
The role of GPCRs in neurodegenerative diseases:avenues for therapeutic interventionYunhong Huang1, Nicholas Todd2 and Amantha Thathiah1,2,3,4
Available online at www.sciencedirect.com
ScienceDirect
Neurodegenerative diseases represent a large group of
neurological disorders with heterogeneous clinical and
pathological profiles. The majority of current therapeutic
strategies provide temporary symptomatic relief but do not
target the underlying disease pathobiology and thus do not
affect disease progression. G protein-coupled receptors
(GPCRs) are among the most successful targets for therapeutic
development of central nervous system (CNS) disorders. Many
current clinical therapeutic agents act by targeting this class of
receptors and downstream signaling pathways. Here, we
review evidence that perturbation of GPCR function
contributes to the pathophysiology of various
neurodegenerative diseases, including Alzheimer’s disease,
Frontotemporal dementia, Vascular dementia, Parkinson’s
disease, and Huntington’s disease.
Addresses1Department of Neurobiology, University of Pittsburgh School of
Medicine, 3501 Fifth Avenue, BST3, Pittsburgh, PA 15213, USA2University of Pittsburgh Brain Institute, University of Pittsburgh School
of Medicine, 3501 Fifth Avenue, BST3, Pittsburgh, PA 15213, USA3Pittsburgh Institute for Neurodegenerative Diseases, University of
Pittsburgh School of Medicine, 3501 Fifth Avenue, BST3, Pittsburgh,
PA 15213, USA4KU Leuven Center for Human Genetics, Leuven 3000, Belgium
Corresponding author: Thathiah, Amantha ([email protected])
Current Opinion in Pharmacology 2017, 32:96–110
This review comes from a themed issue on Neurosciences
Edited by David Chatenet and Terence E. Hebert
For a complete overview see the Issue and the Editorial
Available online 10th March 2017
http://dx.doi.org/10.1016/j.coph.2017.02.001
1471-4892/ã 2017 Elsevier Ltd. All rights reserved.
IntroductionNeurodegenerative diseases are a major cause of disabil-
ity and premature death among the elderly people world-
wide [1]. Alzheimer’s disease (AD), Vascular dementia
(VaD), Frontotemporal dementia (FTD), Parkinson’s
disease (PD), and Huntington’s disease (HD) are the
among the most prevalent neurodegenerative diseases
[2]. AD is the most common neurodegenerative disease
and the predominant cause of dementia in the population
over 65 years of age. AD is characterized by impaired
Current Opinion in Pharmacology 2017, 32:96–110
cognitive function, memory loss, and negative personality
changes [3–5]. The pathological features of AD include
the accumulation of amyloid b (Ab) in amyloid plaques
and hyperphosphorylated aggregates of the microtubule-
associated protein tau in neurofibrillary tangles, which are
first detected in the frontal and temporal lobes and then
slowly progress to the other areas of the neocortex [5].
VaD is the second most common cause of dementia with a
variable age of onset. VaD patients display a disturbance in
frontal executive function [6] and multiple cerebrovascular
pathologies, including vessel occlusion, arteriosclerosis,
hypertensive, aneurysms, and various forms of arteritis
[7�,8�]. Frontotemporal dementia (FTD) is a major cause
of dementia in persons under the age of 65 [9] and
is characterized by neuropsychiatric symptoms and
behavioral, motor, and cognitive impairments [10]. The
pathological features of FTD include the abnormal depo-
sition of three major proteins—tau, transactive response
DNA-binding protein 43 (TDP-43), and fused in sarcoma
(FUS) protein [10] in brain regions such as the hippocam-
pus, frontal cortex, and striatum [11].
Parkinson’s disease (PD) is second most common neuro-
degenerative disease, with an average onset of 50–60
years of age [12]. PD is characterized by motor and
non-motor symptoms. The prominent motor symptoms
in PD patients include bradykinesia, rigidity, tremor, and
gait disorders [13]. Non-motor clinical features include
cognitive impairment and neuropsychiatric symptoms
[13]. The pathological features of PD include deposi-
tion of Lewy bodies and abnormal aggregates of the
a-synuclein protein in several brain regions, such as the
substantia nigra and temporal cortex, and the loss of
dopaminergic neurons in the substantia nigra [13].
Similar to PD, Huntington’s disease (HD) patients suf-
fer from motor and non-motor symptoms. HD patients
suffer from motor symptoms such as chorea, bradykine-
sia, impaired coordination, and rigidity and non-motor
symptoms such as depression and slowed cognitive
function [14]. HD is caused by a CAG trinucleotide
repeat expansion in the Huntingtin (Htt) gene [14].
The CAG repeats vary from 6 to 35 nucleotides in
unaffected individuals. A longer series of CAG repeats
(>36) are present in HD patients and inversely correlate
with the age of onset [15]. The deposition of HTT is
most frequent in the cerebral cortex, and much less in
other brain regions such as striatum, hippocampus, and
cerebellum [16]. Collectively, AD, VaD, FTD, PD,
and HD are neurodegenerative diseases with clinical
www.sciencedirect.com

Role of GPCRs in neurodegenerative diseases Huang, Todd and Thathiah 97
features that include cognitive deficits, motor impair-
ments, and neuropsychiatric symptoms.
G protein-coupled receptors (GPCRs) have been impli-
cated in the pathogenesis of several neurodegenerative
diseases [17,18], including AD, VaD, FTD, PD, and HD.
GPCRs are the largest family of membrane proteins [19].
Over 370 non-sensory GPCRs have been identified of
which more than 90% are expressed in the brain, where
they play important roles in mood, appetite, pain, vision,
immune regulation, cognition, and synaptic transmission
[20]. GPCR ligands include a variety of molecules such as
photons, ions, biogenic amines, peptide, hormones,
growth factors, and lipids [21]. Consequently, GPCRs
represent the most common target for therapeutic drugs.
Here, we mainly focus on the GPCRs that have been
reported in the past 5 years and several well-documented
GPCRs that have been reported to be involved in the
pathophysiology of the neurodegenerative diseases
mentioned above. We review the correlation between
changes in GPCR expression and/or activity with the
neuropathological hallmarks and clinical symptoms
of these neurodegenerative diseases and discuss the
currently available therapeutic strategies targeting the
GPCRs discussed in the text.
Alzheimer’s diseaseGPCRs and cognitive deficits in AD
AD leads to significant degeneration of various brain
regions and the alteration of multiple neurochemical
pathways. Magnetic resonance imaging (MRI) studies
have shown that a reduction in the volume of the hippo-
campus and entorhinal cortex, which are affected early in
disease progression [5,22], and cortical thickness of the
medial temporal, inferior temporal, temporal pole, angu-
lar gyrus, superior parietal, superior frontal, and inferior
frontal cortex correlate with the cognitive deficits
observed in AD patients [3] (Figure 1a). Furthermore,
changes in multiple neurochemical pathways, including
the acetylcholine, serotonin, adenosine pathways have
been shown to be involved in the cognitive impairments
observed in AD.
Currently, there is no effective treatment for AD. Levels
of acetylcholine are reduced in the brains of AD patients
[23]. As such, acetylcholinesterase inhibitors have been
shown to temporarily ameliorate disease symptoms [24]
by decreasing acetylcholine breakdown, which results in
an increase in cholinergic neurotransmission and a mild
improvement in cognitive function. Excitotoxicity due
to overstimulation of glutamatergic neurotransmission
[25] is also associated with the pathophysiology of AD
[26]. Memantine is an N-methyl-D-aspartate (NMDA)
receptor antagonist that inhibits NMDA receptor-medi-
ated calcium influx into neurons [27] and protects exces-
sive glutamate-induced neuronal death and excitotoxicity
[26], providing temporary improvement in cognitive
www.sciencedirect.com
function [28]. Acetylcholinesterase inhibitors and mem-
antine are the only available symptomatic treatments that
slow the decline in cognitive function in individuals with
AD [24]. In this section, we highlight some of the GPCRs
that have been rigorously evaluated in the modulation
of cognitive function in AD mouse models in recent
literature. Additional GPCRs that have been implicated
in the pathophysiology of AD have been included in
Table 1.
Glutamate receptors mediate most of the excitatory neu-
rotransmission in the mammalian brain [29]. The meta-
botropic glutamate receptor (mGluR) family mediate
glutamate neurotransmission. MGluR5 has been shown
to be involved in cognitive function and Ab generation.
Genetic deletion of mGluR5 has been shown to alleviate
cognitive impairment and Ab production in an APPswe/PSEN1DE9 AD mouse model, which overexpresses
human APP harboring the Swedish mutation and human
presenilin 1 lacking exon 9 [30]. Interestingly, pharmaco-
logical inhibition of mGluR5 with 3-[(2-methyl-1,3-thia-
zol-4-yl)ethynyl]-pyridine (MTEP), an antagonist, has
also been shown to alleviate the cognitive deficits in
the same AD mouse model [31]. Similarly, treatment
with the mGluR5 negative allosteric modulator 2-
chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-
1H-imidazol-4-yl)ethynyl) pyridine (CTEP) alleviates
the cognitive deficits and reduces the amyloid plaque
burden in two AD mouse models [32]. These studies
suggest that allosteric modulators of mGluR5 may be an
effective therapeutic strategy for some AD cases.
Extensive serotonergic denervation of the neocortex and
hippocampus has been observed in AD patients. Reduc-
tion in 5-hydroxytryptamine (5-HT, serotonin) and 5-
HT1A, 5-HT2A, 5-HT4, and 5-HT6 receptor levels have
been reported in the hippocampus and/or prefrontal
cortex of AD patients. In rodent models, activation of
5-HT2A and 5-HT4 receptors leads to an improvement in
hippocampal-dependent learning and memory [33,34] via
G protein- or b-arrestin-dependent activation of extracel-
lular signal-regulated kinase (ERK) [35,36]. In contrast,
antagonism of the 5-HT1A and the least studied 5-HT5A
receptors has been shown to ameliorate the memory
deficits in a rat AD model [37,38], possibly through an
inhibition of Gi signaling and activation of protein kinase
A (PKA), which leads to the activation of the NMDA
receptor [39,40]. Interestingly, both 5-HT6 receptor ago-
nists and antagonists enhance learning and memory [41]
through potentially different mechanisms of action. Acti-
vation of the 5-HT6 receptor has been shown to stimulate
Gs protein-dependent brain-derived neurotrophic factor
(BDNF) mRNA expression and Fyn kinase-dependent
activation of ERK1/2 in wild-type rats [42]. Both BDNF
and ERK1/2 have been shown to be associated with
cognitive function [43,44]. In contrast, 5-HT6 receptor
antagonists have been shown to stimulate glutamate and
Current Opinion in Pharmacology 2017, 32:96–110
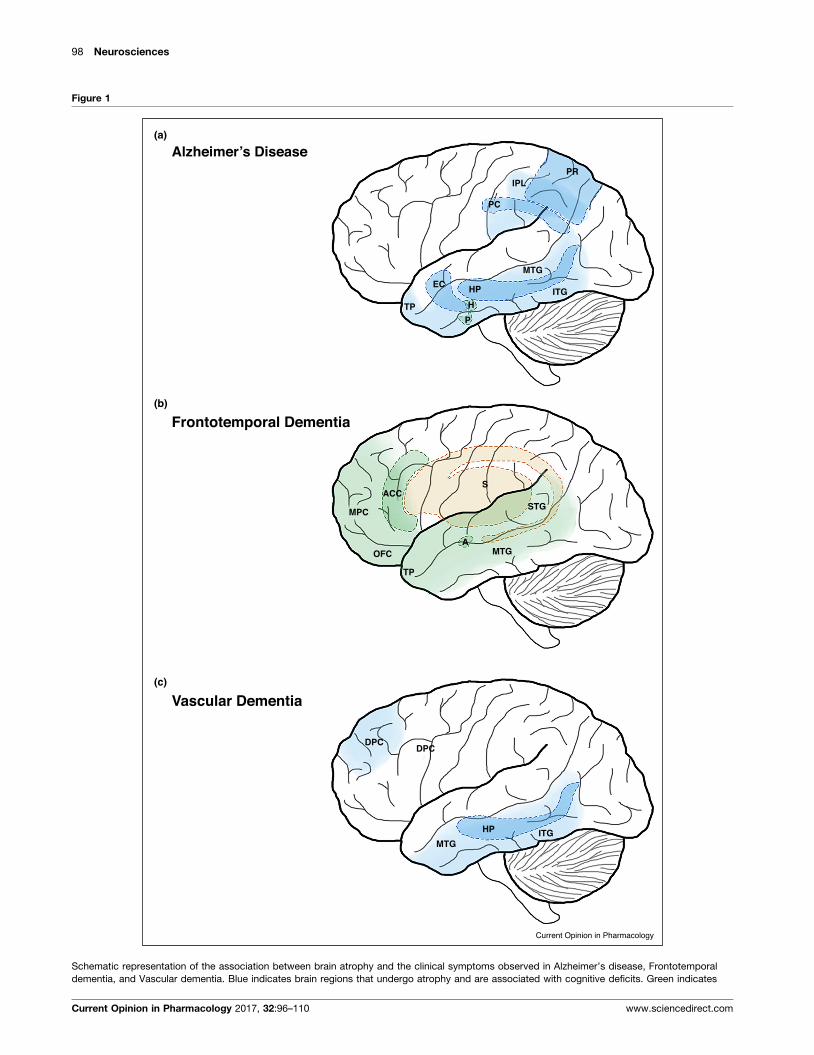
98 Neurosciences
Figure 1
Alzheimer’s Disease(a)
(b)
(c)
Frontotemporal Dementia
PRIPL
PC
EC HP
TP H
P
ITG
MTG
SACC
MPC
OFC
TP
A
STG
MTG
Vascular Dementia
DPC
HP ITGMTG
DPC
Current Opinion in Pharmacology
Schematic representation of the association between brain atrophy and the clinical symptoms observed in Alzheimer’s disease, Frontotemporal
dementia, and Vascular dementia. Blue indicates brain regions that undergo atrophy and are associated with cognitive deficits. Green indicates
Current Opinion in Pharmacology 2017, 32:96–110 www.sciencedirect.com

Role of GPCRs in neurodegenerative diseases Huang, Todd and Thathiah 99
acetylcholine release in rat brains, which has been shown
to improve scopolamine- and MK-801-induced deficits in
associative learning [42]. These studies support the
potential benefit of selective modulation of the 5-HT
receptor subtypes for AD therapy.
Expression of the adenosine A1 and A2A receptors (A1R
and A2AR) has been reported to be elevated in the frontal
cortex of the human AD brain [45]. Caffeine, a nonselec-
tive AR inhibitor, has been shown to enhance memory
consolidation in humans [46] and reduce Ab levels and
improve cognitive function in an AD mouse model [47].
Similarly, caffeine and the A2AR antagonist SCH58261
has been shown to be protective against Ab-inducedcognitive impairment [48]. Interestingly, conditional
deletion of astrocytic A2ARs has been shown to enhance
alleviate the memory deficits in AD transgenic mice
through Gs-coupled signaling [49��], whereas activation
of the Gi-coupled A1R and inhibition of PKA has been
shown enhance long-term depression (LTD) [50]. These
studies potentially suggest that activation of Gs-coupled
receptors, such as the A2AR, which activates PKA, may
suppress LTD and promote long-term plasticity (LTP),
whereas Gi-coupled receptors, such as the A1R may be
involved in the induction of LTD.
In addition to GPCRs with identified ligands, the orphan
GPCR GPR3 has been shown to modulate Ab generation
and cognitive function in vivo. Levels of GPR3 are
elevated in the human AD brain [51,52]. Genetic deletion
of Gpr3 has been shown to alleviate the learning and
memory deficits in an AD mouse model and reduce
amyloid pathology in four AD mouse models [53��].The GPR3-mediated effect on amyloid pathology
involves b-arrestin recruitment, independently of Gs-
coupling [51]. A more comprehensive discussion on the
GPCRs involved in the pathogenesis of AD is the subject
of recent reviews [54,55]. Together with the GPCRs such
as 5-HT receptors, adenosine receptors that are involved
in affected neurochemical pathways in AD suggest viable
therapeutic avenues for the treatment of cognitive deficits
in AD.
GPCRs and neuropsychiatric symptoms in AD
The corticotrophin-releasing hormone (CRH) receptor
1 and 2 (CRHR1 and CRHR2) are GPCRs associated
with depression [56,57��]. Interestingly, a greater density
of amyloid plaques has been observed in the hippocam-
pus of AD patients with a previous history of major
depression [58]. Reports also show that genetic deletion
(Figure 1 Legend Continued) brain regions that undergo atrophy and are a
regions that undergo atrophy and are associated with motor impairments. A
anterior cingulate cortex (ACC), dorsolateral prefrontal cortex (DPC), entorhi
lobule (IPL), inferior temporal gyrus (ITG), medial prefrontal cortex (MPC), m
posterior cingulate (PC), precuneus (PR), striatum (S), superior temporal gyr
www.sciencedirect.com
of Crhr1 in the PSAPP AD mouse model, which over-
express a chimeric mouse/human APP gene with human
APP Swedish mutation and human presenilin 1 lacking
exon 9, leads to a reduction in amyloid pathology [59�].Pharmacological studies in the Tg2576 AD mouse model,
which overexpresses human APP with the Swedish muta-
tion, with the CRHR1 antagonist antalarmin in acutely (7-
days) or in chronically (9-months) stressed mice reduces
Ab production and involves the Gs signaling pathway
[60]; however, pre-treatment with antalarmin failed to
inhibit an increase in Ab levels in acutely (3-hours)
stressed wild-type mice. In vitro cell-free g-secretaseactivity assays with the CRHR1 antagonists astressin,
antalarmin, and NBI-27914 have been shown to modulate
Ab generation in the absence of CRHR1, suggesting that
the compounds tested may have CRHR1-independent
effects on the modulation of g-secretase activity [57��].Treatment of wild-type mice with the CRHR1 antagonist
antalarmin reduces depression-like behaviors, whereas
genetic deficiency of Crhr2 leads to an increase in depres-
sion-like behaviors [61]. Although both receptors have
considerable sequence similarity, the two receptors have
different expression patterns in the brain and affinities for
CRH [62]. Interestingly, CRHR1 is more abundantly
expressed in the pituitary gland, and atrophy of this
region is associated with the neuropsychiatric symptoms
in AD [63]. The in vivo studies suggest that a highly
selective antagonist specific for CRHR1 may be benefi-
cial for the symptoms of depression in AD; however,
careful monitoring of Ab levels would also be necessary
to fully assess the therapeutic potential.
Frontotemporal dementiaFTD is a heterogeneous neurodegenerative disease
caused by degeneration and atrophy of the frontal and
temporal lobes. In general, FTD encompasses a wide
range of neuropathologies associated with mutations in
several genes, including tau, TDP-43, and FUS, which
leads to deterioration in behavior, personality, and motor
functions [10]. MRI and single-photon emission comput-
erized tomography (SPECT) reveal abnormalities and
atrophy in the frontal and temporal lobes of FTD
patients. Further post-mortem examination of FTD
patient brains shows additional degeneration of the stria-
tum [64] (Figure 1b). FTD patients present with a variety
of neuropsychiatric, behavioral, motor, and cognitive
impairments [64,65] including decline in social skills,
depression, compulsive behavior, agitation, bradykinesia,
and/or apathy. Symptom heterogeneity has led to multi-
ple diagnostic clinical categories such as behavioral
ssociated with neuropsychiatric symptoms. Orange indicates brain
bbreviations for the indicated brain regions include: amygdala (A),
nal cortex (EC), hypothalamus (H), hippocampus (HP), inferior parietal
edial temporal gyrus (MTG), orbitofrontal cortex (OFC), pituitary (P),
us (STG), and temporal pole (TP).
Current Opinion in Pharmacology 2017, 32:96–110

100
Neurosciences
Table 1
GPCRs that have been studied in humans and/or animal models
AD FTD VaD PD HD
Cognitive deficits Neuropsychiatric symptoms Neuropsychiatric symptoms Motor impairments Cognitive deficits Motor impairments Cognitive deficits Motor impairments
5-HT1AR [37] 5-HT2AR [149] 5-HT1AR [73,74]a D1R [10,86,87,150]a 5-HT1AR [106] A2AR [131] D1R [117]a A2AR [144,145,151,152]
5-HT2AR [34]a CRHR1 [61]a 5-HT2AR [72–74]a D2R [10,86,87,150]a 5-HT2AR [106] D1R [122] D2R [118�]a D1R [137,140]
5-HT4R [33] CRHR2 [61]a 5-HT2CR [89�]a D1R [98��] D2R [123] D2R [137]
5-HT5AR [38] mGluR5 [79��]a GABABR1 [101�,153] GPR37 [129,131] GPR52 [136�]5-HT6R [41] OXTR [71]a GABABR2 [101�,153] GPR55 [130]a M4R [154]
A1R [48] M1R [94,95] M1R [13] mGluR2 [135]
A2AR [48,49��] M4R [127] mGluR5 [135,155�]BAI1 [156]a mGluR4 [157,158] CB1R [141–143]
CysLT1R [159]
DOR [160]
GABABR [161]a
GIPR [162]a
GPR3 [53��]GPR30 [163]a
GPR48 [164]a
M1R [165]
M3R [166]a
mGluR5 [30]
S1P1 [167]
Abbreviations: 5-HT1AR, 5-hydroxytryptamine receptor 1A; 5-HT2AR, 5-hydroxytryptamine receptor 2A; 5-HT2CR, 5-hydroxytryptamine receptor 2C; 5-HT4R, 5-hydroxytryptamine receptor 4; 5-
HT5AR, 5-hydroxytryptamine receptor 5A; 5-HT6R, 5-hydroxytryptamine receptor 6; A1R, adenosine A1 receptor; A2AR, adenosine A2A receptor; BAI1, brain-specific angiogenesis inhibitor 1; CB1R,
cannabinoid type 1 receptor; CRHR1, corticotrophin-releasing hormone receptor 1; CRHR2, corticotrophin-releasing hormone receptor 2; CysLT1R, cysteinyl leukotriene receptor 1; D1R, dopamine
D1 receptor; D2R, dopamine D2 receptor; DOR, delta-opioid receptor; GABABR, g-Aminobutyric acid B receptor; GIPR, glucose-dependent insulinotropic polypeptide receptor; GPR3, G protein-
coupled receptor 3; GPR30, G protein-coupled receptor 30; GPR37, G protein-coupled receptor 37; GPR48, G protein-coupled receptor 48; GPR52, G protein-coupled receptor 52; GPR55, G
protein-coupled receptor 55; M1R, muscarinic acetylcholine receptor M1; M3R, muscarinic acetylcholine receptor M3;M4R, muscarinic acetylcholine receptor M4; mGluR2, metabotropic glutamate
receptor 2; mGluR4, metabotropic glutamate receptor 4; mGluR5, metabotropic glutamate receptor 5; OXTR, oxytocin receptor; S1P1, sphingosine 1-phosphate receptor.a Studies which were not conducted in animal disease models.
Curre
nt
Opinion
in Pharm
acology
2017,
32:96–110
www.sciencedire
ct.c
om

Role of GPCRs in neurodegenerative diseases Huang, Todd and Thathiah 101
variant FTD (bvFTD), primary progressive aphasia
(PPA), semantic dementia (SD), and FTD with parkin-
sonism-17 (FTDP-17) [10]. FTD variants affect both
distinct and overlapping brain regions and multiple neu-
rochemical pathways, which poses a significant challenge
for studying and treating the disease. Here, we discuss the
GPCRs that are the most prominent candidates for ther-
apeutic treatment of the neuropsychiatric and motor
impairments in FTD. A more comprehensive list of
the GPCRs involved in FTD can be found in Table 1.
GPCRs and neuropsychiatric symptoms in FTD
Deficits in emotion recognition skills are thought to
contribute to deficits in empathy and inappropriate social
behavior in FTD. Several studies suggest that the neuro-
peptide oxytocin is an important mediator of social behav-
ior and neuropsychiatric behaviors in patients with FTD
[66]. In mammals, oxytocin is primarily produced within
the hypothalamic brain regions and is shuttled to the
pituitary for systemic release or projected to various brain
regions for paracrine signaling of the oxytocin receptor
(OXTR) in brain regions such as the amygdala and
anterior cingulate cortex, which have been implicated
in the pathophysiology of FTD [67].
Oxytocin administration has been shown to potentially
improve social interactions [67] and facilitate the devel-
opment of GABAergic synapses, which inhibit signals
that lead to fear and anxiety [68–70]. Interestingly, intra-
nasal administration of oxytocin to FTD patients leads to
improved social interactions, namely patient–caregiver
interactions [71].
Levels of the 5-HT1A and 5-HT2A receptors are reduced
in the anterior cingulate cortex [72] and orbitofrontal and
medial prefrontal cortex of FTD patients [73,74] and the
frontal and temporal cortex of bvFTD patients [72–74].
Clinically, selective serotonin reuptake inhibitors (SSRIs)
such as fluoxetine, fluvoxamine, and sertraline, which
increase 5-HT levels by blocking 5-HT reuptake, have
been used to provide symptomatic relief for depression
and repetitive or compulsive behaviors observed in
patients with multiple variants of FTD [75]. Interest-
ingly, lower levels of the 5-HT1A and 5-HT2A receptors in
the hippocampus and prefrontal cortex of AD patients
have been shown to lead to cognitive deficits in contrast to
the neuropsychiatric symptoms observed in FTD [76].
Collectively, these studies indicate that 5-HT1A and 5-
HT2A receptors are involved in the pathophysiology of
both AD and FTD.
mGluR5 and the NMDA receptors are co-localized in
cortical brain regions and act in a cooperative fashion
[77,78]. Specifically, mGluR5 is involved in the induction
of NMDA receptor-dependent forms of synaptic plastic-
ity and excitotoxicity [78]. Leuzy et al. [79��] recently
showed a decrease in mGluR5 availability in paralimbic
www.sciencedirect.com
cortex and isocortical brain regions of bvFTD patients,
which may precede the neurodegeneration observed in
select frontotemporal brain regions. Electrophysiology
and behavioral studies suggest that mGluR5 activation
enhances NMDA receptor function, whereas mGluR5
inhibition exacerbates the effects of NMDA receptor
blockade [80,81]. These studies suggest that mGluR5
may be in involved in reduced NMDA receptor neuro-
transmission. Interestingly, mGluR5 has also been shown
to play a role in Ab generation, memory, locomotor
function, and anxiety in an AD mouse model, indicating
the multiple functions of the receptor [30].
GPCRs and motor impairments in FTD
The dopamine D1 and D2 receptors (D1R and D2R)
have been reported to play an important role in FTD.
Both the D1R and D2R are most abundantly expressed in
the striatum [82]. Because of disease heterogeneity, both
DR antagonists (antipsychotics) and agonists, which pre-
dominantly target the D2R, have been used to treat FTD.
Clinically, DR antagonists are used to treat behavioral
symptoms such as agitation and disinhibition, and DR
agonists are used to treat motor symptoms such as
rigidity and bradykinesia in bvFTD and FTDP-17,
respectively [10]. Typical antipsychotics such as halo-
peridol and fluphenazine are not commonly used to treat
FTD patients due to neuroleptic side-effects associated
with the high D2R affinity [83]. In contrast, antipsycho-
tics such as olanzapine, quetiapine, and risperidone also
have a high affinity for the D2R, but rapidly dissociate,
resulting in fewer side effects [84]. Dopamine dysfunc-
tion has also been reported to be involved in behavioral
deficits in HD (see below); however, decreased D2R
levels has been reported to be the cause of these impair-
ments [85].
FTDP-17 patients who experience rigidity and brady-
kinesia primarily display a decrease in presynaptic dopa-
minergic nerve terminals and postsynaptic D2R binding
in the striatum. Consequently, FTDP-17 patients are
currently treated with DR agonists such as carbidopa
and levodopa, which have been approved for the treat-
ment of PD [10,86,87] despite potential exacerbation of
the behavioral and psychotic symptoms. Apathy has also
been associated with reduced dopaminergic activity
[88]. Interestingly, a randomized controlled trial with
the 5-HT2C antagonist agomelatine has shown promis-
ing results with improvement in apathy and an indirect
increase in prefrontal dopaminergic tone in FTD
patients [89�]. Clinical trials of agomelatine in AD
and PD patients also indicate a reduction in apathy
and depression in AD patients [90] and a significant
decrease in depression and motor symptoms of PD [91],
indicating the involvement and beneficial effects of
targeting the 5-HT2C receptor in three neurodegenera-
tive disorders.
Current Opinion in Pharmacology 2017, 32:96–110

102 Neurosciences
Vascular dementiaVaD is the second most common cause of dementia and is
associated with multiple cerebrovascular pathologies
[7�,8�]. MRI studies reveal cortical and subcortical micro-
infarcts and atrophy of the frontal and temporal lobes,
hippocampus, and striatum of VAD patients [7�,92](Figure 1c). The M1 muscarinic acetylcholine receptor
(M1R) has been shown to be involved in cognitive
function [93]. In this regard, hippocampal damage caused
by cerebrovascular occlusion leads to a reduction in the
number of M1Rs and reduced [3H]quinuclidinyl benzi-
late binding to all muscarinic acetylcholine receptors in
the hippocampus of a chronic cerebral hypoperfusion
(CCH) rat model of VaD [94,95]. In addition, D1Rs have
been shown to elicit long-term potentiation and enhance
memory storage in the hippocampus [96,97]. A reduction
in D1Rs has been reported in CCH rats. Agonist-induced
activation of the D1R in the dentate gyrus (DG) attenu-
ates the cognitive impairments in CCH rats [98��]. These
studies indicate that both M1Rs and D1Rs are involved in
cognitive function in CCH VaD models.
GABAB receptors in the DG regulate synaptic plasticity,
learning, and memory [99]. Lower levels of GABABR1
and GABABR2 have been reported in the hippocampus of
CCH rats. Administration of the GABABR agonist baclo-
fen to CCH rats leads to an increase in GABABR expres-
sion and an improvement in spatial learning and memory
[100]. In contrast, increased GABABR activity in the
hippocampus of CCH rats has also been observed, and
treatment with the GABABR antagonist saclofen has been
shown to improve spatial learning and memory [101�].
Interestingly, combinatorial treatment with acamprosate,
which reduces glutamatergic neurotransmission, and the
GABABR agonist baclofen has been shown to regulate the
balance between excitatory and inhibitory neuronal sig-
naling, protecting against Ab-induced neurotoxicty and
alleviating cognitive deficits in an AD mouse model
[102�]. The drug PXT864, a combination of baclofen
and acamprosate, is currently in the phase II clinial trials
for the treatment of AD [103]. These reports suggest the
GABABR plays an important role in cognitive function
and that further study of the GABABRs will be necessary
to delineate the role of these GPCRs in VaD.
5-HTRs are abundant in the frontal and temporal cortices
[75,104,105] and have been shown to play an important
role in cognition and memory formation. Increased [(3H)-
WAY 100635] and [(3H)-ketanserin] radioligand binding
has been reported in post-mortem brain samples from
patients to 5-HT1A and 5-HT2A receptors, respectively,
possibly due to decreased 5-HT availability. Additionally,
5-HT1A receptors binding positively correlates with pre-
served cognition based on the mini-mental state exam
[106]. Reduced 5-HT1A receptors in AD brains leads to
cognitive impairments [76], further highlighting the
Current Opinion in Pharmacology 2017, 32:96–110
serotonergic system as a therapeutic target in both VaD
and AD.
Clinical trials of FDA approved AD drugs such as done-
pezil, an acetylcholinesterase inhibitor, galantamine, a
nicotinic acetylcholine receptor agonist, and memantine,
a NMDAR antagonist, have been conducted with VaD
patients with some positive cognitive outcomes [107,108];
however, GPCRs have not been extensively studied in
VaD patients but provide additional neurochemical tar-
gets for therapeutic intervention in VaD.
Parkinson’s diseaseGPCRs and cognitive deficits in PD
PD is a neurodegenerative disease with clinical features
that include motor and non-motor symptoms [13,109].
Approximately 25% of individuals with PD develop mild
cognitive impairment (MCI) [110], including attention,
executive function, episodic memory, visuoperceptual/
visuospatial function, and language deficits [111�]. A 20-
year follow-up study indicates that approximately 80% of
PD patients develop dementia (PDD) [112]. Patholog-
ically, MRI studies of PD patients with MCI have
reduced volume of the nucleus accumbens (NAc)
[113,114�], thalamus [113], and amydala [114�] relative
to cognitively normal individuals with PD (Figure 2a);
however, changes in the volume of the thalamus and the
amydala appear to be PD cohort-dependent.
The dopamine D1 and D2 receptors (D1R and D2R) are
highly expressed in multiple brain regions, including the
striatum, NAc, and substantia nigra [115]. Levels of both
receptors are elevated in PD patients and are associated
with the development of dopamine denervation super-
sensitivity [116]. Infusion of the D1R partial agonist SKF
38393, but not the D2R agonist quinpirole, into the NAc
of wildtype rats enhanced the accuracy of visuospatial
discrimination [117], whereas treatment with a D1R
antagonist SCH 23390 decreased accuracy [117], indicat-
ing that the D1R is involved in visuospatial function.
Treatment with the D2R antagonist sulpiride or D2R
knockdown in the NAc reduced attention performance or
induced attention impairment [118�], respectively, which
suggests that the D2R is involved in the regulation of
attention. Taken together, these studies provide evi-
dence to support a role for the D1R and the D2R involved
in visuospatial and attentional dysfunction in PD-MCI,
respectively.
GPCRs and motor impairments in PD
Dopamine deficiency within the basal ganglia leads to
parkinsonian motor symptoms, including bradykinesia,
muscular rigidity, rest tremor, and postural and gait
impairment [13]. MRI studies have shown that degener-
ation of the putamen nucleus, which is part of the striatum
and basal ganglia, correlate with the motor deficits
observed in PD [119]. Currently, dopamine replacement
www.sciencedirect.com

Role of GPCRs in neurodegenerative diseases Huang, Todd and Thathiah 103
Figure 2
(a)
(b)
Parkinson’s Disease
Huntington’s Disease
T
P
NA
A
CP
Current Opinion in Pharmacology
Schematic representation of the association between brain atrophy and clinical symptoms observed in Parkinson’s disease and Huntington’s
disease, both of which display motor impairments. Blue indicates brain regions that undergo atrophy and are associated with cognitive deficits.
Orange indicates brain regions that undergo atrophy and are associated with motor impairments. Abbreviations for the indicated brain regions
include: amygdala (A), caudate (C), nucleus accumbens (NA), putamen (P), thalamus (T).
therapy with levodopa (L-dopa), a chemical precursor of
dopamine, is the most effective drug for the symptomatic
treatment of PD [120]. However, higher doses of L-dopa
are required to compensate a decline in clinical efficacy
after long-term L-dopa therapy, which results in adverse
effects, such as motor fluctuations and motor complica-
tions such as dyskinesia [120]. Several D1R and D2R
agonists, including rotigotine, bromocriptine and lisuride,
www.sciencedirect.com
have been developed to reduce the adverse effects asso-
ciated with L-dopa therapy [121]. The agonists display
better pharmacokinetic and pharmacodynamic properties
than L-dopa with reduced incidence or delayed onset of
dyskinesia [121]. Currently, the D1R agonist rotigotine
and the D2R agonists bromocriptine and lisuride have
been used as a monotherapy or an adjunctive therapy to
L-dopa for the treatment of PD motor symptoms
Current Opinion in Pharmacology 2017, 32:96–110

104 Neurosciences
[122,123]. However, similar to L-dopa, dopamine agonist
therapy also leads to a decline in efficacy with long-term
treatment, limiting the use of dopaminergic therapy. A
promising non-dopaminergic alternative is the A2AR
antagonist istradefylline, which was recently approved
in Japan as a combination therapy with L-DOPA to treat
motor dysfunction in PD without increasing the risk of
dyskinesia [124�,125]. These studies suggest that a com-
bination therapy could be an alternative approach for the
treatment of parkinsonian motor symptoms.
A balance between the dopaminergic and cholinergic
system is important in PD [126]. Reduced striatal
dopamine in PD leads to overactivity of cholinergic
interneurons and excess acetylcholine release in the
striatum [126]. Anticholinergics such as trihexyphenidyl
and biperiden, which are selective for the M1R, are
effective in reducing tremors in PD patients [13].
Anticholinergics show little effect on bradykinesia
and rigidity, suggesting a specific role for M1Rs in
PD-associated tremor. Genetic deletion of the musca-
rinic acetylcholine receptor 4 (M4R) in mice reduces
antipsychotic-induced catalepsy [127], a PD motor
symptom, supporting a role for the muscarinic acetyl-
choline receptors in PD motor symptoms. Conversely,
dopamine agonists, used to treat motor symptoms, may
worsen cognition in PD patients, thereby complicating
therapeutic options in patients suffering with PD with
dementia (PDD) [128].
Two orphan GPCRs, GPR37 and GPR55, have also been
implicated in motor coordination [129,130]. Interestingly,
in a drug-induced parkinsonian tremor model, genetic
deletion of Gpr37 leads to an attenuation of tremulous jaw
movements (TJMs) in response to the nonselective mus-
carinic acetylcholine receptor agonist pilocarpine [131].
Treatment with the A2AR antagonist SCH-58261 also
attenuates pilocarpine-induced TJMs [131], an effect
which is not observed in GPR37-deficient mice. Collec-
tively, these studies suggest that strategies aimed at the
two orphan GPCRs may represent an alternative thera-
peutic avenue for intervention in PD.
Huntington’s diseaseHD is a progressive neurodegenerative disorder that
presents clinically with involuntary movements, impaired
coordination, depression, and slowed cognitive function.
HD is caused by a CAG trinucleotide repeat expansion in
the first exon of the Huntingtin (Htt) gene. The CAG
repeats vary from 6 to 35 nucleotides in unaffected
individuals. A longer series of CAG repeats (>36) are
present in HD patients and inversely correlate with the
age of onset [15]. Structural MRI studies indicate exten-
sive degeneration of the striatum and, to a lesser extent,
the globus pallidus, thalamus, and hippocampus in HD
patients [14] (Figure 2b).
Current Opinion in Pharmacology 2017, 32:96–110
Chronic glutamate-mediated excitotoxicity has been sug-
gested to contribute to disease progression [132].
mGluRs, including mGluR2 and mGluR5, are widely
expressed in the brain in the neocortical layers, hippo-
campus, striatum, thalamus/hypothalamus, and cerebel-
lum. Activation of presynaptic mGluR2 [133] and block-
ade of postsynaptic mGluR5 [134] inhibit glutamate
release and prevent excitotoxicity. Treatment of R6/2
HD transgenic mice, which express the N-terminally
truncated human HTT with 141–157 CAG repeats,
with either the mGluR2 agonist LY379268 or the
mGluR5 antagonist 2-methyl-6-(phenylethynyl)-pyri-
dine (MPEP) leads to a reduction in hyperactivity
[135]. MPEP treatment also reduces the decline in
motor coordination [135]. Consistent with these findings,
genetic deletion of mGluR5 in HdhQ111/Q111 knock-in
mice, which express a 109 CAG repeat insertion, leads to
an improvement in motor coordination and a reduction
in HTT aggregation [135]. These studies suggests that
mGluRs regulate motor function and HTT protein
aggregation in HD.
Accumulation of the mutant HTT protein is considered
to initiate the cytotoxicity which leads to HD. A recent
study reported that knockdown of the orphan GPCR
GPR52, which is highly expressed in the striatum, re-
duces HTT protein levels in the striatum of HdhQ140/Q140
mice by promoting HTT clearance and suppresses HD
phenotypes in both patient-induced pluripotent stem
cell (iPSC)-derived neurons and in a Drosophila HD
model [136�], suggesting that striatal degradation of
mutant HTT requires the GPR52-mediated upregulation
of cyclic adenosine monophosphate (cAMP) levels.
In both humans and HD animal models, D1R and D2R
expression is reduced in early and late stage HD [137]. In
early stage HD, patients experience hyperkinesia, poten-
tially due to hyperactivity of the dopamine pathway [138].
In contrast to GPR52, activation of the D2R, which
interacts with Gai and negatively regulates cAMP levels,
has been shown to lead to an increase in HTT aggregation
[138] whereas inhibition of the D2R with the antagonist
haloperidol has been shown to reduce HTT aggregation
and protect against striatal cell death, which may be
beneficial in the early stages of HD [139].
D1R antagonists have also been studied in HD. The D1R
antagonist SCH23390 has been shown to prevent dopa-
mine- and glutamate-induced cell death in YAC128 mice
[140], which express multiple copies of the full-length
human mutant HTT protein with 128 glutamine repeats,
mainly composed of CAG repeats and nine interspersed
CAA repeats. These studies suggest that GPR52-, and
D1R-/D2R-specific signaling regulate HTT degradation
and aggregation, respectively, and may serve as potential
therapeutic targets for HD drug discovery. These studies
also suggest that striatal-enriched modulators of HTT
www.sciencedirect.com

Role of GPCRs in neurodegenerative diseases Huang, Todd and Thathiah 105
levels may contribute to the selective vulnerability of
striatal neurons. Further studies will be required to deter-
mine whether the D1Rs, D2Rs, and GPR52 are involved
in the motor impairments observed in HD patients.
The cannabinoid type 1 receptor (CB1R) has been shown
to mitigate HTT aggregation in the R6/2 HD mouse
model. The CB1R is normally highly expressed at syn-
apses in the neocortex, hippocampus, and basal ganglia
[141]; however, CB1R levels are reduced in R6/2 mice
[142]. Moreover, chronic treatment of R6/2 mice with the
CB1R agonist D9-tetrahydrocannabiol (THC) in R6/2
alleviated motor symptoms relative to vehicle-treated
animals [142]. In PC12 cells expressing a mutation in
the HTT [143], activation of the CB1R, which couples to
Gai, with the CB1R agonists HU210 or WIN55212-2 has
been shown to alleviate the cell death associated with
HTT aggregation. Taken together, these studies high-
light CB1R agonists as potential therapeutics for HD and
suggest a complex role for cAMP in HTT aggregation and
degradation.
The A2AR has also been proposed to be a therapeutic
target for HD. A2ARs are localized throughout the brain
but are primarily found in medium spiny neurons in the
striatum [144]. Presynaptically, the A2AR antagonist
SCH58261 in combination with the D1R antagonist
SCH23390 has been shown to play a potentially neuro-
protective role in HD by decreasing glutamate release or
enhancing glutamate uptake [144,145]. In contrast,
SCH58261 and SCH23390 have been shown to promote
neurotoxicity when acting on postsynaptic A2ARs [145].
In addition, the A2AR agonist CGS21680 has been shown
to be neuroprotective by reducing NMDA currents in
stiatal medium spiny neurons [145] and to delay the onset
of motor deterioration in R6/2 mice [146]. Thus, A2AR
agonists and antagonists appear to provide some protec-
tion in animal models of HD. Although the A2AR is clearly
involved in the pathophysiology of HD, further investi-
gation into whether activation or inhibition of the A2AR is
warranted to establish the most advantageous avenue for
therapeutic benefit in HD.
ConclusionCurrent therapeutic strategies provide temporary symp-
tomatic relief but do not target the underlying pathobiol-
ogy of the neurodegenerative diseases discussed here and
thus do not affect disease progression. In the current
work, we summarize studies on several GPCRs that are
expressed in degenerative brain regions involved in AD,
VaD, FTD, PD, and HD and present the current evi-
dence, which supports therapeutic intervention strategies
focused on functional modulation of specific GPCRs.
An increasing number of GPCRs are being identified,
which are involved in modulation of the neuropathologi-
cal changes observed in neurodegenerative diseases.
www.sciencedirect.com
These GPCRs represent potential opportunities for the
development of disease-modifying therapies. Given the
symptom heterogeneity of and the variety of GPCRs
implicated in disease progression of different neurode-
generative disease, a combinatorial therapeutic approach,
targeting multiple GPCRs, may prove to be beneficial to
slow and perhaps halt disease progression. In this regard,
inhibition of both the A2AR and CRHR1 may improve
cognitive function and reduce depression in AD patients.
A combination of of OXTR and DR agonists in FTD
patients may alleviate the neuropsychiatric and motor
symptoms. Furthermore, mechanistic studies to under-
stand the interaction between different neurochemical
pathways are critical to reduce potential side-effects
associated with monotherapies. As such, the combination
therapy of L-DOPA and the A2AR antagonist istradefyl-
line, which target two neurochemical pathways, have
reduced side-effects relative to L-DOPA monotherapy
in PD patients. Abnormal accumulation of the proteins
mentioned in this review also leads to various pathological
changes in the brain, including mitochondrial dysfunc-
tion, oxidative stress, and neuroinflammation. Therefore,
an alternate avenue for therapeutic intervention is to
target the GPCRs involved in neuroprotection. In this
regard, neuropeptides acting on GPCRs, such as vasoac-
tive intestinal peptide pituitary adenylate cyclase-activat-
ing polypeptide have been shown to inhibit mitochon-
drial apoptotic pathways, and protect neurons against
oxidative stress-induced apoptosis and inflammation, pro-
viding an alternate avenue for therapeutic intervention in
neurodegenerative diseases [147,148]. Collectively, the
evidence indicates several viable avenues for therapeutic
intervention in neurodegenerative diseases.
Conflict of interest statementNothing declared.
References and recommended readingPapers of particular interest, published within the period of review,have been highlighted as:
� of special interest�� of outstanding interest
1. Przedborski S, Vila M, Jackson-Lewis V: Neurodegeneration:what is it and where are we? J Clin Invest 2003, 111:3-10.
2. Arlt S: Non-Alzheimer’s disease-related memory impairmentand dementia. Dialogues Clin Neurosci 2013, 15:465-473.
3. Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J,Greve DN, Grodstein F, Wright CI, Blacker D, Rosas HD et al.: Thecortical signature of Alzheimer’s disease: regionally specificcortical thinning relates to symptom severity in very mild tomild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex 2009, 19:497-510.
4. Lopez OL: The growing burden of Alzheimer’s disease. Am JManag Care 2011, 17(Suppl. 13):S339-S345.
5. Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT:Neuropathological alterations in Alzheimer disease. ColdSpring Harb Perspect Med 2011, 1:a006189.
6. Sachdev P, Kalaria R, O’Brien J, Skoog I, Alladi S, Black SE,Blacker D, Blazer DG, Chen C, Chui H et al.: Diagnostic criteria
Current Opinion in Pharmacology 2017, 32:96–110

106 Neurosciences
for vascular cognitive disorders: a VASCOG statement.Alzheimer Dis Assoc Disord 2014, 28:206-218.
7.�
Kalaria RN: Neuropathological diagnosis of vascular cognitiveimpairment and vascular dementia with implications forAlzheimer’s disease. Acta Neuropathol 2016, 131:659-685.
This review highlights the cerebrovascular pathologies of VaD and illus-trates which brain regions are affected by the various pathologies.
8.�
Yassi N, Desmond PM, Masters CL: Magnetic resonanceimaging of vascular contributions to cognitive impairment anddementia. J Mol Neurosci 2016, 60:349-353.
This review highlights the different cerebrovascular pathologies in VaDand highlights the common brain regions that are affected in eachpathology.
9. Warren JD, Rohrer JD, Rossor MN: Frontotemporal dementia.BMJ 2013, 347:f4827.
10. Jicha GA, Nelson PT: Management of frontotemporal dementia:targeting symptom management in such a heterogeneousdisease requires a wide range of therapeutic options.Neurodegener Dis Manag 2011, 1:141-156.
11. Gabryelewicz T, Masellis M, Berdynski M, Bilbao JM, Rogaeva E,St George-Hyslop P, Barczak A, Czyzewski K, Barcikowska M,Wszolek Z et al.: Intra-familial clinical heterogeneity due toFTLD-U with TDP-43 proteinopathy caused by a novel deletionin progranulin gene (PGRN). J Alzheimers Dis 2010,22:1123-1133.
12. Bertram L, Tanzi RE: The genetic epidemiology ofneurodegenerative disease. J Clin Invest 2005, 115:1449-1457.
13. Kalia LV, Lang AE: Parkinson’s disease. Lancet 2015,386:896-912.
14. Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD,Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS et al.:Huntington disease: natural history, biomarkers andprospects for therapeutics. Nat Rev Neurol 2014, 10:204-216.
15. Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J,Adam S, Starr E, Squitieri F, Lin B, Kalchman MA et al.: Therelationship between trinucleotide (CAG) repeat length andclinical features of Huntington’s disease. Nat Genet 1993,4:398-403.
16. Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R,Rye D, Ferrante RJ, Hersch SM, Li XJ: Nuclear and neuropilaggregates in Huntington’s disease: relationship toneuropathology. J Neurosci 1999, 19:2522-2534.
17. Guerram M, Zhang LY, Jiang ZZ: G-protein coupled receptors astherapeutic targets for neurodegenerative andcerebrovascular diseases. Neurochem Int 2016, 101:1-14.
18. Heng BC, Aubel D, Fussenegger M: An overview of the diverseroles of G-protein coupled receptors (GPCRs) in thepathophysiology of various human diseases. Biotechnol Adv2013, 31:1676-1694.
19. Pierce KL, Premont RT, Lefkowitz RJ: Seven-transmembranereceptors. Nat Rev Mol Cell Biol 2002, 3:639-650.
20. Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE,Mortrud MT, Brown A, Rodriguez SS, Weller JR, Wright AC et al.:The G protein-coupled receptor repertoires of human andmouse. Proc Natl Acad Sci U S A 2003, 100:4903-4908.
21. Millar RP, Newton CL: The year in G protein-coupled receptorresearch. Mol Endocrinol 2010, 24:261-274.
22. Du AT, Schuff N, Amend D, Laakso MP, Hsu YY, Jagust WJ,Yaffe K, Kramer JH, Reed B, Norman D et al.: Magneticresonance imaging of the entorhinal cortex and hippocampusin mild cognitive impairment and Alzheimer’s disease. J NeurolNeurosurg Psychiatry 2001, 71:441-447.
23. Colovic MB, Krstic DZ, Lazarevic-Pasti TD, Bondzic AM, Vasic VM:Acetylcholinesterase inhibitors: pharmacology andtoxicology. Curr Neuropharmacol 2013, 11:315-335.
24. Selkoe DJ: The therapeutics of Alzheimer’s disease: wherewe stand and where we are heading. Ann Neurol 2013,74:328-336.
Current Opinion in Pharmacology 2017, 32:96–110
25. Dong XX, Wang Y, Qin ZH: Molecular mechanisms ofexcitotoxicity and their relevance to pathogenesis ofneurodegenerative diseases. Acta Pharmacol Sin 2009,30:379-387.
26. Porsteinsson AP, Cosman KM: Memantine in the treatment ofAlzheimer’s disease. Aging Health 2006, 2:891-904.
27. Danysz W, Parsons CG, Mobius HJ, Stoffler A, Quack G:Neuroprotective and symptomatological action of memantinerelevant for Alzheimer’s disease—a unified glutamatergichypothesis on the mechanism of action. Neurotox Res 2000,2:85-97.
28. Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ,Memantine Study Group: Memantine in moderate-to-severeAlzheimer’s disease. N Engl J Med 2003, 348:1333-1341.
29. Willard SS, Koochekpour S: Glutamate, glutamate receptors,and downstream signaling pathways. Int J Biol Sci 2013,9:948-959.
30. Hamilton A, Esseltine JL, DeVries RA, Cregan SP, Ferguson SS:Metabotropic glutamate receptor 5 knockout reducescognitive impairment and pathogenesis in a mouse model ofAlzheimer’s disease. Mol Brain 2014, 7:40.
31. Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H,Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ et al.:Metabotropic glutamate receptor 5 is a coreceptor forAlzheimer abeta oligomer bound to cellular prion protein.Neuron 2013, 79:887-902.
32. Hamilton A, Vasefi M, Vander Tuin C, McQuaid RJ, Anisman H,Ferguson SS: Chronic pharmacological mGluR5 inhibitionprevents cognitive impairment and reduces pathogenesis inan Alzheimer disease mouse model. Cell Rep 2016,15:1859-1865.
33. Lo AC, De Maeyer JH, Vermaercke B, Callaerts-Vegh Z,Schuurkes JA, D’Hooge R: SSP-002392, a new 5-HT4 receptoragonist, dose-dependently reverses scopolamine-inducedlearning and memory impairments in C57Bl/6 mice.Neuropharmacology 2014, 85:178-189.
34. Zhang G, Asgeirsdottir HN, Cohen SJ, Munchow AH, Barrera MP,Stackman RW Jr: Stimulation of serotonin 2A receptorsfacilitates consolidation and extinction of fear memory inC57BL/6J mice. Neuropharmacology 2013, 64:403-413.
35. Barthet G, Framery B, Gaven F, Pellissier L, Reiter E, Claeysen S,Bockaert J, Dumuis A: 5-Hydroxytryptamine 4 receptoractivation of the extracellular signal-regulated kinase pathwaydepends on Src activation but not on G protein orbeta-arrestin signaling. Mol Biol Cell 2007, 18:1979-1991.
36. Gooz M, Gooz P, Luttrell LM, Raymond JR: 5-HT2A receptorinduces ERK phosphorylation and proliferation throughADAM-17 tumor necrosis factor-alpha-converting enzyme(TACE) activation and heparin-bound epidermal growthfactor-like growth factor (HB-EGF) shedding in mesangialcells. J Biol Chem 2006, 281:21004-21012.
37. Misane I, Ogren SO: Selective 5-HT1A antagonists WAY100635 and NAD-299 attenuate the impairment of passiveavoidance caused by scopolamine in the rat.Neuropsychopharmacology 2003, 28:253-264.
38. Yamazaki M, Okabe M, Yamamoto N, Yarimizu J, Harada K: Novel5-HT5A receptor antagonists ameliorate scopolamine-induced working memory deficit in mice and referencememory impairment in aged rats. J Pharmacol Sci 2015,127:362-369.
39. Francken BJ, Jurzak M, Vanhauwe JF, Luyten WH, Leysen JE: Thehuman 5-ht5A receptor couples to Gi/Go proteins and inhibitsadenylate cyclase in HEK 293 cells. Eur J Pharmacol 1998,361:299-309.
40. Gonzalez R, Chavez-Pascacio K, Meneses A: Role of 5-HT5Areceptors in the consolidation of memory. Behav Brain Res2013, 252:246-251.
41. Woods S, Clarke NN, Layfield R, Fone KC: 5-HT(6) receptoragonists and antagonists enhance learning and memory in aconditioned emotion response paradigm by modulation of
www.sciencedirect.com

Role of GPCRs in neurodegenerative diseases Huang, Todd and Thathiah 107
cholinergic and glutamatergic mechanisms. Br J Pharmacol2012, 167:436-449.
42. Ramirez MJ: 5-HT6 receptors and Alzheimer’s disease.Alzheimer’s Res Ther 2013, 5:15.
43. Lu B, Nagappan G, Lu Y: BDNF and synaptic plasticity,cognitive function, and dysfunction. Handb Exp Pharmacol2014, 220:223-250.
44. Marcos B, Cabero M, Solas M, Aisa B, Ramirez MJ: Signallingpathways associated with 5-HT6 receptors: relevance forcognitive effects. Int J Neuropsychopharmacol 2010,13:775-784.
45. Albasanz JL, Perez S, Barrachina M, Ferrer I, Martin M:Up-regulation of adenosine receptors in the frontal cortex inAlzheimer’s disease. Brain Pathol 2008, 18:211-219.
46. Borota D, Murray E, Keceli G, Chang A, Watabe JM, Ly M,Toscano JP, Yassa MA: Post-study caffeine administrationenhances memory consolidation in humans. Nat Neurosci2014, 17:201-203.
47. Arendash GW, Schleif W, Rezai-Zadeh K, Jackson EK,Zacharia LC, Cracchiolo JR, Shippy D, Tan J: Caffeine protectsAlzheimer’s mice against cognitive impairment and reducesbrain beta-amyloid production. Neuroscience 2006,142:941-952.
48. Dall’Igna OP, Fett P, Gomes MW, Souza DO, Cunha RA, Lara DR:Caffeine and adenosine A(2a) receptor antagonists preventbeta-amyloid (25-35)-induced cognitive deficits in mice.Exp Neurol 2007, 203:241-245.
49.��
Orr AG, Hsiao EC, Wang MM, Ho K, Kim DH, Wang X, Guo W,Kang J, Yu GQ, Adame A et al.: Astrocytic adenosine receptorA2A and Gs-coupled signaling regulate memory. Nat Neurosci2015, 18:423-434.
This is the first study to demonstrate the involvement of an astrocyticGPCR in the modulation of cognitive function. The study also showsincreased level of astrocytic A2A receptors in the human AD brain and inan AD transgenic mouse model.
50. Santschi LA, Zhang XL, Stanton PK: Activation of receptorsnegatively coupled to adenylate cyclase is required forinduction of long-term synaptic depression at Schaffercollateral-CA1 synapses. J Neurobiol 2006, 66:205-219.
51. Thathiah A, Horre K, Snellinx A, Vandewyer E, Huang Y,Ciesielska M, De Kloe G, Munck S, De Strooper B: Beta-arrestin2 regulates Abeta generation and gamma-secretase activity inAlzheimer’s disease. Nat Med 2013, 19:43-49.
52. Thathiah A, Spittaels K, Hoffmann M, Staes M, Cohen A, Horre K,Vanbrabant M, Coun F, Baekelandt V, Delacourte A et al.: Theorphan G protein-coupled receptor 3 modulates amyloid-betapeptide generation in neurons. Science 2009, 323:946-951.
53.��
Huang Y, Skwarek-Maruszewska A, Horre K, Vandewyer E,Wolfs L, Snellinx A, Saito T, Radaelli E, Corthout N, Colombelli Jet al.: Loss of GPR3 reduces the amyloid plaque burden andimproves memory in Alzheimer’s disease mouse models. SciTransl Med 2015, 7:309ra164.
This study extensively scrutinizes the validity of GPR3 as a therapeutictarget for AD in various AD mouse models including two AD knockinmodels. This study also shows increased level of GPR3 in two cohorts ofhuman AD brain samples.
54. Zhao J, Deng Y, Jiang Z, Qing H: G protein-coupled receptors(GPCRs) in Alzheimer’s disease: a focus on BACE1 relatedGPCRs. Front Aging Neurosci 2016, 8:58.
55. Thathiah A, De Strooper B: The role of G protein-coupledreceptors in the pathology of Alzheimer’s disease. Nat RevNeurosci 2011, 12:73-87.
56. Ishitobi Y, Nakayama S, Yamaguchi K, Kanehisa M, Higuma H,Maruyama Y, Ninomiya T, Okamoto S, Tanaka Y, Tsuru J et al.:Association of CRHR1 and CRHR2 with major depressivedisorder and panic disorder in a Japanese population. Am JMed Genet B Neuropsychiatr Genet 2012, 159b:429-436.
57.��
Park HJ, Ran Y, Jung JI, Holmes O, Price AR, Smithson L,Ceballos-Diaz C, Han C, Wolfe MS, Daaka Y et al.: The stressresponse neuropeptide CRF increases amyloid-beta
www.sciencedirect.com
production by regulating gamma-secretase activity. EMBO J2015, 34:1674-1686.
This study demonstrates that CRF increases Ab production throughmodulation of g-secrease function. The effect can be mediated by bothCRF receptor-dependent and independent pathways.
58. Rapp MA, Schnaider-Beeri M, Grossman HT, Sano M, Perl DP,Purohit DP, Gorman JM, Haroutunian V: Increased hippocampalplaques and tangles in patients with Alzheimer disease with alifetime history of major depression. Arch Gen Psychiatry 2006,63:161-167.
59.�
Campbell SN, Zhang C, Roe AD, Lee N, Lao KU, Monte L,Donohue MC, Rissman RA: Impact of CRFR1 ablation onamyloid-beta production and accumulation in a mouse modelof Alzheimer’s disease. J Alzheimer’s Dis 2015, 45:1175-1184.
This study uses a genetic approach to validate the CRF receptor type 1 inan AD transgenic mouse model, demonstrating a reduction in amyloidpathology in the absence of the CRF receptor type 1.
60. Dong H, Wang S, Zeng Z, Li F, Montalvo-Ortiz J, Tucker C,Akhtar S, Shi J, Meltzer HY, Rice KC et al.: Effects ofcorticotrophin-releasing factor receptor 1 antagonists onamyloid-beta and behavior in Tg2576 mice.Psychopharmacology (Berl) 2014, 231:4711-4722.
61. Bale TL, Vale WW: Increased depression-like behaviors incorticotropin-releasing factor receptor-2-deficient mice:sexually dichotomous responses. J Neurosci 2003,23:5295-5301.
62. Reul JM, Holsboer F: On the role of corticotropin-releasinghormone receptors in anxiety and depression. Dialogues ClinNeurosci 2002, 4:31-46.
63. Chalmers DT, Lovenberg TW, De Souza EB: Localization of novelcorticotropin-releasing factor receptor (CRF2) mRNAexpression to specific subcortical nuclei in rat brain:comparison with CRF1 receptor mRNA expression. J Neurosci1995, 15:6340-6350.
64. Snowden JS, Neary D, Mann DM: Frontotemporal dementia.Br J Psychiatry 2002, 180:140-143.
65. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S,Freedman M, Kertesz A, Robert PH, Albert M et al.:Frontotemporal lobar degeneration: a consensus on clinicaldiagnostic criteria. Neurology 1998, 51:1546-1554.
66. Jesso S, Morlog D, Ross S, Pell MD, Pasternak SH, Mitchell DG,Kertesz A, Finger EC: The effects of oxytocin on socialcognition and behaviour in frontotemporal dementia. Brain2011, 134:2493-2501.
67. Donaldson ZR, Young LJ: Oxytocin, vasopressin, and theneurogenetics of sociality. Science 2008, 322:900-904.
68. Baumgartner T, Heinrichs M, Vonlanthen A, Fischbacher U, Fehr E:Oxytocin shapes the neural circuitry of trust and trustadaptation in humans. Neuron 2008, 58:639-650.
69. Sobota R, Mihara T, Forrest A, Featherstone RE, Siegel SJ:Oxytocin reduces amygdala activity, increases socialinteractions, and reduces anxiety-like behavior irrespective ofNMDAR antagonism. Behav Neurosci 2015, 129:389-398.
70. Theodosis DT, Koksma JJ, Trailin A, Langle SL, Piet R, Lodder JC,Timmerman J, Mansvelder H, Poulain DA, Oliet SH et al.: Oxytocinand estrogen promote rapid formation of functional GABAsynapses in the adult supraoptic nucleus. Mol Cell Neurosci2006, 31:785-794.
71. Finger EC, MacKinley J, Blair M, Oliver LD, Jesso S, Tartaglia MC,Borrie M, Wells J, Dziobek I, Pasternak S et al.: Oxytocin forfrontotemporal dementia: a randomized dose-finding study ofsafety and tolerability. Neurology 2015, 84:174-181.
72. Franceschi M, Anchisi D, Pelati O, Zuffi M, Matarrese M,Moresco RM, Fazio F, Perani D: Glucose metabolism andserotonin receptors in the frontotemporal lobe degeneration.Ann Neurol 2005, 57:216-225.
73. Procter AW, Qurne M, Francis PT: Neurochemical features offrontotemporal dementia. Dement Geriatr Cogn Disord 1999,10(Suppl. 1):80-84.
Current Opinion in Pharmacology 2017, 32:96–110

108 Neurosciences
74. Bowen DM, Procter AW, Mann DM, Snowden JS, Esiri MM,Neary D, Francis PT: Imbalance of a serotonergic system infrontotemporal dementia: implication for pharmacotherapy.Psychopharmacology (Berl) 2008, 196:603-610.
75. Pompeiano M, Palacios JM, Mengod G: Distribution and cellularlocalization of mRNA coding for 5-HT1A receptor in the ratbrain: correlation with receptor binding. J Neurosci 1992,12:440-453.
76. King MV, Marsden CA, Fone KC: A role for the 5-HT(1A), 5-HT4and 5-HT6 receptors in learning and memory. TrendsPharmacol Sci 2008, 29:482-492.
77. Alagarsamy S, Marino MJ, Rouse ST, Gereau RWt, Heinemann SF,Conn PJ: Activation of NMDA receptors reversesdesensitization of mGluR5 in native and recombinant systems.Nat Neurosci 1999, 2:234-240.
78. Alagarsamy S, Rouse ST, Junge C, Hubert GW, Gutman D,Smith Y, Conn PJ: NMDA-induced phosphorylation andregulation of mGluR5. Pharmacol Biochem Behav 2002,73:299-306.
79.��
Leuzy A, Zimmer ER, Dubois J, Pruessner J, Cooperman C,Soucy JP, Kostikov A, Schirmaccher E, Desautels R, Gauthier Set al.: In vivo characterization of metabotropic glutamatereceptor type 5 abnormalities in behavioral variant FTD. BrainStruct Funct 2016, 221:1387-1402.
This study is the first to show a decrease in mGluR5 availability in bvFTDpatients, which may lead to NMDAR hypofunction and neurotoxicity.
80. Homayoun H, Moghaddam B: Bursting of prefrontal cortexneurons in awake rats is regulated by metabotropic glutamate5 (mGlu5) receptors: rate-dependent influence and interactionwith NMDA receptors. Cereb Cortex 2006, 16:93-105.
81. Kinney GG, O’Brien JA, Lemaire W, Burno M, Bickel DJ,Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR et al.:A novel selective positive allosteric modulator ofmetabotropic glutamate receptor subtype 5 has in vivo activityand antipsychotic-like effects in rat behavioral models. JPharmacol Exp Ther 2005, 313:199-206.
82. Hurd YL, Suzuki M, Sedvall GC: D1 and D2 dopamine receptormRNA expression in whole hemisphere sections of the humanbrain. J Chem Neuroanat 2001, 22:127-137.
83. Seeman P: Atypical antipsychotics: mechanism of action.Can J Psychiatry 2002, 47:27-38.
84. Kapur S, Seeman P: Does fast dissociation from the dopamined(2) receptor explain the action of atypical antipsychotics? Anew hypothesis. Am J Psychiatry 2001, 158:360-369.
85. Antonini A, Leenders KL, Spiegel R, Meier D, Vontobel P, Weigell-Weber M, Sanchez-Pernaute R, de Yebenez JG, Boesiger P,Weindl A et al.: Striatal glucose metabolism and dopamine D2receptor binding in asymptomatic gene carriers and patientswith Huntington’s disease. Brain 1996, 119(Pt. 6):2085-2095.
86. Kanazawa I, Kwak S, Sasaki H, Muramoto O, Mizutani T, Hori A,Nukina N: Studies on neurotransmitter markers of the basalganglia in Pick’s disease, with special reference to dopaminereduction. J Neurol Sci 1988, 83:63-74.
87. Sperfeld AD, Collatz MB, Baier H, Palmbach M, Storch A,Schwarz J, Tatsch K, Reske S, Joosse M, Heutink P et al.:FTDP-17: an early-onset phenotype with parkinsonism andepileptic seizures caused by a novel mutation. Ann Neurol1999, 46:708-715.
88. Pagonabarraga J, Kulisevsky J, Strafella AP, Krack P: Apathy inParkinson’s disease: clinical features, neural substrates,diagnosis, and treatment. Lancet Neurol 2015, 14:518-531.
89.�
Callegari I, Mattei C, Benassi F, Krueger F, Grafman J, Yaldizli O,Sassos D, Massucco D, Scialo C, Nobili F et al.: Agomelatineimproves apathy in frontotemporal dementia. NeurodegenerDis 2016, 16:352-356.
This study introduces agomelatine, a weak 5-HT2C receptor antagonist,as a potential treatment for apathy in FTD patients.
90. Karaiskos DPD, Katirtzoglou E, Siarkos K, Tzavellas E,Papadimitrious GN, Politis A: EPA-1197-agomelatine for
Current Opinion in Pharmacology 2017, 32:96–110
treating apathy in Alzheimer’s disease. Eur Psychiatry 2014,29 1-1.
91. Avila A, Cardona X, Martin-Baranera M, Leon L, Caballol N,Millet P, Bello J: Agomelatine for depression in Parkinsondisease: additional effect on sleep and motor dysfunction.J Clin Psychopharmacol 2015, 35:719-723.
92. Roman GC, Erkinjuntti T, Wallin A, Pantoni L, Chui HC: Subcorticalischaemic vascular dementia. Lancet Neurol 2002, 1:426-436.
93. Boddeke EW, Enz A, Shapiro G: SDZ ENS 163, a selectivemuscarinic M1 receptor agonist, facilitates the induction oflong-term potentiation in rat hippocampal slices. Eur JPharmacol 1992, 222:21-25.
94. Tanaka K, Ogawa N, Asanuma M, Kondo Y, Nomura M:Relationship between cholinergic dysfunction anddiscrimination learning disabilities in Wistar rats followingchronic cerebral hypoperfusion. Brain Res 1996, 729:55-65.
95. Kondo Y, Ogawa N, Asanuma M, Matsuura K, Nishibayashi K,Iwata E: Preventive effects of bifemelane hydrochloride ondecreased levels of muscarinic acetylcholine receptor and itsmRNA in a rat model of chronic cerebral hypoperfusion.Neurosci Res 1996, 24:409-414.
96. Huang YY, Kandel ER: Age-related enhancement of a proteinsynthesis-dependent late phase of LTP induced by lowfrequency paired-pulse stimulation in hippocampus. LearnMem 2006, 13:298-306.
97. Rossato JI, Bevilaqua LR, Izquierdo I, Medina JH, Cammarota M:Dopamine controls persistence of long-term memory storage.Science 2009, 325:1017-1020.
98.��
Wan P, Wang S, Zhang Y, Lv J, Jin QH: Involvement of dopamineD1 receptors of the hippocampal dentate gyrus in spatiallearning and memory deficits in a rat model of vasculardementia. Pharmazie 2014, 69:709-710.
This study provides evidence that a D1R agonist can attenuate thecognitive impairments observed in a CCH rat model of VD.
99. Shahidi S, Komaki A, Mahmoodi M, Lashgari R: The role ofGABAergic transmission in the dentate gyrus on acquisition,consolidation and retrieval of an inhibitory avoidance learningand memory task in the rat. Brain Res 2008, 1204:87-93.
100. Li CJ, Lu Y, Zhou M, Zong XG, Li C, Xu XL, Guo LJ, Lu Q:Activation of GABAB receptors ameliorates cognitiveimpairment via restoring the balance of HCN1/HCN2 surfaceexpression in the hippocampal CA1 area in rats with chroniccerebral hypoperfusion. Mol Neurobiol 2014, 50:704-720.
101.�
Li G, Lv J, Wang J, Wan P, Li Y, Jiang H, Jin Q: GABAB receptorsin the hippocampal dentate gyrus are involved in spatiallearning and memory impairment in a rat model of vasculardementia. Brain Res Bull 2016, 124:190-197.
This study determined that GABAB receptor activity is elevated in thehippocampus of a VaD rat model. In conjunction with the contradictoryfindings in reference 100, this study highlights the importance of studyinghippocampal GABABRs in VaD models.
102.�
Chumakov I, Nabirotchkin S, Cholet N, Milet A, Boucard A,Toulorge D, Pereira Y, Graudens E, Traore S, Foucquier J et al.:Combining two repurposed drugs as a promising approach forAlzheimer’s disease therapy. Sci Rep 2015, 5:7608.
This study demonstrates that a combination of two FDA-approved drugstargeting excitatory and inhibitory systems alleviate the cognitive deficitsin AD mouse models.
103. Bennys K, Haddad R, Gres CS, Schmitt P, Touchon J: Cognitiveevent-related potentials used as biomarkers in Pleodial-Istudy: first evidence of a neurophysiological effect of PXT864in mild Alzheimer’s disease patients. Alzheimer’s Dement JAlzheimer’s Assoc 2016, 12:P1061.
104. Pompeiano M, Palacios JM, Mengod G: Distribution of theserotonin 5-HT2 receptor family mRNAs: comparison between5-HT2A and 5-HT2C receptors. Brain Res Mol Brain Res 1994,23:163-178.
105. Glikmann-Johnston Y, Saling MM, Reutens DC, Stout JC:Hippocampal 5-HT1A receptor and spatial learning andmemory. Front Pharmacol 2015, 6:289.
www.sciencedirect.com

Role of GPCRs in neurodegenerative diseases Huang, Todd and Thathiah 109
106. Elliott MS, Ballard CG, Kalaria RN, Perry R, Hortobagyi T,Francis PT: Increased binding to 5-HT1A and 5-HT2A receptorsis associated with large vessel infarction and relativepreservation of cognition. Brain 2009, 132:1858-1865.
107. Roman GC, Wilkinson DG, Doody RS, Black SE, Salloway SP,Schindler RJ: Donepezil in vascular dementia: combinedanalysis of two large-scale clinical trials. Dement Geriatr CognDisord 2005, 20:338-344.
108. Baskys A, Hou AC: Vascular dementia: pharmacologicaltreatment approaches and perspectives. Clin Interv Aging 2007,2:327-335.
109. Sveinbjornsdottir S: The clinical symptoms of Parkinson’sdisease. J Neurochem 2016, 139(Suppl. 1):318-324.
110. Aarsland D, Bronnick K, Williams-Gray C, Weintraub D, Marder K,Kulisevsky J, Burn D, Barone P, Pagonabarraga J, Allcock L et al.:Mild cognitive impairment in Parkinson disease: a multicenterpooled analysis. Neurology 2010, 75:1062-1069.
111.�
Wood K-L, Myall DJ, Livingston L, Melzer TR, Pitcher TL,MacAskill MR, Geurtsen JG, Anderson TJ, Dalrymple-Alford JC:Different PD-MCI criteria and risk of dementia in Parkinson’sdisease: 4-year longitudinal study. npj Parkinson’s Dis 2016.
This study presents the criteria from a longitudinal study to predict the riskto develop PDD from PD-MCI and requires at least two impairmentswithin a single cognitive domain.
112. Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG: TheSydney multicenter study of Parkinson’s disease: theinevitability of dementia at 20 years. Mov Disord 2008, 23:837-844.
113. Mak E, Bergsland N, Dwyer MG, Zivadinov R, Kandiah N:Subcortical atrophy is associated with cognitive impairment inmild Parkinson disease: a combined investigation ofvolumetric changes, cortical thickness, and vertex-basedshape analysis. AJNR Am J Neuroradiol 2014, 35:2257-2264.
114.�
Hanganu A, Bedetti C, Degroot C, Mejia-Constain B,Lafontaine AL, Soland V, Chouinard S, Bruneau MA, Mellah S,Belleville S et al.: Mild cognitive impairment is linked with fasterrate of cortical thinning in patients with Parkinson’s diseaselongitudinally. Brain 2014, 137:1120-1129.
This study reveals the correlation between the reduced volume in thenucleus accumbens and amydala in PD with mild cognitive impairment(MCI).
115. Beaulieu JM, Gainetdinov RR: The physiology, signaling, andpharmacology of dopamine receptors. Pharmacol Rev 2011,63:182-217.
116. Hisahara S, Shimohama S: Dopamine receptors andParkinson’s disease. Int J Med Chem 2011, 2011:403039.
117. Pezze MA, Dalley JW, Robbins TW: Differential roles ofdopamine D1 and D2 receptors in the nucleus accumbens inattentional performance on the five-choice serial reactiontime task. Neuropsychopharmacology 2007, 32:273-283.
118.�
Ingallinesi M, Le Bouil L, Biguet NF, Thi AD, Mannoury la Cour C,Millan MJ, Ravassard P, Mallet J, Meloni R: Local inactivation ofGpr88 in the nucleus accumbens attenuates behavioraldeficits elicited by the neonatal administration ofphencyclidine in rats. Mol Psychiatry 2015, 20:951-958.
This study demonstrates the specific role of the dopamine D2 receptor inthe rat nucleus accumbens by reducing receptor expression usingmicroRNAs.
119. Alegret M, Junque C, Pueyo R, Valldeoriola F, Vendrell P, Tolosa E,Mercader JM: MRI atrophy parameters related to cognitive andmotor impairment in Parkinson’s disease. Neurologia 2001,16:63-69.
120. Chase TN, Mouradian MM, Engber TM: Motor responsecomplications and the function of striatal efferent systems.Neurology 1993, 43:S23-S27.
121. Bonuccelli U, Del Dotto P, Rascol O: Role of dopamine receptoragonists in the treatment of early Parkinson’s disease.Parkinsonism Relat Disord 2009, 15(Suppl. 4):S44-S53.
122. Boroojerdi B, Wolff HM, Braun M, Scheller DK: Rotigotinetransdermal patch for the treatment of Parkinson’s disease
www.sciencedirect.com
and restless legs syndrome. Drugs Today (Barc) 2010, 46:483-505.
123. Brooks DJ: Dopamine agonists: their role in the treatment ofParkinson’s disease. J Neurol Neurosurg Psychiatry 2000,68:685-689.
124.�
Mori A, LeWitt P, Jenner P: The story of istradefylline—the firstapproved A2A antagonist for the treatment of Parkinson’sdisease. In The Adenosinergic System: A Non-DopaminergicTarget in Parkinson’s Disease. Edited by Morelli M, Simola N,Wardas J. Springer International Publishing; 2015:273-289.
This work summarizes the development of istradefylline for the treatmentof Parkinson’s disease.
125. Uchida S, Soshiroda K, Okita E, Kawai-Uchida M, Mori A,Jenner P, Kanda T: The adenosine A2A receptor antagonist,istradefylline enhances anti-parkinsonian activity induced bycombined treatment with low doses of L-DOPA and dopamineagonists in MPTP-treated common marmosets. Eur JPharmacol 2015, 766:25-30.
126. Lester DB, Rogers TD, Blaha CD: Acetylcholine-dopamineinteractions in the pathophysiology and treatment of CNSdisorders. CNS Neurosci Ther 2010, 16:137-162.
127. Fink-Jensen A, Schmidt LS, Dencker D, Schulein C, Wess J,Wortwein G, Woldbye DP: Antipsychotic-induced catalepsy isattenuated in mice lacking the M4 muscarinic acetylcholinereceptor. Eur J Pharmacol 2011, 656:39-44.
128. Svenningsson P, Westman E, Ballard C, Aarsland D: Cognitiveimpairment in patients with Parkinson’s disease: diagnosis,biomarkers, and treatment. Lancet Neurol 2012, 11:697-707.
129. Marazziti D, Golini E, Mandillo S, Magrelli A, Witke W, Matteoni R,Tocchini-Valentini GP: Altered dopamine signaling and MPTPresistance in mice lacking the Parkinson’s disease-associated GPR37/parkin-associated endothelin-likereceptor. Proc Natl Acad Sci U S A 2004, 101:10189-10194.
130. Wu CS, Chen H, Sun H, Zhu J, Jew CP, Wager-Miller J, Straiker A,Spencer C, Bradshaw H, Mackie K et al.: GPR55, a G-proteincoupled receptor for lysophosphatidylinositol, plays a role inmotor coordination. PLoS One 2013, 8:e60314.
131. Gandia J, Morato X, Stagljar I, Fernandez-Duenas V, Ciruela F:Adenosine A2A receptor-mediated control of pilocarpine-induced tremulous jaw movements is Parkinson’s disease-associated GPR37 receptor-dependent. Behav Brain Res 2015,288:103-106.
132. Beal MF, Ferrante RJ, Swartz KJ, Kowall NW: Chronic quinolinicacid lesions in rats closely resemble Huntington’s disease.J Neurosci 1991, 11:1649-1659.
133. Trepanier C, Lei G, Xie YF, MacDonald JF: Group II metabotropicglutamate receptors modify N-methyl-D-aspartate receptorsvia Src kinase. Sci Rep 2013, 3:926.
134. Bruno V, Ksiazek I, Battaglia G, Lukic S, Leonhardt T, Sauer D,Gasparini F, Kuhn R, Nicoletti F, Flor PJ: Selective blockade ofmetabotropic glutamate receptor subtype 5 isneuroprotective. Neuropharmacology 2000, 39:2223-2230.
135. Schiefer J, Sprunken A, Puls C, Luesse HG, Milkereit A, Milkereit E,Johann V, Kosinski CM: The metabotropic glutamate receptor5 antagonist MPEP and the mGluR2 agonist LY379268 modifydisease progression in a transgenic mouse model ofHuntington’s disease. Brain Res 2004, 1019:246-254.
136.�
Yao Y, Cui X, Al-Ramahi I, Sun X, Li B, Hou J, Difiglia M, Palacino J,Wu ZY, Ma L et al.: A striatal-enriched intronic GPCR modulateshuntingtin levels and toxicity. Elife 2015, 4.
This study demonstrates that GPR52 stabilizes mutant huntingtin bypreventing degradation. Reduced levels of GPR52 suppress HD pheno-types in both patient iPS-derived neurons and Drosophila HD models.
137. Chen JY, Wang EA, Cepeda C, Levine MS: Dopamine imbalancein Huntington’s disease: a mechanism for the lack ofbehavioral flexibility. Front Neurosci 2013, 7:114.
138. Charvin D, Vanhoutte P, Pages C, Borrelli E, Caboche J:Unraveling a role for dopamine in Huntington’s disease: thedual role of reactive oxygen species and D2 receptorstimulation. Proc Natl Acad Sci U S A 2005, 102:12218-12223.
Current Opinion in Pharmacology 2017, 32:96–110

110 Neurosciences
139. Charvin D, Roze E, Perrin V, Deyts C, Betuing S, Pages C,Regulier E, Luthi-Carter R, Brouillet E, Deglon N et al.: Haloperidolprotects striatal neurons from dysfunction induced bymutated huntingtin in vivo. Neurobiol Dis 2008, 29:22-29.
140. Tang TS, Chen X, Liu J, Bezprozvanny I: Dopaminergic signalingand striatal neurodegeneration in Huntington’s disease.J Neurosci 2007, 27:7899-7910.
141. Katona I, Freund TF: Endocannabinoid signaling as a synapticcircuit breaker in neurological disease. Nat Med 2008,14:923-930.
142. Blazquez C, Chiarlone A, Sagredo O, Aguado T, Pazos MR,Resel E, Palazuelos J, Julien B, Salazar M, Borner C et al.: Loss ofstriatal type 1 cannabinoid receptors is a key pathogenicfactor in Huntington’s disease. Brain 2011, 134:119-136.
143. Scotter EL, Goodfellow CE, Graham ES, Dragunow M, Glass M:Neuroprotective potential of CB1 receptor agonists in an invitro model of Huntington’s disease. Br J Pharmacol 2010,160:747-761.
144. Gomes CV, Kaster MP, Tome AR, Agostinho PM, Cunha RA:Adenosine receptors and brain diseases: neuroprotection andneurodegeneration. Biochim Biophys Acta 1808, 2011:1380-1399.
145. Tebano MT, Pintor A, Frank C, Domenici MR, Martire A, Pepponi R,Potenza RL, Grieco R, Popoli P: Adenosine A2A receptorblockade differentially influences excitotoxic mechanisms atpre- and postsynaptic sites in the rat striatum. J Neurosci Res2004, 77:100-107.
146. Chou SY, Lee YC, Chen HM, Chiang MC, Lai HL, Chang HH,Wu YC, Sun CN, Chien CL, Lin YS et al.: CGS21680 attenuatessymptoms of Huntington’s disease in a transgenic mousemodel. J Neurochem 2005, 93:310-320.
147. Lee EH, Seo SR: Neuroprotective roles of pituitary adenylatecyclase-activating polypeptide in neurodegenerativediseases. BMB Rep 2014, 47:369-375.
148. White CM, Ji S, Cai H, Maudsley S, Martin B: Therapeuticpotential of vasoactive intestinal peptide and its receptors inneurological disorders. CNS Neurol Disord Drug Targets 2010,9:661-666.
149. Price DL, Bonhaus DW, McFarland K: Pimavanserin, a 5-HT2Areceptor inverse agonist, reverses psychosis-like behaviors ina rodent model of Alzheimer’s disease. Behav Pharmacol 2012,23:426-433.
150. Strange PG: Antipsychotic drugs: importance of dopaminereceptors for mechanisms of therapeutic actions and sideeffects. Pharmacol Rev 2001, 53:119-133.
151. Mievis S, Blum D, Ledent C: A2A receptor knockout worsenssurvival and motor behaviour in a transgenic mouse model ofHuntington’s disease. Neurobiol Dis 2011, 41:570-576.
152. Popoli P, Blum D, Martire A, Ledent C, Ceruti S, Abbracchio MP:Functions, dysfunctions and possible therapeutic relevance ofadenosine A2A receptors in Huntington’s disease. ProgNeurobiol 2007, 81:331-348.
153. Bannon NM, Zhang P, Ilin V, Chistiakova M, Volgushev M:Modulation of synaptic transmission by adenosine in layer 2/3of the rat visual cortex in vitro. Neuroscience 2014, 260:171-184.
154. Pancani T, Foster DJ, Moehle MS, Bichell TJ, Bradley E,Bridges TM, Klar R, Poslusney M, Rook JM, Daniels JS et al.:Allosteric activation of M4 muscarinic receptors improvebehavioral and physiological alterations in early symptomaticYAC128 mice. Proc Natl Acad Sci U S A 2015, 112:14078-14083.
Current Opinion in Pharmacology 2017, 32:96–110
155.�
Ribeiro FM, Devries RA, Hamilton A, Guimaraes IM, Cregan SP,Pires RG, Ferguson SS: Metabotropic glutamate receptor5 knockout promotes motor and biochemical alterations in amouse model of Huntington’s disease. Hum Mol Genet 2014,23:2030-2042.
This study demonstrates that the metabotropic glutamate receptor 5 isinvolved in locomotor activity in HD mice by genetic and pharmacologicalapproaches.
156. Zhu D, Li C, Swanson AM, Villalba RM, Guo J, Zhang Z, Matheny S,Murakami T, Stephenson JR, Daniel S et al.: BAI1 regulatesspatial learning and synaptic plasticity in the hippocampus. JClin Invest 2015, 125:1497-1508.
157. Marino MJ, Williams DL Jr, O’Brien JA, Valenti O, McDonald TP,Clements MK, Wang R, DiLella AG, Hess JF, Kinney GG et al.:Allosteric modulation of group III metabotropic glutamatereceptor 4: a potential approach to Parkinson’s diseasetreatment. Proc Natl Acad Sci U S A 2003, 100:13668-13673.
158. Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z,Luo Q, Rodriguez AL, Marlo JE, de Paulis T, Thompson AD et al.:Discovery, characterization, and antiparkinsonian effect ofnovel positive allosteric modulators of metabotropicglutamate receptor 4. Mol Pharmacol 2008, 74:1345-1358.
159. Tang SS, Ji MJ, Chen L, Hu M, Long Y, Li YQ, Miao MX, Li JC, Li N,Ji H et al.: Protective effect of pranlukast on Abeta(1)(�)(4)(2)-induced cognitive deficits associated with downregulation ofcysteinyl leukotriene receptor 1. Int J Neuropsychopharmacol2014, 17:581-592.
160. Teng L, Zhao J, Wang F, Ma L, Pei G: A GPCR/secretasecomplex regulates beta- and gamma-secretase specificity forAbeta production and contributes to AD pathogenesis.Cell Res 2010, 20:138-153.
161. Heaney CF, Kinney JW: Role of GABA(B) receptors in learningand memory and neurological disorders. Neurosci BiobehavRev 2016, 63:1-28.
162. Faivre E, Gault VA, Thorens B, Holscher C: Glucose-dependentinsulinotropic polypeptide receptor knockout mice areimpaired in learning, synaptic plasticity, and neurogenesis.J Neurophysiol 2011, 105:1574-1580.
163. Kubota T, Matsumoto H, Kirino Y: Ameliorative effect ofmembrane-associated estrogen receptor G protein coupledreceptor 30 activation on object recognition memory in mousemodels of Alzheimer’s disease. J Pharmacol Sci 2016,131:219-222.
164. Yi T, Weng J, Siwko S, Luo J, Li D, Liu M: LGR4/GPR48inactivation leads to aniridia-genitourinary anomalies-mental retardation syndrome defects. J Biol Chem 2014,289:8767-8780.
165. Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H,Fisher A, LaFerla FM: M1 receptors play a central role inmodulating AD-like pathology in transgenic mice.Neuron 2006, 49:671-682.
166. Poulin B, Butcher A, McWilliams P, Bourgognon JM, Pawlak R,Kong KC, Bottrill A, Mistry S, Wess J, Rosethorne EM et al.: TheM3-muscarinic receptor regulates learning and memory in areceptor phosphorylation/arrestin-dependent manner.Proc Natl Acad Sci U S A 2010, 107:9440-9445.
167. Asle-Rousta M, Kolahdooz Z, Oryan S, Ahmadiani A, Dargahi L:FTY720 (fingolimod) attenuates beta-amyloid peptide(Abeta42)-induced impairment of spatial learning and memoryin rats. J Mol Neurosci 2013, 50:524-532.
www.sciencedirect.com







