spectroelectrochemical properties of poly(4,4'-dialkyl-2,2'-bithiophenes) and poly(...
TRANSCRIPT

Spectroelectrochemical properties of poly( 4,4’~di~yl~~,2’-bit~ophenes) and poly( 3-~~ylt~ophe~e~)
~~~y~aw bpk~wski a, Malgorzata Zagbrska b, Irma Kulszewicz-Bajer b, Krzysztof KozieX a and Adam Proh ’
’ Institute of Inorganic Chemistry and Technology Siiesian Technical Universi& 44-100 Gliwice (Poland) b Department of Chemistry, Technical University of Warsaw, W-664 Warszawa, Noakowskiego 3 (Paland) ’ Department of Materials and Science and Ceramics, Act&my of Mining and Metallurgy, 30-059 Krakcjw, Mickiewiczu 30 (Fu&md)
fRex.zeiv~ 8 October 1990; in revised form 18 January 1991)
Abstract
The infhrenee of alkyd side chain ~~~budo~ patterns on the s~~~~~~r~~~ prqerties of ~l~~kylt~ophe~~s) has been studied. Two types of compounds were studied as representing different coupling patterns:
(i) poly(4,4’-dialkyl-2,2’-bithiophenes), which represent head-to-head and tail-totail coupled poly(aL kylthiophene) chains; and
(ii) poly(3-alkylthiophenes), which represent head-to-tail coupled poly(alkyhhiophene) chains. Both types of polymers can be obtained chemically or electroehemically by the constant potential
method or the potential soanning method. Poly(4,4’-di~ky~~~~2’“bit~ophenes) differ si~fic~~y from ~iy~3-~~lt~oph~~) in their voltammetric and spectroeIe&rochemical properties. The otidative doping of these compounds occws in a very murow potential range (35 mV as determined by static s~tr~l~tr~he~~ studies) and is ~~f~~~y retarded in dynamic movements. Analysis of the changes in absorption in the 500 nm spectral reejan and the observation of an indu~ion period in the oxidation at constant potenti& seem to indicate that the oxidative doping is preceded by structural changes which facilitate the oxidation.
Atthough in the neutral state pol~4,4’-d~~y~-2,2’-bi~opha~~) absorb at 390 nm, i.e. their s -+ s * transition is blue-shifted as compared to poly(3-alkyhhiophenes), the polaronic states included by doping are located in the same spectral region for both families of p01ymrs. This observation is consistent with the postulate of significant changes induced by doping in poly(4,4’-dialkyl-2,2’-bitbiophenes). The more twisted head-to-head and tail-to-tail coupled poly(alkylthiophene) chains must adopt a more planar structure, similar to poly(%alkyltbiophenes), in order to create polaronic states and accommodate the charge-compensatirtg anions.

58
INTRODUCTION
Poly(heterocyclic) polymers have been the subject of significant research interest in recent years mainly due to their unique electrochemical and electrical properties [l]. Unfortunately, unsubstituted rigid chain polypyrrole and polythiophene are insoluble and infusible. These severe disadvantages make their characterization more difficult and also impede their possible application.
However, in the case of polythiophenes it has been demonstrated that substitu- tion of hydrogen at position 3 by a long alkyl chain substituent leads to polymers which are soluble but retain their electrochemical activity [2].
Soluble poly(3alkylthiophenes) exhibit interesting optical properties. They are thermochromic [3] and solvatochromic [4] in their reduced state. In addition, their oxidation leads to important spectral changes induced by the creation of different charge storage configurations such as polarons or bipolarons [5]. Since the oxidation state of poly(3alkylthiophene) can be controlled electrochemically very conveni- ently, these polymers are very well suited for spectroelectrochemical studies.
One should be aware that the introduction of substituents into the rigid backbone of conjugated polymers may introduce steric effects, non-existent in unsubstituted polymers, which may influence the conjugation length and thus lead to significant changes in the special properties. In particular, depending on the symmetry of the substrate used for the polymerization, a more or less stereoregular chain can be obtained. It has been demonstrated recently by ‘H- and 13C-NMR spectroscopy that oxidative polymerization of 3-substituted thiophenes results in predominantly head- to-tail coupled chains but some coupling defects (head-to-head, tail-to-tail) lowering the regularity of the chain were also detected [6,7].
On the other hand, the application of symmetrically distributed bithiophenes (i.e.
a
150 IJA
b
Fig. 1. Electrochemical polymerization of 3-hexylthiophene (a) and 4.4’~dihexyl-2,2’-bithiophene by potential scanning. Scan rate: 20 mV/s.

59
either 3,3’-dialkyl-2,2’-bithiophenes or 4,4’-dialkyl-2,2’-bithiophenes) for the oxida- tive polymerization should lead to “head-to-head” and “ tail-to-tail” coupled al- kylthiophene rings with better stereoregularity, since in these cases the 5- and 5’-coupling are equivalent [6,7]. Of course, the distribution pattern of the alkyl substituents along the polymer backbone must lead to some changes in the conjuga- tion length and consequently in the optical properties. Indeed, poly(4,4’-dialkyl- 2,2’-bithiophenes) exhibit different electronic spectra from those of poly(3-al- kylthiophenes), In the reduced (neutral) state, the maximum of their absorption peak is shifted towards higher energies, indicating lower conjugation due to the close vicinity of alkyl substituents in the head-to-head coupled rings [6]. These coupling-pattern induced, significant differences between poly(4,4’-dialkyl-2,2’-bi- thiophenes) and poly(3-alkylthiophenes) are further reflected in cyclic voltammetry [8] and in the electrochemical spin response [9]. The oxidation peaks and spin creation peaks are, in the case of poly(4,4’-dialkyl-2,2’-bithiophenes), much sharper and shifted towards higher potentials.
In this paper we present the results of detailed voltammetric and spectroelectro- chemical studies of two poly(4,4’-dialkyl-2,2’-bithiophenes) and the corresponding poly(3alkylthiophenes) trying to expose the similarities and differences between these two groups of conducting polymers.
EXPERIMENTAL
Synthesis of monomers 3-Butylthiophene and 3-hexylthiophene:
R
ii / \
S
were prepared from 3-bromothiophene by Grignard coupling with alkylmagnesium bromide in the presence of Ni(dppp)Cl, catalyst [lo]. 4,4’-Dibutyl-2,2’-bithiophene and 4,4’-dihexyl-2,2’-bithiophene:
were prepared from the corresponding 3-alkylthiophenes by metallation with butyl- lithium in the presence of N, N, N ‘, N ‘-tetramethylethylenediamine, which was added to ensure lithiation in position 5 followed by oxidative coupling with CuCl, at -78°C [ll].

Chemical polymerizations Chemical polymerizations were carried out using a modification of the method of
Sugimoto et al. [12] with FeCl, as oxidant. In particular, for the polymerization of 3-aIkylthiophene the procedure described in ref. 13 was followed, while for 4,4’-dial- kyl-2,2’-bithiophene the preparation presented in ref. 6 was used.
Electrochemical polymerizations Electrochemical polymerizations were carried out in a dry argon atmosphere
using a three-electrode cell with 1 cm’ Pt sheets as the working and counter electrodes and a Luggin capiflary Ag/AgCl/KCl(sat) reference electrode. The anodic and cathodic parts were separated by a frit. All loading operations were carried out in an argon flow. Electropolymerizations were performed either at constant potential or using potential scanning in 0.2 M LiCIO,/acetonitrile electro- lyte. The concentrations of the 3alkylthiophenes and 4,4’-dialkyl-2,2’-bithiophenes were ca. 0.1 and 5 x 10d2 M, respectively.
Cyclic voltammetry studies Cyclic voltammetry measurements were carried out in the same electrochemical
cell as that used for electropolymerization using the same molarity of electrolyte. Chemically prepared films were deposited on the electrode surface by casting from chlorofo~ solutions. El~tr~he~c~ly prepared films were used as grown. They were rinsed repeatedly with pure acetonitrile to remove traces of monomer.
Spectroelectrochemical studies Spectroelectrochemical measurements were carried out in a classical sandwich-
type cell with a Sb-doped SnOz optically transparent electrode. Again the polymer films were deposited on the electrode using an electropolymerization procedure identical to that described above or, in the case of chemically polymerized samples, by casting from chloroform solutions. The spectra were recorded on a Specord UV-Vis (Carl Zeiss Jena) spectrometer.
RESULT!3 AND DISCUSSION
Electropolymerization is a convenient method for the preparation of good quality thin polymer films suitable for spectroelectrochemical studies. In general, three methods can be applied:
(i) the constant current method; (ii) the constant potential method; and (iii) successive layer deposition by potential scanning. Although the constant current method is very popular and has been used
frequently for the polymerization of 3alkylthiophenes [14] it has a significant disadvantage associated with the increase of potential (sometimes uncontrollable) at the end of the pol~e~ation. Since strict control of the potential is crucial for the preparation of structurally well-defined films with reproducible properties [15], the

61
other two methods are favoured. However, in the polymerization of alkylthiophenes that are soluble in acetonitrile, oligomeric products are formed which consume a significant portion of the charge involved in the polymerization. Thus, direct control of the film thickness by coulometry is not very easy. From this point of view the scanning potential method is the most convenient:
(i) it does not lead to overoxidation due to an uncontrollable potential increase as in the constant potential method, and
(ii) the thickness of the film can be determined on the basis of the integrated intensity of the redox peaks associated with the polymer, since the contribution of soluble oligomeric species to this redox couple is negligible.
Constant potential as well as potential scanning methods can be applied to 3alkylthiophenes as well as to 4,4’-dialkyl-2,2’-bithiophenes. In Fig. lb, the poten- tial scanning polymerization of 4,4’-dihexyl-2,2’-bithiophene is presented, whereas in Fig. la the same polymerization of 3-hexylthiophene is shown. A regular step-wise increase of the film thickness is observed after each voltammetric cycle. As has already been stated, oxidative coupling of the former compound leads to head-to-head and tail-to-tail coupled poly(alkylthiophene) chains:
)&g-J&Q R R R
while the latter gives a polymer which is predominantly head-to-tail coupled:
R R R
The geometry of the chains depicted above requires a more detailed discussion. Substituted polythiophenes are poorly crystalline and their structure cannot be elucidated by conventional X-ray diffraction methods. In principle, two chain geometries can be envisaged: (i) a linear structure with alkyl substituents in the truns configuration as depicted schematically above, and (ii) a helical structure with alkyl substituents sticking out of the helix. The crystal structure of the dimers used for the polymerization of poly(4,4’-dialkyl-2,2’-bithiophenes) seems to favour the linear polymer chain. For long substituents such as, for example, the decyl group, the symmetrically substituted dimer is a solid at room temperature which can be easily obtained in the form of single crystals suitable for a detailed X-ray diffraction study. 4,4’-Didecyl-2,2’-bithiophene has a monoclinic structure (space group C,,,) with two sulphur atoms and two alkyl chains in trans configurations and an

62
inversion centre in the middle of the C,-C,? bond. The molecule is planar [16]. It therefore seems reasonable to postulate that 5-5’ coupling of the dimer will lead to a chain with a similar trans configuration. The trans configuration is consistent with CP MAS 13C-NMR results [17]. However, the close vicinity of the alkyl chains in the “head-to-head” coupled alkylthiophene rings must include a significant torsion angle since the rr + Q * transition occurs at 390 run, i.e. it is blue-shifted by ca. 100 nm with respect to poly(3-alkylthiophenes) [6].
Several differences in the polymerization are evident. The onset of polymeriza- tion in the case of 4,4’-dihexyl-2,2’-bithiophene is shifted towards lower potentials by ca. 300 mV as compared to 3-hexylthiophene. It is consistent with the thiophene polymerization mechanism [18], according to which, chain growth is initiated by oxidation of the monomer to the radical cation. Since the potential of this oxidation decreases with the number of rings, i.e. it is lower for dimers than it is for monomers, the use of dimers enables us to carry out the polymerization under milder conditions. In addition, this study confirms that this phenomenon is little dependent on the distribution of the alkyl groups, i.e. a decrease of the oxidation potential, as compared to 3-alkylthiophenes, occurs for 4,4-dialkyl-bithiophenes. The redox couple characteristics of poly(4,4’-dihexyl-2,2’-bithiophene) and poly(3- hexylthiophene) are seen clearly in both figures. In poly(3-hexylthiophene), the oxidation peak is broad and overlaps with the onset of the polymerization peak. On the contrary, the oxidation peak of poly(4,4’-dihexyl-2,2’-bithiophene) is very nar- row and well separated from the polymerization peak by a capacitive plateau usually observed for conducting polymers.
The cyclic voltammograms of the electrochemically prepared poly(4,4’-dialkyl- 2,2’-bithiophenes) obtained in the absence of the monomer as well as the voltammo- grams of chemically prepared polymers are presented in Figs. 2 and 3. For
I a b
115 /JA 116 pA
0
Fig. 2. Cyclic voltammograms of chemically (a) and electrochemically (b) synthesized poly(4,4’-dibutyl- 2,2’-bithiophene). Scan rate: 20 mV/s.
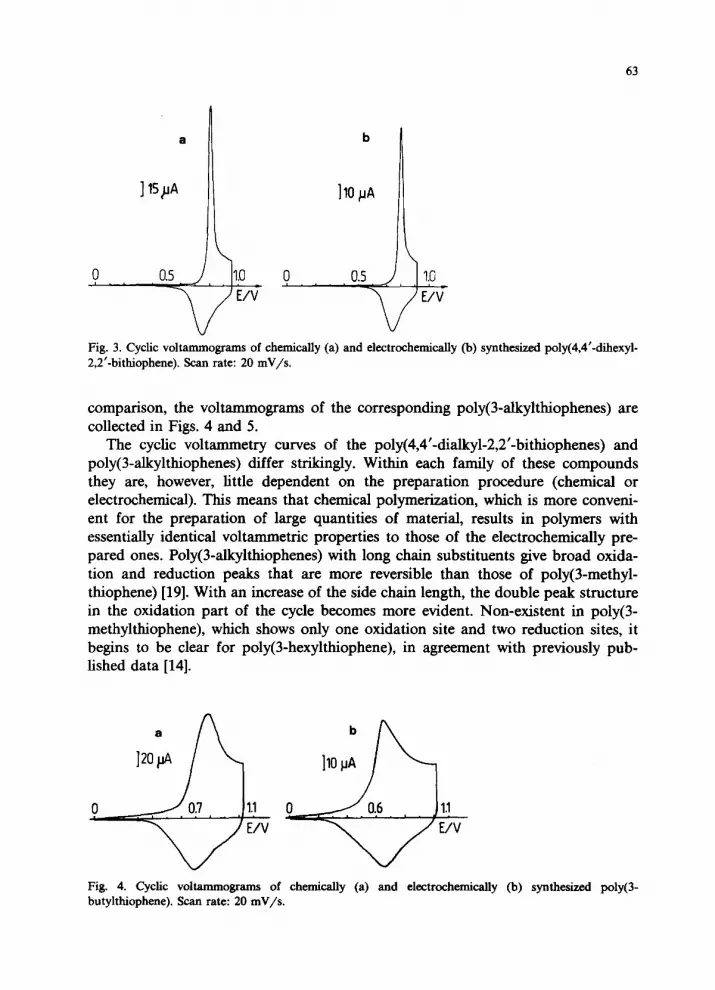
63
b
Fig. 3. Cyclic voltammograms of chemically (a) and electrochemically (b) synthesized poly(4,4’-dihexyl- 2,2’-bithiophene). Scan rate: 20 mV/s.
comparison, the voltammograms of the corresponding poly(3-akylthiophenes) are collected in Figs. 4 and 5.
The cyclic voltammetry curves of the poly(4,4’-dialkyl-2,2’-bithiophenes) and poly(3alkylthiophenes) differ strikingly. Within each family of these compounds they are, however, little dependent on the preparation procedure (chemical or electrochemical). This means that chemical polymerization, which is more conveni- ent for the preparation of large quantities of material, results in polymers with essentially identical voltammetric properties to those of the electrochemically pre- pared ones. Poly(3alkylthiophenes) with long chain substituents give broad oxida- tion and reduction peaks that are more reversible than those of poly(3-methyl- thiophene) [19]. With an increase of the side chain length, the double peak structure in the oxidation part of the cycle becomes more evident. Non-existent in poly(3- methylthiophene), which shows only one oxidation site and two reduction sites, it begins to be clear for poly(3-hexylthiophene), in agreement with previously pub- lished data [14].
Fig. 4. Cyclic voltammograms of chemically (a) and electrochernically (b) synthesized poly(3- butylthiophene). Scan rate: 20 mV/s.

1.l
E/V
Fig. 5. Cyclic voltammograms of chemically (a) and electrochemically (b) synthesized poly(3- hexylthiophene). Scan rate: 20 mV/s.
Poly(4,4’-dialkyl-2,2’-bithiophenes) do not reveal such a distinct double peak structure either in the oxidation or in the reduction part of the cycle. In fact, poly(4,4’-dialkyl-2,2’-bithiophenes) give the narrowest oxidation peaks of all the conducting polymers studied to date, the half width of which is smaller than that predicted theoretically by Feldberg [20].
The position of the oxidation peak is strongly dependent on the potential scan rate, whereas the reduction peak remains practically at the same position for all the scanning rates studied (Fig. 6b). In the case of poly(3alkylthiophenes), the scarming rate-induced shift of the peak position is much smaller (Fig. 6a). The extremely small half-width of the peak and its strong dependence on the scanning rate seem to indicate that in poly(4,4’-dialkyl-2,2’-bithiophenes) the oxidation is retarded, prob- ably for structural reasons. As has already been stated, the oxidation of conjugated
Fig. 6. Cyclic voltammograms of electrochemically synthesized poly(3-hexylthiophene) (a) and poly(4,4’- dihexyl-2,2’-bithiophene) (b).

65
polymers results in a gradual decrease of the B --) rr * transition intensity with the simultaneous creation of a new band associated with polaronic states. One can therefore monitor the oxidation of the polymer spectroscopically by recording the decrease of the a --, II* band intensity and the growth of the polaronic states band. The spectroelectrochemical curves corresponding to the oxidation of poly(3-al- kylthiophene) and poly(4,4’-dialkyl-2,2’-bithiophenes) are presented in Figs. 7a and 7b, respectively. Again it is clear that the oxidation-induced spectral changes in poly(4,4’-dialkyl-2,2’-bithiophenes) occur in a very narrow potential range: the most drastic decrease of the 390 nm band intensity takes place within 35 mV potential range (from 725 to 760 mv). It should be noted that the initiation of the oxidation process is preceded by some structural changes in the polymer since the absorption in the 490-540 nm range for the lightly doped polymer is lower than that for the neutral one (compare the spectroelectrochemical curves in Fig. 7b for the 0 mV potential and the 725 mV one). This behaviour cannot be associated with the chain oxidation, since in conjugated polymers it always results in an increase in the absorption in this spectral range; thus, the change in the extinction coefficient leading to lower absorption must be associated with structural changes rather than with the oxidation. In the beginning, an isosbestic point around 480 run is present, indicating the co-existence of only two optically different phases. For higher oxidation states (above a potential of 775 mv), departure from the isosbestic point takes place, leading to more pronounced “metallic” behaviour. Comparison of the spectroelectrochemical behaviour with cyclic voltammetry shows that, in the spec- troelectrochemical cell, the oxidation of the polymer starts well below the onset of the cyclic voltammetry peak. Since the spectroscopic studies were performed in a “quasi-static” manner (each point was recorded after stabilization of the absorp-
ABS.
Fig. 7. Electronic spectra of poly(3-butylthiophene) (a) recorded at the following potentials: 0.0, 0.45, 0.50, 0.55, 0.60, 0.65, 0.70, 0.75, 0.80 and 0.85 V; and of poly(4,4’-dibutyl-2,2’-bithiophene) (b) recorded at the potentials 0.0, 0.725, 0.74, 0.75, 0.76, 0.725 and 0.80 V.

66
tion), it can be concluded that the redox potential is shifted towards a si~fi~~y lower value in relation to the voltammetric data. This observation is consistent with the previously observed sensitivity of the peak position to the potential scanning rate. In the case of poly(3-alkyl-thiophenes), the oxidation-induced spectral changes occur over a much wider potential range consistent with the observed cyclic voltammetry features. The results are essentially the same as those reported previ- ously for this family of compounds [21-231.
Poly(4,4’-dialkyl-2,2’-bithiophenes) and poly(3-alkylthiophenes) differ strikingly in colour, the former being bright yellow while the latter are deep red in the solid state. The ?T + n * absorption band in poly(4,4’-dialkyl-2,2’-bithiophenes) is located at 390 nm, i.e. it is blue-shifted with respect to the corresponding band of poly(3-~yl~ophenes). This difference can be rationalized on the basis of steric reasons. The close vicinity of the alkyl groups in head-to-head coupled thiophene rings must induce significant torsion of the adjacent rings which in turn lowers the conjugation. The oxidative doping of both “head-to-tail” and “head-to-head” coupled polymers gives rise to an unsymmetrical band which starts at ca. 530 run, reaches its maximum around 790 nm and extends to the near-infrared [6,7,24]. It should be stressed that although the Q + 1~ * absorptions of both types of polymers are separated by ca. 100 nm and exhibit different half-widths, the doping-induced bands exhibit similar widths and their maxima are located essentially at the same position. This means that the chain structure and band structure after doping must be similar in both cases. It may therefore be postulated that in poly(4,4’-dialkyl- 2,2’-bit~ophenes), structural re~~gern~ts of the chains towards higher planarity must occur before the oxidation. The closer si~l~ty between poly(4,4’-di~yl- 2,2’-bithiophenes) and poly(3-~ylt~ophenes) in the oxidized state is also mani- fested by similar conductivities for the corresponding oxidation levels. It is difficult to determine precisely the conductivities of thin films deposited on IT0 electrodes. However, free standing films of both types of polymers, after doping, exhibit conductivities in the range 3-5 S cm-’ depending on minor differences in sample preparation and doping.
The fact that the oxidation of the poly(4,4’-dialkyl-2,2’-bithiophene) chains is blocked by the necessity of structural rearrangements preceding the oxidation is evident from cbronoabsorptiometric studies performed at constant potential. Such studies were carried out for several potentials covering the range of the cyclic voltammetry peak. In particular, the changes in intensity of the a * ?z * band were recorded and taken as a measure of the oxidation rate. The results are summarized in Fig. 8. At all the potentials studied, the oxidation starts after an induction period which decreases with increasing potential but exists even for the highest potentials which coincide with the oxidation peak. For the oxidation potential of 725 mV, this induction period lasts 220 s. The existence of an induction period is consistent with the postulate that the oxidation of the polymer chains is retarded due to the necessity of structural rearrangements. It must be stressed that no such induction period is observed in the chronoabsorptiometric curves of poly(3-alkylthiophenes).
In Figs. 9a and 9b the voltabsorptiometric curves obtained for poly(3-

67
Time/s
Fig. 8. ~on~bso~tiomet~~ curves of ~1~4,4’-dibutyl-2,2’-bit~ophene) at h = 390 mn the following potentials: (a) 725; (b) 740; {c) 750; (d) 760; (e) 770; (f) 780 and (g) 800 mV.
recorded at
butylthiophene) and poly(4,4’-dibutyl-2,2’-bithiophene) are presented together with the corresponding cyclic voltammograms. In the case of poly(4,4’-dialkyl-2,2’-bi- thiophenes), two wavelengths were selected: the maximum of the IT + VT * transition (A = 390 nm) and X = 530 run. The latter was taken from the spectral region in which the changes in the extinction coefficient at the beginning of the oxidation are evident (see above and Fig. 7). In the dynamic measurement the changes of the extinction coefficient can also be seen, since the absorption at 530 nm begins to decrease at the onset of the oxidation peak, despite the fact that it should increase with the oxidation level. This observation ad~tion~y corroborates the structural changes of polymer chains preceding the oxidative doping. The bleaching of the T -+ P* transition coincides with the abrupt increase of the absorption at 530 nm. In fact, these two sharp changes can be correlated with the second oxidation peak
b
a ,.---,___&60 nm I , --_::>_
8’ 8’ ,..390 nm ,/’
: ,,I’ :
Fig. 9. Cyclic voltammetric (solid lines) and voltabsorptiometric (------, + + f +) curves of poly(3- butylthiophene) (a) and poly(4,4’-dibutyl-2,2’-bithiophene) (b) recorded at an optically transparent electrode. Absorbance changes are inversed on the plot. Scan rate: 5 mV/s.

68
which appears in the cyclic vol~o~am after a few cycles, It is plausible that the first sharp peak is associated with changes in the double-layer capacity induced by the structural rearrangement mentioned above. Indeed, the external double-layer capacitance derived from impedance measurements reveals a sharp peak at the same potential [25].
Sharp current peaks due to an abrupt change of the double-layer capacity have previously been observed for small organic molecules adsorbed on an electrode surface which had to undergo reorientation in order to facilitate the electrode reaction [26,27]. The evolution towards a double peak structure is not so pro- nounced in poly(4,4’-dialkyl-2,2’-bithiophenes) with longer substituents, where the higher potential component is present only in the form of d&symmetric broadening of the peak. In poly(3-~lt~ophenes), the bleaching of the Q + W* band follows the cyclic voltammetry curve (Fig. 9a) and of course occurs over a si~fi~~tly broader potential range.
The oxidation of poly(3alkylthiophenes) and poly(4,4’-dialkyl-2,2’-bithiophenes) is a heterogeneous reaction involving the diffusion of the compensating anions in the solid matrix. In the most general manner, its kinetics can be described by the following equations [28]:
k,=k” exp[-arnF/RT(E-E”)]
k,=k” exp[((l-(Y)nF/RT)(E-E”)]
where k, is the rate constant of the forward elementary process. k, is the rate constant ,of the backward elementary process. k o is the standard rate constant, a: is the transfer coefficient, n is the number of electrons, and E * is the formal potential of the electrode reaction.
One can postulate that the standard reaction rate constant k” can be strongly dependent on the geometry of the polymer chain, in particular being very low for high torsion angles between the adjacent rings. The conformation changes of the chain at the beginning of oxidation result in an abrupt increase of the reaction rate constant k ’ and in the next step the whole chain is quickly doped. Thus, the doping proceeds chain by chain. Detailed kinetic studies confirming this model are in progress.
CONCLUSIONS
To summarize, we have studied the influence of the alkyl side chain distribution pattern on the spectroelectrochemical properties of soluble poly(alkylthiophenes). In particular, we found that poly(4,4’-dialkyl-2,2’-bithiophenes) which represent head- to-head and tail-to-tail coupled alkylthiophene rings differ significantly from head- to-tail coupled poly(3-alkylthiophenes). Spectroelectrochemical and cyclic voltam- metry studies indicated that in poly(4,4’-dialkyl-2,2’-bithiophenes) the oxidation is strongly retarded but when initiated it occurs in a very narrow potential range.
The n -+ v * transition in poly(4,4’-dialkyl-2,2’-bithiophenes) is blue-shifted as

69
compared to poly(3alkylthiophenes), indicating a higher torsion angle between adjacent thiophene rings. However, the oxidative doping-induced polaronic states give rise to absorption bands in the same spectral range as that in poly(3-al- kylthiophenes). This seems to indicate that poly(4,4’-dialkyl-2,2’-bithiophenes) must undergo significant conformational changes towards higher planarity which force the chain structure to become similar to that of doped poly(3alkylthiophenes). Dynamic and static spectroelectrochemical measurements indicated that these con- formation changes precede the oxidative doping.
ACKNOWLEDGEMENTS
This work was supported financially by CPBP 01. 15. grant (the spectroelectro- chemical part) and CPBP 01. 14. (the synthetic part).
REFERENCES
1 Proceedings ICSM, Santa Fe, 1988, Synth. Met., 27-29 (1989).
2 R.L. Elsenbaumer, K.Y. Jen and R. Obodi, Synth. Met., 15 (1986) 169; A.O. Patil, Y. Kkenoue, F.
Wudl and A. Heeger, J. Am. Chem. Sot., 109 (1987) 1858.
3 K. Yoshino, S. Nakajima, D.H. Park and R.-I. Sugimoto, Jpn. J. Appl. Phys., 27 (1988) L454; M.J.
Winokur, D. Spiegel, Y. Kim, S. Hotta and A.J. Heeger, Synth. Met., 28 (1989) C419; W.R. SaIaneck,
0. Inganas, J.-O. Nilsson, J.-E. Osterholm, B. Thtmans and J.-L. Bredas, ibid., 28 (1989) C451.
4 S.D.D.V. Rughooputb, S. Hotta, A.J. Heeger and F. Wudl, J. Polym. Sci., Polym. Phys. Ed., 25 (1987)
1071; 0. Inganas, W.R. SaIaneck, J.-E. Osterholm and J. Laaksa, Synth. Met., 22 (1988) 395.
5 M. Colneri, M. Nowak, D. Spiegel, S. Hotta and A.J. Heeger, Phys. Rev. B. 36 (1987) 7964; J.L.
Bredas, F. Wudl and J.A. Heeger, Solid State Commun., 63 (1987) 577; T.-C. Chung, J.H. Kaufman,
A.J. Heeger and F. Wudl, Phys. Rev. B, 30 (1984) 702.
6 M. Zagbrska and B. Krische, Polymer, 31 (1990) 1379.
7 R.M. Suoto Maior, K. Hinkelmann, H. Eckert and F. Wudl, Macromolecules, 23 (1990) 1268.
8 M. Zagorska and B. Krische in H. Kuzmany, M. Mehring and S. Roth @Is.), Electronic Properties of
Conjugated Polymers, Springer Series in Solid State Sciences, Vol. 91, Springer-Verlag, Berlin, 1989,
p. 343.
9 M. Genoud, J. Kruszka, M. Zagorska, I. Kulszewicz, M. Nechtschein and A. Proh, J. Chim. Phys., 87
(1990) 57.
10 K. Tamao, S. Kodama, J. Nakajima and M. Kumada, Tetrahedron, 38 (1982) 3347.
11 F. de Jong and M.T. Janssen, K. Org. Chem., 36 (1971) 1645.
12 R. Sugimoto, S. Takeda, M.B. Gu and K. Yokishito, Chem. Express, 1 (1986) 635.
13 I. Kulszewicz-Bajer, A. PawIicka, J. Plenkiewicz, A. Proh and S. Lefrant, Synth. Meth., 30 (1989) 335.
14 J. Roncah, P. Marque, R. Garreau, F. Gamier and M. Lemaire, Macromolecules, 23 (1990) 1347.
15 A.J. Downard and D. Pletcher, J. EIectroanal. Chem., 206 (1986) 147; L. Laguren-Davidson, C.V.
Pham, H. Zimmer and H.B. Mark, Jr., J. EIectrochem. Sot., 135 (1988) 1406; K. Tanaka, R. Shichiri,
S. Wang and T. Yamabe, Synth. Met., 24 (1988) 203.
16 M. Zagorska, J. Zachara, I. Kulszewicz-Bajer and A. Proh, to be published.
17 M. Zagbrska, L. Firlej, P. Bernier, I. Kulszewicz-Bajer and A. Pro& J. Polym. Sci., Polym. Lett. Ed.,
submitted.
18 E.M. Genies, G. Bidan and A.F. Diaz, J. Electroanal Chem., 149 (1983) 101.
19 P. Marque, J. Ron&i and F. Gamier, J. EIectroanal. Chem., 218 (1987) 107.
20 S.W. Feldberg, J. Am. Chem. Sot., 106 (1984) 4671.

70
21 T. Hattori, W. Hayes, K. Wang, K. Kaneto and K. Yoshino, J. Phys. C, Solid State Phys., 17 (1984) L803.
22 K. Kaneto, Y. Kohno and K. Yoshino, Mol. Cryst. Liq. Crist., 118 (1985) 217. 23 T.-C. Chung, J.H. Kaufman, A.J. Heeger and F. Wudl, Mol. Cryst. Liq. Cryst., 118 (1985) 205. 24 S.N. Hoier, D.S. Ginley and S.M. Park, J. Electrochem. Sot., 135 (1988) 91. 25 J. Tanguy, A. Proh, M. Zagbrska and I. Kulszewicz-Bajer, J. EIectrochem. Sot., submitted. 26 C. Buess-Herman, L. Gierst and N. Vanlaethem-Meuree, J. EIectroanaI. Chem., 123 (1981) 1; C.
Buess-Herman, G. Quarin and L. Gierst, ibid., 148 (1983) 97; C. Buess-Herman, ibid., 186 (1985) 27, 41.
27 M. tapkowski, Bull. EIectrochem., in press. 28 A.J. Bard and L.R. FauIkner, Electrochemical Methods, Fundamentals and Applications. Wiley, New
York, 1980, p. 86.