abnahme des Reibungskoeffizienten.
Dabei wurde der niedrigste Reibungskoeffizient aller mit Graphen gefüllten Proben bei GSiC-
D-4% und GSiC-F-8% mit dem Endwert von 0.09 beobachtet, was einer Reduzierung des
Reibungskoeffizienten um 54 % im Vergleich zur Referenzprobe entspricht (siehe Abbildung
103 unten links). Dieser Effekt konnte mit Hilfe atomistischer Simulationen erklärt werden.
Dabei zeigte sich, dass eine Bedeckung der SiC-Oberflächen mit Graphen in der Lage ist, die
Reibkräfte über einen weiten Bereich hinweg deutlich zu verringern. Diese entstandene
Graphen-Schutzschicht konnte nach dem Tribotest anhand von AFM-Aufnahmen
nachgewiesen werden. Außerdem führte die weitere Zugabe von Graphen zur verbesserten
Verschleißbeständigkeit und zur Einschränkung der Materialabnutzung. Den niedrigsten Wert
aller Proben erreichte ebenfalls die Probe mit 8 Gew.-% GSiC-F, was einer Reduzierung des
Verschleißvolumens um 40 % im Vergleich zum ungefüllten System entspricht (siehe
Abbildung 103 unten rechts). Gegenüber den Arbeiten von BELMONTE et al. zeigen die
Nanokomposite in dieser Arbeit nicht nur eine Verbesserung der Verschleißbeständigkeit,
sondern auch eine Verminderung der Reibungskoeffizienten.[152]
Des Weiteren wurde der technische Nutzen von SiC-Graphen-Nanokompositen insbesondere
im Hinblick auf die mögliche Anwendung in Pumpen bewertet. Dabei wurden Gleitringe aus
den SiC-Graphen-Nanokompositen hergestellt und dessen tribologischen Eigenschaften im
Vergleich zu den kommerziellen Gleitringprodukten der Firma 3M unter anwendungsnahen
Bedingungen untersucht. Die Reibungskoeffizienten aller Graphen-gefüllten SiC-
Nanokomposite wurden in Abbildung 104 aufgetragen.

6 Zusammenfassende Diskussion
- 168 -
Ref
. SiC
Typ
F
Typ
GTR
GO
-600
MG
-Eth
anol
MG
-CO
2-48
hM
G-C
O2-
72h
MG
-CO
2-12
0hG
SiC
-F-4
%G
SiC
-F-8
%G
SiC
-G-2
%G
SiC
-D-4
%Car
bon
Bla
ck
0,00
0,05
0,10
0,15
0,20
0,25 Referenz
TRGO
MG
GSiC
Carbon BlackR
eib
ungskoeffiz
ient
Abbildung 104: Reibungskoeffizienten aller Graphen-gefüllten SiC-Nanokomposite beim
anwendungsnahen Scheibe-Scheibe-Tribotest.
Durch Beimischung von Graphen konnte eine deutliche Verminderung des
Reibungskoeffizienten bei Festkörperreibung gegenüber der Referenzprobe und den
kommerziellen Typ F (0.23) und Typ G (0.20) der Firma 3M, erzielt werden. Des Weiteren
wiesen alle GSiC-Proben wie erwartet die niedrigsten Reibungskoeffizienten mit einer
maximalen Reduzierung von 0.21 bis zu 0.047 auf, was wiederum auf das vorteilhafte
Herstellungsverfahren zurückzuführen ist. Die große Reduzierung des Reibungskoeffizienten
von -78 % wurde erstmals erhalten und übersteigt alle literaturbekannten Beispielen von
Keramik-Graphen-Nanokompositen (-66 %[159] und im Fall SiC -10 %[152]). Darüber hinaus
zeichneten sich die Nanokomposite mit 4 und 8 Gew.-% GSiC-F durch eine sehr hohe
mechanische Belastungsgrenze aus, welche eine tribologische Messung bei 700 statt 250 N
ermöglichte. Damit könnte der Anwendungsbereich von SiC-Werkstoffen ausgeweitet werden.
Weiterhin wurde die Möglichkeit zur Herstellung von SiC-Graphen-Nanokompositen mittels
Spark-Plasma-Sinterverfahren, welches schnellere Sinterzyklen mit verbesserten
Werkstoffeigenschaften ermöglicht, anstatt dem konventionellem drucklosen Sinterverfahren

6 Zusammenfassende Diskussion
- 169 -
untersucht. Aufgrund seines extrem hohen spezifischen Durchgangswiderstands ist die
Verwendung des SPS-Verfahrens bei reinem SiC nicht möglich. Die Beimischung von Graphen
konnte eine elektrische Leitfähigkeit dieser Pulvermischungen bewirken, so konnte das SPS-
Verfahren zur Herstellung der SiC-Nanokomposite eingesetzt werden. Dabei zeigte TRGO in
SPS-Versuchen die beste Eignung. Deshalb wurde eine mit 4 Gew.-% TRGO-600 gefüllte
Keramikprobe hergestellt, die nach erfolgreicher SPS-Verdichtung eine deutlich höhere relative
Dichte (99.1 %), als die konventionell gesinterte Variante (89.9 %) aufwies. Als Folge erzielte
das TRGO-Nanokomposit nach SPS-Sinterung eine höhere Biegefestigkeit (358 MPa, +78 %)
und niedrigeren Reibungskoeffizienten (0.10, -33 %) im Vergleich zu dem aus konventionellen
Sinterverfahren hergestellten Nanokomposit. Weiterhin wurde die elektrische Leitfähigkeit um
8 Größenordnung auf 6 S∙cm-1 gegenüber reinem SiC (3 ∙ 10-8 S∙cm-1) erhöht. Diese hohe
elektrische Leitfähigkeit übersteigt alle literaturbekannte Werte von SiC-Graphen-
Nanokomposite mit ähnlichem Füllstoffgehalt.[127,238] Außerdem wurde die Verarbeitungszeit
im Vergleich zu drucklosem Sintern von 29 h auf 40 min verkürzt.
Zusammenfassend führte die Einarbeitung von Graphen zu tribologisch verstärkten SiC-
Nanokompositen mit verbesserten elektrischen und mechanischen Eigenschaften.
Diesbezüglich veranschaulicht Abbildung 105 die typischen Eigenschaften der ausgewählten
Nanokomposite mit je 2 Gew.-% GSiC-F, TRGO-1000 und MG-CO2-120h sowie
kommerziellem Carbon Black, Graphit und Grade M als Referenz. Dabei ist deutlich, dass die
Variante GSiC bei den SiC-Graphen-Nanokompositen mit Standard-Sinterprozess die besten
Eigenschaften zeigt. Hier wurde das Graphen mittels Präkursor-Verfahren direkt auf die SiC-
Oberfläche aufgebracht und war damit sehr gut in der Matrix verteilt. Die vorteilhafte
Herstellungsmethode von Graphen in GSiC bewirkte eine vollständige Verdichtung, die
höchste Biegefestigkeit und gleichzeitig auch maximale Verbesserung des Reibungs- und
Verschleißverhaltens. Bei der anschließenden Gleitring-Prüfung zeigten die GSiC-Proben
sowohl die niedrigsten Reibungskoeffizienten mit einer maximalen Reduzierung von 0.21 auf
bis zu 0.047 (-78 %) als auch eine sehr hohe mechanische Belastungsgrenze. Diese wesentliche
Senkung des Reibungskoeffizienten des SiC-Werkstoffs könnte zu erheblichen
Energieeinsparungen bei der Anwendung im Maschinenbau, z.B. Gleitringdichtungen und

6 Zusammenfassende Diskussion
- 170 -
94
98
96
100
150
300
450
2,4
2,7
3,010-6
10-3
100
0,18
0,15
0,12
0,24
0,20
0,16
Verschleiß
[mm3]
Reibungskoeffizient
Elektrische Leitfäigkeit [S.cm-1]
Bruchzähigkeit
[MPa.m0.5]
Biegefestigkeit
[MPa]
Relative Dichte [%]
GSiC-F
MG-CO2-120h
TRGO-1000
Ref. SiC
94
98
96
100
150
300
450
2,4
2,7
3,010-6
10-3
100
0,18
0,15
0,12
0,24
0,20
0,16
Verschleiß
[mm3]
Reibungskoeffizient
Elektrische Leitfäigkeit [S.cm-1]
Bruchzähigkeit
[MPa.m0.5]
Biegefestigkeit
[MPa]
Relative Dichte [%]
Carbon Black
Grade M
Graphit
Ref. SiC
Abbildung 105: Eigenschaftsprofil der nach Standardprozess gesinterten SiC-Nanokomposite mit
jeweils 2 Gew.-% GSiC-F, MG-CO2-120h und TRGO-1000 (oben) sowie Carbon Black, Grade M und
Graphit (unten).

6 Zusammenfassende Diskussion
- 171 -
Energieeinsparungen bei der Anwendung im Maschinenbau, z.B. Gleitringdichtungen und
Gleitlagern, führen. Zusätzlich könnte durch die Verbesserung der Verschleißbeständigkeit und
Belastbarkeit die Anlagenverfügbarkeit und die Ressourceneffizienz erhöht, sowie die Gefahr
für Menschen und Umwelt vermindert, werden.
Die anderen Nanokomposite mit TRGO-1000 und MG-CO2-120h sowie kommerziellem
Carbon Black, Graphit und Grade M zeigten ebenfalls verbesserte Reibungs- und
Verschleißverhalten sowie erhöhte elektrische Leitfähigkeit. Trotzdem schienen diese
Füllstoffe weniger für die Herstellung von SiC-Graphen-Nanokompositen geeignet, da sie zu
einer schlechteren Verteilung der Graphen-Partikel in der SiC-Matrix und in Folge dessen zu
schlechten mechanischen Eigenschaften führten.
Für das SPS-Sinterverfahren stellte sich der Füllstoff TRGO-600 in SiC als vielversprechend
heraus. Das erhaltene Keramik-Nanokomposit erreichte wesentlich bessere
Werkstoffeigenschaften als die konventionell gesinterte Variante mit einer verkürzten
Verarbeitungsdauer. Somit besteht durch Zugabe von Graphen die Chance, SiC-Werkstoffe zu
entwickeln, die im SPS-Prozess schneller und effizienter gesintert werden können.

6 Zusammenfassende Diskussion
- 172 -
6.3 Graphen-Nanokomposite auf Basis von Siliciumnitrid
Im Vergleich zu SiC-Werkstoffen wurden Si3N4-Graphen-Nanokomposite bereits intensiver
erforscht. Die Herstellung der Si3N4-Nanokomposite mit reduzierter Porosität konnte durch die
Zugabe von Graphen verwirklicht werden.[154] Darüber hinaus induzierte die Inkorporierung
von Graphen verbesserte mechanischen Eigenschaften.[130,178–181] Außerdem konnten die
tribologischen Eigenschaften durch die Einarbeitung von Graphen optimiert
werden.[131,132,154,162,178,182] Allerdings liegt ein kombinatorischer Anstieg der mechanischen und
tribologischen Eigenschaften mit vollständiger Verdichtung durch Graphen in Si3N4-
Nanokompositen bisher nicht vor und wurde in dieser Kapitel durch die Einarbeitung von
Graphen verwirklicht. Des Weiteren wurde der Einfluss weder von mechanochemisch
funktionalisiertem Multilagen-Graphen (MG) noch von GSi3N4 auf die Eigenschaften von
Si3N4-Werkstoffen bislang untersucht. Als Füllstoffe wurden sowohl die selbst synthetisierten
TRGO (TRGO-600), MG (MG-CO2-48h und MG-Ethanol) und GSi3N4-F als auch die
kommerziellen erhältlichen Graphenvariante (Grade C und Grade M) verwendet. Die meisten
Nanokomposite wurden mit einem Füllstoffgehalt von 3.0 Gew.-% hergestellt, um eine bessere
Vergleichbarkeit mit der Literatur[153,162,250–253] zu gewährleisten. Lediglich das GSi3N4-F-
Nanomoposit wurde aufgrund der unterschiedlichen Herstellungsmethode mit einem
Füllstoffgehalt von 0.5 Gew.-% synthetisiert (siehe Tabelle 20).
Tabelle 20: Übersicht der hergestellten Si3N4-Nanokomposite und ihrer Eigenschaften.
Probe Füllstoffgehalt
[Gew.-%]
Relative Dichte
[%]
Härtea
[GPa]
Bruchzähigkeitb
[MPa·m0.5]
Verschleißc
[mm3]
Ref. Si3N4 0 97.9 15.0 ± 0.5 5.9 ± 0.2 0.335
GSi3N4-F 0.5 97.4 17.5 ± 0.7 8.7 ± 1.3 0.240
MG-CO2-48h 3.0 93.2 11.6 ± 0.7 6.3 ± 0.3 0.194
Grade M 3.0 82.3 8.9 ± 1.7 6.6 ± 1.5 0.388
MG-Ethanol 3.0 79.6 5.8 ± 0.4 7.5 ± 1.3 0.490
TRGO-600 3.0 77.4 7.8 ± 1.5 n. b. 0.397
Grade C 3.0 74.2 4.0 ± 0.3 n. b. 0.472
a Vickers-Härte (HV); b bestimmt nach der Härteeindruckmethode; c Generierte Verschleißvolumina
nach Stift-Scheibe-Tribotest (4 h).

6 Zusammenfassende Diskussion
- 173 -
Die Messung der Dichtewerte zeigte, dass ausschließlich die GSi3N4-F-Probe über eine
ähnliche relative Dichte (97.4 %) wie das Referenzmaterial (97.9 %) verfügte. Im Gegensatz
dazu zeigten die anderen Nanokomposite in Analogie zur Literatur[153,154] niedrigere relative
Dichten im Bereich von 74 bis 93 % auf. Der Unterschied lässt sich vor allem wiederum mit
dem vorteilhaften Herstellungsverfahren von GSi3N4-F und mit der geringeren Füllstoffmenge
erklären. Die Verwendung von GSi3N4-F, bei dem Graphen über eine templatvermittelte
Reaktion direkt auf die Oberfläche von Si3N4 aufgebracht wurde, gewährleistete eine deutlich
homogenere Verteilung von Graphen in der Si3N4-Matrix, eine stärkere Partikel-Matrix-
Anbindungen, was als Folge zu einer höheren Verdichtung des Nanokomposits als unter Zugabe
von anderen Graphenen führte. Die gemessenen Dichtewerte spiegelten sich bei den
morphologischen Untersuchungen von den Nanokompositen exemplarisch mit GSi3N4-F,
TRGO-600, unfunktionalisiertem MG-Ethanol und Grade M wider. Anhand der TEM-
Aufnahmen in Abbildung 106 ist deutlich zu erkennen, dass durch die gewählte
Verarbeitungsmethode lediglich GSi3N4-F-Nanokomposit eine vollständig homogene und
porenfreie Morphologie aufweist. Dagegen beinhalteten alle anderen Nanokomposite große
Poren (helle Bereiche) und Graphen-Agglomerate (mit Pfeilen kenngezeichnet) im µm-Bereich.
Abbildung 106: TEM-Aufnahmen der gesinterten Si3N4-Nanokomposite mit a) GSi3N4-F, b)
TRGO-600, c) unfunktionalisiertem MG-Ethanol und d) Grade M. Die Pfeile deuten auf Graphen-
Agglomerate, während die hellen Bereiche offene Poren darstellen.
Die Einarbeitung von Graphen führte zur Verbesserung der Bruchzähigkeit. Abhängig von
Füllstofftypen wurde die Bruchzähigkeit von 5.9 auf bis zu 8.7 MPa∙m0.5 erhöht, was einer
Verstärkung um 48 % entspricht. Des Weiteren entsprachen die Härtewerte unabhängig von
Füllstofftypen in allen Fällen den relativen Dichten der Nanokomposite (siehe Abbildung 107

6 Zusammenfassende Diskussion
- 174 -
links). So zielte die GSi3N4-F-Probe eine Erhöhung der Härte um 17 % auf 17.5 GPa im
Vergleich zur Referenzprobe (15.0 GPa). Bei den zahlreichen Untersuchungen der Si3N4-
Graphen-Nanokomposite, welche mittels gleichem Sinterverfahren hergestellt
wurden[154,178,179,250,251], wurden schließlich bei den Arbeiten von BALÁZSI[249] eine Erhöhung
der Härtewerte (+6 %) und gleichzeitig eine Verbesserung der Bruchzähigkeit (+43 %) durch
Zugabe von 1 Gew.-% Multilagengraphen erreicht. Jedoch liegen diese Werte sowie die
Porosität (96.6 %)[132] weit unten den Werten von GSi3N4-F-Probe in dieser Arbeit. Im
Gegensatz dazu wiesen die anderen Si3N4-Graphen-Nanokomposite niedrigere Härte im
Bereich von 4 bis 12 GPa gegenüber reinem Si3N4 auf, was auf die entstandene Porosität
zurückzuführen ist und dem Verhalten in Literatur entspricht.[153,154,251]
Ref. Si3N4
GSi3N4-F
MG-CO2-48h
Grade M
TRGO-600
MG-Ethanol
Grade C
4
6
8
10
12
14
16
18 Härte
Relative Dichte
Hä
rte
[G
Pa
]
75
80
85
90
95
100
Re
lati
ve
Dic
hte
[%
]
MG-CO2-48h
GSi3N4-F
Ref. Si3N4
Grade M
TRGO-600
Grade C
MG-Ethanol
0,1
0,2
0,3
0,4
0,5 Verschleißvolumen
Porosität
Ve
rsc
hle
ißv
olu
me
n [
mm
3]
0
5
10
15
20
25
30
Po
ros
itä
t [%
]
Abbildung 107: Härte (links) und Verschleißwerte (rechts) der gesinterten Si3N4-Nanokomposite in
Abhängigkeit der Füllstofftypen, korreliert mit relativer Dichte bzw. Porosität.
Darüber hinaus liefern die Varianten mit MG-CO2-48h und GSi3N4-F reduzierte
Reibungskoeffizienten und verbesserte Verschleißbeständigkeit als die Referenzprobe. Ähnlich
wie die Härtewerte wurden die Verschleißwerte von der relativen Dichte bzw. der Porosität
wesentlich beeinflusst (siehe Abbildung 107 rechts). Dabei nahm das generierte
Verschleißvolumen der MG-CO2-48h-gefüllten Probe um 42 % auf 0.19 mm3 und gefolgt
GSi3N4-F-Probe (28 % bzw. 0.24 mm3) gegenüber der Referenzprobe (0.34 mm3) ab. Im
Vergleich dazu wiesen die Werkstoffvarianten mit Grade M, TRGO-600, Grade C und
MG-Ethanol, welche über eine große Porosität von mind. 17 % verfügten, höhere

6 Zusammenfassende Diskussion
- 175 -
Verschleißvolumina (bis 0.49 mm3) als reines Si3N4 auf, was auf die Verschlechterung der
mechanischen Eigenschaften (reduzierte Härte) und die erhöhten Reibungskoeffizienten
zurückgeführt wurde. Eine Verbesserung der Verschleißbeständigkeit wurde zwar in den
Literaturen[132,153,154,162] beschrieben, aber auf Kosten der relativen Dichte[132,153] und
mechanischen Eigenschaften (Härte[132,153,154,162], E-Modul[162] oder Bruchzähigkeit[154,162]).
80
90
100
8 12 16
6
7
8
9
0,40,30,2Verschleiß [mm3]
Bruchzähigkeit [MPa.m0.5]
Härte [GPa]
Relative Dichte [%] Ref. Si3N4
GSi3N4-F
MG-CO2-48h
Grade M
MG-Ethanol
TRGO-600
Grade C
Abbildung 108: Eigenschaftsprofil der gesinterten Si3N4-Nanokompositemit in Abhängigkeit der
Füllstofftypen.
Zusammenfassend konnten die mechanischen und tribologischen Eigenschaften von Si3N4-
Werkstoffen durch die Beimischung von Graphen wesentlich verbessert werden. Anhand der
Darstellung der erhaltenen Eigenschaften aller Si3N4-Graphen-Nanokomposite in
Abbildung 108 lässt sich erkennen, dass die GSi3N4-F-Probe zu höchstem
Verstärkungspotential in der Si3N4-Matrix führte. Die Einarbeitung des GSi3N4-F führte zur
vollständigen Verdichtung mit höchster Härte (17.5 GPa, +17 %), höchster Bruchzähigkeit
(8.7 MPa∙m0.5, +48 %) und verbesserter Verschleißbeständigkeit bzw. reduziertem

6 Zusammenfassende Diskussion
- 176 -
Verschleißvolumen (0.24 mm3, -28 %), was sich tendenziell mit den Ergebnissen der GSiC-
Nanokomposite in Kapitel 6.2 deckt. Dies beruht auf dem vorteilhaften Herstellungsverfahren
sowie dem geringen Füllstoffgehalt, bei dem das Graphen direkt auf der Oberfläche von Si3N4
erzeugt wurde, was eine homogene Verteilung von Graphen gewährleistet und in einer
porenfreien Struktur, guten mechanischen und tribologischen Eigenschaften resultiert. Durch
diese wesentlich verstärkten mechanischen und tribologischen Eigenschaften könnten
Lebensdauer, Zuverlässigkeit und Leistungsfähigkeit der Si3N4-Komponenten beim Betrieb
beispielsweise für den Anlagenbau (Wälz- & Gleitlager), insbesondere in Pumpen, Mischern
und Rührwerken markant verbessert werden. Die anderen Graphenvarianten hingegen sind
weniger als Füllstoff in Si3N4-Werkstoff geeignet. Diese führten zwar zur erhöhten
Bruchzähigkeit aber meistens auf Kosten der Verdichtung, Härtewerte, Reibungskoeffizienten
und Verschleißbeständigkeit. Die Gründe sind vor allem die ungleichmäßige Verteilung und
Agglomeratbildung, was die Fehlstellen und die hohe Porosität zur Folge hat.
Die Si3N4-Graphen-Nanokomposite wurden zwar intensiv erforscht, allerdings gibt es bisher
kein veröffentlichtes Beispiel, welches über vollständige Verdichtung, verstärkte mechanischen
Eigenschaften und gleichzeitig verbessertes tribologisches Verhalten verfügt. Gegenüber
literaturbekannten Arbeiten wurde das GSi3N4-F-Nanokomposit mit nur sehr geringem
Füllstoffgehalt von 0.5 Gew.-% in dieser Arbeit erfolgreich hergestellt, welches die
unkonventionellen Eigenschaftskombinationen mit vollständiger Verdichtung, steigenden
mechanischen und tribologischen Eigenschaften zeigte.[130–132,153,154,162,178–182,249–251,254]

6 Zusammenfassende Diskussion
- 177 -
6.4 Fazit und Ausblick
In Rahmen dieser Arbeit wurden Keramik-Graphen-Nanokomposite auf Basis von SiC und
Si3N4 hergestellt und untersucht. Es konnte gezeigt werden, dass (funktionalisierte) Graphene
vielversprechende Nanofüllstoffe darstellen, um die Materialeigenschaften der keramischen
Werkstoffe gezielt maßzuschneiden. Durch Einlagerung von Graphen gelang es die
tribologischen, mechanischen und elektrischen Eigenschaften von SiC und Si3N4 zu optimieren.
Die verwendeten Graphenmaterialien wurden in großem Maßstab (10 – 200 g) entweder aus
Graphit mittels Top-down-Methode oder aus einer Kohlenstoffquelle anhand des Bottom-up-
Ansatzes hergestellt und deren Wirkung in Relation zu Benchmark-Kohlenstofffüllstoffen
evaluiert.
Es konnte gezeigt werden, dass eine homogene Verteilung von Graphen in der Matrix von
zentraler Bedeutung für die Herstellung von Keramik-Nanokompositen ist. Das bisher größte
Problem bei der Anwendung von Graphen als Füllstoff in Keramiken ist die Entstehung von
Poren, was zu verschlechterten mechanischen Eigenschaften führte. Aufgrund dessen ist kein
SiC-Graphen-Nanokomposit, welches über reduzierte Reibung und reduzierten Verschleiß ohne
Verschlechterung der mechanischen Eigenschaften verfügt,
literaturbekannt.[127,133,136,152,173,214,238] Im Vergleich zu SiC-Werkstoffen wurden die Si3N4-
Graphen-Nanokomposite zwar intensiver erforscht, aber liegt ein kombinatorischer Anstieg der
mechanischen und tribologischen Eigenschaften mit vollständiger Verdichtung durch Graphen
in Si3N4-Nanokompositen bisher nicht vor.[130–132,153,154,162,178–182,249–251,254] Diese Probleme konnten
durch die Verwendung von graphenbeschichtetem Keramik gelöst werden, somit konnten
literaturunbekannte hervorragende Eigenschaften realisiert werden. Hierzu wurde das Graphen
mit Hilfe der Kohlenstoffquelle mittels Präkursor-Verfahren direkt auf die Keramik-Oberfläche
aufgebracht, wobei graphenbeschichtete SiC (GSiC) und Si3N4 (GSi3N4) erhalten wurden. Dies
begünstigt eine homogene Verteilung und Kompatibilisierung von Graphen in der Matrix und
hat somit eine höhere Verstärkungswirkung zur Folge.
Bei den SiC-Graphen-Nanokompositen mit Standard-Sinterprozess zeigte die Variante GSiC
die vielversprechendsten Eigenschaften. Im Vergleich zu allen anderen Graphenderivaten
bewirkte die vorteilhafte Herstellungsmethode von Graphen in GSiC mit identischem

6 Zusammenfassende Diskussion
- 178 -
Füllstoffgehalt (2 Gew.-%) eine höhere Verdichtung, die höchste Biegefestigkeit und
gelichzeitig auch maximale Verbesserung des Reibungs- und Verschleißverhaltens. Durch
Erhöhung des Füllstoffgehalts in GSiC konnten die tribologischen und elektrischen
Eigenschaften von SiC-Nanokompositen weiterhin verbessert werden aber teilweise auf Kosten
der Mechanik. Bei der anschließenden Bewertung des technischen Nutzens von SiC-
Nanokomposite als Gleitring zeigten die GSiC-Proben sowohl die niedrigsten
Reibungskoeffizienten mit einer maximalen Reduzierung von 0.21 auf bis zu 0.047 (-78 %) als
auch eine sehr hohe mechanische Belastungsgrenze. Diese markante Senkung des
Reibungskoeffizienten wurde erstmals erhalten und könnte zu erheblichen
Energieeinsparungen beim Betrieb der SiC-Komponenten beispielsweise für den Anlagenbau
(Wälz- & Gleitlager), insbesondere in Pumpen, Mischern und Rührwerken, führen. Zusätzlich
könnte der Anwendungsbereich von SiC-Werkstoffen durch erhöhte Belastungsgrenze
ausgeweitet werden, wie z.B. in hochbelasteten mechanischen Pumpen für Treibstoffförderung
in Flüssigkeitsraketentriebwerken. Außerdem könnte durch die Verbesserung der
Verschleißbeständigkeit die Anlagenverfügbarkeit und die Ressourceneffizienz erhöht sowie
die Gefahr für Menschen und Umwelt vermindert werden. Die anderen Nanokomposite mit
TRGO oder MG als Füllstoff zeigten ebenfalls verbesserte Reibungs- und Verschleißverhalten
sowie erhöhte elektrische Leitfähigkeit. Trotzdem schienen diese Füllstoffe weniger für die
Herstellung von SiC-Graphen-Nanokompositen geeignet, da sie zu einer schlechteren
Verteilung der Graphen-Partikel in der SiC-Matrix und in Folge zu Fehlstellen und schlechten
mechanischen Eigenschaften führten. Dieser Effekt des Beschichtungsverfahrens zur
Herstellung von Graphen spiegelt sich bei der Si3N4-Keramik wider. Die besten
Eigenschaftskombinationen von Si3N4-Nanokompositen wurden erstmals durch Einarbeitung
von GSi3N4 mit nur 0.5 Gew.-% Graphengehalt realisiert. Dabei wurde die Härte um 17 %, der
Bruchzähigkeit um 48 % und der Verschleißbeständigkeit um 28 % im Vergleich zu der
Referenzprobe verbessert. Damit könnte die Effizienz, Lebensdauer, Zuverlässigkeit und
Leistungsfähigkeit von Si3N4-Komponenten in den Anwendungen unter Wasser- bzw.
Mangelschmierung, aber auch zur Förderung anderer Medien verbessert werden. Ferne
schränkt sich diese Beschichtungsmethode zur Herstellung von Keramik-Graphen-
Nanokompositen nicht nur auf der SiC oder Si3N4-Matrix. Hier wären weiterführende

6 Zusammenfassende Diskussion
- 179 -
Untersuchungen bei anderen keramischen Materialien wie z.B. WC, B4C, BN, Al2O3, MgO und
ZrO2 denkbar. Im Gegensatz zum Standard-Sinterverfahren der SiC-Werkstoffe kann die
Verwendung von modernem SPS-Sinterverfahren durch die Beimischung von Graphen
realisiert werden, welches deutlich schnellere Sinterzyklen mit verbesserten
Werkstoffeigenschaften ermöglicht. Dabei zeigte TRGO in SPS-Versuchen die beste Eignung.
Das erhaltene SiC-TRGO-Nanokomposit erreichte wesentlich bessere Werkstoffeigenschaften
als die konventionell gesinterte Variante. Somit wäre das SPS-Verfahren auch im industriellen
Maßstab vorteilhaft zur Herstellung von vollständig dichten SiC-Bauteilen mit exzellenten, das
heutige Niveau übertreffende Eigenschaftsprofile einsetzbar. Außerdem wurde die elektrische
Leitfähigkeit bereits mit sehr geringem TRGO-Gehalt (4 Gew.-%) von 3 ∙ 10-8 auf 6 S∙cm-1 nach
erfolgreicher SPS-Sinterung angehoben. Ferner ermöglicht das hohe elektrische Leitvermögen
einerseits die Verwendung von innovativen Bearbeitungsverfahren wie das Funkenerodieren,
was eine Erweiterung der geometrisch darstellbaren Formen und Bauteilgeometrien bedeutet,
andererseits die Anwendung des SiC-Werkstoffs in der Sensorik (z. B. Abgassensorik) oder
Brennstoffzellen. Darüber hinaus könnten durch erhöhte elektrische Leitfähigkeiten der
Keramiken Schutzvorrichtungen zur weiteren Verminderung von Reibung, Verschleiß und
Korrosion entwickelt werden.
Keramik-Graphen-Nanokomposite zeigen das Potenzial zu wesentlich besserer
Schadenstoleranz (durch erhöhte Bruchzähigkeit), Lebensdauer und Ressourcenschonung
(durch erhöhte Verschleißbeständigkeit) sowie verbesserter Energieeffizienz (durch verbesserte
Fertigungsverfahren und verminderte Reibung). Ausblickend verspricht die Entwicklung von
technisch nutzbaren Werkstoffen und Komponenten auf Basis von Keramik-Graphen-
Nanokompositen einen großen technischen und wirtschaftlichen Nutzen auch weit über den
angesprochenen Anwendungsbereich hinaus. Weitere vorteilhafte Anwendungen für diese
neuen Nanokomposite sind zum Beispiel Umform- und Zerspanungswerkzeuge, die geringeren
Verschleiß und einen niedrigeren Kühlschmierstoffbedarf haben, keramische Wälzlager mit
erhöhten Bruchzähigkeit, Dickschichtsysteme als Beschichtungen in Wärmekraftmaschinen,
die zu höheren Betriebstemperaturen und Wirkungsgraden führen und letztlich alle
Maschinenelemente, die hohen tribologischen Beanspruchungen ausgesetzt sind.

7 Experimenteller Teil
- 180 -
7 Experimenteller Teil
7.1 Verwendete Chemikalien
7.1.1 Chemikalien und Lösungsmittel
Alle eingesetzten Chemikalien und Lösungsmittel sind in Tabelle 21 aufgelistet. Alle
Chemikalien wurden ohne weitere Aufreinigung direkt eingesetzt.
Tabelle 21: Verwendete Chemikalien und Lösungsmittel.
Chemikalie Reinheit
[%] Hersteller
Aceton 99.9 Analar Normapur
Argon 99.999 AirLiquide
CO2 99.999 AirLiquide
Dopamin·Hydrochlorid - Sigma-Aldrich
Ethanol ≥ 99.9 Merck
Furfurylalkohol > 98 Merck
D(+)-Glucose wasserfrei > 99.9 VWR Chemicals
Graphit KFL 99.5 99.5 AMG Mining
Kaliumpermanganat 99 Grüssing
N2 99.999 AirLiquide
Natriumhydroxid > 99 Merck
Natriumnitrat 99.5 Merck
2-Propanol (IPA) 99.99 Fisher Scientific
Salpetersäure 65 Merck
Schwefelsäure 95 VWR Chemicals
para-Toluolsulfonsäure > 98 Fluka
Tris(hydroxymethyl)-aminomethan (TRIS) ≥ 99.9 Sigma-Aldrich
Wasserstoffperoxid 30 Roth

7 Experimenteller Teil
- 181 -
7.1.2 Keramik-Rohstoffe & Benchmark Kohlenstofffüllstoffe
In Tabelle 22 sind die verwendeten Keramik-Rohstoffe und Sinterhilfsmittel sowie kommerziell
erhältliche Nanofüllstoffe aufgelistet, welche ohne weitere Aufreinigung direkt eingesetzt
wurden.
Tabelle 22: Verwendete Keramik-Rohstoffe und Sinterhilfsmittel sowie kommerzielle Nanofüllstoffe.
Bezeichnung Kommerzieller Name Hersteller
Aluminiumoxid (Al2O3) A16 Alcoa
Siliciumcarbid (SiC) PRC XF 13 FCT Ingenieurkeramik
α-Siliciumnitrid (α-Si3N4) SN-ESP Ube
Yttriumoxid (Y2O3) grade C H.C. Starck
Carbon Black Derussol N25/L Orion Engineered Carbons
Grade C GnP® Grade C – 750 XG Sciences
Grade M GnP® Grade M – 15 XG Sciences
Nanographit V-HF 98 AMG Mining
7.2 Herstellung von funktionalisiertem Graphen
7.2.1 Herstellung von Graphitoxid und thermisch reduziertem
Graphitoxid
Wie in Abbildung 8 in Kapitel 3.1 darstellt, wurde Graphitoxid (GO) in dieser Arbeit aus
Graphit durch Oxidation nach einer modifizierten HUMMERS-Methode in großem Maßstab
hergestellt[61,183] und nach einer Vorschrift in Anlehnung an das von TÖLLER beschriebene
Aufreinigungsverfahren gereinigt.[184] Dazu wurde Graphit (60 g, KFL 99.5, AMG Milling) in
einem Plastikbecher (5 L) mit konzentrierter Schwefelsäure (95 %, 1.4 L) und Natriumnitrat
(30 g) mit Hilfe eines KPG-Rührers (150 U/min) über Nacht suspendiert. Anschließend wurde
die Probe mit einem Eisbad auf 0 °C abgekühlt. Danach wurde feingepulvertes
Kaliumpermanganat (180 g) innerhalb von 2 h portionsweise zugegeben. Nach Entfernen des

7 Experimenteller Teil
- 182 -
Eisbades erwärmte sich die Dispersion durch Oxidation von selbst auf über 20 °C und wurde
für weitere 2 h weiter gerührt. Nachdem sich die Reaktionsmischung wieder abgekühlt hatte,
wurde die Dispersion langsam auf Eiswasser (1.4 L) gegossen, so dass die Temperatur nicht
über 70 °C stieg. Durch langsame Zugabe von verdünnter H2O2-Lösung (5 %, 200 mL) wurden
das unlösliche Braunstein und weitere Permanganatverbindungen zu löslichen Mn-(II)-Salzen
reduziert. Nach ca. 30 minutiger Gasentwicklung wurde die GO-Dispersion über einem
Büchnertrichter abfiltriert und mit H2O (4 × 5 L) und konzentrierter HNO3 (67 %, 4 × 400 mL)
gewaschen. Die entstandenen Sulfat- und Manganverunreinigungen im GO wurden mittels
Querstromfiltration größtenteils entfernt. Nach der Trocknung wurde ca. 80 g GO erhalten.
Thermisch reduziertes Graphitoxid (TRGO) wird aus Graphitoxid (GO) durch starkes Erhitzen
erzeugt. Zur Vorbereitung wurde das GO 7 d bei 40 °C im Vakuumtrockenschrank getrocknet
und anschließend mit der Kryomühle Cryomill der Firma Retsch unter Stickstoffkühlung
gemahlen. Die Herstellung von TRGO erfolgte durch Erhitzen von GO bei 600, 800 oder
1000 °C unter Stickstoffatmosphäre in einem Drehrohrofen Typ RSR 120/750/11 der Firma
Nabertherm GmbH (Abbildung 31). Der Drehrohrofen besteht aus einer Förderschnecke, einem
drehbaren Quarzglasrohr sowie einem Glasvorratsbehälter. Das gemahlene GO wurde dazu per
Förderschnecke in ein geheiztes Quarzglasrohr gefördert. Zunächst wurde eine hohe
Fördergeschwindigkeit gewählt bis das GO die heiße Zone erreichte. Danach wurde die
Geschwindigkeit deutlich reduziert bis es dem Auslass erreichte, wodurch das GO bzw. TRGO
im Ofen für ca. 2 min verweilte. Die typischen Eigenschaften von GO und TRGO sind in
Kapitel 3.2 zu finden.
Abbildung 109: Herstellung von TRGO aus GO mittels Drehrohrofen Typ RSR 120/750/11.

7 Experimenteller Teil
- 183 -
7.2.2 Herstellung von mechanochemisch funktionalisiertem
Multilagen-Graphen
Die Herstellung von mechanochemisch funktionalisiertem Multilagen-Graphen (MG) erfolgte
durch die Vermahlung von Graphit (KFL 99.5, AMG Mining) in einer Planetenkugelmühle
PM 100 der Firma Retsch mit einem Drehzahlverhältnis von 1 : -2 und bei einer Drehzahl von
250 U/min. Zunächst wurde der Graphit bei 60 °C sowie die Keramik-Mahlkammer
(Y0.05Zr0.95O2, V = 1150 cm3, Retsch) und -Kugeln (Y0.05Zr0.95O2, 96 Stück, Ø = 1 cm, Retsch)
bei 40 °C im Vakuumtrockenschrank (50 mbar) für mindestens 48 h getrocknet. Nach der
Beladung mit Graphit (18.6 g) wurde die Mahlkammer für 10 min am Hochvakuum evakuiert
(10-2 mbar) und mit dem entsprechenden Gas (Argon, CO2 oder N2) befüllt (13 bar). Die Proben
wurden in der Planetenkugelmühle für eine Dauer von 24 bis 120 h gemahlen und anschließend
an Luft-Atmosphäre geöffnet. Die jeweilige Analytik ist aus Kapitel 3.3 zu entnehmen.
7.2.3 Herstellung von graphenbeschichtetem Siliciumcarbid und
Siliciumnitrid
7.2.3.1 Herstellung von graphenbeschichtetem Siliciumcarbid aus Furfurylalkohol
Die Herstellung des graphenbeschichteten Siliciumcarbid aus Furfurylalkohol (GSiC-F)
erfolgte exemplarisch zunächst durch Dispergierung von SiC (152.55 g) in Wasser (200 mL).
Anschließend erfolgte die Zugabe von Furfurylalkohol (15 mL). Nach 10 min Rühren wurde
der Initiator p-TsOH (0.33 g) in Wasser (10 mL) gelöst und zum SiC/FA-Gemisch zugegeben.
Nach weiteren 20 min Rühren wurde das Reaktionsgemisch auf 80 °C erhitzt. Nach 1 h wurde
die Mischung für weitere 6 h bei 100 °C gehalten, bis das Wasser verdampft war. Das
getrocknete Pulver wurde anschließend gemörsert und in einem Rohrofen bei inerter
Atmosphäre (N2) karbonisiert. Dazu wurde bei einer Aufheizrate von 5 °C/min auf 150 °C
erhitzt, die Temperatur für 21 h gehalten und anschließend mit 5 °C/min auf 800 °C erhöht und
für 6 h thermolysiert. Die Variationen des Gewichtsanteils der Kohlenstoffquelle im
Reaktionsgemisch mit SiC sind in Tabelle 23 aufgelistet.

7 Experimenteller Teil
- 184 -
Tabelle 23: Reaktionsbedingungen zur Herstellung von GSiC-F.
Probe SiC FAa Verhältnisb p-TsOHc LMd Ausbeute Graphene 𝐀𝐁𝐄𝐓
𝐟
[g] [mL] [g] [g] [Gew.-%] [m2∙g-1]
GSiC-F-4.4% 152.55 15 90 : 10 0.33 H2O 157.56 4.4 39
GSiC-F-7.6% 90.40 20 80 : 20 0.44 Ethanol 96.33 7.6 53
GSiC-F-14.3% 105.47 40 70 : 30 0.88 Ethanol 114.73 14.3 100
a Furfurylalkohol; b Gewichtsverhältnis von SiC zu FA; c 1 mol% bezogen auf Furfurylalkohol; d Lösungsmittel; e Graphengehalt,
bestimmt durch TGA-Messung. Luft-Atmosphäre, 10 K/min, 50 – 750 °C; f N2-Physisorptionsmessung zur
Oberflächenbestimmung nach Brunauer, Emmett und Teller.
7.2.3.2 Herstellung von graphenbeschichtetem Siliciumcarbid aus Glucose
Die Herstellung des graphenbeschichteten Siliciumcarbid aus Glucose (GSiC-G) erfolgte
exemplarisch zunächst durch Dispergierung von SiC (20 g) in Wasser (90 mL) mittels
Ultraschallbad (2 × 15 min). Anschließend erfolgte die Zugabe der Lösung von Glucose (2.00 g)
in Wasser (10 mL). Nach Vordispergierung mittels Ultraschallbad (2 × 15 min) wurde das
Reaktionsgemisch mittels Ultraschalllanze (2 × 8 min, 40 % Amplitude) unter Eiskühlung
vollständig dispergiert. Danach wurde das Wasser durch Gefriertrocknung entfernt. Das
getrocknete Pulver wurde anschließend gemörsert und in einem Rohrofen bei inerter
Atmosphäre (N2) karbonisiert. Dazu wurde bei einer Aufheizrate von 5 °C/min auf 150 °C
erhitzt, die Temperatur für 21 h gehalten und anschließend mit 5 °C/min auf 800 °C erhöht und
für 6 h thermolysiert. Die Variationen des Gewichtsanteils der Kohlenstoffquelle im
Reaktionsgemisch mit SiC sind in Tabelle 24 aufgelistet.
Tabelle 24: Reaktionsbedingungen zur Herstellung von GSiC-G.
Probe SiC Glucose Verhältnisa Ausbeute Graphenb 𝐀𝐁𝐄𝐓
𝐜
[g] [g] [g] [Gew.-%] [m2∙g-1]
GSiC-G-2.0% 20 2.00 100 : 10 20.06 2.0 20
GSiC-G-5.2% 30 7.50 80 : 20 29.83 5.2 39
GSiC-G-8.8% 30 12.86 70 : 30 30.88 8.8 57
GSiC-G-13.4% 30 20.00 60 : 40 21.85 13.4 76
a Gewichtsverhältnis von SiC zu Glucose; b Graphengehalt, bestimmt durch TGA-Messung. Luft-Atmosphäre, 10 K/min, 50 –
750 °C; c N2-Physisorptionsmessung zur Oberflächenbestimmung nach Brunauer, Emmett und Teller.
7 Experimenteller Teil
- 185 -
7.2.3.3 Herstellung von graphenbeschichtetem Siliciumcarbid aus Dopamin
Die Herstellung des graphenbeschichteten Siliciumcarbid aus Dopamin (GSiC-D) erfolgte
exemplarisch zunächst durch Dispergierung von SiC (18 g) in Tris(hydroxymethyl)-
aminomethan-Lösung (80 mL; 100 mmol/L in H2O) mittels Ultraschallbad (15 min).
Anschließend erfolgte die Zugabe der Lösung von Dopamin (2.00 g) in Tris(hydroxymethyl)-
aminomethan-Lösung (20 mL; 100 mmol/L in H2O). Das Reaktionsgemisch wurde für weitere
72 h bei Raumtemperatur gerührt. Danach wurde das Wasser durch Gefriertrocknung entfernt.
Das getrocknete Pulver wurde anschließend gemörsert und in einem Rohrofen bei inerter
Atmosphäre (N2) karbonisiert. Dazu wurde bei einer Aufheizrate von 5 °C/min auf 150 °C
erhitzt, die Temperatur für 21 h gehalten und anschließend mit 5 °C/min auf 1000 °C erhöht
und für 6 h thermolysiert. Die Variationen des Gewichtsanteils der Kohlenstoffquelle im
Reaktionsgemisch mit SiC sind in Tabelle 25 aufgelistet.
Tabelle 25: Reaktionsbedingungen zur Herstellung von GSiC-D.
Probe SiC Dopamin Verhältnisa Tb Dauerc Ausbeute Graphend σe 𝐀𝐁𝐄𝐓
𝐟
[g] [g] [°C] [h] [g] [Gew.-%] [S/m] [m2∙g-1]
GSiC-D-5.2% 18 2 90 : 10 RT 72 18.53 5.2 5.3 35
GSiC-D-8.4% 16 4 80 : 20 60 96 16.23 8.4 4.7 66
GSiC-D-13.5% 14 6 70 : 30 60 120 16.03 13.5 2.8 106
a Gewichtsverhältnis von SiC zu Dopamin; b Reaktionstemperatur; c Dauer der Polymerisation; d Graphengehalt, bestimmt durch
TGA-Messung. Luft-Atmosphäre, 10 K/min, 50 – 750 °C; e Ermittelt durch Vierpunktmessung; f N2-Physisorptionsmessung
zur Oberflächenbestimmung nach Brunauer, Emmett und Teller.
7.2.3.4 Herstellung von graphenbeschichtetem Siliciumnitrid aus Furfurylalkohol
Die Herstellung des graphenbeschichteten Silciumnitrid aus Furfurylalkohol (GSi3N4-F)
erfolgte exemplarisch zunächst durch Dispergierung von Si3N4 (101.70 g) in Wasser (200 mL).
Anschließend erfolgte die Zugabe von Furfurylalkohol (10 mL). Nach 10 min Rühren wurde
der Initiator p-TsOH (0.22 g) in Wasser (10 mL) gelöst und zum Si3N4/FA-Gemisch zugegeben.
Nach weiten 20 min Rühren wurde das Reaktionsgemisch auf 80 °C erhitzt. Nach 1 h wurde
die Mischung bei 100 °C für weitere 6 h gehalten, bis das Wasser verdampft war. Das
getrocknete Pulver wurde anschließend gemörsert und in einem Rohrofen bei inerter

7 Experimenteller Teil
- 186 -
Atmosphäre (N2) karbonisiert. Dazu wurde bei einer Aufheizrate von 5 °C/min auf 150 °C
erhitzt, die Temperatur für 21 h gehalten und anschließend mit 5 °C/min auf 800 °C erhöht und
für 6 h thermolysiert.
Tabelle 26: Reaktionsbedingungen zur Herstellung von GSi3N4-F.
Probe Si3N4 FAa Verhältnisb p-TsOHc Ausbeute Graphend 𝐀𝐁𝐄𝐓
𝐞
[g] [mL] [g] [g] [Gew.-%] [m2∙g-1]
GSi3N4-F 101.70 10 90 : 10 0.22 99.41 0.5 17
a Furfurylalkohol; b Gewichtsverhältnis von Si3N4 zu FA; c 1 mol% bezogen auf Furfurylalkohol; d Graphengehalt, bestimmt
durch TGA-Messung. Luft-Atmosphäre, 10 K/min, 50 – 750 °C; e N2-Physisorptionsmessung zur Oberflächenbestimmung
nach Brunauer, Emmett und Teller.
7.3 Herstellung von SiC-Graphen-Nanokompositen
7.3.1 Konventioneller Sinter-Ofen
Die Herstellung der SiC-Graphen-Nanokomposite mittels konventionelles Sinter-Ofens
erfolgte in Zusammenarbeit mit M. KNOCH von der Firma FCT Ingenieurkeramik GmbH in
Sonneberg. Hierfür wurde die SiC-Basisformulierung aus reinem SiC und Sinteradditiven
verwendet, welche chemische Zusammensetzung aus patentrechtlichen Gründen hier nicht
näher beschreiben werden kann. Dabei wurden die SiC-Basisformulierung (100 g, 100 Gew.-%)
und Graphen (6 g, 2 Gew.-%, bezogen auf die Pulvermasse) mittels Planetenkugelmühle für 4 h
und mittels Turbula Shaker Mixer für weitere 4 h gemischt und homogenisiert. Dabei wurde
das Graphen in Form einer stabilen Dispersion verwendet, welche durch den in Kapitel 3.1.1
beschriebenen Hochdruckhomogenisierungsprozess in Wasser hergestellt wurde. Nach der
Siebung (Korngröße 63 µm) erfolgte die Rücktrocknung und Granulierung der Probe über eine
Gefriertrocknung. Anschließend wurden aus dem so gewonnenen Granulat (Korngröße
< 180 µm) Probeplatten durch kaltisostatisches Pressen bei 2000 bar Druck verdichtet und
zuletzt im Sinter-Ofen drucklos bei 2050 °C unter Argon für 4 h gesintert. Aus der Platte
wurden Prüfkörper gefräst, die anschließend noch abgeschliffen wurden.

7 Experimenteller Teil
- 187 -
Zur besseren Vergleichbarkeit erfolgte die Herstellung von GSiC-Nanokompositen durch
Mischung von GSiC und reinem SiC, um einen ganzzahligen Graphengehalt zu erhalten.
Beispielsweise wurde das GSiC-F-2%-Nanokomposit aus der SiC-Basisformulierung (170 g,
57 Gew.-%) und GSiC-F-4.4% (136 g, 45 Gew.-%). Tabelle 27 gibt die Zusammensetzungen
der Variationen wieder. Dabei ist der gegebene Graphengehalt bezogen auf die Pulvermasse
von der SiC-Basisformulierung.
Tabelle 27: Zusammensetzungen der Rohstoffe zur Herstellung von GSiC-Nanokompositen.
Nanokomposit
Zusammensetzung
GSiC SiC-Basisformulierung
Typ [g] [Gew.-%] [g] [Gew.-%]
GSiC-F-2% GSiC-F-4.4% 136 45 170 57
GSiC-F-4% GSiC-F-7.6% 158 53 154 51
GSiC-F-8% GSiC-F-14.3% 168 56 156 52
GSiC-G-2% GSiC-G-5.2% 115 38 191 64
GSiC-G-4% GSiC-G-8.8% 136 45 176 59
GSiC-D-2% GSiC-D-5.2% 115 38 191 64
GSiC-D-4% GSiC-D-5.2% 231 77 81 27
7.3.2 Spark Plasma Sintern/Feld-Aktiviertes Sintern
Die Herstellung der SiC-TRGO-600-Nanokomposite mittels Spark Plasma Sintern/Feld-
Aktiviertes Sintern (SPS/FAST-Sintern) erfolgte in Zusammenarbeit mit M. KNOCH von der
Firma FCT Ingenieurkeramik GmbH in Sonneberg. Hierfür wurden die SiC-Basisformulierung
(60.6 g, 100 Gew.-%) und TRGO-600 (2.4 g, 4 Gew.-%, bezogen auf die Pulvermasse) mittels
Planetenkugelmühle für 4 h und mittels Turbula Shaker Mixer für weitere 4 h gemischt und
homogenisiert. Dabei wurde das TRGO-600 in Form einer stabilen Dispersion verwendet,
welche durch den in Kapitel 3.1.1 beschriebenen Hochdruckhomogenisierungsprozess in
wässrigem Medium hergestellt wurde. Nach der Siebung (Korngröße 63 µm) erfolgte die
Rücktrocknung und Granulierung der Probe über eine Gefriertrocknung. Anschließend wurde
das gewonnene Granulat (Korngröße < 180 µm) als Probeplatten in der Spark-Plasma-
Sinteranlage zwischen zwei Graphitstempeln (Ø 60 mm) mit einem Druck von 60 MPa in einer

7 Experimenteller Teil
- 188 -
Si3N4-Matrize verdichtet (Versuchsanlage siehe Abbildung 84 in Kapitel 4.8). Dabei wurde an
den Stempeln eine elektrische Spannung von maximal 10 V angelegt, bis sich die Temperatur
auf 2050 °C erhöhte. Zuletzt wurde die Probe unter demselben Druck bei 2050 °C unter Argon
für 5 min gesintert. Aus der Probe wurden Prüfkörper gefräst, die anschließend noch
abgeschliffen wurden.
7.4 Herstellung von Si3N4-Graphen-Nanokompositen
Die Si3N4-Graphen-Nanokomposite wurden in Zusammenarbeit mit Dr. C. BALÁZSI und Dr. K.
BALÁZSI vom Institute for Technical Physics and Materials Science, Centre for Energy
Research in Budapest synthetisiert. Hierfür erfolgte die Herstellung zunächst durch Mischung
und Homogenisierung der Si3N4-Matrix (72 g, 90 Gew.-%) mit den Sinterhilfsmitteln Al2O3
(3.2 g, 4 Gew.-%) und Y2O3 (4.8 g, 6 Gew.-%) in Ethanol per Attritormühle (01-HD/HDDM
der Firma Union Process) bei 4000 U/min für 5 h. Nach Zugabe von Graphen (1.2 g, 3 Gew.-%,
bezogen auf die Pulvermasse) wurden die Dispersionen für weitere 30 min homogenisiert. Nach
dem Erhalt stabiler Dispersionen erfolgte die Rücktrocknung und Granulierung der Probe.
Anschließend wurde die so gewonnene Granulat-Probe (11 g) nach Siebung (Korngröße
150 µm) bei 220 MPa Druck zwischen zwei Keramikstempeln (Ø 30 mm) verdichtet. Zuletzt
wurde die Sinterung der Probe durch heißisostatisches Pressen (HIP der Firma ABRA) unter
einem Druck von 20 MPa bei 1700 °C unter Stickstoff-Atmosphäre für 3 h durchgeführt. Aus
der Probe wurden Prüfkörper gefräst, die anschließend noch abgeschliffen wurden.

7 Experimenteller Teil
- 189 -
7.5 Charakterisierungsmethoden und verwendete Geräte
7.5.1 Atomkraftmikroskopie (AFM)
Zur AFM Messung von Graphenen wurde eine Graphen-Dispersion (Aceton, 1 mg/mL) mittels
Spin Coating (WS-400B-6NPP/LITE/8K der Firma Laurell) auf ein Glassubstrat aufgebracht.
Die mechanische Abtastung der Oberflächen wurde mit Hilfe eines Atomkraftmikroskops
Nanoscope III der Firma Veeco durchgeführt. Die AFM-Aufnahmen von SiC-Graphen-
Nanokompositen wurden mit Hilfe eines Atomkraftmikroskops Dimension V der Firma Bruker
Ltd. durchgeführt und mit der Software NanoScope Analysis 1.8 ausgewertet.
7.5.2 Biegefestigkeit
Die 4-Punkt-Biegefestigkeit σB der Keramik-Graphen-Nanokomposite wurde in
Zusammenarbeit mit Prof. J. DUSZA von The Institute of Materials Research of SAS (IMR) in
Košice gemäß DIN EN 843-1 mit Hilfe einer Zug/Laden-Maschine LR5K-Plus der Firma Lloyd
Instruments bei Raumtemperatur unter Luft-Atmosphäre ermittelt. Die Charakterisierung
erfolgte unter Verwendung von Stäbchen mit rechteckigem Querschnitt (Länge l: 45 mm, Breite
b: 4 mm, Dicke h: 3 mm), welche mit einer Diamant-Schleifscheibe (Rauheit: 15 µm) poliert
wurden. Dabei wurde die Prüfprobe auf zwei Auflagen (Abstand S1: 40 mm) positioniert und
in der Mitte mit einem Prüfstempel mit zwei Druckpunkten (Abstand S2: 20 mm) belastet. Die
Probe wurde unter Last bei einer Prüfgeschwindigkeit von 0.5 mm/min bis zum Bruch belastet.
Danach wurde die 4-Punkt-Biegefestigkeit σB mit Hilfe der Gleichung 5 aus der beim
Brucheintritt angezeigten Höchstlast P und den geometrischen Abmessungen der Probe und des
Auflagers berechnet. Bei jeder Probe wurden mindestens 7 Messungen durchgeführt und
anschließend gemittelt.
2
21
2
3
bh
)SS(PB
(5)

7 Experimenteller Teil
- 190 -
7.5.3 Bruchzähigkeit
Die Bruchzähigkeit der SiC-Graphen-Nanokomposite wurde in Zusammenarbeit mit Prof. J.
DUSZA von The Institute of Materials Research of SAS (IMR) in Košice nach der Single-Edge
V-Notched-Beam-Methode (SEVNB) mit Hilfe einer Zug/Laden-Maschine LR5K-Plus der
Firma Lloyd Instruments bei Raumtemperatur unter Luft-Atmosphäre ermittelt. Die
Charakterisierung erfolgte unter Verwendung von Stäbchen mit rechteckigem Querschnitt
(Länge l: 45 mm, Breite b: 4 mm, Dicke h: 3 mm), welche mit einer Diamant-Schleifscheibe
(Rauheit: 15 µm) poliert wurden. In der Mitte des Keramikbiegestäbchens wurde eine V-
förmige Kerbe mit möglichst scharfem Spitzenradius und einer Tiefe a (0.8 bis 1.2 mm) mit
einer diamantierten Rasierklinge eingebracht. Für den Kerbvorgang wurde Diamantpaste mit
anfangs großer Korngröße (6 – 3 μm), zum späteren Polieren des Kerbgrunds eine kleinere
Korngröße (1 μm) benutzt. Nach dem Reinigen der einzelnen Proben wurde der
Kerbdurchmesser mittels Mikroskop überprüft. Bei der Messung wurde die Prüfprobe auf zwei
Auflagen (Abstand S1: 40 mm) positioniert und in der Mitte mit einem Prüfstempel mit zwei
Druckpunkten (Abstand S2: 20 mm) belastet. Die Probe wurde unter Last bei einer
Prüfgeschwindigkeit von 0.5 mm/min bis zum Bruch belastet. Danach wurde die
Bruchzähigkeit K1C mit Hilfe der SRAWLEY-Gleichung 6 aus der beim Brucheintritt angezeigten
Höchstlast P und den geometrischen Abmessungen der Probe und des Auflagers berechnet,
wobei Y Geometriefaktor und α der relativen Kerbtiefe entspricht. Bei jeder Probe wurden die
Messungen mindestens 3 Mal wiederholt und anschließend gemittelt.
Y)(h
SS
hb
PYK
.BIC
51
21
12
3
(6)
mit 2
2
1
1351680493326198871
)(
)()...(..Y
und h
a
Aufgrund der kleinen Probemenge erfolgte die Bruchzähigkeitsbestimmung von Si3N4-
Graphen-Nanokompositen in Zusammenarbeit mit Dr. C. BALÁZSI und Dr. K. BALÁZSI vom

7 Experimenteller Teil
- 191 -
Institute for Technical Physics and Materials Science, Centre for Energy Research in Budapest
nach der Härteeindruckmethode mit einem Micro-Vickers Härteprüfgerät MVK-H1 der Firma
Mitutoyo. Dabei wurden zunächst Vickers-Härteeindrücke (10 N, 10 s) in die
Werkstoffoberfläche eingebracht. Aus dem Härtewert (H), der aufgebrachten Eindruckskraft (P)
und der Länge (l) der an den Ecken auftretenden Risse wurde der Bruchzähigkeitswert (KIC)
unter Zuhilfenahme der SHETTY-Gleichung 7 berechnet.[131,179,249,256]
50
408990 .
IC )l
PH(.K (7)
7.5.4 Brunauer-Emmett-Teller Analyse (BET)
Die spezifische Oberfläche der Proben wurde mittels Stickstoff Adsorption-Desorption nach
Brunauer-Emmett-Teller (BET) Theorie mit Hilfe eines Sorptomatic 1990 der Firma
POROTEC GmbH gemessen. Dazu wurden ca. 100 mg Probe verwendet und eine Dichte von
ρ = 0.1 g/cm3 angenommen. Die Auswertung erfolgte durch den 2-Parameter-Fit.
7.5.5 Drehrohrofen
Die thermische Reduktion von GO zu TRGO erfolgte in einem Drehrohrofen Typ RSR
120/750/11 der Firma Nabertherm GmbH. Das gemahlene GO wurde dazu per Förderschnecke
in ein geheiztes Quarzglas-Rohr geleitet und dort unter Stickstoffatmosphäre reduziert.
7.5.6 Elementaranalyse (EA)
Bei der Elementaranalyse wurde der C-, H-, N- und S-Gehalt der Probe durch Verbrennung und
anschließende gaschromatographische Bestimmung der Verbrennungsgase ermittelt. Die
Messungen wurden mit dem Modell VarioEL der Firma Elementaranalysensysteme GmbH
durchgeführt. Der O-Gehalt wurde durch Differenzbildung auf 100 % berechnet.

7 Experimenteller Teil
- 192 -
7.5.7 Energiedispersive Röntgenspektroskopie (EDX)
Zur Bestimmung der elementaren Zusammensetzung wurden bei einigen Proben ergänzend zur
EA die energiedispersive Röntgenspektroskopie verwendet. Dies erfolgte an einem Quanta 250
FEG der Firma FEI mit einem x act Zusatzdetektor von Oxford Instruments. Mit einer
Beschleunigungsspannung von 20 oder 30 kV wurde die Probe angeregt und die
charakteristische gestreute Röntgenstrahlung detektiert.
7.5.8 FT-IR-Spektroskopie
Die Probenpräparation erfolgte durch Mörsern mit wasserfreiem KBr und durch anschließendes
Pressen mit einer Kraft von 10 kN für zwei Minuten. Die erhaltenen Presslinge wurden mit dem
Vektor 22 FT-IR-Spektrometer der Firma Bruker vermessen. Dazu wurden 32 Scans im Bereich
von 4000 – 400 cm-1 bei einer Auflösung von 2 cm-1 durchgeführt. Die resultierenden Daten
wurden mit der Software OPUS (Vers. 5.5) ausgewertet. Zur Hintergrundkorrektur wurde das
Spektrum eines reinen KBr-Presslings verwendet.
7.5.9 Härte
Die Vickershärte (HV) der hergestellten Si3N4-Graphen-Nanokomposite erfolgte in
Zusammenarbeit mit Dr. C. BALÁZSI und Dr. K. BALÁZSI vom Institute for Technical Physics
and Materials Science, Centre for Energy Research in Budapest mit einem Micro-Vickers
Härteprüfgerät MVK-H1 der Firma Mitutoyo. Als Indenter wurde ein Diamant-Eindringkörper
in Form einer Pyramide mit quadratischer Basis und einem Winkel von 136° zwischen
gegenüberliegenden Flächen benutzt. Bei der Prüfung wurde der Indenter mit einer
Anpresskraft von 10 N stoßfrei auf die Probenoberfläche gedrückt. Die Vickershärte wird
gegeben durch den Quotienten aus angewandter Kraft F und der Fläche des Eindrucks.
Gemessen werden die Diagonalen d1 und d2 des Eindruckes. Die Vickershärte HV wird nach

7 Experimenteller Teil
- 193 -
der Gleichung 8 mit d = (d1 + d2)/2 berechnet. Der Messwert wurde durch mehrmaliges Messen
und anschließende Mittelwertbildung erhalten.
2
41854d
F.HV (8)
7.5.10 Hochdruckhomogenisator
Die stabilen und homogenen TRGO- und MG-Dispersionen wurden mit Hilfe eines
Hochdruckhomogenisators Panda NS1001L 2K der Firma GEA Niro Soavi verwendet. Die
Graphen-Dispersionen wurden nach Vordispergierung mittels Ultraschallbehandlung
(2 × 15 min) in den Hochdruckhomogenisator gegeben und bei einem Druck von ungefähr
1000 bar einmal dispergiert.
7.5.11 Kryomühle
Das getrocknete GO wurde mit der Cryomill der Firma Retsch gemahlen. Dazu wurde die
Substanz zusammen mit einer Stahlkugel in eine Edelstahlkammer gegeben. Nach Kühlung mit
flüssigem Stickstoff wurde für 90 Sekunden bei einer Frequenz von 25 s-1 gemahlen.
7.5.12 Leitfähigkeitsmessung
Die elektrischen Messungen der Keramik-Graphen-Nanokomposite erfolgten in
Zusammenarbeit mit M. KNOCH von der Firma FCT Ingenieurkeramik GmbH in Sonneberg mit
Digital-Multimeter 72812 der Firma ELV® an Keramikstäbchen mit rechteckigem Querschnitt
(Länge l: 45 mm, Breite b: 4 mm, Dicke h: 3 mm). Die Kanten der Stäbchen wurden mit
Leitsilber überzogen, um einen besseren Kontakt zwischen dem Probenkörper und der Klemme
zu gewährleisten. Der spezifische Widerstand wurde über die gleichen Formeln wie bei der
Leitfähigkeitsmessung von Graphen bestimmt, mit dem Abstand d zwischen den

7 Experimenteller Teil
- 194 -
Leitsilberkontakten und der Querschnittsfläche A des Probenkörpers. Die spezifische
Leitfähigkeit ergibt sich nach Gleichung 3 und 4 in Kapitel 4.6 aus dem Kehrwert des
spezifischen Widerstandes.
7.5.13 LUMiSizer
Die Stabilitätsanalyse sowie die Bestimmung der Partikelgrößenverteilung der Graphen-
Dispersionen erfolgte mit der analytischen Photozentrifuge LUMiSizer der Firma LUM GmbH.
Die frisch hergestellten Dispersionen (1 mg/mL) wurden in einer Polycarbonat-Küvette
(Durchmesser: 2 mm) vermessen. Die Messung erfolgte bei einer Drehzahl von 1000 U/min
bei einer Temperatur von 25 °C für 100000 s (ca. 28 h) mit Intervallen von 10 s. Zur
Evaluierung der Stabilität wurde das Sedimentationsverhalten der Partikel im Zentrifugalfeld
beobachtet und quantifiziert. Um die Bewegungsprozesse in der Messküvette zu detektieren
wird die Transmission des eingestrahlten Lichts aus einer NIR-Quelle orts- und zeitabhängig
gemessen. Der LUMiSizer® zeichnet ortsaufgelöste Transmissionsprofile im Verlauf eines
Sedimentationsvorgangs auf. Daraus kann der Instabilitätsindex I ermittelt werden
(Gleichung 10). Dieser wurde durch die Aufklarung A als Veränderung der ortsaufgelösten
Transmission der Probe beschrieben (Gleichung 9). Die Berechnung erfolgt über die
Differenzbildung des Transmissionsprofils T zur Zeit ti und des Profils zu Beginn der Messung
T(t0). Die Aufklarung bezieht sich dabei immer auf eine definierte Position r in der Küvette.
)t,r(T)t,r(T)t(A ii 0 (9)
)t,r(T)t,r(T
)t,r(T)t,r(T
A
)t(A)t(I
max
i
max
ii
0
0
(10)
Anschließend wurde der Instabilitätsindex I als dimensionslose Größe aus dem Verhältnis der
zeitlichen Funktion der Aufklarung A(ti) zur maximalen Aufklarung Amax am Ende der Messung
berechnet (Gleichung 10).

7 Experimenteller Teil
- 195 -
Die Partikelgrößenverteilung wurde ebenfalls mittels LUMiSizer nach ISO 13318 bestimmt
(Gleichung 11). Dabei wurde die kumulative Verteilung Q aus dem Verhältnis der örtlichen
Funktion der Extinktion Er zur maximalen Extinktion Emax berechnet. Die Messung beruht auf
dem Lambert-Beerschen Gesetz (Gleichung 12). Dieses beschreibt die Abschwächung der
Intensität T einer Strahlung beim Durchgang eines Mediums mit einer absorbierenden Substanz.
Die Extinktion E errechnet sich hierbei aus dem Produkt des Extinktionskoeffizienten ε, der
Konzentration c der absorbierenden Substanz und der Küvettendicke d.
max
rr
E
EQ (11)
dc)T
Tlg(E
0
(12)
Die Partikelgröße wurde nach der Gleichung 13, basierend auf dem Stokesschen Gesetz,
ermittelt. Gleichung 13 beschreibt die Berechnung der Partikelgröße x in Abhängigkeit der
Startposition r0, einer manuell gewählten konstanten Position rm, der Dichte von Graphen ρp
und des Lösungsmittels ρL, der Viskosität des Lösungsmittels ηL, der Zeit t und der
Winkelgeschwindigkeit ω.
)r
rln(
t)(
m
Lp
L
0
2
18
(13)
7.5.14 Rasterelektronenmikroskopie (SEM)
Die SEM-Aufnahmen wurden mit einem Quanta 250 FEG der Firma FEI aufgenommen. Zur
Detektion der zurück gestreuten Elektronen wurde ein „Low Voltage High Contrast“ Detektor
verwendet. Die Beschleunigungsspannung betrug 20 oder 30 kV.

7 Experimenteller Teil
- 196 -
7.5.15 Thermogravimetrische Analyse (TGA)
Die Analyse des thermischen Abbauverhaltens erfolgte an der Thermowaage STA 409 der Firma
Netzsch GmbH. Zur Probenvorbereitung wurden 10 – 15 mg Substanz in einen Korund-Tiegel
eingewogen. Die Messung wurde entweder bei einer Temperatur von 50 bis 750 °C oder von
50 bis 1500 °C durchgeführt und erfolgte bei einer Aufheizrate von 10 °C/min unter
Stickstoffatmosphäre oder Luft (Durchfluss von 75 cm3/min). Die Auswertung der Daten
erfolgte mit der Software Netzsch Proteus-Thermal Analysis (Vers. 6.1.0). Die Masseverluste
und Restmassen beinhalten einen Fehler von ± 0.5 %.
Die TGA-Messung der in Kapitel 3.5 ausgewählten Proben erfolgte in Zusammenarbeit mit
PD Dr. D. KLIMM vom Institut für Kristallzüchtung (IKZ) in Berlin an der Thermowaage STA
409C in einem Graphit-Ofen der Firma Netzsch GmbH. Dieses Messgerät ist mit einem
Quadrupol-Massenspektrometer gekoppelt, welches die simultane Analyse der entstehenden
Gase ermöglicht. Zur Probenvorbereitung wurden 10 – 15 mg Substanz eingewogen. Die
Messung wurde bei einer Temperatur von 25 bis 2000 °C durchgeführt und erfolgte bei einer
Aufheizrate von 15 °C/min unter Argonatmosphäre.
7.5.16 Transmissionselektronenmikroskopie (TEM)
Die Transmissionselektronenmikroskopie ermöglichte eine direkte Abbildung der Proben in
hoher Vergrößerung. Die TEM-Aufnahmen von Graphenen wurden mit einem Zeiss/LEO 912
Omega Transmissionselektronenmikroskop bei einer Beschleunigungsspannung von 120 kV
gemacht. Zur Untersuchung wurde die Dispersion (1 mg/mL in Aceton) auf ein Cu-Grid
gegeben und getrocknet. Die TEM-Aufnahmen von Si3N4-Graphen-Nanokompositen wurden
in Zusammenarbeit mit Dr. C. BALÁZSI und Dr. K. BALÁZSI vom Institute for Technical Physics
and Materials Science, Centre for Energy Research in Budapest mit einem CM 20 der Firma
Philips bei einer Beschleunigungsspannung von 200 kV gemacht. Des Weiteren wurde die
hochauflösende Transmissionselektronenmikroskopie (HRTEM) von GSiC-Proben in

7 Experimenteller Teil
- 197 -
Abbildung 35 in Kapitel 3.4.4 ebenfalls in Zusammenarbeit mit Dr. C. BALÁZSI und Dr. K.
BALÁZSI vom Institute for Technical Physics and Materials Science, Centre for Energy
Research in Budapest mit einem JEM-3010 der Firma Jeol bei einer Beschleunigungsspannung
von 300 kV durchgeführt.
7.5.17 Tribologie
7.5.17.1 Stift-Scheibe-Tribometer
Die tribologische Prüfung der Keramik-Graphen-Nanokomposite wurde in Zusammenarbeit
mit Dr. C. SCHRÖDER, Dr. B. SCHLÜTER und Dr. A. KAILER vom Fraunhofer-Institut für
Werkstoffmechanik in Freiburg mit Hilfe eines Stift-Scheibe-Tribometers TRM 1000 der Firma
Dr.-Ing. Georg Wazau Meß- + Prüfsysteme GmbH im rotatorischen Bewegungsmodus bei
Raumtemperatur in destilliertem Wasser ermittelt. Als Gegenkörper wurden raupolierte
(Rauheit = 0.65 μm) Gleitringe aus drucklos gesintertem Siliciumcarbid der Firma
EagleBurgmann Germany GmbH & Co. KG verwendet. Abbildung 65 in Kapitel 4.7.1 zeigt
schematisch die verwendeten Probenkörper und die verwendete Testkonfiguration. Die
Charakterisierung erfolgte unter Verwendung eines feinpolierten (Rauheit = 0.01 – 0.02 μm)
Stifts mit rechteckigem Querschnitt (Länge l: 10 mm, Breite b: 4 mm, Dicke h: 4 mm), an
jeweils einer Stirnseite wurde eine Kugelkalotte (r = 5 mm) gefertigt, welche aus Keramik-
Graphen-Nanokompositen in Kooperation mit der Firma FCT Ingenieurkeramik GmbH in
Sonneberg hergestellt wurde. Der Radius der durch die Gleitreibung zwischen Stift und
Gleitring gebildeten Reibspuren war 26.5 mm. Die aufgebrachte Normalkraft betrug 50 N, die
Gleitgeschwindigkeit 0.1 m/s. Die Versuche erfolgten in wässrigem Medium bei
Raumtemperatur für eine Belastungsdauer von 4 oder 20 h. Bei jeder Probe wurden die
Messungen mindestens 3 Mal wiederholt und anschließend gemittelt.
Nach der tribologischen Untersuchung wurden die generierten Verschleißspuren und
Verschleißflächen mit dem Digitalmikroskop VHX-500F der Firma Keyence Deutschland

7 Experimenteller Teil
- 198 -
GmbH analysiert. Das Verschleißvolumen wurde unter Berücksichtigung des Kugelradius
(r = 5 mm) berechnet.
7.5.17.2 Scheibe-Scheibe-Tribometer
Zusätzlich wurde die tribologische Prüfung der Keramik-Graphen-Nanokomposite mit Hilfe
eines Scheibe-Scheibe-Tribometers TRM 2000 der Firma Dr.-Ing. Georg Wazau Meß- +
Prüfsysteme GmbH im rotatorischen Bewegungsmodus bei Raumtemperatur in destilliertem
Wasser ermittelt. Als Prüfkörper wurden die Gleitringpaare aus Keramik-Graphen-
Nanokompositen in Kooperation mit der Firma FCT Ingenieurkeramik GmbH in Sonneberg
hergestellt und von der Firma EagleBurgmann Germany GmbH & Co. KG in Wolfratshausen
durch Politur mit speziellem Diamantkorn auf eine funktionale Oberflächentopografie gebracht
(Rauheit = 0.15 μm). Abbildung 75 in Kapitel 4.7.2 zeigt schematisch die verwendeten
Probenkörper und die verwendete Testkonfiguration. Die Versuche erfolgten unter den von
EagleBurgmann Germany GmbH & Co. KG etablierten standardisierten Bedingungen in
wässrigem Medium bei Raumtemperatur für eine Belastungsdauer von 320 min (siehe
Abbildung 76 in Kapitel 4.7.2). Bei jeder Probe wurden die Messungen mindestens 3 Mal
wiederholt und anschließend gemittelt.
7.5.18 Ultraschalllanze
Zur Homogenisierung wurde die Probe per Ultraschalllanze Sonifier 450 D der Firma Branson
Ultrasonics dispergiert. Die Spitze der Lanze tauchte ca. 1 cm in das Medium ein. Es wurde
mit einer Leistung von 40 % (160 Watt) unter Eisbad-Kühlung für 2 × 8 min (5 min Pause)
dispergiert.

8 Kurzzusammenfassung
- 199 -
8 Kurzzusammenfassung
Zentrales Ziel der vorliegenden Dissertation war die Herstellung und Charakterisierung von neuartigen Keramik-
Nanokompositen auf Basis von Siliciumcarbid (SiC) und Siliciumnitrid (Si3N4) mit bislang unbekannten
Materialeigenschaftskombinationen. Die Nanokomposite wurden entweder aus dem Gemisch von reinem
Keramikrohstoff und Graphen oder aus graphenbeschichtetem Keramikrohstoff durch Sintern hergestellt. Das
bisher größte Problem bei der Anwendung von Graphen als Füllstoff in Keramiken ist die Entstehung von Poren,
was zur Verschlechterung der mechanischen Eigenschaften führt. Um eine homogene Durchmischung von
Graphen und Keramik zu gewährleisten bzw. die Porenvolumina von hergestellten Keramik-Graphen-
Nanokompositen zu minimieren, wurden Graphene mittels Präkursor-Verfahren direkt auf die Oberfläche der
Keramikrohstoffe aufgebracht, wobei graphenbeschichtete SiC (GSiC) und Si3N4 (GSi3N4) erhalten wurden. Im
Vergleich dazu kamen als reine Graphene thermisch reduziertes Graphitoxid (TRGO) und mechanochemisch
funktionalisiertes Multilagen-Graphen (MG) aus trockener Vermahlung von Graphit zum Einsatz, dessen
Eigenschaften (Funktionalität und Morphologie) durch die einfache Variation der Prozessbedingungen
(Reduktionstemperatur bzw. Mahldauer und Gasatmosphäre) gesteuert werden konnten. Außerdem wurden
kommerzielle plättchenförmige Kohlenstofffüllstoffe Graphit, Nanographit, Grade M (Multilagen-Graphen) und
MG-Ethanol (unfunktionalisiertes Multilagen-Graphen), sowie die sphärischen Kohlenstofffüllstoffe Carbon
Black und Grade C als Referenz untersucht. Bei den SiC-Nanokompositen nach Standard-Sinterprozess wurden
die besten Eigenschaften durch die Verwendung von GSiC erreicht. Dies bewirkte bei 2 Gew.-% Graphengehalt
die höchste Sinterdichte (99.9 %) und die höchste Biegefestigkeit (507 MPa). Gleichzeitig wurde eine
Verbesserung der Verschleißbeständigkeit um 32 % erreicht, wobei das Verschleißvolumen sich von 0.22
(Referenzprobe) auf 0.15 mm3 reduzierte. Des Weiteren wurde erstmals eine massive Reduzierung des
Reibungskoeffizienten von 0.19 auf 0.13 erhalten. Durch Erhöhung des Füllstoffgehalts bis zu 8 Gew.-% an GSiC
konnten die tribologischen (Reibung: -60 %; Verschleiß: -40 %) und elektrischen (3 S∙cm-1, +8 Größenordnungen)
Eigenschaften von SiC-Nanokompositen weiterhin verbessert werden, jedoch teilweise auf Kosten der Mechanik.
Bei der anschließenden Bewertung des technischen Nutzens von SiC-Nanokompositen als Gleitring zeigten die
GSiC-Proben sowohl die niedrigsten Reibungskoeffizienten mit einer maximalen Reduzierung von 0.21 auf bis zu
0.047 (-78 %) als auch eine erhöhte mechanische Belastungsgrenze. Diese tribologische Leistung konnte mit Hilfe
atomistischer Simulationen erklärt werden. Dabei zeigte sich, dass eine Bedeckung der SiC-Oberflächen mit
Graphen in der Lage ist, die Reibkräfte über einen weiten Bereich hinweg deutlich zu verringern. Dieser Effekt
des Beschichtungsverfahrens zur Herstellung von Graphen spiegelt sich bei der Si3N4-Keramik wider. Die besten
Eigenschaftskombinationen von Si3N4-Nanokompositen wurden erstmals durch Verwendung von GSi3N4 mit nur
0.5 Gew.-% Graphengehalt realisiert. Dabei wurde die Härte um 17 %, die Bruchzähigkeit um 48 % und die
Verschleißbeständigkeit um 28 % im Vergleich zur Referenzprobe verbessert. Die anderen Keramik-
Nanokomposite nach Standard-Sinterprozess zeigten ebenfalls verbesserte Reibungs- und Verschleißverhalten,
sowie erhöhte elektrische Leitfähigkeit. Trotzdem waren diese Füllstoffe weniger für die Herstellung von Keramik-
Graphen-Nanokompositen geeignet, da sie zu einer schlechteren Verteilung der Graphen-Partikel in der Keramik-
Matrix und in Folge zu Fehlstellen und schlechten mechanischen Eigenschaften führten. Bei der Herstellung von
SiC-Nanokompositen mittels SPS-Verfahren (Spark Plasma Sintern) stellte sich der Füllstoff TRGO als
vielversprechend heraus. Das erhaltene Nanokomposit erreichte wesentlich bessere Werkstoffeigenschaften als die
konventionell gesinterte Variante mit einer verkürzten Verarbeitungsdauer (von 29 h auf 40 min). Dabei wurde die
elektrische Leitfähigkeit bereits mit geringem TRGO-Gehalt (4 Gew.-%) von 3 ∙ 10-8 auf 6 S∙cm-1, nach
erfolgreicher SPS-Sinterung, angehoben.

8 Kurzzusammenfassung
- 200 -
Abstract
The main goal of this dissertation was the fabrication and characterization of novel ceramic
nanocomposites based on silicon carbide (SiC) and silicon nitride (Si3N4) with sofar unknown
combinations of material properties. The nanocomposites were made from either the mixture of pure
ceramic and pure graphene or graphene-coated ceramic by sintering. The biggest problem of using
graphene as a filler in ceramics is the formation of pores, resulting in degraded mechanical properties.
To ensure a homogeneous mix of graphene and ceramics and to minimize the pore volumes of
manufactured ceramic-graphene nanocomposites, graphenes were applied directly to the surface of the
ceramic raw materials using precursor methods to obtain graphene-coated SiC (GSiC) and Si3N4
(GSi3N4). Compared to this, thermally reduced graphite oxide (TRGO) and mechanochemically
functionalized multilayer graphene (MG) from dry grinding of graphite were employed as pure graphene
and there properties (functionality and morphology) were controlled through simple variation of the
process conditions like reduction temperature, grinding time and gas atmosphere. In addition, platelet-
like carbon additives graphite, nanographite, Grade M (multilayer graphene) and MG-Ethanol
(unfunctionalized multilayer graphene), as well as the spherical carbon fillers carbon black and Grade C
served as commercial reference fillers. For the SiC nanocomposites after standard sintering, the best
properties were achieved using GSiC. This resulted in the highest density (99.9 %) and the highest
flexural strength (507 MPa) at 2 wt% graphene content. At the same time, the wear resistance was
improved by 32 % with reduced wear volume from 0.22 (reference sample) to 0.15 mm3. Furthermore,
a massive reduction of the friction coefficient (from 0.19 to 0.13) was obtained for the first time. By
increasing the filler content (up to 8 wt%) of GSiC, the tribological (friction: -60 %; wear: -40 %) and
electrical (3 S∙cm-1, +8 magnitude) properties of SiC nanocomposites could be further improved, but
partly at a cost of the mechanics. In the subsequent evaluation of the technical application of SiC
nanocomposites as a slide ring, the GSiC samples showed the lowest friction coefficients with a
maximum reduction from 0.21 to 0.047 (-78 %) as well as an increased mechanical load limit. This
tribological performance could be explained with atomistic simulations. It was found that a coverage of
the SiC surfaces with graphene layer is able to reduce significantly the friction force over a wide range.
This effect of the graphene coating process is reflected in the Si3N4 ceramic. The best property
combinations of Si3N4 nanocomposites were for the first time realized by using GSi3N4 with only 0.5
wt% graphene content. The hardness was improved by 17 % and the fracture toughness by 48 % at
simultaneously the wear resistance by 28 % compared to the reference sample. The other ceramic
nanocomposites after standard sintering process showed also improved friction and wear behavior, as
well as increased electrical conductivity. Nevertheless, these fillers were less suitable for the production
of ceramic-graphene nanocomposites because of a poor distribution of graphene particles in the ceramic
matrix which consequently led to defects and poor mechanical properties. In the production of SiC
nanocomposites by means of SPS (Spark Plasma Sintering) the filler TRGO turned out to be promising.
The obtained nanocomposite achieved significantly better material properties than the conventionally
sintered variant with a shortened processing time (from 29 h to 40 min). The electrical conductivity was
increased by eight orders of magnitude from 3 ∙ 10-8 to 6 S∙cm-1 with a low TRGO content (4 wt%) after
successful SPS sintering.

9 Lebenslauf
- 201 -
9 Lebenslauf
Die Seite 201 (Lebenslauf) ist aufgrund persönlicher Daten nicht Bestandteil der Online
Veröffentlichung.

10 Literaturverzeichnis
- 202 -
10 Literaturverzeichnis
[1] H. Salmang, H. Scholze, R. Telle, Keramik (German Edition), Springer, Dordrecht, 2007.
[2] Informationszentrum Technische Keramik im Verband der Keramischen Industrie e.V., Brevier
Technische Keramik, Fahner, Lauf, 2003.
[3] W. Kollenberg, Technische Keramik: Grundlagen, Werkstoffe, Verfahrenstechnik, Vulkan-Verlag,
2004.
[4] T. Haase, Keramik, Deutscher Verlag für Grundstoffindustrie, Leipzig, 1968.
[5] https://www.grandviewresearch.com/industry-analysis/ceramics-market. abgerufen am
23.01.2018.
[6] J.-W. An, D.-H. You, D.-S. Lim, Wear 2003, 255, 677.
[7] A. D. MacNaught, A. Wilkinson, Compendium of chemical terminology. IUPAC
recommendations, Blackwell Science, Oxford, 1997.
[8] C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam, A. Govindaraj, Angew. Chem. 2009, 121, 7890.
[9] A. K. Geim, K. S. Novoselov, Nat. Mater. 2007, 6, 183.
[10] C. Soldano, A. Mahmood, E. Dujardin, Carbon 2010, 48, 2127.
[11] C. Lee, X. Wei, J. W. Kysar, J. Hone, Science 2008, 321, 385.
[12] X. Du, I. Skachko, A. Barker, E. Y. Andrei, Nat. Nanotechnol. 2008, 3, 491.
[13] J. S. Bunch, S. S. Verbridge, J. S. Alden, A. M. van der Zande, J. M. Parpia, H. G. Craighead, P.
L. McEuen, Nano Lett. 2008, 8, 2458.
[14] A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao, C. N. Lau, Nano Lett.
2008, 8, 902.
[15] A. B. Kuzmenko, E. van Heumen, F. Carbone, D. van der Marel, Phys. Rev. Lett. 2008, 100,
117401.
[16] R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres,
A. K. Geim, Science 2008, 320, 1308.
[17] B. Trauzettel, Phys. J. 2007, 7, 39.
[18] M. I. Katsnelson, Mater. Today 2007, 10, 20.
[19] K. Balasubramanian, M. Burghard, Chem. unserer Zeit 2011, 45, 240.
[20] G. Ruess, F. Vogt, Monatsh. Chem. verw. Tl. 1948, 78, 222.
[21] H. P. Boehm, A. Clauss, G. O. Fischer, U. Hofmann, Z. anorg. allg. Chem. 1962, 316, 119.
[22] H. P. Boehm, R. Setton, E. Stumpp, Pure Appl. Chem. 1994, 66, 1893.
[23] H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K.
Prud'homme, R. Car, D. A. Saville, I. A. Aksay, J. Phys. Chem. B 2006, 110, 8535.
[24] K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V.
Grigorieva, A. A. Firsov, Science 2004, 306, 666.
[25] J. H. Warner, F. Schaffel, M. Rummeli, A. Bachmatiuk, Graphene. Fundamentals and emergent
applications, Elsevier, Amsterdam, 2013.
[26] X. Li, W. Cai, I. Jung, J. An, D. Yang, A. Velamakanni, R. Piner, L. Colombo, R. S. Ruoff, ECS
Trans. 2009, 19, 41.

10 Literaturverzeichnis
- 203 -
[27] X. Li, C. W. Magnuson, A. Venugopal, J. An, J. W. Suk, B. Han, M. Borysiak, W. Cai, A.
Velamakanni, Y. Zhu et al., Nano Lett. 2010, 10, 4328.
[28] X. Wang, H. You, F. Liu, M. Li, L. Wan, S. Li, Q. Li, Y. Xu, R. Tian, Z. Yu et al., Chem. Vap.
Deposition 2009, 15, 53.
[29] A. W. Robertson, J. H. Warner, Nano Lett. 2011, 11, 1182.
[30] A. Reina, X. Jia, J. Ho, D. Nezich, H. Son, V. Bulovic, M. S. Dresselhaus, J. Kong, Nano Lett.
2009, 9, 30.
[31] Z. Li, L. Zhang, B. Li, Z. Liu, Z. Liu, H. Wang, Q. Li, Chem. Eng. J. 2017, 313, 1242.
[32] A. Kumar, S. Khan, M. Zulfequar, Harsh, M. Husain, Appl. Surf. Sci. 2017, 402, 161.
[33] R. Mo, D. Rooney, K. Sun, H. Y. Yang, Nat. Commun. 2016, 8, 13949.
[34] H. Sakaguchi, S. Song, T. Kojima, T. Nakae, Nat. Chem. 2017, 9, 57.
[35] F. Iacopi, N. Mishra, B. V. Cunning, D. Goding, S. Dimitrijev, R. Brock, R. H. Dauskardt, B.
Wood, J. Boeckl, J. Mater. Res. 2015, 30, 609.
[36] S. F. a. U. Starke, J. Phys. D: Appl. Phys. 2014, 47, 94013.
[37] L. Chen, L. Guo, Y. Wu, Y. Jia, Z. Li, X. Chen, RSC Adv. 2013, 3, 13926.
[38] W. Lu, L. Guo, Y. Jia, Y. Guo, Z. Li, J. Lin, J. Huang, W. Wang, RSC Adv. 2014, 4, 46771.
[39] X. Yang, X. Dou, A. Rouhanipour, L. Zhi, H. J. Räder, K. Müllen, J. Am. Chem. Soc. 2008, 130,
4216.
[40] X. Yan, X. Cui, L.-s. Li, J. Am. Chem. Soc. 2010, 132, 5944.
[41] W. A. Chalifoux, Angew. Chem. Int. Ed. 2017, 56, 8048.
[42] S. Smalley, M. Lahti, K. Pussi, V. R. Dhanak, J. A. Smerdon, J. Chem. Phys. 2017, 146, 184701.
[43] X.-H. Li, S. Kurasch, U. Kaiser, M. Antonietti, Angew. Chem. Int. Ed. 2012, 51, 9689.
[44] M. Beckert, Albert-Ludwigs-Universität Freiburg, Freiburg im Breisgau, 2014.
[45] M. Beckert, M. Menzel, Tolle, F. J., B. Bruchmann, R. Mulhaupt, Green Chem. 2015, 17, 1032.
[46] R. Li, K. Parvez, F. Hinkel, X. Feng, K. Müllen, Angew. Chem. Int. Ed. 2013, 52, 5535.
[47] K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov, A. K. Geim,
Proc Natl Acad Sci U S A 2005, 102, 10451.
[48] X. Jiang, L. T. Drzal, Polym. Compos. 2010, 31, 1091.
[49] W. A. de Heer, C. Berger, X. Wu, P. N. First, E. H. Conrad, X. Li, T. Li, M. Sprinkle, J. Hass, M.
L. Sadowski et al., Solid State Commun. 2007, 143, 92.
[50] M. Hirata, T. Gotou, S. Horiuchi, M. Fujiwara, M. Ohba, Carbon 2004, 42, 2929.
[51] M. H. Wahid, E. Eroglu, X. Chen, S. M. Smith, C. L. Raston, Green Chem. 2013, 15, 650.
[52] P. Kun, F. Wéber, C. Balázsi, Open Chem. 2011, 9, 47.
[53] A. E. Del Rio-Castillo, C. Merino, E. Díez-Barra, E. Vázquez, Nano Res. 2014, 7, 963.
[54] K. Tschoppe, F. Beckert, M. Beckert, R. Mülhaupt, Macromol. Mater. Eng. 2015, 300, 140.
[55] F. Beckert, S. Bodendorfer, W. Zhang, R. Thomann, R. Mülhaupt, Macromolecules 2014, 47,
7036.
[56] V. Leon, M. Quintana, M. A. Herrero, J. L. G. Fierro, A. d. La Hoz, M. Prato, E. Vazquez, Chem.
Commun. 2011, 47, 10936.
[57] I.-Y. Jeon, Y.-R. Shin, G.-J. Sohn, H.-J. Choi, S.-Y. Bae, J. Mahmood, S.-M. Jung, J.-M. Seo, M.-
J. Kim, D. Wook Chang et al., PNAS 2012, 109, 5588.

10 Literaturverzeichnis
- 204 -
[58] T. Xing, J. Sunarso, W. Yang, Y. Yin, A. M. Glushenkov, L. H. Li, P. C. Howlett, Y. Chen,
Nanoscale 2013, 5, 7970.
[59] N.-W. Pu, C.-A. Wang, Y. Sung, Y.-M. Liu, M.-D. Ger, Mater. Lett. 2009, 63, 1987.
[60] C. K. Chua, M. Pumera, Chem. Soc. Rev. 2014, 43, 291.
[61] W. S. Hummers, R. E. Offeman, J. Am. Chem. Soc. 1958, 80, 1339.
[62] D. R. Dreyer, S. Park, C. W. Bielawski, R. S. Ruoff, Chem. Soc. Rev. 2010, 39, 228.
[63] Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts, R. S. Ruoff, Adv. Mater. 2010, 22, 3906.
[64] H.-P. Boehm, W. Scholz, Justus Liebigs Ann. Chem. 1966, 691, 1.
[65] B. C. Brodie, Phil. Trans. R. Soc. Lond. 1859, 149, 249.
[66] A. Lerf, H. He, M. Forster, J. Klinowski, J. Phys. Chem. B 1998, 102, 4477.
[67] D. Pandey, R. Reifenberger, R. Piner, Surf. Sci. 2008, 602, 1607.
[68] T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis, I. Dékány, Chem. Mater.
2006, 18, 2740.
[69] L. B. Casabianca, M. A. Shaibat, W. W. Cai, S. Park, R. Piner, R. S. Ruoff, Y. Ishii, J. Am. Chem.
Soc. 2010, 132, 5672.
[70] O. C. Compton, S. T. Nguyen, Small 2010, 6, 711.
[71] D. Marquardt, F. Beckert, F. Pennetreau, F. Tölle, R. Mülhaupt, O. Riant, S. Hermans, J. Barthel,
C. Janiak, Carbon 2014, 66, 285.
[72] H. Chen, C. Huang, W. Yu, C. Zhou, Polymer 2013, 54, 1603.
[73] B. Dittrich, K.-A. Wartig, D. Hofmann, R. Mülhaupt, B. Schartel, Polym. Degrad. Stab. 2013,
98, 1495.
[74] M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-
Alonso, D. L. Milius, R. Car, R. K. Prud'homme et al., Chem. Mater. 2007, 19, 4396.
[75] P. Steurer, R. Wissert, R. Thomann, R. Mülhaupt, Macromol. Rapid Commun. 2009, 30, 316.
[76] R. Prud'homme, I. Aksay, D. Adamson, A. Abdala, US20070092432 A1, 2007.
[77] F. Cataldo, J. Nanosci. Nanotechnol. 2007, 7, 1446.
[78] T. Ong, H. Yang, Carbon 2000, 38, 2077.
[79] C. I. Smith, H. Miyaoka, T. Ichikawa, M. O. Jones, J. Harmer, W. Ishida, P. P. Edwards, Y.
Kojima, H. Fuji, J. Phys. Chem. C 2009, 113, 5409.
[80] W. Zhao, F. Wu, H. Wu, G. Chen, J. Nanomater. 2010, 2010, 5.
[81] G. Bacon, Acta Crystallogr. 1950, 3, 320.
[82] Y. Yao, Z. Lin, Z. Li, X. Song, K.-S. Moon, C.-p. Wong, J. Mater. Chem. 2012, 22, 13494.
[83] I.-Y. Jeon, H.-J. Choi, M. J. Ju, I. T. Choi, K. Lim, J. Ko, H. K. Kim, J. C. Kim, J.-J. Lee, D. Shin
et al., Sci. Rep. 2013, 3, 2260.
[84] I.-Y. Jeon, H.-J. Choi, S.-M. Jung, J.-M. Seo, M.-J. Kim, L. Dai, J.-B. Baek, J. Am. Chem. Soc.
2013, 135, 1386.
[85] F. Beckert, S. Trenkle, R. Thomann, R. Mülhaupt, Macromol. Mater. Eng. 2014, 299, 1513.
[86] M. Tommasini, C. Castiglioni, G. Zerbi, A. Barbon, M. Brustolon, Chem. Phys. Lett. 2011, 516,
220.
[87] J. Tang, W. Zhao, L. Li, A. U. Falster, W. B. Simmons, W. L. Zhou, Y. Ikuhara, J. H. Zhang, J.
Mater. Res. 1996, 11, 733.

10 Literaturverzeichnis
- 205 -
[88] I.-Y. Jeon, H.-J. Choi, M. Choi, J.-M. Seo, S.-M. Jung, M.-J. Kim, S. Zhang, L. Zhang, Z. Xia,
L. Dai et al., Sci. Rep. 2013, 3, 1810.
[89] J.-M. Seo, I.-Y. Jeon, J.-B. Baek, Chem. Sci. 2013, 4, 4273.
[90] I.-Y. Jeon, S. Zhang, L. Zhang, H.-J. Choi, J.-M. Seo, Z. Xia, L. Dai, J.-B. Baek, Adv. Mater.
2013, 25, 6138.
[91] C. K. Chua, Z. Sofer, B. Khezri, R. D. Webster, M. Pumera, Phys. Chem. Chem. Phys. 2016, 18,
17875.
[92] M.-J. Kim, I.-Y. Jeon, J.-M. Seo, L. Dai, J.-B. Baek, ACS Nano 2014, 8, 2820.
[93] G. Bharath, R. Madhu, S.-M. Chen, V. Veeramani, D. Mangalaraj, N. Ponpandian, J. Mater.
Chem. A 2015, 3, 15529.
[94] F. Beckert, Diessertation, Albert-Ludwigs-Universität Freiburg, Freiburg im Breisgau, 2014.
[95] M. Adel, A. El-Maghraby, O. El-Shazly, E.-W. F. El-Wahidy, M. A. A. Mohamed, J. Mater. Res.
2016, 31, 455.
[96] T. Hallaj, M. Amjadi, J. L. Manzoori, R. Shokri, Microchim. Acta 2015, 182, 789.
[97] L. Tang, R. Ji, X. Li, K. S. Teng, S. P. Lau, Part. Part. Syst. Charact. 2013, 30, 523.
[98] J. Kong, W. A. Yee, L. Yang, Y. Wei, S. L. Phua, H. G. Ong, J. M. Ang, X. Li, X. Lu, Chem.
Commun. 2012, 48, 10316.
[99] R. Liu, S. M. Mahurin, C. Li, R. R. Unocic, J. C. Idrobo, H. Gao, S. J. Pennycook, S. Dai,
Angew. Chem. Int. Ed. 2011, 50, 6799.
[100] Z. Wang, B. Li, X. Ge, F. W. T. Goh, X. Zhang, G. Du, D. Wuu, Z. Liu, T. S. Andy Hor, H.
Zhang et al., Small 2016, 12, 2580.
[101] K. Qu, Y. Zheng, S. Dai, S. Z. Qiao, Nanoscale 2015, 7, 12598.
[102] J. Liebscher, R. Mrówczyński, H. A. Scheidt, C. Filip, N. D. Hădade, R. Turcu, A. Bende, S.
Beck, Langmuir 2013, 29, 10539.
[103] H. Lee, S. M. Dellatore, W. M. Miller, P. B. Messersmith, Science (New York, N.Y.) 2007, 318,
426.
[104] T. Ishii, S. Kashihara, Y. Hoshikawa, J.-i. Ozaki, N. Kannari, K. Takai, T. Enoki, T. Kyotani,
Carbon 2014, 80, 135.
[105] J.-i. Ozaki, N. Kimura, T. Anahara, A. Oya, Carbon 2007, 45, 1847.
[106] C. L. Burket, R. Rajagopalan, H. C. Foley, Carbon 2008, 46, 501.
[107] C. Hu, A. C. Liu, M. Weyland, S. H. Madani, P. Pendleton, F. Rodríguez-Reinoso, K. Kaneko,
M. J. Biggs, Carbon 2015, 85, 119.
[108] J. Guo, J. R. Morris, Y. Ihm, C. I. Contescu, N. C. Gallego, G. Duscher, S. J. Pennycook, M. F.
Chisholm, Small 2012, 8, 3283.
[109] H. E. Hoydonckx, W. M. van Rhijn, W. van Rhijn, D. E. de Vos, P. A. Jacobs in Ullmann's
Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000.
[110] H. W. Hennicke, Ber. Dtsch. Keram Ges. 1967, 44, 209.
[111] G. Roewer, U. Herzog, K. Trommer, E. Müller, S. Frühauf in High Performance Non-Oxide
Ceramics I, Springer Berlin Heidelberg, Berlin, Heidelberg, 2002.
[112] K. A. Schwetz in Handbook of Ceramic Hard Materials, Wiley-VCH Verlag GmbH, 2008.
[113] P. Friedrichs (Ed.) Silicon carbide, Wiley-VCH, Weinheim, 2010.
[114] M. Krupka, A. Kienzle, SAE International, 2000.

10 Literaturverzeichnis
- 206 -
[115] W. Krenkel, F. Berndt, International Conference on Recent Advances in Composite Materials
2005, 412, 177.
[116] J. Fitch, Machinery Lubrication 2016, 1.
[117] H. Czichos, K.-H. Habig (Eds.) Tribologie-Handbuch. Tribometrie, Tribomaterialien,
Tribotechnik, Springer Vieweg, Wiesbaden, 2015.
[118] S. Pintoiu, H. U. V. GmbH, 2017.
[119] H. Spikes, Tribol. Int. 2001, 34, 789.
[120] D. Dowson, History of tribology, Ed. Longman, Londres, 1979.
[121] R. Stribeck, Wesentliche Eigenschaften der Gleit- und Rollenlager, Springer, 1903.
[122] V. L. Popov, Kontaktmechanik und Reibung. Ein Lehr- und Anwendungsbuch von der
Nanotribologie bis zur numerischen Simulation, Springer Berlin Heidelberg, Berlin, Heidelberg,
2009.
[123] P. Miranzo, M. Belmonte, Osendi, M. Isabel, J. Eur. Ceram. Soc. 2017, 37, 3649.
[124] H. Porwal, S. Grasso, M. J. Reece, Adv. Appl. Ceram. 2013, 112, 443.
[125] Y. Wang, G. A. Voronin, T. W. Zerda, A. Winiarski, J. Phys.: Condens. Matter 2006, 18, 275.
[126] N. Saheb, U. Hayat, Ceram. Int. 2017, 43, 5715.
[127] Q. Li, Y. Zhang, H. Gong, H. Sun, T. Li, X. Guo, S. Ai, Ceram. Int. 2015, 41, 13547.
[128] V. M. Candelario, R. Moreno, F. Guiberteau, A. L. Ortiz, J. Eur. Ceram. Soc. 2016, 36, 3083.
[129] https://scifinder.cas.org. abgerufen am 21.07.2017.
[130] L. S. Walker, V. R. Marotto, M. A. Rafiee, N. Koratkar, E. L. Corral, ACS Nano 2011, 5, 3182.
[131] L. Kvetková, A. Duszová, P. Hvizdoš, J. Dusza, P. Kun, C. Balázsi, Scr. Mater. 2012, 66, 793.
[132] P. Hvizdoš, J. Dusza, C. Balázsi, J. Eur. Ceram. Soc. 2013, 33, 2359.
[133] M. Belmonte, A. Nistal, P. Boutbien, B. Román-Manso, M. I. Osendi, P. Miranzo, Scr. Mater.
2016, 113, 127.
[134] A. Centeno, V. G. Rocha, B. Alonso, A. Fernández, C. F. Gutierrez-Gonzalez, R. Torrecillas, A.
Zurutuza, J. Eur. Ceram. Soc. 2013, 33, 3201.
[135] J. Liu, H. Yan, M. J. Reece, K. Jiang, J. Eur. Ceram. Soc. 2012, 32, 4185.
[136] P. Miranzo, C. Ramírez, B. Román-Manso, L. Garzón, H. R. Gutiérrez, M. Terrones, C. Ocal,
Osendi, M. Isabel, M. Belmonte, J. Eur. Ceram. Soc. 2013, 33, 1665.
[137] F. Zeller, C. Müller, P. Miranzo, M. Belmonte, J. Eur. Ceram. Soc. 2017, 37, 3813.
[138] Y. Çelik, A. Çelik, E. Flahaut, E. Suvaci, J. Eur. Ceram. Soc. 2016, 36, 2075.
[139] C. Ramirez, F. M. Figueiredo, P. Miranzo, P. Poza, Osendi, M. Isabel, Carbon 2012, 50, 3607.
[140] K.-S. Kim, H.-J. Lee, C. Lee, S.-K. Lee, H. Jang, J.-H. Ahn, J.-H. Kim, H.-J. Lee, ACS Nano
2011, 5, 5107.
[141] D. Marchetto, C. Held, F. Hausen, F. Wählisch, M. Dienwiebel, R. Bennewitz, Tribol. Lett. 2012,
48, 77.
[142] B. Pan, G. Xu, B. Zhang, X. Ma, H. Li, Y. Zhang, Polym. Plast. Technol. Eng. 2012, 51, 1163.
[143] B. Schlüter, R. Mülhaupt, A. Kailer, Tribol. Lett. 2014, 53, 353.
[144] H.-J. Song, N. Li, Appl. Phys. A 2011, 105, 827.
[145] M. Tabandeh-Khorshid, E. Omrani, P. L. Menezes, P. K. Rohatgi, Eng. Sci. Technol. 2016, 19,
463.
[146] A. Dorri Moghadam, E. Omrani, P. L. Menezes, P. K. Rohatgi, Composites Part B 2015, 77, 402.

10 Literaturverzeichnis
- 207 -
[147] Z. Tai, Y. Chen, Y. An, X. Yan, Q. Xue, Tribol. Lett. 2012, 46, 55.
[148] X.-J. Shen, X.-Q. Pei, S.-Y. Fu, K. Friedrich, Polymer 2013, 54, 1234.
[149] B. Pan, S. Zhang, W. Li, J. Zhao, J. Liu, Y. Zhang, Y. Zhang, Wear 2012, 294, 395.
[150] H. Song, N. Li, Y. Li, C. Min, Z. Wang, J. Mater. Sci. 2012, 47, 6436.
[151] T. Huang, Y. Xin, T. Li, S. Nutt, C. Su, H. Chen, P. Liu, Z. Lai, ACS Appl. Mater. Interfaces
2013, 5, 4878.
[152] J. Llorente, B. Román-Manso, P. Miranzo, M. Belmonte, J. Eur. Ceram. Soc. 2016, 36, 429.
[153] C. Balázsi, Z. Fogarassy, O. Tapasztó, A. Kailer, C. Schröder, M. Parchoviansky, D. Galusek, J.
Dusza, K. Balázsi, J. Eur. Ceram. Soc. 2017, 37, 3797.
[154] M. Maros B., A. K. Németh, Z. Károly, E. Bódis, Z. Maros, O. Tapasztó, K. Balázsi, Tribol. Int.
2016, 93, 269.
[155] C. F. Gutierrez-Gonzalez, A. Smirnov, A. Centeno, A. Fernández, B. Alonso, V. G. Rocha, R.
Torrecillas, A. Zurutuza, J. F. Bartolome, Ceram. Int. 2015, 41, 7434.
[156] Z. Cheng, N. Andy, A. Arvind, Nanomaterials and Energy 2016, 5, 1.
[157] H. Porwal, P. Tatarko, R. Saggar, S. Grasso, M. Kumar Mani, I. Dlouhý, J. Dusza, M. J. Reece,
Ceram. Int. 2014, 40, 12067.
[158] Z. Li, N. W. Khun, X.-Z. Tang, E. Liu, K. A. Khor, J. Mech. Behav. Biomed. Mater. 2017, 65, 77.
[159] R. Sedlák, A. Kovalčíková, J. Balko, P. Rutkowski, A. Dubiel, D. Zientara, V. Girman, E. Múdra,
J. Dusza, Int. J. Refract. Met. Hard Mater 2017, 65, 57.
[160] H. Li, Y. Xie, K. Li, L. Huang, S. Huang, B. Zhao, X. Zheng, Ceram. Int. 2014, 40, 12821.
[161] Y. Xie, H. Li, C. Zhang, X. Gu, X. Zheng, L. Huang, Biomed. Mater. 2014, 9, 25009.
[162] M. Belmonte, C. Ramírez, J. González-Julián, J. Schneider, P. Miranzo, M. I. Osendi, Carbon
2013, 61, 431.
[163] A. Gómez-Gómez, A. Nistal, E. García, Osendi, M. Isabel, M. Belmonte, P. Miranzo, Mater.
Design 2016, 112, 449.
[164] S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D.
Piner, S. T. Nguyen, R. S. Ruoff, Nature 2006, 442, 282.
[165] T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H.
Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff et al., Nat. Nanotechnol. 2008, 3, 327.
[166] X. Huang, X. Qi, F. Boey, H. Zhang, Chem. Soc. Rev. 2012, 41, 666.
[167] H. Kim, A. A. Abdala, C. W. Macosko, Macromolecules 2010, 43, 6515.
[168] V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim, R. Zboril,
Chem. Rev. 2016, 116, 5464.
[169] B. Wicklein, A. Kocjan, G. Salazar-Alvarez, F. Carosio, G. Camino, M. Antonietti, L. Bergström,
Nat. Nanotechnol. 2014, 10, 277.
[170] J. Xu, Y. Wang, S. Hu, Microchim. Acta 2017, 184, 1.
[171] X. Yu, W. Zhang, P. Zhang, Z. Su, Biosens. Bioelectron. 2017, 89, 72.
[172] P. Rutkowski, L. Stobierski, G. Górny, J. Therm. Anal. Calorim. 2014, 116, 321.
[173] B. Román-Manso, Y. Chevillotte, Osendi, M. Isabel, M. Belmonte, P. Miranzo, J. Eur. Ceram.
Soc. 2016, 36, 3987.
[174] C. Dold, T. Amann, A. Kailer, Phys. Chem. Chem. Phys. 2015, 17, 10339.

10 Literaturverzeichnis
- 208 -
[175] E. Ciudad, E. Sánchez-González, O. Borrero-López, F. Guiberteau, M. Nygren, A. L. Ortiz, Scr.
Mater. 2013, 69, 598.
[176] O. Borrero-López, A. L. Ortiz, F. Guiberteau, N. P. Padture, J. Eur. Ceram. Soc. 2007, 27, 3351.
[177] H. Rickert, Angew. Chem. 1978, 90, 38.
[178] J. Balko, P. Hvizdoš, J. Dusza, C. Balázsi, J. Gamcová, J. Eur. Ceram. Soc. 2014, 34, 3309.
[179] L. Kvetková, A. Duszová, M. Kašiarová, F. Dorčáková, J. Dusza, C. Balázsi, J. Eur. Ceram. Soc.
2013, 33, 2299.
[180] C. Ramirez, P. Miranzo, M. Belmonte, M. I. Osendi, P. Poza, S. M. Vega-Diaz, M. Terrones, J.
Eur. Ceram. Soc. 2014, 34, 161.
[181] B. Eszter, T. Orsolya, K. Zoltán, F. Péter, K. Szilvia, K. A. Mária, B. Katalin, S. János, Open
Chem. 2015, 13, 484.
[182] J. Pfeifer, G. Sáfrán, F. Wéber, V. Zsigmond, O. Koszor, P. Arató, C. Balázsi, Mater. Sci. Forum
2010, 659, 235.
[183] F. J. Tölle, M. Fabritius, R. Mülhaupt, Adv. Funct. Mater. 2012, 22, 1136.
[184] F. J. Tölle, Diessertation, Albert-Ludwigs-Universität Freiburg, Freiburg im Breisgau, 2014.
[185] H. He, J. Klinowski, M. Forster, A. Lerf, Chem. Phys. Lett. 1998, 287, 53.
[186] A. Ganguly, S. Sharma, P. Papakonstantinou, J. Hamilton, J. Phys. Chem. C 2011, 115, 17009.
[187] C. Gómez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K.
Kern, U. Kaiser, Nano Lett. 2010, 10, 1144.
[188] H. L. Poh, F. Sanek, A. Ambrosi, G. Zhao, Z. Sofer, M. Pumera, Nanoscale 2012, 4, 3515.
[189] J. Cao, G.-Q. Qi, K. Ke, Y. Luo, W. Yang, B.-H. Xie, M.-B. Yang, J. Mater. Sci. 2012, 47, 5097.
[190] M. D. Stoller, S. Park, Y. Zhu, J. An, R. S. Ruoff, Nano Lett. 2008, 8, 3498.
[191] E. Fuente, J. A. Menéndez, M. A. Díez, D. Suárez, M. A. Montes-Morán, J. Phys. Chem. B 2003,
107, 6350.
[192] J. B. Lambert, Introduction to organic spectroscopy, Macmillan, New York, 1987.
[193] D. Lee, X. Zou, X. Zhu, J. W. Seo, J. M. Cole, F. Bondino, E. Magnano, S. K. Nair, H. Su, Appl.
Phys. Lett. 2012, 101, 21604.
[194] M. Alexandre, P. Dubois, Mater. Sci. Eng., R 2000, 28, 1.
[195] H. Yang, C. Shan, F. Li, D. Han, Q. Zhang, L. Niu, Chem. Commun. 2009, 3880.
[196] L. M. Veca, F. Lu, M. J. Meziani, L. Cao, P. Zhang, G. Qi, L. Qu, M. Shrestha, Y.-P. Sun, Chem.
Commun. 2009, 2565.
[197] S. Edmondson, V. L. Osborne, W. T. S. Huck, Chem. Soc. Rev. 2004, 33, 14.
[198] F. J. Tölle, M. Fabritius, R. Mülhaupt, Adv. Funct. Mater. 2012, 22, 1136.
[199] S. E. McNeil (Ed.) Characterization of Nanoparticles Intended for Drug Delivery, Humana
Press, Totowa, NJ, 2011.
[200] A. Prokop, E. Kozlov, G. Carlesso, J. M. Davidson in Filled Elastomers Drug Delivery Systems,
Springer Berlin Heidelberg, Berlin, Heidelberg, 2002.
[201] R. W. O'Brien, B. R. Midmore, A. Lamb, R. J. Hunter, Faraday Discuss. Chem. Soc. 1990, 90,
301.
[202] B. J. Kirby, Micro- and nanoscale fluid mechanics. Transport in microfluidic devices, Cambridge
Univ. Press, Cambridge, 2010.
[203] D. Mahl, Dissertation, Universität Duisburg-Essen, Essen, 2001.

10 Literaturverzeichnis
- 209 -
[204] D. Li, M. B. Muller, S. Gilje, R. B. Kaner, G. G. Wallace, Nat. Nanotechnol. 2008, 3, 101.
[205] H. Miyagawa, L. T. Drzal, J. A. Carsello, Polym. Eng. Sci. 2006, 46, 452.
[206] Kusmono, M. W. Wildan, Z. A. Mohd Ishak, Int. J. Polym. Sci. 2013, 2013, 7.
[207] M. T. Albdiry, B. F. Yousif, H. Ku, K. T. Lau, J. Compos. Mater. 2013, 47, 1093.
[208] http://www.genizer.com. abgerufen am 23.01.2018.
[209] https://www.microfluidicscorp.com. abgerufen am 23.01.2018.
[210] W. Zhang, R. Mülhaupt, M. Knoch, J. Thelke, DE 10 2017 211 663, 2017.
[211] R. Li, K. Parvez, F. Hinkel, X. Feng, K. Müllen, Angew. Chem. 2013, 125, 5645.
[212] http://xgsciences.com. abgerufen am 23.01.2018.
[213] M. Bousmina, Macromolecules 2006, 39, 4259.
[214] K. Pereira dos Santos Tonello, E. Padovano, C. Badini, S. Biamino, M. Pavese, P. Fino, Mater.
Sci. Eng., A 2016, 659, 158.
[215] M. Shahedi Asl, M. Ghassemi Kakroudi, Mater. Sci. Eng., A 2015, 625, 385.
[216] M. Cadek, O. Vostrowsky, A. Hirsch in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-
VCH Verlag GmbH & Co. KGaA, 2000.
[217] A. Jorio, G. Dresselhaus, M. S. Dresselhaus (Eds.) Topics in Applied Physics, Vol. 111, Springer-
Verlag Berlin/Heidelberg, Berlin, Heidelberg, 2008.
[218] C. T. Herakovich, S. C. Baxter, J. Mater. Sci. 1999, 34, 1595.
[219] B. Sarac, Microstructure-property optimization in metallic glasses. Zugl.: New Haven, Yale
Univ., Dep. of mechanical engineering and materials science, Diss, Springer, Cham, 2015.
[220] P. Rutkowski, L. Stobierski, G. Górny, M. Ziabka, M.Urbanik, Ceram. 2014, 66, 463.
[221] K. Takahashi, A. Yoshikawa, A. Sandhu, Wide Bandgap Semiconductors. Fundamental
Properties and Modern Photonic and Electronic Devices, Springer-Verlag, s.l., 2007.
[222] C. Persson, U. Lindefelt, Phys. Rev. B 1996, 54, 10257.
[223] C. Persson, U. Lindefelt, J. Appl. Phys. 1997, 82, 5496.
[224] J. R. Jenny, M. Skowronski, W. C. Mitchel, H. M. Hobgood, R. C. Glass, G. Augustine, R. H.
Hopkins, J. Appl. Phys. 1995, 78, 3839.
[225] S. Janz, S. Reber, F. Lutz, C. Schetter, Thin Solid Films 2006, 511, 271.
[226] F. Siegelin, H.-J. Kleebe, L. S. Sigl, J. Mater. Res. 2003, 18, 2608.
[227] Y.-W. Kim, K. J. Kim, H. C. Kim, N.-H. Cho, K.-Y. Lim, J. Am. Ceram. Soc. 2011, 94, 991.
[228] K. Wang, Y. Wang, Z. Fan, J. Yan, T. Wei, Mater. Res. Bull. 2011, 46, 315.
[229] Y. Fan, W. Jiang, A. Kawasaki, Adv. Funct. Mater. 2012, 22, 3882.
[230] C. Ramirez, L. Garzón, P. Miranzo, M. I. Osendi, C. Ocal, Carbon 2011, 49, 3873.
[231] J. T. Choi, D. H. Kim, K. S. Ryu, H.-i. Lee, H. M. Jeong, C. M. Shin, J. H. Kim, B. K. Kim,
Macromol. Res. 2011, 19, 809.
[232] B. Wessling, Polym. Eng. Sci. 1991, 31, 1200.
[233] J. N. Bradley, P. D. Greene, Trans. Faraday Soc. 1967, 63, 424.
[234] W. König, D. F. Dauw, G. Levy, U. Panten, CIRP Ann. 1988, 37, 623.
[235] S. Pei, H.-M. Cheng, Carbon 2012, 50, 3210.
[236] R. Rozada, J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso, J. M. D. Tascón, Nano Res. 2013,
6, 216.

10 Literaturverzeichnis
- 210 -
[237] J.-M. Thomassin, C. Jérôme, T. Pardoen, C. Bailly, I. Huynen, C. Detrembleur, Mater. Sci. Eng.,
R 2013, 74, 211.
[238] B. Román-Manso, E. Domingues, F. M. Figueiredo, M. Belmonte, P. Miranzo, J. Eur. Ceram.
Soc. 2015, 35, 2723.
[239] A. Klemenz, L. Pastewka, S. G. Balakrishna, A. Caron, R. Bennewitz, M. Moseler, Nano Lett.
2014, 14, 7145.
[240] J. Tersoff, Phys. Rev. B 1989, 39, 5566.
[241] L. Pastewka, A. Klemenz, P. Gumbsch, M. Moseler, Phys. Rev. B 2013, 87, 205410.
[242] O. Guillon, J. Gonzalez-Julian, B. Dargatz, T. Kessel, G. Schierning, J. Räthel, M. Herrmann,
Adv. Eng. Mater. 2014, 16, 830.
[243] R. Orrù, R. Licheri, A. M. Locci, A. Cincotti, G. Cao, Mater. Sci. Eng., R 2009, 63, 127.
[244] Z. A. Munir, U. Anselmi-Tamburini, M. Ohyanagi, J. Mater. Sci. 2006, 41, 763.
[245] M. Suárez, A. Fernández, J. L. Menéndez, R. Torrecillas, H. U. Kessel, J. Hennicke, R. Kirchner,
T. Kessel in Sintering Applications (Ed.: B. Ertuğ), InTech, Rijeka, 2013.
[246] B. Ertuğ (Ed.) Sintering Applications, InTech, Rijeka, 2013.
[247] J. L. Cour, O. S. Bragstad, Theorie der Wechselströme, Springer Berlin Heidelberg, Berlin,
Heidelberg, s.l., 1910.
[248] J. M. Meek (Ed.) Wiley series in plasma physics, Wiley, Chichester, 1978.
[249] J. Dusza, J. Morgiel, A. Duszová, L. Kvetková, M. Nosko, P. Kun, C. Balázsi, J. Eur. Ceram.
Soc. 2012, 32, 3389.
[250] O. Tapasztó, L. Tapasztó, M. Markó, F. Kern, R. Gadow, C. Balázsi, Chem. Phys. Lett. 2011,
511, 340.
[251] O. Tapasztó, P. Kun, F. Wéber, G. Gergely, K. Balázsi, J. Pfeifer, P. Arató, A. Kidari, S.
Hampshire, C. Balázsi, Ceram. Int. 2011, 37, 3457.
[252] O. Tapasztó, L. Tapasztó, H. Lemmel, V. Puchy, J. Dusza, C. Balázsi, K. Balázsi, Ceram. Int.
2016, 42, 1002.
[253] O. Tapasztó, J. Balko, V. Puchy, P. Kun, G. Dobrik, Z. Fogarassy, Z. E. Horváth, J. Dusza, K.
Balázsi, C. Balázsi et al., Sci. Rep. 2017, 7, 10087.
[254] P. Rutkowski, L. Stobierski, D. Zientara, L. Jaworska, P. Klimczyk, M. Urbanik, J. Eur. Ceram.
Soc. 2015, 35, 87.
[255] Q. Li, Y. Zhang, H. Gong, H. Sun, W. Li, L. Ma, Y. Zhang, J. Mater. Sci. Technol. 2016, 32, 633.
[256] D. K. Shetty, I. G. Wright, P. N. Mincer, A. H. Clauer, J. Mater. Sci. 1985, 20, 1873.