sepsis: etiología, manifestaciones clínicas y diagnóstico
TRANSCRIPT

MEDICRIT Revista de Medicina Interna y Medicina Crítica
Indexada en IMBIOMED y en PERIÓDICA, Base de datos de la
Universidad Nacional Autónoma de México, UNAM.
Revisión
Sepsis: Etiología, Manifestaciones Clínicas y Diagnóstico Indira Briceño M.D. Médica Internista Intensivista, adjunta al Servicio de Emergencia de Adultos del Hospital Universitario de los Andes, Mérida. Venezuela email: [email protected]
Recibido el 14 de Abril de 2005. Aceptado el 30 de Julio de 2005
Medicrit 2005; 2(9): 203–213 ETIOLOGÍA a infección bacteriana es la causa más común de sepsis y shock séptico, siendo los gérmenes gram- negativos los más frecuentemente involucrados, seguidos muy de cerca por los microorganismos grampositivos (1). Los virus también pueden verse invo-lucrados como causa de sepsis, sobre todo en individuos con inmunocompromiso grave existe amplia evidencia de que un cuadro de shock séptico florido puede ser causado por virus del herpes y la infección más grave puede ser por citomega-lovirus en receptores de transplante de médula ósea. En el paciente inmunocomprometido también es habitual el her-pes zoster diseminado con síndrome infeccioso acompañado de erupción cutánea vesicular, a menudo de neumonía difu-sa y con menor frecuencia afección del sistema nervioso central o hepática. El virus del herpes simple también llega a causar síndrome infeccioso evidente en estas personas, raras veces afección visceral; con menor frecuencia este virus ocasiona también un cuadro séptico con CID y hepatitis ful-minante en mujeres embarazadas por lo demás inmunocom-petentes. El virus del dengue y los enterovirus suelen verse también generando cuadros de shock séptico. Otras causas no bacterianas son los parásitos de los cuales el Plasmodium falciparum, las rickettsiosis (2) y los hongos. Los sitios más frecuentes de infección son los pulmones (40%), intraabdominal (30%), tracto urinario (10%), infec-ción de tejidos blandos (5%) e infección de un catéter intra-vascular (5%) (3,4). La bacteriemia aparece en el 40-60% de los pacientes con shock séptico. En un 10-30% de los pa-cientes los microorganismos causales no pueden ser aisla-dos, posiblemente debido a la exposición previa a los anti-bióticos (1). MANIFESTACIONES CLÍNICAS El médico enfrenta la tarea de determinar la causa pro-bable del cuadro clínico para que pueda instituirse un trata-miento apropiado (5). Es muy importante reconocer comor-bilidades asociadas, sobre todo las enfermedades que pue-dan afectar la capacidad inmunológica del huésped como la diabetes, insuficiencia renal, insuficiencia hepática, desnu-trición, tumores malignos, SIDA y el tratamiento con drogas inmunosupresoras y corticoesteroides.
Fiebre o Hipotermia
L Los registros de temperatura deben ser centrales o recta-les. La fiebre se produce por el efecto de las citoquinas IL-1 y TNFα liberados por los macrófagos activados (6,7). Aunque la fiebre y los escalofríos son típicos, algunos pa-cientes que desarrollan infecciones bacterianas sistémicas están debilitados y no exhiben cambios llamativos (por e-jemplo, escalofríos) al comienzo de una infección (4,5). La causa de la hipotermia es menos conocida y su presencia se asocia con mal pronóstico (5). Respecto a la frecuencia de presentación son interesantes los datos de la investigación de Le Gall y colaboradores so-bre 1.130 pacientes con sepsis. En ese estudio presentaron fiebre (mayor de 38°C) el 55% de los pacientes mientras que en el 15% se observó hipotermia (menor de 36 °C) y en el 30% restante la temperatura corporal era normal. Compromiso Cardiovascular Inicialmente los mediadores celulares y toxinas bacteria-nas causan shock circulatorio de tipo distributivo, manifes-tado por disminución de la TA, incremento de la frecuencia cardiaca (FC), disminución de la resistencia vascular sisté-mica (RVS) e incremento del gasto cardiaco (GC); cuando no se incrementa el GC se debe a hipovolemia y la reanima-ción hídrica puede mejorar el GC. Posteriormente se agrega el shock circulatorio de origen cardiogénico que se caracte-riza por disfunción sistólica y diastólica (8) Durante el shock séptico la taquicardia y la reducción de la postcarga incrementan el GC; sólo en caso de hipovole-mia el GC estará disminuido, esto se debe principalmente al mecanismo de Frank-Starling, pero paradójicamente hay de-presión miocárdica intrínseca. Desde 1984 con los trabajos y publicaciones iniciales de Parker, se notó que la disfunción sistólica del ventrículo izquierdo se caracterizaba por dilata-ción de las cavidades del ventrículo izquierdo, incremento de los volúmenes intracavitarios, aumento de la compliance del mismo ventrículo y disminución de la fracción de eyec-ción del ventrículo izquierdo (FEVI). Esta disfunción sistó-lica se inicia en las primeras 24 horas del choque séptico y es reversible en los sobrevivientes en 7 a 10 días (9). Los cambios hemodinámicos de la disfunción sistólica no se pre-sentan en forma importante en aquellos pacientes que no so-breviven al choque séptico (9), por lo que se considera co-mo una medida compensadora del ventrículo izquierdo a la
Indira Briceño Sepsis: etiologia, clínica y diagnóstico
Medicrit 2005; 2(9):203-213 www.medicrit.com 203

depresión miocárdica durante el choque séptico, con el fin de mantener el GC a través del mecanismo de Frank-Star-ling. Por lo tanto los cambios hemodinámicos de la disfun-ción sistólica son considerados también como factor pronós-tico. El índice del volumen latido (IVL) es a menudo nor-mal, el índice del trabajo latido (ITL) está reducido al igual que la FEVI debido a estos cambios hemodinámicos que su-fre el ventrículo izquierdo; así mismo la relación de presión en “cuña” de la arteria pulmonar (PCAP)-ITL se desvía ha-cia la derecha y hacia abajo denotándose la disfunción sis-tólica del ventrículo izquierdo (8). Los cambios sufridos en el ventrículo izquierdo, también son observados en el ventrí-culo derecho; se dilata la cavidad y disminuye la fracción de eyección. Dado lo anterior durante la etapa de reposición de volumen se debería monitorizar la función cardiaca con la relación PCAP-ITL del ventrículo izquierdo (figura 1). Teniendo en mente que el GC o índice cardiaco (IC) da una pobre valoración de la función cardiaca intrínseca, se reco-mienda la monitorización de la FEVI con ecocardiograma (8). Disfunción diastólica No sólo hay disfunción sistólica del ventrículo izquierdo durante el choque séptico, también la hay diastólica, con al-teraciones en la relajación y distensibilidad del ventrículo izquierdo, valorados ecocardiograficamente a nivel de la válvula mitral y es debido principalmente al incremento del volumen intracavitario y disfunción diastólica del VI; en los sobrevivientes estas anormalidades se recuperan. Las altera-ciones diastólicas son por disminución de la onda E, e incre-mento de la onda A, denotando alteración de la relajación del VI, tanto en shock séptico o en la sepsis sin shock. Nor-ton demostró alteraciones de estas ondas en la disfunción diastólica, pero no los correlacionó con la mortalidad. Munt encontró disfunción diastólica del VI manifestado como prolongación del tiempo de desaceleración de la onda E, siendo un predictor independiente de mortalidad en sepsis severa (8). Mecanismos etiológicos Se han postulado diversas hipótesis para tratar de expli-car la disfunción cardíaca en la sepsis, pero ninguna ha lo-grado reunir y satisfacer a plenitud las dudas planteadas. En-tre las hipótesis más aceptables tenemos las siguientes: Isquemia miocárdica: Diversos estudios han demostrado que el flujo sanguí-neo coronario no se encuentra reducido, la extracción de O2 está acortada, comportándose como cualquier vascula-tura sistémica durante la sepsis, la producción de lactato miocárdico no está elevada, ni la producción de fosfatos de alta energía; esta teoría es muy cuestionada.
Disfunción microvascular: Durante el choque de cualquier etiología y durante la re-animación hídrica aparece la disfunción microvascular por acumulación de leucocitos y plaquetas en los capila-res, hay fuga de líquidos causando edema intersticial, hay parálisis de la relajación microvascular y producción de radicales libres de O2 así como producción de sustancias cardiodepresoras, todo esto pudiera causar relativa isque-mia por alteración del flujo sanguíneo.
Sustancias depresoras del miocardio: Se han detectado varias sustancias derivadas o produci-das por bacterias, leucocitos y en el endotelio vascular que han demostrado actividades de depresión miocárdica
en relación directa con sus concentraciones séricas. Estos productos podrían actuar en forma sinérgica para ocasio-nar la depresión miocárdica durante la sepsis, siendo las principales sustancias el TNFα y la IL-1b. Las citoquinas tienen un papel bifásico: uno inicial, con rápida influencia directa sobre el miocardio que se puede bloquear en los experimentos con animales con anticuerpos anti-TNFα y con bloqueadores de receptores celulares de IL-1b. El otro efecto es tardío secundario a la producción de sintetasa del óxido nítrico y sintetasa de la ciclooxigenasa, ambas sustancias producen sustancias depresoras del miocardio. Otra acción de estas citoquinas es de reducir el calcio in-tracelular en el miocito, el cual es importante para la con-tracción miocárdica (10).
Figura 1—Monitoreo de la Función Ventricular Izquierda. Relación entre LVSWI (Indice de Trabajo Sistólico del Ventrículo Izquierdo) y la PWAP (Presión en Cuña de la Arteria Pulmonar) para valorar y monitorizar la función contráctil miocárdica del ventrículo izquierdo
Endotelio vascular: Las células endoteliales participan en los trastornos he-modinámicos durante la sepsis por descarga del factor ac-tivador de plaquetas (PAF), el cual se ha demostrado en modelos animales tiene un efecto inotrópico negativo (10). El endotelio vascular descarga factores cardioactivos con regulación endocrina, tipo paracrina de la función miocárdica; estas son la endotelina y el óxido nítrico. La endotelina es un potente vasoconstrictor 10 veces más potente que la angiotensina II. El óxido nítrico (NO) es una sustancia que proviene de la sintetasa del óxido nitri-co (NOS2), esta enzima normalmente no se encuentra pre-sente en el endotelio vascular coronario, endotelio endo-cárdico y miocitos cardíacos, pero se manifiesta cuando hay exposición a diversas citoquinas durante la sepsis. El NO causa vasodilatación e hipotensión arterial sistémicas; así mismo incremento de la relajación miocárdica e incre-mento de la complianza diastólica que llevaría al incre-mento del volumen diastólico final y aumento de la pre-sión diastólica final del VI. Otro efecto del NO es de ser antagonista de los β-adrenérgicos, lo mismo que induce menor respuesta del miocito al calcio intracelular. Todos estos efectos del NO es por elevación intracelular del 3’5-guanosinmonofosfato cíclico (GMPc). Recientemente se ha aislado una sustancia no identificada aún descargada del endotelio hipóxico, que inhibe el ciclaje de los puentes cruzados miocárdicos, encargados de la contracción del miocito, por inhibición del ATP de la miosina cardiaca activado por la actina (8).
Indira Briceño Sepsis: etiologia, clínica y diagnóstico
Medicrit 2005; 2(9):203-213 www.medicrit.com 204

Alteración de receptores β adrenérgicos: Durante la sepsis es evidente la disminución de la res-puesta del miocardio a la estimulación de las catecolami-nas. Las catecolaminas para poder ejercer su efecto ino-trópico positivo primero forman un complejo integrado por el fármaco β-agonista, el receptor de la membrana ce-lular y de la proteína reguladora nucleótido de guanina (proteína N); este complejo es temporal y desestabilizado rápidamente por la GTP descargando GDP, la molécula formada N-GTP cataliza a la molécula de adenilciclasa, produciéndose AMPc, que es el encargado de los eventos de inotropismo a través de la fosforilización de las proteí-nas intracelulares. La alteración a nivel de los receptores puede llevarse a cabo en cualquiera de los pasos antes ci-tados mediante los siguientes mecanismos:
1. Competencia por la proteína N 2. Fenómeno de taquifilaxia o desensibilización, que de-
pende en forma directa del tiempo y concentración del agonista.
3. Regulación en baja 4. Internalización de receptores β adrenérgicos en la
membrana celular. 5. Alteración del acoplamiento receptor-ciclasa, que
puede ser secundario a la endotoxina y la actividad simpático-adrenal.
Alteración del flujo microvascular La respuesta inflamatoria altera en forma importante la hemostasia circulatoria. La oxigenación tisular depende de la satisfacción de la demanda; es decir, de un equilibrio en-tre la utilización (VO2) y la oferta de O2 (IDO2). El índice de disponibilidad de oxígeno (IDO2), depende de la oxigena-ción sanguínea (CaO2) y el flujo o gasto cardiaco (GC). Cuando cualquiera de estos parámetros se altera, el IDO2 baja y el órgano compensa esto con un incremento de la ex-tracción de oxígeno (ERO2), esto sucede hasta cierto punto en el cual el ERO2 no se incrementa más y baja conforme desciende el IDO2, a esto se le llama disoxia. Durante el es-tado de shock hay diversificación de flujo no sólo entre los órganos, lo hay también dentro de un mismo órgano. Cuan-do el IDO2 disminuye, se sacrifica el flujo sanguíneo a ór-ganos que son capaces de incrementar la ERO2, es decir; no son dependientes del flujo, y se incrementa el flujo hacia ór-ganos con actividad metabólica elevada, como son el cere-bro y el corazón. Todo esto por incremento del tono vaso-motor simpático. Pero este incremento de flujo al órgano no garantiza que la microvasculatura capilar reciba el suficiente flujo sanguíneo. El flujo microvascular también está sujeto al control de metabolitos vasodilatadores (NO) y es determi-nado por flujo corriente y no por gradiente de presión, por los esfínteres pre-capilares. Durante la sepsis el tono vaso-constrictor está alterado (11), y predomina el tono vasodila-tador , este último por descarga del óxido nítrico (NO) por el endotelio vascular. También la hiperemia reactiva es blo-queada en los pacientes sépticos así como la capacidad de la microcirculación del músculo esquelético para la distribu-ción de los eritrocitos y por consiguiente la oxigenación ti-sular (8). Ecocardiografía y función cardíaca La ecocardiografía es un método útil para la valoración integral del funcionamiento del corazón en cualquier tipo de anomalía que sufra este órgano. Con este método podemos valorar la función sistólica y la función diastólica así como anomalías estructurales intrínsecas y extrínsecas del corazón.
Función sistólica: podemos valorar la función sistólica del corazón y a través de la medición de las dimensiones sistólicas y diastólicas en modo M (MM) y bidimensional (BD). Siendo más preciso la utilización BD se utiliza la si-guiente fórmula para calcular la Fracción de Eyección del ventrículo izquierdo (FEVI): FEVI = (Diámetro diastólico)3 - (Diámetro sistólico)3 /(Diámetro diastólico)3 Normal: > 50%. Se puede calcular la fracción de acortamiento del ventrí-culo izquierdo (FRAc) que indica el acortamiento que su-fren las paredes del ventrículo izquierdo de la sístole a la diástole. Calculándose con la siguiente fórmula: Fr Ac = Diámetro diastólico-diámetro sistólico/ Diámetro diastólico Función diastólica: Se valora con Doppler pulsado a ni-vel de la entrada del ventrículo izquierdo, es decir a nivel de la válvula mitral; normalmente se visualizan dos ondas: la onda E que significa el llenado rápido del ventrículo izquier-do y la onda A que significa el llenado del VI con la con-tracción auricular. La relación E: A debe ser de 2:1 normal-mente, así como su velocidad de desaceleración (VDA) de E normal es de 200 ± 40 mseg (figura x). Hay 3 tipos de disfunción diastólica (8): Grado I: donde se invierte la relación E:A (1:2) y la VDA es mayor de 200 m/seg.
Grado II: aquí la relación E:A y la VDA se conservan pero están pseudonormalizadas.
Grado III: la onda E es más pronunciada y la A casi no se manifiesta, siendo la relación E:A más de 2:1 y la VDA es menor de 200 m/seg.
Grado IV: disfunción diastólica irreversible. Trastornos vasculares periféricos Hay alteración de la regulación del tono vasomotor y los pacientes tienen resistencias vasculares sistémicas disminui-das. Esta caída de la resistencia vascular sistémica contribu-ye al desarrollo de hipotensión arterial. El óxido nítrico es el mediador fundamental en este proceso, entre otros factores causales, tales como la deficiencia de hormonas endógenas principalmente la vasopresina y el cortisol, que contribuyen a la disminución del tono vascular arteriolar, la elevación de los niveles séricos del factor natriurético, de la adenosina y de un péptido relacionado con el gen de la calcitonina tam-bién han sido involucrado (12). Todos estos cambios hemo-dinámicos antes descritos, aunados a una resistencia o pobre respuesta a la terapia con drogas vasopresoras caracterizan al shock vasodilatador. La sepsis es la causa más frecuente de shock vasodilata-dor. En todas las formas de shock vasodilatador que se han examinado, las concentraciones plasmáticas de catecolami-nas están incrementadas y el sistema renina-angiotensina es-tá activado. Además, está claro que la vasodilatación y la hi-potensión son debidas a la falla de la constricción en el mús-culo liso vascular. Muchos mecanismos han sido propuestos para explicar esta falla, estos incluyen la muerte de la célula vascular debido a la hipotensión prolongada, inadecuada ex-tracción de oxígeno por los tejidos y un incremento de la ac-tividad de las prostaglandinas con efecto vasodilatador (13). Trabajos recientes han esclarecido los mecanismos res-ponsables de la función normal del músculo liso vascular y han proporcionado una variedad de herramientas fármaco-lógicas para dilucidar la alteración del músculo liso vascular en el shock vasodilatador. Por la importancia clínica de esto, el shock séptico es la forma más estudiada del shock vasodi-latador.
Indira Briceño Sepsis: etiologia, clínica y diagnóstico
Medicrit 2005; 2(9):203-213 www.medicrit.com 205

Tres mecanismos parecen estar involucrados en este síndro-me: Activación de los canales de potasio ATP sensibles en la membrana plasmática del músculo liso vascular.
Activación de la enzima inducible óxido nítrico sintetasa. Deficiencia de la hormona vasopresina.
Activación de los canales de potasio ATP (KATP) sensibles en la membrana plasmática del músculo liso vascular Una vasoconstricción adecuada requiere que los ligandos hormonales y neuronales como la angiotensina II y norepi-nefrina se unan y activen los receptores en la superficie de la célula del músculo liso vascular, por vía de segundos men-sajeros, aumentando la concentración de calcio (Ca++) en el citosol. Este incremento resulta de la liberación de calcio desde los depósitos intracelulares y de la entrada de calcio extracelular a través de los canales de calcio voltaje-depen-dientes. La alta concentración citoplasmática de calcio forma un complejo con la calmodulina y este complejo acti-va la kinasa que regula la fosforilación de la cadena liviana de miosina. La fosforilación de la miosina permite la activa-ción de la miosinATPasa por la actina y el ciclado de los puentes cruzados de miosina por medio de los filamentos de actina, un proceso que contrae al músculo. Conjuntamente los vasodilatadores como el péptido na-triurético auricular y el óxido nítrico activan a la kinasa que por interacción con la miosinfosfatasa, desfosforila la miosi-na y previene la contracción muscular (13). Además de este bien conocido mecanismo que modula el tono vascular, un reciente trabajo indica que el potencial de membrana de la célula del músculo liso vascular puede tener un rol impor-tante en este proceso. Ciertamente la vasodilatación patoló-gica y la resistencia a los fármacos vasopresores, que carac-terizan al shock vasodilatador no pueden entenderse sin una apreciación detallada del rol del potencial de membrana en la regulación del tono vascular (13). El potencial de reposo de la membrana de la célula del músculo liso vascular oscila entre -30 y -60mV, dependien-do del tipo de células. Un potencial más positivo (despola-rización) abre los canales de calcio voltaje-dependientes, au-mentando la concentración de calcio citoplasmático e indu-ciendo vasoconstricción. Consecuentemente la hiperpolari-zación cierra estos canales, disminuyendo la concentración de calcio en el citoplasma e induciendo vasodilatación. Ade-más las sustancias vasoconstrictoras requieren que el calcio extracelular entre a la célula, la hiperpolarización de la mem-brana previene la vasoconstricción en presencia de los ligan-dos vasoconstrictores. Una variedad de transportadores de iones y canales, par-ticularmente canales de potasio, contribuyen con el poten-cial de membrana de la célula del músculo liso vascular. De los cuatro tipos de canales de potasio conocidos en la mem-brana plasmática de la célula del músculo liso vascular, el canal de KATP es el mejor conocido y tiene un rol crítico en la patogénesis del shock vasodilatador. La apertura de los canales KATP permite un eflujo de potasio, esto hiperpo-lariza la membrana plasmática y previene la entrada de cal-cio a la célula. Esto explica porque la activación farmacoló-gica de los canales K-ATP con diazóxido por ejemplo, inhibe la vasoconstricción inducida por las catecolaminas o angiotensina II. Los canales K-ATP son fisiológicamente activados por la disminución de la concentración de ATP en la célula y un aumento de las concentraciones celulares de hidrógeno (H+) y de lactato, un mecanismo que enlaza el metabolismo celular con el tono vascular y el flujo sanguí-
neo. Esto se evidencia bajo condiciones normales de reposo, donde los canales K-ATP son cerrados y sus inhibidores, tales como las sulfonilureas no causan vasoconstricción. Ba-jo condiciones de aumento del metabolismo tisular o hipo-xia tisular, sin embargo, la activación de estos canales causa vasodilatación, la cual puede ser revertida con una sulfonilu-rea. La activación de los canales K-ATP en las arteriolas es un mecanismo crítico en la hipotensión y vasodilatación carac-terísticas del shock vasodilatador. Es por esta razón, que la administración de sulfonilureas aumenta la presión arterial y la resistencia vascular en el shock vasodilator debido a hi-poxia, shock séptico posterior a la administración de lipo-polisacáridos y durante la fase final vasodilatadora del shock hemorrágico. Los canales K-ATP son también probablemen-te activados en el shock hemorrágico moderado y en el shock cardiogénico, contribuyendo a la ya tan baja resisten-cia vascular periférica observada en estas condiciones. Los activadores neurohormonales de los canales K-ATP pueden ser también involucrados en algunas formas de shock vasodilatador. Por ejemplo el péptido natriurético auricular, calcitonina y adenosina pueden activar los canales K-ATP. La concentración plasmática de estas sustancias está marca-damente elevada en el shock séptico, también las concen-traciones plasmáticas del péptido natriurético atrial está aumentado en la fase final vasodilatadora del shock hemo-rrágico. El canal K-ATP puede ser activado por un incre-mento del óxido nítrico a través de guanosinmonofosfatoci-clasa (GMPc). En resumen, muchas condiciones que comprometen la o-xigenación tisular y conllevan a acidosis láctica probable-mente activan los canales K-ATP en el músculo liso vascu-lar causando shock vasodilator (13). Incremento de la síntesis de óxido nítrico El incremento del óxido nítrico contribuye a la hipoten-sión y resistencia a las drogas vasopresoras que ocurre en el shock vasodilatador. El aumento de su producción se debe a un incremento de la expresión de la sintesa inducible del óxido nítrico, sin embargo su acción vasodilatadora en el shock séptico es mediada por la activación de la miosinfos-fatasa de cadena liviana. No obstante, el óxido nítrico puede causar vasodilatación por activación de los canales K en la membrana de la célula del músculo liso vascular, que son sensibles al calcio citoplasmático (canales KCa ), ya que en condiciones normales una de las funciones de este canal es mitigar los efectos vasonconstrictores. El incremento de la concentración citoplasmática de calcio en las células del músculo liso vascular, que es inducido por sustancias vaso-constrictoras como la norepinefrina, abre los canales KCa, lo cual hiperpolariza la membrana plasmática, previniendo la vasoconstricción. El óxido nítrico puede activar los cana-les KCa por dos mecanismos a saber: nitrosilación directa del canal y por activación de GMPc dependiente de la pro-teinkinasa. En humanos y en modelos animales la administración de inhibidores de la síntesis del óxido nítrico, durante el shock séptico incrementan la presión arterial y disminuyen las do-sis de catecolaminas necesarias para mantener la presión ar-terial (13). Deficiencia de vasopresina La conservación del agua es la acción más notable de la vasopresina, la cual es una hormona secretada bajo control osmótico, para regular la permeabilidad del túbulo colector
Indira Briceño Sepsis: etiologia, clínica y diagnóstico
Medicrit 2005; 2(9):203-213 www.medicrit.com 206

renal al agua. Sin embargo la vasopresina está también indo-lucrada en la homeostasis cardiovascular y es también secre-tada bajo control de los barorreceptores, produciendo vaso-constricción. Mientras la regulación del túbulo colector re-nal requiere de niveles plasmáticos de vasopresina de 1-7 pg/ml (0,9 a 6,5 pmol/L) sus efectos vasoconstrictores ocu-rren con concentraciones más altas de aproximadamente 10 a 200 pg/ml (9 a 187pmol/L) (13). Los efectos periféricos de la vasopresina son mediados por diferentes receptores, de-nominados V1(V1a), V2 y V3 (V1b). Los receptores V1 han sido encontrados en los vasos sanguíneos arteriales e indu-cen vasoconstricción, por incremento del calcio iónico cito-plasmático a través de la vía del fosfatidil-inositol-bisfos-fonato (12). Normalmente la vasopresina juega un papel me-nor en la regulación de la presión arterial, pero en respuesta a la hipotensión, tal como ocurre en el shock séptico o he-morrágico, la vasopresina es liberada por la neurohipófisis y su concentración en plasma aumenta considerablemente. Durante la fase inicial del shock hemorrágico y shock séptico la vasopresina contribuye al igual que otros vaso-constrictores al mantenimiento de la presión arterial (14). Además los agentes que bloquean el receptor de la vaso-presina en el músculo liso vascular, disminuyen la presión arterial tanto en el shock hemorrágico como en el shock séptico; y animales con diabetes insípida toleran muy mal el shock (13). Cuando la hipotensión persiste por más de una hora, las altas concentraciones iniciales de vasopresina, disminuyen en el plasma, el mecanismo responsable de esta disminución todavía está por determinarse, pero se sabe que los depósitos de vasopresina en la neurohipófisis pueden depletarse des-pués de una marcada estimulación osmótica, también esta depleción ocurre como consecuencia de una estimulación sostenida de los barorreceptores. Un soporte de esta hipó-tesis es el análisis inmunohistoquímico de la neurohipófisis de perros, que reveló que la vasopresina desaparece después de una hora de hipotensión secundaria a hemorragia severa. En contraste con las catecolaminas los efectos de la vasopre-sina son preservados durante la hipoxia y la acidosis severa. El efecto vasopresor es mayor en la vasculatura esplácnica, muscular y cutánea (12). En el shock vasodilatador se han descrito otros mecanis-mos de acción de la vasopresina independientes del receptor V1 para generar vasoconstricción, entre los cuales podemos mencionar (12,13): El bloqueo de la actividad de los canales K-ATP, que fa-cilita la despolarización del miocito y produce vasocons-tricción.
La vasopresina atenúa la estimulación de la producción de NO por parte de la endotoxina e IL-1β, inhibiendo la va-sodilatación excesiva.
Ella disminuye además las concentraciones intracelulares del GMPc, el cual es el segundo mensajero del NO.
Puede revertir la regulación en baja delos receptores adre-nérgicos.
Induce a través de la estimulación de los receptores V3 (V1b) la producción de hormona adrenocorticotropa (AC TH) y cortisol, quienes se encuentran disminuidos en un número importante de pacientes en shock séptico. El in-cremento de los niveles plasmáticos de cortisol se ha rela-cionado con reducción del tiempo necesario para realizar el cese de la terapia con vasopresina, así como también se ha asociado con una resolución más temprana de la dis-función orgánica secundaria a la sepsis.
Estimula la síntesis de endotelina I, el más potente vaso-
constrictor que se ha encontrado. La corrección de las bajas concentraciones plasmáticas de vasopresina en el shock vasodilator, se hace mediante la administración continua de la hormona a dosis que varían entre 2-6UI/h, incrementando significativamente la presión arterial (aproximadamente 25-50mmHg). Además la res-puesta vasopresora de la vasopresina ocurre en pacientes con shock séptico y shock hemorrágico severo que no responden al reemplazo de volumen y a la administración de catecola-minas, así como también en el shock vasodilatador después de bypass cardiopulmonar (12,15). Compromiso del Sistema Nervioso Central Los pacientes con sepsis pueden tener trastornos del ni-vel de conciencia, que pueden variar desde confusión a de-lirio, obnubilación y coma (4,5,9). Estos cambios del es-tado mental pueden ser atribuidos a hipotensión arterial o hipoxemia, pero una vez que estos parámetros han sido nor-malizados, la persistencia de la disfunción cerebral en au-sencia de otras causas se denomina encefalopatía secundaria a sepsis o más correctamente encefalopatía séptica, cuya pa-togénesis probablemente multifactorial, no es del todo clara. Esta alteración es habitualmente reversible. De acuerdo a Young y Bolton, la encefalopatía séptica es la manifestación más común de la sepsis severa, con una incidencia anual de 200.000 casos, siendo sin embargo su incidencia variable según los diferentes trabajos de acuerdo a la definición utili-zada. Estos mismos autores encontraron en un trabajo pre-sentado en 1990 que la encefalopatía séptica era un predic-tor independiente de mortalidad. La presencia de signología neurológica focal y de con-vulsiones es excepcional en la disfunción encefálica séptica. Cabe mencionar también dentro de las alteraciones del siste-ma nervioso la polineuropatía séptica, caracterizada por una polineuropatía axonal distal a predominio motor (16–18). Manifestaciones Pulmonares La taquipnea es muy frecuente al comienzo de la sepsis (4). El monitoreo continuo en las unidades de cuidados in-tensivos indicó que el hallazgo clínico más temprano es la aprehensión y la hiperventilación; siendo la alcalosis respi-ratoria la alteración metabólica más temprana del síndrome séptico ocasionado por bacilos gramnegativos. Por tanto en los pacientes graves la presencia de hiperventilación debe inducir a obtener hemocultivos y a una evaluación cuidado-sa de la posibilidad de infección (5). Al inicio del cuadro la hipoxemia no suele ser importante, pero los pacientes que progresan a sepsis severa o shock séptico tienen mayores posibilidades de desarrollar injuria pulmonar aguda (IPA) y presentar el Síndrome de distress respiratorio agudo (SD RA) (19). La reunión de Consenso Americano-Europeo de 1992 (20) propuso los siguientes criterios para definir el SDRA: Lesión pulmonar aguda: Perturbaciones pulmonares de comienzo agudo. PaO2/FiO2 <300 mmHg (indepedientemente de la presión teleinspiratoria).
Infiltrados bilaterales en la radiografía frontal de tórax. Presión de oclusión de la arteria pulmonar <18mmHg o ningún dato de hipertensión de aurícula izquierda
Síndrome de distress respiratorio agudo: Igual que los elementos de lesión pulmonar. PaO2/FiO2 <200mmHg.
Indira Briceño Sepsis: etiologia, clínica y diagnóstico
Medicrit 2005; 2(9):203-213 www.medicrit.com 207

El TNFα e IL-1 parecen ser fundamentales en los dife-rentes procesos que conducen a la lesión del endotelio. El li-popolisacárido y la trombina inducen la producción de IL-1 en la superficie del endotelio, ésta a su vez induce la síntesis endotelial de metabolitos del ácido araquidónico y factor ac-tivador plaquetario (PAF). El lipopolisacárido activa el complemento y sus productos de degradación inducen la formación de TNFα, el cual estimula también la producción de metabolitos del ácido araquidónico y PAF, los cuales ini-cian la lesión pulmonar (5). Ambas citoquinas (TNFα e IL-1) inducen la expresión de moléculas de adhesión en la superficie de los neutrófilos y células endoteliales, facilitando así su interacción, produ-ciendo leucoestasis y atrapamiento de granulacitos en el le-cho pulmonar, ocasionando lesión del endotelio vascular. No obstante, la presencia de neutrófilos adheridos al endote-lio pulmonar o atrapados en vasos no es esencial para el de-sarrollo de edema pulmonar no cardiogénico, pues incluso en pacientes neutropénicos se presenta el SDRA, quizás de-bido al efecto directo de las citoquinas inducidas por el lipo-polisácarido sobre la integridad de la superficie endotelial. El TNFα también estimula la angiogénesis y la neovas-cularización, mientras que la IL-1 incrementa la liberación de aniones superóxido de las células endoteliales, éstos úl-timos junto con los intermediarios de oxígeno derivados de los neutrófilos incrementan la lesión endotelial (5). Debido a que el TNFα puede inducir hipotensión, la lesión pulmonar aguda puede contribuir con la hipotensión y el daño adicio-nal de las células pulmonares. Este daño que tiene como resultado una hipotensión sostenida así como una disminu-ción de la resistencia vascular sistémica, puede conducir al aumento de la presión hidrostática en el pulmón y del edema no cardiogénico. Se ha demostrado niveles aumentados de citoquinas en el parénquima pulmonar, líquido de lavados broncoalveolares y durante la lesión pulmonar experimental, así como en pacientes con SDRA. Otra citoquina que induce el síndrome de pérdida vascu-lar pulmonar es IL-2, la cual atrae elementos linfoides al te-jido pulmonar y promueve el desarrollo de los linfocitos, és-tos pueden adherirse a la superficie del endotelio microvas-cular. Por lo que IL-2 y las células natural Killer activadas por linfoquinas también pueden mediar en la lesión del endotelio vascular pulmonar. La patogénesis del SDRA se caracteriza por (20–22): Fase aguda Exudativa: Injuria de la membrana alveolo-capilar (endotelio-epitelio) fluido rico en proteínas dentro del espacio alveolar a consecuencia del incremento de la permeabilidad alveolo-capilar. El epitelio alveolar normal está compuesto por 2 tipos de células, Tipo I plano, constituyen 90% del área de la su-perficie alveolar y se dañan fácilmente. Tipo II neuma-cito que constituye el 10% restante y son más resistentes a la lesión, sus funciones incluyen producción de surfac-tante, transporte de iones, proliferación y diferenciación para la reabsorción de las células después de la lesión. Los neutrófilos-macrófagos alveolares intervienen Princ.-palmente en esta fase. además de otras citoquinas proin-flamatorias y mediadores de la inflamación.
Fase alveolitis fibrosante o fibroproliferativa: El espa-cio alveolar se llena de las células del intersticio y sus productos junto con los nuevos vasos sanguíneos. Existe acumulación de colágeno y fibronectina.
Fase Resolución o Fibrosis: El edema alveolar es resuelto por al transporte de sodio y cloro en la parte distal del in-tersticio pulmonar. El agua ingresa pasivamente a través
de sus canales, aquaporinas localizadas principalmente en las células Tipo I. La proteína insoluble es removida por endocitosis por las células epiteliales alveolares y por la fagocitosis de los macrófagos, la proteína soluble sale por difusión entre las células. El epitelio alveolar neumocito tipo II es la progenitora para la reepitelización. La apoptó-sis (muerte celular programada) es el mecanismo por el cual son removidos los neutrófilos.
Tradicionalmente, el fracaso respiratorio en la sepsis se ha atribuido a este daño pulmonar anteriormente descrito, que da lugar a un deterioro del intercambio de gases, una disminución de la distensibilidad pulmonar y un aumento del cortocircuito (shunt) pulmonar. Sin embargo, se ha de-mostrado recientemente que el shock séptico también se asocia al fracaso de la bomba ventilatoria. En este sentido Burke y colaboradores observaron la existencia de insufi-ciencia respiratoria hipercápnica con normoxemia en los pa-cientes con sepsis fulminante. En otro estudio se describió la existencia de una disfunción muscular diafragmática, tanto clínica como electromiográfica, en pacientes con sepsis se-vera que no podían ser desconectados de la ventilación mecánica. Este hallazgo se basa en las observaciones des-critas por Friman, quien observó que tanto la fuerza máxima como la resistencia de varios músculos periféricos estaban disminuidas durante el curso de infecciones agudas. Dentro de los factores implicados en el desarrollo de disfunción muscular a nivel respiratorio se encuentran factores ventila-torios, metabólicos y hemodinámicos. Este grupo de facto-res son la consecuencia del desequilibrio existente entre las necesidades metabólicas aumentadas de los músculos venti-latorios (debido al incremento de la ventilación, la hipoxe-mia y las elevadas resistencias pulmonares) y la pobre ex-tracción de oxígeno y nutrientes por parte del músculo es-quelético. Las demandas ventilatorias aumentadas en la sep-sis, así como el hecho de que el flujo sanguíneo muscular está en función de la presión arterial sistémica, hacen pre-decir que este flujo muscular será insuficiente para satis-facer sus necesidades metabólicas, dando lugar a un incre-mento del metabolismo anaeróbico y la consiguiente pro-ducción de ácido láctico. Diversos estudios han demostrado que el daño del mús-culo esquelético inducido por la sepsis está en gran parte mediado por un incremento en la producción de radicales li-bres de oxígeno. Los efectos de los radicales libres de oxíge-no sobre la función del músculo esqulético se asocian sien-pre a ciertas condiciones fisiopatológicas musculares como la fatiga tras el ejecicio intenso, el daño por isquemia-reper-fusión, las enfermedades inflamatorias musculares y otras miopatías. Sin embargo, se ha comprobado recientemente que los radicales libres de oxígeno endógenos también regu-lan la función contráctil del músculo sano, produciéndose en baja concentración en el músculo en reposo, siendo esen-ciales para la producción normal de fuerza muscular y sus valores se incrementan progresivamente en respuesta a la activación muscular. Los valores de radicales libres de oxí-geno se mantiene relativamente bajos gracias a la acción de los sistemas antioxidantes intracelulares como las superóxi-do dismutasas, cuya acción se ve disminuida durante la sep-sis, debido a que la excesiva cantidad de óxido nítrico, como ocurre en los procesos activos inmunoinflamatorios, compi-te con estas enzimas celulares por el peróxido (reacción que es tres veces más rápida que la de aquellas enzimas con el peróxido), favoreciendo la formación del más potente y tó-xico oxidante el peroxinitrito (23).
Indira Briceño Sepsis: etiologia, clínica y diagnóstico
Medicrit 2005; 2(9):203-213 www.medicrit.com 208
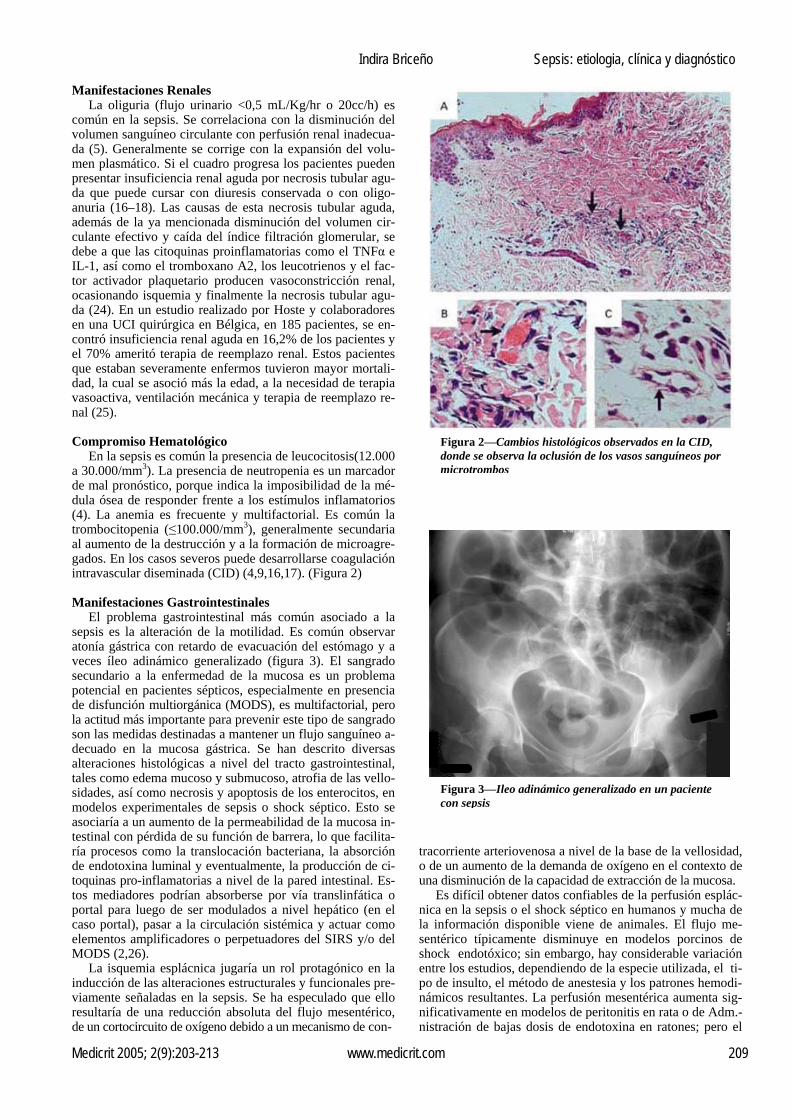
Manifestaciones Renales La oliguria (flujo urinario <0,5 mL/Kg/hr o 20cc/h) es común en la sepsis. Se correlaciona con la disminución del volumen sanguíneo circulante con perfusión renal inadecua-da (5). Generalmente se corrige con la expansión del volu-men plasmático. Si el cuadro progresa los pacientes pueden presentar insuficiencia renal aguda por necrosis tubular agu-da que puede cursar con diuresis conservada o con oligo-anuria (16–18). Las causas de esta necrosis tubular aguda, además de la ya mencionada disminución del volumen cir-culante efectivo y caída del índice filtración glomerular, se debe a que las citoquinas proinflamatorias como el TNFα e IL-1, así como el tromboxano A2, los leucotrienos y el fac-tor activador plaquetario producen vasoconstricción renal, ocasionando isquemia y finalmente la necrosis tubular agu-da (24). En un estudio realizado por Hoste y colaboradores en una UCI quirúrgica en Bélgica, en 185 pacientes, se en-contró insuficiencia renal aguda en 16,2% de los pacientes y el 70% ameritó terapia de reemplazo renal. Estos pacientes que estaban severamente enfermos tuvieron mayor mortali-dad, la cual se asoció más la edad, a la necesidad de terapia vasoactiva, ventilación mecánica y terapia de reemplazo re-nal (25). Compromiso Hematológico En la sepsis es común la presencia de leucocitosis(12.000 a 30.000/mm3). La presencia de neutropenia es un marcador de mal pronóstico, porque indica la imposibilidad de la mé-dula ósea de responder frente a los estímulos inflamatorios (4). La anemia es frecuente y multifactorial. Es común la trombocitopenia (≤100.000/mm3), generalmente secundaria al aumento de la destrucción y a la formación de microagre-gados. En los casos severos puede desarrollarse coagulación intravascular diseminada (CID) (4,9,16,17). (Figura 2) Manifestaciones Gastrointestinales El problema gastrointestinal más común asociado a la sepsis es la alteración de la motilidad. Es común observar atonía gástrica con retardo de evacuación del estómago y a veces íleo adinámico generalizado (figura 3). El sangrado secundario a la enfermedad de la mucosa es un problema potencial en pacientes sépticos, especialmente en presencia de disfunción multiorgánica (MODS), es multifactorial, pero la actitud más importante para prevenir este tipo de sangrado son las medidas destinadas a mantener un flujo sanguíneo a-decuado en la mucosa gástrica. Se han descrito diversas alteraciones histológicas a nivel del tracto gastrointestinal, tales como edema mucoso y submucoso, atrofia de las vello-sidades, así como necrosis y apoptosis de los enterocitos, en modelos experimentales de sepsis o shock séptico. Esto se asociaría a un aumento de la permeabilidad de la mucosa in-testinal con pérdida de su función de barrera, lo que facilita-ría procesos como la translocación bacteriana, la absorción de endotoxina luminal y eventualmente, la producción de ci-toquinas pro-inflamatorias a nivel de la pared intestinal. Es-tos mediadores podrían absorberse por vía translinfática o portal para luego de ser modulados a nivel hepático (en el caso portal), pasar a la circulación sistémica y actuar como elementos amplificadores o perpetuadores del SIRS y/o del MODS (2,26). La isquemia esplácnica jugaría un rol protagónico en la inducción de las alteraciones estructurales y funcionales pre-viamente señaladas en la sepsis. Se ha especulado que ello resultaría de una reducción absoluta del flujo mesentérico, de un cortocircuito de oxígeno debido a un mecanismo de con-
Figura 2—Cambios histológicos observados en la CID,
donde se observa la oclusión de los vasos sanguíneos por microtrombos
Figura 3—Ileo adinámico generalizado en un paciente con sepsis
tracorriente arteriovenosa a nivel de la base de la vellosidad, o de un aumento de la demanda de oxígeno en el contexto de una disminución de la capacidad de extracción de la mucosa. Es difícil obtener datos confiables de la perfusión esplác-nica en la sepsis o el shock séptico en humanos y mucha de la información disponible viene de animales. El flujo me-sentérico típicamente disminuye en modelos porcinos de shock endotóxico; sin embargo, hay considerable variación entre los estudios, dependiendo de la especie utilizada, el ti-po de insulto, el método de anestesia y los patrones hemodi-námicos resultantes. La perfusión mesentérica aumenta sig-nificativamente en modelos de peritonitis en rata o de Adm.-nistración de bajas dosis de endotoxina en ratones; pero el
Indira Briceño Sepsis: etiologia, clínica y diagnóstico
Medicrit 2005; 2(9):203-213 www.medicrit.com 209

flujo mesentérico típicamente disminuye cuando el insulto séptico es más agudo o masivo. Los estudios clínicos han demostrado que el flujo hepa-toesplácnico y el consumo de oxígeno aumentan significati-vamente en pacientes sépticos, pese a lo cual puede haber hipoxia de la mucosa. Mediciones tonométricas simultáneas en estómago, intestino delgado y sigmoides, muestran una disminución del pH intramucoso (pHi) durante la bacteria-mia. El pHi declina invariablemente antes que la presión ar-terial o el pH sistémico se deterioren. El pHi disminuye en la sepsis pese a un consumo de oxígeno esplácnico manteni-do o aumentado. Esta isquemia esplácnica se asocia experi-mentalmente a aumentos en la permeabilidad de la mucosa. La isquemia esplácnica inducida por oclusión parcial de la arteria mesentérica superior no provoca tanto aumento de la permeabilidad. En la sepsis habría por tanto, factores adicio-nales a la isquemia contribuyendo a los cambios de permea-bilidad. Experimentos recientes demuestran que el pHi cae rápidamente después de la inducción de peritonitis fecal, a pesar de mantener el aporte de oxígeno o incluso de aumen-tar el consumo de oxígeno. Ello sugiere que el metabolismo celular de oxígeno puede estar perturbado en la sepsis, evi-denciándose isquemia esplácnica tonométrica pese a una presión tisular de oxígeno normal. En el shock séptico ocurren también severas alteraciones microcirculatorias siendo el endotelio un blanco preferencial para diversas citoquinas. A ese nivel las citoquinas inducen acciones procoagulantes y proinflamatorias, resultando en cambios trombóticos, adhesión leucocitaria e hipermeabili-dad de la mucosa. La activación de enzimas endoteliales produce gran cantidad de óxido nítrico y prostaciclina, que pueden contribuir a la vasoplejía. El flujo esplácnico está deprimido en el shock séptico no reanimado, pero puede estar preservado o incluso aumenta-do durante el shock hiperdinámico reanimado con fluidos. En un estudio clínico en sepsis hiperdinámica, el flujo es-plácnico se encontró elevado en comparación con volunta-rios sanos o pacientes con trauma. El consumo de oxígeno sin embargo aumentó en forma desproporcionada por sobre el aumento del transporte de oxígeno, exponiendo a la mu-cosa al riesgo de isquemia. Más aún las alteraciones micro-vasculares de la sepsis disminuyen la perfusión de la muco-sa, y por tanto, reducen el transporte de oxígeno a la porción metabólicamente más activa del intestino (26). Manifestaciones Hepáticas En la sepsis severa es frecuente observar trastornos de la función hepática, manifestados por un incremento leve o moderado de las enzimas hepáticas y la bilirrubina. En los casos severos los pacientes pueden progresar a insuficiencia hepática franca con caída de los niveles de protrombina, ic-tericia e hipoglicemia (16,17). Manifestaciones Cutáneas Pueden verse en las infecciones bacterianas, virales, mi-cóticas e incluso parasitarias. En las infecciones bacterianas por estafilococos y estreptococos se puede producir una in-fección metastásica en la piel, permitiendo la posibilidad de un diagnóstico e iniciación de una antibióticoterapia especí-fica temprana. Dentro de estas manifestaciones se puede men-cionar la eritrodermia difusa, la cual se debe a la acción fisio-patológica de toxinas pirógenas o eritrogénicas. Las manifestaciones cutáneas ocasionadas por la bacte-riemia de los gérmenes gramnegativos se deben en su gran mayoría Pseudomonas aeruginosa y reciben el nombre de
ectima gangrenoso. Este último se caracteriza por ser una le-sión redondeada u ovalada de tamaño variable (1 a 5 cm), con un halo o borde elevado de eritema que rodea una zona central que puede empezar como una vesícula, y que por lo general evoluciona a una úlcera necrótica, por lo cual tam-bién se les ha denominado “lesiones en ojo de buey” (figura 4). Se cree que estas lesiones son producidas por los pro-ductos extracelulares de las bacterias, tales como proteasas o exotoxinas. No obstante, algunos trabajos atribuyen este fe-nómeno a una zona localizada de coagulopatía por consumo en la que la trombosis venosa, activada por endotoxinas es la iniciadora de la lesión tisular (fenómeno de Shwartzman). Aunque las lesiones de tipo ectima sugieren con firmeza in-fección por Pseudomona aeruginosa, estos no son los únicos agentes infecciosos que pueden originarla, también las Ae-romonas hydrophila, Echerichia coli, Klebsiella, Enterobac-ter y Serratia pueden ocasionarlas. Los microorganismos gram negativos pueden ocasionar también reacciones erite-matosas difusas muy similares a la escarlatina, y lesiones pe-tequiales no muy diferentes a la de la meningococcemia (5). ENFOQUE CLÍNICO DEL PACIENTE La evaluación inicial del paciente debe empezar con la elaboración de una historia clínica en forma detallada y cui-dadosa, prestando especial atención a la evaluación de cual-quier trastorno subyacente o predisponente (operaciones, tras-plante, quimioterapia o traumatismos) (Tabla 1), así como de los tratamientos aplicados para dichos trastornos y de su fase clínica. Se deberá hacer hincapié en antecedentes de in-fecciones anteriores y antibioticoterapia empleada, junto con cualquier dato de estudios microbiológicos realizados sema-nas previas, que pueda estar disponible, son a menudo úti-les. Además, los síntomas y signos como dolor, eritema, ede-ma o dolor de cabeza pueden ser pistas importantes que in-dican la fuente y la magnitud de un proceso infeccioso. Una historia exacta de la alimentación, los viajes y las exposi-ciones con especial atención al contacto con agentes infec-ciosos o presentes en el medio ambiente, puede ser muy va-liosa para que el médico identifique un proceso infeccioso. Figura 4—Ectima gangrenoso
Indira Briceño Sepsis: etiologia, clínica y diagnóstico
Medicrit 2005; 2(9):203-213 www.medicrit.com 210

Es probable que un proceso infeccioso sistémico, como una bacteriemia o un síndrome séptico con fiebre y otras manifestaciones de enfermedad sistémica, tengan una fuente focal inicial. Sin embargo, una cantidad de infecciones bac-teriémicas no tienen ninguna fuente conocida o identificable con facilidad, por lo que han sido clasificadas por el Centro de Control y Prevención de Enfermedades como ¨bacterie-mias primarias¨. Es probable que en muchos pacientes in-munosuprimidos, estas bacteriemias primarias se originen en el aparato gastrointestinal. El síndrome séptico puede ori-ginarse en un foco localizado de infección antes de que se desarrolle bacteriemia y si los hemocultivos se toman en e-tapa temprana en el curso del proceso infeccioso, la bacte-riemia puede no ser detectable. La administración de trata-miento antimicrobiano puede exacerbar la fiebre en forma paradójica, como consecuencia de un efecto bacteriolítico que produce liberación de endotoxinas o de pirógenos. El enfoque al lado de la cama del paciente involucra una integración de los datos recolectados en la anamnesis con los hallazgos de un examen físico completo. Los pacientes inmunosuprimidos y neutropénicos pueden tener una res-puesta inflamatoria disminuida y los signos típicos de infec-ción, como induración, fluctuaciones, calor local, linfadeno-patía reactiva y producción de pus pueden estar ausentes, en estos pacientes el mecanismo de la tos puede estar supri-mido y no producir esputo purulento u otros exudados; in-cluso en la infección del aparato urinario, estos individuos pueden no presentar los síntomas clásicos de localización ni piuria. Así mismo, el signo de rigidez de nuca puede faltar en los pacientes inmunosuprimidos con meningitis, pero quedan dos pistas importantes: el dolor de cabeza y el dete-rioro del estado mental. El eritema junto con el dolor puede ser más útil para la detección de un proceso infeccioso lo-calizado que el edema y el calor en inmunosuprimidos. La elección de los estudios de laboratorio debe basarse en los hallazgos físicos y en las manifestaciones clínicas ge-nerales. Deben hacerse todos los esfuerzos posibles para ob-tener secreciones o líquidos corporales infectados o aspirar una zona que se sospeche que alberga una infección, como el borde de avance de una celulitis. Si el paciente tiene dia-rrea el examen de heces en busca de leucocitos fecales y los coprocultivos pueden ser apropiados, asi como los estudios para detectar toxina de Clostridium difficile. En el paciente con alteración del estado mental o signos más específicos de infección del sistema nervioso central debe realizarse una pun-
Tabla 1—Factores que Agravan la Infección Bacteriana Sistémica
Cuadros subyacentes Neutropenia Hipogammaglobulinemia Diabetes Alcoholismo (cirrosis) Insuficiencia renal Insuficiencia respiratoria Complicaciones del episodio infeccioso al comienzo del tratamiento (p.ej., shock, anuria) Quimioterapia antimicrobiana Magnitud (gravedad) de la bacteriemia (bacteriemia polimicrobiana) Fuente de la infección Intervalo hasta el comienzo del tratamiento Edad
ción lumbar, siempre y cuando no haya evidencia de aumen-to de la presión intracraneana o de una lesión supratentorial focalizada. En vista de que el síndrome séptico implica enfermedad sistémica deben obtenerse por lo menos dos hemocultivos tomados de sitios diferentes para detectar una bacteriemia. Si la situación clínica está en deterioro y el paciente luce grave, es prudente obtener con rapidez tantos estudios diag-nósticos como sea posible y comenzar un tratamiento anti-microbiano empírico, el cual puede modificarse si es nece-sario en los pacientes con hemocultivos positivos. La pre-sencia de un síndrome séptico con posibilidad de shock y otras complicaciones graves, por lo general obligan al trata-miento inmediato. Es claro que el nivel de los mecanismos de defensa del huésped y la capacidad de mantener la fun-ción de los órganos vitales son los factores que determinan el resultado de cualquier infección del torrente circulatorio. Aunque los hemocultivos cuantitativos se realizan con poca frecuencia en la actualidad, varios estudios indican que la mortalidad es mayor con bacteriemias de alto grado y poli-microbianas. La ausencia de un foco discernible de infec-ción también puede asociarse con peor pronóstico. La edad también es importante, ya que se observa peor pronóstico en las edades extremas de la vida. La selección del tratamiento antimicrobiano y la rapidez con la que se administra tam-bién puede afectar los resultados (5). MARCADORES DIAGNÓSTICOS Y PRONÓSTICOS En los pacientes críticos, los mismos síntomas y signos característicos de sepsis pueden aparecer durante la inflama-ción sistémica de origen no infeccioso, por lo que el diag-nóstico y la definición de la severidad del proceso séptico pueden ser dificultosos. Durante los últimos años se ha bus-cado un marcador clínico o de laboratorio capaz de identifi-car a los pacientes con sepsis, diferenciándolos de los porta-dores de otras patologías que también cursan con SIRS. Entre ellos podemos mencionar: Procalcitonina (PCT) La procalcitonina ha sido señalada por muchas publica-ciones como un posible marcador de SIRS en respuesta a in-fección. La procalcitonina es un propéptido de calcitonina producido en la glándula tiroides, de vida media prolongada (>24 horas). En individuos normales los niveles plasmáticos
Indira Briceño Sepsis: etiologia, clínica y diagnóstico
Medicrit 2005; 2(9):203-213 www.medicrit.com 211

son muy bajos (<0,1 ng/mL), pero en los pacientes sépticos se observa un significativo aumento. Su síntesis también puede ser inducida por injurias no infecciosas, pero los nive-les no son tan elevados como en sepsis y shock séptico (ni-veles mayores de 10 ng/mL y a veces superiores a 100 ng/ mL) (27–29). Proteína C reactiva (PCR) Es una proteína de fase aguda liberada por el hígado des-pués del comienzo de la reacción inflamatoria o del daño ti-sular (30). Los niveles plasmáticos aumentan significativa-mente en los pacientes con sepsis. Es un marcador sensible pero tardío y de baja especificidad. No sólo está aumentado en las injurias agudas, sino que también está elevado en los procesos inflamatorios crónicos (enfermedades autoinmunes y reumáticas) y en el infarto agudo de miocardio. Fiebre El registro de la temperatura corporal es muy fácil de de-terminar y es con frecuencia el primer signo de infección. Es muy sensible, pero carece de especificidad (31). Recuento leucocitario y diferencial La leucocitosis se interpreta habitualmente como eviden-cia de posible infección, pero no es un marcador sensible ni específico. El recuento de glóbulos blancos puede elevarse por ejemplo después de una hemorragia digestiva, de una transfusión de sangre o después de una cirugía. La neutrofi-lia es muy limitada como marcador de inflamación sistémi-ca. Parámetros de coagulación La activación de la coagulación es un hecho común en el curso de la sepsis (32,33), con consumo de factores, aumen-to del dímero D y sobre todo disminución de la actividad de los anticoagulantes naturales. Diversos estudios han mostra-do que los niveles plasmáticos de proteína C (PC) están dis-minuidos en los pacientes con sepsis (33–35). Se ha de-mostrado que más del 85% de los pacientes con sepsis seve-ra (tres o cuatro criterios de SIRS más uno de disfunción) presentan déficit adquirido de PC y que esta disminución persiste en el tiempo, por lo que podría transformarse en un marcador útil de sepsis. Citoquinas proinflamatorias Varias citoquinas proinflamatorias, sobre todo los niveles plasmáticos de la IL-6 e IL-8, han mostrado correlación con el pronóstico en diversos estudios efectuados en pacientes críticos (7). Pero su determinación tiene varios inconve-nientes: alto costo, la vida media de las citoquinas es muy corta y las concentraciones varían rápidamente por lo que es difícil su interpretación. No se sugiere su utilización en la práctica clínica. Niveles plasmáticos de endotoxina (LPS), Fosfolipasa A2, Elastasa de neutrófilos, HLA-DR de monocitos Son diferentes determinaciones que se han planteado co-mo marcadores diagnósticos y pronósticos de sepsis, pero no se ha determinado todavía su utilidad clínica. REFERENCIAS 1. Mark E Astiz, Eric C Rackow. Septic shock. Lancet.
1998.351:1501-1505. 2. Hall, Schmidt and Wood.Insuficiencia Orgánica Múltiple:
Manifestaciones clínicas, patogenia y tratamiento. Cuidados Intensivos. Editorial McGraw-Hill Interamericana. México. Segunda Edición. 1998.Capítulo 17:243-270.
3. Brun-Buisson C, Doyon F, Carlet J, et al. Incidencie, risk factor and outcome of severe sepsis and septic shock in adult. JAMA.1995. 274:968-974.
4. Polly E. Parsons, Jeanine P. Wiener-Kronish.Sepsis. Secretos de los Cuidados Intensivos. Editorial McGraw-Hill Interamericana. México. Segunda Edición. 1998. Capítulo 39:231-236.
5. Mandell.. Síndrome de Sepsis. Tratado de Infectología. Capítulo 63. 973-987.
6. Alberto Dougnac L. Sepsis y Shock Séptico. Apuntes de Medicina Intensiva. Pontificia Universidad Católica de Chile.2000;1-9.
7. Shoemaker, Ayres, Grenvik and Holbrook. Citoquinas. Tratado de Medicina Crítica y Terapia Intensiva. Editorial Panamericana.Buenos Aires-Argentina. 3a Edición. 1998.Capítulo 21:154-160.
8. Hugo Zetina Tun, María del Carmen Rentería Arellano y Luis Carlos Bonilla Rivera. Sepsis, corazón e inotrópicos. Revista de la Asociación Mexicana de Medicina Crítica y Terapia Intensiva. 2000.16;3:102-110.
9. Carlos Lovesio. Sepsis, septicemia y shock séptico. Medicina Intensiva. Editorial El Ateneo. Buenos Aires-Argentina. Cuarta Edición. Capítulo 38:557-565.
10. Grocott-Mason R, Shah A. Cardiac dysfunction in sepsis: New theories and clinical implications. Intensive Care Med. 1998.24:286-295.
11. Ulevitch RJ, Tobias PS: Recognition of endotoxin by cells leading to transmembrane signalling. Curr Opin Inmunol. 1994;6:125-130,1994.
12. Martin W Dünser, Volker Wenzel, Andreas J. Mayr and Walter R. Hasibeder. Manegement of Vasodilatory Shock. Drug. 2003.63;3:237-252.
13. Donald W. Landry and Juan A. Oliver. The Pathogenesis of Vasodilatory Shock. N Engl J Med. 2001.345;8:588-594.
14. Tsuneyoshi I, Yamada H, Kakihana Y, Nakamura M, et al. Hemodynamic and metabolic effects of low-dose vasopressin infusions in vasodilatory shock. Crit Care Med. 2001.29:487-493.
15. Dunser MW, Mayr AJ, Ulmer H, Ritsch N, Knotzer H, et al. The effects of vasopressin on systemic hemodynamics in catecholamines-resistance postcardiotomy shock: A retrospective analysis. Anesth Analg. 2001. 93:7-13.
16. Marshall JC, Cook DJ, Christou NV, et al. Multiple organ dysfunction Score: A reliable descriptor of complex clinical outcome. Crit Care Med. 1995.23:1638-1652.
17. Vincent JL, de Mendonca, Cantraine F, et al. Use of the SOFA score to asses the incidence of organ dysfunction/failure in intensive care units: Results of a multicenter, prospective study. Crit Care Med. 1998.26:1793-1800.
18. Jean-Roger Le Gall, Janelle Klar, Stanley Lemeshow, et al. The Logistic Organ Dysfunction System: A new way to asses organ dysfunction in the Intensive Care Unit. JAMA. 1996.276:802-810.
19. [47] Alan L Beal, Frank B. Cerra. Multiple organ failure syndrome in the 1990s: Systemic Inflammatory Response and Organ Dysfunction. JAMA. 1994.271:226-233.
20. Ramona L. Doyle, Nancy Szaflarski, Gunnard W. Modin, Jeanine P.Wiener-Kronish, and Michael A. Matthay. Identification of patients with Acute Lung Injury: Predictor of Mortality. Am J Respir Crit Care Med. 1995.152: 1818-1824.
21. Kollef MH, Shuster DP. The acute respiratory distress syndrome (ARDS). N Engl J Med. 1995.332:27.37.
22. Dantzker, Scharf. Patogenia de la lesión pulmonar aguda. Cuidados Intensivos Cardiopulmonares. Editorial McGraw-Hill Interamericana. México. Tercera Edición. 1998. Capítulo 1:3-21.
23. E Barreiro, SNA Hussain. Fracaso de los músculos
Indira Briceño Sepsis: etiologia, clínica y diagnóstico
Medicrit 2005; 2(9):203-213 www.medicrit.com 212

respiartorios en la sepsis. Archivos de Bronconeumología.2002.38;5:226-235.
24. Van der Poll T., Van Deventer SJH. Cytokines and anticytokines in the patogénesis of sepsis. Infect Dis Clin North Am. 1999.13:413-425.
25. Eric A.J. Hoste. Norbert H. Lameire. Raymond C. Vanholder. Dominique D. Benoit Johan M. A. Decruyenaere and Francis A. Colardyn. Acute Renal Failure in Patients with sepsis in a surgical ICU: Predictive Factor, Incidence, Comorbidity and Outcome. J Am Soc Nephrol. 2003. 14 (4): 1022-1030. Abstract.
26. S M Pastores. Drotrecogin alfa (activated): a novel therapeutic strategy for severe sepsis. Postgraduate Medical Journal. 2003.79:5-10. Abstract.
27. Glenn Hernández P, Vinko Tomicic F. Efectos de las catecolaminas sobre la perfusión esplácnica en la sepsis. Apuntes de Medicina Intensiva. Pontificia Universidad Católica de Chile. 1998;1-11.
28. Wanner GA, Keel M, Steckholzer U, et al. Relationship between procalcitonin plasma levels and severity of injury, sepsis organ failure and mortality in injure patients. Crit Care Med. 2000.28:950-957.
29. Urgate H, Eliezer Silva, Mercan Dany, et al. Procalcitonin used as a marker of infection in intensive Care Unit. Crit Care Med. 1999.27:498-504.
30. Giamarellos.Bourboulis E, Mega A, Grecka P, Scarpa N, Korat Thomopoulos G, Giamarelluo H. Procalcitonin: a marker to clearly differentiate systemic inflammatory response syndrome and sepsis in the patients?. Crit Care Med. 2002.28:1351-1355.
31. Yentis SM, Soni N, Sheldon J. C-reactive protein as an indicator of resolution of sepsis in intensive care unit. Intensive Care Med. 1995.21:602-605.
32. O’Grady NP, Barie PS, Bartlett JG, et al. Practice guidelines for evaluating new fever in critically ill adult patients. Crit Care Med. 1998.26:1042-1059.
33. Lorente JA and et al. Time course of hemostatic abnormalities in sepsis and its relation to outcome. Chest. 1993.103:1536-1542.
34. Yan SB, Helterbrand JD, Hartman DL, et al. Low levels of protein C are associated with poor outcome in severe sepsis. Chest.2001.120:699-701.
35. Bolt j, Papsdorf M, Rothe A, Kumle B, et al. Change of the hemostatic network in critical ill patients. Is there a difference between sepsis, trauma and neurosurgery patients?. Crit Care Med.2000.28:2209-2216.
Correspondencia: Dra Indira Briceño. Servicio de Emergencia General de Adultos. Instituto Autónomo Hospital Universitario de Los Andes. Avenida 16 de Septiembre, Sector Campo de Oro. Mérida 5101, Venezuela. Para comentarios sobre este artículo favor dirigirse a: [email protected] [email protected]
Indira Briceño Sepsis: etiologia, clínica y diagnóstico
Medicrit 2005; 2(9):203-213 www.medicrit.com 213