synthesis of 2,6-dialkyl-1,2,5,6-tetrahydropyridines and their applications in total synthesis
TRANSCRIPT

Current Organic Synthesis, 2004, 1, 83-109 83
1570-1794/04 $45.00+.00 © 2004 Bentham Science Publishers Ltd.
Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines and Their Applicationsin Total Synthesis
François-Xavier Felpin and Jacques Lebreton*
Laboratoire de Synthèse Organique, CNRS UMR 6513, Faculté des Sciences et desTechniques, 2 rue de la Houssinière, BP 92208, 44322 Nantes Cedex 3, France
Abstract: This review focuses on the different approaches to construct the 2,6-dialkyl-1,2,5,6-tetrahydropyridine skeleton and their applications in total synthesis ofalkaloids.
Keywords: Total Synthesis, Alkaloids, Tetrahydropyridines, Piperidines.
It is Our Pleasure to Dedicate this Review to Our Friend and Colleague Dr AndréGuingant in Thanks of his Help and Guidance
INTRODUCTION
The piperidine ring system is one of the most commonmotifs found in numerous natural products, drugs and drugcandidates. It was recently pointed out by Watson et al. [1]that the piperinidinic substructure was mentioned in over12000 compounds in clinical or pre-clinical studies fromJuly 1988 through December 1998. Accordingly, theimportant bioactivities of piperidines have stimulated thedevelopment of new approaches and considerable syntheticeffort has been devoted to the preparation of thesecompounds [2], in particular with respect to enantiomericallyselective synthesis [3]. 2,6-Disubstituted piperidinesrepresent a subclass of naturally occurring alkaloids that havealso been the targets of many synthetic efforts due to theirwide range of pharmacological activities [4]. Notableexamples include (-)-solenopsin A, a constituent of fire-antvenom Solenopsis species, which is characterized by apredominance of 2-alkyl-6-methylpiperidines [5], and (+)-himbacine, a piperidine alkaloid isolated from the bark ofGalbulimina baccata of the magnolia family. These haveappeared as novel drug candidates for the treatment ofAlzheimer’s disease [6] (see Figure 1).
NCH3 C11 H2 3
H
O
O
CH3
H
H
H
H
NCH3
CH3
(-)-Solenopsin A (+)-Himbacine
2 6
Fig. (1).
*Address correspondence to this author at the Laboratoire de SynthèseOrganique, CNRS UMR 6513, Faculté des Sciences et des Techniques, 2rue de la Houssinière, BP 92208, 44322 Nantes Cedex 3, France; Tel: + 332 51 12 54 03; Fax: + 33 2 51 12 54 02; E-mail: [email protected]
Compared to this class of alkaloid, the 2,6-disubstituted-1,2,5,6-tetrahydropyridine skeleton is present in only a fewnatural products [7]. (+)-Cannabisativine is an unusualmacrocyclic spermidine alkaloid containing a trans-2,6-disubstituted-1,2,5,6-tetrahydropyridine ring, which wasisolated in 1975 from the roots and leaves of the commonmarijuana plant, Cannabis sativa L (see Figure 2) [8].Another member of this class of alkaloids, (-)-palustrine [9],a toxic component of the horsetail plant Equisetum palusterL. found in the meadows of Europe, has been isolated as has(-)-sedacrine [10] a sedum alkaloid.
N
HO
HO
H
OHN
HN
NH H
OHO NH
NH
N
CH3
PhHH
OHO
NR1 R2
H
(+)-Cannabisativine (-)-Palustrine
62
3
4
5
H
(-)-Sedacrine 2,6-disubstituted-1,2,5,6-tetrahydropyridine
Fig. (2).
It is not surprising that these natural products,particularly cannabisativine and palustrine, have been thetarget of different syntheses and have served as usefulmodels to develop a new strategy to construct the 2,6-disubstituted tetrahydropyridine skeleton. Moreover, the 2,6-disubstituted tetrahydropyridine framework should beregarded as a valuable basic unit since the possibility ofmodification and functionalization of the double bondenables the preparation of polysubstituted piperidines.
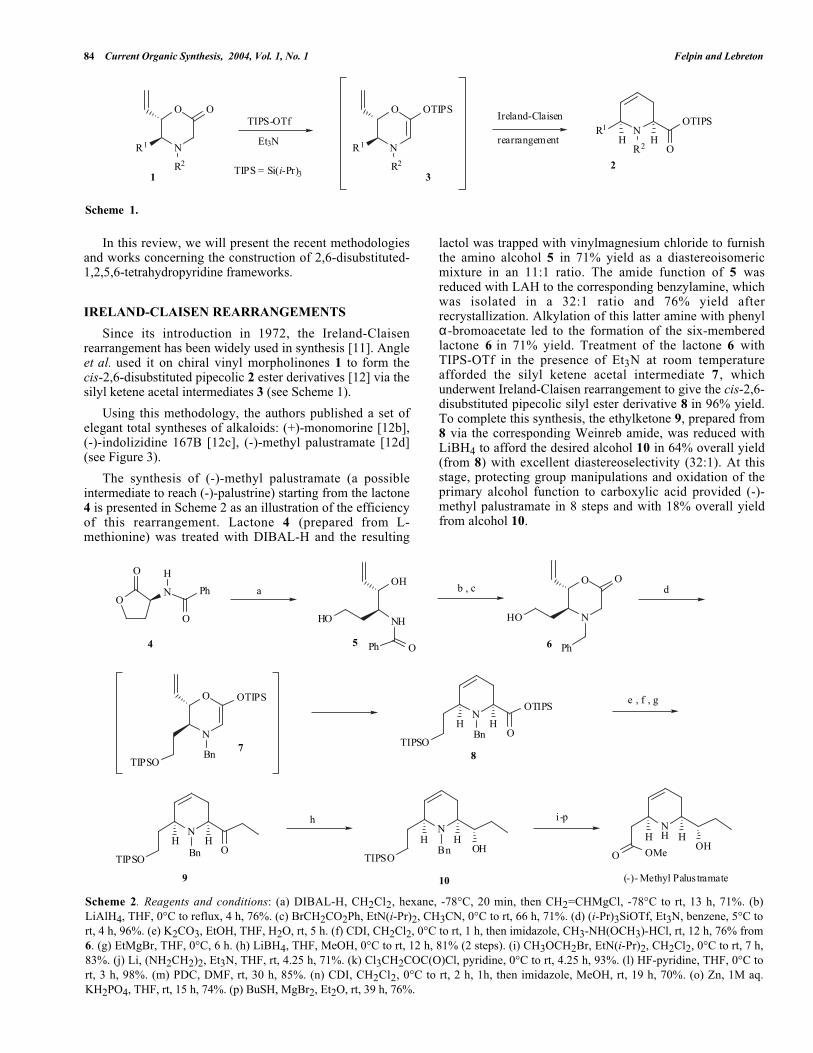
84 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
N
O
R1
R2
OTIPS-OTf
N
O
R1
R2
OTIPS
Et3NNR1
R2
OTIPS
OH H
TIPS = Si(i-Pr)312
3
Ireland-Claisen
rearrangement
Scheme 1.
O
O
N
O
Ph
H
N
Bn OH H
TIPSO
N
O
BnTIPSO
OTIPS
HO
OH
NH
OPh
N
BnH H
TIPSOOH
N
Bn OH H
OTIPS
TIPSO
HO N
O O
Ph
NHH H
OOH
OMe
a b , c d
e , f , g
h i-p
4 5 6
78
9 10 (-)- Methyl Palus tramate
Scheme 2. Reagents and conditions: (a) DIBAL-H, CH2Cl2, hexane, -78°C, 20 min, then CH2=CHMgCl, -78°C to rt, 13 h, 71%. (b)LiAlH4, THF, 0°C to reflux, 4 h, 76%. (c) BrCH2CO2Ph, EtN(i-Pr)2, CH3CN, 0°C to rt, 66 h, 71%. (d) (i-Pr)3SiOTf, Et3N, benzene, 5°C tort, 4 h, 96%. (e) K2CO3, EtOH, THF, H2O, rt, 5 h. (f) CDI, CH2Cl2, 0°C to rt, 1 h, then imidazole, CH3-NH(OCH3)-HCl, rt, 12 h, 76% from6. (g) EtMgBr, THF, 0°C, 6 h. (h) LiBH4, THF, MeOH, 0°C to rt, 12 h, 81% (2 steps). (i) CH3OCH2Br, EtN(i-Pr)2, CH2Cl2, 0°C to rt, 7 h,83%. (j) Li, (NH2CH2)2, Et3N, THF, rt, 4.25 h, 71%. (k) Cl3CH2COC(O)Cl, pyridine, 0°C to rt, 4.25 h, 93%. (l) HF-pyridine, THF, 0°C tort, 3 h, 98%. (m) PDC, DMF, rt, 30 h, 85%. (n) CDI, CH2Cl2, 0°C to rt, 2 h, 1h, then imidazole, MeOH, rt, 19 h, 70%. (o) Zn, 1M aq.KH2PO4, THF, rt, 15 h, 74%. (p) BuSH, MgBr2, Et2O, rt, 39 h, 76%.
In this review, we will present the recent methodologiesand works concerning the construction of 2,6-disubstituted-1,2,5,6-tetrahydropyridine frameworks.
IRELAND-CLAISEN REARRANGEMENTS
Since its introduction in 1972, the Ireland-Claisenrearrangement has been widely used in synthesis [11]. Angleet al. used it on chiral vinyl morpholinones 1 to form thecis-2,6-disubstituted pipecolic 2 ester derivatives [12] via thesilyl ketene acetal intermediates 3 (see Scheme 1).
Using this methodology, the authors published a set ofelegant total syntheses of alkaloids: (+)-monomorine [12b],(-)-indolizidine 167B [12c], (-)-methyl palustramate [12d](see Figure 3).
The synthesis of (-)-methyl palustramate (a possibleintermediate to reach (-)-palustrine) starting from the lactone4 is presented in Scheme 2 as an illustration of the efficiencyof this rearrangement. Lactone 4 (prepared from L-methionine) was treated with DIBAL-H and the resulting
lactol was trapped with vinylmagnesium chloride to furnishthe amino alcohol 5 in 71% yield as a diastereoisomericmixture in an 11:1 ratio. The amide function of 5 wasreduced with LAH to the corresponding benzylamine, whichwas isolated in a 32:1 ratio and 76% yield afterrecrystallization. Alkylation of this latter amine with phenylα-bromoacetate led to the formation of the six-memberedlactone 6 in 71% yield. Treatment of the lactone 6 withTIPS-OTf in the presence of Et3N at room temperatureafforded the silyl ketene acetal intermediate 7 , whichunderwent Ireland-Claisen rearrangement to give the cis-2,6-disubstituted pipecolic silyl ester derivative 8 in 96% yield.To complete this synthesis, the ethylketone 9, prepared from8 via the corresponding Weinreb amide, was reduced withLiBH4 to afford the desired alcohol 10 in 64% overall yield(from 8) with excellent diastereoselectivity (32:1). At thisstage, protecting group manipulations and oxidation of theprimary alcohol function to carboxylic acid provided (-)-methyl palustramate in 8 steps and with 18% overall yieldfrom alcohol 10.
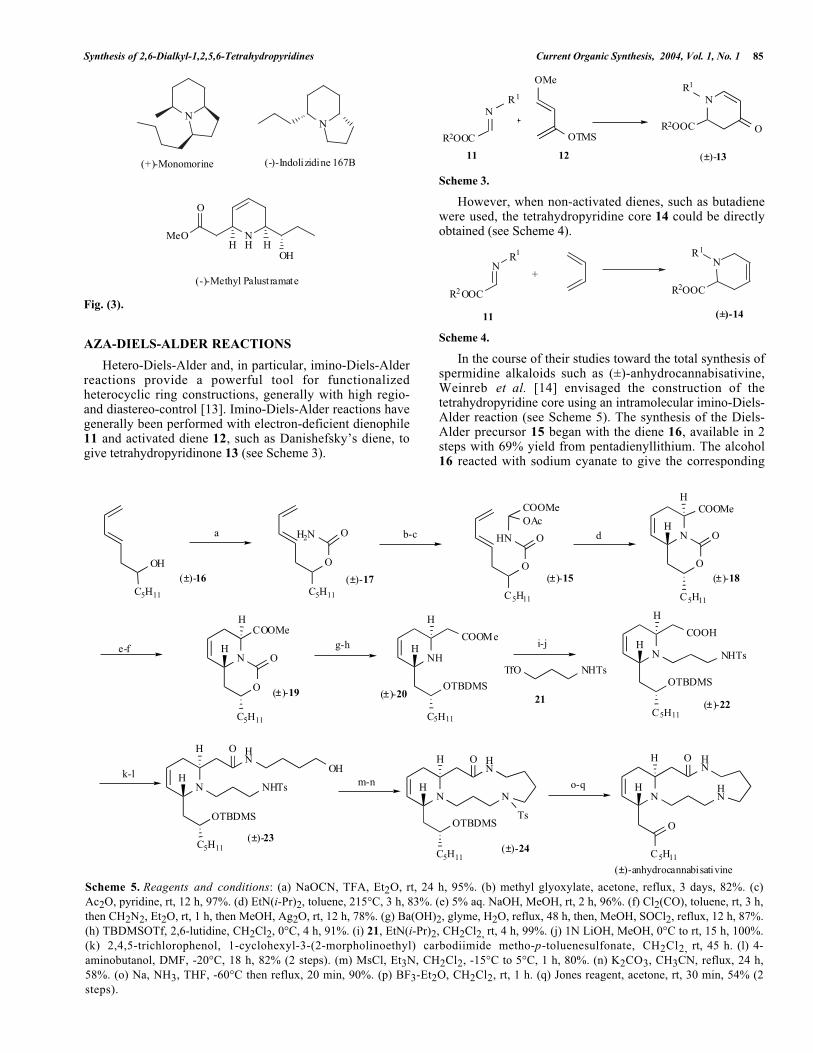
Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 85
OH
C5H11
O
N O
C5H11
H
COOMeH
TfO NHTs
N N
HN
OTBDMS
C5H11
H O
Ts
OTBDMS
N
H
H
C5H11
COOH
NHTs
O
H2N O
C5H11
O
N O
COOMeH
H
C5H11
O
HN O
C5H11
COOMeOAc
NHN
HN
O
C5H11
H
H O
OTBDMS
NH
H
H
C5H11
COOMe
N NHTs
HN
OH
H O
OTBDMS
C5H11
(±)-15
a b-c d
e-f g-h i-j
k-lm-n o-q
(±)-17(±)-16 (±)-18
(±)-19
(±)-23
(±)-20(±)-22
(±)-24
(±)-anhydrocannabisativine
21
HH
Scheme 5. Reagents and conditions: (a) NaOCN, TFA, Et2O, rt, 24 h, 95%. (b) methyl glyoxylate, acetone, reflux, 3 days, 82%. (c)Ac2O, pyridine, rt, 12 h, 97%. (d) EtN(i-Pr)2, toluene, 215°C, 3 h, 83%. (e) 5% aq. NaOH, MeOH, rt, 2 h, 96%. (f) Cl2(CO), toluene, rt, 3 h,then CH2N2, Et2O, rt, 1 h, then MeOH, Ag2O, rt, 12 h, 78%. (g) Ba(OH)2, glyme, H2O, reflux, 48 h, then, MeOH, SOCl2, reflux, 12 h, 87%.(h) TBDMSOTf, 2,6-lutidine, CH2Cl2, 0°C, 4 h, 91%. (i) 21, EtN(i-Pr)2, CH2Cl2, rt, 4 h, 99%. (j) 1N LiOH, MeOH, 0°C to rt, 15 h, 100%.(k) 2,4,5-trichlorophenol, 1-cyclohexyl-3-(2-morpholinoethyl) carbodiimide metho-p-toluenesulfonate, CH2Cl2, rt, 45 h. (l) 4-aminobutanol, DMF, -20°C, 18 h, 82% (2 steps). (m) MsCl, Et3N, CH2Cl2, -15°C to 5°C, 1 h, 80%. (n) K2CO3, CH3CN, reflux, 24 h,58%. (o) Na, NH3, THF, -60°C then reflux, 20 min, 90%. (p) BF3-Et2O, CH2Cl2, rt, 1 h. (q) Jones reagent, acetone, rt, 30 min, 54% (2steps).
NH
OH
MeO
O
H H
NN
(-)-Methyl Palustramate
(+)-Monomorine (-)-Indolizidine 167B
Fig. (3).
AZA-DIELS-ALDER REACTIONS
Hetero-Diels-Alder and, in particular, imino-Diels-Alderreactions provide a powerful tool for functionalizedheterocyclic ring constructions, generally with high regio-and diastereo-control [13]. Imino-Diels-Alder reactions havegenerally been performed with electron-deficient dienophile11 and activated diene 12, such as Danishefsky’s diene, togive tetrahydropyridinone 13 (see Scheme 3).
OMe
OTMS
N
R2OOC
R1 N
O
R1
R2OOC
(±)-131211
Scheme 3.
However, when non-activated dienes, such as butadienewere used, the tetrahydropyridine core 14 could be directlyobtained (see Scheme 4).
N
R2OOC
R1
+
11 (±)-14
N
R2 OOC
R1
Scheme 4.
In the course of their studies toward the total synthesis ofspermidine alkaloids such as (±)-anhydrocannabisativine,Weinreb et al. [14] envisaged the construction of thetetrahydropyridine core using an intramolecular imino-Diels-Alder reaction (see Scheme 5). The synthesis of the Diels-Alder precursor 15 began with the diene 16, available in 2steps with 69% yield from pentadienyllithium. The alcohol16 reacted with sodium cyanate to give the corresponding

86 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
OBn
N
BuOOC
Ts
OBnBnO
C5 H1 1
N
BuOOC
Ts
N
COOBu
Ts
OBn
H
H
N
COOBu
Ts
OBnBnO
H
C5H11
H
+
+
a
b
25
26
27
27
28
29
Scheme 6. Reagents and conditions: (a) neat, 40°C, 48 h, 83%; (b) neat, 40°C, 6 days, 81%.
carbamate 17 in high yield (95%). This carbamate 17 wastreated with methyl glyoxylate and the generated methylolacetate was then acetylated with acetic anhydride to affordthe precursor for the Diels-Alder reaction 15 in 97% yield forthis process. Upon heating in a sealed tube at 215°C,compound 15 in toluene solution containing an equivalentof Hünig’s base was converted stereo- and regioselectively totetrahydropyridine 18 in 83% yield with a trans relationshipof hydrogens α to nitrogen. After mild saponification of themethyl ester, the resulting acid was homologated by theArndt-Eistert procedure to yield ester 19 in 75% yield overtwo steps. The cyclic carbamate 19 was opened with bariumhydroxide then the resulting amino acid was re-esterified andthe hydroxyl function was protected as silyl ether to giveamine 20 in good yield (79%, two steps). The secondaryamine 20 was alkylated with compound 21 and the esterfunction was saponified to give the corresponding acid 22 inquantitative yield over two steps. After activation of thecarboxylic acid function as the 2,4,5-trichlorophenyl ester,the latter was treated with 4-aminobutanol to afford amide-alcohol 23 in 82% yield over two steps. The activation ofthe alcohol function of 23 as a mesylate under classicalconditions and the subsequent treatment with K2CO3 inacetonitrile afforded the expected cyclized product 24 (46%,two steps). The final steps resulted in cleavage of the N-tosyl and the O-TBDMS protecting groups followed by theJones oxidation to give (±)-anhydrocannabisativine (49%,three steps).
In their initial investigation of the synthesis of naturalproducts related to palustrine and cannabisativine, Hamada etal. [15, 16] reported the formation of the functionalizedtetrahydropyridine core by an aza-Diels-Alder reaction.Starting from dienes 25 or 26 (the latter chiral diene isprepared in 12 steps and 21% yield from n-hexanal), theDiels-Alder reaction with the imino-ester dienophile 27 gavewith good yields (83% and 81% respectively) the expectedcyclo-adducts 28 and 29 in a regio- and stereoselectivemanner (see Scheme 6).
With cycloadduct 29 in hand, these authors [15] reportedthe total synthesis of (-)-cannabisativine (see Scheme 7). Thehomologation of the ester function of 29 via a Wittigolefination of the aldehyde intermediate withmethoxymethylene phosphorane gave, in a few steps, the
tetrahydropyridine 30 (62%, four steps). From thisintermediate 30, subsequent cleavage of the N-tosyl group,acylation of the liberated amine function with acid chloride31, and reduction of the resulting amide with LAH furnishedcompound 32 in 76% yield over three steps. The tosylaminogroup of 32 was alkylated with N - (4-bromobuty l )phthalimide and the phthaloyl group of the primary aminewas replaced with a trifluoroacetyl-protecting group tofurnish 33 in 78% yield over three steps. The acetal functionwas subjected to acidic media to give the correspondingaldehyde, which was readily epimerized by treatment withK2CO3 in MeOH. This aldehyde intermediate was thenoxidized with sodium chlorite and immediately esterifiedwith diazomethane to afford 34 (68%, four steps). Treatmentof 34 with Ba(OH)2 resulted in the hydrolysis of the methylester and N-trifluoroacetyl group to give the expected aminoacid, which was cyclized using the Mukaiyama reagent (2-chloro-1-methylpyridinium iodide) and DMAP to afford 35(60%, two steps). Finally, removal of the protecting groupsof 35 with sodium-naphthalenide afforded (-)-cannabisativine(non-natural enantiomer) in 83% yield. The synthesis of thisenantiomer assigned the absolute configuration of (+)-cannabisativine.
Bailey et al. [17] have developed an aza-Diels-Alderreaction using a chiral imino-ester 36 as dienophile with thechiral auxiliary branched on the imino nitrogen. The aza-Diels-Alder carried out with the latter dienophile 36 and 1-methylbutadiene gave the expected cis cyclo-adduct 37 withmodest yield and asymmetric induction (see Scheme 8).
To improve their results, these authors [18] studied thereactivity of the dienophile 38 (prepared in three steps fromfumaric acid), which combined two chiral auxiliaries,namely 1-phenylethylamine on the imino nitrogen and 8-phenylmenthol on the ester function (see Scheme 9). Due tothe presence of the two chiral auxiliaries, they distinguisheda matched and a mismatched process depending on theconfiguration of each one. As an example of the efficiency ofthis process, when matched auxiliaries were used with 1-methylbutadiene in the cycloaddition reaction, the resultingcyclo-adduct 39 was isolated with complete diastereo-selectivity. As a limitation, 8-phenylmenthol is expensiveand only one enantiomer is readily accessible (prepared from(R)-pulegone).
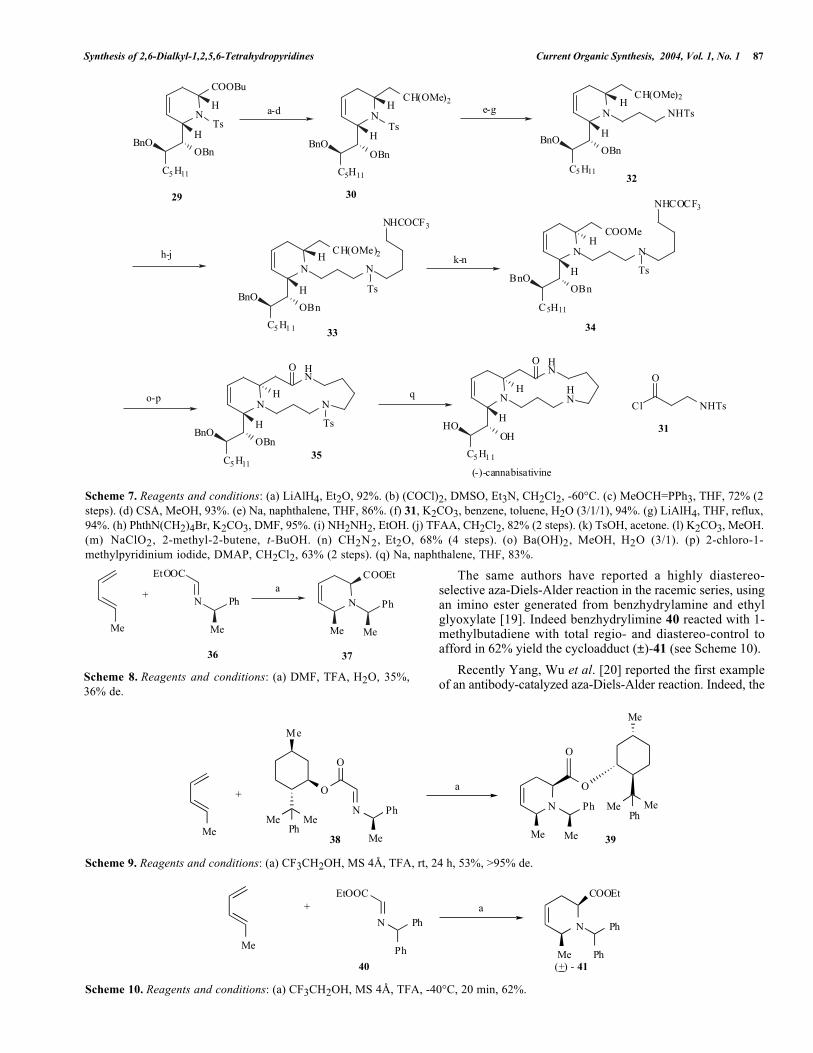
Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 87
N
COOBu
Ts
OBnBnO
H
C5 H11
HN
OBnBnO
H
C5 H11
CH(OMe)2
NHTsH
N
OBnBnO
H
C5H11
COOMe
N
Ts
NHCOCF3
H
N
OHHO
H
C5 H1 1
HN
O
HN
H
NTs
OBnBnO
C5H11
CH(OMe)2
H
H
Cl NHTs
O
N
OBnBnO
H
C5 H11
HN
O
N
Ts
H
N
OBnBnO
H
C5 H1 1
CH(OMe)2
N
Ts
NHCOCF3
H
a-d e-g
29 30
32
33 34
35
(-)-cannabisativine
31
h-j k-n
o-p q
Scheme 7. Reagents and conditions: (a) LiAlH4, Et2O, 92%. (b) (COCl)2, DMSO, Et3N, CH2Cl2, -60°C. (c) MeOCH=PPh3, THF, 72% (2steps). (d) CSA, MeOH, 93%. (e) Na, naphthalene, THF, 86%. (f) 31, K2CO3, benzene, toluene, H2O (3/1/1), 94%. (g) LiAlH4, THF, reflux,94%. (h) PhthN(CH2)4Br, K2CO3, DMF, 95%. (i) NH2NH2, EtOH. (j) TFAA, CH2Cl2, 82% (2 steps). (k) TsOH, acetone. (l) K2CO3, MeOH.(m) NaClO2, 2-methyl-2-butene, t-BuOH. (n) CH2N2, Et2O, 68% (4 steps). (o) Ba(OH)2, MeOH, H2O (3/1). (p) 2-chloro-1-methylpyridinium iodide, DMAP, CH2Cl2, 63% (2 steps). (q) Na, naphthalene, THF, 83%.
Me
N
Me
Ph
O
O
Me
Me MePh
N
Me
Ph
Me
O
Me MePh
Me
+a
38 39
O
Scheme 9. Reagents and conditions: (a) CF3CH2OH, MS 4Å, TFA, rt, 24 h, 53%, >95% de.
Me
+EtOOC
N Ph
Ph
N
COOEt
Ph
PhMe40
a
(+) - 41
Scheme 10. Reagents and conditions: (a) CF3CH2OH, MS 4Å, TFA, -40°C, 20 min, 62%.
Me
N
EtOOC
Me
Ph N
COOEt
Me
Ph
Me
+a
36 37
Scheme 8. Reagents and conditions: (a) DMF, TFA, H2O, 35%,36% de.
The same authors have reported a highly diastereo-selective aza-Diels-Alder reaction in the racemic series, usingan imino ester generated from benzhydrylamine and ethylglyoxylate [19]. Indeed benzhydrylimine 40 reacted with 1-methylbutadiene with total regio- and diastereo-control toafford in 62% yield the cycloadduct (±)-41 (see Scheme 10).
Recently Yang, Wu et al. [20] reported the first exampleof an antibody-catalyzed aza-Diels-Alder reaction. Indeed, the
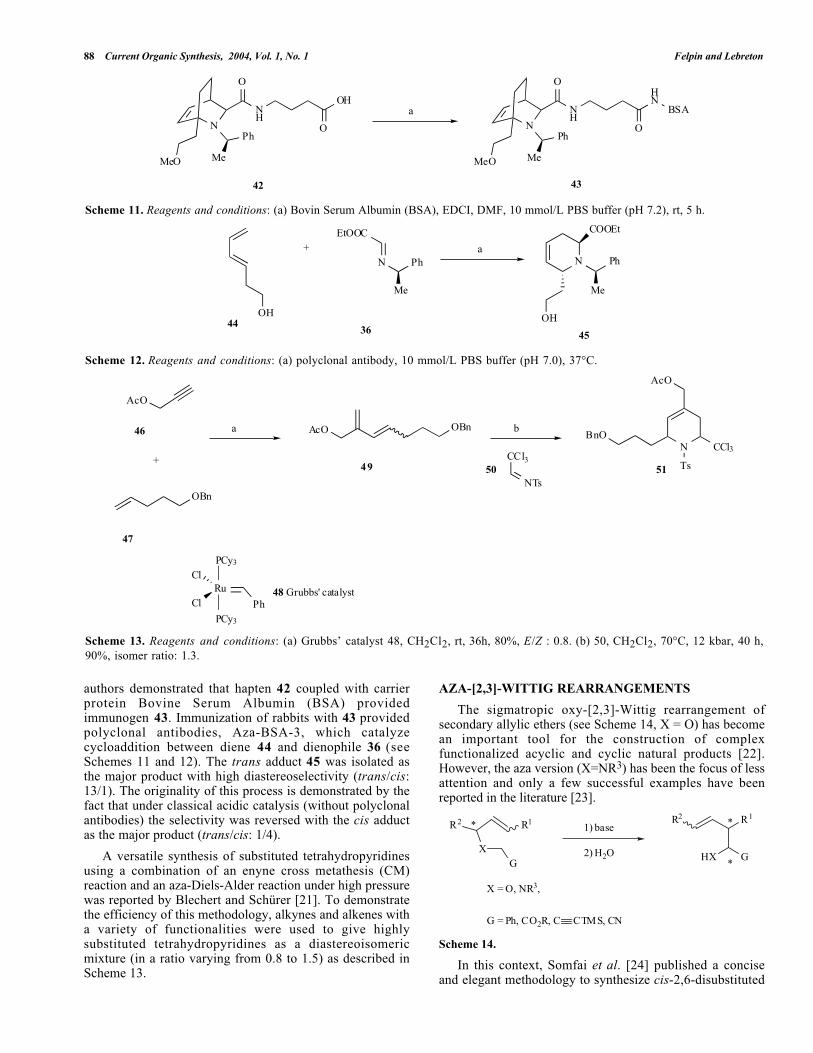
88 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
N
MeO
NH
OH
O
O
Me
PhN
MeO
NH
HN
O
O
Me
Ph
BSAa
42 43
Scheme 11. Reagents and conditions: (a) Bovin Serum Albumin (BSA), EDCI, DMF, 10 mmol/L PBS buffer (pH 7.2), rt, 5 h.
+EtOOC
N Ph
Me
N
COOEt
Ph
Me
36
a
OH OH
4544
Scheme 12. Reagents and conditions: (a) polyclonal antibody, 10 mmol/L PBS buffer (pH 7.0), 37°C.
AcO
OBn
Ru
PCy3
PCy3
Ph
Cl
Cl
OBnAcO
CCl3
NTs
N
AcO
Ts
CCl3BnO
+
a b46
47
4 9 50 51
48 Grubbs' catalyst
Scheme 13. Reagents and conditions: (a) Grubbs’ catalyst 48, CH2Cl2, rt, 36h, 80%, E/Z : 0.8. (b) 50, CH2Cl2, 70°C, 12 kbar, 40 h,90%, isomer ratio: 1.3.
authors demonstrated that hapten 42 coupled with carrierprotein Bovine Serum Albumin (BSA) providedimmunogen 43. Immunization of rabbits with 43 providedpolyclonal antibodies, Aza-BSA-3, which catalyzecycloaddition between diene 44 and dienophile 36 (seeSchemes 11 and 12). The trans adduct 45 was isolated asthe major product with high diastereoselectivity (trans/cis:13/1). The originality of this process is demonstrated by thefact that under classical acidic catalysis (without polyclonalantibodies) the selectivity was reversed with the cis adductas the major product (trans/cis: 1/4).
A versatile synthesis of substituted tetrahydropyridinesusing a combination of an enyne cross metathesis (CM)reaction and an aza-Diels-Alder reaction under high pressurewas reported by Blechert and Schürer [21]. To demonstratethe efficiency of this methodology, alkynes and alkenes witha variety of functionalities were used to give highlysubstituted tetrahydropyridines as a diastereoisomericmixture (in a ratio varying from 0.8 to 1.5) as described inScheme 13.
AZA-[2,3]-WITTIG REARRANGEMENTS
The sigmatropic oxy-[2,3]-Wittig rearrangement ofsecondary allylic ethers (see Scheme 14, X = O) has becomean important tool for the construction of complexfunctionalized acyclic and cyclic natural products [22].However, the aza version (X=NR3) has been the focus of lessattention and only a few successful examples have beenreported in the literature [23].
XG
R1R2
HX
R1
G
R2
1) base
2) H2O
X = O, NR3,
G = Ph, CO2R, C CTMS, CN
* *
*
Scheme 14.
In this context, Somfai et al. [24] published a conciseand elegant methodology to synthesize cis-2,6-disubstituted
Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 89
NR1
CO2tBu
R2
1) LDA
2) H2O NR1
H
R2
CO2tBuNR1
CO2tBu
1) LDA
2) H2O NR1
H
R2
CO2tBu
R2
(E) (Z)
Scheme 15.
NR1
CO2 tBu
R2N
LiO
OtBu
H
R2
R1
H
H
H2O
NR1
H
R2
CO2tBu
LDA
Scheme 16.
OH
C5H11
O
OTBDMS
C5H11
N
CO2 tBu
NH
Me
C5H11 CO2tBu
NH
Me
C5H11
CO2Et
OH
C5H11 OH
N3
C5H11
N
CO2tBu
Me
OH
C5 H1 1 N3
HO
N
Me
C5 H1 1
O
NH
Me
C5H11
OH
OTBDMS
C5H11
NH
NH
Me
C5H11 CO2tBu
NH
Me
C5H11
CO2Et
N
Me
C5H11
a b, c d
(-)-indolizidine 209B
e, f, g h
j
52 53 55
56
54
+
57
i k
58
l m n
59 60 61
62 63
23
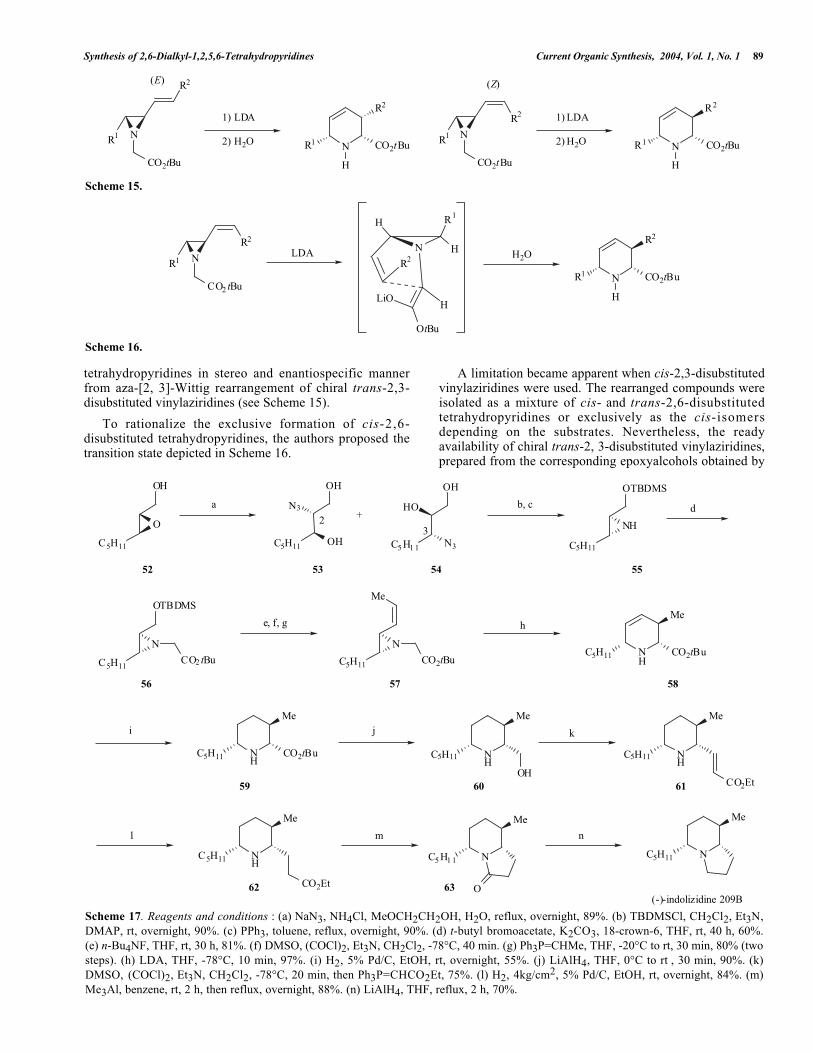
Scheme 17. Reagents and conditions : (a) NaN3, NH4Cl, MeOCH2CH2OH, H2O, reflux, overnight, 89%. (b) TBDMSCl, CH2Cl2, Et3N,DMAP, rt, overnight, 90%. (c) PPh3, toluene, reflux, overnight, 90%. (d) t-butyl bromoacetate, K2CO3, 18-crown-6, THF, rt, 40 h, 60%.(e) n-Bu4NF, THF, rt, 30 h, 81%. (f) DMSO, (COCl)2, Et3N, CH2Cl2, -78°C, 40 min. (g) Ph3P=CHMe, THF, -20°C to rt, 30 min, 80% (twosteps). (h) LDA, THF, -78°C, 10 min, 97%. (i) H2, 5% Pd/C, EtOH, rt, overnight, 55%. (j) LiAlH4, THF, 0°C to rt , 30 min, 90%. (k)DMSO, (COCl)2, Et3N, CH2Cl2, -78°C, 20 min, then Ph3P=CHCO2Et, 75%. (l) H2, 4kg/cm2, 5% Pd/C, EtOH, rt, overnight, 84%. (m)Me3Al, benzene, rt, 2 h, then reflux, overnight, 88%. (n) LiAlH4, THF, reflux, 2 h, 70%.
tetrahydropyridines in stereo and enantiospecific mannerfrom aza-[2, 3]-Wittig rearrangement of chiral trans-2,3-disubstituted vinylaziridines (see Scheme 15).
To rationalize the exclusive formation of cis-2,6-disubstituted tetrahydropyridines, the authors proposed thetransition state depicted in Scheme 16.
A limitation became apparent when cis-2,3-disubstitutedvinylaziridines were used. The rearranged compounds wereisolated as a mixture of cis- and trans-2,6-disubstitutedtetrahydropyridines or exclusively as the cis-isomersdepending on the substrates. Nevertheless, the readyavailability of chiral trans-2, 3-disubstituted vinylaziridines,prepared from the corresponding epoxyalcohols obtained by

90 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
N
TMS
Me
Ph
O
N
TMS
Me
Ph
O
TMS
A B
N
TMS
Me
Ph
O
TMS
NMe Ph
OH
NMe Ph
OH
+
(±)-anti-64 (±)-cis-65 (±)-trans-66
a
Scheme 18. Reagents and conditions : (a) 1.0 eq TMSOTf, CH2Cl2, -40°C, 8 h, 91%.
C
N
TMSO
Me Ph
TMS
N
TMS
Me
Ph
OTMSD
E
F
N
TMSO
Me Ph
TMS
N
TMS
Me
Ph
OTMS
+
+
trans-66
cis -65
Scheme 19.
Sharpless epoxidation of allylic alcohols, makes thismethodology very attractive. Somfai et al. [25] haveexploited this aza-[2,3]-Wittig rearrangement as a key step toconstruct the piperidine ring in the synthesis of indolizidinealkaloids. For example, starting from chiral 2, 3-epoxyalcohol 52, they achieved a 14-step synthesis of (-)-indolizidine 209B [25b] as presented in Scheme 17.
Bimolecular nucleophilic ring-opening of the chiral 2,3-epoxyalcohol 52 with excess of sodium azide afforded amixture of substituted compounds at the C-2 and C-3positions, 53 and 54 respectively. The use of the mixture ofazidoalcohols 53 and 54 has no consequence on theformation of the chiral aziridine 55 and no attempt was madeto separate these compounds [26]. After selective silylationof the primary alcohol, the resulting mixture was treatedwith triphenylphosphine in refluxing toluene to give thedesired aziridine 55 as a single isomer in 72% overall yieldfrom 52. N-Alkylation of the aziridine 55 with t-butylbromoacetate was carried out with potassium carbonate inthe presence of 18-crown-6 to furnish the N-alkylatedcompound 56 in 60% yield. The aza-Wittig substrate with(Z) double bond 57 was synthesized in 65% yield fromcompound 56 following a three-step sequence involving thecleavage of the silyl protecting group, a Swern oxidation ofthe resulting primary alcohol and a Wittig olefination. Thetreatment of the aziridine 57 with LDA at low temperatureled to the rapid formation of the cis-2,6-disubstitutedtetrahydropyridine 58 as the sole diastereoisomer in 97%yield. The cis-2,6-disubstituted tetrahydropyridine 58 wassuccessfully transformed to (-)-indolizidine 209B via a 6-step sequence using known procedures.
INTRAMOLECULAR ALLYLSILANE-NITRONECYCLOADDITIONS
A comparative study between thermal and TMSOTf-induced intramolecular allylsilane-nitrone cycloaddition has
been reported by Wuts et al. [27]. This work has beensuccessfully used to build either racemic cis- or trans-2,6-dialkyl tetrahydropyridine motifs.
Thus, treatment of the racemic anti-nitrone 64 with oneequivalent of trimethylsilyl triflate (TMSOTf) in methylenechloride at low temperature afforded a mixture of the N-hydroxylamines cis-65 and trans-66 in 91% yield as a 1 to 1mixture, without formation of [3+2] dipolar cycloadducts(see Scheme 18). The anti-nitrone 64 was prepared byreaction of benzaldehyde with the corresponding anti-hydroxylamine, which was isolated from an anti/syn (3.8/1)mixture obtained by condensation of acetaldehyde oximewith γ-(trimethylsilyl)allylboronate.
The formation of the racemic tetrahydropyridines cis-65and trans-66 could be explained by the fact that thestoichiometric amount of TMSOTf leads to the formation ofan electrophilic N-siloxyiminium intermediate A, whichreacts in intramolecular fashion with the allylsilane moietyto generate a cation B stabilized by the neighboring Si-Cbond (see Scheme 18). The formation of cis-65 and trans-66adducts arises from the boat C and chair D transition states,in which the silyl group adopts an axial position to allow aσ-π stabilization of the β-silyl cation intermediates E and F(Scheme 19).
This methodology was used for the synthesis of racemicsolenopsin A from the anti-nitrone 67 as depicted in Scheme20.
In contrast with the results above, thermal intramolecularallylsilane-nitrone [3+2] dipolar cycloaddition of anti-nitrone 64 conducted in refluxing toluene proceeded in theusual way to afford a diastereoisomeric mixture of bicyclicisoxazolidines 70, 71 and 72 in 79/17/4 ratio (compounds70 and 71 are inseparable but could be separated from 72)(see Scheme 21).
Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 91
N
TMS
Me
C11H23
O NMe C11H23
OH
NMe C1 1H23
OH
NMe C11H23
H
+
(±)-anti-67 (±)-trans-68
trans-68/cis-69 : 68/32
(±)-cis-69 (±)-Solenopsin A
a b, c
Scheme 20. Reagents and conditions : (a) 1.0 eq. TMSOTf, CH2Cl2, -40°C, 8 h, 58% of trans-68, 27% of cis-69. (b) Zn, AcOH, H2O,60°C, 4 h, 97%. (c) PtO2, H2, EtOH, rt, 100%.
N
TMS
Me
Ph
O
N
O
Me Ph
TMS
N
O
PhTMS
Me
N
O
Me
TMS Ph
(±)-anti-64
70
+ +
71 72
70/71/72 : 79/17/4
a
Scheme 21. Reagents and conditions : (a) Toluene, reflux, 2 h, 87%.
N
O
Me Ph
TMS
N
O
PhTMS
Me
N
O
Me
TMS PhNH
Me Ph
N
OH
Me Ph NH
Me Ph
(±)-70
+
(±)-71
(±)-72
+a b
a, b
(±)-75 (±)-cis-73
(±)-trans-74
(±)-71
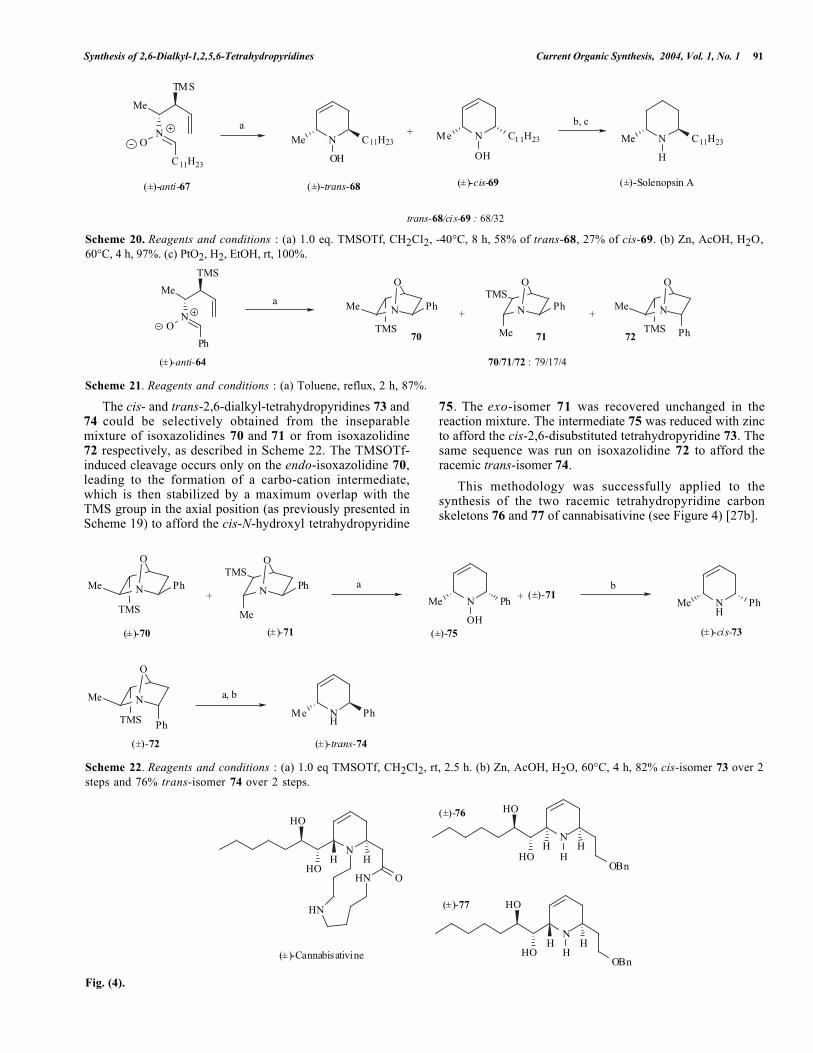
Scheme 22. Reagents and conditions : (a) 1.0 eq TMSOTf, CH2Cl2, rt, 2.5 h. (b) Zn, AcOH, H2O, 60°C, 4 h, 82% cis-isomer 73 over 2steps and 76% trans-isomer 74 over 2 steps.
N
HO
HO
H H
OHN
HN
N
HHO
HO
H H
OBn
N
HHO
HO
HH
OBn
(±)-Cannabisativine
(±)-77
(±)-76
Fig. (4).
The cis- and trans-2,6-dialkyl-tetrahydropyridines 73 and74 could be selectively obtained from the inseparablemixture of isoxazolidines 70 and 71 or from isoxazolidine72 respectively, as described in Scheme 22. The TMSOTf-induced cleavage occurs only on the endo-isoxazolidine 70,leading to the formation of a carbo-cation intermediate,which is then stabilized by a maximum overlap with theTMS group in the axial position (as previously presented inScheme 19) to afford the cis-N-hydroxyl tetrahydropyridine
75. The exo-isomer 71 was recovered unchanged in thereaction mixture. The intermediate 75 was reduced with zincto afford the cis-2,6-disubstituted tetrahydropyridine 73. Thesame sequence was run on isoxazolidine 72 to afford theracemic trans-isomer 74.
This methodology was successfully applied to thesynthesis of the two racemic tetrahydropyridine carbonskeletons 76 and 77 of cannabisativine (see Figure 4) [27b].
92 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
H11 C5 N
TMS
O
O
H OH
N
O
TMS
OBn
O
O
H1 1C5
OBn
OHC
N
OTMS
OBn
H1 1C5
O
O
H11C5
NO
O
OBn
O
TMS
H
N
OHH H
H11C5
O
OOBn
NH H
H11C5
OO
OHTMS
H
OBn
NH
H11C5
HO
HO
H H
OHTMS
OBn
NH
H1 1C5
HO
HO
H H
OBn
H11C5
NO
O O
TMS
H
OBn
NH
H11C5
HO
HO
H H
OBn
+
+
a
+
e f
b
c, d
(±)-78 79 (±)-80
(±)-85 (±)-77
(±)-76(±)-84(±)-83
(±)-81 (±)-82
(±)-80
(±)-78
14.8:1
Scheme 23. Reagents and conditions : (a) toluene, CaCl2, -20°C, 8 h, then reflux, 2 h, 100%. (b) Zn, AcOH, H2O, 60°C, 4 h. (c) APTS,toluene, reflux, 3 h. (d) TFA, THF, H2O, rt, 36 h, 61% (three steps). (e) 79, CH2Cl2, CaCl2, -20°C, 14 h, then 1.0 eq TMSOTf, -40°C, 18 h,79%. (f) Zn, AcOH, H2O, 80°C, 4 h, then TFA, THF, rt, 20 h, 73%.
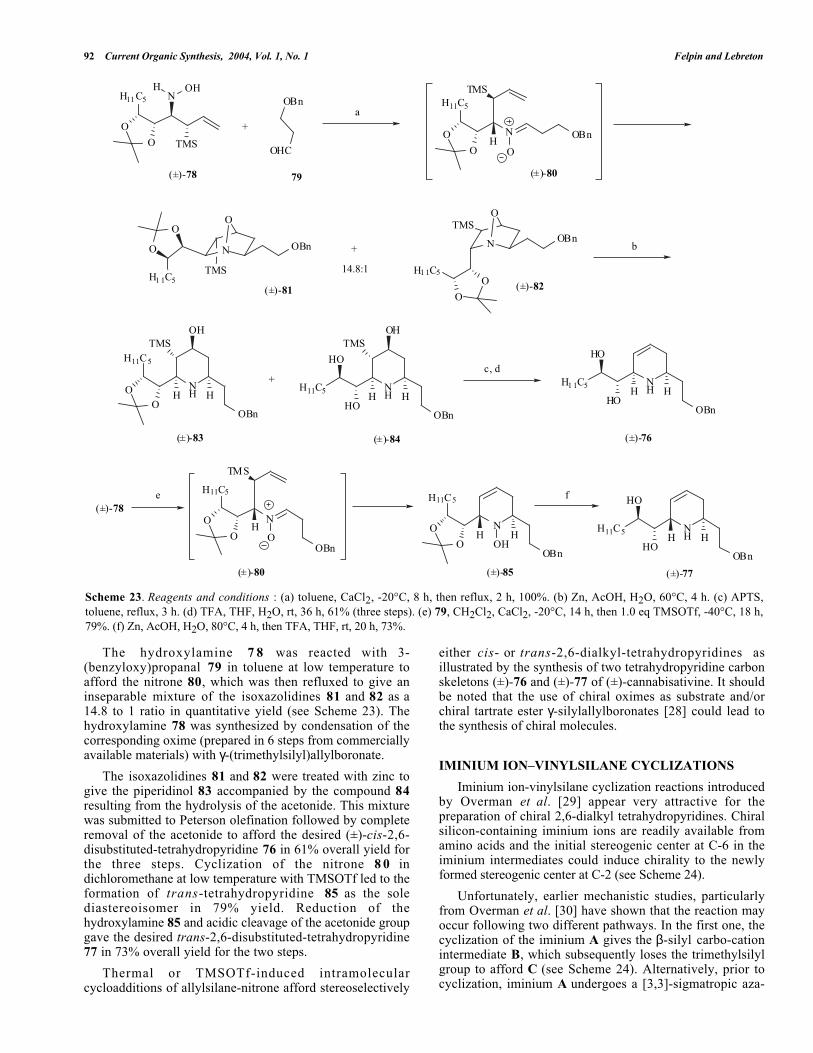
The hydroxylamine 7 8 was reacted with 3-(benzyloxy)propanal 79 in toluene at low temperature toafford the nitrone 80, which was then refluxed to give aninseparable mixture of the isoxazolidines 81 and 82 as a14.8 to 1 ratio in quantitative yield (see Scheme 23). Thehydroxylamine 78 was synthesized by condensation of thecorresponding oxime (prepared in 6 steps from commerciallyavailable materials) with γ-(trimethylsilyl)allylboronate.
The isoxazolidines 81 and 82 were treated with zinc togive the piperidinol 83 accompanied by the compound 84resulting from the hydrolysis of the acetonide. This mixturewas submitted to Peterson olefination followed by completeremoval of the acetonide to afford the desired (±)-cis-2,6-disubstituted-tetrahydropyridine 76 in 61% overall yield forthe three steps. Cyclization of the nitrone 8 0 indichloromethane at low temperature with TMSOTf led to theformation of trans-tetrahydropyridine 85 as the solediastereoisomer in 79% yield. Reduction of thehydroxylamine 85 and acidic cleavage of the acetonide groupgave the desired trans-2,6-disubstituted-tetrahydropyridine77 in 73% overall yield for the two steps.
Thermal or TMSOTf-induced intramolecularcycloadditions of allylsilane-nitrone afford stereoselectively
either cis- or trans-2,6-dialkyl-tetrahydropyridines asillustrated by the synthesis of two tetrahydropyridine carbonskeletons (±)-76 and (±)-77 of (±)-cannabisativine. It shouldbe noted that the use of chiral oximes as substrate and/orchiral tartrate ester γ-silylallylboronates [28] could lead tothe synthesis of chiral molecules.
IMINIUM ION–VINYLSILANE CYCLIZATIONS
Iminium ion-vinylsilane cyclization reactions introducedby Overman et al. [29] appear very attractive for thepreparation of chiral 2,6-dialkyl tetrahydropyridines. Chiralsilicon-containing iminium ions are readily available fromamino acids and the initial stereogenic center at C-6 in theiminium intermediates could induce chirality to the newlyformed stereogenic center at C-2 (see Scheme 24).
Unfortunately, earlier mechanistic studies, particularlyfrom Overman et al. [30] have shown that the reaction mayoccur following two different pathways. In the first one, thecyclization of the iminium A gives the β-silyl carbo-cationintermediate B, which subsequently loses the trimethylsilylgroup to afford C (see Scheme 24). Alternatively, prior tocyclization, iminium A undergoes a [3,3]-sigmatropic aza-
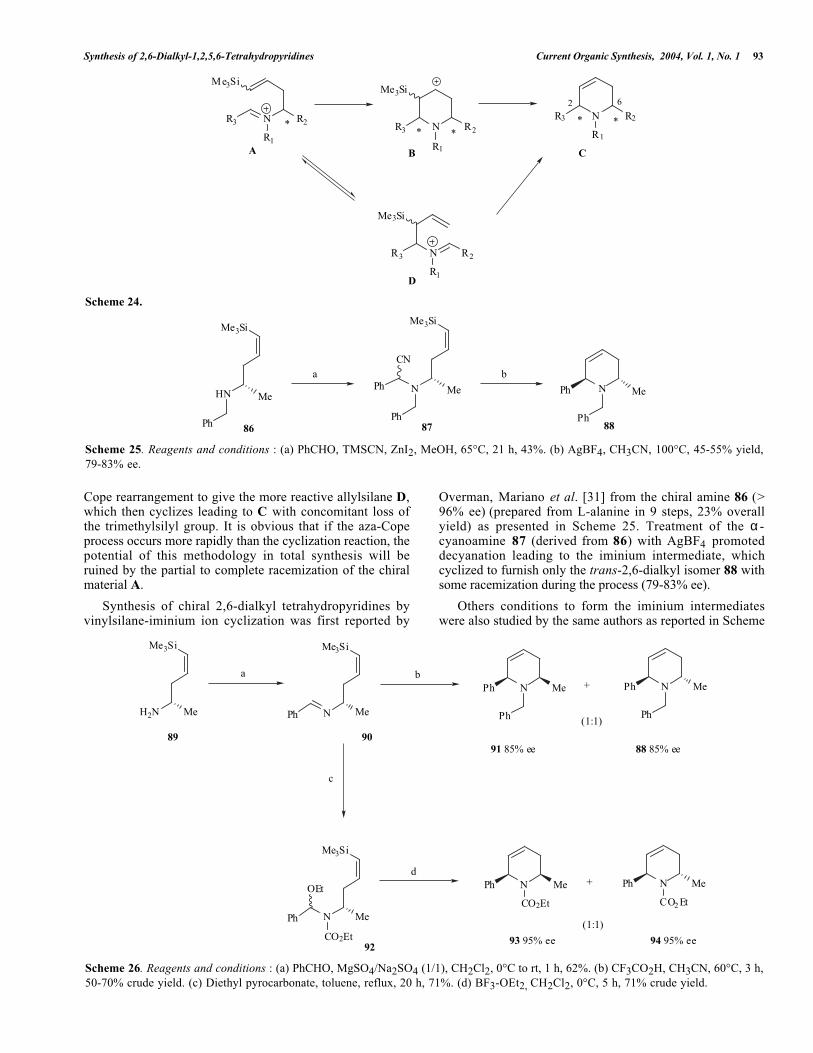
Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 93
D
A
N R2R3
Me3Si
R1
N R2R3
Me3Si
R1
B
N R2R3
Me3Si
R1 C
N R2R3
R1
***
* *
2 6
Scheme 24.
Me3Si
HN
Ph
Me
Me3Si
N
Ph
MePh
CN
N
Ph
MePh
86 87 88
a b
Scheme 25. Reagents and conditions : (a) PhCHO, TMSCN, ZnI2, MeOH, 65°C, 21 h, 43%. (b) AgBF4, CH3CN, 100°C, 45-55% yield,79-83% ee.
H2N
Me3Si
Me N
Me3Si
MePh
N
Me3Si
MePh
OEt
CO2Et
N Me
Ph
Ph
N Me
CO2Et
Ph N Me
CO2 Et
Ph
N Me
Ph
Ph+
(1:1)
+
(1:1)
a b
c
d
89 90
92
91 85% ee
93 95% ee
88 85% ee
94 95% ee
Scheme 26. Reagents and conditions : (a) PhCHO, MgSO4/Na2SO4 (1/1), CH2Cl2, 0°C to rt, 1 h, 62%. (b) CF3CO2H, CH3CN, 60°C, 3 h,50-70% crude yield. (c) Diethyl pyrocarbonate, toluene, reflux, 20 h, 71%. (d) BF3-OEt2, CH2Cl2, 0°C, 5 h, 71% crude yield.
Cope rearrangement to give the more reactive allylsilane D,which then cyclizes leading to C with concomitant loss ofthe trimethylsilyl group. It is obvious that if the aza-Copeprocess occurs more rapidly than the cyclization reaction, thepotential of this methodology in total synthesis will beruined by the partial to complete racemization of the chiralmaterial A.
Synthesis of chiral 2,6-dialkyl tetrahydropyridines byvinylsilane-iminium ion cyclization was first reported by
Overman, Mariano et al. [31] from the chiral amine 86 (>96% ee) (prepared from L-alanine in 9 steps, 23% overallyield) as presented in Scheme 25. Treatment of the α -cyanoamine 87 (derived from 86) with AgBF4 promoteddecyanation leading to the iminium intermediate, whichcyclized to furnish only the trans-2,6-dialkyl isomer 88 withsome racemization during the process (79-83% ee).
Others conditions to form the iminium intermediateswere also studied by the same authors as reported in Scheme
94 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
HN
Me3Si
Ph
OBn
Ph N
Me3Si
CN
Ph
OBn
NC6 H13
Ph
OBn
NPh
Ph
OBn
a
b
c
(±)-95 (±)-96
(±)-97 (±)-98
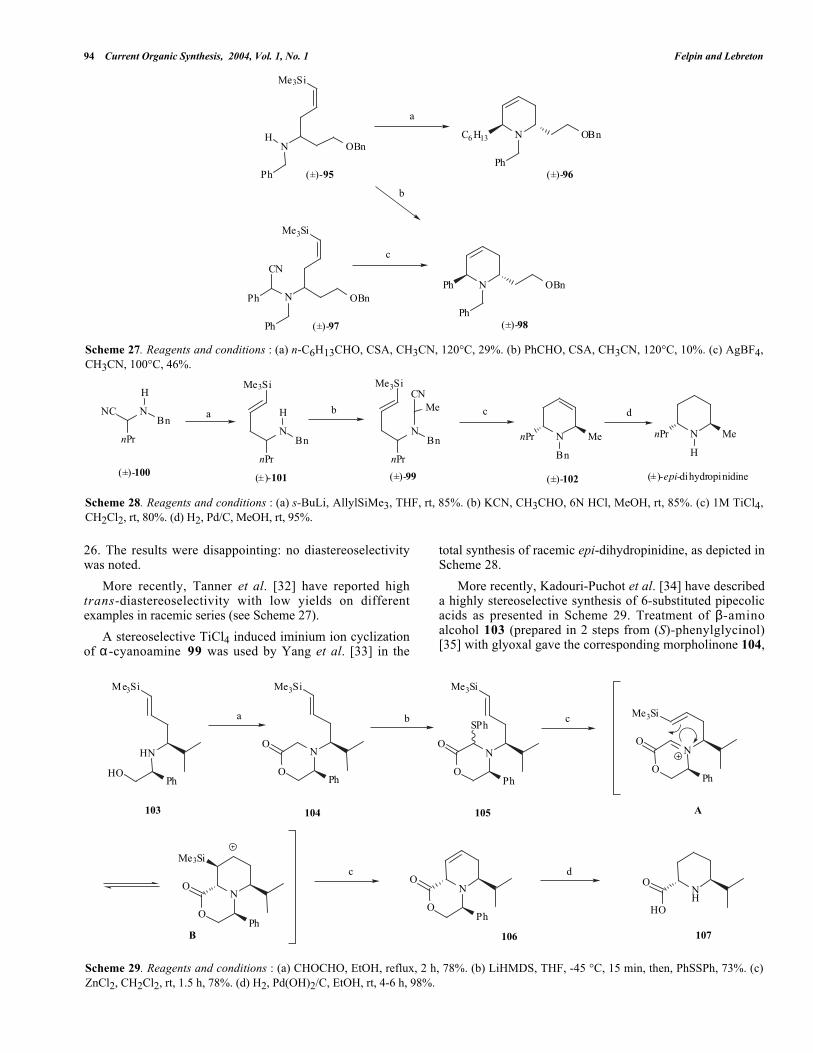
Scheme 27. Reagents and conditions : (a) n-C6H13CHO, CSA, CH3CN, 120°C, 29%. (b) PhCHO, CSA, CH3CN, 120°C, 10%. (c) AgBF4,CH3CN, 100°C, 46%.
NC NBn
nPr
H
NBn
nPr
H
Me3Si
NBn
nPr
Me3Si
MeCN
NnPr Me
H
NnPr Me
Bn
(±)-100 (±)-101 (±)-99 (±)-102 (±)-epi-dihydropinidine
a b c d
Scheme 28. Reagents and conditions : (a) s-BuLi, AllylSiMe3, THF, rt, 85%. (b) KCN, CH3CHO, 6N HCl, MeOH, rt, 85%. (c) 1M TiCl4,CH2Cl2, rt, 80%. (d) H2, Pd/C, MeOH, rt, 95%.
HN
HOPh
Me3Si
N
O
O
Ph
Me3Si
B
N
Me3Si
O
O
Ph
N
O
O
Ph
N
Me3Si
O
O
SPh
Ph
NH
O
HO
N
O
O
Ph
Me3Si
A
a b c
d
103 104 105
107
c
106
Scheme 29. Reagents and conditions : (a) CHOCHO, EtOH, reflux, 2 h, 78%. (b) LiHMDS, THF, -45 °C, 15 min, then, PhSSPh, 73%. (c)ZnCl2, CH2Cl2, rt, 1.5 h, 78%. (d) H2, Pd(OH)2/C, EtOH, rt, 4-6 h, 98%.
26. The results were disappointing: no diastereoselectivitywas noted.
More recently, Tanner et al. [32] have reported hightrans-diastereoselectivity with low yields on differentexamples in racemic series (see Scheme 27).
A stereoselective TiCl4 induced iminium ion cyclizationof α -cyanoamine 99 was used by Yang et al. [33] in the
total synthesis of racemic epi-dihydropinidine, as depicted inScheme 28.
More recently, Kadouri-Puchot et al. [34] have describeda highly stereoselective synthesis of 6-substituted pipecolicacids as presented in Scheme 29. Treatment of β-aminoalcohol 103 (prepared in 2 steps from (S)-phenylglycinol)[35] with glyoxal gave the corresponding morpholinone 104,

Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 95
BocN
SiMe3
H
N
H
a
108 109
Scheme 30. Reagents and conditions : (a) 2-methylpropanal, TiCl4, CH2Cl2, 0°C to rt, then addition of 108, 3 min, rt, 51%.
A
B
N
R2
R1
H
N R2
H
R1N
R2
SiMe3
R1
H
A
(ii)(i) protodesilylation
Scheme 31.
NH
OH
Ph
SiMe3
N
O
O
Ph
A
NO
Ph
Me3Si
OH
NH
HO2 C
ONPh
HMe3Si
OH
B
N
O OH
Ph
a
b
111 112
110 (S)-Baikiain acid
Scheme 32. Reagents and conditions : (a) CHOCHO, THF/H2O (1/1), rt, 48 h, 71%. (b) DMSO, (COCl)2, CH2Cl2, - 60°C, 10 min., then112, -60 °C, 30 min, NEt3, -60 °C to rt, 1 h, 70%.
which was alkylated with PhSSPh in the presence of base toafford the thioether derivative 105. Treatment of the lattercompound 105 with ZnCl2 induced the formation of theiminium ion A, which underwent cyclization with attack tothe allylsilane moiety from the less hindered face (oppositephenyl) to give the trans-2,6-disubstituted-tetrahydro-pyridine system 106 with an excellent diastereoselectivity(up to 95%). Upon treatment with H2 in the presence ofPearlman catalyst, the compound 106 was converted into 6-substituted pipecolic acid 107.
IMINIUM ION-ALLYLSILANE CYCLIZATIONS
A highly diastereoselective intramolecular iminium ion-allylsilane cyclization was reported by Taddei et al. [36].The chiral allylsilane 108 (prepared in 4 steps from N-Bocleucine) was added to a solution of 2-methylpropanal andTiCl4 to afford the cis-2,6-dialkyl-tetrahydropyridine 109 in51% yield (see Scheme 30). Under these conditions, the Bocprotecting group was cleaved and the Lewis acid promotedthe formation of the iminium species A (see Figure 31). Theauthors proposed two possible mechanisms via the iminiumA: (i) intramolecular attack to the allylic carbon close to thesilicon, (ii) a protodesilylation to give intermediate Bfollowed by an intramolecular ene-iminium reaction.
In connection with their work on iminium ion-vinylsilane cyclization (see above), Kadouri-Puchot et al.[34] reported a stereoselective synthesis of a protected trans-6-substituted baikiain acid 110. The condensation of the β-amino alcohol 111 (prepared in two steps from (S) -phenylglycinol) [35] with glyoxal led to the formation of theiminium ion A, which then cyclized leading to the stabilizedβ-silyl cation intermediate B to afford, after elimination ofthe axial TMS group, the hemiketal 112 in 71% yield. Thislatter compound 112 was converted into the lactone 110using Swern oxidation conditions.
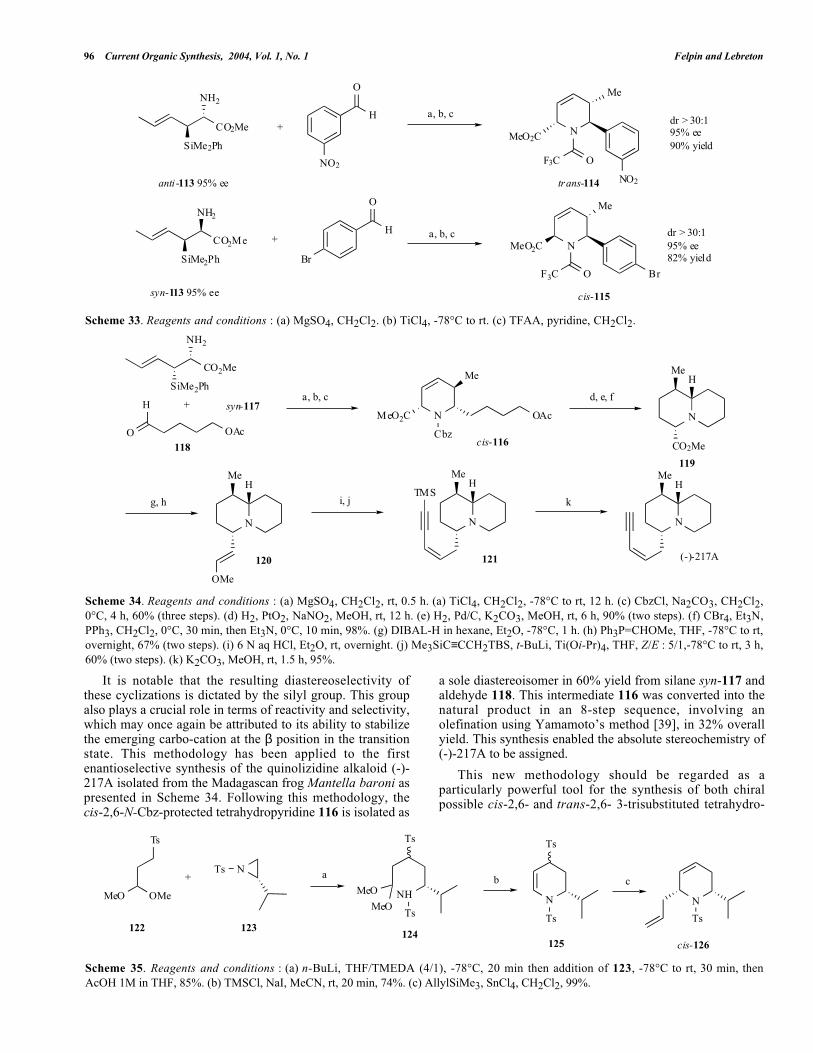
Very recently, Panek et al. [37] have reported a highlystereoselective methodology for the preparation of bothpossible cis-2,6- and trans-2,6-trisubstituted tetrahydro-pyridines. In a one-pot procedure, the silanes anti-113 orsyn-113 were condensed with different aldehydes to form thecorresponding imines. These were treated with an excess ofTiCl4 to afford, in a highly diastereo- and enantioselectiveprocess, the tetrahydropyridines (isolated as trifluoro-acetamides) as outlined by the two representative examplespresented in Scheme 33. It should be emphasized that thepreparation of both enantiomers of the silanes anti-113 andsyn-113 have previously been described by Panek et al. [38],giving access to the four possible cis-2,6- and trans-2,6-tetrahydropyridine stereoisomers.
96 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
CO2Me
NH2
SiMe2Ph
H
O
NO2
CO2Me
NH2
SiMe2Ph
H
O
Br
NMeO2C
Me
F3C O
NO2
NMeO2C
Me
F3C O Br
+dr > 30:195% ee90% yield
+dr > 30:195% ee82% yield
anti-113 95% ee
syn-113 95% ee
trans-114
cis-115
a, b, c
a, b, c
Scheme 33. Reagents and conditions : (a) MgSO4, CH2Cl2. (b) TiCl4, -78°C to rt. (c) TFAA, pyridine, CH2Cl2.
CO2Me
NH2
SiMe2Ph
O OAc
H
N
MeH
OMe
N
MeH
TMS
NMeO2C
Me
Cbz
OAc
N
MeH
N
MeH
CO2Me
+ syn-117
cis-116
a, b, c d, e, f
g, h i, j k
(-)-217A
118
119
121120
Scheme 34. Reagents and conditions : (a) MgSO4, CH2Cl2, rt, 0.5 h. (a) TiCl4, CH2Cl2, -78°C to rt, 12 h. (c) CbzCl, Na2CO3, CH2Cl2,0°C, 4 h, 60% (three steps). (d) H2, PtO2, NaNO2, MeOH, rt, 12 h. (e) H2, Pd/C, K2CO3, MeOH, rt, 6 h, 90% (two steps). (f) CBr4, Et3N,PPh3, CH2Cl2, 0°C, 30 min, then Et3N, 0°C, 10 min, 98%. (g) DIBAL-H in hexane, Et2O, -78°C, 1 h. (h) Ph3P=CHOMe, THF, -78°C to rt,overnight, 67% (two steps). (i) 6 N aq HCl, Et2O, rt, overnight. (j) Me3SiC≡CCH2TBS, t-BuLi, Ti(Oi-Pr)4, THF, Z/E : 5/1,-78°C to rt, 3 h,60% (two steps). (k) K2CO3, MeOH, rt, 1.5 h, 95%.
OMe
Ts
MeO
NTs
Ts
N
Ts
N
Ts
Ts
MeO
MeONH
Ts
a b+
122 123
c
124125 cis-126
Scheme 35. Reagents and conditions : (a) n-BuLi, THF/TMEDA (4/1), -78°C, 20 min then addition of 123, -78°C to rt, 30 min, thenAcOH 1M in THF, 85%. (b) TMSCl, NaI, MeCN, rt, 20 min, 74%. (c) AllylSiMe3, SnCl4, CH2Cl2, 99%.
It is notable that the resulting diastereoselectivity ofthese cyclizations is dictated by the silyl group. This groupalso plays a crucial role in terms of reactivity and selectivity,which may once again be attributed to its ability to stabilizethe emerging carbo-cation at the β position in the transitionstate. This methodology has been applied to the firstenantioselective synthesis of the quinolizidine alkaloid (-)-217A isolated from the Madagascan frog Mantella baroni aspresented in Scheme 34. Following this methodology, thecis-2,6-N-Cbz-protected tetrahydropyridine 116 is isolated as
a sole diastereoisomer in 60% yield from silane syn-117 andaldehyde 118. This intermediate 116 was converted into thenatural product in an 8-step sequence, involving anolefination using Yamamoto’s method [39], in 32% overallyield. This synthesis enabled the absolute stereochemistry of(-)-217A to be assigned.
This new methodology should be regarded as aparticularly powerful tool for the synthesis of both chiralpossible cis-2,6- and trans-2,6- 3-trisubstituted tetrahydro-
Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 97
HN CO2Me
Ns
o
OBn
N
Ns
CO2Me
B
NH
CO2 H
N CO2 Me
Ns
BnO
N
Ns
CO2Me N
Ns
CO2Me
N
Ns
CO2MeBnO
a b+
127 128
c
129 130
+
d, e
132131
133
98:2
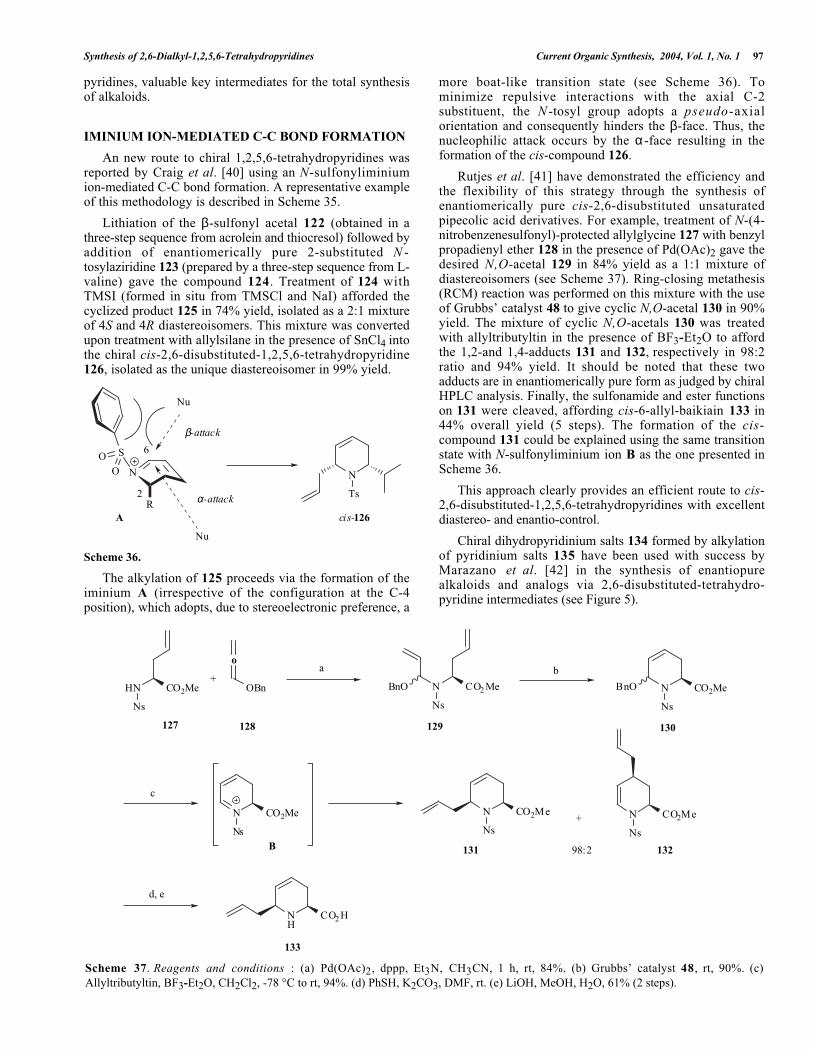
Scheme 37. Reagents and conditions : (a) Pd(OAc)2, dppp, Et3N, CH3CN, 1 h, rt, 84%. (b) Grubbs’ catalyst 48, rt, 90%. (c)Allyltributyltin, BF3-Et2O, CH2Cl2, -78 °C to rt, 94%. (d) PhSH, K2CO3, DMF, rt. (e) LiOH, MeOH, H2O, 61% (2 steps).
pyridines, valuable key intermediates for the total synthesisof alkaloids.
IMINIUM ION-MEDIATED C-C BOND FORMATION
An new route to chiral 1,2,5,6-tetrahydropyridines wasreported by Craig et al. [40] using an N-sulfonyliminiumion-mediated C-C bond formation. A representative exampleof this methodology is described in Scheme 35.
Lithiation of the β-sulfonyl acetal 122 (obtained in athree-step sequence from acrolein and thiocresol) followed byaddition of enantiomerically pure 2-substituted N -tosylaziridine 123 (prepared by a three-step sequence from L-valine) gave the compound 124. Treatment of 124 withTMSI (formed in situ from TMSCl and NaI) afforded thecyclized product 125 in 74% yield, isolated as a 2:1 mixtureof 4S and 4R diastereoisomers. This mixture was convertedupon treatment with allylsilane in the presence of SnCl4 intothe chiral cis-2,6-disubstituted-1,2,5,6-tetrahydropyridine126, isolated as the unique diastereoisomer in 99% yield.
N
Ts
A
N
R
S
OO
cis-126
2
6
Nu
Nu
β-attack
α-attack
Scheme 36.
The alkylation of 125 proceeds via the formation of theiminium A (irrespective of the configuration at the C-4position), which adopts, due to stereoelectronic preference, a
more boat-like transition state (see Scheme 36). Tominimize repulsive interactions with the axial C-2substituent, the N -tosyl group adopts a pseudo-axialorientation and consequently hinders the β-face. Thus, thenucleophilic attack occurs by the α -face resulting in theformation of the cis-compound 126.
Rutjes et al. [41] have demonstrated the efficiency andthe flexibility of this strategy through the synthesis ofenantiomerically pure cis-2,6-disubstituted unsaturatedpipecolic acid derivatives. For example, treatment of N-(4-nitrobenzenesulfonyl)-protected allylglycine 127 with benzylpropadienyl ether 128 in the presence of Pd(OAc)2 gave thedesired N,O-acetal 129 in 84% yield as a 1:1 mixture ofdiastereoisomers (see Scheme 37). Ring-closing metathesis(RCM) reaction was performed on this mixture with the useof Grubbs’ catalyst 48 to give cyclic N,O-acetal 130 in 90%yield. The mixture of cyclic N,O-acetals 130 was treatedwith allyltributyltin in the presence of BF3-Et2O to affordthe 1,2-and 1,4-adducts 131 and 132, respectively in 98:2ratio and 94% yield. It should be noted that these twoadducts are in enantiomerically pure form as judged by chiralHPLC analysis. Finally, the sulfonamide and ester functionson 131 were cleaved, affording cis-6-allyl-baikiain 133 in44% overall yield (5 steps). The formation of the cis-compound 131 could be explained using the same transitionstate with N-sulfonyliminium ion B as the one presented inScheme 36.
This approach clearly provides an efficient route to cis-2,6-disubstituted-1,2,5,6-tetrahydropyridines with excellentdiastereo- and enantio-control.
Chiral dihydropyridinium salts 134 formed by alkylationof pyridinium salts 135 have been used with success byMarazano et al. [42] in the synthesis of enantiopurealkaloids and analogs via 2,6-disubstituted-tetrahydro-pyridine intermediates (see Figure 5).
98 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
C12H25SO3
N
OHPh
H
NMe
OPh
HCl
NH
Me C11H23
NMe
OPh
N
OMgClPh
H
MeH2O
N
OHPh
H
Me C11H23
N
OHPh
H
Me
134 (R=Me)
N
OHPh
H
Me C11H23
a
+ +
(+)-solenopsin A
b
c
135
136 137138 139
90:10 66:34
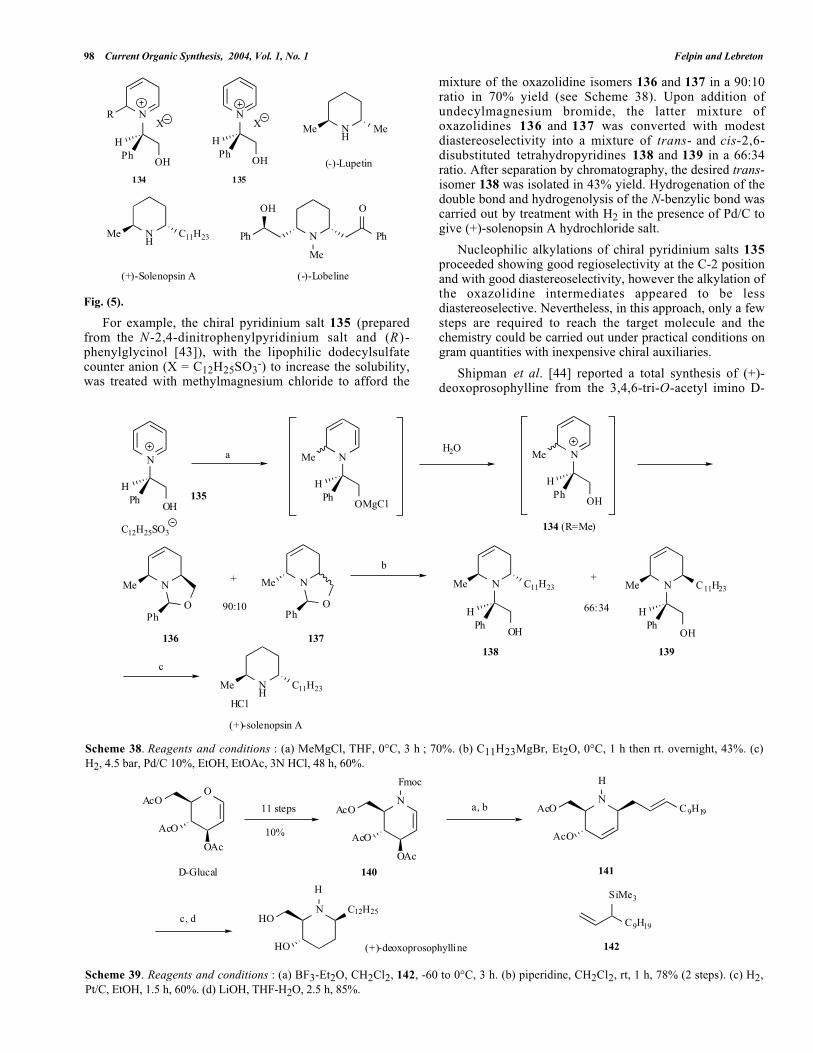
Scheme 38. Reagents and conditions : (a) MeMgCl, THF, 0°C, 3 h ; 70%. (b) C11H23MgBr, Et2O, 0°C, 1 h then rt. overnight, 43%. (c)H2, 4.5 bar, Pd/C 10%, EtOH, EtOAc, 3N HCl, 48 h, 60%.
O
OAc
AcO
AcO
N
HO
HO
H
C12H25
N
OAc
AcO
AcO
Fmoc
C9H19
SiMe3
N
AcO
AcO
H
C9H19
D-Glucal 140
11 steps a, b
(+)-deoxoprosophylline
c, d
141
142
10%
Scheme 39. Reagents and conditions : (a) BF3-Et2O, CH2Cl2, 142, -60 to 0°C, 3 h. (b) piperidine, CH2Cl2, rt, 1 h, 78% (2 steps). (c) H2,Pt/C, EtOH, 1.5 h, 60%. (d) LiOH, THF-H2O, 2.5 h, 85%.
N
OHPh
H
R
NH
Me C11H23
NH
Me Me
N
Me
Ph Ph
OOH
N
OHPh
H
X X
(+)-Solenopsin A
(-)-Lupetin
(-)-Lobeline
134 135
Fig. (5).
For example, the chiral pyridinium salt 135 (preparedfrom the N -2,4-dinitrophenylpyridinium salt and (R) -phenylglycinol [43]), with the lipophilic dodecylsulfatecounter anion (X = C12H25SO3
-) to increase the solubility,was treated with methylmagnesium chloride to afford the
mixture of the oxazolidine isomers 136 and 137 in a 90:10ratio in 70% yield (see Scheme 38). Upon addition ofundecylmagnesium bromide, the latter mixture ofoxazolidines 136 and 137 was converted with modestdiastereoselectivity into a mixture of trans- and cis-2,6-disubstituted tetrahydropyridines 138 and 139 in a 66:34ratio. After separation by chromatography, the desired trans-isomer 138 was isolated in 43% yield. Hydrogenation of thedouble bond and hydrogenolysis of the N-benzylic bond wascarried out by treatment with H2 in the presence of Pd/C togive (+)-solenopsin A hydrochloride salt.
Nucleophilic alkylations of chiral pyridinium salts 135proceeded showing good regioselectivity at the C-2 positionand with good diastereoselectivity, however the alkylation ofthe oxazolidine intermediates appeared to be lessdiastereoselective. Nevertheless, in this approach, only a fewsteps are required to reach the target molecule and thechemistry could be carried out under practical conditions ongram quantities with inexpensive chiral auxiliaries.
Shipman et al. [44] reported a total synthesis of (+)-deoxoprosophylline from the 3,4,6-tri-O-acetyl imino D-

Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 99
NO
O
AcOOAc
A
NO
O
OAc
OAc
B
6
Nu
6
Scheme 40.
glucal 140 prepared from D-glucal in 11 steps using animinium ion-mediated C-C bond formation as outlined inScheme 39.
Reaction of the imino glucal 140 with trimethyl-(1-nonyl-allyl)-silane 142 in the presence of BF3-Et2O affordedafter cleavage of the Fmoc group the cis-2,6-disubstituted-1,2,5,6-tetrahydropyridine 141 as the major diastereoisomer(cis/trans in a 9 to 1 ratio). The alkylation of 140 proceedsvia the formation of an intermediate N-acyliminium ion,which is in conformational equilibrium between A and B(see Scheme 40). Also, steric repulsion between the C-6acetoxymethyl group and the N-Fmoc group disfavours Arelative to B. Stereoelectronically controlled axial attack ofthe nucleophile occurs from the top face to give the cis-isomer 1 4 1 . From this latter intermediate, (+)-deoxoprosophylline was obtained by hydrogenation of thedouble bonds and saponification of the acetate groups.
OLEFIN METATHESIS REACTIONS
During this last decade, RCM reaction has emerged as apowerful method for C-C bond formation as demonstratedby the total synthesis of numerous natural products [45]. Aspart of a continuing program directed toward thedevelopment of an efficient and novel strategy for theconstruction of piperidinic and pyrrolidinic alkaloids [46],we have developed a novel access to enantio-enriched trans-2,6-disubstituted-1,2,5,6-tetrahydropyridines [47]. In thiscontext, it appeared to us that the double bond of thetetrahydropyridine motif A could be generated by an RCMreaction from the corresponding diethylenic amine B (seeScheme 41).
R1 N
PG
R2
RCM
A
R1 N
PG
R2
B
Scheme 41.
Our synthetic route started from the commerciallyavailable Garner aldehyde 143 on which a stereoselectiveolefination, via a Horner-Wadsworth-Emmons (HWE)reaction, introduced the first olefin required for the RCMstep (see Scheme 42) to furnish 144. The cleavage of bothoxazolidine and N-Boc protecting groups of 144 in the samestep afforded the expected amino alcohol 145 in quantitativeyield (97%). Introduction of the second olefin was carriedout by diastereoselective allylation of an imine generated by
condensation of 145 with an appropriate aldehyde. Asexpected, the stereoselectivity in favor of the trans isomer(separated from the cis by flash chromatography) could beexplained by the generally accepted transition state underchelation control (see Figure 6).
OMgBr
NH
R
Disfavored
Favored
Fig. (6).
Next, we turned our attention toward cyclization byRCM. However, it was well established that the rutheniumcatalyst was incompatible with free amine [44e, 44f].Consequently, we decided to transform the amino alcoholfunction of 146 , 148 and 150 as its correspondingoxazolidinone 152-154. Thus, the RCM reaction on theoxazolidinone derivative 152-154 afforded the expectedtetrahydropyridines 155-157 in good yields. Thismethodology was applied to the synthesis of the non-natural(-)-3-epi-deoxoprosopinine, an analog of the Prosopisalkaloid family. Epoxidation of the double bond of 157 withmCPBA afforded a separable mixture of exo-158 and endo-epoxide 159 with good selectivity (respectively 1/9). Thestructure was confirmed by X-ray crystallographic analysis.Finally, the regioselective reduction of 159 with Super-Hydride® and the subsequent hydrolysis of oxazolidinone inbasic media afforded the desired (-)-3-epi-deoxoprosopinine[48].
Recently, Couty et al. [49] have described the synthesisof optically active cis and trans-2,6-disubstituted-1,2,5,6-tetrahydropyridines. Starting from ketone 160, readilyavailable from (S)-phenylalaninol in 3 steps, a stereoselectivereduction with NaBH4/EtOH (76% de) or under Lucheconditions, NaBH4/CeCl3/EtOH (58% de) gave bothstereoisomers 161 and 162, respectively (see Scheme 43).Treatment of 161 with NaH led to an intramolecularcyclization to furnish oxazolidinone 163 . This latterintermediate 163 was reacted with allyltrimethylsilane in thepresence of TiCl4 to afford stereoselectively the transoxazolidinone 164. Next, the oxidation of alcohol withDess-Martin periodinane followed by Wittig olefination gavethe diethylenic amine 1 6 5 . The formation of the
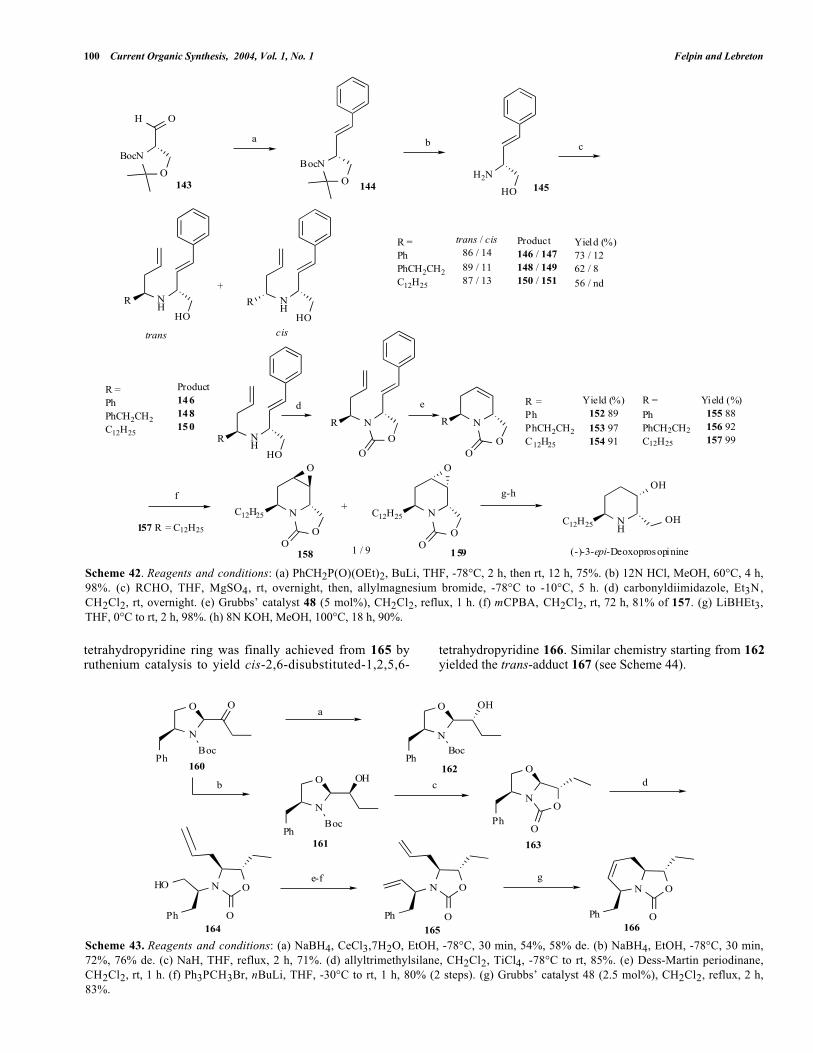
100 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
H O
BocN
O
NH
HO
R
C12H25 N
OO
O
NH
HO
R
NH
HO
R
BocN
O
C12H25 N
OO
O
NR
O
O
H2N
HO
NH
OH
OHC12H25
R N
OO
+
1 / 9
+
(-)-3-epi-Deoxoprosopinine
a b c
d e
f g-h
143 144 145
158 1 59
R = PhPhCH2CH2C12H25
trans / cis86 / 14
89 / 1187 / 13
Yield (%) 73 / 1262 / 8
56 / nd
157 R = C12H25
Yield (%) 152 89
153 97154 91
Yield (%)
155 88156 92157 99
Product 146 / 147148 / 149150 / 151
R = PhPhCH2CH2C12H25
Product 14 614 815 0
R = PhPhCH2CH2C12H25
R =
PhPhCH2CH2
C12H25
trans cis
Scheme 42. Reagents and conditions: (a) PhCH2P(O)(OEt)2, BuLi, THF, -78°C, 2 h, then rt, 12 h, 75%. (b) 12N HCl, MeOH, 60°C, 4 h,98%. (c) RCHO, THF, MgSO4, rt, overnight, then, allylmagnesium bromide, -78°C to -10°C, 5 h. (d) carbonyldiimidazole, Et3N,CH2Cl2, rt, overnight. (e) Grubbs’ catalyst 48 (5 mol%), CH2Cl2, reflux, 1 h. (f) mCPBA, CH2Cl2, rt, 72 h, 81% of 157. (g) LiBHEt3,THF, 0°C to rt, 2 h, 98%. (h) 8N KOH, MeOH, 100°C, 18 h, 90%.
N
O O
PhBoc
N O
O
HO
Ph
N
O OH
PhBoc
N O
OPh
N
O OH
PhBoc
N
O
Ph
O
O
N O
OPh
a
b c d
e-f g
160
161
162
163
164 165 166
Scheme 43. Reagents and conditions: (a) NaBH4, CeCl3,7H2O, EtOH, -78°C, 30 min, 54%, 58% de. (b) NaBH4, EtOH, -78°C, 30 min,72%, 76% de. (c) NaH, THF, reflux, 2 h, 71%. (d) allyltrimethylsilane, CH2Cl2, TiCl4, -78°C to rt, 85%. (e) Dess-Martin periodinane,CH2Cl2, rt, 1 h. (f) Ph3PCH3Br, nBuLi, THF, -30°C to rt, 1 h, 80% (2 steps). (g) Grubbs’ catalyst 48 (2.5 mol%), CH2Cl2, reflux, 2 h,83%.
tetrahydropyridine ring was finally achieved from 165 byruthenium catalysis to yield cis-2,6-disubstituted-1,2,5,6-
tetrahydropyridine 166. Similar chemistry starting from 162yielded the trans-adduct 167 (see Scheme 44).
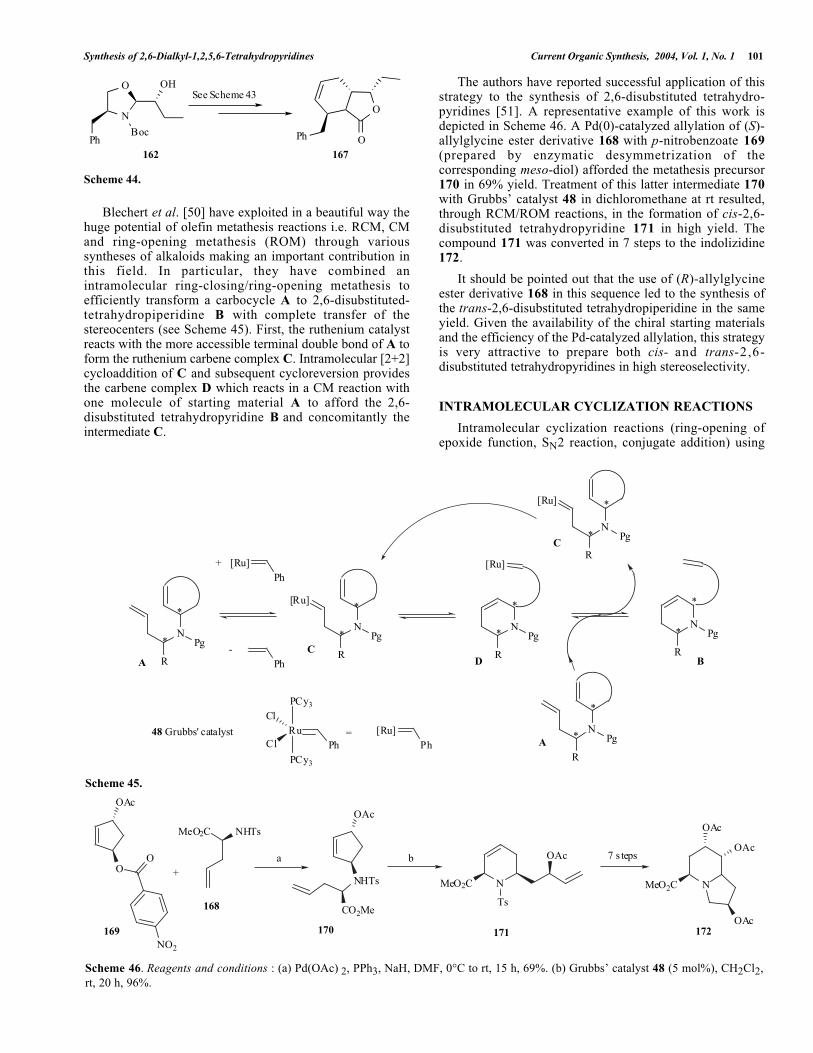
Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 101
N
O OH
PhBoc
O
OPh
See Scheme 43
162 167
Scheme 44.
N
R
Pg
[Ru]Ph
N
[Ru]
R
Pg
[Ru]
N
R
Pg
C
N
[Ru]
R
Pg
Ph
N
R
Pg
AC
D
ARu
PCy3
PCy3
Ph
Cl
Cl[Ru]
Ph
N
R
Pg
B
+
-*
*
*
*
*
*
*
*
*
*
*
*
48 Grubbs' catalyst =
Scheme 45.
OAc
O
NO2
O
MeO2C NHTs
OAc
NHTs
CO2Me
NMeO2C
OAc
Ts
N
OAc
OAc
OAc
MeO2C+
a 7 s tepsb
168
169 170 171 172
Scheme 46. Reagents and conditions : (a) Pd(OAc) 2, PPh3, NaH, DMF, 0°C to rt, 15 h, 69%. (b) Grubbs’ catalyst 48 (5 mol%), CH2Cl2,rt, 20 h, 96%.
Blechert et al. [50] have exploited in a beautiful way thehuge potential of olefin metathesis reactions i.e. RCM, CMand ring-opening metathesis (ROM) through varioussyntheses of alkaloids making an important contribution inthis field. In particular, they have combined anintramolecular ring-closing/ring-opening metathesis toefficiently transform a carbocycle A to 2,6-disubstituted-tetrahydropiperidine B with complete transfer of thestereocenters (see Scheme 45). First, the ruthenium catalystreacts with the more accessible terminal double bond of A toform the ruthenium carbene complex C. Intramolecular [2+2]cycloaddition of C and subsequent cycloreversion providesthe carbene complex D which reacts in a CM reaction withone molecule of starting material A to afford the 2,6-disubstituted tetrahydropyridine B and concomitantly theintermediate C.
The authors have reported successful application of thisstrategy to the synthesis of 2,6-disubstituted tetrahydro-pyridines [51]. A representative example of this work isdepicted in Scheme 46. A Pd(0)-catalyzed allylation of (S)-allylglycine ester derivative 168 with p-nitrobenzoate 169(prepared by enzymatic desymmetrization of thecorresponding meso-diol) afforded the metathesis precursor170 in 69% yield. Treatment of this latter intermediate 170with Grubbs’ catalyst 48 in dichloromethane at rt resulted,through RCM/ROM reactions, in the formation of cis-2,6-disubstituted tetrahydropyridine 171 in high yield. Thecompound 171 was converted in 7 steps to the indolizidine172.
It should be pointed out that the use of (R)-allylglycineester derivative 168 in this sequence led to the synthesis ofthe trans-2,6-disubstituted tetrahydropiperidine in the sameyield. Given the availability of the chiral starting materialsand the efficiency of the Pd-catalyzed allylation, this strategyis very attractive to prepare both cis- and trans-2,6-disubstituted tetrahydropyridines in high stereoselectivity.
INTRAMOLECULAR CYCLIZATION REACTIONS
Intramolecular cyclization reactions (ring-opening ofepoxide function, SN2 reaction, conjugate addition) using
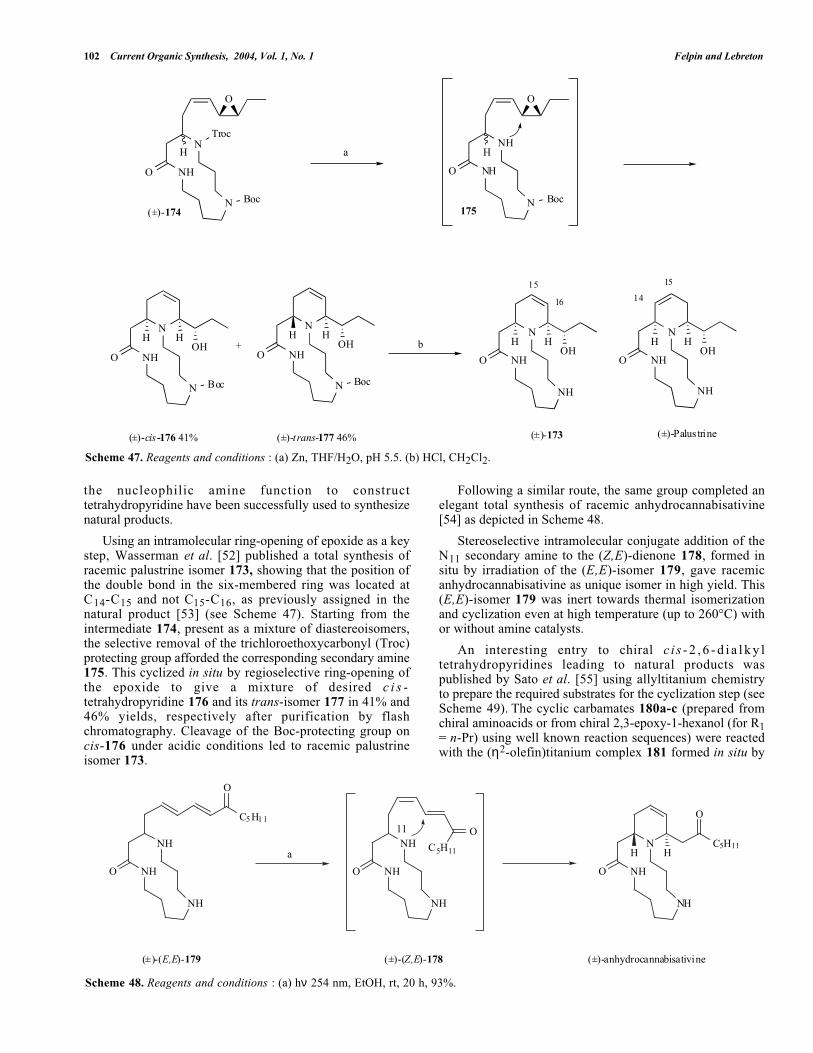
102 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
N
O NH
N Boc
H
Troc
O
NH
O NH
N Boc
H
O
NH
OHO NH
NH
H
NH
OHO NH
N
H
Boc
NH H
O NH
N Boc
OHN
H HOH
O NH
NH
(±)-173
+
(±)-Palus trine
15
1416
15
(±)-174
(±)-cis -176 41% (±)-trans-177 46%
a
b
175
Scheme 47. Reagents and conditions : (a) Zn, THF/H2O, pH 5.5. (b) HCl, CH2Cl2.
NH
O NH
NH
C5 H1 1
O
NH
O NH
NH
O
C5H11N
H
O NH
NH
HC5H11
O
(±)-(E,E)-179
11
(±)-(Z,E)-178 (±)-anhydrocannabisativine
a
Scheme 48. Reagents and conditions : (a) hν 254 nm, EtOH, rt, 20 h, 93%.
the nucleophilic amine function to constructtetrahydropyridine have been successfully used to synthesizenatural products.
Using an intramolecular ring-opening of epoxide as a keystep, Wasserman et al. [52] published a total synthesis ofracemic palustrine isomer 173, showing that the position ofthe double bond in the six-membered ring was located atC14-C15 and not C15-C16, as previously assigned in thenatural product [53] (see Scheme 47). Starting from theintermediate 174, present as a mixture of diastereoisomers,the selective removal of the trichloroethoxycarbonyl (Troc)protecting group afforded the corresponding secondary amine175. This cyclized in situ by regioselective ring-opening ofthe epoxide to give a mixture of desired c i s -tetrahydropyridine 176 and its trans-isomer 177 in 41% and46% yields, respectively after purification by flashchromatography. Cleavage of the Boc-protecting group oncis-176 under acidic conditions led to racemic palustrineisomer 173.
Following a similar route, the same group completed anelegant total synthesis of racemic anhydrocannabisativine[54] as depicted in Scheme 48.
Stereoselective intramolecular conjugate addition of theN11 secondary amine to the (Z,E)-dienone 178, formed insitu by irradiation of the (E,E)-isomer 179, gave racemicanhydrocannabisativine as unique isomer in high yield. This(E,E)-isomer 179 was inert towards thermal isomerizationand cyclization even at high temperature (up to 260°C) withor without amine catalysts.
An interesting entry to chiral c i s - 2 , 6 - d i a l k y ltetrahydropyridines leading to natural products waspublished by Sato et al. [55] using allyltitanium chemistryto prepare the required substrates for the cyclization step (seeScheme 49). The cyclic carbamates 180a-c (prepared fromchiral aminoacids or from chiral 2,3-epoxy-1-hexanol (for R1= n-Pr) using well known reaction sequences) were reactedwith the (η2-olefin)titanium complex 181 formed in situ by
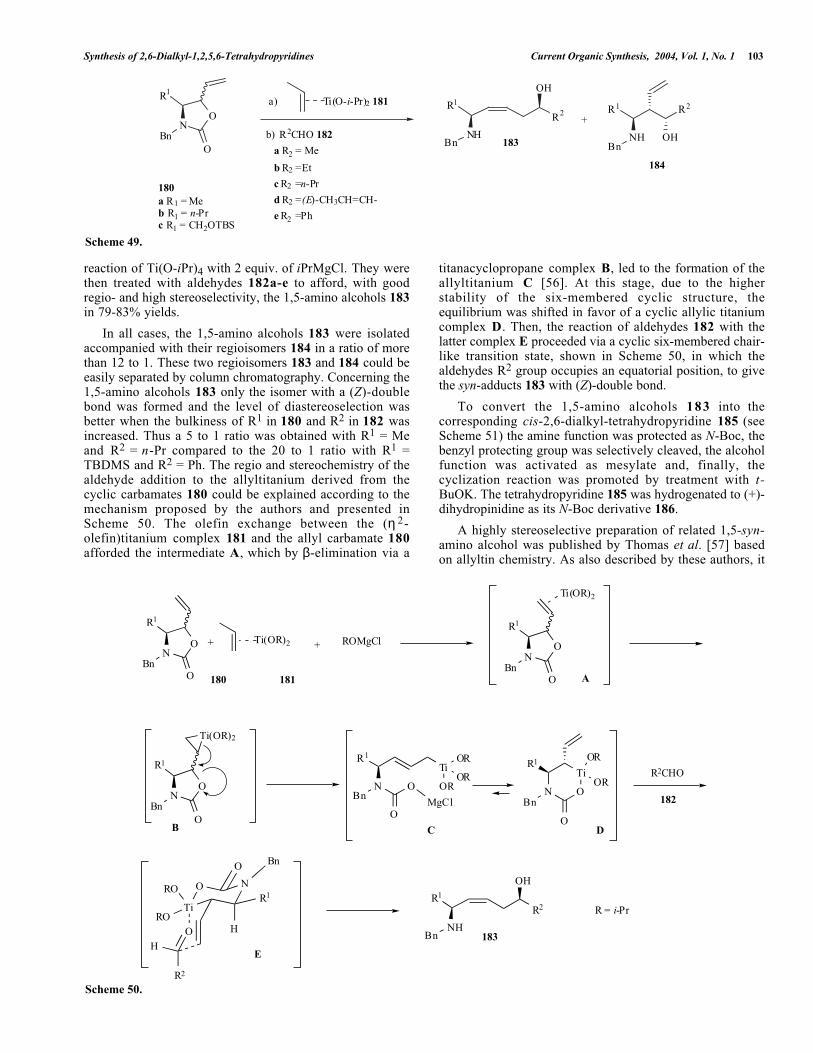
Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 103
NO
OBn
R1
R1
R2
NH
OH
Bn NH OHBn
R1 R2Ti(O-i-Pr)2 181a)
b) R2CHO 182
a R2 = Me
b R2 =Et
c R2 =n-Pr
d R2 =(E)-CH3CH=CH-
e R2 =Ph
+
180a R1 = Me b R1 = n-Pr c R1 = CH2OTBS
183
184
Scheme 49.
NO
OBn
R1
NO
OBn
R1
Ti(OR)2
Ti(OR)2 ROMgCl
ON
O
Bn
R1
MgCl
TiOR
OROR
N O
Ti
O
OR
OR
R1
Bn
Ti(OR)2
A
O
Ti
N
O
R2
H
O
H
R1RO
RO
Bn
R1
R2
NH
OH
Bn
B C D
R2CHO
183
180
+
181
+
182
R = i-Pr
E
NO
OBn
R1
Scheme 50.
reaction of Ti(O-iPr)4 with 2 equiv. of iPrMgCl. They werethen treated with aldehydes 182a-e to afford, with goodregio- and high stereoselectivity, the 1,5-amino alcohols 183in 79-83% yields.
In all cases, the 1,5-amino alcohols 183 were isolatedaccompanied with their regioisomers 184 in a ratio of morethan 12 to 1. These two regioisomers 183 and 184 could beeasily separated by column chromatography. Concerning the1,5-amino alcohols 183 only the isomer with a (Z)-doublebond was formed and the level of diastereoselection wasbetter when the bulkiness of R1 in 180 and R2 in 182 wasincreased. Thus a 5 to 1 ratio was obtained with R1 = Meand R2 = n-Pr compared to the 20 to 1 ratio with R1 =TBDMS and R2 = Ph. The regio and stereochemistry of thealdehyde addition to the allyltitanium derived from thecyclic carbamates 180 could be explained according to themechanism proposed by the authors and presented inScheme 50. The olefin exchange between the (η 2-olefin)titanium complex 181 and the allyl carbamate 180afforded the intermediate A, which by β-elimination via a
titanacyclopropane complex B, led to the formation of theallyltitanium C [56]. At this stage, due to the higherstability of the six-membered cyclic structure, theequilibrium was shifted in favor of a cyclic allylic titaniumcomplex D. Then, the reaction of aldehydes 182 with thelatter complex E proceeded via a cyclic six-membered chair-like transition state, shown in Scheme 50, in which thealdehydes R2 group occupies an equatorial position, to givethe syn-adducts 183 with (Z)-double bond.
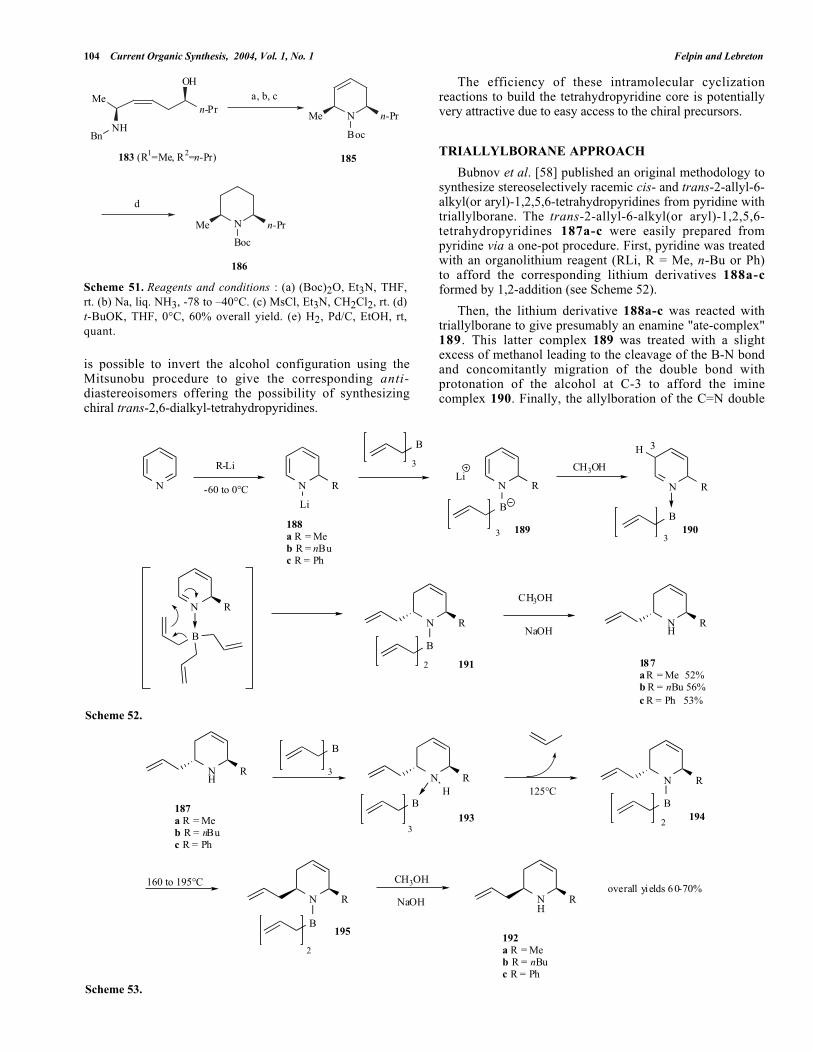
To convert the 1,5-amino alcohols 183 into thecorresponding cis-2,6-dialkyl-tetrahydropyridine 185 (seeScheme 51) the amine function was protected as N-Boc, thebenzyl protecting group was selectively cleaved, the alcoholfunction was activated as mesylate and, finally, thecyclization reaction was promoted by treatment with t-BuOK. The tetrahydropyridine 185 was hydrogenated to (+)-dihydropinidine as its N-Boc derivative 186.
A highly stereoselective preparation of related 1,5-syn-amino alcohol was published by Thomas et al. [57] basedon allyltin chemistry. As also described by these authors, it
104 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
Men-Pr
NH
OH
Bn
NMe n-Pr
Boc
NMe n-Pr
Boc
183 (R1=Me, R2=n-Pr)
a, b, c
d
185
186
Scheme 51. Reagents and conditions : (a) (Boc)2O, Et3N, THF,rt. (b) Na, liq. NH3, -78 to –40°C. (c) MsCl, Et3N, CH2Cl2, rt. (d)t-BuOK, THF, 0°C, 60% overall yield. (e) H2, Pd/C, EtOH, rt,quant.
N N R
Li
B
N R
BN R
B
LiN R
B
CH3OH
NaOH
CH3OH
NH
R
N R
B
H
R-Li
-60 to 0°C
188a R = Meb R = nBuc R = Ph
3
33
2 187a R = Me 52%b R = nBu 56%c R = Ph 53%
189
3
190
191
Scheme 52.
NH
R
B
N RH
B
NH
R
CH3OH
NaOHN R
B
N R
B
3
32
125°C
160 to 195°C
2
overall yields 60-70%
187a R = Me b R = nBu c R = Ph
192a R = Me b R = nBu c R = Ph
193 194
195
Scheme 53.
is possible to invert the alcohol configuration using theMitsunobu procedure to give the corresponding anti-diastereoisomers offering the possibility of synthesizingchiral trans-2,6-dialkyl-tetrahydropyridines.
The efficiency of these intramolecular cyclizationreactions to build the tetrahydropyridine core is potentiallyvery attractive due to easy access to the chiral precursors.
TRIALLYLBORANE APPROACH
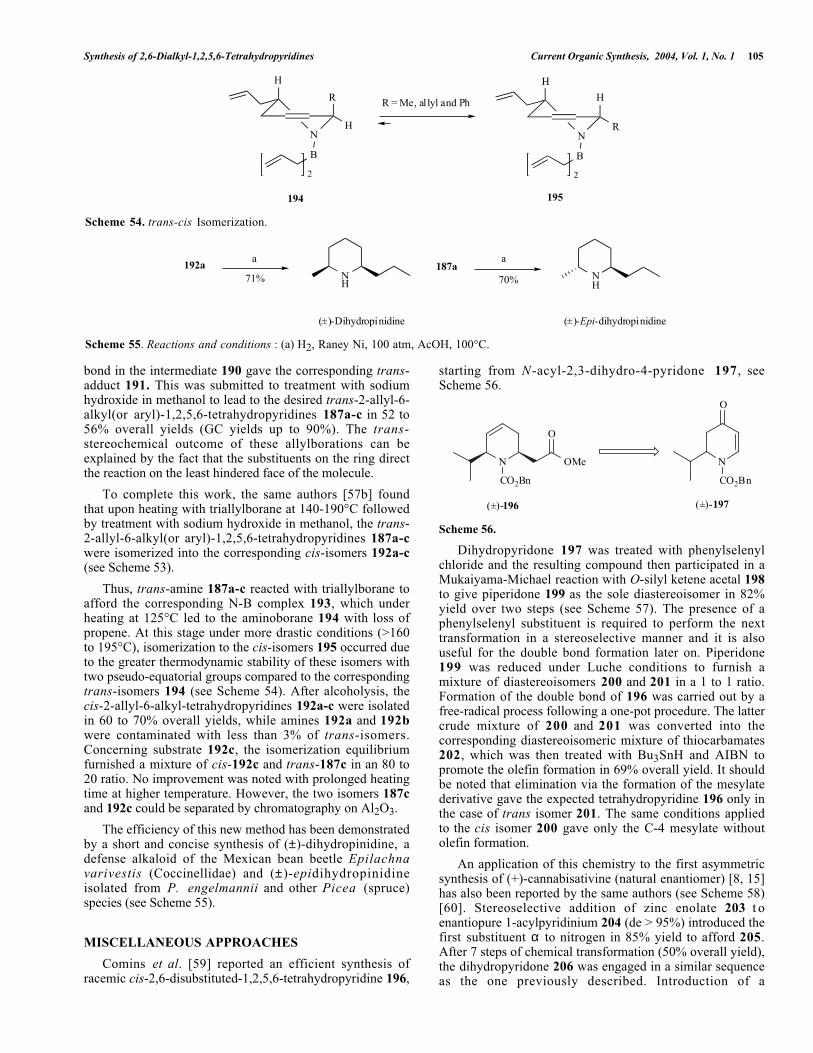
Bubnov et al. [58] published an original methodology tosynthesize stereoselectively racemic cis- and trans-2-allyl-6-alkyl(or aryl)-1,2,5,6-tetrahydropyridines from pyridine withtriallylborane. The trans-2-allyl-6-alkyl(or aryl)-1,2,5,6-tetrahydropyridines 187a-c were easily prepared frompyridine via a one-pot procedure. First, pyridine was treatedwith an organolithium reagent (RLi, R = Me, n-Bu or Ph)to afford the corresponding lithium derivatives 188a-cformed by 1,2-addition (see Scheme 52).
Then, the lithium derivative 188a-c was reacted withtriallylborane to give presumably an enamine "ate-complex"189. This latter complex 189 was treated with a slightexcess of methanol leading to the cleavage of the B-N bondand concomitantly migration of the double bond withprotonation of the alcohol at C-3 to afford the iminecomplex 190. Finally, the allylboration of the C=N double
Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 105
N
H
H
R
B
N
H
R
H
B
2 2
R = Me, al lyl and Ph
194 195
Scheme 54. trans-cis Isomerization.
NH
(±)-Epi-dihydropinidine
NH
(±)-Dihydropinidine
192a 187aa a
71% 70%
Scheme 55. Reactions and conditions : (a) H2, Raney Ni, 100 atm, AcOH, 100°C.
bond in the intermediate 190 gave the corresponding trans-adduct 191. This was submitted to treatment with sodiumhydroxide in methanol to lead to the desired trans-2-allyl-6-alkyl(or aryl)-1,2,5,6-tetrahydropyridines 187a-c in 52 to56% overall yields (GC yields up to 90%). The trans-stereochemical outcome of these allylborations can beexplained by the fact that the substituents on the ring directthe reaction on the least hindered face of the molecule.
To complete this work, the same authors [57b] foundthat upon heating with triallylborane at 140-190°C followedby treatment with sodium hydroxide in methanol, the trans-2-allyl-6-alkyl(or aryl)-1,2,5,6-tetrahydropyridines 187a-cwere isomerized into the corresponding cis-isomers 192a-c(see Scheme 53).
Thus, trans-amine 187a-c reacted with triallylborane toafford the corresponding N-B complex 193, which underheating at 125°C led to the aminoborane 194 with loss ofpropene. At this stage under more drastic conditions (>160to 195°C), isomerization to the cis-isomers 195 occurred dueto the greater thermodynamic stability of these isomers withtwo pseudo-equatorial groups compared to the correspondingtrans-isomers 194 (see Scheme 54). After alcoholysis, thecis-2-allyl-6-alkyl-tetrahydropyridines 192a-c were isolatedin 60 to 70% overall yields, while amines 192a and 192bwere contaminated with less than 3% of trans-isomers.Concerning substrate 192c, the isomerization equilibriumfurnished a mixture of cis-192c and trans-187c in an 80 to20 ratio. No improvement was noted with prolonged heatingtime at higher temperature. However, the two isomers 187cand 192c could be separated by chromatography on Al2O3.
The efficiency of this new method has been demonstratedby a short and concise synthesis of (±)-dihydropinidine, adefense alkaloid of the Mexican bean beetle Epilachnavarivestis (Coccinellidae) and (±)-epidihydropinidineisolated from P. engelmannii and other Picea (spruce)species (see Scheme 55).
MISCELLANEOUS APPROACHES
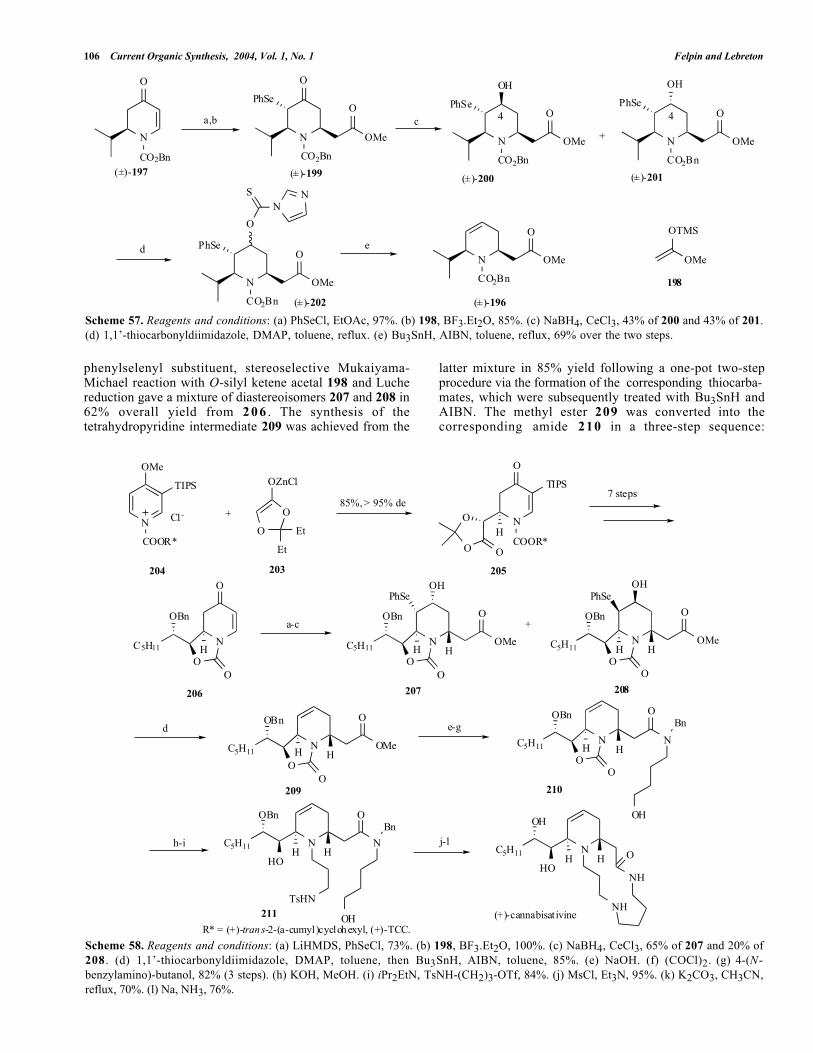
Comins et al. [59] reported an efficient synthesis ofracemic cis-2,6-disubstituted-1,2,5,6-tetrahydropyridine 196,
starting from N-acyl-2,3-dihydro-4-pyridone 197, seeScheme 56.
N OMe
O
CO2Bn
N
CO2Bn
O
(±)-196 (±)-197
Scheme 56.
Dihydropyridone 197 was treated with phenylselenylchloride and the resulting compound then participated in aMukaiyama-Michael reaction with O-silyl ketene acetal 198to give piperidone 199 as the sole diastereoisomer in 82%yield over two steps (see Scheme 57). The presence of aphenylselenyl substituent is required to perform the nexttransformation in a stereoselective manner and it is alsouseful for the double bond formation later on. Piperidone199 was reduced under Luche conditions to furnish amixture of diastereoisomers 200 and 201 in a 1 to 1 ratio.Formation of the double bond of 196 was carried out by afree-radical process following a one-pot procedure. The lattercrude mixture of 200 and 201 was converted into thecorresponding diastereoisomeric mixture of thiocarbamates202, which was then treated with Bu3SnH and AIBN topromote the olefin formation in 69% overall yield. It shouldbe noted that elimination via the formation of the mesylatederivative gave the expected tetrahydropyridine 196 only inthe case of trans isomer 201. The same conditions appliedto the cis isomer 200 gave only the C-4 mesylate withoutolefin formation.
An application of this chemistry to the first asymmetricsynthesis of (+)-cannabisativine (natural enantiomer) [8, 15]has also been reported by the same authors (see Scheme 58)[60]. Stereoselective addition of zinc enolate 203 t oenantiopure 1-acylpyridinium 204 (de > 95%) introduced thefirst substituent α to nitrogen in 85% yield to afford 205.After 7 steps of chemical transformation (50% overall yield),the dihydropyridone 206 was engaged in a similar sequenceas the one previously described. Introduction of a
106 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
N
CO2Bn
O
N
CO2Bn
O
PhSe
OMe
O
N
CO2Bn
OH
PhSe
OMe
O
N
CO2Bn
OH
PhSe
OMe
O
N
CO2Bn
OMe
O OTMS
OMe
N
CO2Bn
O
PhSe
OMe
O
NS N
+
a,b c
d e
(±)-197
198
(±)-199 (±)-200 (±)-201
(±)-202 (±)-196
4 4
Scheme 57. Reagents and conditions: (a) PhSeCl, EtOAc, 97%. (b) 198, BF3.Et2O, 85%. (c) NaBH4, CeCl3, 43% of 200 and 43% of 201.(d) 1,1’-thiocarbonyldiimidazole, DMAP, toluene, reflux. (e) Bu3SnH, AIBN, toluene, reflux, 69% over the two steps.
N
OMe
TIPS
COOR*
Cl-
O
O
OZnCl
Et
Et
N
O
OO
C5H11
OBn
H
N
OO
C5H11
OBn
OMe
O
H H
N N
O
OH
Bn
C5H11
OBn
HO
TsHN
H H
N
OH
OO
C5H11
OBn
OMe
O
PhSe
H H
N
O
TIPS
COOR*
O
O O
H
N
OO
C5H11
OBn
N
O
OH
Bn
H H
NC5H11
OH
HO
NH
NH
OH H
N
OH
OO
C5H11
OBn
OMe
O
PhSe
H H
+
(+)-cannabisat ivine
a-c
d e-g
h-i j-l
203
207 208
205
206
+
7 steps85%, > 95% de
204
209 210
211
R* = (+)-trans-2-(a-cumyl)cyclohexyl, (+)-TCC.
Scheme 58. Reagents and conditions: (a) LiHMDS, PhSeCl, 73%. (b) 198, BF3.Et2O, 100%. (c) NaBH4, CeCl3, 65% of 207 and 20% of208. (d) 1,1’-thiocarbonyldiimidazole, DMAP, toluene, then Bu3SnH, AIBN, toluene, 85%. (e) NaOH. (f) (COCl)2. (g) 4-(N-benzylamino)-butanol, 82% (3 steps). (h) KOH, MeOH. (i) iPr2EtN, TsNH-(CH2)3-OTf, 84%. (j) MsCl, Et3N, 95%. (k) K2CO3, CH3CN,reflux, 70%. (l) Na, NH3, 76%.
phenylselenyl substituent, stereoselective Mukaiyama-Michael reaction with O-silyl ketene acetal 198 and Luchereduction gave a mixture of diastereoisomers 207 and 208 in62% overall yield from 2 0 6 . The synthesis of thetetrahydropyridine intermediate 209 was achieved from the
latter mixture in 85% yield following a one-pot two-stepprocedure via the formation of the corresponding thiocarba-mates, which were subsequently treated with Bu3SnH andAIBN. The methyl ester 209 was converted into thecorresponding amide 210 in a three-step sequence:

Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 107
N
Ts
MeCH3(CH2)10N
Me
(CH2)10CH3
Ts
212 (±)-213
a
Scheme 59. Reagents and conditions : (a) 3 mol% Pd(PPh3) 4, DMSO, 25 min, 50°C, 91%.
NTs
o
CO2EtN
CN
CO2Et
H
214 215
+a
(±)-216 98:2 dr
Scheme 60. Reagents and conditions : (a) 20 mol% PBu3, CH2Cl2, rt, 99%.
saponification and subsequent formation of acid chlorideenabled the addition of the required aminoalcohol underSchotten-Bauman conditions (82% yield over three steps).The oxazolidinone ring of 210 was hydrolyzed under basicconditions and the liberated secondary amine was alkylatedwith the appropriate triflate-amine to give 211 in 74% yieldover 2 steps. Formation of the macrocycle by activation ofthe primary alcohol followed by treatment in basic media(67% yield, two steps) and cleavage of benzyl and tosylgroups using sodium in liquid ammonia achieved the firsttotal synthesis of (+)-cannabisativine (19 steps, 9% overallyield from 204).
A palladium(0) catalyzed rearrangement of 1,3-butadienylazetidines, activated by N-tosyl group, to 2,6-disubstituted-1,2,5,6-tetrahydropyridines has been publishedby Oshima et al. [61]. The 1,3-butadienylazetidine 212
(prepared in a five-step sequence from γ-butyrolactone) wastreated with Pd(PPh3)4 in DMSO at 50°C to furnishtetrahydropyridine 213 in high yield but without anydiastereoselectivity (cis/trans :47/53) (see Scheme 59) .
Very recently a phosphine-catalyzed [4+2] annulationreaction of N-tosylbenzaldimines 214 with 2-benzyl-2,3-butadienoates 215 leading to, in good to excellentdiastereoselectivity, the cis-2,6-disubstituted functionalizedtetrahydropyridines 216 has been published by Kwon et al.[62]. A typical example is described in Scheme 60; this newmethod was exemplified by the preparation of 11 adducts.
Recently, Liebeskind et al. [63] described a particularlyoriginal and concise highly stereo-controlled asymmetricsynthesis of alkaloids based on the chemistry of chiraldihydropyridinyl-molybdenum complexes. Bothenantiomers of molybdenum complex 217 are prepared inexcellent enantiopurity from racemic starting material 218following an eight-step sequence including a lipase-catalyzedtransformation (28% overall yield) (see Scheme 61). Thesequential functionalization of (-)-217 was carried out bymethoxy abstractions and nucleophilic additions: treatmentwith Ph3CPF6 followed by addition of Grignard reagent 219gave the intermediate 220 in high regioselectivity (up to49:1). This sequence was repeated on 220 with HBF4/n-PrMgBr to furnish the (2,3,6-trisubstituted dihydropyri-
dinyl)-molybdenum complex 221 in 67% yield. At thisstage the molybdenum complex 221 offered the possibilityof affording in good to excellent regio and stereoselectivitythe trisubstituted tetrahydropyridines 222, 223 and 224following the 3 different demetalation procedures: (i)reductive demetalation by replacing CO with NO+ followedby nucleophilic attack, (ii) protodemetalation or (iii)photolytic protodemetalation. From the trisubstitutedtetrahydropyridine 224, the completion of the total synthesisof (-)-indolozidine 209B was effected in 2 steps (6% overallyield from 218).
Similar chemistry was successfully applied to chiralmolybdenum complex (-)-225 by the same authors for thetotal synthesis of (-)-dihydropinidine and (-)-andrachcinidine(see Scheme 62) [64].
CONCLUSION
This review has attempted to summarize themethodologies available for the synthesis of 2,6-dialkyl-1,2,5,6-tetrahydropyridines developed by different groupssince 1980’s, as well as their application in total synthesis.2,6-Dialkyl-1,2,5,6-tetrahydropyridine frameworks areimportant synthetic building blocks that allow access tomore complex substituted piperidines and this fact mayprovide a driving force for future development in this field.As widely reported, a particular challenge remaining to besolved is the cis/trans stereocontrol for achieving moreefficient syntheses.
ACKNOWLEDGEMENTS
We thank the ‘Conseil Général de la Loire Atlantique’,the ‘Centre National de la Recherche Scientifique’, the‘Ministère de l’Education Nationale de la Recherche et de laTechnologie’ for a doctoral fellowship for F.-X. F. and the‘Université de Nantes’ for supporting our work in the fieldof total synthesis of alkaloids. We would like to warmlythank Prof. Yannick Landais (Laboratoire de ChimieOrganique et Organométallique, UMR-CNRS 5802, TalenceFrance) for fruitful discussions. We also express our
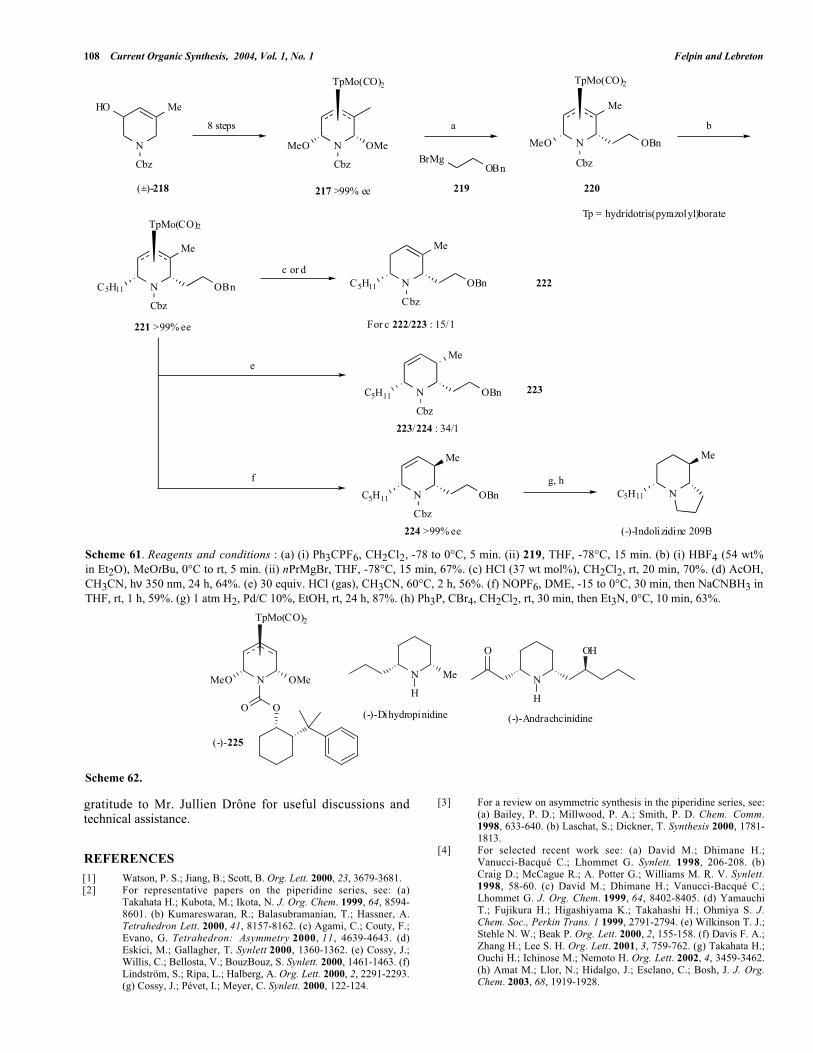
108 Current Organic Synthesis, 2004, Vol. 1, No. 1 Felpin and Lebreton
N
HO
Cbz
Me
N
Cbz
Me
C5H11 OBn
N
Cbz
MeO OMe
TpMo(CO)2
N
Cbz
Me
C5H11 OBn
N
Cbz
Me
C5H11 OBn
N
Cbz
Me
C5H11 OBn
BrMgOBn
N
Cbz
Me
MeO OBn
N
Me
C5H11
a b
(±)-218 217 >99% ee 219
8 steps
221 >99% ee
(-)-Indolizidine 209B
c or d
f
e
222
223
Tp = hydridotris(pyrazolyl)borate
For c 222/223 : 15/1
223/224 : 34/1
g, h
224 >99% ee
220
TpMo(CO)2
TpMo(CO)2
Scheme 61. Reagents and conditions : (a) (i) Ph3CPF6, CH2Cl2, -78 to 0°C, 5 min. (ii) 219, THF, -78°C, 15 min. (b) (i) HBF4 (54 wt%in Et2O), MeOtBu, 0°C to rt, 5 min. (ii) nPrMgBr, THF, -78°C, 15 min, 67%. (c) HCl (37 wt mol%), CH2Cl2, rt, 20 min, 70%. (d) AcOH,CH3CN, hν 350 nm, 24 h, 64%. (e) 30 equiv. HCl (gas), CH3CN, 60°C, 2 h, 56%. (f) NOPF6, DME, -15 to 0°C, 30 min, then NaCNBH3 inTHF, rt, 1 h, 59%. (g) 1 atm H2, Pd/C 10%, EtOH, rt, 24 h, 87%. (h) Ph3P, CBr4, CH2Cl2, rt, 30 min, then Et3N, 0°C, 10 min, 63%.
NMeO OMe
O O
TpMo(CO)2
N Me
HN
H
OHO
(-)-Dihydropinidine
(-)-225
(-)-Andrachcinidine
Scheme 62.
gratitude to Mr. Jullien Drône for useful discussions andtechnical assistance.
REFERENCES[1] Watson, P. S.; Jiang, B.; Scott, B. Org. Lett. 2000, 23, 3679-3681.[2] For representative papers on the piperidine series, see: (a)
Takahata H.; Kubota, M.; Ikota, N. J. Org. Chem. 1999, 64, 8594-8601. (b) Kumareswaran, R.; Balasubramanian, T.; Hassner, A.Tetrahedron Lett. 2000, 41, 8157-8162. (c) Agami, C.; Couty, F.;Evano, G. Tetrahedron: Asymmetry 2000, 11, 4639-4643. (d)Eskici, M.; Gallagher, T. Synlett 2000, 1360-1362. (e) Cossy, J.;Willis, C.; Bellosta, V.; BouzBouz, S. Synlett. 2000, 1461-1463. (f)Lindström, S.; Ripa, L.; Halberg, A. Org. Lett. 2000, 2, 2291-2293.(g) Cossy, J.; Pévet, I.; Meyer, C. Synlett. 2000, 122-124.
[3] For a review on asymmetric synthesis in the piperidine series, see:(a) Bailey, P. D.; Millwood, P. A.; Smith, P. D. Chem. Comm.1998, 633-640. (b) Laschat, S.; Dickner, T. Synthesis 2000, 1781-1813.
[4] For selected recent work see: (a) David M.; Dhimane H.;Vanucci-Bacqué C.; Lhommet G. Synlett. 1998, 206-208. (b)Craig D.; McCague R.; A. Potter G.; Williams M. R. V. Synlett.1998, 58-60. (c) David M.; Dhimane H.; Vanucci-Bacqué C.;Lhommet G. J. Org. Chem. 1999, 64, 8402-8405. (d) YamauchiT.; Fujikura H.; Higashiyama K.; Takahashi H.; Ohmiya S. J.Chem. Soc., Perkin Trans. 1 1999, 2791-2794. (e) Wilkinson T. J.;Stehle N. W.; Beak P. Org. Lett. 2000, 2, 155-158. (f) Davis F. A.;Zhang H.; Lee S. H. Org. Lett. 2001, 3, 759-762. (g) Takahata H.;Ouchi H.; Ichinose M.; Nemoto H. Org. Lett. 2002, 4, 3459-3462.(h) Amat M.; Llor, N.; Hidalgo, J.; Esclano, C.; Bosh, J. J. Org.Chem. 2003, 68, 1919-1928.

Synthesis of 2,6-Dialkyl-1,2,5,6-Tetrahydropyridines Current Organic Synthesis, 2004, Vol. 1, No. 1 109
[5] (a) Jones T. H.; Blum M. S.; Fales, H. M. Tetrahedron 1982, 38,1949-1958. (b) Kumareswaran R.; Hassner A. Tetrahedron:Asymmetry 2001, 12, 2269-2276.
[6] Takadoi M.; Yamaguchi K., Terashima S. Bioorg. Med. Chem.2003, 11, 1169-1186.
[7] For some leading references, see: (a) Angle, S. R.;Breitenbucher, J. G. In Studies in Natural Products Chemistry:Stereoselective Synthesis; Atta-ur Rahman, Ed.; Elsevier: NewYork, 1995 ; Vol. 16 , Part J, pp. 453-502. (b) Wang, C. J.;Wuonola, M. A. Org. Prep. Proced. Int. 1992, 24, 583-621.
[8] Lotter, H. L.; Abraham, D. J.; Tuner, C. E.; Knapp, J. E., Schiff,P. L.; Slatkin, D. J. Tetrahedron Lett. 1975, 7, 2815-2818.
[9] Karrer, Von P.; Eugster, C. Helv. Chim. Acta 1948, 31, 1062-1066.
[10] Bates, R. W.; Sa-Ei, K. Tetrahedron 2002, 58, 5957-5978.[11] For a recent review in this field, see: Chai, Y.; Hong, S.; Lindsay,
H. A.; McFarland, C.; McIntosh, M. C. Tetrahedron 2002, 58,2905-2928.
[12] (a) Angle, S. R.; Breitenbucher, J. G.; Arnaiz, D. O. J. Org.Chem. 1992, 57, 5947-5955. (b) Angle, S. R.; Breitenbucher, J. G.Tetrahedron Lett. 1993, 34, 3985-3988. (c) Angle, S. R.; Henry, R.M. J. Org. Chem. 1997, 62, 8549-8552. (d) Angle, S. R.; Henry,R. M. J. Org. Chem. 1998, 63, 7490-7497.
[13] (a) Waldmann, H. Synthesis 1994, 535-551. (b) Buonora, P.;Olsen, J.-C.; Oh, T. Tetrahedron 2001, 57, 6099-6138.
[14] Bailey, T. R.; Garigipati, R. S.; Morton, J. A.; Weinreb, S. M. J.Am. Chem. Soc. 1984, 106, 3240-3245.
[15] Hamada, T.; Zenkoh, T.; Sato, H.; Yonemitsu, O. TetrahedronLett. 1991, 32, 1649-1652.
[16] Hamada, T.; Sato, H.; Hikota, M.; Yonemitsu, O. TetrahedronLett. 1989, 30, 6405-6408.
[17] Bailey, P. D.; Brown, G. R.; Korber, F.; Reed, A., Wilson, R. D.Tetrahedron: Asymmetry 1991, 2, 1263-1282.
[18] Bailey, P. D.; Londesbrough, D. J.; Hancox, T. C.; Heffernan, J.D.; Holmes, A. B. Chem. Comm. 1994, 2543-2544.
[19] Bailey, P. D.; Smith, P. D., Pederson, F.; Clegg, W.; Rosair, G. M.;Teat, S. J. Tetrahedron Lett. 2002, 43, 1067-1070.
[20] Shi, Z.-D.; Yang, B.-H.; Wu, Y.-L.; Pan, Y.-J.; Ji, Y.-Y.; Yeh, M.Bioorg. Med. Chem. Lett. 2002, 12, 2321-2324.
[21] Schürer, S. C.; Blechert, S. Tetrahedron Lett. 1999, 40, 1877-1880.[22] (a) Marshall, J. A. In Comprehensive Organic Synthesis; Trost, B.
M., Fleming, I., Eds. Pergamon: Oxford, 1991; Vol. 3, Chapter3.11, p 975-1014. (b) Nakai, T.; Tomooka, K. Pure and Appl.Chem. 1997, 69, 595-600.
[23] Anderson, J. C.; Roberts, C. A. Tetrahedron Lett. 1998, 39, 159-162.
[24] (a) Åhman, J.; Somfai, P. J. Am. Chem. Soc. 1994, 116, 9781-9782. (b) Åhman, J.; Jarevång, T.; Somfai, P. J. Org. Chem. 1996,61, 8148-8159.
[25] (a) Åhman, J.; Somfai, P. Tetrahedron Lett. 1995, 36, 303-306. (b)Åhman, J.; Somfai, P. Tetrahedron 1995, 51, 9747-9756.
[26] Legters, J.; Thijs, L.; Zwanenburg, B. Tetrahedron Lett. 1989, 30,4881-4884.
[27] (a) Wuts, P. G. M.; Jung, Y.-W. J. Org. Chem. 1988, 53, 1957-1965. (b) Wuts, P. G. M.; Jung, Y.-W. J. Org. Chem. 1988, 53,5989-5994.
[28] Roush, W. R.; Pinchuk, A. N.; Micalizio, G. C. Tetrahedron Lett.2000, 41, 9413-9417.
[29] (a) Overman, L. E.; Malone, T. C.; Meier, G. P. J. Am. Chem.Soc. 1983, 105, 6993-6994. (b) Blumenkopf, T. A.; Overman, L.E. Chem. Rev. 1986, 86, 857-873.
[30] Daub, G. W.; Heerding, D. A.; Overman, L. E. Tetrahedron 1988,44, 3919-3930.
[31] Castro, P.; Overman, L. E.; Zhang, X.; Mariano, P. S. TetrahedronLett. 1993, 34, 5243-5246. It should be noted that the resultsdescribed in this paper are in contrast to an earlier report (see ref30) mentioning total racemization in the cyclization reaction.
[32] Kværnø, L.; Norrby, P.-O.; Tanner, D. Org. Biomol. Chem. 2003,1, 1041-1048.
[33] Yang, T.-K.; Teng, T.-F.; Lin, J.-H.; Lay, Y.-Y. Tetrahedron Lett.1994, 35, 3581-3582.
[34] Agami, C.; Comesse, S.; Kadouri-Puchot, C. J. Org. Chem. 2002,67, 2424-2428.
[35] (a) Agami, C.; Comesse, S.; Kadouri-Puchot, C. Tetrahedron Lett.2000, 41, 6059-6062. (b) Agami, C.; Comesse, S.; Kadouri-Puchot, C. J. Org. Chem. 2002, 67, 1496-1500.
[36] Franciotti, M.; Mann, A.; Mordini, A.; Taddei, M. TetrahedronLett. 1993, 34, 1355-1358.
[37] Huang, H.; Spande, T. F.; Panek J. S. J. Am. Chem. Soc. 2003,125, 626-627.
[38] (a) Sparks, M. A.; Panek J. S. J. Org. Chem. 1991, 56, 3431-3438.(b) See also: Mohamed, M.; Brook, M. A. Tetrahedron Lett. 2001,42, 191-193.
[39] Furuta, K.; Ishiguro, M.; Ikera, N.; Yamamoto, H. J. Am. Chem.Soc. 1981, 103, 5568-5570.
[40] Craig, D.; McCague, R.; Potter, G. A.; Williams, M. R. V. Synlett.1998, 55-57.
[41] Tjen, K. C. M. F.; Kinderman, S. S.; Schoemaker, H. E.; Hiemstra,H.; Rutjes, F. P. J. T. Chem. Comm. 2000, 699-700.
[42] (a) Compère, D.; Marazano, C.; Das, B. C. J. Org. Chem. 1999,64, 4528-4532. (b) Guilloteau-Bertin, B.; Compère, D.; Gil, L.;Marazano, C.; Das, B. C. Eur. J. Org. Chem. 2000, 1391-1399.
[43] Genisson, Y.; Marazano, C.; Mehmandoust, M.; Gnecco, D.; Das,B. C. Synlett. 1992, 431-434.
[44] (a) Dransfield, P. J.; Gore, P. M.; Shipman, M.; Slawin, A. M. Z.Chem Comm. 2002, 150-151. (b) Dransfield, P. J.; Gore, P. M.;Prokes,
>
I.; Shipman, M.; Slawin, A. M. Z. Org. Biomol. Chem.2003, 1, 2723-2733.
[45] For general review of RCM, see: (a) Fürstner, A. Angew. Chem.,Int. Ed. Engl. 2000, 39, 3013-3043. (b) Grubbs, R. H.; Chang, S.Tetrahedron 1998, 54, 4413-4450. (c) Armstrong, S. K. J. Chem.Soc., Perkin Trans. 1 1998, 371-388. (d) Grubbs, R. H.; Miller, S.J.; Fu, G. C. Acc. Chem. Res. 1995, 28, 446-552. For generalreview of RCM reactions in the alkaloids field, see: (e) Phillips,A. J.; Abell, A. D. Aldrichimica Acta 1999, 32, 75-89. (f) Felpin,F.-X.; Lebreton J. Eur. J. Org. Chem. 2003, 3693-3712.
[46] (a) Felpin, F.-X.; Girard, S.; Vo-Thanh, G.; Robins, R. J.;Villiéras, J.; Lebreton, J. J. Org. Chem. 2001, 66, 6305-6312. (b)Felpin, F.-X.; Bertrand, M.-J.; Lebreton, J. Tetrahedron 2002, 58,7381-7389. (c) Felpin, F.-X.; Lebreton, J. Tetrahedron Lett. 2002,43, 225-227. (d) Felpin, F.-X.; Lebreton, J. J. Org. Chem. 2002,67, 9192-9199.
[47] Felpin, F.-X.; Lebreton J. Tetrahedron Lett. 2003, 44, 527-530.[48] Felpin, F.-X.; Lebreton, J. Eur. J. Org. Chem, in press.[49] Agami, C.; Couty, F.; Rabasso, N. Tetrahedron 2001, 57, 5393-
5401.[50] (a) Bushmann, N.; Rückert, A.; Blechert, S. J. Org. Chem. 2002,
67, 4325-4329. (b) Blechert, S.; Stapper, C. Eur. J. Org. Chem.2002, 2855-2858. (c) Blechert, S.; Stapper, C. J. Org. Chem.2002, 67, 6456-6460.
[51] Voigtmann, U.; Blechert, S. Synthesis 2000, 893-898.[52] Wassermann, H. H.; Leadbetter, M. R.; Kopka, I. E. Tetrahedron
Lett. 1984, 25, 2391-2394.[53] (a) Mayer, C.; Green, C. L.; Treub, W.; Wälchli, P. C.; Eugster,
C. H. Helv. Chem. Acta 1978, 61, 905-921. (b) Wälchli, P.;Eugster, C. H. Helv. Chem. Acta 1978, 61, 885-898.
[54] Wassermann, H. H.; Pierce, B. C. Tetrahedron 1988, 44, 3365-3372
[55] Xin, T.; Okamoto, S.; Sato, F. Tetrahedron Lett. 1998, 39, 6927-6930.
[56] Kasatkin, A.; Nakagawa, T.; Okamoto, S.; Sato, F. J. Am. Chem.Soc. 1995, 117, 3881-3882.
[57] Stanway, S. J.; Thomas, E. J. Chem. Comm. 1994, 285-286[58] (a) Bubnov, Y. N.; Klimkina, E. V.; Ignatenko, A. V.; Gridnev, I.
D. Tetrahedron Lett. 1996, 37, 1317-1320. (b) Bubnov, Y. N.;Klimkina, E. V.; Ignatenko, A. V.; Gridnev, I. D. TetrahedronLett. 1997, 38, 4631-4634.
[59] Kuethe, J. T.; Comins, D. L. Org. Lett. 1999, 1, 1031-1033.[60] Kuethe, J. T.; Comins, D. L. Org. Lett. 2000, 2, 855-857.[61] Fugami, K.; Miura, K.; Morizawa, Y.; Oshima, K.; Utimoto, K.;
Nozaki, H. Tetrahedron 1989, 45, 3089-3098.[62] Zhu, X.-F.; Lan, J.; Kwon, O. J. Am. Chem. Soc. 2003, 125, 4716-
4717.[63] Shu, C.; Alcudia, A.; Yin, J.; Liebeskind, L. S. J. Am. Chem. Soc.
2001, 123, 12477-12487.[64] Shu, C.; Liebeskind, L. S. J. Am. Chem. Soc. 2003, 125, 2878-
2879.