targeting the human dead-box polypeptide 3 (ddx3) rna helicase as a novel strategy to inhibit viral...
TRANSCRIPT

Current Medicinal Chemistry, 2011, 18, 3015-3027 3015
0929-8673/11 $58.00+.00 © 2011 Bentham Science Publishers Ltd.
Targeting the Human DEAD-Box Polypeptide 3 (DDX3) RNA Helicase as a Novel
Strategy to Inhibit Viral Replication
A. Garbelli1, M. Radi
2, F. Falchi
2, S. Beermann
3, S. Zanoli
1, F. Manetti
2, U. Dietrich
3, M. Botta
2,4 and
G. Maga*,1
1Institute of Molecular Genetics IGM-CNR, via Abbiategrasso 207, I-27100 Pavia, Italy
2Dipartimento Farmaco Chimico Tecnologico, University of Siena,Via Alcide de Gasperi 2, I-53100 Siena, Italy
3Georg-Speyer-Haus, Paul-Ehrlich-Str. 42-44 60596 Frankfurt, Germany
4Sbarro Institute for Cancer Research and Molecular Medicine, Center for Biotechnology, College of Science and Technology,
Temple UniVersity, BioLife Science Building, Suite 333, 1900 N 12th Street, Philadelphia, PA 19122, USA
Abstract: Compounds currently used for the treatment of HIV-1 Infections are targeted to viral proteins. However, the high intrinsic
mutation and replication rates of HIV-1 often lead to the emergence of drug resistant strains and consequent therapeutic failure. On this
basis, cellular cofactors represent attractive new targets for HIV-1 chemotherapy, since targeting a cellular factor that is required for viral
replication should help to overcome the problem of viral resistance. We and others have recently reported the identification of
compounds suppressing HIV-1 replication by targeting the cellular DEAD-box helicase DDX3. These results provide a proof-of-principle
for the feasibility of blocking HIV-1 infection by rendering the host cell environment less favorable for the virus. The rationale for such
an approach and its implications in potentially overcoming the problem of drug resistance related to drugs targeting viral proteins will be
discussed in the context of the known cellular functions of the DEAD-box helicase DDX3.
Keywords: Antiviral therapy, cancer, DEAD-box, drug design, HIV-1, RNA helicase, viral replication.
INTRODUCTION
Viruses (from the Latin virus meaning toxin or poison) are obligate intracellular parasites and, as such, they exploit the metabolic mechanisms of their hosts in order to replicate. To this aim, viruses encode proteins able to interact with host proteins and to redirect their functions at the advantage of the pathogen. Thus, host cellular proteins are becoming very popular as novel targets for the treatment of viral infections. The main point behind such a rationale, is that targeting a host factor instead of a viral protein should make it more difficult for the virus to develop resistance against the drug. On the other side, it has to be always recalled that inhibiting a cellular function could lead to serious complications, including the development of cancer. Thus, it is of paramount importance to identify host cell factors that, while absolutely essential for the virus, are dispensable for the host cell metabolism. One protein that has recently attracted much attention is the human DEAD-box RNA helicase DDX3. It has been reported that four different viruses, Hepatitis C virus (HCV), Hepatitis B virus (HBV), HIV-1 and Poxviruses encode proteins that can interact with DDX3 to modulate its functions (for a recent review see also [1]). For this reason, DDX3 seems a prime target for viral manipulation. In this review, the current knowledge about DDX3 in cellular and viral metabolism will be presented, with special emphasis on its roles in the HIV-1 viral cycle, along with the first successful example of the development of small molecule DDX3 inhibitors, able to suppress HIV-1 replication.
THE DEAD-BOX FAMILY OF RNA HELICASES
Helicases are nucleic acid-dependent ATPases that are capable of unwinding DNA or RNA duplex substrates. RNA helicases belong to an abundant protein family that is conserved from bacteria to humans. The Saccharomyces cerevisiae genome, for example, encodes 39 putative helicases that have been implicated in all aspects of RNA metabolism (SGD and YPD database)[2], from transcription, mRNA splicing and export, to rRNA processing, RNA degradation and translation.
*Address correspondence to this author at the Institute of Molecular Genetics IGM-
CNR, via Abbiategrasso 207, I-27100 Pavia, Italy; Tel: (+39) 0382546354; Fax: (+39) 0382422286; E-mail: [email protected]
The current classification divides helicases into six
superfamilies (SF1-6), based on the sequence similarities within conserved Motifs. However, it is important to consider that
helicases share these “distinctive” Motifs with a large group of proteins not actually able to unwind nucleic acids, called
translocases for their ability to move directionally along nucleic acids. Helicases are therefore only a subgroup of translocases.
Most of the RNA helicases belong to the SF2 superfamily that include the DEAD box, DEAH box and Ski2-like proteins [2-7].
The DEAD box family, is the largest subgroup identified so far within SF2. DEAD-box proteins are often referred to as RNA
helicases, due to the ability of some members to unwind RNA duplexes, but in most cases this activity is not processive because it
is limited to short duplexes. The reason could be that efficiency and processivity of recombinant proteins is not the same as that proteins
in their biological context [4].
DEAD-box RNA helicases show conserved sequence Motifs, located in two different domains: Motif I, Ia, Ib, II, and III in
domain 1 and Motifs IV, V, and VI in domain 2. Based on genetic, biochemical, and structural data, specific functions have been
assigned to most of these Motifs.
Motif I
The amino-terminal Motif I Gly-Ser-Gly-Lys-Thr (G-S-G-K-T), also known as Walker A Motif, has been found in all helicases
as well as in many general nucleotide triphosphate hydrolases (NTPases). Walker A is probably the Motif whose function has
been best characterized [8, 9]. It is known that the amino group of the Lys interacts with the phosphate groups of MgATP/ADP, while
the hydroxyl of the Thr or Ser coordinates the Mg2+
ion, thus contributing to NTP binding [3, 10]. Mutations of either the
conserved Lys or the last Thr dramatically decrease the ATPase activity by reducing the affinity for ATP and the rate of hydrolysis
[11-15]. These results suggest that Motif I is crucial for the ATPase and helicase activities. The Walker A Motif is structurally
equivalent in SF1 and SF2 enzymes and its sequence is highly conserved.
Crystal structures of some DEAD-box proteins show that the
phosphate binding loop, or P-loop, of Motif I is found in two

3016 Current Medicinal Chemistry, 2011 Vol. 18, No. 20 Garbelli et al.
different conformations [10, 14]: the “open” conformation [10, 16,
17] and the “closed” conformation in the absence of a bound nucleotide [10, 18, 19].
Motif I plays a crucial role in helicases and, beside interacting
with the phosphates of the nucleotide, it makes contacts with the Q Motif, Motif II and Motif III (see below).
Motif II
The DEAD box family takes its name from the characteristic
Asp-Glu-Ala-Asp (D-E-A-D) sequence of Motif II, also known as the Walker B Motif [5]. Along with Walker A, the Walker B Motif
has been shown to be required for the ATPase activity and is present in many NTPase families. The carboxyl group of the first
Asp residue coordinates the Mg2+
ion of MgATP/ADP, whereas the highly conserved Glu is suggested to act as the probable catalytic
residue in ATP hydrolysis [10, 20, 21] and to activate the , -phosphoanhydride bond of ATP through a coordinated water
molecule, in association with a conserved His residue of Motif VI and a bound magnesium ion [22]. Site-directed mutagenesis in the
Motif II decreases or abolishes ATPase and helicase activities without altering RNA binding [23, 24].
Motif III
Motif III is defined by the sequence Ser–Ala–Thr (S-A-T) in
the DEAD-box family. However, Motif III is not universally conserved in SF2 families or even in the DEAD-box family. In fact,
other proteins have other sequences, like the herpes simplex virus UL9 protein (D-A-T) and the cyanobacteria cold shock DEAD-box
protein CrhC (F-A-T) [25, 26].
Mutation experiments of the consensus sequence showed that
the Ala and the Thr in the second and third positions of Motif III are more important than the Ser, but that mutations of all three
residues have strong phenotypes [27]. Previous studies in Motif III of eIF4A and in related DEAH-box proteins, showed that mutations
at these positions cause a dramatic loss of helicase activity without affecting ATP binding and hydrolysis in vitro [28, 29]. The
substitution of Ser with Leu in the S-A-T helicase Motif of DDX3 reduces ATPase and helicase activities [30]. The growth and RNA
unwinding defects resulting from alteration of Motif III in the DEAH box helicase Prp22 can be suppressed by single amino acid
changes within the core ATPase/helicase domain. These mutations could affect the conformation and/or flexibility of Prp22, thus
restoring mRNA release and RNA unwinding [29, 31].
Mutations in Motif III also reduce the affinity for single-stranded RNA and subsequently reduce the ability to disrupt
duplexes [27]. Accordingly, mutations in Motif III have been shown to uncouple NTP hydrolysis from the unwinding activity of
DEAD-box and DEAH-box proteins. In summary, the S-A-T region seems essential for RNA unwinding.
Motif IV
Motif IV has been poorly studied in DEAD-box proteins [32]
and its function has not been elucidated so far. The minimal consensus sequence of this Motif is L/V-I-F, although the sequence
could encompass as many as eight residues [22].
In the solved crystal structure of the NS3 helicase, Motif IV is located at the bottom of domain 2, where NS3 binds ssDNA [33].
DEAD-box proteins, as well as SF2 helicases in general, are thought to bind RNA through the ribose-phosphate backbone, and
the last two Arg of Motif IV of eIF4A (V-I-F-C-N-T-R-R) were proposed to bind ssRNA [3]. Behind its role in RNA binding, Motif
IV was shown to function in ATP hydrolysis [28].
Motif V
Motif V (A-R-G-L-D) is proposed to be an RNA-binding Motif in association with Motif Ia, Ib and IV. However, this Motif could also play a role in coupling ATP binding to general protein conformational changes, and may be implicated in transmitting the signal of RNA binding to the ATPase domain and consequently in regulating the hydrolysis of ATP [34]. In eIF4A, the conserved Arg of Motif V was proposed to interact with Motif II or with the bound oligonucleotide [34]. Moreover, the last Asp residue could interact directly with the ribose of the ATP in a manner similar to the Glu of Motif V of PcrA [3, 34]. Mutations in Motif V of the DEAD-box protein Prp28 have a negative effect on yeast growth, indicating that the conserved Arg and Asp play an important role in the in vivo activity of Prp28[35]. In addition, three amino acids in Motif V of Prp22 (T757, I764, and T765) have been identified that are important for mRNA release and for helicase activity in vitro [36].
Motif VI
The C-terminal H/Q-R-I-G-R-x-x-R region (Motif VI) is one of the most conserved regions in helicase superfamily II and is located at the interface between domains 1 and 2. It was shown to be involved in ATP hydrolysis-dependent RNA interaction during unwinding [28, 37]. It was shown to be involved in ATP hydrolysis-dependent RNA interaction during unwinding [28, 37]. It has been suggested that this region is responsible for the protein interaction with RNA, because of the presence of three conserved arginines [5]. Mutations in any one of the three Arg in the H-R-I-G-R-x-x-R sequence, demonstrated a drastic reduction of eIF-4A cross-linking to RNA. In addition, all these mutations abrogate RNA helicase activity and some, but not all, of these mutations affect ATP binding and ATPase activity. The first and second Arg of Motif VI could bind the -phosphate of ATP in the DEAD-box proteins. In addition, the second Arg could function like an “arginine finger” that would stabilize the water-Mg2+
- / phosphate intermediate during ATP hydrolysis [36]. It was also shown that mutations of the Gln and of the second Arg into Ala in the DEAH-box protein Prp22 drastically reduced ATPase activity [38].
The generation of a double mutant of the last Asp of Motif II into a His and of the His of Motif VI to Gln in eIF4A resulted in an increased binding of ATP and increased ATPase activity, but with reduced helicase activity [23]. These results are an evidence for the interaction of the highly conserved His residue of Motif VI with the Glu of Motif II [20, 22]. In summary, these data support an involvement of residues of this Motif in both ATP hydrolysis and coupling of ATP binding or hydrolysis to RNA binding and helicase activity.
Motif Q
A few years ago, an additional Motif was identified that occurs ~17 amino acids (aa) upstream of Motif I [14], including an almost invariant Gln that is present in more than 99% of the sequences. This Motif contains the sequence G-F-c-c-P-T-P-I-Q, where “c” is a charged group and the Gln is invariant. Consequently, this element was called the Q Motif. Although an additional conserved Gln residue was found ~17 aa upstream of the Q Motif in most helicases, other characteristic residues of the Q Motif are conserved only among DEAD box proteins and, therefore, the Q Motif is thought to be specific to DEAD box RNA helicases.
The crystal structure of DEAD-box proteins [16, 20, 34] and the secondary structure of the Q-Motif, suggested that it is part of a highly conserved structure consisting of a loop–helix–loop, where the isolated conserved aromatic group interacts with the conserved central Pro of the Q Motif. These two elements regulate ATP binding, and also the RNA substrate affinity and helicase activity of DEAD-box proteins [15]. Both the Gln and the Ser/Thr from the Q-

Targeting the Human DEAD-Box Polypeptide 3 (DDX3) RNA Helicase Current Medicinal Chemistry, 2011 Vol. 18, No. 20 3017
Motif form hydrogen bonds with conserved residues of Motif I [14]. The interaction between the Q-Motif and Motif I is important for both ATP and RNA binding and the regulation of ATPase activity [15]. Similar to Motif III, the Q Motif may not always be essential for function of the protein, though this may depend on assay conditions [39, 40].
FUNCTIONS OF DEAD-BOX PROTEINS IN GENERAL
RNA METABOLISM
The DEAD-box family comprises several members, but only few of them have been characterized biochemically. They all share an ATPase activity, as expected from the presence of Motifs I and II, conserved in all NTPases. This activity is generally stimulated by the presence of RNA, but an RNA-indipendent ATPase activity has also been reported [22, 41].
As mentioned above, DEAD-box proteins have been described to be necessary for, or involved in, many different processes mediated by RNA. Below, we summarize our current understanding of the roles of DEAD-box helicases in general and DDX3 in particular, in RNA metabolism.
Pre-mRNA Splicing
Proteomic analysis of the spliceosome complexes in eukaryotic cells, suggested that over 200 proteins are involved in the splicing of pre-mRNAs. Among those, it was shown that many DEAD-box and DEAH-box proteins participate in different steps of the splicing process. In general, DEAD-box proteins are involved in the early steps of splicing and DEAH-box in the later steps [42-44]. In yeast, three DEAD-box proteins have been shown to be required for in vivo splicing (Prp5, Prp28 and Sub2) [45]. Prp5 is able to bind the U1 and U2 small nuclear ribonucleoproteins (snRNPs) that link the 5’ splice site and the branch site [46]. Prp28 is important for the ATP-dependent destabilization of U1 snRNA and its replacement by U6 snRNA at the splice site [45] and strengthens the interaction between U1C protein and U1 snRNA [47]. This suggests that Prp28 may be required for the dissociation of U1C protein from U1 snRNA. Sub2 is a DECD-box protein able to associate with pre-mRNA in the early step of spliceosome assembly and it’s involved in splicing [48] and in nuclear export. In higher eukaryotes the p68 protein is involved in constitutive and alternative mRNA splicing [49, 50], and its homolog p72 participates in alternative splicing [51]. Higher eukaryotes contain other DEAD-box proteins important for the making of a functional spliceosome, such as eIF4AIII, DDX35 and DDX9 [52]. However their role in this process remains still unclear. DDX3 is an element of spliceosome and preferentially binds spliced mRNAs [53, 54] [55]. Recent evidence showed that DDX3 directly interacts with the Tip-associated protein (TAP) and associates with messenger ribonucleoproteins (mRNPs) [56] (see below).
Ribosome Biogenesis
Ribosome biogenesis is a multistep process that involves several RNA species, ribosomal proteins, and a variety of trans-acting factors. Many NTPases are required for ribosome biogenesis. Saccharomyces cerevisiae contains 17 RNA helicases implicated in ribosome biogenesis and genetic experiments showed that 14 of them are DEAD-box proteins which are required for the formation of 40S and 60S ribosomal subunits [2, 39, 40, 57]. Most of these DEAD-box proteins from S. cerevisiae present homologs in higher eukaryotes, indicating that their function is conserved. In E. coli there are at least three DEAD-box proteins involved in ribosome biogenesis: SrmB [58], CsdA [59] and probably DbpA [60, 61]. In general, ribosome biogenesis, both in eukaryotes and prokaryotes, involves a large number of DEAD-box proteins that are associated with rRNA maturation [2, 57]. However, a role of DDX3 in
ribosome biogenesis is still unclear and further investigation is needed. In fact, experiments of in vivo knockdown of DDX3 showed no influence on ribosome biogenesis [56].
Nuclear Export
The transport of the mature mRNA from nucleus to cytoplasm in eukaryotic cells is an important step requiring specialized proteins able to bind mRNAs [62, 63, 64]. DEAD-box proteins are involved in different steps of this process. The yeast DEAD-box protein Dbp5 binds RNA cotranscriptionally and functionally interacts with the transcription machinery [65]. Experiments in yeast have shown that a defect in the yeast Dbp5/Rat8 gene results in accumulation of poly(A) mRNA in the nucleus [66, 67]. Recently, other DEAD-box like proteins, Uap56/Bat1 in higher eukaryotes and Sub2 in yeast, were found to be required for mRNA export. Sub2 is also involved in pre-mRNA splicing [68-71] and in the export of fully spliced mRNAs.
Recent evidence suggests that DDX3 is involved in mRNA nuclear export in association with other two shuttle proteins CRM1 and TAP. DDX3 interacts with the TAP protein with its C-terminal sequence (aa 536-661). DDX3 binds TAP mainly into the nucleus, but the interaction might also occur in the cytoplasm. The proposed mechanism is that DDX3 binds either mRNAs and TAP in the nucleus and after that it helps to facilitate mRNPs export to the cytoplasm [56]. The interaction with CRM1 seems to be important only for the export of incompletely spliced RNAs of HIV-1 [30], see also below), while the physiological role of these interactions in the context of uninfected cells is not clear. Probably DDX3 is not a general cofactor of CRM1.
Translation Initiation
The translation initiation factor eIF4a was the first DEAD-box protein described to have a RNA-dependent ATPase activity [72]. Recently, it was discovered that also the S. cerevisiae homologs of DDX3, Ded1p and Dbp1p, function in translation initiation [73]. The eIF4A protein participates in the initiation of translation as part of the eIF4F complex [37, 74]. Probably, it’s able to directly interact with eIF4B, an RNA-binding protein, and this interaction increases the binding efficiency to the target RNA [75]. The protein Ded1 is also required for translation initiation in vivo and in vitro [76-78], facilitating the unwinding of 5’ untranslated regions. In D. melanogaster, the Vasa protein has been shown to play an important role in translation initiation via its interaction with IF2 [79]. Genetic complementation studies demonstrated that human DDX3 can functionally replace yeast Ded1p, suggesting that DDX3 is involved in translation initiation. DDX3 is able to interact with several translation initiation factors: eIF4e [80], eIF4a, eIF2a, PABP [56] and eIF3 [81]. Most studies suggest that DDX3 has a positive influence in protein translation but it was also shown that DDX3 acted as repressor of cap-dependent protein translation [80], because its interaction with eIF4e prevented the recruitment of eIF4G, an important translation activating factor. Thus, in spite of the robust evidence that DDX3 is involved in protein translation, current data do not allow to distinguish between a positive or negative role of DDX3 in this process.
Transcription Regulation
Only a few DEAD-box proteins are found to be associated with transcription. Human DDX20/DP103 was shown to be essential for repression of transcription [82, 83]. DP103 probably mediates repression of the steroidogenic factor 1 (SF-1) transcriptional activity, enhancing its sumoylation [84]. Human proteins p68 and p72 are able to interact with different transcription factors and nuclear receptors and operate as co-repressors or co-activators of transcription [85, 86] [87]. DHX9, DDX5 and DDX17 mediate the
3018 Current Medicinal Chemistry, 2011 Vol. 18, No. 20 Garbelli et al.
interactions between transcription factors and co-activators and RNA polymerase II [88]. DDX3 is able to interact with the E-Cadherin promoter, downregulating its expression, and with the interferon (IFN)b promoter, upregulating its activity. Currently, it is clear that the DDX3 effect on IFNb promoter is independent of its ATPase activity or unwinding function, in fact the K230E mutant of DDX3, that has lost both functions, can substitute for the wild type. DDX3 is also able to interact with the p21waf promoter and to enhance its activity, in this case the ATPase, but not unwinding activity, is required. Probably, DDX3 is able to interact with other promoters with different effects and also in different ways, for example in an helicase-dependent manner [89].
THE HUMAN DDX3 GENE
The human DDX3 gene is located in the X chromosomal region Xp11.4 [90, 91] and codes for a transcript of 5.3 kb in size, corresponding to a polypeptide of 662 amino acids, rich in serines (11.3%) and glycines (11.4%).
The human DDX3 gene has an homolog in the non-recombining region of the Y chromosome [92]. The DDX3Y gene, also named DBY, is located on AZFa chromosomal region on Yq11.21 and expressed only in the male germ line [92, 93]. DDX3Y is essential for spermatogenesis, because its deletion leads to a significant reduction in germ cells (oligozoospermia) or even to their complete absence (azospermia) [94, 95]. The X chromosomal gene DDX3 is highly homologous (91.9% identity, 97.7% similar) to DDX3Y, however, it is ubiquitously expressed in a wide range of tissues. Human Multiple Tissue Northern blots exhibit DDX3 expression in testis, colon, lung, liver, skeletal muscle, and kidney [91].
DDX3 has orthologous candidates in many eukaryotic
organisms, from yeast and plants to animals. Analysis of the protein sequence encoded by DDX3, reveals amino-acid sequence
homology to various members of the DEAD-box RNA helicase family with a high level of identity with the mouse PL10 (95%)
[96], mouse DDX3 (98%) [97], human DDX3Y (92%) and Xenopus An3 (80%) proteins, whereas the level of identity with
yeast Ded1p (50%) and Ded1 (50%) proteins is lower. Phylogenetic analysis indicates that the DDX3 proteins from invertebrates,
urochordates and lower vertebrates seem to be the ancestral forms
of both mammalian DDX3 and DDX3Y proteins [95].
BLAST analysis of the conserved domains demonstrated that human DDX3 and DDX3Y are most closely related to DDX5 (94%
identity) and DDX4 (92% identity), but the alignments of multiple amino acid sequences, indicate differences outside the conserved
regions between these proteins (Table 1).
THE DDX3 PROTEIN
The amino acid sequence of DDX3 contains all nine conserved
Motifs that characterize the well known member of the RNA helicase superfamily (Fig. 1) [6].
Also, DDX3 contains a nuclear export signal (NES) within its
N-terminal 22 amino acids. The crystal structure of DDX3 shows that these conserved motifs are found in two subdomains connected
via a short flexible linker [98]. The amino-terminal domain 1 contains the ATP binding Motifs Q, I (Walker A) and II (Walker
B), as well as the RNA-binding Motifs Ia, Ib and the Motif III. Whereas the RNA-binding Motifs IV, V and Motif VI, which may
coordinate ATPase and unwinding activities, are found in the carboxyl-terminal domain 2 [22]. Both domains 1 and 2 have a fold
typical to the RecA superfamily, with five -strands surrounded by five -helices. Sequence alignment of all different human DExD-
box helicases shows a DDX3-specific insertion between Motifs I and Ia. The crystal structure of DDX3 revealed that this inserted
sequence of ten residues (250–259, E-A-L-R-A-M-K-E-N-G) forms a helix that positions a positively charged loop in close proximity to
the putative RNA ligand. This unique insertion is not generally found in other human DExD-box helicases [98] and could be an
important determinant in RNA–substrate recognition/activation by DDX3 (Fig. 2). DDX3 contains at its C-terminal domain an
arginine-serine (RS) dipeptide rich segment. RS domains are typical modules of the SR family of proteins, generally involved in the
recruitment of components of the spliceosome complex [99]. It has been shown that the RS-Motif of DDX3 is involved in binding the
Tip-associated protein (TAP), a fundamental factor for mRNA export [56, 100, 101].
Table 1. Sequence Alignment and Identity Between the Conserved Domains of Human DDX3 and other DEAD-Box Helicases
Protein Conserved domains Identity
I Ia Ib II III IV V VI
hDDX3 AQTGSGKT PTRELAVQ TPGR DEAD SAT LVF ARGLD YVHRIGRTGRVG -
hDDX3Y AQTGSGKT PTRELAVQ TPGR DEAD SAT LVF ARGLD YVHRIGRTGRVG 100%
hDDX4 AQTGSGKT PTRELVNQ TPGR DEAD SAT MVF ARGLD YVHRIGRTGRCG 91%
hDDX5 AQTGSGKT PTRELAQQ TPGR DEAD SAT IVF SRGLD YVHRIGRTGRVG 94%
hDDX17 AQTGSGKT PTRELAQQ TPGR DEAD SAT IIF SRGLD YVHRIGRTARST 85%
hDDX42 AQTGSGKT PTRELCQQ TPGR DEAD SAT LLF ARGLD HTHRIGRTGRAG 87%
caDDX3 AQTGSGKT PTRELAVQ TPGR DEAD SAT LVF ARGLD YVHRIGRTGRVG 100%
mmp110 AQTGSGKT PTRELAVQ TPGR DEAD SAT LVF ARGLD YVHRIGRTGRVG 100%
rnDDX3 AQTGSGKT PTRELAVQ TPGR DEAD SAT LVF ARGLD YVHRIGRTGRVG 100%
maDDX3 AQTGSGKT PTRELAVQ TPGR DEAD SAT LVF ARGLD YVHRIGRTGRVG 100%
xlAn3 AQTGSGKT PTRELAVQ TPGR DEAD SAT LVF ARGLD YVHRIGRTGRVG 100%
zfDDX3 AQTGSGKT PTRELALQ TPGR DEAD SAT LVF ARGLD YVHRIGRTGRVG 98%
scDBP1 AQTGSGKT PTRELATQ TPGR DEAD SAT LIF ARGLD YVHRIGRTGRAG 94%
scDED1 AQTGSGKT PTRELATQ TPGR DEAD SAT LIF ARGLD YVHRIGRTGRAG 94%
spDED1 AQTGSGKT PTRELVCQ TPGR DEAD SAT LIF SRGLD YVHRIGRTGRAG 89%
Consensus AxxGxGKT PTRELAxQ TPGR DEAD SAT LVF ARGLD YVHRxGRxGRxG

Targeting the Human DEAD-Box Polypeptide 3 (DDX3) RNA Helicase Current Medicinal Chemistry, 2011 Vol. 18, No. 20 3019
Fig. (1). A. Sequence alignment of hDDX3 and its orthologs from S. cerevisiae, S. pombe, M. musculus and X. laevis. Conserved sequences are shaded in blue.
Equivalent amino acids are shaded in green. Helicase Motifs are in bold. B. Schematic representation of the conserved helicase boxes in hDDX3 along with
their sequences.

3020 Current Medicinal Chemistry, 2011 Vol. 18, No. 20 Garbelli et al.
Fig. (2). Sequence alignment of 40 different human DExD-box helicases covering the conserved region between Motifs I and Ia (amino acid residues 229–279
in DDX3X) and showing the DDX3X/Y-specific insertion between residues 250 and 259 (red box). Conserved residues are shaded in cyan.
CELLULAR LOCALIZATION OF DDX3
DDX3 has been described as a nucleo-cytoplasmic shuttling protein, which binds CRM1 and localizes to the outer side of nuclear membrane pores [30, 102]. In situ confocal images with detergent extraction, suggest that DDX3 ends up on the cytoplasmic side of the nuclear pore. DDX3 shuttles from the nucleus through the nuclear pore and releases the binding partners from the cytoplasmic side of the pore. This is facilitated by the enzymatic unwinding action of the DDX3 RNA helicase [30]. Inhibition of CRM1 function, results in nuclear accumulation of DDX3. The subcellular localization of DDX3, however, is still controversial. Two studies found HeLa cell DDX3 predominantly nuclear, in the absence of any treatment. In addition, DDX3 was found to be a nuclear protein in healthy primary epidermoid cells, but mainly cytoplasmic in squamous cell carcinoma skin cells. Thus, it seems that the nuclear-cytoplasmic shuttling of DDX3 is under multiple levels of regulation and could, possibly, be cell type-specific. Fractionation of HeLa cells extracts showed DDX3 mainly in the nucleoplasmic fraction, with a chromatin-bound slower migrating form, suggestive of possible post-translational modifications (Maga G., personal communication).
SPECIFIC FUNCTIONAL ROLES OF DDX3 IN NORMAL AND TUMOR CELLS
DDX3 has been involved in many cellular metabolic pathways. The known cellular proteins interacting with DDX3 are listed in
Table 2. Below, the various proposed functions of DDX3 in the context of cellular metabolism are briefly summarized.
Cellular Proliferation
Besides its functions in RNA metabolism (see above), DDX3
has been suggested to play additional roles in cellular proliferation.
DDX3 has been shown to upregulate the promoter of the cell cycle
regulatory protein p21, together with the transcription factor Sp-1
[89]. Knockdown of DDX3, along with overexpression of the
oncogene v-Ras, led to premature S-phase entry and enhanced the
cellular transformation phenotype of murine fibroblast NIH3T3
cells [103]. However, in a different study, it was found that
exposure of human breast epithelial MCF10A cells to a
carcinogenic compound enhanced DDX3 expression [104].
Elevated DDX3 levels were also found to correlate with a more
aggressive phenotype of breast cancer cell lines [104]. DDX3 overexpression and knock down experiments showed that DDX3
negatively regulates the promoter of E-cadherin, a protein important
for cell-cell contact and adhesion, while no effects were noted on
the levels of p21 [103]. Similarly, analysis of the expression levels
of DDX3 in tumor cells led to conflicting results. One group
showed that DDX3 expression is downregulated in human
hepatocellular carcinoma (HCC) cells derived from HBV-infected
patients [103]. On the contrary, another group found elevated
DDX3 mRNA levels in the majority (64%) of a representative set

Targeting the Human DEAD-Box Polypeptide 3 (DDX3) RNA Helicase Current Medicinal Chemistry, 2011 Vol. 18, No. 20 3021
of HCC samples [105]. Thus, whether DDX3 acts as an oncogene
or a tumor suppressor is still debated.
DDX3 expression knock down experiments in HeLa [30] (Debyser Z., personal communication), HEK293 and Ghost [106] cell lines, consistently failed to reveal any deleterious effect on cell proliferation or vitality. These results, suggest that DDX3 functions in cell proliferation are either not essential, or they take place in the context of altered cell metabolism only, such as tumor transformation or viral infections [107, 108].
Innate Immunity
The innate immune system is responsible for early detection of viral infection, by sensing cytoplasmic viral nucleic acids through the combined action of the endosomal subset of toll-like receptors and of the DExH-box proteins RIG-like helicases. These two pathways converge in activating the transcription factor NF-kB and the Interferon (IFN) regulatory factors IRF3 and IRF7, thus stimulating IFN- production. DDX3 has been recently shown to interact with the protein kinases IKK and TBK-1, involved in IRF3 and IRF7 activation and contributes in augmenting IFN- production [102, 109, 110]. Interestingly, this function is ATPase-independent. In addition, DDX3 has been found to be a component of the multiprotein complex IPS-1, which is involved in the regulation of IKK and TBK-1 kinases, thus placing DDX3 action further upstream in the IFN- expression regulatory cascade [110, 111].
SPECIFIC ROLES OF DDX3 IN VIRAL INFECTIONS
It has been reported that four major human viruses, namely Hepatitis C virus (HCV), Hepatitis B virus (HBV), Human Immunodeficiency Virus (HIV-1) and Poxviruses, encode proteins that can interact with DDX3 to modulate its function (Table 2). These host-pathogen interactions, however, have different functional consequences, suggesting that DDX3 performs a wide range of diverse functions in the context of viral infections. Here we will summarize the current knowledge about DDX3 functions in viral replication, with particular focus on its role in the HIV-1 life cycle.
DDX3 and Hepatitis C Virus Replication
The first viral protein to be recognized as an interactor of human DDX3, was the HCV structural core protein, which is used by the virus to build its nucleocapsid. The interaction domain between DDX3 and the HCV core protein has been mapped to the C-ter part of DDX3 [112, 113, 114]. HCV core and DDX3 co-localize in discrete cytoplasmic speckles, while the nuclear localization of DDX3 is not affected. Functional experiments suggested that the HCV core protein might enhance the transcriptional co-activator function of DDX3, while repressing its role in enhancing capped RNA translation. Knock down of DDX3 expression in the hepatocyte cell line HuH-7 significantly suppressed HCV replication [115]. However, DDX3 mutants unable to interact with the HCV core protein, still allowed efficient HCV replication [116]. Thus, the HCV core-DDX3 interaction seems not to be required for HCV RNA replication.
DDX3 and Hepatitis B Virus Replication
Another viral protein found to interact with human DDX3 is the DNA polymerase/reverse transcriptase of HBV (HBV pol) [117]. HBV pol is an enzyme essential for the duplication of the viral genetic information. It has been shown that DDX3 is incorporated into the viral nucleocapsids, binds to HVB pol and inhibits its activity. These findings, along with the observation that DDX3 expression is downregulated in HCC cell lines derived from HBV-positive patients, suggest that DDX3 might directly restrict HBV replication in hepatocytes. Another recent study, however, showed that HBV pol is involved itself in the inhibition of the IFN-response pathway. In particular, it was shown that sequestration of DDX3 by HBV pol inhibits the TBK1/IKK pathway [118, 119] see also above). Thus, the HBV pol-DDX3 interaction might have a functional significance similar to the one of the Vaccinia virus K7 protein (see below).
DDX3 and Vaccinia Virus Replication
The K7 protein of Vaccinia virus has been shown to interact with the N-terminal domain of human DDX3 [110, 120]. This interaction is essential for the inhibition of IFN-mediated cellular
Table 2. Known Cellular and Viral Interactors of Human DDX3
Interactors Function Ref.
Cellular Proteins
Tip-associated protein (TAP) mRNA export 56
CRM1 mRNA export 30
eIF4a translation 56
eIF4e translation 80
eIF2a translation 56
eIF3 translation 81
PABP translation 56
IKKe signal transduction
(IFN response)
102
TBK-1 signal transduction
(IFN response)
102
Viral Proteins
HCV core protein unclear
(regulation of subcellular localization)
112-114
HBV DNA polymerase inhibition of IFN response 117-119
Vaccinia virus K7 protein inhibition of IFN response 110, 120
HIV-1 Rev viral RNA export 30

3022 Current Medicinal Chemistry, 2011 Vol. 18, No. 20 Garbelli et al.
response. Sequestration of DDX3 by K7 leads to inhibition of the TBK1/IKKe-dependent activation of the IRF3 and IRF7, thus reducing IFN- production.
In summary, it appears that a common strategy used by different viruses, such as HBV and Vaccinia virus, to reduce IFN-mediated response, is to target human DDX3. However, viral targeting of DDX3 can have also different functional roles, as shown by the case of HIV-1.
SPECIFIC ROLES OF DDX3 IN THE CONTEXT OF HIV-1
REPLICATION
The Rev/RRE-Dependent HIV-1 RNA Export Pathway
All nucleo-cytoplasmic transport processes take place through the nuclear pore complex (NPC), a large dynamic multi-protein assembly that acts as the passageway for transport [121]. This active transport can also occur against a concentration gradient, and is mediated by soluble transport factors, that in turn shuttle between the nucleus and the cytoplasm. Importantly, even molecules that are theoretically small enough for passive diffusion (e.g., HIV-1 Rev) are actively and selectively transported, since regulated transport appears to be more efficient and more amendable for specific regulation [122].
HIV-1 has a total of nine genes that are expressed by alternative splicing of a single, initial proviral transcript that also forms the RNA genome. Importantly, HIV-1 replication requires the nuclear export and translation of unspliced, singly-spliced and multiply-spliced derivatives of this initial transcript. Fully spliced mRNAs encode the viral regulatory proteins Tat, Rev and Nef, whereas incompletely spliced HIV-1 mRNAs primarily encode viral auxiliary (Vif, Vpr, Vpu) and structural proteins. The HIV-1 Rev is a sequence-specific nuclear mRNA-export factor. In the absence of
Rev function, the incompletely spliced HIV-1 mRNAs that encode the viral structural proteins are retained in the cell nucleus, whereas nuclear export of fully-spliced HIV-1 mRNAs, including the mRNA encoding Rev itself, is independent of Rev function. Nuclear export of unspliced HIV-1 mRNAs also requires a structured cis-acting RNA sequence, called the Rev response element (RRE), which is specifically bound by Rev. The nuclear retention of the incompletely spliced viral mRNAs in the absence of Rev results from the fact that splice sites present in the retained introns are recognized by cellular mRNA processing factors, termed splicing commitment factors, that normally prevent cellular pre-mRNAs (i.e. incompletely spliced cellular mRNAs) from exiting the nucleus [123]. The HIV-1 Rev protein has two distinct functional domains, an N-terminal sequence required for RRE binding and Rev multimerization and a 10-amino acid leucine-rich domain near the C-terminus that serves as the Rev nuclear export signal (NES). This signal mediates the nuclear export of the Rev/RNA complex, through interaction with the cellular export receptor CRM1 and proposed additional cellular cofactors [30].
The DDX3/Rev Axis
DDX3 expression was found to be induced in HIV-1 infected cells by the viral transcriptional activator Tat, but it apparently plays no role in HIV-1 transcription. Instead, DDX3 was shown to be an RNA-dependent ATPase/helicase which functions in the Rev-RRE/CRM1 pathway for the export of unspliced/partially spliced HIV-1 transcripts [30]. DDX3 is a nucleo-cytoplasmic shuttling protein that binds CRM1 and associates to the cytoplasmic side of nuclear pores (Fig. 3).
Its enzymatic activity, as well as its physical interaction with both Rev and CRM1, is necessary for the Rev/RRE mediated nuclear export of viral RNAs. Downregulation of DDX3 expression
Fig. (3). Schematic representation of the role of DDX3 in mediating the Rev-dependent nuclear export of unspliced HIV-1 mRNAs. A. In the early phase of
proviral DNA transcription, only fully spliced mRNAs (encoding Tat and Rev) are exported from the nucleus. B-C. The Rev protein is imported into the
nucleus and binds to the Rev-responsive element (RRE) present on HIV-1 transcribed RNAs. Rev binding inhibits splicing, allowing accumulation of
unspliced (B) or partially spliced (C) viral mRNAs. Interaction of Rev with DDX3 and Crm1 allows nuclear export of these incompletely spliced mRNAs into
the cytoplasm.

Targeting the Human DEAD-Box Polypeptide 3 (DDX3) RNA Helicase Current Medicinal Chemistry, 2011 Vol. 18, No. 20 3023
through antisense or dominant negative mutant approaches, led to a reduction of the export of unspliced viral transcripts from the nucleus to the cytoplasm, hence inhibiting HIV-1 replication at a post-integration level. However, the molecular details of DDX3 role(s) in this pathway are yet to be fully elucidated. For example, DDX3 was not required for the CRM1-dependent export of endogenous transcripts, raising the intriguing possibility that the role of DDX3 might be specific for HIV-1 RNAs.
Another DEAD-box helicase, DDX1 was also shown to have probably a similar role than DDX3 in the Rev-mediated RNA export [124, 125]. Moreover, gene-profiling analysis showed that two additional genes encoding DEAD-box proteins, DDX18, a DEAD-box protein induced by Myc and Max and DDX39, (or URH49), a DEAD-box protein induced by growth stimulation or protein synthesis inhibition thought to be involved in splicing and nuclear export, with homology to the yeast Sub2p protein, were differentially expressed during viral latency (DDX39, DDX18) and at early times (DDX18) after induction into active replication [126]. Since some DEAD-box proteins are important for viral RNA nuclear export and active viral replication, it may be reasonable to consider that other members of this family may have roles in maintaining HIV-1 latency.
DDX3 AS A TARGET FOR HIV-1 CHEMOTHERAPY
Therapies currently used for the treatment of HIV-1 Infections are targeted to viral proteins, but the problem of drug resistance, correlated to the high intrinsic mutation and replication rates of HIV-1, implies the need to develop new drugs. The possibility of using a cellular protein as an antiviral target represents an attractive solution, since it is known that human proteins are less prone to mutate than the viral ones. In addition, any "escape" mutant, that is any variant of the cellular target protein escaping inhibition, will be counterselected, since it will not confer any replicative advantage to that particular cell lineage, but rather will render the cells more susceptible to viral Infection and death.
The recently unveiled roles of DDX3 in HIV-1 replication made this protein an attractive target for the development of an alternative anti-HIV-1 strategy [30, 127, 128]. A random-screening approach, led to discovery of a series of ring-expanded nucleoside analogs, able to inhibit the RNA helicase activity of DDX3 and to suppress HIV-1 replication [129].
At the same time, our group decided to target the ATPase activity of DDX3, which seemed to be dispensable for most of the cellular functions of DDX3 but essential for its role in HIV-1 replication, through a rational-based drug design approach. This was the first example of rational drug design applied to a DEAD-box protein.
A RATIONAL-BASED DRUG DESIGN APPROACH FOR
THE IDENTIFICATION OF DDX3 INHIBITORS
Chemical feature-based pharmacophoric models have been established as state of the art technique for virtual screening. The feature-based pharmacophore recognition derived from a set of bio-active ligands was for long time one of the most used and valuable molecular modeling techniques. In the last years, the use of structure-based pharmacophores obtained from ligand-protein complexes has represented an important improvement of virtual screening procedures because of the possibility to include into the pharmacophore important information directly correlated to the interaction between proteins and ligands [130]. Structure-based virtual screening usually involves docking of compounds into a protein binding site using docking algorithms, followed by the application of ranking of selected compounds to identify potential hits. However, several recent studies have shown that pharmacophore-based search in combination with docking-based
virtual screening can significantly improve the probability of identifying putative candidates [131]. In fact, the use of pharmacophore-based searches before docking studies can significantly narrow down the number of compounds to be analyzed, thus allowing a more exhaustive docking together with a more accurate investigation of compound binding modes and interactions with the target.
The availability of two different crystallographic structures for the human DDX3 helicase (PDB entries code 2JGN and 2I4I) [98, 107] allowed us to plan a classical structure-based approach for the identification of potential inhibitors. Since the 2JGN structure possesses the helicase domain only (the ATP binding domain is missing), the 2I4I protein was selected as a starting point for a virtual screening protocol even if a few residues were missing (namely, residues 407-410; 535-536 and 581-582).
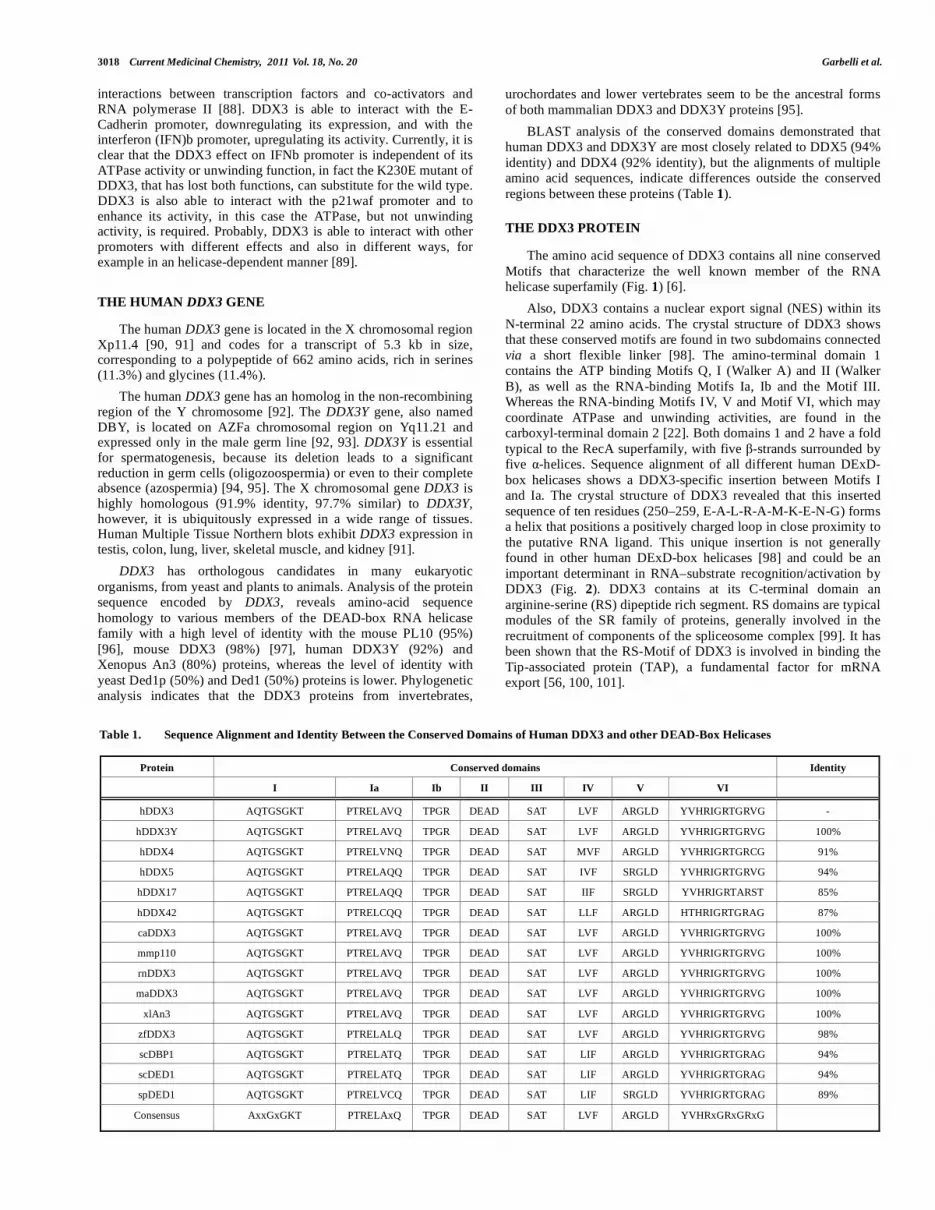
The LigandScout software [Further details at the web page http://www.inteligand.com.] has been used for the detection and interpretation of the crucial interaction pattern between the DDX3 protein and its cocrystallized ligand (AMP) (PDB code: 2I4I): the chemical features responsible for AMP-enzyme interactions were thus codified into a structure-based pharmacophoric model (Fig. 4) [127].
The adenine moiety of the nucleoside interacts with amino acids in the Q-Motif, whereas residues in the P-loop in Motif I interact with the phosphate group. The C6-amino group of the purine interacts with the backbone carbonyl group of Arg202 and with O
of Gln207 in the Q-Motif. N of Gln207 also interacts with
adenine N7. In addition, the pyrimidine ring of adenine makes - interactions with the aromatic portion of Tyr200, whose hydroxyl group gives a hydrogen bond contact with the 2`-OH of the ribose. The phosphate group gives polar interactions with the hydroxyl group of Thr231 and with backbone nitrogen atoms of residues 227-231 in the P-loop in Motif I as well as with several water molecules (see Fig. 4A).
The pharmacophore built with LigandScout was automatically simplified for its interpretation and use in Catalyst (see Fig. 4b-c), and then further simplified by removing two hydrogen bond features (HBA3 and HBD2), in order to retrieve more hits (Fig. 4d). The resulting model consisted of four pharmacophoric features (one hydrogen bond donor, two hydrogen bond acceptors, and an aromatic ring) that accounted for the interactions between AMP and DDX3, and 10 excluded volumes, corresponding to regions of AMP binding site occupied by DDX3 portions and, thus, forbidden to any ligand portion.
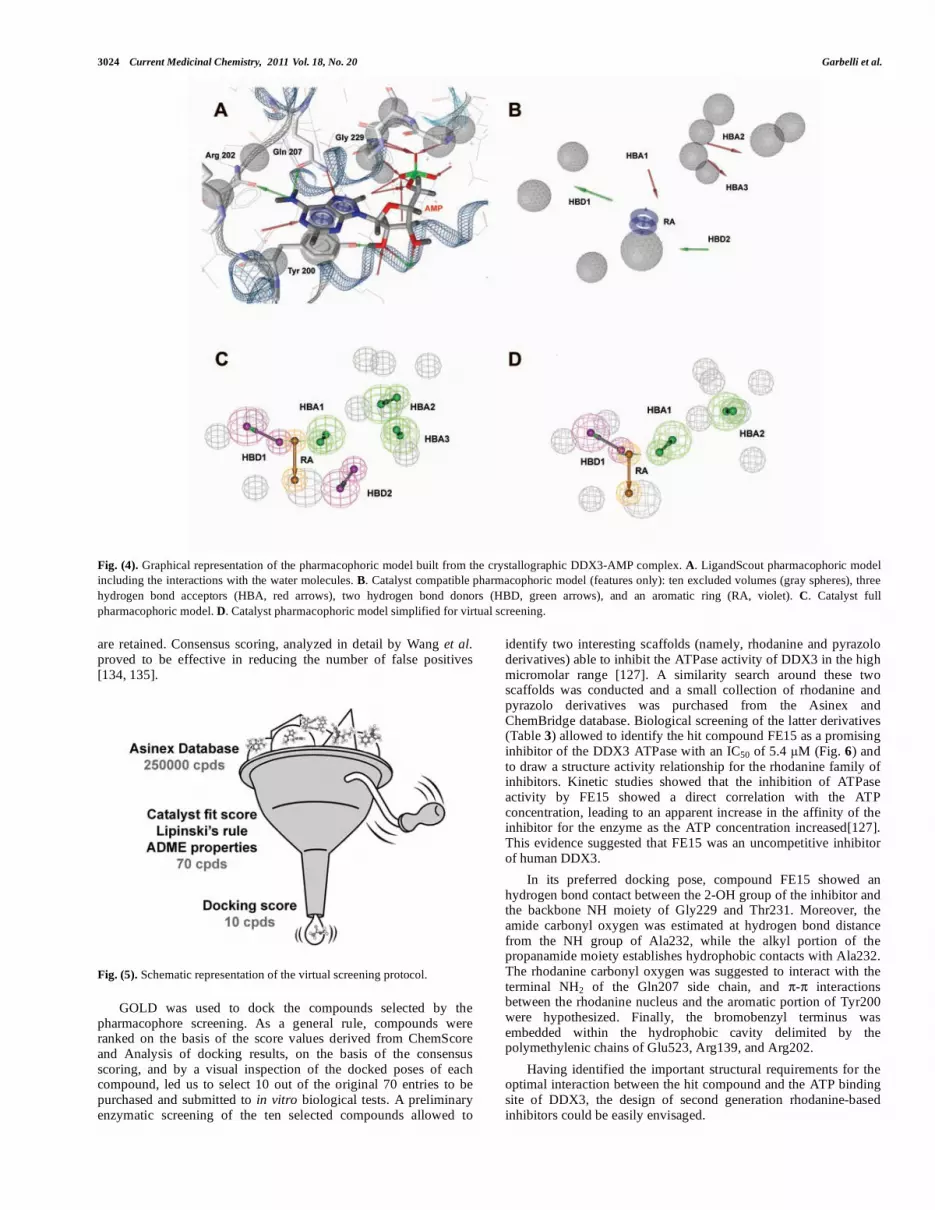
The model thus defined was used as a three-dimensional query in a virtual screening approach to filter databases of commercially available compounds from Asinex Gold and Synergy collections (about 250000 compounds) to identify chemical scaffolds with putative affinity toward the DDX3 ATP binding site, leading to a ranking list of 100 compounds.
These compounds were further processed by Cerius2 4.10L software in order to apply a second filter based on Lipinski's rule of five, allowing the retrieval of the most drug-like ones [132]. According to these criteria, 70 molecules were selected (Fig. 5).
The molecules selected via the in silico screening approach were submitted to geometry optimizations and their ionization states were set at a physiological pH. The compounds were then docked into the DDX3 ATP binding site, to analyze their ability to have profitable interactions with the enzyme, and a consensus scoring based on the two scoring functions available in GOLD was applied to improve the enrichment of true positives.
The approach, called 'consensus scoring', was pioneered by Charifson and colleagues [133]: two or more scoring functions are applied to the same set of poses, and only structures that perform well in more than a predefined share of the said scoring functions
3024 Current Medicinal Chemistry, 2011 Vol. 18, No. 20 Garbelli et al.
Fig. (4). Graphical representation of the pharmacophoric model built from the crystallographic DDX3-AMP complex. A. LigandScout pharmacophoric model
including the interactions with the water molecules. B. Catalyst compatible pharmacophoric model (features only): ten excluded volumes (gray spheres), three
hydrogen bond acceptors (HBA, red arrows), two hydrogen bond donors (HBD, green arrows), and an aromatic ring (RA, violet). C. Catalyst full
pharmacophoric model. D. Catalyst pharmacophoric model simplified for virtual screening.
are retained. Consensus scoring, analyzed in detail by Wang et al. proved to be effective in reducing the number of false positives [134, 135].
Fig. (5). Schematic representation of the virtual screening protocol.
GOLD was used to dock the compounds selected by the pharmacophore screening. As a general rule, compounds were ranked on the basis of the score values derived from ChemScore and Analysis of docking results, on the basis of the consensus scoring, and by a visual inspection of the docked poses of each compound, led us to select 10 out of the original 70 entries to be purchased and submitted to in vitro biological tests. A preliminary enzymatic screening of the ten selected compounds allowed to
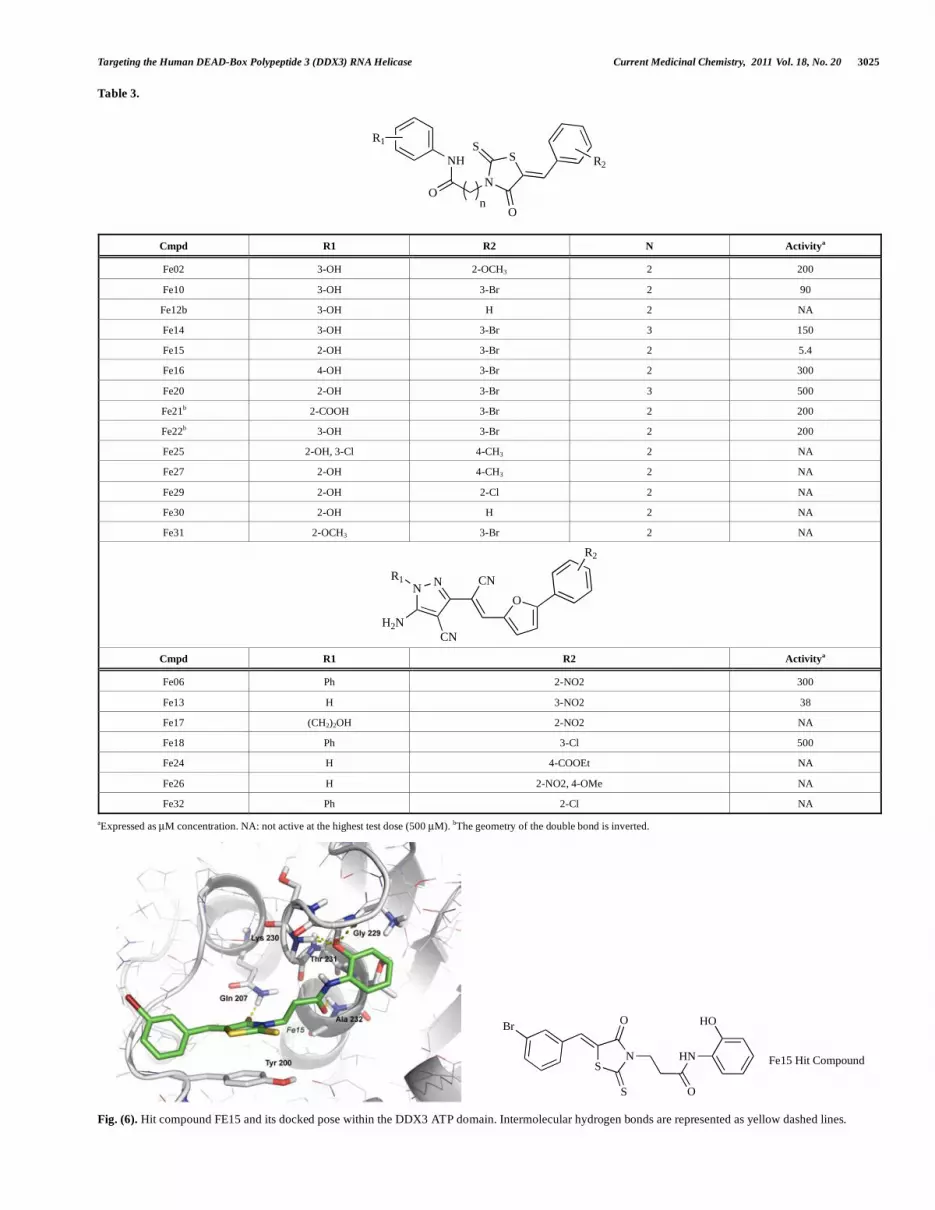
identify two interesting scaffolds (namely, rhodanine and pyrazolo derivatives) able to inhibit the ATPase activity of DDX3 in the high micromolar range [127]. A similarity search around these two scaffolds was conducted and a small collection of rhodanine and pyrazolo derivatives was purchased from the Asinex and ChemBridge database. Biological screening of the latter derivatives (Table 3) allowed to identify the hit compound FE15 as a promising inhibitor of the DDX3 ATPase with an IC50 of 5.4 M (Fig. 6) and to draw a structure activity relationship for the rhodanine family of inhibitors. Kinetic studies showed that the inhibition of ATPase activity by FE15 showed a direct correlation with the ATP concentration, leading to an apparent increase in the affinity of the inhibitor for the enzyme as the ATP concentration increased[127]. This evidence suggested that FE15 was an uncompetitive inhibitor of human DDX3.
In its preferred docking pose, compound FE15 showed an hydrogen bond contact between the 2-OH group of the inhibitor and the backbone NH moiety of Gly229 and Thr231. Moreover, the amide carbonyl oxygen was estimated at hydrogen bond distance from the NH group of Ala232, while the alkyl portion of the propanamide moiety establishes hydrophobic contacts with Ala232. The rhodanine carbonyl oxygen was suggested to interact with the terminal NH2 of the Gln207 side chain, and - interactions between the rhodanine nucleus and the aromatic portion of Tyr200 were hypothesized. Finally, the bromobenzyl terminus was embedded within the hydrophobic cavity delimited by the polymethylenic chains of Glu523, Arg139, and Arg202.
Having identified the important structural requirements for the optimal interaction between the hit compound and the ATP binding site of DDX3, the design of second generation rhodanine-based inhibitors could be easily envisaged.
Targeting the Human DEAD-Box Polypeptide 3 (DDX3) RNA Helicase Current Medicinal Chemistry, 2011 Vol. 18, No. 20 3025
Table 3.
N
SS
O
O
NH
R1
R2
n
Cmpd R1 R2 N Activitya
Fe02 3-OH 2-OCH3 2 200
Fe10 3-OH 3-Br 2 90
Fe12b 3-OH H 2 NA
Fe14 3-OH 3-Br 3 150
Fe15 2-OH 3-Br 2 5.4
Fe16 4-OH 3-Br 2 300
Fe20 2-OH 3-Br 3 500
Fe21b 2-COOH 3-Br 2 200
Fe22b 3-OH 3-Br 2 200
Fe25 2-OH, 3-Cl 4-CH3 2 NA
Fe27 2-OH 4-CH3 2 NA
Fe29 2-OH 2-Cl 2 NA
Fe30 2-OH H 2 NA
Fe31 2-OCH3 3-Br 2 NA
NN
CN
H2N
R1 CN
O
R2
Cmpd R1 R2 Activitya
Fe06 Ph 2-NO2 300
Fe13 H 3-NO2 38
Fe17 (CH2)2OH 2-NO2 NA
Fe18 Ph 3-Cl 500
Fe24 H 4-COOEt NA
Fe26 H 2-NO2, 4-OMe NA
Fe32 Ph 2-Cl NA
aExpressed as μM concentration. NA: not active at the highest test dose (500 μM). bThe geometry of the double bond is inverted.
Fig. (6). Hit compound FE15 and its docked pose within the DDX3 ATP domain. Intermolecular hydrogen bonds are represented as yellow dashed lines.
SN
O
S
Br
HN
O
HO
Fe15 Hit Compound

3026 Current Medicinal Chemistry, 2011 Vol. 18, No. 20 Garbelli et al.
CONCLUSIONS AND PERSPECTIVES
The problem of drug resistance development due to mutations in HIV-1 proteins targeted by antiviral drugs, could be overcome by the development of novel anti-HIV agents targeting host factors essential for viral replication. One of such host factors is the DEAD-box RNA helicase/ATPase DDX3. However, a more detailed knowledge of its functions in the normal vs. infected cell context is needed in order to rationally design safe and effective drugs for patient treatment. In addition, DDX3 might reveal a useful target also for anticancer and immunomodulatory therapies. Thus, the knowledge gained in the antiviral drug development process might as well be expanded to other pharmaceutically relevant issues.
ACKNOWLEDGEMENTS
Work in Authors' laboratories has been partially supported by the EU-FP6 Project LSHP-CT-2006-037257-ExCellENT-HIT to G.M, U.D. and M.B. and by the Italian Cancer Research Association IG 4538 and National AIDS Programme 40H26 grant to G.M.
REFERENCES
[1] Schroder, M. Biochem. Pharmacol., 2010, 79(3), 297-306. [2] de la Cruz, J.; Kressler, D.; Linder, P. Trends Biochem. Sci., 1999, 24(5),
192-198. [3] Caruthers, J.M.; McKay, D.B. Curr. Opin. Struc. Biol., 2002, 12(1), 123-133.
[4] Linder, P. Nucleic Acids Res., 2006, 34(15), 4168-4180. [5] Linder, P.; Lasko, P.F.; Ashburner, M.; Leroy, P.; Nielsen, P.J.; Nishi, K.;
Schnier, J.; Slonimski, P.P. Nature, 1989, 337(6203), 121-122. [6] Tanner, N.K.; Linder, P. Mol. Cell, 2001, 8(2), 251-262.
[7] Wassarman, D.A.; Steitz, J.A. Nature, 1991, 349(6309), 463-464. [8] Walker, J.E.; Saraste, M.; Runswick, M.J.; Gay, N.J. Embo J., 1982, 1(8),
945-951.
[9] Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. Nature,
1988, 333(6168), 22.
[10] Shi, H.; Cordin, O.; Minder, C.M.; Linder, P.; Xu, R.M. Proc. Natl. Acad.
Sci. U. S. A., 2004, 101(51), 17628-17633.
[11] Rozen, F.; Pelletier, J.; Trachsel, H.; Sonenberg, N. Mol. Cell Biol., 1989, 9(9), 4061-4063.
[12] Blum, S.; Schmid, S.R.; Pause, A.; Buser, P.; Linder, P.; Sonenberg, N.; Trachsel, H. Proc. Natl. Acad. Sci. U. S. A., 1992, 89(16), 7664-7668.
[13] Pugh, G.E.; Nicol, S.M.; Fuller-Pace, F.V. J. Mol. Biol., 1999, 292(4), 771-778.
[14] Tanner, N.K.; Cordin, O.; Banroques, J.; Doere, M.; Linder, P. Mol. Cell,
2003, 11(1), 127-138.
[15] Cordin, O.; Tanner, N.K.; Doere, M.; Linder, P.; Banroques, J. Embo J.,
2004, 23(13), 2478-2487. [16] Benz, J.; Trachsel, H.; Baumann, U. Structure, 1999, 7(6), 671-679.
[17] Zhao, R.; Shen, J.; Green, M.R.; MacMorris, M.; Blumenthal, T. Structure,
2004, 12(8), 1373-1381.
[18] Johnson, E.R.; McKay, D.B. Rna, 1999, 5(12), 1526-1534. [19] Carmel, A.B.; Matthews, B.W. Rna, 2004, 10(1), 66-74.
[20] Story, R.M.; Li, H.; Abelson, J.N. Proc. Natl. Acad. Sci. U. S. A., 2001, 98(4), 1465-1470.
[21] Dillingham, M.S.; Soultanas, P.; Wigley, D.B. Nucleic Acids Res., 1999, 27(16), 3310-3317.
[22] Cordin, O.; Banroques, J.; Tanner, N.K.; Linder, P. Gene, 2006, 367, 17-37. [23] Pause, A.; Sonenberg, N. Embo J., 1992, 11(7), 2643-2654.
[24] Iost, I.; Dreyfus, M.; Linder, P. J. Biol. Chem., 1999, 274(25), 17677-17683.
[25] Martinez, R.; Shao, L.; Weller, S.K. J. Virol., 1992, 66(11), 6735-6746. [26] Chamot, D.; Magee, W.C.; Yu, E.; Owttrim, G.W. J. Bacteriol., 1999,
181(6), 1728-1732. [27] Banroques, J.; Doere, M.; Dreyfus, M.; Linder, P.; Tanner, N.K. J. Mol.
Biol., 2009. [28] Pause, A.; Methot, N.; Sonenberg, N. Mol. Cell Biol., 1993, 13(11), 6789-
6798. [29] Schwer, B.; Meszaros, T. Embo J., 2000, 19(23), 6582-6591.
[30] Yedavalli, V.S.; Neuveut, C.; Chi, Y.H.; Kleiman, L.; Jeang, K.T. Cell, 2004, 119(3), 381-392.
[31] Campodonico, E.; Schwer, B. Genetics, 2002, 160(2), 407-415. [32] Banroques, J.; Cordin, O.; Doere, M.; Linder, P.; Tanner, N.K. Mol. Cell
Biol., 2008, 28(10), 3359-3371.
[33] Kim, J.L.; Morgenstern, K.A.; Griffith, J.P.; Dwyer, M.D.; Thomson, J.A.; Murcko, M.A.; Lin, C.; Caron, P.R. Structure, 1998, 6(1), 89-100.
[34] Caruthers, J.M.; Johnson, E.R.; McKay, D.B. Proc. Natl. Acad. Sci. U. S. A.,
2000, 97(24), 13080-13085.
[35] Chang, T.H.; Latus, L.J.; Liu, Z.; Abbott, J.M. Nucleic Acids Res., 1997,
25(24), 5033-5040. [36] Schneider, S.; Campodonico, E.; Schwer, B. J. Biol. Chem., 2004, 279(10),
8617-8626. [37] Rogers, G.W. Jr.; Komar, A.A.; Merrick, W.C. Prog. Nucleic Acid Res. Mol.
Biol., 2002, 72, 307-331. [38] Schneider, S.; Schwer, B. J. Biol. Chem., 2001, 276(24), 21184-21191.
[39] Bernstein, K.A.; Granneman, S.; Lee, A.V.; Manickam, S.; Baserga, S.J.
Mol. Cell Biol., 2006, 26(4), 1195-1208. [40] Granneman, S.; Bernstein, K.A.; Bleichert, F.; Baserga, S.J. Mol. Cell. Biol.,
2006, 26(4), 1183-1194. [41] Franca, R.; Belfiore, A.; Spadari, S.; Maga, G. Proteins, 2007, 67(4), 1128-
1137. [42] Luking, A.; Stahl, U.; Schmidt, U. Crit. Rev. Biochem. Mol. Biol., 1998,
33(4), 259-296. [43] Silverman, E.; Edwalds-Gilbert, G.; Lin, R.J. Gene, 2003, 312, 1-16.
[44] Rocak, S.; Linder, P. Nat. Rev. Mol. Cell Biol., 2004, 5(3), 232-241. [45] Staley, J.P.; Guthrie, C. Mol. Cell, 1999, 3(1), 55-64.
[46] Xu, Y.Z.; Newnham, C.M.; Kameoka, S.; Huang, T.; Konarska, M.M.; Query, C.C. Embo J., 2004, 23(2), 376-385.
[47] Chen, J.Y.; Stands, L.; Staley, J.P.; Jackups, R.R. Jr.; Latus, L.J.; Chang,
T.H. Mol. Cell, 2001, 7(1), 227-232. [48] Libri, D.; Graziani, N.; Saguez, C.; Boulay, J. Genes Dev., 2001, 15(1), 36-
41. [49] Liu, Z.R. Mol. Cell Biol., 2002, 22(15), 5443-5450.
[50] Guil, S.; Gattoni, R.; Carrascal, M.; Abian, J.; Stevenin, J.; Bach-Elias, M. Mol. Cell Biol., 2003, 23(8), 2927-2941.
[51] Honig, A.; Auboeuf, D.; Parker, M.M.; O'Malley, B.W.; Berget, S.M. Mol.
Cell Biol., 2002, 22(16), 5698-5707.
[52] Jurica, M.S.; Moore, M.J. Mol. Cell, 2003, 12(1), 5-14. [53] Stevens, S.W.; Ryan, D.E.; Ge, H.Y.; Moore, R.E.; Young, M.K.; Lee, T.D.;
Abelson, J. Mol. Cell, 2002, 9(1), 31-44. [54] Zhou, Z.; Licklider, L.J.; Gygi, S.P.; Reed, R. Nature, 2002, 419(6903), 182-
185.
[55] Merz, C.; Urlaub, H.; Will, C.L.; Luhrmann, R. Rna, 2007, 13(1), 116-128. [56] Lai, M.C.; Lee, Y.H.; Tarn, W.Y. Mol. Biol. Cell, 2008, 19(9), 3847-3858.
[57] Kressler, D.; Doere, M.; Rojo, M.; Linder, P. Mol. Cell Biol., 1999, 19(12), 8633-8645.
[58] Charollais, J.; Pflieger, D.; Vinh, J.; Dreyfus, M.; Iost, I. Mol. Microbiol.,
2003, 48(5), 1253-1265.
[59] Charollais, J.; Dreyfus, M.; Iost, I. Nucleic Acids Res., 2004, 32(9), 2751-2759.
[60] Fuller-Pace, F.V.; Nicol, S.M.; Reid, A.D.; Lane, D.P. Embo J., 1993, 12(9), 3619-3626.
[61] Kossen, K.; Uhlenbeck, O.C. Nucleic Acids Res., 1999, 27(19), 3811-3820. [62] Linder, P.; Stutz, F. Curr. Biol., 2001, 11(23), R961-963.
[63] Hieronymus, H.; Silver, P.A. Nat. Genet., 2003, 33(2), 155-161.
[64] Keene, J.D. Nat. Genet., 2003, 33(2), 111-112. [65] Estruch, F.; Cole, C.N. Mol. Biol. Cell, 2003, 14(4), 1664-1676.
[66] Tseng, S.S.; Weaver, P.L.; Liu, Y.; Hitomi, M.; Tartakoff, A.M.; Chang, T.H. Embo J., 1998, 17(9), 2651-2662.
[67] Snay-Hodge, C.A.; Colot, H.V.; Goldstein, A.L.; Cole, C.N. Embo J., 1998, 17(9), 2663-2676.
[68] Gatfield, D.; Le Hir, H.; Schmitt, C.; Braun, I.C.; Kocher, T.; Wilm, M.; Izaurralde, E. Curr. Biol., 2001, 11(21), 1716-1721.
[69] Jensen, T.H.; Boulay, J.; Rosbash, M.; Libri, D. Curr. Biol., 2001, 11(21), 1711-1715.
[70] Luo, M.L.; Zhou, Z.; Magni, K.; Christoforides, C.; Rappsilber, J.; Mann,
M.; Reed, R. Nature, 2001, 413(6856), 644-647. [71] Strasser, K.; Hurt, E. Nature, 2001, 413(6856), 648-652.
[72] Grifo, J.A.; Tahara, S.M.; Leis, J.P.; Morgan, M.A.; Shatkin, A.J.; Merrick, W.C. J. Biol. Chem., 1982, 257(9), 5246-5252.
[73] Tarn, W.Y.; Chang, T.H. RNA Biol., 2009, 6(1), 17-20. [74] Sonenberg, N.; Dever, T.E. Curr. Opin. Struct. Biol., 2003, 13(1), 56-63.
[75] Grifo, J.A.; Abramson, R.D.; Satler, C.A.; Merrick, W.C. J. Biol. Chem.,
1984, 259(13), 8648-8654.
[76] Chuang, R.Y.; Weaver, P.L.; Liu, Z.; Chang, T.H. Science, 1997, 275(5305), 1468-1471.
[77] de la Cruz, J.; Iost, I.; Kressler, D.; Linder, P. Proc. Natl. Acad. Sci. U. S. A.,
1997, 94(10), 5201-5206.
[78] Linder, P. Biol. Cell, 2003, 95(3-4), 157-167.
[79] Carrera, P.; Johnstone, O.; Nakamura, A.; Casanova, J.; Jackle, H.; Lasko, P. Mol. Cell, 2000, 5(1), 181-187.
[80] Shih, J.W.; Tsai, T.Y.; Chao, C.H.; Wu Lee, Y.H. Oncogene, 2008, 27(5), 700-714.
[81] Lee, C.S.; Dias, A.P.; Jedrychowski, M.; Patel, A.H.; Hsu, J.L.; Reed, R. Nucleic Acids Res., 2008, 36(14), 4708-4718.
[82] Yan, X.; Mouillet, J.F.; Ou, Q.; Sadovsky, Y. Mol. Cell Biol., 2003, 23(1), 414-423.
[83] Gillian, A.L.; Svaren, J. J. Biol. Chem., 2004, 279(10), 9056-9063. [84] Lee, M.B.; Lebedeva, L.A.; Suzawa, M.; Wadekar, S.A.; Desclozeaux, M.;
Ingraham, H.A. Mol. Cell. Biol., 2005, 25(5), 1879-1890. [85] Endoh, H.; Maruyama, K.; Masuhiro, Y.; Kobayashi, Y.; Goto, M.; Tai, H.;
Yanagisawa, J.; Metzger, D.; Hashimoto, S.; Kato, S. Mol. Cell Biol., 1999,
19(8), 5363-5372.

Targeting the Human DEAD-Box Polypeptide 3 (DDX3) RNA Helicase Current Medicinal Chemistry, 2011 Vol. 18, No. 20 3027
[86] Watanabe, M.; Yanagisawa, J.; Kitagawa, H.; Takeyama, K.; Ogawa, S.;
Arao, Y.; Suzawa, M.; Kobayashi, Y.; Yano, T.; Yoshikawa, H.; Masuhiro, Y.; Kato, S. Embo J., 2001, 20(6), 1341-1352.
[87] Wilson, B.J.; Bates, G.J.; Nicol, S.M.; Gregory, D.J.; Perkins, N.D.; Fuller-Pace, F.V. BMC Mol. Biol., 2004, 5, 11.
[88] Fuller-Pace, F.V. Nucleic Acids Res., 2006, 34(15), 4206-4215. [89] Chao, C.H.; Chen, C.M.; Cheng, P.L.; Shih, J.W.; Tsou, A.P.; Lee, Y.H.
Cancer Res., 2006, 66(13), 6579-6588.
[90] Park, S.H.; Lee, S.G.; Kim, Y.; Song, K. Cytogenet. Cell Genet., 1998, 81(3-4), 178-179.
[91] Kim, Y.S.; Lee, S.G.; Park, S.H.; Song, K. Mol. Cells, 2001, 12(2), 209-214. [92] Lahn, B.T.; Page, D.C. Science, 1997, 278(5338), 675-680.
[93] Ditton, H.J.; Zimmer, J.; Kamp, C.; Rajpert-De Meyts, E.; Vogt, P.H. Hum.
Mol. Genet., 2004, 13(19), 2333-2341.
[94] Foresta, C.; Ferlin, A.; Moro, E. Hum. Mol. Genet., 2000, 9(8), 1161-1169. [95] Rosner, A.; Rinkevich, B. Curr. Med. Chem., 2007, 14(23), 2517-2525.
[96] Leroy, P.; Alzari, P.; Sassoon, D.; Wolgemuth, D.; Fellous, M. Cell, 1989, 57(4), 549-559.
[97] Gee, S.L.; Conboy, J.G. Gene, 1994, 140(2), 171-177. [98] Hogbom, M.; Collins, R.; van den Berg, S.; Jenvert, R.M.; Karlberg, T.;
Kotenyova, T.; Flores, A.; Karlsson Hedestam, G.B.; Schiavone, L.H. J. Mol.
Biol., 2007, 372(1), 150-159. [99] Ma, X.; He, F. Genom. Proteom. Bioinf., 2003, 1(1), 2-8.
[100] Katahira, J.; Strasser, K.; Podtelejnikov, A.; Mann, M.; Jung, J.U.; Hurt, E. Embo J., 1999, 18(9), 2593-2609.
[101] Stutz, F.; Izaurralde, E. Trends Cell Biol., 2003, 13(6), 319-327. [102] Schroder, M.; Baran, M.; Bowie, A.G. Embo J., 2008, 27(15), 2147-2157.
[103] Chang, P.C.; Chi, C.W.; Chau, G.Y.; Li, F.Y.; Tsai, Y.H.; Wu, J.C.; Wu Lee, Y.H. Oncogene, 2006, 25(14), 1991-2003.
[104] Botlagunta, M.; Vesuna, F.; Mironchik, Y.; Raman, A.; Lisok, A.; Winnard, P. Jr.; Mukadam, S.; Van Diest, P.; Chen, J.H.; Farabaugh, P.; Patel, A.H.;
Raman, V. Oncogene, 2008. [105] Huang, J.S.; Chao, C.C.; Su, T.L.; Yeh, S.H.; Chen, D.S.; Chen, C.T.; Chen,
P.J.; Jou, Y.S. Biochem. Biophys. Res. Commun., 2004, 315(4), 950-958.
[106] Ishaq, M.; Hu, J.; Wu, X.; Fu, Q.; Yang, Y.; Liu, Q.; Guo, D. Mol.
Biotechnol., 2008.
[107] Fukumura, J.; Noguchi, E.; Sekiguchi, T.; Nishimoto, T. J. Biochem., 2003, 134(1), 71-82.
[108] Lai, M.C.; Chang, W.C.; Shieh, S.Y.; Tarn, W.Y. Mol. Cell Biol., 2010, 30(22), 5444-5453.
[109] Soulat, D.; Burckstummer, T.; Westermayer, S.; Goncalves, A.; Bauch, A.; Stefanovic, A.; Hantschel, O.; Bennett, K.L.; Decker, T.; Superti-Furga, G.
Embo J., 2008, 27(15), 2135-2146.
[110] Mulhern, O.; Bowie, A.G. Eur. J. Immunol., 2010, 40(4), 933-935.
[111] Oshiumi, H.; Sakai, K.; Matsumoto, M.; Seya, T. Eur. J. Immunol., 2010, 40(4), 940-948.
[112] Mamiya, N.; Worman, H.J. J. Biol. Chem., 1999, 274(22), 15751-15756. [113] Owsianka, A.M.; Patel, A.H. Virology, 1999, 257(2), 330-340.
[114] You, L.R.; Chen, C.M.; Yeh, T.S.; Tsai, T.Y.; Mai, R.T.; Lin, C.H.; Lee, Y.H. J. Virol., 1999, 73(4), 2841-2853.
[115] Ariumi, Y.; Kuroki, M.; Abe, K.; Dansako, H.; Ikeda, M.; Wakita, T.; Kato,
N. J. Virol., 2007, 81(24), 13922-13926. [116] Angus, A.G.; Dalrymple, D.; Boulant, S.; McGivern, D.R.; Clayton, R.F.;
Scott, M.J.; Adair, R.; Graham, S.; Owsianka, A.M.; Targett-Adams, P.; Li, K.; Wakita, T.; McLauchlan, J.; Lemon, S.M.; Patel, A.H. J. Gen. Virol.,
2010, 91(Pt 1), 122-132. [117] Wang, H.; Kim, S.; Ryu, W.S. J. Virol., 2009, 83(11), 5815-5824.
[118] Wang, H.; Ryu, W.S. PLoS Pathog., 2010, 6(7), e1000986. [119] Yu, S.; Chen, J.; Wu, M.; Chen, H.; Kato, N.; Yuan, Z. J. Gen. Virol., 2010,
91(Pt 8), 2080-2090. [120] Oda, S.; Schroder, M.; Khan, A.R. Structure, 2009, 17(11), 1528-1537.
[121] Pante, N. Dev. Cell, 2004, 7(6), 780-781. [122] Pemberton, L.F.; Paschal, B.M. Traffic, 2005, 6(3), 187-198.
[123] Cullen, B.R. J. Cell Sci., 2003, 116(Pt 4), 587-597.
[124] Fang, J.; Kubota, S.; Yang, B.; Zhou, N.; Zhang, H.; Godbout, R.; Pomerantz, R.J. Virology, 2004, 330(2), 471-480.
[125] Fang, J.; Acheampong, E.; Dave, R.; Wang, F.; Mukhtar, M.; Pomerantz, R.J. Virology, 2005, 336(2), 299-307.
[126] Krishnan, V.; Zeichner, S.L. Retrovirology, 2004, 1, 42. [127] Maga, G.; Falchi, F.; Garbelli, A.; Belfiore, A.; Witvrouw, M.; Manetti, F.;
Botta, M. J. Med. Chem., 2008, 51(21), 6635-6638. [128] Chen, C.Y.; Yedavalli, V.R.; Jeang, K.T. Methods Mol. Biol., 2010, 587,
281-289. [129] Yedavalli, V.S.; Zhang, N.; Cai, H.; Zhang, P.; Starost, M.F.; Hosmane, R.S.;
Jeang, K.T. J. Med. Chem., 2008, 51(16), 5043-5051. [130] Wolber, G.; Langer, T. J. Chem. Inf. Model, 2005, 45(1), 160-169.
[131] Polgar, T.; Keseru, G.M. J. Med. Chem., 2005, 48(11), 3749-3755.
[132] Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Adv. Drug. Deliv.
Rev., 2001, 46(1-3), 3-26.
[133] Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. J. Med. Chem.,
1999, 42(25), 5100-5109.
[134] Wang, R.; Wang, S. J. Chem. Inf. Comput. Sci., 2001, 41(5), 1422-1426. [135] Verdonk, M.L.; Berdini, V.; Hartshorn, M.J.; Mooij, W.T.; Murray, C.W.;
Taylor, R.D.; Watson, P. J. Chem. Inf. Comput. Sci., 2004, 44(3), 793-806.
Received: February 19, 2011 Revised: May 13, 2011 Accepted: May 15, 2011