treatment of hela cells with bacterial water extracts inhibits shigella flexneri invasion
TRANSCRIPT

ELSEVIER FEMS Immunology and Medical Microbiology 15 (1996) 149-158
/$vlD~~f;OGY AND
MICROBIOLOGY
Treatment of HeLa cells with bacterial water extracts inhibits Shigella JZexneri invasion
Bonny Breckinridge DiNovo a, Richard Doan a, Roy B. Dyer a, Samuel Baron a, Norbert K. Herzog aYbTc, David W. Niesel a3c, *
a Department of Microbiology and Immunology, The University of Texas Medical Branch, Galveston, TX 77555-1019, USA b Department of Pathology, The University of Texas Medical Branch, Galveston, TX 775550609, USA
’ The WHO Collaborating Centerfor Tropical Diseases, The University of Texas Medical Branch Galweston, TX 77555-0609, USA
Received 16 January 1996; revised 6 June 1996; accepted 7 June 1996
Abstract
Pathogenesis mediated by Shigella jlexneri requires invasion of the gastrointestinal epithelium. It has been previously shown that HeLa cells challenged with S. flexneri show alterations in their phosphotyrosine-containing protein profile. In this report, we demonstrated that bacterial water extracts (WE) abrogated the invasion of HeLa cells by S. flexneri in a dose-dependent manner. ,4 proteinaceous component of S. jlexneri was shown to be responsible for this inhibitory activity. Proteins encoded on the L40-MDa plasmid were not responsible for the observed inhibition. WE from other Gram-negative bacteria also inhibited Shigella invasion of HeLa cells. HeLa cells pretreated with WE showed changes in the profile and the intensity of phosphotyrosine-containing protein bands. These data were consistent with a surface protein component in WE which initiated aberrant host cell signaling at the membrane which may account for the inhibition of bacterial entry.
Keywords: Shigella flexneri; Cellular invasion; Cell signaling
1. Introduction
The Gram-negative bacterium, Shigellu jlexneri,
is the etiological agent of bacillary dysentery in humans and primates. Critical to the disease process are interactions between the bacterium and the ep- ithelial cell which result in bacterial entry, intra- cellular residence and multiplication in cells of the colonic epithelium [ 11. Cellular invasion proceeds via an induced uptake mechanism, requiring active par-
* Corresponding author. Tel: + 1 (409) 772-4996; Fax: + 1
(409) 772-5065.
ticipation by both the bacterium [2-41 and the host cell [4-61. For example, kanamycin-treated or forma- lin-treated bacteria fail to invade cultured epitbelial cells [zs]. Epithelial cells treated with agents which alter the cytoskeleton, or lead to depolarization of the membrane are refractory to invasion by S. flexneri [2,4]. More recently, actin polymerization and myosin accumulation has been demonstrated at contact points between the bacterium and the host cells [4].
A number of genes encoded on a 140-MDa viru- lence plasmid are required for the invasion of epithe- lial cells by S. jlexneri [7]. The ipuBCD genes, which encode outer membrane proteins, have been shown to be critical for bacterial uptake into epithe-
0928-8244/96/$15.00 Copyright 0 1996 Federation of European Microbiological Societies. Published by Elsevier Science B.V.
PI1 SO928-8244(96)00057-O

150 B. Breckinridge DiNouo et al. / FEMS Immunology and Medical Microbiology 15 (1996) 149-158
lial cells and responsible for the induction of actin polymerization at the point of bacterial attachment [8,9]. Additional studies have shown that IpaB, IpaC and IpaD proteins are secreted from the surface of the bacterium through the action of accessory pro-
teins encoded by the spa and mxi loci [lo,1 I]. The mechanism by which secreted Ipa proteins induce Shigella uptake into epithelial cells remains un- known.
Recently, information has become available which indicates that various host cell signaling processes are activated following interaction with S. jlexneri. The DNA binding activities of NF-KB and other cellular transcription factors are increased within 35 min of challenge of HeLa cells [12]. Alterations in the profiles of tyrosine phosphorylated HeLa cell
proteins can be observed within 5 min of S. jlexneri challenge [ 131. Recently, the phosphorylation of cort- actin by the cytoskeleton-associated pp60”~“” signal- ing pathway following challenge of HeLa cells has been reported [ 141. Further, transient overexpression of pp60’+” promoted membrane ruffling and stimu- lated uptake of non-invasive Shigellu strains [14]. While these studies suggest an important role for host cellular signaling processes in the invasion pro- cess, the Shigellu proteins involved and the mecha- nism by which the signaling mechanism is initiated remains unknown.
Previous studies have shown that water extraction
of S. jlexneri releases a number of different highly immunogenic proteins including the IpaB, IpaC, and IpaD proteins [15]. Convalescent sera from infected monkeys and convalescent children have been shown to exhibit a significant immune response to these water extract antigens after oral S. jZexneri challenge [15]. While these studies identified the principle protein antigens which were observed following Shigellu infection, putative biological properties of the proteins present in WE had not been determined. We are interested in S. flexneri proteins which may initiate host cell responses to bacterial challenge. It is likely that at least some of these proteins are associ- ated with the bacterial cell surface. In this study, we use water extraction of intact S. jlexneri as a source of bacterial cell surface components.
In this report, we demonstrated that WE of Shigellu abrogated the invasion of HeLa cells by S. flexneri. The inhibitory activity showed dose-depen-
dent kinetics, did not alter bacterial adherence to the monolayers, and had a protein component necessary for activity. Further, the mechanism of inhibition did not involve loss of bacterial growth or viability. WE did not result in generalized inhibition to cellular entry since infection of WE-treated cells with en-
veloped and non-enveloped viruses was not affected. The mechanism of WE action may be related to the initiation or disruption of signaling across the host cell membrane.
2. Materials and methods
2.1. Bacteria and cells
SAlOO, an invasive S. flexneri 2A strain, and BS176, a plasmidless strain of S. flexneri have been previously described [16,17]. S. ji’exneri stains 28, 52, and 95 were derived from SAlOO and harbor TnPhoA insertions in ipuB, ipuC, and uirG respec- tively. E. coli Y 1090, Sulmonellu typhimurium TML, and Yersiniu enterocoliticu WA have been described previously [ 18,191. HeLa cells were grown as mono- layers (2 X lo5 cells/well) in 24 well plates or in suspension (8 X lo5 cells/ml) in supplemented
RPM1 1640 (5% fetal bovine serum (FBS), 2 mM glutamine, 50 U/ml of penicillin G, 50 kg/ml streptomycin (Pen-Strep)) at 37°C in a 5% CO,- humidified atmosphere. Antibiotic-free medium of the same composition was used to wash cells prior to bacterial challenge. Monkey VERO cells (6 X lo5 cells/ml) and the HEp-2 cells (3 X lo5 cells/ml) were maintained as monolayers in supplemented EMEM (Earles minimal essential medium) 0.22% HCO,, 50 U penicillin and 50 pg streptomycin, and 10% FBS) at 37°C in a 5% CO, humidified atmo-
sphere for the virus inhibition assay.
2.2. WE preparations
Water extraction of whole S. flexneri cells was performed as previously described by Oaks et al. [15]. Bacterial strains were grown overnight in L- broth with vigorous aeration. The cells were har- vested by centrifugation (5520 X g, 15 min), and the bacterial pellet was resuspended in 0.1 X volume of distilled water. The bacterial suspension was incu-

B. Breckinridge DiNovo et al./ FEMS Immunology and Medical Microbiology 15 (1996) 149-158 151
bated for 2 h with vigorous shaking at 37°C and whole cells were removed from the WE by centrifu- gation ( 18 200 X g , 2 X 20 min). Prior to lyophiliza- tion, the WE was centrifuged at 220000 X g for 1 h. The supematant was lyophilized and stored at - 20°C. The protein concentration of WE prepara-
tions was 25-35 p,g pmtein/mg of dried lyophilized WE. Protein levels were determined using Coomassie blue binding as described by the manufacturer (Bio- Rad, Hercules, CA).
2.3. HeLa cell invasion assay
S. flexneri invasion of HeLa cells was performed as previously described [20]. Prior to S. jlexneri challenge, 24 well plates containing HeLa cell mono- layers were treated with 2 mg/well WE (in RPMI) or the concentration noted in the figure legend. The plates were then incubated at 37°C for 15 min prior to challenge with S. jkxneri. Viability of the HeLa cells was not significantly altered by the WE over the time course of the experiment (data not shown). In some experiments, HeLa cell monolayers were pretreated with WE or Shigella LPS (Sigma, St. Louis, MO; 20 Ii,g/ml) for 15 min prior to chal- lenge. In some experiments, the WE was removed from the wells, and the monolayers were washed with RPM1 prior to bacterial challenge. For Shigella challenge, SAlOO was diluted in RPM1 and 2 X lo6
cfu placed in each well (bacteria:HeLa cell; 2O:l). The plates were incubated for 60 min at 37°C to allow for bacterial entry. Media containing non-inter- nalized bacteria were aspirated from the monolayers followed by 5 X washing (67% Hanks balanced salt solution, 33% RPM1 1640 supplemented with 2% FBS and 100 kg/ml gentamicin). Monolayers were gently agitated for 1 rnin between washes. A final solution of RPM1 supplemented with 100 pg/ml gentamicin and 5% FBS was applied to the monolay- ers and incubated at 37°C to counter-select non-inter- nalized bacteria. After 90 min, this solution was aspirated from the monolayers, and each well was
overlaid with 0.5 ml of 1% agarose in distilled water and 0.5 ml 2 X L-agar. The plates were incubated overnight at 37”C, and the number of infected HeLa cells was determined b,y counting the number of cfu on the bottom of triplicate wells. Results are ex- pressed as average cfu,/well f S.E.
2.4. Stability tests
Resistance to digestion by RNase (Sigma, St.
Louis, MO) was examined by adding 10 pg/ml of ribonuclease to the WE samples (20 mg). Samples were then incubated for 30 min at 25°C. A similar procedure was used to determine DNase resistance (DNase, 30 kg/ml, Sigma, St. Louis, MO). Appro- priate nucleic acids in the same buffer and the enzymes alone were included as controls (data not shown).
Resistance of the activity to digestion with trypsin was determined by adding TPCK-treated trypsin im-
mobilized on DITC glass beads (Sigma, St. Louis, MO). Samples were incubated with 200 mg/ml of beads (trypsin activity: 8400 U/g> for 6 h, and the beads were removed by centrifugation at 15 000 rpm in a microcentrifuge. Resistance to glycohydrolases
was assessed by adding a cocktail of 3.26 U (Y- galactosidase from E. coli (Boehringer Mannheim, Indianapolis, IN), 2.18 U B-galactosidase from E. coli (Boehringer Mannheim, Indianapolis, IN), 4.6 U B-glucosidase (Boehringer Mannheim, Indianapolis, IN), 6.24 U B-N-acetylglucosaminidase (Sigma, St. Louis, MO), 2.1 U endoglycosidase from Flauobac- terium meningosepticum (Sigma, St. Louis, MO), 2.1 U/ml neuraminidase from Clostridium perjringens (Sigma, St. Louis, MO) and 49 U/ml a-mannosi- dase (Sigma, St. Louis, MO) to the samples. The WE samples were incubated for 12- 14 h at 37°C then
heated to 80°C for 6 min to inactivate the glycohy- drolases after treatment. Commercial reagents sup- plied with the enzymes functioned as controls.
2.5. Growth curve
Distilled H,O or WE samples (4 mg/ml) were added to L-broth (pH 7.0) and vortexed. An overnight culture of S. jlexneri SAlOO (200 l_~l) was added, and the optical density was followed at 600 nm. The samples were incubated at 37°C and the optical density was determined at 30 min intervals.
2.6. Adherence assay
HeLa cells were pretreated with RPM1 or SAlOO WE and incubated at 37°C for 30 min. The bacterial suspension was washed once with ice cold PBS,

152 B. Breckinridge DiNouo et al./ FEMS Immunology and Medical Microbiology 15 (1996) 149-158
resuspended in RPMI, then applied to the HeLa cell monolayers. The plates were incubated for 4 h at 4°C. Non-adhered bacteria were removed by gentle aspiration, and the monolayers were washed once with PBS. The wells were treated with 1 ml of 0.1% Triton X-100 solution, and the plates were gently agitated for 15 min to solubilize the monolayers to release the attached bacteria. Samples were diluted in PBS and cfu determined by plating on L-agar in
triplicate.
2.7. Virus inhibitor assay
The virus inhibitory activity was quantified by
plaque assay [21]. The following viruses were used in our assay: Newcastle disease virus (NDV) or Mengo virus. EMEM (2% FBS, 0.1% Pen-Strep) was added to the VERO and HEp-2 cells. For all assays, SAlOO WE samples and the controls (8316 (UTI-a) [22], UTI-B [22,23], and albumin) were added to the wells and serially diluted. UTI-a and
UTI-B are broad spectrum viral inhibitors which have been described previously [22,23]. The viruses were then applied to the wells. In the treatment assay, the plates were incubated for 1 h at 37°C in a CO, incubator following viral challenge, and the cells were overlaid with 1% methylcellulose (Fisher, Pittsburgh, PA) followed by incubation at 37°C overnight. For the removed assay, the plates were incubated for 30 min at 37°C then decanted to remove free virus, and then overlaid with 1% meth- ylcellulose. Cells were incubated overnight at 37°C and pfu determined after crystal violet staining [21].
2.8. Identification of tyrosine phosphorylated pro- teins
A suspension of 8 X lo5 HeLa cells/ml were pretreated with RPM1 or 2 mg/ml of SAIOO WE for 15 min at 37°C. Washed bacterial cells (1.6 X lo7 cfu) from a mid-logarithmic culture were prepared and added to the HeLa cell suspensions as previously described [13,20]. Cells were gently mixed and then pelleted by microcentrifugation. Next, the cells were incubated at 37°C for 5 min. Cells were harvested by microcentrifugation and lysed by boiling in SDS- sample buffer for 10 min [24]. Microcentrifugation
(13 000 X g, 5 min) clarified the protein extract prior
to SDS-PAGE [24]. HeLa cell proteins were trans- ferred to nitrocellulose and blocked in 5% non-fat dry milk in Tris-buffered saline-Tween-20 pH 7.6
(TBS-T) (20 mM Tris, 137 mM NaCl, 0.1% Tween- 20) overnight. The membrane was then washed twice for 5 min in TBS-T at 25°C and reacted with mono- clonal anti-phophotyrosine antibody (1 pg/ml in TBS-T (PY20, Transduction Labs Inc, Albany, NY)) for 1 h. Membranes were then washed four times as previously described. Anti-mouse IgG horseradish peroxidase-conjugated secondary antibody
(Calbiochem, La Jolla, CA) was next used at a concentration of 0.25 p,g/ml in TBS-T for 1 h. Following four washes as described above, a final wash in TBS was performed. Enhanced Chemilumi- nescence (ECL) Substrate (Amersham, UK) was used as described by the manufacturer to detect tyrosine- phosphorylated proteins.
Table 1
Treatment of HeLa cells with water extracts (WE) from S. flexneri inhibits cellular invasion
Condition a Infected
cell count
(cfu/welll b
% Reduction
in infected
HeLa cells
Experiment 1
none
0 min WE pretreatment
5 min WE pretreatment
1.5 min WE pretreatment
30 min WE pretreatment
Experiment 2
none
WE treatment
WE pretreatment/removal
Experiment 3
none
WE treatment
Shigeila LPS (ZOpg/ml)
57.0+ 1.8
50.0+2.6
31.3dz6.5
19.3 + 5.0
19.7 f 5.5
75.7+9.7
14.8 k 2.8
80.3 f 6.7
63.5f11.2
21.3k2.2
61.5k7.5
12
45
66
65
81
0
66
3
a HeLa cell monolayers were treated with WE (2 mg/well) for 15
mm or as indicated prior to challenge with S. flexneri SAlOO
(20: 1; bacteria:HeLa cell). WE! were left on the monolayers during
the 60 mm challenge period, or removed after the 15 min pretreat-
ment period. Shigella LPS (20 pg/ml) was added to the mono- layer 15 min prior to treatment and left on during the challenge
period.
b Counts of infected HeLa cells represent the average of at least
triplicate wells, Error is expressed as the standard error.
B. Breckinridge DiNovo et al./ FEMS Immunology and Medical Microbiology 15 (1996) 149-158 153
2.9. Densitometric analysis
The appropriate bands on the autoradiographs fol- lowing enhanced chemiluminescence detection were
scanned with a ScanMaster 3 + (Scanalytics, a divi- sion of CSPI, Billerica, MA) and quantitated by One-Dscan software (Scanalytics, a division of CSPI, Billerica, MA). Several scans were performed on multiple exposures to ensure film linearity. The data were presented as percent of control values. The range of densitometric: values ranged from 0.51 to 26.86 (data not shown). The data presented were from a representative ekxperiment.
3. Results
3.1. Treatment of HeLa cells with water extracts (WE) from S. jlexneri reduces the number of infected HeLa cells
We investigated whether S. jlexneri WE would alter the number of infected HeLa cells following S.
Jlexneri challenge. Initial experiments with WE pre- treatment showed a time-dependent inhibition in the number of Shigella-infected HeLa cells. The WE was left on the monolayers during Shigella chal- lenge. As shown in Table 1, WE added concurrently with Shigella resulted .in a modest 12% inhibition of infected HeLa cells. ‘The level of inhibition was
shown to increase at 5 min pretreatment (45%) reaching a maximal inhibition at 15 min (66%). Pretreatment times longer than 15 min did not in- crease inhibition. Addition of the WE to wells did not result in a detectable pH change or loss of viability of the HeLa cells over the course of the assay (data not shown).
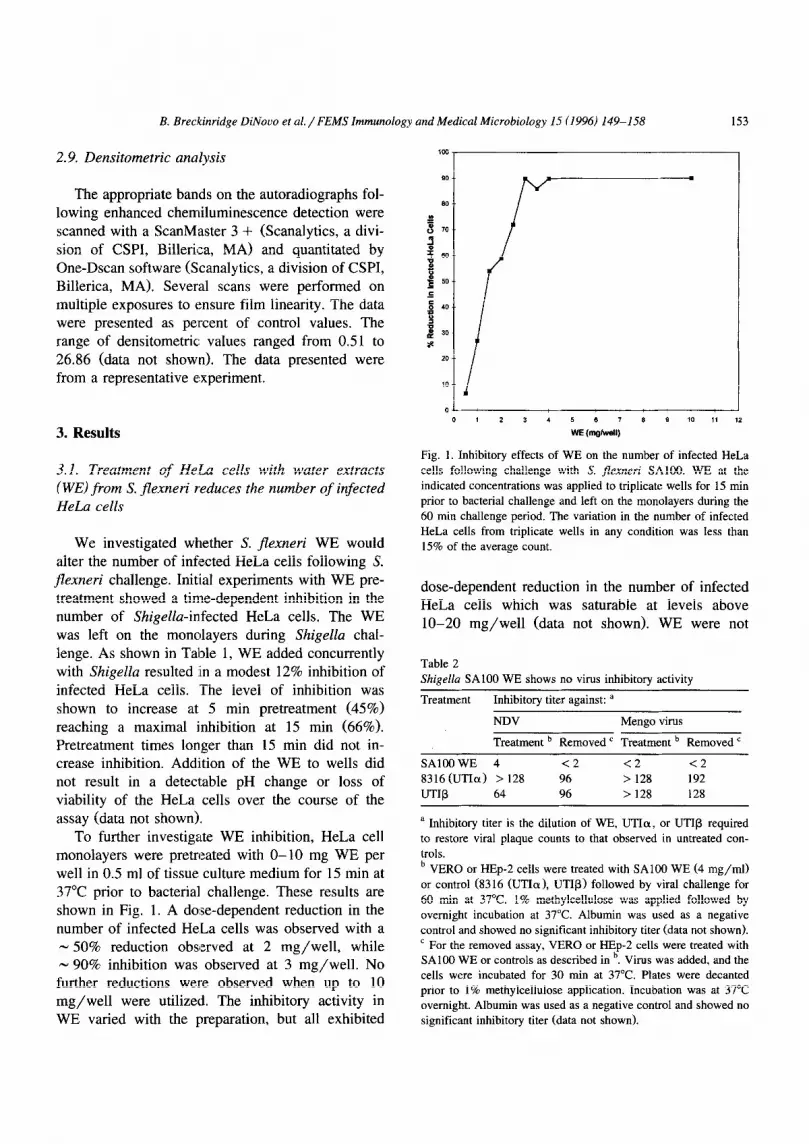
To further investigate WE inhibition, HeLa cell monolayers were pretreated with O-10 mg WE per well in 0.5 ml of tissue culture medium for 15 min at 37°C prior to bacterial challenge. These results are shown in Fig. 1. A do:se-dependent reduction in the number of infected HeLa cells was observed with a - 50% reduction observed at 2 mg/well, while - 90% inhibition was observed at 3 mg/well. No further reductions were observed when up to 10 mg/well were utilized. The inhibitory activity in WE varied with the preparation, but all exhibited
Fig. 1. Inhibitory effects of WE on the number of infected HeLa
cells following challenge with S. flexnerr SAlOO. WE at the
indicated concentrations was applied to triplicate wells for 15 min
prior to bacterial challenge and left on the monolayers during the
60 mitt challenge period. The variation in the number of infected
HeLa cells from triplicate wells in any condition was less than
15% of the average count.
dose-dependent reduction in the number of infected HeLa cells which was saturable at levels above lo-20 mg/well (data not shown). WE were not
Table 2
Shigella SAlOO WE shows no virus inhibitory activity
Treatment Inhibitory titer against: a
NDV Mengo virus
Treatment b Removed ’ Treatment b Removed ’
SAlOOWE 4 <2 <2 <2
8316(LJTIa) > 128 96 > 128 192
UTIR 64 96 > 128 128
a Inhibitory titer is the dilution of WE, UTIa, or UTIR required
to restore viral plaque counts to that observed in untreated con-
trols.
b VERO or HEp-2 cells were treated with SAlOO WE (4 mg/ml)
or control (8316 (UTIo), UTI@) followed by viral challenge for
60 min at 37°C. 1% methylcellulose was applied followed by
overnight incubation at 37°C. Albumin was used as a negative
control and showed no significant inhibitory titer (data not shown).
’ For the removed assay, VERO or HEp-2 cells were treated with SAlOO WE or controls as described in b. Virus was added, and the
cells were incubated for 30 min at 37°C. Plates were decanted
prior to 1% methylcellulose application. Incubation was at 37°C
overnight. Albumin was used as a negative control and showed no
significant inhibitory titer (data not shown).

154 B. Breckinridge DiNouo et al. / FEM.5 Immunology and Medical Microbiology 15 (1996) 149-158
Table 3
Enzymatic stability of the WE inhibitory activity
Treatment a % Reduction in
Shigella-infected
HeLa cells
Experiment 1
Shigella WE
Shigella WE + RNAse
RNAse alone
Experiment 2
Shigella WE
Shigella WE + DNAse
DNAse alone
Experiment 3
Shigella WE
Shigella WE + glycohydrolase
Glycohydrolase alone
Experiment 4
Shigella WE Shigella WE + trypsin
Trypsin alone
15
62
20
76
76
5
59
71
18
62 2
16
a RNAse (10 pg/ml) was added to SAlOO WE for 30 mitt at
25°C. DNAse (30 pg/ml) was added to WE for 30 min at 25°C.
Glycohydrolases were added to the samples and incubated for
12-14 h at 37°C. then heated to 80°C for 6 min to inactivate the
activity. TPCK-treated insoluble trypsin attached to DITC glass
beads (200 mg) was added to the WE and incubated for 6 h at
37°C. Treated WE (4 mg/ml) were applied to HeLa cells for 15
mitt at 37°C prior to and during S. flexneri challenge. Results are
from triplicate wells and are representative of three experiments.
effective at reducing the number of infected HeLa cells if removed from the monolayers prior to bacte- rial challenge. Pretreatment of monolayers followed by removal of the WE before bacterial challenge resulted in no inhibitory activity while WE left on the monolayers during the challenge period resulted in a 81% reduction in infected HeLa cells (Table 1). Also, inhibitory activity was not observed if Shigella LPS was used to treat monolayers. Shigella WE showed little or no inhibitory activity against NDV or Mengovirus in an in vitro assay (Table 2).
3.2. Enzymatic stability of the inhibitory activity of S. jl’exneri WE
To further characterize the inhibitory activity of WE, the WE were assessed for stability to a number of degradative enzymes. As seen in Table 3, treat- ment of WE with RNase, DNase, or glycohydrolases did not significantly reduce the inhibitory activity of
the WE. However, treatment of WE with trypsin immobilized on glass beads virtually abrogated the inhibitory activity of the WE. These experiments demonstrated that a polymeric nucleic acid or carbo- hydrate was not required for the inhibitory activity, and that the inhibitory activity was associated with a
protein component.
3.3. WE does not inhibit Shigella growth or adher- ence to HeLa cells
It is possible that the reduction in the number of infected HeLa cells results from an inhibitory effect
of the WE directly on Shigella growth or via a reduction in the efficiency of S. jlexneri binding to cell monolayers. To address the possibility of direct action on the bacterium, we investigated the capacity of WE to inhibit the growth of S. jlexneri. WE at 4 mg/ml was added to L-broth inoculated with S. jlexneri. Controls received the same volume of dis- tilled water. The optical densities of the cultures were recorded at 30 min intervals during the 6 h incubation at 37°C. As observed in Fig. 2, the WE had no effect on the growth of S. JZexneri.
The effect of WE on Shigella adherence to HeLa cells was also determined. As shown in Fig. 3, WE treatment did not lead to alterations in the ability of
Fig. 2. WE shows no inhibitory effects on the growth of S.
flexneri SAlOO. L-broth was supplemented with the same volume
of WE (4 mg/ml) or distilled water prior to inoculation (200 ~1)
with an overnight culture of SAlOO. Optical density at 600 nm
was determined for duplicate cultures every 30 min during growth
at 37°C with aeration.

B. Breckinridge DiNouo et al./ FEMS Immunology and Medical Microbiology I5 (1996) 149-1.58 1.55
2wJ
40
20
0
rhllml SA1W WE
Fig. 3. SAlOO WE does no1 inhibit S. flexneri SAlOO adherence
to HeLa cells. HeLa cell monolayers were treated with WE (4
mg/ml) for 30 min at 37°C prior to challenge with S. flexneri and
left on during the 4 h challenge period. HeLa cells were gently
washed then solubilized from the plates using 0.1% Triton X-100
in PBS. Solubilized extracts were serially diluted and plated on
L-agar. Following overnight incubation at 37°C cfu were deter-
mined. Results are from triplicate plating.
Shigellu to bind to the monolayers as determined by the recovery of Shigellu from monolayers following incubation at 4°C. Additional experiments using 35S- labeled bacteria also showed little effect of WE on the ability of Shigella to adhere to monolayers (data
not shown).
3.4. Inhibitory activity of WE is not the product of a 140-MDa plasmid-enc<oded gene
We next investigated whether the inhibitory activ-
ity of WE originated from a 140-MDa plasmid gene. WE were prepared from strains harboring TnPhoA insertions in 140-MDa virulence-related genes (ipa& ipaC, icsA) and a plasmidless strain, BS176 [18]. As shown in Table 4, WE from all TnPhoA strains reduced the number of infected HeLa cells to ap- proximately the same level as that observed for the virulent parental strain, SAlOO. In addition, WE from the plasmidless BS176 strain also showed in- hibitory activity of approximately equivalent po- tency. These results suggested that the inhibitory activity in Shigella WE was not dependent on genes encoded on the 140~MDa plasmid.
3.5. SAZOO WE induces tyrosine phosphorylation of HeLa cell proteins
Shigellu challenge of HeLa cells has been shown to result in the phosphorylation of HeLa cell proteins on tyrosine [13,14] and may constitute one of the initial events which leads to bacterial entry and new host cell gene expression. Since WE inhibited S. jlexneri invasion of HeLa cells, we investigated whether WE could also induce tyrosine phosphoryla- tion and/or inhibit SAlOO-induced alterations. Ex-
tracts were prepared from cells challenged with WE, cultures of S. jlexneri SAlOO, or left untreated. Proteins were separated by SDS-PAGE and analyzed
by immunoblotting with a phosphotyrosine-specific monoclonal antibody (PY20). This is shown in Fig. 4 along with a densitometric analysis to quantitate the amounts of the indicated protein bands. In compari- son to the protein bands observed in unchallenged HeLa cells, a significant increase in the phosphoryla- tion of u 78-80-kDa protein doublet (4.1), 84-kDa (10.6), 90-kDa (1.4), and a 102~kDa doublet protein (7.9) was observed from extracts of Shigella-chal- lenged HeLa cells @AlOO). Increases in the tyrosine phosphorylation of these proteins following Shigella
Table 4
Treatment of HeLa cells with mutant S. flexneri WE inhibits
cellular invasion following SAlOO challenge
Bacterial strains % Reduction in infected HeLa cells
Experiment 1
SAlOO 59
28 52
Experiment 2
SAlOO 58
52 44
Experiment 3
SAlOO 59
95 64
Experiment 4
SAlOO 71
BS176 68
WE were prepared as previouly described using S. Jexneri mu-
tants, 28 (ipaC::TnphoA), 52 (ipaB::TnphoA), 95 (icsA::TnphoA), BS176 (140-MDa plasmid cured). WE were applied to triplicate wells for 15 min prior to S. flexneri SAlOO challenge and left on
the monolayers during the 60 min challenge period. Results are
from triplicate wells and representative of three experiments.
Variation in the number of infected HeLa cells for all conditions
was less than 15% of the average count.

156 B. Breckinridge DiNouo et al. / FEMS Immunology and Medical Microbiology 15 (1996) 149-158
WE+ SAIOO SAlOO WE C
Protein Band WdtOCpDfml
NW COlltml WE SAlOO wE+sA100
102 1.M) 3.4 7.9 7.9 90 1.M) 1.9 1.4 2.9 84 1.00 4.8 106 14.3
78-80 1.00 1.4 4.1 4.5
Fig. 4. SAlOO WE induces changes in HeLa cell phospho-
tyrosine-containing proteins. HeLa cells in suspension were un-
treated, treated with SAlOO WE (2 mg), or challenged with S.
jlexneri SAlOO for 5 min. Also, HeLa cells were pretreated with
SAlOO WE then challenged with S. flexneri for 5 min. Equal
amounts of phosphotyrosine containing proteins were immuno-
blotted as previously described. Following immunoblotting, pro-
tein bands were visualized using chemiluminescence. Protein bands
showing an increase in tyrosine phosphorylation are indicated by
arrowheads, and the molecular weights are marked in kDa on the
right. Densitometric values scanned from the various bands are
shown below and recorded as optical densities (density per unit
area). Results are representative of three experiments.
challenge have been reported previously [13]. Ex- tracts from WE-treated cells also exhibited increases over control-treated cells for these tyrosine phospho- rylated proteins. Increases in the 78-80-kDa, 84-kDa, 10ZkDa doublet bands, however, were consistently
less than that observed following challenge with SAlOO. The 80-kDa band was only slightly induced (1.4 X over control) by WE, while the 7%kDa band was not observed. The 78-SO&Da doublet band showed a N 3 X higher intensity for SAlOO-treated samples versus WE-treated samples. The absence of the 7%kDa band probably accounted for some of this difference. The intensities of the 102~kDa and 84-kDa bands were N 2 X higher for the SAlOO compared to the WE-treated samples. In contrast, the 90-kDa band showed a similar intensity for both WE- and SAlOO-treated groups (1.9 X vs. 1.4 X 1. These re- sults demonstrated that WE alone could induce the tyrosine phosphorylation of HeLa cell proteins. However, these data also demonstrated that, while a
similar profile of tyrosine phosphorylated bands was induced, the intensity of the phosphotylated proteins induced by WE was distinct from that observed from SAlOO.
Additionally, we considered whether treatment with WE altered the phosphorylation of HeLa cell proteins induced by SAlOO. As shown in Fig. 4, all four phosphorylated proteins were observed follow- ing concurrent challenge with SAlOO and WE. In- creases in the 84-kDa and 90-kDa bands were addi-
tive over that observed for SAlOO and WE treatment alone. The 102~kDa and 80-kDa doublet bands were at similar levels to that observed following SAlOO
challenge alone. It should be noted that phosphoryla- tion changes induced by WE occurred with similar kinetics to that induced by the intact bacteria. These results may suggest that treatment of HeLa cells with WE may lead to alterations in HeLa cell signaling which may contribute to the inhibition of bacterial invasion.
3.6. WE from other enteric bacteria reduce the num- ber of infected cells following S. jlexneri challenge
We next investigated whether pretreatment of HeLa cell monolayers with WE prepared from other Gram-negative enteric bacteria also inhibited S. fiexneri invasion. Using the same protocol, WE were prepared from an avirulent E. coli strain and viru- lence-attenuated S. typhimurium and Y. enterocolit- ica. WE from all strains showed comparable potency in inhibiting the infection of HeLa cells by S. jlexneri (Table 5). These results suggested that the inhibitory component might be a common component present on the surface of Gram-negative enteric bacteria.
Table 5
Enteric bacterial WE inhibit the number of infected HeLa cells
following S. Jexneri challenge
Bacteria ’ % Reduction in infected HeLa cells
E. coli Y1090 72
S. typhimurium TML 87
Y. enterocolitica WA 89
a WE were prepared as previously described. HeLa cell monolay-
ers in 24 well plates were treated in triplicate with WE (2
mg/well) for 15 min prior to challenge with S. Jexneri SAlOO.
Protein levels for the WE preparations were 25-30 p,g/ml. WE
were left on the monolayers during the 60 min challenge period.

B. Breckinridge DiNouo et al./ FEMS Immunology and Medical Microbiology 15 (1996) 149-158 157
4. Discussion from subversion or modification of these events.
In this study, we have shown that treatment of monolayers with water extracts (WE) from S. jlexneri inhibits bacterial entry into host cells. The inhibitory activity was shown not to be mediated by LPS or other carbohydrate or nucleic acid containing materi- als but rather had a proteinaceous component as WE treated with trypsin wasi greatly reduced in inhibitory activity. The relatively high levels of WE required for inhibitory activity may reflect the low concentra-
tions of the active protein component in the extracts. Alternatively, the active component may be partially destroyed during the demanding multi-step prepara- tion of WE.
The mechanism by which WE inhibited the inva- sion of HeLa cells was not due to a reduction in the growth of the bacterium or loss of adherence of the bacterium to the monolayers. Interestingly, inhibition increased with pretreatment times and required the continued presence of WE. These data suggest a requirement that host cells respond to the WE treat- ment and possibly indicate a short ‘half-life’ of the
inhibitor or its action. Further, this inhibition did not extend to the HeLa cells challenged with Mengo virus. This indicates that receptor-mediated viral en-
try is unaffected by WE treatment. Analogously, infection with NDV, which proceeds via fusion of the viral envelope with the cell membrane, is also
not altered by WE treatment. Therefore, inhibition does not appear to be lthe result of generalized host cell membrane alterations.
It is clear that elements of the cellular signaling pathway are induced as a consequence of bacterial challenge of epithelial cells. S. jlexneri challenge of HeLa cells alters the activity of a number of tran- scription factors including NF-KB [12]. We and oth- ers have shown that S. jlexneri induces tyrosine phosphorylation of HeLa cell proteins following
challenge [ 13,141. In the experiments described above, we showed that WE treatment of HeLa cells induces tyrosine phosphorylation of proteins with similar kinetics as observed with intact bacteria. These results complemented our earlier studies in which we demonstrated that Shigella invasion was
not prerequisite for bacterially induced alterations in host cell tyrosine phosphorylated proteins. However, changes in the intensity and profile of these proteins may indicate a disruption of the normal host cell signaling induced by S. flexneri. It is possible that inhibition of S. flexneri invasion of HeLa cells by WE may arise from this aberrant or incomplete signaling at the membrane. Optimal inhibition of invasion by WE requires a 15 min pretreatment,
suggesting that downstream host cell events which are a consequence of tyrosine phosphorylation, are required to abrogate bacterial invasion.
It is well established that the interaction of S. flexneri with epithelial cells leads to significant changes at the surface of these cells. Actin poly- merization and myosin accumulation at the point of
bacterial contact have been demonstrated [4]. These actin filaments, in association with plastin are in high concentrations in the membrane ruffles which accu- mulate and eventually engulf the invading bacterium [25]. Dehio et al. [14] have shown Shigellu-stimu- lated tyrosine phosphorylation of cortactin by pp60”~“‘“. Cortactin phosphorylation is believed to be involved in the observed membrane ruffling. How- ever, the mechanism by which Shigellu induces these changes and how these changes result in bacterial uptake remains unknown. It is possible that WE inhibition of ShigeZZa infection of HeLa cells results
Studies have demonstrated that water extraction of S. Jlexneri leads to the release of a number of highly immunogenic polypeptides which are encoded on the large 140-MDa non-conjugative plasmid [15]. These proteins include the IpaB, IpaC, and IpaD proteins as well as IcsA [15]. IpaB and IpaC have been shown to be required for the entry of ShigeZZa
into epithelial cells and for the lysis of the phagoso- ma1 vesicle [8,9]. A role for IpaBCD in the induction of host cell signaling induced by the bacteria has yet to be determined. However, ipaB and @UC strains of S. jlexneri failed to induce NF-KB DNA binding activity [12] but weakly induced changes in tyrosine phosphorylation of host cell proteins [ 131. In this study, we showed that WE from a plasmidless strain and ipuB and ipaC strains effectively inhibited ShigeZZu infection of HeLa cells. In addition, WE from an avirulent E. coli strain, and other Gram- negative enteric pathogens were also able to inhibit ShigeZZu infection of HeLa cells. These data may suggest that the source of the inhibition is not related to virulence products unique to ShigeZZa and further

158 B. Breckinridge DiNovo et al./ FEMS Immunology and Medical Microbiology 15 (1996) 149-158
appears to involve a common extractable component of these Gram-negative bacteria. However, these studies demonstrated that soluble surface proteins could modulate host cell responses which may lead to the inhibition of bacterial entry into treated cells. Induced changes in tyrosine phosphorylation by ShigelZu cell surface proteins appear to be an early host cell event which may initiate a signaling cas-
cade which is exploited by the bacterium to facilitate its entry into the host cell. Identification of the component(s) of the water extract that stimulate
changes in host cell tyrosine phosphorylation and/or inhibit bacterial entry is currently under investiga- tion.
Acknowledgements
This work was supported by the John Sealy Memorial Endowment for Biomedical Research.
References
[l] LaBrec, E.H., Schneider, H., Magnani, T.J. and Formal, S.B.
(1964) Epithelial cell penetration as an essential step in the
pathogenesis of bacillary dysentery. J. Bacterial. 88, 1503-
1518.
[2] Hale, T.L. and Bonventre, P.F. (19791 Shigella infection of Henle intestinal epithelial cells: role of the bacterium. Infect.
Immun. 24, 879-886.
[3] Hale, T.L. and Formal, S.B. (1986) Genetics of virulence in
Shigella. Microb. Pathog. 1, 511-518.
[4] Clerc, P. and Sansonetti, P.J. (1987) Entry of Shigellaflexneri into HeLa cells: evidence for directed phagocytosis involving
actin polymerization and myosin accumulation. Infect. Im-
mun. 55, 2681-2688.
[5] Hale, T.L., Morris, R.E. and Bonventre, F.P. (1979) Shigella infection of Henle intestinal epithelial cells: role of the host
cell. Infect. Immun. 24, 887-894.
[6] Polotsku, Y.E., Snigirevskaya, E.S. and Dragunskaya, E.M. (1974) Electron-microscopic data on method of penetration
of Shigella into the intestinal epithelial cells. Bulletin Exper-
imental de Biologie et Medicine 77, 202-205.
[7] Sansonetti, P.J., Kopecko, D.J. and Formal, S.B. (1982) Involvement of a plasmid in the invasive ability of Shigella flexneri. Infect. Immun. 35, 852-860.
[S] High, N., Mounier, J., Prevost, M.-C. and Sansonetti, P.J.
(1992) IpaB of Shigella j7exneri causes entry into epithelial
cells and escape from the phagocytic vacuole. EMBO J. 11,
1991-1999.
[9] Menard, R., Sansonetti, P.J. and Parsot, C. (1993) Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD
as effecters of S. flexneri entry into epithelial cells. J. Bacterial. 175, 5899-5906.
1101
1111
1121
[I31
1141
1151
[161
[I71
El81
[I91
La
ml
WI
1231
b41
La
Venkatessan, M., Buysse, J.M., and Oaks, E.W. (1992)
Surface presentation of Shigella flexneri invasion plasmid
antigens requires the products of the spa locus. J. Bacterial.
174, 1990-2001.
Andrews, G.P., Hromockyj, A.E., Coker, C. and Maurelli,
A.T. (1991) Two novel virulence loci, mxiA and mxiB, in
Shigella ji’exneri 2a facilitate excretion of invasion plasmid
antigens. Infect. Immun. 59, 1997-2005.
Dyer, R.B., Collaco, C.R., Niesel, D.W. and Herzog, N.K.
(1993) Shigella flexneri invasion of HeLa cells induces
NF-KB DNA-binding activity. Infect. Immun. 61, 4427-
4433. Collaco, C., Dyer, R.B., Doan, R., Herzog, N.K. and Niesel,
D.W. (1995) Shigellaflexneri-HeLa cell interactions: a puta-
tive role for host cell protein kinases. FEMS Immun. Med.
Microbial. 10, 93-100.
Dehio, C., Prevost, M. and Sansonetti, P.J. (1995) Invasion of epithelial cells by Shigella flexneri induces tyrosine phos-
phorylation of cortactin by a pp60c-src-mediated signalling
pathway. EMBO J. 14, 2471-2482.
Oaks, E.V., Hale, T.L. and Formal, S.B. (19861 Serum
immune response to Shigella protein antigens in rhesus
monkeys and humans infected with Shigella spp. Infect.
Immun. 53, 57-63.
Payne, S.M., Niesel, D.W. Peiotto, S. and Lawlor, K.M.
(1983) Expression of hydroxamate and phenolate
siderophores by Shigellaflexneri. J. Bacterial. 155, 949-955.
Sansonetti, P.J., Ryter, A., Clerc, P., Maurelli, A.T. and
Mounier, J. (1986) Multiplication of Shigella flexneri within
HeLa cells: lysis of the phagocytic vacuole and plasmid-
mediated contact hemolysis. Infect. Immun. 5 1, 461-469.
Gianella, R.A., Formal, S.B., Dammin, G.J. and Collins, H.
(1973) Pathogenesis of Salmonellosis studies on fluid secre-
tion, mucosal invasion and morphologic reaction in the rabbit ileum. J. Clin. Invest. 52, 441-453.
Gemksi, P., Lazere, J.R. and Casey, T. (1980) Plasmid
associated with pathogenicity and calcium dependency of Yersinia enterocolitica. Infect. Immun. 27, 682-685.
Niesel, D.W., Chambers, C.E. and Stockman, L.S. (1985)
Quantitation of HeLa cell monolayer invasion by Shigella and Salmonella species. J. Clinical. Microbial. 22, 897-902.
Baron, S. and McKerlie, L. (1981) Broadly active inhibitor
of viruses spontaneously produced by many cell types in
culture. Infect. Immun. 32, 449-453.
Baron, S., Niesel, D., Singh, LP., McKerlie, L., Poast, J.,
Chopra, A., Antonelli, G., Dianzani, F. and Coppenhaver,
D.H. (1989) Recently described innate broad spectrum virus
inhibitors. Microb. Pathogen 7, 237-247.
Singh, I.P., Coppenhaver, D.H., Chopra, A.K. and Baron, S.
(1992) Further characterization of a broad-spectrum antiviral
substance in human serum. Viral Immunol. 5, 293-303.
Laemmli, U.K. (1970) Cleavage of structural proteins during
the assembly of the head of bacteriophage T4. Nature 227,
680-686.
Adam, T., Arpin, M., Prevost, M.-C., Gounon, P. and San-
sonetti, P.J. (19951 Cytoskeletal rearrangement and the func- tional role of T-plastin during entry of Shigella Jexneri into
HeLa cells. J. Cell Biol. 129. 367-381.