fdli february 11, 2008 bradley merrill thompson managing and complying with clinical trial quality...
TRANSCRIPT

FDLIFebruary 11, 2008
Bradley Merrill Thompson
Managing and Complying with Clinical Trial Quality Obligations
for Sponsors

Topics
1. Quality assurance concept
2. Risk management approach
3. Good Laboratory Practices
4. Good Clinical Practices
5. Basic FDA GCP Elements
6. GCP experience

3
Importance of Quality Assurance
o More studies; more sites; greater volume
o Expansion and fluidity of clinical investigator pool
o New players in new roles (CROs, SMOs)
o New technologies (electronic recordkeeping)
o More participation by vulnerable subjects
o Global expansion (areas new to GCP)

4
The end product of clinical research
o A product is an output from a process
o The output from the clinical research process is information
trial protocol
collectionof data
poolingof datain the
database
analysis trialreport
Slide Originally Developed by Dorothy Switula

5
Customers of clinical research
o Society / consumers
o Research subjects
o Sponsors
o Regulatory authorities worldwide
o Hospitals / institutions
o Ethics committees
Slide Originally Developed by Dorothy Switula

6
Clinical research customers’ requirements
Law & Regulations:EU Directives
US CFRlocal legislation
Ethical Standards:Declaration of Helsinki Good Practice Standards:
GXPs:ICH GCP, ICH GMP, OECD GLP
Slide Originally Developed by Dorothy Switula

Topics1. Quality assurance concept
2. Risk management approach
3. Good Laboratory Practices
4. Good Clinical Practices
5. Basic FDA GCP Elements
6. GCP experience

8
Risk Management
oProduct Risk– FDA does, and you should, consider
the intrinsic risk of the product you are investigating, when deciding the level of sophistication and elegance of your quality system.
– Product risk is a function of:• Technology• Disease or condition• Setting• User/Patient

9
Risk Management
o Regulatory Risk– FDA does, and you should, consider the
commercial complexity of the structure of the clinical trial you are organizing when deciding the level of sophistication and elegance of your quality system and the extent of training needed
• Use of CROs, sub-investigators, and so forth
– The more complexity, the greater the burden to manage it well

10
Risk Management
Drive it with data and study--1. Your products
2. The commercial arrangements to identify risks and weaknesses
3. FDA experience inspecting clinical trials
4. Litigation brought against sponsors
5. Reports investigating clinical trial quality

11
Why? If you don’t, you might …
o Harm patientso Liability to patientso Data from expensive studies
disqualified from use in FDA submissions– Or expensive fixes required
o Civil and criminal liability to federal and state regulators– Such as OIG, FDA, and state
agencies

12
FDA Regulatory Actions
oRejection of dataoDeficiency letteroIDE withdrawaloUntitled lettersoWarning lettersoConsent
AgreementoDisqualification
– CI, IRB, GLP
o IRB restrictions– No new studies
or subjectso Application Integrity
Policyo Civil Money
Penaltieso Seizure / Detentiono Injunctiono Criminal
Prosecution

13
Investigator Sanctions
o FDA– Restricted (and formerly restricted)
• Informal agreements and corrective actions
– Disqualified (NIDPOE, NOOH process)– Debarred per § 306 of the Act
o HHS—Office of Human research Protections– Assurances of compliance– Corrective action that includes
debarment

14
More Investigator Sanctions
o HHS—Office of Research Integrity– Assurances– Corrective actions
o HHS—Office of Inspector General– Agreements and CIAs
o Potentially States

Topics
1. Quality assurance concept
2. Risk management approach
3. Good Laboratory Practices
4. Good Clinical Practices
5. Basic FDA GCP Elements
6. GCP experience

16
Good Laboratory Practices
o Serve as the quality system prior to GCP
o Scope: The GLPs prescribe good laboratory practices for conducting nonclinical laboratory studies that support applications for research or marketing permits for products regulated by the FDA. Part 58
Copyright 2004 Robin Guy

17
GLPs do not cover:
o Human subjects
o Basic exploratory studies to determine:
–Potential utility
–Physical or chemical characteristics of a test article

18
GLPs Elements
1. General Provisions
2. Organization & Personnel
3. Facilities
4. Equipment
5. Testing Facilities Operations
6. Test and Control Articles
7. Protocol for and Conduct of a Non-clinical Laboratory Study
8. Records and Reports
9. Disqualification

19
Primary FDA GLP Elements
o Testing Facility Management
o Study Director
o Quality Assurance Unit
o Equipment requires scheduled maintenance and SOP’s
o Archived, orderly storage for all records, documentation and feasible specimens for expedient retrieval

Topics
1. Quality assurance concept
2. Risk management approach
3. Good Laboratory Practices
4. Good Clinical Practices
A. What are they conceptually?
B. Where do you find them?
5. Basic FDA Elements
6. GCP experience

21
What are Good Clinical Practices?
o The rules—and self-determined standards—governing clinical research
o Promote scientific and ethical quality in the conduct of clinical trials
o Set standards for:1. Design 2. Conduct 3. Monitoring4. Auditing5. Recording 6. Analysis7. Reporting

22
What are they?
1. Ethics: Clinical trials should be conducted in
accordance with the ethical principles that
have their origin in the Declaration of Helsinki,
and that are consistent with GCP and the
applicable regulatory requirements.
2. Risk/Benefit: Before a trial is initiated,
foreseeable risks and inconveniences should
be weighed against the anticipated benefit for
the individual trial subject and society. A trial
should be initiated and continued only if the
anticipated benefits justify the risks.

23
What are they?
3. Protection: The rights, safety, and well-being of the trial subjects are the most important considerations and should prevail over interests of science and society.
4. Well-Supported: The available nonclinical and clinical information on an investigational product should be adequate to support the proposed clinical trial.
5. Scientifically sound: Clinical trials should be scientifically sound, and described in a clear, detailed protocol.

24
What are they?
6. IRB Review: A trial should be conducted in compliance with the protocol that has received prior intuitional review board (IRB)/independent ethics committee (IEC) approval/favorable opinion.
7. Doctor Managed: The medical care given to, and medical decisions made on behalf of, subjects should always be the responsibility of a qualified physician or, when appropriate, of a qualified dentist.

25
What are they?
8. Well-staffed: Each individual involved in conducting a trial should be qualified by education, training, and experience to perform his or her respective task(s).
9. Informed Consent: Freely given informed consent should be obtained from every subject prior to clinical trial participation.
10. Record keeping: All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation, and verification.

26
What are they?
11. Confidentiality: The confidentiality of records that could identify subjects should be protected, respecting the privacy and confidentiality rules in accordance with the applicable regulatory requirement(s).
12. Products: Investigational products should be manufactured, handled, and stored in accordance with applicable good manufacturing practice (GMP). They should be used in accordance with the approved protocol.

27
What are they?
13. SOP Driven: Systems with procedures that assure the quality of every aspect of the trial should be implemented.
14. Others: Country specific requirements also must be factored in, such as financial conflict of interest and investigator payment rules

28
Where do you find them in US?
A Tale of Two Systems
o In the US, the standards actually vary depending on the purpose of the clinical trial. – FDA rules for trials that will be conducted
on articles subject to regulation by FDA and for possible submission to FDA
– HHS (common rule) for research conducted or supported by HHS and those who voluntarily agree to be bound (federal-wide assurance)
– Both sets of standards can apply
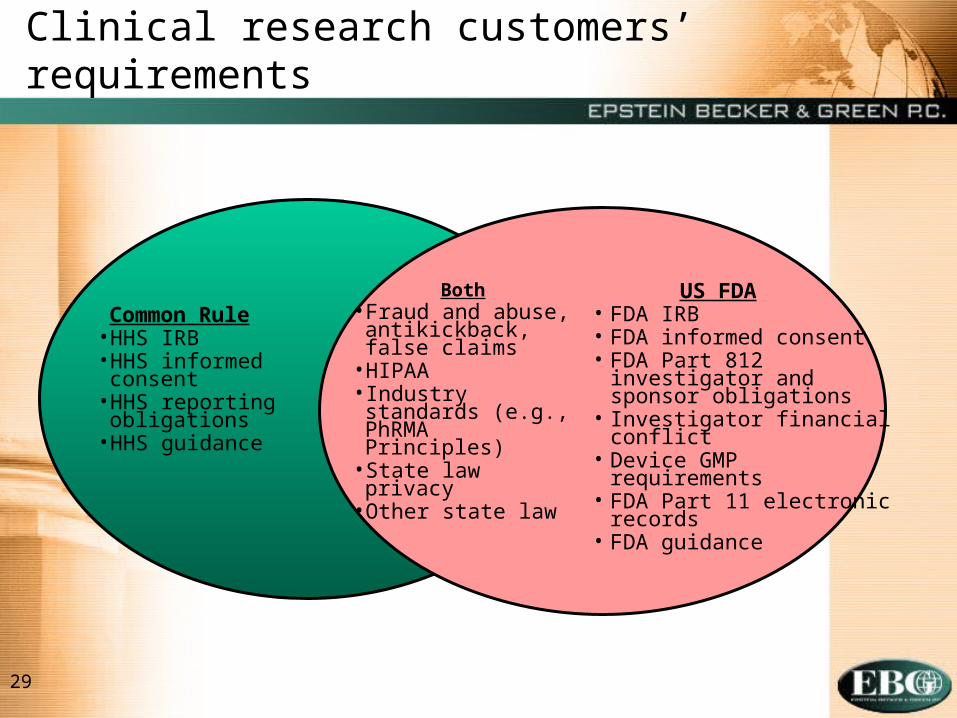
29
Clinical research customers’ requirements
Common Rule• HHS IRB• HHS informed
consent• HHS reporting
obligations• HHS guidance
US FDA• FDA IRB• FDA informed consent• FDA Part 812 investigator
and sponsor obligations• Investigator financial conflict• Device GMP requirements• FDA Part 11 electronic
records• FDA guidance
Both• Fraud and abuse,
antikickback, false claims
• HIPAA• Industry standards
(e.g., PhRMA Principles)
• State law privacy• Other state law

Topics
1. Quality assurance concept
2. Risk management approach
3. Good Laboratory Practices
4. Good Clinical Practices
5. Basic FDA GCP ElementsA. FDA Structure
B. Sponsor Obligations
C. Investigator Obligations
D. Adverse Event Reporting
E. Monitoring
F. Future Changes
6. GCP experience

31
FDA’s GCP Program
o Focal point within FDA for GCP issues arising in human research trials regulated by FDA
o FDA’s liaison to the Department of Health and Human Services (HHS) Office for Human Research Protection (OHRP)
o Small office located within the Office of Science Coordination and Communication
o Focus on quality assurance and quality improvement
o There are also enforcement offices located in the centers, such as DSI at CDER.

32
Basic US sponsor obligations
o Select qualified investigatorso Provide sufficient information to
investigators to conduct the trialo Oversee the trial and enforce complianceo Monitor trial progresso Conduct safety analyses, provide
notification, and submit reports (when appropriate)
o Keep appropriate recordso Review and evaluate safety and
effectivenessPart 812

33
Basic US sponsor obligations
o Sponsors may transfer regulatory obligations to a CRO– Describe in writing– If not all are transferred, specify which ones
are and which ones aren’t– Any obligation not addressed in writing is
deemed not to have been transferred
o CROs that assume obligations must comply with regulatory requirements and are subject to same regulatory actions as a sponsor

34
Basic US investigator obligations
o Ensure IRB review and complianceo Protect the rights and welfare of trial
subjectso Obtain informed consento Follow the protocolo Personally supervise the study and clinical
staffo Report adverse eventso Submit appropriate financial disclosureso Keep appropriate records
Part 812

37
Adverse Event Reporting
o Safety monitoring is required in all clinical research studies– Data safety monitoring plan should be
incorporated in the protocol– Plan should be consistent with the
degree of risk involved with the studyo Remember we’re focusing here on FDA
requirements – there are likely other safety reporting requirements that apply.

38
Safety reporting requirements
Investigator IRB
Investigator Sponsor
Sponsor Investigator
Sponsor FDA

39
All adverse events
Report to sponsor and IRB in 10 daysRecord and document
Serious
adverse effects
CFR 812.150(a)(ii)
Device-related adverse effects
Unanticipated adverse effects
Unreasonable
risk
CFR 812.140(a)(3)(ii)
Inve
sti
ga
tor
req
uir
em
en
ts

40
All adverse events
Record Document and evaluate unanticipated serious device effects; report in 10 days to FDA, other IRBs, and participating investigators
Terminate in five days, and no later than 15 days after learning
Device-related adverse effects
Unanticipated adverse effects
CFR 812.140(b)(5)
CFR 812.46(b)
CFR 812.150 (b)(1)
CFR 812.48(b)(2)
Sp
on
sor
req
uir
emen
ts
Serious adverse effects
Unreasonable
risk

41
Unanticipated Adverse Device Effect (UADE)
o Any serious adverse effect on health or safety or any life-threatening problem or death caused by, or associated with, a device, if that effect, problem, or death was not previously identified in nature, severity, or degree of incidence in investigational plan
o Or any other unanticipated serious problem associated with a device that relates to the rights, safety, or welfare of the subjects. (Sec. 812.3(s))

42
“Anticipated” Adverse Events
o No definition in IDE regulation; Those that are prospectively identified in the investigational plan in the IDE
o Reported by sponsor to FDA, investigators, and IRBs in progress reports (Section 812.150(b)(5))

46
Site Monitoring
o One of the most important roles in the process
o Evaluates the site as QC, and does not supplement the site (e.g. complete CRFs)
o Monitors everything, including—– Product handling– Protocol compliance– Reporting– Subject recruitment and
informed consent– Etc.

47
Future Changes: FDA’s BIMO Initiative
o Draft Guidance on AE Reporting to IRBs (April 2007)
o Concept Paper on data quality and May 2007 meeting
o E-Clinical trials
o Industry monitoring practices
o Clinical trial compliance, perhaps accreditation of sites
o BIMO drug inspectorate

1. Quality assurance concept2. Risk management approach 3. Good Laboratory Practices4. Good Clinical Practices5. Basic FDA Elements6. GCP experience
A. FDA’s experience inspecting themB. FDA’s experience enforcing themC. Sponsor’s experience defending themD. Studies examining weaknessesE. FDA’s international experience
Topics
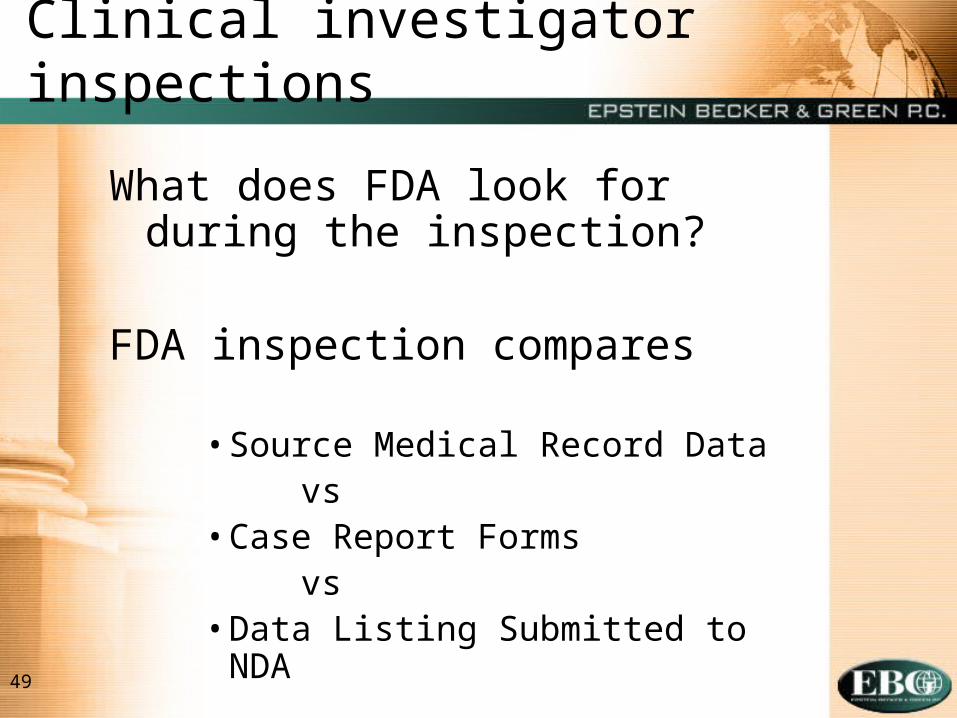
49
Clinical investigator inspections
What does FDA look for during the inspection?
FDA inspection compares
• Source Medical Record Datavs
• Case Report Formsvs
• Data Listing Submitted to NDA
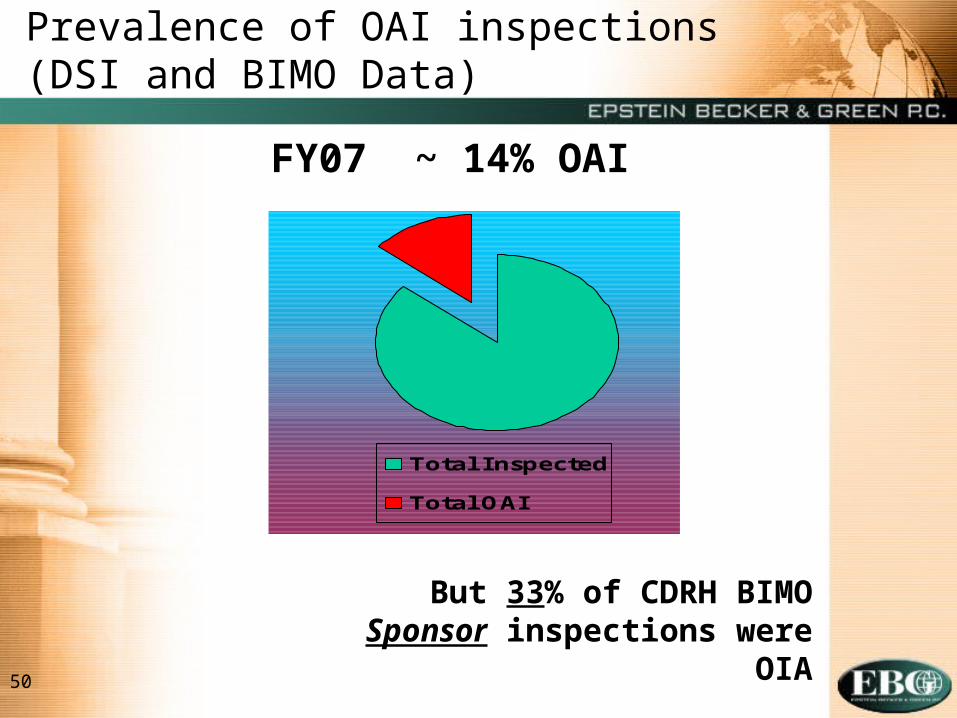
50
Prevalence of OAI inspections (DSI and BIMO Data)
Total Inspected
Total OAI
FY07 ~ 14% OAI
But 33% of CDRH BIMO Sponsor inspections were OIA

51
Prevalence of OAI inspections (DSI Data)
Routine FDA inspections
FY04 – FY06* = 936
Total OAI cases = 2
Percent OAI = .6%
Directed FDA inspections
FY04 – FY06* - 239
Total OAI cases = 25
Percent OAI = 11%
Routine Inspections Total OAI
Directed Inspections Total OAI
*FY06 to date

52
What are they complaining about?
o Failure to follow the protocol (25%)
o Falsification (24%)o Informed Consent
Issues (19%) o Failure to report
adverse events (14%)o Inadequate Recordso Qualifications of
persons performing physicals
o Failure to get IRB approval, report changes in research
o Failure to follow FDA regulations
o Drug accountability (8%)
o Recruitment Practiceso Poor Supervisiono No active INDo Violations of GLP regso Monitoring practiceso Blindingo Charging for the test
articleo Misleading
advertisements

53
Complaints received: 1992-2006
0
50
100
150
200
250
300
350
4001992
1993
1994
1995
1996
1997
1998
1999
2000
2001
2002
2003
2004
2005
2006

54
CDRH BIMO INSPECTIONSFiscal Years 2002 - 2006
357 353 350332 336
100
200
300
400
500
FY02 FY03 FY04 FY05 FY06

55
CDRH BIMO INSPECTIONSFiscal Years 2002 - 2006
Inspected Entity 2002 2003 2004 2005 2006
Sponsor 72 81 73 70 53
CI 151 170 183 183 200
IRB 128 85 73 48 59
GLP 6 9 19 31 24
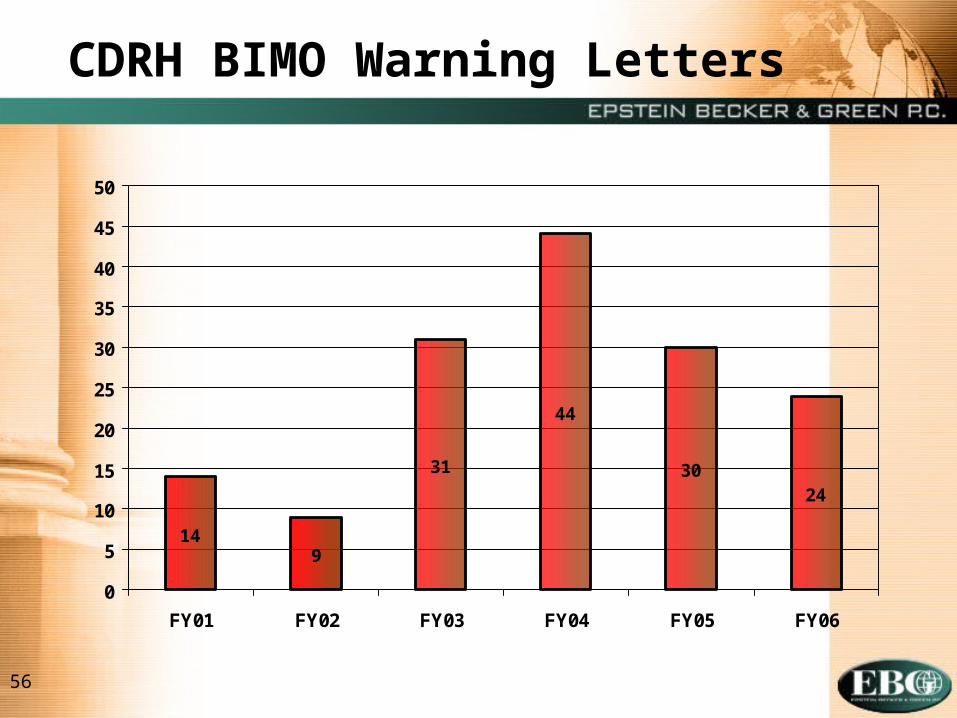
56
14
44
3024
9
31
0
5
10
15
20
25
30
35
40
45
50
FY01 FY02 FY03 FY04 FY05 FY06
CDRH BIMO Warning Letters
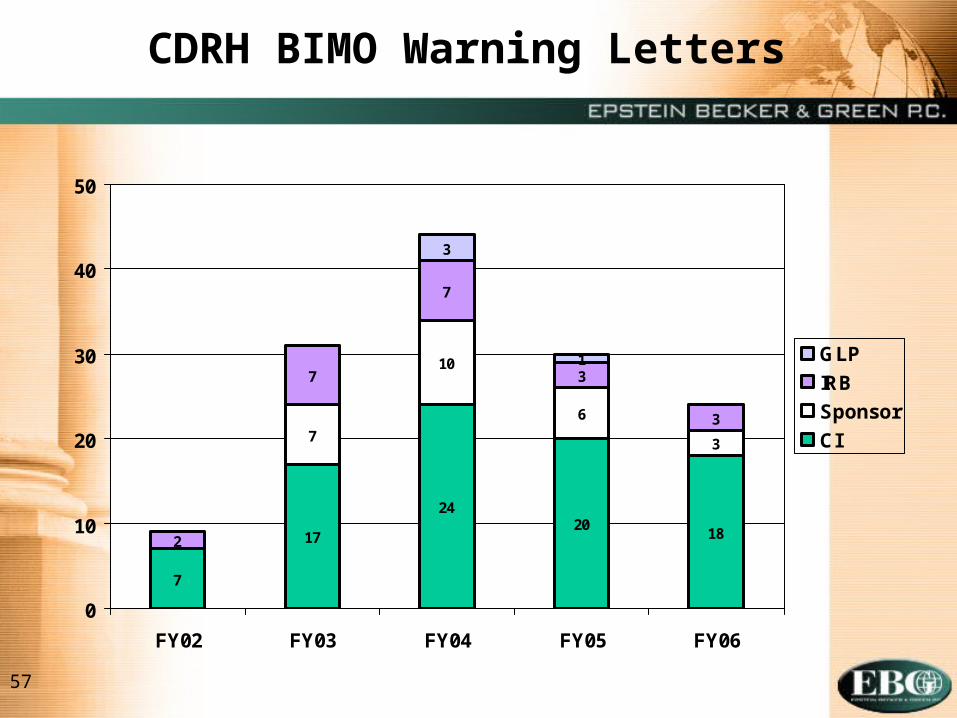
57
7
2018
10
6
3
7
3
3
1
24
17
7
2
7
3
0
10
20
30
40
50
FY02 FY03 FY04 FY05 FY06
GLP
IRB
Sponsor
CI
CDRH BIMO Warning Letters
58
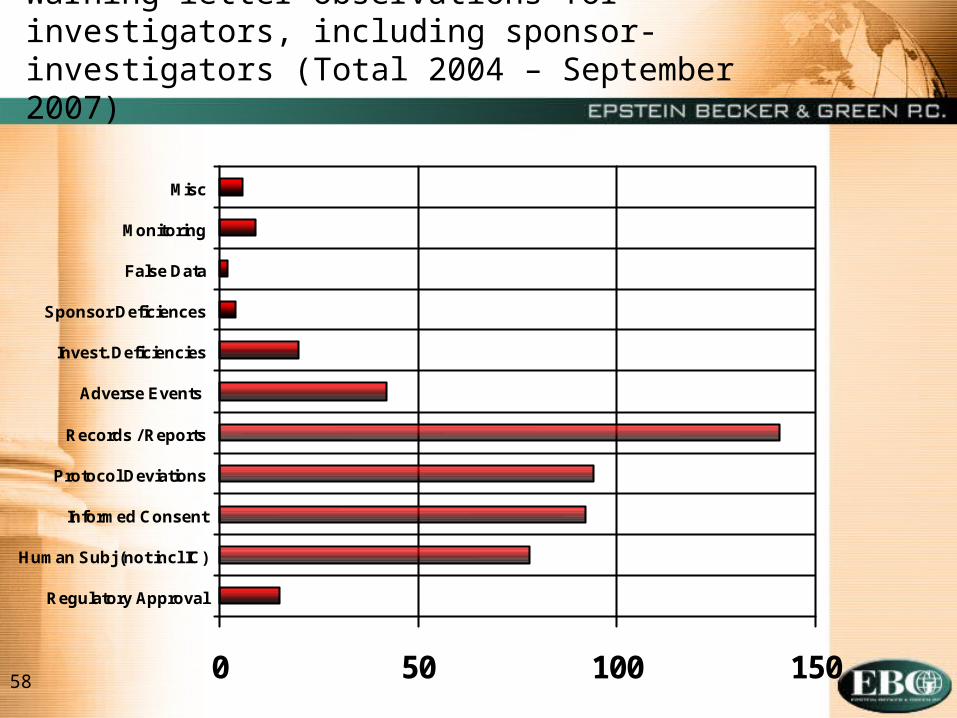
Warning letter observations for investigators, including sponsor-investigators (Total 2004 – September 2007)
0 50 100 150
Regulatory Approval
Human Subj (not incl IC)
Informed Consent
Protocol Deviations
Records / Reports
Adverse Events
Invest. Deficiencies
Sponsor Deficiences
False Data
Monitoring
Misc

59
Common Investigator Deficiencies (Per BIMO)
o Failure to follow investigational plan, investigator agreement, or protocol
o Protocol deviations
o Inadequate subject protection or informed consent
o Inadequate device accountability
o Lack of FDA or IRB approval

60
Warning letter observations for sponsors (Total 2004 – September 2007)
0 10 20 30 40
Regulatory Approval
Human Subj (not incl IC)
Informed Consent
Protocol Deviations
Records / Reports
Adverse Events
Invest. Deficiencies
Sponsor Deficiences
False Data
Monitoring
Misc

61
Most Common CDRHSponsor Deficiencies (Per BIMO)
o Inadequate monitoring
o Failure to secure investigator compliance
o Inadequate device accountability
o Failure to obtain FDA/IRB approval

62
Studies examining weaknesses
o 1996 GAO study suggesting that Federal oversight is lax
o 1999 NYT “Research for Hire. A Doctor’s Drug Studies Turn into Fraud.”
o 2000 Washington Post “The Body Hunters.”o 1999-2001 National Bioethics Advisory
Commission Report on research compliance
o 2001 GAO report on progress in strengthening protections
o 2005 Office of Research Integrity/HHS Annual Report
o 2007 OIG Report on FDA’s Oversight of Clinical Trials

63
Summary of hot spots
o Investigator financial conflicts and interests, including consulting relationships
o Reporting requirements, including adverse event reporting
o Enforcement focusing on device studies– Device regulatory environment
becoming more like drugs

Questions and Discussion
Arguing with a lawyer is like mud wrestling with a pig: after a while you realize
that the pig actually enjoys it.