structural properties of gelatin—pectin gels. part i: effect of ethylene glycol
TRANSCRIPT

Food Hydrocolloids
-Vol. 11 no. 3 pp. 271-279, 1997
Structural properties of gelatin-pectin gels. Part I: Effect of ethylene
glycol
Joannis S.Chronakis, Stefan Kasapis! and Rukmal Abeysekera>
Department of Food Research and Technology, Cranfield University, SilsoeCollege, Silsoe,Bedfordshire MK45 4DT and2Institute for Applied Biology, University of York, Yorkshire YOISOD, UK
ITowhom correspondence should be addressed
AbstractThe role of ethylene glycol (EG) in the gelation mechanisms of acid pigskin gelatin and high methoxypectin has been monitored and used as a baseline for the investigation of mixed gelatin-pectin gels invarious mixed ethylene glycol-water solvents. The addition ofEG did not alter the gelation (tgelr::! 14°C)and melting (tmel r::! 28°C) temperatures ofan aqueous gelatin network, the strength of which, however,was first increased, and then reduced at concentrations of co-solute higher than 30%. The reduction invalues of storage modulus (G) was attributed to a decrease in the proportion of polypeptide chainsinvolved in the formation ofjunction zones. By contrast, increasing levels of ethylene glycol encouragedformation ofpectin gels at high temperatures (e.g. tgel was 63°C at 800/0 EG) which largely retained theirstructure upon subsequent heating. The network strength increased rapidly and peaked at 60% co-solutefollowed by a subsequent reduction in storage moduli at conditions of low water activity (60-800/0 EG).On the basis of a model for gel formation involving a two-step process, it was proposed that ethyleneglycol promotes the ordered structure of contiguous pectin chains (first stage) but 'dissolves' the hydrophobic clusterings ofmethyl groups (second stage). Differential scanning calorimetry demonstrated thatthermodynamic incompatibility between the two polymers is the driving force behind the phase behaviourof mixed preparations. Based on the mechanical properties of single components, it was argued thatincreasing amounts of EO, within the 0-25% range, promote pectin's conformational ordering, whichbecomes more and more effective in excluding, concentrating up and strengthening the continuous gelatinphase. At higher levels of co-solute (from 30 to 70%), pectin can form a thermally stable network andduring cooling it does so before gelatin's gelation at lower temperatures. Light microscopy work stronglysuggests that gelatin also forms a continuous network throughout the body of the sample. Therefore, thelatter mixtures can be described as phase-separated, bicontinuous arrangements.
Introduction
Mixtures of protein and polysaccharide are used increasinglyin the manufacturing of food products with a low calorificvalue and novel textural properties (e.g. low-fat spreads andconfectioneries). Mixing of two different biopolymers insolution commonly results in heterotypic complexation orsteric exclusion, depending on the balance between theentropy of mixing plus mutual energetic associations and theenthalpy of self-interaction (1). Favourable interactionsoccur, for example, between anionic polysaccharides andproteins below their isoelectric point. Thus the sulphategroups of carrageenan can form electrostatic bonds with the
© Oxford University Press
positively charged amino groups of the basic sweet protein,thaumatin, leading to complex formation and a subsequentreduction in the sweetness intensity of thaumatin (2).Alternatively, polyanion-polycation interactions canproduce an insoluble precipitate, like the acidic complex ofgelatin and gellan in deionized water (3), behaviour widelyused in encapsulation technology.
In the absence of favourable interactions, concentratedsolutions of two biopolymers will often immediately becomecloudy, then resolve gradually into two clear layers, eachcontaining most of one polymer and little of the other. Upon

272 IS. Chronakis. S.Kasapis and R.Abeysekera
cooling the mixture before bulk phase separation, compositegels are formed, with the network of the faster gellingbiopolymer forming the continuous, supporting phase andthe second species confined to discontinuous inclusions.Work on a number of biopolymer mixtures of relevance tothe food industry has shown that the thermal regime dictatesthe extent of phase separation, the development ofmechanical properties and the polymer composition at whichphase inversion occurs in the blend (4). Slower cooling rates,e.g. a scan rate of l°/min as opposed to quenching, reducethe overall levels of polymer needed for phase separation in amixture, which can be rationalized on the basis of physicalproperties of the individual components (5).
The gelatin-polysaccharide-water system has attracted theattention of food scientists and it has been demonstrated for>80 mixtures that steric exclusion or heterotypic interactionscould be induced by changes in bulk concentration, pH,temperature and ionic strength (6). At relatively low polymerconcentrations, thermodynamic incompatibility is theoverriding influence in mixtures of gelatin with neutralpolysaccharides. Thus for composite gels of 1%agar and 1%gelatin, light microscopy shows a continuous polysaccharidephase surrounding the protein inclusions, with the systemphase inverting to a protein continuous network at -3%gelatin (7). On the other hand, steric exclusion between thepositively charged gelatin chains (pH below its isoelectricpoint) and an anionic polysaccharide occurs only when theimportant electrostatic interactions between the twopolymers are suppressed. Work on mixed systems of gelatinand gellan, a polysaccharide with a carboxyl group perrepeat sequence, has documented the striking transformationfrom a precipitating coacervate in the absence of salt (3) to aclear composite gel in the presence of sodium cations (addedto stoichiometric levels and beyond), which screen thenegative charge of the gellan chain (8).
Recently, the above reasoning was used to provide abaseline for the development of high solids confectioneryproducts using gelatin-gellan formulations at levels ofco-solute (sucrose plus corn syrup) of 60-85% in the mixture(9,10). Steric incompatibility between the two polymersresulted in a stable, two-phase system and a rubbery gellannetwork was obtained at temperatures close to the boilingpoint, which on subsequent cooling started transforminginto a body with a glass-like consistency. In the present studywe replaced gellan with pectin, a polysaccharide traditionallyused for structuring high solids products (l I). To extend ourknowledge of the effect of polyhydric compounds on themechanical properties of mixed gels, ethylene glycol (EG)was used as the co-solute in part I of this investigation, withpart II being a full account of the work on gelatinpectin-sugar-corn syrup mixtures.
MaterialsandmethodsThe gelatin sample used was kindly supplied by SystemsBio-Industries (Baupte, France). It was the acid pretreatment
product of pigskin with an isoelectric point of pH 8. Stocksolutions (25% polymer) were prepared by soaking thegranules at room temperature overnight and then heating to60°C. Before each experiment, the pH of the single gelatinpreparations (3% protein plus 0-70% EG) was adjusted to 3with 2 mol/dm! HCl. The citrus peel pectin used was acommercial product kindly supplied by Hercules,LilIe-Skensved, Denmark (GENU B). It is a rapid set varietywith a galacturonate content (esterified or nonesterified) of83% and a high degree of esterification (70%). Since thesecommercial materials are 'standardized' to specific gelproperties by blending with sucrose, they were dialysedextensively against distilled water (four changes) andfreeze-dried to produce pure polysaccharide samples. The pHvalue in 1%pectin with 0-80% EG solutions was adjusted to3 (2 mol/dm! HCl). For preparation of binary systems,appropriate amounts of gelatin and pectin solutions weremixed at 75°C followedby addition of EG and the remainingdistilled water to bring the sample to the requiredcomposition (stilI at pH 3).
Differential scanning calorimetry (DSC) experiments werecarried out using a Setaram batch and flow microcalorimeter. Single or mixed polymer preparations withethylene glycolwere weighed directly (-0.8 g) on a pan whichwas then sealed hermetically. A second pan containing thesame amount of EG-water solution was used as a reference.Samples were equilibrated at 5°C for 2 h and then heated to90°C at a scan rate of 0.2°/min.
Small deformation oscillatory measurements were madeusing a parallel plate geometry (40 mm diameter; I mmgap) on a controlled stress Carri-Med CSL 500 rheometer.The hot single or mixed solutions (75°C) were loaded on theplaten of the machine preset at the same temperature. Theexposed edges of samples were covered with a light siliconeoil (50 cs) to prevent evaporation. Temperature wascontrolled by a Pt 100 thermometer in a Peltier device.Network development was monitored on cooling to 5°C at ascan rate of l°/min and frequency of 1.6 Hz (-10 rad/s). Theimposed strain was fixed at 1%. Strain sweeps on a fewselected gelsdemonstrated that the working deformation waswell within the linear viscoelastic region (extending up to20% strain). Cooling was followed by an isothermal scan(5°C) for 2 h and a frequency sweep between 0.01 and 10 Hz.The rheological routine was completed with a heating runfrom 5 to 95°C, thus generating a complete picture of changein storage modulus (G), loss modulus (G') and dynamicviscosity (n").
For the microscopy work, samples were allowed to set upover 12 h at 5°C. Pieces of dimension 10 x 10 x 0.5 mm werecut from the composite gels using a sharp scalpel. Unstainedsamples were placed on a microscope slide and a cover slipwas lowered gently onto the gel surface, excluding anytrapped air, and examined immediately. For the stainedspecimen, a drop of 0.05% w/w aqueous Sirius red (containssulphonic acid groups that can react with basic groups ofcollagen) or Toluidine blue (a metachromatic dye that stains

Mixed gels of gelatin-pectin in the presence of ethylene glycol 273
- 0.2 - 1.4 - 3.6a
~- 0.3 - 1.5
- 3.7S--~ -0.4 - 1.6Q
G:~ - 3.8Q,I
= - 0.5 - 1.7
d- 0.6 - 1.8 - 3.9
10 30 SO 70 10 30 50 10
- 3.4 - 2.6 - 2.4
~· 2.5
§ - 3.6
~ ·2.8 - 2.6;:s - 3.8
= - 2.7
e- 4.0 -3.0 - 2.8
10 30 50 70 10 30 50 10
- 1.6 - 2.0
C
~ - 1.7 - 2.05
E--~- 2.1Q
- 1.8;:....~= - 1.9 - 2.15
- 2.2f
- 2.010 30 50 70 10 30 50 70
Temperature (0C) Temperature (OC)
Figure 1 Differential scanning calorimetry work on: 3% gelatin with 0 (a; left y axis) and 70% (a; right y axis) ethylene glycol; 1% pectin with30 (b; left y axis) and 50% (b; righty axis) EG; 1% pectin with 70% EG(c); and 3% gelatin plus 1% pectin with 30 (d), 50 (e) and 70%(f) EO.Heating scan rate of O.2°/min.
274 I.S. Chronakis. SiKasapis and RAbeysekera
pectin) solution was placed on the upper surface of thecomposite gel. Ten minutes were allowed for staining to takeplace and then the samples were processed like theirunstained counterparts. Images were acquired on TMax 100photographic film using a Nikon inverted microscopeDiaphot-TMD with bright field optics.
Results and discussion
Steric incompatibility versus heterotypic interactions
Mixing of clear gelatin and pectin solutions (pH 3) results ina turbid blend which indicates microscopic phase separationand formation of discontinuous inclusions capable ofscattering visible light. Mixtures remain cloudy uponaddition of ethylene glycol. Centrifugation of 3% gelatin-l%pectin with 40 or 50% EG at 45°C (4600 g; 30 min) results inbulk phase separation with two discrete layers. The bottomlayer is rich in pectin and has a characteristic creamy colour,whereas the top one is relatively colourless, as is typical forgelatin-rich preparations.
DSC (Fig . la) provides thermodynamic information aboutthe melting profiles of gelatin gels prepared in the absenceand maximum level of ethylene glycol (70%). The presence ofco-solute stabilizes the gelatin helices, with the temperatureat maximum heat flow (Tmax) being shifted to 28°C from-23°C for an aqueous gelatin gel. The heating thermogramsof pectin gels denote a less cooperative order-to-disordertransition which proceeds over the majority of theexperimental temperature range (Fig. lb and c). The broadendotherms make it difficult to define accurately themid-point of a transition but there is an apparent shift ofTmax values from -38 and 44 to 49°C at ethylene glycolconcentrations of 30, 50 and 70% respectively. For the same
amounts of co-solute (F ig. ld-f) the endothermic spectra ofmixed gels reproduce in terms of Tmax position and generalband form the sharp deconvolution of gelatin chains and thebroader loss of order seen for individual pectin assemblies athigher temperatures.
To recap, centrifugation shows evidence of phaseseparation in solution, and in the gel state calorimetryindicates that the two polymeric components form separatemolecular associations in the manner observed for the singlepreparations. By contrast, heterotypic interactions betweentwo biopolymers (e.g. in x-carrageenan-konjac mannanblends) have been shown to either generate a newendothermic peak or to distort the characteristic positionand form of the single component transitions (12). A 70%ester pectin has a pKa of 3.6 (13), and on this basis Morrisand co-workers (14) estimated that only 5.6% of the totaluronate residues are ionized at pH 3. Furthermore, thereduction in gel strength at lower or higher degrees ofesterification suggested that 70% is the ideal ester content formaximum stabilization of pectin's interchain junctions. Theabove findings support our experimental evidence ofpolymer exclusion and the ensuing homotypic intermolecular associations at the expense of potentialelectrostatic interactions between gelatin and pectin.
Effect of ethylene glycol on single-component gelatin gels
Recording the mechanical properties of gelatin and pectinnetworks as a function of increasing ethylene glycolconcentration constitutes the most informative baseline forunderstanding the macromolecular organization of themixed system. Although the first priority of this work was toexamine the phase behaviour of blends, the same exercisegives some indications of the nature of the intermolecularinteractions responsible for network formation.
3.5 80 3.5a b
70
3 3
~- 60 - -~ o ~c:l.o °;;:; so '-' •~ 2.5
ll.I 12.SL. •::I
40..
e E •'-' 8-'-'
~ 2 30 ~ 2= e...:l ll.I ...:l •Eo<
20~~1.5 1.5
TEMP. 10 ~c0 1Ie.!Vi 1"1
0 3000 6000 9000 12000 0 10 20 30 40
Time (sec) Temperature ('C)
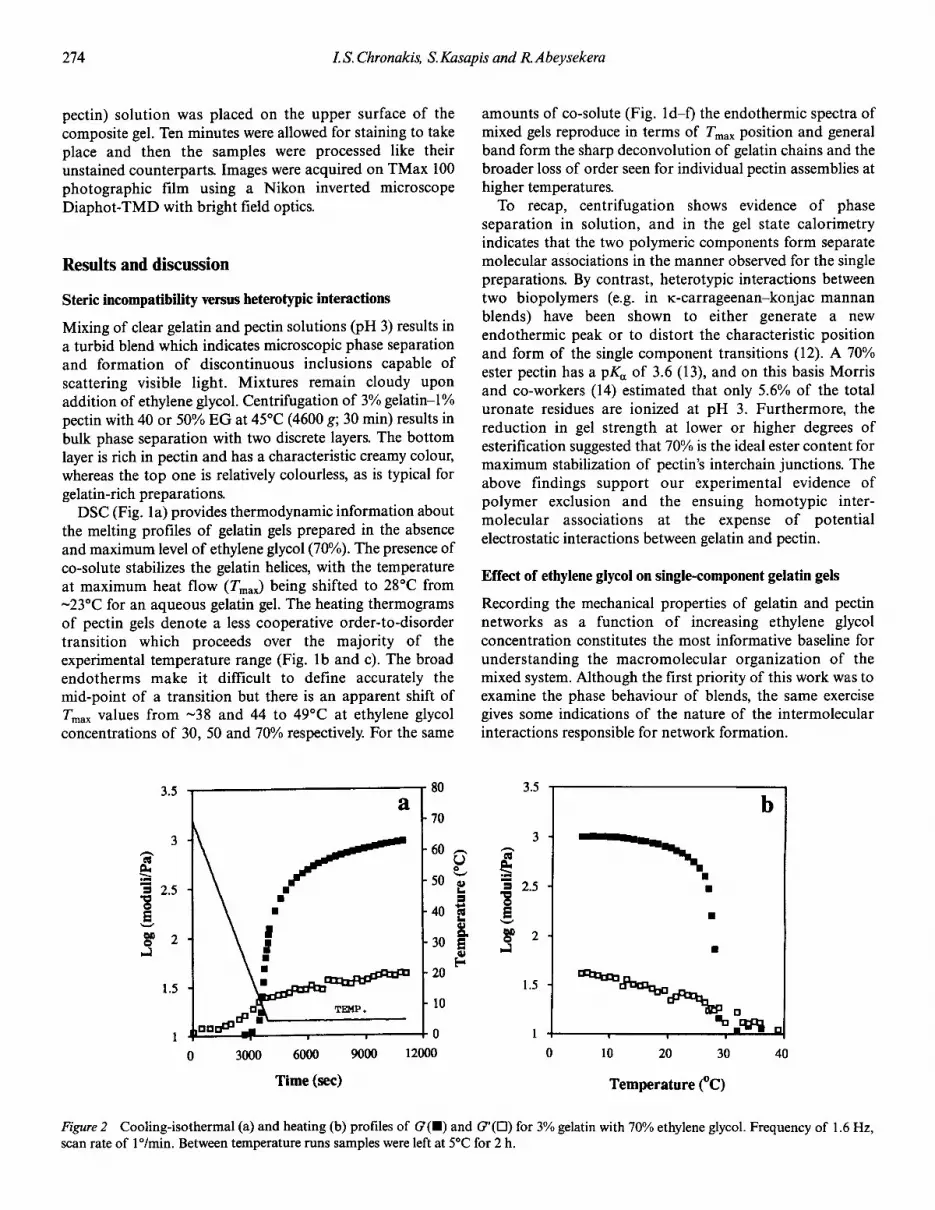
Figure2 Cooling-isothermal (a) and heating (b) profiles of G(.) and G'(O) for 3% gelatin with 70% ethylene glycol. Frequency of 1.6 Hz,scan rate of l°/min. Between temperature runs samples were left at SoCfor 2 h.

Mixed gels ofgelatin-pectin in the presence ofethylene glycol 275
Figure 4 Storage modulus (G) development with increasing levelsof ethylene glycol for 3% gelatin (--), 1% pectin (-), and 3%gelatin plus 1% pectin (.). Values of G were taken at the end of the2 h isothermal sweep (SoC).
•• •• ••••
•• •• •---
o 10 20 30 40 50 60 70 80
Ethylene Glycol (% w/w)
4
3
4.5
2.5
Timasheff, which demonstrated that addition of severaldifferent proteins to an aqueous glycerol solvent makes thepolymer-solvent interactions thermodynamically unfavourable and increases the chemical potential of glycerol (16).They proposed that glycerol enhances the self-association ofprotein molecules by being preferentially excluded from theirimmediate domain. On the basis of the non-specific natureof the aforementioned process, it was suggested that thesame mechanism determines the interactions of a number ofpolyolswith gelatin (17). Therefore, a possible explanation ofthe effect of EG on gelatin is that the co-solute is excludedfrom the domain of the protein, which results in an increasein the chemical potential of ethylene glycol. Consequently,the surface of contact between gelatin and solvent isminimized, the polymer-EG interaction becomes thermodynamically unfavourable and the polypeptide chain foldsincreasingly in the helical form which creates extra junctionzones and results in the initial rise of the storage modulusseen in Figure 4.
Cramer and Truhlar (18), using quantum chemicalconformational analysis, have looked at the prevailing typeof conformer and hydrogen bond in ethylene-glycol-watersolutions. Calculated relative solvation free energies showedthat 92% of the rotamers were in the gauche form and 77%of the hydrogen bonds were internal. They concluded thatthe intramolecular hydrogen bonds found between vicinalhydroxyl groups in the gas phase are only slightly disruptedin aqueous solutions in order to permit additionalintermolecular hydrogen bonding with foreign hydroxylgroups. Obviously, a limited extent of intermolecular
Figure 2a shows combined cooling and isothermal runs for3% gelatin in the presence of 70% ethylene glycol. Networkformation is indicated by a sharp increase in G, the onset ofwhich has been taken as the gelation temperature (tgel).Samples were left at 5°C for 2 h, thus allowing G to approachasymptotically a constant value (i.e. attainment of a'pseudoequilibrium' modulus). The mechanical changes arereversible on heating, albeit shifted to higher tem- peratures,thus producing thermal hysteresis (Fig. 2b). As an index ofnetwork liquefaction, we have considered the temperature atwhich G' >G (tmel)' Figure 3 shows that the onset of a steeprise in G is not affected by increasing amounts of co-solute;tgel remains at 14 ± I"C. Similarly, the temperature oncompletion of structure melting remains constantthroughout the experimental range of EG (tmel is 28 ± I"C).With increasing EG concentration, however, there is initiallyan increase in the G values at the end of the 2 h time sweep,with a substantial fall when the co-solute level rises to >30%w/w. As shown in Figure 4, the storage modulus of gelatingels increases from 1.7 to 2.2 kPa at 0 and 30% EG in theblend, and then it drops to just below I kPa at 70% of theco-solute.
To describe qualitatively the effect of ethylene glycolmolecules on gelatin's supramolecular structure, one maystart with the conception of an infinite network of triplehelicesstabilized by neighbouring water molecules (15). Thenone can consider the densimetric investigation of Gekko and
100
90 ->:80-U 70
"L- 4U5 60...~... 508-!4O 4
30
20~_':~-~~--
10
0
0 10 20 30 40 50 60 70 80 90
Ethylene Glycol (% w/w)
Figure 3 Gelation (tgel) and melting (tmel) temperatures as afunction of ethylene glycol content for: 3% gelatin with tgel and tmel
values at 14 and 28°C respectively (--); 1% pectin with the tgeldepicted as a solid-line curve and the tmel as a long arrow; 3% gelatinplus 1% pectin showing the tgel as (~) and the tmel as (A), with theshort arrow indicating that mixed gels become thermally irreversiblelike their pectin counterparts.
276 IS. Chronakis, S.Kasapis and RAbeysekera
4,-------------...,
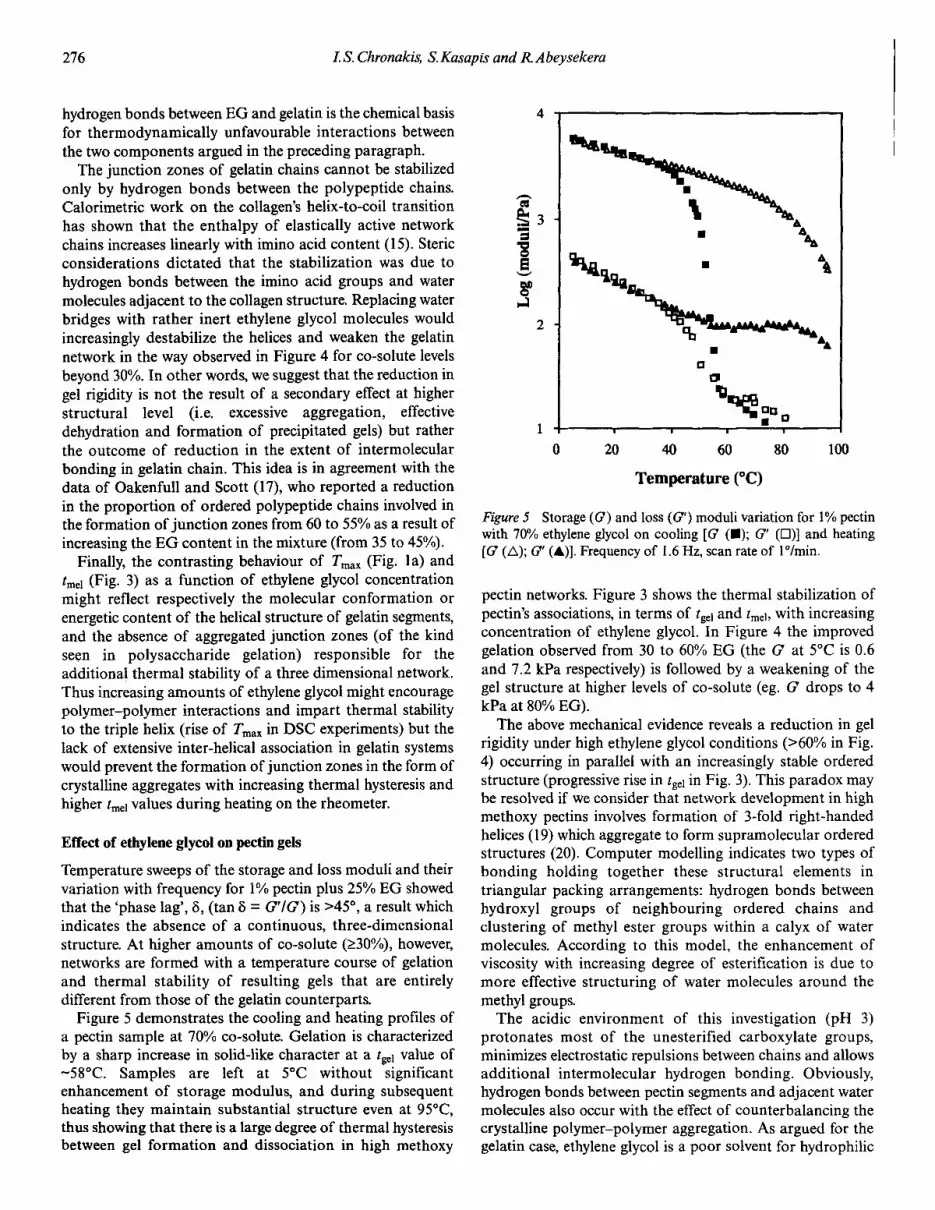
Figure 5 Storage (G) and loss (G') moduli variation for 1% pectinwith 70% ethylene glycol on cooling [G (.); G' (D)] and heating(G (L); G' (A)]. Frequency of 1.6 Hz, scan rate of IOlmin.
tOO8060
•ao~
~aa• a
40
Temperature (DC)
20o
pectin networks. Figure 3 shows the thermal stabilization ofpectin's associations, in terms of tgel and tme!> with increasingconcentration of ethylene glycol. In Figure 4 the improvedgelation observed from 30 to 60% EG (the G at 5°C is 0.6and 7.2 kPa respectively) is followed by a weakening of thegel structure at higher levels of co-solute (eg. G drops to 4kPa at 80% EG).
The above mechanical evidence reveals a reduction in gelrigidity under high ethylene glycol conditions (>60% in Fig.4) occurring in parallel with an increasingly stable orderedstructure (progressiverise in tgel in Fig. 3). This paradox maybe resolved if we consider that network development in highmethoxy pectins involves formation of 3-fold right-handedhelices (19) which aggregate to form supramolecular orderedstructures (20). Computer modelling indicates two types ofbonding holding together these structural elements intriangular packing arrangements: hydrogen bonds betweenhydroxyl groups of neighbouring ordered chains andclustering of methyl ester groups within a calyx of watermolecules. According to this model, the enhancement ofviscosity with increasing degree of esterification is due tomore effective structuring of water molecules around themethyl groups.
The acidic environment of this investigation (pH 3)protonates most of the unesterified carboxylate groups,minimizes electrostatic repulsions between chains and allowsadditional intermolecular hydrogen bonding. Obviously,hydrogen bonds between pectin segments and adjacent watermolecules also occur with the effect of counterbalancing thecrystalline polymer-polymer aggregation. As argued for thegelatin case, ethylene glycol is a poor solvent for hydrophilic
hydrogen bonds between EG and gelatin is the chemical basisfor thermodynamically unfavourable interactions betweenthe two components argued in the preceding paragraph.
The junction zones of gelatin chains cannot be stabilizedonly by hydrogen bonds between the polypeptide chains.Calorimetric work on the collagen's helix-to-coil transitionhas shown that the enthalpy of elastically active networkchains increases linearly with imino acid content (15). Stericconsiderations dictated that the stabilization was due tohydrogen bonds between the imino acid groups and watermolecules adjacent to the collagen structure. Replacing waterbridges with rather inert ethylene glycol molecules wouldincreasingly destabilize the helices and weaken the gelatinnetwork in the way observed in Figure 4 for co-solute levelsbeyond 30%. In other words, we suggest that the reduction ingel rigidity is not the result of a secondary effect at higherstructural level (i.e. excessive aggregation, effectivedehydration and formation of precipitated gels) but ratherthe outcome of reduction in the extent of intermolecularbonding in gelatin chain. This idea is in agreement with thedata of Oakenfull and Scott (17), who reported a reductionin the proportion of ordered polypeptide chains involved inthe formation of junction zones from 60 to 55% as a result ofincreasing the EG content in the mixture (from 35 to 45%).
Finally, the contrasting behaviour of Tmax (Fig. la) andtmel (Fig. 3) as a function of ethylene glycol concentrationmight reflect respectively the molecular conformation orenergetic content of the helical structure of gelatin segments,and the absence of aggregated junction zones (of the kindseen in polysaccharide gelation) responsible for theadditional thermal stability of a three dimensional network.Thus increasing amounts of ethylene glycol might encouragepolymer-polymer interactions and impart thermal stabilityto the triple helix (rise of Tmax in DSC experiments) but thelack of extensive inter-helical association in gelatin systemswould prevent the formation of junction zones in the form ofcrystalline aggregates with increasing thermal hysteresis andhigher tmel values during heating on the rheometer.
Effect of ethylene glycol on pectin gels
Temperature sweeps of the storage and loss moduli and theirvariation with frequency for 1% pectin plus 25% EG showedthat the 'phase lag', 8, (tan 8 =G'IG) is >45°, a result whichindicates the absence of a continuous, three-dimensionalstructure. At higher amounts of co-solute (~30%), however,networks are formed with a temperature course of gelationand thermal stability of resulting gels that are entirelydifferent from those of the gelatin counterparts.
Figure 5 demonstrates the cooling and heating profiles ofa pectin sample at 70% co-solute. Gelation is characterizedby a sharp increase in solid-like character at a tgel value of-58°C. Samples are left at 5°C without significantenhancement of storage modulus, and during subsequentheating they maintain substantial structure even at 95°C,thus showing that there is a large degree of thermal hysteresisbetween gel formation and dissociation in high methoxy

Mixed gels ofgelatin-pectin in the presence of ethylene glycol 277
polymer parts. Thus, the co-solute reduces the water activityand enhances the polymer-polymer hydrogen bonding , withthe concomitant increase in network strength seen for pectingels with an EG content of up to 60% (Fig. 4). However, theexistence of methyl substituents introduces a seconddimension in the effect of ethylene glycol on pectin gelationsince the co-solute is a good solvent for hydrocarbons andrelated hydrophobic assemblies (21). High levels of EG,therefore, will facilitate disruption of the water cages anddissolution of the methyl clusterings which are a requisite ofgel strength, and this is therefore reduced beyond 60%co-solute (Fig. 4). Overall ethylene glycol promotes helix
formation and antagonizes hydrophobic aggregation . Itsdirect involvement in the gelation process of pectin mightexplain the continuous stabilization of the ordered structure,judged by the progressive increase in temperature at which itforms, as compared with the non-specific exclusion withgelatin that generates a constant t gel value.
Phase phenomena in gelatin-pectin-ethyIene glycol gels
The experimental approach of the investigation in singlegelatin and pectin systems was also employed in the phasecharacterization of their mixed gels. Figure 6 reproduces the
3.5 ..- 80 3.5a b
~..... 703 I
,-... 3 •,-... 60 U ,-... •ell 0 ell'-'g., • ~ ~ •~ • 50~ 2.5 • = i 2.5• - •Q • ca::JCP"
,mCCa:J E
~.s .~40 8- s
'-' '-'OIl 2 lID S ~ 2Q 30 <U..J ItI Eo- ..J
C •• c20
~1.5 1.510 ItI
TEMP. lIgi c •0
0 3000 6000 9000 12000 0 10 20 30 40
Time (sec) Temperature ~C)
Figure 6 Cooling-isothermal (a) and heating (b) profiles of G (_) and 0" (D) for 3% gelatin plus 1% pectin with 20% ethylene glycol.Frequency of 1.6 Hz, scan rate of la/min . Between temperature runs samples were left at SoC for 2 h.
4.5 a 80 4.5b
4 --- 70 4 ••••••- 60 - ~ •CIl 3.5 U 3.5 ••••g.,0 •::.
50 '-' ;;;. ••"3 e "3'C 3 'g 3 •Q = Cc •S 40 - S •E ccc •'-' i 2.5 DC~ 2.5 <U •
30Cl. C Cc •..J S ..J cCaa C a •
2 ~ 2 CC~ \20
1.5 10 1.5 ,TEMP.
IlJ0
0 3000 6000 9000 12000 0 20 40 60 80 100
Time (sec) Temperature ~C)
Figure 7 Cooling-isothermal (a) and heating (b) profiles of G (_) and G' (D) for 3% gelatin plus 1% pectin with 70% ethylene glycol.Frequency of 1.6 Hz, scan rate of la/min . Between temperature runs samples were left at 5°C for 2 h.

278 IS. Chronakis, S.Kasapis and R Abeysekera
progression of gelation and melting with decreasing,constant and increasing temperature at 20% ethylene glycol.The temperatures for the onset of the sol-gel process andcompletion of the gel-sol transition (17 and 28°Crespectively) are similar to tgel and tmel values of single gelatinpreparations. Increasing concentrations of co-solute,however, alter dramatically the time-temperature profile ofthe gelatin-pectin blend. As shown in Figure 7a, the sol-geltransition moves to higher temperatures (tgel is 58°C at 70%EG) and a second 'wave' of structure appears at -15°C.During subsequent heating (Fig. 7b) there is a partial loss ofstructure at low temperatures but then a broad endothermicprocess takes over which sees a predominant elasticbehaviour even at the highest temperature available to therheometer (i.e. tmel is >95°C). The immediate conclusionfrom this evidence is that a high ethylene glycol content in theblend promotes the formation of a pectin network andgelation of gelatin at the temperature ranges associated withthe structuring of the individual components. Similarly, thebimodal heating profile comprises a melting step of thethermally metastable gelatin structure and a gradualweakening of the pectin network.
Figures 3 and 4 map out the effectof ethylene glycolon thegelation temperature, melting process and elastic modulus at5°C for our system. At low levelsof co-solute (0-25%), wherepectin shows no evidence of network formation, the tgel andtmel values of the mixtures remain close to those of singlegelatin gels. The rigidity of the blends, however, divergesfrom the mechanical strength of gelatin networks withincreasing ethylene glycol concentration. Taking intoaccount that thermodynamic incompatibility determines thephase behaviour of this mixture, increasing amounts of EGshould promote pectin's conformational ordering whichbecomes more and more effectivein excluding gelatin, hencecreating a more concentrated and stronger proteincontinuous phase. The mechanical analogue of thiscomposite system is that of an isostrain arrangement (5),where a strong continuous phase (gelatin) is penetrated bymuch weaker discontinuous inclusions (pectin).
The structuring and melting characteristics of sampleswith a higher EG content (from 30 to 70%) follow the onsetof gelation and the extent of thermal hysteresis observed forsingle pectin gels. The change in the pattern of tgel and tmel
values in Figure 3 is coincident with a sharp increase in gelstrength from 3.4 to 12.3 kPa occurring at 25 and 30%co-solute respectively (Fig. 4). Obviously, gelation of pectinat 30% EG transforms the filler inclusions of pectin, arguedfor lower levels of co-solute, into a continuous network. Ofcourse, the topology of gelatin in these mixtures comes intoquestion. A continuous gelatin structure that interpenetratesthe pectin network, with both polymers not 'seeing' eachother, will create a type of mixed gel known asinterpenetrating networks (22). In this case, both networksspan the entire system, with each one effectively having aphase volume equal to one. The modulus of the blend shouldbe estimated from the sum of the moduli of the individual
components. Scanning through the data of Figure 4,however, reveals that for our system this is not the case; forexample, the mechanical strength of gelatin, pectin and themixed preparation at 50% EG is 1.5, 6.3 and 15.5 kParespectively. Therefore, thermodynamic incompatibilitybetween the two polymers prevents the formation of twoseparate interpenetrating networks, a result which is inagreement with the steric exclusion phenomena in mixedsolutions of pectin and gelatin, observed in the form ofmicro- and bulk phase separation.
Steric incompatibility in the second range of EG samples(30-70%) may result in phase separation in the form of agelatin filler surrounded by a continuous pectin phase(composite gel) or of a bicontinuous arrangement. In eithercase, each component tends to exclude the other from itsdomain, the solvent is partitioned between the two phasesand the effective concentrations of both polymers are raised.As a result the sum of moduli of the effective concentrations,scaled down by the phase volume of each network, canaccommodate the high values of the storage modulusrecorded for the binary gels of Figure 4. Ouantitativeanalysis of mechanical properties for phase-separatedbiopolymer gels has been attempted in the past (7,23), basedon the isostress and isostrain equations of the blending laws(5) and the concentration-dependence of the modulus of thesingle gelling components (24). In doing so, a computerizedprocedure was devised to assess every possible distributionof water between the two phases. However, the introductionof a fourth component (ethylene glycol) to the analysis willrequire a rather complicated computerization to address theproblem of solvent partition, and a combined polymerconcentration-ethylene glycol-storage modulus relationshipWhich, in our opinion, may prove difficult to achieve and tointerpret.
Qualitatively, our understanding of the macromolecularorganization at high levels of co-solute was refined by usinghistochemical stains to identify the gelatin and pectin phasesin conventional transmission microscopy. Gross differencesin images were detected either as a reduction in the intensityof monochromatic light on passing through optically denseregions of the unstained specimen or by the coloursassociated with different structures when Sirius red(gelatin-specific) and Toluidine blue (which labels pectin)were used. The emerging picture of the microscopy work is asfollows: individual polymer gels under conditions of low orhigh ethylene glycol show homogeneous networks across thewhole sample, which undoubtedly constitute a single phase.Mixed polymer gels at low co-solute concentrations showstructures which are similar to the molecular organizationsof single gelatin-EG preparations. The absence of astructured pectin image is, of course, in accordance with therheological evidence that pectin does not form a gel at levelsof co-solute of <30%.
Greater amounts of co-solute promote gelation of pectin,large assemblies of which are clearly detectable in mixedsystems. Figure 8 shows the pattern of phase separation at

Mixed gels ofgelatin-pectin in the presence of ethylene glycol 279
Figure 8 Light microscopy picture of a mixed gel at 3% gelatin plus1% pectin and 60% ethylene glycol. The magnification is x200 .
60% ethylene glycol, with the Toluidine blue staining theirregularly shaped particle-like structures of pectin (with anaverage size of Sum). The unstained continuous backgroundis due to the gelatin structure. Treatment of the sample withonly Sirius red stains this continuous phase exclusively.Therefore, the light microscopy observations strongly suggestthat there is a continuous gelatin network in addition to thethree-dimensional structure of pectin detected from themechanical measurements. Furthermore, we have learnt byexperience that composite systems of a well defined proteinfiller and a continuous polysaccharide assembly, that bindclearly to specific stains, should show an intense stainingcontrast. However, in the case of gelatin and pectin thecolour contrast across the sample, from one region toanother, was clearly quite faint, supporting the argument fortwo closely intertwined networks in the form of abicontinuous system.
Acknowledgements
We are grateful to Dr Claus Rolin/Copenhagen PectinDivision of Hercules Inc. for providing analytical values forthe citrus pectin samples, and to our colleagues Prof.E.R.Morris and Dr M.WN.Hember for stimulatingdiscussions.
References
1. Jones,R. (1995) Physics World, March, 47-51.2. Ohashi,S., Ura ,F., Takeuchi,M., Iida,H., Sakaue,K.,
Ochi,T., Ukai,S. and Hiramatsu.K. (1990) FoodHydro coll., 4, 323-333 .
3. Chilvers,G.R. and Morris,Y.l (1987) Carbohydr. Polym.,7, 111-119.
4. Kasapis,S., Alevisopoulos,S., Abeysekera,R., Manoj,P.,Chronakis,I.S. and Papageorgiou,M. (1996) InPhillips,G.O., Wedlock,D.l and Williams,P.A. (eds),Gums and Stabilisers for the Food Industry, 8. OxfordUniversity Press, Oxford, pp. 195-206.
5. Takayanagi,M., Harima,H. and Iwata,Y. (1963) Mem.Fac. Eng. Kyusha Univ. , 23, 1-13 .
6. Tolstoguzov.Vb. (1990) In Phillips,G.O., Wedlock,D.land Williams,P.A. (eds), Gums and Stabilisers for the FoodIndustry, 5. Oxford University Press, Oxford, pp.157-175.
7. Clark,A.H., Richardson,R.K., Ross-Murphy,S.B. andStubbs,lM. (1983) Macromolecules, 16, 1367-1374.
8. Papageorgiou,M., Kasapis,S. and Richardson,R.K.(1994) Food Hydro coli., 8, 97-112.
9. Papageorgiou,M. and Kasapis,S. (1995) Food Hydrocoll.,9,211-220.
10. Papageorgiou,M., Kasapis,S. and Richardson,R.K.(1994) Carbohydr. Polym., 25, 101-109.
11. Rolin,C. (1994) The European Food and Drink Review,Autumn, 61-65.
12. Williams, P.A., Clegg,S.M., Langdon,M.l, Nishinari,K.and Piculell,L. (1993) Macromolecules, 26,5441-5446.
13. Plaschina,I.G. , Braudo,E.E. and Tolstoguzov.VB. (1978)Carbohydr. Res. , 60, 1-8.
14. Morris,E.R., Gidley,M.l, Murray,E.l, Powell,D.A. andRees,D.A. (1980) Int. 1 Bioi. Macromol., 2, 327-330.
15. Privalov,P.L. and Tiktopulo,E.I. (1970) Biopolymers, 9,127-139.
16. Gekko,K. and Timasheff,S.N. (1981) Biochemistry, 20,4667-4676.
17. Oakenfull,D. and Scott,A. (1986) Food Hydrocoll., 1,163-175.
18. Cramer,C.l and Truhlar,D.G. (1994) 1 Am. Chem. Soc.,116, 3892-3900.
19. Walkinshaw,M.D. andArnott,S. (1981) 1 Mol. Bioi., 153,1075-1085.
20. Rolin,C. (1993) In Whistler,R.L. and BeMiller,lN. (eds),Industrial Gums. Academic Press, London, pp. 257-293.
21. Tanford,C. (1980) The Hydrophobic Effect. Formation ofMicelles and Biological Membranes. 1 Wiley and SonsInc., New York.
22. Morris,V.I (1986) In Phillips,G.O., Wedlock,D.l andWilliams,P.A. (eds), Gums and Stabilisers for the FoodIndustry, 3. Elsevier, London, pp. 87-99.
23. Kasapis,S. (1995) In Harding,S.E., Hill,S.E. andMitchell,lR. (eds), Biopolymer Mixtures. NottinghamUniversity Press, Nottingham, pp. 193-224.
24. Ross-Murphy,S.B. and McEvoy,H. (1986) Br. Biopolym.1 ,18,2-7.
Accepted on December 4, 1995