thiazolidinediones are partial agonists for the glucocorticoid receptor
TRANSCRIPT

Thiazolidinediones Are Partial Agonists for theGlucocorticoid Receptor
L. Matthews, A. Berry, M. Tersigni, F. D’Acquisto, A. Ianaro,* and D. Ray*
Endocrine Sciences Research Group (L.M., A.B., D.R.), Division of Cardiovascular and Endocrine Science, University ofManchester, Manchester M13 9PT, United Kingdom; Department of Experimental Pharmacology (M.T., A.I.), Universityof Naples Federico II, 80131 Naples, Italy; and Research Centre in Biochemical Pharmacology (F.D.), William HarveyResearch Institute, John Vane Science Centre, London EC1M 6BQ, United Kingdom
Although thiazolidinediones were designed as specific peroxisome proliferator-activated receptor(PPAR)-�-ligands, there is evidence for some off-target effects mediated by a non-PPAR� mecha-nism. Previously we have shown that rosiglitazone has antiinflammatory actions not explicable byactivation of PPAR�,but possibly by the glucocorticoid receptor (GR). Rosiglitazone induces nucleartranslocation both of GR-green fluorescent protein, and endogenous GR in HeLa and U20S cells butwith slower kinetics than dexamethasone. Rosiglitazone also induces GR phosphorylation (Ser211),a GR ligand-binding-specific effect. Rosiglitazone drives luciferase expression from a simple glu-cocorticoid-response element containing reporter gene in a GR-dependent manner (EC50 4 �M),with a similar amplitude response to the partial GR agonist RU486. Rosiglitazone also inhibitsdexamethasone-driven reporter gene activity (IC50 2.9 �M) in a similar fashion to RU486, suggestingpartial agonist activity. Importantly we demonstrate a similar effect in PPAR�-null cells, suggestingboth GR dependence and PPAR� independence. Rosiglitazone also activates a GAL4-GR chimera,driving a upstream activating sequence promoter, demonstrating DNA template sequence inde-pendence and furthermore enhanced steroid receptor coactivator-1-GR interaction, measured bya mammalian two-hybrid assay. Both ciglitazone and pioglitazone, structurally related to rosigli-tazone, show similar effects on the GR. The antiproliferative effect of rosiglitazone is increased inU20S cells that overexpress GR, suggesting a biologically important GR-dependent component ofrosiglitazone action. Rosiglitazone is a partial GR agonist, affecting GR activation and traffickingto influence engagement of target genes and affect cell function. This novel mode of action mayexplain some off-target effects observed in vivo. Additionally, antagonism of glucocorticoid actionmay contribute to the antidiabetic actions of rosiglitazone. (Endocrinology 150: 75–86, 2009)
Peroxisome proliferator-activated receptors (PPARs) are nu-clear receptors activated by binding endogenously produced
fatty acids to regulate lipid homeostasis (1). They are membersof the nuclear receptor superfamily and heterodimerize on DNAwith retinoid X receptor to regulate target gene expression (2).PPAR� is highly expressed in adipocyte and macrophage inwhich it acts to influence differentiation, lipid storage, and glu-cose homeostasis (2–5). It is this activity that is exploited bysynthetic PPAR� ligands, notably the thiazolidinediones, e.g.rosiglitazone, and pioglitazone, to improve insulin sensitivity(6–10). However, even though these drugs are in routine clinical
use, their precise mode of action remains elusive because muscleis the major tissue responsible for insulin mediated glucose dis-posal, and PPAR� expression is very low in muscle (11). A fur-ther mechanism of action is the repression of secreted moleculesfrom fat, including TNF�, which is known to induce insulinresistance (12–14).
Thiazolidinediones, acting through PPAR�, also act to influ-ence differentiation of stem cells to direct differentiation intoadipocytes at the expense of osteoblasts (2, 5, 15–18). This re-sults in loss of mineralized bone mass, and there are reports ofincreased fracture risk in women. In one report there was a strik-
ISSN Print 0013-7227 ISSN Online 1945-7170Printed in U.S.A.Copyright © 2009 by The Endocrine Societydoi: 10.1210/en.2008-0196 Received February 11, 2008. Accepted September 8, 2008.First Published Online September 18, 2008* A.I. and D.R. are joint corresponding authors.
Abbreviations: DAC, Deacylcortivazol; DAPI, 4�,6�-diamino-2-phenylindole; FCS, fetal calfserum; Gc, glucocorticoid; GFP, green fluorescent protein; GR, glucocorticoid receptor;GRE, glucocorticoid-response element; hGR, human GR; LBD, ligand-binding domain;M2H, mammalian two hybrid; NCoR, nuclear receptor corepressor; PFA, paraformalde-hyde; PPAR, peroxisome proliferator-activated receptor; SRC, steroid receptor coactivator.
D I A B E T E S - I N S U L I N - G L U C A G O N - G A S T R O I N T E S T I N A L
Endocrinology, January 2009, 150(1):75–86 endo.endojournals.org 75

ing 6% annual rate of bone loss, similar to that seen with glu-cocorticoids (19, 20).
Although thiazolidinediones were designed as specific li-gands for PPAR�, there is evidence for some effects beingmediated by a nonclassical, non-PPAR� mechanism (21–23).In particular, there is evidence that high concentrations ofPPAR� ligands are required to inhibit expression of proin-flammatory cytokines and yet modulation of lipid metabolicgenes, and differentiation of preadipocytes to adipocytes oc-
curs with concentrations of ligand orders of magnitude lower,close to the measured Kd for binding PPAR� (23). Further-more, in embryonic stem cells null for PPAR� induced to dif-ferentiate down the macrophage lineage treatment withPPAR� ligands, both endogenous (prostaglandin J2) and ex-ogenous (thiazolidinediones) suppressed expression of proin-flammatory cytokines including TNF, IL-1�, IL-6, and en-zymes inducible nitric oxide synthase and cyclooxygenase-2(23). This effect is also typical of the response these cells have
after glucocorticoid exposure (24, 25). Glucocor-ticoids (Gcs) modulate cellular events after bind-ing and activation of the ubiquitously expressedintracellular glucocorticoid receptor (GR) (24).The GR is also a member of the nuclear hormonereceptor superfamily of ligand-activated tran-scription factors (24).
In an inactive state, the GR resides in the cyto-plasm as part of a multiprotein complex, whichincludes chaperone proteins and immunophilins(26–28). Ligand-activated GR is released from thiscomplex and is then free to initiate nongenomiceffects within the cytoplasm and can also translo-cate to the cell nucleus where it dimerizes and bindspalindromic glucocorticoid-response elements(GREs). Once in the nucleus, the GR-GRE complexhas the capacity to recruit either coactivator orcorepressor molecules that can modify chromatinand either facilitate or inhibit transcription initia-tion (29–33).
Recently we showed that the antiinflammatoryeffects of the PPAR� ligands rosiglitazone and cigli-tazone in vivo could be reversed by the GR antag-onist RU486 and also that in vitro their actions incell lines was dependent on expression of GR butnot PPAR� (34). To further analyze the potentialactivation of GR by exposure to PPAR� ligands, wehave undertaken a detailed series of studies to shownuclear translocation of GR in response to rosigli-tazone, accompanied by GR Ser211 phosphoryla-tion, a posttranslational modification seen rapidlyafter ligand activation of the GR. This was accom-panied by rosiglitazone-mediated regulation of aGR reporter gene and recruitment of the steroidreceptor coactivator (SRC)-1, in a GR expression-specific and PPAR�-independent manner. We werealso able to show GR-dependent inhibition of osteo-blast cell proliferation by rosiglitazone. Taken togetherthis is strong evidence for rosiglitazone actionthroughGRandmayexplain someof the spectrumofrosiglitazone activities observed in vivo.
Materials and Methods
Anti-GR (hGR) (clone 41) from BD Biosciences (Oxford,UK); anti-GR (M20) from Santa Cruz Biotechnology(Santa Cruz, CA); antiphospho-(Ser 211)-GR from Cell
A
Control
Dex 30m
inD
ex 120min
anti GR GR-GFP DAPI merge
Rosi30m
in(2)R
osi120min
Rosi30m
in (1)
100
80
60
40
20
0
Num
ber o
f cel
ls (%
)
B
Control Dex 30min Rosi 30minDex 120min Rosi 120min
C>NN>C N=C C>NN>C N=C C>NN>C N=C C>NN>C N=C C>NN>C N=C
**
**
**
** **
**
FIG. 1. GR-GFP translocates to the nucleus in the presence of rosiglitazone. Aftertransfection with GR-GFP, serum-starved U20S cells were incubated with 100 nM
dexamethasone (Dex) or 10 �M rosiglitazone (Rosi; 30 or 120 min, as indicated), fixedwith PFA, and analyzed for subcellular GR localization (A) using a GR-specific antibody(red) and also by the localization of the GFP tag (green). Nuclei were counterstainedusing DAPI (blue). Images are representative of three independent experiments (for 30min rosiglitazone treatment, two representative images are shown). In each case, at least100 cells/experiment were scored for GR distribution and represented graphically (B). N,Nucleus; C, cytosol. *, Significant difference from control treatment where P � 0.05.
76 Matthews et al. Rosiglitazone Activates the GR Endocrinology, January 2009, 150(1):75–86
Signaling Technology (Beverly, MA); horseradish peroxi-dase conjugated antimouse and antirabbit from GE Health-care (Buckinghamshire, UK); Alexa 546 conjugated anti-mouse from Invitrogen (Paisley, UK); dexamethasonefrom Sigma (Dorset, UK); rosiglitazone, ciglitazone, andpioglitazone from IDS Ltd. (Tyne and Wear, UK). TAT3-Luc a kind gift of Dr. J. Lluihi-Ineguez (University of Cal-ifornia, San Francisco, San Francisco, CA). hGR pcDNA3and hGR-green fluorescent protein (GFP) pcDNA3 are akind gift of Dr. M. Norman (University of Bristol, Bristol,UK).
Cell culture and maintenanceHuman epithelial lung and cervical carcinoma cells
(A549 and HeLa; ECACC, Wiltshire, UK) and mouse em-bryonic fibroblasts from PPAR� knockout mice [PPAR(�/
�)]; a generous gift of Prof. B. Spiegelman (Dana-FarberCancer Institute and Harvard Medical School, Boston,MA) were cultured in DMEM containing glutamax sup-plemented with 10% charcoal dextran-stripped fetal calfserum (FCS; Invitrogen) in a humidified atmosphere of 5%carbon dioxide at 37 C. The human osteosarcoma cellline, (U20S; American Type Culture Collection, Manas-sas, VA) was cultured in McCoy’s 5A medium (Invitro-gen) containing glutamax supplemented with 10% char-coal dextran-stripped FCS in a humidified atmosphere of5% carbon dioxide at 37 C.
ImmunofluorescenceAfter 24 h serum withdrawal, HeLa, PPAR(�/�), or
U20S cells transfected (Fugene 6; Roche Diagnostics, Bur-gess Hill, UK) with hGR-GFP were treated were treated asspecified in Results. Cells were fixed with 4% paraformal-dehyde (PFA) for 30 min at 4 C and then permeabilized(0.02% Triton X-100 in PBS) for 30 min at room temper-ature. Fixed cells were blocked (1% FCS in PBS) for 4 h atroom temperature with agitation and then in primary an-tibody (1:200, diluted in blocking buffer) overnight at 4 C.After three 10-min washes in PBS, cells were incubated insecondary antibody (1:500, diluted in PBS) for 2 h. Afterthree further 10-min washes, coverslips were mounted us-ing vectashield hard set mountant containing the nuclear4�,6�-diamino-2-phenylindole (DAPI) stain (Vector Labo-ratories, Peterborough, UK). Fluorescent cells were vi-sualized using a C1 confocal microscope (Nikon, Tokyo,Japan; an upright 90i microscope with a �60/1.40 PlanApo objective; pinhole 30 �m, scan speed 400 Hz uni-directional, format 1024 � 1024). Images for DAPI, Al-exa488, and Alexa546 were excited with the 405-, 488-,and 543-nm laser lines, respectively, as previously de-scribed (35). One hundred cells from three independentexperiments were scored for GR localization, whetherGR was localized principally to the nucleus, cytoplasm,or distributed evenly between the two compartments.
Immunoblot analysisCell lysates (30 �g protein) were electrophoresed on
sodium dodecyl sulfate/acrylamide gels and transferred to0.2-�m nitrocellulose membranes (Bio-Rad Laboratories,Hertfordshire, UK) overnight at 4 C. Membranes wereblocked for 6 h (0.15 M NaCl, 1% dried milk, 0.1% Tween20) and incubated with primary antibodies (diluted inblocking buffer) overnight at 4 C. After three 10-minwashes [88 mM Tris (pH 7.8), 0.25% dried milk, 0.1%Tween 20], membranes were incubated with a species-spe-cific horseradish peroxidase-conjugated secondary anti-body (diluted in wash buffer) for 1 h at room temperature
A
Control
Dex 30m
inD
ex 120min
anti GR DAPI merge
Rosi120m
inR
osi30min(2)
Rosi30m
in(1)
100
80
60
40
20
0
Control Dex 30min Rosi 30minDex 120min Rosi 120min
Num
ber o
f cel
ls (%
)
B
C>NN>C N=C C>NN>C N=C C>NN>C N=C C>NN>C N=C C>NN>C N=C
**
**
**
** **
FIG. 2. Endogenous GR translocates to the nucleus in the presence of rosiglitazone.Serum-starved HeLa cells were incubated with 100 nM dexamethasone (Dex) or 10 �M
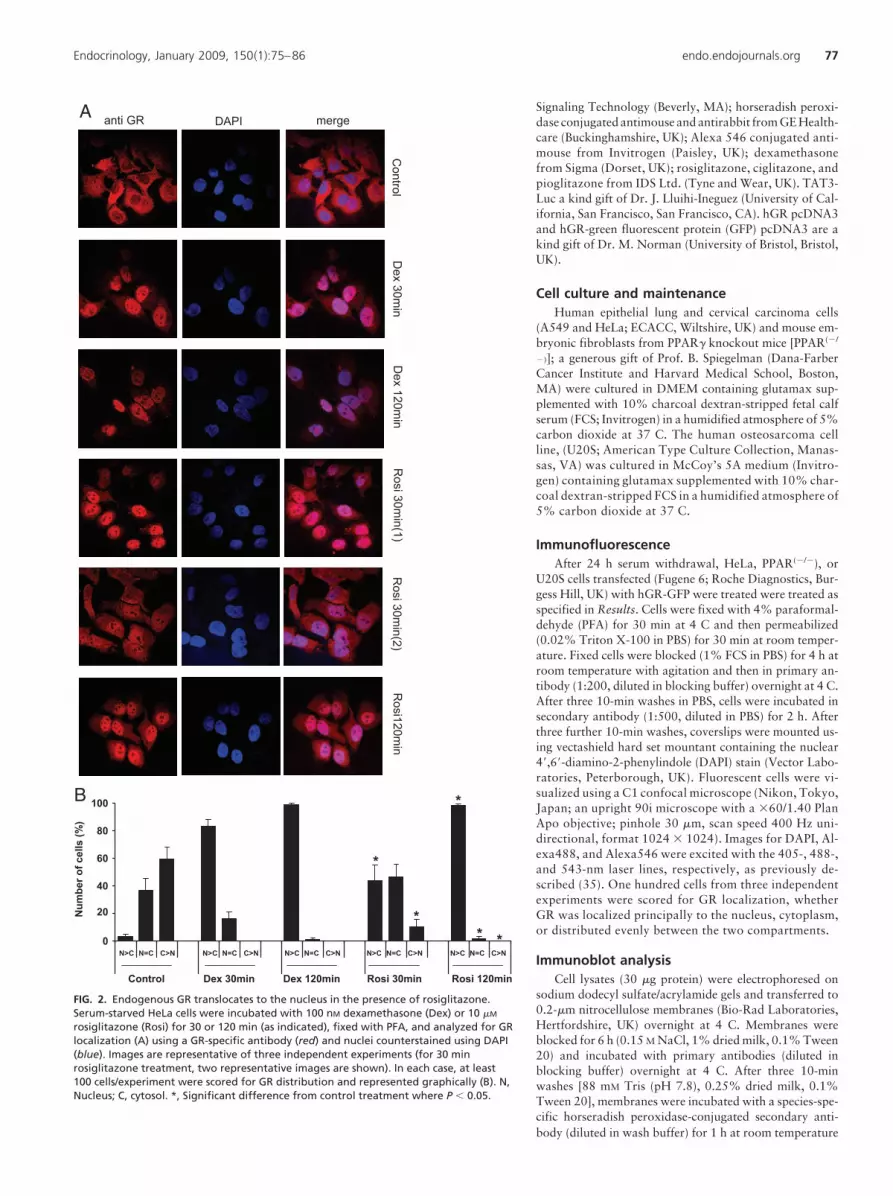
rosiglitazone (Rosi) for 30 or 120 min (as indicated), fixed with PFA, and analyzed for GRlocalization (A) using a GR-specific antibody (red) and nuclei counterstained using DAPI(blue). Images are representative of three independent experiments (for 30 minrosiglitazone treatment, two representative images are shown). In each case, at least100 cells/experiment were scored for GR distribution and represented graphically (B). N,Nucleus; C, cytosol. *, Significant difference from control treatment where P � 0.05.
Endocrinology, January 2009, 150(1):75–86 endo.endojournals.org 77

and washed a further three times, each for 10 min. Immunoreactiveproteins were visualized using enhanced chemiluminescence (ECL Ad-vance; GE Healthcare), as previously described (35, 36).
Reporter gene assayCells were cotransfected with 1 �g TAT3-luciferase reporter gene
construct together with 0.1 �g cytomegalovirus-renilla luciferase (tocorrect for transfection efficiency) using Fugene 6. After 24 h serumwithdrawal, cells were treated as specified in results before lysis andthen assayed for luciferase activity using a dual-luciferase reporterassay system following the manufacturer’s instructions (Promega,Southampton, UK) (37).
Mammalian one- and two-hybrid assaysUsing a previously described method (29, 38), cells were cotrans-
fected with 5 �g each of pG5-luc and either pBIND or pBINDGR-LBD(mammalian one hybrid assay) or 5 �g each of pG5-luc and pACTGR-LBD together with pBIND, pBIND-SRC-1, or pBIND-NCoR [mamma-lian two hybrid (M2H) assay]. After 18 h ligand treatment (details spec-ified in Results) cells were lysed, harvested, and assayed for luciferaseactivity using a dual-luciferase reporter assay system (Promega) follow-ing the manufacturer’s instructions.
Proliferation assayCells were grown in DMEM supplemented with vehicle, dexameth-
asone, and/or rosiglitazone as indicated. Five days later, cells weretrypsinized, aliquots seeded in 96-well plates, and cells allowed to adhereovernight. Cell numbers were calculated using a CellTiter96 AQueouscell proliferation assay system (Promega) following the manufacturer’sinstructions (36).
Modeling of rosiglitazone dockingBinding modes of rosiglitazone in the GR-ligand-binding domain
(LBD) were investigated using molecular docking calculations. The GR-LBD docking model was derived from the GR/deacylcortivazol (DAC)complex crystal structure (PDB code 3bqd) that was recently reported bySuino-Powell et al. (39). The MOE software package was used to preparethe GR-LBD docking model (MOE 2007.0902; Chemical ComputingGroup Inc., Montreal, Canada): DAC and water oxygen atoms weredeleted, hydrogen atoms were added to the protein residues, and theatomistic structure subjected to energy minimization using the Amber94force-field option with constraints placed on nonhydrogen atoms. Mo-lecular docking calculations of rosiglitazone were performed with theGOLD software package (GOLD 4.0; Cambridge Crystallographic DataCenter, Cambridge, UK).
Results
GR translocates to the nucleus in the presence ofrosiglitazone
GR undergoes ligand-dependent nuclear translocation to en-gage target genes. To assess nuclear translocation in response torosiglitazone treatment the U20S cell line, which expresses lowlevels of GR detectable by immunofluoresence (Fig. 1), wastransfected with GR-GFP. After incubation with vehicle, 100 nM
dexamethasone or 10 �M rosiglitazone (times indicated), cellswere analyzed for GR localization using a GR-specific antibodyand also by the colocalization of the GFP tag. In untreated cells,the GR localizes predominantly to the cytoplasm (82 � 3%) orthroughout the entire cell (16 � 3%), with few cells showingnuclear GR accumulation (2 � 1%) (Fig. 1, A and B). Treatmentwith 100 nM dexamethasone induces near-complete nuclear
translocation (98 � 2%) of GR by 30 min, which is sustainedover the 120-min assay period. After incubation with rosiglita-zone for 30 min, GR translocates into the nucleus in a significantproportion of cells (31 � 8% nuclear GR) and is found diffuselydistributed throughout the cell in others (54 � 10%). The mag-nitude of GR translocation is further increased after 120 minrosiglitazone treatment (95 � 2% nuclear GR, Fig. 1, A and B).GR translocation in response to 120 min treatment with rosigli-tazone is almost as efficient as that seen in response to dexa-methasone (rosiglitazone 95 � 2%; dexamethasone 98 � 2%nuclear GR). Vehicle treatment is without effect at each timepoint (data not shown).
Although overexpression of GFP-tagged GR is a very sensitiveand specific bioassay for analysis of GR trafficking, it is recognizedthat overexpression of the GR can alter intracellular compartmentsegregation. To exclude this possibility, these studies were repeatedin HeLa cells that express only endogenous GR (Fig. 2). After 30min exposure, dexamethasone treatment promotes near-completetranslocationofGR(98�1%nuclearGR),androsiglitazone treat-ment promotes a heterogeneous GR distribution (n � c, 44 � 11%;n � c, 46 � 10%). Within 120 min, the rosiglitazone effect is close
A549A549
A
1010μμM M RosiRosi
PP--GRGR
GRGR
100nM Dex100nM Dex
1010μμM M RosiRosi 100nM Dex100nM Dex
PP--GRGR
GRGR
HeLaHeLa
0 30 60 90 120 0 30 60 90 120
0 30 60 90 120 0 30 60 90 120
min
min
A549A549
B
1010μμM M RosiRosi
PP--GRGR
GRGR
100nM Dex100nM Dex
hr0 1 2 3 4 0 1 2 3 4
1010μμM M RosiRosi 100nM Dex100nM Dex
0 1 2 3 4 0 1 2 3 4
PP--GRGR
GRGR
hr
HeLaHeLa
FIG. 3. GR is phosphorylated after treatment with rosiglitazone. HeLaand A549 cells were serum starved and after treatment with 100 nM
dexamethasone (Dex) or 10 �M rosiglitazone (Rosi) for up to 120 min (A)or 4 h (B), cells were lysed in assay buffer containing phosphataseinhibitors. Whole-cell extracts were analyzed by immunoblotting for GRabundance and phosphorylation on Ser211 (as indicated). Images arerepresentative of three independent experiments.
78 Matthews et al. Rosiglitazone Activates the GR Endocrinology, January 2009, 150(1):75–86

tothatseenwithdexamethasone(Fig.2,AandB,rosiglitazone98�
2%; dexamethasone 99 � 1% nuclear GR).
GR is phosphorylated after treatment with rosiglitazoneWithin the cytoplasm, GR undergoes rapid ligand-dependent
phosphorylation on Ser211 (40). To examine whether GR isphosphorylated after rosiglitazone treatment, HeLa and A549cells were serum starved and treated with dexamethasone orrosiglitazone for times ranging from 30 min (Fig. 3A) to 4 h (Fig.3B). Cell extracts were analyzed for GR (Ser211) phosphorylationand abundance by immunoblotting. Images in Fig. 3 demonstratethat GR is Ser211 phosphorylated in response to treatment withdexamethasone and rosiglitazone in both HeLa and A549 cells.
Although GR is rapidly phosphorylated by either rosiglitazoneor dexamethasone, the magnitude of effect after rosiglitazone is
reduced incomparison. Interestingly,whereas treatmentwithdexa-methasone clearly promotes both phosphorylation and down-reg-ulation of GR (more evident in HeLa cells), rosiglitazone treatmentonly promotes GR Ser211 phosphorylation (Fig. 3, A and B).
Rosiglitazone-mediated TAT3-luc activation is dependenton GR expression
After nuclear translocation the GR binds to recognition siteson DNA to regulate transcription. Dexamethasone promotes arobust and easily measured induction of reporter gene expressionin both HeLa and A549 cells (Fig. 4, A and B). Rosiglitazone alsoinduces target gene expression, although to a lesser extent. Incomparison with dexamethasone, the dose-response curve forrosiglitazone is markedly right shifted. There is still, however, ahighly significant induction of TAT3-luc in HeLa cells (34-fold
induction, EC50 �60 �M) and a much smaller in-crease in A549 cells (4-fold induction, EC50 24 �M).The decreased induction seen in A549 cells whencompared with HeLa cells is compatible with thereduced induction of reporter genes seen with dexa-methasone in these cell models (HeLa cells 127-foldinduction, EC50 0.6 nM; A549 cells 36-fold induc-tion, EC50 0.6 nM). This is also compatible with themuch lower induction of GR Ser211 phosphorylationin A549 cells (Fig. 3). To gain additional evidence forthe dependence of this transcriptional induction onthe presence of the GR, the GR-deficient U20S cellline was used. In U20S cells, there is no induction ofthe transfected reporter gene until 1 nM dexameth-asone and no induction of the reporter gene seen atall with rosiglitazone (Fig. 4C). Higher concentra-tions of dexamethasone did induce the reporter gene,probably due to activation of low-level endogenousGR. After transfection of U20S cells with the humanGR expression vector, there is a significant inductionof reporter gene activity by 0.01 nM dexamethasone,indicating that this effect is dependent on transfectedGR. In addition, transfected GR confers rosiglita-zone responsiveness to the reporter gene (Fig. 4C;U20S pcDNA3 EC50 �8 mM; U20S pcDNA3-hGREc50 4 �M).
Rosiglitazone and RU486 inhibitdexamethasone-induced TAT3-luc activation
RU486 is a high-affinity partial agonist of the GR,causingnuclear translocation,Ser211 phosphorylation,DNAbinding,andinhibitingtheactionsoffullagonistssuch as dexamethasone. Figure 5A demonstrates thatRU486 effectively inhibits dexamethasone- but notrosiglitazone-induced TAT3-luc activity.
Graphs presented in Fig. 5, A and B, further suggestthat both rosiglitazone and RU486 activate the re-porter gene to a similar extent, perhaps suggesting par-tial agonist activity for rosiglitazone.
Indeed, experiments demonstrate that rosiglita-zone effectively inhibits the response to a saturatingconcentration of dexamethasone (Fig. 5C; dexa-
Dex EC50; 0.6nMRosi EC50; 23.6µM
Dex EC50; 0.6nMRosi EC50; >60µM
A BHeLaHeLa A549A549
RLU
(x10
6 )
90
80
70
60
50
40
30
20
10
0
20
15
10
5
0
RLU
(x10
3 )
0 0.01 0.1 1 10 100 0 0.01 0.1 1 10 100
nM DexμM Rosi
nM DexμM Rosi
Dose Dose
**
**
C
1800
1600
1400
1200
1000
800
600
400
200
0
2000
RLU
(x10
3 )
Dex EC50; 0.061nMRosi EC50; 4.0µM
Dex EC50; 3.9nM
U20S pcDNA3 GRU20S pcDNA3 GRU20S pcDNA3U20S pcDNA3
0 0.01 0.1 1 10 100 0 0.01 0.1 1 10 100
nM DexμM Rosi
Dose Dose
** **
FIG. 4. Rosiglitazone-mediated TAT3-luc activation is enhanced by GR overexpression.HeLa (A), A549 (B), and U20S cells transiently overexpressing hGR (C) were transfected withTAT3-luc and after serum starvation were treated with various concentrations ofdexamethasone (Dex) or rosiglitazone (Rosi). Cells were then lysed and subjected to analysisby luciferase assay. Data for U20S and U20SGR cells were obtained from the sameexperiments and are represented on different axes to aid comparison. Graphs (mean � SD)show one of three representative experiments performed in triplicate. EC50 values weredetermined from pooled data. *, Significant difference from control treatment where P � 0.05.
Endocrinology, January 2009, 150(1):75–86 endo.endojournals.org 79

methasone, 9971 � 106 RLU; rosiglitazone � dexamethasone,2948 � 106 RLU). More detailed studies show concentration-dependent rosiglitazone antagonism of 1 nM dexamethasone(Fig. 5D; IC50 2.9 �M) and 10 nM dexamethasone (Fig. 5E; IC50
26 �M).
GR activation by rosiglitazone is PPAR� independentBecause PPAR� is the target receptor for rosiglitazone, it is
possible that activation of GR is an indirect downstream effectafter PPAR� activation. To determine the requirement of PPAR�
for the effect of rosiglitazone on GR, studies were also conductedin cells isolated from PPAR� knockout mice [PPAR�(�/�)]. Inline with experiments presented in Figs. 1–3, data shown in Fig.6 demonstrate nuclear translocation of endogenous GR (Fig. 6A)and Ser211 phosphorylation (Fig. 6B) in response to treatmentwith rosiglitazone .
Reporter gene assays in PPAR�(�/�) cells (Fig. 6C) producedrobust induction of TAT3-luc in response to rosiglitazone, whichnot only shows PPAR� independence but also GR expressiondependence. Pretreatment with rosiglitazone also significantlyimpaired the response of PPAR�(�/�) cells to dexamethasone(Fig. 6, D and E), further suggesting partial agonist activity.
Rosiglitazone also activates transcription through aGAL4-GR LBD chimera
As an indication of rosiglitazone binding, the ability of ros-iglitazone to activate isolated GR LBD was explored using amammalian one-hybrid assay system.
Rosiglitazone activates transcription through an isolated GRLBD, fused to a GAL4 DNA binding domain. In comparison
with the 23.5-fold induction caused by dexamethasone treat-ment, RU486 and rosiglitazone produced only modest but sig-nificant induction at their respective maximal doses (Fig. 7A;RU486, 2.9-fold; rosiglitazone, 3.1-fold). Because data were col-lected in PPAR�(�/�) cells, this once again suggests PPAR� in-dependence. Importantly, because only the GR-LBD and notfull-length GR was used, this suggests that only the LBD is re-quired for interaction with rosiglitazone and also that the ros-iglitazone effect is independent of the DNA response elementsequence.
Rosiglitazone recruits SRC-1 and nuclear receptorcorepressor (NCoR) to the GR ligand binding domain
Recruitment of cofactors to GR is ligand specific. Using amammalian two-hybrid (M2H) system, differential recruitmentof coactivators after rosiglitazone treatment was examined.
M2H assays were conducted in HeLa cells using GR-LBD fusedto VP16 and the nuclear receptor interaction domain of SRC-1 orNCoR fused to Gal4. Figure 7 shows that dexamethasone but notRU486 recruits SRC-1 to the GR-LBD (Fig. 7B) and also thatRU486 and not dexamethasone recruits NCoR to the GR-LBD(Fig. 7C). Interestingly, treatment with rosiglitazone recruited bothSRC-1 and NCoR to the GR, perhaps suggesting a function distinctfrom either dexamethasone or RU486. In both cases, vehicle treat-ment was without effect (data not shown).
M2H assays were also conducted in PPAR�(�/�) cells (Fig. 7D).Interestingly, the magnitude of response to rosiglitazone wasgreater in the PPAR�(�/�) cells than the HeLa cells (HeLa, 3.3-fold;PPAR�(�/�), 6.7-fold over vehicle treated controls), as was the re-sponse to dexamethasone (HeLa, 12.4-fold; PPAR�(�/�), 36.8-
A
0 1 10 100
450400350300250200150100
500
500
RLU
(x10
3 )
Vehicle + nM Dex 100nM RU + nM Dex Vehicle + µM Rosi 100nM RU + µM Rosi
0 1 10 100 0 1 10 100 0 1 10 100
********
B350
300
250
200
150
100
50
0
RLU
(x10
3 )
nM Dex µM Rosi nM RU
0 1 10 100 0 1 10 100 0 1 10 100
C
Dex RosiRUDexDex
Con
120
100
80
60
40
20
0
RLU
(x10
6 )
****
****
D
RLU
(x10
6 )
140
12010080
60
40
20
0
RLU
(x10
6 )
70
6050
40
30
20
10
0
ERosi IC50; 2.9μM Rosi IC50; 26μM
1 nM Dex µM Rosi0 0 0.01 0.1 1 10 100
- + + + + + + 10 nM Dex µM Rosi0 0 0.01 0.1 1 10 100
- + + + + + +
****
** **
**
****
FIG. 5. Rosiglitazone inhibits dexamethasone-mediated TAT3-luc activation. HeLa cells were transfected with TAT3-luc. Twenty-four hours aftertransfection, cells were treated with 1–100 nM dexamethasone (Dex) or 1–100 �M rosiglitazone (Rosi) for 18 h after 1 h pretreatment with 100 nM RU486(A) or 1–100 nM RU486 independently (B). Cells were also treated with dexamethasone for 18 h after pretreatment with RU486 or rosiglitazone (C–E).Cells were then lysed and subjected to analysis by luciferase assay. Graphs (mean � SD) show one of three representative experiments performed intriplicate. IC50 values were determined from pooled data. Significant difference from treatment with dexamethasone alone is indicated (*, P � 0.05;**, P � 0.01).
80 Matthews et al. Rosiglitazone Activates the GR Endocrinology, January 2009, 150(1):75–86

fold), These data suggest not only that the isolated GR-LBD is suf-ficient to recruit coactivators in response to rosiglitazone but alsothat this effect does not require PPAR�.
The antiproliferative effect of rosiglitazone is enhancedby GR overexpression
Both rosiglitazone and dexamethasone are potent inhibitorsof cellular proliferation. To determine whether rosiglitazone-mediated antiproliferative effects are attributable to activationof GR, cells were treated with various concentrations of dexa-methasone or rosiglitazone or a combination of both for 5 d andanalyzed for proliferation by MTS assay.
In HeLa (Fig. 8A) and A549 (Fig. 8B) cells, treatment with ros-iglitazone reduces rates of proliferation. The effect is smaller thanthatobservedwithdexamethasone inbothcases.Cotreatmentwithrosiglitazone alters the antiproliferative profile of dexamethasone,suggesting a modest inhibitory effect on Gc action.
In GR-deficient U20S cells, rosiglitazone has an antiprolif-erative effect (Fig. 8C). However, because overexpression of GR
increases this effect, this suggests that at leasta component of the antiproliferative effect ofrosiglitazone may be GR dependent. Onceagain, cotreatment with rosiglitazone im-pairs the antiproliferative effect comparedwith dexamethasone alone (Fig. 8C). Be-cause neither dexamethasone nor rosiglita-zone had an antiproliferative effect inPPAR�(�/�) cells, even after overexpressionof GR (data not shown), the requirement ofPPAR� for the antiproliferative effect of ros-iglitazone was not explored.
The structurally related compoundsciglitazone and pioglitazone alsoactivate GR
The thiazolidinediones ciglitazone andpioglitazone share structural similarity withrosiglitazone. The effect of treatment withciglitazone and pioglitazone on GR activitywas examined. To also demonstrate PPAR�
independence, experiments were conductedin the PPAR�(�/�) cells. Figure 9A shows sig-nificant nuclear GR accumulation after expo-sure to 10 �M ciglitazone. In contrast, piogli-tazone shows only marginal effects on GRnuclear translocation (Fig.9A)during the120-min assay period. Immunoblots demonstrateSer211 phosphorylation of endogenous GR inresponse to both ciglitazone (1–100 �M) andpioglitazone (1–100 �M) with comparable ef-fects with rosiglitazone (Fig. 9B).
In PPAR�(�/�) cells, treatment with eitherciglitazone (Fig. 9C) or pioglitazone (Fig. 9D)induced a significant increase in luciferase ex-pression, with similar effects observed afterrosiglitazone treatment.ExpressionofGRwasrequired to generate this response and once
again demonstrate both GR dependence and PPAR� independenceforactivationofTAT3-luc.Mammalianone-hybridassays(Fig.9E)and M2H assays (Fig. 9F) in PPAR�(�/�) cells demonstrate thatboth ciglitazone and pioglitazone promote recruitment of cofactorsincluding SRC-1 to GR-LBD. Whereas the effects are significantlylower than the response to dexamethasone, they are comparablewith the rosiglitazone effect. Vehicle treatment was without effect(data not shown).
Modeling rosiglitazone binding to the GRTo explore the possible mode of rosiglitazone binding to the
GR, a series of modeling experiments were performed. The high-est ranked docking pose of rosiglitazone shares some similaritieswith the binding of DAC as observed in the crystal structure (Fig.10). In particular, the pyridine ring of rosiglitazone is positionedsimilarly to the phenyl ring of DAC, and the thiazole ring ofrosiglitazone is positioned similarly to the hydroxyacetyl moietyof DAC to allow the carbonyl oxygen atom to form hydrogenbonding interaction with Asn564. In common with DAC, ros-
Control
A
Rosi120m
inD
ex 120min
anti mGR (1) anti mGR (2) anti mGR (3)
B
0 10 30 60 120 0 10 30 60 120 min
PP--GRGR
GRGR
tubulintubulin
PPARPPARγγ((--//--))
1010μμM M RosiRosi100nM Dex100nM Dex 0 0.01 0.1 1 10 100 0 0.01 0.1 1 10 100
C
Dex EC50; 6.1nM
RLU
(x10
3 )
50
40
30
20
10
0
Dex EC50; 0.02nM Rosi EC50; 12.6µM
PPARPPARγγ((--//--)) pcDNA3pcDNA3 PPARPPARγγ((--//--)) pcDNA3GRpcDNA3GR
nM DexμM Rosi
Dose Dose
**
****
D
RLU
(x10
3 )
70605040302010
01 nM Dex µM Rosi0 0.01 0.1 1 10 100
RLU
(x10
3 )
140120
160
100806040200
E
Rosi IC50; 0.08μM
Rosi IC50; 0.2μM
PPARγ(-/-) pcDNA3PPARγ(-/-) pcDNA3GR
PPARγ(-/-) pcDNA3PPARγ(-/-) pcDNA3GR
+ + + + + +
10 nM Dex µM Rosi0 0.01 0.1 1 10 100
+ + + + + +
** ** **
****
** ** **
****
**
FIG. 6. Activation of GR by rosiglitazone is PPAR� independent. PPAR�(�/�) cells were serumstarved, treated with 100 nM dexamethasone (Dex) or 10 �M rosiglitazone (Rosi) for the timesindicated, and subjected to immunoblot analysis to assay for GR phosphorylation (A) andimmunofluorescent microscopy to examine subcellular trafficking of endogenous GR (B; threerepresentative fields are shown). PPAR�(�/�) cells transiently cotransfected with hGR and TAT3-luc were serum starved, treated with 0.01–100 nM dexamethasone or 0.01–100 �M rosiglitazonefor 18 h, and subjected to luciferase assay as an indicator of GR activation (C). Data for PPAR�(�/�)
and PPAR�(�/�) GR cells were obtained from the same experiment and are represented ondifferent axes to aid comparison. PPAR�(�/�) and PPAR�(�/�) GR cells were pretreated with 0.01–100 �M rosiglitazone and then coincubated with 1 nM (D) or 10 nM (E) dexamethasone beforeanalysis by luciferase assay. Graphs (mean � SD) show one of three representative experimentsperformed in triplicate. EC50 (C) and IC50 (D and E) values were determined from pooled data.Significant difference from control treatment (C) or treatment with dexamethasone alone (D andE) is indicated (*, P � 0.05; **, P � 0.01).
Endocrinology, January 2009, 150(1):75–86 endo.endojournals.org 81

iglitazone is in van der Waals contact with atoms of the followingresidues: Asn564, Gln642, Thr739, Tyr735, Arg611, Ala607,Leu608, Phe623, Gln570, Leu566, Gly567, Leu563, Met646,Phe749, Cys736, Ile747. and Met604. We note that in additionto the rosiglitazone binding mode that has just been described,we also observed lower ranked docking poses in which the ros-iglitazone is reversed (e.g. in which the thiazole group of rosigli-tazone overlaps with the phenyl ring of DAC), but these reversemodes do not appear to exhibit any specific hydrogen bondinginteractions with the protein.
Discussion
Thiazolidinedione drugs are a significant advance in the treat-ment of diabetes and are thought principally to improve insulinsensitivity by activating PPAR� (11). However, they also exert anumber of other effects including inhibition of expression ofproinflammatory cytokines, an apparent beneficial effect in in-flammatory disease (including inflammatory bowel disease), in-
hibition of cell proliferation, and chemoprevention of cancer incarcinogen-treated animals but also induction of osteoporosis(19, 21, 23, 41). In addition, there is concern that fluid retentionmediated by thiazolidinediones may exacerbate congestive heartfailure (42). Understanding the spectrum of thiazolidinedioneaction may lead to either new molecules with increased speci-ficity of action or the better use of those that already exist. To thisend we comprehensively explored the potential role of the GR asmediator of some thiazolidinedione effects.
In a previous study, we investigated the possibility thatPPAR� ligands may exert some effects via the GR based on ob-servations that activation of both GR and PPAR� results in in-hibition of similar spectrum of proinflammatory cytokines (34).In our earlier work, we were able to show that thiazolidinedio-nes exert a suppressive activity on proinflammatory cytokinesin cells that lack PPAR� expression. Moreover, the effect ap-peared to be dependent on GR expression. We found similarresults using ciglitazone, a different molecular structure in thesame class, suggesting that it is a property of the thiazo-lidinedione class of drugs (34). Accordingly, we set out to
A
Cor
rect
ed R
LU (f
old
indu
ctio
n)
pBIND pBIND GR-LBD
00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100
nM Dex nM RU μM Rosi nM Dex nM RU μM Rosi
30
25
20
15
10
5
0
****
********
** **
12
16
14
10
8
6
4
2
0
Cor
rect
ed R
LU (F
old
Indu
ctio
n)
********
****
**
pACT GR-LBD + pBIND pACT GR-LBD + pBIND SRC-1
00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100
nM Dex nM RU μM Rosi nM Dex nM RU μM Rosi
B
C
pACT GR-LBD + pBIND pACT GR-LBD + pBIND NCoR
00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100
nM Dex nM RU μM Rosi nM Dex nM RU μM Rosi
Cor
rect
ed R
LU (F
old
Indu
ctio
n)
************
**
7
9
8
6
5
4
3
2
1
0
10
D
pACT GR-LBD + pBIND pACT GR-LBD + pBIND SRC-1
00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100
nM Dex nM RU μM Rosi nM Dex nM RU μM Rosi
35
45
40
30
25
20
15
10
5
0
Cor
rect
ed R
LU (F
old
Indu
ctio
n)
**
****
**
FIG. 7. Rosiglitazone recruits SRC-1 to GR. PPAR�(�/�) cells were transfected with 5 �g each of pG5-luc and either pBIND or pBIND-GRLBD (A) and thentreated with dexamethasone (Dex), RU486, or rosiglitazone (Rosi) and analyzed for luciferase activity. HeLa (B and C) and PPAR�(�/�) (D) cells weretransfected with 5 �g each of pG5-luc, pACT-GRLBD, and pBIND, pBIND-SRC-1 (B and D), or pBIND-NCoR (C) and then treated with dexamethasone,RU486, or rosiglitazone and analyzed for luciferase activity using a dual-luciferase assay system. Data represent fold induction over control treatment.Graphs (mean � SD) show one of at least two representative experiments performed in triplicate. Significant difference from control treatment isindicated (*, P � 0.05; ** P � 0.01).
82 Matthews et al. Rosiglitazone Activates the GR Endocrinology, January 2009, 150(1):75–86

explore a potential mediating role of the GR for some of theactions of thiazolidinediones.
Initial studies used both genetically modified cells that over-express a fluorophore-tagged form of the GR or HeLa cells thatexpress endogenous GR. Both show that rosiglitazone promotessignificant nuclear GR translocation. Nuclear translocation ofthe GR occurs very rapidly after ligand activation and is a pre-requisite for subsequent engagement of the GR with the genomeand therefore regulation of target genes. Consistently we foundthat the rate of nuclear translocation in response to rosiglitazonewas significantly slower when compared with those observedafter dexamethasone treatment. After 30 min there was a het-erogeneous appearance with approximately even distribution ofthe GR between nuclear and cytoplasmic compartments. By 120min, GR compartmentalization after rosiglitazone treatmentwas indistinguishable from that seen after treatment with dexa-methasone. Because both models of overexpressed GR and endog-enous GR gave comparable data, this increases the confidence thatthe resultsare in factbiologicallymeaningful. Indeed,wepreviouslyfound an induction in GR content in nuclear extracts prepared bysubcellular fractionation and analyzed by immunoblotting, provid-
ing additional reassurance that the rosiglitazone-dependent trans-location is a consistent and robust phenomenon (34).
The most rapid observable phenomenon associated with ac-tivation of the GR is induction of phosphorylation of Ser211 inthe GR N-terminal domain (36, 43, 44). Whereas in both celllines studied, rosiglitazone caused clear induction of phosphor-ylation of the GR, the magnitude of induction seen with rosigli-tazone is clearly greater in the HeLa cells compared with theA549 cells. This exactly parallels the increased induction ofphosphor Ser211 GR seen in response to dexamethasone betweenthe two cell lines. Ligand binding to GR also results in reducedsteady-state GR protein concentration. This was very readilyseen and of rapid onset in the HeLa cells, and although seenconsistently in the A549 cells, the effect was of a delayed onset.In neither cell type did rosiglitazone have an effect on GR ex-pression level. Taken together this suggests that HeLa cells aremore sensitive to the effects of glucocorticoids than A549 cells asmanifest by the greater induction of phospho Ser211 GR and thegreater suppression of total GR protein level. This is compatiblewith the greater rosiglitazone effect seen in HeLa cells comparedwith A549 cells.
HeLa cells also showed a greater fold change in reportergene activity after dexamethasone, fitting with their increasedglucocorticoid sensitivity. Rosiglitazone activated the re-porter gene in both cell lines, although the effect seen wasagain much greater in the HeLa cells. To determine whetherthe transcriptional effect was dependent on GR, we used U20Scells, which are deficient in GR expression. In these cells wewere able to show that transfection of the GR resulted in amarked left shift of the dose response to dexamethasone andalso that overexpression of GR was absolutely required topermit any rosiglitazone regulation of the reporter gene. It isalso important to note that the reporter gene harbors idealizedconsensus GREs and no potential PPAR� binding sites and istherefore not regulated by PPAR�.
Further analysis of the transcriptional effects of rosiglitazonemediated by the GR showed that whereas RU486 efficiently an-tagonized transactivation of the reporter gene by dexametha-sone, it had no discernible effect on the rosiglitazone induction,although the maximal transactivation seen with the two mole-cules was similar. This suggested that they may have a similarmode of action as partial agonists. Indeed, both rosiglitazone andRU486 similarly inhibited the reporter gene activation seen withdexamethasone. This is important because opposition to glu-cocorticoid action in the liver is known to increase insulin sen-sitivity (45).
Because previous reports suggested that rosiglitazone andother PPAR� ligands exert an antiproliferative effect, a phe-nomenon frequently observed in epithelial cells for glucocor-ticoids, we also examined the actions of rosiglitazone on pro-liferation using HeLa, A549, and U20S cells (2, 36, 41,46 – 48). These data show a significant inhibition of prolifer-ation seen in both HeLa and A549 cells, although the mag-nitude of effect is less than that seen with dexamethasone. InU20S cells, the inhibition of proliferation seen with glucocor-
A549A5491.8
1.6
1.4
1.2
1.0
0.8
0.6
Form
azan
leve
ls (O
D)
HeLaHeLa0.8
0.7
0.6
0.5
0.4
Form
azan
leve
ls (O
D)
A B
0 0.01 0.1 1 10 100 0 0.01 0.1 1 10 100
μM Rosi100μM Rosi + nM Dex
nM DexμM Rosi100μM Rosi + nM Dex
nM Dex
Dose Dose
C
Form
azan
leve
ls (O
D)
U20S pcDNA3 GRU20S pcDNA3 GRU20S pcDNA3U20S pcDNA31.75
1.5
1.25
1.0
0.75
0.50 0.01 0.1 1 10 100 0 0.01 0.1 1 10 100
μM Rosi100μM Rosi + nM Dex
nM Dex
Dose Dose
FIG. 8. Rosiglitazone-mediated antiproliferative effect is enhanced by GRoverexpression. HeLa (A), A549 (B), and U20S cells transientlyoverexpressing hGR (C) were serum starved, treated with variousconcentrations of dexamethasone (Dex) or rosiglitazone (Rosi) or acombination of both and analyzed for proliferation by MTS assay. Datafor U20S and U20SGR cells were obtained from the same experiment andare represented on different axes to aid comparison. Graphs (mean � SD)are representative of three independent experiments performed intriplicate.
Endocrinology, January 2009, 150(1):75–86 endo.endojournals.org 83

ticoids was exclusively dependent on expression of the GR,and we were also able to show that GR expression enhancedthe antiproliferative effects of rosiglitazone. Although therewas an inhibitory effect seen with rosiglitazone in the non-GR-expressing U20S cells, presumably mediated by a non-GR-dependent mechanism, this effect is clearly greater in cellstransfected with the GR expression vector, indicating a me-diating role for GR.
Studies using cells derived from PPAR�-deficient mice[PPAR�(�/�)] demonstrate that GR nuclear translocation,ser211 phosphorylation and TAT3-luc transactivation are bothPPAR�-independent and GR-dependent.
The effect of rosiglitazone on GR activation appeared greaterin the PPAR�(�/�) cells compared with either HeLa or A549cells, which is perhaps suggestive of competition between PPAR�
and GR for rosiglitazone binding.Glucocorticoids had no inhibitory effect on PPAR�(�/�) cells,
even when GR was overexpressed, indicating that this cell typedoes not couple GR activation to the antiproliferative program.Consistent with the view that the antiproliferative effects of ros-iglitazone are dependent on the GR, there was no inhibitoryeffect seen with rosiglitazone either.
We go on to show that two structurally related compoundsciglitazone and pioglitazone also promote nuclear GR translo-cation, ser211 phosphorylation, TAT3-luc transactivation, andSRC-1 recruitment in a GR-dependent and PPAR�-independentmanner, which suggests that GR activation is a property of theentire class of drugs and not a characteristic specific for ros-iglitazone. Interestingly, the overall potency of each com-pound across the series of experiments broadly correlates withtheir reported affinity for PPAR�.
We show that only the isolated GR-LBD is sufficient for therosiglitazone effect, suggesting direct binding to the GR-LBD.In our previous paper, we were unable to detect high-affinitybinding of rosiglitazone to the GR (34). However, at highconcentrations rosiglitazone did compete with labeled dexa-methasone for binding to the GR, suggesting a low-affinityinteraction. These data, together with that from the M2Hassays (which used only isolated GR-LBD) suggest it is pos-sible that the mode of action of the PPAR� ligand rosiglitazonemay be through direct interaction with GR. In further supportof this hypothesis, we provide theoretical modeling evidenceof rosiglitazone binding to the GR-LBD structure, which fur-ther supports direct binding of rosiglitazone to the GR. How-ever, we cannot exclude the possibility that rosiglitazone anddrugs of its class can activate the GR by an indirect mecha-nism: either inducing local synthesis of a GR ligand or beingmetabolized to an active molecule. But the effect observed iscertainly independent of the conventional rosiglitazone re-ceptor, PPAR�.
Taken together, we now provide evidence that thiazo-lidinediones promote partial activation of the GR. Althoughrosiglitazone clearly promotes GR nuclear translocation andSer211 phosphorylation and weakly activates a simple reportergene, it also powerfully inhibits Gc-mediated gene induction.Therefore, we propose that, like RU486, rosiglitazone is apartial GR agonist.
We have identified an unexpected mechanism for rosiglita-zone action in human cells that may explain some of the activitiesof the drug observed in vivo that do not appear to be attributableto action via PPAR�. We propose that this mechanism may beimportant for the design of new, more specific PPAR� ligandsand also potentially may be exploited for new therapeutics tar-geting the GR itself.
Pi 120 m
inC
igli120min
Control
Aanti mGR (1) anti mGR (2) anti mGR (3)
0 1 10 100
B
PP--GRGR
GRGR
tubulintubulin
PPARPPARγγ((--//--))
μμM M RosiglitazoneRosiglitazone μμM M CiglitazoneCiglitazone μμM M PioglitazonePioglitazone
0 1 10 100 0 1 10 100
FIG. 9. Ciglitazone and pioglitazone activate GR. PPAR�(�/�) cells wereserum starved, treated with 1–100 �M rosiglitazone, 1–100 �M ciglitazone(Cigli), or 1–100 �M pioglitazone (Pi) for 120 min and subjected toimmunoblot analysis to assay for GR phosphorylation (A) andimmunofluorescent microscopy to examine subcellular trafficking ofendogenous GR (B; three representative fields are shown). PPAR�(�/�) cellstransiently cotransfected with hGR and TAT3-luc were serum starved;treated with rosiglitazone (Rosi), ciglitazone (Cigli), or pioglitazone (Pi;0.01–100 �M) for 18 h; and subjected to luciferase assay as an indicator ofGR activation (C and D). Data for PPAR�(�/�) and PPAR�(�/�) GR cells wereobtained from the same experiment and are represented on differentaxes to aid comparison. PPAR�(�/�) cells were transfected with 5 �g eachof pG5-luc and either pBIND or pBIND-GRLBD (E) or with 5 �g each ofpG5-luc, pACT-GRLBD, and either pBIND or pBIND-SRC-1 (F); then treatedwith 1–100 �M rosiglitazone, 1–100 �M ciglitazone, or 1–100 �M
pioglitazone for 18 h; and analyzed for luciferase activity using a dual-luciferase kit. Data represent fold induction over control treatment.Graphs (mean � SD) show one of at least two representative experimentsperformed in triplicate. EC50 values were determined from pooled data.Significant difference from control treatment is indicated (*, P � 0.05; **,P � 0.01).
84 Matthews et al. Rosiglitazone Activates the GR Endocrinology, January 2009, 150(1):75–86

Acknowledgments
We gratefully acknowledge Professor Spiegelman for donation ofPPAR�(�/�) cells and Drs. J. Lluihi-Ineguez and M. Norman for the gift ofTAT3-luc and GR-GFP constructs, respectively. We also thank to RobertFernandez (BioImaging Facility, University of Manchester, Manchester,
UK) for technical assistance. BioImaging Facility microscopes used in thisstudy were purchased with grants from Biotechnology and Biological Sci-ences Research Council, Wellcome Trust, and the University of ManchesterStrategic Fund. Modeling services were provided by Evotec (UK) Ltd.(Abingdon, UK).
Address all correspondence and requests for reprints to: David Ray,Endocrine Sciences Research Group, AV Hill Building, University ofManchester, Oxford Road, Manchester M13 9PT, United Kingdom.E-mail: [email protected]; or Angela Ianaro, Departmentof Experimental Pharmacology, University of Naples Federico II, ViaDomenico Montesano 49, 80131 Naples, Italy. E-mail: [email protected].
This work was supported by the Wellcome Trust.Disclosure Statement: The authors have nothing to declare.
References
1. Evans RM 1988 The steroid and thyroid hormone receptor superfamily. Sci-ence 240:889–895
2. Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CD, BrunRP, Mueller E, Altiok S, Oppenheim H, Evans RM, Spiegelman BM 1997Terminal differentiation of human liposarcoma cells induced by ligands forperoxisome proliferator-activated receptor � and the retinoid X receptor. ProcNatl Acad Sci USA 94:237–241
3. Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM 1998 PPAR� pro-motes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell93:241–252
ROSIDAC
FIG. 10. Docked binding mode of rosiglitazone (Rosi) in the GR-LBDbinding pocket (pink atoms). The protein surface of the GR ligand bindingpocket is shown along with the conformation of DAC taken from thecrystal structure (green atoms). Arg611, Asn564, and Thr739 side-chainatoms have been highlighted for reference.
0 0.01 0.1 1 10 100
PPARPPARγγ((--//--)) pcDNA3pcDNA3
RLU
(x10
3 )
30
20
10
0
PPARPPARγγ((--//--)) pcDNA3GRpcDNA3GR
Dose Dose
0 0.01 0.1 1 10 100
μM Pi
μM Rosi
Rosi EC50; 12.6µMPi EC50; 26.8µM
****
****
D
E
3
4
2
1
0
Cor
rect
ed R
LU (F
old
Indu
ctio
n)
pBIND pBIND GR-LBD
00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100
μM Rosi μM RosiμM Cigli μM CigliμM Pi μM Pi
****
** ** **
FC
orre
cted
RLU
(Fol
d In
duct
ion)
6
8
7
5
4
3
2
1
0
9
pACT GR-LBD + pBIND pACT GR-LBD + pBIND SRC-1
00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100 00 11 1010 100
100
μM Rosi μM RosiμM Cigli μM CigliμM Pi μM Pi
****
** **
**
0 0.01 0.1 1 10 100
PPARPPARγγ((--//--)) pcDNA3pcDNA3
RLU
(x10
3 )
C30
20
10
0
PPARPPARγγ((--//--)) pcDNA3GRpcDNA3GR
Dose Dose
0 0.01 0.1 1 10 100
μM Rosi
μM Cigli
Rosi EC50; 12.6µMCigli EC50; 2.3µM
****
**
**
**
**
FIG. 9. Continued
Endocrinology, January 2009, 150(1):75–86 endo.endojournals.org 85

4. Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM 2001 PPAR-�dependent and independent effects on macrophage-gene expression in lipidmetabolism and inflammation. Nat Med 7:48–52
5. Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM 199515-Deoxy-12, 14-prostaglandin J2 is a ligand for the adipocyte determina-tion factor PPAR�. Cell 83:803–812
6. Willson TM, Lambert MH, Kliewer SA 2001 Peroxisome proliferator-acti-vated receptor � and metabolic disease. Annu Rev Biochem 70:341–367
7. Brown KK, Henke BR, Blanchard SG, Cobb JE, Mook R, Kaldor I, Kliewer SA,Lehmann JM, Lenhard JM, Harrington WW, Novak PJ, Faison W, Binz JG,Hashim MA, Oliver WO, Brown HR, Parks DJ, Plunket KD, Tong WQ,Menius JA, Adkison K, Noble SA, Willson TM 1999 A novel N-aryl tyrosineactivator of peroxisome proliferator-activated receptor-� reverses the diabeticphenotype of the Zucker diabetic fatty rat. Diabetes 48:1415–1424
8. Parks DJ, Tomkinson NC, Villeneuve MS, Blanchard SG, Willson TM 1998Differential activity of rosiglitazone enantiomers at PPAR�. Bioorg Med ChemLett 8:3657–3658
9. Willson TM, Wahli W 1997 Peroxisome proliferator-activated receptor ago-nists. Curr Opin Chem Biol 1:235–241
10. Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, KliewerSA 1995 An antidiabetic thiazolidinedione is a high affinity ligand for peroxisomeproliferator-activated receptor � (PPAR�). J Biol Chem 270:12953–12956
11. Lee CH, Olson P, Evans RM 2003 Minireview: lipid metabolism, metabolicdiseases, andperoxisomeproliferator-activated receptors.Endocrinology144:2201–2207
12. Miles PD, Romeo OM, Higo K, Cohen A, Rafaat K, Olefsky JM 1997 TNF-�-induced insulin resistance in vivo and its prevention by troglitazone. Dia-betes 46:1678–1683
13. Peraldi P, Spiegelman B 1998 TNF-� and insulin resistance: summary andfuture prospects. Mol Cell Biochem 182:169–175
14. Peraldi P, Xu M, Spiegelman BM 1997 Thiazolidinediones block tumor necrosisfactor-�-induced inhibition of insulin signaling. J Clin Invest 100:1863–1869
15. Brun RP, Tontonoz P, Forman BM, Ellis R, Chen J, Evans RM, SpiegelmanBM 1996 Differential activation of adipogenesis by multiple PPAR isoforms.Genes Dev 10:974–984
16. Tontonoz P, Hu E, Spiegelman BM 1995 Regulation of adipocyte gene ex-pression and differentiation by peroxisome proliferator activated receptorgamma. Curr Opin Genet Dev 5:571–576
17. Tontonoz P, Hu E, Spiegelman BM 1994 Stimulation of adipogenesis in fibro-blasts by PPAR�2, a lipid-activated transcription factor. Cell 79:1147–1156
18. Soroceanu MA, Miao D, Bai XY, Su H, Goltzman D, Karaplis AC 2004Rosiglitazone impacts negatively on bone by promoting osteoblast/osteocyteapoptosis. J Endocrinol 183:203–216
19. Schwartz AV, Sellmeyer DE, Vittinghoff E, Palermo L, Lecka-Czernik B,Feingold KR, Strotmeyer ES, Resnick HE, Carbone L, Beamer BA, Park SW,Lane NE, Harris TB, Cummings SR 2006 Thiazolidinedione use and bone lossin older diabetic adults. J Clin Endocrinol Metab 91:3349–3354
20. Murphy CE, Rodgers PT 2007 Effects of thiazolidinediones on bone loss andfracture (December). Ann Pharmacother 41:2014–2018
21. Gardner OS, Dewar BJ, Graves LM 2005 Activation of mitogen-activatedprotein kinases by peroxisome proliferator-activated receptor ligands: an ex-ample of nongenomic signaling. Mol Pharmacol 68:933–941
22. Gardner OS, Dewar BJ, Earp HS, Samet JM, Graves LM 2003 Dependence ofperoxisome proliferator-activated receptor ligand-induced mitogen-activatedprotein kinase signaling on epidermal growth factor receptor transactivation.J Biol Chem 278:46261–46269
23. Koeffler HP 2003 Peroxisome proliferator-activated receptor gamma and can-cers. Clin Cancer Res 9:1–9
24. Rhen T, Cidlowski JA 2005 Antiinflammatory action of glucocorticoids—newmechanisms for old drugs. N Engl J Med 353:1711–1723
25. Adcock IM, Ito K 2000 Molecular mechanisms of corticosteroid actions.Monaldi Arch Chest Dis 55:256–266
26. Morishima Y, Murphy PJ, Li DP, Sanchez ER, Pratt WB 2000 Stepwise as-sembly of a glucocorticoid receptor.hsp90 heterocomplex resolves two se-quential ATP-dependent events involving first hsp70 and then hsp90 in open-ing of the steroid binding pocket. J Biol Chem 275:18054–18060
27. Pratt WB, Silverstein AM, Galigniana MD 1999 A model for the cytoplasmic
trafficking of signalling proteins involving the hsp90-binding immunophilinsand p50cdc37. Cell Signal 11:839–851
28. Gustafsson JA, Carlstedt-Duke J, Poellinger L, Okret S, Wikstrom AC, BronnegardM, Gillner M, Dong Y, Fuxe K, Cintra A 1987 Biochemistry, molecular biology,and physiology of the glucocorticoid receptor. Endocr Rev 8:185–234
29. Garside H, Stevens A, Farrow S, Normand C, Houle B, Berry A, Maschera B,Ray D 2004 Glucocorticoid ligands specify different interactions with NF-�Bby allosteric effects on the glucocorticoid receptor DNA binding domain. J BiolChem 279:50050–50059
30. Nissen RM, Yamamoto KR 2000 The glucocorticoid receptor inhibits NF�Bby interfering with serine-2 phosphorylation of the RNA polymerase II car-boxy-terminal domain. Genes Dev 14:2314–2329
31. Han SJ, Demayo FJ, Xu J, Tsai SY, Tsai MJ, O’Malley BW 2006 Steroidreceptor coactivator (SRC)-1 and SRC-3 differentially modulate tissue-specificactivation functions of the progesterone receptor. Mol Endocrinol 20:45–55
32. McKenna NJ, Xu J, Nawaz Z, Tsai SY, Tsai MJ, O’Malley BW 1999 Nuclearreceptor coactivators: multiple enzymes, multiple complexes, multiple func-tions. J Steroid Biochem Mol Biol 69:3–12
33. Nawaz Z, O’Malley BW 2004 Urban renewal in the nucleus: is protein turn-over by proteasomes absolutely required for nuclear receptor-regulated tran-scription? Mol Endocrinol 18:493–499
34. Ialenti A, Grassia G, Di Meglio P, Maffia P, Di Rosa M, Ianaro A 2005Mechanism of the anti-inflammatory effect of thiazolidinediones: relationshipwith the glucocorticoid pathway. Mol Pharmacol 67:1620–1628
35. Garside H, Waters C, Berry A, Rice L, Ardley HC, White A, Robinson PA, RayD 2006 UbcH7 interacts with the glucocorticoid receptor and mediates re-ceptor autoregulation. J Endocrinol 190:621–629
36. Sommer P, Le Rouzic P, Gillingham H, Berry A, Kayahara M, Huynh T, WhiteA, Ray DW 2007 Glucocorticoid receptor overexpression exerts an antisur-vival effect on human small cell lung cancer cells. Oncogene 26:7111–7121
37. Kudrin A, Scott M, Martin S, Chung CW, Donn R, McMaster A, Ellison S, RayD, Ray K, Binks M 2006 Human macrophage migration inhibitory factor: aproven immunomodulatory cytokine? J Biol Chem 281:29641–29651
38. Stevens A, Garside H, Berry A, Waters C, White A, Ray D 2003 Dissociationof steroid receptor coactivator 1 and nuclear receptor corepressor recruitmentto the human glucocorticoid receptor by modification of the ligand-receptorinterface: the role of tyrosine 735. Mol Endocrinol 17:845–859
39. Suino-Powell K, Xu Y, Zhang C, Tao YG, Tolbert WD, Simons Jr SS, Xu HE2008 Doubling the size of the glucocorticoid receptor ligand binding pocket bydeacylcortivazol. Mol Cell Biol 28:1915–1923
40. Matthews L, Berry A, Ohanian V, Ohanian J, Garside H, Ray D 2008 Caveolinmediates rapid glucocorticoid effects, and couples glucocorticoid action to theantiproliferative programme. Mol Endocrinol 22:1320–1330
41. Weng JR, Chen CY, Pinzone JJ, Ringel MD, Chen CS 2006 Beyond peroxisomeproliferator-activated receptor � signaling: the multi-facets of the antitumoreffect of thiazolidinediones. Endocr Relat Cancer 13:401–413
42. Lago RM, Singh PP, Nesto RW 2007Congestiveheart failureandcardiovasculardeath in patients with prediabetes and type 2 diabetes given thiazolidinediones: ameta-analysis of randomised clinical trials. Lancet 370:1129–1136
43. Krstic MD, Rogatsky I, Yamamoto KR, Garabedian MJ 1997 Mitogen-activatedand cyclin-dependent protein kinases selectively and differentially modulate tran-scriptionalenhancementbytheglucocorticoidreceptor.MolCellBiol17:3947–3954
44. Wang Z, Frederick J, Garabedian MJ 2002 Deciphering the phosphorylation“code” of the glucocorticoid receptor in vivo. J Biol Chem 277:26573–26580
45. Zinker B, Mika A, Nguyen P, Wilcox D, Ohman L, von Geldern TW,Opgenorth T, Jacobson P 2007 Liver-selective glucocorticoid receptor antag-onism decreases glucose production and increases glucose disposal, amelio-rating insulin resistance. Metabolism 56:380–387
46. Wang JC, Shah N, Pantoja C, Meijsing SH, Ho JD, Scanlan TS, Yamamoto KR2006 Novel arylpyrazole compounds selectively modulate glucocorticoid re-ceptor regulatory activity. Genes Dev 20:689–699
47. Rogatsky I, Hittelman AB, Pearce D, Garabedian MJ 1999 Distinct glucocor-ticoid receptor transcriptional regulatory surfaces mediate the cytotoxic andcytostatic effects of glucocorticoids. Mol Cell Biol 19:5036–5049
48. Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, Fletcher C,Singer S, Spiegelman BM 1998 Terminal differentiation of human breast can-cer through PPAR�. Mol Cell 1:465–470
86 Matthews et al. Rosiglitazone Activates the GR Endocrinology, January 2009, 150(1):75–86