experimental studies of bmp signalling in neuronal cells
TRANSCRIPT

Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1259
Experimental Studies of BMPSignalling in Neuronal Cells
BY
SUSANNA ALTHINI
ACTA UNIVERSITATIS UPSALIENSISUPPSALA 2003

List of papers
This thesis is based on the following papers referred to in the text by their Roman numerals:
I Targeted Deletion of GDF10 has no Effect on Long Term Potentiation, Contextual Learning Ability or Gene Transcription in the Hippocampus Susanna Althini, Magnus Åbrink, Stine Söderström, Jonas Lindeberg, Annika Kylberg, Vidar Jensen, Öivind Hvalby and Ted Ebendal. Manuscript, 2003
II Normal Nigrostriatal Innervation but Dopamine Dysfunction in
Mice Carrying Hypomorphic Tyrosine Hydroxylase Alleles Susanna Althini, Henrik Bengtsson, Dmitry Usoskin, Stine Söderström, Annika Kylberg, Eva Lindqvist, Susana Chuva de Sousa Lopes, Lars Olson, Jonas Lindeberg and Ted Ebendal. Journal of Neuroscience Research, 2003
III Selective Blocking of MAP Kinase Activity is Enhancing
Neurotrophic Growth Responses Susanna Althini, Dmitry Usoskin, Annika Kylberg, Paul L. Kaplan, and Ted Ebendal. Manuscript, 2003
IV BMP signalling in NGF-stimulated PC12 cells
Susanna Althini, Dmitry Usoskin, Annika Kylberg, Peter ten Dijke & Ted Ebendal. Manuscript, 2003
Reprint of paper II was made with permission from the publisher.

”Sluta inte göra saker du ångrar, sluta ångra saker du gör”

Contents
Introduction.....................................................................................................1 The transforming growth factor beta superfamily......................................2
Ligands ..................................................................................................2 Receptors ...............................................................................................4 Smad as an intracellular signalling mediator.........................................7 BMP target genes ..................................................................................9 Biological actions of BMPs.................................................................10
Neurotrophic factors.................................................................................14 The family of neurotrophins ................................................................14 Neurotrophin induced intracellular signalling pathways .....................16 The GDNF family................................................................................19
Nerve cells examined ...............................................................................21 Hippocampus.......................................................................................21 Catecholaminergic neurons .................................................................23
Present investigation .....................................................................................27 Aims .........................................................................................................27 Results ......................................................................................................28
Paper I..................................................................................................28 Paper II ................................................................................................28 Paper III ...............................................................................................33 Paper IV...............................................................................................34
Discussion.....................................................................................................36 Knockout without phenotypical alterations..............................................36 Dopamine dysfunction in TH-hypomorphic mice ...................................36 Crosstalk...................................................................................................37
Concluding remarks......................................................................................38 Paper I..................................................................................................38 Paper II ................................................................................................38 Paper III ...............................................................................................39 Paper IV...............................................................................................39

Materials and methods ..................................................................................40 Targeted gene disruption/alteration (Paper I and II) ................................40
Generation of truncated ALK2 and BMPRII by PCR .........................40 Screening for genomic DNA clones and targeting vector construction.............................................................................................................40 Culturing of embryonic stem (ES) cells ..............................................41 DNA-preparation and screening of ES-cell clones..............................41 Blastocyst injection and germ line transmission .................................42 Housing and Breeding .........................................................................42 Tail biopsies and DNA-preparation.....................................................43 Genotyping ..........................................................................................43
Behavioural testing (Paper I and II) .........................................................44 A protocol for primary phenotypic screening......................................44 Spontaneous movement (Paper I and II) .............................................45 Spatial learning ability (Paper I)..........................................................45
Electrophysiological recordings (Paper I)................................................45 LTP in the CA1-region of the hippocampus and the medial lateral perforant paths of the dentate gyrus ....................................................45
Biological assays (Paper III) ....................................................................46 Neurite outgrowth assays.....................................................................46 Survival assay......................................................................................47 Pharmacological inhibitors of MAPK pathways (Paper III) ...............47
Biochemical and molecular studies..........................................................48 Determination of catecholamine and 5HT levels (Paper II) ................48 Immunofluorescence (Paper II) ...........................................................48 In situ hybridisation (paper I and II)....................................................49 RNA preparation and microarray analysis (Paper I) ...........................50 Western blot (Papers III and IV) .........................................................51
Cell culture ...............................................................................................51 PC12 cells ............................................................................................51 Luciferase assay (Papers II and IV).....................................................52 Crosslinking (Paper II, supplementary data) .......................................53
Acknowledgements.......................................................................................54
References.....................................................................................................59

Abbreviations
ALK Activin Like Kinase ART Artemin BAMBI BMP & Activin Membrane Bound Inhibitor BDNF Brain Derived Neurotrophic Factor BMP Bone Morphogenetic Protein BMPRII BMP type II Receptor CA Catecholamine CNS Central Nervous System DA Dopamine DBH Dopamine β Hydroxylase DG Dentate Gyrus Erk Extracellular Regulated Kinase GDF Growth/Differentiation Factor GDNF Glial cell-line Derived Neurotrophic Factor GFLs GDNF Family Ligands GFRα GDNF Family Receptor-α GSK3 Glycogen Synthase Kinase 3 Id Inhibitor of differentiation JNK Jun N-terminal Kinase NGF Nerve Growth Factor NT Neurotrophin NTN Neurturin PNS Peripheral Nervous System PSP Persephin PI3K Phosphatidylinostisol-3 Kinase SARA Smad Anchor for Receptor Activation Smad Sma/Mad homologue Smurf Smad ubiquitination regulating factor TGFβ Transforming Growth Factor beta

Introduction
More than 100 billions of neurons in the brain are joined together into functional networks by trillions of synaptic connections in a remarkably well-controlled manner. This network controls everything we do. Walking, talking, sleeping, eating, feeling, perceiving, or perhaps writing a thesis… How can the fertilized egg develop into a person with body, brain and mind? How do brain and nerves know how to organize themselves into a functional system? How can the brain produce the individuality of human action? What is consciousness? Where and how are our memories stored? What goes wrong when a person suffers from depression, schizophrenia, Parkinson’s or Alzheimer’s disease?
Molecular biology, neurophysiology, genetics, cell biology, anatomy, developmental neurobiology and psychology are some of the disciplines contributing to modern neuroscience research dealing with these mysteries, attempting to link genes and proteins to mind, behaviour and disease.
This thesis is focused on how growth factors belonging to the transforming growth factor beta (TGFβ) superfamily affect neuronal cells. I have used different genetic and cell biological strategies to study the molecular mechanisms and the effects of BMP-signalling on neuronal morphology in cell cultures, as well as on neuronal function and behaviour in mouse models.
1

The transforming growth factor beta superfamily The TGFβ superfamily of secreted cytokines is extensive. In this introduction, I will give an overview of different aspects of TGFβ superfamily signalling. I will focus on the biological functions of the subgroups that constitute Bone Morphogenetic Proteins (BMPs) and especially the action of these cytokines in neuronal cells.
Crosstalk between the BMP/TGFβ pathways and other intracellular signalling cascades is a way for cells to adjust their behaviour to the environment by integrating input from various extracellular cues. Papers III and IV describe apparent crosstalk between BMPs and neurotrophic factors resulting in potentiation of neurite formation. I will therefore also make a summary of pathways activated upon neurotrophic factor stimulation and their biological actions.
The hippocampus, a brain area involved in memory formation, as well as the catecholaminergic parts of the nervous system regulating behaviours like affect, emotion, memory storage, drives, motivation and movement will also be introduced as a background to papers I and II respectively.
Ligands The cytokines of the TGFβ superfamily are dimeric proteins with a broad range of biological effects, that control actions such as cellular growth, differentiation, chemotaxis, apoptosis, and secretion of extracellular matrix components, both during development and in the adult (Kawabata et al., 1998a). Over 30 members of this superfamily have been described in mammals, and in addition, a number of homologues in Drosophila melanogaster, Xenopus laevis and Caenorhabitis elegans (FIG. 1; reviewed by Kawabata et al., 1998a; Miyazawa et al., 2002). All factors are synthesized as large precursor proteins with an N-terminal pro-peptide, which is proteolytically cleaved off from the biologically active C-terminal peptide upon secretion. The active peptide contains seven conserved cystein residues, characteristic for all superfamily members (Kingsley, 1994), except GDF3 and GDF9 with only six cystines in conserved positions (McPherron and Lee, 1993). Disulfide bonds between six of the cysteins in the monomeric form of the proteins have been shown to form the core cystine knot, essential for their three-dimensional structure (Daopin et al., 1992; Griffith et al., 1996; Schlunegger and Grutter, 1992). The seventh cysteine has, together with hydrophobic contact surfaces, been implicated in the formation of the biologically active dimeric factors (Kingsley, 1994).
2

FIG. 1: TGFβ superfamily members. (d) Drosophila, (h) human, (m) mouse, (x) Xenopus (adapted from Miyazawa et al., 2002).
Typically, this superfamily is divided into the following subgroups, based on sequence homology in the c-terminal region: the TGFβs (TGFβs and nodal),
3

the BMPs (all BMPs and some of the GDFs), the activin/inhibin group, and anti-Müllerian hormone (AMH) forming its own subgroup. To exert action, the factors bind hetero-tetrameric typeI/typeII-serine/threonine kinase receptor complexes, transmitting the signal through the cytoplasm via the Smad-pathways to the nucleus, where transcription of target genes is regulated (see below). The two groups can be functionally grouped in two subgroups, depending on which intracellular pathway they use. One group is signalling via the activin/TGFβ activated Smads (Smad2/3) and the other signalling via the BMP activated Smads (Smad1/5/8; FIG. 2). The fact that a factor is related to BMPs by its sequence does, however, not necessarily mean that it signals via the BMP-Smads, as exemplified by BMP3, shown to use the Smad2/3 pathway to inhibit bone formation (Bahamonde and Lyons, 2001; Daluiski et al., 2001). Glial cell-line derived neurotrophic factor (GDNF), neurturin (NTN), artemin (ART) and persephin (PSP) form another subgroup, sharing the cystein knot structure with the TGFβ related proteins. These proteins do however signal via other pathways and will be discussed separately.
Receptors All factors belonging to the TGFβ superfamily, except the GDNF-subgroup, signal via tetrameric typeI/typeII serine/threonine-kinase-receptor complexes. In mammals, seven type I and five type II receptors have been described (Miyazono, 2000a). Theoretically, more than 30 combinations of type I and type II receptors can be formed (one for each ligand). However, under physiological conditions some combinations are favoured as some type I receptors tend to interact only with certain type II receptors. Hence, the number of ligands is larger than the number of functional receptor complexes, causing the variety of ligands to converge to a lower degree of variability at the receptor level. The type I receptors, termed activin receptor-like kinase (ALK) 1-7 consist of (i) an extracellular ligand-binding domain, (ii) a hydrophobic trans-membrane region, (iii) a characteristic glycine-serine rich GS-domain and (iv) a serine/threonine kinase. The type II receptors display a similar arrangement, with an extracellular ligand-binding domain, a transmembrane region and an intracellular kinase, but differ in that they lack the GS-domain. The kinase domain of the type II receptor is constitutively active even without ligand stimulation. Upon ligand binding, hetero-tetrameric complexes, containing two type I and two type II receptor molecules, are formed and the type II kinases transphosphorylate the GS-domain of the type I receptors, thereby activating them to transmit the signal to specific intracellular substrates (Kirsch et al., 2000; Qin et al., 2002).
4

The seven ALKs differ in ligand specificity and in the use of intracellular substrates. The choice of intracellular substrate is determined by the L45 loop of the type I receptors (Feng and Derynck, 1997). ALK1 is only expressed in endothelial cells, where it acts as a type I receptor for TGFβ. It is structurally very similar to ALK2. However, ALK2 has been shown to act as a BMP receptor, responding to BMP6 and BMP7 (ten Dijke et al., 1994; Macias-Silva et al., 1998; Ebisawa et al., 1999; Aoki et al., 2001). ALK3 and ALK6 are also acting as BMP receptors, however less restricted in their choice of ligand, transmitting signals from all BMPs. Another difference between ALK2 on one hand, and ALK3 and ALK6 on the other is that ALK2 recruits only Smad1/5 while ALK3/6 can signal to Smad1/5/8 (Ebisawa et al., 1999; Aoki et al., 2001). ALK5 binds TGFβs and the ALK5-relatives; ALK4 responds to activins and nodal, while ALK7 has nodal as its only known ligand (Reissmann et al., 2001). The activin/TGFβ/nodal/GDF8-responding receptors; ALK4, ALK5 and ALK7 have Smad2/3 as intracellular targets while ALK2, ALK3 and ALK6 transmit BMP2-8/GDF5-7/AMH-induced signals to Smad1/5/8. The endothelial specific TGFβ receptor, ALK1, was recently shown to use Smad1/5 to induce the BMP-target gene IdI and thereby to counteract TGFβ signalling via ALK5 to Smad2/3 in the same cells (Oh et al., 2000; Goumans et al., 2002). In other words, ALK1 and ALK2 use Smad1/5/8 as intracellular substrate, irrespective of their closer relation, based on amino-acid sequence comparisons, to ALK4/5/7 than to ALK3/6. The receptor and intracellular signalling pathway remains to be shown for some ligands including BMP9-11, GDF1,3,9 and GDF10 (paper I). GDF10 is also called BMP3b, and is highly homologous to BMP3. One could therefore speculate that it is likely that GDF10 uses the Smad2/3 pathway, as BMP3 does. However, it would not be a big surprise if the opposite is shown, considering the many exceptions to the concept that sequence homology indicates similar functions, as exemplified above.
Of the five type II receptors present in mammals, the TGFβ type II receptor (TβRII) is specific for TGFβs. Activin type II receptors (ActRII and ActRIIB) can in addition to responding to activin, also transmit signals from other TGFβ superfamily members, including BMPs and nodal. The BMP type II receptor (BMPRII) is specific for BMPs and AMH type II receptor (AMHRII) for AMH (FIG. 2; Mishina et al., 1997). Many of the type1/typeII receptors are expressed in the nervous system and some of them are also present in the adult brain (Bengtsson et al., 1995; Söderström et al., 1996).
5

FIG. 2: TGFβ superfamily signalling.
A pseudo-receptor, structurally similar to type I receptors in the extracellular ligand-binding domain, but lacking the intracellular kinase, can antagonize BMP/TGFβ signalling by forming heteromeric complexes with the serine/threonine kinase receptors. This type of pseudo-receptor was first shown in Xenopus and named BAMBI (BMP and Activin membrane-bound
6

inhibitor; (Onichtchouk et al., 1999). The mammalian BAMBI-homologue is the nma gene product. Other membrane-anchored factors, including β-glycan endoglin and crypto seem to facilitate ligand binding to the TGFβ superfamily receptors. Some of these play dual roles, exemplified by β-glycan, which is facilitating TGFβ signalling as well as the binding of the activin inhibitor α-inhibin to activin receptors, decreasing the cellular response to activin (Lopez-Casillas et al., 1993; Massagué, 1998; Lewis et al., 2000)
Smad as an intracellular signalling mediator Smad, the name for the mammalian intracellular serine/threonine kinase-substrates, was formed from the name of two homologues; Sma from the C. elegans Small-gene and mad from the Drosophila mad (mothers against decapentaplegic)-gene (Raftery et al., 1995; Sekelsky et al., 1995; Derynck et al., 1996; Hoodless et al., 1996; Savage et al., 1996). The eight known mammalian Smads are divided into three groups based on their functions. One group of Smads is recruited to and activated by type I receptors and is hence denoted receptor-activated Smads (R-Smads). This group constitutes of the already mentioned Smad2/3 and Smad1/5/8, classically known to respond to activin/TGFβs and BMPs respectively. The second group, with Smad4 (Hahn et al., 1996) as its only member, functions as a common-mediator Co-Smad, forming complexes with the activated R-Smads. These R-Smad/Co-Smad complexes can then be translocated to the nucleus where they participate in transcriptional regulation of target genes. Smad6/7, belonging to the third group of Smads, acts as a negative regulator of Smad-signalling in a negative feedback loop. These inhibitory I-Smads are produced in response to BMP or TGFβ signalling and can interfere with the Smad-pathways at different levels to exert their negative feedback effect. The I-Smads can also be induced by other signals, which in this way can modulate the amount of I-Smads present and hence also the magnitude and duration of TGFβ superfamily signalling (Heldin et al., 1997; Itoh et al., 2000; Moustakas et al., 2001).
Smads have two mad homology domains, MH1 and MH2, connected by a linker region. The N-terminal MH1 domain, which is responsible for DNA-binding to specific sequences present in target gene promoters, is conserved in the R-Smad and Co-Smad subgroups but not in I-Smads. The C-terminal MH2 domain, on the other hand, is conserved in all three subclasses. The R-Smads have a characteristic Serine-Serine-X-Serine (SSXS) motif at the extreme C-terminal end of MH2, which is phosphorylated by type I receptors. The amino acid sequence of the linker region is quite divergent between the different Smads. Some Smads (Smad1, Smad2 and Smad5)
7

have been shown to carry a number of sites in the linker region which, when phosphorylated by Erk, can prevent nuclear translocation of the R-Smad/Co-Smad complexes (Kretzschmar et al., 1997 and 1999; paper III and IV). I will come back to this topic when describing crosstalk between Smads and other intracellular signalling pathways.
The MH1 and MH2 domains of inactive R-Smads are physically associated to each other and at least R-Smad2/3 are anchored as dimers to the plasma membrane through SARA (Smad anchor for receptor activation) and possibly other molecules (Tsukazaki et al., 1998; Qin et al., 2002) via their MH2 domains. Upon receptor activation and phosphorylation of the SSXS motifs, the interactions between the MH-domains as well as between the MH domains and the anchor proteins are disrupted. The R-Smads form hetero-oligomers with Co-Smad through the MH2-motifs. The number of Smads involved in each oligomer is a subject for controversy, since both hetero-dimer and hetero-trimer models have been proposed (Kawabata et al., 1998b; Qin et al., 2001; Wu et al., 2001). R-Smad/Co-Smad complexes translocate into the nucleus, where they interact with various DNA-binding proteins and target gene promoters in order to regulate transcription of these genes. Somewhat generalised; the activin/TGFβ responsive Smads work by binding a CAGA-promoter motif, while BMP-Smads have been shown to use CG-rich promoter sequences in stead in order to transactivate target genes (Dennler et al., 1998; Ishida et al., 2000; Itoh et al., 2000; Kusanagi et al., 2000; Miyazono et al., 2001; Zawel et al., 1998). Surprisingly, a reporter construct (Smad Binding Element x4 - SBEx4-luc) containing repeated CAGAC sequences from the junB gene could unexpectedly respond strongly to BMP signalling as well as to TGFβ signalling (Jonk et al., 1998; this reporter was used in paper IV). R-Smad/Co-Smad complexes can also interact with transcriptional co-activators and co-repressors to promote acetylation and de-acetylation of histones, respectively, and in this way influence the transcription machinery of the cell (Massagué and Wotton, 2000; Miyazono, 2000a; ten Dijke et al., 2000).
The I-Smads can, as already stated above, inhibit Smad-signalling at different levels. Both Smad6 and Smad7 can interact with activated type I receptors and in this way prevent phosphorylation of R-Smads. In addition, Smad6 can bind to already activated R-Smads and thus interfere with the complex formation between Co-Smad. Smad7 has broader action-spectrum and inhibits TGFβ, activin and BMP-signalling, while Smad6 acts mostly on BMP-signalling (Bai et al., 2000). Smad6 has also been reported to act as a transcriptional repressor in the nucleus by interaction with Hoxc8, a homeobox gene (Bai et al., 2000). I-Smad expression is induced by TGFβ superfamily members themselves (Nakao et al., 1997), and by for example Interferon gamma (IFNγ) and Tumour Necrosis Factor alfa (TNFα).
8

Smad activity is also regulated by mechanisms causing its degradation. Smad1/5 (but not Smad8) have a PY motif (PPAY) in their linker region, which is recognized by a HECT-class E3 ubiquitin ligase called Smurf1 (Smad ubiquitination regulatory factor). Smurf1 recognition of Smad1/5 leads to ubiquitin-mediated proteasomal degradation of these Smads. Smad8 does not have the PY-motif and cannot be degraded by the Smurf1-mechanism unlike the other BMP-Smads. Interestingly, the Smurf1 mediated degradation of Smad1/5 occurs independently of BMP receptor activation, indicating that Smurf1 does not function downstream of activated Smads to turn off BMP signals, but rather adjusts the basal level of Smads (Heldin and ten Dijke, 1999; Zhu et al., 1999; Suzuki et al., 2002)
BMP target genes Only a few DNA-binding partners for the BMP Smads 1/5/8 have been identified, in contrast to the long list of interactors for the activin/TGFβ- Smads 2/3 (Miyazawa et al., 2002). In addition, the transcriptional co-activators p300 and CBP, as well as the co-repressors cSki and SnoN interact strongly with Smad2/3 but only weakly with Smad1/5/8. p300 and CBP have histone-acetyl transferase activity, suggesting that they may help transcription of Smad-target genes by chromatin remodelling, and hence make the promoter more accessible to the transcription machinery. With their many partners, Smad2/3 have the ability to transactivate many target genes, including plasminogen activator inhibitor-1 (PAI-1), type I collagen, the junB transcription factor, Smad7, Mix.2, and the cell cycle regulators p21 and p15, to influence cellular processes such as extracellular matrix formation and growth inhibition (reviewed by Massagué and Wotton, 2000). In addition, Smad2/3 inhibits transcription of the c-Myc gene. In consistence with the small number of DNA-binding partners, only a few BMP target genes have been found, including inhibitor of differentiation (Id) 1-3, Smad6, Vent-2 and Tlx-2 (Ogata et al., 1993; Onichtchouk et al., 1996; Norton et al., 1998; Afrakhte et al., 1998; Tang et al., 1998; Takase et al., 1998; Hollnagel et al., 1999; Ishisaki et al., 1999; Lopez-Rovira et al., 2002). The question of how BMPs exert their multiple cellular actions is thus still unanswered. Recent reports have raised interest for the Id-proteins as important mediators of BMP induced biological activities (Miyazawa et al., 2002).
Id proteins have a Helix-Loop-Helix (HLH) dimerisation structure, which enables them to negatively regulate the actions of other bHLH transcription factors, as well as members of the retinoblastoma (Rb) and Ets families (Norton et al., 1998; Miyazono and Miyazawa, 2002; Yokota and Mori, 2002). However, they lack a basic DNA binding structure and by physically
9

interfering with ubiquitously expressed bHLH transcription factors, normally forming heteromers with tissue specific bHLH proteins, they block transcription of genes that have an E-box motif in their promoters. In other words, Id proteins act as dominant negative antagonists of bHLH transcription factors. Four mammalian Id proteins (Id1-4) with partially overlapping expression patterns have been identified. Generally, Id proteins block differentiation because most bHLH proteins positively regulate differentiation. Id proteins (at least Id2, possibly also in Id4) promote cell cycle progression by inhibiting the anti-proliferative actions of Rb-family proteins. Other Id proteins can also regulate the cell cycle, by suppression of bHLHs leading to induced expression of cyclin dependent kinase (CDK) inhibitors, including p21 (Yokota and Mori, 2002). Having these two major tasks, Id-proteins play an important role in regulating the balance between cell proliferation and differentiation during embryogenesis. For example, Id1/Id3 double knockout mice die at embryonic day 13.5. Their brains are very small due to premature withdrawal of neuroblasts from the cell cycle, instead causing them to differentiate and express neuron-specific markers, with too few neurons as result (Lyden et al., 1999). During neurogenesis, the expression of the Id family members has unique patterns along the dorsoventral axis of the neural tube. Later on, Id1 and Id3 are found in dividing neuroblasts, while Id2 and Id4 are expressed by maturing neurons (Jen et al., 1997). This indicates that different Ids may have different physiological tasks, contributing in unique ways in neuronal differentiation.
Biological actions of BMPs The first indication of a BMP was found over 30 years ago in experiments done by Urist et al., showing the existence of a diffusible substance with bone inducing properties. In 1973, he identified a secreted factor, a protein that he named bone morphogenetic protein (Urist, 1965; Urist et al., 1979). Since then, many more BMPs have been isolated and shown to control a plethora of cellular functions, such as proliferation, differentiation, apoptosis, survival and even cell fate. Their activity is critical for regulating numerous developmental and homeostatic processes and they play pivotal roles in morphogenesis of various tissues and organs, including the nervous system (Reviewed by Hogan, 1996). Spontaneous mutations in GDF8 in cattle have for example resulted in double-muscled Belgian Blue breed (Kambadur et al., 1997). We have shown, that BMPs cooperate with neurotrophic factors in inducing neuronal differentiation and survival both in sympathetic and sensory neurons using explanted ganglia from embryonic day 9 chickens (Paper III; Bengtsson et al., 1998).
10

Gene targeting experiments in the mouse have revealed that BMP signalling is strictly required for early embryonic developmental events, such as primitive streak formation and epiblast proliferation during gastrulation (Goumans and Mummery, 2000; Zhao, 2003). GDF1, nodal and its antagonists lefty1 and lefty2, as well as ActRIIa, ActRIIb, Smad2 and Smad5 appear to play key roles in the establishment of left-right asymmetry (Meno et al., 1997; Nomura and Li, 1998; Chang et al., 2000; Rankin et al., 2000; Lowe et al., 2001; Whitman and Mercola, 2001). However, the relationship between these ligands, receptors and downstream Smads remains to be determined.
Due to early embryonic lethality resulting from conventional targeted deletions of many of the molecules involved in BMP signalling in the mouse, it has been hard to show their contribution in mammalian neuronal development. Data obtained from other animal models, for example embryonic Xenopus and chick assays (Hartley et al., 2001; Wilson et al., 2001; Stern, 2002), have however revealed that BMP-signalling is essential for the proper formation of the peripheral and central nervous systems, through effects on primary neuronal induction, dorso-ventral patterning of the neural tube, regionalisation of the brain, eye development and lineage determination in the peripheral nervous system (Hemmati-Brivanlou et al., 1994; Hemmati-Brivanlou and Melton, 1994; Xu et al., 1995; Furuta et al., 1997; Hemmati-Brivanlou and Melton, 1997; Wilson and Hemmati-Brivanlou, 1997; Baker et al., 1999; Harland, 2000). Tissue-specific and inducible knockouts and mutants (i.e. the Cre/LoxP and Tet on/off systems) will be highly valuable in the mapping of BMP-functions in the nervous system (Chytil et al., 2002; Higashi et al., 2002; Huang et al., 2002; Kulessa and Hogan, 2002; Mishina et al., 2002).
Many of the ligands, receptors and Smads are expressed in the nervous system, both during development and in the adult (Bengtsson et al., 1995; Lorentzon et al., 1996; Rydén et al., 1996; Söderström et al., 1996; Tsuchida et al., 1996; Ebendal et al., 1998; Söderström and Ebendal, 1999). The expression of various BMP-signalling components is regulated in response to traumatic brain injury, global cerebral ischemia and kainic acid induced seizures (Lewén et al., 1997; Charytoniuk et al., 2000; Wang et al., 2001).
In the developing PNS, selected BMPs play an instructive role in programming the elaboration of the neuronal lineage from neural crest stem cells. BMPs, secreted from the dorsal aorta, are sensed by migrating, Mash1-expressing neural crest cells, leading to the induction of Phox2a, Phox2b and eventually tyrosine hydroxylase (TH) and dopamine β-hydroxylase (DBH) gene expression, pushing these cells to develop into sympathetic noradrenergic neurons (Schneider et al., 1999; Ernsberger et al., 2000; McPherson et al., 2000). The same gene products, of which Phox and GATA
11

represent transcription factors and TH and DBH enzymes needed for catecholamine synthesis, are also involved in the development of other noradrenergic neurons (in locus coeruleus and other NA-nuclei). It is not clear, however, how the molecular cascade of the BMP signal transduction connects to the action of Phox2 transcription factors and the expression of TH and DBH (Reissmann et al., 1996; Varley and Maxwell, 1996; Schneider et al., 1999; Ernsberger et al., 2000; Pattyn et al., 2000; Stull et al., 2001;).
Peripheral sensory neurons do, like sympathetic neurons, emanate from the neural crest. They develop from committed precursors that differentiate within two days after emigration from the neural tube. These precursors are different from the once differentiating into sympathetic neurons and the presence of BMPs cannot force these precursors into a sympathetic neuronal fate, nor prevent expression of sensory neuronal markers, such as neurogenin-1 and -2, and NeuroD. The mechanism that induces the resistance to BMP signalling and hence the commitment for a sensory fate is still unknown (Greenwood et al., 1999; Christiansen et al., 2000).
In addition to involvement in determination of the catecholaminergic neuronal phenotype, BMPs have been shown to affect the production of other neurotransmitters. BMP9 induces the expression of genes required for a cholinergic neuronal phenotype in the CNS. Primary embryonic cells from the septal area treated with BMP9 respond by increased levels of choline acetyltransferase (ChAT) and the vesicular acetylcholine transporter (VAChT), leading to up-regulation of acetylcholine synthesis. Indications for a direct effect through a BMP9 responsive element within the cholinergic gene locus were also shown (Lopez-Coviella et al., 2000, 2002).
A human neuroblastoma cell line (SH-SY5Y) expresses catecholamine features upon TGFβ1 treatment, an effect that is counteracted by BMP2. In addition, BMP2 could, under certain conditions, induce the cholinergic marker ChAT in these cells, suggesting that TGFβ1 and BMP2 contribute in opposite ways to the differentiation of neurotransmitter phenotype (Gomez-Santos et al., 2002). BMPs (2, 4 and 6) can transiently induce TH expression in mouse embryonic striatal neurons in culture, during a time window from E13 to E16, making them potentially important for the specification of a dopaminergic phenotype (Stull et al., 2001). Additionally, BMP6 has been shown to have neurotrophic effect on developing striatal neuron; this effect is however in part mediated by astroglia (Gratacos et al., 2002). Indirect trophic effects of BMPs have also been noted before; BMP2 and BMP6 are highly expressed in the developing rat midbrain and protect dopaminergic neurons against MPP+ toxicity by promoting astroglial differentiation, probably leading to production and/or release of glial derived neurotrophic factor (GDNF, see below; Jordan et al., 1997).
12

At the earliest stages of the development of the nervous system, BMPs actually inhibit neural fate and have to be antagonized by other factors (e.g. noggin, chordin) for normal gastrulation and neurulation to occur. This happens much earlier than the instructive effects BMPs have on peripheral sympathetic neuron differentiation. In addition, embryonic telencephalic neuroblasts, treated with BMP2, choose an astrocytic fate, rather than a neuronal. BMP2 induces Smad-dependent expression of Id1 and Id3 and Hes5, which in turn inhibits neurogenic transcription factors (Mash1, neurogenin, NeuroD etc.) and hence also neurogenesis (Nakashima et al., 2001). Adult neuronal stem cells in the subventricular zone (SVZ) continuously undergo neuronal differentiation. The BMP inhibitor Noggin is expressed by ependymal cells adjacent to the SVZ, and has been shown to create a microenvironment that permits neurogenesis by blocking BMP-induced glial differentiation (Lim et al., 2000). This is again different to the BMP-effects on neural crest cells. In conclusion, depending on developmental stage and cell type, the effects of BMPs can be contradictive, at some stages inhibiting neuronal fate and at others driving neuronal differentiation.
13

Neurotrophic factors “Neurotrophic factors are endogenous soluble proteins regulating survival, growth, morphological plasticity, or synthesis of proteins for differentiated functions of neurons” (Hefti et al., 1993)
FIG. 3: Target derived neurotrophic factors.
The family of neurotrophins The “NGF saga” started in the middle of the last century when Rita Levi-Montalcini, Victor Hamburger and colleagues first discovered trophic effects of mouse sarcomas grafted to chick embryos with drastic enlargement of sympathetic and dorsal root ganglia as a result (Bueker, 1948; Levi-Montalcini and Hambuger 1951 and 1953). Stanley Cohen made some critical experiments to identify the active substance, and showed how it could be isolated from male mouse submandibular glands and by chance, that it was present at a high concentration in moccasin snake venom (Cohen et al., 1954; Cohen 1959 and 1960). The substance was termed Nerve Growth Factor or NGF and Rita Levi-Montalcini and Stanley Cohen were awarded the Nobel Prize in medicine or physiology in 1986 for their discoveries (reviewed by Cowan, 2001). The isolation and characterisation of NGF led to the formulation of the “neurotrophic theory,” which states that target tissues produce limited amounts of secreted neurotrophic factors and that neurons, sending out their axons towards the target, compete for the support given by the trophic factors, in order to avoid programmed cell death, causing death of the least successful neurons (Thoenen and Barde, 1980). To date, four murine members of the family of neurotrophins are known; NGF, BDNF (brain derived neurotrophic factor) NT3 and NT4 (NT is short for neurotrophin) The amount of research done on these factors is voluminous and has been extensively reviewed over the years and recently by Segal (2003).
14

FIG. 4: The neurotrophins and their receptors
The neurotrophins bind to and activate Trk tyrosine kinase receptors and can in addition bind p75NTR, a member of the tumour-necrosis-factor receptor family (Radeke et al., 1987). NGF binds to TrkA (Cordon-Cardo et al., 1991; Hempstead et al., 1991; Kaplan et al., 1991a; Kaplan et al., 1991b), BDNF and NT4 to TrkB (Soppet et al., 1991; Klein et al., 1992) and NT3 preferentially to TrkC (Lamballe et al., 1991) but also to TrkA and TrkB.
The neurotrophins are found both in the in peripheral and central nervous systems, as well as in target tissues. They have different expression patterns, which also changes during embryogenesis and in the development of the adult nervous system. The neurotrophins are involved in the sculpting of the functional nervous system due to their ability to regulate neuronal survival, programmed cell death, axon guidance, axon and dendrite arborisation and activity dependent neuronal plasticity. Most of these effects are mediated through Trk-signalling, and one role of p75NTR may be to induce apoptosis. Balancing the expression of Trks, p75NTR and neurotrophins create a means for eliminating excessive neurons, or neurons sending projections in wrong directions (Miller and Kaplan, 2001). Gene targeting studies of several of the Trks and neurotrophins have revealed essential roles for neurotrophic signalling during development of the peripheral nervous system (Snider, 1994). Signalling downstream of the different Trks can have remarkably different outcomes in the same cells, as exemplified in differentiating avian neurons. In these cells signalling from NT3/TrkC promote cholinergic differentiation, whereas NGF/TrkA induce an adrenergic phenotype (Bibel and Barde, 2000; Brodski et al., 2000).
In the adult nervous system, the function of neurotrophins is less clear than during the development, since they are not needed for acute neuronal survival. They have however been shown to be involved in the maintenance
15

and the continuous sculpting of the nervous system during for example learning and memory storage. To function as molecular mediators of synaptic and morphological plasticity, the expression of the neurotrophins and their receptors must be activity dependent and their action must induce changes in neuronal circuits through altered synaptic function, membrane excitability or neuronal morphology and the number of synapses. Numerous experiments have indeed demonstrated neurotrophin mediated effects on synaptic plasticity but the mechanisms involved are still intriguingly elusive (McAllister et al., 1999).
Neurotrophin induced intracellular signalling pathways The different intracellular signalling pathways activated by Trk tyrosine kinases regulate survival, differentiation, growth and apoptosis. Survival is a particularly potent activity of neurotrophins in developing sympathetic and sensory neurons, which to a high degree depend on constant neurotrophic support. This fact has made it problematic to study the mechanisms mediating other effects of neurotrophins, such as neurite outgrowth. Recently, experiments using apoptosis resistant sensory neurons from Bax-/- mice have given some insight into the mechanism of neurotrophin induced morphological changes (Markus et al., 2002). These experiments show that NGF/TrkA signalling induces a greater activation of Raf, while NT3/TrkC preferentially activate Akt. In addition, they show that Raf-dependent signalling primarily induces axonal elongation, whereas Akt seems to be critical for branching and increasing axonal calibre. Thus, the morphology of growing axons could, at least in part, depend on the relative strengths of Akt and Raf signalling. Bax-/- mice have also been crossed to generate double mutants with TrkA-/- and NGF-/-. These mutants show a drastic reduction in the number of sensory axons at birth, suggesting the requirement of NGF for either elongation or maintenance of peripheral axons in vivo (Patel et al., 2000)
The small GTP-binding protein Ras has been shown to be crucial for most neurons to survive. It functions by translating and directing neurotrophin-induced signals into multiple intracellular signalling pathways. The major Ras activated pathways are the PI3K/Akt and the MEK/Erk cascades. For Trk-induced activation of PI3K, the action of Ras is combined with that of adapter protein Gab1. Crowder and Freeman (1998) showed that blocking of the PI3K pathway in sympathetic neurons with the inhibitor LY294002 could counteract the NGF-mediated survival. PI3K activates many signalling proteins, among which the serine/threonine kinase Akt (also named protein kinase B) was one of the first to be shown to function as a survival mediator (Dudek et al., 1997; Andjelkovic et al., 1998; Ashcroft et al., 1999). In
16

FIG. 5: Trk-induced intracellular signalling pathways.
addition, Akt has been shown to be involved in depolarisation-elicited survival, giving Akt a status as a convergence point for different survival-signals (Crowder and Freeman, 1999; Vaillant et al., 1999; Brunet et al., 2001a). Akt phosphorylation, and thereby inhibition, of a number of different pro-apoptotic substrates has been suggested to be involved in rescuing neurons from apoptosis (Brunet et al., 2001a; Miller and Kaplan, 2001; Patapoutian and Reichardt, 2001). A prime actor downstream of Akt is FKHRL1, a member of a forkhead box subfamily (FOXO) of transcription factors. Phosphorylation of FKHRL1 locates it to the cytoplasm in complex with 14-3-3 docking proteins, thus preventing transcription of cell death genes like FasL (Brunet et al., 2001b). Other possible substrates for Akt
17

include Bad and glycogen synthase kinase-3β (GSK3) (Brunet et al., 2001a; Patapoutian and Reichardt, 2001). Akt inhibits GSK3 and this will prevent its apoptotic effects, as shown in cultured neurons using the GSK3 inhibitor SB216763 on dissociated sensory chicken neurons (Cross et al., 2001). Targets, for PI3K signalling other than Akt, are members of the IAP (inhibitor of apoptosis) family of caspase inhibitors (Wiese et al., 1999), also implicated as anti-apoptotic actors.
The second well characterized pathway for the Trk receptors, that also starts with activation of Ras, continues with Raf/MEK1/Erk1,2 in succession. Downstream actors of the MAP kinase Erk2 (p42) include ternary complex factors like Elk and the Rsk kinase that under some circumstances activate further transcription factors including CREB (Kaplan and Miller, 2000). CREB interacts with the transcription machinery and induces transcription through the cAMP responsive elements, present in the promoter of some anti-apoptotic genes, including for example Bcl2, and is thus thought to be important for neuronal survival (Aloyz et al., 1998; Bonni et al., 1999). Evidence for the contribution of this pathway to neuronal survival is, however, conflicting (Kaplan and Miller, 2000). A major role for the Raf/MEK1/Erk1,2-pathway is to mediate neuronal growth. MEK-induced survival pathways have, in contrast to the PI3K/Akt pathway, also been shown to protect from death due to injury or toxicity, rather than trophic factor withdrawal. This pathway may also serve other functions in neuronal differentiation, such as modulating neurite outgrowth (paper III).
Other MAP kinase pathways including p38, c-Jun N-terminal kinase (JNK) and MEK5/Erk5 exist in neurons in parallel with the Raf/MEK/Erk signalling cascade (Watson et al., 2001). Removal of growth factors leads to inactivation of Erk activation of the JNK and p38 pathways (Xia et al., 1995) and in the case of JNK, further activation of c-jun mediated transcription of apoptotic genes. In addition, JNK has been demonstrated to serve functions in neurite outgrowth (Kita et al., 1998), presumably regulated by Trk-receptor activated PI3K, engaging the small G proteins Rac or Cdc42 to activate JNK to reorganise actin in the cytoplasm (Otto et al., 2000). Horstmann et al., (1998) found that p38 inhibitors SB203580 and SB202190 promoted survival in sympathetic, ciliary and sensory neurons from the chicken embryo, confirming that activation of p38 pathway in these neurons leads to neuronal death. When distal axons of sensory or sympathetic neurons are stimulated by neurotrophins, MEK1/Erk1,2 are activated in the nerve terminal whereas MEK5/Erk5 activation occurs in the remote cell soma. The activation of MEK5/Erk5 is therefore dependent on retrograde signalling in the axon (Watson et al., 2001), and because there are different substrates for the different Erks, this compartmentation may contribute to the neurotrophic outcome.
18

Trk elicited signalling from phospho lipase C gamma (PLCγ) releases Ca2+ from intracellular stores, hence increasing cytosolic Ca2+ concentration with various consequences. It also activates protein kinase C (PKC) may affect neurite outgrowth in neuronal cells (Friedman and Greene, 1999). PLCγ has been implicated as an important mediator of short pulses of neurotrophin signalling, likely reflecting a special role for PLCγ in mediating effects on synaptic plasticity (Minichiello et al., 1999; Segal, 2003).
Interestingly, the p75 receptor elicits pathways different from the ones induced by Trks. This receptor belongs to the TNFR/Fas superfamily and possesses a so-called death domain in its intracellular part. P75 appears to increase the sensitivity of Trks to the neurotrophins, but in the absence of their cognate Trk-receptor, the neurotrophins can induce caspase dependent apoptosis through the p75 receptor, which then probably exerts its action via the JNK/p53/Bax cascade (Kaplan and Miller, 2000). In sympathetic neurons, TrkA signalling, which inhibits the p75-mediated apoptosis, silences this JNK/p53/Bax cascade pathway via Ras and perhaps PI3K/Akt (Mazzoni et al., 1999).
Many of the pathways described here as intracellular mediators of Trks, are also likely to be activated after GDNF family ligand (GFL)-activation of the Ret tyrosine kinase receptor (see next section). Creedon et al. (1997) showed that neurturin (NTN) stimulated survival as well as PI3K activity in cultured neurons from the superior cervical ganglion. Moreover, these authors showed that NTN induced phosphorylation of Erk1/2 to an extent similar to NGF in these sympathetic neurons. Ret has also been shown to signal through JNK, an effect mediated by Rac/Cdc42 and not dependent on Erk activation (Chiariello et al., 1998).
The GDNF family Glial cell line derived neurotrophic factor (GDNF) was first purified from a rat glioma cell-line supernatant as a trophic agent for embryonic midbrain dopaminergic neurons (Lin et al., 1993). It has since then been shown to be involved in many developmental processes, both within and outside the nervous system. Knockout studies revealed its critical role in for example establishment of the enteric nervous system, kidney development, and spermatogenesis. It promotes survival of many types of neurons including subpopulations of peripheral autonomic and sensory, as well as central motor, dopamine and noradrenergic neurons (Lin et al., 1993; Arenas et al., 1995; Buj-Bello et al., 1995; Trupp et al., 1995; Hearn et al., 1998; Heuckeroth et al., 1998; Airaksinen et al., 1999; Airaksinen and Saarma, 2002). Because of its protective and regenerative effects on dopaminergic
19

neurons, much effort has over the last decade been put into developing restorative treatment for Parkinson’s disease using GDNF (Reviewed by Grondin and Gash, 1998). Unfortunately, no breakthrough leading to controlled clinical trials has so far been made.
GDNF is the founder member
of the GDNF-family of ligands (GFLs), with three additional members; neurturin (NTN; Kotzbauer et al., 1996), artemin (ART; Baloh et al., 1998) and persephin (PSP; Milbrandt et al., 1998). They are, as stated above related to the TGFβ superfamily, despite low amino-acid sequence homology, by their cystine knot structure, involved in protein stabilization and dimer formation. All GFLs signal through the RET tyrosine kinase receptor (Re-arranged in transformation; (Takahashi et al., 1985). To be able to do so, they must first bind to their corresponding GDNF family receptor-α (GFR α1-4), linked to lipid rafts in the plasma membrane by glycosyl phosphatidylinostisol (GPI) anchors. GDNF binds preferentially to GFR α1, NTN to GFR α2, ART to GFR α3 and PSP only to GFR α4. RET has four extracellular cadherin-like repeats, one transmembrane region and a typical intracellular tyrosine kinase domain. Upon GFL-GFRα binding and recruitment of RET to the lipid raft, the association of the cytoplasmic protein Src is triggered, leading to the transmission of the signal further into the cell via additional adaptor proteins. Soluble forms of GFR α exist and trigger different intracellular signals than the GPI-anchored ones. Like neurotrophins signalling via Trk-tyrosine kinases, GFL activation of RET triggers two major intracellular routes; the phosphatidylinostisol-3 kinase (PI3K; van Weering and Bos, 1997) and the mitogen-activated protein kinase (MAPK; Santoro et al., 1994; Worby et al., 1996) pathways, both contributing to neuronal survival. In addition, the Jun N-terminal kinase (JNK; Chiariello et al., 1998; Xing et al., 1998) and the phospholipase Cγ (PLCγ) pathways can also be activated to mediate survival upon RET-signalling. MAPK-activation is classically known to be crucial for neuritogenesis and PI3K and PLCγ have been implicated as potentiators of neurotransmission upon neurotrophin activation (Kaplan and Miller, 2000).
FIG. 6: GDNF family ligands and their receptors.
20

Nerve cells examined
Hippocampus Named the seahorse, because of its shape, the hippocampus is the main relay station that determines whether a new memory goes into long-term storage or is deleted after its short-term usefulness is over. It is clear that the hippocampus is not the “hard drive” of the brain, although it has some capacity for memory storage, this storage is transient, and hence the function of hippocampus appears to be to prepare contents for long-term storage in the cortex (Albright et al., 2000a,b). Much information about the significance of the hippocampus have arisen from the studies of a famous patient named H.M. who had most of his medial temporal lobes, including the hippocampus, removed during an epilepsy surgery in 1953. After surgery, he formed no new declarative memories, i.e. he could not memorize people, places, objects or how to find the way home etc. He remembered things that happened before the surgery and had the ability to form procedural memories, like learning to play tennis or solving a puzzle. He could have a conversation with a person, but if H.M. left the room for a moment, he met a “new person” when he re-entered the room (Kandel et al., 2000). Furthermore, the hippocampus is known to be important for spatial learning, or learning how to find the way based on landmarks in the environment. London taxi drivers are for example known for their ability to navigate the vast network of London streets, and have indeed been shown to have enlarged hippocampi (Maguire et al., 1996). Spatial learning is also often used in research as a marker for hippocampal function. In paper III of this thesis, I used a well-established model for studies of spatial learning in rats and mice, the Morris water maze (Morris et al., 1981; Morris, 1984), in which the rat/mouse is placed in a circular pool with a platform hidden just beneath the water surface. After repeated trials, a mouse with intact hippocampal function will learn to locate the platform by using visual cues around the pool.
The hippocampus consists of multiple interconnected areas, including CA1, CA3 and the dentate gyrus (DG), each containing a unique set of neurons composing a distinct cellular network. There is essentially a one-way flow of information entering the hippocampus via the perforant path from the entorhinal cortex into the dentate gyrus, where the entorhinal axons synapse on cells, which in turn project their axons, called mossy fibres to the CA3 region. The neurons in CA3 send axons called Schaffer collaterals to CA1, which sends yet another set of fibres exiting the hippocampus to the
21

FIG. 7: Schematic drawing of the hippocampus. Sites for electrophysiological stimulation and recording are indicated. (DG dentate gyrus, EC entorhinal cortex, GC granule cells, PP peforant path, LPP lateral PP, MMP medial PP, MFP mossy fibre path, SCP Schaffer collateral path)
subiculum. The subiculum is responsible for the hippocampal output and has neurons that project to the hypothalamus and mammillary bodies via the fornix and neurons that project back to the entorhinal cortex or to the sensory cortex.
The hippocampal mechanisms for long-term memory formations are not entirely understood. However, it is generally believed that a memory is stored in a neuronal network with a specific pattern of long-lasting alterations in synaptic strength. The synaptic strength can either be increased, as in long-term potentiation (LTP), or decreased, as in long-term depression. Whether both LTP and LTD contribute to memory storage is still under dispute. LTP is a mechanism by which individual neurons and sets of neurons increase their responsiveness to particular stimuli (Kandel, 2001). The changes in synaptic strength, mediated by electric neuronal activity, or by the action of growth factors or other substances, are collectively termed synaptic plasticity. If an axon signals to a particular dendrite repeatedly, the receiving dendrite will change its structure so that it responds with greater sensitivity to that input. Activation of a synapse leads to release of neurotransmitters from the presynaptic cell in the synaptic cleft where receptors on the postsynaptic cell bind the transmitter, in turn leading to opening of ion channels and propagation of the electric signal in the postsynaptic neuron. Frequent
22

activation of the synapse also leads to increased synthesis of cAMP and protein kinase signalling, eventually resulting in synthesis of new proteins contributing to growth and strengthening of the synapse (Greengard, 2001).
FIG. 8: Brain areas involved in memory processing (adapted from Kandel et al., 2000)
Catecholaminergic neurons In the 1950’s Arvid Carlsson in Göteborg made some critical experiments, which led to the suggestion that dopamine is an important transmitter of neuronal signals in the brain. He injected rabbits with a drug called reseperine that empties the dopamine stores in the brain. The rabbits presented with symptoms such as akinesia and rigidity, much like patients with Parkinson’s disease. After treatment with L-dopa, the recovered from their symptoms, and the same treatment was then tested on Parkinson patients with very good result. This discovery was awarded the Nobel Prize in medicine or physiology 2000 (Benes, 2001; Carlsson, 2001a; Carlsson, 2001b). The prize was shared with Paul Greengard and Eric Kandel, who both substantially have contributed to the knowledge about the underlying molecular mechanisms of synaptic plasticity and learning and memory. The catecholamines belong to the monoamine class of neurotransmitters, and are synthesized from the essential amino acid, tyrosine. The step-wise biosynthesis of catecholamines is initialised by the conversion of tyrosine to an inactive intermediate, L-dihydroxy phenylalanine (L-dopa), by tyrosine hydroxylase (TH; Nagatsu et al., 1964; Levitt et al., 1965). This enzyme is required for the synthesis of all catecholamines and represents the rate-limiting step in this pathway. In turn, L-aromatic amino acid decarboxylase (AADC) converts L-dopa to dopamine, the first active neurotransmitter produced in this process. Therefore, all catecholaminergic neurons contain TH and AADC, whereas only noradrenergic and adrenergic cells express the enzymes converting dopamine into noradrenaline and then noradrenaline
23

into adrenaline. These enzymes are dopamine β-hydroxylase (DBH) and phenylethanolamine-N-methyl transferase respectively. TH and AADC are often used as markers for catecholaminergic neurons/cells whereas DBH can be used to distinguish between dopaminergic and noradrenergic neurons.
In the adult, TH expression can be detected in all catecholaminergic neurons. During the development, TH shows a scattered pattern of expression in cells in the developing gut (Cochard et al., 1978; Teitelman et al., 1979; Jonakait et al., 1989) and a transient expression in cranial sensory ganglia, dorsal root ganglia, pancreas and kidney (Teitelman et al., 1981; Jonakait et al., 1984). Developing dopaminergic, noradrenergic and sympathetic systems do also express TH. CNS catecholaminergic neurons arise from precursors in the floor plate (Wang et al., 1995a; Wang et al., 1995b; Stull et al., 2001) and peripheral sympathetic neurons are derived from the neural crest (Shah et al., 1996). BMP-signalling influences both central and peripheral catecholaminergic neuronal development via Mash-, Phox-, Hand- and Gata- transcription factors (Shah et al., 1996; Ernsberger, 2000; Pattyn et al., 2000). In 1995, two groups independently presented targeted deletions of TH (Kobayashi et al., 1995; Zhou and Palmiter, 1995; Zhou et al., 1995) and showed that homozygous inactivation of TH is embryo lethal because of cardiovascular defects, while heterozygous mice survive to adulthood and apparently have a normal phenotype. The homozygous embryos can be rescued by a human TH-transgene or treatment of pregnant females with L-dopa. However, the pups in the latter case died before weaning, indicating that catecholamines are essential for both embryogenesis and postnatal survival. The number of catecholaminergic neurons was not altered by the lack of TH, and is therefore probably not dependent on the presence of catecholamines for their development and survival. Even when the level of TH was lowered to ~50% in the heterozygotes as compared to wild-type littermates, were the levels of catecholamines only moderately decreased. This indicates the presence of regulatory mechanisms to compensate for the lack of TH.
Dopaminergic neurons in the adult brain are mainly found in three midbrain nuclei; substantia nigra (SN), the ventral tegmental area (VTA) and the retrorubral field (Dahlström and Fuxe, 1964), as well as in several hypothalamic nuclei, the retina and the olfactory bulb (Dark grey areas in FIG. 9; Moore and Bloom, 1978). Three major tracts arise in the midbrain dopaminergic nuclei and project to various brain areas including the basal ganglia (striatum and globus pallidus), cortex and the limbic system. The nigrostriatal tract does, as the name implies, arise in SN and project to the
24

FIG. 9: Catecholamines in the nervous system of the mouse Dopaminergic neurons are found in the midbrain areas substantia nigra and ventral tegmental area (VTA), in some hypothalamic nuclei, the olfactory bulb and in the retina. The main site for noradrenergic neurons in the brain is locus coeruleus, and in the periphery, they are found in sympathetic ganglia. Dashed and drawn lines represent noradrenergic and dopaminergic projections, respectively.
striatum. It is involved in the translation of motor plans into actual motor commands and is therefore important for control of movement. Parkinson’s disease, a condition associated with muscle tremor and difficulty in initiating and sustaining locomotion, is caused by degeneration of the nigrostriatal path (Lang and Lozano, 1998a; Lang and Lozano, 1998b). Neurons in the VTA and retrorubral field provide a major ascending input to frontal and temporal cortices and to the limbic system in the basal forebrain via the mesocortical and mesolimbic tracts, respectively (Ungerstedt, 1971). The mesolimbic tract modulates information from various brain regions in the nucleus accumbens and can in this way influence behaviours like affect, emotion, memory storage, drives and motivation. Some symptoms of schizophrenia result from over-activity in the mesolimbic system (Carlsson et al., 2001), and in addition, neurons of the mesolimbic system are involved in abuse of drugs such as cocaine, amphetamine and nicotine, which are increasing the levels of dopamine released from VTA neurons in the nucleus accumbens. The fourth dopaminergic tract arises in hypothalamus and
25

projects to the pituitary gland, where it controls hormone release and participates in autonomic and endocrine regulation.
The main site for noradrenergic function in the brain is locus coeruleus (LC) that send projections diffusely to every major region of the brain throughout cortex, cerebellum and the spinal cord. LC maintains vigilance and responsiveness to novel stimuli and hence influences both arousal at the level of the forebrain and sensory perception and motor tone in the brain stem and spinal cord. There are also some other noradrenergic nuclei in pons and medulla oblongata, controlling cardiovascular and endocrine functions, as well as autonomic reflexes and pain sensation. The central noradrenergic nerve-populations are involved in control of feelings such as fear, motivation and pleasure. Disturbances here and in the serotonergic system cause anxiety and other mood disorders (Kandel et al., 2000).
In the periphery, neural crest derivatives give rise to sympathetic ganglia, housing postganglionic sympathetic neurons and the chromaffin cells in the adrenal medulla, releasing adrenaline and noradrenaline into the blood (Cochard et al., 1978; Teitelman et al., 1979; Jonakait et al., 1989). The sympathetic division of the autonomic nervous system controls many physiological functions that are fundamental for survival and mediates the fight or flight response to stressful situation.
26

Present investigation
Aims The aims of this thesis are to investigate functions of BMPs in neuronal cells in general and more specifically to: • evaluate the role of GDF10 in the brain by conventional gene targeting
(paper I) • study impaired BMP-signalling in catecholaminergic neurons by cell-type
specific gene targeting (paper II) • untangle the pathways of synergistic induction of neurite outgrowth by
BMPs and neurotrophic factors in primary embryonic neurons and PC12 cells (papers III and IV)
27

Results
Paper I “Absence of evidence is not evidence of absence”
Paper I describes the successful generation of a GDF10 knockout strain. Normally GDF10 is expressed in the granule cells of the dentate gyrus. However, we fail to prove that this targeted deletion has any impact on the phenotype. The mice appear normal regarding body weight, reproduction, age etc. Focusing the studies on the hippocampus, showed that lack of GDF10 does not affect long term potentiation of synaptic strength, nor spatial learning ability but that a few genes are differently regulated in the knockout, as studied by microarray analysis. Among these genes, the upregulation of CC10 and POD1 was most prominent. Real-time PCR of these genes did however not confirm the upregulation and we conclude that the impact on gene regulation by the GDF10 deficiency in the adult hippocampus is negligible.
Paper II In Paper II, dominant negative ALK2- and BMPRII-constructs (A2 and B2 respectively) were introduced in catecholaminergic cells in the mouse by gene targeting in the 3’ untranslated region of the TH locus, in order to impair BMP-signalling to these cells. The A2 and B2 constructs were produced by PCR using 3-primer strategies preserving the extracellular domain, truncating the receptors just inside the transmembrane region and replacing the intracellular kinase domain with a tag for future detection of the mutated receptors. The PCR-fragments were sequenced to confirm that no unexpected mutations had been introduced during the PCR, and then cloned into pcDNA3, a CMV-promoter driven expression vector for over-expression in cell culture. The dominant negative properties of A2 are shown below while B2 has been described earlier (Henrik Bengtsson, 2001, thesis).
To test whether A2 was expressed on the cell surface, a crosslinking experiment was performed. HA-tagged A2, ALK2 and BMPRII were over-expressed in COS-cells. Iodine labelled BMP7 was added after two days. Receptor-ligand complexes were fixated by crosslinking and subjected to immunoprecipitation with a HA-tag specific antibody. The expected size of A2 is ~25kD, so the BMP7/A2 complex corresponds to the size of the indicated band on the gel (FIG. 10A). In addition, a Smad-responsive luciferase reporter (SBEx4-luc) and the A2-HA-pcDNA3 plasmid were
28

transfected into HepG2 cells by calcium phosphate precipitation. The cells were then stimulated over night with BMP7, Activin A or TGFβ and harvested the next day for quantification of luciferase activity. As shown in FIG. 10B, the A2 construct efficiently inhibited Smad-activity in all three cases, as compared to mock-transfected cells. This result show that the truncated ALK2 receptors indeed have dominant negative properties.
FIG. 10: (A) Crosslinking of 125I labelled BMP7 with overexpressed BMP receptors, including the dominant negative A2-construct. (B) Luciferase assay in HepG2 cells transfected with A2 and the Smad-signalling responsive SBE4x-luc reporter and stimulated with BMP7, ActivinA or TGFβ.
A2 and B2 were cloned downstream of IRES and targeted to the 3’ untranslated region of the TH gene to generate bicistronic mRNAs, with the IRES mediating the production of both A2 (or B2) and TH proteins. Also included in the targeting vector, was a neor cassette for selection of ES-cell clones in which homologous recombination had occurred. The neor cassette was flanked with frt-sites to enable its removal in vivo. The promoter driving the transcription of neor runs in opposite orientation to that of the TH-gene, leading to the production of two complementary mRNAs. If they hybridise with each other, cellular mechanisms will recognise the fault and degrade the duplexed RNA, ultimately preventing protein production.
29

We show in paper II that the production of TH is severely hampered in the B2+ animals, leading to reduced catecholamine production. The reduction of TH-production was seen both on mRNA and protein levels. Using AADC (L-aromatic amino acid decarboxylase) as a second marker for dopaminergic neurons of the substantia nigra and DAT (membrane-bound dopamine transporter) for dopaminergic nerve endings in the striatum, the presence of these neurons is shown and we conclude that the impaired TH-production is due to hypomorphic behaviour of the TH-locus, rather than neuronal death.
FIG. 11: TH-immunofluorescence in the substantia nigra and the striatum of A2- and wt mice.
Removing the neor cassette from the altered TH-loci as in the A2- and B2- strains, restored the levels of TH in the substantia nigra of A2- and B2- as well as in the striatum of A2- but not in the striatum of B2- mice (A2- see FIG. 11; B2- see Fig. 4 in paper II). This discrepancy between the different strains with respect to TH-production is reflected in the results from extensive measurements of catecholamine levels in the corresponding brain areas of all strains. We have created a spectrum of strains with various degrees of dopamine deficiency in substantia nigra and striatum (FIG. 12). Double copies of the B2+-allele leads to severe TH-hypomorphism and concomitant dopamine deficiency, most prominent in the striatum where the
30

dopamine level drops to approximately 3% of that of the wild-type control. In the substantia nigra of these mice, the level is around 20% and for homozygous A2+-mice, the corresponding levels are 15% and 50% in substantia nigra and striatum, respectively. In heterozygous A2+ and B2+ mice, the presence of one wild-type TH-allele compensates for the effect of the hypomorphic one giving a less penetrating effect. However, in substantia nigra of B2+-heterozygouts and in striatum of B2+-heterozygouts, a smaller but significant decrease of dopamine was scored. This could possibly be ascribed to impaired BMP-signalling in the presence of the dominant negative ALK2 and BMPRII receptors.
FIG. 12: Dopamine levels in the substantia nigra and the striatum of B2 mice (A and B) and of A2 mice (C and D).
31

Looking at the A2-- and B2-- strains, (in which the neor cassette was removed and hence also one possible explanation of TH-hypomorphism) it is clear that the effect of the B2- locus is more prominent than that of the A2- locus. In all of the studied B2- mice, a decrease in dopamine levels was seen, both in the substantia nigra and in the striatum with the most severe effect seen in homozygotes. The only significant decrease in dopamine levels in A2- mice was in the substantia nigra of the homozygotes. However it was only a ~20% decrease with a p-value of <0.05 to be compared to ~40% (p<0.001) decrease in the substantia nigra of homozygous B2- mice. To conclude, we see that the effect of B2 is stronger than that of A2; both in neor -positive and -negative mice and that homozygotes are more severely affected than heterozygotes.
FIG. 13: Spontaneous movement (A) and body weight (B) of B2+ compared to A2+ mice.
The tag sequences were shown to work when the constructs were overexpressed in cell cultures; however, we failed to detect A2-HA and B2-cMyc in vivo by immunofluorescence. On the other hand, the bicistronic mRNAs are shown by in situ hybridisation, and taken together, these facts give us a reason to believe that the constructs actually are expressed as proteins on the cell surface of catecholaminergic neuron. The A2- and B2-
32

probe signals in the substantia nigra of neor negative heterozygotes is quite strong while virtually absent in neor positive heterozygotes indicating that the presence of the neor cassette in the bicistronic mRNA leads to partial degradation thereof. We have not looked at the mRNA level in homozygous. However, I suspect that the level of the bicistronic mRNA in neor positive homozygotes is low and that the resulting TH-deficiency is the cause of impaired catecholamine biosynthesis rather than the expression of A2 or B2 and their dominant negative effects on BMP-signalling. In the case of neor negative homozygous mice, it seems likely that the bicistronic mRNA-expression more closely follows that of the wt TH-allele, so that rather high levels of dominant negative receptors are reached on TH-positive neurons. In this case, the effects seen might be a result of impaired BMP-signalling affecting aspects of the well being of catecholaminergic neurons. A critical experiment will be to cross our mouse-strains with a BMP-reporter mouse to get proof of what happens with BMP-signalling in TH-neurons. The levels of catecholamine deficiency have to pass some threshold level to give significant impact on explorative behaviour and body weight (FIG. 13). Consequently, the greatest effects are seen in homozygous B2+ mice.
Paper III BMP7 has previously been shown to potentiate the action of NT3 and GDNF on neurite outgrowth in embryonic chick sympathetic ganglia (Bengtsson et al., 1998). Here we extend the range of neurotrophic factors tested with BMPs to also include the GDNF-related neurturin (NTN). Moreover, we tested BMP6, belonging to the BMP7-subfamily of BMPs, and the more distantly related BMP4. We did not find any variation in how the different BMPs affected the ganglia and conclude that the potentiation of neurotrophic factor induced neurite outgrowth is mediated through a general BMP-induced mechanism. We could also show the potentiation effects in ciliary and nodose ganglia in addition to the previously shown sympathetic ganglia, indicating a general mechanism, also regarding neuronal subtype. As an attempt to find critical intracellular signalling pathways involved in the studied synergistic effects, we tried a number of commercially available kinase inhibitors. PI3K and JNK inhibitors partially and completely blocked neurite outgrowth induced by NT3 and potentiated by BMP4. P38, PKC, mTOR and GSK3 inhibitors did not affect ganglia treated with the same factors. Surprisingly, a MEK inhibitor seemed to act as an additional potentiator, which led us to test it together with NT3 alone. Inhibition of MEK strongly potentiates NT3 induced neurite outgrowth in the different ganglia tested and can also act to potentiate GDNF and NTN. We also show that 4h priming with either BMP-stimulation or MEK-inhibition followed by
33

washing before addition of NT3 alone to sympathetic ganglia was enough to potentiate neurite outgrowth. However, if the ganglia were first primed with NT3 for 4h and then treated with either BMP4 or a MEK-inhibitor, no neurites grew from the explants. This indicates that neurotrophic signalling must be maintained during the culture period whereas the potentiating effects by BMP-activation or MEK-inhibition requires only a priming period to become evident.
Biologically, BMP-stimulation and MEK-inhibition show strikingly similar effects. We wanted to know if they also work by similar molecular mechanisms and therefore went on to measure phosphorylation of Erk, acting downstream of MEK and phosphorylation at the C-terminal of Smad1/5 in the BMP induced pathway. Our first hypothesis that activation of intracellular signals by BMPs would interfere with the signalling activated by NTs at the level of Erk-phosphorylation was disproved. Nor did inhibition of MEK interfere with BMP4 induced Smad-activation. It is however known from the literature that Smads have sites in their linker region, which if Erk phosphorylates them, prevent nuclear translocation of Smad and hence counteract BMP stimulation. In order to test if MEK-inhibition leads to decreased phosphorylation of Smad in the linker region, releasing the restrain on Smad nuclear translocation and concomitant gene regulation, real time PCR was run on IdI and Smad6 known as BMP-target genes. BMP4 gives a 5-fold induction of Id1, but MEK-inhibition fails to induce a significant increase in Id1-expression. Smad6 expression is increased by a factor of 1.5 after 4h BMP-stimulation. MEK-inhibition does not significantly alter Smad6 expression. Hence, it seems that MEK-inhibition does not lead to a generally increased Smad-activated gene transcription. We have also monitored the levels of TrkA and TrkC mRNAs after stimulation with BMP or MEK-inhibition in order to check if the improved effect of NT3 was caused through upregulation of its receptor. This mechanism was ruled out since the levels of TrkA and TrkC were unaffected by the BMP or MEK-inhibitor treatment.
Paper IV PC12 cells grown on collagen send out neurites when stimulated with NGF. Here we describe a potentiating effect on neurite outgrowth from NGF-stimulated PC12 cells when BMP4 or BMP6 is added to the cultures together with NGF in analogy with the findings in paper III. Erk-phosphorylation was not affected by the addition of BMP6 as measured in a western blot using phospho-Erk antibodies. Luciferase reporters responding to BMP signalling or activation of Gal4-fusion proteins were transfected into NGF stimulated cells, which were then further stimulated in different ways.
34

To confirm that NGF treated PC12 cells do respond to BMP stimulation by activation of the BMP-Smad pathway, an SBE4x-luciferase reporter was used. This reporter was also co-transfected with caALK2, Smad1/wt, Smad7/wt, Smad5/wt and Smad5 with five mutated Erk-phosphorylation sites in the linker region. As expected, caALK2 efficiently induced a reporter response, which was enhanced by co-expression with Smad1 and reduced by Smad7. The Smad5 mutant, designated Smad5/5SA, with the defective Erk1/2-phosphorylation sites in the linker region was shown to give a stronger response than Smad5/wt, indicating a possible mechanism for Erk1/2 to down regulate Smad5-signalling. Fusion constructs of the Gal4 DNA binding domain and the Smad5 variants were also tested in luciferase assays, using a Gal4-trans-reporter system. Smad5/5SA was shown to give a stronger response than the wt and the dominant negative Smad5/2SA, presumably caused by a release in Erk mediated Smad inhibition.
35

Discussion
Knockout without phenotypical alterations Aspects of targeted gene disruption, commonly regarded as contributing to the phenotypical alterations, are for instance genetic background, compensatory mechanisms at different levels and redundancy among members of a gene family. We have backcrossed our GDF10 mice for 10 generations but the penetrance of the GDF10 null allele does not seem to increase in a more homogeneous background. GDF10 is one of many ligands belonging to the TGFβ superfamily, of which at least some are co-expressed with GDF10. One could speculate that redundancy within this gene family could account for functional compensation for the lack of GDF10.
Our attempts to characterise the GDF10 null mice have so far not resulted in any clearcut phenotypical alterations but; “Inability to observe a phenotypical change may not mean that there is no phenotypical change to observe” (Gerlai et al., 2001). One possibility could be that the knockouts are more susceptible to injury, and in an ongoing project we are testing this hypothesis in a traumatic brain injury model.
Dopamine dysfunction in TH-hypomorphic mice DA and NA are products in the synthesis pathway in which TH is the first and rate-limiting enzyme. Measuring the concentration of these neurotransmitters in the TH hypomorphic mice gives insight into the effects of our different genotypes with respect to the catecholaminergic systems. B2+ mice have only 3% of normal levels of dopamine in striatum. Even with this severe dopamine deficiency, the mice survive to adulthood. Given that the dopamine deficiency results from TH hypomorphism, it will be of interest to see if treatment of these mice with L-dopa will restore the dopamine levels, as would be expected.
36

Crosstalk The topic of papers III and IV is crosstalk between BMP and neurotrophin/GFL signalling pathways, leading to potentiated neurite outgrowth and increased survival rate in primary neurons and in PC12 cells, respectively.
Paper III demonstrates synergistic effects in a range of different neuronal populations, including sympathetic, ciliary and nodose ganglia giving credibility to the idea that the observed reaction is widespread among neurons. An extensive study of the effects of pharmacological inhibitors of various cascades involved in the apparent crosstalk showed a general requirement of PI3K and JNK for the synergistic effects. The most intriguing finding was that inhibition of MEK gave a robust potentiation of Trk/Ret induced neurite growth, very similar to that elicited by BMP signalling in neurons stimulated by NT3 and NTN. We did not obtain any suggestions of crosstalk between the NT3 and BMP pathways at the level of phosphorylation of activation sites in Erk and Smad. Nor did stimulation with BMP or MEK inhibition lead to increased expression of TrkA or TrkC receptors. Instead, the synergistic effects are likely to arise in the nucleus by transcriptional co-regulation of target genes, presumably involving Smads, Erk-substrates such as Elk1 or Rsk, and various co-regulators. Id1 and Smad6, known as BMP target genes, were confirmed to respond to BMP signalling but were not induced by MEK-inhibition. On the contrary, the expression of the neurotrophin-responsive immediate early gene Egr1 was reduced after BMP stimulation as well as after MEK inhibition. The relevance of the observed downregulation of Egr1 for the potentiation of neurite growth and survival remains to be proven. To further evaluate the underlying mechanisms of the apparent synergy in the BMP stimulated and MEK-inhibited ganglia, a comparison of transcription profiles by microarray would be of great interest.
In paper IV, the suggested ihibiting influence of Erk activity on BMP
signalling by phophorylation of the linker region of Smad5 was tested. We show that Erk-activity and hence Smad5-linker phosphorylation, helps keeping the background levels of BMP signalling low, while activation of the Smad5 pathway by constituively active ALK2 is strong enough to override the hampering effect of Erk. The system used is however artificial and the physiological relevance of Smad5-linker phosphorylation for enhanced neurite outgrowth is not demonstrated by these experiments.
37

Concluding remarks
Paper I Describes the successful generation of a targeted GDF10 null allele and thereby the elimination of the transcription of this gene. The knock out has an apparently normal neuronal morphology in the hippocampus and the cerebellum where GDF10 expression normally is high during development and persists at a lower level in the adult. We have not been able to pinpoint any clear phenotypical alterations of the hippocampus on the level of gross morphology, neurophysiology or cognitive function. Nor could we show any alterations in the gene transcription profile of the adult hippocampus by cDNA microarray, followed by real-time PCR.
Paper II Describes the establishment of four different tyrosine hydroxylase alleles in the mouse. These mice represent a novel spectrum of phenotypes, showing graded TH hypomorphism and reduction of catecholamines. We have made efforts to gather evidence for inhibited BMP signalling caused by the expression of truncated receptors on catecholaminergic neurons, driven by the TH locus. Despite the dominant negative properties of A2 and B2, confirmed in cell culture experiments, no clear evidence was obtained for neuromodulatory effects of BMPs in the nigro-striatal system using the presented approach. The created mice will nevertheless provide new tools for the research on the catecholaminergic system having relevance for Parkinson’s disease and L-dopa responsive dystonia.
38

Paper III Describes a striking similarity between BMPs and MEK inhibitors in potentiating neurotrophic activities of NT3, GDNF and NTN in embryonic autonomic and sensory neurons, seen as enhanced neurite outgrowth and survival. It is shown that inhibition of MEK does not result in activation of known BMP-induced genes, indicationg that convergence points involve novel gene activations.
Paper IV Describes that BMPs potentiate the neurite formation response of PC12 cells stimulated NGF. BMPs do however not induce neuronal differentiation in PC12 on their own. This is reminiscent of other instances where BMPs have been shown to enhance neurotrophic activities of other classes of growth factors.
We show here that elimination of the linker-region Erk-phosphorylation sites in Smad5 is not functionally equivalent to reducing Erk activity by MEK inhibition. This is in agreement with real-time PCR data in primary neurons (Paper III) where MEK inhibition did not activate the BMP-regulated genes Id1 and Smad6.
It is apperent that further genetic models are needed to firmly establish a physiological role for BMPs to enhance neurotrophic factors in vivo. The synergy between BMPs and neurotrophic factors seems to be regulated at the transcriptional level and to involve target genes that remains to be identified.
39

Materials and methods
Targeted gene disruption/alteration (Paper I and II)
Generation of truncated ALK2 and BMPRII by PCR The following primers were used: 5′-primer for A2 was 5′-TATGAATTCG TTATACCATGGTCGATGGAGTA-3′ (designated SAL1b; nucleotides 487-510 of GenBank Acc. No. L15436) and for B2, 5′-TATGAATTCCAG CCATGGCTTCCTCGCTGCATCG-3′ (designated HB22b; nucleotides 375-394 of GenBank Acc. No. AF003942). The “intermediate” 3’-primers used, were for A2; 5′-ACCATACTCCACGTCTCTGGGGTT-3′ (SAL2, antisense to nucleotides 966-990) and for B2; 5′-ACGCTGTTTCCGGTCTCCTGT CAACATTC-3′ (HB23, antisense to nucleotides 892-917). Tagged 3’-primers (tag sequence underlined) were 5′-CAGTCTAGATCATGCATA ATCTGGAACGTCATATGGATATCCACCATACTCCA-3′ (SAL3) for A2 and 5′-CTGTCTAGATCAGAGGTCCTCTTCGGAAATAAGTTTCTGTT CACGCTGTTT C-3′ (HB24) for B2. The 3’-Tag-primers do also introduce a translational TGA-stop codon just after the tag in the construct. (See paper II for further details.)
Screening for genomic DNA clones and targeting vector construction A SVJ129 mouse genomic λ-phage library (Stratagene, CA, USA) was screened using the cDNA for GDF10 (paper I) and a fragment covering exon 13 (including stop codon and poly-A signal of the TH-gene (paper II) as probes. A 15 kb GDF10 and a 16 kb TH clone were isolated and further characterized by Southern blot and DNA sequencing. Based on the physical maps of the clones, targeting vectors were constructed. For most clonings and subclonings, pMOS (Amersham) and pBluescriptKS/SK (Stratagene) vectors were used. λ-phage library screening and all DNA manipulations were done according to standard protocols and suggestions from
40

manufacturers. Sequencing of DNA was achieved using fluorescent dye-terminator chemistry and automated gel- or capillary-based ABI prism systems (Perkin Elmer).
Culturing of embryonic stem (ES) cells 30-40µg of the targeting constructs were linearised and electroporated (260V; 500µF) into 6x106 ES cells (GSI-1; derived from 129SVJ; Genome Systems Inc., S:t Louis, MO, USA), which were then left to recover in DMEM culture media supplemented with 16% ES-qualified fetal calf serum (FCS; Gibco-BRL), non essential amino acids, 2mM L-glutamine, 1000 units/ml recombinant leukaemia inhibitory factor, 1:100 000 2-mercaptoethanol and 10mM HEPES on neomycin resistant feeder cells (semi-confluent, embryonic fibroblasts from Genome Systems inc.) for 24h, after which G-418 was added for 5-6 days of selection. Single ES-cell clones were picked and dissociated with trypsin (0.5mg/ml and 1mM EDTA) for further culturing on feeder cells in 96-well plates under G-418 pressure. Subcultivation to replica plates was performed when sub-confluency was reached. The original plates (containing 2/3 of the cells) were stored frozen at –80°C with 26% FCS and 10% DMSO in the medium. The embryonic fibroblasts were, prior to use as feeders, mitotically arrested by a 3h-treatment with 10mg/ml mitomycin. Care was taken to preserve undifferentiated morphology of ES-cell colonies throughout the procedure.
DNA-preparation and screening of ES-cell clones ES cell DNA was prepared from the replica plates through lysis of the cells over night in a humified chamber at 60°C in 50 µl lysis buffer containing 10mM Tris pH 7.7, 10mM EDTA, 10mM NaCl, 0.5% Sarcosyl, and 1mg/ml freshly added proteinase K. Precipitation of genomic DNA with 100 µl 96 % ethanol containing 75 mM NaCl and the following washing (70 % ethanol) step was performed in the 96-well plates with the DNA stuck to the bottom. Residual ethanol was allowed to evaporate and BglII restriction enzyme reactions in a total volume of 35 µl (1x appropriate buffer, 1 mM spermidin, 0.1 µg/µl BSA and 0.1 µg/µl RNase; 37°C overnight) were prepared for Southern blot analysis. The digested DNA was run on a 0.7 % agarose gel in 1x TAE buffer. The gel was then soaked in 0.2 M HCl for 20 min and in 0.4 M NaOH for 20 min before blotting over night onto Hybond N+ membranes (Amersham) using 0.4M NaOH as transfer buffer. Neutralization of the membrane in 2x SSC, 0.2 M Tris pH 7.4 preceded hybridisation, in 10 % PEG 6000, 7 % SDS, 1.5x SSPE, 10 µg/ml salmon sperm DNA at 65°C over night with the external probes, described above. The probes were labelled
41

with 32P using the RediPrime kit (Amersham). After hybridisation, the membranes were washed at high stringency in 0.1x SSC, 0.1% SDS at 65°C for 2x 30 min and exposed to PhosphoImager screens (ABI).
Blastocyst injection and germ line transmission ES-cell clones (129X1), shown to be positive for the targeted gene alterations in the screening procedure, were thawed, DMSO containing media removed and the cells were expanded on feeder cells for microinjection into B6 blastocysts under a Zeiss Diaphot 135 DIC inverted microscope in a chamber containing ES-medium with 5% FCS. 10-15 ES-cells were injected per blastocyst and 8-12 re-expanded blastocysts were transferred to one of the uterine horns of pseudopregnant foster mothers (CBA), which, 4 days earlier, had been mated with vasectomised males. Successful transfer of injected blastocysts resulted in the birth of chimeric mice, easily recognized by their speckled (brown-black-white) coat colour. The chimera were mated to B6 dams and transmission of the mutation through the germline to coming generations, which depends on whether the manipulated ES-cells contribute to gonad formation in the chimera, resulted in brown pups.
Housing and Breeding All mice were housed at constant room temperature (20°C) with free access to food and water. They were kept on a 12:12h light/dark cycle with all experiments, except the Morris water-maze tasks (see below) run during the light period. All experiments were run with the approval from the local ethical committee.
The GDF10 knockout strain has been backcrossed to B6 for 10 generations and can therefore be considered to be congenic. The frt flanked neor cassette was removed from the TH-IRES-A2+/B2+ loci to generate the neor-negative strains (TH-IRES-A2- and TH-IRES-B2-) through breeding of the neor-positive strains (TH-IRES-A2+ and TH-IRES-B2+) with Flp-deletor mice. A2+ and B2+ males were crossed with Flp-deletor dams to generate offspring carrying both the A2+, or the B2+, and the Flp-deletor allele. Mice with these genotypes were then backcrossed to B6. The second round of crossings resulted (with low efficiency; 4 out of 65 for A2) in A2- and B2- mice, which were used as founders of the neor-negative strains and further backcrossed to B6.
42
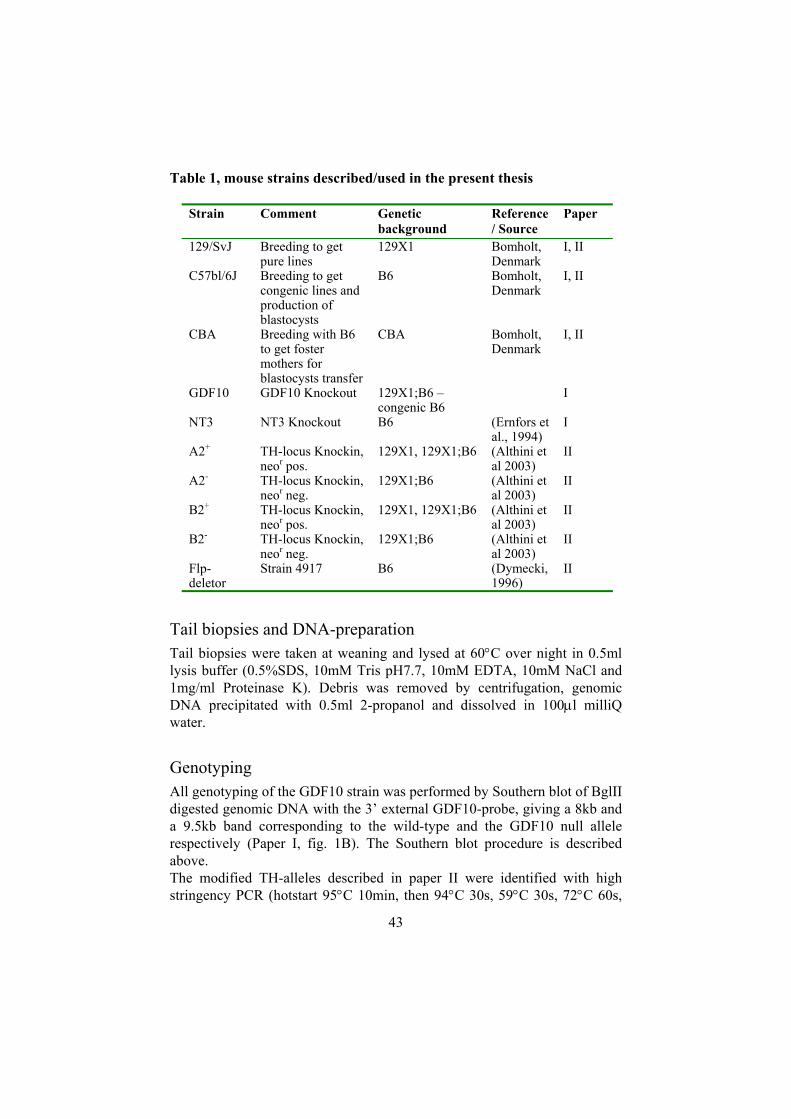
Table 1, mouse strains described/used in the present thesis
Strain Comment
Genetic background
Reference / Source
Paper
129/SvJ Breeding to get pure lines
129X1 Bomholt, Denmark
I, II
C57bl/6J Breeding to get congenic lines and production of blastocysts
B6 Bomholt, Denmark
I, II
CBA Breeding with B6 to get foster mothers for blastocysts transfer
CBA Bomholt, Denmark
I, II
GDF10 GDF10 Knockout 129X1;B6 – congenic B6
I
NT3 NT3 Knockout B6 (Ernfors et al., 1994)
I
A2+ TH-locus Knockin, neor pos.
129X1, 129X1;B6 (Althini et al 2003)
II
A2- TH-locus Knockin, neor neg.
129X1;B6 (Althini et al 2003)
II
B2+ TH-locus Knockin, neor pos.
129X1, 129X1;B6 (Althini et al 2003)
II
B2- TH-locus Knockin, neor neg.
129X1;B6 (Althini et al 2003)
II
Flp-deletor
Strain 4917 B6 (Dymecki, 1996)
II
Tail biopsies and DNA-preparation Tail biopsies were taken at weaning and lysed at 60°C over night in 0.5ml lysis buffer (0.5%SDS, 10mM Tris pH7.7, 10mM EDTA, 10mM NaCl and 1mg/ml Proteinase K). Debris was removed by centrifugation, genomic DNA precipitated with 0.5ml 2-propanol and dissolved in 100µl milliQ water.
Genotyping All genotyping of the GDF10 strain was performed by Southern blot of BglII digested genomic DNA with the 3’ external GDF10-probe, giving a 8kb and a 9.5kb band corresponding to the wild-type and the GDF10 null allele respectively (Paper I, fig. 1B). The Southern blot procedure is described above. The modified TH-alleles described in paper II were identified with high stringency PCR (hotstart 95°C 10min, then 94°C 30s, 59°C 30s, 72°C 60s,
43

36 cycles, finally 72°C 7 min) using the SAL12-SAL3 and HB22b-HB24 primers for the A2- and B2-strains respectively. In both cases, 600bp fragments were amplified. The wild-type TH allele was identified, in the same reaction, with the 5TH-3TH primer pair giving a 280bp fragment. These primers did not amplify the IRES-A2/B2 cassette introduced between them in the mutated alleles. In case of heterozygosity, both the 600bp and the 280bp bands were amplified (Paper II, fig. 1C). A Southern blot strategy was used to confirm the deletion of the neor cassette in the A2- strain. The 5’ probe hybridised to BglII-digested DNA gives 5kb, 7kb and 14kb bands, corresponding to wild-type, A2+ and A2- respectively (see fig. 1A and 1C in paper II).
Behavioural testing (Paper I and II)
A protocol for primary phenotypic screening General status of the mice was scored according to the SHIRPA primary observation screen (Rogers et al., 1997), during which a total of 40 separate measurements are recorded for each animal. Undisturbed behaviour was first observed in a cylindrical clear viewing jar. The mouse was then transferred to an arena (32x56 cm) that was marked into even sized squares (8×8 cm) for observation of motor behaviour startle response and the tendency to escape upon touch. This was followed by a sequence of tail suspension manipulations where measurements of positional passivity, visual acuity, grip strength, body tone, various reflexes (eye closure, ear twitch and toe pinch) and negative geotaxis were recorded. To complete the assessment, the animals were restrained in a supine position to record the skin colour, the body temperature and the heart rate. Estimation of limb tone, salivation and provoked biting responses were also recorded. Throughout this procedure, any incidences of abnormal behaviour, fear, irritability, aggression or vocalization were recorded. All parameters were individually scored to provide a quantitative measure of behaviours.
The protocol was used to get a general picture of how a normal mouse behaves. It demands an experienced person and can be criticized in many ways. I do not show any data from my screens but I do think it is worth mentioning that this has been done since it has given me a lot of experience in judging mouse behaviour.
44

Spontaneous movement (Paper I and II) Spontaneous movement was monitored in activity field arenas (20x20 cm, beam spacing 1.5 cm) using TruScan software (Coulbourn Instruments, PA, USA) during 15 min sessions. The average distance moved by different groups of mice was compared and tested for significance using ANOVA or a student t-test.
Spatial learning ability (Paper I) Mice, provided with secret codes, were housed separately with free access to food and water in the water maze room with reversed light/dark 12 h cycle (experiments were run during the dark period in dim lighting). The water maze tests were carried out in a circular open field pool (1.5m in diameter, 24-26°C; Morris, 1984) with prominent extra-maze visual cues. A platform (10cm in diameter) was hidden ~0.5 cm beneath the water surface. Trials lasted for up to 120 s and each mouse went through 18 trials (6 per day) of place navigation, during which the platform was hidden at a (for each mouse) constant position. After 3 days of learning (the acquisition phase), the platform was moved to the opposite quadrant and reversal training was monitored for 2 more days (6 trials/day, the reversal phase). To test visual capacity of each mouse, a cued test with 6 trials was performed (Minichiello et al., 1999). In this case, a clearly visible object was placed on the platform, and moved together with it, to a new position for each trial. Video tracking of water maze experiments was done using the 2020 tracker and software from HVS Image ltd. UK. The accumulated data was then processed in Wintrack, public domain windows software for off-line path analysis (http://www.dpwolfer.ch/Wintrack/).
Electrophysiological recordings (Paper I)
LTP in the CA1-region of the hippocampus and the medial lateral perforant paths of the dentate gyrus The brains from wild type and mutant mice, sacrificed with halothane, were dissected out and cooled to 0-4oC in artificial cerebrospinal fluid (ACSF). Transverse slices from the middle portion of each hippocampus were cut with a vibroslicer. The slices were then placed in an interface chamber exposed to humidified gas at 28-32oC and perfused with ACSF. Orthodromic synaptic stimulation (0.2 Hz) was delivered alternately through two
45

monopolar tungsten electrodes placed in the outer and middle molecular layer in the dentate or in the stratum radiatum and stratum oriens in the CA1. Extracellular synaptic responses were monitored by two glass electrodes situated in the corresponding layers. After stable synaptic responses had been obtained in both pathways for at least 15 min, one pathway was tetanised (100 Hz, 1 sec), the other pathway served as an untetanised control pathway. The synaptic efficacy was assessed by measuring the slope of the field epsp in the middle third of its rising phase.
Biological assays (Paper III)
Neurite outgrowth assays E9 chicken (White Leghorn) sympathetic (paravertebral trunk), dorsal root (sensory) nodose (from the vagus nerve, rostral to the heart) and ciliary (from the orbit; parasympathetic) ganglia were dissected and embedded in 100µl transparent collagen gel (Ebendal, 1989). The collagen was prepared from rat-tail tendons and dissolved in 0.5M acetic acid (Ebendal and Jacobson, 1977; Elsdale and Bard, 1972). The cultures were incubated under standard tissue-culture (Eagle’s Basal Medium, 0.5% fetal calf serum) conditions for two days with the addition of neurotrophic factors, BMPs and inhibitors in different combinations in 100 µl additional media added on top of the collagen gel (Bengtsson et al., 1998). Ganglia were examined under dark-field illumination and two experienced observers, using a scale from 0-5, scored neurite outgrowth in blinded cultures, where 0 is absence of neurites and 5 resembles a dense fibre halo. Each determination was repeated at least twice with 4-20 ganglia scored in each group. The mean +/- SEM was calculated and significance was tested with a Student T-test.
FIG. 12: E9 chick embryo with the locationof ganglia used in Paper III indicated
46

Survival assay Sympathetic (paravertebral) and ciliary ganglia were dissected from E9 chick embryos, dissociated with 0.25% trypsin, pre-plated for 90 min to remove many non-neuronal cells and cultured in a thin collagen gel in BME 1%FCS plus combinations of growth factors and inhibitors (concentration range) (Ebendal et al., 1980). Surviving neurons, identified, by their size, rounded shape, phase-bright appearance and frequent possession of neuritis, were counted in a strip across the gel after two days under a phase-contrast microscope. Results are expressed as percent survival as compared to control cultures stimulated with factors known to be almost 100% protective for the different neurons (NGF 5ng/ml for sympathetic and extract from the choroid coat from the day 18 chicken embryo for ciliary ganglia). (Survival is shown relative to the number of cells initially seeded). Several hundred neurons were counted and the assays repeated at least twice. (Ebendal, 1989; Ebendal et al., 1982)
Pharmacological inhibitors of MAPK pathways (Paper III) Mitogen activated protein kinase pathways (MAPK) are major information highways from the cell surface to the nucleus. In order to map the synergistic effects seen between neurotrophic factors signalling via Trk and Ret tyrosine kinase receptors and BMPs signalling via type I and type II serine/threonine kinases, a number of pharmacological inhibitors were tested.
Table 2, Pharmacological inhibitors
Inhibitors of: Final concentration
Inhibitors of: Final concentration
MEK PI3Kinase PD98059 50µM LY294002 50µM U0126 1µM PD184352 2µM mTOR/FRAP Rapamycin 0.2 or 1µM p38 SB203580 10µM PKC SB202190 50µM Gö6850 10(50)µM PD169316 10µM GSK-3 SB216763 10 (5-50)µM Control substances
RNA synthesis
U0124 5µM Actinomycin D 1µg/ml SB202474 50µM
47

Biochemical and molecular studies
Determination of catecholamine and 5HT levels (Paper II) Endogenous levels of noradrenaline, dopamine and 5-HT were determined in substantia nigra, striatum, hippocampus and the submandibular gland by high-pressure liquid chromatography (HPLC) with electrochemical detection according to (Luthman et al., 1993). Shortly: Brains and submandibular glands were rapidly taken out from mice, sacrificed by decapitation, and chilled in saline (on ice). The cooled brains were further dissected in order to isolate the striatum, hippocampus and substantia nigra. The samples were immediately frozen on dry ice and stored at –70°C until homogenisation by sonication. After centrifugation of the homogenates, the supernatant was separated by HPLC on a reverse phase column (SupelcosilTM, LC-18, 75mm x 4.6mm, 3µm particle diameter). A buffer containing 0.05M sodium phosphate, 0.03M citric acid, 0.1mM EDTA and various amounts methanol and sulphonic acid was used as mobile phase, carrying the samples at a flow rate of 0.4ml/min through the column. Noradrenaline, dopamine and 5-HT were electrochemically detected using a glassy-carbon electrode detector and the resulting peak-heights were scored and compared to standard curves for the respective substance.
Immunofluorescence (Paper II) Animals were anaesthetised with Dormicum/Hypnorm and fixated by perfusion. A butterfly needle coupled to a peristaltic pump was introduced into the apex of the heart and an incision was made in the left heart ventricle. The blood vessels were flushed with 10ml Tyrode before perfusion with 50ml 4% paraformaldehyd. The brain was dissected out and immersed in 4% paraformaldehyd for 1h postfixation and then moved to a large volume tyrode containing 10% sucrose for storage at 4°C. Sucrose saturated brains were rapidly frozen on dry ice and mounted for Cryo sectioning. 12mm sections were collected on cold chromalun (Chrome Potassium Sulphate)/gelatine coated microscope slides. The sections were rinsed 3x10 min in PBS prior to incubation with primary antibodies (diluted in PBS containing 0.3% Triton-x) over night at 4°C in a humified chamber. Fluorescein isothiocyanate (FITC)-labelled secondary antibodies were applied for 1h at room temperature. The slides were washed 3x10 min in PBS before and after incubation with the secondary antibodies. Finally, the slides were mounted in 90% glycerine containing the anti-fade substance 1,4-phenylenediamine for evaluation by fluorescence microscopy (Zeiss,
48

Axioplan 2). The following antibodies were used; anti-rat TH (1:500; Pel-Freeze, Rogers, AR, USA), anti-bovine AADC (1:200; Calbiochem, La Jolla, CA, USA), anti-human DAT (1:40 000; Chemicon International, Temecula, CA, USA). FITC-labelled secondary anti-bovine, -rat, or -human antibodies were used to detect primary antibodies.
In situ hybridisation (paper I and II) Single stranded oligo nucleotide probes (~50 nucleotides; table 2) were designed, based on sequence information available in the literature, to cover unique parts of the coding regions of the genes of interest. The probes for A2 and B2 (described in paper II) cover partly the truncated ALK2 and BMPRII receptors and partly the tag-sequence replacing the intracellular domain of these receptors in the constructs. All probes were synthesized by Scandinavian Gene Synthesis, Sweden.
Table 3, Oligo nucleotide probes
Probe Probe sequence Reference GDF10 (mouse)
5’-TCT GGA TGG TGG CAT GGT TGG ATG GGC GGA CAA TCT TGG GCA TGG GGA-3’
(Cunningham et al., 1995) Paper I
TrkC (rat)
5’-GAA GTG GCC GTT G ATG GTC TGG TTG GCT GTG CCC AGG GCA TTC TTA GCA AT-3’
(Merlio et al., 1993) Paper I
TH (rat)
5’-TGC GTG GGC CAG GGT GTG CAG CTC ATC CTG GAC CCC CTC CAA GGA GCG-3’
(Grima et al., 1985) Paper II
AADC (rat)
5’-AGC ATC AAT GTG CAG CCA TAC ACC CTC CTG GTT GCA GAT GGG ACC CAC-3’
(Tanaka et al., 1989)
A2 (mouse)
Truncated ALK2 and HA-tag
5’-ATA TCC ACC ATA CTC CAC GTC TCT GGG GTT CAG GCG CTC TTG ATT G-3’
Paper II
B2 (mouse)
Truncated BMPRII and cMyc-tag
5’-AGT TTC TGT TCA CGC TGT TTC CGG TCT CCT GTC AAC ATT CTG TAT CCA-3’
Paper II
49

Adult brains were rapidly dissected, frozen on dry ice, and serially sectioned on a Leitz Digital 1702 cryostat. 12 µm coronal sections from the hippocampal formation and the cerebellum were mounted onto SuperFrost+ slides and stored at -80°C until use. Oligonucleotide probes were 3′-end labelled with deoxyadenosine 5′-[α-35S]thiotriphosphate (α-35S-dATP; NEN) to a specific activity of approximately 1 106 cpm/ng, using terminal deoxynucleotidyl transferase (TdT; Intl. Biotechnologies, Inc.) and purified using G50 micro columns (Amersham Pharmacia). Hybridisation was performed at 42°C for 16 h in a formamide-humified chamber [Bengtsson, 1995 #2]. The hybridisation cocktail contained 50 % formamide, 4x SSC, 1x Denhardt’s, 1 % sarcosyl, 20mM phosphate buffer pH 7.0, 0.1 g/ml dextrane sulphate, 0.27 µg/µl yeast tRNA, 0.5 µg/µl salmon sperm DNA and 1x107 cpm/ml labelled probe. Sections were rinsed and washed at 55°C 4 times in 1 SSC (once in 1x SSC + 0.05 % sarcosyl, and then 3 times in 1x SSC). Thereafter, the slides were rinsed quickly in ice-cold RNase-free water, dehydrated in ethanol (70 % 5 min and 99 % 5 min) and left to air-dry. The sections were mounted for exposure to X-ray films (Kodak BioMax MR) for 1 week at room temperature.
RNA preparation and microarray analysis (Paper I) Hippocampi were dissected and put in RNAlater solution (Qiagen Inc.), which efficiently inhibits RNA degradation. Homogenisation of the tissue using a polytron was followed by preparation of total RNA with the Qiagen RNeasy kit. The final concentration and A260/A280 ratio was determined and the RNA samples were stored at –80°C. 10-20 µg of total RNA was labelled with Cy3-green and Cy5-red fluorescent dyes (Amersham Biosciences, Uppsala, Sweden) during reverse transcription into cDNA using oligo-dT primers. The labelled probes were hybridised with the micro-array spots (spotted slides were purchased from Toronto University, Canada) under a microscope slide cover slip at 65°C for 18 hours in a high-humidity chamber. Dye swap hybridisations were performed for parallel sample-pairs. After hybridisation, the arrays were washed repeatedly at increasing stringency, dried and thereafter immediately scanned using an Affymetrix ® 418 Scanner. Two consecutive scans were done, first with a green laser (532 nm) and subsequently with a red laser (635 nm). For quantification of array results, the scanned (16-bit TIFF) files were analysed using a commercial software package called Array Vision ®. Template grids were produced for all spots. Integrated background-subtracted spot intensities were transferred to a spreadsheet file (Excel) for statistical evaluation.
50

Western blot (Papers III and IV) Chick ganglia or PC12-cell cultures were lysed in 20mM Tris pH 7.4, 150mM NaCl, 10% glycerol, 1% Triton X-100, 1mM PMSF and 10µg/ml aprotenin. Protein concentration was determined with the Bio-Rad Protein Assay kit, (Cat #500-0002). Loading buffer (100mM Tris pH 6.8, 5% glycerol, 2% SDS, 0.4M β-mercaptoethanol and bromophenol blue) was added to the samples, which were then boiled and run on a 10% SDS polyacrylamide gel. Separated proteins were transferred to a Hybond-P PVDF membrane (Amersham, RPN2020F) that was then blocked using dry milk powder in Tris buffered saline (TBS) at room temperature for >1h and subsequently incubated with a mouse monoclonal phospho-Erk antibody (recognizing activation by phosphorylation at Thr203/Tyr205 of rat Erk1 and Thr183/Tyr185 of rat Erk2; Cell Signalling Technology, diluted 1:10,000 in TBS) overnight at 4oC. The membrane was then washed in TBS-tween (3x10min) and incubated for 1h at room temperature with a secondary anti-mouse antibody conjugated with horseradish peroxidase for visualization of specific band intensities with enhanced chemiluminescense detection on X-ray film. The filter was stripped in 60mM Tris pH 6.8, 0.2% SDS, 1:400 β-mercaptoethanol for 30min at 55ºC and re-probed with a rabbit polyclonal MAP kinase antibody (1:5000) recognizing the total Erk1/2 proteins.
Cell culture
PC12 cells In 1976, Greene and Tischler established a cell line, PC12, responding to NGF with ceased proliferation and neurite extension, from a rat adrenal medulla derived phaeochromocytoma. This cell line has since then been used in thousands of neurobiological and neurochemical studies. Upon NGF treatment, PC12 cells differentiate into sympathetic like neurons, synthesizing and storing dopamine and noradrenaline, but not adrenaline. The PC12 cells used in paper IV were obtained from the European Collection of Cell Cultures, CAMR, Salisbury, Wiltshire, UK (ECACC, ref. No. 88022401) and routinely grown in RPMI 1640 with 2mM L-glutamine, 10% horse serum and 5% fetal bovine serum (FBS). For differentiation in NGF-containing medium, the cells were seeded on a thin film of collagen type I. For luciferase assays (see below), PC12 cells were transfected using Lipofectamine 2000 according to recommendations from the manufacturer. HepG2 cells (Paper II) were cultured in DMEM supplemented with %FCS
51

and transfected by the calcium phosphate precipitation method described previously by (ref).
Luciferase assay (Papers II and IV) Various luciferase reporters, a β-gal reporter and expression-vectors were used (Table 4) for transfection of HepG2 (paper II; supplementary data, shown in the results section of this thesis) or PC12 (paper IV) cells in order to measure transcriptional activity caused by activation of different intracellular signalling pathways. Transfected cells were lysed in reporter lysis buffer (#E397A; Promega, Inc., Madison, WI, USA) after 16-48 hours of stimulation. Following centrifugation, luciferase activity in the lysates was measured using a Wallac Victor luminometer (Model 1420 Multilabel Counter) and a luciferase assay substrate (Promega, #E151A). To normalize the transfection efficiency, β-gal activity was measured in a fluorometer (Dynatech MicroFluor reader) as the amount of the resulting fluorescent product of b-gal mediated breakdown of 4-methylumbelliferyl-β-galactoside (0.2 mM) in a β-galactosidase buffer. Each experiment is based on the analysis of three wells and repeated at least twice. Statistical analyses of normalized values were carried out using the SigmaStat software (SPSS, Chicago, IL, USA).
Table 4, Expression-plasmids and luciferase reporters
Description Vector name Expression vector pcDNA3 Dominant negative ALK2 A2-HA-pcDNA3 Dominant negative BMPRII B2-cMyc-pcDNA3 Wild-type ALK2 wtALK2-HA-pcDNA3 Wild-type BMPRII wtBMPRII-HA-pcDNA3 Constitutively active ALK2 caALK2-pcDNA3 Wild-type Smad1 FLAG-wtSmad1-pCMV5 Wild-type Smad5 FLAG-wtSmad5-pcDNA3 Wild-type Smad7 FLAG-wtSmad7-pcDNA3 Dominant negative Smad5 FLAG-Smad52S/A-pcDNA3 Linker region mutated Smad5 FLAG-Smad55S/A-pcDNA3 Gal4 DNA-binding domain pFC2-dbd Gal4-wtSmad5 fusion wtSmad5-pFC2-dbd Gal4-Smad5 2S/A fusion Smad52S/A-pFC2-dbd Gal4-Smad5 5S/A fusion Smad55S/A-pFC2-dbd Gal4-Elk1 fusion pFA2-Elk1 Constitutively active MEK1 pFC-MEK1 Smad Binding Element- luciferase reporter SBE4x-luc Gal4-luciferace reporter pFR-luc βgal reporters PCMV-βgal and pCH110
52

Crosslinking (Paper II, supplementary data) Recombinant BMP7 was iodinated according to the chloramines T method, as described by (ten Dijke et al., 1994a). COS cells were transfected for overexpression of the dominant negative ALK2-construct (A2) or wild-type tALK2 in combination with wild-type tBMPRII (A2-HA-pcDNA3 plus wtALK2-HA-pcDNA3, wtBMPRII-HA-pcDNA3, pcDNA3 etc.). After two days, the cells were incubated on ice for 3h with 0.2-0.5nM of 125I-labelled BMP7 in a binding buffer composed of phosphate buffered saline (PBS), 0.9mM CaCl2, 0.49mM MgCl2 and 1mg/ml bovine serum albumin (BSA). Thereafter, the cells were washed in binding buffer without BSA prior to crosslinking for 15 min on ice, by the addition of binding buffer containing 0.27mM disuccinyl substrate (Pierce Chemical Co., Rockford, IL, USA). The cells were then washed in 10mM Tris pH 7.4, 1mM EDTA, 10% glycerol, and 0.3mM phenylmethylsulphonyl fluoride (PMSF) and incubated on ice for 1h in lysis buffer (20mM Tris pH 7.4, 150mM NaCl, 10mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, supplemented with 1.5% Trasylol and 1mM PMSF just before use). Lysates were cleared by centrifugation and the supernatants were then incubated at 4°C overnight with anti HA-tag antibodies. Immune complexes were bound to A-sepharose (Pharmacia Biotech, Uppsala, Sweden) for 1h at 4°C, washed by repeated centrifugations (twice in washing buffer; 20mM Tris pH 7.5, 500mM NaCl, 1% Triton X-100, 1% sodium deoxycholate and 0.2% SDS; and once in MilliQ water) and finally diluted in sample buffer (100mM Tris-HCl pH 8.8, 0.01% bromphenol blue, 36% glycerol, 4% SDS and 10mM dithiothreitol) for SDS-polyacrylamide gel electrophoresis (PAGE) on 8.5% gels. The gel was fixed, dried and exposed to a Fuji BAS 2000 Bio-image analyser (Fuji Photo Film, Tokyo, Japan).
53

Acknowledgements
Five years of struggle would not have led to this thesis without encouragement and support from many friend and colleagues around me. I am grateful to all of you and in particular, I would like to thank the following persons: Ted Ebendal; my supervisor, for accepting me as a graduate student and helping me to navigate on the scientific ocean. Your support during the years has been invaluable. Stine Söderström; for all hilarious conversations we had while working together, making even “mini-prepping” enjoyable, and for being such a good listener and encourager! Henrik Bengtsson; my co-struggler with the TH-IRES-A2/B2 project. The paper is out there now… Thank you for all good times in the lab, at conferences in LA and NO, pub-nights and parties. Jonas Lindeberg; for being a bright scientist and party-animal in one, and for introducing the now legendary fire-breathing tradition on the Fågelmara crayfish parties. Mitya Usoskin; for working hard and for driving like in Russia… passing the annoyingly long line on the verge… Magnus Åbrink; for always telling me that research is not “that bad”… and for exploring things like “negative geo-taxis and positional righting reflexes” in the animal house. Annika Kylberg; for dissecting ganglia for the hundreds of assays included in the work of this thesis and for sharing interest in TV programs like doctor Phil and documentaries about how to clean a bathroom with a tooth brush. Nestor Carri; for advise on cryosectioning and immunostaining. Charlotte Israelsson; for renewing the group and for hitting my GDF10 mice in the head. Good luck with your work!!! Karin Nilsson; for sharing office with me during your time in the lab. Marianne Jonsson; for all “uppdok-certificates” etc. you have provided me with over the years and for taking time to chat. The ten Dijke group at NKI, the Netherlands Cancer Institute in Amsterdam: Peter ten Dijke; for your generous welcoming attitude, creating a positive
54

creative atmosphere in your group. Thank you also for listening to my frustrations… and for understanding and support!! Last but not least, to you and Midory, for your hospitality and for nice dinner-parties during my stay in Amsterdam. Gudrun, Marie-José, Fumiko, Susumu, Sylviane, Martejn, Sascha and Midory; thank you for providing a nice environment to work in! I really felt welcomed in your group and had a great time!! Special thanx to Gudrun and Sylviane for letting me stay with you in the “Johannes Hilverdink palace”. Carl-Henrik Heldin; my co-supervisor, at the Ludwig Institute for Cancer Research in Uppsala for helpful comments on my manuscripts and for introducing me to Peter and his group. Anita and other helpful people at Ludwig; for providing me with plasmids and helping me out with various technical matters.
Öivind Hvalby and Vidar Jensen, my co-authors in Oslo; for running the electrophysiology in paper I. Lars Olsson, Eva Lindqvist and Karin Pernold, my co-authors in Stockholm; for your contributions to paper II. Finn Hallböök; for believing in me and for being supportive when needed. Miriam Karlsson; “Det är ju för tokigt allting” Thank you for all brilliant times in and outside the lab, for a completely crazy trip to NO including “fluff”, voodoo experiences, fortune-tellers etc… Niclas Lindqvist; my former office-companion and NNN-colleague, who will also present his thesis in May. Thank you for arranging pub-nights and for being a step ahead of me in the process of writing a thesis. Ulla, So Joeng, Robert, Ulrika and PH, as well as past members; Raquel, Ulrika B. and others, of the Retina-group; for company in the lab and the lunchroom. Special thanks to So Joeng for helping me out with my “last-minute-experiment”. Dan Lindholm; for your expertise in neurobiology, always coming up with interesting questions. Laura, Helena, Lotta, Inga, Karin, Beat, Rodrigo, Staffan, Angelika, Håkan, Cecilia, Yuming; for coffee break and lunch discussions on all kinds of topics. Susanne Frykman; for the Highway 1 adventure, for being a great companion in the lab and for all good times at parties, including your wedding. Ylva Skoglösa; for proof-reading my thesis, a superb midsummer party in Vittsjö, and inspiring “wine drinking” discussions about things in life.
55

Alex, P-A, Cesare, Sandra, Johanna, Carolina S, Ulrika, Carolina R and all other past members of the Dan-group for good times in and outside the lab.
Håkan Aldskogius and Elena Kozlowa Aldskogius; for support during the NBB course, and for interesting meetings about neuro-courses on the “new MD-education”. Ing-Marie; for help with histological stainings. Anna and Johan Ledin + Agnes and Karin; for ”vardagsmiddagar”, summer days on Runmarö, and for all the work we put into the SNiB99 project together with Johan Aa and Marika (HTML for dummies, pyssel å knåp, creative grey boxes etc). Anna, thank you also for good suggestions on my thesis. Johanna H; for company at BMC, “happy-love-film” nights, skiing-, ice-skating and kayak-tours, laughing therapy and rockadooodle… Lisa A; for “allt-jag-rör-vid-blir-till-sten” and “Nobel-Prize-winning-days” in the lab and for being my “career-mentor”. Anna G; my first friend in Uppsala, for being my “fitness enforcer”… for pre-spinning chats, long walks, shopping-tours, etc. Fredrik Å; for the long discussions and laughs we’ve had over a beer and a snus or two… or many… at “Snerran”, in NO and at NNN-meetings… Karin; my cousin and neighbour, for all the fun we’ve had since… as long as I can remember… including discussions on how long time one spend doing different things in a life-time… Henrik; for all the good times we’ve shared this last year!! The Molbio-girls: Karin, Hanna, Sara, Linda, Erika; for biking tours in Denmark and on Åland, “pyssel and gourmet” weekends in Hästerum, crayfish parties, etc... Special thanx to Hanna “ditt halmpynt” for hours and hours on the phone… and Karin for ”SHOPPAAAAA” and for kayak and sailing adventures... Emma; for falling into the same hole as I did 1993 and for being my dear sensible crazy friend ever since... soon 10 years! Vips så fort det gick! + Mattias, Love and the whole Goksör/Heimann clan, for being my stand-in family in Göteborg Eva, Mathias, Linn and Ebba plus gosenosen Felix. Jag kommer å busar och plockar blåbär i sommar!!
56

Mamma Elisabet and pappa Peter; for our “big potty family”, and for supporting me throughout my “never-ending” education… (Only four years to go now…) For welcoming me and my friends in Fågelmara and for just being my dear parents!! Frida “söstra mi” and Stefan; for “painting the world in happy colours” together!! Philip, my one and only brother; for smoooke, rainbow & strobben!! Ebba, my metropolitan sister; for Vivvilona, Fiffianne, Zannette…!!
The work of this thesis has been performed at the Unit of Developmental Neuroscience, Department of Neuroscience, Uppsala University, Sweden and was funded by NNN- the National Network in Neuroscience and SSF, Swedish Foundation for Strategic Research. STINT, the Swedish Foundation for International Cooperation in Research and Higher Education, sponsored my time at NKI, the Netherlands Cancer Institute in Amsterdam. I would in addition like to thank NNN for providing a network for interesting scientific discussions, for enjoyable annual meeting and for supporting my participation in the EMBO/FENS course on “Mouse transgenics and behaviour” in Zürich 2001.
57

Men ought to know that from the brain, and from the brain only, arise our pleasures, joys, laughters and jests, as well as our sorrows, pains, griefs and tears. Through it, in particular, we think, see, hear and distinguish the ugly from the beautiful, the bad from the good, the pleasant from the unpleasant… Attributed to Hippocrates The fifth century, BC.
58

References
Afrakhte, M., Moren, A., Jossan, S., Itoh, S., Sampath, K., Westermark, B., Heldin, C. H., Heldin, N. E., and ten Dijke, P. (1998). Induction of inhibitory Smad6 and Smad7 mRNA by TGF-beta family members. Biochem Biophys Res Commun 249: 505-511.
Airaksinen, M. S., and Saarma, M. (2002). The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3: 383-394.
Airaksinen, M. S., Titievsky, A., and Saarma, M. (1999). GDNF family neurotrophic factor signaling: four masters, one servant? Mol Cell Neurosci 13: 313-325.
Albright, T.D., Jessell, T.M., Kandel, E.R. and Posner, M.I. (2000a). Neural science: a century of progress and the mysteries that remain. Cell 100:1-55
Albright, T.D., Kandel, E.R. and Posner, M.I. (2000b). Cognitive neuroscience. Curr Opin in Neurobiol 10:612-624
Aloyz, R. S., Bamji, S. X., Pozniak, C. D., Toma, J. G., Atwal, J., Kaplan, D. R., and Miller, F. D. (1998). p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol 143: 1691-1703.
Andjelkovic, M., Suidan, H. S., Meier, R., Frech, M., Alessi, D. R., and Hemmings, B. A. (1998). Nerve growth factor promotes activation of the alpha, beta and gamma isoforms of protein kinase B in PC12 pheochromocytoma cells. Eur J Biochem 251: 195-200.
Aoki, H., Fujii, M., Imamura, T., Yagi, K., Takehara, K., Kato, M., and Miyazono, K. (2001). Synergistic effects of different bone morphogenetic protein type I receptors on alkaline phosphatase induction. J Cell Sci 114: 1483-1489.
Arenas, E., Trupp, M., Akerud, P., and Ibanez, C. F. (1995). GDNF prevents degeneration and promotes the phenotype of brain noradrenergic neurons in vivo. Neuron 15: 1465-1473.
Ashcroft, M., Stephens, R. M., Hallberg, B., Downward, J., and Kaplan, D. R. (1999). The selective and inducible activation of endogenous PI 3-kinase in PC12 cells results in efficient NGF-mediated survival but defective neurite outgrowth. Oncogene 18: 4586-4597.
Bahamonde, M. E., and Lyons, K. M. (2001). BMP3: to be or not to be a BMP. J Bone Joint Surg Am 83-A: S56-62.
Bai, S., Shi, X., Yang, X., and Cao, X. (2000). Smad6 as a transcriptional corepressor. J Biol Chem 275: 8267-8270.
Baker, J. C., Beddington, R. S., and Harland, R. M. (1999). Wnt signaling in Xenopus embryos inhibits bmp4 expression and activates neural development. Genes Dev 13: 3149-3159.
Baloh, R. H., Tansey, M. G., Lampe, P. A., Fahrner, T. J., Enomoto, H., Simburger, K. S., Leitner, M. L., Araki, T., Johnson, E. M., Jr., and Milbrandt, J. (1998).
59

Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRalpha3-RET receptor complex. Neuron 21: 1291-1302.
Benes, F. M. (2001). Carlsson and the discovery of dopamine. Trends Pharmacol Sci 22: 46-47.
Bengtsson, H., Soderstrom, S., and Ebendal, T. (1995). Expression of activin receptors type I and II only partially overlaps in the nervous system. Neuroreport 7: 113-116.
Bengtsson, H., Soderstrom, S., Kylberg, A., Charette, M. F., and Ebendal, T. (1998). Potentiating interactions between morphogenetic protein and neurotrophic factors in developing neurons. J Neurosci Res 53: 559-568.
Bibel, M., and Barde, Y. A. (2000). Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev 14: 2919-2937.
Bonni, A., Brunet, A., West, A. E., Datta, S. R., Takasu, M. A., and Greenberg, M. E. (1999). Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286: 1358-1362.
Brodski, C., Schnurch, H., and Dechant, G. (2000). Neurotrophin-3 promotes the cholinergic differentiation of sympathetic neurons. Proc Natl Acad Sci U S A 97: 9683-9688.
Brunet, A., Datta, S. R., and Greenberg, M. E. (2001a). Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11: 297-305.
Brunet, A., Park, J., Tran, H., Hu, L. S., Hemmings, B. A., and Greenberg, M. E. (2001b). Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol 21: 952-965.
Bueker, E.D. (1948). Implantation of tumors in the hind limb field of the embryonic chick and the developmental response of the lumbosacral nervous system. Anat Rec 102:369-390
Buj-Bello, A., Buchman, V. L., Horton, A., Rosenthal, A., and Davies, A. M. (1995). GDNF is an age-specific survival factor for sensory and autonomic neurons. Neuron 15: 821-828.
Carlsson, A. (2001a). A half-century of neurotransmitter research: impact on neurology and psychiatry. Nobel lecture. Biosci Rep 21: 691-710.
Carlsson, A. (2001b). A paradigm shift in brain research. Science 294: 1021-1024. Carlsson, A., Waters, N., Holm-Waters, S., Tedroff, J., Nilsson, M., and Carlsson,
M. L. (2001). Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol 41: 237-260.
Chang, H., Zwijsen, A., Vogel, H., Huylebroeck, D., and Matzuk, M. M. (2000). Smad5 is essential for left-right asymmetry in mice. Dev Biol 219: 71-78.
Charytoniuk, D. A., Traiffort, E., Pinard, E., Issertial, O., Seylaz, J., and Ruat, M. (2000). Distribution of bone morphogenetic protein and bone morphogenetic protein receptor transcripts in the rodent nervous system and up- regulation of bone morphogenetic protein receptor type II in hippocampal dentate gyrus in a rat model of global cerebral ischemia. Neuroscience 100: 33-43.
Chiariello, M., Visconti, R., Carlomagno, F., Melillo, R. M., Bucci, C., de Franciscis, V., Fox, G. M., Jing, S., Coso, O. A., Gutkind, J. S., Fusco, A., and Santoro, M. (1998). Signalling of the Ret receptor tyrosine kinase through the c-Jun NH2- terminal protein kinases (JNKS): evidence for a divergence of the ERKs and JNKs pathways induced by Ret. Oncogene 16: 2435-2445.
60

Christiansen, J. H., Coles, E. G., and Wilkinson, D. G. (2000). Molecular control of neural crest formation, migration and differentiation. Curr Opin Cell Biol 12: 719-724.
Chytil, A., Magnuson, M. A., Wright, C. V., and Moses, H. L. (2002). Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis 32: 73-75.
Cochard, P., Goldstein, M., and Black, I. B. (1978). Ontogenetic appearance and disappearance of tyrosine hydroxylase and catecholamines in the rat embryo. Proc Natl Acad Sci U S A 75: 2986-2990.
Cohen, S., Levi-Montalcini, R., Hamburger, V. (1954). A nerve growth stimulating factor isolated from sarcomas 37 and 180. Proc Natl Acad Sci USA 40:1014-1018
Cohen, S. (1959). Purification and metabolic effects of a nerve growth promoting protein from snake venom. J Biol Chem 234:1129-1142
Cohen, S. (1960). Purification of a nerve growth promoting protein from the mouse salivary gland and its neurocytotoxic antiserum. Proc Natl Acad Sci USA 46:302-311
Cordon-Cardo, C., Tapley, P., Jing, S. Q., Nanduri, V., O'Rourke, E., Lamballe, F., Kovary, K., Klein, R., Jones, K. R., Reichardt, L. F., and et al. (1991). The trk tyrosine protein kinase mediates the mitogenic properties of nerve growth factor and neurotrophin-3. Cell 66: 173-183.
Cowan, W. M. (2001). Viktor Hamburger and Rita Levi-Montalcini: the path to the discovery of nerve growth factor. Annu Rev Neurosci 24: 551-600.
Creedon, D. J., Tansey, M. G., Baloh, R. H., Osborne, P. A., Lampe, P. A., Fahrner, T. J., Heuckeroth, R. O., Milbrandt, J., and Johnson, E. M., Jr. (1997). Neurturin shares receptors and signal transduction pathways with glial cell line-derived neurotrophic factor in sympathetic neurons. Proc Natl Acad Sci U S A 94: 7018-7023.
Cross, D. A., Culbert, A. A., Chalmers, K. A., Facci, L., Skaper, S. D., and Reith, A. D. (2001). Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J Neurochem 77: 94-102.
Crowder, R. J., and Freeman, R. S. (1998). Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci 18: 2933-2943.
Crowder, R. J., and Freeman, R. S. (1999). The survival of sympathetic neurons promoted by potassium depolarization, but not by cyclic AMP, requires phosphatidylinositol 3- kinase and Akt. J Neurochem 73: 466-475.
Cunningham, N. S., Jenkins, N. A., Gilbert, D. J., Copeland, N. G., Reddi, A. H., and Lee, S. J. (1995). Growth/differentiation factor-10: a new member of the transforming growth factor-beta superfamily related to bone morphogenetic protein-3. Growth Factors 12: 99-109.
Daluiski, A., Engstrand, T., Bahamonde, M. E., Gamer, L. W., Agius, E., Stevenson, S. L., Cox, K., Rosen, V., and Lyons, K. M. (2001). Bone morphogenetic protein-3 is a negative regulator of bone density. Nat Genet 27: 84-88.
Daopin, S., Piez, K. A., Ogawa, Y., and Davies, D. R. (1992). Crystal structure of transforming growth factor-beta 2: an unusual fold for the superfamily. Science 257: 369-373.
Dennler, S., Itoh, S., Vivien, D., ten Dijke, P., Huet, S., and Gauthier, J. M. (1998). Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in
61

the promoter of human plasminogen activator inhibitor-type 1 gene. Embo J 17: 3091-3100.
Derynck, R., Gelbart, W. M., Harland, R. M., Heldin, C. H., Kern, S. E., Massague, J., Melton, D. A., Mlodzik, M., Padgett, R. W., Roberts, A. B., Smith, J., Thomsen, G. H., Vogelstein, B., and Wang, X. F. (1996). Nomenclature: vertebrate mediators of TGFbeta family signals. Cell 87: 173.
Dudek, H., Datta, S. R., Franke, T. F., Birnbaum, M. J., Yao, R., Cooper, G. M., Segal, R. A., Kaplan, D. R., and Greenberg, M. E. (1997). Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275: 661-665.
Dymecki, S. M. (1996). Flp recombinase promotes site-specific DNA recombination in embryonic stem cells and transgenic mice. Proc Natl Acad Sci U S A 93: 6191-6196.
Ebendal, T. 1989. Use of collagen gels to bioassay nerve growth factor activity. In Nerve growth factors. IBRO Handbook series: Methods in the Neurosciences. Vol. 12. R. A. Rush, editor. John Wiley, Chichester. 81-93.
Ebendal, T., Bengtsson, H., and Soderstrom, S. (1998). Bone morphogenetic proteins and their receptors: potential functions in the brain. J Neurosci Res 51: 139-146.
Ebendal, T., Hedlund, K. O., and Norrgren, G. (1982). Nerve growth factors in chick tissues. J Neurosci Res 8: 153-164.
Ebendal, T., and Jacobson, C. O. (1977). Tissue explants affecting extension and orientation of axons in cultured chick embryo ganglia. Exp Cell Res 105: 379-387.
Ebendal, T., Olson, L., Seiger, A., and Hedlund, K. O. (1980). Nerve growth factors in the rat iris. Nature 286: 25-28.
Ebisawa, T., Tada, K., Kitajima, I., Tojo, K., Sampath, T. K., Kawabata, M., Miyazono, K., and Imamura, T. (1999). Characterization of bone morphogenetic protein-6 signaling pathways in osteoblast differentiation. J Cell Sci 112: 3519-3527.
Elsdale, T., and Bard, J. (1972). Collagen substrata for studies on cell behavior. J Cell Biol 54: 626-637.
Ernfors, P., Lee, K. F., Kucera, J., and Jaenisch, R. (1994). Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell 77: 503-512.
Ernsberger, U. (2000). Evidence for an evolutionary conserved role of bone morphogenetic protein growth factors and phox2 transcription factors during noradrenergic differentiation of sympathetic neurons. Induction of a putative synexpression group of neurotransmitter-synthesizing enzymes. Eur J Biochem 267: 6976-6981.
Ernsberger, U., Reissmann, E., Mason, I., and Rohrer, H. (2000). The expression of dopamine beta-hydroxylase, tyrosine hydroxylase, and Phox2 transcription factors in sympathetic neurons: evidence for common regulation during noradrenergic induction and diverging regulation later in development. Mech Dev 92: 169-177.
Feng, X. H., and Derynck, R. (1997). A kinase subdomain of transforming growth factor-beta (TGF-beta) type I receptor determines the TGF-beta intracellular signaling specificity. Embo J 16: 3912-3923.
Friedman, W. J., and Greene, L. A. (1999). Neurotrophin signaling via Trks and p75. Exp Cell Res 253: 131-142.
62

Furuta, Y., Piston, D. W., and Hogan, B. L. (1997). Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development 124: 2203-2212.
Gomez-Santos, C., Ambrosio, S., Ventura, F., Ferrer, I., and Reiriz, J. (2002). TGF-beta1 increases tyrosine hydroxylase expression by a mechanism blocked by BMP-2 in human neuroblastoma SH-SY5Y cells. Brain Res 958: 152-160.
Goumans, M. J., and Mummery, C. (2000). Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int J Dev Biol 44: 253-265.
Goumans, M. J., Valdimarsdottir, G., Itoh, S., Rosendahl, A., Sideras, P., and ten Dijke, P. (2002). Balancing the activation state of the endothelium via two distinct TGF- beta type I receptors. Embo J 21: 1743-1753.
Gratacos, E., Gavalda, N., and Alberch, J. (2002). Bone morphogenetic protein-6 is a neurotrophic factor for calbindin- positive striatal neurons. J Neurosci Res 70: 638-644.
Greene, L. A., and Tischler, A. S. (1976). Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A 73: 2424-2428.
Greengard (2001) The neurobiology of slow synaptic transmission. Science 294:1024-30.
Greenwood, A. L., Turner, E. E., and Anderson, D. J. (1999). Identification of dividing, determined sensory neuron precursors in the mammalian neural crest. Development 126: 3545-3559.
Griffith, D. L., Keck, P. C., Sampath, T. K., Rueger, D. C., and Carlson, W. D. (1996). Three-dimensional structure of recombinant human osteogenic protein 1: structural paradigm for the transforming growth factor beta superfamily. Proc Natl Acad Sci U S A 93: 878-883.
Grima, B., Lamouroux, A., Blanot, F., Biguet, N. F., and Mallet, J. (1985). Complete coding sequence of rat tyrosine hydroxylase mRNA. Proc Natl Acad Sci U S A 82: 617-621.
Grondin, R., and Gash, D. M. (1998). Glial cell line-derived neurotrophic factor (GDNF): a drug candidate for the treatment of Parkinson's disease. J Neurol 245: 35-42.
Hahn, S. A., Schutte, M., Hoque, A. T., Moskaluk, C. A., da Costa, L. T., Rozenblum, E., Weinstein, C. L., Fischer, A., Yeo, C. J., Hruban, R. H., and Kern, S. E. (1996). DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 271: 350-353.
Harland, R. (2000). Neural induction. Curr Opin Genet Dev 10: 357-362. Hartley, K. O., Hardcastle, Z., Friday, R. V., Amaya, E., and Papalopulu, N. (2001).
Transgenic Xenopus embryos reveal that anterior neural development requires continued suppression of BMP signaling after gastrulation. Dev Biol 238: 168-184.
Hearn, C. J., Murphy, M., and Newgreen, D. (1998). GDNF and ET-3 differentially modulate the numbers of avian enteric neural crest cells and enteric neurons in vitro. Dev Biol 197: 93-105.
Hefti, F., Denton, T.L., Knusel, B., Lapchak, P.A. (1993). Neurotrophic factors: what are they and what are they doing? In Neurotrophic factors (Loughlin, S.E. and Fallon, J.H., eds.), pp 25-50. Academic Press, Inc., San Diego.
Heldin, C. H., Miyazono, K., and ten Dijke, P. (1997). TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390: 465-471.
63

Heldin, C. H., and ten Dijke, P. (1999). SMAD destruction turns off signalling. Nat Cell Biol 1: E195-197.
Hemmati-Brivanlou, A., Kelly, O. G., and Melton, D. A. (1994). Follistatin, an antagonist of activin, is expressed in the Spemann organizer and displays direct neuralizing activity. Cell 77: 283-295.
Hemmati-Brivanlou, A., and Melton, D. (1997). Vertebrate neural induction. Annu Rev Neurosci 20: 43-60.
Hemmati-Brivanlou, A., and Melton, D. A. (1994). Inhibition of activin receptor signaling promotes neuralization in Xenopus. Cell 77: 273-281.
Hempstead, B. L., Martin-Zanca, D., Kaplan, D. R., Parada, L. F., and Chao, M. V. (1991). High-affinity NGF binding requires coexpression of the trk proto- oncogene and the low-affinity NGF receptor. Nature 350: 678-683.
Heuckeroth, R. O., Lampe, P. A., Johnson, E. M., and Milbrandt, J. (1998). Neurturin and GDNF promote proliferation and survival of enteric neuron and glial progenitors in vitro. Dev Biol 200: 116-129.
Higashi, Y., Maruhashi, M., Nelles, L., Van de Putte, T., Verschueren, K., Miyoshi, T., Yoshimoto, A., Kondoh, H., and Huylebroeck, D. (2002). Generation of the floxed allele of the SIP1 (Smad-interacting protein 1) gene for Cre-mediated conditional knockout in the mouse. Genesis 32: 82-84.
Hogan, B. L. (1996). Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev 10: 1580-1594.
Hollnagel, A., Oehlmann, V., Heymer, J., Ruther, U., and Nordheim, A. (1999). Id genes are direct targets of bone morphogenetic protein induction in embryonic stem cells. J Biol Chem 274: 19838-19845.
Hoodless, P. A., Haerry, T., Abdollah, S., Stapleton, M., O'Connor, M. B., Attisano, L., and Wrana, J. L. (1996). MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell 85: 489-500.
Horstmann, S., Kahle, P. J., and Borasio, G. D. (1998). Inhibitors of p38 mitogen-activated protein kinase promote neuronal survival in vitro. J Neurosci Res 52: 483-490.
Huang, S., Tang, B., Usoskin, D., Lechleider, R. J., Jamin, S. P., Li, C., Anzano, M. A., Ebendal, T., Deng, C., and Roberts, A. B. (2002). Conditional knockout of the Smad1 gene. Genesis 32: 76-79.
Ishida, W., Hamamoto, T., Kusanagi, K., Yagi, K., Kawabata, M., Takehara, K., Sampath, T. K., Kato, M., and Miyazono, K. (2000). Smad6 is a Smad1/5-induced smad inhibitor. Characterization of bone morphogenetic protein-responsive element in the mouse Smad6 promoter. J Biol Chem 275: 6075-6079.
Ishisaki, A., Yamato, K., Hashimoto, S., Nakao, A., Tamaki, K., Nonaka, K., ten Dijke, P., Sugino, H., and Nishihara, T. (1999). Differential inhibition of Smad6 and Smad7 on bone morphogenetic protein- and activin-mediated growth arrest and apoptosis in B cells. J Biol Chem 274: 13637-13642.
Itoh, S., Itoh, F., Goumans, M. J., and Ten Dijke, P. (2000). Signaling of transforming growth factor-beta family members through Smad proteins. Eur J Biochem 267: 6954-6967.
Jen, Y., Manova, K., and Benezra, R. (1997). Each member of the Id gene family exhibits a unique expression pattern in mouse gastrulation and neurogenesis. Dev Dyn 208: 92-106.
Jonakait, G. M., Markey, K. A., Goldstein, M., and Black, I. B. (1984). Transient expression of selected catecholaminergic traits in cranial sensory and dorsal root ganglia of the embryonic rat. Dev Biol 101: 51-60.
64

Jonakait, G. M., Rosenthal, M., and Morrell, J. I. (1989). Regulation of tyrosine hydroxylase mRNA in catecholaminergic cells of embryonic rat: analysis by in situ hybridization. J Histochem Cytochem 37: 1-5.
Jonk, L. J., Itoh, S., Heldin, C. H., ten Dijke, P., and Kruijer, W. (1998). Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J Biol Chem 273: 21145-21152.
Jordan, J., Bottner, M., Schluesener, H. J., Unsicker, K., and Krieglstein, K. (1997). Bone morphogenetic proteins: neurotrophic roles for midbrain dopaminergic neurons and implications of astroglial cells. Eur J Neurosci 9: 1699-1709.
Kambadur, R., Sharma, M., Smith, T. P., and Bass, J. J. (1997). Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res 7: 910-916.
Kandel, E.R., Schwartz, J.H., Jessel, T.M. (2000). Principles of Neuroscience, 4th ed., McGraw-Hill
Kandel, E.R. (2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science. 294:1024-30.
Kaplan, D. R., Hempstead, B. L., Martin-Zanca, D., Chao, M. V., and Parada, L. F. (1991a). The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science 252: 554-558.
Kaplan, D. R., Martin-Zanca, D., and Parada, L. F. (1991b). Tyrosine phosphorylation and tyrosine kinase activity of the trk proto- oncogene product induced by NGF. Nature 350: 158-160.
Kaplan, D. R., and Miller, F. D. (2000). Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381-391.
Kawabata, M., Imamura, T., and Miyazono, K. (1998a). Signal transduction by bone morphogenetic proteins. Cytokine Growth Factor Rev 9: 49-61.
Kawabata, M., Inoue, H., Hanyu, A., Imamura, T., and Miyazono, K. (1998b). Smad proteins exist as monomers in vivo and undergo homo- and hetero- oligomerization upon activation by serine/threonine kinase receptors. Embo J 17: 4056-4065.
Kingsley, D. M. (1994). The TGF-beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev 8: 133-146.
Kirsch, T., Nickel, J., and Sebald, W. (2000). BMP-2 antagonists emerge from alterations in the low-affinity binding epitope for receptor BMPR-II. Embo J 19: 3314-3324.
Kita, Y., Kimura, K. D., Kobayashi, M., Ihara, S., Kaibuchi, K., Kuroda, S., Ui, M., Iba, H., Konishi, H., Kikkawa, U., Nagata, S., and Fukui, Y. (1998). Microinjection of activated phosphatidylinositol-3 kinase induces process outgrowth in rat PC12 cells through the Rac-JNK signal transduction pathway. J Cell Sci 111: 907-915.
Klein, R., Lamballe, F., Bryant, S., and Barbacid, M. (1992). The trkB tyrosine protein kinase is a receptor for neurotrophin-4. Neuron 8: 947-956.
Kobayashi, K., Morita, S., Sawada, H., Mizuguchi, T., Yamada, K., Nagatsu, I., Hata, T., Watanabe, Y., Fujita, K., and Nagatsu, T. (1995). Targeted disruption of the tyrosine hydroxylase locus results in severe catecholamine depletion and perinatal lethality in mice. J Biol Chem 270: 27235-27243.
Kotzbauer, P. T., Lampe, P. A., Heuckeroth, R. O., Golden, J. P., Creedon, D. J., Johnson, E. M., Jr., and Milbrandt, J. (1996). Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature 384: 467-470.
65

Kretzschmar, M., Doody, J., and Massague, J. (1997). Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 389: 618-622.
Kretzschmar, M., Doody, J., Timokhina, I., and Massague, J. (1999). A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev 13: 804-816.
Kulessa, H., and Hogan, B. L. (2002). Generation of a loxP flanked bmp4loxP-lacZ allele marked by conditional lacZ expression. Genesis 32: 66-68.
Kusanagi, K., Inoue, H., Ishidou, Y., Mishima, H. K., Kawabata, M., and Miyazono, K. (2000). Characterization of a bone morphogenetic protein-responsive Smad- binding element. Mol Biol Cell 11: 555-565.
Lamballe, F., Klein, R., and Barbacid, M. (1991). trkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3. Cell 66: 967-979.
Lang, A. E., and Lozano, A. M. (1998a). Parkinson's disease. First of two parts. N Engl J Med 339: 1044-1053.
Lang, A. E., and Lozano, A. M. (1998b). Parkinson's disease. Second of two parts. N Engl J Med 339: 1130-1143.
Lapchak, P. A., Gash, D. M., Jiao, S., Miller, P. J., and Hilt, D. (1997). Glial cell line-derived neurotrophic factor: a novel therapeutic approach to treat motor dysfunction in Parkinson's disease. Exp Neurol 144: 29-34.
Lewen, A., Soderstrom, S., Hillered, L., and Ebendal, T. (1997). Expression of serine/threonine kinase receptors in traumatic brain injury. Neuroreport 8: 475-479.
Levi-Montalcini, R. and Hamburger, V. (1951). Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J Exp Zool 116:321-361
Levi-Montalcini, R. and Hamburger, V. (1953). A diffusible agent of mouse sarcoma producing hyperplasia of sympathetic ganglia and hyperneurotization of the chick embryo. J Exp Zool 123:233-252
Lewis, K. A., Gray, P. C., Blount, A. L., MacConell, L. A., Wiater, E., Bilezikjian, L. M., and Vale, W. (2000). Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 404: 411-414.
Levitt, M., Spector, S., Sjoerdsma, A., Udenfried, S. (1965). Elucidation of the rate-limiting step in norepinephrine biosynthesis in the perfused guinea-pig heart. J Pharmacol Exp Ther 148:1-8
Lim, D. A., Tramontin, A. D., Trevejo, J. M., Herrera, D. G., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (2000). Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron 28: 713-726.
Lin, L. F., Doherty, D. H., Lile, J. D., Bektesh, S., and Collins, F. (1993). GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260: 1130-1132.
Lopez-Casillas, F., Wrana, J. L., and Massague, J. (1993). Betaglycan presents ligand to the TGF beta signaling receptor. Cell 73: 1435-1444.
Lopez-Coviella, I., Berse, B., Krauss, R., Thies, R. S., and Blusztajn, J. K. (2000). Induction and maintenance of the neuronal cholinergic phenotype in the central nervous system by BMP-9. Science 289: 313-316.
Lopez-Coviella, I., Berse, B., Thies, R. S., and Blusztajn, J. K. (2002). Upregulation of acetylcholine synthesis by bone morphogenetic protein 9 in a murine septal cell line. J Physiol Paris 96: 53-59.
66

Lopez-Rovira, T., Chalaux, E., Massague, J., Rosa, J. L., and Ventura, F. (2002). Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. J Biol Chem 277: 3176-3185.
Lorentzon, M., Hoffer, B., Ebendal, T., Olson, L., and Tomac, A. (1996). Habrec1, a novel serine/threonine kinase TGF-beta type I-like receptor, has a specific cellular expression suggesting function in the developing organism and adult brain. Exp Neurol 142: 351-360.
Lowe, L. A., Yamada, S., and Kuehn, M. R. (2001). Genetic dissection of nodal function in patterning the mouse embryo. Development 128: 1831-1843.
Luthman, J., Friedemann, M., Bickford, P., Olson, L., Hoffer, B. J., and Gerhardt, G. A. (1993). In vivo electrochemical measurements and electrophysiological studies of rat striatum following neonatal 6-hydroxydopamine treatment. Neuroscience 52: 677-687.
Lyden, D., Young, A. Z., Zagzag, D., Yan, W., Gerald, W., O'Reilly, R., Bader, B. L., Hynes, R. O., Zhuang, Y., Manova, K., and Benezra, R. (1999). Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature 401: 670-677.
Macias-Silva, M., Hoodless, P. A., Tang, S. J., Buchwald, M., and Wrana, J. L. (1998). Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J Biol Chem 273: 25628-25636.
Maguire, E. A., Frackowiak, R. S., and Frith, C. D. (1996). Learning to find your way: a role for the human hippocampal formation. Proc R Soc Lond B Biol Sci 263: 1745-1750.
Markus, A., Zhong, J., and Snider, W. D. (2002). Raf and akt mediate distinct aspects of sensory axon growth. Neuron 35: 65-76.
Massague, J. (1998). TGF-beta signal transduction. Annu Rev Biochem 67: 753-791.
Massague, J., and Wotton, D. (2000). Transcriptional control by the TGF-beta/Smad signaling system. Embo J 19: 1745-1754.
Mazzoni, I. E., Said, F. A., Aloyz, R., Miller, F. D., and Kaplan, D. (1999). Ras regulates sympathetic neuron survival by suppressing the p53- mediated cell death pathway. J Neurosci 19: 9716-9727.
McAllister, A. K., Katz, L. C., and Lo, D. C. (1999). Neurotrophins and synaptic plasticity. Annu Rev Neurosci 22: 295-318.
McPherron, A. C., and Lee, S. J. (1993). GDF-3 and GDF-9: two new members of the transforming growth factor-beta superfamily containing a novel pattern of cysteines. J Biol Chem 268: 3444-3449.
McPherson, C. E., Varley, J. E., and Maxwell, G. D. (2000). Expression and regulation of type I BMP receptors during early avian sympathetic ganglion development. Dev Biol 221: 220-232.
Meno, C., Ito, Y., Saijoh, Y., Matsuda, Y., Tashiro, K., Kuhara, S., and Hamada, H. (1997). Two closely-related left-right asymmetrically expressed genes, lefty-1 and lefty-2: their distinct expression domains, chromosomal linkage and direct neuralizing activity in Xenopus embryos. Genes Cells 2: 513-524.
Merlio, J. P., Ernfors, P., Kokaia, Z., Middlemas, D. S., Bengzon, J., Kokaia, M., Smith, M. L., Siesjo, B. K., Hunter, T., Lindvall, O., and et al. (1993). Increased production of the TrkB protein tyrosine kinase receptor after brain insults. Neuron 10: 151-164.
67

Milbrandt, J., de Sauvage, F. J., Fahrner, T. J., Baloh, R. H., Leitner, M. L., Tansey, M. G., Lampe, P. A., Heuckeroth, R. O., Kotzbauer, P. T., Simburger, K. S., Golden, J. P., Davies, J. A., Vejsada, R., Kato, A. C., Hynes, M., Sherman, D., Nishimura, M., Wang, L. C., Vandlen, R., Moffat, B., Klein, R. D., Poulsen, K., Gray, C., Garces, A., Johnson, E. M., Jr., and et al. (1998). Persephin, a novel neurotrophic factor related to GDNF and neurturin. Neuron 20: 245-253.
Miller, F. D., and Kaplan, D. R. (2001). Neurotrophin signalling pathways regulating neuronal apoptosis. Cell Mol Life Sci 58: 1045-1053.
Minichiello, L., Korte, M., Wolfer, D., Kuhn, R., Unsicker, K., Cestari, V., Rossi-Arnaud, C., Lipp, H. P., Bonhoeffer, T., and Klein, R. (1999). Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24: 401-414.
Mishina, Y., Hanks, M. C., Miura, S., Tallquist, M. D., and Behringer, R. R. (2002). Generation of Bmpr/Alk3 conditional knockout mice. Genesis 32: 69-72.
Mishina, Y., Tizard, R., Deng, J. M., Pathak, B. G., Copeland, N. G., Jenkins, N. A., Cate, R. L., and Behringer, R. R. (1997). Sequence, genomic organization, and chromosomal location of the mouse Mullerian-inhibiting substance type II receptor gene. Biochem Biophys Res Commun 237: 741-746.
Miyazawa, K., Shinozaki, M., Hara, T., Furuya, T., and Miyazono, K. (2002). Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells 7: 1191-1204.
Miyazono, K. (2000a). Positive and negative regulation of TGF-beta signaling. J Cell Sci 113: 1101-1109.
Miyazono, K. (2000b). TGF-beta signaling by Smad proteins. Cytokine Growth Factor Rev 11: 15-22.
Miyazono, K., Kusanagi, K., and Inoue, H. (2001). Divergence and convergence of TGF-beta/BMP signaling. J Cell Physiol 187: 265-276.
Miyazono, K., and Miyazawa, K. (2002). Id: a target of BMP signaling. Sci STKE 2002: E40.
Moore, R. Y., and Bloom, F. E. (1978). Central catecholamine neuron systems: anatomy and physiology of the dopamine systems. Annu Rev Neurosci 1: 129-169.
Morris, R. (1984). Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11: 47-60.
Moustakas, A., Souchelnytskyi, S., and Heldin, C. H. (2001). Smad regulation in TGF-beta signal transduction. J Cell Sci 114: 4359-4369.
Morris, R.G.M. (1981). Spatial Localization does not require the presence of local cues. Learning and motivation 12:239-260
Nagatsu, T., Levitt, M., and Udenfriend, S. (1964). Conversion of L-tyrosine to 3,4-dihydroxyphenylalanine by cell-free preparations of brain and sympathetically innervated tissues. Biochem Biophys Res Commun 14: 543-549.
Nakao, A., Afrakhte, M., Moren, A., Nakayama, T., Christian, J. L., Heuchel, R., Itoh, S., Kawabata, M., Heldin, N. E., Heldin, C. H., and ten Dijke, P. (1997). Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 389: 631-635.
Nakashima, K., Takizawa, T., Ochiai, W., Yanagisawa, M., Hisatsune, T., Nakafuku, M., Miyazono, K., Kishimoto, T., Kageyama, R., and Taga, T. (2001). BMP2-mediated alteration in the developmental pathway of fetal mouse brain cells from neurogenesis to astrocytogenesis. Proc Natl Acad Sci U S A 98: 5868-5873.
68

Nomura, M., and Li, E. (1998). Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 393: 786-790.
Norton, J. D., Deed, R. W., Craggs, G., and Sablitzky, F. (1998). Id helix-loop-helix proteins in cell growth and differentiation. Trends Cell Biol 8: 58-65.
Ogata, T., Wozney, J. M., Benezra, R., and Noda, M. (1993). Bone morphogenetic protein 2 transiently enhances expression of a gene, Id (inhibitor of differentiation), encoding a helix-loop-helix molecule in osteoblast-like cells. Proc Natl Acad Sci U S A 90: 9219-9222.
Oh, S. P., Seki, T., Goss, K. A., Imamura, T., Yi, Y., Donahoe, P. K., Li, L., Miyazono, K., ten Dijke, P., Kim, S., and Li, E. (2000). Activin receptor-like kinase 1 modulates transforming growth factor- beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A 97: 2626-2631.
Olson, L. (1997). The coming of age of the GDNF family and its receptors: gene delivery in a rat Parkinson model may have clinical implications. Trends Neurosci 20: 277-279.
Onichtchouk, D., Chen, Y. G., Dosch, R., Gawantka, V., Delius, H., Massague, J., and Niehrs, C. (1999). Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 401: 480-485.
Onichtchouk, D., Gawantka, V., Dosch, R., Delius, H., Hirschfeld, K., Blumenstock, C., and Niehrs, C. (1996). The Xvent-2 homeobox gene is part of the BMP-4 signalling pathway controlling [correction of controling] dorsoventral patterning of Xenopus mesoderm. Development 122: 3045-3053.
Otto, I. M., Raabe, T., Rennefahrt, U. E., Bork, P., Rapp, U. R., and Kerkhoff, E. (2000). The p150-Spir protein provides a link between c-Jun N-terminal kinase function and actin reorganization. Curr Biol 10: 345-348.
Patapoutian, A., and Reichardt, L. F. (2001). Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11: 272-280.
Patel, T. D., Jackman, A., Rice, F. L., Kucera, J., and Snider, W. D. (2000). Development of sensory neurons in the absence of NGF/TrkA signaling in vivo. Neuron 25: 345-357.
Pattyn, A., Goridis, C., and Brunet, J. F. (2000). Specification of the central noradrenergic phenotype by the homeobox gene Phox2b. Mol Cell Neurosci 15: 235-243.
Qin, B. Y., Chacko, B. M., Lam, S. S., de Caestecker, M. P., Correia, J. J., and Lin, K. (2001). Structural basis of Smad1 activation by receptor kinase phosphorylation. Mol Cell 8: 1303-1312.
Qin, B. Y., Lam, S. S., Correia, J. J., and Lin, K. (2002). Smad3 allostery links TGF-beta receptor kinase activation to transcriptional control. Genes Dev 16: 1950-1963.
Radeke, M. J., Misko, T. P., Hsu, C., Herzenberg, L. A., and Shooter, E. M. (1987). Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature 325: 593-597.
Raftery, L. A., Twombly, V., Wharton, K., and Gelbart, W. M. (1995). Genetic screens to identify elements of the decapentaplegic signaling pathway in Drosophila. Genetics 139: 241-254.
Rankin, C. T., Bunton, T., Lawler, A. M., and Lee, S. J. (2000). Regulation of left-right patterning in mice by growth/differentiation factor-1. Nat Genet 24: 262-265.
Reissmann, E., Ernsberger, U., Francis-West, P. H., Rueger, D., Brickell, P. M., and Rohrer, H. (1996). Involvement of bone morphogenetic protein-4 and bone
69

morphogenetic protein-7 in the differentiation of the adrenergic phenotype in developing sympathetic neurons. Development 122: 2079-2088.
Reissmann, E., Jornvall, H., Blokzijl, A., Andersson, O., Chang, C., Minchiotti, G., Persico, M. G., Ibanez, C. F., and Brivanlou, A. H. (2001). The orphan receptor ALK7 and the Activin receptor ALK4 mediate signaling by Nodal proteins during vertebrate development. Genes Dev 15: 2010-2022.
Rogers, D. C., Fisher, E. M., Brown, S. D., Peters, J., Hunter, A. J., and Martin, J. E. (1997). Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome 8: 711-713.
Ryden, M., Imamura, T., Jornvall, H., Belluardo, N., Neveu, I., Trupp, M., Okadome, T., ten Dijke, P., and Ibanez, C. F. (1996). A novel type I receptor serine-threonine kinase predominantly expressed in the adult central nervous system. J Biol Chem 271: 30603-30609.
Santoro, M., Wong, W. T., Aroca, P., Santos, E., Matoskova, B., Grieco, M., Fusco, A., and di Fiore, P. P. (1994). An epidermal growth factor receptor/ret chimera generates mitogenic and transforming signals: evidence for a ret-specific signaling pathway. Mol Cell Biol 14: 663-675.
Savage, C., Das, P., Finelli, A. L., Townsend, S. R., Sun, C. Y., Baird, S. E., and Padgett, R. W. (1996). Caenorhabditis elegans genes sma-2, sma-3, and sma-4 define a conserved family of transforming growth factor beta pathway components. Proc Natl Acad Sci U S A 93: 790-794.
Schlunegger, M. P., and Grutter, M. G. (1992). An unusual feature revealed by the crystal structure at 2.2 A resolution of human transforming growth factor-beta 2. Nature 358: 430-434.
Schneider, C., Wicht, H., Enderich, J., Wegner, M., and Rohrer, H. (1999). Bone morphogenetic proteins are required in vivo for the generation of sympathetic neurons. Neuron 24: 861-870.
Segal, R. A. (2003). Selectivity in Neurotrophin Signaling: Theme and Variations. Annu Rev Neurosci 18: 18.
Sekelsky, J. J., Newfeld, S. J., Raftery, L. A., Chartoff, E. H., and Gelbart, W. M. (1995). Genetic characterization and cloning of mothers against dpp, a gene required for decapentaplegic function in Drosophila melanogaster. Genetics 139: 1347-1358.
Shah, N. M., Groves, A. K., and Anderson, D. J. (1996). Alternative neural crest cell fates are instructively promoted by TGFbeta superfamily members. Cell 85: 331-343.
Snider, W. D. (1994). Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell 77: 627-638.
Soderstrom, S., Bengtsson, H., and Ebendal, T. (1996). Expression of serine/threonine kinase receptors including the bone morphogenetic factor type II receptor in the developing and adult rat brain. Cell Tissue Res 286: 269-279.
Soderstrom, S., and Ebendal, T. (1999). Localized expression of BMP and GDF mRNA in the rodent brain. J Neurosci Res 56: 482-492.
Soppet, D., Escandon, E., Maragos, J., Middlemas, D. S., Reid, S. W., Blair, J., Burton, L. E., Stanton, B. R., Kaplan, D. R., Hunter, T., and et al. (1991). The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell 65: 895-903.
Stern, C. D. (2002). Induction and initial patterning of the nervous system - the chick embryo enters the scene. Curr Opin Genet Dev 12: 447-451.
70

Stull, N. D., Jung, J. W., and Iacovitti, L. (2001). Induction of a dopaminergic phenotype in cultured striatal neurons by bone morphogenetic proteins. Brain Res Dev Brain Res 130: 91-98.
Suzuki, C., Murakami, G., Fukuchi, M., Shimanuki, T., Shikauchi, Y., Imamura, T., and Miyazono, K. (2002). Smurf1 regulates the inhibitory activity of Smad7 by targeting Smad7 to the plasma membrane. J Biol Chem 277: 39919-39925.
Takahashi, M., Ritz, J., and Cooper, G. M. (1985). Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 42: 581-588.
Takase, M., Imamura, T., Sampath, T. K., Takeda, K., Ichijo, H., Miyazono, K., and Kawabata, M. (1998). Induction of Smad6 mRNA by bone morphogenetic proteins. Biochem Biophys Res Commun 244: 26-29.
Tanaka, T., Horio, Y., Taketoshi, M., Imamura, I., Ando-Yamamoto, M., Kangawa, K., Matsuo, H., Kuroda, M., and Wada, H. (1989). Molecular cloning and sequencing of a cDNA of rat dopa decarboxylase: partial amino acid homologies with other enzymes synthesizing catecholamines. Proc Natl Acad Sci U S A 86: 8142-8146.
Tang, S. J., Hoodless, P. A., Lu, Z., Breitman, M. L., McInnes, R. R., Wrana, J. L., and Buchwald, M. (1998). The Tlx-2 homeobox gene is a downstream target of BMP signalling and is required for mouse mesoderm development. Development 125: 1877-1887.
Teitelman, G., Baker, H., Joh, T. H., and Reis, D. J. (1979). Appearance of catecholamine-synthesizing enzymes during development of rat sympathetic nervous system: possible role of tissue environment. Proc Natl Acad Sci U S A 76: 509-513.
Teitelman, G., Gershon, M. D., Rothman, T. P., Joh, T. H., and Reis, D. J. (1981). Proliferation and distribution of cells that transiently express a catecholaminergic phenotype during development in mice and rats. Dev Biol 86: 348-355.
ten Dijke, P., Miyazono, K., and Heldin, C. H. (2000). Signaling inputs converge on nuclear effectors in TGF-beta signaling. Trends Biochem Sci 25: 64-70.
ten Dijke, P., Yamashita, H., Ichijo, H., Franzen, P., Laiho, M., Miyazono, K., and Heldin, C. H. (1994a). Characterization of type I receptors for transforming growth factor- beta and activin. Science 264: 101-104.
ten Dijke, P., Yamashita, H., Sampath, T. K., Reddi, A. H., Estevez, M., Riddle, D. L., Ichijo, H., Heldin, C. H., and Miyazono, K. (1994b). Identification of type I receptors for osteogenic protein-1 and bone morphogenetic protein-4. J Biol Chem 269: 16985-16988.
Thoenen, H., and Barde, Y. A. (1980). Physiology of nerve growth factor. Physiol Rev 60: 1284-1335.
Trupp, M., Ryden, M., Jornvall, H., Funakoshi, H., Timmusk, T., Arenas, E., and Ibanez, C. F. (1995). Peripheral expression and biological activities of GDNF, a new neurotrophic factor for avian and mammalian peripheral neurons. J Cell Biol 130: 137-148.
Tsuchida, K., Sawchenko, P. E., Nishikawa, S., and Vale, W. W. (1996). Molecular cloning of a novel type I receptor serine/threonine kinase for the TGF beta superfamily from rat brain. Mol Cell Neurosci 7: 467-478.
Tsukazaki, T., Chiang, T. A., Davison, A. F., Attisano, L., and Wrana, J. L. (1998). SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell 95: 779-791.
Ungerstedt, U. (1971). Stereotaxic mapping of the monoamine pathways in the rat brain. Acta Physiol Scand Suppl 367: 1-48.
71

Urist, M. R. (1965). Bone: formation by autoinduction. Science 150: 893-899. Urist, M. R., Mikulski, A., and Lietze, A. (1979). Solubilized and insolubilized
bone morphogenetic protein. Proc Natl Acad Sci U S A 76: 1828-1832. Vaillant, A. R., Mazzoni, I., Tudan, C., Boudreau, M., Kaplan, D. R., and Miller, F.
D. (1999). Depolarization and neurotrophins converge on the phosphatidylinositol 3- kinase-Akt pathway to synergistically regulate neuronal survival. J Cell Biol 146: 955-966.
van Weering, D. H., and Bos, J. L. (1997). Glial cell line-derived neurotrophic factor induces Ret-mediated lamellipodia formation. J Biol Chem 272: 249-254.
Wang, M. Z., Jin, P., Bumcrot, D. A., Marigo, V., McMahon, A. P., Wang, E. A., Woolf, T., and Pang, K. (1995a). Induction of dopaminergic neuron phenotype in the midbrain by Sonic hedgehog protein. Nat Med 1: 1184-1188.
Wang, Y., Chang, C. F., Morales, M., Chou, J., Chen, H. L., Chiang, Y. H., Lin, S. Z., Cadet, J. L., Deng, X., Wang, J. Y., Chen, S. Y., Kaplan, P. L., and Hoffer, B. J. (2001). Bone morphogenetic protein-6 reduces ischemia-induced brain damage in rats. Stroke 32: 2170-2178.
Wang, Y. Q., Sizeland, A., Wang, X. F., and Sassoon, D. (1995b). Restricted expression of type-II TGF beta receptor in murine embryonic development suggests a central role in tissue modeling and CNS patterning. Mech Dev 52: 275-289.
Varley, J. E., and Maxwell, G. D. (1996). BMP-2 and BMP-4, but not BMP-6, increase the number of adrenergic cells which develop in quail trunk neural crest cultures. Exp Neurol 140: 84-94.
Watson, F. L., Heerssen, H. M., Bhattacharyya, A., Klesse, L., Lin, M. Z., and Segal, R. A. (2001). Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci 4: 981-988.
Whitman, M., and Mercola, M. (2001). TGF-beta superfamily signaling and left-right asymmetry. Sci STKE 2001: RE1.
Wiese, S., Digby, M. R., Gunnersen, J. M., Gotz, R., Pei, G., Holtmann, B., Lowenthal, J., and Sendtner, M. (1999). The anti-apoptotic protein ITA is essential for NGF-mediated survival of embryonic chick neurons. Nat Neurosci 2: 978-983.
Wilson, P. A., and Hemmati-Brivanlou, A. (1997). Vertebrate neural induction: inducers, inhibitors, and a new synthesis. Neuron 18: 699-710.
Wilson, S. I., Rydstrom, A., Trimborn, T., Willert, K., Nusse, R., Jessell, T. M., and Edlund, T. (2001). The status of Wnt signalling regulates neural and epidermal fates in the chick embryo. Nature 411: 325-330.
Worby, C. A., Vega, Q. C., Zhao, Y., Chao, H. H., Seasholtz, A. F., and Dixon, J. E. (1996). Glial cell line-derived neurotrophic factor signals through the RET receptor and activates mitogen-activated protein kinase. J Biol Chem 271: 23619-23622.
Wu, J. W., Fairman, R., Penry, J., and Shi, Y. (2001). Formation of a stable heterodimer between Smad2 and Smad4. J Biol Chem 276: 20688-20694.
Xia, Z., Dickens, M., Raingeaud, J., Davis, R. J., and Greenberg, M. E. (1995). Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270: 1326-1331.
Xing, S., Furminger, T. L., Tong, Q., and Jhiang, S. M. (1998). Signal transduction pathways activated by RET oncoproteins in PC12 pheochromocytoma cells. J Biol Chem 273: 4909-4914.
72

Xu, R. H., Kim, J., Taira, M., Zhan, S., Sredni, D., and Kung, H. F. (1995). A dominant negative bone morphogenetic protein 4 receptor causes neuralization in Xenopus ectoderm. Biochem Biophys Res Commun 212: 212-219.
Yokota, Y., and Mori, S. (2002). Role of Id family proteins in growth control. J Cell Physiol 190: 21-28.
Zawel, L., Dai, J. L., Buckhaults, P., Zhou, S., Kinzler, K. W., Vogelstein, B., and Kern, S. E. (1998). Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell 1: 611-617.
Zhao, G. Q. (2003). Consequences of knocking out BMP signaling in the mouse. Genesis 35: 43-56.
Zhou, Q. Y., and Palmiter, R. D. (1995). Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell 83: 1197-1209.
Zhou, Q. Y., Quaife, C. J., and Palmiter, R. D. (1995). Targeted disruption of the tyrosine hydroxylase gene reveals that catecholamines are required for mouse fetal development. Nature 374: 640-643.
Zhu, H., Kavsak, P., Abdollah, S., Wrana, J. L., and Thomsen, G. H. (1999). A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 400: 687-693.
73