solid phase speciation of arsenic by sequential extraction in standard reference materials and...
TRANSCRIPT

Solid phase speciation of arsenic by sequential extractionin standard reference materials and industrially contaminated
soil samples
Samuel Van Herreweghea,*, Rudy Swennena, Carlo Vandecasteeleb, Valerie Cappuynsa
aPhysico-Chemical Geology, K.U. Leuven, Celestijnenlaan 200C, B-3001 Heverlee, BelgiumbDepartment of Chemical Engineering, K.U. Leuven, de Croylaan 46, B-3001 Heverlee, Belgium
Received 29 March 2002; accepted 26 August 2002
‘‘Capsule’’: Leaching experiments, a mineralogical survey and larger samples are preferred when arsenic is present asdiscrete mineral phases.
Abstract
Availability, mobility, (phyto)toxicity and potential risk of contaminants is strongly affected by the manner of appearance of
elements, the so-called speciation. Operational fractionation methods like sequential extractions have been applied for a long timeto determine the solid phase speciation of heavy metals since direct determination of specific chemical compounds can not always beeasily achieved. The three-step sequential extraction scheme recommended by the BCR and two extraction schemes based on the
phosphorus-like protocol proposed by Manful (1992, Occurrence and Ecochemical Behaviours of Arsenic in a GoldsmelterImpacted Area in Ghana, PhD dissertation, at the RUG) were applied to four standard reference materials (SRM) and to a batchof samples from industrially contaminated sites, heavily contaminated with arsenic and heavy metals. The SRM 2710 (Montanasoil) was found to be the most useful reference material for metal (Mn, Cu, Zn, As, Cd and Pb) fractionation using the BCR
sequential extraction procedure. Two sequential extraction schemes were developed and compared for arsenic with the aim toestablish a better fractionation and recovery rate than the BCR-scheme for this element in the SRM samples. The major part of arsenicwas released from the heavily contaminated samples after NaOH-extraction. Inferior extraction variability and recovery in the heavily
contaminated samples compared to SRMs could be mainly contributed to subsample heterogeneity.# 2002 Elsevier Science Ltd. All rights reserved.
Keywords: Heavy metals; Fractionation; Mineralogy; Certified reference materials; Three-step BCR extraction
1. Introduction
Arsenic is relatively scarce in the natural environment(20th position in elemental abundance in the continentalcrust), but it is widely distributed. It can be found indetectable amounts in nearly all rocks and soils. Arsenicin soils relates to the geological substratum and conse-quently a rather wide range of arsenic levels have beenreported in soils around the world with an average of 5–10 mg/kg in uncontaminated soils (Fergusson, 1990).However, arsenic may accumulate in soils due to humanactivities such as waste discharges of metal processingplants, burning of fossil fuels, mining of arsenic con-
taining ores and agricultural use of arsenical pesticides.In this respect it is important to know that arsenic hasdifferent degrees of toxicity depending on the speciationof arsenic, route and dose of exposure, and individualand local tissue susceptibilities (Nriagu, 1994).Arsenic is a semimetallic element and its behavior in
soils, resembles more that of group V A elements thanthat of heavy metals. Oxidation state of arsenic isimportant since it affects movement, persistence and(phyto)toxicity of arsenic in soils and sediments. Inoxygen-rich environments and well-drained soils, arse-nate (+V) species dominate (H2AsO4
� in acidic soils andHAsO4
2� in alkaline ones). Under reducing conditions,such as regularly flooded soils, arsenite (+III) is thestable oxidation state, but elemental arsenic and arsine(�III) can also be present in strongly reducing environ-ments. Arsenic may also exist in organometallic forms
0269-7491/03/$ - see front matter # 2002 Elsevier Science Ltd. All rights reserved.
PI I : S0269-7491(02 )00332-9
Environmental Pollution 122 (2003) 323–342
www.elsevier.com/locate/envpol
* Corresponding author. Fax: +32-16-327981.
E-mail address: [email protected] (S. Van Her-
reweghe).

such as monomethylarsenic acid, dimethylarsenic acidand the more volatile methylarsines. Iron, aluminiumand calcium arsenates are, like the corresponding phos-phates, virtually insoluble in water and arsenate exhibitsa phosphate-like specific adsorption (inner sphere com-plexation) on oxide and clay surfaces (Jacobs et al.,1970). Arsenites, however, are 10 times more solubleand mobile and are furthermore more toxic thanarsenates.The aim of this paper is to assess the applicability of
sequential extractions on highly contaminated soilswhere arsenic is also present as discrete As-bearingminerals. Sequential extractions are mostly developed tofractionate heavy metals occurring in trace amounts andtheir applicability on highly contaminated samplesremains insufficiently studied. Therefore, both con-taminated soils as well as slag material were analyzed inthis study.There is, furthermore, a need to evaluate sequential
extraction schemes specifically focussing on metalloidelement extraction such as arsenic. Therefore, CertifiedReference Materials or CRMs [also referred to as’Standard Reference Materials’ (SRMs) by the NationalInstitute of Standards and Technology, USA] wereanalyzed. Based on these reference samples, the appliedmethods have been validated for arsenic recovery, inter-method variability and reagent selectivity prior to theiruse on highly contaminated samples. Subsequently con-taminated soil and slag samples from two sites inBelgium were analyzed.All samples were first subjected to the standardized
three-step BCR extraction scheme to evaluate the suit-ability of this method to fractionate arsenic and to getinformation on the heavy element speciation in thesamples. This scheme was selected based on its wide-spread use and easiness of application. Subsequently,two different sequential schemes for selective arsenicextraction were applied that were adapted from a‘Chang and Jackson (1957) like’ protocol as developedby Manful (1992). One scheme comprehended onlyminor changes while the second scheme adopted chan-ges such as the use of anion exchange membrane (AEM)strips according to Hedley et al. (1982). Advantages anddisadvantages of the latter labor intensive sequentialextraction scheme will be evaluated with respect to themore confined BCR scheme and conclusions will bedrawn with respect to interpretation of the results. Forthis purpose, some mineralogical analysis was carriedout in addition.
2. Speciation and sequential extractions: a literature
overview
Chemical speciation analysis in soils and sediments isdefined by the BCR of the European Community (Ure
et al., 1993) as: the process of identification and quantifi-cation of the different defined species, forms or phases inwhich an element occurs in the material. Speciation alsorepresents the actual description of the quantity andvariety of species. Chemical speciation can be sub-divided in three classes: (i) functionally defined species(e.g. plant available species) where the species aredefined by their role or as (ii) operationally defined spe-cies, characterized by the procedure of isolation andidentification (e.g. acid ammonium oxalate extractablefraction) or finally as (iii) specific chemical compounds oroxidation states (e.g. methyl-mercury, AsH3, . . .). How-ever, according to the recent recommendations of theIUPAC (International Union for Pure and AppliedChemistry) regarding the terminology of chemical spe-ciation (Templeton et al., 2000), the term ‘speciation’ ismore or less restricted to the latter class of species.Accordingly, the former two speciation classes will bereferred to as fractionation, which is the process ofclassification of an analyte or a group of analytes from acertain sample according to physical (e.g. size, solubi-lity) or chemical (e.g. bonding, reactivity) properties.Speciation analysis is of major importance in envir-
onmental research since it can provide crucial informa-tion about the ecotoxicological characteristics(reactivity, bioavailability, toxicity) of contaminants.Furthermore, mobility and transport of contaminantsbetween different environmental compartments is alsospecies-dependent.Trace elements occur in soils in certain ‘pools’ or
‘sinks’ of different solubility and mobility, of which ingeneral six categories can be differentiated in the solidphase (McLean and Bledsoe, 1992): (i) occupyingexchangeable sites as diffuse ion or as outer-spherecomplexes; (ii) specific adsorption as inner-sphere com-plexes; (iii) associated with insoluble organic matter; (iv)(co)precipitated as pure or mixed solids; and finallypresent in the structure of (v) secondary or (vi) primaryminerals. Trace element speciation analysis in soils mustalmost always be performed by operational fractiona-tion methods like chemical extractions since directdetermination of specific chemical compounds is mostlyimpossible. Chemical extraction is, however, compli-cated by the fact that no chemical solution uniquelyextracts trace elements out of one of these pools.Sequential extractions, in which a soil sample is reactedwith a series of carefully selected chemical solutions ofincreasing strengths, were developed to increase extrac-tion selectivity of the distinct geochemical fractions(Pickering, 1981). Today, sequential extractions are themost frequently applied analytical method for studyingthe solid phase speciation of environmental relevantheavy metals (Tack and Verloo, 1995). However,sequential extractions determine only an operationaldefined speciation (i.e. a fractionation), which dependson different factors like choice and order of extraction
324 S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342

agents, the extraction duration, solid/liquid ratio, andsample preparation and conservation (Rapin et al.,1986; Martin et al., 1987). Sample preparation andconservation are very important, especially for samplestaken from reducing environments, since (labile) phasescan actually be transformed into other phases duringsample preparation. Some significant differences inoperational speciation are even encountered when thesame scheme is applied by different analysts (Davidsonet al., 1999). However, lack of precision (especially atconcentrations close to the detection limit) and selectiv-ity are the main problems that occur among sequentialextractions (Tessier et al., 1979; Tipping et al., 1985;Shuman, 1985). Lack of selectivity is manifested byincomplete dissolution of the intended phase by theextraction agents for example when the chemical systembecomes overburdened, in (partial) dissolution of unde-sired phases resulting in overrating the targeted phase,and/or in readsorption- and redistribution phenomena.Validation tests of sequential extractions have been a
matter of debate for a long time and can be accom-plished either by treating synthetic and/or pure miner-alogical phases or by applying some kind of standardadditions (e.g. spiked samples) (Kheboian and Bauer,1987 & 1988; Tessier and Campbell, 1988, 1991; Belzileet al., 1989; Nirel and Morel, 1990).It has been suggested that extraction efficiency could
be improved by monitoring some parameters. Forexample, incomplete dissolution of acid-soluble miner-als due to strong acid/base buffering could be checkedby measuring the pH of the solution before and afterextraction (Dodd et al., 2000). Rauret et al. (1989) sug-gested therefore a multiple successive extraction proce-dure with monitoring the mandatory parameters (pH,Fe and Mn concentration, and Eh) in metal release todetermine the number of extractions required. Thisshould lead, according to these authors, to a more reli-able distribution pattern especially when dealing withheavily contaminated samples. Keon et al. (2001) sug-gested using low sediment-to-extractant ratios (1/100) toensure that each extractant did not become exhausted(e.g. to prevent that iron oxide dissolution is limited byoxalate concentration).The extraction scheme for trace elements developed
by Tessier et al. (1979) together with many adaptedschemes such as those proposed by Kersten and For-stner (1986), Li et al. (1995a, b) and Kirby and Rimstidt(1993) are the most accepted and popular ones. Thescheme proposed by Tessier et al. (1979) includes fivefractions: (i) exchangeable, (ii) extracted by acetic-ace-tate buffer, (iii) extracted by hydroxylamine, (iv)extracted by hydrogen peroxide in nitric acid and (v)residual. Complex schemes give usually a more detailedimage of the chemical speciation [e.g. more specific iso-lation of Fe and Mn (hydr)oxide phases], but in generalthey are more labor-intensive and have a decreasing
precision as a result of accumulation of errors. Becauseof a lack of harmonization between the different proce-dures and the consequential impossibility to comparemutual results, the Community Bureau of Reference(BCR, now Measurements and Testing Programme)formulated a standardized, three-stage, sequentialextraction protocol (Ure et al., 1993; Quevauviller et al.,1994). This protocol uses: (i) 0.11 mol l�1 HAc; (ii) 0.1mol l�1 NH2OH.HCl (adjusted to pH=2 with nitricacid); (iii) 8.8 mol l�1 H2O2 (adjusted to pH=2–3 withnitric acid) followed by NH4Ac (adjusted to pH=2 withnitric acid) to sequentially extract the (i) carbonate (ii)Mn and Fe oxide and (iii) organic and sulfide fraction.A Tessier-like protocol was for example applied to
arsenic contaminated lake sediment by Lum and Edgar(1983), to arsenic contaminated soil samples by Voigt etal. (1996) and by Roussel et al. (2000) to arsenic-bearingsuspended material derived from a mine waste drain.The latter authors found that the most important phasebinding arsenic were Mn–Fe oxides and organic matteror sulfides. Dhoum and Evans (1998) applied an adap-ted BCR protocol to evaluate arsenic retention by soil.Maher (1984) investigated arsenic fractionation in sedi-ments by the application of two different sequentialextraction procedures, one similar to the Tessier-proto-col and one based on a phosphate fractionation scheme.Arsenic appeared here to be mainly associated with theFe-sesquioxide phase.These commonly used sequential extractions are,
however, developed within the framework of trace ele-ments or heavy metals that are occurring in cationicforms in soils and sediments. The uncritical applicationof these extraction schemes on metalloids like arsenicthat are prevailing in anionic form in soils, however,should be avoided (Gruebel et al., 1988). Consequently,many extraction protocols for arsenic were based uponsequential extraction schemes of phosphorus because ofthe similar chemical behavior in soils. They both com-monly form oxyanions (arsenate and phosphate) in the+V oxidation state in soils. However, the soil chemistryof arsenic is much more diverse because phosphate isstable over a much wider range of Eh and pH conditionsthan is arsenate. Furthermore As can form bonds with Sand C much more readily than does P (O’Neill, 1995).Additionally, organic As is of less importance thanorganic P and do not appear to compose an appreciableproportion of total soil As (Johnson and Hiltbold, 1969).A widely used sequential extraction method of phos-
phorus was developed by Chang and Jackson (1957)with many later modifications such as those of Williamset al. (1967). The sequential extraction procedureemploys NH4Cl to extract labile P followed by NH4F todissolve Al associated P. This is followed by NaOH toextract Fe-bound P, by DCB (dithionite citrate bicar-bonate) for ‘‘occluded’’ P forms and by HCl or H2SO4
for Ca-bound P.
S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342 325

Arsenic in soils has been characterized in a similarway by various authors (Johnson and Hiltbold, 1969;Jacobs et al., 1970; Woolson et al., 1971, 1973; Manful,1992; Onken and Adriano, 1997). An alternativeP-fractionation scheme (Hedley et al., 1982) is alsowidely used in soil P studies and therefore also used tofractionate As in soils (McLaren et al., 1998). This pro-tocol differentiates ‘‘anion exchange membrane-extrac-table’’; ‘‘NaHCO3-extractable’’; ‘‘NaOH-extractable’’;‘‘sonicated/NaOH-extractable’’; ‘‘HCl-extractable’’ and‘‘residual’’ arsenic. However, the development of allthese extraction schemes and the link of the extractedfractions with the geochemical arsenic fractions wasmainly based on pure theoretical grounds of analogousphosphorus behavior and can therefore be questioned.Gleyzes et al. (2001) criticized this scheme and applied iton industrially contaminated soils. Johnston and Bar-nard (1979) found that the quantities of As removedfrom the four western New York soils by solutionscontaining acetate, sulfate, fluoride, bicarbonate,hydroxide, and hydrogen ions are consistent with thethesis that As and P react similarly when treated bythese solutions. Although the soils investigated in thatstudy were selected to reflect differences in soil type andland use, they finally found that As, sequentiallyextracted in the P-like protocol, demonstrated the sameorder of As-extraction for all soils: NaOH >> H2SO4
> NH4F > NH4Cl. Il’yin and Konarbayeva (1996)applied an adapted Chang and Jackson-like sequentialextraction protocol on some West Siberian soils withelevated concentrations of native arsenic. They sequen-tially used (i) NH4Cl (soluble arsenic), (ii) (NH4)2CO3 atpH9 (to dissolve sulfides and arsenic bound to alumi-nium compounds), (iii) Trilon B (enhanced dissolutionof carbonates), (iv) NaOH (organic matter and ironhydroxides), (v) H2SO4 (low soluble arsenic) and finally(vi) residual arsenic. They found that most arsenic waspresent in the residual form. Jackson and Miller (2000)found that hydroxide was the most effective extractantfor desorption of all arsenic species except As(III) fromboth goethite and amorphous Fe oxide, and arsenitewas extracted most efficiently by 0.5 mol l�1 PO4
3- at lowpH. Onken and Adriano (1997) found that arsenite,freshly added to the soil, was predominantly extractedby NH4Cl as a result of the neutral (non-ionic) char-acter of arsenite not being sorbed to anionic or cationicexchange sites in the soil while arsenate was present inthe more recalcitrant forms (NH4F- and NaOH-extrac-table). Hence, the occurrence of As in more than oneoxidation state, each of which having different adsorp-tive behavior, further complicates the application of astraightforward and unambiguous sequential extractionscheme for As. Keon et al. (2001) partitioned As withthe following extraction protocol: 1 mol l�1 MgCl2(loosely adsorbed As), 1 mol l�1 NaH2PO4 (stronglyadsorbed As), 1 mol l�1 HCl (acid volatile sulfides and
amorphous metal oxides), 0.2 mol l�1 oxalic acid (Asincorporated in amorphous iron and aluminium oxi-des), 0.05 mol l�1 Ti-citrate-EDTA (As incorporated incrystalline iron oxides), 10 mol l�1 HF (As oxides andAs in silicate minerals), 16 mol l�1 HNO3 (As in pyrite),and hot concentrated HNO3 and H2O2 (crystalline Assulfides). Notice that this method is rather circum-stantial and embodies some hazardous reagents [e.g.hydrofluoric acid or Ti(III) chloride]. Amacher andKotuby-Amacher (1994) developed a two-step methodthat was intended to selectively extract As associatedwith metal oxides (0.25 mol l�1 NH2OH.HCl/0.25 moll�1 HCl/0.025 mol l�1 H3PO4) and metals sulfides (aquaregia and 8.8 mol l�1 H2O2). During reductive dissolu-tion of amorphous iron oxide by acidified warm hydro-xylamine hydrochloride (H2NOH.HCl), arsenicassociated with the dissolved reductive phase is notalways found in solution, due to readsorption to goe-thite or inversely arsenic associated with nontargetedsolids (e.g. montmorillonite) may be desorbed (Gruebelet al., 1988). Addition of 0.1 mol l�1 PO4 to the extrac-tant used for reductive dissolution could prevent thereadsorption of As(V) to goethite (Jackson and Miller,2000).
3. Materials and methods
3.1. Reconaissance research
In a reconnaissance research, a number of purearsenic minerals were subjected to a spectrum ofextractions in order to examine their extraction effi-ciency. Following solutions were used based upon theKirby and Rimstidt (1993) protocol: water, neutral salt,acid, oxidizing, reducing and 3-acid solutions. Bothnatural As minerals (realgar, orpiment, lollingiet), com-mercially available minerals (arsenolite), organicallybound As in moss taken on a contaminated site(Reppel) and synthetic arsenate minerals (Na-, Ca-, Al-and Fe-arsenate) were subjected to these extractions.This reconnaissance research was only carried out tointerpret the extraction of samples containing arsenicwith discrete phases (e.g. minerals).
3.2. Sample collection and characterization
Soil and slag samples were collected on two sites inBelgium, severely contaminated with heavy metals andarsenic due to former industrial activities. The soil onthe first site (the former arsenic refinery in Reppel) wasextremely polluted with As, Cu (both in percentagelevel), Co and Ni (both >0.5%). During a first generalsurvey of the Reppel site (Cappuyns et al., 2002), threedifferent areas were sampled, collecting both slags, sur-face soil and soil profile samples. The profile pits were
326 S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342

excavated to a depth of 120 cm. Most profiles, however,did not exhibit the normal characteristics of a classicalsoil and could better be described as made-up ground orUrbic Anthropic Regosols. Soil samples from a secondcontaminated site (the former fertilizer plant in St-Amands) became contaminated with so-called burnedpyrite (sulfuric acid production waste) containing highconcentrations of Pb, Cu, As and Zn (>0.1%). Theselected soil samples were dried at 40 �C, desaggregatedin a agate mortar, homogenized and splitted into sub-samples by the manual cone-and-quarter method toreduce bias in sub-sampling. A subsample was thensieved retaining the <2 mm fraction for the chemicalanalysis. An unsieved subsample was put aside for grainsize analysis. A fraction (�25 g) of the sieved subsamplewas ground to a fine powder of <150 mm with a mill(spex mixer/Mill Nr 8000) for the total chemical analy-sis. The size of the stony samples (slags and so on) wasfirstly reduced with a hammer on a chopping board. Apaper packaging around the sample was utilized to pre-vent metal contamination from the hammer or chop-ping board. These stony fragments were subsequentlyfurther reduced with the mill (<150 mm).Total element analysis was analyzed by AAS (Al, Cd,
Ca, Co, Cu, Fe, Pb, Mg, Mn, Ni and Zn) after 3-aciddigestion (4 ml HClconc, 2 ml HNO3conc and 2 mlHFconc). Arsenic was analyzed according to the HG–AAS (Hydride Generation–AAS) method: 0.25 g ofsample was melted with 1 g K2S2O7, leached with 10 ml1 M HCl at 85 �C (45 min) and reduced with NaBH4 togenerate AsH3, which was measured with AAS (Var-ian1 Techtron). On a number of representative samples,general soil characteristics like grain size, effectivecation exchange capacity (ECEC), organic carbon,phosphate and sulfur were also quantified. Grain sizewas determined by laser diffraction analysis (Malvern1
Mastersizer S long bed). ECEC was analyzed applyingthe ‘silver thiourea method’ (Van Reeuwijk, 1992) andorganic carbon was determined according to Walkley &Black (Nelson and Sommers, 1982). PO4
3- was deter-mined colorimetrically (spectrophotometer Varian1
Techtron model 635) according to methods described byBoltz (1958) and by Peachey et al. (1973). Sulfur wasanalyzed by the so-called Strohlein apparatus: combus-tion at 1350 �C, absorption of produced SO2 in H2O2
and titration of H2SO4 formed, with a standard solutionof NaOH (methyl orange indicator).Total concentrations and soil parameters of the selec-
ted samples are given in Table 1. The first three samples(Rep-sl_1, Rep-sl_2 and Rep-sl_3) are slag samples,Rep-so_1 to Rep-so_5 are surficial soil samples (0–20cm) and Rep-cl_1a to Rep-cl_3 are soil samples taken inthe most contaminated layer of some vertical soil pro-files on the Reppel-site. Rep-cl_1b is a subsample takenin a colorful spot in the soil layer where sample Rep-cl_1a was taken. Rep-slu is a sludge sample taken in a
drainpipe that was found below one of the excavatedprofiles. Rep-sP1 to Rep-sP7 are soil samples, all takenin a vertical profile on the Reppel site. Although thisprofile was situated in a highly contaminated sector,there was still plenty vegetation (mainly grass) present,suggesting the existence of a less contaminated topsoil.In this profile, above a colorful antropogenetic, highlycontaminated layer at�30 cm depth, a topsoil was pre-sent that was different from the rest of the profile (e.g.neutral soil-pH and high organic carbon content)(Fig. 1). A more detailed description of the Reppel sitecan be found in Cappuyns et al. (2002). Stam-so_1 toStam-so_3 are soil samples containing remainders of theburned pyrite taken on the St-Amands site. Thus, in thisstudy a diverse set of soil samples with variable As andassociated heavy metal concentrations, with differentorigins of contamination and soil characteristics andsamples that are non-soil material (slag material andsludge) were studied.
3.3. Mineralogy
Mineralogy was studied combining optical petro-graphy, X-ray diffraction (XRD; Philips1, Co-target,
Fig. 1. Picture of the excavated soil profile pit (Reppel site). Sample
Rep-sP3 is taken in the highly contaminated horizon.
S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342 327
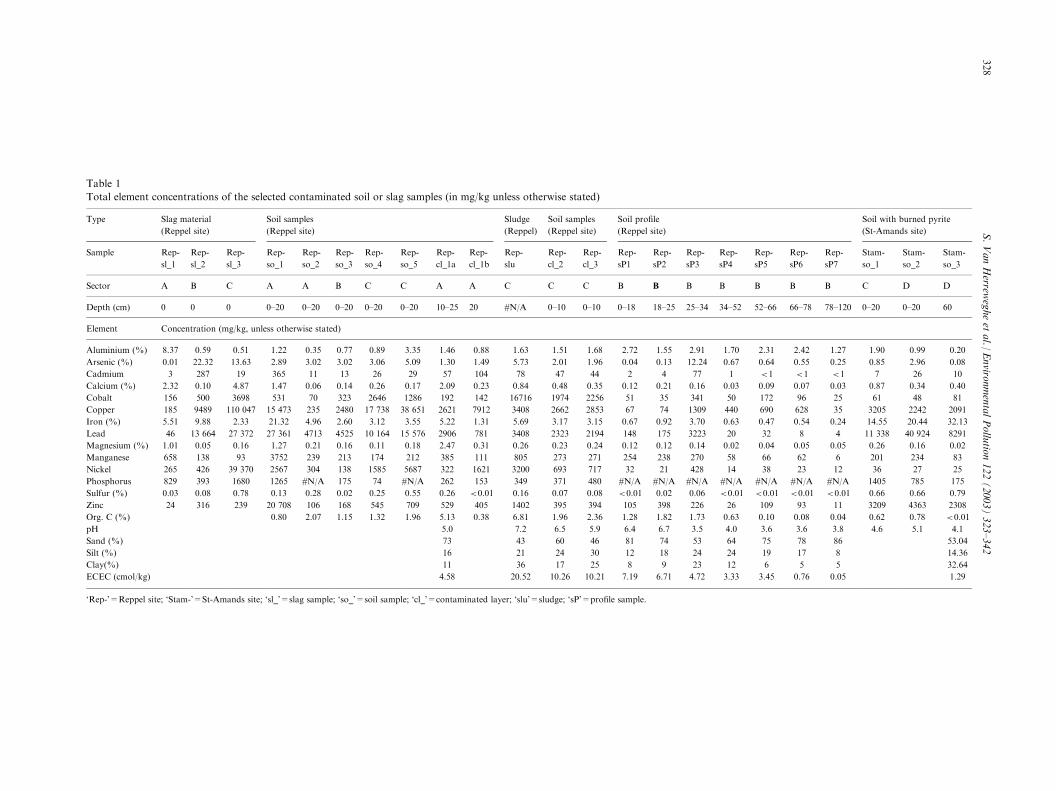
Table 1
Total element concentrations of the selected contaminated soil or slag samples (in mg/kg unless otherwise stated)
Type Slag material
(Reppel site)
Soil samples
(Reppel site)
Sludge
(Reppel)
Soil samples
(Reppel site)
Soil profile
(Reppel site)
Soil with burned pyrite
(St-Amands site)
Sample Rep-
sl_1
Rep-
sl_2
Rep-
sl_3
Rep-
so_1
Rep-
so_2
Rep-
so_3
Rep-
so_4
Rep-
so_5
Rep-
cl_1a
Rep-
cl_1b
Rep-
slu
Rep-
cl_2
Rep-
cl_3
Rep-
sP1
Rep-
sP2
Rep-
sP3
Rep-
sP4
Rep-
sP5
Rep-
sP6
Rep-
sP7
Stam-
so_1
Stam-
so_2
Stam-
so_3
Sector A B C A A B C C A A C C C B B B B B B B C D D
Depth (cm) 0 0 0 0–20 0–20 0–20 0–20 0–20 10–25 20 #N/A 0–10 0–10 0–18 18–25 25–34 34–52 52–66 66–78 78–120 0–20 0–20 60
Element Concentration (mg/kg, unless otherwise stated)
Aluminium (%) 8.37 0.59 0.51 1.22 0.35 0.77 0.89 3.35 1.46 0.88 1.63 1.51 1.68 2.72 1.55 2.91 1.70 2.31 2.42 1.27 1.90 0.99 0.20
Arsenic (%) 0.01 22.32 13.63 2.89 3.02 3.02 3.06 5.09 1.30 1.49 5.73 2.01 1.96 0.04 0.13 12.24 0.67 0.64 0.55 0.25 0.85 2.96 0.08
Cadmium 3 287 19 365 11 13 26 29 57 104 78 47 44 2 4 77 1 <1 <1 <1 7 26 10
Calcium (%) 2.32 0.10 4.87 1.47 0.06 0.14 0.26 0.17 2.09 0.23 0.84 0.48 0.35 0.12 0.21 0.16 0.03 0.09 0.07 0.03 0.87 0.34 0.40
Cobalt 156 500 3698 531 70 323 2646 1286 192 142 16716 1974 2256 51 35 341 50 172 96 25 61 48 81
Copper 185 9489 110 047 15 473 235 2480 17 738 38 651 2621 7912 3408 2662 2853 67 74 1309 440 690 628 35 3205 2242 2091
Iron (%) 5.51 9.88 2.33 21.32 4.96 2.60 3.12 3.55 5.22 1.31 5.69 3.17 3.15 0.67 0.92 3.70 0.63 0.47 0.54 0.24 14.55 20.44 32.13
Lead 46 13 664 27 372 27 361 4713 4525 10 164 15 576 2906 781 3408 2323 2194 148 175 3223 20 32 8 4 11 338 40 924 8291
Magnesium (%) 1.01 0.05 0.16 1.27 0.21 0.16 0.11 0.18 2.47 0.31 0.26 0.23 0.24 0.12 0.12 0.14 0.02 0.04 0.05 0.05 0.26 0.16 0.02
Manganese 658 138 93 3752 239 213 174 212 385 111 805 273 271 254 238 270 58 66 62 6 201 234 83
Nickel 265 426 39 370 2567 304 138 1585 5687 322 1621 3200 693 717 32 21 428 14 38 23 12 36 27 25
Phosphorus 829 393 1680 1265 #N/A 175 74 #N/A 262 153 349 371 480 #N/A #N/A #N/A #N/A #N/A #N/A #N/A 1405 785 175
Sulfur (%) 0.03 0.08 0.78 0.13 0.28 0.02 0.25 0.55 0.26 <0.01 0.16 0.07 0.08 <0.01 0.02 0.06 <0.01 <0.01 <0.01 <0.01 0.66 0.66 0.79
Zinc 24 316 239 20 708 106 168 545 709 529 405 1402 395 394 105 398 226 26 109 93 11 3209 4363 2308
Org. C (%) 0.80 2.07 1.15 1.32 1.96 5.13 0.38 6.81 1.96 2.36 1.28 1.82 1.73 0.63 0.10 0.08 0.04 0.62 0.78 <0.01
pH 5.0 7.2 6.5 5.9 6.4 6.7 3.5 4.0 3.6 3.6 3.8 4.6 5.1 4.1
Sand (%) 73 43 60 46 81 74 53 64 75 78 86 53.04
Silt (%) 16 21 24 30 12 18 24 24 19 17 8 14.36
Clay(%) 11 36 17 25 8 9 23 12 6 5 5 32.64
ECEC (cmol/kg) 4.58 20.52 10.26 10.21 7.19 6.71 4.72 3.33 3.45 0.76 0.05 1.29
‘Rep-’=Reppel site; ‘Stam-’=St-Amands site; ‘sl_’=slag sample; ‘so_’=soil sample; ‘cl_’=contaminated layer; ‘slu’=sludge; ‘sP’=profile sample.
328
S.Van
Herrew
egheet
al./
Enviro
nm
enta
lPollu
tion
122
(2003)
323–342
l=1.79 A), scanning electron microscopy with energydispersive X-ray analysis (SEM-EDX; JSM-6400) andelectron microprobe analysis (EMPA; Cameca FranceSX50) on imbedded and polished slag fragments and onpre-concentrated soil samples. Pre-concentration wasachieved by separating different soil fractions accordingto magnetic properties (Frantz isodynamic separator),density (bromoform [�=2.89 g/cm3]) and grain size.This should facilitate mineralogical identification ofparticular minerals that otherwise would be untraceablein a bulk sample due to high background counts of theXRD technique.Three types of phases were detected in the waste
material of the Reppel site. Firstly, primary ore miner-als such as sulfides [CuS, Cu2S, FeAsS, PbS, PbSbS,Ni(Sb,As)S], arsenides [(Ni,Fe,Co)As2, Cu5As2, Cu3As,CoAs] and quartz were detected together with a num-ber of secondary minerals such as FeAsO4
.2H2O,Cu3(AsO4)2.6H2O and Pb2O(OH). Furthermoreremainders of the production process were also presentin oxide minerals such as As2O3, PbO2, FeAl2O4, Fe2O3
and Al2O3syn and in large quantity of metallurgical slag,glass and ash phases of mineralogical indeterminablecomposition.Mineralogy of the St-Amands samples showed a
dominant presence of hematite [Fe2O3], which origi-nates from roasting of pyrite ore. Many sulfate specieshave also been found, in particular CaSO4
.2H2O,CuSO4
.5H2O, PbSO4 and the jarosite-type mineralbeudantite [PbFe3(AsO4)(SO4)(OH)6]. These mineralsare secondary in origin; i.e. they are weathering
products. Furthermore, remainders of the original pyr-ite were also detected.
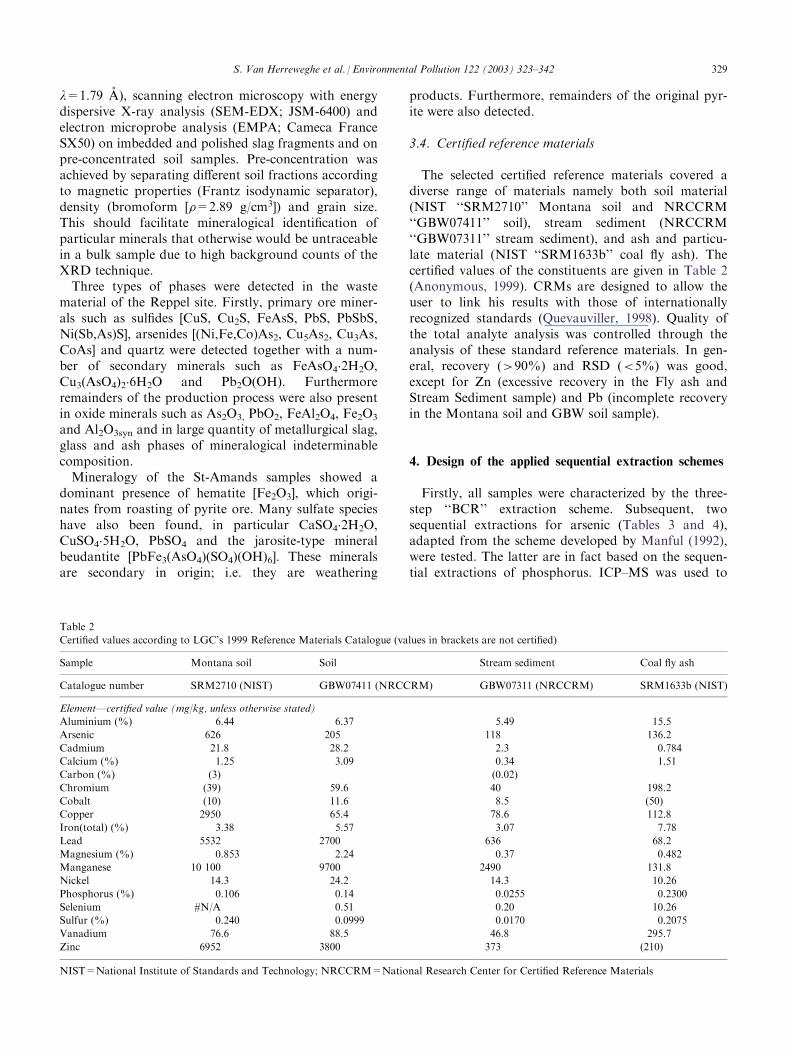
3.4. Certified reference materials
The selected certified reference materials covered adiverse range of materials namely both soil material(NIST ‘‘SRM2710’’ Montana soil and NRCCRM‘‘GBW07411’’ soil), stream sediment (NRCCRM‘‘GBW07311’’ stream sediment), and ash and particu-late material (NIST ‘‘SRM1633b’’ coal fly ash). Thecertified values of the constituents are given in Table 2(Anonymous, 1999). CRMs are designed to allow theuser to link his results with those of internationallyrecognized standards (Quevauviller, 1998). Quality ofthe total analyte analysis was controlled through theanalysis of these standard reference materials. In gen-eral, recovery (>90%) and RSD (<5%) was good,except for Zn (excessive recovery in the Fly ash andStream Sediment sample) and Pb (incomplete recoveryin the Montana soil and GBW soil sample).
4. Design of the applied sequential extraction schemes
Firstly, all samples were characterized by the three-step ‘‘BCR’’ extraction scheme. Subsequent, twosequential extractions for arsenic (Tables 3 and 4),adapted from the scheme developed by Manful (1992),were tested. The latter are in fact based on the sequen-tial extractions of phosphorus. ICP–MS was used to
Table 2
Certified values according to LGC’s 1999 Reference Materials Catalogue (values in brackets are not certified)
Sample Montana soil Soil Stream sediment Coal fly ash
Catalogue number SRM2710 (NIST) GBW07411 (NRCCRM) GBW07311 (NRCCRM) SRM1633b (NIST)
Element—certified value (mg/kg, unless otherwise stated)
Aluminium (%) 6.44 6.37 5.49 15.5
Arsenic 626 205 118 136.2
Cadmium 21.8 28.2 2.3 0.784
Calcium (%) 1.25 3.09 0.34 1.51
Carbon (%) (3) (0.02)
Chromium (39) 59.6 40 198.2
Cobalt (10) 11.6 8.5 (50)
Copper 2950 65.4 78.6 112.8
Iron(total) (%) 3.38 5.57 3.07 7.78
Lead 5532 2700 636 68.2
Magnesium (%) 0.853 2.24 0.37 0.482
Manganese 10 100 9700 2490 131.8
Nickel 14.3 24.2 14.3 10.26
Phosphorus (%) 0.106 0.14 0.0255 0.2300
Selenium #N/A 0.51 0.20 10.26
Sulfur (%) 0.240 0.0999 0.0170 0.2075
Vanadium 76.6 88.5 46.8 295.7
Zinc 6952 3800 373 (210)
NIST=National Institute of Standards and Technology; NRCCRM=National Research Center for Certified Reference Materials
S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342 329

determine both arsenic and heavy metals in all sequen-tial extraction solutions.
4.1. BCR-scheme
A detailed description of the BCR-protocol isdescribed by Quevauviller et al. (1994). Briefly, thesequential extraction was performed by sequentiallyextracting 0.5 grams of the soil samples in a 50 ml cen-trifuge tube at room temperature using a mechanicalshaker with:
Step 1: 20 ml 0.11 mol l�1 acetic acid (16 h shakingperiod).Step 2: 20 ml 0.1 mol l�1 hydoxylamine hydrochloride(16 h shaking period).Step 3: a two-fold 5 ml 8.8 mol l�1 H2O2 (pH 2–3)digestion at 85 �C. Subsequently extraction with 25ml 1 mol l�1 ammonium acetate (16 h shakingperiod).
After shaking, the soil suspension was each time cen-trifuged at 3000 rpm and the supernatant solution wasdecanted, filtered through a Millipore filter (pore size0.45 mm) and put aside for analysis. After each of thefirst two steps, the soil residue was washed every timewith 10 ml H2O (15 min). After centrifuging, the wash-ing solution was carefully decanted and discarded.
4.2. Arsenic extraction scheme I
In a first arsenic extraction scheme, the existing Man-ful (1992)-scheme was only slightly changed. An over-view of this scheme is given in Table 3. Here thesaturated NaCl washing solution was substituted bywater since NaCl leads to serious operational problems(e.g. matrix effects) when solutes are analyzed by ICP–MS. Keon et al. (2001) found that less than 5% of theAs liberated in a previous step was additionally releasedby a PO4
3- wash and therefore also considered a waterwash between each step to be sufficient, thereby mini-mizing matrix effects in the measurements of thewashes.As a second adaption, a HClconc/ HNO3conc/ HFconc
mixture was used to dissolve the residual fraction forcomparison reasons, since this acid mixture was routi-nely used in our research to determine total concentra-tions. The sequential extraction was performed bysequentially attacking 0.5 g of the soil samples with30ml of following agents: NH4Cl, NH4F, NaOH,dithionite-citrate-bicarbonate [DCB] and 0.25 mol l�1
H2SO4 (Table 3) in a 50ml centrifuge tube. After con-tinuous shaking using a mechanical shaker, the soilsuspension was each time centrifuged at 3000 rpm for 10min and the supernatant solution was decanted, filteredthrough a Millipore filter (pore size 0.45 mm), which isrecommended for ICP-determinations, and put aside for
Table 3
Sequential extraction scheme I sligthly modified from Manful (1992)
Fraction Chemical agents added to residue Duration Fraction extracted
Fraction 1 30 ml 1 mol l�1 NH4Cl 2h shaking Easily soluble fraction
Fraction 2 30 ml 0.5 mol l�1 NH4F (pH 8.2) 15h shaking NH4F-extractable fraction
Fraction 3 30 ml 0.1 mol l�1 NaOH 17h shaking NaOH- extractable fraction
Fraction 4 30 ml 0.5 mol l�1 sodium citrate and 2.5ml 1 mol l�1 NaHCO3 while adding 0.5g Na2S2O4.2H2O 15 min heating (85 �C) Reducible fraction
Fraction 5 30 ml 0.25 mol l�1 H2SO4 12h shaking Acid soluble fraction
Fraction 6 4 ml HClconc, 2 ml HNO3conc and 2 ml HFconc mixture; gently heated until half dry, subsequently
reattacked with these three acids, heated until completely dry, redissolved with 20 ml 2.5N HCl
and diluted to 50 ml
Residual fraction
Table 4
Sequential extraction scheme II modified from Manful (1992)
Fraction Chemical agents added to residue Duration Fraction extracted
Fraction 1 30 ml H2O 12 h shaking Water soluble fraction Easily soluble fraction
Fraction 2 2 anion exchange membrane (AEM) stripsa Exchangeable anions
Fraction 3 30 ml 0.5 mol l�1 NH4F (pH 8.2) 12 h shaking NH4F-extractable fraction
Fraction 4 30 ml 0.1 mol l�1 NaOH 12 h shaking NaOH- extractable fraction
Fraction 5 30 ml 0.5 mol l�1 sodium citrate and 2.5ml 1 mol l�1 NaHCO3 (85�C)
while adding 0.5g Na2S2O4.2H2O
15 min heating
(85 �C)
Reducible fraction
Fraction 6 20 ml 8.8 mol l�1 H2O2 (acidified to pH 2 with HNO3); heat (85�C) until
completely dry; redissolve with 30 ml HNO3 (0.02M)
Evaporating
+12 h shaking
Oxidizable fraction
Fraction 7 4 ml HClconc, 2 ml HNO3conc and 2 ml HFconc mixture; gently heated
until half dry, subsequently reattacked with these three acids, heated until
completely dry, redissolved with 20 ml 2.5N HCl and diluted to 50 ml
Residual fraction
a The AEM strips are regenerated, for reuse after the extraction, by washing them for 3 days with 6 batches of 0.5 mol l�1 HCl and 3 days with 6 batches of 0.5 mol l�1
NaHCO3 and then rinsing them well with distilled water.
330 S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342

analysis. After centrifuging and decanting, soil residuesof fractions 2–5 were washed once with 20 ml H2O,which was also centrifuged, filtered and added to theextracted solution of the respective fraction. The residuefrom step 5 was finally transferred into a Teflon beakerwith a small amount of water. After the addition of 4 mlHClconc[37%], 2 ml HNO3conc[65%] and 2 mlHFconc[48%], the mixture was gently heated on a hotplate until almost dry and subsequently reattacked withthe same amounts of these three acids and heated todryness at about 220 �C. This residue was redissolved bygently heating with 20 ml of 2.5 mol l�1 HCl, trans-ferred into volumetric flasks and diluted to 50 ml.
4.3. Arsenic extraction scheme II
In a second version, additional changes to Manful(1992) were made with the aim to improve the fractio-nation of arsenic. Firstly, the easily soluble fraction wasdivided in two parts according to the phosphorussequential extraction scheme of Hedley et al. (1982) asdiscussed by Tiessen and Moir (1993), i.e. water soluble,anionic arsenic species extracted out of the suspensionby means of adsorption on anion exchange membrane(AEM) strips and secondly the water soluble fractionnot extracted by these AEM strips. Both exchangeableas well as some of the soluble anionic arsenic species inprecipitates will enter the solution and be absorbed bythe AEM. The superiority of the extraction of anionicelements by AEM strips relates to the fact that theAEM provides a sink without exerting destructiveinfluence on the soil, as is in the case of several chemicalextractants (Van Raij et al., 1986).Additionally, Manful (1992) expected to dissolve Ca-
bound arsenic in one of the last extraction steps (H2SO4
extraction, i.e. step five in scheme I) as is the case forphosphorus. However, Ca-bound arsenic is much moresoluble than the corresponding Ca-phosphate. It is alsocommonly known that Fe- and Al-arsenates are farmore stable than Ca-arsenates, the latter being onlystable in well-oxidized and alkaline environments. ThelogKsp of the three compounds are, respectively, �20.2,�15.8 and �18.2, but due to their stoichiometries, Al-and Fe-arsenates are less soluble than Ca-arsenate(Fergusson, 1990). Consequently, Ca-arsenates willprobably already dissolve in one of the extraction solu-tions (e.g. easily soluble or NH4F-extraction) precedingthe H2SO4-extraction. The fact that Ca3(AsO4)2 is onlystable at pH>10 was also confirmed by a thermo-dynamical calculation with MinteqA2 (Allison et al.,1991). The H2SO4-extracted As probably arises fromarsenic bound to iron oxides, which are dissolved by theDCB treatment, yet it was most likely not removedfrom the residue during the DCB-extraction for exam-ple due to redistribution phenomena (Jacobs et al.,1970). Therefore, we decided to replace the acid
‘‘H2SO4’’ extraction step by an oxidizing extraction (8.8mol l�1 H2O2).The following steps were implemented (Table 4): a
sample of soil (0.5 g) and two 9 mm by 62 mm strips ofAEM (BDH Product 55164), converted to bicarbonateform, were suspended in 30 ml deionized water in a 50ml centrifuge tube and shaken overnight (12 h). TheAEM strips were then carefully removed from the sus-pension, rinsed with deionized water to remove anyadhering soil particles and then placed into a clean 50ml tube containing 20 ml of 0.5 mol l�1 HCl. The rinsewater was discarded. The tube containing the AEMswas then set aside for 1 h to allow gas to escape, capped,shaken overnight (12 h) and the solution was put asidefor the As determination. Following the removal of theAEM strips, the soil suspension was centrifuged at 3000rpm, the supernatant solution was decanted through aMillipore filter (pore size 0.45 mm) and the solution wasput aside for analysis. The soil residue was subsequentlyreattacked (Table 4: fraction 3–6) with following agents:NH4F, NaOH, dithionite-citrate-bicarbonate [DCB]and H2O2-digestion/HNO3-extraction. After continuousshaking using a mechanical shaker, the soil suspensionwas centrifuged at 3000 rpm and the solution was dec-anted through a Millipore filter (pore size 0.45 mm).After centrifuging and decanting, the soil residues offractions 3–5 were washed once with 20 ml H2O whichwas also centrifuged, filtered and added to the extractedsolution of the respective fraction. The residue fromextraction 6 was finally transferred into a Teflon beakerwith a small amount of water and was treated in asimilar way as fraction 6 of arsenic extraction scheme I.
4.4. Leachate analysis
Extracted samples were stored under cool (4 �C) anddark conditions prior to analysis. Multi-element analy-sis of the samples diluted with 5% HNO3 (ultrapure)(Mg, Al, Ca, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, As, Se, Cd,and Pb) by ICP–MS (HP 4500 series) was carried outwithin a week after the end of the experiment. Standardseries were made up starting from the ‘‘10 ppm Multi-Element Calibration Standard-2A in 5% HNO3’’ fromHewlett Packard1, with the appropriate extractingsolution, equally diluted with 5% HNO3 as the samplesto achieve an identical background. An Indium (In)internal standard was applied to both samples andstandards. Each ICP–MS measurement was carried outwith five repetitions. Relative standard deviationsremained below five percent. Accuracy was checked bymeasuring one of the standard solutions as unknownsample (bias generally <10%). The spectroscopic inter-ference of ArCl, which has the same m/z as As (75), wascorrected according to recommendations of the EPA(method 200.8) by following equation: corrected Assignal=[m/z 75 signal]�3.127([m/z 77 signal]�0.815 [m/z
S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342 331
82 signal]). This coincides with following reasoning: (i)estimate 77Se based on 82Se, (ii) estimate 75ArCl basedon 77ArCl, and (iii) consider the remaining m/z countsdue to As.
5. Results and discussion
5.1. Reconnaissance research
The results of the reconnaissance research on purearsenic containing minerals resulted in the followingobservations: sodium arsenate (Na2HAsO4
.7H2O) wascompletely soluble in water, calcium arsenate(Ca3(AsO4)2.xH2O) was soluble at pH<8, aluminiumarsenate (AlAsO4
.xH2O) at pH>8 and iron arsenate(FeAsO4
.xH2O) under reducing conditions. Arsenolite(As2O3) was released in more than one extraction step(both in neutral salt-, acid-, alkaline- and oxidizing-solutions) which is an indication that it is difficult toselectively extract arsenolite. Voigt et al. (1996) founda similar behavior for the mineral hoernesite(Mg3(AsO4)2.8H2O) that dissolved in both neutral salt,acid and reducing solutions. The arsenic sulfides realgar(AsS) and orpiment (As2S3) were mainly present in theresidual fraction although oxidation with H2O2 is ableto release a considerable arsenic amount from thesesulfides. Oxidation with Na4P2O7 released less orpimentbut was on the other hand a better extractant fororganic bound arsenic. Lollingite (FeAs2) is also mainlypresent in the residual fraction but a considerable part isalso released during reducing extractions. Terashimaand Taniguchi (1997) also found that only a small frac-tion of the arsenic bearing sulfides arsenopyrite andloellingite in ores could be dissolved under oxidizing
circumstances (heating in 0.7 mol l�1 NaOCl) and con-sequently arsenic was mainly present in the residualphase. According to Keon et al. (2001), orpiment(As2S3) is dissolved by hot concentrated HNO3/H2O2,amorphous As-sulfide by 1 mol l�1 HCl, 50 mmol l�1
Ti-citrate-EDTA and 16 mol l�1 HNO3, and arseno-pyrite by 16 mol l�1 HNO3. They also found thatarsenic trioxide (As2O3) was set free by 10 mol l�1
HF and As adsorbed on goethite by 1 mol l�1
NaH2PO4.The approach of attacking pure and non-natural
minerals will be only an approximation of the reality forseveral reasons. The main reason is that these extractionschemes are not developed to extract pure minerals sothat the solid/liquid ratio and extraction duration willbe inadequate. Nevertheless, this information can beuseful to interpret to a certain extent the extractions ofsamples containing arsenic that is present in discreteminerals.
5.2. BCR-extraction
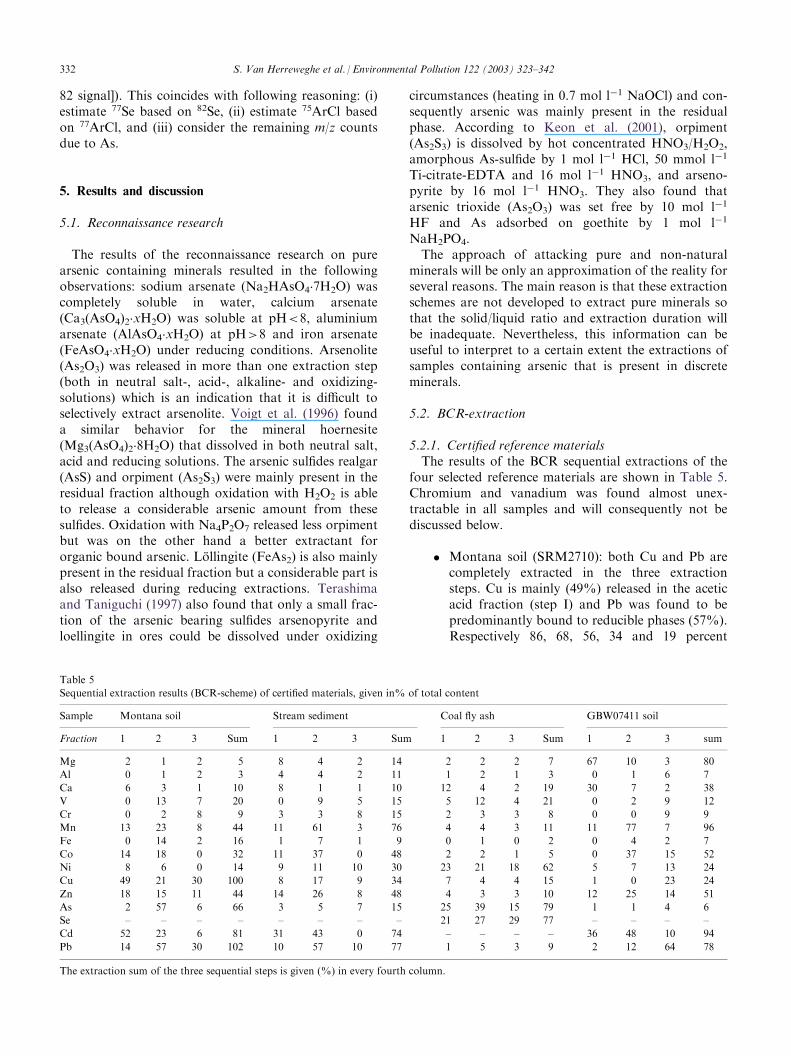
5.2.1. Certified reference materialsThe results of the BCR sequential extractions of the
four selected reference materials are shown in Table 5.Chromium and vanadium was found almost unex-tractable in all samples and will consequently not bediscussed below.
� Montana soil (SRM2710): both Cu and Pb arecompletely extracted in the three extractionsteps. Cu is mainly (49%) released in the aceticacid fraction (step I) and Pb was found to bepredominantly bound to reducible phases (57%).Respectively 86, 68, 56, 34 and 19 percent
Table 5
Sequential extraction results (BCR-scheme) of certified materials, given in% of total content
Sample Montana soil Stream sediment Coal fly ash GBW07411 soil
Fraction 1 2 3 Sum 1 2 3 Sum 1 2 3 Sum 1 2 3 sum
Mg 2 1 2 5 8 4 2 14 2 2 2 7 67 10 3 80
Al 0 1 2 3 4 4 2 11 1 2 1 3 0 1 6 7
Ca 6 3 1 10 8 1 1 10 12 4 2 19 30 7 2 38
V 0 13 7 20 0 9 5 15 5 12 4 21 0 2 9 12
Cr 0 2 8 9 3 3 8 15 2 3 3 8 0 0 9 9
Mn 13 23 8 44 11 61 3 76 4 4 3 11 11 77 7 96
Fe 0 14 2 16 1 7 1 9 0 1 0 2 0 4 2 7
Co 14 18 0 32 11 37 0 48 2 2 1 5 0 37 15 52
Ni 8 6 0 14 9 11 10 30 23 21 18 62 5 7 13 24
Cu 49 21 30 100 8 17 9 34 7 4 4 15 1 0 23 24
Zn 18 15 11 44 14 26 8 48 4 3 3 10 12 25 14 51
As 2 57 6 66 3 5 7 15 25 39 15 79 1 1 4 6
Se – – – – – – – – 21 27 29 77 – – – –
Cd 52 23 6 81 31 43 0 74 – – – – 36 48 10 94
Pb 14 57 30 102 10 57 10 77 1 5 3 9 2 12 64 78
The extraction sum of the three sequential steps is given (%) in every fourth column.
332 S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342

remained unextracted for Ni, Co, Zn, As and Cd.Cd is mainly (52%) released in the acetic acidfraction (step I) and As was found to be pre-dominantly bound to reducible phases (57%)(step II). In Table 6 our results are comparedwith the results of the BCR-extraction of theMontana soil (SRM 2710) executed by Ho andEvans (1997). Our results match very well for Znand Cd. Cu is released to a greater extent in ourwork, especially in the 1 and 3rd extraction step.In our experiment, Pb is extracted ten times morein the reducible step and conversely lower con-centrations were detected in the third step. Hoand Evans (1997) also reported lower Pb extrac-tion in the first two fractions and elevated con-centrations in the third compared with resultsfrom other authors [e.g. the Tessier et al. (1979)-like sequential extraction of SRM 2710 executedby Li et al. (1995a)]. They interpreted these dif-ferences as readsorption effects from theexchangeable and reducible fractions onto anoxidizable fraction. Apparently, such possiblereadsorption effects were less important in ourexperiment.
� Stream sediment (GBW07311): metal contentextracted in this sample by the three-step BCR-scheme are low. Pb and Cd were extracted for,respectively, 77 and 74%, mainly in the reduciblefraction (respectively 57 and 43%). For Cd, 31and 43% were released in the first and secondstep, respectively, yet none was detected in thethird step. Although Zn, Cu and Ni were foundrather equally distributed in all three steps, only,respectively, 48, 34 and 30% of their total con-tent was extracted. Mn was mainly released dur-ing the reducible step (61%). As remained for85% unextracted.
� Coal fly ash (SRM1933b): the BCR-schemeextracted the metalloids As and Se well, i.e. 79and 77%, respectively. Both elements were fairlydistributed between all three fractions, whereby
Se was somewhat more bound to the oxidizablefraction compared to As, which was recordedmore in the reducible fraction. Except for Ni(62%), extraction of heavy metals was low andremained below 20%.
� Soil (GBW07411): Cd was almost completely(94%) extracted (respectively 36, 48 and 10% inBCR-step 1, 2 and 3) from this sample. Pb wasmainly (64%) extracted in the third step. Half ofthe Zn and Co amount was extracted whereasthe extraction of Ni, Cu and especially Asremained low. The first fraction apparentlyreleased a major part of Ca (30%) and Mg(67%) whereas 77% of Mn was released duringthe reducible extraction.
5.2.2. Highly contaminated soil samplesThe results of the BCR sequential extractions of the
highly contaminated soil samples are shown in Table 7.The St-Amands samples (Stam-so_x in Table 7) were
iron-rich due to the presence of the burned pyrite-wasteand therefore, an important fraction of arsenic is occlu-ded within the mineral structure of the secondary Fe-(oxi)hydroxides (such as goethite and hematite). How-ever, the acid hydroxylamine hydrochloride reagentonly releases elements bound to easily reducible phasesso that much of the heavy metals remained unextractedduring the BCR-extraction of these samples, explainingthe low recovery. The heavy metals in the Reppel sam-ples were generally better extracted during this protocol.In general, much of Mn in the samples remained
unextracted, while extracted Mn was mostly pro-portionally distributed between the first two extractionsteps. Co was completely extracted from samples Rep-sl_2, Rep-slu, Rep-cl_2 and Rep-cl_3 and almost notextracted from samples Rep-sl_1, Rep-sl_3 and Rep-so_1. In most samples more than 20% of the totalcobalt content was HAc-soluble, somewhat less wasreleased during the reducing step. Much of Ni wasfound unextracted, while Cu was almost always com-pletely extracted (except Rep-sl_1, Rep-so_1, Rep-so_2). Zn displayed an analogous leaching behavior asCu although in absolute terms it was generally some-what less extracted. Ni, Cu and Zn were released in allthree-extraction solutions. Conversely, Pb remainedfairly unextracted or released after oxidation (>10%HAc soluble Pb only in samples Rep-sl_1 and-so_2).Note that the sum of extracted fractions sometimesexceedes the total element concentration. This observa-tion will be discussed in succeeding sections.On average 50% of As was extracted and most of the
extracted As was found to be bound to reducible pha-ses. However, with this extraction protocol it is impos-sible to discriminate between arsenic adsorbed andcoprecipitated to iron hydroxides. Since no adsorption
Table 6
Comparison of the extraction results (BCR-scheme) of the Montana
soil, given in% of total content
1 2 3 sum
Cu This work 49 21 30 100
Ho and Evans (1997) 34 29 13 76
Zn This work 18 15 11 44
Ho and Evans (1997) 17 16 7 41
Cd This work 51 23 6 80
Ho and Evans (1997) 54 23 6 83
Pb This work 14 57 30 102
Ho and Evans (1997) 14 6 46 66
The extraction sum of the three sequential steps is given (%) in every
fourth column.
S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342 333
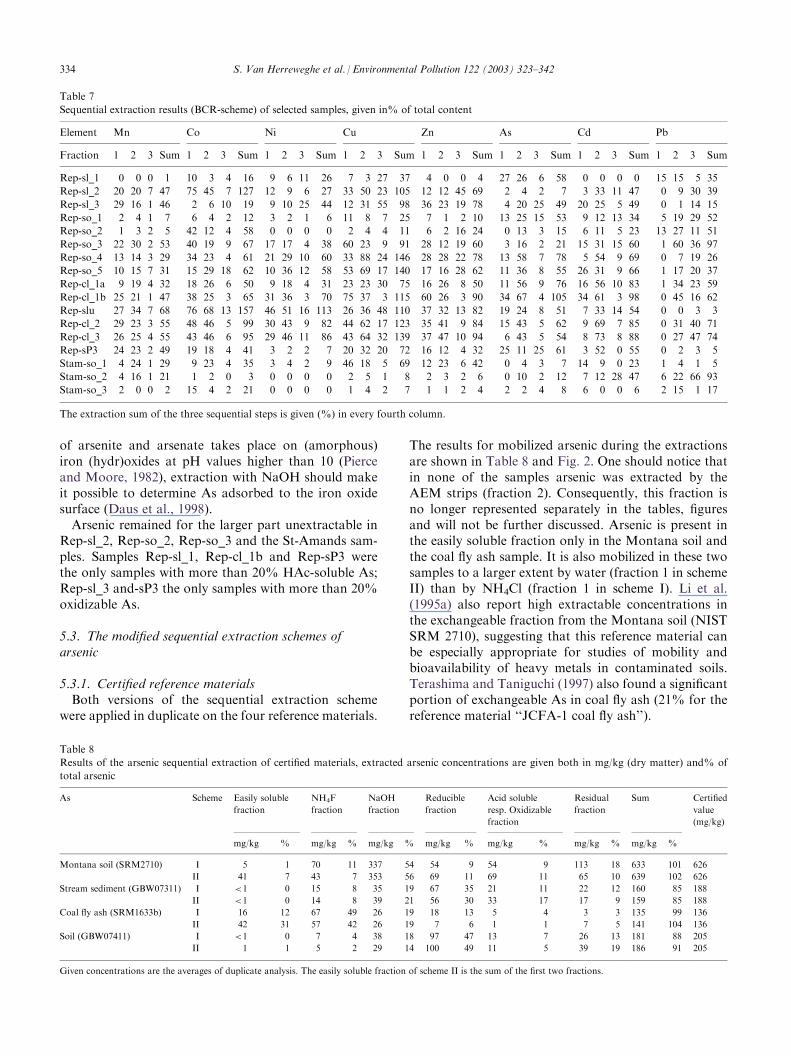
of arsenite and arsenate takes place on (amorphous)iron (hydr)oxides at pH values higher than 10 (Pierceand Moore, 1982), extraction with NaOH should makeit possible to determine As adsorbed to the iron oxidesurface (Daus et al., 1998).Arsenic remained for the larger part unextractable in
Rep-sl_2, Rep-so_2, Rep-so_3 and the St-Amands sam-ples. Samples Rep-sl_1, Rep-cl_1b and Rep-sP3 werethe only samples with more than 20% HAc-soluble As;Rep-sl_3 and-sP3 the only samples with more than 20%oxidizable As.
5.3. The modified sequential extraction schemes ofarsenic
5.3.1. Certified reference materialsBoth versions of the sequential extraction scheme
were applied in duplicate on the four reference materials.
The results for mobilized arsenic during the extractionsare shown in Table 8 and Fig. 2. One should notice thatin none of the samples arsenic was extracted by theAEM strips (fraction 2). Consequently, this fraction isno longer represented separately in the tables, figuresand will not be further discussed. Arsenic is present inthe easily soluble fraction only in the Montana soil andthe coal fly ash sample. It is also mobilized in these twosamples to a larger extent by water (fraction 1 in schemeII) than by NH4Cl (fraction 1 in scheme I). Li et al.(1995a) also report high extractable concentrations inthe exchangeable fraction from the Montana soil (NISTSRM 2710), suggesting that this reference material canbe especially appropriate for studies of mobility andbioavailability of heavy metals in contaminated soils.Terashima and Taniguchi (1997) also found a significantportion of exchangeable As in coal fly ash (21% for thereference material ‘‘JCFA-1 coal fly ash’’).
Table 8
Results of the arsenic sequential extraction of certified materials, extracted arsenic concentrations are given both in mg/kg (dry matter) and% of
total arsenic
As Scheme Easily soluble
fraction
NH4F
fraction
NaOH
fraction
Reducible
fraction
Acid soluble
resp. Oxidizable
fraction
Residual
fraction
Sum Certified
value
(mg/kg)
mg/kg % mg/kg % mg/kg % mg/kg % mg/kg % mg/kg % mg/kg %
Montana soil (SRM2710) I 5 1 70 11 337 54 54 9 54 9 113 18 633 101 626
II 41 7 43 7 353 56 69 11 69 11 65 10 639 102 626
Stream sediment (GBW07311) I <1 0 15 8 35 19 67 35 21 11 22 12 160 85 188
II <1 0 14 8 39 21 56 30 33 17 17 9 159 85 188
Coal fly ash (SRM1633b) I 16 12 67 49 26 19 18 13 5 4 3 3 135 99 136
II 42 31 57 42 26 19 7 6 1 1 7 5 141 104 136
Soil (GBW07411) I <1 0 7 4 38 18 97 47 13 7 26 13 181 88 205
II 1 1 5 2 29 14 100 49 11 5 39 19 186 91 205
Given concentrations are the averages of duplicate analysis. The easily soluble fraction of scheme II is the sum of the first two fractions.
Table 7
Sequential extraction results (BCR-scheme) of selected samples, given in% of total content
Element Mn Co Ni Cu Zn As Cd Pb
Fraction 1 2 3 Sum 1 2 3 Sum 1 2 3 Sum 1 2 3 Sum 1 2 3 Sum 1 2 3 Sum 1 2 3 Sum 1 2 3 Sum
Rep-sl_1 0 0 0 1 10 3 4 16 9 6 11 26 7 3 27 37 4 0 0 4 27 26 6 58 0 0 0 0 15 15 5 35
Rep-sl_2 20 20 7 47 75 45 7 127 12 9 6 27 33 50 23 105 12 12 45 69 2 4 2 7 3 33 11 47 0 9 30 39
Rep-sl_3 29 16 1 46 2 6 10 19 9 10 25 44 12 31 55 98 36 23 19 78 4 20 25 49 20 25 5 49 0 1 14 15
Rep-so_1 2 4 1 7 6 4 2 12 3 2 1 6 11 8 7 25 7 1 2 10 13 25 15 53 9 12 13 34 5 19 29 52
Rep-so_2 1 3 2 5 42 12 4 58 0 0 0 0 2 4 4 11 6 2 16 24 0 13 3 15 6 11 5 23 13 27 11 51
Rep-so_3 22 30 2 53 40 19 9 67 17 17 4 38 60 23 9 91 28 12 19 60 3 16 2 21 15 31 15 60 1 60 36 97
Rep-so_4 13 14 3 29 34 23 4 61 21 29 10 60 33 88 24 146 28 28 22 78 13 58 7 78 5 54 9 69 0 7 19 26
Rep-so_5 10 15 7 31 15 29 18 62 10 36 12 58 53 69 17 140 17 16 28 62 11 36 8 55 26 31 9 66 1 17 20 37
Rep-cl_1a 9 19 4 32 18 26 6 50 9 18 4 31 23 23 30 75 16 26 8 50 11 56 9 76 16 56 10 83 1 34 23 59
Rep-cl_1b 25 21 1 47 38 25 3 65 31 36 3 70 75 37 3 115 60 26 3 90 34 67 4 105 34 61 3 98 0 45 16 62
Rep-slu 27 34 7 68 76 68 13 157 46 51 16 113 26 36 48 110 37 32 13 82 19 24 8 51 7 33 14 54 0 0 3 3
Rep-cl_2 29 23 3 55 48 46 5 99 30 43 9 82 44 62 17 123 35 41 9 84 15 43 5 62 9 69 7 85 0 31 40 71
Rep-cl_3 26 25 4 55 43 46 6 95 29 46 11 86 43 64 32 139 37 47 10 94 6 43 5 54 8 73 8 88 0 27 47 74
Rep-sP3 24 23 2 49 19 18 4 41 3 2 2 7 20 32 20 72 16 12 4 32 25 11 25 61 3 52 0 55 0 2 3 5
Stam-so_1 4 24 1 29 9 23 4 35 3 4 2 9 46 18 5 69 12 23 6 42 0 4 3 7 14 9 0 23 1 4 1 5
Stam-so_2 4 16 1 21 1 2 0 3 0 0 0 0 2 5 1 8 2 3 2 6 0 10 2 12 7 12 28 47 6 22 66 93
Stam-so_3 2 0 0 2 15 4 2 21 0 0 0 0 1 4 2 7 1 1 2 4 2 2 4 8 6 0 0 6 2 15 1 17
The extraction sum of the three sequential steps is given (%) in every fourth column.
334 S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342
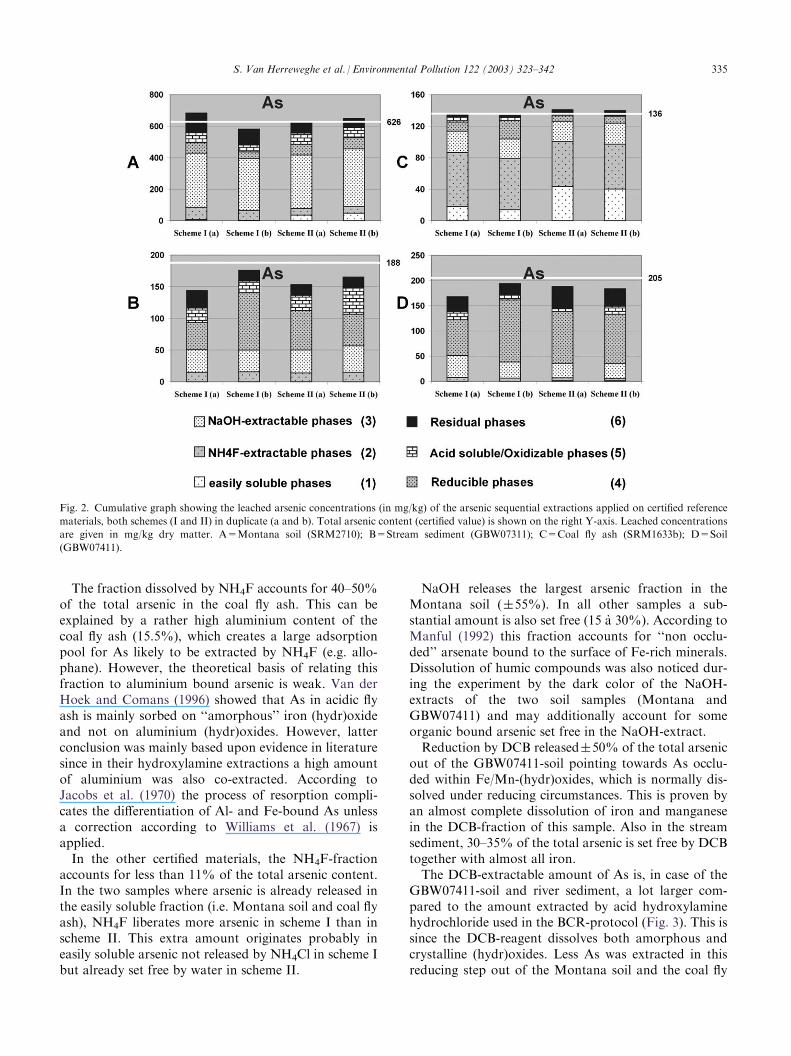
The fraction dissolved by NH4F accounts for 40–50%of the total arsenic in the coal fly ash. This can beexplained by a rather high aluminium content of thecoal fly ash (15.5%), which creates a large adsorptionpool for As likely to be extracted by NH4F (e.g. allo-phane). However, the theoretical basis of relating thisfraction to aluminium bound arsenic is weak. Van derHoek and Comans (1996) showed that As in acidic flyash is mainly sorbed on ‘‘amorphous’’ iron (hydr)oxideand not on aluminium (hydr)oxides. However, latterconclusion was mainly based upon evidence in literaturesince in their hydroxylamine extractions a high amountof aluminium was also co-extracted. According toJacobs et al. (1970) the process of resorption compli-cates the differentiation of Al- and Fe-bound As unlessa correction according to Williams et al. (1967) isapplied.In the other certified materials, the NH4F-fraction
accounts for less than 11% of the total arsenic content.In the two samples where arsenic is already released inthe easily soluble fraction (i.e. Montana soil and coal flyash), NH4F liberates more arsenic in scheme I than inscheme II. This extra amount originates probably ineasily soluble arsenic not released by NH4Cl in scheme Ibut already set free by water in scheme II.
NaOH releases the largest arsenic fraction in theMontana soil (�55%). In all other samples a sub-stantial amount is also set free (15 a 30%). According toManful (1992) this fraction accounts for ‘‘non occlu-ded’’ arsenate bound to the surface of Fe-rich minerals.Dissolution of humic compounds was also noticed dur-ing the experiment by the dark color of the NaOH-extracts of the two soil samples (Montana andGBW07411) and may additionally account for someorganic bound arsenic set free in the NaOH-extract.Reduction by DCB released�50% of the total arsenic
out of the GBW07411-soil pointing towards As occlu-ded within Fe/Mn-(hydr)oxides, which is normally dis-solved under reducing circumstances. This is proven byan almost complete dissolution of iron and manganesein the DCB-fraction of this sample. Also in the streamsediment, 30–35% of the total arsenic is set free by DCBtogether with almost all iron.The DCB-extractable amount of As is, in case of the
GBW07411-soil and river sediment, a lot larger com-pared to the amount extracted by acid hydroxylaminehydrochloride used in the BCR-protocol (Fig. 3). This issince the DCB-reagent dissolves both amorphous andcrystalline (hydr)oxides. Less As was extracted in thisreducing step out of the Montana soil and the coal fly
Fig. 2. Cumulative graph showing the leached arsenic concentrations (in mg/kg) of the arsenic sequential extractions applied on certified reference
materials, both schemes (I and II) in duplicate (a and b). Total arsenic content (certified value) is shown on the right Y-axis. Leached concentrations
are given in mg/kg dry matter. A=Montana soil (SRM2710); B=Stream sediment (GBW07311); C=Coal fly ash (SRM1633b); D=Soil
(GBW07411).
S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342 335
ash, at least in comparison to the other samples as wellas compared to the BCR-extraction of these samples.This most likely relates to NH4F- and/or NaOH-extractable arsenic already set free in preceding steps.The acid or oxidizing fraction is only of minor
importance (>10%) in the stream sediment sample.Some iron is still mobilized in the acid dissolution frac-tion, probably accounting for a further removal of(reduced) iron compounds that were not completelyextracted in the previous extraction step.Finally, some 10–20% of the total arsenic was liber-
ated in the last step (residual fraction) in the two soilsamples (Montana Soil and GBW07411) and the streamsediment while the residual fraction was negligible in thecoal fly ash. The ‘overall arsenic recovery rates’, whichis a indication of the method accuracy, are very goodin the sequential extractions (both schemes) for theMontana soil and coal fly ash sample (�100%), andrather good for the GBW07411 soil and stream sedi-ment samples (85–90%).
5.3.2. Highly contaminated soil samples and slagmaterialThe arsenic sequential extraction was applied on
soil and slag material (highly) contaminated witharsenic. Both slag material (Rep-sl_1, Rep-sl_2 andRep-sl_3), surface soil samples (Rep-so_1 to Rep-cl_3and Stam-so_1 to Stam-so_3) and profile soil samples(Rep-sP1 to Rep-sP7) were subjected to the extractionscheme I. Scheme II was also applied on the soil samplesexcept the soil profile samples. In the soil profile, onlythe highly contaminated layer was subjected to schemeII.
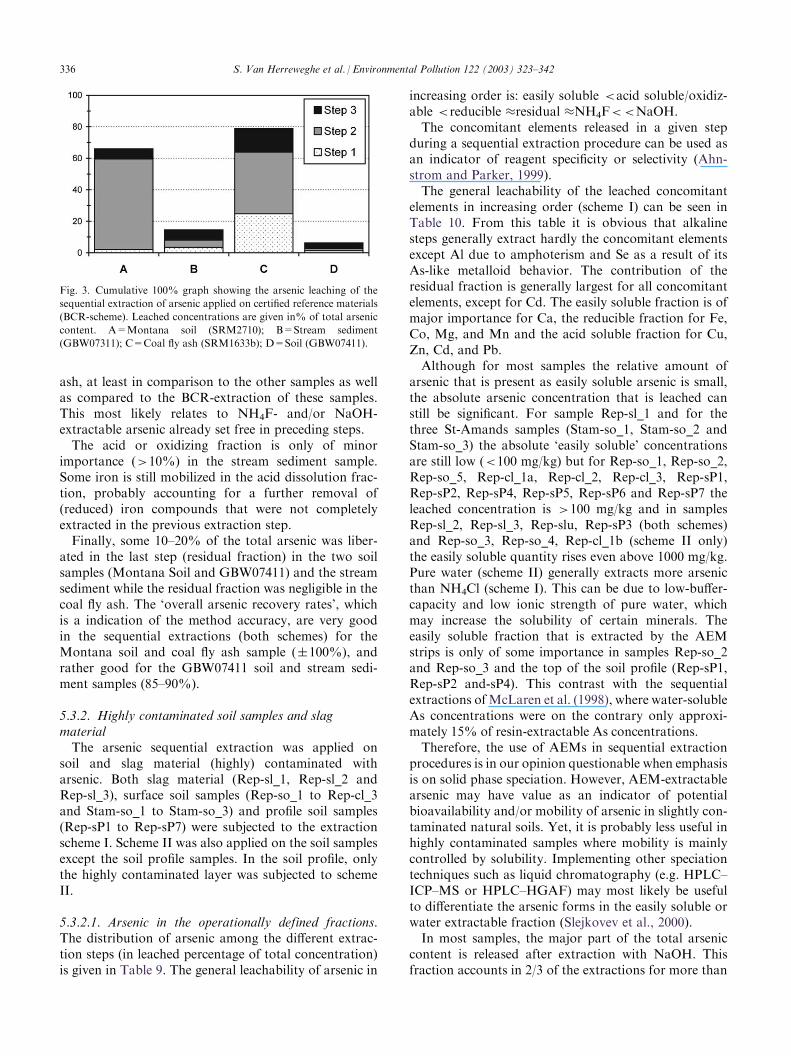
5.3.2.1. Arsenic in the operationally defined fractions.The distribution of arsenic among the different extrac-tion steps (in leached percentage of total concentration)is given in Table 9. The general leachability of arsenic in
increasing order is: easily soluble <acid soluble/oxidiz-able <reducible �residual �NH4F<<NaOH.The concomitant elements released in a given step
during a sequential extraction procedure can be used asan indicator of reagent specificity or selectivity (Ahn-strom and Parker, 1999).The general leachability of the leached concomitant
elements in increasing order (scheme I) can be seen inTable 10. From this table it is obvious that alkalinesteps generally extract hardly the concomitant elementsexcept Al due to amphoterism and Se as a result of itsAs-like metalloid behavior. The contribution of theresidual fraction is generally largest for all concomitantelements, except for Cd. The easily soluble fraction is ofmajor importance for Ca, the reducible fraction for Fe,Co, Mg, and Mn and the acid soluble fraction for Cu,Zn, Cd, and Pb.Although for most samples the relative amount of
arsenic that is present as easily soluble arsenic is small,the absolute arsenic concentration that is leached canstill be significant. For sample Rep-sl_1 and for thethree St-Amands samples (Stam-so_1, Stam-so_2 andStam-so_3) the absolute ‘easily soluble’ concentrationsare still low (<100 mg/kg) but for Rep-so_1, Rep-so_2,Rep-so_5, Rep-cl_1a, Rep-cl_2, Rep-cl_3, Rep-sP1,Rep-sP2, Rep-sP4, Rep-sP5, Rep-sP6 and Rep-sP7 theleached concentration is >100 mg/kg and in samplesRep-sl_2, Rep-sl_3, Rep-slu, Rep-sP3 (both schemes)and Rep-so_3, Rep-so_4, Rep-cl_1b (scheme II only)the easily soluble quantity rises even above 1000 mg/kg.Pure water (scheme II) generally extracts more arsenicthan NH4Cl (scheme I). This can be due to low-buffer-capacity and low ionic strength of pure water, whichmay increase the solubility of certain minerals. Theeasily soluble fraction that is extracted by the AEMstrips is only of some importance in samples Rep-so_2and Rep-so_3 and the top of the soil profile (Rep-sP1,Rep-sP2 and-sP4). This contrast with the sequentialextractions ofMcLaren et al. (1998), where water-solubleAs concentrations were on the contrary only approxi-mately 15% of resin-extractable As concentrations.Therefore, the use of AEMs in sequential extraction
procedures is in our opinion questionable when emphasisis on solid phase speciation. However, AEM-extractablearsenic may have value as an indicator of potentialbioavailability and/or mobility of arsenic in slightly con-taminated natural soils. Yet, it is probably less useful inhighly contaminated samples where mobility is mainlycontrolled by solubility. Implementing other speciationtechniques such as liquid chromatography (e.g. HPLC–ICP–MS or HPLC–HGAF) may most likely be usefulto differentiate the arsenic forms in the easily soluble orwater extractable fraction (Slejkovev et al., 2000).In most samples, the major part of the total arsenic
content is released after extraction with NaOH. Thisfraction accounts in 2/3 of the extractions for more than
Fig. 3. Cumulative 100% graph showing the arsenic leaching of the
sequential extraction of arsenic applied on certified reference materials
(BCR-scheme). Leached concentrations are given in% of total arsenic
content. A=Montana soil (SRM2710); B=Stream sediment
(GBW07311); C=Coal fly ash (SRM1633b); D=Soil (GBW07411).
336 S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342
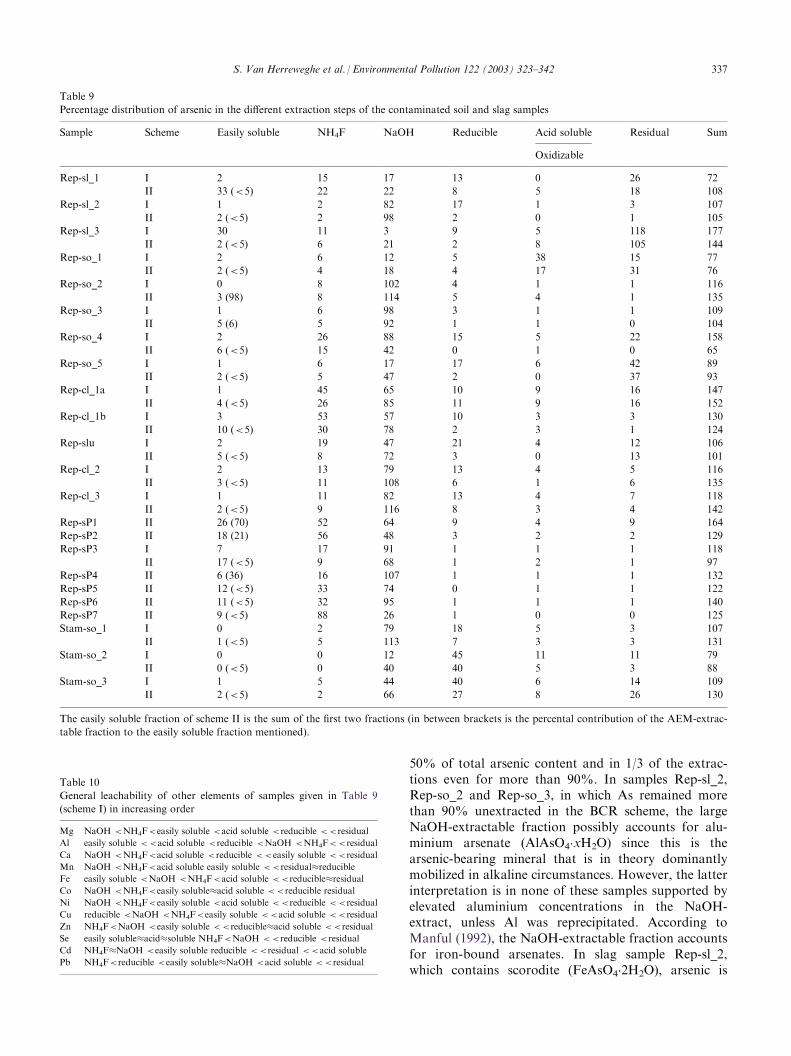
50% of total arsenic content and in 1/3 of the extrac-tions even for more than 90%. In samples Rep-sl_2,Rep-so_2 and Rep-so_3, in which As remained morethan 90% unextracted in the BCR scheme, the largeNaOH-extractable fraction possibly accounts for alu-minium arsenate (AlAsO4
.xH2O) since this is thearsenic-bearing mineral that is in theory dominantlymobilized in alkaline circumstances. However, the latterinterpretation is in none of these samples supported byelevated aluminium concentrations in the NaOH-extract, unless Al was reprecipitated. According toManful (1992), the NaOH-extractable fraction accountsfor iron-bound arsenates. In slag sample Rep-sl_2,which contains scorodite (FeAsO4
.2H2O), arsenic is
Table 10
General leachability of other elements of samples given in Table 9
(scheme I) in increasing order
Mg NaOH <NH4F<easily soluble <acid soluble <reducible <<residual
Al easily soluble <<acid soluble <reducible <NaOH <NH4F<<residual
Ca NaOH <NH4F<acid soluble <reducible <<easily soluble <<residual
Mn NaOH <NH4F<acid soluble easily soluble <<residual�reducible
Fe easily soluble <NaOH <NH4F<acid soluble <<reducible�residual
Co NaOH <NH4F<easily soluble�acid soluble <<reducible residual
Ni NaOH <NH4F<easily soluble <acid soluble <<reducible <<residual
Cu reducible <NaOH <NH4F<easily soluble <<acid soluble <<residual
Zn NH4F<NaOH <easily soluble <<reducible�acid soluble <<residual
Se easily soluble�acid�soluble NH4F<NaOH <<reducible <residual
Cd NH4F�NaOH <easily soluble reducible <<residual <<acid soluble
Pb NH4F<reducible <easily soluble�NaOH <acid soluble <<residual
Table 9
Percentage distribution of arsenic in the different extraction steps of the contaminated soil and slag samples
Sample Scheme Easily soluble NH4F NaOH Reducible Acid soluble Residual Sum
Oxidizable
Rep-sl_1 I 2 15 17 13 0 26 72
II 33 (<5) 22 22 8 5 18 108
Rep-sl_2 I 1 2 82 17 1 3 107
II 2 (<5) 2 98 2 0 1 105
Rep-sl_3 I 30 11 3 9 5 118 177
II 2 (<5) 6 21 2 8 105 144
Rep-so_1 I 2 6 12 5 38 15 77
II 2 (<5) 4 18 4 17 31 76
Rep-so_2 I 0 8 102 4 1 1 116
II 3 (98) 8 114 5 4 1 135
Rep-so_3 I 1 6 98 3 1 1 109
II 5 (6) 5 92 1 1 0 104
Rep-so_4 I 2 26 88 15 5 22 158
II 6 (<5) 15 42 0 1 0 65
Rep-so_5 I 1 6 17 17 6 42 89
II 2 (<5) 5 47 2 0 37 93
Rep-cl_1a I 1 45 65 10 9 16 147
II 4 (<5) 26 85 11 9 16 152
Rep-cl_1b I 3 53 57 10 3 3 130
II 10 (<5) 30 78 2 3 1 124
Rep-slu I 2 19 47 21 4 12 106
II 5 (<5) 8 72 3 0 13 101
Rep-cl_2 I 2 13 79 13 4 5 116
II 3 (<5) 11 108 6 1 6 135
Rep-cl_3 I 1 11 82 13 4 7 118
II 2 (<5) 9 116 8 3 4 142
Rep-sP1 II 26 (70) 52 64 9 4 9 164
Rep-sP2 II 18 (21) 56 48 3 2 2 129
Rep-sP3 I 7 17 91 1 1 1 118
II 17 (<5) 9 68 1 2 1 97
Rep-sP4 II 6 (36) 16 107 1 1 1 132
Rep-sP5 II 12 (<5) 33 74 0 1 1 122
Rep-sP6 II 11 (<5) 32 95 1 1 1 140
Rep-sP7 II 9 (<5) 88 26 1 0 0 125
Stam-so_1 I 0 2 79 18 5 3 107
II 1 (<5) 5 113 7 3 3 131
Stam-so_2 I 0 0 12 45 11 11 79
II 0 (<5) 0 40 40 5 3 88
Stam-so_3 I 1 5 44 40 6 14 109
II 2 (<5) 2 66 27 8 26 130
The easily soluble fraction of scheme II is the sum of the first two fractions (in between brackets is the percental contribution of the AEM-extrac-
table fraction to the easily soluble fraction mentioned).
S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342 337

indeed mainly dissolved with NaOH. Some iron wassolubilized during the NaOH-extraction of this samplebut not enough to stochiometrically explain the arsenicsolubilization, which was more than 300 times larger,unless iron was reprecipitated as oxides or hydroxides ofcourse. In sample Rep sP3, large amounts of lead wereco-released during the NaOH-extraction suggesting thepresence of Pb-arsenate. Both elements are also releasedfrom this sample in other extraction steps, although inmuch smaller amounts, whereby a good correlationbetween Pb and As leaching throughout the wholeextraction scheme was observed (r=0.97 and corre-lation is significant at P<0.01 if r>0.83). Latter obser-vation confirms the presence of lead arsenate but thenagain it also indicates that the extraction of lead arse-nate by NaOH is not completely selective. This illus-trates the usefulliness to analyse also the concomitantleached elements but it is obvious that, even with theinformation of the concomitant elements, it is in mostsamples very difficult to link the NaOH leached arsenicwith particular arsenic-bearing phases. Most likely, thesituation is complicated by the presence of complexmineral phases and solid solutions in the waste material.A large part of arsenic is, in most samples, also
released during the NH4F extraction (>25% of total Ascontent in one fourth of the extractions). However, thisfraction is nearly always relatively smaller than theamount set free during the NaOH-extraction. The largeextent of both alkaline extractions in all samples, whichalso corroborates the general phenomena of increasingarsenic mobility with increasing pH, suggests that itmay be interesting to split up these extractions intovarious different alkaline extraction steps of differentstrengths and perhaps skipping one of more of the otherextraction steps to make the protocol more specific.The reducing step of the arsenic sequential extractions
is relatively less important, except in the St-Amandssamples, where arsenic was probably occluded within Fe-(oxi)hydroxides. The amount of arsenic extracted duringthe reducing step is generally larger when applying theBCR-protocol than in case of the modified arsenicextraction protocols. This can again be explained bymobilization of a part of the arsenic bound to the redu-cible phases in the BCR-scheme by the precedingNH4F- and NaOH-extractions. The opposite is true incase of the St-Amands samples since DCB extracts morestrongly Fe-(oxi)hydroxides (and As occluded therein)than hydroxylamine. Therefore, the amount of arsenicextracted from the iron-rich St-Amands samples in thereducing step was larger in the modified arsenic extrac-tion protocols than in the BCR-scheme.Only Rep-so_1 contains a relatively large arsenic
amount in the oxidizable or acid soluble fraction. Alarge amount of Ca and Mg is also released in this stepand their overall leaching behavior shows a good corre-lation with that of arsenic (r=0.94 resp. r=0.96; and
correlation is significant at P<0.01 if r>0.83). Thissuggests that As is bound to Ca/Mg-bearing phases inthis sample. Although Rep-sl_3 and Rep-sP3 containedmore than 20% As in the oxidizable fraction after theBCR-extraction, this is not manifested in the modifiedarsenic extraction protocols.The largest arsenic fraction in slag sample Rep-sl_3
was only released after the final 3-acid treatment.Mineralogical analysis demonstrated that arsenic is inthis sample mainly present in arsenides (CoAs, Cu5As2,(Ni,Fe,Co)As, Cu3As), which are furthermore occluded,together with several sulfide minerals such as Cu2S andPbS, in metallurgical or glass phases. These glass andsilicate phases protect the arsenides and sulfides to thetreatments such as the oxidizing step that normallywould dissolve these minerals. Therefore, arsenic will befinally released by HF in the final 3-acid treatment.Nevertheless, some considerable arsenic quantities, yetrelatively small compared to the residual fraction, arealso released from this sample in other extraction steps.In scheme I, large amounts of As are released in the firststep together with large amounts of Cu and Ca. Inaddition, a good correlation between As and Cu (andCa) (r=0.95 resp. r=0.90; and correlation is significantat P<0.01 if r>0.83) in this scheme exists, which pointstowards the presence of a Cu–As mineral (e.g. copperarsenate) with possibly a certain degree of substitutionof Cu by Ca. In the first step of the second scheme,almost no Cu and Ca and substantially less As isreleased, which is for the most part compensated bylarger leaching during NaOH and oxidizing extractions.A possible explanation for this difference between thetwo schemes is complex-formation reactions (such asCuCl+) by the NH4Cl solution, which enhance thesolubility of copper minerals such as copper arsenate.Soil Rep-so_5, which was collected in the same sectorwhere slag sample Rep-sl_3 was found, did also containarsenides and sulfides (such as the clinosafflorite-likemineral [(Fe,Ni,Co,Cu)(As,Sb)2], ullmanite [Ni(Sb,As)S]and arsenopyrite), which were probably responsible forthe relatively large residual fraction and the presence ofarsenic in the oxidizable fraction (scheme I). [(Fe,Cu,-Co,Ni)3(AsO4)2.2H2O], which is a weathering mineralof arsenopyrite, and arsenolite [As2O3] were also detec-ted in this sample and probably account for arsenicreleased in the other fractions.Based on these results it is concluded here that in
highly contaminated soil samples such as those underinvestigation, the influence of the mineralogical compo-sition will be of greater importance with respect to As-mobilization than other soil properties since no sig-nificant correlation between the quantities of arsenicextracted by the various solutions and the different soilproperties (e.g. O.M. content) could be drawn.When the distribution of arsenic is considered
through a whole soil profile, some conclusions can be
338 S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342

drawn apart from the general observations discussedabove. Because the total As concentration decreasessubstantially downwards from the highly contaminatedlayer (Rep-sP3), the leached concentrations in the dif-ferent fractions decline (Fig. 4), except the NH4F-extractable fraction, which increases down the profile.Latter effect indicates leaching of fairly soluble arsenicNH4F-extractable phases in this profile, whereby thesephases are depleted in the upper horizons and progres-sively accumulate bottomwards.Arsenic is in the two top samples (above the heavily
contaminated layer) relatively (in percentage of totalcontent) more present in stable phases (reducible, oxi-dizable and residual fraction) than the rest of the profilesuggesting a different arsenic source in these layerscompared to the rest of the profile. The arsenic source inthe cover layer was most likely airborne dust particlesfrom the rest of the site. It may be so that the source ofthe airborne dust particles is, relatively, more enrichedwith waste material treated by high temperature pro-cesses where the elements are present in quite stablephases, instead of the dumped material, which contains,relatively, more ‘‘unstable’’ phases such as discardedprimary minerals (e.g. sulfides). Latter observationsemphasizes the importance to study subsurfacial (below20 cm) soil samples on a contaminated site and notrestricting speciation research to the topsoil since, like-
wise, the origin, source and fate of the contaminants canbe elucidated.
5.3.2.2. Arsenic recovery, inter-method variability andreagent selectivity. Only about 30% of the performedsequential extractions has a good to very good recoveryof the total arsenic present in the samples (e.g. Rep-sl_2,Rep-so_3 and Rep-slu, both schemes). In more than50% of the extractions, the sum of extracted arsenic ishigher than the total arsenic determined by total analy-sis. A possible explanation of the worse ‘overall recov-ery rates’ in comparison to the certified materialsprobably relates to subsample heterogeneity, which is atypical and unavoidable characteristic of ‘‘made-upground’’, frequently encountered on industrial sites(Davidson et al., 1998, 1999). This is also affirmed bythe replicate total arsenic analysis of the samples. Thereplicate (n=5) total arsenic analysis of the CRMs isvery consistent, namely: relative standard deviations(and recovery rates in%) of 2.7% (96% recovery), 2.1%(94% recovery), 2.4% (98% recovery), and 2.1% (94%recovery) for, respectively, SRM2710 Montana soil,GBW07311 stream sediment, SRM1633b coal fly ash,and GBW07411 soil, which confirms the reliability ofthe used analytical method. Conversely, replicate (n=3)total arsenic analysis of five highly contaminated sam-ples exhibit a larger variation, namely: relative standard
Fig. 4. Sequential extraction of arsenic (scheme II) applied on a soil profile on the Reppel site. Leached arsenic concentrations are given in mg/kg
dry matter. Rep-sP3 respresents the most contaminated layer (12.24% As).
S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342 339

deviations of 17, 20, 11, 11, and 19% for, respectively,Rep-sl_3, Rep-so_4, Rep-cl-3, Rep-sP3, and Stam-so_1.The heterogeneous nature of the contamination on
the Reppel site is mainly due to the presence of largequantities of anthropogenic material (ash particles, slagfragments and ore remainders) that is mixed up with thesoil. It is obvious that the presence of such contaminantenriched particles in a subsample already dramaticallyalters its total contaminant concentration. Therefore,leaching experiments applied on larger amounts ofsample (in the order of degree of 100 g or more)focussing on risk analysis and/or a throughout miner-alogical survey are probably more preferable. A>100% recovery may also be attributable to a cumula-tive error in each of the extraction measurements(Onken and Hossner, 1996).Application of both schemes on the same samples
resulted in a profound difference in distribution pattern(Table 9). This is most acute in the extractions of sampleRep-so_4 where there are substantial differences in sev-eral extraction steps. These differences resulted here in amassive accumulated recovery variability (65% Asrecovery for scheme II versus 158% for scheme I). Thedifference in procedure cannot be entirely responsiblefor this large recovery difference in sample Rep-so_4since the leached arsenic concentrations in extractionsteps where both schemes differ (mainly step 1 and 5)are relatively small. This shows in our opinion thatsubsample heterogeneity could be very significant. Keonet al. (2001) draw the same conclusion. In other samplesextraction difference between the two schemes wasrestricted to only one step (e.g. easily soluble fraction insample Rep-sl_1 and Rep-sl_3). However, the extractiondifferences between the corresponding steps of the twoschemes did not always lead to an accumulation ofdivergence in the recovery rate (e.g. sample Rep-sl_2,Rep-so_1, Rep-so_3, Rep-so_5, Rep-cl_1a, Rep-cl_1band Rep-slu) which consequently indicates lack ofselectivity and redistribution or readsorption effects.The largest differences between the two schemes areobserved in the NaOH-extractable fraction, which ismostly the largest extracted fraction.Leaching variation between different extraction
schemes can be matrix dependent as well. This has beendemonstrated in the sequential extraction of sampleRep-sl_3 where the presence of copper minerals causedlarge differences in the easily soluble fraction due to thecomplex-formation reactions in the NH4Cl extract(scheme I) compared to pure water (scheme II).
6. Conclusions
The sequential extractions of standard referencematerials (SRM’s) showed that the extraction schemesbased upon the phosphor sequential extraction proto-
cols gave a better fractionation and recovery of arsenicthan the three-step BCR scheme. Since oxide phasesplay a major role in the arsenic retention, this betterrecovery could partly be accredited to the differentreducing agent. Hence, DCB extracts these oxide phasesto a greater extent (both amorphous and crystallineoxides) than hydoxylamine hydrochloride (amorphousoxides only). Furthermore, arsenic extracted from theamorphous oxides in the BCR scheme was found to bereleased in the phosphor-like protocols before the redu-cible DCB-step either by NaOH (Montana soil) orNH4F (coal fly ash). No arsenic was extracted with theanion exchange membranes (AEM’s).Based on its high heavy metal concentrations, its
good extraction, the nice spreading between the differ-ent extraction steps with large concentrations in the firststep (exchangeable fraction), reference materialSRM2710 (Montana soil) is considered here as the mostuseful reference material if metal (Mn, Cu, Zn, As, Cdand Pb) fractionation of contaminated soils is studiedusing the three-step BCR sequential extraction. Li et al.(1995a) comes to a similar conclusion, however, theseauthors applied a Tessier-like sequential extraction.Nevertheless, the comparison with the results Ho andEvans (1997) indicates that considerable variability ordiscrepancies exist in the application of sequentialextractions on reference materials. In addition, applica-tion of the phosphor-like arsenic protocols validated theappropriateness of SRM2710 as reference materialwhen using these schemes for studying arsenic specia-tion and bioavailability.The major part of arsenic was released from the
heavily contaminated samples after NaOH-extraction.In some samples, considerable amounts of arsenic werealso found in NH4F-extractable, reducible and residualphases. Normally, arsenic was found less in the reduci-ble fraction of the phosphorus-like arsenic sequentialextractions compared to the BCR-extractions, whichconfirms that arsenic bound to easily reducible phasesare extracted by the preceding alkaline extractions,except in the St-Amands samples where arsenic is pre-sent in more crystalline oxides that only could beextracted by DCB. Oxidizable or acid soluble and easilysoluble phases were mostly only of relative minorimportance, although in many cases large absoluteeasily soluble amounts evidenced a serious environ-mental risk on the site of the former arsenic refinery inReppel. It was concluded that the use of anion exchangemembranes (AEMs) was also not useful in sequentialextractions of highly contaminated samples and shouldbe skipped in future research, which should simplify theprotocol.The analysis of concomitant elements was found to be
useful in identifying solubility-controlling minerals suchas lead arsenate in sample Rep-sP3, As-bearing Ca–Mg-phases in sample Rep-so_1 or the Cu-As mineral in slag
340 S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342

sample Rep-sl_3. The usefulness of complementarymineralogical research was exemplified for example inslag sample Rep-sl_3, where the presence of glass phasesexplained the large residual arsenic fraction. Sequentialextraction of a vertical profile revealed different originsof contaminants in different profile horizons and showedthe mobilization of NH4F-extractable As-species.Inferior overall recovery rates when extracting highly
contaminated samples compared to the extraction ofSRMs could be attributed to subsample heterogeneity.The leaching differences between the different adaptedarsenic extraction protocols were also mainly dependingon subsample heterogeneity. The adapted steps weremostly less important. Consequently, the problem ofsubsample heterogeneity causes a major drawback whenusing sequential extractions on soil samples where heavymetals and arsenic are present in a variety of particularminerals. Therefore, leaching experiments applied onlarger amounts of sample (in the order of degree ofhundred grams or more) focussing on risk analysis and/or a throughout mineralogical survey are probablymore preferable.
Acknowledgements
We acknowledge Danny Coutermans for hisassistance with the experiments and to prof. Dr N.Vandenberghe for the use of the Malvern Laser Mas-tersizer. This research was financially supported by theFund for Scientific Research-Flanders (FWO-Vlaanderen;Onderzoeksproject G.0327.98N).
References
Ahnstrom, Z.S., Parker, D.R., 1999. Development and assessment of a
sequential extraction procedure for the fractionation of soil cad-
mium. Soil Sci. Soc. Am. J. 63, 1650–1658.
Allison, J.D., Brown, D.S., Novo-Gradac, K.J., 1991. MINTEQA/
PRODEFA2, A Chemical Assessment Model for Environmental
Systems: Version 3.0 User’s Manual. Environmental research
laboratory office of research and development. US-Environmental
Protection Agency, Athens.
Amacher, M.C., Kotuby-Amacher, J., 1994. Selective extraction of
arsenic from minespoils, soils, and sediments. Agronomy abstracts
86, 256.
Anonymous, 1999. Reference Materials Catalogue. LGC’s Office of
Reference Materials, Teddington, UK.
Belzile, N., Lecomte, P., Tessier, A., 1989. Testing readsorption of
trace elements during partial chemical extractions of bottom sedi-
ments. Environ. Sci. Technol. 23, 1015–1020.
Boltz, D.F., 1958. Colorimetric Determination of Non-metals. Inter-
science Publishers, New York.
Cappuyns, V., Van Herreweghe, S., Swennen, R., Ottenburgs, R.,
Deckers, J., 2002. Arsenic pollution at the industrial site of Reppel-
Bocholt (North-Belgium). Sci. Total Environ. 295, 217–240.
Chang, S.C., Jackson, M.L., 1957. Fractionation of soil phosphorus.
Soil Sci. 84, 133–144.
Daus, B., Weib, H., Wennrich, R., 1998. Arsenic speciation in iron
hydroxide precipitates. Talanta 46, 867–873.
Davidson, C.M., Duncan, A.L., Littlejohn, D., Ure, A.M., Garden,
L.M., 1998. A critical evaluation of the three-stage BCR sequential
extraction procedure to assess the potential mobility and toxicity of
heavy metals in industrially-contaminated land. Anal. Chim. Acta
363, 45–55.
Davidson, C.M., Ferreira, P.C.S., Ure, A.M., 1999. Some sources of
variability in application of the three-stage sequential extraction
procedure recommended by BCR to industrially-contaminated soil.
Fresen. J. Anal. Chem. 363, 446–451.
Dhoum, R.T., Evans, G.J., 1998. Evalution of uranium and arsenic
retention by soil from a low level radioactive waste management site
using sequential extraction. Appl. Geochem. 13, 415–420.
Dodd, J., Large, D.J., Fortey, N.J., Milodowski, A.E., Kemp, S.,
2000. A petrographic investigation of two sequential extraction
techniques applied to anaerobic canal bed mud. Environ. Geochem.
Health 22, 281–296.
Fergusson, J.E., 1990. The Heavy Elements: Chemistry, Environ-
mental Impact and Health Effects. Pergamon Press, Oxford.
Gleyzes, C., Tellier, S., Sabrier, R., Le Cloirec, P., 2001. Arsenic
characterisation in industrial soils by chemical extractions. Environ.
Technol. 22, 27–39.
Gruebel, K.A., Davis, J.A., Leckie, J.O., 1988. The feasibility of using
sequential extraction techniques for arsenic and selenium in soils
and sediments. Soil Sci. Soc. Am. J. 52, 390–397.
Hedley, M.J., Stewart, J.W.B., Chauchan, B.S., 1982. Changes in
inorganic and organic soil phosphorus fractions induced by culti-
vation practices and by laboratory incubations. Soil Sci. Soc. Am.
Proc. 46, 970–976.
Ho, M.D., Evans, G.J., 1997. Operational speciation of cadmium,
copper, lead and zinc in the NIST Standard Reference Materials
2710 and 2711 (Montana Soil) by the BCR sequential extraction
procedure and flame atomic absorption spectrometry. Anal. Com-
mun. 34, 363–364.
Il’yin, B.V., Konarbayeva, G.A., 1996. Arsenic in soils of West Siberia
as related to the regional environmental monitoring. Eurasian Soil
Sci. 28, 144–150.
Jackson, B.P., Miller, W.P., 2000. Effectiveness of phosphate and
hydroxide for desorption of arsenic and selenium species from iron
oxides. Soil Sci. Soc. Am. J. 54, 1616–1622.
Jacobs, L.W., Seyers, J.K., Keeney, D.R., 1970. Arsenic sorption by
soils. Soil Sci. Soc. Am. Proc. 34, 750–754.
Johnson, L.R., Hiltbold, A.E., 1969. Arsenic content of soil and crops
following use of methanearsenate herbicides. Soil Sci. Am. Proc. 33,
279–282.
Johnston, S.E., Barnard, W., 1979. Comparative effectiveness of four-
teen solutions for extracting arsenic from four Western New York
soils. Soil Sci. Soc. Am. J. 43, 304–308.
Keon, N.E., Swartz, C.H., Brabander, D.J., Harvey, C., Hemond,
H.F., 2001. Validation of an arsenic sequential extraction method
for evaluating mobility in sediments. Environ. Sci. Technol. 35,
2778–2784.
Kersten, M., Forstner, U., 1986. Chemical fractionation of heavy
metals in anoxic estuarine and coastal sediments. Wat. Sci. Technol.
18, 121–130.
Kheboian, C., Bauer, C.F., 1987. Accurancy of selective extraction
procedures for metal speciation in model aquatic sediments. Anal.
Chem. 59, 1417–1423.
Kheboian, C., Bauer, C.F., 1988. Response to comments on the test-
ing of the accurancy of an extraction procedure for determining the
partioning of trace metals in sediments. Anal. Chem. 60, 1477.
Kirby, C.S., Rimstidt, J.D., 1993. Mineralogy and surface properties
of municipal solid waste ash. Environ. Sci. Technol. 27, 652–660.
Li, X., Coles, B.J., Ramsey, M.H., Thornton, I., 1995a. Chemical
partitioning of the new national institute of standards and technol-
ogy standard reference materials by sequential extraction using
S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342 341

inductively coupled plasma atomic emission spectrometry. Analyst
120, 1415–1419.
Li, X., Coles, B.J., Ramsey, M.H., Thornton, I., 1995b. Sequential
extraction of soils for multielemental analysis. Chem. Geol. 124, 88–94.
Lum, K.R., Edgar, D.G., 1983. The determination of arsenic by flame
AAS using the Zeeman effect and its application to the analysis of
sediments extracts. Intern. J. Environ. Anal. Chem. 15, 241–248.
Maher, W.A., 1984. Mode of occurrence and speciation of arsenic in
some pelagic and estuarine sediments. Chem. Geol. 47, 333–345.
Manful, G., 1992. Occurrence and Ecochemical Behaviour of Arsenic
in a Goldsmelter Impacted Area in Ghana. PhD dissertation, Cen-
trum voor milieusaneringen aan de RUG.
Martin, J.M., Nirel, P.M., Thomas, A.J., 1987. Sequential extraction
techniques: promises and problems. Mar. Chem. 22, 313–341.
McLaren, R.G., Naidu, R., Smith, J., Tiller, K.G., 1998. Fractiona-
tion and distribution of arsenic in soils contaminated by cattle dip.
J. Environ. Qual. 27, 348–354.
McLean, J.E., Bledsoe, B.E., 1992. Ground Water Issue: Behaviour of
Metals in Soils. Superfound Technology Support Center for
Ground Water (US EPA/540/S-92/018), Washington, DC.
Nelson D.W., Sommers L.E., 1982. Total carbon, organic carbon and
organic matter. In: Methods of Soil Analysis, Part 2: Chemical and
Microbiological Properties, second ed. pp. 561–593.
Nirel, P.M.V., Morel, F.M.M., 1990. Pitfalls of sequential extractions.
Water Res. 24, 1055–1056.
Nriagu, J.O., 1994. Arsenic in the Environment. Part II: Human
Health and Ecosystem Effects. John Wiley & Sons, New York.
O’Neill, P., 1995. Arsenic. In: Alloway, B.J. (Ed.), Heavy Metals in
Soils, second ed. Blackie Academic & Professional, Glasgow.
Onken, B.M., Adriano, D.C., 1997. Arsenic availability in soil with
time under saturated and subsaturated conditions. Soil Sci. Soc.
Am. J. 61, 746–752.
Onken, B.M., Hossner, L.R., 1996. Determination of arsenic species in
soil solution under flooded conditions. Soil Sci. Soc. Am. J. 60,
1385–1392.
Peachey, D., Roberts, J.L., Scot-Baker, J., 1973. Rapid colorimetric
determination of phosphorus in geochemical survey samples. J.
Geochem. Explor. 2, 115–120.
Pickering, W.F., 1981. Selective chemical extractions of soil compo-
nents and bound metal species. Crit. Rev. Anal. Chem. 12, 233–266.
Pierce, M.L., Moore, C.B., 1982. Adsorption of arsenite and arsenate
on amorphous iron hydroxide. Water Res. 16, 1247–1253.
Quevauviller, Ph., Rauret, G., Muntau, H., Ure, A.M., Rubio, R.,
Lopez-Sanchez, J.F., Fiedler, H.D., Griepink, B., 1994. Evaluation
of sequential extraction procedure for the determination of extrac-
table trace metal contents in sediments. Fresen. J. Anal. Chem. 349,
808–814.
Quevauviller, Ph., 1998. Requirements for production and use of cer-
tified reference materials for speciation analysis: a European Com-
mission perspective. Spectrochim. Acta Part B 53, 1261–1279.
Rapin, F., Tessier, A., Campbell, P.G.C., Carigan, R., 1986. Potential
artefacts in the determination of metal partitioning in sediments by a
sequential extraction procedure. Environ. Sci. Technol. 20, 836–840.
Rauret, G., Rubio, R., Lopez-Sanchez, J.F., Casassas, E., 1989. Spe-
cific procedure for metal solid speciation in heavily polluted river
sediments. Intern. J. Environ. Anal. Chem. 35, 89–100.
Roussel, C., Bril, H., Fernandez, A., 2000. Arsenic speciation: invol-
vement in evaluation of envrionmental impact caused by mine
wastes. J. Environ. Qual. 29, 182–188.
Shuman, L.M., 1985. Fractionation method for soil microelements.
Soil Sci. 140, 11–22.
Slejkovec, Z., Salma, I., Van Elteren, J.T., Zemplen-Papp, e., 2000.
Speciation of arsenic in coarse and fine urban aerosols using
sequential extraction combined with liquid chromatography and
atomic fluorescence detection. Fresen. J. Anal. Chem. 366, 830–834.
Tack, F.M.G., Verloo, M.G., 1995. Chemical speciation and fractio-
nation in soil and sediment heavy metal analysis: a review. Intern. J.
Environ. Anal. Chem. 59, 225–238.
Templeton, D.M., Ariese, F., Cornelis, R., Danielsson, L.-G., Mun-
tau, H., Van Leeuwen, H.P., Lobinski, R., 2000. Guidelines for
terms related to chemical speciation and fractionation of elements.
Definitions, structural aspects, and methodological approaches
(IUPAC Recommendations 2000. Pure Appl. Chem. 72, 1453–1470.
Terashima, S., Taniguchi, M., 1997. Mineralogical associations of
arsenic and antimony in thirty five geochemical reference materials
by sequential extraction with hydride generation and atomic
absorption spectrometry. Geostandard. Newslett. 22, 103–112.
Tessier, A., Campbell, P.G.C., Bisson, M., 1979. Sequential extraction
procedure for the speciation of particulate trace metals. Anal.
Chem. 51, 844–851.
Tessier, A., Campbell, P.G.C., 1988. Comments on the testing of the
accurancy of an extraction procedure for determining the partioning
of trace metals in sediments. Anal. Chem. 60, 1475–1476.
Tessier, A., Campbell, P.C.G., 1991. Comment on ‘‘Pitfalls of
sequential extractions’’ by P.M.V. Nirel and F.M.M. Morel, Wat
Res 24: 1055–1056. Water Res. 25: 115–117.
Tiessen, H., Moir, J.O., 1993. Characterisation of available P by
sequential extraction. In: Carter, M.R. (Ed.), Soil Sampling and
Methods of Analysis. Canadian Society of Soil Science, Lewis Pub-
lishers, pp. 75–86.
Tipping, E., Hetherington, N.B., Hilton, J., Thompson, D.W., Bowles,
E., Hamilton-Taylor, J., 1985. Artifacts in the use of selective che-
mical extraction to determine the distribution of metals between
oxides of manganese and iron. Anal. Chem. 57, 1944–1946.
Ure, A., Quevauviller, Ph, Muntau, H., Griepink, B., 1993. Improve-
ments in the determination of extractable contents of trace metals in
soil and sediment prior to certification. Report EUR 14763 EN,
CEC, Brussels.
Van der Hoek, E.E., Comans, R.N.J., 1996. Modeling arsenic and
selenium leaching from acidic fly ash by sorption on iron (hydr)ox-
ide in the fly ash matrix. Environ. Sci. Technol. 30, 517–523.
Van Raij, B., Quaggio, J.A., da Silva, N.M., 1986. Extraction of phos-
phorus, potassium, calcium, and magnesium from soils by an ion-
exchange resin procedure. Commun. Soil Sci. Plant Anal. 17, 547–566.
Van Reeuwijk, L.P., 1992. Procedures for Soil Analysis, third ed.
International Soil Reference and Information Center, Wageningen,
The Netherlands.
Voigt, D.E., Brantley, S.L., Hennet, R.J.-C., 1996. Chemical fixation
of arsenic in contaminated soils. Appl. Geochem. 11, 633–643.
Williams, J.D.H., Seyers, J.K., Walker, T.W., 1967. Fractionation of
soil inorganic phosphate by a modification of Chang and Jackson’s
procedure. Soil Sci. Soc. Am. Proc. 31, 736–739.
Woolson, E.A., Axley, J.H., Kearney, P.C., 1971. The chemistry and
phytotoxicity of arsenic in soils: I Contaminated field soils. Soil Sci.
Soc. Am. Proc. 35, 938–943.
Woolson, E.A., Axley, J.H., Kearney, P.C., 1973. The chemistry and
phytotoxicity of arsenic in soils: II Effects of time and phosphorus.
Soil Sci. Soc. Am. Proc. 37, 254–259.
342 S. Van Herreweghe et al. / Environmental Pollution 122 (2003) 323–342