therapeutic lentivirus-mediated neonatal in vivo gene therapy in hyperbilirubinemic gunn rats
TRANSCRIPT

ARTICLE doi:10.1016/j.ymthe.2005.06.482
Therapeutic Lentivirus-Mediated Neonatal in Vivo GeneTherapy in Hyperbilirubinemic Gunn Rats
Tuan Huy Nguyen,1,* Marta Bellodi-Privato,2,* Dominique Aubert,2 Virginie Pichard,2
Anne Myara,3 Didier Trono,1,4 and Nicolas Ferry2,y
1Department of Microbiology and Molecular Medicine, CMU, University of Geneva, CH-1211 Geneva, Switzerland2Biotherapies Hepatiques, CIC-INSERM 04, CHU Hotel Dieu, 44093 Nantes Cedex 01, France
3Service de Biochimie, Hopital Saint Joseph, 75014 Paris, France4Ecole Polytechnique Federale de Lausanne, CH-1015 Lausanne, Switzerland
*These authors equally contributed to this study.
yTo whom correspondence and reprint requests should be addressed. Fax: (33) 2 40 08 75 06. E-mail: [email protected].
Available online 2 September 2005
852
Crigler–Najjar type 1 disease (CN-1) is a genetic disorder characterized by high levels ofunconjugated bilirubin due to the absence of hepatic UDPglucuronosyltransferase (UGT1) activity.Here we show that in vivo neonatal hepatocyte transduction with a lentiviral vector expressing thedefective enzyme resulted in long-term correction in Gunn rats, a model of CN-1. Lentiviral vectorsharboring the human UGT1 cDNA (approved symbol UGT1A1) under the control of a liver-specifictransthyretin promoter were produced. Two-day-old Gunn rats were injected with 50 Ml of vector.Bilirubinemia was monitored at 6 weeks and monthly thereafter. At 6 weeks, bilirubinemia wascompletely normalized in treated animals, whereas it remained around 100 MM in control rats. Thelevel of correction remained stable for up to 42 weeks. Large amounts of bilirubin conjugates werepresent in the bile of corrected animals. PCR and Western blots confirmed the presence andexpression of UGT1 in liver. The estimated proportion of transduced hepatocytes was 40% andtransduced cells were not detected in extrahepatic tissues except bone marrow in some animals.This work represents the first demonstration of a complete and permanent correction ofhyperbilirubinemia in Gunn rats using lentiviral vectors.
Key Words: Crigler–Najjar syndrome, bilirubin, glucuronosyltransferase, gene therapy,lentivirus, Gunn rat
INTRODUCTION
Crigler–Najjar type I disease (CN-1) is a rare recessiveinherited disorder of metabolism caused by completeinactivation of the enzyme bilirubin glucuronosyltrans-ferase (UGT1; EC 2.4.1.17). Bilirubin is a by-product inthe degradation pathway of hemoglobin and is conju-gated to glucuronic acid in liver microsomes by UGT1 toform a water-soluble derivative that is eliminated in thebile. In CN-1 patients, unconjugated bilirubin chroni-cally accumulates to very high levels in most tissues,resulting in severe jaundice as well as a life-threateningneurotoxic injury referred to as kernicterus. Bilirubin maybe degraded by exposure to UV light and prolonged dailysessions of UV phototherapy are required in CN-1patients. The only curative alternative to phototherapyis liver transplantation.
CN-1 disease is an attractive candidate disease for genetherapy for a number of reasons. (i) CN-1 results from asingle genetic defect that accounts for the whole clinicalspectrum of symptoms, (ii) the histology of the liverparenchyma remains strictly normal throughout thecourse of the disease, (iii) the liver is a unique organharboring a fenestrated endothelium that makes it easilyaccessible to many types of gene transfer vectors deliveredto the blood stream, and (iv) the existence of a mutantjaundiced rat first identified by Gunn [1] and whichreplicates the human CN-1 disease has facilitated in vivoevaluation of gene therapy strategies aimed at correctingbilirubin conjugation deficiencies. For unknown reasons,bilirubinemia in Gunn rats rarely exceeds 200 AM and theanimals do not develop kernicterus. Therefore, althoughcerebellar lesions as well as deafness may be present, there
MOLECULAR THERAPY Vol. 12, No. 5, November 2005
Copyright C The American Society of Gene Therapy
1525-0016/$30.00
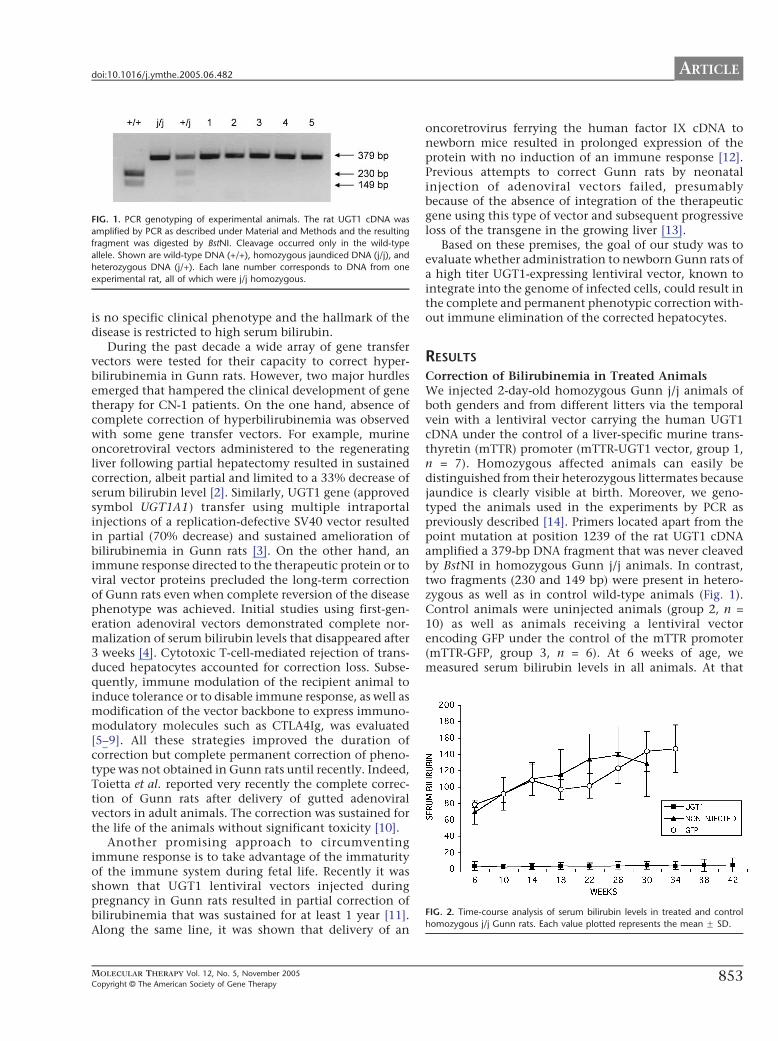
FIG. 1. PCR genotyping of experimental animals. The rat UGT1 cDNA was
amplified by PCR as described under Material and Methods and the resulting
fragment was digested by BstNI. Cleavage occurred only in the wild-type
allele. Shown are wild-type DNA (+/+), homozygous jaundiced DNA (j/j), and
heterozygous DNA (j/+). Each lane number corresponds to DNA from one
experimental rat, all of which were j/j homozygous.
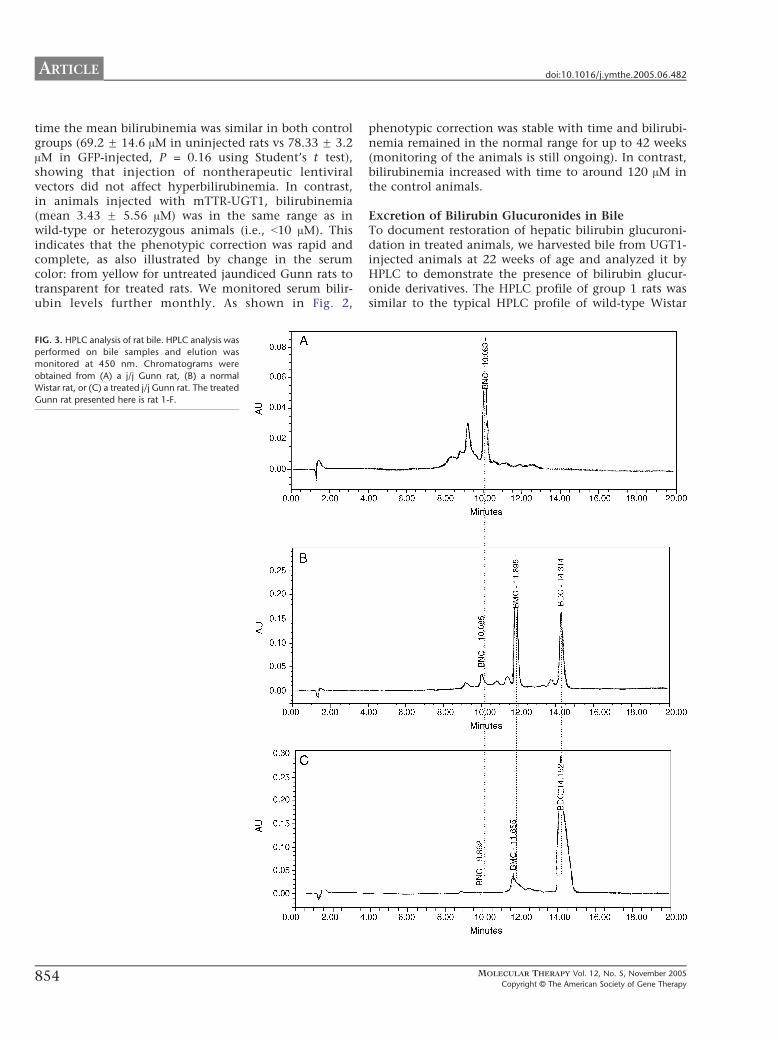
IG. 2. Time-course analysis of serum bilirubin levels in treated and control
omozygous j/j Gunn rats. Each value plotted represents the mean F SD.
ARTICLEdoi:10.1016/j.ymthe.2005.06.482
is no specific clinical phenotype and the hallmark of thedisease is restricted to high serum bilirubin.
During the past decade a wide array of gene transfervectors were tested for their capacity to correct hyper-bilirubinemia in Gunn rats. However, two major hurdlesemerged that hampered the clinical development of genetherapy for CN-1 patients. On the one hand, absence ofcomplete correction of hyperbilirubinemia was observedwith some gene transfer vectors. For example, murineoncoretroviral vectors administered to the regeneratingliver following partial hepatectomy resulted in sustainedcorrection, albeit partial and limited to a 33% decrease ofserum bilirubin level [2]. Similarly, UGT1 gene (approvedsymbol UGT1A1) transfer using multiple intraportalinjections of a replication-defective SV40 vector resultedin partial (70% decrease) and sustained amelioration ofbilirubinemia in Gunn rats [3]. On the other hand, animmune response directed to the therapeutic protein or toviral vector proteins precluded the long-term correctionof Gunn rats even when complete reversion of the diseasephenotype was achieved. Initial studies using first-gen-eration adenoviral vectors demonstrated complete nor-malization of serum bilirubin levels that disappeared after3 weeks [4]. Cytotoxic T-cell-mediated rejection of trans-duced hepatocytes accounted for correction loss. Subse-quently, immune modulation of the recipient animal toinduce tolerance or to disable immune response, as well asmodification of the vector backbone to express immuno-modulatory molecules such as CTLA4Ig, was evaluated[5–9]. All these strategies improved the duration ofcorrection but complete permanent correction of pheno-type was not obtained in Gunn rats until recently. Indeed,Toietta et al. reported very recently the complete correc-tion of Gunn rats after delivery of gutted adenoviralvectors in adult animals. The correction was sustained forthe life of the animals without significant toxicity [10].
Another promising approach to circumventingimmune response is to take advantage of the immaturityof the immune system during fetal life. Recently it wasshown that UGT1 lentiviral vectors injected duringpregnancy in Gunn rats resulted in partial correction ofbilirubinemia that was sustained for at least 1 year [11].Along the same line, it was shown that delivery of an
MOLECULAR THERAPY Vol. 12, No. 5, November 2005
Copyright C The American Society of Gene Therapy
oncoretrovirus ferrying the human factor IX cDNA tonewborn mice resulted in prolonged expression of theprotein with no induction of an immune response [12].Previous attempts to correct Gunn rats by neonatalinjection of adenoviral vectors failed, presumablybecause of the absence of integration of the therapeuticgene using this type of vector and subsequent progressiveloss of the transgene in the growing liver [13].
Based on these premises, the goal of our study was toevaluate whether administration to newborn Gunn rats ofa high titer UGT1-expressing lentiviral vector, known tointegrate into the genome of infected cells, could result inthe complete and permanent phenotypic correction with-out immune elimination of the corrected hepatocytes.
RESULTS
Correction of Bilirubinemia in Treated AnimalsWe injected 2-day-old homozygous Gunn j/j animals ofboth genders and from different litters via the temporalvein with a lentiviral vector carrying the human UGT1cDNA under the control of a liver-specific murine trans-thyretin (mTTR) promoter (mTTR-UGT1 vector, group 1,n = 7). Homozygous affected animals can easily bedistinguished from their heterozygous littermates becausejaundice is clearly visible at birth. Moreover, we geno-typed the animals used in the experiments by PCR aspreviously described [14]. Primers located apart from thepoint mutation at position 1239 of the rat UGT1 cDNAamplified a 379-bp DNA fragment that was never cleavedby BstNI in homozygous Gunn j/j animals. In contrast,two fragments (230 and 149 bp) were present in hetero-zygous as well as in control wild-type animals (Fig. 1).Control animals were uninjected animals (group 2, n =10) as well as animals receiving a lentiviral vectorencoding GFP under the control of the mTTR promoter(mTTR-GFP, group 3, n = 6). At 6 weeks of age, wemeasured serum bilirubin levels in all animals. At that
F
h
853
ARTICLE doi:10.1016/j.ymthe.2005.06.482
time the mean bilirubinemia was similar in both controlgroups (69.2 F 14.6 AM in uninjected rats vs 78.33 F 3.2AM in GFP-injected, P = 0.16 using Student’s t test),showing that injection of nontherapeutic lentiviralvectors did not affect hyperbilirubinemia. In contrast,in animals injected with mTTR-UGT1, bilirubinemia(mean 3.43 F 5.56 AM) was in the same range as inwild-type or heterozygous animals (i.e., b10 AM). Thisindicates that the phenotypic correction was rapid andcomplete, as also illustrated by change in the serumcolor: from yellow for untreated jaundiced Gunn rats totransparent for treated rats. We monitored serum bilir-ubin levels further monthly. As shown in Fig. 2,
FIG. 3. HPLC analysis of rat bile. HPLC analysis was
performed on bile samples and elution was
monitored at 450 nm. Chromatograms were
obtained from (A) a j/j Gunn rat, (B) a normal
Wistar rat, or (C) a treated j/j Gunn rat. The treated
Gunn rat presented here is rat 1-F.
854
phenotypic correction was stable with time and bilirubi-nemia remained in the normal range for up to 42 weeks(monitoring of the animals is still ongoing). In contrast,bilirubinemia increased with time to around 120 AM inthe control animals.
Excretion of Bilirubin Glucuronides in BileTo document restoration of hepatic bilirubin glucuroni-dation in treated animals, we harvested bile from UGT1-injected animals at 22 weeks of age and analyzed it byHPLC to demonstrate the presence of bilirubin glucur-onide derivatives. The HPLC profile of group 1 rats wassimilar to the typical HPLC profile of wild-type Wistar
MOLECULAR THERAPY Vol. 12, No. 5, November 2005
Copyright C The American Society of Gene Therap
y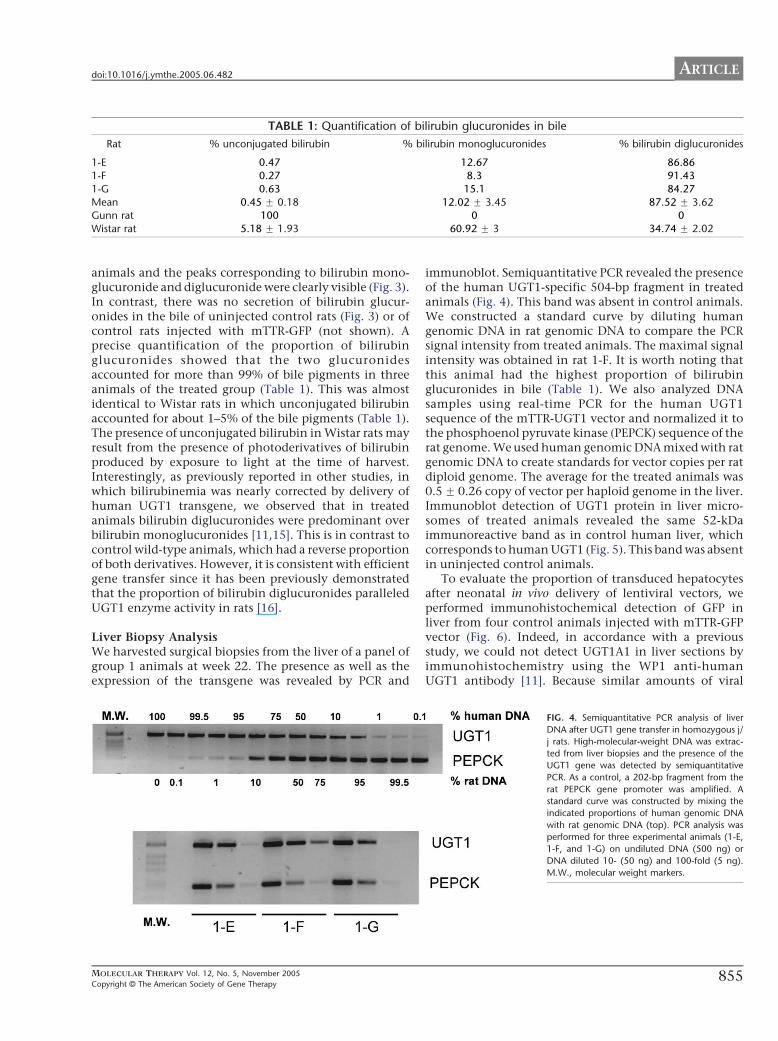
TABLE 1: Quantification of bilirubin glucuronides in bile
Rat % unconjugated bilirubin % bilirubin monoglucuronides % bilirubin diglucuronides
1-E 0.47 12.67 86.86
1-F 0.27 8.3 91.43
1-G 0.63 15.1 84.27
Mean 0.45 F 0.18 12.02 F 3.45 87.52 F 3.62Gunn rat 100 0 0
Wistar rat 5.18 F 1.93 60.92 F 3 34.74 F 2.02
ARTICLEdoi:10.1016/j.ymthe.2005.06.482
animals and the peaks corresponding to bilirubin mono-glucuronide and diglucuronide were clearly visible (Fig. 3).In contrast, there was no secretion of bilirubin glucur-onides in the bile of uninjected control rats (Fig. 3) or ofcontrol rats injected with mTTR-GFP (not shown). Aprecise quantification of the proportion of bilirubinglucuronides showed that the two glucuronidesaccounted for more than 99% of bile pigments in threeanimals of the treated group (Table 1). This was almostidentical to Wistar rats in which unconjugated bilirubinaccounted for about 1–5% of the bile pigments (Table 1).The presence of unconjugated bilirubin in Wistar rats mayresult from the presence of photoderivatives of bilirubinproduced by exposure to light at the time of harvest.Interestingly, as previously reported in other studies, inwhich bilirubinemia was nearly corrected by delivery ofhuman UGT1 transgene, we observed that in treatedanimals bilirubin diglucuronides were predominant overbilirubin monoglucuronides [11,15]. This is in contrast tocontrol wild-type animals, which had a reverse proportionof both derivatives. However, it is consistent with efficientgene transfer since it has been previously demonstratedthat the proportion of bilirubin diglucuronides paralleledUGT1 enzyme activity in rats [16].
Liver Biopsy AnalysisWe harvested surgical biopsies from the liver of a panel ofgroup 1 animals at week 22. The presence as well as theexpression of the transgene was revealed by PCR and
MOLECULAR THERAPY Vol. 12, No. 5, November 2005
Copyright C The American Society of Gene Therapy
immunoblot. Semiquantitative PCR revealed the presenceof the human UGT1-specific 504-bp fragment in treatedanimals (Fig. 4). This band was absent in control animals.We constructed a standard curve by diluting humangenomic DNA in rat genomic DNA to compare the PCRsignal intensity from treated animals. The maximal signalintensity was obtained in rat 1-F. It is worth noting thatthis animal had the highest proportion of bilirubinglucuronides in bile (Table 1). We also analyzed DNAsamples using real-time PCR for the human UGT1sequence of the mTTR-UGT1 vector and normalized it tothe phosphoenol pyruvate kinase (PEPCK) sequence of therat genome. We used human genomic DNA mixed with ratgenomic DNA to create standards for vector copies per ratdiploid genome. The average for the treated animals was0.5 F 0.26 copy of vector per haploid genome in the liver.Immunoblot detection of UGT1 protein in liver micro-somes of treated animals revealed the same 52-kDaimmunoreactive band as in control human liver, whichcorresponds to human UGT1 (Fig. 5). This band was absentin uninjected control animals.
To evaluate the proportion of transduced hepatocytesafter neonatal in vivo delivery of lentiviral vectors, weperformed immunohistochemical detection of GFP inliver from four control animals injected with mTTR-GFPvector (Fig. 6). Indeed, in accordance with a previousstudy, we could not detect UGT1A1 in liver sections byimmunohistochemistry using the WP1 anti-humanUGT1 antibody [11]. Because similar amounts of viral
FIG. 4. Semiquantitative PCR analysis of liver
DNA after UGT1 gene transfer in homozygous j/
j rats. High-molecular-weight DNA was extrac-
ted from liver biopsies and the presence of the
UGT1 gene was detected by semiquantitative
PCR. As a control, a 202-bp fragment from the
rat PEPCK gene promoter was amplified. A
standard curve was constructed by mixing the
indicated proportions of human genomic DNA
with rat genomic DNA (top). PCR analysis was
performed for three experimental animals (1-E,
1-F, and 1-G) on undiluted DNA (500 ng) or
DNA diluted 10- (50 ng) and 100-fold (5 ng).
M.W., molecular weight markers.
855

FIG. 5. Western blot analysis of rat liver after UGT1 gene transfer. Microsome
were prepared from liver biopsies of three animals (1-E, 1-F, 1-G) 22 week
after gene transfer. After immunoblotting, the presence of UGT1 protein wa
detected in all treated animals but was absent in uninjected control (Ctrl)
Human liver, control human microsomes.
FIG. 6. Immunohistochemical detection of transduced hepatocytes in GFP
injected animals. The presence of GFP-positive hepatocytes was detected by
immunohistochemistry as described under Material and Methods. This picture
was obtained in an experimental animal with 40% GFP-positive hepatocytes
Hematoxylin counterstained. Original magnification �220.
ARTICLE doi:10.1016/j.ymthe.2005.06.482
856
s
s
s
.
particles were infused in UGT1-treated and GFP controlanimals, the proportion of GFP-transduced hepatocytesshould reflect the proportion of lentivirally transducedhepatocytes in UGT1-injected animals [11]. Lentivirallytransduced hepatocytes were distributed throughout theliver parenchyma. The mean number of transducedhepatocytes was 36.15 F 29% (minimum 10.3%, max-imum 70%, data not shown) and was in agreement withthe PCR experiments. GFP-positive cells were notdetected in uninjected rats. We did not detect non-parenchymal cells stained by immunohistochemistryusing anti-GFP antibodies, which is in accordance withthe liver specificity of the mTTR promoter.
Tissue Distribution of the TransgeneBecause delivery of viral vectors to the bloodstream shouldresult in transduction of multiple organs, we performedsemiquantitative PCR detection of human UGT1 cDNA invarious organs of two treated animals. We observed apositive signal in the liver of both animals. A faint bandwas also present in the bone marrow DNA in one animal.All other tested organs were negative, indicating that
FIG. 7. Determination of vector dissemination by PCR. High-molecular-weigh
genomic DNA was prepared from various organs of two mTTR-UGT1-injected
j/j Gunn rat at 3 months after gene delivery. The presence of the UGT1 cDNA
was detected by PCR. As a control, a 202-bp fragment from the rat PEPCK
gene promoter was amplified. M.W., molecular weight markers.
-
.
transduction of organs with a continuous endotheliumeither was absent or occurred at a low level (Fig. 7).
DISCUSSION
CN-1 disease represents a paradigm for the gene therapy ofinherited liver disorders for a number of reasons. It is amonogenic disease that does not injure the liver paren-chyma. Because the toxic product that accumulates duringthe course of the disease, unconjugated bilirubin, islipophilic, it can easily be taken up by a small proportionof corrected cells and there is no need to transduce thewhole liver to get a therapeutic effect. The disease isdetected early after birth and CN-1 patients are amenableto various strategies of gene delivery as well as to celltherapy [17,18]. Finally the existence of an animal modelmimicking the human disease has made it possible todevise preclinical approaches of gene therapy.
A wide array of gene transfer vectors harboring thehuman UGT1 cDNA has been designed and their efficacyhas been evaluated in Gunn rats. All these studiesconclusively demonstrated the validity of gene replace-ment therapy for improving the hallmarks of the diseasein this model although no complete and permanentcorrection was achieved until recently. As an example,first-generation adenoviral vector-mediated gene deliv-ery to the liver resulted in a transient complete correc-tion that was rapidly abrogated by a strong immunerejection of transduced cells [4]. Various strategies tocircumvent the immune response triggered by thishighly immunogenic vector type were designed andtested in Gunn rats [6,7,9]. An encouraging studyreported that adenovirus-mediated gene transfer to new-born animals decreased rejection of transduced cells [13].However, hyperbilirubinemia eventually resumed in all
MOLECULAR THERAPY Vol. 12, No. 5, November 2005
Copyright C The American Society of Gene Therap
t
y

ARTICLEdoi:10.1016/j.ymthe.2005.06.482
cases after several weeks. Very recently, Toietta et al.reported the complete and permanent correction ofGunn rats after a single injection of gutted adenoviralvectors in adult animals [10]. However, the clinicalapplication of this approach is still questionable. Theabsence of toxicity of adenoviral vectors in rat does notpreclude potential toxicity in human. This issue is ofparticular importance since a previous clinical trial ofgene therapy for OTC deficiency using adenoviralvectors resulted in the death of a young patientattributed to innate immunity immediately after injec-tion [19]. The dose of vector injected into the patient(6 � 1011 viral particles/kg) was lower than the doserequired to treat the Gunn rats (3 � 1012 particles/kg).
Retroviral vectors govern integration of the trans-gene into the genome of target cells. Therefore, thecorrection level achieved after infection is constantover time whatever the number of subsequent divisionsof the corrected cell. This is particularly importantwhen gene therapy is aimed at treating young children.Such sustained correction was reported in studies inwhich lentiviral vectors were delivered in utero as wellas when oncoretroviral vectors were delivered afterpartial hepatectomy in adult Gunn rats [2,11]. In bothstudies, correction was sustained for prolonged periodsof time (up to 1 year). However, since oncoretroviralvectors are less efficient than adenoviral vectors attransducing hepatocytes, a complete correction wasnever achieved. In our present study we demonstratedthat when high-titer integrative vectors are delivered tonewborn animals, a sufficient amount of hepatocytes istargeted and a complete correction is achieved. More-over, this complete correction is sustained even duringgrowth of treated animals.
We previously reported that when high-titer retroviralvectors were delivered to the liver of adult rats followingpartial hepatectomy, the decrease of serum bilirubin levelwas blunted by an immune response that eliminatedtransduced cells in less than 4 weeks [20]. It has recentlybeen reported that in vivo delivery of recombinant retro-viruses to newborn mice could induce tolerance to nonselfproteins [12]. Such a mechanism may account for theabsence of immune response against UGT1 in our study. Inone animal PCR analysis revealed the presence of thetransgene in bone marrow cells and therefore transductionof bone marrow stem cells is likely as it has been previouslyreported after neonatal delivery of oncoretroviruses [21].However, it must be stressed that the development kineticsof the immune system may differ in humans and inrodents. Therefore, prenatal delivery of the vectors (e.g.,in utero) may be required to achieve tolerance in humans.
Tolerance may also result from the restricted expres-sion of UGT1 in hepatocytes and not in professionalantigen-presenting cells, avoiding the development oftransgene-specific cellular and humoral immuneresponses [22]. More generally, liver-specific promoters
MOLECULAR THERAPY Vol. 12, No. 5, November 2005
Copyright C The American Society of Gene Therapy
would be preferable to target transgene expression inhepatocytes and could be superior to ubiquitouspromoters with regard to level and very long termtransgene expression in liver.
Hyperbilirubinemia is easily detected early after birth.Although the genetic diagnosis for CN-1 disease may notbe established in the very first days of life, patientsamenable to gene therapy are not older than 2 to 3months. Our study warrants that strong consideration begiven to a clinical trial for CN-1 based on the in vivodelivery of lentiviral vectors harboring the human UGT1cDNA under control of a liver-specific promoter.
MATERIAL AND METHODS
Description and production of lentiviral vectors. High-titer lentiviral
vector stocks were generated as previously described by calcium
phosphate-mediated transient transfection of three plasmids: the
transfer vector plasmid, the packaging plasmid pCMVDR8.91, and the
VSVG envelope protein-coding plasmid pMD.G [23]. These self-inacti-
vating transfer vectors harbored the human UGT1 cDNA under control
of a promoter that specifically directs transcription to the liver, the
murine transthyretin promoter fused to a synthetic hepatocyte-specific
enhancer [24,25]. They also harbored the cis-acting cPPT/CTS from
HIV-1, which facilitates the nuclear translocation of preintegrative
vector complexes [26,27] and the posttranscriptional regulatory ele-
ment from the woodchuck hepatitis virus, which increases transgene
expression [28] and was inserted downstream of the transgene. Control
viruses containing the GFP gene under control of the same promoter
were also produced and used as controls. The titers of the viral
preparations were determined on HeLa cells by real-time quantitative
PCR using primers and probe specific for 5V-untranslated lentiviral
vectors: GAG-F, GGAGCTAGAACGATTCGCAGTTA; GAG-R, GGTTGTA-
GCTGTCCCAGTATTTGTC; and GAG-P, 5V-(FAM)-ACAGCCTTCTGATG-
TTTCTAACAGGCCAGG-(Eclipse Dark Quencher)-3V. For normalization
of the amounts of genomic DNA, primers and probe specific for the a-
actin gene were used: HB2-F, TCCGTGTGGATCGGCGGCTCCA; HB2-R,
CTGCTTGCTGATCCACATCTG; and 5V-(Yakima yellow)-CCTGGCCTC-
GCTGTCCACCTTCCA-(Eclipse Dark Quencher)-3V. Reactions were per-
formed and analyzed using an ABI Prism 7700 sequence detection
system (RE-Applied Biosystems). The titer routinely reached 1010 HeLa
transducing units/ml.
Animal experiments. All animals were housed at the animal facilities of
Nantes University Medical school and received human care according to
the guidelines of the French Ministere de l’Agriculture. Homozygous
Gunn j/j rats of both genders were used in this study. They were obtained
from our breeding colony by mating homozygous Gunn j/j males with
heterozygous j/o females.
At birth, homozygous icteric pups were easily distinguished from
heterozygous littermates by simple examination of the yellow coloration of
the tegument. Moreover, a genetic confirmation of homozygosity was
achieved in adulthood by the enzymatic restriction of PCR fragments
encompassing the point mutation in the common exon of the UGT1 gene
family [29,30]. To this end high-molecular-weight DNA was extracted from
primary blood mononuclear cells and a portion was subjected to
amplification by PCR using primers (sense, 5V-GGGATTCTCAGAATCTA-
GACATT-3V; antisense, 5V-GTGTGTGGTATAAATGCTGTAGG-3V). The sam-
ples were submitted to 32 cycles of amplification (948C for 30 s for
denaturation, 558C for 30 s for annealing, and 728C for 1 min for
elongation). The amplicon that was 379 bp in size was analyzed with or
without digestion with the restriction enzyme BstNI [14]. The amplifica-
tion product of homozygous j/j Gunn rat DNA lacks the restriction site for
BstNI and, therefore, yields a single band (379 bp). The restriction pattern
of wild-type controls yields two bands (230 and 149 bp). In heterozygous j/
857

ARTICLE doi:10.1016/j.ymthe.2005.06.482
o littermates both patterns are visible. At day 2 of life, lentiviral vectors (5�108 infectious particles in 50 Al PBS containing 8 Ag/ml Polybrene) were
injected via the temporal vein.
Bilirubin assays. Six weeks after gene delivery and monthly thereafter,
blood samples were drawn from the retro-orbital sinus for serum analysis.
Serum total bilirubin levels were measured spectrophotometrically on a
Hitachi 717 apparatus using the Bil-T kit (Boehringer Mannheim France,
Meylan, France). Bile was harvested by laparotomy and cannulation of the
main bile duct using a fine polyethylene catheter (PE 10). The presence of
monoglucuronide and diglucuronide bilirubin conjugates in the bile was
assessed using alkaline methanolysis as previously described [31].
Western blot analysis. Total microsomes (100,000g supernatant) were
isolated from liver biopsies by ultracentrifugation. Protein contents were
determined using the Bio-Rad protein assay kit. For immunoblot analysis,
proteins (40 Ag/lane) were resolved by electrophoresis on a 10%
polyacrylamide gel. After electrophoretic transfer onto nitrocellulose
membranes, immunoblots were incubated sequentially with 5% low-fat
milk blocking solution and the human WP1 primary antibody diluted
1:5000. The WP1 antibody interacts with the common carboxy-terminal
domains of human UGT1 isoforms [32]. The secondary antibody was
biotinylated conjugated anti-mouse Ig FVab fragment that was detected by
using the ECL Western blotting detection system (Amersham Bioscien-
ces). Human hepatic microsomes were used as positive control.
PCR analysis. High-molecular-weight DNA (500 ng) was subjected to
amplification by PCR using primers (5V-TCTGCTATGCTTTTGTCTGG-3V
and 5V-GGATAGTGGATTTTGGTGAA-3V) specific for human UGT1. Semi-
quantitative PCR amplification was performed by denaturation for 5 min
at 948C, followed by 28 cycles of amplification (948C for 20 s, 608C for 30
s, 728C for 40 s), and a final extension for 10 min at 728C. The resulting
amplified fragment was 504 bp. We carried out a positive control by
amplification of a 202-bp fragment of the rat PEPCK gene promoter using
the two primers 5V-GTCATATTTCTTCAGCTTGCG-3V and 5V-ATAATGGT-
CTGGACTTCTCTG-3V. Serial dilutions of human genomic DNA in rat
genomic DNA were used as a standard curve. Amplified products were
separated by gel electrophoresis on 2% agarose gel and DNA bands were
revealed by ethidium bromide staining.
Real-time quantitative PCR was performed using Sybr Green I dye
(Perkin–Elmer Biosystems) with an ABI Prism 7700 sequence detection
system. The values were normalized for the amount of DNA using
amplification of a PEPCK gene as an internal standard. The sequence of
the forward primer was 5V-GTGATGATGCCCTTGTTTGGT-3V and that of
the reverse primer was 5V-AAACTCCACCCAGAACACGG-3V. A 96-well
optical tray and caps were used with a final reaction mixture
containing 50 ng of DNA, 7.5 pmol (each) of the forward and reverse
primers, and Sybr Green PCR Master Mix (PE Biosystems). Reaction
conditions were 508C for 2 min, 958C for 10 min, followed by 40
cycles at 958C for 15 s and 608C for 1 min. The amplicon generated
was 228 bp. A standard scale was constructed using human genomic
DNA diluted in various proportions with rat genomic DNA. Results are
expressed as Ct values for each sample.
Immunohistochemistry. The presence of GFP-positive hepatocytes was
assessed by immunohistochemistry on formalin-fixed/paraffin-embed-
ded sections (5 Am). Sections were deparaffinized and endogenous
peroxidase activity was inhibited by incubation for 30 min in a 3%
H2O2 solution in PBS. Monoclonal primary mouse anti-GFP antibody
diluted 1:100 in PBS containing BSA (2% w/v) and Tween 20 (0.1%
v/v) was applied for 2 h at room temperature. GFP-positive cells were
revealed with biotinylated goat anti-mouse immunoglobulin and
streptavidin–peroxidase using diaminobenzidine as a chromogenic
substrate. Slides were counterstained with hematoxylin and the
GFP-positive cell index was calculated as the percentage of positively
stained cells in 10 fields at 40� magnification (i.e., at least 3000
cells).
Statistical analysis. Statistical analysis was performed using the Student t
test. A P value of less than 0.05 was considered to be statistically
significant.
858
ACKNOWLEDGMENTS
This work was supported by grants from the Association Francaise contre les
Myopathies and the Swiss National Science Foundation. We thank Wilbert H.
M. Peters for the gift of WP1 antibodies, Daniele Paumier for technical help with
HPLC, Andrew Simmons for the mTTR promoter, Patrick Salmon for helpful
discussions on real-time PCR, and the Lentiviral Vector Production Unit of the
University of Geneva, supported by the Association Francaise contre les
Myopathies, for providing some of the lentiviral vector stocks (http://www.
medecine.unige.ch/%7Esalmon/lvpu/).
RECEIVED FOR PUBLICATION MAY 2, 2005; REVISED JUNE 13, 2005;
ACCEPTED JUNE 27, 2005.
REFERENCES1. Gunn, C. K. (1939). Hereditary acholuric jaundice in a new mutant strain of rat. J. Hered.
29: 137 – 139.
2. Tada, K., et al. (1998). Long-term reduction of serum bilirubin levels in Gunn rats by
retroviral gene transfer in vivo. Liver Transplant. Surg. 4: 78 – 88.
3. Sauter, B. V., et al. (2000). A replication-deficient rSV40 mediates liver-directed gene
transfer and a long-term amelioration of jaundice in Gunn rats. Gastroenterology 119:
1348 – 1357.
4. Askari, F. K., Hitomi, Y., Mao, M., and Wilson, J. M. (1996). Complete correction of
hyperbilirubinemia in the Gunn rat model of Crigler–Najjar syndrome type I following
transient in vivo adenovirus-mediated expression of human bilirubin UDP-glucurono-
syltransferase. Gene Ther. 3: 381 – 388.
5. Ilan, Y., et al. (1996). Induction of central tolerance by intrathymic inoculation of
adenoviral antigens into the host thymus permits long-term gene therapy in Gunn rats.
J. Clin. Invest. 98: 2640 – 2647.
6. Ilan, Y., et al. (1997). Insertion of the adenoviral E3 region into a recombinant viral
vector prevents antiviral humoral and cellular immune responses and permits long-
term gene expression. Proc. Natl. Acad. Sci. USA 94: 2587 – 2592.
7. Ilan, Y., et al. (1997). Transient immunosuppression with FK506 permits long-term
expression of therapeutic genes introduced into the liver using recombinant
adenoviruses in the rat. Hepatology 26: 949 – 956.
8. Ilan, Y., et al. (1997). Oral tolerization to adenoviral antigens permits long-term gene
expression using recombinant adenoviral vectors. J. Clin. Invest. 99: 1098 – 1106.
9. Thummala, N. R., et al. (2002). A non-immunogenic adenoviral vector, coexpressing
CTLA4Ig and bilirubin-uridine-diphosphoglucuronateglucuronosyltransferase permits
long-term, repeatable transgene expression in the Gunn rat model of Crigler–Najjar
syndrome. Gene Ther. 9: 981 – 990.
10. Toietta, G., et al. (2005). Lifelong elimination of hyperbilirubinemia in the Gunn rat
with a single injection of helper-dependent adenoviral vector. Proc. Natl. Acad. Sci. USA
102: 3930 – 3935.
11. Seppen, J., et al. (2003). Long-term correction of bilirubin UDPglucuronyltransferase
deficiency in rats by in utero lentiviral gene transfer. Mol. Ther. 8: 593 – 599.
12. Xu, L., et al. (2003). Neonatal or hepatocyte growth factor-potentiated adult gene
therapy with a retroviral vector results in therapeutic levels of canine factor IX for
hemophilia B. Blood 101: 3924 – 3932.
13. Li, Q., Murphree, S. S., Willer, S. S., Bolli, R., and French, B. A. (1998). Gene therapy
with bilirubin-UDP-glucuronosyltransferase in the Gunn rat model of Crigler–Najjar
syndrome type 1. Hum. Gene Ther. 9: 497 – 505.
14. Roy-Chowdhury, J., et al. (1991). Molecular basis for the lack of bilirubin-specific and 3-
methylcholanthrene-inducible UDP-glucuronosyltransferase activities in Gunn rats: the
two isoforms are encoded by distinct mRNA species that share an identical single base
deletion. J. Biol. Chem. 266: 18294 – 18298.
15. Takahashi, M., et al. (1996). Long term correction of bilirubin-UDP-glucuronosyltrans-
ferase deficiency in Gunn rats by administration of a recombinant adenovirus during
the neonatal period. J. Biol. Chem. 271: 26536 – 26542.
16. Van Steenbergen, W., and Fevery, J. (1990). Effects of uridine diphosphate
glucuronosyltransferase activity on the maximal secretion rate of bilirubin conjugates
in the rat. Gastroenterology 99: 488 – 499.
17. Fox, I. J., et al. (1998). Treatment of the Crigler–Najjar syndrome type I with hepatocyte
transplantation. N. Engl. J. Med. 338: 1422 – 1426.
18. Nguyen, T. H., and Ferry, N. (2004). Liver gene therapy: advances and hurdles. Gene
Ther. 11: S76 – S84.
19. Raper, S. E., et al. (2003). Fatal systemic inflammatory response syndrome in an
ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol.
Genet. Metab. 80: 148 – 158.
20. Aubert, D., et al. (2002). Cytotoxic immune response blunts long-term transgene
expression after efficient retroviral-mediated hepatic gene transfer in rat. Mol. Ther. 5:
388 – 396.
21. Xu, L., et al. (2004). In vivo transduction of hematopoietic stem cells after neonatal
intravenous injection of an amphotropic retroviral vector in mice. Mol. Ther. 10: 37 – 44.
22. Follenzi, A., et al. (2003). Targeting lentiviral vector expression to hepatocytes limits
MOLECULAR THERAPY Vol. 12, No. 5, November 2005
Copyright C The American Society of Gene Therapy

ARTICLEdoi:10.1016/j.ymthe.2005.06.482
transgene-specific immune response and establishes long-term expression of human
antihemophilic factor IX in mice. Blood 30: 30.
23. Bovia, F., et al. (2003). Efficient transduction of primary human B lymphocytes and
nondividing myeloma B cells with HIV-1-derived lentiviral vectors. Blood 101:
1727 – 1733.
24. Costa, R. H., and Grayson, D. R. (1991). Site-directed mutagenesis of hepatocyte
nuclear factor (HNF) binding sites in the mouse transthyretin (TTR) promoter reveals
synergistic interactions with its enhancer region. Nucleic Acids Res. 19: 4139 – 4145.
25. Nguyen, T. H., Khakhoulina, T., Simmons, A., Morel, P., Trono D. (in press). A simple
and highly effective method for the stable transduction of uncultured porcine
hepatocytes using lentiviral vector. Cell Transplant.
26. Follenzi, A., Ailles, L., Bakovic, S., Geuna, M., and Naldini, L. (2000). Gene transfer by
lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol
sequences. Nat. Genet. 25: 217 – 222.
27. Zennou, V., et al. (2000). HIV-1 genome nuclear import is mediated by a central DNA
flap. Cell 10: 173 – 185.
MOLECULAR THERAPY Vol. 12, No. 5, November 2005
Copyright C The American Society of Gene Therapy
28. Zufferey, R., Donello, J. E., Trono, D., and Hope, T. J. (1999). Woodchuck hepatitis virus
posttranscriptional regulatory element enhances expression of transgenes delivered by
retroviral vectors. J. Virol. 73: 2886 – 2892.
29. Sato, H., Aono, S., Kashiwamata, S., and Koiwai, O. (1991). Genetic defect of bilirubin
UDP-glucuronyltransferase in the hyperbilirubinemic Gunn rat. Biochem. Biophys. Res.
Commun. 169: 260 – 264.
30. Sato, H., Koiwai, O., Tanabe, K., and Kashiwamata, S. (1990). Isolation and
sequencing of rat liver bilirubin UDP-glucuronyl transferase cDNA: a possible
alternate splicing of a common primary transcript. Biochem. Biophys. Res. Commun.
169: 260 – 264.
31. Muraca, M., and Blanckaert, N. (1983). Liquid-chromatographic assay and identifica-
tion of mono- and diester conjugates of bilirubin in normal serum. Clin. Chem. 29:
1767 – 1771.
32. Peters, W. H., Allebes, W. A., Jansen, P. L., Poels, L. G., and Capel, P. J. (1987).
Characterization and tissue specificity of a monoclonal antibody against human uridine
5V-diphosphate-glucuronosyltransferase. Gastroenterology 93: 162 – 169.
859