smad3-dependent and -independent pathways are involved in peritoneal membrane injury
TRANSCRIPT

Smad3-dependent and -independent pathwaysare involved in peritoneal membrane injuryPranali Patel1, Yoshimi Sekiguchi1, Kook-Hwan Oh2, Sarah E. Patterson1, Martin R.J. Kolb1
and Peter J. Margetts1
1Department of Medicine, McMaster University, Hamilton, Canada and 2Department of Internal Medicine, Seoul NationalUniversity Hospital, Seoul, Korea
Transition of peritoneal mesothelial cells to a mesenchymal
phenotype plays an integral role in the angiogenic
and fibrotic changes seen in the peritoneum of patients
receiving long-term peritoneal dialysis. While signaling by
transforming growth factor (TGF)-b through Smad proteins
likely causes these changes, it is possible that non-Smad
pathways may also play a role. Here, we found that
Smad3-deficient mice were protected from peritoneal fibrosis
and angiogenesis caused by adenovirus-mediated gene
transfer of active TGF-b1 to mesothelial cells; however,
mesothelial transition occurred in this setting, suggesting
involvement of non-Smad mechanisms. The phosphatidyl
inositol 3 kinase (PI3K) target, Akt, was upregulated in both
Smad-deficient and wild-type mice after exposure to TGF-b1.
In vivo inhibition of the mammalian target of rapamycin
(mTOR) by rapamycin completely abrogated the transition
response in Smad3-deficient but not in wild-type mice.
Rapamycin blocked nuclear localization of b-catenin
independent of glycogen synthase kinase 3b activity.
Further, in Smad3-deficient mice rapamycin reduced
the expression of a-smooth muscle actin, which is an
epithelial-to-mesenchymal transition-associated gene.
Hence, we conclude that TGF-b1 causes peritoneal injury
through Smad-dependent and Smad-independent pathways;
the latter involves redundant mechanisms inhibited by
rapamycin, suggesting that suppression of both pathways
may be necessary to abrogate mesothelial transition.
Kidney International (2010) 77, 319–328; doi:10.1038/ki.2009.436;
published online 2 December 2009
KEYWORDS: adenovirus; angiogenesis; fibrosis; mTOR; peritoneum; rapamycin
During long-term peritoneal dialysis treatment, the peri-toneal membrane undergoes structural and functionalalterations. The structural alterations include fibrosis andangiogenesis of the peritoneal tissue.1 The functional changesresults in an increased rate of solute transport across theperitoneal membrane and ultrafiltration dysfunction. Thesechanges are associated with adverse outcome, techniquefailure, and death.2 We have previously shown that trans-forming growth factor (TGF)-b1 induces transition ofthe lining peritoneal mesothelial cells to myofibroblasts.3
This cellular transition is observed in patients in peritonealdialysis patients4 and is an integral component of bothfibrosis and angiogenesis.5
TGF-b binds to its receptors to activate the Smad familyof signaling proteins.6 Upon ligand binding and TGF-b1receptor activation, phosphorylation of receptor-regulatedSmad2 and Smad3 occurs, which leads to the formation ofcomplexes with Smad4. These complexes then translocateinto the nucleus, in which they regulate expression of TGF-b-responsive genes.
TGF-b1 signaling through Smad3 seems to be a crucialelement in the signal transduction pathways involved inwound healing and fibrosis. Smad3�/� mice were protectedagainst radiation-induced fibrosis of the skin,7 renal inter-stitial fibrosis,8 and TGF-b1-induced pulmonary fibrosis.9
TGF-b1 also induces epithelial-to-mesenchymal transition(EMT), a process involved in many physiological andpathological processes.10 EMT seems to be an integralcomponent of injury response and tissue fibrosis in a varietyof organs, and inhibition of EMT ameliorates fibrosis;11
however, it is uncertain whether EMT is necessary for organfibrosis.12 EMT involves a series of distinct processes,including loss of cell polarity, loss of intercellular adhesionand epithelial markers, cytoskeletal reorganization andexpression of mesenchymal markers, extracellular matrixdegradation, and cellular mobilization.13 The role of Smad3signaling in TGF-b1-induced EMT is controversial. Somestudies have invoked a requirement for Smad3 signaling inthe induction of EMT,8,14,15 whereas others have found amodified EMT to occur in the absences of Smad3.16,17
Clearly, other non-Smad pathways are involved in TGF-b1-induced EMT.18,19
http://www.kidney-international.org o r i g i n a l a r t i c l e
& 2010 International Society of Nephrology
Received 8 June 2009; revised 19 August 2009; accepted 22 September
2009; published online 2 December 2009
Correspondence: Peter J. Margetts, Department of Medicine, McMaster
University, Division of Nephrology, St Joseph’s Hospital, 50 Charlton Avenue
East, Hamilton, Ontario, Canada L8P 4A6.
E-mail: [email protected]
Kidney International (2010) 77, 319–328 319

Among the potential non-Smad mechanisms involvedin EMT, the phosphatidyl inositol 3 kinase (PI3K)/Akt andwnt/b-catenin pathways have been extensively studied,20,21
and we have focused on these pathways in our experimentalmodel. PI3K is activated by several growth factors includingTGF-b.22 PI3K activates Akt through phosphorylation atserine 473 (p-Akt). p-Akt has multiple actions includingupregulation of the mammalian target of rapamycin(mTOR).23 Through the downstream kinase p70 S6, mTORmodulates mRNA translation and protein expression and isinvolved in cell growth and proliferation.24 Wnt signalingleads to the downstream suppression of glycogen synthasekinase (GSK)-3b and accumulation of b-catenin thattranslocates to the nucleus and exerts an effect as atranscriptional regulator.25
RESULTSSmad3�/� mice are protected from AdTGF-b1-inducedfibrosis and angiogenesis
Smad3þ /þ and Smad3�/� mice received an intraperitonealinjection of an adenovirus expressing TGF-b1 (AdTGF-b1),vascular endothelial growth factor (AdVEGF), or control adeno-virus (AdDL). AdDL did not induce any observable change inthe peritoneum at day 7 and there were no differences bet-ween Smad3þ /þ (Figure 1a) and Smad3�/� mice (Figure 1b).We observed significant histological changes in the parietalperitoneal tissue of Smad3þ /þ mice treated with TGF-b1(Figure 1c). The obvious changes included a fibroproliferativeresponse with increased submesothelial thickness, increasedcellularity of the submesothelium, and angiogenesis. Quantita-tive histology of von-Willebrand factor-stained sections(Figure 2) showed that maximum submesothelial thickness inSmad3þ /þ animals occurred at day 14 and started to decreaseby day 21 (Figure 2a). Submesothelial vascularization also in-creased in Smad3þ /þ mice treated with AdTGF-b1 (Figure 2b).These changes were maximal at day 14 and declined there-after. In contrast, Smad3�/� mice treated with TGF-b1 showedonly minor histological changes in the peritoneal membrane(Figure 1d) with no significant submesothelial thickening orangiogenic response (Figure 2a and b).
To show that the angiogenic difference between Smad3þ /þ
and Smad3�/� mice was TGF-b1 specific, we also assessed theresponse to a different angiogenic signal. Both Smad3þ /þ andSmad3�/� mice showed robust but transient peritonealangiogenesis in response to AdVEGF (Figures 1e, f, and 2b).
Smad3�/� mice do not show changes in fibrogenic andangiogenic gene expression after AdTGF-b1 infection
We confirmed that Smad3�/� mice do not develop fibrosis inresponse to TGF-b1 (Figure 2). Total birefringent area (red þgreen) from picrosirius red-stained sections indicates totalcollagen, whereas the green birefringent area is suggestive ofnew collagen deposition.26 There was a significant increase inthe amount of existing (Figure 2c) and newly depositedcollagen (Figure 2d) in Smad3þ /þ mice treated withAdTGF-b1. There was a minor increase in collagen deposi-
tion in Smad3�/� animals treated with AdTGF-b comparedwith control adenovirus-treated animals. This was significantat day 7 for total collagen only (Figure 2c).
We extracted RNA from the parietal peritoneal membrane,as has been previously described,3 and carried out quantitativereverse transcriptase-polymerase chain reaction for fibrosis-associated gene expression. There was a significant transientincrease in plasminogen activator inhibitor 1 gene expressionin Smad3þ /þ animals treated with AdTGF-b1 (Figure 2e).These mice also showed a progressive increase in VEGF geneexpression after infection with AdTGF-b1 (Figure 2f).Smad3�/� mice treated with AdTGF-b1 showed no significantincreases in either plasminogen activator inhibitor 1 or VEGF.
Smad3þ /þ and Smad3�/� mice develop EMT in responseto TGF-b1
Smad3þ /þ mice developed EMT as expected after over-expression of TGF-b1. In Figure 3, there is evidence of
Figure 1 | Histology of the anterior abdominal wall 7 daysafter intraperitoneal adenovirus administration. (a) Smad3þ /þ
and (b) Smad3�/� mice treated with control adenovirus (AdDL)show normal peritoneal histology with a mesothelial cell layer(thin arrow), a thin submesothelial layer (thick arrow) overlyingthe abdominal musculature. (c) Smad3þ /þ mice treated withadenovirus expressing transforming growth factor-b1 (AdTGF-b1)show a strong fibroproliferative response (arrow). (d) Smad3�/�
mice treated with AdTGF-b1 show a moderate proliferativeresponse without significant fibrosis or angiogenesis. (e) Smad3þ /þ
and (f) Smad3�/� mice treated with adenovirus expressingvascular endothelial growth factor (AdVEGF) show a similarangiogenic response (thick arrows). Masson’s trichrome stain,original magnification � 200.
320 Kidney International (2010) 77, 319–328
o r i g i n a l a r t i c l e P Patel et al.: Smad and peritoneal membrane injury

epithelial cells (cytokeratin positive) that co-expressa-smooth muscle actin (SMA). At 4 days after infection,these cells exist in a single superficial mesothelial celllayer (Figure 3a–c). Smad3�/� mice treated with AdTGF-b1showed similar but attenuated changes to those observedin the Smad3þ /þ animals (Figure 3d–f).
By day 7, cytokeratin-positive cells are observed in thesubmesothelial zone of Smad3þ /þ mice treated withAdTGF-b1 (Figure 4a–c, Supplementary Figure S2), bothdual and single labeled. This is suggestive of an invasivephenotype of these cells undergoing transition. Again, thesechanges were observed in the Smad3�/� mice but in anattenuated form (Figure 4d–f, Supplementary Figure S2).There were no changes suggestive of EMT observed in eitherSmad3þ /þ or Smad3�/� animals treated with the controladenovirus (Figures 3g–i and 4g–i).
We analyzed the expression of EMT-associated genes.We observed a significant and similar increase in a-SMA geneexpression in Smad3þ /þ animals treated with AdTGF-b1(Figure 5a), with no differences between the Smad3þ /þ andSmad3�/� mice. Snail, a zinc-finger regulatory protein impor-tant in EMT,27 was induced by TGF-b1 in Smad3þ /þ animals(Figure 5b). Snail mRNA was significantly higher in AdTGF-b1-treated Smad3þ /þ animals compared with Smad3�/�
animals. Despite this difference in SNAIL expression, weobserved a markedly lower level of E-cadherin protein thatwas similarly suppressed in both Smad3�/� and Smad3þ /þ
mice treated with AdTGF-b1 compared with controladenovirus-treated animals (Figure 5d).
EMT was quantified by counting the number of dual-labeled cells in the peritoneal tissue (Figure 5e) andby studying the amount of EMT-related cellular invasion(Figure 5f). EMT occurred, but at a significantly lowerfrequency in the Smad3�/� mice treated with AdTGF-b1compared with the Smad3þ /þ mice. Cellular invasion wasprevalent in the Smad3þ /þ mice, but was transient in theSmad3�/� mice treated with AdTGF-b1.
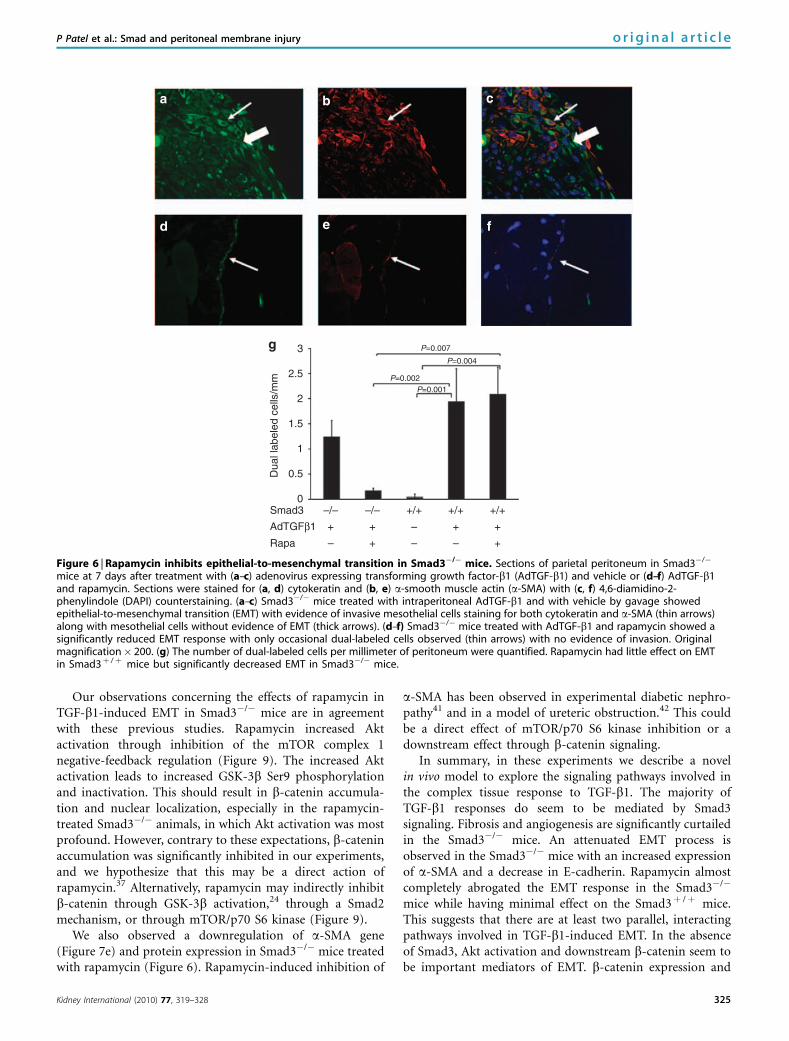
Rapamycin blocks EMT response in Smad3�/� mice
After transfer of TGF-b1 to the peritoneum of Smad3þ /þ
and Smad3�/� mice, we treated them with the mTORinhibitor rapamycin or vehicle by daily gavage. Rapamycinalmost completely blocked evidence of EMT response toTGF-b1 in the Smad3�/� mice (Figure 6). Invasive dual-labeled cells were observed in the Smad3�/� animals treatedwith AdTGF-b1 (Figure 6a–c), but only occasional dual-labeled cells were observed in the Smad3�/� animals treatedwith rapamycin. We quantified these cells (Figure 6g) andshowed that rapamycin significantly reduced the EMT res-ponse in Smad3�/�mice but had no effect on the Smad3þ /þ
mice. Animals treated with rapamycin and AdTGF-b1 hadsimilar peritoneal effluent levels of TGF-b1 compared tovehicle-treated animals (Supplementary Figure S1). Therewere very rare dual-labeled cells observed in the Smad3þ /þ
animals exposed to the control adenovirus. There was noobserved EMT-related submesothelial invasion in theSmad3þ /þ animals treated with AdDL or Smad3�/� micetreated with rapamycin.
Akt activity is increased by TGF-b1. To measure PI3K/Aktpathway activation, serine 473 phosphorylation of Akt wasassessed using western blot (Figure 7b). Overall, TGF-b1increased p-Akt concentration in the peritoneal tissue
25
20
15
10
5
0
454035302520151050
VE
GF
:GA
PD
H m
RN
A
14121086420
16
Sub
mes
othe
lial
thic
knes
s (m
icro
ns) P<0.004 P=0.004
Ves
sels
/mm
Per
cent
gre
en a
rea
Per
cent
red
+ g
reen
area
PA
I1:G
AP
DH
mR
NA
P<0.001
P=0.005
P=0.016
P=0.008 P=0.002
P<0.001
P<0.001
P=0.009
P=0.05
P=0.022
P=0.002
P=0.002
P=0.006
P=0.007
P=0.003
P=0.02
P=0.011P<0.001 P=0.005
P=0.027
6050403020100
908070605040302010
0
876543210
Days after infection Days after infection
Days after infection
Days after infection
Smad3–/– AdVEGF
Smad3–/– AdTGFβ1Smad3+/+ AdTGFβ1Control adenovirus
Smad3+/+ AdVEGF
Days after infection
Days after infection
4 7 14 21 4 7 14 21
4 7 144 7 14
214 7 14 214 7 14
Figure 2 | Quantification of submesothelium thickness andvascularization. (a) Adenovirus expressing transforming growthfactor-b1 (AdTGF-b1)-induced submesothelial thickening is Smad3dependent. Smad3þ /þ animals treated with AdTGF-b1 showedincreased submesothelial thickening compared with controladenovirus (AdDL)-treated animals and with Smad3�/� animalstreated with AdTGF-b1. (b) Angiogenesis measured as vessels permillimeter length of peritoneum. Animals treated with adenovirusexpressing vascular endothelial growth factor (AdVEGF) areincluded in this analysis. AdTGF-b1 induced angiogenesisin Smad3þ /þ but not in Smad3�/� animals. VEGF inducedsignificant but transient angiogenesis in both Smad3þ /þ
and Smad3�/� animals. (c) Sections were stained with picrosiriusred and evaluated using polarized light microscopy. Totalbirefringence (red þ green area) represents total collagen and(d) green birefringent area represents newly deposited collagen.Smad3þ /þ animals treated with AdTGF-b1 showed more totaland new collagen deposition compared with control AdDL-treated animals and Smad3�/� animals treated with AdTGF-b1.(c) There was a transient but significant increase in totalcollagen deposited at day 7 in the Smad3�/� animal treatedwith AdTGF-b1 compared with control AdDL-treated animals.mRNA was extracted from parietal peritoneal tissues and analyzedusing quantitative real-time polymerase chain reaction. There wasa strong upregulation of (e) plasminogen activator inhibitor 1(PAI-1) and (f) VEGF in Smad3þ /þ mice exposed to TGF-b1.Smad3�/� mice did not show a significant change in theexpression of these genes.
Kidney International (2010) 77, 319–328 321
P Patel et al.: Smad and peritoneal membrane injury o r i g i n a l a r t i c l e

(Po0.001, AdTGF-b1 vs AdDL). In AdTGF-b1-treatedanimals, p-Akt was increased in Smad3�/� mice comparedwith Smad3þ /þ mice (P¼ 0.011). Rapamycin treatment alsoincreased p-Akt in AdTGF-b1-treated mice (P¼ 0.024).Smad3�/� mice infected with AdTGF-b1 and treated withrapamycin showed the greatest increase in p-Akt.
Rapamycin increased GSK-3b phosphorylation
GSK-3b is constitutively active and is inactivated throughserine 9 (Ser9) phosphorylation. Akt is known to phospho-rylate GSK-3b and we found that Smad3�/� mice treatedwith AdTGF-b1 and rapamycin, which showed the greatestAkt activation, also showed the highest level of Ser9 GSK-3bphosphorylation (Figure 7c). GSK-3b protein concentrationand Ser9 phosphorylation were not significantly regulated inSmad3þ /þ mice.
Rapamycin blocks b-catenin protein, nuclear accumula-tion, and a-SMA expression. Overall, when both Smad3�/�
and Smad3þ /þ mice are combined, rapamycin significantlydownregulated total b-catenin protein concentration(P¼ 0.02; Figure 7d). We further analyzed b-catenin protein
expression and cellular localization using immunohisto-chemistry (Figure 8). There was an increase in b-cateninin the peritoneal tissue of Smad3�/� (Figure 8a and b) andSmad3þ /þ (Figure 8g and h) mice treated with AdTGF-b1compared with control adenovirus-treated animals (Figure 8eand f). Rapamycin treatment reduced b-catenin expres-sion and inhibited nuclear localization in both Smad3�/�
(Figure 8c and d) and Smad3þ /þ (Figure 8i and j) micetreated with AdTGF-b1.
Gene (Figure 7e) and protein (Figure 6) expression ofa-SMA was blocked by rapamycin in the animals treatedwith Smad3�/�. Rapamycin had no effect on a-SMA geneexpression in the Smad3þ /þ mice.
DISCUSSION
In these experiments, we analyzed the signaling pathwaysinvolved in peritoneal injury, EMT, and peritoneal fibrosisafter exposure to the profibrotic cytokine TGF-b1. We obser-ved that Smad3�/� mice are protected from fibrosis, angio-genesis, and the associated peritoneal membrane functionalchanges. The lack of extracellular matrix expansion in the
Figure 3 | Sections of parietal peritoneal tissue were stained for cytokeratin (green) and a-SMA (red) with 4,6-diamidino-2-phenylindole(DAPI) counterstaining. At 4 days after infection with adenovirus expressing transforming growth factor-b1 (AdTGF-b1), Smad3þ /þ miceshowed mesothelial cells that stained for both cytokeratin and a-smooth muscle actin (a-SMA; a–c, thin arrows) along with mesothelialcells without evidence of epithelial-to-mesenchymal transition (thick arrows). Smad3�/� animals (d–f) showed a similar response at thistime point with AdTGF-b1-induced dual-labeled cells (thin arrows) and non-transformed mesothelium (thick arrows). (g–i) Control adenovirus-treated Smad3þ /þ animals did not show any cellular transition or migration. Original magnification � 200.
322 Kidney International (2010) 77, 319–328
o r i g i n a l a r t i c l e P Patel et al.: Smad and peritoneal membrane injury

Smad3�/� mice after intraperitoneal administration ofAdTGF-b1 is in agreement with a study of lung injury byBonniaud et al.9 We have previously shown that TGF-b1overexpression leads to an increased expression of VEGF inthe peritoneum.28 This process is clearly Smad3 dependent,as there was no increase in VEGF mRNA in the Smad3�/�
mice treated with AdTGF-b1 (Figure 2f). We also showedthat the lack of angiogenic response is specific for TGF-b1as both Smad3�/� and Smad3þ /þ mice have robust buttransient angiogenic responses to adenovirus-mediated genetransfer of VEGF (Figures 1 and 2).
EMT is an integral element of wound repair, as iselaboration of extracellular matrix and angiogenesis. Wefound that EMT occurred in the absence of significant fibrosisin the Smad3�/� mice treated with AdTGF-b1, suggestingthat EMT and fibrosis are independent events and fibrosisrequires further Smad3-dependent process to occur. TheEMT response in the Smad3�/� mice was attenuated and thecellular invasion was transient. This suggests that Smad3 isimportant for some aspects of persisting EMT, and Banh
et al.17 have suggested that Smad3 may inhibit apoptosisof newly generated myofibroblasts. Further, we show thatinhibition of mTOR signaling completely inhibited the EMTresponse in Smad3�/� mice but had no effect on EMT inSmad3þ /þ . Specifically, in Smad3�/� mice, rapamycinblocked a-SMA expression and evidence of cellular transi-tion. Rapamycin also eliminated cellular invasion, whichis a feature of TGF-b1-induced EMT in this peritonealinjury model.3
Smad3 signaling has been implicated in TGF-b-inducedEMT in several in vitro studies.15,29 However, the evidence fora role of Smad3 in EMT in vivo is contradictory. In a modelof unilateral ureteric obstruction, Sato et al.8 found thatSmad3�/� mice were protected against EMT and tubuloin-terstitial fibrosis. Similarly, in a study of ocular lensepithelium, Saiko et al.14 found that EMT did not occur inthe absence of Smad3. In contrast, Banh et al.17 found anattenuated EMT response in the lens epithelium of animalsoverexpressing TGF-b1 on a Smad3-null background.17 Inour experiments with overexpression of TGF-b1 in the
Figure 4 | Sections of parietal peritoneal tissue taken at day 7 were stained for cytokeratin (green) and a-smooth muscle actin(a-SMA; red) with 4,6-diamidino-2-phenylindole (DAPI) counterstaining. (a–c) In Smad3þ /þ animals, transforming growth factor-b1(TGF-b1) induced cellular transition and submesothelial invasion with both dual-labeled (thin arrows) and cytokeratin-labeled (thick arrows)cells intermingled in the submesothelial zone. (d–f) Smad3�/� mice showed similar but attenuated epithelial-to-mesenchymal transitionresponses 7 days after treatment with adenovirus expressing TGF-b1 (AdTGF-b1). (g–i) Control adenovirus-treated Smad3�/� animalsdid not show any cellular transition or migration. Original magnification � 200.
Kidney International (2010) 77, 319–328 323
P Patel et al.: Smad and peritoneal membrane injury o r i g i n a l a r t i c l e

peritoneum, E-cadherin was suppressed in both Smad3þ /þ
and Smad3�/� mice. Snail gene expression was mostly Smad3dependent. Snail is a key regulator of E-cadherin gene expres-sion and our data suggest that other factors are involved inE-cadherin downregulation in the Smad3�/� mice.
The novel and most important finding from our study isthat there seems to be a separate but interacting pathwayinvolved in TGF-b1-induced EMT and that dual inhibition of
Smad3 and mTOR is necessary to completely abrogate theEMT response. Our observation that rapamycin did not affectEMT in the Smad3þ /þ mice is supported by recent work byLamouille et al.20. They found that TGF-b activated mTOR,and mTOR inhibition affected cell growth, invasion, andmigration but did not affect the overall EMT response. Otherin vitro work supports an inhibiting effect of rapamycin onEMT in peritoneal mesothelial30 or renal tubular epithelialcells.31 How the proposed switch to an alternate PI3K/AktEMT pathway in the absence of Smad3 is made is not clear.There is evidence that PI3K interacts with Smad3, but in apositive manner, so that PI3K or Akt can activate Smad3.20,22
The possibility that PI3K activity is increased in the absenceof Smad3 has not been observed previously. This suggeststhat Smad3 may have an inhibitory effect on PI3K/Aktsignaling (Figure 9).
Rapamycin is known to inhibit mTOR complex 1 that isactivated downstream by the PI3K/Akt pathway.23 We founda significant increase in Akt phosphorylation after TGF-b1exposure in both Smad3þ /þ and Smad3�/� mice in keepingwith previous observations.22 We also found that Akt phos-phorylation was increased in rapamycin-treated animals. Thisis in agreement with recent observations that mTOR complex1 negatively feeds back to inhibit Akt activation and rapamy-cin inhibits this negative-feedback mechanism (Figure 9).32,33
The regulation of GSK-3b is complex. GSK-3b is aninhibitory kinase that is constitutively present and targetsa variety of proteins for ubiquitination. GSK-3b is involvedin the control of glycogen metabolism, cell cycle, mobility,and survival.34 Akt is able to directly phosphorylate GSK-3bat Ser9 and inhibit its kinase activity.23 GSK-3b is alsophosphorylated and inhibited by the canonical wnt signalingpathway,35 and recently, p70 S6 kinase, a downstream effectorof mTOR complex 1, has been shown to inhibit GSK-3b.33,34
Interestingly, rapamycin has been shown to increase GSK-3bactivity through a mechanism independent of Ser9 phospho-rylation.24 GSK-3b has multiple targets including b-catenin.b-catenin forms a scaffold with E-cadherin at the inner cellmembrane and is quickly marked for degradation in thecytosol through phosphorylation by GSK-3b. If GSK-3bis inhibited, primarily through Akt or wnt signaling, thenb-catenin will accumulate, transport to the nucleus, andinitiate transcription of genes involved in cell growth, proli-feration, and survival.36
Although inhibition of GSK-3b is the classic pathwayfor cytoplasmic b-catenin accumulation and its nucleartranslocation, there is evidence that b-catenin can accumu-late even in the presence of functionally active GSK-3b.34
Interestingly, several recent studies have shown that b-cateninexpression37,38 and nuclear localization37 are directly inhibi-ted by rapamycin. The exact mechanism of this inhibitionremains unclear. In the Smad3�/� animals, Smad2 is stillfunctional39 and recently, Smad2 has been shown to directlyinteract with b-catenin in the nucleus.40 Persisting Smad2activity in the Smad3�/� mice may therefore be having a rolein the b-catenin-dependent EMT that we have observed.
P=
0.00
6
P=0.001P=0.009 P=0.036
P=0.008P=0.008
P=0.003P=0.001
P=0.042
P=0.043
P=0.013
P=0.037P=0.008
P=0.021
P=0.036P=0.002
P=0.007
P=0.006
P=0.017P=0.014
E-Cadherin
Rel
ativ
e de
nsity
Dua
l lab
eled
cel
ls/m
m
Are
as o
f inv
asio
n/m
m
1.61.41.2
10.80.60.40.2
0
0.80.70.60.50.40.30.20.1
0
43.5
32.5
21.5
10.5
0
β-Actin
4 7 14 21
Smad3–/–
Smad3–/– AdTGFβ1
Smad3+/+ AdTGFβ1
Control adenovirus
Smad3+/+
01020304050607080
Days after infection
AdTGFβ1AdTGFβ1 AdDL AdDL
4 7 14 21Days after infection
4 7 14 21Days after infection
4 7 14 21Days after infection
4 7 14Days after infection
3.53
2.5
1.51
0.50
2
Alp
ha-S
MA
:GA
PD
Hm
RN
A
Sna
il:G
AP
DH
mR
NA
Figure 5 | Epithelial-to-mesenchymal transition occurs inthe absence of Smad3. Quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) analysis of (a) a-smoothmuscle actin (a-SMA) and (b) SNAIL. Transforming growthfactor-b1 (TGF-b1) induced a significant increase in a-SMAgene expression in Smad3þ /þ animals compared with controltreated animals. (b) TGF-b1 also induced a significant increase inSNAIL gene expression in Smad3þ /þ mice. (c, d) E-cadherinprotein expression is downregulated in mice treated withadenovirus expressing TGF-b1 (AdTGF-b1). A representativewestern blot is shown in (c) and quantified in (d), which confirmeda significant downregulation in both Smad3þ /þ and Smad3�/�
mice treated with AdTGF-b1. (e) Epithelial-to-mesenchymaltransition (EMT) was quantified by counting the number of dual-labeled a-SMA/cytokeratin-positive cells in the peritoneal tissues.Both Smad3�/� and Smad3þ /þ had a quantifiable increase indual-labeled cells, and Smad3þ /þ mice had significantly moredual-labeled cells than Smad3�/� mice treated with AdTGF-b1.(f) The degree of cellular invasion was also quantified. Smad3þ /þ
mice treated with AdTGF-b1 had prolonged evidence ofsubmesothelial invasion with dual-labeled cells, whereasSmad3�/� mice treated with AdTGF-b1 showed only transientEMT-associated invasion. No evidence of migration of dual-labeled cells was observed in the control adenovirus-treatedanimals.
324 Kidney International (2010) 77, 319–328
o r i g i n a l a r t i c l e P Patel et al.: Smad and peritoneal membrane injury
Our observations concerning the effects of rapamycin inTGF-b1-induced EMT in Smad3�/� mice are in agreementwith these previous studies. Rapamycin increased Aktactivation through inhibition of the mTOR complex 1negative-feedback regulation (Figure 9). The increased Aktactivation leads to increased GSK-3b Ser9 phosphorylationand inactivation. This should result in b-catenin accumula-tion and nuclear localization, especially in the rapamycin-treated Smad3�/� animals, in which Akt activation was mostprofound. However, contrary to these expectations, b-cateninaccumulation was significantly inhibited in our experiments,and we hypothesize that this may be a direct action ofrapamycin.37 Alternatively, rapamycin may indirectly inhibitb-catenin through GSK-3b activation,24 through a Smad2mechanism, or through mTOR/p70 S6 kinase (Figure 9).
We also observed a downregulation of a-SMA gene(Figure 7e) and protein expression in Smad3�/� mice treatedwith rapamycin (Figure 6). Rapamycin-induced inhibition of
a-SMA has been observed in experimental diabetic nephro-pathy41 and in a model of ureteric obstruction.42 This couldbe a direct effect of mTOR/p70 S6 kinase inhibition or adownstream effect through b-catenin signaling.
In summary, in these experiments we describe a novelin vivo model to explore the signaling pathways involved inthe complex tissue response to TGF-b1. The majority ofTGF-b1 responses do seem to be mediated by Smad3signaling. Fibrosis and angiogenesis are significantly curtailedin the Smad3�/� mice. An attenuated EMT process isobserved in the Smad3�/� mice with an increased expressionof a-SMA and a decrease in E-cadherin. Rapamycin almostcompletely abrogated the EMT response in the Smad3�/�
mice while having minimal effect on the Smad3þ /þ mice.This suggests that there are at least two parallel, interactingpathways involved in TGF-b1-induced EMT. In the absenceof Smad3, Akt activation and downstream b-catenin seem tobe important mediators of EMT. b-catenin expression and
P=0.007
P=0.004
P=0.002P=0.001
Dua
l lab
eled
cel
ls/m
m
3
2.5
2
1.5
1
0.5
0Smad3
AdTGFβ1
Rapa – –
–/– –/–
–
–
+
++
+/+ +/+ +/+
+
+
+
Figure 6 | Rapamycin inhibits epithelial-to-mesenchymal transition in Smad3�/� mice. Sections of parietal peritoneum in Smad3�/�
mice at 7 days after treatment with (a–c) adenovirus expressing transforming growth factor-b1 (AdTGF-b1) and vehicle or (d–f) AdTGF-b1and rapamycin. Sections were stained for (a, d) cytokeratin and (b, e) a-smooth muscle actin (a-SMA) with (c, f) 4,6-diamidino-2-phenylindole (DAPI) counterstaining. (a–c) Smad3�/� mice treated with intraperitoneal AdTGF-b1 and with vehicle by gavage showedepithelial-to-mesenchymal transition (EMT) with evidence of invasive mesothelial cells staining for both cytokeratin and a-SMA (thin arrows)along with mesothelial cells without evidence of EMT (thick arrows). (d–f) Smad3�/� mice treated with AdTGF-b1 and rapamycin showed asignificantly reduced EMT response with only occasional dual-labeled cells observed (thin arrows) with no evidence of invasion. Originalmagnification� 200. (g) The number of dual-labeled cells per millimeter of peritoneum were quantified. Rapamycin had little effect on EMTin Smad3þ /þ mice but significantly decreased EMT in Smad3�/� mice.
Kidney International (2010) 77, 319–328 325
P Patel et al.: Smad and peritoneal membrane injury o r i g i n a l a r t i c l e

nuclear localization was inhibited by rapamycin. Furtherwork is required to delineate the exact mechanism of theSmad3/PI3K pathway interaction, and the mechanism ofrapamycin inhibition of b-catenin.
MATERIALS AND METHODSRecombinant adenovirusesThe construction of the adenovirus vectors AdTGF-b143 has beenpreviously described. AdTGF-b1 was created with TGF-b1 cDNAmutated at residues 223 and 225, so that the transgene productdoes not bind to the latency-associated protein and is thereforebiologically active. AdVEGF was created from the human splicevariant cDNA VEGF165 (gift from Dr D. Anthony, University ofOxford, UK). A null adenovirus (AdDL) was used for control.
Animal studiesSmad3�/� mice were generously provided by Dr A. Roberts. Thesemice were generated by removal of exon 8 of the Smad3 gene inthe mice of background 127SV/EV � C57BL/6 as per Yang et al.39
Animals were treated in accordance with the guideline of theCanadian Council on Animal Care. Smad3�/� and littermateSmad3þ /þ mice were infected with AdTGF-b1, AdVEGF, or AdDLat a dose of 1.5� 108 plaque-forming units diluted in 100 ml inphosphate-buffered saline by intraperitoneal injection, and groupsof 5–7 animals were killed on days 4, 7, 14, and 21 after infection.Before the mice were killed, 5 ml of 4.25% Dianeal (BaxterHealthcare, McGaw Park, IL, USA) was administered by intra-peritoneal injection and then recovered at the time of killing. Theentire anterior abdominal wall was removed and half was taken forhistology and half for RNA extraction. Omental tissue was takenand frozen in liquid nitrogen for protein extraction. Rapamycin(generously provided by Wyeth, Markham, ON, Canada) or vehiclewas administered by oral gavage starting on day 3 and continuingdaily until day 7 at a dose of 2.5 mg/kg.
Immunohistochemical analysisParaffin-embedded tissues of anterior abdominal wall weresectioned and immunohistochemical analysis was performed usingvon-Willebrand factor-related antigen (Dako, Carpenteria, CA,USA). Peritoneum-associated blood vessels and submesothelialthickness of the peritoneum were analyzed using Northern Eclipse
p-Akt P=0.018
P=0.002
P<0.001
P=0.021P=0.001
P=0.004P=0.05
0.90.80.70.60.50.40.30.20.1
0
00.20.40.60.8
11.21.41.61.8
2
–/–
––– –
–/– +/+++
++++
+/+ +/+–/–
––– –
–/– +/+++
++++
+/+ +/+
–/–
––– –
–/– +/+++
++++
+/+ +/+
–/–
––– –
–/– +/+++
++++
+/+ +/+
–/–
––– –
–/– +/+++
++++
+/+ +/+
pAkt
/Akt
Akt
p-GSK3β
pGS
K3β
/GS
K3β
GSK3β
Smad3
Rapa
43.5
32.5
21.5
10.5
0
α-S
MA
:GA
PD
H m
RN
A 3.5
3
2.5
2
1.5
1
0.5
0
AdTGFβ1
Smad3
RapaAdTGFβ1
Smad3
RapaAdTGFβ1
Smad3
RapaAdTGFβ1
Smad3
RapaAdTGFβ1
β-Catenin
β-C
aten
in/β
-act
in
β-Actin
Figure 7 | Protein was extracted from peritoneal tissues andanalyzed for p-Akt, glycogen synthase kinase (GSK)-3b, andb-catenin expression. (a) Representative western blots. (b) p-Akt/Akt was upregulated by transforming growth factor-b1 (TGF-b1),which was more evident in Smad3�/� mice. (c) pGSK-3b/GSK-3bwas increased in the Smad3�/� mice treated with rapamycin(Rapa). (d) b-catenin total protein concentration was increasednon-significantly in Smad3�/� treated with adenovirus expressingTGF-b1 (AdTGF-b1) and this effect was reversed by rapamycin.(e) a-smooth muscle actin (a-SMA) gene expression was foundto be significantly inhibited by rapamycin.
Smad3–/– AdTGFβ1 Vehicle
Smad3–/– AdTGFβ1 Rapa
Smad3+/+ AdDL Vehicle
Smad3+/+ AdTGFβ1 Vehicle
Smad3+/+ AdTGFβ1 Rapa
Figure 8 | Anterior abdominal wall sections were stainedfor b-catenin (green) and counterstained with 4,6-diamidino-2-phenylindole (DAPI; blue). (a, b) Smad3�/� mice treated withadenovirus expressing transforming growth factor-b1 (AdTGF-b1)and vehicle show increased b-catenin expression in the peritonealtissues with evidence of both cytoplasmic and nuclear staining(arrows). (c, d) Smad3�/� mice treated with AdTGF-b1 and rapamycin(Rapa) show minimal b-catenin staining and no evidence of nuclearlocalization. (e, f) A similar pattern is observed in Smadþ /þ micetreated with control adenovirus (AdDL) and vehicle. (g, h) Smadþ /þ
mice treated with AdTGF-b1 and vehicle show increased b-cateninexpression with evidence of nuclear localization (arrow).(i, j) b-catenin expression is present in Smad3þ /þ mice treatedwith AdTGF-b1 (arrow), but nuclear localization seems to beinhibited by treatment with rapamycin.
326 Kidney International (2010) 77, 319–328
o r i g i n a l a r t i c l e P Patel et al.: Smad and peritoneal membrane injury

image analysis software (Empix Imaging, Mississauga, ON, Canada).Sections were stained for a-SMA (Dako) followed by a secondaryrabbit anti-mouse antibody labeled with Texas Red (MolecularProbes, Eugene, OR, USA) and fluorescein isothiocyanate-conjugatedanti-cytokeratin (Sigma Chemicals, Oakville, ON, Canada). b-cateninwas assessed in formalin-fixed tissue using b-catenin antibody(Cell Signaling Technology, Inc, MA, USA) followed by a secondaryfluorescein isothiocyanate-labeled antibody (Jackson Immuno-Research, West Grove, PA, USA). Sections were also stained with1% picrosirius red stain (Sigma). These sections were viewed underpolarized light and digitized images were taken. Using NorthernEclipse software, relative orange–red or green birefringent areaswere calculated as a percentage of the total submesothelial tissue.Previous studies have suggested that green birefringent areas repre-sent type III collagen and is indicative of newly deposited collagen.26
Quantitative polymerase chain reactionmRNA was extracted from the peritoneum of the killed animalsby immersing the parietal peritoneal surface in Trizol reagent(Invitrogen, Burlington, Ontario, Canada) for 20 min. RNA wasextracted from Trizol according to the manufacturer’s instruction.RNA (1mg) was then reverse-transcribed using the standardprotocol (Invitrogen). Quantitative real-time polymerase chainreaction for plasminogen activator inhibitor 1, VEGF, a-SMA, andSNAIL was performed on mRNA using an ABI Prism 7700 SequenceDetector (Applied Biosystems, Foster City, CA, USA). Glyceralde-hyde-3-phosphate dehydrogenase was used as housekeeping gene.
Protein analysisProtein analysis was carried out using western blot for E-cadherin(BD Biosciences, Mississauga, ON, Canada), pAkt (Cell SignalingTechnology, MA, USA; specific for Ser 473 phosphorylation), totalAkt (Cell Signaling), GSK-3b, pGSK-3b (Cell Signaling; specific forSer 9 phosphorylation), b-catenin (Cell Signaling), and b-actin
(Sigma). Band density was measured using Scion Image Software(Scion, Frederick, MD, USA).
Statistical analysisWe did not identify any substantial differences between Smad3�/�
and Smad3þ /þ animals treated with the control adenovirus. Forthe purposes of data analysis, these two groups were combined.Differences between groups were compared using analysis ofvariance with Bonferroni’s post hoc analysis.
DISCLOSUREPJM has received research funding from Wyeth Canada and BaxterHealthcare, USA. The other authors declared no competing interests.
ACKNOWLEDGMENTSThis research was supported by the Canadian Institutes ofHealth Research, and St Joseph’s Healthcare, Hamilton, Ontario.MRJK holds a CIHR Scientist award and PJM is a CIHR ClinicianScientist.
SUPPLEMENTARY MATERIALFigure S1.Figure S2.Supplementary material is linked to the online version of the paper athttp://www.nature.com/ki
REFERENCES1. Margetts PJ, Bonniaud P. Basic mechanisms and clinical implications of
peritoneal fibrosis. Perit Dial Int 2003; 23: 530–541.2. Brimble KS, Walker M, Margetts PJ et al. Meta-analysis: peritoneal
membrane transport, mortality, and technique failure in peritonealdialysis. J Am Soc Nephrol 2006; 17: 2591–2598.
3. Margetts PJ, Bonniaud P, Liu L et al. Transient overexpression ofTGF-{beta}1 induces epithelial mesenchymal transition in the rodentperitoneum. J Am Soc Nephrol 2005; 16: 425–436.
4. Yanez-Mo M, Lara-Pezzi E, Selgas R et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N Engl J Med 2003; 348:403–413.
5. Aroeira LS, Aguilera A, Sanchez-Tomero JA et al. Epithelial tomesenchymal transition and peritoneal membrane failure in peritonealdialysis patients: pathologic significance and potential therapeuticinterventions. J Am Soc Nephrol 2007; 18: 2004–2013.
6. Massague J. TGF-beta signal transduction. Annu Rev Biochem 2007; 67:753–791.
7. Flanders KC, Sullivan CD, Fujii M et al. Mice lacking Smad3 are protectedagainst cutaneous injury induced by ionizing radiation. Am J Pathol 2002;160: 1057–1068.
8. Sato M, Muragaki Y, Saika S et al. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis inducedby unilateral ureteral obstruction. J Clin Invest 2003; 112: 1486–1494.
9. Bonniaud P, Kolb M, Galt T et al. Smad3 null mice develop airspaceenlargement and are resistant to TGF-beta-mediated pulmonary fibrosis.J Immunol 2004; 173: 2099–2108.
10. Savagner P. Leaving the neighborhood: molecular mechanisms involvedduring epithelial-mesenchymal transition. Bioessays 2001; 23: 912–923.
11. Zeisberg M, Bottiglio C, Kumar N et al. Bone morphogenic protein-7inhibits progression of chronic renal fibrosis associated with two geneticmouse models. Am J Physiol Renal Physiol 2003; 285: F1060–F1067.
12. Lin SL, Kisseleva T, Brenner DA et al. Pericytes and perivascular fibroblastsare the primary source of collagen-producing cells in obstructive fibrosisof the kidney. Am J Pathol 2008; 173: 1617–1627.
13. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and itsimplications for fibrosis. J Clin Invest 2003; 112: 1776–1784.
14. Saika S, Kono-Saika S, Ohnishi Y et al. Smad3 signaling is required forepithelial-mesenchymal transition of lens epithelium after injury. Am JPathol 2004; 164: 651–663.
15. Cho HJ, Baek KE, Saika S et al. Snail is required for transforming growthfactor-beta-induced epithelial-mesenchymal transition by activating PI3kinase/Akt signal pathway. Biochem Biophys Res Commun 2007; 353: 337–343.
16. Shirai K, Saika S, Tanaka T et al. A new model of anterior subcapsularcataract: involvement of TGFbeta/Smad signaling. Mol Vis 2006; 12: 681–691.
TGFβ
Smad3
mTOR
Akt
GSK3b
EMTEMT
β-Catenin
Rapamycin
P70 S6kinase
Figure 9 | The observations from these experiments suggest thatthere are at least two parallel pathways involved in transforminggrowth factor-b1 (TGF-b1)-mediated epithelial-to-mesenchymaltransition (EMT). In the absence of Smad3, the phosphatidylinositol 3 kinase (PI3K)/Akt pathway dominates. The increasedexpression of Akt in the absence of Smad3 suggests an inhibitorymechanism (dotted line). Smad3-independent EMT is blockedby rapamycin and the main effect occurs through decreasedexpression and nuclear localization of b-catenin. This mayoccur directly or indirectly through glycogen synthase kinase(GSK)-3b or mammalian target of rapamycin (mTOR)/p70 S6kinase (dotted lines).
Kidney International (2010) 77, 319–328 327
P Patel et al.: Smad and peritoneal membrane injury o r i g i n a l a r t i c l e

17. Banh A, Deschamps PA, Gauldie J et al. Lens-specific expression of TGF-beta induces anterior subcapsular cataract formation in the absence ofSmad3. Invest Ophthalmol Vis Sci 2006; 47: 3450–3460.
18. Cho HJ, Yoo J. Rho activation is required for transforming growth factor-beta-induced epithelial-mesenchymal transition in lens epithelial cells.Cell Biol Int 2007; 31: 1225–1230.
19. Sebe A, Leivonen SK, Fintha A et al. Transforming growth factor-inducedalpha-smooth muscle cell actin expression in renal proximal tubular cellsis regulated by p38 mitogen-activated protein kinase, extracellular signal-regulated protein kinase1,2 and the Smad signalling during epithelialmyofibroblast transdifferentiation. Nephrol Dial Transplant 2008; 23:1537–1545.
20. Lamouille S, Derynck R. Cell size and invasion in TGF-beta-inducedepithelial to mesenchymal transition is regulated by activation of themTOR pathway. J Cell Biol 2007; 178: 437–451.
21. Fujishita T, Aoki K, Lane HA et al. Inhibition of the mTORC1 pathwaysuppresses intestinal polyp formation and reduces mortality inApcDelta716 mice. Proc Natl Acad Sci USA 2008; 105: 13544–13549.
22. Runyan CE, Schnaper HW, Poncelet AC. The phosphatidylinositol 3-kinase/Akt pathway enhances Smad3-stimulated mesangial cell collagen Iexpression in response to transforming growth factor-beta1. J Biol Chem2004; 279: 2632–2639.
23. Jiang BH, Liu LZ. PI3K/PTEN signaling in tumorigenesis and angiogenesis.Biochim Biophys Acta 2008; 1784: 150–158.
24. Dong J, Peng J, Zhang H et al. Role of glycogen synthase kinase 3beta inrapamycin-mediated cell cycle regulation and chemosensitivity. CancerRes 2005; 65: 1961–1972.
25. Conacci-Sorrell M, Zhurinsky J, Ben Ze’ev A. The cadherin-cateninadhesion system in signaling and cancer. J Clin Invest 2002; 109: 987–991.
26. Zhang H, Sun L, Wang W et al. Quantitative analysis of fibrosis formationon the microcapsule surface with the use of picro-sirius red staining,polarized light microscopy, and digital image analysis. J Biomed Mater ResA 2006; 76: 120–125.
27. Carver EA, Jiang R, Lan Y et al. The mouse snail gene encodes a keyregulator of the epithelial-mesenchymal transition. Mol Cell Biol 2001; 21:8184–8188.
28. Margetts PJ, Kolb M, Galt T et al. Gene transfer of transforming growthfactor-beta1 to the rat peritoneum: effects on membrane function. J AmSoc Nephrol 2001; 12: 2029–2039.
29. Valcourt U, Kowanetz M, Niimi H et al. TGF-beta and the Smad signalingpathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell 2005; 16: 1987–2002.
30. Aguilera A, Aroeira LS, Ramirez-Huesca M et al. Effects of rapamycin onthe epithelial-to-mesenchymal transition of human peritonealmesothelial cells. Int J Artif Organs 2005; 28: 164–169.
31. Copeland JW, Beaumont BW, Merrilees MJ et al. Epithelial-to-mesenchymal transition of human proximal tubular epithelial cells:effects of rapamycin, mycophenolate, cyclosporin, azathioprine, andmethylprednisolone. Transplantation 2007; 83: 809–814.
32. LoPiccolo J, Granville CA, Gills JJ et al. Targeting Akt in cancer therapy.Anticancer Drugs 2007; 18: 861–874.
33. Zhang HH, Lipovsky AI, Dibble CC et al. S6K1 regulates GSK3 underconditions of mTOR-dependent feedback inhibition of Akt. Mol Cell 2006;24: 185–197.
34. Luo J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis andcancer chemotherapy. Cancer Lett 2009; 273: 194–200.
35. Inoki K, Ouyang H, Zhu T et al. TSC2 integrates Wnt and energy signals viaa coordinated phosphorylation by AMPK and GSK3 to regulate cellgrowth. Cell 2006; 126: 955–968.
36. Huang D, Du X. Crosstalk between tumor cells and microenvironment viaWnt pathway in colorectal cancer dissemination. World J Gastroenterol2008; 14: 1823–1827.
37. Liu H, Remedi MS, Pappan KL et al. Glycogen synthase kinase-3 andmammalian target of rapamycin pathways contribute to DNA synthesis,cell cycle progression, and proliferation in human islets. Diabetes 2009;58: 663–672.
38. Segrelles C, Moral M, Lara MF et al. Molecular determinants of Akt-induced keratinocyte transformation. Oncogene 2006; 25: 1174–1185.
39. Yang X, Letterio JJ, Lechleider RJ et al. Targeted disruption of SMAD3results in impaired mucosal immunity and diminished T cellresponsiveness to TGF-beta. EMBO J 1999; 18: 1280–1291.
40. Clifford RL, Deacon K, Knox AJ. Novel regulation of vascular endothelialgrowth factor-A (VEGF-A) by transforming growth factor (beta)1:requirement for Smads, (beta)-CATENIN, AND GSK3(beta). J Biol Chem2008; 283: 35337–35353.
41. Lloberas N, Cruzado JM, Franquesa M et al. Mammalian target ofrapamycin pathway blockade slows progression of diabetic kidneydisease in rats. J Am Soc Nephrol 2006; 17: 1395–1404.
42. Wu MJ, Wen MC, Chiu YT et al. Rapamycin attenuates unilateralureteral obstruction-induced renal fibrosis. Kidney Int 2006; 69:2029–2036.
43. Sime PJ, Xing Z, Graham FL et al. Adenovector-mediated gene transfer ofactive transforming growth factor-beta1 induces prolonged severefibrosis in rat lung. J Clin Invest 1997; 100: 768–776.
328 Kidney International (2010) 77, 319–328
o r i g i n a l a r t i c l e P Patel et al.: Smad and peritoneal membrane injury