thiol reducing compounds prevent human amylin-evoked cytotoxicity
TRANSCRIPT

Thiol reducing compounds prevent human amylin-evokedcytotoxicityBarbara Konarkowska1,2, Jacqueline F Aitken1,2, Joerg Kistler1, Shaoping Zhang1,2 andGarth J S Cooper1,2,3
1 The School of Biological Sciences, University of Auckland, New Zealand
2 Centre for Research Excellence in Molecular Biodiscovery, Faculty of Science, University of Auckland, New Zealand
3 Department of Medicine, Faculty of Medical and Health Sciences, University of Auckland, New Zealand
Human amylin (hA) is a small protein that is cosecreted
with insulin from pancreatic islet b-cells upon stimula-
tion by glucose or other chemical signals [1,2]. It is the
main proteinaceous constituent of the amyloid deposits
commonly found in the islets of human subjects with
type-2 diabetes mellitus (T2Dm) [3,4], as well as in dia-
betic cats and primates [5,6], and has also been
referred to as islet amyloid polypeptide (IAPP) [5]. The
toxicity of hA towards cultured pancreatic b-cells is a
well-documented phenomenon [7–10]. Post mortem
examination of pancreatic tissue from T2Dm patients
has shown a correlation between the presence of amy-
loid deposits in pancreatic islets and b-cell loss [11–13].Evidence from longitudinal studies in spontaneously
diabetic primates has also indicated an inverse rela-
tionship between b-cell number and the extent of islet
amyloidosis [6]. Studies of hA transgenic animals
have shown that the occurrence of islet amyloid is
Keywords
amylin; b-cell apoptosis; diabetes; oxidative
stress; reactive oxygen species; N-acetyl-
L-cysteine
Correspondence
G. J. S. Cooper, School of Biological
Sciences, University of Auckland, Private
Bag 92–019, Auckland, New Zealand
Fax: +64 9 373 7045
Tel. +64 9 373 7599 ext. 87394
E-mail: [email protected]
(Received 7 June 2005, revised 1 August
2005, accepted 4 August 2005)
doi:10.1111/j.1742-4658.2005.04903.x
Human amylin (hA) is a small fibrillogenic protein that is the major con-
stituent of pancreatic islet amyloid, which occurs in most subjects with
type-2 diabetes mellitus (T2Dm). There is growing evidence that hA toxic-
ity towards islet b-cells is responsible for their gradual loss of function in
T2Dm. Preventing hA-mediated cytotoxicity has been proposed as a route
to halt the progression of this disease, although this has not yet been dem-
onstrated in vivo. The aim of our studies, in which we show that a small
number of hA-treated cells exhibit intracellular accumulation of reactive
oxygen species (ROS), was to evaluate the role of oxidative stress in the
mechanism of hA-mediated cytotoxicity. Here we report that catalase and
n-propyl gallate, antioxidants that are thought to act mainly as free radical
scavengers, afford RINm5F cells only limited protection against hA-medi-
ated toxicity. By contrast, the thiol antioxidants, N-acetyl-L-cysteine
(NAC), GSH and dithiothreitol, which not only react with ROS, but also
modulate the cellular redox potential by increasing intracellular levels of
GSH and ⁄or by acting as thiol reducing agents, afford almost complete
protection and inhibit the progression of hA-evoked apoptosis. We also
show that hA treatment is not associated with changes in intracellular
GSH levels and that inhibition of GSH biosynthesis has no effect on either
hA-mediated cytotoxicity or NAC-mediated protection. These results indi-
cate that, in addition to the induction of oxidative stress, hA appears to
mediate cytotoxicity through signalling pathways that are sensitive to the
actions of thiol antioxidants.
Abbreviations
BSO, buthionine-(S,R)-sulfoximine; carboxy-H2DCFDA, 5-(and-6)-carboxy-2¢,7¢-dichlorodihydrofluorescein diacetate; EthD-1, ethidium
homodimer-1; hA, human amylin; JNK, c-Jun NH2-terminal kinase; KRH, Hepes-balanced Krebs-Ringer bicarbonate buffer; NAC, N-acetyl-
L-cysteine; n-PG, n-propyl gallate; rA, rat amylin; RIN, rat insulinoma; ROS, reactive oxygen species; T2DM, type-2 diabetes mellitus.
FEBS Journal 272 (2005) 4949–4959 ª 2005 FEBS 4949

associated with the degeneration of pancreatic b-cells[14–16]. To summarise, there is strong support for the
hypothesis that hA-mediated toxicity towards pancre-
atic b-cells occurs in vivo and plays a role in the patho-
physiology of T2Dm.
Preventing hA-induced loss of pancreatic b-cells has
been proposed as a route to attenuate the gradual
decline in endogenous production of insulin in T2Dm
[17]. However, at this time knowledge of the mecha-
nisms by which hA evokes pancreatic b-cell death is
not complete.
Electron microscopic examination of hA-treated
RINm5F cells provided morphological evidence of cell
death through apoptosis [18]. In addition, DNA frag-
mentation, a hallmark of apoptosis, as well as the
up-regulation of the apoptosis-specific genes, p53 and
p21WAF1 ⁄CIP1, were detected in RINm5F cells follow-
ing treatment with hA [9]. Recent data point towards
the role of c-Jun, caspases 8 and 3, and the c-Jun
NH2-terminal kinase (JNK) signalling pathway in the
hA-induced apoptosis of RINm5F cells [19–21].
The signalling pathways found to be implicated in
the apoptosis of pancreatic b-cells are generally
believed to include phenomena such as (a) increases
in intracellular calcium [22,23] (b) overproduction of
reactive oxygen species (ROS) [24] (c) increased pro-
duction of ceramide [25], and (d) activation of mito-
gen-activated protein kinases [26]. Previous results
from our laboratory excluded the elevation of intracel-
lular calcium as a signalling mechanism in the
hA-mediated death of RINm5F cells [27]. Reports in
the literature implicating hA-mediated induction of
oxidative stress in pancreatic b-cells are contradictory
[7,8,28]. Lorenzo et al. [7] reported that hA toxicity
towards primary cultures of b-cells could not be pre-
vented with the antioxidant vitamin E. However,
Janciauskiene and Ahren [8,28] reported hA treatment
of RINm5F cells to be associated with elevated levels
of cellular lipid peroxidation, and increased activity of
glutathione reductase and membrane NADPH oxidase.
These authors interpreted their findings as indicative of
the occurrence of oxidative stress in hA-treated
RINm5F cells. In support of this proposition, they
correlated the sensitivity of RINm5F cells to hA-medi-
ated toxicity with sensitivity to hydrogen peroxide
(H2O2), while another pancreatic b-cell line, hamster
insulinoma HIT-T15, was found to be resistant to both
hA and H2O2 [8].
Furthermore, there is growing evidence for the
involvement of oxidative stress in the neurotoxicity of
amyloidogenic peptides thought to be responsible for
neuronal cell loss in various neurodegenerative dis-
orders [29]. In addition, it has been shown that the
cytotoxic effects of fibril-forming peptides, which are
not implicated in any amyloid diseases, are also associ-
ated with increased ROS production [30,31]. Indeed, it
has been proposed that oxidative stress represents a
common mechanism of toxicity shared by all amyloido-
genic peptides, including human amylin [29,30,32].
Much of this work has been performed on neuronal
cells, but little with b-cells.Here, our aim was to determine whether oxidative
stress might play a role in hA-mediated b-cell death.
We report that thiol antioxidants act on hA-induced
toxicity to prevent apoptosis. While we show that hA
treatment is associated with a certain degree of intra-
cellular oxidative activity, our results do not support
induction of oxidative stress as the only mechanism
through which hA mediates its cytotoxicity in these
cells. Instead our data indicate that toxicity of hA also
involves a signalling pathway that is regulated by the
redox status of intracellular thiol-containing com-
pounds.
Results
Antioxidants protect pancreatic b-cells against
hA-mediated toxicity
Several antioxidants were evaluated for their protect-
ive potential against hA-mediated toxicity of b-cells:catalase, n-propyl gallate (n-PG), N-acetyl-L-cysteine
(NAC), reduced glutathione (GSH) and dithiothreitol.
These compounds have diverse antioxidant actions
and their protective effects against oxidative damage
have been reported in other cell systems [33–37]. Our
selection of antioxidants included those that act enzy-
matically (catalase [38]) or as cofactors of endo-
genous antioxidant enzymes (NAC and GSH [39]),
and others that are thought to act nonenzymatically
(n-PG and dithiothreitol). Catalase acts in the extra-
cellular space, while the other antioxidants are cell
membrane permeable (n-PG [40], NAC and dithio-
threitol [41]; GSH ethyl ester [42]). In addition, these
antioxidants scavenge different types of ROS, e.g.
catalase decomposes H2O2 [38], n-PG scavenges
superoxide anion [34,43], NAC and GSH scavenge
hydroxyl radical [44]. They are also thought to pre-
vent different types of oxidative damage, e.g. n-PG
prevents lipid peroxidation [45], whereas NAC and
GSH prevent thiol oxidation [46]. Catalase and n-PG
are thought to act purely as free radical scavengers,
while the thiol antioxidants, NAC, GSH and dithio-
threitol, can also act by replenishing intracellular
stores of the endogenous antioxidant, GSH, or as
thiol-reducing agents [46,47].
Thiol antioxidants suppress hA cytotoxicity B. Konarkowska et al.
4950 FEBS Journal 272 (2005) 4949–4959 ª 2005 FEBS
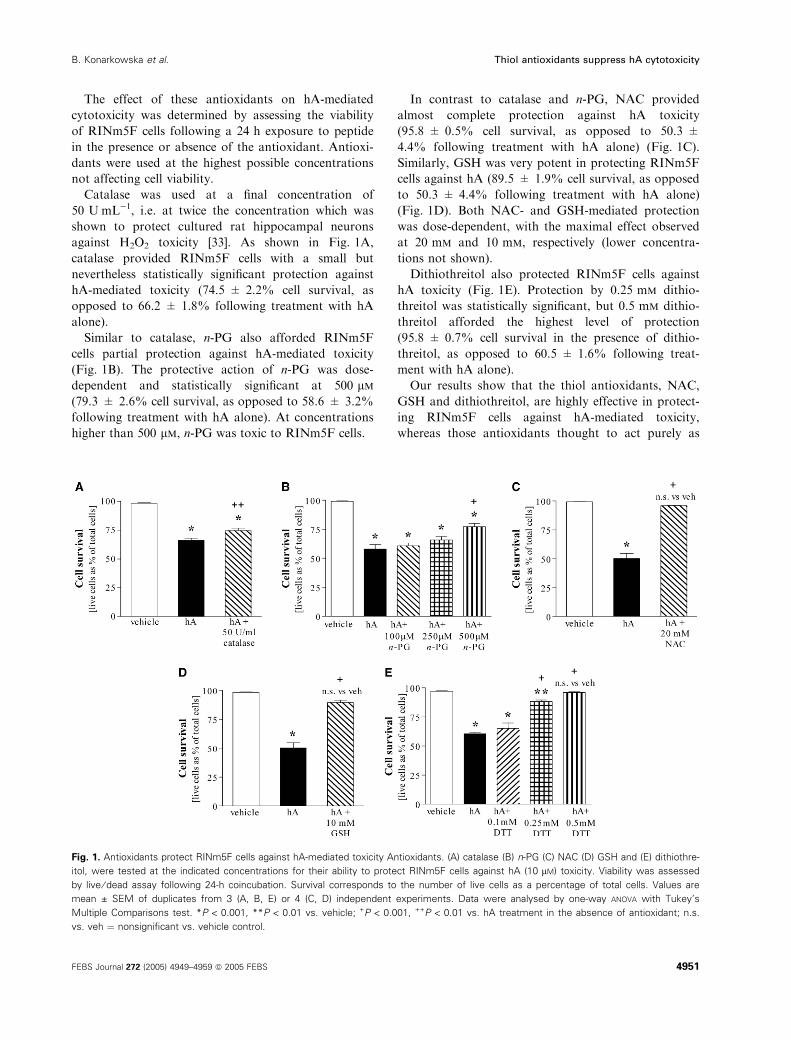
The effect of these antioxidants on hA-mediated
cytotoxicity was determined by assessing the viability
of RINm5F cells following a 24 h exposure to peptide
in the presence or absence of the antioxidant. Antioxi-
dants were used at the highest possible concentrations
not affecting cell viability.
Catalase was used at a final concentration of
50 UÆmL)1, i.e. at twice the concentration which was
shown to protect cultured rat hippocampal neurons
against H2O2 toxicity [33]. As shown in Fig. 1A,
catalase provided RINm5F cells with a small but
nevertheless statistically significant protection against
hA-mediated toxicity (74.5 ± 2.2% cell survival, as
opposed to 66.2 ± 1.8% following treatment with hA
alone).
Similar to catalase, n-PG also afforded RINm5F
cells partial protection against hA-mediated toxicity
(Fig. 1B). The protective action of n-PG was dose-
dependent and statistically significant at 500 lm(79.3 ± 2.6% cell survival, as opposed to 58.6 ± 3.2%
following treatment with hA alone). At concentrations
higher than 500 lm, n-PG was toxic to RINm5F cells.
In contrast to catalase and n-PG, NAC provided
almost complete protection against hA toxicity
(95.8 ± 0.5% cell survival, as opposed to 50.3 ±
4.4% following treatment with hA alone) (Fig. 1C).
Similarly, GSH was very potent in protecting RINm5F
cells against hA (89.5 ± 1.9% cell survival, as opposed
to 50.3 ± 4.4% following treatment with hA alone)
(Fig. 1D). Both NAC- and GSH-mediated protection
was dose-dependent, with the maximal effect observed
at 20 mm and 10 mm, respectively (lower concentra-
tions not shown).
Dithiothreitol also protected RINm5F cells against
hA toxicity (Fig. 1E). Protection by 0.25 mm dithio-
threitol was statistically significant, but 0.5 mm dithio-
threitol afforded the highest level of protection
(95.8 ± 0.7% cell survival in the presence of dithio-
threitol, as opposed to 60.5 ± 1.6% following treat-
ment with hA alone).
Our results show that the thiol antioxidants, NAC,
GSH and dithiothreitol, are highly effective in protect-
ing RINm5F cells against hA-mediated toxicity,
whereas those antioxidants thought to act purely as
Fig. 1. Antioxidants protect RINm5F cells against hA-mediated toxicity Antioxidants. (A) catalase (B) n-PG (C) NAC (D) GSH and (E) dithiothre-
itol, were tested at the indicated concentrations for their ability to protect RINm5F cells against hA (10 lM) toxicity. Viability was assessed
by live ⁄ dead assay following 24-h coincubation. Survival corresponds to the number of live cells as a percentage of total cells. Values are
mean ± SEM of duplicates from 3 (A, B, E) or 4 (C, D) independent experiments. Data were analysed by one-way ANOVA with Tukey’s
Multiple Comparisons test. *P < 0.001, **P < 0.01 vs. vehicle; +P < 0.001, ++P < 0.01 vs. hA treatment in the absence of antioxidant; n.s.
vs. veh ¼ nonsignificant vs. vehicle control.
B. Konarkowska et al. Thiol antioxidants suppress hA cytotoxicity
FEBS Journal 272 (2005) 4949–4959 ª 2005 FEBS 4951
free radical scavengers, catalase and n-PG, afford only
limited protection.
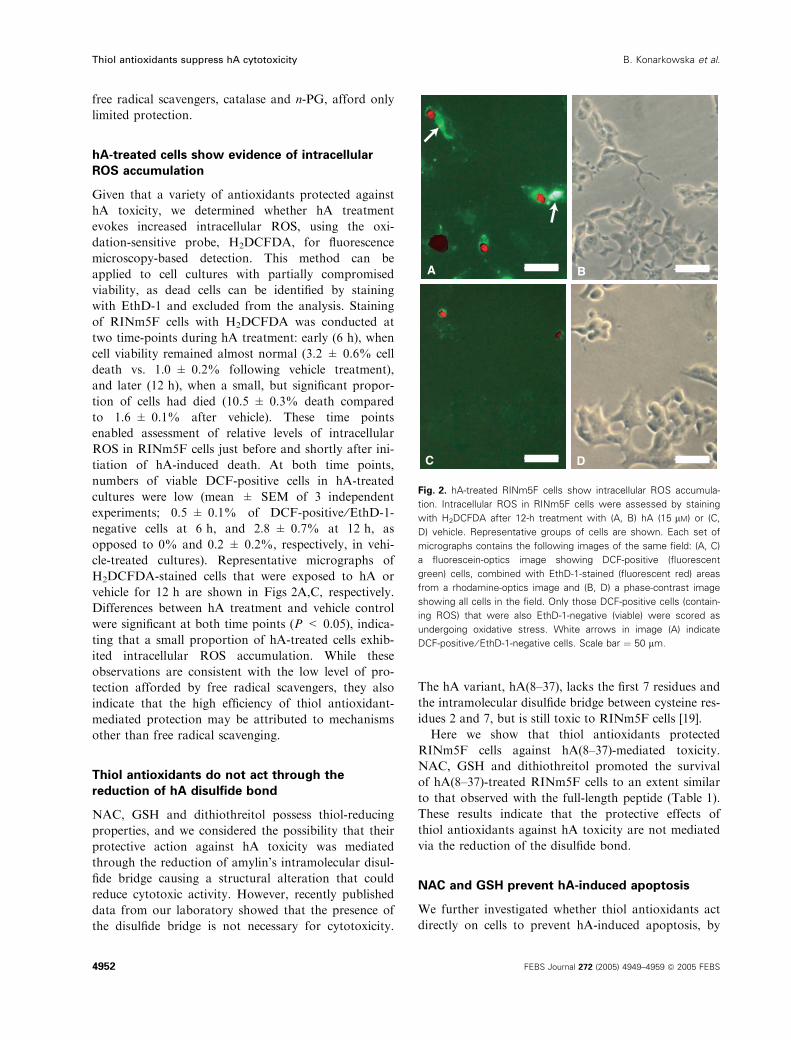
hA-treated cells show evidence of intracellular
ROS accumulation
Given that a variety of antioxidants protected against
hA toxicity, we determined whether hA treatment
evokes increased intracellular ROS, using the oxi-
dation-sensitive probe, H2DCFDA, for fluorescence
microscopy-based detection. This method can be
applied to cell cultures with partially compromised
viability, as dead cells can be identified by staining
with EthD-1 and excluded from the analysis. Staining
of RINm5F cells with H2DCFDA was conducted at
two time-points during hA treatment: early (6 h), when
cell viability remained almost normal (3.2 ± 0.6% cell
death vs. 1.0 ± 0.2% following vehicle treatment),
and later (12 h), when a small, but significant propor-
tion of cells had died (10.5 ± 0.3% death compared
to 1.6 ± 0.1% after vehicle). These time points
enabled assessment of relative levels of intracellular
ROS in RINm5F cells just before and shortly after ini-
tiation of hA-induced death. At both time points,
numbers of viable DCF-positive cells in hA-treated
cultures were low (mean ± SEM of 3 independent
experiments; 0.5 ± 0.1% of DCF-positive ⁄EthD-1-
negative cells at 6 h, and 2.8 ± 0.7% at 12 h, as
opposed to 0% and 0.2 ± 0.2%, respectively, in vehi-
cle-treated cultures). Representative micrographs of
H2DCFDA-stained cells that were exposed to hA or
vehicle for 12 h are shown in Figs 2A,C, respectively.
Differences between hA treatment and vehicle control
were significant at both time points (P < 0.05), indica-
ting that a small proportion of hA-treated cells exhib-
ited intracellular ROS accumulation. While these
observations are consistent with the low level of pro-
tection afforded by free radical scavengers, they also
indicate that the high efficiency of thiol antioxidant-
mediated protection may be attributed to mechanisms
other than free radical scavenging.
Thiol antioxidants do not act through the
reduction of hA disulfide bond
NAC, GSH and dithiothreitol possess thiol-reducing
properties, and we considered the possibility that their
protective action against hA toxicity was mediated
through the reduction of amylin’s intramolecular disul-
fide bridge causing a structural alteration that could
reduce cytotoxic activity. However, recently published
data from our laboratory showed that the presence of
the disulfide bridge is not necessary for cytotoxicity.
The hA variant, hA(8–37), lacks the first 7 residues and
the intramolecular disulfide bridge between cysteine res-
idues 2 and 7, but is still toxic to RINm5F cells [19].
Here we show that thiol antioxidants protected
RINm5F cells against hA(8–37)-mediated toxicity.
NAC, GSH and dithiothreitol promoted the survival
of hA(8–37)-treated RINm5F cells to an extent similar
to that observed with the full-length peptide (Table 1).
These results indicate that the protective effects of
thiol antioxidants against hA toxicity are not mediated
via the reduction of the disulfide bond.
NAC and GSH prevent hA-induced apoptosis
We further investigated whether thiol antioxidants act
directly on cells to prevent hA-induced apoptosis, by
A B
C D
Fig. 2. hA-treated RINm5F cells show intracellular ROS accumula-
tion. Intracellular ROS in RINm5F cells were assessed by staining
with H2DCFDA after 12-h treatment with (A, B) hA (15 lM) or (C,
D) vehicle. Representative groups of cells are shown. Each set of
micrographs contains the following images of the same field: (A, C)
a fluorescein-optics image showing DCF-positive (fluorescent
green) cells, combined with EthD-1-stained (fluorescent red) areas
from a rhodamine-optics image and (B, D) a phase-contrast image
showing all cells in the field. Only those DCF-positive cells (contain-
ing ROS) that were also EthD-1-negative (viable) were scored as
undergoing oxidative stress. White arrows in image (A) indicate
DCF-positive ⁄ EthD-1-negative cells. Scale bar ¼ 50 lm.
Thiol antioxidants suppress hA cytotoxicity B. Konarkowska et al.
4952 FEBS Journal 272 (2005) 4949–4959 ª 2005 FEBS
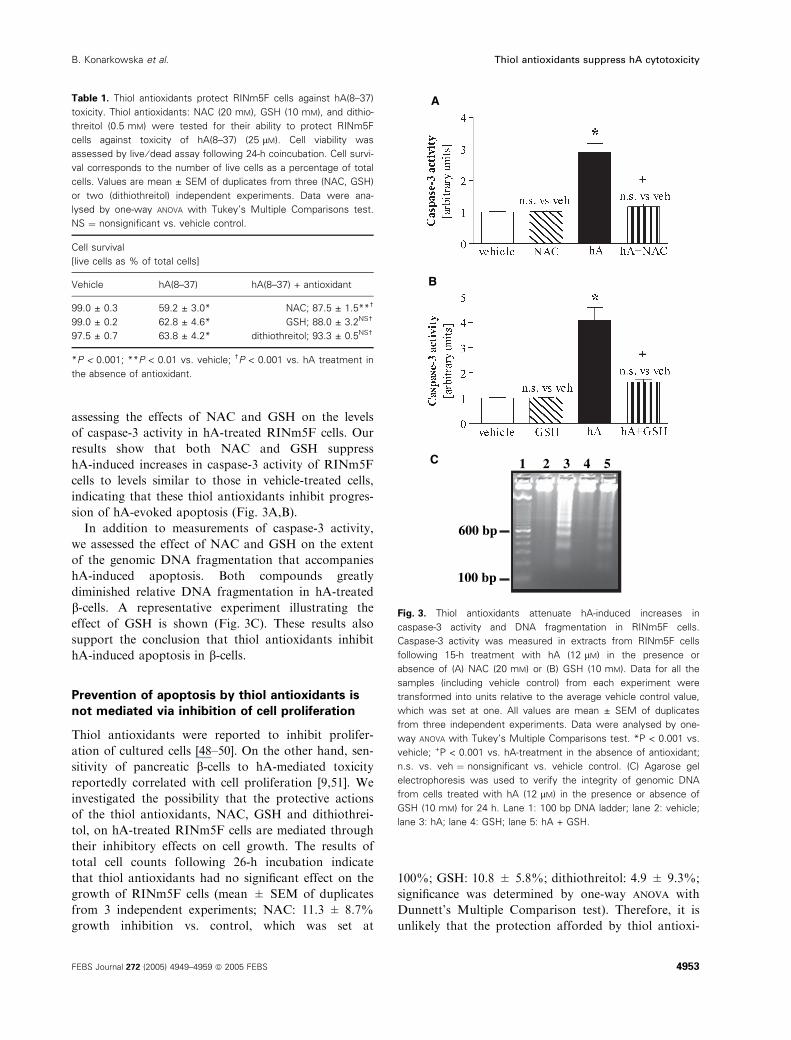
assessing the effects of NAC and GSH on the levels
of caspase-3 activity in hA-treated RINm5F cells. Our
results show that both NAC and GSH suppress
hA-induced increases in caspase-3 activity of RINm5F
cells to levels similar to those in vehicle-treated cells,
indicating that these thiol antioxidants inhibit progres-
sion of hA-evoked apoptosis (Fig. 3A,B).
In addition to measurements of caspase-3 activity,
we assessed the effect of NAC and GSH on the extent
of the genomic DNA fragmentation that accompanies
hA-induced apoptosis. Both compounds greatly
diminished relative DNA fragmentation in hA-treated
b-cells. A representative experiment illustrating the
effect of GSH is shown (Fig. 3C). These results also
support the conclusion that thiol antioxidants inhibit
hA-induced apoptosis in b-cells.
Prevention of apoptosis by thiol antioxidants is
not mediated via inhibition of cell proliferation
Thiol antioxidants were reported to inhibit prolifer-
ation of cultured cells [48–50]. On the other hand, sen-
sitivity of pancreatic b-cells to hA-mediated toxicity
reportedly correlated with cell proliferation [9,51]. We
investigated the possibility that the protective actions
of the thiol antioxidants, NAC, GSH and dithiothrei-
tol, on hA-treated RINm5F cells are mediated through
their inhibitory effects on cell growth. The results of
total cell counts following 26-h incubation indicate
that thiol antioxidants had no significant effect on the
growth of RINm5F cells (mean ± SEM of duplicates
from 3 independent experiments; NAC: 11.3 ± 8.7%
growth inhibition vs. control, which was set at
100%; GSH: 10.8 ± 5.8%; dithiothreitol: 4.9 ± 9.3%;
significance was determined by one-way anova with
Dunnett’s Multiple Comparison test). Therefore, it is
unlikely that the protection afforded by thiol antioxi-
Table 1. Thiol antioxidants protect RINm5F cells against hA(8–37)
toxicity. Thiol antioxidants: NAC (20 mM), GSH (10 mM), and dithio-
threitol (0.5 mM) were tested for their ability to protect RINm5F
cells against toxicity of hA(8–37) (25 lM). Cell viability was
assessed by live ⁄ dead assay following 24-h coincubation. Cell survi-
val corresponds to the number of live cells as a percentage of total
cells. Values are mean ± SEM of duplicates from three (NAC, GSH)
or two (dithiothreitol) independent experiments. Data were ana-
lysed by one-way ANOVA with Tukey’s Multiple Comparisons test.
NS ¼ nonsignificant vs. vehicle control.
Cell survival
[live cells as % of total cells]
Vehicle hA(8–37) hA(8–37) + antioxidant
99.0 ± 0.3 59.2 ± 3.0* NAC; 87.5 ± 1.5**†
99.0 ± 0.2 62.8 ± 4.6* GSH; 88.0 ± 3.2NS†
97.5 ± 0.7 63.8 ± 4.2* dithiothreitol; 93.3 ± 0.5NS†
*P < 0.001; **P < 0.01 vs. vehicle; †P < 0.001 vs. hA treatment in
the absence of antioxidant.
A
B
C 1 2 3 4 5
100 bp
600 bp
Fig. 3. Thiol antioxidants attenuate hA-induced increases in
caspase-3 activity and DNA fragmentation in RINm5F cells.
Caspase-3 activity was measured in extracts from RINm5F cells
following 15-h treatment with hA (12 lM) in the presence or
absence of (A) NAC (20 mM) or (B) GSH (10 mM). Data for all the
samples (including vehicle control) from each experiment were
transformed into units relative to the average vehicle control value,
which was set at one. All values are mean ± SEM of duplicates
from three independent experiments. Data were analysed by one-
way ANOVA with Tukey’s Multiple Comparisons test. *P < 0.001 vs.
vehicle; +P < 0.001 vs. hA-treatment in the absence of antioxidant;
n.s. vs. veh ¼ nonsignificant vs. vehicle control. (C) Agarose gel
electrophoresis was used to verify the integrity of genomic DNA
from cells treated with hA (12 lM) in the presence or absence of
GSH (10 mM) for 24 h. Lane 1: 100 bp DNA ladder; lane 2: vehicle;
lane 3: hA; lane 4: GSH; lane 5: hA + GSH.
B. Konarkowska et al. Thiol antioxidants suppress hA cytotoxicity
FEBS Journal 272 (2005) 4949–4959 ª 2005 FEBS 4953
dants towards hA-treated RINm5F cells is mediated
through inhibition of proliferation.
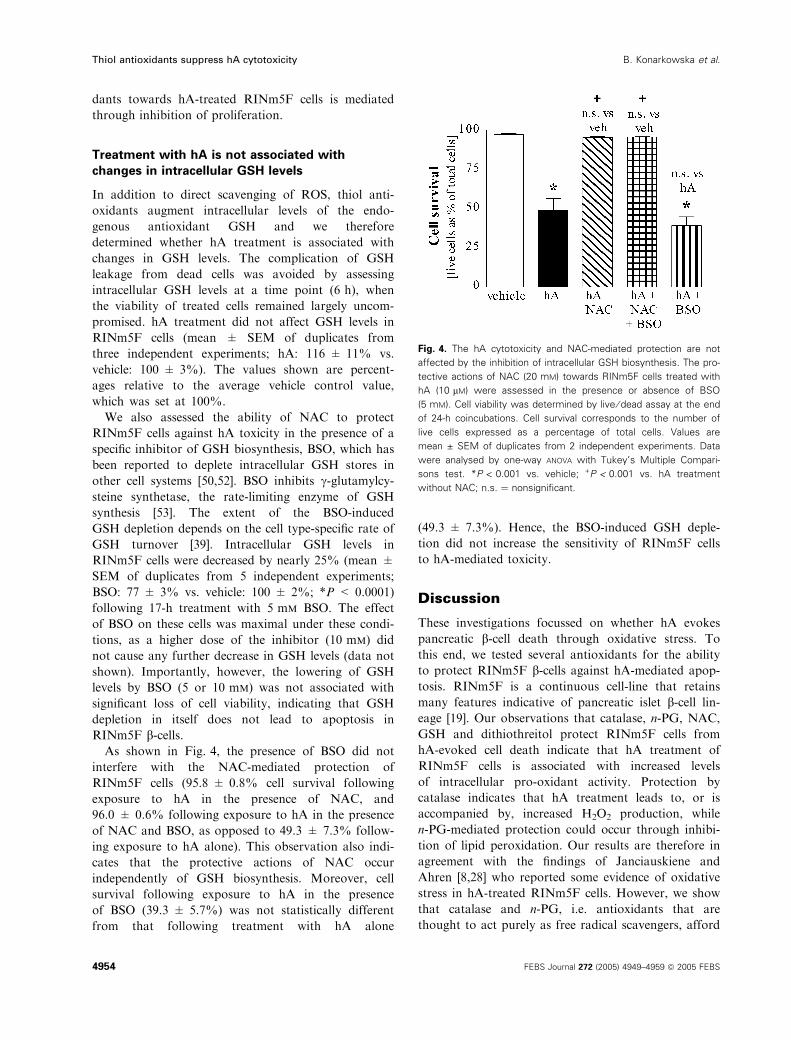
Treatment with hA is not associated with
changes in intracellular GSH levels
In addition to direct scavenging of ROS, thiol anti-
oxidants augment intracellular levels of the endo-
genous antioxidant GSH and we therefore
determined whether hA treatment is associated with
changes in GSH levels. The complication of GSH
leakage from dead cells was avoided by assessing
intracellular GSH levels at a time point (6 h), when
the viability of treated cells remained largely uncom-
promised. hA treatment did not affect GSH levels in
RINm5F cells (mean ± SEM of duplicates from
three independent experiments; hA: 116 ± 11% vs.
vehicle: 100 ± 3%). The values shown are percent-
ages relative to the average vehicle control value,
which was set at 100%.
We also assessed the ability of NAC to protect
RINm5F cells against hA toxicity in the presence of a
specific inhibitor of GSH biosynthesis, BSO, which has
been reported to deplete intracellular GSH stores in
other cell systems [50,52]. BSO inhibits c-glutamylcy-
steine synthetase, the rate-limiting enzyme of GSH
synthesis [53]. The extent of the BSO-induced
GSH depletion depends on the cell type-specific rate of
GSH turnover [39]. Intracellular GSH levels in
RINm5F cells were decreased by nearly 25% (mean ±
SEM of duplicates from 5 independent experiments;
BSO: 77 ± 3% vs. vehicle: 100 ± 2%; *P < 0.0001)
following 17-h treatment with 5 mm BSO. The effect
of BSO on these cells was maximal under these condi-
tions, as a higher dose of the inhibitor (10 mm) did
not cause any further decrease in GSH levels (data not
shown). Importantly, however, the lowering of GSH
levels by BSO (5 or 10 mm) was not associated with
significant loss of cell viability, indicating that GSH
depletion in itself does not lead to apoptosis in
RINm5F b-cells.As shown in Fig. 4, the presence of BSO did not
interfere with the NAC-mediated protection of
RINm5F cells (95.8 ± 0.8% cell survival following
exposure to hA in the presence of NAC, and
96.0 ± 0.6% following exposure to hA in the presence
of NAC and BSO, as opposed to 49.3 ± 7.3% follow-
ing exposure to hA alone). This observation also indi-
cates that the protective actions of NAC occur
independently of GSH biosynthesis. Moreover, cell
survival following exposure to hA in the presence
of BSO (39.3 ± 5.7%) was not statistically different
from that following treatment with hA alone
(49.3 ± 7.3%). Hence, the BSO-induced GSH deple-
tion did not increase the sensitivity of RINm5F cells
to hA-mediated toxicity.
Discussion
These investigations focussed on whether hA evokes
pancreatic b-cell death through oxidative stress. To
this end, we tested several antioxidants for the ability
to protect RINm5F b-cells against hA-mediated apop-
tosis. RINm5F is a continuous cell-line that retains
many features indicative of pancreatic islet b-cell lin-
eage [19]. Our observations that catalase, n-PG, NAC,
GSH and dithiothreitol protect RINm5F cells from
hA-evoked cell death indicate that hA treatment of
RINm5F cells is associated with increased levels
of intracellular pro-oxidant activity. Protection by
catalase indicates that hA treatment leads to, or is
accompanied by, increased H2O2 production, while
n-PG-mediated protection could occur through inhibi-
tion of lipid peroxidation. Our results are therefore in
agreement with the findings of Janciauskiene and
Ahren [8,28] who reported some evidence of oxidative
stress in hA-treated RINm5F cells. However, we show
that catalase and n-PG, i.e. antioxidants that are
thought to act purely as free radical scavengers, afford
Fig. 4. The hA cytotoxicity and NAC-mediated protection are not
affected by the inhibition of intracellular GSH biosynthesis. The pro-
tective actions of NAC (20 mM) towards RINm5F cells treated with
hA (10 lM) were assessed in the presence or absence of BSO
(5 mM). Cell viability was determined by live ⁄ dead assay at the end
of 24-h coincubations. Cell survival corresponds to the number of
live cells expressed as a percentage of total cells. Values are
mean ± SEM of duplicates from 2 independent experiments. Data
were analysed by one-way ANOVA with Tukey’s Multiple Compari-
sons test. *P < 0.001 vs. vehicle; +P < 0.001 vs. hA treatment
without NAC; n.s. ¼ nonsignificant.
Thiol antioxidants suppress hA cytotoxicity B. Konarkowska et al.
4954 FEBS Journal 272 (2005) 4949–4959 ª 2005 FEBS

only partial protection against hA-mediated toxicity.
Moreover, intracellular ROS accumulation is observed
only in a small number of hA-treated RINm5F cells,
although it is possible that hA-induced increase in
ROS levels is either transient or occurs shortly before
cell death. The suboptimal efficiency with which cat-
alase and n-PG suppress hA-mediated cytotoxicity, is
consistent with the findings of Lorenzo et al. [7], who
did not observe any protection of hA-treated human
and mouse islet b-cells by another free radical scaven-
ger, vitamin E. While our results provide evidence in
support of hA-induced oxidative stress, they also indi-
cate that it is not the only mechanism through which
hA evokes b-cell death. This conclusion is similar to
that drawn by other researchers who showed that the
occurrence of oxidative stress in cells exposed to other
amyloidogenic peptides such as b-amyloid or HypF-N
did not play a major role in the induction of cell death
[31,54,55].
In contrast to catalase and n-PG, the thiol antioxi-
dants, NAC, GSH and dithiothreitol, afford RINm5F
cells almost complete protection against the toxic hA
stimulus. Furthermore, these compounds hinder the
progression of hA-induced apoptosis, as indicated by
their inhibitory effects on hA-induced increase in ca-
spase-3 activity and internucleosomal DNA fragmenta-
tion.
In addition to their ROS-scavenging capabilities,
thiol antioxidants can also modulate the redox poten-
tial of cells by augmenting intracellular GSH levels
and ⁄or by acting as thiol reducing agents [46,47].
However, our experiments indicate that these com-
pounds do not act via the reduction of the hA disulfide
bridge, or through effects on cell proliferation. More-
over, intracellular GSH levels in hA-treated cells were
unchanged. Our observations that the BSO-induced
depletion of GSH failed to affect viability of RINm5F
cells or to increase the severity of hA-mediated toxicity
do not support the proposition that changes in GSH
levels play a decisive role in hA-evoked cell death, and
we conclude that the protective actions of NAC are
independent of GSH biosynthesis. Rather, it appears
that the protective actions of thiol antioxidants may be
mediated through effects on the redox status of cellular
thiols other than GSH.
We also investigated the possibility that thiol
antioxidants exerted their protective action towards
hA-treated RINm5F cells by affecting aggregate for-
mation. However, the combined results of our thiofla-
vin-T fluorescence and circular dichroism experiments
showed that there was only a small inhibitory effect of
thiol antioxidants on peptide aggregation, thus making
this proposal unlikely (data not shown).
In summary, we conclude that ROS are not the only
mediators of hA cytotoxicity. Instead hA-induced cell
death can be prevented by agents that affect the redu-
cing potential of the cell. An important component of
the hA-activated cell death signalling pathway is thus
likely to be regulated via its thiol redox status. There
is growing evidence in the literature that thiol-reduc-
tants can alter the expression and activity of various
cell death-regulating transcription factors [56–59].
Redox-regulated proteins include JNK, a pro-apop-
totic factor, and extracellular signal-regulated kinase
(ERK), which reportedly has antiapoptotic activity
[60]. Both these proteins have been recently implicated
in hA-mediated apoptosis [19,21]. NAC itself, has been
reported to activate ERK [61] and inhibit JNK activity
[58]. Perhaps NAC with its dual ability to activate
ERK and suppress JNK activity may influence the sig-
nalling pathway leading to hA-mediated apoptosis.
Thus, inclusion of thiol antioxidants in the therapy of
T2Dm patients might be beneficial in the prevention of
hA-mediated loss of pancreatic islet b-cells.
Experimental procedures
Peptides and chemicals
hA and hA(8–37) were lyophilized preparations (Bachem
California, Torrance, CA, USA; hA, #0542559 and 0538994;
hA(8–37), #0523544). Aqueous stock solutions were pre-
pared immediately before use, by dissolution in water (18
MW; MilliQ, Millipore) to a final concentration of 500 lm.Catalase (from bovine liver), n-propyl gallate (n-PG), N-ace-
tyl-l-cysteine (NAC), reduced glutathione ethyl ester (GSH)
and buthionine-(S,R)-sulfoximine (BSO) were from Sigma
(St Louis, MO, USA), and dithiothreitol was from Biochemi-
ca (AppliChem GmbH, Germany). Catalase was dissolved
(final concentration 55 UÆmL)1; one unit decomposes
1.0 lmol of H2O2 per min at pH 7.0 at 25 �C) in RPMI 1640
and supplemented with fetal bovine serum [FBS, 10% (v ⁄ v)].Stocks of n-PG, NAC, GSH, dithiothreitol and BSO in water
were prepared (n-PG, 10 mm; NAC, 1000 mm; GSH,
500 mm; dithiothreitol, 100 mm; BSO, 225 mm) and aliquots
used to prepare antioxidant-containing media. Proteinase K
was from Life Technologies (Auckland, New Zealand), and
5-(and-6)-carboxy-2¢,7¢-dichlorodihydrofluorescein diacetate
(carboxy-H2DCFDA) from Molecular Probes (Sunnyvale,
CA, USA). Caspase-3 substrate Ac-DEVD-AFC (Bio-Rad,
Hercules, CA, USA) was dissolved in DMSO. General cell
culture materials were from Invitrogen (Carlsbad, CA,
USA). All other chemicals were of analytical grade or better.
Hepes-balanced Krebs-Ringer bicarbonate buffer (KRH)
was comprised of 119 mm NaCl, 4.74 mm KCl, 2.54 mm
CaCl2, 1.19 mm MgCl2, 1.19 mm KH2PO4, 25 mm NaHCO3,
and 10 mm Hepes, pH 7.4.
B. Konarkowska et al. Thiol antioxidants suppress hA cytotoxicity
FEBS Journal 272 (2005) 4949–4959 ª 2005 FEBS 4955

RINm5F cell culture
RINm5F cells were gifted by H K Oie (NIH, Bethesda,
MD) and maintained as previously described [9,27]. Here,
cells between passages 28 and 39 were grown in 24- or
6-well cell dishes (Greiner, Germany). The mechanism of
hA-evoked islet b-cell apoptosis in RINm5F cells closely
resembles that in human b-cells [19].
Cytotoxicity evoked by hA
Cells were cultured (48 h, 24-well dishes, initially 1.5 · 105
per well); washed (NaCl ⁄Pi, once); then preincubated (fresh
medium with antioxidant or relevant vehicle) for 30 min
(catalase) or 3–4 h (n-PG, NAC, GSH or dithiothreitol). In
experiments to assess the effect of BSO on hA toxicity or
the protective potential of NAC, treatment with 5 mm BSO
(or relevant vehicle) was initiated 2 h before incubation with
hA or preincubation with NAC, respectively. Next, aliquots
of either hA or hA(8–37) stock solution (or vehicle) were
added to final concentrations as follows: hA, 10 lm;hA(8–37), 25 lm; catalase, 50 UÆmL)1; n-PG, 100, 250 and
500 lm; NAC, 20 mm; GSH, 10 mm; and dithiothreitol, 0.1,
0.25 and 0.5 mm. Following hA treatment (24 h), viability
was determined [calcein-AM ⁄ ethidium homodimer-1 (EthD-
1)], as described previously [17,18].
Detection of intracellular ROS accumulation
Intracellular ROS were detected by carboxy-H2DCFDA
staining wherein fresh stock solutions (10 mm in DMSO)
were used for each experiment. RINm5F cells were plated
(24-well dishes, 1.2 · 105 cells per well), grown (48 h), then
treated [6 or 12 h, hA (15 lm) or vehicle]. Three independ-
ent experiments were performed, each of which included
two wells per condition: cells in one well were stained with
carboxy-H2DCFDA and the other well treated with DMSO.
After hA treatment, cells were rinsed (KRH), then serum-
free phenol red-free RPMI 1640 and carboxy-H2DCFDA
(100 lm final) or an equivalent amount of DMSO were
added. Cells were incubated (1 h, 37 �C, dark), rinsed
twice (serum-free phenol red-free RPMI 1640), visualized
[2¢,7¢-dichlorofluorescein, (DCF), Zeiss Axiovert S100] (Carl
Zeiss International, Oberkochen, Germany), and images
digitized (25 · , Zeiss AxioCam). In each experiment, six
fields from each well were recorded (with both fluorescein
optics and phase-contrast). EthD-1 (final concentration
2 lm) was added and each field re-photographed (rhodam-
ine optics for dead cells). DCF-positive cells (i.e. exhibiting
intracellular ROS accumulation) that were also EthD-1-neg-
ative (i.e. viable), were scored as cells undergoing oxidative
stress; numbers were expressed as percentages of total cells.
No fluorescence was observed in control or hA-treated cells
without carboxy-H2DCFDA-staining.
Caspase-3 activity
Caspase-3 activity in cell extracts was quantified by measur-
ing cleavage of a synthetic oligopeptide substrate for
caspase-3, Ac-DEVD-AFC, according to established proto-
cols [19] with the following modifications: (a) cells were
grown (plating density 1.6 · 106 cells per well, 6-well plates)
for 48 h before treatment; (b) following hA-treatment
(15 h, 12 lm), cells were lysed in buffer (100 lL per well,
or 85 lL per well when 2–3 wells were combined), then
combined with lysates (20 lL) of floating cells; (c) following
freeze-thaw cycles, samples were incubated (ice, 15 min)
and centrifuged; (d) 40–60 lL of extracts were added to
reaction mixtures [100 lL final volume; Hepes, 15 mm,
pH 7.4; EDTA, 3 mm; CHAPS, 0.15% (w ⁄ v); dithiothrei-
tol, 9.5 mm; DMSO, 3% (v ⁄ v); Ac-DEVD-AFC, 55 lm];and (e) mixtures were incubated (2 h, 37 �C) before fluores-
cence measurement (emission, 540 nm; excitation, 400 nm)
of duplicates for each condition. Data were trans-
formed into units relative to the average control value, set
at one.
Genomic DNA integrity
RINm5F cells were cultured (48 h; plated at 1.6 · 106 cells
per well in 6-well plates); preincubated [4 h; GSH (10 mm)
or vehicle]; then treated with hA (12 lm) or vehicle
(± GSH, 24 h). Cells were then harvested by trypsiniza-
tion, resuspended (fresh lysis buffer, 10 mm Tris.HCl,
pH 8.0, 10 mm NaCl, 10 mm EDTA, 0.5% SDS,
3 mgÆmL)1 proteinase K), and incubated (24 h, 55 �C).Genomic DNA was extracted (buffer-saturated phenol,
pH 8.0 – chloroform) in three steps: (a) phenol; (b)
phenol ⁄ chloroform ⁄ isoamyl alcohol (25 : 24 : 1; v ⁄ v ⁄ v);and (c) chloroform ⁄ isoamyl alcohol (24 : 1; v ⁄ v). DNA was
precipitated (0.3 mm sodium acetate in ethanol, on solid
CO2, 30 min) or at )20 �C for 24 h. Precipitated DNA was
washed [70% (v ⁄ v) aqueous ethanol], air-dried and resus-
pended (water). Following RNase A treatment (20–45 min,
37 �C), DNA was re-purified using the same procedures for
extraction, precipitation and purification as described
above, except that the first extraction step was carried out
with phenol ⁄ chloroform ⁄ isoamyl alcohol (25 : 24 : 1;
v ⁄ v ⁄ v), and then resuspended [10 mm Tris ⁄HCl
(pH 8.0) ⁄ 1 mm EDTA]. DNA samples (15 lg) were electro-
phoresed (1.5% agarose), stained (ethidium bromide) and
UV-visualized with a 100 bp DNA ladder (Invitrogen).
RINm5F cell growth
RINm5F cells were grown (48 h; 24-well plates initially at
1.5 · 105 cells per well), then treated with antioxidants
(20 mm NAC or 10 mm GSH or 0.5 mm dithiothreitol)
for 26 h. Cells were harvested (trypsinization), combined
Thiol antioxidants suppress hA cytotoxicity B. Konarkowska et al.
4956 FEBS Journal 272 (2005) 4949–4959 ª 2005 FEBS

with the floating cells, centrifuged and counted (haemo-
cytometer), and analysed in duplicate. Counts were
expressed as percentages of control (i.e. vehicle-treated)
numbers.
Changes in intracellular GSH
Intracellular GSH was determined (Glutathione Apoptosis
Detection Kit; Oncogene, San Diego, CA, USA) according
to the manufacturer’s instructions, with minor modifica-
tions. Cells were cultured (48 h; plating density 1.2 · 105
per well in 24-well dishes; or at 6.0 · 105 per well, 6-well
dishes). Experiments were performed in duplicate or tripli-
cate and cells from either one (6-well plate) or two wells (24-
well plate) were used for sample preparation. Following hA-
(15 lm) or vehicle-treatment (6 h), cells were trypsinized
and resuspended (lysis buffer, 60 lL per well in 6-well
plates, or 25 lL per two wells in 24-well plates). Following
debris removal (centrifugation), reaction mixtures were pre-
pared: extract (50 lL) from a 6-well dish was mixed with
monochlorobimane solution (1 lL) and GST reagent
(1 lL); or extract (16 lL) from a 24-well dish, monochloro-
bimane solution (0.5 lL), and glutathione-S-transferase rea-
gent (0.5 lL). Fluorescence values for ‘treated’ and ‘vehicle
control’ samples were normalized to protein concentrations.
Results are expressed as percentages relative to the average
vehicle control value, which was set at 100%.
Statistical analysis
Statistical analysis was conducted using either unpaired
two-tailed t-test (to compare two conditions), or one-way
anova with Dunnett’s or Tukey’s Multiple Comparisons
test (to analyse more than two conditions). Statistical signi-
ficance was determined at P < 0.05.
Acknowledgements
This research was supported by Grants from the Foun-
dation for Research, Science and Technology, New
Zealand, Endocore Research Trust, Lottery Health
(NZ), Maurice & Phyllis Paykel Trust, and the Univer-
sity of Auckland Graduate Research Fund. We thank
J.Z. Bai for technical assistance.
References
1 Cooper GJ (1994) Amylin compared with calcitonin
gene-related peptide: structure, biology, and relevance
to metabolic disease. Endocr Rev 15, 163–201.
2 Moore CX & Cooper GJ (1991) Co-secretion of amylin
and insulin from cultured islet b-cells: modulation by
nutrient secretagogues, islet hormones and hypoglycemic
agents. Biochem Biophys Res Comm 179, 1–9.
3 Bell ET (1959) Hyalinization of the islet of Langerhans
in non-diabetic individuals. Am J Pathol 35, 801–805.
4 Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB
& Reid KB (1987) Purification and characterization of
a peptide from amyloid-rich pancreases of type 2
diabetic patients. Proc Natl Acad Sci USA 84, 8628–
8632.
5 Lukinius A, Wilander E, Westermark GT, Engstrom U
& Westermark P (1989) Colocalization of islet amyloid
polypeptide and insulin in the B cell secretory granules
of the human pancreatic islets. Diabetologia 32, 240–
244.
6 de Koning EJ, Bodkin NL, Hansen BC & Clark A
(1993) Diabetes mellitus in Macaca mulatta monkeys is
characterised by islet amyloidosis and reduction in beta-
cell population. Diabetologia 36, 378–384.
7 Lorenzo A, Razzaboni B, Weir GC & Yankner BA
(1994) Pancreatic islet cell toxicity of amylin associated
with type-2 diabetes mellitus. Nature 368, 756–760.
8 Janciauskiene S & Ahren B (1998) Different sensitivity
to the cytotoxic action of IAPP fibrils in two insulin-
producing cell lines, HIT-T15 and RINm5F cells.
Biochem Biophys Res Comm 251, 888–893.
9 Zhang S, Liu J, Saafi EL & Cooper GJ (1999) Induction
of apoptosis by human amylin in RINm5F islet b-cellsis associated with enhanced expression of p53 and
p21WAF1 ⁄ CIP1. FEBS Lett 455, 315–320.
10 Janson J, Ashley RH, Harrison D, McIntyre S & Butler
PC (1999) The mechanism of islet amyloid polypeptide
toxicity is membrane disruption by intermediate-sized
toxic amyloid particles. Diabetes 48, 491–498.
11 Westermark P (1973) Fine structure of islets of Langer-
hans in insular amyloidosis. Virchows Arch-a: Pathol-
Pathologische Anat 359, 1–18.
12 Westermark P & Wilander E (1978) The influence of
amyloid deposits on the islet volume in maturity onset
diabetes mellitus. Diabetologia 15, 417–421.
13 Garg A, Chandalia M & Vuitch F (1996) Severe islet
amyloidosis in congenital generalized lipodystrophy.
Diabetes Care 19, 28–31.
14 Wang F, Hull RL, Vidal J, Cnop M & Kahn SE (2001)
Islet amyloid develops diffusely throughout the pancreas
before becoming severe and replacing endocrine cells.
Diabetes 50, 2514–2520.
15 Couce M, Kane LA, O’Brien TD, Charlesworth J, Soel-
ler W, McNeish J, Kreutter D, Roche P & Butler PC
(1996) Treatment with growth hormone and dexametha-
sone in mice transgenic for human islet amyloid poly-
peptide causes islet amyloidosis and b-cell dysfunction.Diabetes 45, 1094–1101.
16 Soeller WC, Janson J, Hart SE, Parker JC, Carty MD,
Stevenson RW, Kreutter DK & Butler PC (1998) Islet
amyloid-associated diabetes in obese A(vy) ⁄ a mice
expressing human islet amyloid polypeptide. Diabetes
47, 743–750.
B. Konarkowska et al. Thiol antioxidants suppress hA cytotoxicity
FEBS Journal 272 (2005) 4949–4959 ª 2005 FEBS 4957

17 Aitken JF, Loomes KM, Konarkowska B & Cooper GJ
(2003) Suppression by polycyclic compounds of the
conversion of human amylin into insoluble amyloid.
Biochem J 374, 779–784.
18 Saafi EL, Konarkowska B, Zhang S, Kistler J & Cooper
GJ (2001) Ultrastructural evidence that apoptosis is the
mechanism by which human amylin evokes death in
RINm5F pancreatic islet b-cells. Cell Biol Internat 25,339–350.
19 Zhang S, Liu J, Dragunow M & Cooper GJ (2003)
Fibrillogenic amylin evokes islet b-cell apoptosisthrough linked activation of a caspase cascade and
JNK1. J Biol Chem 278, 52810–52819.
20 Zhang S, Liu J, MacGibbon G, Dragunow M &
Cooper GJ (2002) Increased expression and activation
of c-Jun contributes to human amylin-induced apop-
tosis in pancreatic islet b-cells. J Mol Biol 324, 271–
285.
21 Rumora L, Hadzija M, Barisic K, Maysinger D & Gru-
biic TZ (2002) Amylin-induced cytotoxicity is associated
with activation of caspase-3 and MAP kinases. Biol
Chem 383, 1751–1758.
22 Efanova IB, Zaitsev SV, Zhivotovsky B, Kohler M,
Efendic S, Orrenius S & Berggren PO (1998) Glucose
and tolbutamide induce apoptosis in pancreatic b-cells.A process dependent on intracellular Ca2+ concentra-
tion. J Biol Chem 273, 33501–33507.
23 Wang L, Bhattacharjee A, Zuo Z, Hu F, Honkanen
RE, Berggren PO & Li M (1999) A low voltage-acti-
vated Ca2+ current mediates cytokine-induced pancre-
atic b-cell death. Endocrinology 140, 1200–1204.
24 Dypbukt JM, Ankarcrona M, Burkitt M, Sjoholm A,
Strom K, Orrenius S & Nicotera P (1994) Different
prooxidant levels stimulate growth, trigger apoptosis,
or produce necrosis of insulin-secreting RINm5F cells.
The role of intracellular polyamines. J Biol Chem 269,
30553–30560.
25 Shimabukuro M, Zhou YT, Levi M & Unger RH
(1998) Fatty acid-induced b cell apoptosis: a link
between obesity and diabetes. Proc Natl Acad Sci USA
95, 2498–2502.
26 Mandrup-Poulsen T (2001) b-cell apoptosis: stimuli and
signaling. Diabetes 50 (Suppl. 1), S58–S63.
27 Bai JZ, Saafi EL, Zhang S & Cooper GJ (1999) Role of
Ca2+ in apoptosis evoked by human amylin in pancre-
atic islet b-cells. Biochem J 343, 53–61.
28 Janciauskiene S & Ahren B (2000) Fibrillar islet amy-
loid polypeptide differentially affects oxidative mechan-
isms and lipoprotein uptake in correlation with
cytotoxicity in two insulin-producing cell lines. Biochem
Biophys Res Comm 267, 619–625.
29 Stefani M & Dobson CM (2003) Protein aggregation
and aggregate toxicity: new insights into protein folding,
misfolding diseases and biological evolution. J Mol Med
81, 678–699.
30 Schubert D, Behl C, Lesley R, Brack A, Dargusch R,
Sagara Y & Kimura H (1995) Amyloid peptides are
toxic via a common oxidative mechanism. Proc Natl
Acad Sci USA 92, 1989–1993.
31 Bucciantini M, Calloni G, Chiti F, Formigli L, Nosi D,
Dobson CM & Stefani M (2004) Prefibrillar amyloid
protein aggregates share common features of cytotoxi-
city. J Biol Chem 279, 31374–31382.
32 Mattson MP & Goodman Y (1995) Different amyloido-
genic peptides share a similar mechanism of neurotoxi-
city involving reactive oxygen species and calcium.
Brain Res 676, 219–224.
33 Lockhart BP, Benicourt C, Junien JL & Privat A (1994)
Inhibitors of free radical formation fail to attenuate
direct b-amyloid 25)35 peptide-mediated neurotoxicity in
rat hippocampal cultures. J Neurosci Res 39, 494–505.
34 Reddan JR, Giblin FJ, Sevilla M, Padgaonkar V,
Dziedzic DC, Leverenz VR, Misra IC, Chang JS &
Pena JT (2003) Propyl gallate is a superoxide dismutase
mimic and protects cultured lens epithelial cells from
H2O2 insult. Exp Eye Res 76, 49–59.
35 Wu TW, Fung KP, Zeng LH, Wu J & Nakamura H
(1994) Propyl gallate as a hepatoprotector in vitro and
in vivo. Biochem Pharmacol 48, 419–422.
36 Liu L, Trimarchi JR & Keefe DL (1999) Thiol oxida-
tion-induced embryonic cell death in mice is prevented
by the antioxidant dithiothreitol. Biol Reprod 61, 1162–
1169.
37 Starke PE & Farber JL (1985) Endogenous defenses
against the cytotoxicity of hydrogen peroxide in cul-
tured rat hepatocytes. J Biol Chem 260, 86–92.
38 Halliwell B & Gutteridge JMC (1989) Free Radicals in
Biology and Medicine. Oxford University Press, Oxford,
UK.
39 Meister A & Anderson ME (1983) Glutathione. Annu
Rev Biochem 52, 711–760.
40 Takami M, Preston SL, Toyloy VA & Behrman HR
(1999) Antioxidants reversibly inhibit the spontaneous
resumption of meiosis. Am J Physiol 276, E684–E688.
41 Newton GL, Aguilera JA, Kim T, Ward JF & Fahey
RC (1996) Transport of aminothiol radioprotectors into
mammalian cells: passive diffusion versus mediated
uptake. Radiat Res 146, 206–215.
42 Anderson ME, Powrie F, Puri RN & Meister A (1985)
Glutathione monoethyl ester: preparation, uptake by
tissues, and conversion to glutathione. Arch Biochem
Biophys 239, 538–548.
43 Deeble DJ, Parsons BJ & Phillips GO (1987) Evidence
for the addition of the superoxide anion to the anti-
oxidant n-propyl gallate in aqueous solution. Free Radic
Res Commun 2, 351–358.
44 Aruoma OI, Halliwell B, Hoey BM & Butler J (1989) The
antioxidant action of N-acetylcysteine: its reaction with
hydrogen peroxide, hydroxyl radical, superoxide, and
hypochlorous acid. Free Radic Biol Med 6, 593–597.
Thiol antioxidants suppress hA cytotoxicity B. Konarkowska et al.
4958 FEBS Journal 272 (2005) 4949–4959 ª 2005 FEBS

45 Zhu J, Johnson WJ, Sevilla CL, Herrington JW &
Sevilla MD (1990) An electron spin resonance study of
the reactions of lipid peroxyl radicals with antioxidants.
J Phys Chem 94, 7185–7190.
46 Cotgreave IA (1997) N-acetylcysteine: pharmacological
considerations and experimental and clinical applica-
tions. Adv Pharmacol 38, 205–227.
47 Meister A (1988) Glutathione metabolism and its select-
ive modification. J Biol Chem 263, 17205–17208.
48 Burdon RH, Alliangana D & Gill V (1994) Endogen-
ously generated active oxygen species and cellular glu-
tathione levels in relation to BHK-21 cell proliferation.
Free Radic Res 21, 121–133.
49 Ferrari G, Yan CY & Greene LA (1995) N-acetylcys-
teine (d- and l-stereoisomers) prevents apoptotic death
of neuronal cells. J Neurosci 15, 2857–2866.
50 Yan CY, Ferrari G & Greene LA (1995) N-acetylcys-
teine-promoted survival of PC12 cells is glutathione-
independent but transcription-dependent. J Biol Chem
270, 26827–26832.
51 Ritzel RA & Butler PC (2003) Replication increases
b-cell vulnerability to human islet amyloid polypeptide-
induced apoptosis. Diabetes 52, 1701–1708.
52 Oda T, Iwaoka J, Komatsu N & Muramatsu T (1999)
Involvement of N-acetylcysteine-sensitive pathways in
ricin-induced apoptotic cell death in U937 cells. Biosci
Biotechnol Biochem 63, 341–348.
53 Griffith OW, Anderson ME & Meister A (1979) Inhibi-
tion of glutathione biosynthesis by prothionine sulfoxi-
mine (S-n-propyl homocysteine sulfoximine), a selective
inhibitor of c-glutamylcysteine synthetase. J Biol Chem
254, 1205–1210.
54 Pike CJ, Ramezan-Arab N & Cotman CW (1997)
b-amyloid neurotoxicity in vitro: evidence of oxidative
stress but not protection by antioxidants. J Neurochem
69, 1601–1611.
55 Zhang Z, Rydel RE, Drzewiecki GJ, Fuson K, Wright
S, Wogulis M, Audia JE, May PC & Hyslop PA (1996)
Amyloid b-mediated oxidative and metabolic stress in
rat cortical neurons: no direct evidence for a role for
H2O2 generation. J Neurochem 67, 1595–1606.
56 Sato N, Iwata S, Nakamura K, Hori T, Mori K &
Yodoi J (1995) Thiol-mediated redox regulation of
apoptosis. Possible roles of cellular thiols other than
glutathione in T cell apoptosis. J Immunol 154, 3194–
3203.
57 Abate C, Patel L, Rauscher FJ 3rd & Curran T (1990)
Redox regulation of Fos and Jun DNA-binding activity
in vitro. Science 249, 1157–1161.
58 Park DS, Stefanis L, Yan CY, Farinelli SE & Greene
LA (1996) Ordering the cell death pathway. Differential
effects of BCL2, an interleukin-1-converting enzyme
family protease inhibitor, and other survival agents on
JNK activation in serum ⁄ nerve growth factor-deprived
PC12 cells. J Biol Chem 271, 21898–21905.
59 Meyer M, Schreck R & Baeuerle PA (1993) H2O2 and
antioxidants have opposite effects on activation of
NF-kappa B and AP-1 in intact cells: AP-1 as second-
ary antioxidant-responsive factor. EMBO J 12, 2005–
2015.
60 Xia Z, Dickens M, Raingeaud J, Davis RJ & Greenberg
ME (1995) Opposing effects of ERK and JNK-p38
MAP kinases on apoptosis. Science 270, 1326–1331.
61 Muller JM, Cahill MA, Rupec RA, Baeuerle PA &
Nordheim A (1997) Antioxidants as well as oxidants
activate c-fos via Ras-dependent activation of extracellu-
lar-signal-regulated kinase 2 and Elk-1. Eur J Biochem
244, 45–52.
B. Konarkowska et al. Thiol antioxidants suppress hA cytotoxicity
FEBS Journal 272 (2005) 4949–4959 ª 2005 FEBS 4959