small molecule antagonists of integrin receptors
TRANSCRIPT

Current Medicinal Chemistry, 2010, 17, 2371-2392 2371
0929-8673/10 $55.00+.00 © 2010 Bentham Science Publishers Ltd.
Small Molecule Antagonists of Integrin Receptors
A. Perdih1 and M. Sollner Dolenc*,2
1National Institute of Chemistry, Hajdrihova 19, 1001 Ljubljana, Slovenia
2Faculty of Pharmacy, University of Ljubljana, A ker eva 7, 1000 Ljubljana, Slovenia
Abstract: The complex and widespread family of integrin receptors is involved in numerous physiological processes, such as tissue remodeling, angiogenesis, development of the immune response and homeostasis. In addition, their key role has been elucidated in important pathological disorders such as cancer, cardiovascular diseases, osteoporosis, autoimmune and inflammatory diseases and in the pathogenesis of infectious diseases, making them highly important targets for mod-ern drug design campaigns.
In this review we seek to present a concise overview of the small molecule antagonists of this diverse and highly complex receptor family. Integrin antagonists are classified according to the targeted integrin receptor and are discussed in four sections. First we present the fibrinogen IIb 3 and the vitronectin V 3 receptor antagonists. The remaining selective in-tegrin antagonists are examined in the third section. The final section is dedicated to molecules with dual or multiple in-tegrin activity. In addition, the use of antibodies and peptidomimetic approaches to modulate the integrin receptors are discussed, as well providing the reader with an overall appreciation of the field.
Keywrds: Integrin receptors, fibrinogen ( IIb 3) receptor, vitronectin ( V 3) receptor, fibronectin ( 5 1) receptor, small mole-cule antagonists, dual/multiple antagonists.
1. INTRODUCTION
In 1987 the term "integrin" was used to describe a family of structurally, immunochemically, and functionally related cell-surface heterodimeric receptors, which integrated the extracellular matrix with the intracellular cytoskeleton in order to mediate cell migration and adhesion [1]. Integrins comprise a large family of highly glycosylated, heterodi-meric transmembrane receptors composed of non-covalently associated - and -subunits. The latter function as receptor elements for the extracellular matrix components and also bind to the counter receptors located on the surfaces of other cells [1]. Each subunit contains a large extracellular domain, a single transmembrane domain and, except for integrin 4, a short cytoplasmic tail [1]. Integrin receptors modulate nu-merous critical cellular processes like adhesion, spreading, migration, gene expression and differentiation. These proc-esses play an important role in growth and development of angiogenesis, hemostasis, immune response, tissue remodel-ing and numerous other physiological processes [1].
The integrin family consists of receptors with different numbers and types of - and - subunits that interact with each other via non-covalent association. [1] 18 and 8 subunits have been identified that assemble into at least 24 different integrin receptors (Fig. 1) [1]. Furthermore, in-tegrins can be divided, as one of the possible division schemes in Fig. (1) depicts, into several subfamilies based on evolutionary relationships and ligand specificity. Half the integrin subunits contain the inserted I domain, present in
1, 2, D, E, L, M and X subunits, which is involved in ligand binding to these integrins [2].
Several integrin receptors, together with the pathological states in which they play a vital role, are listed in Table 1. Fibrinogen receptor IIb 3 has been widely investigated due
*Address correspondence to this author at the Faculty of Pharmacy, Univer-sity of Ljubljana, Asker eva 7, 1000 Ljubljana, Slovenia; Tel: +386-1-4769-572; Fax: +386-1-4258-031; E-mail: [email protected]
to its important role in blood platelet aggregation, and its blocking reduces thrombotic complications after percutane-ous coronary interventions [3]. The vitronectin v 3 integrin receptor is expressed in various cell types, such as endothe-lial cells, melanoma, osteoclast and smooth muscle cells and plays an important role in angiogenesis and tumor cell mi-gration [4, 5]. The 5 1 fibronectin integrin is presumed to regulate the function of integrin v 3 on endothelial cells during their migration in vitro or during angiogenesis in vivo. Activation of 5 1 potentiates v 3-mediated migration on vitronectin [6, 7]. There are many examples where two or more integrins (for example V 3/ 5 1, V 3/ V 5 and
V 3/ 5 1 integrin pairs) modulate the same process and concurrent modulation of these integrins could be beneficial [8].
The integrin family of receptors binds a large number of diverse extracellular ligands and, interestingly, different in-tegrins can recognize the same ligand [9]. Ligand binding is regulated by the activation state of the cell [1]. All integrins bind their ligands in a divalent cation dependent manner [2]. Manganese and magnesium ions usually promote binding, in contrast to calcium which has different effects depending on its concentration and the receptor involved [2]. The ligands indentified so far are extremely diverse structurally and were systematically presented by Plow et al. (Table 2) [9].
Extracellular matrix and cell surface protein ligands all contain the most important (Arg-Gly-Asp) RGD tripeptide recognition sequence. They include fibrinogen, vitronectin, fibronectin, osteopontin, thrombospondin and von Wille-brand factor, which are recognized by the IIb 3, the v in-tegrins ( 1, 3, 5, 6, 8) and by the 1 integrins ( 5 1, 8 1) [9]. A second group, the most important examples being
4 1 and 4 7 integrins, is RGD sequence independent. The most important recognition sequences for the integrin recep-tors are listed in Table 3 as reported by Plow et al. [9].
Ligand binding can activate the receptor, resulting in ac-tivation of different intracellular signaling pathways (out-side-in signaling) [10, 11]. The signals for activation of

2372 Current Medicinal Chemistry, 2010 Vol. 17, No. 22 Perdih and Dolenc
Fig. (1). General overview of the integrin receptor family (adapted from [1,8]).
Table 1. Members of the Integrin Receptor Family and Potential Indications for the Therapeutic Use of Antagonists Associated
with the Corresponding Receptor [8]
Integrin Pathological State
b 3 Thrombolytic disorders
V 3
Metastatic melanoma
Breast and prostate tumors
Late stage glioblastoma
Angiogenic vessels
Atherosclerotic lesions
Osteoporosis
V 5 Lung carcinoma
Osteoporosis
4 1
Asthma
Arthritis
Multiple sclerosis
4 7
Crohn`s disease
Ulcerative colitis
Hepatitis C
2 1 Thrombolytic disorders
5 1 Angiogenesis
Infectious disorders
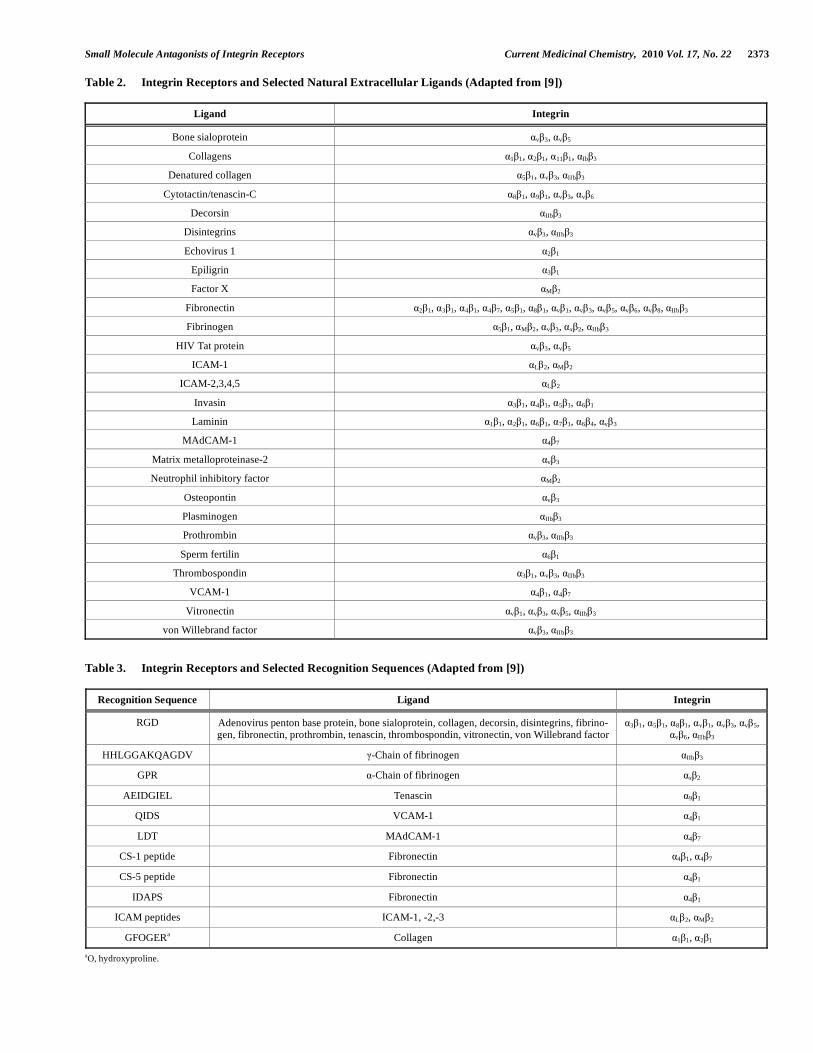
Small Molecule Antagonists of Integrin Receptors Current Medicinal Chemistry, 2010 Vol. 17, No. 22 2373
Table 2. Integrin Receptors and Selected Natural Extracellular Ligands (Adapted from [9])
Ligand Integrin
Bone sialoprotein v 3, v 5
Collagens 1 1, 2 1, 11 1, Ib 3
Denatured collagen 5 1, v 3, IIb 3
Cytotactin/tenascin-C 8 1, 9 1, v 3, v 6
Decorsin IIb 3
Disintegrins v 3, IIb 3
Echovirus 1 2 1
Epiligrin 3 1
Factor X M 2
Fibronectin 2 1, 3 1, 4 1, 4 7, 5 1, 8 1, v 1, v 3, v 5, v 6, v 8, IIb 3
Fibrinogen 5 1, M 2, v 3, x 2, IIb 3
HIV Tat protein v 3, v 5
ICAM-1 L 2, M 2
ICAM-2,3,4,5 L 2
Invasin 3 1, 4 1, 5 1, 6 1
Laminin 1 1, 2 1, 6 1, 7 1, 6 4, v 3
MAdCAM-1 4 7
Matrix metalloproteinase-2 v 3
Neutrophil inhibitory factor M 2
Osteopontin v 3
Plasminogen IIb 3
Prothrombin v 3, IIb 3
Sperm fertilin 6 1
Thrombospondin 3 1, v 3, IIb 3
VCAM-1 4 1, 4 7
Vitronectin v 1, v 3, v 5, IIb 3
von Willebrand factor v 3, IIb 3
Table 3. Integrin Receptors and Selected Recognition Sequences (Adapted from [9])
Recognition Sequence Ligand Integrin
RGD Adenovirus penton base protein, bone sialoprotein, collagen, decorsin, disintegrins, fibrino-gen, fibronectin, prothrombin, tenascin, thrombospondin, vitronectin, von Willebrand factor
3 1, 5 1, 8 1, v 1, v 3, v 5, v 6, IIb 3
HHLGGAKQAGDV -Chain of fibrinogen IIb 3
GPR -Chain of fibrinogen x 2
AEIDGIEL Tenascin 9 1
QIDS VCAM-1 4 1
LDT MAdCAM-1 4 7
CS-1 peptide Fibronectin 4 1, 4 7
CS-5 peptide Fibronectin 4 1
IDAPS Fibronectin 4 1
ICAM peptides ICAM-1, -2,-3 L 2, M 2
GFOGERa Collagen 1 1, 2 1
aO, hydroxyproline.

2374 Current Medicinal Chemistry, 2010 Vol. 17, No. 22 Perdih and Dolenc
integrins can also come from the cytoplasm - the signals re-ceived by other receptors activate intracellular signaling pathways which make the extracellular domain of integrins competent for ligand binding (inside-out signaling) [10, 11]. Inside-out and outside-in signaling are associated with dis-tinct tertiary and quaternary conformational changes in the extracellular segment of the integrins [1, 10].
The integrin receptors are thought to equilibrate between at least two boundary conformations: bent, low affinity and straightened, high affinity conformations. The bent confor-mation is considered to be inactive and the extended to be the integrin active state. The state of the receptor is highly dependent on the nature of the ions present at the cation binding sites, ligation by the natural ligands, and the ability of the ligands to induce clustering of receptors and on vari-ous intracellular signals [12, 13]. In this respect, a switch-blade-like mechanism of integrin activation, based on obser-vations of conformational changes of the integrins V 3 and
IIb 3 after ligand binding, was proposed [10]. The mecha-nism suggests that the bent conformation present on the cell surface usually has low affinity for the ligand. Activation of the integrin by Mn2+ ions, or by other high affinity ligand mimetics, results in unbending of the integrin to an extended structure [10]. The switchblade model, however, is not able to explain the full complexity of the integrin family, and much more research will be required to fully understand the sophisticated integrin biochemistry. For example, it was shown that the V shaped bent conformation exists on both resting and activated platelets [14].
Integrins are difficult to characterize at the molecular level, which would provide crucial structural information to enable further insights into their function. The structure and binding sites of a few integrin proteins have been defined by electron microscopy, mutagenesis and monoclonal antibody studies [15, 16]. The first crystal structure of the extracellular portion of the V 3 integrin was published in 2001 [17] fol-lowed by the structure of this integrin segment in complex with cyclo(Arg-Gly-Asp-{D-Phe}-{N-methyl}-Val) (9) [18].
The next structural breakthrough came in 2004 with the structure of IIb 3 extracellular fragments in complex with fibrinogen-mimetic drugs 1 and 2, which enabled a deeper insight into the atomic structure and conformational behavior [19].
In the vitronectin V 3 crystal structure (Fig. 2, left), 12 domains are assembled into an ovoid "head" and two "tails" [17, 18]. In the crystal, V 3 is severely bent at a defined
region in its tails, reflecting a flexibility that is presumably linked to integrin regulation. The main inter-subunit inter-face lies within the head, between a seven-bladed ß-propeller from
V and a A domain from 3, and bears a striking re-semblance to the G /G interface present in G proteins. A metal ion dependent adhesion site (MIDAS) in the A do-main is located at a ligand-binding interface formed of loops from the propeller and A domains. MIDAS lies adjacent to a calcium-binding site with a prospective regulatory function [17, 18].
The IIb 3 receptor crystal structures consist of the IIb -propeller and the 3 A, hybrid, and PSI domains (Fig. 2, right) [19]. The structures revealed an open, presumably high-affinity, conformation containing a bound ligand (fi-brinogen-mimetic therapeutics 1 and 2). IIb and 3 subunits are separated by an angle of 62°, due in part to a 10 Å downward movement of the 7 helix of the A domain rela-tive to the hybrid and the PSI domain. Reorganization of the hydrogen bonds in the interface between the 7 helix and C strand of the hybrid domain allows the hybrid domain and the rigidly connected PSI domain to swing out, causing a 70 Å separation of the IIb and 3 domains. This structural fea-ture was also observed in electron microscopy (EM) images of active forms of IIb 3 integrin in the presence and absence of ligand [19, 21].
The use of molecular electron microscopy enabled the three-dimensional structure of the ligand-binding headpiece of integrin 5 1, complexed with fragments of its physio-logical ligand (fibronectin), to be solved. The density map for the unliganded 5 1 headpiece showed a 'closed' confor-
Fig. (2). Crystal structures of the extracellular segment of V 3 (PDB: 1L5G) and fibrinogen IIb 3 headpiece (PDB: 1TY5). The -subunits are depicted in gray and the ß-subunits in black. Ions and bound ligands are omitted [17-20].

Small Molecule Antagonists of Integrin Receptors Current Medicinal Chemistry, 2010 Vol. 17, No. 22 2375
mation, similar the one seen in the V 3 structure. In con-trast, binding to the fibronectin-induced conformation result-ing in an 'open' conformation with a dramatic, 80° change in the angle of the hybrid domain of the ß subunit relative to its I-like domain. The fibronectin fragment is bound to the inter-face between the ß-propeller and I-like domains in the in-tegrin headpiece, through the RGD-containing module [22].
Despite all available attempts to clarify integrin structure and function, it should be emphasized that the exact struc-tural characteristics of integrin receptors in different physio-logical surroundings are extremely diverse and have not yet been fully characterized. The results of the EM and atomic force microscopy (AFM) experiments have only partially revealed the activation/deactivation mechanism and cannot provide the full topology of the binding site. The resolution of the current crystal structures is not fully optimal. All these facts make rational structure-based drug design, by utilizing available integrin structural data, a major challenge [8].
Due to its wide range of functions in physiological and pathological processes, the integrin receptor family is con-stantly utilized in drug design projects aimed at producing novel effective integrin receptor antagonists [8, 23-28]. In addition to the huge amount of small molecule design, much success has been achieved with the family of proteins known as disintegrins, that occur naturally in several snake venoms. These sequences of 60-80 amino acids contain a RGD se-quence and are interesting leads for the development of dual or even triple integrin antagonists [29]. Several humanized monoclonal antibodies have been identified as effective in-tegrin antagonists and are described in succeeding chapters.
The development of small molecule antagonists is based predominantly on the recognition sequences of the integrin physiological ligands. In this respect drug design strategies are generally divided into two groups, based on their ligand recognition sequence: either designing molecules that mimic the RGD sequence or taking other integrin recognition se-quences as starting points. The first strategy has resulted predominantly in antagonists of the IIb 3, V 3, V 5 and
5 1 integrins [23-25]. The most important integrin targets for the second strategy are the 4 1 and 4 7 receptors, in which the key sequence recognized by 4 1 is Ile-Asp-Ser (IDS), whereas 4 7 recognizes and binds the Leu-Asp-Thr (LDT) sequence [26, 27]. They are both able to recog-nize Leu-Asp-Val (LDV) which is found in an alternatively spliced connecting segment of fibronectin (CS-1).
The aim of this review is to present a concise overview of the small molecule antagonists of this diverse and highly complex receptor family. In recent years a number of diverse integrin modulators have been reported in the scientific and patent literature. The present review, along with other over- views of the integrin antagonists field, [23-28] is aimed at highlighting the advances in utilizing these important protein targets for designing new therapeutic agents. The small molecule integrin antagonists are classified according to the targeted integrin receptor and are divided into four large groups. First we present the fibrinogen IIb 3 and vitronectin
V 3 receptor antagonists. The remaining selective integrin antagonists are presented in the third chapter. The final sec- tion is dedicated to molecules with dual or even multiple integrin activity. Other classifications are also possible, es-
pecially on the basis of their mechanism of action, by which antagonists fall into three different classes: / I-like com- petitive antagonists ( IIb 3, V 3, V 5 5 1 4 1 and 4 7 receptors) , / I-like allosteric antagonists and I allosteric antagonists (e.g. M 2, X 2, and D 2. L 2 receptors) [23].
2. SMALL MOLECULE ANTAGONISTS OF INTE-
GRIN RECEPTORS
2.1. Fibrinogen IIb 3 Receptor Antagonists
The fibrinogen IIb 3 integrin receptor was initially the most exploited drug target within the integrin family [30]. The development of antagonists of this receptor has been based predominantly on the RGD sequence. Numerous drug design techniques have been utilized, among them cycliza-tion of RGD-containing peptides and design of conforma-tionally constrained peptidomimetics and non-peptide small molecules, and has resulted in many effective antagonists [30].
The first successful IIb 3 antagonist, which also showed clinical effectiveness, was a human-murine chimeric Fab fragment of a monoclonal antibody (abciximab). Several of the specific properties of abciximab, such as its long half-life, lack of receptor-blocking specificity, and tendency for antigenicity, have prompted the development of alternative
IIb 3 antagonists with distinctive pharmacologic profiles [31].
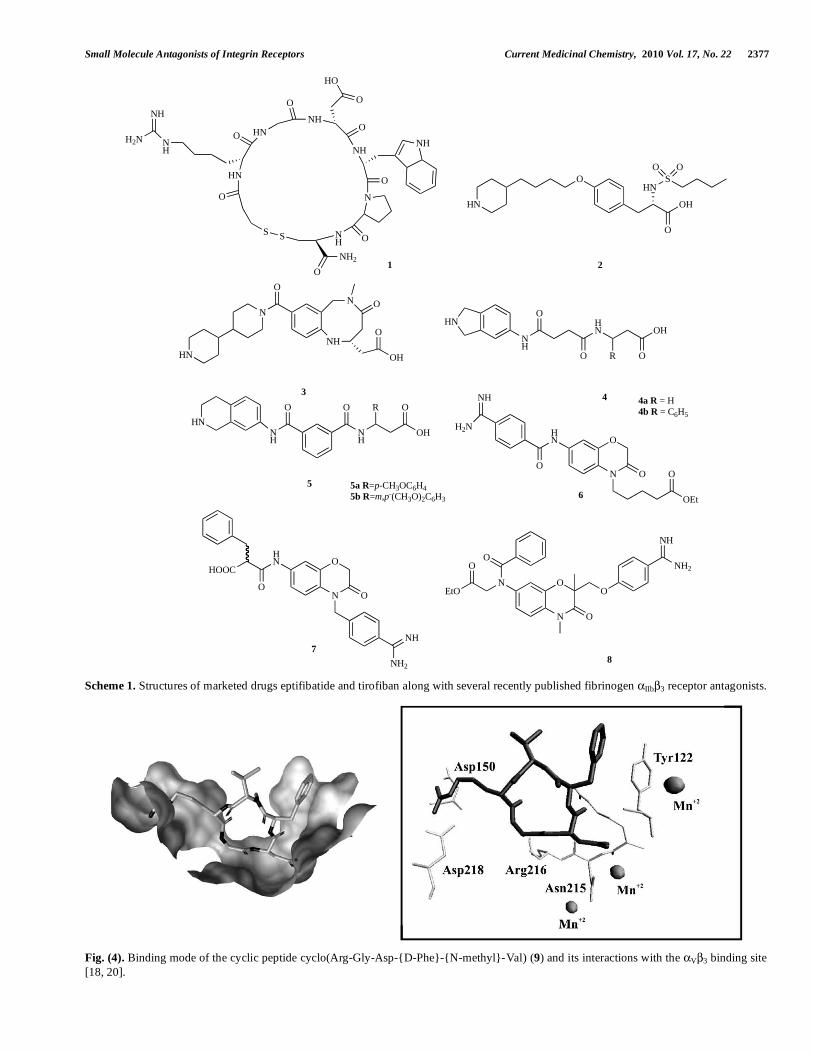
Starting from the naturally occurring compound bar-bourin (disintegrin) [29] – an IIb 3 antagonist found in the venom of the south-eastern pigmy rattlesnake – eptifibatide 1 (Integrilin®), a small cyclic heptapeptide, was designed. It showed high specificity and high affinity for the IIb 3 in-tegrin receptor, a short plasma half-life, and rapid onset of antiplatelet action followed by rapid reversibility of the platelet inhibition once the treatment was stopped [32, 33]. Integrilin® is indicated clinically for the treatment of pa-tients with acute coronary syndrome (unstable angina/non-ST-segment elevation myocardial infarction) and has been shown to decrease mortality and new myocardial infarction [33].
Further development of non-peptide low molecular weight antagonists led to the discovery of tirofiban (Aggra-stat®), 2, another effective IIb 3 antagonist approved for intravenous application in 1999 [34]. Tirofiban (2) is consid-ered one of the first drug candidates whose origins can be traced back to a pharmacophore-based virtual screening lead. Aggrastat®, in combination with heparin, is indicated for the treatment of acute coronary syndrome. The crystal structures of the extracellular segment of IIb 3 integrin in complex with eptifibatide (1) and tirofiban (2) were solved, providing for the first time structural data for further drug design (Fig. 3) [19, 30]. Small molecule antagonists, such as tirofiban, bind into a small pocket at the top of the integrin head formed by the loops from the -propeller of the II subunit and the 3 I domain. The crystal structure with bound tirofi-ban showed that the basic ligand-mimetic side chain can reach deeper into the -propeller pocket of the II subunit compared to the v integrin subunit. The basic group of 2 forms a strong interaction with Asp 224. The intermolecular interactions between tirofiban and its binding site are shown in Fig. (3) [19].

2376 Current Medicinal Chemistry, 2010 Vol. 17, No. 22 Perdih and Dolenc
Further development of fibrinogen antagonists that could be administered orally led to a large number of active agents like lotrafiban 3 (Scheme 1) [35]. Unfortunately, lotrafiban, and several other developed compounds, failed in clinical phase II or III, due to the partial agonism of these agents that led to increased mortality as a consequence of increased ag-gregation [35]. Additional reasons for the failure can be at-tributed to poor pharmacokinetic properties – modest bioavailability and relatively short half-life – resulting in large variation of the plasma drug concentration, and thus preventing the sustained inhibition required for a beneficial pharmacological effect [35].
In the recent search for novel active IIb 3 receptor an-tagonists, higher affinity for the fibrinogen receptor and, consequently, more favorable pharmacokinetic properties, constitute a crucial benchmark for validation [34]. Krysko and co-workers have reported several promising IIb 3 recep-tor antagonists obtained by developing new Arg-Gly frag-ments, enabling the creation of new RGD(F) mimetics. Arg-Gly surrogates – 4-(isoindoline-5-yl)amino-4-oxobutyric acid (e.g. compound 4), 4-(1,2,3,4-tetrahydro-isoquinoline-7-yl)amino-4-oxo-butyric acid, 7-amino-1,2,3,4-tetrahydro-isoquinoline (e.g. compound 5) and isophthalic acids – have been synthesized, and joined mostly to -substituted -alanines, acting as surrogates of RGDF Asp-Phe moiety [36, 37].
2H-1,4-benzoxazine-3(4H)-one, a popular peptidomi- metic building block, has enabled a range of substitution options that allow different spatial positions of the RGD pharmacophore functional groups to be studied. Resulting compounds, like 6, displayed rather unexpected behavior, ethyl esters being more active than free acids against ADP- induced human platelet aggregation. Consquently it was suggested that compounds bearing small alkyl ester moieties are full antagonists of the IIb 3 receptor, while the corre- sponding free acids act as partial agonists. This proposal offers a new insight into the reasons why several IIb 3 antagonists fail to elicit appropriate therapeutic activity [38].
An innovative concept in the development of IIb 3 an-tagonists was recently proposed. Since a combination of low doses of IIb 3 antagonists and direct thrombin inhibitor has been reported to be successful during thrombolysis, active
agents possessing dual action – acting on both targets – would be highly advantageous. In an initial attempt to de-velop low-molecular weight peptidomimetic antithrombotic drugs possessing both thrombin inhibitory and IIb 3 antago-nistic activity, the D-Phe-Pro-Arg (thrombin binding motif) and the RGD fibrinogen pharmacophore were successfully merged into one molecule, using the previously introduced 1,4-benzoxazin-3(4H)-one scaffold, to give non-peptide low-molecular weight derivatives 7 [39]. Subsequent optimiza-tion of the initial leads led to novel molecules 8 with submi-cromolar inhibition constants (Ki = 0.38 μM) for thrombin and submicromolar IC50 = 1.45 μM for inhibition of binding of fibrinogen [40]. Within this framework Bi and coworkers designed peptide-based, dual acting compounds that exhib-ited antithrombin and antiplatelet activity, by combining large peptide recombinant hirudin variants (rHV2-K47), act-ing as thrombin inhibitors, with an RGD binding motif re-lated to platelet aggregation [41].
2.2. Vitronectin V 3 Receptor Antagonists
Integrin v 3 receptor shares a common 3 subunit with the platelet fibrinogen receptor and, like the latter, binds to extracellular matrix proteins through the RGD sequence. Along with v 5 integrin, v 3 is associated with various pa-thologies, including osteoporosis, [42] arthritis, [43] and metastatic cancer, [44] and there are numerous identified
v 3 integrin antagonists, among them Cilengitide (9) [45, 46] and compound SB273005 (10) [47] that are undergoing clinical evaluation for the treatment of cancer and osteoporo-sis. To date, various therapeutic candidates, including mono-clonal antibodies, small molecule peptidomimetics and cy-clic peptides, have been evaluated clinically and shown to modulate v 3-mediated processes successfully. We will discuss some of the molecules disclosed recently pursuing different drug designing strategies.
The major step forward in terms of structure-based drug design was the determination of the crystal structure of the
V 3 integrin in complex with a cyclic peptide cyclo(Arg-Gly-Asp-{D-Phe}-{N-methyl}-Val) (9) [18]. The structure revealed that the cyclic ligand binds at the major interface between the
V and 3 subunits, forming extensive contacts with both. The tertiary and quaternary structure of the
Fig. (3). The binding pocket and bound conformation of tirofiban (2), and interactions of 2 with the IIb 3 binding site as determined by high resolution X-ray spectroscopy [19, 20].
Small Molecule Antagonists of Integrin Receptors Current Medicinal Chemistry, 2010 Vol. 17, No. 22 2377
O
NH
O
HO
HNO
HN
NH
NH
H2N
O
S S NH
O
NH2
O
N
O
O
NHNH
1
HNS
O OO
HN
O
OH
2
NH
N O
O
OH
N
O
HN
3
NH
O
O
HN
HN
R O
OH
4a R = H4b R = C6H5
4O
NH
O
OH
O
NH
HN
R
5a R=p-CH3OC6H45b R=m,p-(CH3O)2C6H3
5
6
N
O
O
HN
OO
OEt
NH
H2N
8
N
O
O
NO
NH
NH2
OO
EtO
7
N
O
O
NH
NH2
HN
O
HOOC
Scheme 1. Structures of marketed drugs eptifibatide and tirofiban along with several recently published fibrinogen IIb 3 receptor antagonists.
Fig. (4). Binding mode of the cyclic peptide cyclo(Arg-Gly-Asp-{D-Phe}-{N-methyl}-Val) (9) and its interactions with the V 3 binding site [18, 20].

2378 Current Medicinal Chemistry, 2010 Vol. 17, No. 22 Perdih and Dolenc
vitronectin receptor were observed to have changed from that of the unligated V 3 structure. The tertiary rearrange-ments took place in A, the ligand-binding domain of the 3 subunit. In the ligand-protein complex, A acquires two cations (Mn+2 ions), one of which contacts the ligand’s Asp moiety directly and the other stabilizes the ligand-binding surface. Quaternary changes were observed in the integrin head region, especially in the interface between A and the
V propeller as the two domains move closer at the peptide-binding site [18].
More precisely, the cyclic peptide 9 is located in a cleft between the -propeller and the A domain on the integrin head. Its Arg and Asp side chains point in opposite direc-tions. The Arg moiety is inserted into a narrow groove at the top of the -propeller domain, where its guanidinium group forms a bidentate bridge to Asp218 at the bottom of the groove and an additional salt bridge to Asp150. Most of the upper portion of the Arg side chain is exposed to the solvent. Asp protrudes into a cleft in the A domain, where one Asp carboxylate contacts the metal ion dependent adhesion site, (MIDAS) Mn2+, while the second Asp carboxylate makes hydrogen bonds with the backbone amides of Tyr122 and Asn215 and contacts the aliphatic portion of the Arg214 side chain. Unlike the arginine moiety of 9 the Asp part is com-pletely buried in the V 3 complex. Gly lies at the interface between the two subunits and makes several hydrophobic contacts with the V subunit (the contact with the carbonyl oxygen of Arg216) [18].
After publication of these seminal structural results, mo-lecular models were developed that would ease the further structure-based development of effective v 3 antagonists. A molecular model of the preferential binding of non-peptide ligands to the integrin v 3 over IIb 3 was developed at Merck [48]. Four different chemical classes were investi-gated and the results of docking analyses into the crystal structure of unliganded V 3 integrin led to the identification of a novel binding interaction for selective antagonists of the
v 3 integrin [48]. All the classes investigated were shown to bind in similar fashions, with the basic nitrogen interacting with Asp150 of v and a carboxylic acid interacting with Arg214 of 3. Selective v 3 antagonists adopted an extended conformation with little ligand-protein induced internal strain, while the cup-shaped conformation was characteristic of inactive compounds – the selective IIb 3 antagonists. All molecules exhibited an additional, energetically favorable “exo-site” - interaction with Tyr178 of v. Additionally, an important difference between IIb 3 and V 3 active sites is the critical distance between the charged residues. In the binding model for IIb 3 it is 15.3 Å ( IIb: Glu117 – 3: Arg214), while in the binding model for V 3 it is 12.9 Å ( v: Asp150 – 3: Arg214) [48].
Marinelli and coworkers performed docking studies us-ing the crystal structure of liganded V 3, devising a phar-macophore model for the ligand binding to V 3. The fol-lowing interactions were then proposed to govern the ligand–receptor recognition process: (i) coordination of Ca2+ ion at the MIDAS from one carboxylate oxygen of the substrate; (ii) a salt bridge between the ( )-Asp218 or the ( )-Asp150 and the guanidine(-like) moiety of the bound ligand; (iii) a
-shaped interaction between the ( )-Tyr122 side chain and
the aromatic ring of the ligand; (iv) a hydrogen bond donated by one ligand NH to the carbonyl oxygen of ( )-Arg216 backbone; (v) a hydrogen bond linking a carbonyl group in the ligand to the ( )-Arg214 guanidinium group in the pro-tein [49].
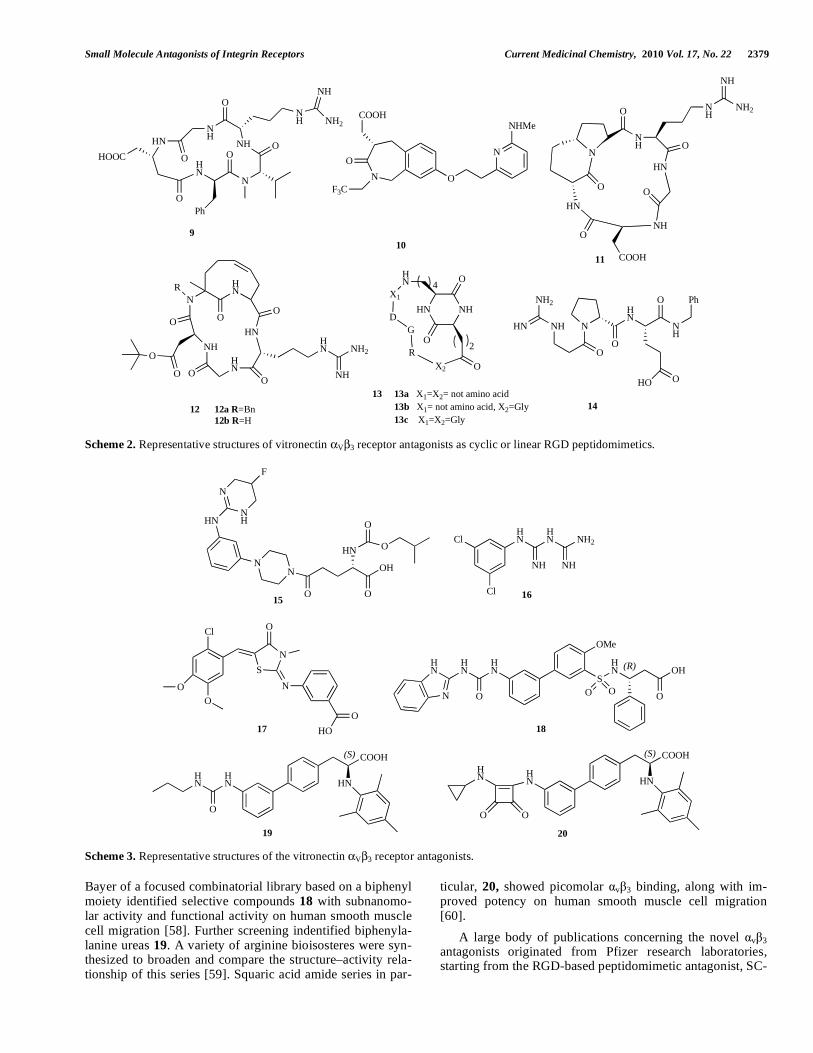
Using peptidomimetic approaches, Belvisi and coworkers synthesized a small library of cyclic pentapeptide (9) mimet-ics incorporating stereoisomeric 5,6- and 5,7-fused bicyclic lactams. Some were found to be high-affinity v 3 ligands. Compound 11 – ST1646 – was a highly selective v 3/ v 5 integrin antagonist [50]. Further investigations of 11 by structure determination of the cyclic RGD pentapeptide mimics, performed by a combination of NMR spectroscopy and computational approaches, revealed its greatest ability within the library to adopt the RGD orientation required for binding to the v 3 integrin, as found in the crystal structure [50]. Other interesting cyclic pentapeptides incorporating the RGD motif (compound 12) were synthesized, incorporating a tetrahydroazoninone scaffold. Binding to the v 5 receptor was weaker, indicating a certain degree of selectivity be-tween the two receptors [51]. Solid-phase synthesis of two different families of rigidified RGD mimetics, containing either diketopiperazine (compound 13) or hydantoin struc-tures, resulted in leads suitable for constructing potent v 3 receptor antagonists [52]. A selected library of heterochiral D-Pro-containing RGD-peptidomimetics was described and utilized for studies of inhibition of fibronectin adhesion to SK-MEL-24 tumor cells. Compound 14 showed an IC50 in the nanomolar range of activity and is presumed to adopt a folded conformation in a polar environment [53].
Small molecule v 3 antagonists constitute a vast area of research on integrin receptor antagonists [23, 25]. A new scaffold search, based on Kessler's early v 3 ligand-binding model, [54] identified the 3-aminophenyl-piperazine moiety as a constrained scaffold for the non-peptide v 3 integrin antagonists. Of these derivatives, the fluorine-substituted compound 15 showed strong inhibitory activity and high selectivity for v 3 integrin receptor (IC50 = 0.055 nM). In vivo evaluation of the antistenotic effects of 15 indicated that this compound significantly inhibits neointima formation in the rat balloon injury model [55].
Phenylbiguanide antagonists, e.g. compound 16, were identified by application of a virtual screening protocol based on the crystal structure of integrin v 3 in complex with an RGD ligand. The results indicated that the substi-tuted phenylbiguanides might be involved in the inhibition of bivalent cation-mediated ligand binding of integrin v 3 [56]. Computational drug design tools were used for pharma-cophore screening of an in-house database of small-molecule drug-like compounds. 3D pharmacophore models were gen-erated and validated, utilizing a set of known integrin v 3 antagonists. The screening procedure and molecular docking calculations successfully retrieved structurally diverse novel compounds (e.g. compound 17) with higher potency [57].
As already stressed, a critical element in the design of the RGD mimetics is correct spatial presentation of the arginine and aspartic acid residues around a central core. The crystal structure of cyclic peptide 9 bodes well for the use of beta-turn mimetic templates in the development of vitronectin receptor antagonists [18]. With this premise, screening at
Small Molecule Antagonists of Integrin Receptors Current Medicinal Chemistry, 2010 Vol. 17, No. 22 2379
N
COOH
O
N
NHMe
O
F3C
NH
NH
O
N
HN
O
NH
NH
NH2
OO
HN
O
HOOC
Ph
9
10
11
13 13a X1=X2= not amino acid
13b X1= not amino acid, X2=Gly
13c X1=X2=Gly
O
NH
NH
NH2
NH
HN
N
O
HN
O
O
O
NH
COOH
HN NH
O
O2
HN
X1
DG
R
X2 O
4
12 12a R=Bn 12b R=H
N
O
HNR
HN
OO
NH
O
O
OHN
O
HN
NH
NH2
NH2
HN NH
O
N
O
HN
O
14
HO O
NH
Ph
Scheme 2. Representative structures of vitronectin V 3 receptor antagonists as cyclic or linear RGD peptidomimetics.
18
HN
O
HN
HN
N
S
HN
O O
(R)
O
OH
OMe
HN
NH
HN
NH
NH2
Cl
Cl
16
NN
O O
OH
HN
O
O
HNNH
N
F
15
S
N
O
N
O
HO
O
O
Cl
17
19
HN
O
HN
(S) COOH
HNHN
(S) COOH
HN
OO
HN
20
Scheme 3. Representative structures of the vitronectin V 3 receptor antagonists.
Bayer of a focused combinatorial library based on a biphenyl moiety identified selective compounds 18 with subnanomo-lar activity and functional activity on human smooth muscle cell migration [58]. Further screening indentified biphenyla-lanine ureas 19. A variety of arginine bioisosteres were syn-thesized to broaden and compare the structure–activity rela-tionship of this series [59]. Squaric acid amide series in par-
ticular, 20, showed picomolar v 3 binding, along with im-proved potency on human smooth muscle cell migration [60].
A large body of publications concerning the novel v 3
antagonists originated from Pfizer research laboratories, starting from the RGD-based peptidomimetic antagonist, SC-

2380 Current Medicinal Chemistry, 2010 Vol. 17, No. 22 Perdih and Dolenc
68448 (21) [61] which demonstrated anti-tumor efficacy in a mouse Leydig cell tumor model. Next, a series of conforma-tionally restricted cinnamic acid analogues, in which the glycine-3-aminopropionic acid functionality was replaced by the bioisosteric 4-aminocinnamic acid moiety, was synthe-sized (compound 22) [62]. This work was also extended to additional isosteres (3-phenylpropionic acids, phenoxyacetic acids and 2-phenylcyclopropylcarboxylic acids). Further work from this laboratory reported oxadiazoles- (23) [63], thiazoles- (24) [64], pyrazoles- (25) [65] and isooxalozes- (26) [65] containing analogues as selective v 3 antagonists with acceptable pharmacokinetics.
Other research started from compound 27 which showed excellent anti-angiogenic properties, including its suppres-sion of tumor growth in animal models. It was designed through depeptidization and optimization of the RGD se-quence [66]. When examining the influence of the stereo-chemistry of the incorporated -amino acids it became ap-parent that not only S- isomers but also R- isomers are rela-tively potent and, in some cases (e.g. compound 28), even more selective v 3 antagonists [67]. In addition, optimizing compound 27 yielded novel molecules 29 that, despite lack of the amide bond, still mimic the RGD sequence [68].
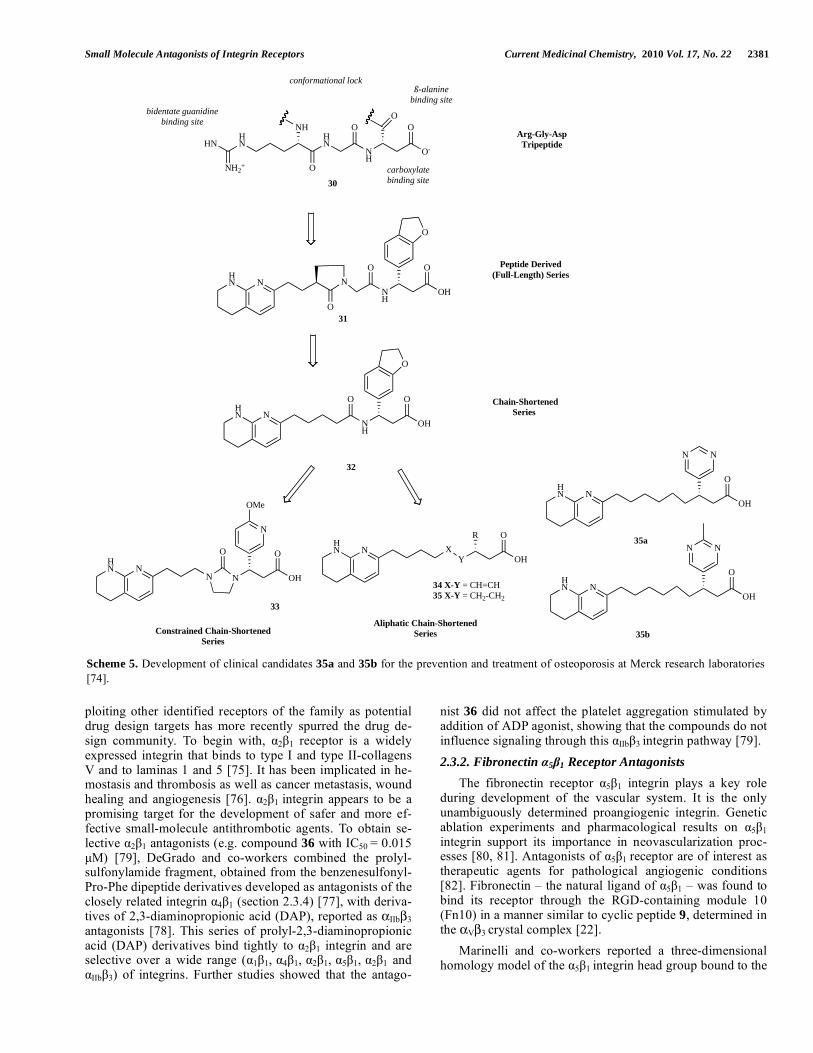
The design and synthesis of clinical candidates 35a and 35b was reported from Merck research laboratories. In initial reports on their evaluation in the treatment of osteoporosis it was demonstrated that “chain-shortened” v 3 antagonists
are potent antagonists with improved pharmacokinetics [69]. The RGD peptide (30) mimetics, such as compound 32, were two atoms shorter in chain length than the extended tripep-tide or full-length antagonists (e.g. compound 31). Although this modification was associated with enhanced pharmaco-kinetics in 32 (dog pharmacokinetics: F = 99%; Cl = 1.2 mL/min/kg) versus 31 (F = 6%; Cl = 33 mL/min/kg), the latter chain-shortened antagonists were less potent than con-strained, full-length antagonists, 31 [70]. Various efforts were made to improve receptor potency within this series, while maintaining acceptable pharmacokinetics, focusing on the incorporation of cyclic constraints, [71] N-terminus modifications, [72] C-terminus modifications, [73] and am-ide deletion. The chain-shortened v 3 antagonist 33, with an imidazolidone central constraint, was a high-affinity ligand that inhibited bone resorption in vivo and proved an v 3 antagonist whose preclinical pharmacokinetic parameters predicted use of a once-daily dosing regimen. This program concluded by finding v 3 antagonists with general structures 34 and 35. The most promising compounds of the series, 35a
and 35b, were selected for clinical studies [74].
2.3. Antagonists of Other Integrin Receptors
2.3.1. Integrin 2 1 Receptor Antagonists
In addition to the V 3 and IIb 3 integrin receptors used in various drug design campaigns, increasing interest in ex-
HN N
NO
N
O
OHAr
HN N
NO
O
OHAr
26
HN N O
O
OH29
OH
O
NH
O
HN
HN
HN
N OH
BrCl
O OH
28
F
NH
O
O
HN
Cl Cl
COOH
HN
NH
H2N
HN N
S
N
O
OHAr
HN N
NN
O
OHAr
25
2122
2324
NH
O
O
OHHN
HN
N
OH
O
NH
O
HN
HN
HN
N OH
BrCl
O OH
27
HO
Scheme 4. Representative structures of the vitronectin V 3 receptor antagonists from Pfizer research laboratories. The Ar substituent in compounds 23-26 denotes the various aromatic substituents introduced [63-65].
Small Molecule Antagonists of Integrin Receptors Current Medicinal Chemistry, 2010 Vol. 17, No. 22 2381
ploiting other identified receptors of the family as potential drug design targets has more recently spurred the drug de-sign community. To begin with, 2 1 receptor is a widely expressed integrin that binds to type I and type II-collagens V and to laminas 1 and 5 [75]. It has been implicated in he-mostasis and thrombosis as well as cancer metastasis, wound healing and angiogenesis [76]. 2 1 integrin appears to be a promising target for the development of safer and more ef-fective small-molecule antithrombotic agents. To obtain se-lective 2 1 antagonists (e.g. compound 36 with IC50 = 0.015 μM) [79], DeGrado and co-workers combined the prolyl-sulfonylamide fragment, obtained from the benzenesulfonyl-Pro-Phe dipeptide derivatives developed as antagonists of the closely related integrin 4 1 (section 2.3.4) [77], with deriva-tives of 2,3-diaminopropionic acid (DAP), reported as IIb 3
antagonists [78]. This series of prolyl-2,3-diaminopropionic acid (DAP) derivatives bind tightly to 2 1 integrin and are selective over a wide range ( 1 1, 4 1, 2 1, 5 1, 2 1 and
IIb 3) of integrins. Further studies showed that the antago-
nist 36 did not affect the platelet aggregation stimulated by addition of ADP agonist, showing that the compounds do not influence signaling through this IIb 3 integrin pathway [79].
2.3.2. Fibronectin 5 1 Receptor Antagonists
The fibronectin receptor 5 1 integrin plays a key role during development of the vascular system. It is the only unambiguously determined proangiogenic integrin. Genetic ablation experiments and pharmacological results on 5 1 integrin support its importance in neovascularization proc-esses [80, 81]. Antagonists of 5 1 receptor are of interest as therapeutic agents for pathological angiogenic conditions [82]. Fibronectin – the natural ligand of 5 1 – was found to bind its receptor through the RGD-containing module 10 (Fn10) in a manner similar to cyclic peptide 9, determined in the V 3 crystal complex [22].
Marinelli and co-workers reported a three-dimensional homology model of the 5 1 integrin head group bound to the
HN N
N N
O O
OH
N
OMe
HN N
YX
O
OH
R
HN N
HN N N
NH
Constrained Chain-Shortened
Series
Aliphatic Chain-Shortened
Series
Chain-Shortened
Series
34 X-Y = CH=CH35 X-Y = CH2-CH2
Peptide Derived
(Full-Length) Series
O
O
O
O
OH
NH
O
O
O
OH
NH2+
HNHN
O
HN
O
NH
O
O-
NHO
Arg-Gly-Asp
Tripeptide
conformational lock
bidentate guanidine
binding site
ß-alanine
binding site
carboxylate
binding site
HN N
O
OH
HN N
O
OH
NN
NN
31
32
33
35a
35b
30
Scheme 5. Development of clinical candidates 35a and 35b for the prevention and treatment of osteoporosis at Merck research laboratories [74].

2382 Current Medicinal Chemistry, 2010 Vol. 17, No. 22 Perdih and Dolenc
selective 5 1 ligand SJ749 (37). It was established that, within the 5 1 binding pocket, other regions, different from those that bind Arg and Asp of the RGD sequence, have to be targeted to achieve fibronectin selectivity. This is in con-trast to the V 3/ IIb 3 case where selectivity can be achieved by changing the distance between the basic and acid moie-ties. The selective 5 1 compound, SJ749, possesses an addi-tional chain, represented by a carbamate group. According to the derived model, this functional group was inserted be-tween strategically located loops, each of which bears a ser-ine residue. Thus, residues Ser224 ( 5) and Ser221 ( 1) are accessible attachment points for antagonist H-bond accep-tors. The presence of Gln221 ( 5) and the reduced basicity of the D3 A3 loop are valuable for selective variation of the guanidine group. Furthermore, a wide pocket along the di-mer border, which consists of some 5 1 atypical amino ac-ids, was found to be a special feature of such this integrin – and a further structural element for achieving potency and selectivity [83].
Taking this model into account, the synthesis of a novel class of selective 5 1 antagonists (e.g. compound 38) has been reported. Selectivity was controlled by switching from a sulfonamide to a mesitylene amide moiety [84]. In addi-tion, the 5 1 integrin homology model was recently applied in designing, not only dual V 3 / 5 1 integrin antagonists, but also a selective 5 1 antagonist 39 (IC50( 5 1) = 0.7 nM) [85].
2.3.3. L 2 and M 2 Integrin Antagonists/Agonists
Integrin L 2 (LFA-1, lymphocyte function associated antigen 1) is a member of the leukocyte integrin subfamily that shares the 2 subunit and is an important target for blocking rejection in organ transplantation and for treating autoimmune diseases [86]. M 2, X 2, D 2 and L 2 are all members of the inserted I domain integrins. A humanized antibody to L 2
that blocks its binding to the ligand ICAM-1 has been approved by the FDA for treatment of psoriasis, a T cell-mediated autoimmune disease of the skin [87]. Several low molecular weight L 2 antagonists are under develop-ment as anti-inflammatory agents and are classified into two groups. One group binds underneath the C-terminal 7 helix of the L I domain (e.g. compounds 40-42), blocking the downward axial displacement of the C-terminal helix, and inhibits ligand binding of L 2 allosterically by stabilizing the I domain in the low-affinity conformation. This class is often referred to as I allosteric antagonists [88, 89]. The other group of antagonists appears to bind to the 2 I domain
near a key regulatory interface with the L I domain, block-ing communication of the conformational change to the L I domain while, at the same time, activating conformational rearrangements elsewhere in the integrins. These antagonists, such as compound 43
[90, 91] and compound 44 [92, 93], are
called / I-like allosteric antagonists. All these compounds also inhibit binding to M 2 integrin [23].
The latest findings suggest that not only the antagonistic but also the agonistic behavior of small molecules acting on
L 2 integrin receptors might prove valuable in therapy. The small molecule 43, previously reported as an antagonist of integrin L 2, acts as an agonist of this receptor in the pres-ence of Ca2+ and Mg2+ ions and as an antagonist with Mn2+
ions. Although it stimulated ligand binding, compound 43
nonetheless inhibited lymphocyte transendothelial migration. This opened up completely new therapeutic possibilities, in addition to its earlier reported anti-inflammatory potential [94]. Since immune recognition of tumor cells is LFA-1 de-pendent, it was speculated that application of LFA-1 agonist would enhance immune responses, including cytotoxic kill-ing of tumor cells.
2.3.4. Integrin 4- Receptor Antagonists
The main members of the 4- integrin subfamily are 4 1 integrin, also known as very late heterodimeric antigen-4 (VLA-4), and 4 7 integrin. Ligands bind to 4 1 and 4 7 integrins in an RGD-independent manner. The 4 1 integrin is expressed on many leukocytes, including eosinophils, ba-sophils, and monocytes. The 4 7 integrin is found primarily on mucosal lymphocytes, and may be beneficial in the treat-ment of inflammatory bowel disease. The binding of 4 1 to ligands such as the vascular cell vascular cell adhesion mole-cule-1 (VCAM-1), expressed on endothelial cells, is recog-nized as a key step in the processes of recruitment to sites of inflammation [95, 96]. It was proposed that concurrent
4 1/ 4 7 antagonism may be advantageous to achieve maximum efficacy (discussed further in section 2.4.4).
The therapeutic potential of 4 1 integrin antagonists has been demonstrated in the treatment of multiple sclerosis by natalizumab (marketed as Tysabri®), a humanized mono-clonal antibody that binds to the 4 integrin subunit [97]. In November 2004, Tysabri® received accelerated approval from the FDA, but shortly after was voluntarily withdrawn due to reports of rare but serious adverse events of progres-sive multifocal leukoencephalopathy (PML). The drug was returned to the US market in 2006 under a special prescrip-tion program. In 2008, further incidents of PML were re-ported. There are many publications of small molecule 4 antagonists, due to its great therapeutic potential in the treatment of inflammatory and autoimmune diseases [98-100]. The antagonists are classified into several groups, ranging from monoclonal antibody antagonists, peptide an-tagonists, LDV peptides, RGD-like peptides, peptidomimetic and proteomimetic compounds and small molecule antago-nists [100].
The substituted biphenyl group and functionalized N-acyl phenylalanines (e.g. compound 45) [101, 102] can be found in many of the antagonists in this class. Okigami and co-workers reported the identification of an 4 1 antagonist, (2S)-3-(2 ,5'-dichlorobiphenyl-4-yl)-2-({[1-(2-methoxyben-zoyl)piperidin-3yl]carbonyl} amino) propanoic acid, 46, and characterized the antagonist activities of this molecule in cell-based assays and in an animal model of eosinophil mi-gration. The results supported the use of 46 in the treatment of asthma, atopic dermatitis, and allergic rhinitis [103]. In addition to its normal function in regulating inflammatory and autoimmune responses, the activated form of integrin
4 1 is present in lymphomas, and regulates tumor growth, metastasis, and angiogenesis. It also promotes the dissemina-tion of tumor cells to distal organs while facilitating tumor cell extravasation [104]. Starting from a bisaryl urea pepti-domimetic antagonist, [105] a highly soluble 2-arylaminobenzimidazole antagonist 47 was developed that is strongly prospective in diagnosis and treatment of lympho-mas [106]. In addition to its therapeutic utility for treating
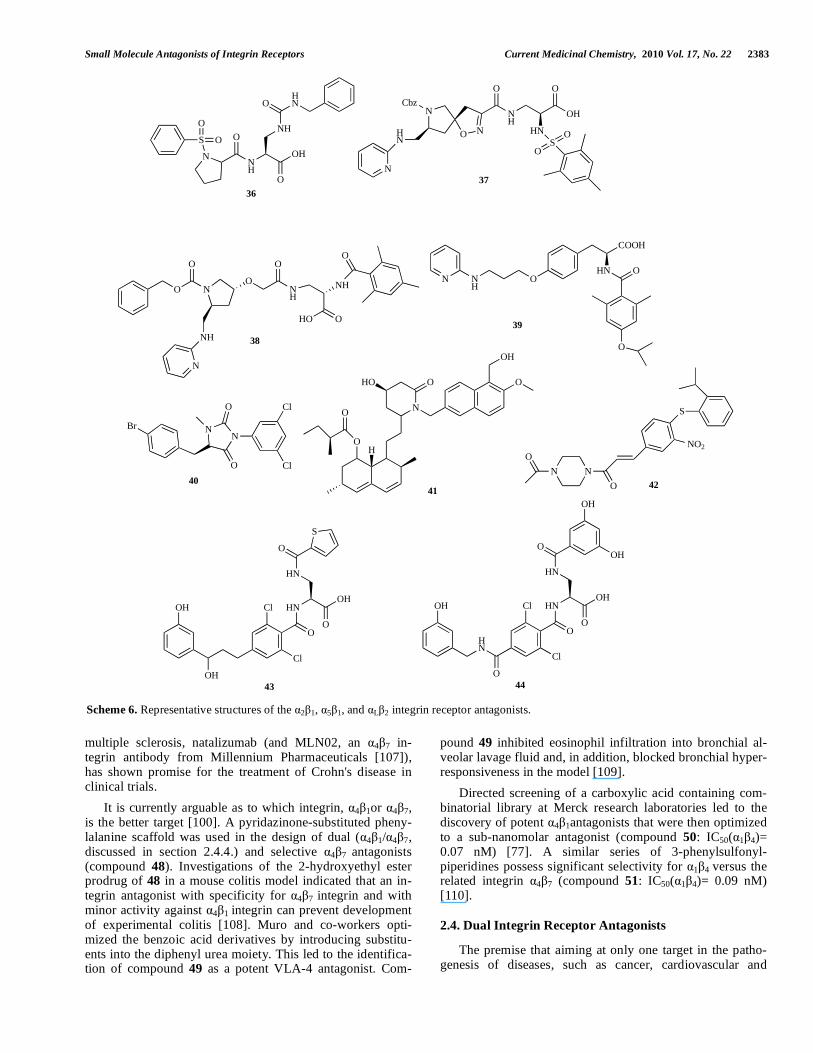
Small Molecule Antagonists of Integrin Receptors Current Medicinal Chemistry, 2010 Vol. 17, No. 22 2383
multiple sclerosis, natalizumab (and MLN02, an 4 7 in-tegrin antibody from Millennium Pharmaceuticals [107]), has shown promise for the treatment of Crohn's disease in clinical trials.
It is currently arguable as to which integrin, 4 1or 4 7, is the better target [100]. A pyridazinone-substituted pheny-lalanine scaffold was used in the design of dual ( 4 1/ 4 7, discussed in section 2.4.4.) and selective 4 7 antagonists (compound 48). Investigations of the 2-hydroxyethyl ester prodrug of 48 in a mouse colitis model indicated that an in-tegrin antagonist with specificity for 4 7 integrin and with minor activity against 4 1 integrin can prevent development of experimental colitis [108]. Muro and co-workers opti-mized the benzoic acid derivatives by introducing substitu-ents into the diphenyl urea moiety. This led to the identifica-tion of compound 49 as a potent VLA-4 antagonist. Com-
pound 49 inhibited eosinophil infiltration into bronchial al-veolar lavage fluid and, in addition, blocked bronchial hyper-responsiveness in the model [109].
Directed screening of a carboxylic acid containing com-binatorial library at Merck research laboratories led to the discovery of potent 4 1antagonists that were then optimized to a sub-nanomolar antagonist (compound 50: IC50( 1 4)= 0.07 nM) [77]. A similar series of 3-phenylsulfonyl-piperidines possess significant selectivity for 1 4 versus the related integrin 4 7 (compound 51: IC50( 1 4)= 0.09 nM) [110].
2.4. Dual Integrin Receptor Antagonists
The premise that aiming at only one target in the patho-genesis of diseases, such as cancer, cardiovascular and
NNH
S O
O
O
O
OH
NH
HNO
NO
O
NH
N
O
O
NH
OHO
NH
O
36
37
39
N
O N
O
NH
O
OH
HN
Cbz
HN
N
SO
O
NH
ON
COOH
HN O
O38
BrN
N
O
O Cl
Cl
40
N
O
O
H
HO O
OH
O
41
N N
O
O
S
NO2
42
OH
OH
Cl
Cl
HN
O
OH
HN
S
O
O
43
OH
HN
Cl
Cl
HN
O
OH
HN
O
O
44
O
OH
OH
Scheme 6. Representative structures of the 2 1, 5 1, and L 2 integrin receptor antagonists.
2384 Current Medicinal Chemistry, 2010 Vol. 17, No. 22 Perdih and Dolenc
N
N
N
N
O OH
OMeNH
O
HO
HN
O
NCl
Cl
45
N
OOO
HN
O
HO
Cl
Cl
46
49
NH
NH
ON
F
Cl
F
O
NH2
HNO
NH
O
HOO
NH NHO
NHN
N
NH
O
47
Cl
Cl SO2
NO
HN COOH
OCH3
H3CO
50
O
HN COOH
OCH3
H3CO
51
NH
SO2
N
N
O
H3CO
O
OHNH
OH
O
O
48
O
COOH
Scheme 7. Representative structures of antagonists to the subfamily of 4 integrins.
rheumatic disorders, may be inferior or even inadequate compared to a multiple target approach is supported by ex-perimental evidence [8]. Drugs with dual or multiple target action should provide superior treatment by avoiding the pharmacokinetic problems connected with applying more than one drug at the same time, and thus improving patient compliance [8]. The idea of dual targeting is gaining in popularity in the drug design field. For example, some recent examples of dual targeting include a platelet activating factor receptor antagonist with thromboxane synthase inhibitors [111], lipoxygenase–cyclooxygenase inhibitors, [112] and the already discussed combination of thrombin inhibitor and
IIb 3 antagonist (7 and 8) [39, 40].
The targets suitable for dual or multiple targeting are ei-ther different proteins in the same biochemical pathway, with topologically similar active sites, or proteins involved in different pathways, playing a significant role in one patho-logical state. The target combination should be as near to unique for the disease as possible and hence provide the re-quired selective toxicity, given that the main danger of such promiscuous compounds is off-target activity. Dual integrin receptor antagonists and the requirements for successful de-sign have been discussed [8]. Several integrin pairs whose dual action could be beneficial are listed in Table 4. [8] In these cases both proteins play an important role in the same
pathological disorder. Dual antagonists of the IIb 3/ V 3, V 3/ V 5, V 3/ 5 1 and 4 1/ 4 7 integrin receptor pairs
will be discussed in more detail.
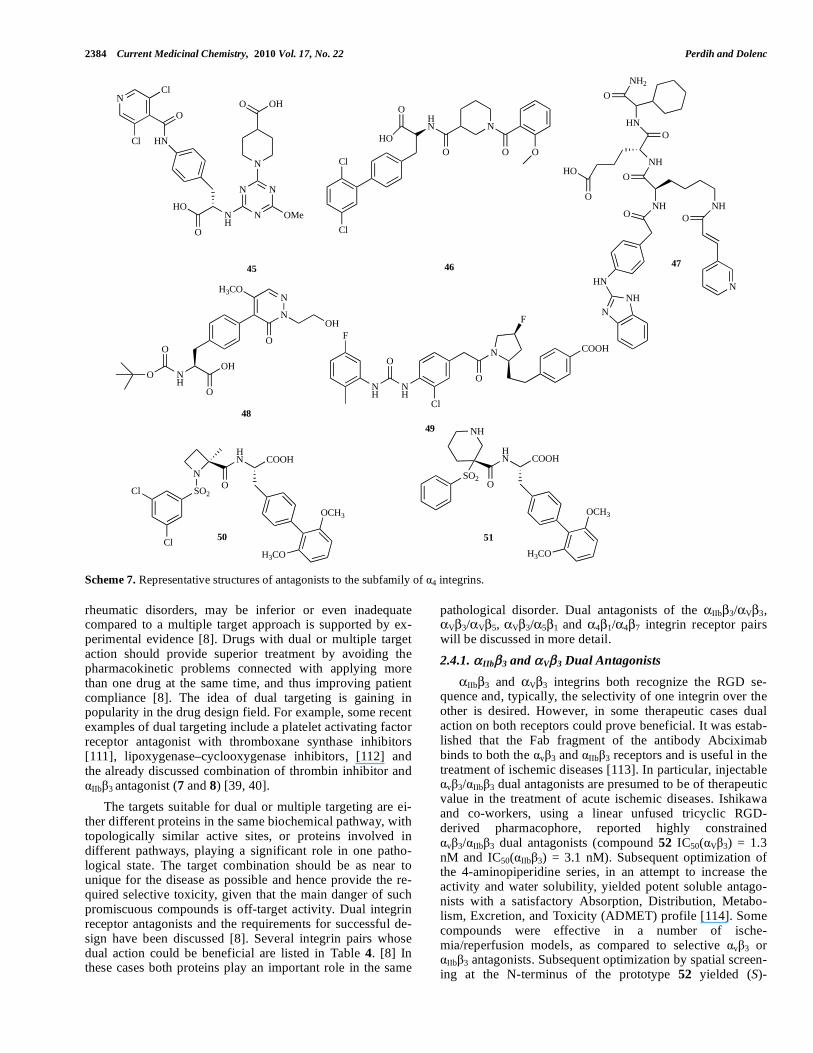
2.4.1. IIb 3 and V 3 Dual Antagonists
IIb 3 and V 3 integrins both recognize the RGD se-quence and, typically, the selectivity of one integrin over the other is desired. However, in some therapeutic cases dual action on both receptors could prove beneficial. It was estab-lished that the Fab fragment of the antibody Abciximab binds to both the v 3 and IIb 3 receptors and is useful in the treatment of ischemic diseases [113]. In particular, injectable
v 3/ IIb 3 dual antagonists are presumed to be of therapeutic value in the treatment of acute ischemic diseases. Ishikawa and co-workers, using a linear unfused tricyclic RGD-derived pharmacophore, reported highly constrained
v 3/ IIb 3 dual antagonists (compound 52 IC50( V 3) = 1.3 nM and IC50( IIb 3) = 3.1 nM). Subsequent optimization of the 4-aminopiperidine series, in an attempt to increase the activity and water solubility, yielded potent soluble antago-nists with a satisfactory Absorption, Distribution, Metabo-lism, Excretion, and Toxicity (ADMET) profile [114]. Some compounds were effective in a number of ische-mia/reperfusion models, as compared to selective v 3 or
IIb 3 antagonists. Subsequent optimization by spatial screen-ing at the N-terminus of the prototype 52 yielded (S)-
Small Molecule Antagonists of Integrin Receptors Current Medicinal Chemistry, 2010 Vol. 17, No. 22 2385
aminopiperidine-based dual antagonists (compound 53: IC50( V 3) = 0.48 nM and IC50( IIb 3) = 0.56 nM). The v 3 antagonistic activity differed, depending on the space that was occupied by the N-terminus, but high potency against
IIb 3 was maintained. The (3S)-aminopiperidine analogue had the strongest activity against v 3, and the (S) isomer at the piperidine moiety was more potent than the correspond-ing (R) isomer [115, 116]. Later it was also shown that selec-tivity for v 3 over IIb 3 in this class of compounds can be achieved by meta-substitution of the central benzene ring (compound 54: IC50( V 3) = 18 nM and IC50( IIb 3) = 2000 nM) [117].
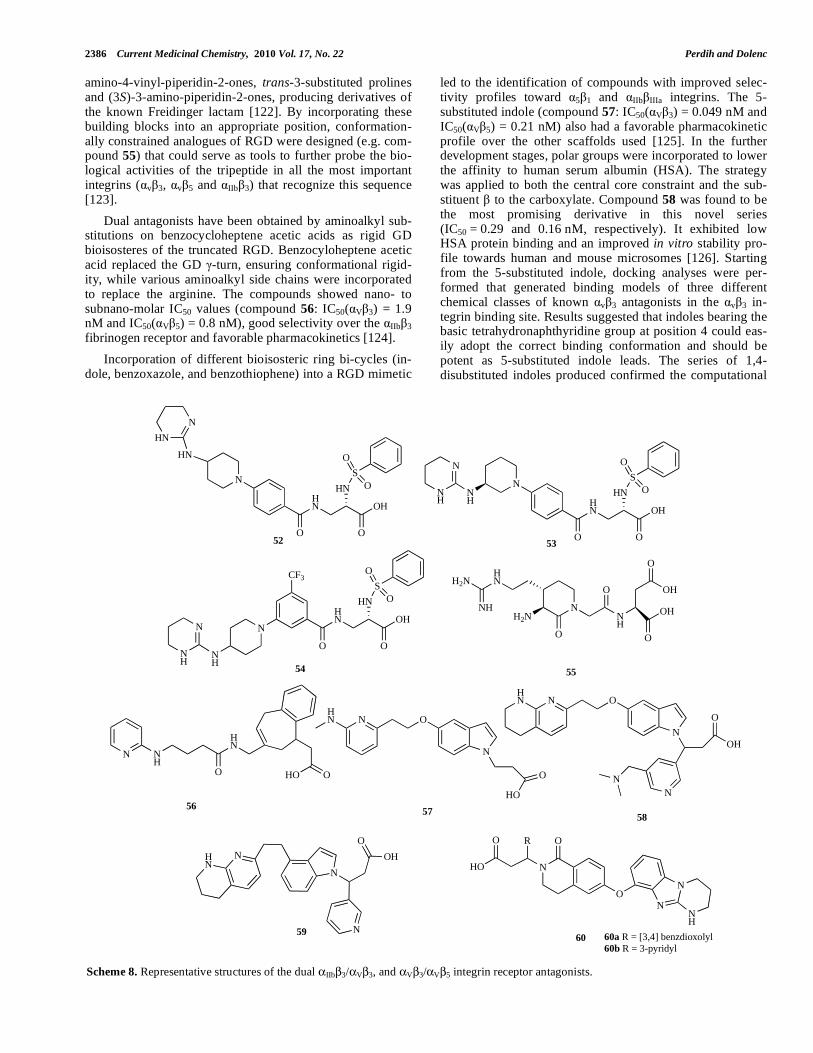
2.4.2. V 3 and V 5 Dual Antagonists
The extracellular domains of v 3 and v 5 both recog-nize the RGD sequence. In fact, most of the developed V 3 antagonists already described in section 2.2 would display little selectivity towards the V 5 integrins. The latest under-standing of the role of integrins in angiogenesis (the forma-tion of new blood vessels) supports the development of
v 3/ v 5 dual antagonists for the treatment of cancer, be-cause of their ability to prevent the “angiogenic switch” that allows endothelial cells to migrate, proliferate and invade the surrounding matrix [118]. v 3/ v 5 dual antagonists have, like the v 3 antagonists, potential in the treatment of osteo-porosis, since the RGD constraining peptides and non-peptide v 3/ v 5 antagonists have been shown to inhibit bone loss in the ovariectomized rat model [119]. Since both
receptors share the V subunit, most of the differences are in the acidic part of the antagonists, which binds to the A do-main of the subunit [8]. Marnelli and coworkers con-structed a 3D structural model of a v 5 receptor, using ho-mology modeling based on the crystal coordinates of v 3
with bound RGD-containing peptide as a template. The ini-tial model was refined using energy minimization and mo-lecular dynamics simulations in the explicit solvent. The refined v 5 model was used to explore the interactions be-tween this integrin and v 3/ v 5 dual and v 3-selective ligands to provide rationalization, at the molecular level, of the ligand selectivity towards the two v integrins. The in-tegrins differ, especially in the region above the MIDAS, due to the diversity of the Specificity Determining Loop (SDL). A partial “roof”, composed mainly of the SDL residues ( 5)-Tyr179 and ( 5)-Lys180, is present in the v 5 receptor and hampers the binding of compounds containing bulky sub-stituents in the proximity of the carboxylate group [120].
The cycloRGDfV ligands, like peptide 9, are among the most active and selective compounds for the V 3 and V 5 integrin subtype receptors and they offer a good starting point for small molecule design. Recently, a regioselective derivatization (introduction of an aminooxy or glyoxylalde-hyde group) on the lysine side of 9 yielded various conju-gates for biological and conformational investigations [121]. Another exploration of the bioactive RGD conformation involved the synthesis of the N-functionalized (3S,4S)-3-
Table 4. Involvement of Integrin Pairs in Diseases [8]
Receptor pair Disease/pathological process
thromboembolic diseases
hantavirus entry
IIb 3 V 3
acute ischemic disease
restenosis after angioplasty infection
angiogenesis
osteoporosis
V 3 V 5
adenovirus infection
angiogenesis
Lyme disease
tumor metastasis
V 3 5 1
reactive astrogliosis
atherosclerosis
tumor cell growth and invasion
angiogenesis
V 3 2 1
rotavirus infection
inflammatory diseases
atherosclerosis
V 3 4 1
angiogenesis
Crohn's disease
rheumatoid arthritis
4 1 4 7
other autoimmune diseases
2386 Current Medicinal Chemistry, 2010 Vol. 17, No. 22 Perdih and Dolenc
amino-4-vinyl-piperidin-2-ones, trans-3-substituted prolines and (3S)-3-amino-piperidin-2-ones, producing derivatives of the known Freidinger lactam [122]. By incorporating these building blocks into an appropriate position, conformation-ally constrained analogues of RGD were designed (e.g. com-pound 55) that could serve as tools to further probe the bio-logical activities of the tripeptide in all the most important integrins ( v 3, v 5 and IIb 3) that recognize this sequence [123].
Dual antagonists have been obtained by aminoalkyl sub-stitutions on benzocycloheptene acetic acids as rigid GD bioisosteres of the truncated RGD. Benzocyloheptene acetic acid replaced the GD -turn, ensuring conformational rigid-ity, while various aminoalkyl side chains were incorporated to replace the arginine. The compounds showed nano- to subnano-molar IC50 values (compound 56: IC50( V 3) = 1.9 nM and IC50( V 5) = 0.8 nM), good selectivity over the IIb 3 fibrinogen receptor and favorable pharmacokinetics [124].
Incorporation of different bioisosteric ring bi-cycles (in-dole, benzoxazole, and benzothiophene) into a RGD mimetic
led to the identification of compounds with improved selec-tivity profiles toward 5 1 and b a integrins. The 5-substituted indole (compound 57: IC50( V 3) = 0.049 nM and IC50( V 5) = 0.21 nM) also had a favorable pharmacokinetic profile over the other scaffolds used [125]. In the further development stages, polar groups were incorporated to lower the affinity to human serum albumin (HSA). The strategy was applied to both the central core constraint and the sub-stituent to the carboxylate. Compound 58 was found to be the most promising derivative in this novel series (IC50 = 0.29 and 0.16 nM, respectively). It exhibited low HSA protein binding and an improved in vitro stability pro-file towards human and mouse microsomes [126]. Starting from the 5-substituted indole, docking analyses were per-formed that generated binding models of three different chemical classes of known v 3 antagonists in the v 3 in-tegrin binding site. Results suggested that indoles bearing the basic tetrahydronaphthyridine group at position 4 could eas-ily adopt the correct binding conformation and should be potent as 5-substituted indole leads. The series of 1,4-disubstituted indoles produced confirmed the computational
O
HN
O
OH
HN
S
O
ON
N
O
NH
O
OH
O
OH
O
H2N
HN
NH
H2N
HN
N
N
O
OH
N
52
55
59
N NH
O
HN
OHO
56
HN N O
N
O
HO
57
N
O
HO
O
ON
N
NH
R
60 60a R = [3,4] benzdioxolyl60b R = 3-pyridyl
O
HN
O
OH
HN
S
O
ON
NH
N
NH
53
54
HN
N
HN
O
HN
O
OH
HN
S
O
O
CF3
N
NH
N
NH
58
HN N O
N
O
OH
N
N
Scheme 8. Representative structures of the dual IIb 3/ V 3, and V 3/ V 5 integrin receptor antagonists.
Small Molecule Antagonists of Integrin Receptors Current Medicinal Chemistry, 2010 Vol. 17, No. 22 2387
results (compound 59: IC50( V 3) = 0.5 nM and IC50( V 5) = 0.96 nM) with excellent selectivity over IIb 3, although also expressing high HSA binding (96.5–97.3 %) [127].
High-throughput screening of compound libraries, fol-lowed by subsequent optimization, led to the discovery of compounds 60a and 60b with reduced polar surface area and improved potency for v 3/ v 5 receptors. Both compounds showed activity in functional cellular assays, and compound 60a (IC50( V 3) = 1.0 nM and IC50( V 5) = 5.0 nM) exhibited a promising Caco-2 permeability profile [128].
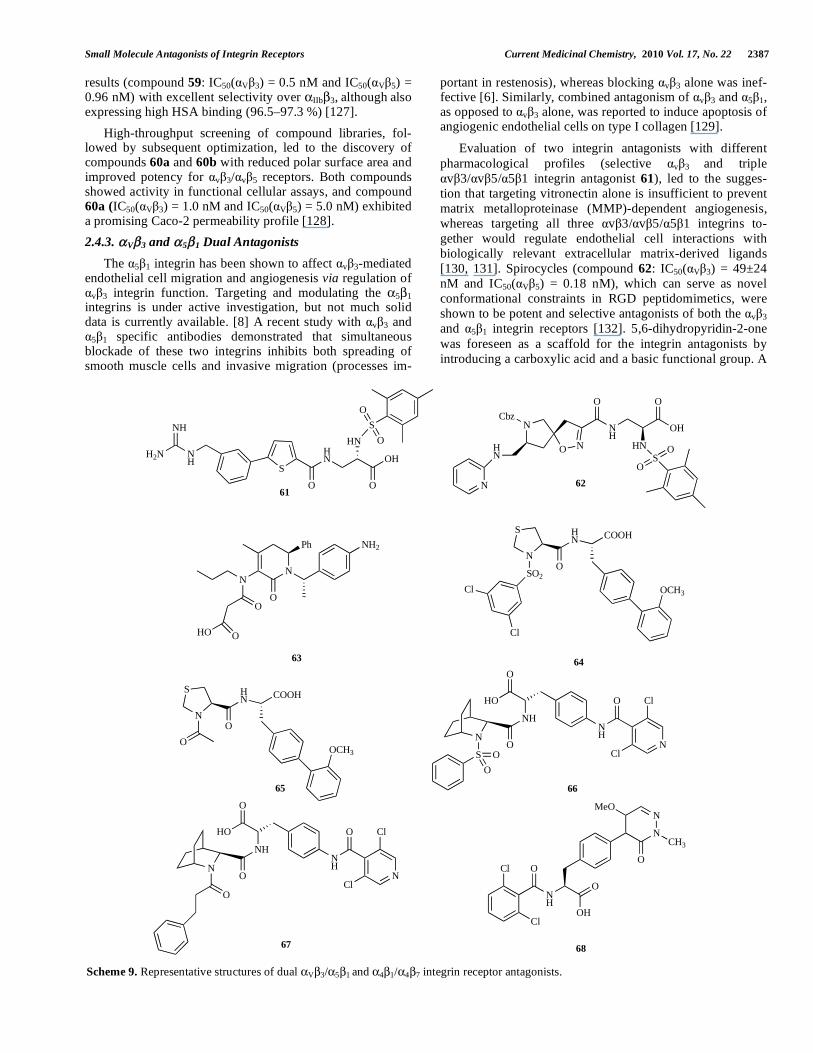
2.4.3. V 3 and 5 1 Dual Antagonists
The 5 1 integrin has been shown to affect v 3-mediated endothelial cell migration and angiogenesis via regulation of
v 3 integrin function. Targeting and modulating the 5 1 integrins is under active investigation, but not much solid data is currently available. [8] A recent study with v 3 and
5 1 specific antibodies demonstrated that simultaneous blockade of these two integrins inhibits both spreading of smooth muscle cells and invasive migration (processes im-
portant in restenosis), whereas blocking v 3 alone was inef-fective [6]. Similarly, combined antagonism of v 3 and 5 1, as opposed to v 3 alone, was reported to induce apoptosis of angiogenic endothelial cells on type I collagen [129].
Evaluation of two integrin antagonists with different pharmacological profiles (selective v 3 and triple
v 3/ v 5/ 5 1 integrin antagonist 61), led to the sugges-tion that targeting vitronectin alone is insufficient to prevent matrix metalloproteinase (MMP)-dependent angiogenesis, whereas targeting all three v 3/ v 5/ 5 1 integrins to-gether would regulate endothelial cell interactions with biologically relevant extracellular matrix-derived ligands [130, 131]. Spirocycles (compound 62: IC50( V 3) = 49±24 nM and IC50( V 5) = 0.18 nM), which can serve as novel conformational constraints in RGD peptidomimetics, were shown to be potent and selective antagonists of both the v 3 and 5 1 integrin receptors [132]. 5,6-dihydropyridin-2-one was foreseen as a scaffold for the integrin antagonists by introducing a carboxylic acid and a basic functional group. A
S
NH
NH
H2N
O
HN
O
OH
HN
S
O
O
NH
O
HO
ON
S O
O
NH
N
Cl
Cl
O
61
66
N
O N
O
NH
O
OH
HN
Cbz
HN
N
SO
O
62
N
NMeO
CH3
O
OH
ONH
OCl
Cl
68
N
S HN
O
COOH
OCH3
65
NH
O
HO
ON
O
NH
N
Cl
Cl
O
67
N
Ph
O
NH2
N
O
HO O
63
N
S
SO2
Cl
Cl
HN
O
COOH
OCH3
64
O
Scheme 9. Representative structures of dual V 3/ 5 1 and 4 1/ 4 7 integrin receptor antagonists.

2388 Current Medicinal Chemistry, 2010 Vol. 17, No. 22 Perdih and Dolenc
small library of peptidomimetics created using this strategy (e.g. compound 63: IC50( V 3) = 71 ± 11 nM and IC50( V 5) = 57 ± 17 nM) was able to recognize and bind to v 3 and
5 1 integrins [133].
2.4.4. 4 1 and 4 7 Dual Antagonists
Lin and coworkers first proposed a binding model for se-lective 4 1 or dual 4 1/ 4 7 antagonists based on the struc-ture-activity relationship (SAR) of the N-substituted thiopro-lyl biarylalanine derivatives. The main difference between the binding models for the selective 4 1 or dual 4 1/ 4 7 antagonists was in the size of the pocket that can accommo-date the bulky phenyl ring. In the 4 1 integrin this pocket is considerably smaller than in the 4 7 integrin. This led to the superior selectivity of compound 64 (IC50( 4 1) = 0.3 nM and IC50( 4 7) = 306 nM) over compound 65 (IC50( 4 1) = 0.2 nM and IC50( 4 7) = 2.1 nM) [134] series of aza-bicyclic amino acid sulfonamides (compound 66: IC50( 4 1) = 12 ± 4 nM and IC50( 4 7) = 13 ± 8 nM) and aza-bicyclic amino acid carboxamides (compound 67: IC50( 4 1) = 1 ± 0.5 nM and IC50( 4 7) = 7 ± 2 nM) were reported as dual 4 1/ 4 7 an-tagonists. Several compounds had low or sub- nanomolar potency. Compound 66 was effective in the antigen-sensitized sheep model of asthma, and in vivo administration of 67 produced significant elevation in circulating lympho-cytes and total white cells [135]. Furthermore, one optimiza-tion route of the pyridazinone-substituted phenylalanine scaffold of compound 48 [108] discussed previously yielded promising results. Compound 68 (IC50( 4 1) = 31 ± 8 nM and IC50( 4 7) = 3 ± 1 nM) demonstrated dose-responsive effects on the elevation of circulating leukocytes in a mouse leukocytosis study [136].
CONCLUSIONS
The physiological importance of integrin receptors in a wide range of pathological states like thrombosis, inflamma-tion, atherosclerosis, osteoporosis, cancer and infectious dis-eases, makes this receptor family an attractive collection of targets for drug design campaigns. Despite intensive research in this field, the mechanistic aspects of integrin receptor function still pose many complex questions that will have to be addressed in order to fully understand these fascinating and important heterodimeric transmembrane receptors.
Due to the great complexity of integrin biochemistry, caution is always necessary when designing new small molecule integrin antagonists, in order to avoid undesirable off-target effects. Currently, the fibrinogen receptor IIb 3 and the vitronectin receptor V 3 stand at the forefront of attempts to design integrin receptor drugs. The fibronectin receptor 5 1 and the 4-integrin antagonists are gaining in-creasing importance. The potential of creating molecules that target multiple integrin receptors shows great promise for future drug design.
The work done in the integrin field has captured the imagination and stimulated the creativity of many research groups in academia and industry. Several drugs are currently undergoing clinical trials, so the true potential of integrin-based drug therapy should soon be revealed. The initial suc-
cess stories in the field of low molecular weight fibrinogen antagonists like Aggrastat® and Integrilin®, already intro-duced into the clinic, provide a lot of optimism for further, fruitful development of the field.
ACKNOWLEDGEMENT
The authors wish to thank Prof. Roger Pain for critical reading of the manuscript.
REFERENCES
[1] Hynes, R.O. Integrins: bidirectional, allosteric signaling machines. Cell, 2002, 110, 673-687.
[2] Fernandez, C.; Clark, K.; Burrows, L.; Schofield, N.R.; Humphries, M.J. Regulation of the extracellular ligand binding activity of in-tegrins. Front. Biosci., 1998, 3, 684-700.
[3] Coller, B.S.; Shattil, S.J. The GPIIb/IIIa (integrin alphaIIbbeta3) odyssey: a technology-driven saga of a receptor with twists, turns, and even a bend. Blood, 2008, 112, 3011-3025.
[4] Silva, R.; D'Amico, G.; Hodivala-Dilke, K.M.; Reynolds, L.E. Integrins: the keys to unlocking angiogenesis. Arterioscler.
Thromb. Vasc. Biol., 2008, 10, 1703-1713. [5] Mousa, S.A. v vitronectin receptors in vascular-mediated disor-
ders. Med. Res. Rev., 2003, 23, 190-199. [6] Kim, S.; Harris, M.; Varner, J.A. Regulation of integrin v 3-
mediated endothelial cell migration and angiogenesis by integrin 5 1 and protein kinase A. J. Biol. Chem., 2000, 275, 33920-33928.
[7] Sudhakar, A.; Sugimoto, H.; Yang, C.; Lively, J.; Zeisberg, M.; Kalluri, R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by v 3 and 5 1 integrins. Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4766-4771.
[8] Nadrah, K.; Sollner Dolenc, M. Dual Antagonists of Integrins. Curr. Med. Chem., 2005, 12, 1449-1466.
[9] Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem., 2000, 275, 21785-21788.
[10] Takagi, J.; Petre, B.M.; Walz, T.; Springer, T.A. Global conforma-tional rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell, 2002, 110, 599-611.
[11] Ginsberg, M.H.; Partridge, A.; Shattil, S.J. Integrin regulation. Curr. Opin. Cell. Biol., 2005, 17, 509-516.
[12] Luo, B.-H.; Springer, T.A. Integrin structures and conformational signaling Curr. Opin. Cell Biol., 2006, 18, 579-586.
[13] Liddington, R.C.; Ginsberg, M.H. Integrin activation takes shape. J. Cell. Biol., 2002, 158, 833-839.
[14] Calzada, M.J.; Alvarez, M.V.; Gonzalez-Rodriguez, J. Agonist-specific structural rearrangements of integrin alpha IIbbeta 3. Con-firmation of the bent conformation in platelets at rest and after acti-vation. J. Biol. Chem., 2002, 277, 39899-39908.
[15] Humphries M.J.; Symonds, E.J.; Mould, A.P. Mapping functional residues onto integrin crystal structures. Curr. Opin. Struct. Biol., 2003, 13, 236-243.
[16] Humphries M.J. Insights into integrin-ligand binding and activation from the first crystal structure. Arthritis Res., 2002, 4, (Suppl 3), 69-78.
[17] Xiong, J.P.; Stehle, T.; Diefenbach, B.; Zhang, R.; Dunker, R.; Scott, D.L.; Joachimiak, A.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin V 3. Science, 2001, 294, 339-345.
[18] Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellu-lar segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science, 2002, 296, 151-155.
[19] Xiao, T.; Takagi, J.; Coller, B.S.; Wang, J.H.; Springer, T.A. Struc-tural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature, 2004, 432, 59-67.
[20] Figure was made using LigandScout software: Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Mo-
del., 2005, 45, 160-169.

Small Molecule Antagonists of Integrin Receptors Current Medicinal Chemistry, 2010 Vol. 17, No. 22 2389
[21] Takagi, J., Springer, T.A. Integrin activation and structural rear-rangement. Immunol. Rev., 2002, 186, 141-163.
[22] Takagi, J.; Strokovich, K.; Springer, T.A.; Walz, T. Structure of integrin alpha5beta1 in complex with fibronectin. EMBO J, 2003, 22, 4607-4615.
[23] Shimaoka, M.; Springer, T.A. Therapeutic antagonists and confor-mational regulation of integrin function. Nat. Rev. Drug Discov. 2003, 2, 703-716.
[24] Ferguson, J. Low-molecular-weight heparins and glycoprotein IIb/IIIa antagonists in acute coronary syndromes. J. Invasive. Car-
diol., 2004, 16, 136-144. [25] Mousa, S.A. Alpha nu vitronectin receptors in vascular-mediated
disorders. Med. Res. Rev., 2003, 23, 190-199. [26] Tilley, J.W.; Chen, L.; Sidduri, A.; Fotouhi, N. The discovery of
VLA-4 antagonists. Curr. Top. Med. Chem., 2004, 4, 1509-1523. [27] Singh, J.; Adams, S.; Carter, M.B.; Cuervo, H.; Lee, W.C.; Lobb,
R.R.; Pepinsky, R.B.; Petter, R.; Scott, D. Rational design of potent and selective VLA-4 inhibitors and their utility in the treatment of asthma. Curr. Top. Med. Chem., 2004, 4, 1497-1507.
[28] Scarborough, R.M. Structure-activity relationships of beta-amino acid-containing integrin antagonists. Curr. Med. Chem., 1999, 6, 971-981.
[29] Ojima, I.; Chakravarty, S.; Dong, Q. Antithrombotic agents: from RGD to peptide mimetics. Bioorg. Med. Chem., 1995, 3, 337-360.
[30] Andronati, S.A.; Karaseva, T.L.; Krysko, A.A. Peptidomimetics - antagonists of the fibrinogen receptors: molecular design, structu-res, properties and therapeutic applications. Curr. Med. Chem., 2004, 11, 1183-1211.
[31] Tamhane, U.U.; Gurm, H.S. The chimeric monoclonal antibody abciximab: a systematic review of its safety in contemporary prac-tice. Expert Opin. Drug Saf., 2008, 7, 809-819.
[32] Scarborough, R.M. Development of eptifibatide. Am. Heart J.,
1999, 138, 1093-1104. [33] Harrington, R.A.; and collaborators. Inhibition of platelet glycopro-
tein IIb/IIIa with eptifibatide in patients with acute coronary syn-dromes. N. Engl. J. Med., 1998, 339, 436-443.
[34] Bazzino, O.; Barrero, C.; Garre, L.; Sosa, A.; Aylward, P.; Slany, J.; Beaudry, P.; Bedard, J.; DeLarochelliere, R.; Nguyen, M.; Bogaty, P.; Boudreault, J.R.; Diodati, J.G.; Dupuis, J.; Fitchett, D.; Fung, A.; Gervais, P.; Gossard, D.; Grandmont, D.; Huynh, T.; Kouz, S.; Langer, A.; Laramee, P.; Lemay, M.; Maillette, S.; Ma-randa, C.; Nasmith, J.; Pesant, Y.; Phanuef, D.; Proulx, G.; Ruel, M.; Thompson, C.; Corbalan, R.; Botero, R.; Husted, S.; Heikkila, J.; Charbonnier, B.; Moguel, R.; Commerford, P.; Landless, P.; Weich, D.; Maritz, F.; Marx, J.; Fernandes, F.; Lidon, R.; Sanz, G.; Bosch, X.; Moccetti, T.; Ali, N.; Anderson, H.V.; Bajwa, T.; Bor-zak, S.; Boyek, T.; Brewer, D.; Cambier, P.; Chivukula, S.; Corder, C.; Ezekowitz, M.; Feldman, R.; Glickman, L.; Jang, I.K.; Klein, M.; Korn, D.; Kraus, S.; Kwan, T.; Mann, J.T.; Marshall, J.; Mar-tin, J.; Monrad, E.S.; Mueller, H.; Penny, W.; Schmedtje, J.; Stro-ny, J.; Armstrong, P.; DeCani, J.; Hirsh, J.; Pepine, C.; Ryan, T. J.; Theroux, P.; Catella-Lawson, F.; Diodati, J.; Roy, L.; Willerson, J.T.; Yusuf, S.; Zhao, X.; Sax, F.L.; Harris, K.H.; Pelletier, G.; Da-vies, R.; Flather, M.; Gosselin, G.; Herrmann, H.; Kells, C.; Knudtson, M.; Thadani, U.; Snapinn, S.M.; Ghannam, A.; Haggert, B.; Watson, A.; Lis, J.; Brancato, C.; Fong, D. Inhibition of the platelet glycoprotein IIb/IIIa receptor with tirofiban in unstable an-gina and non-Q-wave myocardial infarction. Platelet receptor inhi-bition in ischemic syndrome management in patients limited by un-stable signs and symptoms (PRISM-PLUS) study investigators. N. Engl. J. Med., 1998, 338, 1488-1497.
[35] Cannon, C.P. Oral platelet glycoprotein IIb/IIIa receptor inhibitors-part I. Clin. Cardiol., 2003, 26, 358-364.
[36] Krysko, A.A.; Chugunov, B.M.; Malovichko, O.L.; Andronati, S.A.; Kabanova, T.A.; Karaseva, T.L.; Kiriyak, A.V. Novel fi-brinogen receptor antagonists. RGDF mimetics, derivatives of 4-(isoindoline-5-yl)amino-4-oxobutyric acid. Bioorg. Med. Chem.
Lett., 2004, 14, 5533-5535. [37] Malovichko, O.L.; Petrus, A.S.; Krysko, A.A.; Kabanova, T.A.;
Andronati, S.A.; Karaseva, T.L.; Kiriyak, A.V. Derivatives of 7-amino-1,2,3,4-tetrahydroisoquinoline and isophthalic acids as novel fibrinogen receptor antagonists. Bioorg. Med. Chem. Lett., 2006, 16, 5294-5297.
[38] Anderluh, M.; Cesar, J.; Stefanic, P.; Kikelj, D.; Janes, D.; Murn, J.; Nadrah, K.; Tominc, M.; Addicks, E.; Giannis, A.; Stegnar, M.; Dolenc, M.S. Design and synthesis of novel platelet fibrinogen re-ceptor antagonists with 2H-1,4-benzoxazine-3(4H)-one scaffold. A systematic study. Eur. J. Med. Chem., 2005, 40, 25-49.
[39] Stefanic Anderluh, P.; Anderluh, M.; Ilas, J.; Mravljak, J.; Sollner Dolenc, M.; Stegnar, M.; Kikelj, D. Toward a novel class of anti-thrombotic compounds with dual function. Discovery of 1,4-benzoxazin-3(4H)-one derivatives possessing thrombin inhibitory and fibrinogen receptor antagonistic activities. J. Med. Chem., 2005, 48, 3110-3113.
[40] Ilas, J.; Jakopin, Z.; Borstnar, T.; Stegnar, M.; Kikelj, D. 3,4-Dihydro-2H-1,4-benzoxazine derivatives combining thrombin in-hibitory and glycoprotein IIb/IIIa receptor antagonistic activity as a novel class of antithrombotic compounds with dual function. J.
Med. Chem., 2008, 51, 5617-5629. [41] Bi, Q.; Zhou, X.; Cen, X.; Qu, H.; Luo, J.; Huang, Y.; Zhu, S.
Efficient targeted anticoagulant with active RGD motif. Thromb.
Res., 2007, 120, 541-547. [42] Lark, M.W.; Stroup, G.B.; Hwang, S.M.; James, I.E.; Rieman, D.J.;
Drake, F.H.; Bradbeer, J.N.; Mathur, A.; Erhard, K.F.; Newlander, K.A.; Ross, S.T.; Salyers, K.L.; Smith, B.R.; Miller, W.H.; Huff-man, W.F.; Gowen, M. Design and characterization of orally active Arg-Gly-Asp peptidomimetic vitronectin receptor antagonist SB 265123 for prevention of bone loss in osteoporosis. J. Pharmacol.
Exp. Ther., 1999, 291, 612-617. [43] Storgard, C.M.; Stupack, D.G.; Jonczyk, A.; Goodman, S.L.; Fox,
R.I.; Cheresh, D.A. Decreased angiogenesis and arthritic disease in rabbits treated with an alphavbeta3 antagonist. J. Clin. Invest., 1999, 103, 47-54.
[44] Hood, J.D.; Cheresh, D.A. Role of integrins in cell invasion and migration. Nat. Rev. Cancer, 2002, 2, 91-100.
[45] Mitjans, F.; Meyer, T.; Fittschen, C.; Goodman, S.; Jonczyk, A.; Marshall, J.F.; Reyes, G.; Piulats, J. In vivo therapy of malignant melanoma by means of antagonists of alphav integrins. Int. J. Can-
cer., 2000, 87, 716-723. [46] Sorbera, L.A.; Graul, A.; Castaner, J. Cilengitide. Drugs Fut.,
2000, 25, 674-678. [47] Badger, A.M.; Blake, S.; Kapadia, R.; Sarkar, S.; Levin, J.; Swift,
B. A.; Hoffman, S.J.; Stroup, G.B.; Miller, W.H.; Gowen, M.; Lark, M.W. Disease-modifying activity of SB 273005, an orally ac-tive, nonpeptide alphavbeta3 (vitronectin receptor) antagonist, in rat adjuvant-induced arthritis. Arthritis Rheum., 2001, 44, 128-137.
[48] Feuston, B.P.; Culberson, J.C.; Duggan, M.E.; Hartman, G.D.; Leu, C.T.; Rodan, S.B. Binding model for nonpeptide antagonists of al-pha(v)beta(3) integrin. J. Med. Chem., 2002, 45, 5640-5648.
[49] Marinelli, L.; Lavecchia, A.; Gottschalk, K.E.; Novellino, E.; Kes-sler, H. Docking studies on alphavbeta3 integrin ligands: pharma-cophore refinement and implications for drug design. J. Med.
Chem., 2003, 46, 4393-4404. [50] Belvisi, L.; Bernardi, A.; Colombo, M.; Manzoni, L.; Potenza, D.;
Scolastico, C.; Giannini, G.; Marcellini, M.; Riccioni, T.; Castori-na, M.; LoGiudice, P.; Pisano, C. Targeting integrins: insights into structure and activity of cyclic RGD pentapeptide mimics contain-ing azabicycloalkane amino acids. Bioorg. Med. Chem., 2006, 14, 169-180.
[51] Banfi, L.; Basso, A.; Damonte, G.; De Pellegrini, F.; Galatini, A.; Guanti, G.; Monfardini, I.; Riva, R.; Scapolla, C. Synthesis and biological evaluation of new conformationally biased integrin ligands based on a tetrahydroazoninone scaffold. Bioorg. Med.
Chem. Lett., 2007, 17, 1341-1345. [52] Royo, M.; Van Den Nest, W.; del Fresno, M.; Frieden, A.; Yaha-
lom, D.; Rosenblatt, M.; Chorev, M.; Albericio F. Solid-phase syn-theses of constrained RGD scaffolds and their binding to the v 3 integrin receptor. Tetrahedron Lett., 2001, 42, 7387-7391.
[53] Gentilucci, L.; Cardillo, G.; Squassabia, F.; Tolomelli, A.; Spampi-nato, S.; Sparta, A.; Baiula, M. Inhibition of cancer cell adhesion by heterochiral Pro-containing RGD mimetics. Bioorg. Med. Chem.
Lett., 2007, 17, 2329-2333. [54] Haubner, R.; Gratias, R.; Diefenbach, B.; Goodman, S.L.; Jonczyk,
A.; Kessler, H. Structural and Functional Aspects of RGD-Containing Cyclic Pentapeptides as Highly Potent and Selective In-tegrin V 3 Antagonists. J. Am. Chem. Soc., 1996, 118, 7461-7472.

2390 Current Medicinal Chemistry, 2010 Vol. 17, No. 22 Perdih and Dolenc
[55] Iwama, S.; Kitano, T.; Fukuya, F.; Honda, Y.; Sato, Y.; Notake, M.; Morie, T. Discovery of a potent and selective alpha v beta 3 in-tegrin antagonist with strong inhibitory activity against neointima formation in rat balloon injury model. Bioorg. Med. Chem. Lett.,
2004, 14, 2567-2570. [56] Zhou, Y.; Peng, H.; Ji, Q.; Qi, J.; Zhu, Z.; Yang, C. Discovery of
small molecule inhibitors of integrin v 3 through structure-based virtual screening. Bioorg. Med. Chem. Lett., 2006, 16, 5878-5882.
[57] Dayam, R.; Aiello, F.; Deng, J.; Wu, Y.; Garofalo, A.; Chen, X.; Neamati, N. Discovery of small molecule integrin alphavbeta3 an-tagonists as novel anticancer agents. J. Med. Chem., 2006, 49, 4526-4534.
[58] Urbahns, K.; Härter, M.; Albers, M.; Schmidt, D.; Stelte-Ludwig, B.; Brüggemeier, U.; Vaupel, A.; Gerdes, C. Biphenyls as potent vitronectin receptor antagonists Bioorg. Med. Chem. Lett., 2002, 12, 205-208.
[59] Urbahns, K.; Härter, M.; Vaupel, A.; Albers, M.; Schmidt, D.; Brüggemeier, U.; Stelte-Ludwig, B.; Gerdes, C.; Tsujishita, H. Biphenyls as potent vitronectin receptor antagonists. Part 2: biphe-nylalanine ureas. Bioorg. Med. Chem. Lett., 2003, 13, 1071-1074.
[60] Urbahns, K.; Härter, M.; Albers, M.; Schmidt, D.; Stelte-Ludwig, B.; Brüggemeier, U.; Vaupel, A.; Keldenich, J.; Lustig, K.; Tsu-jishita, H.; Gerdes, C. Biphenyls as potent vitronectin receptor an-tagonists. Part 3: Squaric acid amides. Bioorg. Med. Chem. Lett.,
2007, 17, 6151-6154. [61] Carron, C.P.; Meyer, D.M.; Pegg, J.A.; Engleman, V.W.; Nickols,
M.A.; Settle, S.L.; Westlin, W.F.; Ruminski, P.G.; Nickols, G.A. Peptidomimetic antagonist of the integrin (v) 3 inhibits leydig cell tumor growth and the development of hypercalcemia of malig-nancy. Cancer Res., 1998, 58, 1930-1935.
[62] Penning, T.D.; Russell, M.A.; Chen, B.B.; Chen, H.Y.; Desai, B.N.; Docter, S.H.; Edwards, D.J.; Gesicki, G.J.; Liang, C.D.; Malecha, J.W.; Yu, S.S.; Engleman, V.W.; Freeman, S.K.; Han-neke, M.L.; Shannon, K.E.; Westlin, M.M.; Nickols, G.A. Synthe-sis of cinnamic acids and related isosteres as potent and selective
v 3 receptor antagonists. Bioorg. Med. Chem. Lett., 2004, 14,
1471-1476. [63] Boys, M.L.; Schretzman, L.A.; Chandrakumar, N.S.; Tollefson,
M.B.; Mohler, S.B.; Downs, V.L.; Penning, T.D.; Russell, M.A.; Wendt, J.A.; Chen, B.B.; Stenmark, H.G.; Wu, H.; Spangler, D.P.; Clare, M.; Desai, B.N.; Khanna, I.K.; Nguyen, M.N.; Duffin, T.; Engleman, V.W.; Finn, M.B.; Freeman, S.K.; Hanneke, M.L.; Keene, J.L.; Klover, J.A.; Nickols, G.A.; Nickols, M.A.; Stein-inger, C.N.; Westlin, M.; Westlin, W.; Yu, Y.X.; Wang, Y.; Dalton, C.R.; Norring, S.A. Convergent, parallel synthesis of a series of -substituted 1,2,4-oxadiazole butanoic acids as potent and selective
v 3 receptor antagonists. Bioorg. Med. Chem. Lett., 2006, 16, 839-844.
[64] Wendt, J.A.; Wu, H.; Stenmark, H.G.; Boys, M.L.; Downs, V.L.; Penning, T.D.; Chen, B.B.; Wang, Y.; Duffin, T.; Finn, M.B.; Keene, J.L.; Engleman, V.W.; Freeman, S.K.; Hanneke, M.L.; Shannon, K.E.; Nickols, M.A.; Steininger, C.N.; Westlin, M.; Klover, J.A.; Westlin, W.; Nickols, G.A.; Russell, M.A. Synthesis of 2,5-thiazole butanoic acids as potent and selective v 3 integrin receptor antagonists with improved oral pharmacokinetic proper-ties. Bioorg. Med. Chem. Lett., 2006, 16, 845-849.
[65] Penning, T.D.; Khilevich, A.; Chen, B.B.; Russell, M.A.; Boys, M.L.; Wang, Y.; Duffin, T.; Engleman, V.W.; Finn, M.B.; Free-man, S.K.; Hanneke, M.L.; Keene, J.L.; Klover, J.A.; Nickols, G.A.; Nickols, M.A.; Rader, R.K.; Settle, S.L.; Shannon, K.E.; Steininger, C.N.; Westlin, M.M.; Westlin, W.F. Synthesis of pyra-zoles and isoxazoles as potent alpha(v)beta3 receptor antagonists. Bioorg. Med. Chem. Lett., 2006, 16, 3156-3161.
[66] Rogers, T.E.; Ruminski, P.G.; Rico, J.G.; Lu, H.-F.; Colson, P.J.; Awasthi, A.K.; Nagarajan, S.R.; Devadas, B.; Malecha, J.W.; Chandrakumar, N.S.; Nickols, G.A.; Westlin, W.F.; Meyer, D.M.; Keene, J.L.; Settle, S.L.; Carron, C.P.; Engleman, V.W.; Freeman, S.K.; Shannon, K.E.; Knoerzer, D.L.; Westlin, M.M.; Nickols, M.A. In Abstracts of Papers, 221st ACS National Meeting, San Di-
ego, 2001; MEDI-225. [67] Nagarajan, S.R.; Devadas, B.; Malecha, J.W.; Lu, H.F.; Ruminski,
P.G.; Rico, J.G.; Rogers, T.E.; Marrufo, L.D.; Collins, J.T.; Kleine, H.P.; Lantz, M.K.; Zhu, J.; Green, N.F.; Russell, M.A.; Landis, B.H.; Miller, L.M.; Meyer, D.M.; Duffin, T.D.; Engleman, V.W.;
Finn, M.B.; Freeman, S.K.; Griggs, D.W.; Williams, M.L.; Nick-ols, M.A.; Pegg, J.A.; Shannon, K.E.; Steininger, C.; Westlin, M.M.; Nickols, G.A. Discovery of a potent and selective alpha v beta 3 integrin antagonist with strong inhibitory activity against neointima formation in rat balloon injury model. Bioorg. Med.
Chem. Lett., 2004, 14, 2567-2570. [68] Nagarajan, S.R.; Lu, H.F.; Gasiecki, A.F.; Khanna, I.K.; Parikh,
M.D.; Desai, B.N.; Rogers, T.E.; Clare, M.; Chen, B.B.; Russell, M.A.; Keene, J.L.; Duffin, T.; Engleman, V.W.; Finn, M.B.; Free-man, S.K.; Klover, J.A.; Nickols, G.A.; Nickols, M.A.; Shannon, K.E.; Steininger, C.A.; Westlin, W.F.; Westlin, M.M.; Williams, M.L. Discovery of +(2-{4-[2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethoxy]phenyl}-cyclopropyl)acetic acid as potent and selective alphavbeta3 inhibitor: design, synthesis, and optimization. Bioorg.
Med. Chem., 2007, 15, 3390-3412. [69] Coleman, P.J.; Askew, B.C.; Hutchinson, J.H.; Whitman, D.B.;
Perkins, J.J.; Hartman, G.D.; Rodan, G.A.; Leu, C.T.; Prueksarita-nont, T.; Fernandez-Metzler, C.; Merkle, K.M.; Lynch, R.; Lynch, J.J.; Rodan, S.B.; Duggan, M.E. Non-peptide alpha(v)beta(3) an-tagonists. Part 4: potent and orally bioavailable chain-shortened RGD mimetics. Bioorg. Med. Chem. Lett., 2002, 12, 2463-2465.
[70] Coleman, P.J.; Brashear, K.M.; Hunt, C.A.; Hoffman, W.F.; Hutchinson, J.H.; Breslin, M.J.; McVean, C.A.; Askew, B.C.; Hartman, G.D.; Rodan, S.B.; Rodan, G.A.; Leu, C.T.; Prueksarita-nont, T.; Fernandez-Metzler, C.; Ma, B.; Libby, L.A.; Merkle, K.M.; Stump, G. L.; Wallace, A.A.; Lynch, J.J.; Lynch, R.; Dug-gan, M.E. Non-peptide alpha(v)beta(3) antagonists. Part 3: identifi-cation of potent RGD mimetics incorporating novel beta-amino ac-ids as aspartic acid replacements. Bioorg. Med. Chem. Lett., 2002, 12, 31-34.
[71] Hutchinson, J.H.; Halczenko, W.; Brashear, K.M.; Breslin, M.J.; Coleman, P.J.; Duong, le T.; Fernandez-Metzler, C.; Gentile, M.A.; Fisher, J.E.; Hartman, G.D.; Huff, J.R.; Kimmel, D.B.; Leu, C.T.; Meissner, R.S.; Merkle, K.; Nagy, R.; Pennypacker, B.; Perkins, J.J.; Prueksaritanont, T.; Rodan, G.A.; Varga, S.L.; Wesolowski, G.A.; Zartman, A.E.; Rodan, S.B.; Duggan, M.E. Nonpeptide al-phavbeta3 antagonists. 8. In vitro and in vivo evaluation of a potent alphavbeta3 antagonist for the prevention and treatment of osteopo-rosis. J. Med. Chem., 2003, 46, 4790-4798.
[72] Wang, J.; Breslin, M.J.; Coleman, P.J.; Duggan, M.E.; Hunt, C.A.; Hutchinson, J.H.; Leu, C.-T.; Rodan, S.B.; Rodan, G.A.; Hartman, G.D. Non-peptide alpha v beta 3 antagonists. Part 7: 3-Substituted tetrahydro-naphthyridine derivatives. Bioorg. Med. Chem. Lett.,
2004, 14, 1049-1052. [73] Brashear, K.M.; Hunt, C.A.; Kucer, B.T.; Duggan, M.E.; Hartman,
G.D.; Rodan, G.A.; Rodan, S.B.; Leu, C.-T.; Prueksaritanont, T.; Fernandez-Metzler, C.; Barrish, A.; Homnick, C.F.; Hutchinson, J.H.; Coleman, P.J. Non-peptide alpha(v)beta(3) antagonists. Part 5: identification of potent RGD mimetics incorporating 2-aryl beta-amino acids as aspartic acid replacements. Bioorg. Med. Chem.
Lett., 2002, 12, 3483-3486. [74] Coleman, P.J.; Brashear, K.M.; Askew, B.C.; Hutchinson, J.H.;
McVean, C.A.; Duong le, T.; Feuston, B.P.; Fernandez-Metzler, C.; Gentile, M.A.; Hartman, G.D.; Kimmel, D.B.; Leu, C.T.; Lipfert, L.; Merkle, K.; Pennypacker, B.; Prueksaritanont, T.; Rodan, G.A.; Wesolowski, G.A.; Rodan, S.B.; Duggan, M.E. Nonpeptide al-phavbeta3 antagonists. Part 11: discovery and preclinical evalua-tion of potent alphavbeta3 antagonists for the prevention and treat-ment of osteoporosis. J. Med. Chem., 2004, 47, 4829-4837.
[75] Holtkötter, O.; Nieswandt, B.; Smyth, N.; Müller, W.; Hafner, M.; Schulte, V.; Krieg, T.; Eckes, B. Integrin alpha 2-deficient mice develop normally, are fertile, but display partially defective platelet interaction with collagen. J. Biol. Chem., 2002, 277, 10789-10794.
[76] Guo, W.; Giancotti, F.G. Integrin signalling during tumour pro-gression. Nat. Rev. Mol. Cell Biol., 2004, 5, 816-826.
[77] Hagmann, W.K.; Durette, P.L.; Lanza, T.; Kevin, N.J.; de Laszlo, S.E.; Kopka, I.E.; Young, D.; Magriotis, P.A.; Li, B.; Lin, L. S.; Yang, G.; Kamenecka, T.; Chang, L.L.; Wilson, J.; MacCoss, M.; Mills, S.G.; Van Riper, G.; McCauley, E.; Egger, L.A.; Kidambi, U.; Lyons, K.; Vincent, S.; Stearns, R.; Colletti, A.; Teffera, J.; Tong, S.; Fenyk-Melody, J.; Owens, K.; Levorse, D.; Kim, P.; Schmidt, J.A.; Mumford, R.A. The discovery of sulfonylated dipeptides as potent VLA-4 antagonists. Bioorg. Med. Chem. Lett., 2001, 11, 2709-2713.

Small Molecule Antagonists of Integrin Receptors Current Medicinal Chemistry, 2010 Vol. 17, No. 22 2391
[78] Xue, C.-B.; Rafalski, M.; Roderick, J.; Eyermann, C.J.; Mousa, S.; Olson R.E.; DeGrado, W.F. Design, synthesis and in vitro activities of a series of benzimidazole/benzoxazole glycoprotein IIb/IIIa in-hibitors. Bioorg. Med. Chem. Lett., 1996, 6, 339-344.
[79] Choi, S.; Vilaire, G.; Marcinkiewicz, C.; Winkler, J.D.; Bennett, J.S.; DeGrado, W.F. Small molecule inhibitors of integrin al-pha2beta1. J. Med. Chem., 2007, 50, 5457-5462.
[80] Francis, S.E.; Goh, K.L.; Hodivala-Dilke, K.; Bader, B.L.; Stark, M.; Davidson, D.; Hynes, R.O. Central roles of alpha5beta1 in-tegrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arterioscler. Thromb. Vasc. Biol., 2002, 22, 927-933.
[81] Kim, S.; Bell, K.; Mousa, S.A.; Varner, J.A. Regulation of angio-genesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am. J. Pathol., 2000, 156, 1345-1362.
[82] Brower, V. Tumor angiogenesis--new drugs on the block. Nat.
Biotechnol., 1999, 17, 963-968. [83] Marinelli, L.; Meyer, A.; Heckmann, D.; Lavecchia, A.; Novellino,
E.; Kessler, H. Ligand binding analysis for human alpha5beta1 in-tegrin: strategies for designing new alpha5beta1 integrin antago-nists. J. Med. Chem., 2005, 48, 4204-4207.
[84] Stragies, R.; Osterkamp, F.; Zischinsky, G.; Vossmeyer, D.; Kalk-hof, H.; Reimer, U.; Zahn, G. Design and synthesis of a new class of selective integrin alpha5beta1 antagonists. J. Med. Chem., 2007, 50, 3786-3794.
[85] Heckmann, D.; Meyer, A.; Marinelli, L.; Zahn, G.; Stragies, R.; Kessler, H. Probing integrin selectivity: rational design of highly active and selective ligands for the alpha5beta1 and alphavbeta3 in-tegrin receptor. Angew. Chem. Int. Ed. Engl., 2007, 46, 3571-3574.
[86] Harris, E.S.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. The leukocyte integrins. J. Biol. Chem., 2000, 275, 23409-23412.
[87] Gottlieb, A.B.; Bos, J.D. Recombinantly engineered human pro-teins: transforming the treatment of psoriasis. Clin. Immunol., 2002, 105, 105-116.
[88] Shimaoka, M.; Salas, A.; Yang, W.; Weitz-Schmidt, G.; Springer, T.A. Small molecule integrin antagonists that bind to the beta2 subunit I-like domain and activate signals in one direction and block them in the other. Immunity, 2003, 19, 391-402.
[89] Kallen, J.; Welzenbach, K.; Ramage, P.; Geyl, D.; Kriwacki, R.; Legge, G.; Cottens, S.; Weitz-Schmidt, G.; Hommel, U. Structural basis for LFA-1 inhibition upon lovastatin binding to the CD11a I-domain. J. Mol. Biol., 1999, 292, 1-9.
[90] Burdick, D.J.; Gadek, T.R.; Mcdowell, R.S.; Marsters, J.; Oare, D.A.; Reynolds, M.E.; Stanley, M.S. Weese, K.J. Antagonists for treatment of CD11/CD18 adhesion receptor mediated disorders. PCT Int. Appl. WO 9949856, October 7, 1999.
[91] Burdick, D.J.; Gadek, T.R.; Marsters, J.; Oare, D.A.; Reynolds, M.E.; Stanley, M.S. LFA-1 antagonist compounds. PCT Int. Appl. WO 02059114, August 01, 2002.
[92] Fotouhi, N.; Gillespie, P.; Guthrie, R.; Pietranico-Cole, S.; Yun, W. Diaminopropionic acid derivatives. PCT Int. Appl. WO 0021920, April 20, 2000.
[93] Fotouhi, N.; Gillespie, P.; Guthrie, R.; Pietranico-Cole, S.; Yun, W. Dehydroamino acids. PCT Int. Appl. WO 0158853, August 16, 2001.
[94] Yang, W.; Carman, C.V.; Kim, M.; Salas, A.; Shimaoka, M.; Springer, T.A. A Small Molecule Agonist of an Integrin, L 2. J.
Biol. Chem., 2006, 281, 37904-37912. [95] Yusuf-Makagiansar, H.; Anderson, M.E.; Yakovleva, T.V.;
Murray, J.S.; Siahaan, T.J. Inhibition of LFA-1/ICAM-1 and VLA-4/VCAM-1 as a therapeutic approach to inflammation and autoim-mune diseases. Med. Res. Rev., 2002, 22, 146-167.
[96] von Andrian, U.H.; Engelhardt, B. 4 integrins as therapeutic targets in autoimmune disease. N. Engl. J. Med., 2003, 348, 68-72.
[97] Miller, D.H.; Khan, O.A.; Sheremata, W.A.; Blumhardt, L.D.; Rice, G.P.; Libonati, M.A.; Willmer-Hulme, A.J.; Dalton, C.M.; Miszkiel, K.A.; O'Connor, P.W. A controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med., 2003, 348, 15-23.
[98] Tilley, J.W.; Sidduri, A. VLA-4 antagonists. Drugs Future, 2001, 26, 985-998.
[99] Yang, G.X.; Hagmann, W.K. VLA-4 antagonists: potent inhibitors of lymphocyte migration. Med. Res. Rev., 2003, 23, 369-392.
[100] Jackson, D.Y. Alpha 4 integrin antagonists. Curr. Pharm. Des., 2002, 8, 1229-1253.
[101] Porter, J.R.; Archibald, S.C.; Brown, J.A.; Childs, K.; Critchley, D.; Head, J.C.; Hutchinson, B.; Parton, T.A.; Robinson, M.K.; Shock, A.; Warrellow, G.J.; Zomaya, A. Discovery and evaluation of N-(triazin-1,3,5-yl) phenylalanine derivatives as VLA-4 integrin antagonists. Bioorg. Med. Chem. Lett., 2002, 12, 1591-1594.
[102] Porter, J.R.; Archibald, S.C.; Brown, J.A.; Childs, K.; Critchley, D.; Head, J.C.; Parton, T.A.; Robinson, M.K.; Shock, A.; Taylor, R.J.; Warrellow, G.J. Dehydrophenylalanine derivatives as VLA-4 integrin antagonists. Bioorg. Med. Chem. Lett., 2003, 13, 805-808.
[103] Okigami, H.; Takeshita, K.; Tajimi, M.; Komura, H.; Albers, M.; Lehmann, T.E.; Rölle, T.; Bacon, K.B. Inhibition of eosinophilia in
vivo by a small molecule inhibitor of very late antigen (VLA)-4. Eur. J. Pharmacol., 2007, 559, 202-209.
[104] Vincent, A.M.; Cawley, J.C.; Burthem, J. Integrin function in chronic lymphocytic leukemia. Blood, 1996, 87, 4780-4788.
[105] Peng, L.; Liu, R.; Marik, J.; Wang, X.; Takada, Y.; Lam, K.S. Combinatorial chemistry identifies high-affinity peptidomimetics against alpha4beta1 integrin for in vivo tumor imaging. Nat. Chem.
Biol., 2006, 2, 381-389. [106] Carpenter, R.D.; Andrei, M.; Lau E.Y.; Lightstone, F.C.; Liu, R.;
Lam, K.S.; Kurth, M.J. Highly Potent, Water Soluble Benzimida-zole Antagonist for Activated 4 1 Integrin. J. Med. Chem., 2007, 50, 5863-5867.
[107] Feagan, B.G.; Greenberg, G.R.; Wild, G.; Fedorak, R.N.; Paré, P.; McDonald, J.W.; Dubé, R.; Cohen, A.; Steinhart, A.H.; Landau, S.; Aguzzi, R.A.; Fox, I.H.; Vandervoort, M.K. Treatment of ulcera-tive colitis with a humanized antibody to the alpha4beta7 integrin. N. Engl. J. Med., 2005, 352, 2499-2507.
[108] Gong, Y.; Barbay, J.K.; Dyatkin, A.B.; Miskowski, T.A.; Kimball, E.S.; Prouty, S.M.; Fisher, M.C.; Santulli, R.J.; Schneider, C.R.; Wallace, N.H.; Ballentine, S.A.; Hageman, W.E.; Masucci, J.A.; Maryanoff, B.E.; Damiano, B.P.; Andrade-Gordon, P.; Hlasta, D.J.; Hornby, P.J.; He, W. Synthesis and biological evaluation of novel pyridazinone-based 4 integrin receptor antagonists. J. Med.
Chem., 2006, 49, 3402-3411. [109] Muro, F.; Iimura, S.; Yoneda, Y.; Chiba, J.; Watanabe, T.; Setogu-
chi, M.; Iigou, Y.; Takayama, G.; Yokoyama, M.; Takashi, T.; Na-kayama, A.; Machinaga, N. Identification of 4-[1-[3-chloro-4-[N’-(5-fluoro-2-methylphenyl)ureido]phenylacetyl]-(4S)-fluoro-(2S)-pyrrolidinylmethoxy]benzoic acid as a potent, orally active VLA-4 antagonist. Bioorg. Med. Chem., 2008, 16, 9991-10000.
[110] Gutteridge, C.E.; de Laszlo, S.E.; Kamenecka, T.M.; McCauley, E.; van Riper, G.; Mumford, R.A.; Kidambi, U.; Egger, L.A.; Tong, S.; Hagmann, W.K. N-(3-Phenylsulfonyl-3-piperidinoyl)-phenylal-anine derivatives as potent, selective VLA-4 antagonists. Bioorg.
Med. Chem. Lett., 2003, 13, 885-890. [111] Fujita, M.; Seki, T.; Inada, H.; Shimizu, K.; Takahama, A.; Sano,
T. Novel Agents Combining Platelet Activating Factor (PAF) Re-ceptor Antagonist with Thromboxane Synthase Inhibitor (TxSI). Bioorg. Med. Chem. Lett., 2002, 12, 771-774.
[112] Rotondo, S.; Dell'Elba, G.; Manarini, S.; Cerletti, C.; Evangelista, V. The lipoxygenase–cyclooxygenase inhibitor licofelone prevents thromboxane A2-mediated cardiovascular derangement triggered by the inflammatory peptide fMLP in the rabbit. Eur. J. Pharm., 2006, 546, 95-101.
[113] Tam, S.H.; Sassoli, P.M.; Jordan, R.E.; Nakada, M.T. Abciximab (ReoPro, Chimeric 7E3 Fab) Demonstrates Equivalent Affinity and Functional Blockade of Glycoprotein IIb/IIIa and vß3 Integrins. Circulation, 1998, 98, 1085-1091.
[114] Kubota, D.; Ishikawa, M.; Yamamoto, M.; Murakami, S.; Hachisu, M.; Katano, K.; Ajito, K. Tricyclic pharmacophore-based mole-cules as novel integrin v 3 antagonists. Part 1: Design and synthe-sis of a lead compound exhibiting v 3/ IIb 3 dual antagonistic ac-tivity. Bioorg. Med. Chem., 2006, 14, 2089-2108.
[115] Ishikawa, M.; Kubota, D.; Yamamoto, M.; Kuroda, C.; Iguchi, M.; Koyanagi, A.; Murakami, S.; Ajito, K. Tricyclic pharmacophore-based molecules as novel integrin v 3 antagonists. Part 2: Synthe-sis of potent v 3/ IIb 3 dual antagonists. Bioorg. Med. Chem.,
2006, 14, 2109-2130. [116] Ishikawa, M.; Hiraiwa, Y.; Kubota, D.; Tsushima, M.; Watanabe,
T.; Murakami, S.; Ouchi, S.; Ajito, K. Tricyclic pharmacophore-based molecules as novel integrin v 3 antagonists. Part III: Syn-

2392 Current Medicinal Chemistry, 2010 Vol. 17, No. 22 Perdih and Dolenc
thesis of potent antagonists with v 3/ IIb 3 dual activity and im-proved water solubility. Bioorg. Med. Chem., 2006, 14, 2131-2150.
[117] Kubota, D.; Ishikawa, M.; Ishikawa, M.; Yahata, N.; Murakami, S.; Fujishima, K.; Kitakaze, M.; Ajito, K. Tricyclic pharmacophore-based molecules as novel integrin v 3 antagonists. Part IV: Pre-liminary control of v 3 selectivity by meta-oriented substitution Bioorg. Med. Chem., 2006, 14, 4158-4181.
[118] Max, R.; Gerritsen, R.R.; Nooijen, P.T.; Goodman, S.L.; Sutter, A.; Keilholz, U.; Ruiter, D.J.; De Waal, R.M. Immunohistochemical analysis of integrin v 3 expression on tumor-associated vessels of human carcinomas. Int. J. Cancer., 1997, 71, 320-324.
[119] Henry, C.; Moitessier, N.; Chapleur, Y. Vitronectin receptor - V 3 integrin- antagonists: chemical and structural requirements for ac-tivity and selectivity. Mini Rev. Med. Chem., 2002, 2, 531-542.
[120] Marinelli, L.; Gottschalk, K.E.; Meyer, A.; Novellino, E.; Kessler, H. Human integrin v 5: Homology modeling and ligand binding. J. Med. Chem., 2004, 47, 4166-4177.
[121] Boturyn, D.; Dumy, P. A convenient access to V 3/ V 5 integrin ligand conjugates: regioselective solid-phase functionalisation of an RGD based peptide. Tetrahedron Lett., 2001, 42, 2787-2790.
[122] Perdih, A.; Kikelj, D. The Application of Freidinger Lactams and their Analogs in the Design of Conformationally Constrained Pep-tidomimetics. Curr. Med. Chem., 2006, 13, 1525-1556.
[123] Kumar, S.; Wang, Q.; Sasaki, N.A. Synthesis of conformationally constrained analogues of RGD tripeptide. Tetrahedron, 2007, 63, 2084-2094.
[124] 124 Perron-Sierra, F.; Saint Dizier, D.; Bertrand, M.; Genton, A.; Tucker, G.C.; Casara, P. Substituted benzocyloheptenes as potent and selective v integrin antagonists. Bioorg. Med. Chem. Lett.
2002, 12, 3291-3296. [125] Marugán, J.J.; Manthey, C.; Anaclerio, B.; Lafrance, L.; Lu, T.;
Markotan, T.; Leonard, K.A.; Crysler, C.; Eisennagel, S.; Das-gupta, M.; Tomczuk, B. Design, synthesis, and biological evalua-tion of Novel Potent and Selective v 3/ v 5 Integrin dual inhibi-tors with improved bioavailability. Selection of the molecular core. J. Med. Chem., 2005, 48, 926-934.
[126] Raboisson, P.; Manthey, C.L.; Chaikin, M.; Lattanze, J.; Crysler, C.; Leonard, K.; Pan, W.; Tomczuk, B.E.; Marugán, J. J. Novel po-tent and selective v 3/ v 5 integrin dual antagonists with reduced binding affinity for human serum albumin. Eur. J. Med. Chem.,
2006, 41, 847-861. [127] Raboisson, P.; Desjarlais, R.L.; Reed, R.; Lattanze, J.; Chaikin, M.;
Manthey, C.L.; Tomczuk, B.E.; Marugán, J.J. Identification of novel short chain 4-substituted indoles as potent v 3 antagonist us-
ing structure-based drug design. Eur. J. Med. Chem., 2007, 42, 334-343.
[128] Letourneau, J.J.; Liu, J.; Ohlmeyer, M.H.; Riviello, C.; Rong, Y.; Li, H.; Appell, K.C.; Bansal, S.; Jacob, B.; Wong, A.; Webb, M.L.. Synthesis and initial evaluation of novel, non-peptidic antagonists of the v-integrins v 3 and v 5. Bioorg. Med. Chem. Lett., 2009, 19, 353-355.
[129] Ikari, Y.; Yee, K.O.; Schwartz, S.M. Role of 5 1 and v 3 in-tegrins on smooth muscle cell spreading and migration in fibrin gels. Thromb. Haemost., 2000, 84, 701-705.
[130] Bubenik, M.; Meerovitch, K.; Bergeron, F.; Attardo, G.; Chan, L. Thiophene-based vitronectin receptor antagonists. Bioorg. Med.
Chem. Lett., 2003, 13, 503-506. [131] Meerovitch, K.; Bergeron, F.; Leblond, L.; Grouix, B.; Poirier, C.;
Bubenik, M.; Chan, L.; Gourdeau, H.; Bowlin, T.; Attardo, G. A novel RGD antagonist that targets both v 3 and 5 1 induces apoptosis of angiogenic endothelial cells on type I collagen. Vasc.
Pharm., 2003, 40, 77-89. [132] Smallheer, J.M.; Weigelt, C.A.; Woerner, F.J.; Wells, J.S.; Dane-
ker, W.F.; Mousa, S. A.; Wexler, R.R.; Jadhav, P.K. Synthesis and biological evaluation of nonpeptide integrin antagonists containing spirocyclic scaffolds. Bioorg. Med. Chem. Lett., 2004, 14, 383-387.
[133] Benfatti, F.; Cardillo, G.; Fabbroni, S.; Galzerano, P.; Gentilucci, L.; Juris, R.; Tolomelli, A.; Baiula, M.; Spartà, A.; Spampinato, S. Synthesis and biological evaluation of non-peptide v 3/ 5 1 in-tegrin dual antagonists containing 5,6-dihydropyridin-2-one scaf-folds. Bioorg. Med. Chem., 2007, 15, 7380-7390.
[134] Lin, L.S.; Lanza, T.; McCauley, E.; Van Riper, G.; Kidambi, U.; Cao, J.; Egger, L.A.; Mumford, R.A.; Schmidt, J.A.; MacCoss, M.; Hagmann, W.K. Specific and dual antagonists of 4 1 and 4 7 in-tegrins. Bioorg. Med. Chem. Lett., 2002, 12, 133-136.
[135] Dyatkin, A.B.; Gong, Y.; Miskowski, T.A.; Kimball, E.S.; Prouty, S.M.; Fisher, M.C.; Santulli, R.J.; Schneider, C.R.; Wallace, N.H.; Hornby, P.J.; Diamond, C.; Kinney, W.A.; Maryanoff, B.E.; Dami-ano, B.P.; He, W. Aza-bicyclic amino acid carboxamides as
4 1/ 4 7 integrin receptor antagonists. Bioorg. Med. Chem., 2005, 13, 6693-6702.
[136] Gong, Y.; Kent Barbay, J.; Kimball, E.S.; Santulli, R.J.; Carolyn Fisher, M., Dyatkin A.B.; Miskowski, T.A.; Hornby, P.J.; He, W. Synthesis and SAR of pyridazinone-substituted phenylalanine am-ide 4 integrin antagonists. Bioorg. Med. Chem. Lett., 2008, 18, 1331-1335.
Received: February 06, 2010 Revised: May 07, 2010 Accepted: May 09, 2010