the stability of niobium-silica catalysts in repeated liquid-phase epoxidation tests: a comparative...
TRANSCRIPT

Inorganica Chimica Acta 431 (2015) 190–196
Contents lists available at ScienceDirect
Inorganica Chimica Acta
journal homepage: www.elsevier .com/locate / ica
The stability of niobium-silica catalysts in repeated liquid-phaseepoxidation tests: A comparative evaluation of in-framework andgrafted mixed oxides
http://dx.doi.org/10.1016/j.ica.2015.01.0480020-1693/� 2015 Elsevier B.V. All rights reserved.
⇑ Corresponding author. Tel.: +39 02 5031 4428.E-mail address: [email protected] (M. Guidotti).
Cristina Tiozzo a, Chiara Palumbo a, Rinaldo Psaro a, Chiara Bisio a,b, Fabio Carniato b, Antonella Gervasini c,Paolo Carniti c, Matteo Guidotti a,⇑a CNR-Istituto di Scienze e Tecnologie Molecolari, Via C. Golgi 19, Milano, Italyb Dipartimento di Scienze e Innovazione Tecnologica, Centro Interdisciplinare NanoSistemi, Università del Piemonte Orientale, Viale T. Michel 11, Alessandria, Italyc Dipartimento di Chimica Università degli Studi di Milano, Via C. Golgi 19, Milano, Italy
a r t i c l e i n f o
Article history:Received 29 October 2014Received in revised form 25 January 2015Accepted 29 January 2015Available online 16 February 2015
Keywords:NiobiumHeterogeneous catalysisMesoporous silicaHydrogen peroxideAlkene epoxidation
a b s t r a c t
Two types of niobium-containing silica catalysts, (i) in-framework mixed oxide, obtained by co-precipita-tion type synthesis, and (ii) grafted niobium sites on silica supports, obtained via post-synthesis deposi-tion from niobocene dichloride, were prepared. Both solids were tested in a series of five repeated batch-wise liquid-phase epoxidation tests of limonene with 50% aqueous hydrogen peroxide.
Nb/SiO2 prepared by co-precipitation synthesis kept fully its epoxidation activity during the first tworecycles, with a gradual decrease in activity in the following runs. Conversely, Nb/SiO2 prepared by depo-sition suffered from a more marked loss of activity since the first reuses. Most of the isolated Nb siteswere maintained and no large Nb2O5 domains were formed during the recovery and regeneration tests.The distribution, geometry and coordination state of the Nb sites was therefore preserved, especially forthe Nb/SiO2 sample prepared via co-precipitation. The presence of small Nb2O5 domains, even formedduring repeated recycles, was not detrimental for the epoxidation reaction and, in general, Nb(V) sitesinserted in a silica matrix proved to be a rather robust epoxidation catalyst for alkenes in the presenceof aqueous hydrogen peroxide.
� 2015 Elsevier B.V. All rights reserved.
1. Introduction Hydrogen peroxide is a sought-after oxidant according to green
Heterogeneous niobium-based catalytic systems, in particularmixed oxides and silicates, have recently emerged from the transi-tion metal set, since they showed useful properties which can beexploited in several reactions [1,2], for their marked acid proper-ties [3,4] as well as for their oxidation properties [5], such as inthe oxidation of alkenes [6–12], phenols [13], alcohols [14–16] orsulfides [17,18]. The high robustness towards hydrolysis and metalleaching is one of the reasons for the major attention paid to Nb-containing silica solids in the field of heterogeneous oxidation cat-alysis. Such peculiar feature is ascribed by some authors to therelatively low surface mobility of niobium atoms and to the opti-mal geometry of Nb–O–Si bond angles [19]. These factors likelyrender Nb species less prone to hydrolysis/solvolysis in proticmedia and make them suitable to be used in aqueous media, inwater-containing liquid phases and in the presence of aqueoushydrogen peroxide, as a direct oxidant.
chemistry guidelines and is also able to selectively epoxidise alke-nes via radical-free heterolytic oxidation mechanisms [20]. V, Ta orZr-containing solids have previously been proposed for alkeneepoxidations, but they often suffer from metal leaching problems[21,22]. Widely-studied Ti-silica systems, which have shown, forthis reaction, very good catalytic performances, often requirewater-free conditions and anhydrous organic hydroperoxides, asoxidants, especially when the reaction is performed over highlyhydrophilic nanostructured Ti-silicate solids [23]. Therefore, theuse of Nb-silicate/H2O2 oxidizing catalytic systems is promisingand highly desirable, although it has been only partially exploredin the recent literature.
Prompted by these observations and the promising resultsrecently achieved with the Nb–SiO2/H2O2 system, we focused ourattention onto the following question: how can the method ofpreparation of a solid niobium-silica catalyst affect itsperformance?
Some previous studies have investigated the relationshipsbetween structural and/or morphological features and catalytic

C. Tiozzo et al. / Inorganica Chimica Acta 431 (2015) 190–196 191
properties of niobium oxide species on silica [24–26]. But, to ourbest knowledge, only one direct comparison has been reported sofar, between the performances in the selective oxidation of alkenesof in-framework niobia-silica mixed oxides and the ones of graftedniobium(V)-silica catalysts obtained by post-synthesis modifica-tions [27] and no reports are present on grafted catalysts preparedfrom organometallic niobium precursors. In addition, in mostpapers, the robustness and stability of Nb-silica catalysts wereevaluated after two catalytic runs maximum (i.e. 1 recycle). It is,on the contrary, essential to evaluate the catalyst behavior afterrepeated cycles under stress conditions, as evidenced by someworks on niobia-silica [10,28–30] and widely-studied titanium-silica epoxidation catalysts [17,31]. In fact, in the presence of water(both as a medium for the oxidant and as a product from hydrogenperoxide consumption), some isolated Nb(V) sites may aggregateand form NbOx domains.
In order to shed some light onto these points, we have here com-pared two types of niobium(V)-silica catalysts: a mixed oxide,obtained by co-precipitation type synthesis, and a grafted niobi-um-silica catalyst, obtained via post-synthesis deposition from aniobium organometallic precursor. The amount of Nb was in anycase low (<2 wt.%) to exploit the catalytic properties typical of dis-persed Nb phases and because such loading proved to be the optimalloading for the epoxidation of a large variety of alkenes [10,32]. Bothsystems were tested in the batch-wise liquid-phase epoxidation oflimonene with aqueous hydrogen peroxide and their behavior wasalso studied in repeated use and recovery catalytic tests.
2. Experimental
2.1. Materials
Niobium-silica catalysts were prepared by co-precipitationusing ammonium niobate(V) oxalate hydrate complex, ANBO (Sig-ma Aldrich, 99.99%), as niobium precursor, and tetraethy-lorthosilicate, TEOS (Sigma Aldrich, purity >99.0%), as silicasource, and by grafting bis(cyclopentadienyl)niobium(IV) dichlo-ride (Nb(Cp)2Cl2; 95%, Aldrich) onto Grace Davison, Dav SiO2, (Dav-isil SiO2 LC60A, 60–200 lm). Dav silica is commercially availablefrom the manufacturer.
The Nb-silica catalyst obtained via co-precipitation (Nb/SiO2-CP) was prepared as described in Ref. [26]. Briefly, the Si-font(TEOS) was hydrolyzed in acid-aqueous solution and then ANBOwas added. Then, pH was increased by ammonia solution thatcaused the precipitation of the unripe solid. The Nb-silica catalystobtained via dry impregnation (Nb/SiO2-DI) was prepared asdescribed in detail previously [34]. Briefly, the silica support wasfirst hydrated with high purity deionized water (MilliQ Academic,Millipore, 18 MX cm) for 2 h, dried in a rotary evaporator thenheated in air at 300 �C for 1 h and under vacuum overnight at300 �C. Nb(Cp)2Cl2 was finely ground and mixed, under an inertatmosphere in the solid phase, with the silica. The mixture wasstirred overnight under vacuum at room temperature. The result-ing light brown solid were calcined in dry oxygen at 500 �C for2 h to obtain the final white Nb/SiO2-DI catalyst.
Bulk hexagonal-phase Nb2O5 (Aldrich, 99.99%) was used assuch, for comparison.
2.2. Characterization
The content of Nb in the prepared samples was determined byinductively coupled plasma optical emission spectroscopy ICP-OES(ICAP 6300 Duo, Thermo Fisher Scientific) after mineralization ofthe samples in a microwave digestion apparatus (Milestone MLS
1200; maximum power 500 W) with a mixture of hydrofluoric(aq. 40 wt.%) and fuming nitric acid.
Diffuse reflectance UV–Vis (DR UV–Vis) spectra were measuredby using a Perkin Elmer Lambda 900 spectrometer equipped withan integrating sphere accessory and using a hand-made quartz cellthat allows analysis both under vacuum conditions (residual pres-sure 10�5 mbar) and under controlled gas atmosphere. Prior to theanalysis, the samples were dispersed in anhydrous BaSO4 (10 wt%).DR-UV–Vis spectra were collected in air atmosphere. Spectra weremeasured in absorbance mode and converted in Kubelka–Munkfunction in the 500–200 nm range.
N2 physisorption measurements were carried out at �196 �C inthe relative pressure range from 1 � 10�6 P/P0 to 1 using a Quan-tachrome Autosorb 1MP/TCD instrument. Prior to the analysis,the samples were outgassed at 100 �C for 3 h (residual pressure10�6 mbar). Specific surface area values were determined usingthe Brunauer–Emmett–Teller (BET) equation. Pore size distribu-tions were obtained by applying the NLDFT method.
Thermogravimetric analysis (TGA) was performed on a high-temperature thermal balance (Perkin Elmer; 7HT – Pyris Manager)in the temperature range between 50 and 1000 �C, under dry airwith a temperature rate of 5 �C min�1.
2.3. Catalytic tests
All catalysts were pretreated under dry air at 500 �C and cooleddown to room temperature under vacuum prior to use. Theepoxidation tests on (R)-(+)-limonene (97% Aldrich; 98% e.e.) wereperformed in a round-bottom glass batch reactor in an oil bath at90 �C with magnetic stirring (ca. 800 rpm) under inert atmosphere.Acetonitrile (Aldrich, HPLC grade) and aqueous hydrogen peroxide(H2O2; Aldrich, 50%) were used as solvent and oxidant, respective-ly, with an oxidant to substrate molar ratio of 2:1. The total volumeof the mixture was 5 mL. Samples were taken at regular intervalsand analyzed on GC chromatogram (Agilent 6890 Series; HP-5 col-umn, 30 m � 0.25 mm; FID detector) with mesitylene (Fluka, pur-iss. P99%) as internal standard. GC-peaks were identified bycomparison with peaks of genuine samples of reference standards.A standard deviation of ±2%, ±4% and ±2 h�1 has to be consideredon average for the conversion, selectivity and specific activity val-ues, respectively. The distribution of stereoisomers in the reactionsamples was determined by 1H NMR spectroscopy (Bruker,300 MHz), evaluating the diagnostic peak ratios. The formation ofperoxyimidic acid, due to the concomitant presence of acetonitrileand hydrogen peroxide, can be excluded under these conditions.After all tests, the presence of residual hydrogen peroxide waschecked and confirmed by iodometric assays. Through the quan-tification of residual hydrogen peroxide, it was possible to evaluatethe average oxidant efficiency, that is the global amount of oxi-dized products obtained per amount of consumed oxidant.
In order to check the leaching of niobium species, the solid cata-lyst was removed from the liquid mixture by centrifugation at thetemperature of the reaction (‘‘hot filtration’’ test) and the resultingsolution was tested for further reaction [33]. In the tests for therecovery and reuse of the catalyst, the solid was separated by filtra-tion at room temperature and thoroughly washed with fresh ace-tonitrile and then with methanol (Fluka, HPLC grade). The filteredsolid was dried at 100 �C, weighed, activated again at 500 �C in dryair and then reused up to five times in a new test as described above.
3. Results and discussion
3.1. Preparation of the catalysts
Two types of Nb/SiO2 catalysts were prepared following twodifferent synthesis strategies. Nb/SiO2-CP was obtained by co-pre-
Scheme 1. Flow-sheet for the sol–gel synthesis of Nb/SiO2-CP.
Table 1Textural properties and Nb content of the catalysts.
SBETa(m2 g�1)a
PVb
(cm3 g�1)PDc (nm) Nb contentd
(wt.%)
Nb/SiO2-CP 278 0.59 4.5 1.00Nb/SiO2-DI 444 0.78 4.0–10.0 1.52SiO2
e 580 0.97 4.0–10.0 –
a BET specific surface areab specific pore volume.c Mean pore diameter.d Obtained by ICP-AES analysis.e Pristine silica support from which Nb/SiO2-DI catalyst was obtained.
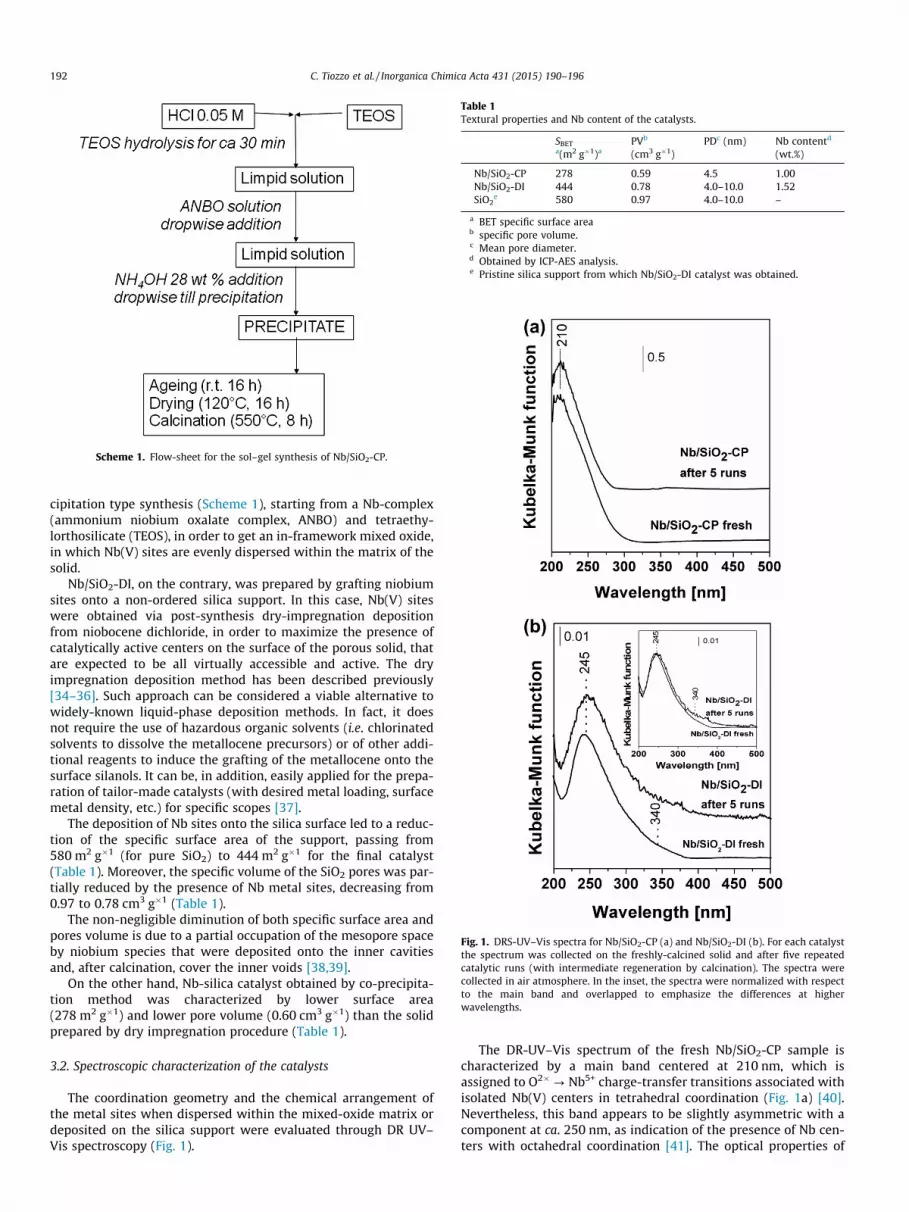
Fig. 1. DRS-UV–Vis spectra for Nb/SiO2-CP (a) and Nb/SiO2-DI (b). For each catalystthe spectrum was collected on the freshly-calcined solid and after five repeatedcatalytic runs (with intermediate regeneration by calcination). The spectra werecollected in air atmosphere. In the inset, the spectra were normalized with respectto the main band and overlapped to emphasize the differences at higherwavelengths.
192 C. Tiozzo et al. / Inorganica Chimica Acta 431 (2015) 190–196
cipitation type synthesis (Scheme 1), starting from a Nb-complex(ammonium niobium oxalate complex, ANBO) and tetraethy-lorthosilicate (TEOS), in order to get an in-framework mixed oxide,in which Nb(V) sites are evenly dispersed within the matrix of thesolid.
Nb/SiO2-DI, on the contrary, was prepared by grafting niobiumsites onto a non-ordered silica support. In this case, Nb(V) siteswere obtained via post-synthesis dry-impregnation depositionfrom niobocene dichloride, in order to maximize the presence ofcatalytically active centers on the surface of the porous solid, thatare expected to be all virtually accessible and active. The dryimpregnation deposition method has been described previously[34–36]. Such approach can be considered a viable alternative towidely-known liquid-phase deposition methods. In fact, it doesnot require the use of hazardous organic solvents (i.e. chlorinatedsolvents to dissolve the metallocene precursors) or of other addi-tional reagents to induce the grafting of the metallocene onto thesurface silanols. It can be, in addition, easily applied for the prepa-ration of tailor-made catalysts (with desired metal loading, surfacemetal density, etc.) for specific scopes [37].
The deposition of Nb sites onto the silica surface led to a reduc-tion of the specific surface area of the support, passing from580 m2 g�1 (for pure SiO2) to 444 m2 g�1 for the final catalyst(Table 1). Moreover, the specific volume of the SiO2 pores was par-tially reduced by the presence of Nb metal sites, decreasing from0.97 to 0.78 cm3 g�1 (Table 1).
The non-negligible diminution of both specific surface area andpores volume is due to a partial occupation of the mesopore spaceby niobium species that were deposited onto the inner cavitiesand, after calcination, cover the inner voids [38,39].
On the other hand, Nb-silica catalyst obtained by co-precipita-tion method was characterized by lower surface area(278 m2 g�1) and lower pore volume (0.60 cm3 g�1) than the solidprepared by dry impregnation procedure (Table 1).
3.2. Spectroscopic characterization of the catalysts
The coordination geometry and the chemical arrangement ofthe metal sites when dispersed within the mixed-oxide matrix ordeposited on the silica support were evaluated through DR UV–Vis spectroscopy (Fig. 1).
The DR-UV–Vis spectrum of the fresh Nb/SiO2-CP sample ischaracterized by a main band centered at 210 nm, which isassigned to O2�? Nb5+ charge-transfer transitions associated withisolated Nb(V) centers in tetrahedral coordination (Fig. 1a) [40].Nevertheless, this band appears to be slightly asymmetric with acomponent at ca. 250 nm, as indication of the presence of Nb cen-ters with octahedral coordination [41]. The optical properties of
0
10
20
30
40
50
60
70
80
0 1 2 3 4
Conv
ersi
on (%
)
Time (h)
C. Tiozzo et al. / Inorganica Chimica Acta 431 (2015) 190–196 193
Nb/SiO2-DI sample deserve additional comments. In fact, Nb siteswith octahedral coordination are mainly evident in the grafted cat-alyst, as testified by the presence of the maximum of absorption atca. 245 nm (Fig. 1b). Furthermore, the solid prepared by dryimpregnation shows a weak shoulder centered at 340 nm (absentin the DR-UV–Vis spectrum of Nb/SiO2-CP) assigned to the pres-ence of Nb2O5-like nano-aggregates formed during the dry impreg-nation process [43]. This means that the co-precipitation approachled to a better distribution of Nb(V) sites throughout the frame-work, thanks to the more uniform mixing of the Nb precursor(ANBO) with the silica species (TEOS) since the start of the synthe-sis procedure.
For all Nb-containing materials prepared via both dry impreg-nation and co-precipitation methodology, however, no absorptionbands are detected at ca. 350–400 nm, indicating that no largethree-dimensional Nb2O5 domains were present on the catalystssurface (Fig. 1) [26].
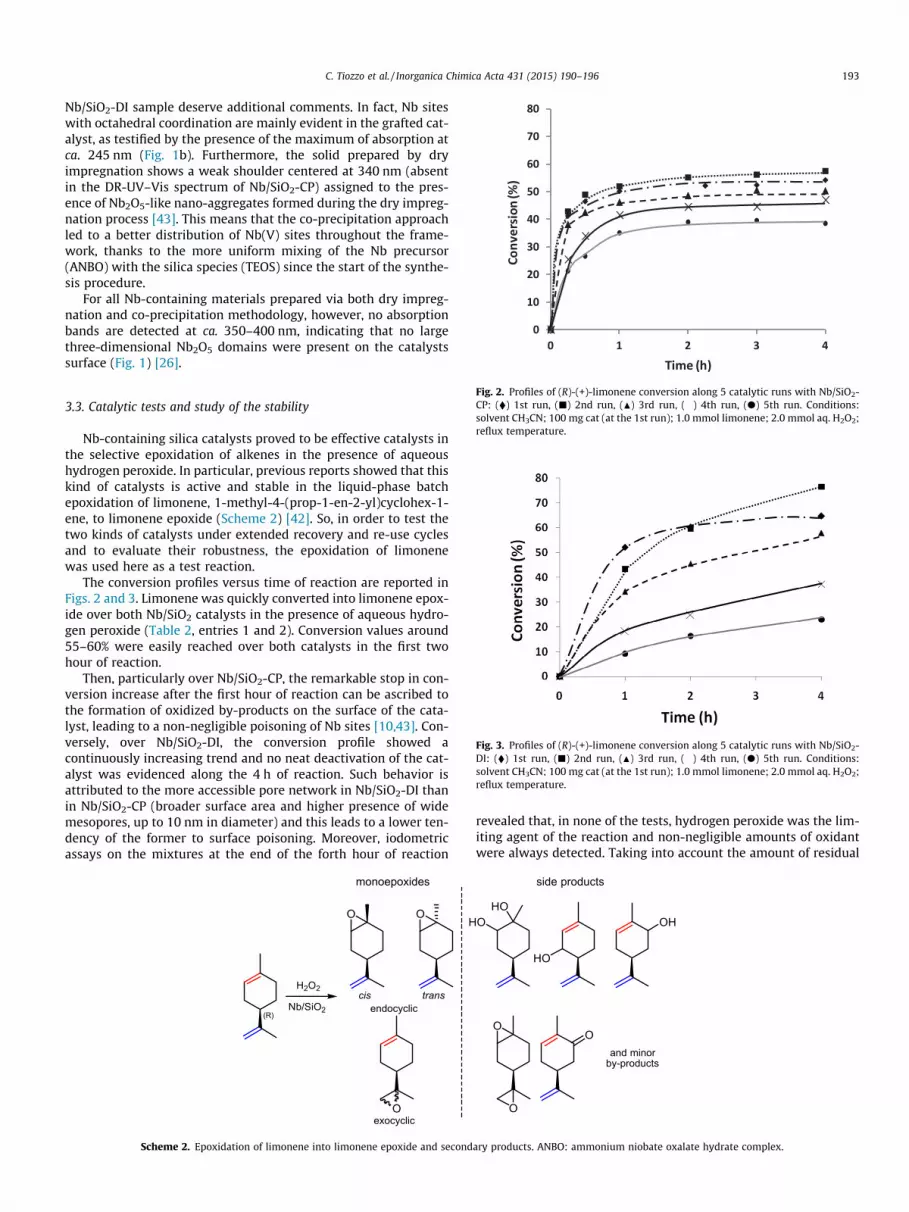
Fig. 2. Profiles of (R)-(+)-limonene conversion along 5 catalytic runs with Nb/SiO2-CP: (�) 1st run, (j) 2nd run, (N) 3rd run, (�) 4th run, (d) 5th run. Conditions:solvent CH3CN; 100 mg cat (at the 1st run); 1.0 mmol limonene; 2.0 mmol aq. H2O2;reflux temperature.
Fig. 3. Profiles of (R)-(+)-limonene conversion along 5 catalytic runs with Nb/SiO2-DI: (�) 1st run, (j) 2nd run, (N) 3rd run, (�) 4th run, (d) 5th run. Conditions:solvent CH3CN; 100 mg cat (at the 1st run); 1.0 mmol limonene; 2.0 mmol aq. H2O2;reflux temperature.
3.3. Catalytic tests and study of the stability
Nb-containing silica catalysts proved to be effective catalysts inthe selective epoxidation of alkenes in the presence of aqueoushydrogen peroxide. In particular, previous reports showed that thiskind of catalysts is active and stable in the liquid-phase batchepoxidation of limonene, 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene, to limonene epoxide (Scheme 2) [42]. So, in order to test thetwo kinds of catalysts under extended recovery and re-use cyclesand to evaluate their robustness, the epoxidation of limonenewas used here as a test reaction.
The conversion profiles versus time of reaction are reported inFigs. 2 and 3. Limonene was quickly converted into limonene epox-ide over both Nb/SiO2 catalysts in the presence of aqueous hydro-gen peroxide (Table 2, entries 1 and 2). Conversion values around55–60% were easily reached over both catalysts in the first twohour of reaction.
Then, particularly over Nb/SiO2-CP, the remarkable stop in con-version increase after the first hour of reaction can be ascribed tothe formation of oxidized by-products on the surface of the cata-lyst, leading to a non-negligible poisoning of Nb sites [10,43]. Con-versely, over Nb/SiO2-DI, the conversion profile showed acontinuously increasing trend and no neat deactivation of the cat-alyst was evidenced along the 4 h of reaction. Such behavior isattributed to the more accessible pore network in Nb/SiO2-DI thanin Nb/SiO2-CP (broader surface area and higher presence of widemesopores, up to 10 nm in diameter) and this leads to a lower ten-dency of the former to surface poisoning. Moreover, iodometricassays on the mixtures at the end of the forth hour of reaction
(R)
O
O
O
H2O2
Nb/SiO2 endocyclic
exocyclic
cis
monoepoxides
trans
H
Scheme 2. Epoxidation of limonene into limonene epoxide and second
revealed that, in none of the tests, hydrogen peroxide was the lim-iting agent of the reaction and non-negligible amounts of oxidantwere always detected. Taking into account the amount of residual
side products
O
O
OH
HO
O
OHO
and minorby-products
ary products. ANBO: ammonium niobate oxalate hydrate complex.

Table 2Catalytic performance of the catalysts in limonene epoxidation.
Entry Catalyst Ca 1 h (%) Ca 4 h (%) SAb 1 h (h�1) Sepoxc 1 h (%) Sepox
c 4 h (%)
1 Nb/SiO2-CP 52 57 48 78 702 Nb/SiO2-DI 51 62 31 77 683 Nb2O5 36 48 0.5 58 634 No catalyst 4 10 – n.d.d n.d5 SiO2 5 9 – n.d. n.d
Conditions: Glass batch reactor; solvent CH3CN; 100 mg cat; 1.0 mmol limonene; 2.0 mmol aq. H2O2; reflux temperature.a Limonene conversion.b Specific activity ([mol converted limonene]/[mol total Nb h]).c Selectivity to limonene monoepoxide.d Not determined.
194 C. Tiozzo et al. / Inorganica Chimica Acta 431 (2015) 190–196
H2O2, the oxidant efficiency (the global amount of oxidized prod-ucts obtained per amount of consumed oxidant) was, on average,moderately good with hydrogen peroxide efficiency values around50–55%. Anyway, the decomposition of hydrogen peroxide after4 h was ca. 10 mol% and 15 mol%, in the absence or in the presenceof (Nb-free) silica, respectively. This result suggests that only a partof the oxidant loss was due to thermal decomposition and that apossible Nb-catalyzed decomposition of H2O2 cannot be ruled out.
Hot-filtration tests to ascertain the leaching of catalyticallyactive Nb species into the solution proved that no solid releasedspecies were able to act as homogeneous catalysts [33]. Neithersignificant auto oxidation nor support-catalyzed contributions toepoxidation were recorded with niobium-free siliceous supports,limonene conversion being ca. 10% after 4 h with no remarkableepoxide formation (Table 2, entries 4 and 5).
In terms of specific activity (i.e. the amount of converted limo-nene per amount of Nb in the whole catalyst, per unit of time), Nb/SiO2-CP was more active than Nb/SiO2-DI. Nevertheless, if onetakes into account that Nb/SiO2-CP had a lower content of totalniobium and that a part of niobium centers were likely locatedwithin the bulk structure of the solid (and, hence, were not acces-sible to the reactant molecules), it is evident that each Nb(V) site atthe surface of Nb/SiO2-CP was far more active than the Nb site inNb/SiO2-DI obtained through grafting. Such considerations comefrom previous spectroscopic observations performed on analogoustitanium-silica systems [44], according to which titanium-graftedsilica solids have a larger fraction of exposed and accessible Ti(IV)sites than Ti-SiO2 mixed oxides.
With regard to selectivity, limonene monoepoxide (as sum ofendo- and exocyclic epoxides) was the major product (Scheme 2).The formation of the diepoxide was indeed detected for longerreaction time (after 6 h), when most of the pristine limonene hadalready been consumed. Side-products (ca. 30 mol% of the totalproducts) were mainly composed by compounds derived fromthe allylic oxidation of the C@C bond (i.e. carveol and its isomers),due to free radicals generated through homolytic cleavage of H2O2
[45] or from the opening of the epoxide ring (i.e. menthanediols; cf.Scheme 2), due to the acid character of the Nb(V) sites in siliceousmatrices [46], for both catalysts. Under these conditions, no rele-vant differences in selectivity were evidenced as a consequenceof the different surroundings of the Nb(V) sites in Nb/SiO2-CPand Nb/SiO2-DI.
As far as regio- and stereo-selectivity are concerned, at ca. 50%conversion, the two catalyst showed a marked preferential forma-tion of the exocyclic epoxide with a exo/endo ratio of 72:28 and78:22 mol/mol for Nb/SiO2-CP and Nb/SiO2-DI, respectively. Suchunexpected regioselectivity towards the less electron-rich, butmore sterically accessible alkene bond is consistent with some pre-vious observations by some of us on a similar Nb-SiO2 system [8].Analogously, the cis/trans ratio for the endocyclic monoepoxide(Scheme 2) was 63:37 and 57:43 mol/mol for Nb/SiO2-CP and
Nb/SiO2-DI, respectively. These results suggest that the catalytical-ly active Nb sites in the two solids show a comparable behaviorand they are likely surrounded by a similar steric environment thatfavors the oxygen transfer from the oxidant to the alkene in a fullysimilar way.
In order to study the solids under repeated recycle tests, the cat-alysts underwent five catalytic runs overall (Figs. 2 and 3) and wereeasily recovered by filtration, washed, calcined in order to recoverthe pristine activity and remove the organic deposits on the cata-lyst surface (2.3 wt.% and 3.8 wt.%, for Nb/SiO2-CP and Nb/SiO2-DI, respectively, according to TGA measurements). If one considersthe specific surface area for the two solids (278 and 444 m2 g�1, forNb/SiO2-CP and Nb/SiO2-DI, respectively), the specific amount offouling agents per surface area unit was thus comparable: 83 and85 lg m�2, respectively.
Initial conversion rate (within the first hour) decreases mildlyon Nb/SiO2-CP and more markedly on Nb/SiO2-DI from first to fifthrun. In particular, Nb/SiO2-CP kept its epoxidation activity duringthe first two recycles, whereas Nb/SiO2-DI suffered from a moremarked loss of initial rate value since the first reuses. Peculiarly,the conversion value after 4 h of reaction in the second run washigher than the one in the first run. Such behavior suggests alsothat Nb/SiO2-DI is less sensitive to surface poisoning than Nb/SiO2-CP.
Selectivity to epoxide is practically constant throughout therecycles. This suggests a gradual diminution of active Nb sites,but without an intrinsic change of their nature and the reactivityoccurring on them.
In terms of yield to limonene epoxide, the behavior is depictedin Fig. 4, where the tests were conducted with 2.5 times theamount of catalyst, substrate and oxidant with respect to the pre-vious tests. The conversion and selectivity values of the set of testsin Fig. 4 are fully comparable with the results in Figs. 2 and 3. Atthe first run, both catalysts gave an epoxide yield of ca. 40%. Then,at the fifth run, the decrease in epoxide yield was more marked forthe grafted catalyst than for the in-framework one (35% and 75%diminution in yield for Nb/SiO2-CP and Nb/SiO2-DI, respectively).Interestingly, for Nb/SiO2-CP a slight increase in limonene epoxideyield was recorded on the second run (Fig. 4), whereas it was notthe case for Nb/SiO2-DI.
Aiming at understanding the reasons for such a large differencebetween the two catalysts, the dispersion of Nb sites was evaluatedby DRS-UV–Vis at the end of the five runs (Fig. 1). The DR-UV–Visspectrum of both catalysts was substantially preserved after fiverepeated catalytic runs (with intermediate regeneration by calcina-tion). A good fraction of isolated Nb sites was kept and no absorp-tion bands around 400 nm were detected for both samples,indicating that no large Nb2O5 domains were formed during therecovery and regeneration tests (Fig. 1). The distribution, geometryand coordination state of the Nb sites were therefore preserved.Nevertheless, it is important to note that the fraction of Nb2O5-like

Fig. 4. Yield to limonene epoxide after 1 h over Nb/SiO2-CP and Nb/SiO2-DI along 5catalytic runs. Conditions: solvent CH3CN; 250 mg cat (at the 1st run); 2.5 mmollimonene; 5.0 mmol aq. H2O2; reflux temperature.
C. Tiozzo et al. / Inorganica Chimica Acta 431 (2015) 190–196 195
nano-domains (band at ca. 340 nm) tends to increase in the spentNb/SiO2-DI catalyst, in comparison to the fresh solid (see inset inFig. 1,b). Additionally, any formation of large Nb2O5 domain isnot to be considered detrimental for the epoxidation activity, asit happens with Ti-containing silica epoxidation catalysts. In fact,large anatase-like TiO2 domains on Ti-silica systems usually leadto the useless decomposition of H2O2 into water and molecularoxygen [23,47], whereas bulk Nb2O5 is indeed an effective epoxida-tion catalyst [48,49]. Actually, under these reaction conditions,bulk commercial hexagonal-phase Nb2O5 proved to be an activecatalyst for limonene epoxidation too with good performances,yet with slightly lower selectivity values to the desired epoxide(Table 2, entry 3).
The niobium content of the two catalysts was also checked atthe end of the fifth catalytic run. In both cases a non-negligible lossof active metal was recorded (from 1.00 wt.% to 0.83 wt.% and from1.52 wt.% to 1.27 wt.%, for Nb/SiO2-CP and Nb/SiO2-DI, respective-ly). This means a value of ca. 17% of leaching for both catalysts dueto a gradual leaching of Nb out of the solid. Such behavior is con-sistent to the values found for Ti-silica solid which underwentrepeated recycles in a previous work [39]. Such diminution ofactive species was remarkable and likely due to the complexationactivity by polyols and highly polar species formed in situ and inthe proximity of the catalyst surface [5,50]. However, such leachingdid not give rise to the formation of catalytically active homoge-neous soluble species in solution, as seen above. Moreover, sinceapproximately the same relative loss of Nb was found for thetwo solids, the different extent of deactivation observed for Nb/SiO2-CP and Nb/SiO2-DI cannot be ascribed to a different loss ofactive metal.
Merging these observations together, it appears that the deacti-vation process is thus due to a combination of surface poisoning,pore plugging and site blocking. Nb/SiO2-CP suffered from deacti-vation more markedly than Nb/SiO2-DI. Nevertheless, as a mixedoxide, Nb/SiO2-CP was intrinsically more robust along repeatedrecycles than the grafted solid and it showed a less evident modifi-cation of the catalytically active Nb sites. On the contrary, Nb/SiO2-DI sample showed a partial alteration of Nb sites, with a slightlyincrease of the Nb2O5 nano-domains in the spent catalyst.
4. Conclusions
Niobium-containing silica catalysts, either prepared via in-framework co-precipitation or via post-synthesis depositionapproach, provide interesting results in the epoxidation of limo-nene to the related epoxide with good selectivity and
moderate-high activities. Both catalysts keep their epoxidationactivity also in repeated recovery and reuse tests. Nb(V) sites insuch systems do not suffer from a marked change in geometryand coordination state. Moreover, the small amount of Nb2O5-likedomains that are formed in minor amounts during the recycles arenot detrimental to the epoxidation reaction, as they are indeedactive as oxidation systems. In terms of robustness, Nb/SiO2-CPshows the most promising performance with respect to Nb/SiO2-DI. Even though the former is more prone than the latter to surfacepoisoning after 1 h of reaction, because of its higher sensitivity tothe presence of surface-covering organic by-products, the insertionof Nb sites within the silica matrix makes Nb/SiO2-CP a catalystthat can be recycled several times, after it undergoes a short calci-nation pre-treatment.
For the future, the development of a novel class of Nb-contain-ing mesoporous silicates obtained by co-precipitation, but showinga higher pore volume and broader mesopore accessibility, may be apromising topic of investigation in the search for effective, selec-tive and robust heterogeneous epoxidation catalysts in the pres-ence of aqueous hydrogen peroxide.
Acknowledgments
C.P. thanks Regione Lombardia for a fellowship within the pro-ject ‘‘SusChem-Lombardia’’. C.T. thanks the Italian Ministry ofEducation, University and Research through the Project ‘‘I-talNanoNet’’ (prot. no. RBPR05JH2P) for a fellowship.
References
[1] I. Nowak, M. Ziolek, Chem. Rev. 99 (1999) 3603.[2] V. Lacerda Jr., D. Araujo dos Santos, L.C. da Silva-Filho, S.J. Greco, R. Bezerra dos
Santos, Aldrichim. Acta 45 (2012) 19.[3] (a) J.M. Jehng, I.E. Wachs, Catal. Today 8 (1990) 37;
(b) T. Onfroy, G. Clet, M. Houalla, J. Phys. Chem. B 109 (2005) 14588;(c) X. Gao, I.E. Wachs, M.S. Wong, J.Y. Ying, J. Catal. 203 (2001) 18.
[4] J.M.R. Gallo, S. Teixeira, U. Schuchardt, Appl. Catal. A 311 (2006) 199.[5] W. Yan, A. Ramanathan, M. Ghanta, B. Subramaniam, Sci. Technol. 4 (2014)
4433.[6] D. Hoegaerts, B.F. Sels, D.E. De Vos, F. Verpoort, P.A. Jacobs, Catal. Today 60
(2000) 209.[7] I. Nowak, B. Kilos, M. Ziolek, A. Lewandowska, Catal. Today 78 (2003) 487.[8] C. Tiozzo, C. Bisio, F. Carniato, M. Guidotti, Catal. Today 235 (2014) 49.[9] B. Subramaniam, A. Ramanathan, M. Ghanta, W. Yan, Patent WO2014004768
A2, (2014).[10] F. Somma, A. Puppinato, G. Strukul, Appl. Catal. A 309 (2006) 115.[11] N. Marin-Astorga, J.J. Martinez, G. Borda, J. Cubillos, D.N. Suarez, H. Rojas, Top.
Catal. 55 (2012) 620.[12] Y. Liu, K. Murata, M. Inaba, Chem. Lett. 32 (2003) 992.[13] M. Selvaraj, D.-W. Park, I. Kim, S. Kawi, C.S. Ha, Dalton Trans. 41 (2012) 9633.[14] M. Ziolek, P. Decyk, I. Sobczak, M. Trejda, J. Florek, H. Golinska, W. Klimas, A.
Wojtaszek, Appl. Catal. A 391 (2011) 194.[15] M. Trejda, A. Tuel, J. Kujawa, B. Kilos, M. Ziolek, Microporous Mesoporous
Mater 110 (2008) 271.[16] X. Gao, I.E. Wachs, M.S. Wong, J.Y. Ying, J. Catal. 203 (2001) 18.[17] M.V. Barmatova, I.D. Ivanchikova, O.A. Kholdeeva, A.N. Shmakov, V.I.
Zaikovskii, M.S. Mel’gunov, J. Mater. Chem. 19 (2009) 7332.[18] F. Somma, P. Canton, G. Strukul, J. Catal. 229 (2005) 490.[19] M. Ziolek, Catal. Today 78 (2003) 47.[20] R. Noyori, M. Aoki, K. Sato, Chem. Commun. (2003) 1977.[21] K.K. Kang, W.S. Ahn, J Mol. Catal. A 159 (2000) 403.[22] Y. Deng, C. Lettmann, W.F. Maier, Appl. Catal. A 214 (2001) 31.[23] M. G. Clerici, Titanium silicalite-1. In: Metal oxide catalysis, S. D. Jackson, J. S. J.
Hargreaves (eds), Weinheim, Wiley-VCH, (2009), pp. 705-754.[24] H. Yoshida, T. Tanaka, T. Yoshida, T. Funabiki, S. Yoshida, Catal. Today 28 (1–2)
(1996) 79.[25] R. Brayner, F. Bozon-Verduraz, Phys. Chem. Chem. Phys. 5 (2003) 1457.[26] P. Carniti, A. Gervasini, M. Marzo, J. Phys. Chem. C 112 (36) (2008) 14064.[27] M. Trejda, J. Kujawa, M. Ziolek, J. Mrowiec-Bialon, Catal. Today 139 (2008) 196.[28] A. Aronne, M. Turco, G. Bagnasco, G. Ramis, E. Santacesaria, M. Di Serio, E.
Marenna, M. Bevilacqua, C. Cammarano, E. Fanelli, Appl. Catal. A 347 (2008)179.
[29] J.V. Coelho, L.C.A. Oliveira, F.C.C. Moura, P.P. de Souza, C.A. Silva, K.B. Batista,M.J. da Silva, Appl. Catal. A 419–420 (2012) 215.
[30] M. Selvaraj, S. Kawi, D.W. Park, C.S. Ha, J. Phys. Chem. C 113 (2009) 7743.[31] N.V. Maksimchuk, M.S. Melgunov, J. Mrowiec-Białon, A.B. Jarzebski, O.A.
Kholdeeva, J. Catal. 235 (2005) 175.

196 C. Tiozzo et al. / Inorganica Chimica Acta 431 (2015) 190–196
[32] L.J. Burcham, J. Datka, I.E. Wachs, J. Phys. Chem. B 103 (1999) 6015.[33] R.A. Sheldon, M. Wallau, I.W.C.E. Arends, V. Schuchardt, Acc. Chem. Res. 31
(1998) 485.[34] A. Gallo, C. Tiozzo, R. Psaro, F. Carniato, M. Guidotti, J. Catal. 298 (2013) 77.[35] V. Dal Santo, F. Liguori, C. Pirovano, M. Guidotti, Molecules 15 (6) (2010)
3829.[36] V. Meille, Appl. Catal. A 315 (2006) 1.[37] V. Dal Santo, M. Guidotti, R. Psaro, L. Marchese, F. Carniato, C. Bisio, Proc. R. Soc.
A 468 (2012) 1904.[38] C. Tiozzo, C. Bisio, F. Carniato, L. Marchese, A. Gallo, N. Ravasio, R. Psaro, M.
Guidotti, Eur. J. Lipids Technol. 115 (2013) 86.[39] M. Guidotti, N. Ravasio, R. Psaro, G. Ferraris, G. Moretti, J. Catal. 214 (2) (2003)
242.[40] A.M. Prakash, L. Kevan, J. Am. Chem. Soc. 120 (1998) 13148.
[41] C. Tiozzo, C. Bisio, F. Carniato, A. Gallo, S.L. Scott, R. Psaro, M. Guidotti, Phys.Chem. Chem. Phys. 15 (2013) 13354.
[42] M.A. Feliczak-Guzik, I. Nowak, Catal. Today 142 (2009) 288.[43] J.M.R. Gallo, H.O. Pastore, U. Schuchardt, J. Non Cryst. Solids 354 (2008) 1648.[44] E. Gianotti, L. Marchese, M. Guidotti, N. Ravasio, R. Psaro, S. Coluccia, Stud. Surf.
Sci. Catal. 155 (2005) 311.[45] J.M. Fraile, J.I. Garcia, J.A. Mayoral, E. Vispe, J. Catal. 204 (2001) 146.[46] I. Nowak, Catal. Today 192 (2012) 80.[47] O.A. Kholdeeva, N.N. Trukhan, Russ. Chem. Rev. 75 (5) (2006) 411.[48] M. Ziolek, I. Sobczak, P. Decyk, L. Wolski, Catal. Commun. 37 (2013) 85.[49] H. Shima, M. Tanaka, H. Imai, T. Yokoi, T. Tasumi, J.N. Kondo, J. Phys. Chem. C
113 (2009) 21693.[50] L.J. Davies, P. McMorna, D. Bethell, P.C.P. Page, F. King, F.E. Hancock, G.J.
Hutchings, J. Mol. Catal. A: Chem. 165 (2001) 243.