transforming growth factor-β stimulates β amyloid uptake by microglia through smad3-dependent...
TRANSCRIPT

Transforming Growth Factor-b Stimulatesb Amyloid Uptake by Microglia ThroughSmad3-Dependent Mechanisms
Juan E. Tichauer and Rommy von Bernhardi*
Department of Neurology, Faculty of Medicine, Pontificia Universidad Catolica de Chile, Santiago, Chile
Inflammatory cytokines and b amyloid (Ab) induce acti-vation of glial cells, leading to both protective and dele-terious changes that are relevant for the pathogenesisof Alzheimer disease (AD). We have shown that astro-cytes downregulate microglial cell cytotoxic activationthrough secretion of transforming growth factor-b(TGFb1), and there is evidence that TGFb1 modifies Abremoval through the modulation of microglia. However,inflammatory activation of microglia is increased andAb clearance is reduced in AD patients, regardless ofthe fact that TGFb1 is increased in their nervous sys-tem. We propose that changes in TGFb Smad3 signaltransduction could modify the regulation mediated byTGFb1. Here we evaluated the participation of theTGFb Smad3 pathway in regulation of the expressionpattern of scavenger receptors (SR) and activation ofmicroglia through nitric oxide (NO�) secretion and phag-ocytosis of Ab. We found that TGFb1 increased SR-Aby 2.4-fold and decreased SR-BI expression by 79% at48 hr, whereas it did not change SR-MARCO or CD36expression. In addition, we observed a 51% increaseof Ab uptake and an 83% decrease of NO� productioninduced by lipopolysaccharide in microglial cell cul-tures. Increased expression of SR-A, phagocytosis, anddownregulation of NO� by TGFb1 were prevented bythe inhibition of the TGFb Smad3 pathway. Our resultsindicate that the modulation of microglial cell activationby TGFb1, leading to increased clearance of Ab andreduced cytotoxicity, is at least partially mediated bythe Smad pathway. VVC 2012 Wiley Periodicals, Inc.
Key words: Alzheimer’s disease; cytokines; glia;neuroinflammation; SR-A; transforming growth factor-b
Alzheimer’s disease (AD) anatomopathology ischaracterized by the presence of extracellular amyloidplaques, with b amyloid (Ab) as their main component(Haass et al., 1992), intracellular neurofibrillary tangles,and dystrophic neurites surrounded by activated astro-cytes and microglia in neocortex and hippocampus(Hardy and Selkoe, 2002). Microglia are the residentmacrophages of the central nervous system. These cellsof mesodermal origin migrate into all regions, dissemi-nate through the brain parenchyma, and acquire aspecific ramified morphological phenotype termed ‘‘sur-
veillance microglia.’’ Even in the normal brain, microgliahave highly motile processes by which they scan theirterritorial domains (Kettenmann, 2011). Together withastrocytes, microglia have the ability to bind and take upAb in vitro and in vivo (Wyss-Coray et al., 2003; Alar-con et al., 2005), being important players for the deposi-tion and removal of Ab (Block et al., 2007), althoughthere are also reports suggesting that Ab formationoccurs independently of microglial cells (Grathwohlet al., 2009). Microglial cells express several pattern-rec-ognition receptors, which allow them to remove poten-tially toxic molecules such as Ab. These receptorsinclude scavenger receptors (SR) class A (SR-A; Kriegerand Herz 1994), class B type I (Husemann et al., 2002),low-density lipoprotein receptor-related protein (LRP;Marzolo et al., 2000), cluster of differentiation 36(CD36; (Coraci et al., 2002), receptor for end glycationproduct (Yan et al., 1996), mannose receptor (Marzoloet al., 1999), and SR-MARCO (Alarcon et al., 2005).
SR changes can be especially relevant, because theycan affect the activation and phagocytic function of glialcells. Furthermore, because glial cells are also major pro-ducers of inflammatory factors in the brain (Paresceet al., 1996), changes in glial cell activation can result inchanges in their inflammatory status. In fact, microglialcells secrete multiple cytokines such as interleukin-1b(IL1b), tumor necrosis factor (TNFa), interleukin 10(IL10), and transforming growth factor-b (TGFb) aswell as short-lived cytotoxic factors such as superoxide(O2�2) and NO�, which contribute to neurotoxicity(Meda et al., 2001).
We have shown that TGFb1 produced by astro-cytes and hippocampal cells prevents induction of reac-
Contract grant sponsor: VRAID of the Pontificia Universidad Catolica
de Chile; Contract grant number: PUENTE 01/08; Contract grant spon-
sor: FONDECYT; Contract grant number: 1090353 (to R.v.B.).
*Correspondence to: Rommy von Bernhardi, MD, PhD, Department of
Neurology, Faculty of Medicine, Pontificia Universidad Catolica de
Chile, Marcoleta 391, Santiago, Chile. E-mail: [email protected]
Received 29 January 2012; Revised 25 March 2012; Accepted 13 April
2012
Published online 20 June 2012 in Wiley Online Library
(wileyonlinelibrary.com). DOI: 10.1002/jnr.23082
Journal of Neuroscience Research 90:1970–1980 (2012)
' 2012 Wiley Periodicals, Inc.

tive oxygen species (ROS) and NO� production byinflammatory mediators (lipopolysaccharide [LPS] 1interferon-g [IFNg]) and neurotoxicity in vitro (Her-rera-Molina and von Bernhardi, 2005; Uribe-San Martınet al., 2009) and that the modulation of microglia is atleast partially mediated by the activation of the ERKpathway (Herrera-Molina and von Bernhardi, 2005;Saud et al., 2005). TGFb superfamily signaling plays im-portant roles in a diverse set of cellular responses, includ-ing cell proliferation, differentiation, extracellular matrixremodeling, and embryonic development. TGFb1 sig-naling is mediated by cell surface type I and type IIreceptors, which phosphorylates the two R-Smad pro-teins Smad2 and Smad3 downstream. Phospho-Smad2/3forms a complex with the common mediator Smad4 thatbinds to a Smad-binding element (SBE) in the nucleuswith a large number of transcription coregulators to acti-vate gene promoter (Wrighton et al., 2009; Zhang,2009).
There is controversy regarding whether TGFb1has a protective or a deleterious effect in AD. On onehand, TGFb1 appears to be involved in the promotionof amyloid deposits in blood vessels and meninges(Wyss-Coray et al., 1997) and has been shown to induceproduction of Ab by astrocytes in transgenic hAPP/TGFb mice overexpressing TGFb1 selectively by astro-cytes (Lesne et al., 2003). On the other hand, studiessuggest that TGFb1 has an antiamyloidogenic role,decreasing the total load of Ab and of neuritic plaquesin brain parenchyma, an effect that appears to be medi-ated by activated microglia (Wyss-Coray et al., 2001).
AD patients have increased levels of TGFb1 in cere-brospinal fluid (CSF) and plasma (Rota et al., 2006; Mottaet al., 2007). However, the expression of mRNA encod-ing for Smad3, the main effector pathway of TGFb1, isdecreased in the hippocampus (Colangelo et al., 2002).Furthermore, hyperphosphorilation of tau, one of the ADhallmarks, affects Smad2/3 translocation (Baig et al.,2009), and neurofibrillary tangles could interfere withSmad2/3 signaling (Lee et al., 2006; Chalmers and Love,2007). Because of that evidence, we decided to examinethe relevance of the Smad3 pathway for TGFb1-inducedmodulation of cell activation, SR expression pattern, andAb uptake in rat neonatal microglia. We propose thatmodifications of the TGFb1 transduction pathwaysinduce changes in the activation patterns of microglia andreduce their capability to clear Ab.
MATERIALS AND METHODS
Plasmid pacEF-MycSmad7 was a kind gift from Dr.Kenji Okazaki (Biomolecular Engineering, Research Institute,Osaka, Japan). TGFb1 was purchased from R&D Systems(Minneapolis, MN); LPS, polyinosinic acid (polyI), polycyti-dylic acid (polyC), Alk5 inhibitor SB431542, MEK inhibitorPD98059, p38 inhibitor SB203580, mouse anti-b-tubulin,Alexa Flour 568-lectin (Griffonnia simplicifolia), and Lidocainewere from Sigma (St. Louis, MO); and Smad3 inhibitor SIS3was from Calbiochem (La Jolla, CA). Rat anti-SR-A and -
SR-MARCO were purchased from Serotec (Bicester, UnitedKingdom), rabbit anti-SR-BI from Novus (Littleton, CO),GAPDH from Chemicon (Temecula, CA), and Alexa-conju-gated secondary antibodies from Molecular Probes (Eugene,OR). Fluorescent mounting medium was from Dako(Glostrup, Denmark). Lipofectamine 2000 was from Invitro-gen (Carlsbad, CA). Cell culture media, antibiotics, and serumwere purchased from Gibco Life Technologies (Grand Island,NY). EOC20 cell line was from ATCC (Manasas, VA). Ani-mals were obtained from the institutional animal facility. Theywere anesthetized with ether before sacrifice, and all proce-dures followed the animal handling and bioethical require-ments defined by the Pontificia Universidad Catolica de ChileEthics Committee, in accordance with NIH guidelines.
Glial Cultures
We obtained mixed glial cultures from cerebral cortexof 1–2-day-old Sprague-Dawley rats as previously described(Giulian and Baker 1986; von Bernhardi and Eugenın, 2004).We rinsed cortices with HBSS, removed meninges, mincedthe tissue, and incubated it with HBSS containing 0.25% tryp-sin-EDTA at 378C for 10 min. We mechanically disruptedthe tissue and seeded cells in 75-cm2 cell culture flasks coatedwith poly-L-lysine (one brain per flask) in supplementedDMEM/F12 (10% fetal bovine serum [FBS], 100 U/ml peni-cillin, and 100 lg/ml streptomycin) and incubated in a water-saturated, 5% CO2 atmosphere at 378C.
After 14 days in culture, microglia were purified by dif-ferential adherence. Conditioned culture media was obtainedfrom the glial culture flasks by centrifugation (200g for 10min) and later was used to seed the purified microglial cells.Glial cultures were incubated in DMEM-F12 containing 6mM lidocaine for 10 min (von Bernhardi and Eugenin, 2004).Detached microglia were collected by centrifugation (200g for10 min), resuspended in conditioned media, and plated at adensity of 2.5 3 106 cells in six-well plates for Western blotassays, at a density of 5 3 105 cells on glass coverslips in 24-well plates for immunofluorescence and uptake assays, respec-tively, and at density of 3 3 104 cells in 96-well plates for ni-trite (NO22�) determination. After 15 min of seeding, me-dium was changed with fresh supplemented DMEM/F-12.Purity of microglial cell cultures was assessed by lectin (Grif-fonnia simplicifolia; Sigma) labeling, and over 99% of cells werepositive for lectin labeling (data not shown). The microglialcell line EOC20 was seeded in 24-well plates onto glass coversfor transfection assays.
TGFb Pathway Inhibition Assays
To evaluate the participation of TGFb1 signaling path-ways in SR expression, NO22� secretion, and Ab uptake, weexposed microglial cell cultures to 2 ng/ml TGFb1 for 24,48, or 96 hr with or without pretreating cell cultures with 10lM SIS3 (Smad3 inhibitor) or 10 lM SB431542 (Alk5 inhibi-tor) for 1 hr or with 30 lM SB203580 (p38 inhibitor) or 20lM PD98059 (ERK inhibitor) for 30 min. Microglia wereexposed to DMSO as a vehicle control condition. As a posi-tive control for the induction of NO� secretion and phagocy-tosis, cultures were stimulated with 1 lg/ml LPS.
TGFb Regulates Ab Uptake by Microglia 1971
Journal of Neuroscience Research

Western Blot
Cells were lysed with 1:1 ice-cold lysis buffer (50 mMTris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1%SDS, and protease inhibitors) and loading buffer (50 mMTris-HCl, pH 6.8, 2% SDS, 10%, glycerol, 1% b-mercapto-ethanol, 2.5 mM EDTA, 0.02% bromophenol blue). Cellsamples were denatured in boiling water for 5 min, electro-phoretically separated on 12% polyacrylamide gels, and trans-ferred to a nitrocellulose membrane. After transference, mem-branes were treated with blocking buffer (PBS plus 0.05%Tween 20, 5% milk) and then incubated with the primaryantibodies in blocking buffer: rat anti-SR-A (1:500; Serotec),rabbit anti-SR-BI (1:1,000; Novus Biological), rabbit anti-CD36 (1:1,000; Novus Biological), rat anti-SR-MARCO(1:1,000; Serotec), rabbit anti-pSmad3 and anti-Smad3(1:1,000; Cell Signalling, Beverly, MA), mouse anti-a-tubulin(1:1,000; Sigma), and mouse anti-GADPH (1:1,000; Chemi-con). The primary antibody was rinsed, and the membranewas incubated with the corresponding horseradish peroxidase-labeled secondary antibody: donkey anti-rat IgG, goat anti-rabbit IgG, or goat anti-mouse IgG, in blocking buffer. Signalswere detected by enhanced chemiluminescence (AmershamBiosciences, Arlington Heights, IL) in accordance with themanufacturer’s instructions. The molecular mass was estimatedwith a molecular mass marker kit (Invitrogen). Densitometrywas performed with the ImageJ program (NIH).
Immunolabeling of Live Cells
To determine SRs surface expression levels exclusively,live microglial cells were labeled with antibodies against vari-ous SRs: rat anti-SR-A (1:50), rat anti-SR-MARCO (1:100),and rabbit anti-SR-BI (1:100) in DMEM-F12 for 1 hr. Afterlabeling with the primary antibody, cells were washed withPBS Ca21 and fixed with 100% methanol at 2208C for 5min to permeabilize them or with 4% p-formaldehyde (PFA)for 15 min to minimize permeabilization. Fixed cells were la-beled with the corresponding fluorescein-conjugated second-ary antibodies and with Alexa Flour 568-conjugated lectin(Griffonnia simplicifolia; microglial identity marker) at 48C over-night. As a control to evaluate membrane integrity, cells werelabeled for the intracellular antigen Lamp-1. Nuclei werestained with Hoechst (Molecular Probes). Finally, cells werewashed and mounted with fluorescence mounting medium(Dako). Preparations were visualized with an inverted epifluo-rescence microscope (Leica).
Determination of NO22�NO22�, a stable downstream product of NO� released
to the cell culture medium, was determined via the Griessassay (Pfeiffer et al., 1997). For the determination of NO22�,3 3 104 microglia were cultured for 72 hr. Cultures weretreated with 1 lg/ml LPS, 2 ng/ml TGFb1, or their combi-nation for 96 hr. To evaluate the participation of specificpathways activated by TGFb1 in microglial modulation, cul-tures were pretreated with 10 lM SIS3 (Smad3 inhibitor) for1 hr or with 20 lM PD98059 (ERK inhibitor) or 30 lMSB203580 (p38 inhibitor) for 30 min. For the Griess assay, 50ll medium was mixed with 10 ll EDTA:H2O 1:1 (0.5 M,
pH 8.0) and 60 ll freshly prepared Griess reagent (20 mg N-[1-naphtyl]-ethylendiamine and 0.2 g sulphanilamide dissolvedin 20 ml of 5% phosphoric acid, w/v). Calibration curveswere established with 1–80 lM NaNO2. Absorbency wasmeasured at 570 nm in a microplate autoreader (Anthos 2010;Anthon Lactic Instrument).
Uptake Assays
To determine the ability of microglia to phagocytosesoluble Ab, microglial cell cultures were incubated with 2 ng/ml TGFb1 with and without pretreatment with signalingpathway inhibitors; 10 lM SIS3 (Smad3 inhibitor), 20 lMPD98059 (ERK inhibitor), or 30 lM SB203580 (p38 inhibi-tor) for 1 hr. After 48 hr, cells were rinsed and incubatedwith Fluo III-conjugated Ab1–40 in DMEM/F12 for 3 hr. Toevaluate the robustness of SR-mediated uptake, cultures werecoincubated with 25 and 100 lg/ml polyI, a polyanionicligand molecule that competes for scavenger receptor bindingsites. PolyC was used as negative control. Cells were washedtwice with 1 mM Ca21 in PBS and fixed with 4% parafor-maldehyde at room temperature for 15 min. Fixed cells werepermeabilized with 0.03% Triton X-100 in PBS for 15 min,and the nucleus was stained with Hoechst for 10 min. Finally,glass covers were washed and mounted with mounting me-dium (Dako). We acquired 10 fields at 340 magnification byrandom sampling from each cell culture and assessed the per-centage of cells containing Ab1–40 Fluo III.
Cell Line Transfection
The microglial cell line EOC20 was seeded in 24-wellplates with a glass cover at a low density of 1 3 104 cells/wellfor transfection assays. Briefly, for each well, 1 lg DNA plas-mid pactEF-MycSmad7 and 10 ll Lipofectamine 2000 (Invi-trogen) were separately diluted in 100 ll growth mediumwithout serum. After 5 min, plasmid DNA and Lipofectaminewere combined, mixed, and incubated at room temperaturefor 20 min. The mixture (200 ll) was added to the culturescontaining 250 ll culture medium. Cells were incubated at378C in a CO2 incubator overnight. Then, growth mediumwas removed and replaced by fresh media with 10% fetal bo-vine serum (FBS) in the different treatments, 2 ng/ml TGFbor 10 lM SIS3 1 2 ng/ml TGFb, and incubated for an addi-tional 24 hr. After incubation, Ab phagocytosis was evaluatedas previously described. Finally, cells were fixed with 4% para-formaldehyde (PFA), and Smad7 overexpression was evaluatedby immunofluorescence. As a control for transfection assay,plasmid expressing green fluorescent protein (GFP) wascotransfected.
Statistical Analysis
Statistical analyses were performed in GraphPad Prism4.0 software (GraphPad Software, San Diego, CA) and GB-Stat statistical software (Dynamic Microsystems, Inc.). Westernblot and uptake assays were analyzed using a one-wayANOVA with Tukey-Kramer post hoc test. For those statisti-cal analyses, a value of P < 0.05 was considered significant.For NO22� assays, an initial evaluation was performed with aone-way ANOVA with a Kruskal Wallis H-test. For those
1972 Tichauer and von Bernhardi
Journal of Neuroscience Research
groups with significant differences in the variance analysis, aWilcoxon rank sum/Mann-Whitney U-test was used forcomparisons of NO� production between control cultures andthose treated with the cytokines and signaling pathways inhib-itors. To correct the level of significance for multiple compar-isons, a was corrected according the number of inhibitor con-ditions (five), and significance was defined as P < 0.01.
RESULTS
TGFb1 Changed the Pattern of ScavengerReceptors Expression in Microglia
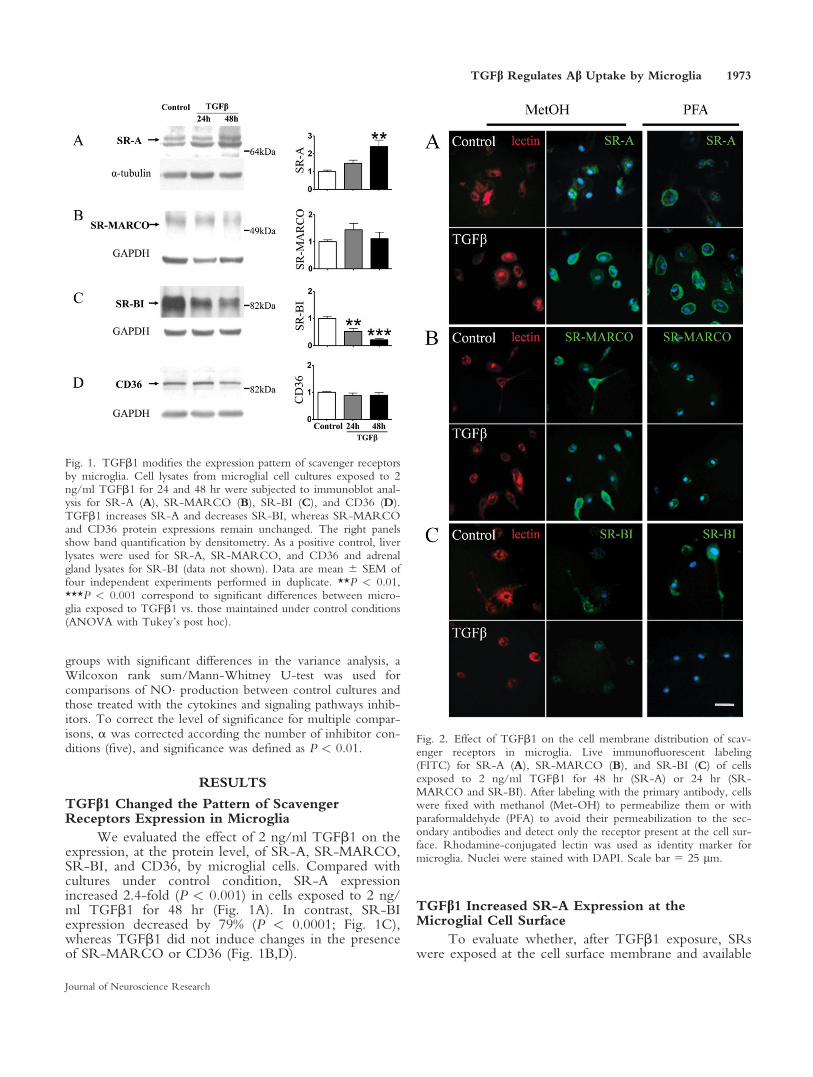
We evaluated the effect of 2 ng/ml TGFb1 on theexpression, at the protein level, of SR-A, SR-MARCO,SR-BI, and CD36, by microglial cells. Compared withcultures under control condition, SR-A expressionincreased 2.4-fold (P < 0.001) in cells exposed to 2 ng/ml TGFb1 for 48 hr (Fig. 1A). In contrast, SR-BIexpression decreased by 79% (P < 0.0001; Fig. 1C),whereas TGFb1 did not induce changes in the presenceof SR-MARCO or CD36 (Fig. 1B,D).
TGFb1 Increased SR-A Expression at theMicroglial Cell Surface
To evaluate whether, after TGFb1 exposure, SRswere exposed at the cell surface membrane and available
Fig. 1. TGFb1 modifies the expression pattern of scavenger receptorsby microglia. Cell lysates from microglial cell cultures exposed to 2ng/ml TGFb1 for 24 and 48 hr were subjected to immunoblot anal-ysis for SR-A (A), SR-MARCO (B), SR-BI (C), and CD36 (D).TGFb1 increases SR-A and decreases SR-BI, whereas SR-MARCOand CD36 protein expressions remain unchanged. The right panelsshow band quantification by densitometry. As a positive control, liverlysates were used for SR-A, SR-MARCO, and CD36 and adrenalgland lysates for SR-BI (data not shown). Data are mean 6 SEM offour independent experiments performed in duplicate. **P < 0.01,***P < 0.001 correspond to significant differences between micro-glia exposed to TGFb1 vs. those maintained under control conditions(ANOVA with Tukey’s post hoc).
Fig. 2. Effect of TGFb1 on the cell membrane distribution of scav-enger receptors in microglia. Live immunofluorescent labeling(FITC) for SR-A (A), SR-MARCO (B), and SR-BI (C) of cellsexposed to 2 ng/ml TGFb1 for 48 hr (SR-A) or 24 hr (SR-MARCO and SR-BI). After labeling with the primary antibody, cellswere fixed with methanol (Met-OH) to permeabilize them or withparaformaldehyde (PFA) to avoid their permeabilization to the sec-ondary antibodies and detect only the receptor present at the cell sur-face. Rhodamine-conjugated lectin was used as identity marker formicroglia. Nuclei were stained with DAPI. Scale bar 5 25 lm.
TGFb Regulates Ab Uptake by Microglia 1973
Journal of Neuroscience Research
to bind ligands or remained in an intracellular compart-ment, SRs immunolabeling was performed with livecells. Later, we fixed the cells either with PFA to mini-mize permeabilization or with 100% methanol to perme-abilize them to the secondary antibody.
TGFb1 induced morphological changes in micro-glia. Cells became round, whereas control cells weremore elongated. Labeling for SR-A at the cell mem-brane increased after treatment with TGFb1 comparedwith control cells. The increment was similar both in
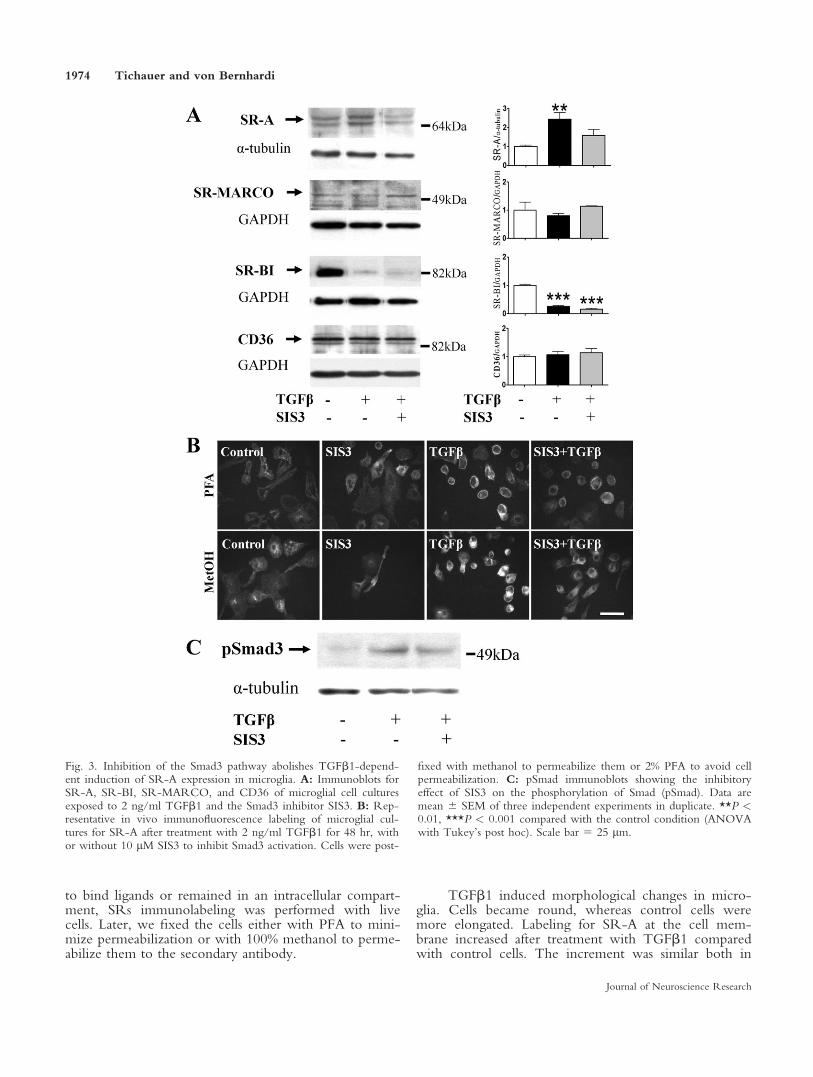
Fig. 3. Inhibition of the Smad3 pathway abolishes TGFb1-depend-ent induction of SR-A expression in microglia. A: Immunoblots forSR-A, SR-BI, SR-MARCO, and CD36 of microglial cell culturesexposed to 2 ng/ml TGFb1 and the Smad3 inhibitor SIS3. B: Rep-resentative in vivo immunofluorescence labeling of microglial cul-tures for SR-A after treatment with 2 ng/ml TGFb1 for 48 hr, withor without 10 lM SIS3 to inhibit Smad3 activation. Cells were post-
fixed with methanol to permeabilize them or 2% PFA to avoid cellpermeabilization. C: pSmad immunoblots showing the inhibitoryeffect of SIS3 on the phosphorylation of Smad (pSmad). Data aremean 6 SEM of three independent experiments in duplicate. **P <0.01, ***P < 0.001 compared with the control condition (ANOVAwith Tukey’s post hoc). Scale bar 5 25 lm.
1974 Tichauer and von Bernhardi
Journal of Neuroscience Research

MetOH- and in PFA-fixed cells (Fig. 2A), suggestingthat SR-A was located mainly at the cell surface inTGFb1-treated cultures. For SR-MARCO (Fig. 2B)and SR-BI (Fig. 2C), immunolabeling was distributedthroughout the whole cell. TGFb1 did not change SR-MARCO (Fig. 2B) and decreased SR-BI expression(Fig. 2C) compared with control cells. Labeling of thelisosomal membrane-associated glycoprotein Lamp-1 wasassayed as a control for cell permeabilization. Lamp-1was detected in methanol-fixed cells but not in PFA-fixed cells (data not shown).
TGFb1-Induced Expression of SR-A by MicrogliaDepends on Activation of the Smad3 Pathway
Because Smad is the main signaling pathway effec-tor for TGFb1 signaling and is involved in the inductionof the transcription of specific genes, we assessedwhether inhibition of Smad3 by SIS3, a specific Smad3inhibitor, affected TGFb1-induced changes in proteinexpression of SRs (Fig. 3A,C). Inhibition of the TGFb1Smad3 pathway resulted in a 59% decrease of the SR-Aexpression induced by TGFb1 (Fig. 3A). Inhibition ofSmad3 had no effect on basal SR-A expression, but itconspicuously decreased TGFb1-induced expression atthe membrane (Fig. 3B). In contrast, TGFb1-inducedreduction of SR-BI expression was not prevented by in-hibition of Smad3, and expressions of SR-MARCO andCD36 were not affected by TGFb1 or Smad3 inhibition(Fig. 3B).
Downregulation of LPS-Induced NO� Secretion byTGFb1 Is Mediated by the Smad3 Pathway
Activated microglia produce short-lived cytotoxicspecies such as ROS and NO�. LPS is a known inducerof NO� secretion, an effect that we have shown to bedownregulated by TGFb1 (Herrera-Molina and vonBernhardi, 2005). However, the identity of the TGFb1signaling pathway involved in such downregulation hasnot been clearly defined. To study signaling pathwaysinvolved in the downregulation of NO� production byTGFb1, microglial cell cultures were exposed to LPSalone or in combination with TGFb1, with or withoutpretreatment with the Smad3 inhibitor SIS3, the Alk5inhibitor SB431542, the ERK inhibitor PD98059, orthe p38 inhibitor SB203580. TGFb1 reduced LPS-induced NO� production by 83% (P 5 0.0023). Smad3inhibition did not prevent NO� secretion induced byLPS, but TGFb1 downregulation of LPS-induced NO�production was reduced by 60% (compare Fig. 4A,B).Alk5 inhibition resulted in a reduction of NO� produc-tion by LPS by 54% (P 5 0.01), without further effectof TGFb1 on LPS-induced NO� production (P 50.0019; Fig. 4C). In contrast, ERK and p38 inhibition(Fig. 4D,E) decreased LPS-induced NO� production by66% (P 5 0.0027) and 60% (P 5 0.0015), respectively.TGFb1 inhibited LPS-induced NO� production by 90%in the presence of MAPK inhibitors, similar to its effectin their absence.
Induction of Ab Uptake by TGFb1 Is Mediated bya Smad3-Dependent Mechanism
To test whether TGFb1 played a role in Abuptake and whether modification of phagocytotic activ-ity depended on the activation of Smad3 or otherTGFb1 signaling pathways, microglial cell cultures wereexposed to TGFb1 for 48 hr with or without pretreat-ment with the Smad3 inhibitor SIS3, the ERK inhibitorPD98059, or the p38 inhibitor SB203580. After treat-ment of cells with these inhibitors, uptake of Ab1–40-FluoIII was evaluated in a phagocytosis assay.
Fig. 4. Effect of TGFb pathway inhibitors on the induction ofnitrites by LPS in microglia. A: Microglial cells were cultured undercontrol conditions or were exposed to 1 lg/ml LPS, 2 ng/mlTGFb1, or combinations for 96 hr with or without preincubationwith inhibitors for Smad3 (B; 10 lM SIS3), TGFb1 receptor (C; 10lM SB431542), ERK (D; 20 lM PD98059), or p38 (E; 30 lMSB203580). NO22� content was determined by the Griess assay.NO22� levels were expressed as fold over control for each experi-mental condition. Data are mean 6 SEM of three to six independentexperiments performed in triplicate. Statistical analysis revealed highlysignificant differences for the four inhibitors tested, with P < 0.0001(ANOVA with Kruskal Wallis H-test). *P < 0.05, **P < 0.01,***P < 0.001 for comparisons with control condition and #P <0.05, ##P < 0.01, ###P < 0.001 for comparisons with the corre-sponding LPS condition; &P < 0.05, &&P < 0.01 show the signifi-cance when a condition was compared with the modulator effect ofTGFb1 on LPS-induced NO� production (Wilcoxon rank sum/Mann-Whitney U-test).
TGFb Regulates Ab Uptake by Microglia 1975
Journal of Neuroscience Research

Microglial cell cultures exposed to TGFb1 showeda 51% increase in the number of cells that took up Abcompared with the control condition (P < 0.001). Inhi-bition of the Smad3 pathway reduced induction of phag-ocytosis by TGFb1 by 84% (P < 0.001). In contrast, in-hibition of non-Smad transduction pathways, ERK andp38, did not significantly reduce the uptake of Ab1–40
induced by TGFb1 (Fig. 5A). Ab-loaded vesicles colo-calized with Lamp-1, an identity marker for lysosomesin the endocytic pathway. To corroborate these results,we overexpressed Smad7, the endogenous antagonist ofthe TGFb1 signaling pathway, using transfection assaysin the microglial cell line EOC20, which showed atransfection efficacy better than that in cultures obtainedfrom glial primary cultures (transfection efficacy 15%).We observed that the overexpression of Smad7 resultedin a clear reduction of TGFb1-mediated induction ofphagocytosis, which was correlated with an increasedimmunolabeling for Smad7 (Fig. 6).
Induction of Ab Uptake by TGFb1 Dependson SR Activity
To determine whether TGFb1-induced phagocy-tosis depended on the increased expression of SR-A, weused the SR ligand polyI as a control for SR-mediateduptake, evaluating the effect of the SR ligand polyI onthe phagocytosis of Ab. PolyI prevented Ab1–40 uptakeinduced by TGFb1 in a concentration-dependent man-ner, by 44.3% at 25 lg/ml and by 63.4% at 100 lg/ml.In fact, cells incubated with polyI showed a reduction ofphagocytosis, with uptake levels that were 30.6% and54.4% lower than those observed under control andTGFb1-induced conditions, respectively (Fig. 7).
DISCUSSION
Amyloid accumulation, glial cell activation, andincreased inflammatory signaling have been consideredpart of the molecular mechanisms underlying AD (Hardyand Selkoe, 2002; von Bernhardi, 2007). Here, we showthat the TGFb1 Smad3 pathway is involved in modula-tion of microglial cell activity through its effects on
Fig. 5. Participation of different TGFb1 signaling pathways inTGFb1-induced Ab uptake by microglia. A: Microglial cell culturesmaintained under control conditions or exposed to 2 ng/ml TGFb1for 48 hr with or without preincubation with inhibitors: 10 lM SIS3(Smad3 inhibitor), 20 lM PD98059 (ERK inhibitor), or 30 lMSB203580 (p38 inhibitor). LPS (1 lg/ml) was used as positive con-trol for the induction of phagocytosis. After stimulation with the var-ious experimental conditions for 48 hr, Ab uptake by microglia wasassessed with a phagocytosis assay using Fluo III-conjugated Ab for 3hr. The percentage of cells that took up Ab was evaluated. B:FITC-conjugated Ab together with immunofluorescence against thelissome marker Lamp-1 allowed assessing colocalization of Ab in thelysosomes. Data correspond to the mean 6 SEM of three to fiveindependent experiments in duplicate *P < 0.05, ***P < 0.001compared with control condition; ###P < 0.001 compared with thecorresponding LPS condition (ANOVA with Tukey’s post hoc test).Scale bars 5 25 lm.
1976 Tichauer and von Bernhardi
Journal of Neuroscience Research

expression of SRs, NO� secretion, and phagocytosis. Wefound a conspicuous increase in the expression of SR-Aat the cell membrane in neonatal rat microglia exposedto TGFb1; induction that was partially prevented by theinhibition of the Smad3 signaling pathway.
There is evidence that increased microglial cell ac-tivity specifically correlates with decrease of parenchymaAb plaques, suggesting that Ab clearance could be medi-ated by microglial cells (Wyss-Coray et al., 2001). Acti-vated microglia express different scavenger receptorssuch as SR-A, SR-MARCO, SR-BI, and CD36, all ofwhich appear to be capable of binding and taking up Ab(Rigotti et al., 1995; El Khoury et al., 1996; Husemannet al., 2001; Coraci et al., 2002; Alarcon et al., 2005)and also inducing secretion of NO� secondary to theinduction of inducible nitric oxide synthase (iNOS) tran-scription and expression. Microglial cell activation canbe initiated by various signal transduction pathways,including nuclear factor-jB, JAK/STAT, and p38 path-ways, which are activated by several inflammatory medi-ators and Ab (Kleinert et al., 2003; Ramirez et al.,
2008). The relevance of SR-A has been previously chal-lenged in studies with double-transgenic animal models,including the transgenic AD model, hAPP mice, andSR-A KO mice. In those double-transgenic animals,elimination of SR-A did not affect amyloid plaque for-mation (Huang et al., 1999). However, elimination ofSR-A could have been compensated by an increase inother receptors such as SR-MARCO, which have beenreported to be involved in the adhesion of astrocytes andmicroglia to Ab, as well as in Ab uptake (Alarcon et al.,2005). Furthermore, binding of SR ligands inducesinflammatory activation of glial cells (Godoy et al.,2012), an effect that is potentiated by Ab (Murgas et al.,2012).
We found that TGFb1 increased SR-A expression,decreased SR-BI expression, and did not modify SR-MARCO or CD36 expression. Only changes of SR-Aappeared to be dependent on Smad3 activation. Paradoxi-cally, even though in our model TGFb1 also inducedexpression of SR-A, it decreased LPS-induced NO� secre-tion through activation of the Smad3 pathway. It has beendescribed that polyI, a known SR-A ligand, inducesERK, JNK, and p38 phosphorylation and NO� produc-tion in RAW 264.7 macrophages and microglia (Campaet al., 2005; Murgas et al., 2012; Godoy et al., 2012),results that could be contradictory to our findings showingincreased SR-A expression and decreased LPS-induced
Fig. 7. Identification of the scavenger receptor on TGFb-mediateduptake of Ab by microglia. Microglial cell cultures were maintainedunder control conditions or exposed to 2 ng/ml TGFb1 for 48 hr,with or without coincubation with 25 lg/ml and 100 lg/ml polyI,SR ligands capable of competing for SR-mediated Ab uptake. Cellswere stimulated for 48 hr, and Ab uptake by microglial cells wasassessed by the phagocytosis of Fluo III-conjugated Ab for 3 hr,assessing the percentage of cells that took up Ab. Data are mean 6SEM of three or four independent experiments performed in dupli-cate. *P < 0.05, **P < 0.01 correspond to significance comparedwith control condition; ##P < 0.01, ###P < 0.001 correspond tosignificance compared with the corresponding LPS condition(ANOVA with Tukey’s post hoc test).
Fig. 6. Effect of Smad7 overexpression on TGFb1-mediated uptakeof Ab. The microglial cell line EOC20 was transfected with pactEF-MycSmad7 to overexpress Smad7 and block the TGFb Smad3 path-way (right; Smad7 plasmid). After transfection, microglial cells weretreated with TGFb for 24 hr, and Ab uptake by microglial cells wasassessed by a phagocytosis assay of Fluo III-conjugated Ab for 3 hr.Lower panels: Immunofluorescence labeling showing expression ofSmad7 in nontransfected and transfected cells as evidence of transfec-tion success. Scale bar 5 25 lm.
TGFb Regulates Ab Uptake by Microglia 1977
Journal of Neuroscience Research

NO� production. However, TGFb also induces MAPKphosphatase MKP1,2 expression through a Smad3 pathwaythat signals for the dephosphorylation of ERK and p38(Xiao et al., 2002; Eljaschewitsch et al., 2006; Rameshet al., 2008). In that sense, depending on the cellular con-text, TGFb could signal for both potentiation and inhibi-tion of NO� production. In our hands, both Alk5 andSmad3 blockers inhibited to a similar extent the downreg-ulation of TGFb1 over LPS-induced NO� production,suggesting that the effect depended mainly on an Smad-dependent pathway. Nevertheless, Alk5 blockers alsoaffected the magnitude of LPS-dependent induction ofNO�, an effect that depended on mechanisms that willrequire additional work for elucidation.
Increased SR-A expression was correlated withincreased Ab uptake, which was also dependent on theactivation of Smad3. The effect of the Smad3 inhibitorSIS3 was different on phagocytosis (reduction of 85%)compared with the reversal of NO� release (reduction of30%), indicating that Smad3 plays a more robust role inpromoting phagocytosis than in inhibiting NO� produc-tion. In contrast, induction of phagocytosis was not sig-
nificantly modified by ERK or p38 inhibition, two addi-tional signaling pathways that are also modulated byTGFb1 (Yu et al., 2002; Lee et al., 2007). SRs can beblocked by polyanionic molecules such as polyI thathave been shown to prevent Ac-LDL uptake in mouseliver (Van Berkel et al., 1998). The competition for SRwhen using polyI not only prevented the uptake of Abinduced by TGFb1 but decreased Ab uptake bellowcontrol levels, suggesting the presence of additional SRsbinding Ab. However, this effect is not specific for SR-A b ut also appears to affect different receptors such asSR-BI and SR-MARCO. On the other hand, the pro-tein levels of SR-BI decreased, whereas CD36 and SR-MARCO did not change, suggesting that these SRs ful-fill a minor role, if any, in the microglial induction ofTGFb.
In contrast to our results showing that TGFb1induced microglial cells responses that potentially coulddecrease accumulation of Ab, there is evidence thatincreased levels of TGFb1 mRNA and certain TGFb1polymorphisms found in the frontal cortex of ADbrains are associated with increased amyloid angiopathy
Fig. 8. Schematic model of the mechanism by which TGFb1 modu-lates NO� induction and Ab uptake by microglia. TGFb1 secretedby astrocytes activates its canonical pathway Smad3 in microglia,increasing SR-A at the plasma membrane, in turn inducing the
uptake of Ab and downregulating LPS-induced NO� secretion.Downregulation by TGFb1 of LPS-induced NO� secretion anduptake of Ab was decreased by Smad3-specific inhibition. Uptakewas prevented by blocking SRs with their ligand polyI.
1978 Tichauer and von Bernhardi
Journal of Neuroscience Research

(Wyss-Coray et al., 1997; Hamaguchi et al., 2005). Fur-thermore, hAPP/TGFb transgenic mice overexpressingTGFb1 and APP selectively in astrocytes develop anaccelerated deposition of Ab around blood vessels buthave decreased parenchyma Ab burden and reducednumbers of dystrophic neurites. In contrast, recent stud-ies in Tg2576 mice crossed with a dominant negativeTGFb1 receptor II that blocks TGFb1 Smad2/3 signal-ing show a marked attenuation of amyloid deposits inbrain parenchyma (Town et al., 2008). Although thismay seem contradictory to an antiamyloidogenic role forTGFb1, a possible explanation for the contradictoryeffect of TGFb1 on macrophage cells is that TGFb1Smad2/3 signaling appeared to be blocked only in pe-ripheral macrophages and not in microglia. In fact, pe-ripheral and brain TGFb1 levels were not determined,and the TGFb1 concentration could have been modifiedby the transgene, modulating microglial activation, butstill allowing macrophage infiltration. In that scenario,microglial cells, being the first line of defense in thebrain, could still have a relevant participation in Ab re-moval (Sastre et al., 2006). It is noteworthy that passiveimmunotherapy in AD mouse models shows decreasedAb plaques in parenchyma but increased cerebral amy-loid angiopathy (Pfeifer et al., 2002), as well as inhAPP/TGFb1 transgenic mice (Wyss-Coray et al.,2001), suggesting that, in mice treated by immunother-apy, an immunomodulation mediated by TGFb1 couldchange the microglial cell activation profile to a morephagocytic phenotype with reduced cytotoxic activity.
Our results are relevant for the understanding ofAD mechanisms, because there is evidence that TGFb1is increased in plasma and CSF of AD patients, whereastheir Smad3 mRNA levels are decreased in the hippo-campus (Colangelo et al., 2002; Rota et al., 2006). Inour in vitro study, we assessed the effect of TGFb1Smad3 pathway stimulation on microglial cell activation,the precise pathway that appears to be downregulated byaging and AD. Impairment of Ab deposit removal,which can result in increased inflammatory stimulationand induce a cytotoxic microglial phenotype, can lead toneurodegenerative processes, triggering a vicious circleleading to microglial cell cytotoxic activation (von Bern-hardi et al., 2007; Ramirez et al., 2008).
In summary, we have shown that TGFb1decreased LPS-induced NO� secretion and inducedphagocytosis mediated at least partially by the increasedexpression of SR-A. Both effects depended on Smad3activation, a pathway that appears to be impaired in ADbrains and could be one of the mechanisms favoring theaccumulation of Ab and inflammatory activation ofmicroglia (Fig. 8). As a next step, we are evaluating theactivation pattern induced by TGFb1 and inflammatorystimulus in microglial cell cultures derived from adultand aged mice.
ACKNOWLEDGMENTS
Plasmid pacEF-MycSmad7 was a kind gift fromDr. Kenji Okazaki, Biomolecular Engineering, Research
Institute, Osaka, Japan. The authors declare that theyhave no conflicts of interest.
REFERENCES
Alarcon R, Fuenzalida C, Santibanez M, von Bernhardi R. 2005.
Expression of scavenger receptors in glial cells. Comparing the adhesion
of astrocytes and microglia from neonatal rats to surface-bound beta-
amyloid. J Biol Chem 280:30406–30415.
Baig S, van Helmond Z, Love S. 2009. Tau hyperphosphorylation affects
Smad 2/3 translocation. Neurosci 163:561–570.
Block ML, Zecca L, Hong JS. 2007. Microglia-mediated neurotoxicity:
uncovering the molecular mechanisms. Nat Rev 8:57–69.
Campa VM, Iglesias JM, Carcedo MT, Rodriguez R, Riera J, Ramos S,
Lazo PS. 2005. Polyinosinic acid induces TNF and NO production as
well as NF-kappaB and AP-1 transcriptional activation in the monocy-
temacrophage cell line RAW 264.7. Inflamm Res 54:328–337.
Chalmers KA, Love S. 2007. Neurofibrillary tangles may interfere with
Smad 2/3 signaling in neurons. J Neuropathol Exp Neurol 66:158–167.
Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ.
2002. Gene expression profiling of 12633 genes in Alzheimer hippo-
campal CA1: transcription and neurotrophic factor down-regulation
and up-regulation of apoptotic and pro-inflammatory signaling. J Neu-
rosci Res 70:462–473.
Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella
GK, Luster AD, Silverstein SC, El-Khoury JB. 2002. CD36, a class B
scavenger receptor, is expressed on microglia in Alzheimer’s disease
brains and can mediate production of reactive oxygen species in
response to beta-amyloid fibrils. Am J Pathol 160:101–112.
El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike
JD. 1996. Scavenger receptor-mediated adhesion of microglia to beta-
amyloid fibrils. Nature 382:716–719.
Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S,
Hoertnagl H, Raine CS, Schneider-Stock R, Nitsch R, Ullrich O.
2006. The endocannabinoid anandamide protects neurons during CNS
inflammation by induction of MKP-1 in microglial cells. Neuron
49:67–79.
Godoy B, Murgas P, Tichauer J, von Bernhardi R. 2012. Scavenger
receptors class A ligands induce secretion of IL1b and exert a modula-
tory effect on inflammatory activation of astrocytes in culture. J Neuro-
immunol (accepted).
Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser
SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M,
Mathews PM, Wolburg H, Heppner FL, Jucker M. 2009. Formation
and maintenance of Alzheimer’s disease beta-amyloid plaques in the ab-
sence of microglia. Nat Neurosci 12:1361–1363.
Gugan D, Baker TJ. 1986. Characterization of ameboid microglia isolated
from devezo PIN6 mammalian brain. J Neurosci 8:2163–2178.
Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A,
Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, et al.
1992. Amyloid beta-peptide is produced by cultured cells during nor-
mal metabolism. Nature 359:322–325.
Hamaguchi T, Okino S, Sodeyama N, Itoh Y, Takahashi A, Otomo E,
Matsushita M, Mizusawa H, Yamada M. 2005. Association of a poly-
morphism of the transforming growth factor-beta1 gene with cerebral
amyloid angiopathy. J Neurol Neurosurg Psychiatry 76:696–699.
Hardy J, Selkoe DJ. 2002. The amyloid hypothesis of Alzheimer’s disease:
progress and problems on the road to therapeutics. Science 297:
353–356.
Herrera-Molina R, von Bernhardi R. 2005. Transforming growth factor-
beta 1 produced by hippocampal cells modulates microglial reactivity in
culture. Neurobiol Dis 19:229–236.
Huang F, Buttini M, Wyss-Coray T, McConlogue L, Kodama T, Pitas
RE, Mucke L. 1999. Elimination of the class A scavenger receptor does
not affect amyloid plaque formation or neurodegeneration in transgenic
TGFb Regulates Ab Uptake by Microglia 1979
Journal of Neuroscience Research

mice expressing human amyloid protein precursors. Am J Pathol
155:1741–1747.
Husemann J, Loike JD, Kodama T, Silverstein SC. 2001. Scavenger re-
ceptor class B type I (SR-BI) mediates adhesion of neonatal murine
microglia to fibrillar beta-amyloid. J Neuroimmunol 114:142–150.
Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. 2002.
Scavenger receptors in neurobiology and neuropathology: their role on
microglia and other cells of the nervous system. Glia 40:195–205.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. 2011. Physiology
of microglia. Physiol Rev 91:461–553.
Kleinert H, Schwarz PM, Forstermann U. 2003. Regulation of the expres-
sion of inducible nitric oxide synthase. Biol Chem 384:1343–1364.
Krieger M, Herz J. 1994. Structures and functions of multiligand lipopro-
tein receptors: macrophage scavenger receptors and LDL receptor-
related protein (LRP). Annu Rev Biochem 63:601–637.
Lee HG, Ueda M, Zhu X, Perry G, Smith MA. 2006. Ectopic expres-
sion of phospho-Smad2 in Alzheimer’s disease: uncoupling of the trans-
forming growth factor-beta pathway? J Neurosci Res 84:1856–1861.
Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith
SM, Derynck R. 2007. TGF-beta activates Erk MAP kinase signalling
through direct phosphorylation of ShcA. EMBO J 26:3957–3967.
Lesne S, Docagne F, Gabriel C, Liot G, Lahiri DK, Buee L, Plawinski L,
Delacourte A, MacKenzie ET, Buisson A, Vivien D. 2003. Transform-
ing growth factor-beta 1 potentiates amyloid-beta generation in astro-
cytes and in transgenic mice. J Biol Chem 278:18408–18418.
Marzolo MP, von Bernhardi R, Inestrosa NC. 1999. Mannose receptor
is present in a functional state in rat microglial cells. J Neurosci Res
58:387–395.
Marzolo MP, von Bernhardi R, Bu G, Inestrosa NC. 2000. Expression of
alpha2-macroglobulin receptor/low density lipoprotein receptor-related
protein (LRP) in rat microglial cells. J Neurosci Res 60:401–411.
Meda L, Baron P, Scarlato G. 2001. Glial activation in Alzheimer’s dis-
ease: the role of Abeta and its associated proteins. Neurobiol Aging
22:885–893.
Motta M, Imbesi R, Di Rosa M, Stivala F, Malaguarnera L. 2007.
Altered plasma cytokine levels in Alzheimer’s disease: correlation with
the disease progression. Immunol Lett 114:46–51.
Murgas P, Godoy B, von Bernhardi R. 2012. Ab potentiates inflamma-
tory activation of glial cells induced by Scavenger receptor ligands in
culture. Neurotox Res DOI: 10.1007/s12640–011-9306-3.
Paresce DM, Ghosh RN, Maxfield FR. 1996. Microglial cells internalize
aggregates of the Alzheimer’s disease amyloid beta-protein via a scav-
enger receptor. Neuron 17:553–565.
Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel
M, Mathews PM, Jucker M. 2002. Cerebral hemorrhage after passive
anti-Abeta immunotherapy. Science 298:1379.
Pfeiffer S, Gorren AC, Schmidt K, Werner ER, Hansert B, Bohle DS,
Mayer B. 1997. Metabolic fate of peroxynitrite in aqueous solution.
Reaction with nitric oxide and pH-dependent decomposition to nitrite
and oxygen in a 2:1 stoichiometry. J Biol Chem 272:3465–3470.
Ramesh S, Qi XJ, Wildey GM, Robinson J, Molkentin J, Letterio J,
Howe PH. 2008. TGF beta-mediated BIM expression and apoptosis
are regulated through SMAD3-dependent expression of the MAPK
phosphatase MKP2. EMBO Rep 9:990–997.
Ramirez G, Rey S, von Bernhardi R. 2008. Proinflammatory stimuli are
needed for induction of microglial cell-mediated AbetaPP_{244-C}
and Abeta-neurotoxicity in hippocampal cultures. J Alzheimers Dis
15:45–59.
Rigotti A, Acton SL, Krieger M. 1995. The class B scavenger receptors
SR-BI and CD36 are receptors for anionic phospholipids. J Biol Chem
270:16221–16224.
Rota E, Bellone G, Rocca P, Bergamasco B, Emanuelli G, Ferrero P.
2006. Increased intrathecal TGF-beta1, but not IL-12, IFN-gamma and
IL-10 levels in Alzheimer’s disease patients. Neurol Sci 27:33–39.
Sastre M, Klockgether T, Heneka MT. 2006. Contribution of inflamma-
tory processes to Alzheimer’s disease: molecular mechanisms. Int J Dev
Neurosci 24:167–176.
Saud K, Herrera-Molina R, Von Bernhardi R. 2005. Pro- and anti-
inflammatory cytokines regulate the ERK pathway: implication of the
timing for the activation of microglial cells. Neurotox Res 8:277–287.
Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman
RS, Flavell RA. 2008. Blocking TGF-beta-Smad2/3 innate immune
signaling mitigates Alzheimer-like pathology. Nat Med 14:681–687.
Uribe-San Martın R, Herrera-Molina R, Olavarrıa L, Ramırez G, von
Bernhardi R. 2009. Reduction of b-amyloid-induced neurotoxicity on
hippocampal cell cultures by moderate acidosis is mediated by TGFb.
Neurosci 158:1338–1347.
Van Berkel TJ, Van Velzen A, Kruijt JK, Suzuki H, Kodama T. 1998.
Uptake and catabolism of modified LDL in scavenger-receptor class A
type I/II knock-out mice. Biochem J 331:29–35.
von Bernhardi R. 2007. Glial cell dysregulation: a new perspective on
Alzheimer disease. Neurotox Res 12:215–232.
von Bernhardi R, Eugenın J. 2004. Microglial reactivity to beta-amyloid
is modulated by astrocytes and proinflammatory factors. Brain Res
1025:186–193.
von Bernhardi R, Ramirez G, Toro R, Eugenın J. 2007. Pro-inflamma-
tory conditions promote neuronal damage mediated by amyloid precur-
sor protein and decrease its phagocytosis and degradation by microglial
cells in culture. Neurobiol Dis 26:153–164.
Wrighton KH, Lin X, Feng XH. 2009. Phospho-control of TGF-beta
superfamily signaling. Cell Res 19:8–20.
Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood
K, Lin C, Mucke L. 1997. Amyloidogenic role of cytokine TGF-beta1
in transgenic mice and in Alzheimer’s disease. Nature 389:603–606.
Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Mas-
liah E, Mucke L. 2001. TGF-beta1 promotes microglial amyloid-beta
clearance and reduces plaque burden in transgenic mice. Nat Med
7:612–618.
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Sil-
verstein SC, Husemann J. 2003. Adult mouse astrocytes degrade amy-
loid-beta in vitro and in situ. Nat Med 9:453–457.
Xiao YQ, Malcolm K, Worthen GS, Gardai S, Schiemann WP, Fadok
VA, Bratton DL, Henson PM. 2002. Cross-talk between ERK and p38
MAPK mediates selective suppression of pro-inflammatory cytokines by
transforming growth factor-beta. J Biol Chem 277:14884–14893.
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L,
Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt
AM. 1996. RAGE and amyloid-beta peptide neurotoxicity in Alzhei-
mer’s disease. Nature 382:685–691.
Yu L, Hebert MC, Zhang YE. 2002. TGF-beta receptor-activated p38
MAP kinase mediates Smad-independent TGF-beta responses. EMBO J
21:3749–3759.
Zhang YE. 2009. Non-Smad pathways in TGF-b signaling. Cell Res
19:128–139.
1980 Tichauer and von Bernhardi
Journal of Neuroscience Research