unmarked gene integration into the chromosome of mycobacterium smegmatisvia precise replacement of...
TRANSCRIPT

PLASMID 37, 129–140 (1997)ARTICLE NO. PL971286
Unmarked Gene Integration into the Chromosome of Mycobacteriumsmegmatis via Precise Replacement of the pyrF Gene
Nancianne Knipfer, Anjali Seth, and Thomas E. Shrader1
Department of Biochemistry, Albert Einstein College of Medicine,1300 Morris Park Avenue, Bronx, New York 10461
Received October 14, 1996; revised February 7, 1997
After integration into the bacterial chromosome an exogenous gene may be stably expressedwithout continued selection for the recombinant locus. However, chromosomal integrationevents occur infrequently, requiring the concomitant integration of a drug resistance markerin order to identify colonies of recombinant cells. The generation of a drug-resistant recombi-nant strain can both reduce the in vivo applicability of the strain and preclude the use ofrecombinant vectors which use the same drug resistance marker. We have constructed aplasmid, pINT-D, which allows recombination of exogenous genes onto the Mycobacteriumsmegmatis chromosome. The exogenous gene completely replaces the pyrF gene and theresultant strain lacks any exogenous drug resistance marker. The methodologies describedherein are general and applicable even to those bacteria for which extrachromosomal plasmidsare not available. Using pINT-D we integrated the lacZ gene into the M. smegmatis chromo-some via a precise exchange of lacZ and pyrF. The resultant strain was used to demonstratethat the expression of genes integrated at the pyrF locus is repressed twofold by inclusionof uracil in the growth medium. In addition, we used pINT-D to construct an M. smegmatisstrain with a precise deletion of its pyrF locus. This strain, TSm-627, grows normally in richmedium but does not grow in medium lacking uracil. TSm-627 cells allow the pyrF gene tobe used as a selectable marker for growth on medium lacking uracil. In TSm-627 cells, thepyrF gene is also useful as a counterselectable marker on complete medium containing 5*-fluoroorotic acid and uracil. Two pyrF-containing plasmids, designed to exploit the newDpyrF strain, have been constructed and their possible applications to problems in mycobac-teriology are discussed. q 1997 Academic Press
The acid fast bacterium Mycobacterium smegmatis is now the most genetically tracta-ble species among the Gram-positive bacteriasmegmatis has become an attractive model or-of the order Actinomycetales. The order Acti-ganism in which to study the basic physiologynomycetales contains several important patho-of the mycobacteria. Unlike its pathogenic rel-gens, such as species of Actinomyces and No-atives, M. tuberculosis, M. leprae, and M.cardia, as well as commercially important or-avium; M. smegmatis cells grow rapidly andganisms such as the Corynebacteria. Thecells divide every 2–3 h under optimal condi-comparatively close evolutionary relationshiptions (Jacobs et al., 1991). This rapid genera-of M. smegmatis with the other Actinomycet-tion time provides such an enormous advan-ales adds to the overall importance of resultstage that questions concerning the physiologyobtained in M. smegmatis, as the Actinomy-of the pathogenic mycobacteria are often an-cetales are evolutionarily very distant fromswered first in M. smegmatis followed by con-other well-studied model organisms such asfirmation in the slow growing pathogenicEscherichia coli and the Gram-positive Bacil-strains (Banerjee et al., 1993). In addition tolus subtilis. These factors conspire to makeits role as a model mycobacterial cell, M.future genetic analyses of M. smegmatis rele-vant to researchers specifically interested in a1 To whom correspondence should be addressed. E-
mail: [email protected]. Fax: 718-430-8565. wide range of organisms.
129 0147-619X/97 $25.00Copyright q 1997 by Academic PressAll rights of reproduction in any form reserved.
AID Plasmid 1286 / 660c$$$121 04-03-97 10:16:53 plasa AP: Plasmid

130 KNIPFER, SETH, AND SHRADER
Only a single autonomously replicatinggenetic element has been described that isgenerally useful in the M. smegmatis labora-tory strain mc2-155 (Jacobs et al., 1991).This oriM element was originally isolatedfrom M. fortuitum and is included in allcommonly used mycobacterial vectors(Ranes et al., 1990). The applicability of asingle oriM element is in contrast to E. coli,where the same cell may stably maintainplasmids based on both the ColE1 and p15Aorigins of replication (Chang and Cohen,1978). This shortcoming makes more so- FIG. 1. Scheme for replacing the M. smegmatis pyrFphisticated genetic analyses, requiring two gene with a ‘‘new’’ gene. This procedure is carried outindependent recombinant genes, less tracta- in two steps. Step I: identification of cells containing an
insertion of the nonreplicating plasmid pINT-D (a) intoble and places increased importance on thethe chromosome via single recombination between thedevelopment of integrating vectors for strainpyrF-flanking sequences in the chromosome and thosemc2-155. Current integrating vectors for the contained on pINT-D. Step II: resolution of the resulting
mycobacteria, which integrate via an attP chromosomal tandem repeat (b) to either the original wild-phage attachment site, result in a drug-resis- type locus (not shown) or the desired recombinant locus
(new, c). Also isolated by this method are point mutationstant bacterium (Stover et al., 1991). We de-in the pyrF gene that render the unresolved tandem dupli-scribe here a plasmid-based system whichcation (b) resistant to 5*-FOA but KmR (d).allows integration of exogenous genes into
the M. smegmatis chromosome without re-sulting in a drug-resistant strain. In addition,the integrated genes may be expressed at are generated by first isolating M. smegmatis
cells containing an insertion of our nonrepli-two levels from the chromosome. The toolspresented are also applicable as an alterna- cating plasmid (pINT-D, described below)
near the pyrF gene. In M. smegmatis, thesetive to current methodologies to delete insuccession multiple unlinked genes from the insertions occur overwhelmingly via a single
homologous recombination event between theM. smegmatis chromosome (Pelicic et al.,1996a,b; Sander et al., 1995). Finally, an M. chromosome and pyrF-flanking sequences
contained on pINT-D (Fig. 1a) (Kalpana etsmegmatis strain containing a precise dele-tion of its chromosomal pyrF gene but lack- al., 1991; Husson et al., 1990). Cells con-
taining plasmid insertions may be selecteding any exogenous chromosomal drugmarker is described. This strain allows use from the population of wild type cells via the
aminoglycoside phosphotransferase markerof two new vectors whose continued resi-dence in these DpyrF cells may be selected (kanR) on the vector. The resulting cells con-
tain tandem repeats of the pyrF chromosomaleither for or against.loci, separated by vector-derived sequences(Fig. 1b). These tandem repeats are generallyRESULTS AND DISCUSSIONunstable as all the intervening vector se-
General Approachquences are removed by a second homologousrecombination event between the tandem re-Our procedure for constructing insertions
of a recombinant gene (new in Fig. 1) into the peats. This second recombination results ineither recreation of the wild-type locus (notM. smegmatis chromosome without the con-
comitant creation of a drug-resistant strain is shown) or the desired unmarked insertionevent (Fig. 1c). Cells that have potentially un-carried out in two steps (Fig. 1). In this
scheme, unmarked chromosomal insertions dergone a second recombination to yield the
AID Plasmid 1286 / 660c$$$122 04-03-97 10:16:53 plasa AP: Plasmid

131GENE INTEGRATION VIA pyrF REPLACEMENT
desired replacement of the pyrF gene are iden- chromosomal region containing the pyrFgene from M. smegmatis strain mc2-155 (Fig.tified by selecting against cells containing an
active pyrF gene (Husson et al., 1990). Also 2) and determined its complete nucleotide se-quence (Fig. 3, GenBank Accession No.isolated by this method were cells containing
point mutations in the active copy of the pyrF U91572). As expected, the M. smegmatispyrF gene is more closely related to the pyrFgene (Fig. 1d).gene of Mycobacterium bovis strain BCG(Bacillus Calmette Guerin) than it is to anyThe pyrF Geneof the other pyrF genes sequenced to date
To date, three genes have been described (Ç70% identity) (Aldovini et al., 1993). Inthat may be selected against in mycobacte- addition, the pyrF gene is the final gene inrial cells. The first gene identified as count- an operon that also contains the pyrAB gene.erselectable in the mycobacteria was the The pyrAB gene encodes the enzyme carbam-pyrF gene encoding the enzyme orotidine oylphosphate synthetase, an enzyme essential5*-monophosphate decarboxylase (Husson for the biosynthesis of both pyrimidines andet al., 1990). This enzyme converts 5*-flu- arginine. The pyrAB and pyrF genes haveoroorotic acid (5*-FOA)2 into a toxic nucle- overlapping initiation and termination co-otide analog that irreversible inactivates the dons; thus, any independent promoter for theessential enzyme thymidylate synthetase. pyrF gene must lie entirely within the pyrABTherefore, wild-type cells are killed by 5*- coding region (Fig. 3). Below we describe anFOA (Boeke et al., 1984). Conversely, M. M. smegmatis strain in which the lacZ gene,smegmatis pyrF mutants are not susceptible encoding b-galactosidase, completely re-to 5*-FOA and grow on medium containing places the pyrF gene. In this strain (TSm-the drug. The pyrF mutants are uracil axo- 628), expression of b-galactosidase is regu-trophs; hence this pyrimidine must be pro- lated by uracil levels in the growth medium.vided in the medium. Subsequently, the This result suggests that the pyrF gene, andrpsL/ and sacB genes, that render cells sus- probably an entire operon of pyrimidineceptible to streptomycin and sucrose, re- biosynthetic genes, is regulated by its endspectively, have been incorporated into my- product.cobacterial vectors (Pelicic et al., 1996a,b;Sander et al., 1995). In each case, these vec-
Design of the pINT-D Vectortors have been utilized to improve currentmethodologies for the deletion of mycobac- The pINT-D vector that allows unmarkedterial genes. For our purposes, the pyrF insertion into the M. smegmatis chromosomegene is the most useful as this gene is con- is schematized in Fig. 2. pINT-D is a deriva-tained within the wild-type chromosome of tive of E. coli cloning vector pSP72 (Promega)M. smegmatis and may be utilized as both that lacks oriM sequences and therefore doesa selectable and counterselectable marker in not replicate in mycobacteria. pINT-D con-a DpyrF genetic background. tains a polylinker cloning region flanked by
The pyrF gene from M. smegmatis was 1.37 kb of chromosomal DNA from upstreamisolated several years ago and the utility of of the pyrF gene and 0.45 kb from down-5*-FOA selection established (Husson et stream of the pyrF gene. The plasmid alsoal., 1990). However, no sequence data were contains all of the translation signals for pyrFreported precluding genetic manipulations but no coding sequence. In other words, pINT-at the nucleotide level. We recloned the D contains a copy of the pyrF chromosomal
region with the pyrF gene cleanly excised andreplaced by a polylinker. The vector also con-2 Abbreviations used: 5*FOA, 5*-fluoroorotic acid; LB-tains unique recognition sites for the BglII andkan, Luria medium containing 15 mg/ml kanamycin;
ONPG, 2-nitrophenyl-b-D-galactopyranoside. XhoI restriction endonucleases. These sites
AID Plasmid 1286 / 660c$$$122 04-03-97 10:16:53 plasa AP: Plasmid

132 KNIPFER, SETH, AND SHRADER
FIG. 2. Schematic of pINT-D and the corresponding region of the M. smegmatis chromosome containedin plasmid pPyrF-1. (Upper) Restriction map of the pyrF region of the M. smegmatis chromosome containedin the insert of plasmid pPyrF-1. Restriction sites referred to in the text are indicated, as are the initiationand stop codon (*) of the pyrF gene. The region of the insert of pPyrF-1 that was sequenced on bothstrands (see Fig. 3) is indicated. (Lower) Schematic of plasmid pINT-D indicating the chromosomalfragments that are used to target the plasmid for insertion, via homologous recombination, into the M.smegmatis chromosome. The polylinker region of the plasmid is shown at nucleotide resolution. Restrictionsites shown in the polylinker are unique. These sites allow cloning exogenous genes into pINT-D forsubsequent recombination onto the M. smegmatis chromosome. Pairs of restriction sites in parenthesesindicate junctions formed during plasmid construction; neither site remains in pINT-D. The PstI sitedestroyed via a silent mutation is indicated as PstI* (see Experimental Procedures).
precede and follow the upstream and down- plasmid pMC1871 (Pharmacia) and clonedinto the BamHI site of pINT-D. This step cre-stream M. smegmatis-derived fragments, re-
spectively. Finally, a silent mutation was in- ated an in-frame fusion of the lacZ gene withthe initiation codon of the pyrF gene. Subse-troduced into the extreme 5*-region of the
pyrAB gene that destroys a chromosomal PstI quently, the kanR marker was isolated as aBamHI fragment from vector pUC4K and in-site without altering any amino acids in the
pyrAB gene product. This mutation leaves the troduced into the unique BglII site of this in-termediate. The kanR marker allows for thePstI site of the polylinker as a unique cloning
site in the pINT-D vector. A silent mutation initial selection of single recombination eventsbetween the M. smegmatis chromosome andwas introduced because carbamoylphosphate
synthetase is essential for both arginine and the pINT-D vector (see Fig. 1a), but is re-moved in the second recombination (Fig. 1c).pyrimidine biosynthesis.
To construct the pINT-Z vector, a lacZ gene To expedite other applications employing thepINT-D vector, introduction of the kanRblock was excised as a BamHI fragment from
AID Plasmid 1286 / 660c$$$122 04-03-97 10:16:53 plasa AP: Plasmid

133GENE INTEGRATION VIA pyrF REPLACEMENT
FIG. 3. Nucleotide sequence of the pyrF gene of M. smegmatis. The complete nucleotide sequence of the pyrFgene and inferred amino acid sequence of orotidine 5*-monophosphate decarboxylase are shown. The extreme 3*-end of the pyrAB gene and the overlapping termination/initiation codons between the two genes are also shown.Restriction sites mentioned in the text are underlined and identified in bold. (GenBank Accession No. U91572).
marker was made as flexible as possible. For Recombination of Unmarked Genes onto theM. smegmatis Chromosomeexample, the kanR gene block, derived from
plasmid pUC4K, can be excised with eitherWe used the pINT-D plasmid to constructBamHI or SalI and inserted into the unique
unmarked insertions of a polylinker and theBglII or XhoI sites of pINT-D, respectively.lacZ gene into the M. smegmatis chromosomeThe occurrence of internal restriction sitesto generate strains TSm-627 and TSm-628,within the gene to be inserted will dictate bothrespectively. The initial steps in the construc-the optimal site for insertion of the kanR
tion of M. smegmatis strains TSm-627 andmarker and the order in which insertions areTSm-628 were the integration of vectorsmade during plasmid construction. AlthoughpINT-D and pINT-Z into the M. smegmatispINT-D is directly applicable only to M.chromosome, respectively. This integrationsmegmatis, analogous plasmid systems couldwas carried out by electroporation of strainbe constructed for other less well character-
ized bacteria. mc2-155 with the desired nonreplicating plas-
AID Plasmid 1286 / 660c$$$122 04-03-97 10:16:53 plasa AP: Plasmid

134 KNIPFER, SETH, AND SHRADER
FIG. 4. Characterization of M. smegmatis DpyrF and DpyrF::lacZ strains. Southern hybridization wasused to characterize the clean deletion of the pyrF locus of strain TSm-627 and the unmarked insertion ofthe lacZ gene into the pyrF locus of strain TSm-628. (A) Chromosomal DNA of strains mc2-155 (wildtype), TSm-627.1 through TSm-627.5, and TSm-628 digested with BamHI and probed with the M. smeg-matis-derived BamHI fragment of plasmid pPyrF-1 (see Fig. 2). (B) Identical chromosomal DNA samplesas above probed with the entirety of the pyrF gene, isolated from plasmid pPyrF-21 as a ClaI–XbaIfragment. (C) Chromosomal DNA of strains mc2-155 and TSm-628 digested with BamHI and probed withthe lacZ gene. The lacZ gene is derived as a BamHI fragment from plasmid pMC1871.
mid followed by selection for cells containing 627.1 through TSm-627.5 are all strains inwhich the pyrF gene was completely deletedplasmid integrations by plating on solid Luria
medium containing 15 mg/ml kanamycin (LB- and replaced by the polylinker [mc2-155DpyrF]. In the last strain, TSm-628, the pyrFkan). Colonies of transformed cells were col-
ony purified by restreaking for single colonies gene was completely deleted and replaced bylacZ [mc2-155 DpyrF::lacZ]. Figure 4Aon LB-kan plates. From these plates, colonies
of kanamycin-resistant (KmR) cells were shows that in each of the final strains, theBamHI fragment encoding 1.72 kb at the 3*-patched on solid medium containing 1.0 mg/
ml 5*-FOA and the plates incubated for 5–7 end of the pyrAB gene through the first 0.56kb at the 5*-end of the pyrF gene has been lostdays at 377C. Single colonies of cells that grew
on 5*-FOA were subsequently tested for loss and replaced by the shorter BamHI fragmentcreated by integration of the polylinker re-of the kanR marker by streaking on LB-kan.
Cells that lost the kanR marker in response to gions of plasmids pINT-D and pINT-Z. Fig-ure 4B shows that in each of the TSm-627selection against the pyrF marker were candi-
dates for having undergone a second homolo- and TSm-628 strains, no hybridization signalfor the pyrF gene was observed, suggestinggous recombination event (Fig. 1c). This pro-
cedure does not require nonselective growth, complete deletion of all pyrF-derived se-quences during the second recombination stepon medium lacking kanamycin, prior to the
5*-FOA selection step. In different experi- (see Fig. 1; in addition, all strains are KmS).Finally, Fig. 4C shows that a strong hybridiza-ments 10–50% of the cells that survived 5*-
FOA selection became susceptible to kanamy- tion signal is seen for the lacZ gene for strainTSm-628 but not for wild-type mc2-155 cells.cin (KmS). Cells that survived 5*-FOA selec-
tion without concomitant reversion to KmS
probably arose via point mutations in their Expression at the pyrF LocuspyrF gene (Fig. 1d). Cells from 5*-FOAR, KmS
colonies were used to prepare chromosomal Very little is known about the transcrip-tional control of biosynthetic genes in the my-DNA for confirmation of the desired chro-
mosomal locus via Southern hybridization cobacteria. In other bacterial species, the pyrFgene is regulated in response to the level of(Fig. 4).
The first five strains characterized, TSm- uracil in the culture (Turnbough et al., 1987;
AID Plasmid 1286 / 660c$$$122 04-03-97 10:16:53 plasa AP: Plasmid
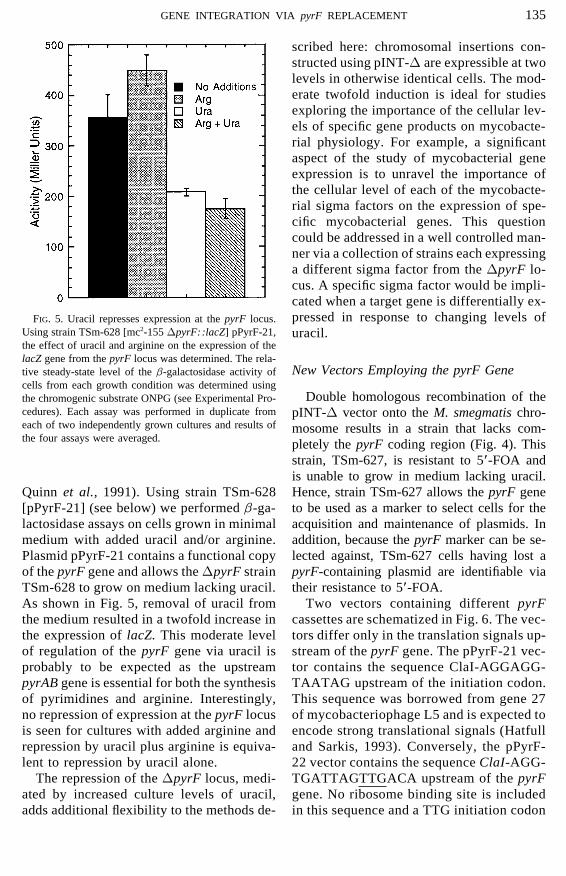
135GENE INTEGRATION VIA pyrF REPLACEMENT
scribed here: chromosomal insertions con-structed using pINT-D are expressible at twolevels in otherwise identical cells. The mod-erate twofold induction is ideal for studiesexploring the importance of the cellular lev-els of specific gene products on mycobacte-rial physiology. For example, a significantaspect of the study of mycobacterial geneexpression is to unravel the importance ofthe cellular level of each of the mycobacte-rial sigma factors on the expression of spe-cific mycobacterial genes. This questioncould be addressed in a well controlled man-ner via a collection of strains each expressinga different sigma factor from the DpyrF lo-cus. A specific sigma factor would be impli-cated when a target gene is differentially ex-pressed in response to changing levels ofFIG. 5. Uracil represses expression at the pyrF locus.
Using strain TSm-628 [mc2-155 DpyrF::lacZ] pPyrF-21, uracil.the effect of uracil and arginine on the expression of thelacZ gene from the pyrF locus was determined. The rela-
New Vectors Employing the pyrF Genetive steady-state level of the b-galactosidase activity ofcells from each growth condition was determined using
Double homologous recombination of thethe chromogenic substrate ONPG (see Experimental Pro-cedures). Each assay was performed in duplicate from pINT-D vector onto the M. smegmatis chro-each of two independently grown cultures and results of mosome results in a strain that lacks com-the four assays were averaged. pletely the pyrF coding region (Fig. 4). This
strain, TSm-627, is resistant to 5*-FOA andis unable to grow in medium lacking uracil.Hence, strain TSm-627 allows the pyrF geneQuinn et al., 1991). Using strain TSm-628
[pPyrF-21] (see below) we performed b-ga- to be used as a marker to select cells for theacquisition and maintenance of plasmids. Inlactosidase assays on cells grown in minimal
medium with added uracil and/or arginine. addition, because the pyrF marker can be se-lected against, TSm-627 cells having lost aPlasmid pPyrF-21 contains a functional copy
of the pyrF gene and allows the DpyrF strain pyrF-containing plasmid are identifiable viatheir resistance to 5*-FOA.TSm-628 to grow on medium lacking uracil.
As shown in Fig. 5, removal of uracil from Two vectors containing different pyrFcassettes are schematized in Fig. 6. The vec-the medium resulted in a twofold increase in
the expression of lacZ. This moderate level tors differ only in the translation signals up-stream of the pyrF gene. The pPyrF-21 vec-of regulation of the pyrF gene via uracil is
probably to be expected as the upstream tor contains the sequence ClaI-AGGAGG-TAATAG upstream of the initiation codon.pyrAB gene is essential for both the synthesis
of pyrimidines and arginine. Interestingly, This sequence was borrowed from gene 27of mycobacteriophage L5 and is expected tono repression of expression at the pyrF locus
is seen for cultures with added arginine and encode strong translational signals (Hatfulland Sarkis, 1993). Conversely, the pPyrF-repression by uracil plus arginine is equiva-
lent to repression by uracil alone. 22 vector contains the sequence ClaI-AGG-TGATTAGTTGACA upstream of the pyrFThe repression of the DpyrF locus, medi-
ated by increased culture levels of uracil, gene. No ribosome binding site is includedin this sequence and a TTG initiation codonadds additional flexibility to the methods de-
AID Plasmid 1286 / 660c$$$123 04-03-97 10:16:53 plasa AP: Plasmid

136 KNIPFER, SETH, AND SHRADER
(underlined) followed by an ACA threonine expression signals, located in the vector-de-rived sequences, and render TSm-627codon (rarely used in M. smegmatis) have
also been included. Neither vector contains [pPyrF-21] cells 5*-FOAR. Terminator clon-ing vectors of this type have been describeda known mycobacterial promoter and it was
originally envisioned that the pPyrF-21 vec- for the eukaryote Saccharomyces cerevisiae(Snyder et al., 1988), but we are unaware oftor would be useful for cloning weak pro-
moters, while pPyrF-22 would allow clon- their use in bacteria. Identification of strongtranscriptional terminators are expected toing of strong promoters.
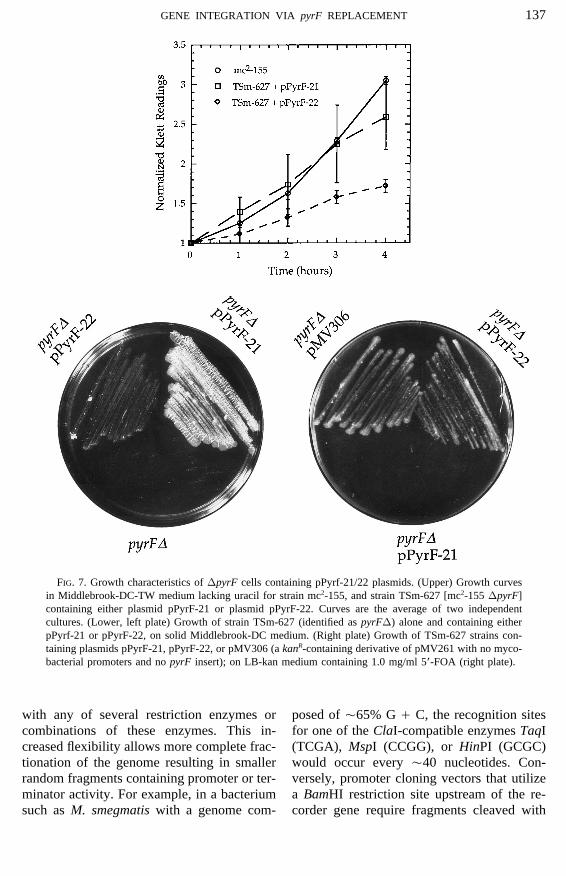
As shown in Fig. 7, TSm-627 cells con- improve the yields of mycobacterial expres-sion plasmids as has been described for E.taining the pPyrF-21 vector are able to grow
in medium lacking uracil and are suscepti- coli expression plasmids (Shatzman andRosenberg, 1987). In addition, attempts toble to 5*-FOA. We conclude that cryptic
promoter sequences encoded by the vector- map the expression signals of complex my-cobacterial operons will benefit from thisderived sequences of this plasmid, coupled
with strong translational signals, produce simple method to identify functional termi-nator elements.sufficient orotidine 5*-monophosphate de-
carboxylase activity to render TSm-627 TSm-627 cells containing the pPyrF-22plasmid are 5*-FOAR (Fig. 6) and grow verycells URA/. These conditions allow this
strain and plasmid combination to be used slowly in minimal medium (Fig. 7). Thisallows pPyrF-22 in conjunction with strainin synthetic lethal screens and other genetic
manipulations that require the identification TSm-627 to be used in the identification ofDNA fragments encoding mycobacterialof cells which have lost a recombinant plas-
mid. In addition, the pPyrF-21 plasmid promoters. This promoter detection systemallows selection for DNA fragments encod-could be useful for the identification of tran-
scriptional terminators active in mycobacte- ing promoter activity rather than requiringscreening for promoter activity. In additionrial cells. Terminators could be identified
through via their ability to isolate the pyrF pPyrF-22 is smaller than existing promotercloning vectors that monitor the expressiongene of plasmid pPyrF-21 from the crypticof the large lacZ gene (Barletta et al., 1992).Therefore, the generation of complex librar-ies of potential promoters is more straight-forward. Finally, the pPyrF-22 vector couldbe used to identify promoters with differen-tial expression patterns. For example, by re-quiring cells to behave as URA/ under cer-tain growth conditions but 5*-FOAR underalternate growth conditions, populations ofrandom DNA sequences can be screened fordifferentially active promoters. In light ofthe importance of the identification of my-cobacterial genes expressed during differentgrowth conditions, such as the onset of bac-terial stationary phase or during oxygen-FIG. 6. Schematic of vectors pPyrF-21/22. The two
vectors pPyrF-21/22 differ only in the translational sig- limiting conditions, a tool for screening en-nals immediately upstream of the pyrF gene. Unique tire genomes for differentially active pro-ClaI and XbaI restriction sites found in both plasmids moters is potentially valuable.are indicated. The origin of replication of each vector
The unique ClaI restriction sites located up-is derived from pAL5000 and therefore is expected tostream of the pyrF genes in the pPyrF-21/22result in Çfive copies of the plasmid per cell (Stover
et al., 1991). vectors allow insertion of fragments generated
AID Plasmid 1286 / 660c$$$123 04-03-97 10:16:53 plasa AP: Plasmid
137GENE INTEGRATION VIA pyrF REPLACEMENT
FIG. 7. Growth characteristics of DpyrF cells containing pPyrf-21/22 plasmids. (Upper) Growth curvesin Middlebrook-DC-TW medium lacking uracil for strain mc2-155, and strain TSm-627 [mc2-155 DpyrF]containing either plasmid pPyrF-21 or plasmid pPyrF-22. Curves are the average of two independentcultures. (Lower, left plate) Growth of strain TSm-627 (identified as pyrFD) alone and containing eitherpPyrf-21 or pPyrF-22, on solid Middlebrook-DC medium. (Right plate) Growth of TSm-627 strains con-taining plasmids pPyrF-21, pPyrF-22, or pMV306 (a kanR-containing derivative of pMV261 with no myco-bacterial promoters and no pyrF insert); on LB-kan medium containing 1.0 mg/ml 5*-FOA (right plate).
with any of several restriction enzymes or posed of Ç65% G / C, the recognition sitesfor one of the ClaI-compatible enzymes TaqIcombinations of these enzymes. This in-
creased flexibility allows more complete frac- (TCGA), MspI (CCGG), or HinPI (GCGC)would occur every Ç40 nucleotides. Con-tionation of the genome resulting in smaller
random fragments containing promoter or ter- versely, promoter cloning vectors that utilizea BamHI restriction site upstream of the re-minator activity. For example, in a bacterium
such as M. smegmatis with a genome com- corder gene require fragments cleaved with
AID Plasmid 1286 / 660c$$$123 04-03-97 10:16:53 plasa AP: Plasmid

138 KNIPFER, SETH, AND SHRADER
the Sau3AI restriction enzyme to be generated ml of 10 mg/ml lysozyme (Sigma) followedby incubation at 377C for 1 h. To this solution,in order to be inserted upstream of the recorder
gene (Barletta et al., 1992). A Sau3AI site 750 ml of 10% SDS and 80 ml of 10 mg/mlproteinase K (Boehringer) were added fol-occurs on average every Ç300 nucleotides in
the same G/C-rich genome. lowed by incubation at 557C for 15 min. Celldebris was precipitated by addition of 1.0 mlof 5.0 M NaCl and 800 ml CTAB/NaCl solu-EXPERIMENTAL PROCEDUREStion (0.274 M hexacetylmethylammonium
Bacterial Strains, Media, and bromide (Sigma), 0.7 M NaCl), incubation atTransformation 557C for 10 min, extraction with 0.7 vol of
CHCl3 , and centrifugation at 18,000g. NucleicE. coli strain MC1061 [hsdR mcrB araD139acids were precipitated from the supernatantD(araABC -leu)7679 galU galK rpsL thi] wasby the addition of 0.6 vol of isopropanol andused for plasmid construction. Where appro-centrifugation at 18,000g. The nucleic acidpriate, E. coli cells were grown in Luria brothpellet was resuspended in TE. RNA was re-containing antibiotics at the following concen-moved by adding RNase to the restriction en-trations: Amp, 100 mg/ml; Kan, 25 mg/ml. M.donuclease digests. Southern hybridization in-smegmatis strain mc2155 was treated as wildvolved electrophoresis of restriction digeststype in this study, although this strain ison 0.7% agarose gels, transfer of DNA to aknown to contain mutations that result in anGenescreen Plus membrane (DuPont), and hy-increased efficiency of transformation by plas-bridization of 32P-labeled probe DNA in 61mids (mc2155 was derived from M. smegmatisSSPE, 51Denhart’s reagent, 50% formamide,strain ATCC 607) (Jacobs et al., 1991). M.100 mg/ml salmon sperm DNA. The finalsmegmatis cells were grown in Luria broth,wash step was 0.11 SSC, 1% SDS for 30 minMiddlebrook-DC-TW medium (Jacobs et al.,at 427C.1991), or TSGUT medium (Sander et al.,
1995). Plasmids purified from E. coli wereintroduced into M. smegmatis by electropora- Characterization of the pyrF Genetion using standard procedures (Jacobs et al.,
The pyrF gene of M. smegmatis was isolated1991). To construct growth curves, TSm-627essentially as described previously (Husson etcells were electroporated with various plas-al., 1990). Briefly, M. smegmatis chromosomalmids, grown to mid-log phase in LB-kan me-DNA was isolated and partially digested withdium, washed with an equal volume of M-the Sau3AI restriction endonuclease. The diges-DC-TW medium, and resuspended in 50 mltion reaction was fractionated using a 0.7% agar-of fresh M-DC-TW medium. Cells were incu-ose gel in TAE buffer (40 mM Tris–acetate, 1.0bated with shaking at 377C and the opticalmM EDTA). DNA fragments in the size rangedensity of the culture was measured hourly3.0–5.0 kb were excised from the gel and puri-using a Klett–Summerson colorimeter. Thefied away from the gel matrix using a Gen-data reported are the average of two experi-eClean kit (Bio 101; manufacturer’s suggestedments.procedures). These M. smegmatis chromosomalDNA fragments were cloned into the pUC19Chromosomal DNA Isolation and Southerncloning vector that had been digested withAnalysisBamHI. The ligation mix was transformed intoMC1061 cells using standard procedures andLarge-scale chromosomal DNA isolation
for Southern hybridization analysis involved amplified by 8 h of growth in liquid LB mediumcontaining 100 mg/ml ampicillin. The final li-growing 50 ml of M. smegmatis cells to satu-
ration in TSGUT medium. The culture was brary contained Ç5000 independent clones. Li-brary clones expressing orotidate-5*-monophos-pelleted and resuspended in 5.0 ml of TE (100
mM Tris (pH 8.0), 1.0 mM EDTA) plus 500 phate decarboxylase activity were isolated by
AID Plasmid 1286 / 660c$$$123 04-03-97 10:16:53 plasa AP: Plasmid

139GENE INTEGRATION VIA pyrF REPLACEMENT
transforming the amplified library into E. coli ing primers (initiation codons are under-lined): 21-sense Å 5*-GGCACTGCAT-strain DB6656 [lacZ624(Am), l0, trp-49(Am)
pyrF::Mu, rpsL179, hsdR27] and plating on C GA TA GG A GG TA AT A GA TG AC G-GGATTCGGGCAGCGG-3*; 22-sense Åminimal medium lacking uracil. Using this strat-
egy, three colonies were isolated, all of which 5*-GGCACTGCATCGATAGGTGATTA-GTTGACAGGATTCGGGCAGCGG-3*;contained the same M. smegmatis chromosomal
fragment (named plasmid pPyrF-1, see Fig. 2 antisense Å 5*-CGTTCAAGAGCTCCGG-3*. These PCR products were digested withfor a restriction map of the insert). Upon further
sequence analysis, these fragments were found ClaI / SacI and cloned into vector pSP72digested with the same enzymes. These in-to encode a pyrF homolog (Fig. 3). The nucleo-
tide sequence shown in Fig. 3 was obtained on termediates were digested with SacI / SmaIand used to clone the SacI–ApaI fragmentboth strands. Additional sequence from only a
single strand was obtained for the remainder of containing the remainder of the pyrF gene(see Fig. 3). From these pSP72 clones, thethe fragment depicted in Fig. 2 (data not shown
but available upon request). It should be noted ClaI–XbaI fragments, containing the en-tirety of the pyrF genes, were cloned intothat previous authors reported cloning the M.
smegmatis pyrF gene via complementation of a the E. coli-mycobacterial shuttle vectorpMV261 (Stover et al., 1991), which hadpyrF::Tn5 strain for its uracil auxotrophy (Hus-
son et al., 1990). In our hands this strategy is been digested with the same enzymes, toform the final vectors pPyrF-21/22. Digest-less straightforward than that described here due
to the high frequency of precise excision of the ing expression vector pMV261 with ClaI /XbaI removed the strong mycobacterial pro-Tn5 that results in very high background cell
growth. moter (PHSP60 of BCG). Therefore, no knownmycobacterial promoters are contained in ei-ther of the plasmids pPyrF-21/22.Additional Construction Details for pINT-D,
pINT-Z, and pPyrF-21/22b-Galactosidase Assays
Plasmid pINT-D was constructed in twosteps. The 1.5-kb M. smegmatis chromosomal To quantitate the expression of the lacZ
gene in mycobacterial cells, TSm-628 cellsfragment from the first PvuII site upstream ofthe pyrF gene to the PstI site immediately containing pPyrF-21 were grown in M-DC-
TW or M-DC-TW supplemented with eitherupstream of pyrF was isolated from plasmidpPyrF-1. This fragment was cloned in a three- uracil (0.1 mM) or arginine (0.6 mM) or both,
until the absorbance of the culture at 600 nmpart ligation into the pSP72 cloning vector(Promega) that had been digested with EcoRV reached 0.8–1.2 (Fig. 5). Thirty milliliters of
these late-log growths was pelleted and resus-and EcoRI. The third DNA fragment in thisthree-part ligation was a PstI*–EcoRI oligo- pended in 5 ml of Z buffer (100 mM K3PO4
(pH 7.0), 10 mM KCl, 1 mM MgSO4, 50 mMnucleotide of sequence: top strand Å 5*-AGAACTGCACAGCGAACTGGGGTCG- b-mercaptoethanol) and the cells were lysed
by sonication. The lysate was centrifuged andCGCCGGTCATG-3* and bottom strand Å5*-AATTCATGACCGGCGCGACCCCAG- 0.1 ml of the supernatant was added to 0.9 ml
of Z buffer and 0.2 ml of 2-nitrophenyl-b-D-TTCGCTGTGCAGTTCTTGCA-3*. This in-termediate vector was digested with PvuII and galactopyranoside (ONPG, Boehringer). The
reaction mixture was incubated at room tem-used to clone the ApaI–SmaI fragment down-stream of the pyrF gene. perature until the reactions showed sufficient
color development and then stopped by theThe pyrF gene blocks of plasmids pPyrF-21/22 were constructed in two pieces. The addition of 0.5 ml of 1.0 M Na2CO3. The ex-
tent of the reaction was determined by mea-region of pyrF, 5* to the internal SacI site,was constructed using PCR and the follow- suring the absorbance of the reaction mixture
AID Plasmid 1286 / 660c$$$123 04-03-97 10:16:53 plasa AP: Plasmid

140 KNIPFER, SETH, AND SHRADER
Gene replacement and expression of foreign DNA inat 420 nm (A420). The activity was calculatedmycobacteria. J. Bacteriol. 172, 519–524.using the equation: (A420 1 1000)/(A595 1 Jacobs, W. R., Kalpana, G. V., Cirillo, J. D., Pascopella,
Time of reaction (hours)). A595 is proportional L., Snapper, S. B., Udani, R. A., Jones, W., Barletta,to the total amount of soluble protein released R. G., and Bloom, B. R. (1991). Genetic systems for
mycobacteria. Methods Enzymol. 204, 537–555.during sonication of cells as determined byKalpana, G. V., Bloom, B. R., and Jacobs, W. R. (1991).dye binding (BioRad protein assay; manufac-
Insertional mutagenesis and illegitimate recombinationturer’s procedures). in mycobacteria. Proc. Natl. Acad. Sci. USA 88, 5433–5437.
Pelicic, V., Reyrat, J., and Gicquel, B. (1996a). Expres-ACKNOWLEDGMENTS sion of the Bacillus subtilis sacB gene confers sucrose
sensitivity on mycobacteria. J. Bacteriol. 178, 1197–1199.We thank Dr. W. R. Jacobs Jr. for M. smegmatis strain
Pelicic, V., Reyrat, J., and Gicquel, B. (1996b). Genera-mc2-155 and plasmid pMV261 and Drs. A. Brown, M.tion of unmarked directed mutations in mycobacteria,Pavelka, and S. Bardarov for helpful advice on workingusing sucrose counter-selectable suicide vectors. Mol.with mycobacteria.Microbiol. 20, 919–925.
Quinn, C. L., Stephenson, B. T., and Switzer, R. L.(1991). Functional organization and nucleotide se-
REFERENCES quence of the Bacillus subtilis pyrimidine biosyntheticoperon. J. Biol. Chem. 266, 9113–9127.
Ranes, M. G., Rauzier, J., Lagranderie, M., Gheorghiu,Aldovini, A., Husson, R. N., and Young, R. A. (1993).M., and Gicquel, B. (1990). Functional analysis ofThe uraA locus and homologous recombination in My-pAL5000, a plasmid from Mycobacterium fortuitum:cobacterium bovis BCG. J. Bacteriol. 175, 7282–7289.construction of a ‘‘mini’’ mycobacterium-EscherichiaBanerjee, A., Dubnau, E., Quemard, A., Balasubraman-coli shuttle vector. J. Bacteriol. 172, 2793–2797.ian, V., Um, K. S., Wilson, T., Collins, D., de Lisle,
Sander, P., Meier, A., and Bottger, E. C. (1995). rpsL/:G., and Jacobs, W. R., Jr. (1993). inhA, a gene encodingA dominant selectable marker for gene replacement ina target for isoniazid and ethionamide in Mycobacte-mycobacteria. Mol. Microbiol. 16, 991–1000.rium tuberculosis. Science 263, 227–230.
Shatzman, A. R., and Rosenberg, M. (1987). Expression,Barletta, R. G., Kim, D. D., Snapper, S. B., Bloom, B. R.,identification, and characterization of recombinant geneand Jacobs, W. R., Jr. (1992). Identification of expres-products in Escherichia coli. Methods Enzymol. 152,sion signals of the mycobacteriophage Bxb1, L1 and661–673.TM4 using Escherichia-Mycobacterium shuttle plas-
Snyder, M., Sapolsky, R. J., and Davis, R. W. (1988).mids pYUB75 and pYUB76 designed to create transla-Transcription interferes with elements important fortional fusions to the lacZ gene. J. Gen. Microbiol. 138,chromosome maintenance in Saccharomyces cerevis-23–30.iae. Mol. Cell. Biol. 8, 2184–2194.Boeke, J. D., LaCroute, F., and Fink, G. R. (1984). A
Stover, C. K., de la Cruz, V. F., Fuerst, T. R., Burlein,positive selection for mutants lacking orotidine-5*-J. E., Benson, L. A., Bennett, L. T., Bansal, G. P.,phosphate decarboxylase activity in yeast: 5-fluorooro-Young, J. F., Lee, M. H., Hatfull, G. F., Snapper, S. B.,tic acid resistance. Mol. Gen. Genet. 197, 345–346.Barletta, R. G., Jacobs, W. R., and Bloom, B. R. (1991).Chang, A. C. Y., and Cohen, S. N. (1978). ConstructionNew use for BCG recombinant vaccines. Nature 351,and characterization of amplifiable multicopy DNA456–460.
cloning vehicles derived from the p15A cryptic mini-Turnbough, C. I., Kerr, K. H., Funderburg, W. H., Do-
plasmid. J. Bacteriol. 134, 1141–1156.nahue, J. P., and Powell, F. E. (1987). Nucleotide se-
Hatfull, G. F., and Sarkis, G. J. (1993). DNA sequence,quence and characterization of the pyrF operon of
structure and gene expression of mycobacteriophageEscherichia coli K12. J. Biol. Chem. 262, 10239–
L5: A phage system for mycobacterial genetics. Mol. 10245.Microbiol. 7, 395–405.
Communicated by D. J. LeBlancHusson, R. N., James, B. E., and Young, R. A. (1990).
AID Plasmid 1286 / 660c$$$124 04-03-97 10:16:53 plasa AP: Plasmid