risk factors for metastasis in retinoblastoma
TRANSCRIPT

1
MAJOR REVIEW
SURVEY OF OPHTHALMOLOGY
VOLUME 47
•
NUMBER 1
•
JANUARY–FEBRUARY 2002
© 2002 by Elsevier Science Inc. 0039-6257/02/$–see front matterAll rights reserved. PII S0039-6257(01)00279-X
Risk Factors for Metastasis in Retinoblastoma
Paul T. Finger, MD,
1
J. William Harbour, MD,
2
and Zeynel A. Karcioglu, MD
3
1
The New York Eye Cancer Center, New York, New York;
2
Department of Ophthalmology and Visual Sciences, Washington University School of Medicine, St. Louis, Missouri; and
3
Department of Ophthalmology, Tulane University MedicalSchool and Cancer Center, New Orleans, Louisiana, USA
Abstract.
Children with retinoblastoma typically survive their cancer due to advances in early diagno-sis and treatment. Despite this success, risk factors persist for metastasis that are thought to be relatedto patient age, sex, laterality, treatment, genetics, histopathology, and extraocular extension. Thisreview has found that invasion of the uvea, orbit, and optic nerve continue to be the most importantpredictors of metastatic retinoblastoma. Bilaterality and delays in diagnosis are also important factors.We examine molecular and genetic studies that offer the potential of predicting which tumors arelikely to metastasize, which will recur within the eye, and which will undergo senescence. In this review,we describe which clinical evaluations, genetic studies, and histopathologic evaluations of retrievedspecimens are currently used widely. This review has been performed to help those caring for patientswith retinoblastoma and to aid informed consent. (
Surv Ophthalmol 47:
1–16, 2002. © 2002 byElsevier Science Inc. All rights reserved.)
Key words.
choroid
•
extrascleral
•
metastasis
•
optic nerve
•
orbit
•
pathology
•
retinoblastoma
Treatment of retinoblastoma (Rb) is a modernday medical success story.
25
Advances in diagnosis(aided by indirect ophthalmoscopy, ophthalmic ultra-sound, and computed tomographic imaging) followedby removal of the Rb-containing eye has decreased theincidence of metastases to less than 10% in developedcountries.
1–3,5,17,19
In cases where the tumor has locallyspread or systemically metastasized, advances in chemo-therapy have increased survival.
22,27
Early recognition of risk factors for metastasis mayoffer the potential to improve survival. Clinical, ge-netic, and histopathologic features have been identi-fied as risk factors for metastatic disease. Early recog-nition of these risk factors followed by prompttreatment may reduce the incidence of metastaticdeath. Defining these factors allows physicians to fo-cus systemic evaluations on those at risk.
Herein we review established and potential clini-cal, genetic, and histopathologic risk factors for met-astatic retinoblastoma.
I. Clinical Risk Factors
Clinical risk factors include demography, lateral-ity, orbital extension, and delay in diagnosis.
A. DEMOGRAPHY
1. Age at Diagnosis
Retinoblastoma is the most common primary in-traocular tumor in children, affecting 11 per millionunder the age of 5 years, or approximately 350 chil-dren in the United States each year.
2,85,103,117
Most pa-tients (60–70%) will have a unilateral tumor, whereas30–40% will have bilateral involvement. Over 90%have no family history of Rb. In Tamboli’s series, 95%

2 Surv Ophthalmol 47 (1) January–February 2002
FINGER ET AL
of 220 patients with Rb were diagnosed before theage of 5, and 40% were diagnosed before the age of1.
20,117
There is a trend toward less metastatic diseasein children diagnosed at a younger age.
26
Most chil-dren with Rb are diagnosed before their third yearof life and new tumors can develop until they are 7years old. Children discovered to have Rb in botheyes (bilateral cases) tend to be younger than unilat-eral cases.
62
2. Age at Treatment
Bilateral tumors are discovered earlier, and, there-fore, patients are treated at a younger age than chil-dren with unilateral Rb.
4,26,62,74,97
Leukocoria (whitepupil) and strabismus (deviated eyes) are the mostcommon findings associated with Rb, and althoughneither is thought to be a risk factor for metastases,these findings are likely to motivate a parent to seekmedical attention for their child. These findings andpoor vision are more common in bilateral cases.
3. Race and Sex Distribution
Although multiple studies suggest that there is norace or sex-based predisposition to the developmentof Rb, access to health care has been associated withdelay in diagnosis, which is a risk factor for me-tastases.
62
Similarly, other circumstances that delaydiagnosis (cataract, failure to diagnose) are associ-ated with optic nerve invasion, orbital extension,and metastatic disease.
62,73
The presence of endemicareas (around the world) has been suggested.
B. LATERALITY
1. Unilateral Versus Bilateral
Patients with unilateral Rb have only one eye at riskfor optic nerve and orbital extension, making themless likely to develop metastasis. Despite this, manystudies suggest that unilateral tumor patients are atequal risk for metastatic Rb. This led researchers toconsider that the delay in diagnosis of unilateral tu-mors increased the risk (as to be equivalent) to that ofthe anatomic risk of having two eyes affected.
14,115
Kopelman et al performed a multivariate analysisfocusing on laterality of Rb.
62
They found that if pa-tients with concurrent optic nerve and orbital exten-sion were removed from the analysis, patients with bi-lateral Rb were more likely to develop metastaticdisease (p
�
0.0029).
62
This could be due to a geneticdifference between these tumors, or it could simplybe due to the fact that bilateral Rb patients have moretumors (larger tumor volumes). Thus, the chancethat they will develop metastatic ability increases.
2. Trilateral Disease
About 5–7% of bilateral patients (and a few unilat-eral patients) will develop so-called
trilateral retino-
blastoma
, in which the eye tumors are accompaniedby a midline intracranial primitive neuroectodermaltumor (PNET).
9,11,59,68
These tumors were previouslycalled
pinealomas
because they arise in the region ofthe pineal gland. PNETs are not considered to becentral nervous system extension (metastases) fromintraocular tumors, rather a concurrent or consecu-tive primary intracranial malignancy. Radiographicevaluation to screen for PNET should be part of acomplete work up for extraocular disease (Fig. 1).
46
C. LOCAL EXTENSION
Although clinical evaluation can suggest opticnerve and extraocular extension, the ocular patholo-gist plays a crucial role in determining whether ornot the tumor has broken out of its ocular confines.Reported risk factors for metastasis include choroi-dal invasion, transscleral migration, and intraneuralinvasion. Whereas clinical findings can suggest suchlocal extension, ocular or orbital histopathology istypically required to reveal local extension and meta-static risk.
48,57,62,67,73,95
1. Optic Nerve Invasion
The presence of tumor beyond the lamina crib-rosa (in the neural parenchyma, cerebral spinalfluid, and neural blood vessels) is a risk factor formetastatic disease (Table 1).
62,104
Clearly, access tothe subarachnoid space allows Rb cells to spread tothe spinal fluid and central nervous system (Fig. 2).
48
In the clinical setting it can be difficult to determinewhether a Rb is extending into the nerve or merelyoverhanging the optic disk. When the posterior sur-face of the tumor cannot be seen, ultrasonography,
Fig. 1. A sagittal section of the brain by magnetic reso-nance imaging. A midline intracranial primitive neuroec-todermal tumor (PNET) also known as a pinealoma is re-vealed as an ovoid mass in the center of the scan.

RISK FACTORS FOR Rb METASTASIS
3
computed axial tomography, and magnetic reso-nance imaging studies have been reported to be use-ful.
6,37,38,47
Glaucoma has been reported to be associated withmetastasis related to optic nerve invasion.
104
Clinicalevidence of optic nerve invasion is typically an indi-cation for enucleation, and histopathologic confir-mation of tumor beyond the surgical margin necessi-
tates additional treatment (i.e., radiation and/orchemotherapy).
27
2. Uveal Invasion
Choroidal invasion has been considered an impor-tant risk factor for metastatic disease because it allowsfor access to the sclera and emissary vessels.
62,73,90,98,105,115
Although histopathologic evidence of choroidal inva-sion is often considered to be a risk factor for metasta-sis, it is not clear if choroidal invasion alone necessi-tates the use of systemic chemotherapy for presumedsubclinical metastatic disease.
27,62
Choroidal invasion is difficult to quantify by clini-cal and histopathologic evaluations. Logically, exo-phytic and plaque-like (rather than endophytic) ret-inoblastomas are more likely to extend to the choroid.Once in the choroid, the next step is scleral invasion,and then extrascleral extension. Certainly, extras-cleral extension indicates that the tumor has traveledthrough the choroid. The multivariate analysis doneby Kopelman et al supports the concept that choroi-dal invasion (in itself) was not a significant risk factorfor metastasis (Table 1).
62
Clearly there is some con-troversy as to whether choroidal involvement (by it-self) is a significant risk factor for metastasis.
The presence of Rb seeds on the iris and trabecu-lar meshwork indicates vitreous and/or ciliary bodyinvolvement (Fig. 3).
36
In general, the larger the Rbthe more likely that the choroid, iris, and ciliarybody are involved. In multivariate analysis, Rb seed-ing and tumor size by themselves have not beenproven to be risk factors for Rb metastasis.
62
3. Orbital Invasion
When a tumor extends through the sclera, it gainsaccess to the vascular and lymphatic channels outsidethe eye (Fig. 4).
24,43,62,95
Orbital invasion is a significantrisk factor for metastatic disease and an indication forsystemic treatment (Table 1).
62
Although orbital inva-sion is typically treated with combinations of radiationand chemotherapy, relapses do occur (typically withinthe first year). New chemotherapeutic approaches haveincreased survival. Svenberg-Winholt et al reported esti-mated 3-year survival of 46.7% in 16 patients with or-bital Rb without clinically evident metastasis.
58,116
Neovascular glaucoma should be considered a rel-ative risk factor for orbital extension but has not (initself) been shown to be a risk factor for Rb metasta-sis. High intraocular pressures can cause thinning ofthe sclera and cornea, facilitating tumor extensioninto the optic nerve, choroid, and orbit.
Microscopic orbital extension is impossible to dis-cover by clinical examination. Larger extensions intothe orbit can be imaged by ultrasound, computed to-mography, and magnetic resonance imaging.
6,47
Grossorbital extension can be difficult to miss, and may sim-
TABLE 1
Significant Risk Factors as Determined by Multiple Stepwise Logistical Regression of 15 Variables Based upon
361 Cases
Risk Factor Multivariate Odds Ratio p
Invasion ofChoroid 1.8
�
0.05Sclera 3.9
�
0.05Orbit 21.6
�
0.01Optic Nerve
Resected 3.8
�
0.01Unresected 8.7
�
0.01Cataract 2.2
�
0.05Bilaterally 2.9
�
0.01Clinically 2.5
�
0.05Undiagnosed
Not significant (p
�
0.05) factors (variables) includedthe following: age of patient, photoreceptor differentia-tion, size of tumor, angle closure, rosette formation, race,pseudohypopyon, year of enucleation, and those notedabove. Table modified From Kopelman et al, MultivariateAnalysis of Risk Factors For Metastasis in RetinoblastomaTreated by Enucleation.
62
Fig. 2. Histopathologic evaluation of the optic nerve(ON) reveals retinoblastoma in the subarachnoid space(RB) (hematoxylin and eosin � 40).

4 Surv Ophthalmol 47 (1) January–February 2002
FINGER ET AL
ulate orbital cellulitis, result in a palpable mass, and in-duce exophthalmos (Fig. 4).
63,79,93,110
D. TIMING OF DIAGNOSIS AND TREATMENT
1. Delay in Diagnosis
Factors that delay Rb diagnosis can be responsiblefor metastasis (e.g., cataract obscuring the tumor,misdiagnosis, lack of access to medical care).
19,39,92,110,113
Prior to the widespread availability of indirect oph-thalmoscopy, ultrasound, and radiographic imagingtechniques, Stafford et al reported 15% clinical mis-diagnoses in the analysis of 618 histopathologicallyconfirmed Rb cases.
113
In 6.6% of their cases, the in-correct clinical diagnosis led to a delay in enucle-ation. In a more recent study, Kopelman et al reportedthat the chances of metastasis and death were 2.5
times greater in patients in whom the clinical diagnosisof Rb was delayed.
62
Messmer quantified delays of 120days or more to be statistically significant for the devel-opment of metastatic Rb.
73
2. Eye Conservation
Although most unilateral Rbs continue to be curedby enucleation, early diagnosis has allowed for eyeand vision-sparing alternative treatments for children(primarily) with bilateral disease.
1,76
Radiation ther-apy was the first commonly used alternative treatmentoffering a sight and eye-sparing alternative to enucle-ation.
3,5,88
A more recent approach has been the useof chemotherapy (chemoreduction) to reduce thesize of intraocular tumors followed by multiple alter-native local therapies (laser, cryotherapy, and plaqueirradiation) to destroy the tumor residua.
27,28,32,80
Fig. 3. Left: Tumor seeding of the iris in a case of unilateral retinoblastoma without extraocular extension. This patientdeveloped metastasis 6 months after enucleation. Right: White clumps of retinoblastoma cells “seed” the vitreous humor(RetCam image courtesy Barrett Haik, MD).
Fig. 4. Left: Proptosis, chemosis, and moderate displacement of the eye inferiorly characterized this patient with orbitalextension of his retinoblastoma. Right: Computed tomography suggests what is later revealed to be extraocular extensionof retinoblastoma along the medial aspect of the left globe.

RISK FACTORS FOR Rb METASTASIS
5
Unlike enucleation, all conservative treatments resultin the presence of residual (destroyed or sterilized) tu-mor within the eye. While there exists good long-termexperience with local control after external beam radi-ation therapy, primary chemotherapy rarely eliminatesor sterilizes the intraocular Rb. There is no proof thatthe presence of live residual tumor presents a signifi-cant risk of metastasis, but this question deserves study.
II. Genetics and Molecular Pathophysiology of Metastasis
Retinoblastoma occurs in both a hereditary andnonhereditary form. Even though less than 10% ofnew retinoblastoma patients have a positive family his-tory, 30–40% of new patients will have the hereditary(or germline) form of the disease due to sporadic germ-line mutations. Hereditary retinoblastoma is charac-terized by autosomal dominant inheritance (pheno-type), a high risk of multiple bilateral tumors, and alifelong predisposition to cancers throughout thebody. Nonhereditary (or somatic) retinoblastoma oc-curs in 60–70% of cases and consists of a unifocal reti-nal tumor and no increased risk of other cancers.Knudson’s “two hit hypothesis” explains this clinicalobservation by pointing out that Rb occurs when bothcopies of the Rb gene are mutationally inactivated inthe same developing retinoblast.
60,61
The Rb gene onchromosome 13q14 is now known to encode an im-portant tumor suppressor protein that normally func-tions to inhibit cancer not only in the eye but alsothroughout the body.
10,30,31
In the hereditary form,one Rb mutation is already present in the germline soonly one subsequent mutation is needed. The “sec-ond hit” that inactivates the other copy of the Rbgene occurs frequently enough during retinal devel-opment that multiple tumors often occur. Becausethe inherited mutation is also present throughout thebody, cancers are more likely to arise in other tissues.In contrast, nonhereditary Rb occurs only in the un-likely event that somatic mutations occur in both al-leles of the Rb gene in the same retinoblast.
A. TUMOR INVASION AND METASTASIS
Tumor invasion and metastasis are the most com-mon causes of death from cancer, and yet the molec-ular events underlying these processes are still poorlyunderstood. The evolution of the malignant cellfrom initial neoplastic transformation to distant me-tastasis is a result of successive molecular events thatprovide the tumor cell with a growth and survival ad-vantage with respect to adjacent cells. Neoplastictransformation involves an accumulation of geneticmutations that allows the tumor to usurp control ofthe cell cycle, overcome growth inhibitory signals,circumvent apoptosis, avoid senescence, and recruita blood supply. Many of the tumor suppressor genes
and proto-oncogenes involved in this transformationhave now been identified.
40
Once established as aprimary neoplasm, the tumor’s continued successmay become threatened by limited space, nutrients,or oxygen. In response to these limitations, the tu-mor may acquire the ability to colonize new siteswhere it can continue to grow. This ability requiresadditional molecular alterations that allow the tumorto invade surrounding tissue, gain access to bloodvessels and other conduits, survive while travelingthrough the body, implant within a distant tissue, in-teract with the new microenvironment, evade theimmune system, recruit a blood supply, and continueto proliferate (Fig. 5).
40
While our understanding oftumor invasion and metastasis is still very limited, someof the genetic events associated with Rb metastasisare starting to emerge, which may allow the develop-ment of new diagnostic and therapeutic strategies tocounteract the development of metastatic disease.
B. GENETIC EVENTS IN THE PATHOGENESIS OF Rb
Although the actual sequence of genetic alter-ations that lead to Rb are not known with certainty,inferences about early versus late genetic events canbe made based on their frequency in tumors andtheir association with metastatic disease.
1. Initiating Event: Mutation of the Rb Gene
Retinoblastoma is unique in that all cases arethought to involve the same initiating genetic event—biallelic mutational inactivation of the Rb gene. Al-though it remains controversial whether mutations inother genes are also needed for Rb to develop, it isclear from the autosomal inheritance of familial Rbthat mutational inactivation of both alleles of the Rbgene is both necessary and rate-limiting in tumorigene-sis.
60
In familial Rb, one inherited Rb gene mutation ispresent in most or all cells in the body, and Rb devel-ops when the second Rb allele is mutated in a somaticretinal cell. In contrast, nonhereditary Rb occurs in theunlikely event that two somatic mutations occur in thesame retinoblast.
The Rb gene is located on chromosome 13q14and mutational inactivation of this gene has beenobserved in a large number of retinoblastomas.
30,31,65
The Rb protein is a ubiquitous cell cycle regulatorand arrests cells in the G
1
phase of the cell cycle,thereby inhibiting cellular proliferation.
10
Loss of Rbdisrupts cell-cycle control and sends the developingRb toward uncontrolled growth and neoplastic trans-formation. Other genetic mutations may contribute tofurther malignant progression.
2. Early Events
Several chromosomal abnormalities are found withhigh frequency in retinoblastomas and presumably

6 Surv Ophthalmol 47 (1) January–February 2002
FINGER ET AL
provide a growth or survival advantage for the evolv-ing tumor. No specific genes from the altered chro-mosomal regions have been associated specificallywith Rb, however, and the pathophysiologic conse-quences of these alterations remain speculative.
a. Increased Copy Number of Chromosome 6p
Additional copies of chromosome 6p are found inover half of retinoblastomas.
13,112
This abnormality
can occur by formation of an isochromosome 6p(65%), translocation of 6p to other chromosomes(14%), retention of additional copies of chromo-some 6 (14%), and other mechanisms (7%).
13
Addi-tional copies of 6p are acquired early in the patho-genesis of Rb and may provide a growth advantagefor the evolving tumor.
34,83
b. Increased Copy Number of Chromosome 1q
Most retinoblastomas also have a gain in copies ofchromosome 1q, and this abnormality appears to oc-cur relatively early.
33,83
3. Late Events
Several genetic alterations occur at lower frequencyin retinoblastomas and presumably are not requiredfor neoplastic transformation; however, these abnor-malities may contribute to the development of an in-vasive or metastatic phenotype.
a. Telomerase Activity
The telomeres (distal ends of chromosomes) of anormal cell become shorter with every cell division,and the cell eventually becomes senescent when theends of the telomeres shorten to a critical length.This is an important mechanism for limiting cellproliferation and avoiding the accumulation of po-tentially harmful mutations.
121
Tumors commonlycircumvent this protective mechanism by upregulat-ing telomerase, the enzyme that restores telomerelength. Up to half of retinoblastomas express highlevels of telomerase, and this may be associated withthe development of metastatic disease.
35
b. Loss of Chromosome 1p
Loss of part or all of chromosome 1p occurs inabout 20% of retinoblastomas and is statistically asso-ciated with metastatic disease.
23
Several known tumorsuppressor genes reside on chromosome 1p, butnone have yet been associated specifically with Rb.
c. N-myc Amplification
The myc family of proto-oncogenes promotes cellu-lar proliferation and has been linked to a large num-ber of cancers.
8
N-myc amplification was one of thefirst genetic abnormalities identified in Rb and was ini-tially thought to be an early and important step in tu-morigenesis.
66
Now it is clear that this alteration occursin only a minority of tumors and may be a late eventthat can provide an additional growth advantage.
23
d. p53 Inactivation
The p53 tumor suppressor protein is a critical regu-lator of cell growth and genomic stability, and muta-tion of the p53 gene is the most common geneticevent in cancer.
41
There is strong selection for muta-tion of p53, since deregulation of this pathway pre-
Fig. 5. Retinoblastoma develops as a result of mutationalinactivation of the Rb gene, which, along with other ge-netic events, allows the tumor to overcome cellular safe-guards against neoplastic transformation. The tumor even-tually encounters limited space, nutrients, and oxygen,which can select for further genetic mutations that allowthe tumor to recruit a blood supply, invade other tissues,spread to distant sites, and set up new tumor colonies.Some of the genetic events involved in this process havenow been identified as risk factors for metastatic Rb.

RISK FACTORS FOR Rb METASTASIS
7
vents the cell from undergoing apoptosis in responseRb loss and other accumulating mutations. Interest-ingly though, p53 mutations are uncommon in Rb.16
In fact, the apoptotic pathway in Rb appears to be atleast partially intact, as p53 can induce apoptosis inthese tumors.82 Mutational loss of p53 may be a lateevent in Rb associated with more advanced tumors.16
C. PATHOPHYSIOLOGIC STEPS TOWARD METASTASIS
The mechanisms by which the genetic changesoutlined above contribute to metastasis are still poorlyunderstood. However, we now have some understand-ing of specific pathophysiologic events involved inmetastasis, as well as their underlying molecular causes.
1. Angiogenesis
Tumor angiogenesis (the development of new bloodvessels) may result from limited oxygen or nutrients.Angiogenesis can often be detected in primary tumorsand is virtually required for the acquisition of meta-static ability, presumably because it allows the tumoraccess to the bloodstream. One important molecularmechanism leading to angiogenesis in Rb and othertumors is expression of vascular endothelial growthfactor (VEGF). In Rb cells, it has been shown thathypoxia (which is common within retinoblastomas)can induce abnormally high levels of VEGF expres-sion.64 The mechanism for this deregulated VEGFexpression is still unclear.
2. Tissue Invasion
The pattern of tissue invasion and metastasis in Rbsuggests that the most common sequence of eventsis local invasion into the choroid and optic nervehead, followed by direct extension down the opticnerve into the brain or through the sclera into theorbit. Distant metastasis occurs later as the tumorgains access to the bloodstream or cerebrospinalfluid. This sequence involves molecular events thatprogressively disrupt the adhesion of tumor cells toother cells and to the extracellular matrix.40
3. Deregulation of Cell-to-Cell Adhesion Molecules
Cell–cell adhesion molecules (CAMs) such as thecadherins are an important class of cell surface proteinsthat are altered in metastatic tumor cells. Couplingof adjacent cells by cadherins causes antiprolifera-tive signals to be sent to the cells through �-cateninand other downstream effectors.15 Deregulation ofthis system can allow cells inappropriately to surviveand proliferate when dissociated from their normaltissue environment. Cell adhesion molecules suchas N-cadherin have been shown to be deregulatedin Rb.100
4. Changes in Integrins
Successful invasion and metastasis requires that thetumor cell adapt to a changing extracellular environ-ment. Contacts between the cell and adjacent extra-cellular components are regulated by integrins ex-pressed on the cell surface.40 Tumor cells adapt tochanges in the extracellular environment as it invadesand metastasizes by shifting the � and � integrin sub-units displayed on the cell surface. Altered expressionof integrin subunits has been observed in Rb, but adetailed understanding of how these alterations occurand how they promote metastasis is still lacking.111
5. Other Steps in Metastasis
We now have a number of clues to the molecularpathophysiology of metastatic Rb, but our understand-ing of this process is still very limited. There are manyother steps in the metastatic process about which weknow very little in Rb, including activation of proteasesto invade adjacent tissues, and changes in cell surfaceproteins that allow adherence to distant tissues andevasion of the immune system. As we understand morefully the genetic events that occur in the progression ofRb from initial neoplastic transformation to late meta-static disease, we may be more able to recognize high-risk tumors and to design a more effective treatment.
III. Histopathologic ParametersIn reviewing the histopathologic parameters asso-
ciated with metastatic Rb, two fundamental ques-tions should be critically assessed (Table 2). First,what occasions enable us access to tissue material,and second, how does the histopathologic examina-tion contribute to diagnosis and management of thisdisease? When one views histopathology within theframework of these questions, it is realized that whilethe monumental amounts of literature concerningthe morphology of Rb helps us understand the dis-ease process, it has limited usefulness in the clinicalmanagement of the patient.
In Rb patients, access to tissue material is gainedthrough enucleation/exenteration, orbital biopsy,anterior chamber paracentesis, fine needle aspira-tion biopsy (FNAB), vitrectomy, lumbar puncture,and bone marrow biopsy/aspirate. The utilization ofthe information obtained from these specimens issummarized in this section.
A. ENUCLEATION SPECIMENS
1. Tumor Morphology
Most tumor tissue in Rb becomes available fromenucleated specimens. Although early detection hasallowed for the diagnosis to be made before local ex-tension and metastasis, it does not occur until the sizeof the tumor necessitates enucleation. Therefore,

8 Surv Ophthalmol 47 (1) January–February 2002 FINGER ET AL
post enucleation tumor measurements contribute lit-tle to the staging and clinical management.27,108,109
On the other hand, study of tumor growth pat-terns may be useful. Classically, four growth patternsare recognized in Rb and include endophytic; exo-phytic; mixed endophytic and exophytic; and diffuseplaque-like tumors.69 The most common growth pat-tern, however, occurs as combinations of endophyticand exophytic tumors.
a. Endophytic Rb
Endophytic Rb grows from the retina towards thevitreous (Fig. 6). Thus, on indirect ophthalmoscopicexamination the tumor is viewed directly as a pro-truding mass into the vitreous chamber. Rb is a fria-ble necrotic tumor, in which small clusters of tumorcells are detached from the main mass and form sat-ellite tumor nodules within the vitreous, known as“vitreous seeding,” and is readily observed by oph-thalmoscopy and documented by digital photogra-phy (Fig. 3). These detached tumor clusters tend toform deposits on other sites of the retina, as well asonto the ciliary body, iris, and lens (Fig. 3). In theposterior and anterior chambers, they may followthe outflow of aqueous humor evidenced by seedsfound on the posterior surface of the cornea andwithin the trabecular meshwork. When tumor frag-ments invade the retina, it can be difficult to distin-guish between secondary seeding and multifocal Rb.
Histopathologic patterns of endophytic growthand tumor seeding do not typically contribute toclinical management. These growth patterns and ev-idence of tumor seeding are discovered by ophthal-
moscopy or imaging techniques prior to enucle-ation. On the other hand, because multiple tumorsindicate a germinal mutation, detection of multicen-tric Rb (histologically distinguishable from retinalseeding) is useful.
b. Exophytic Rb
Exophytic retinoblastomas typically grow from theouter retinal layers and extend underneath the de-tached retina toward the choroid. As these tumorsgrow larger, they detach the retina, and many tumorcells detach into the subretinal fluid. Dislodged massesof Rb may implant onto the retinal pigment epithe-lium and erode through Bruch’s membrane into thechoroid. Because choroidal involvement has beenthought to increase the risk of systemic dissemina-tion and exophytic growth patterns lead to an earlierinvolvement of the choroid, it may be logical to as-sume that exophytic growth patterns increase therisk of metastasis. This, however, has not beenproven.
c. Plaque-like Rb
Diffuse plaque-like Rb is the least common mor-phologic growth pattern, and the most difficult to di-agnose in the clinical setting.75,81 Plaque-like retino-blastomas grow diffusely and insidiously within theretina without ever forming a detectable mass. Mostof these tumors are anteriorly located in eyes thatmaintain good vision. This growth pattern can presentconfusing clinical pictures; for example, when tumorcells are discharged into the vitreous and anteriorchamber, they can masquerade an inflamed eye. Someother authors suggest that diffuse Rb carries a morefavorable prognosis due to its slow growth, while others
TABLE 2
Metastatic Sites of Retinoblastoma in Patients with Positive LP and BM Cytology*
Age(yrs)/Sex
Laterality(U/B) Stage
MetastaticSite LP BM
7.3/M U IV Bone ND 1.6/F U IV Bone 1.8/F U IV Brain ND 2.3/F B IV Brain 3.2/M U IV Lymph node
brainND ND
1.5/F U III None 4.6/M U IV Brain ND 1.1/M B IV Other 1.2/M B II None 1.8/F U IV Lymph node ND 2.3/F B IV Brain ND 2.8/M U IV Bone ND ND8.3/F B IV Lymph node 2.8/M U IV Brain
ND � test not done; U � unilateral; B � bilateral; LP �lumbar puncture; BM � bone marrow aspiration/biopsy.
*Modified from Karcioglu et al—14 cases48
Fig. 6. An endophytic retinoblastoma extends into thevitreous and away from the subjacent choroid (digital im-age obtained by RetCam).

RISK FACTORS FOR Rb METASTASIS 9
have noted that this type of slow growth pattern ismore commonly encountered in older children.50,106
When Rb presents with a plaque-like pattern and theclinical diagnosis is in doubt, enucleation usually is notthe first choice of treatment. Unfortunately, in othercases, the eye may be enucleated even without con-firmed clinical diagnosis, but as a result of suspectedRb. In these instances, histopathologic examination ofthe globe becomes extremely important to rule in orout the diagnosis of Rb. This unusual growth patternand the confusion it creates delays diagnosis in certaincases, which is a risk factor for metastatic Rb.
d. The Unexpected Retinoblastoma
With the advent of heightened awareness, indirectophthalmoscopy, and radiographic imaging tech-niques, the primary histopathologic diagnosis of unsus-pected Rb is much less common in modern practice.With the best clinical techniques currently available, in-accurate diagnoses of Rb are still encountered.113 Inthese cases the histopathological examination of theenucleated globe will give the diagnosis and providecrucial information for patient management.
When retinoblastomas become very large and dif-fusely necrotic, tumor byproducts can seep into sur-rounding orbital soft tissues resulting in an orbitalcellulitis-like picture (swollen soft tissues and prop-tosis without actual extraocular extension of the tu-mor).77,110 In necrotic Rb, the radiographic appear-ance of the eye changes and calcification may bemasked. If this occurs in an eye previously treatedfor Rb the diagnosis is not difficult, but if this occursin the clinical situation where the patients presentsde novo, these findings may lead the ophthalmolo-gist away from the diagnosis.
The Rb cells lack cohesion. Therefore, detachedfragments of tumor are easily separated from themain mass, spread throughout the globe and seedthemselves onto other internal structures includingthe lens capsule, zonules, ciliary body, iris, and cor-nea. The tumor deposits in certain parts of the globeinfluence the clinical behavior. Tumor cells on thelens capsule may lead to secondary cataract, tumorcells within the anterior chamber and the trabecularmeshwork may lead to secondary glaucoma, and ag-gregates of tumor within the anterior chamber form-ing a pseudohypopyon may lead to a uveitis-likesymptoms. These findings complicate the clinical di-agnosis and are typically related to the necrotic Rbupon histopathologic evaluation.
2. Local Invasion
Histopathologic features of eyes enucleated dueto Rb influence the management and prognosis ofthe patient. These features include choroidal andscleral tumor invasion, infiltration of the optic nerve,
and extraocular extension. These features are “riskfactors” for metastasis because exposure to extraocu-lar vasculature allows dissemination, while direct in-filtration of the optic nerve is a gateway to the cen-tral nervous system (Fig. 7). Messmer et al reportedthree significant risk factors for metastasis in Rb.Two significant risk factors were histopathologicallydetermined (choroidal and optic nerve involvement)and one was management related (late enucleation).73
Rubin et al agreed with the Messmer et al but addedrubeosis iridis as a risk factor.98 In multivariate analy-sis, Kopelman et al found histopathologic documen-tation of tumor within the optic nerve and orbit washighly predictive of metastatic disease (Table 1).62
a. Choroidal Involvement
There has been controversy about choroidal in-volvement as a risk for metastasis. When the tumoraccesses the choroid, it spreads rapidly, both in lat-eral and vertical directions. It is able to exploit therich choroidal vascular network and gain access tothe sclera and emissary vascular channels.
Although most authors agree that the involvementof the choroid is a risk factor for metastatic Rb, histo-pathologic criteria quantifying the degree of involve-ment has been insufficient. For example, Rootmanhas stated that “significant invasion of the choroid” in-creases the chance of Rb dissemination but does notquantify “significant invasion.”94 Schilling et al recentlyreported histologic staging criteria for choroidal inva-sion in 297 Rb eyes and attempted a correlation be-tween the extent of invasion and metastatic rate.101
Choroidal involvement ranged from stage I (infiltra-tion of the retinal pigment epithelium without involve-ment of Bruch’s membrane) to stage IV (complete in-filtration of choroid with Rb cells extending laterally).No metastases were noted in 48% of cases in which noRPE or choroid changes were detected. The meta-static rate increased from 0.4% in stages I–III to 12%in stage IV. They concluded that the staging of choroi-dal invasion was clinically significant and advised eyesenucleated for Rb that were found to have choroidalinvolvement at stages II or III should be further exam-ined with serial sections (to rule out the possibility ofstage IV choroidal invasion).101
The correlation between choroidal involvementand metastatic disease was reported by Karcioglu et alin a series of 261 Rb patients.48 A correlation betweenchoroidal involvement, bone marrow aspirates, andradionuclide bone scans was statistically significant.In this work, choroidal involvement was defined asthe presence or absence of Rb cells in the choroid.
b. Optic Nerve Involvement
The optic nerve itself and the surrounding meningesshould be examined with multiple sections for the
10 Surv Ophthalmol 47 (1) January–February 2002 FINGER ET AL
presence or absence of Rb. If tumor is present withinthe nerve, the extent of its involvement should bedetermined. The most common route of extensionfor Rb to the central nervous system is through theoptic nerve. Retinoblastoma invasion through theoptic nerve and leptomeninges can lead to tumorspread on the basilar surface of the brain.18,96,123 Thetumor may also spread posteriorly along the nervebundles toward the optic chiasm, or it may spill intothe subarachnoid space. If the optic nerve extensionis more than 10 mm, the tumor usually spreads notonly into the subarachnoid space but also into theperimeningeal orbital soft tissues. These are thecases with a high incidence of central nervous systemand distant metastasis.18,26,71
In most cases, when retinoblastomas invade theoptic nerve, they extend no more than a few milli-meters beyond the optic nerve head with or withoutinvolvement of lamina cribrosa. When the extensionis beyond lamina cribrosa, and if the optic nerve isinvolved posterior to the surgical transsection, the riskof metastatic Rb increases.52 Whereas some believe thatif any Rb is found within the optic nerve it representsa risk factor for metastasis, most believe it must extendbeyond the line of surgical transsection.73,98
In the majority of cases, if an 8–10 mm optic nervestump is removed during enucleation, the surgicaltranssection margin is clear of tumor.91 However, ifthe case merits a clinical or radiologic suspicion ofextensive tumor invasion of the nerve, the surgicalresection margin can be examined with a frozen sec-tion preparation at the time of surgery (Fig. 8).53
This can be done by obtaining a transverse sectionfrom the enucleation specimen’s most distal portionof the optic nerve. This posterior margin is typicallyorienting as to mark its distal surface. Frozen sec-tions only take a few minutes to process, and arecompleted while pressure is being applied to the or-bit for hemostasis. If tumor cells are identified at thecut margin, another segment of optic nerve can beremoved as to obtain a free margin. Such frozen sec-tion preparations commonly display a crush artifact,which does not interfere with the purpose of theprocedure. This is because the objective is not tohave cytologically perfect-appearing Rb cells, but todetermine the presence or absence of tumor at thesurgical margin.
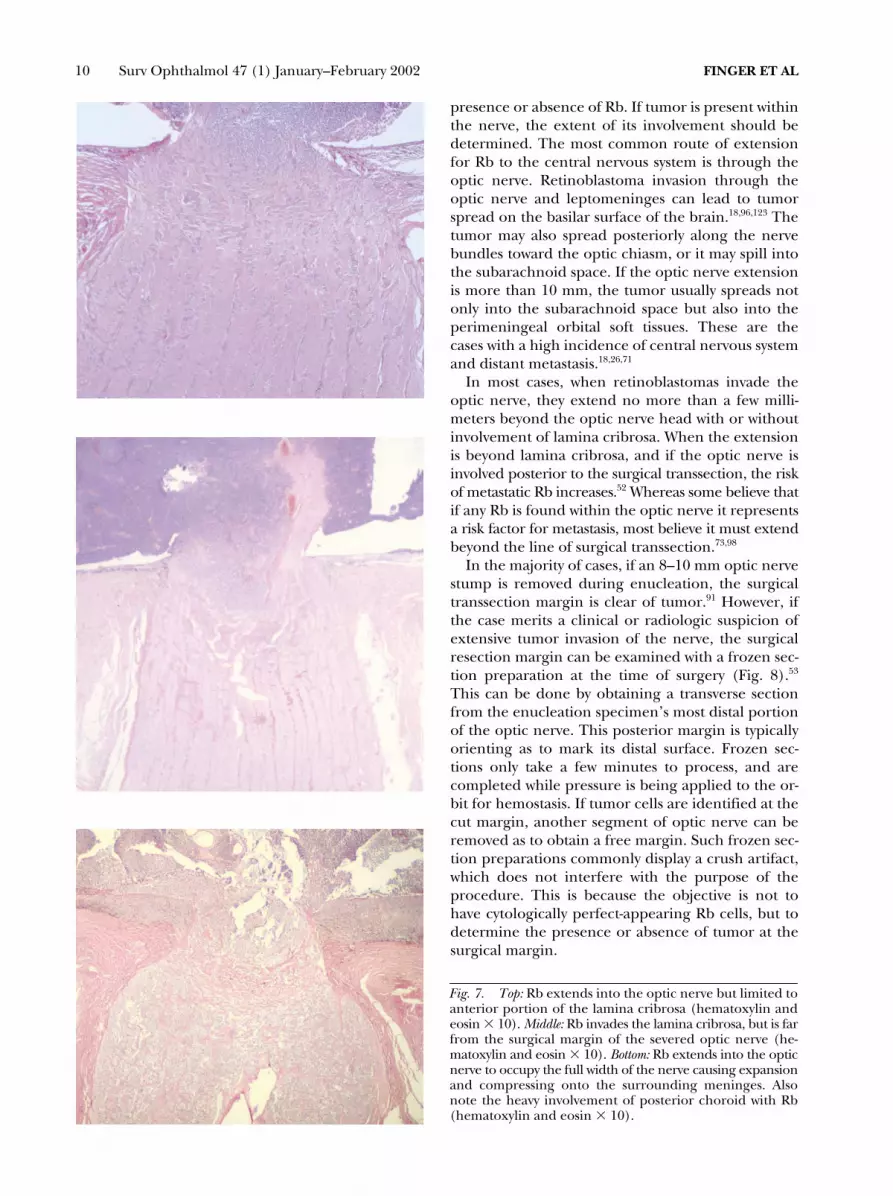
Fig. 7. Top: Rb extends into the optic nerve but limited toanterior portion of the lamina cribrosa (hematoxylin andeosin � 10). Middle: Rb invades the lamina cribrosa, but is farfrom the surgical margin of the severed optic nerve (he-matoxylin and eosin � 10). Bottom: Rb extends into the opticnerve to occupy the full width of the nerve causing expansionand compressing onto the surrounding meninges. Alsonote the heavy involvement of posterior choroid with Rb(hematoxylin and eosin � 10).

RISK FACTORS FOR Rb METASTASIS 11
Frozen sections are not commonly performed atmost centers. Alternatively, paraffin sections may beprepared overnight. With this technique, the opticnerve margin should be removed before the globe isopened (for tumor tissue sampling). Opening theglobe before sampling the optic nerve can result inartifactitious tumor spread onto the optic nerve mar-gin and raise concern about metastasis.
Optic nerve involvement can become very impor-tant when Rb is found in a phthisical eye. BecauseRb is not typically present in phthisical eyes andclinical evaluation is typically impeded by cornealopacities and ectopic calcifications, histopathologi-cal examination of the globe and the nerve are in-valuable. Mullaney et al have reported 10 cases ofRb presenting as phthisis bulbi where all enucle-ated phthisical globes had residual viable-appear-ing tumor cells.78 Optic nerve extension was identi-fied in two patients, both of whom subsequentlydied of metastatic Rb.
c. Orbital Extension
Orbital tumor extension may be the most impor-tant risk factor for Rb metastasis.44,45,67,115 This route ofextraocular extension is not as dependent on the histo-pathological examination of the enucleated globe. Inthe case of orbital Rb, clinical examination–togetherwith modern imaging techniques–may be diagnostic.Clearly, fine needle aspiration or open biopsies oforbital tissues are possible. Limited amounts of epis-cleral extraocular extension are most commonly re-vealed by histopathologic examination of the enu-cleated eyes (Fig. 9).
McLean et al compared metastases and deathrates of 514 patients in the United States to 460 Ger-man patients; all patients were seen prior to 1963.
Invasion of extraocular tissues was the most signifi-cant risk factor in both series.70
d. Summary
Extraocular extension of Rb is estimated to takeplace in less than 10% of the patients in NorthAmerica. The classic findings of choroidal invasion,optic nerve extension, and orbital invasion by Rbhave been associated with metastatic disease. Whilenew studies of Rb cell differentiation may allow forimprovements in treatment, tumor vascularity maybe found to affect metastatic potential. These studiesunderscore the importance of careful histopatho-logic examination aimed at detection of choroidaland optic nerve involvement as well as microscopicextraocular extension for treatment planning.
3. Histopathologic Evaluation
a. Retinoblastoma Histopathology
Retinoblastoma is a poorly differentiated, malig-nant neuroectodermal tumor with significant ne-crotic/apoptotic changes. The predominant celltype is a round cell with basophilic nucleus of vari-able size and relatively little cytoplasm. Mitotic fig-ures and pleomorphic changes of the nuclei arecommon. Typically, ribbons of viable-appearing tu-mor cells alternate with zones of necrosis; the viableareas are characteristically present around the bloodvessels.12 Calcification is a very common feature thatprimarily develops within the areas of necrosis. Inbetter differentiated areas of Rb, Flexner–Winter-steiner and Homer–Wright rosettes can be identi-fied.7,102 These rosettes, which are pathognomonic ofRb, are made of cuboidal cells, the apical ends ofwhich are joined by terminal bars forming the bor-der of the lumen. Ultrastructural studies have deter-
Fig. 8. Frozen cross-section of the optic nerve showingtwo distinct clusters of Rb cells in the center of the nerve.(hematoxylin and eosin � 40).
Fig. 9. Episcleral Rb revealed by histopathologic evalua-tion of an enucleated globe (hematoxylin and eosin � 10).

12 Surv Ophthalmol 47 (1) January–February 2002 FINGER ET AL
mined that the tumor cells forming these rosettesoriginate from photoreceptors and the lining of thelumen of the rosette is analogous to the outer limit-ing membrane of the retina.118,119 Photoreceptor dif-ferentiation may even be better in certain areas inwhich the tumor cells present an attempt to formouter segments of rods and cones; these structuresare known as florettes.
Although cytological details of the tumor maytheoretically affect the tumor’s clinical behavior,management of decisions is rarely based on tumorhistology. Unlike choroidal melanoma, cell type,degree of differentiation, and necrosis have notbeen defined (in themselves) as risk factors for me-tastasis. On the other hand, it is claimed that poorlydifferentiated tumors are more likely to respond toradiation. Unfortunately, this information is typi-cally not available at the time of radiation treat-ment (prior to enucleation).120 There is a clinicalsituation where radiation sensitivity could affect lo-cal control, metastasis, and radiation side effects.That is, in cases of residual tumor in the opticnerve, orbit, or metastatic sites, the dose of externalbeam irradiation or brachytherapy could be modu-lated to reflect the degree of differentiation. In cur-rent practice, radiation dose and dose rates arelargely empirical and the ports of delivery are posi-tioned based on the location and number of the le-sions. The same is true for brachytherapy, in whichthe dose is based primarily on the size and the loca-tion of the tumor(s).
b. Vascular Density
The vascular patterns within Rb may prove to haveprognostic significance. In some malignant neoplasms,a direct association has been reported between tumorvascularity and prognosis.122 It is postulated that poorlydeveloped tumor blood vessels provide increased num-bers of entry sites to the systemic vasculature, therebyenabling tumor dissemination. This hypothesis has beeninvestigated in Rb.84
Karcioglu et al studied the relationship betweenthe number of blood vessels and prognosis in twogroups of eyes enucleated for Rb. The first groupconsisted of 11 patients who developed recurrent ordisseminated disease. The second group consisted of11 patients with tumors limited to their eyes. Eachset of eyes were examined for “blood vessel counts”calculated as percentage of squares including ablood vessel. The average vascularity percentage was29.64% (SD � 13.77) versus 12.73% (SD � 9.34) inthe first and second groups, respectively (p � 0.05).This study revealed that intraocular retinoblastomasthat had subsequently recurred within the orbit ormetastasized were more vascular compared to the tu-mors that exhibited no subsequent recurrence or
metastasis. Karcioglu et al concluded that the num-ber of tumor vessels in the enucleated eye may be auseful predictor for metastasis in Rb.56
The same group also studied the association be-tween tumor vessel counts and other prognostic deter-minants in 38 Rb patients. The relationship betweenclinically assessed features such as age, sex, laterality,size, location and histopathologic features, growth pat-tern, differentiation, necrosis, calcification, anteriorchamber involvement, optic nerve involvement, andchoroid involvement were examined. Significant cor-relations were established between tumor size andblood vessel count and between the degree of necro-sis and clinically assessed size (p � 0.025).54
B. ORBITAL BIOPSY SPECIMENS
Most cases of Rb in North America do not presentwith gross orbital extension. It is more common tohave orbital masses of recurrent Rb requiring biopsyfollowing orbital irradiation and chemotherapy.43
These tumors initially regress, but later recur at thesame location or elsewhere in the orbit. Karcioglu et alhave described cases of recurrent retinoblastomas lead-ing to extrusion and exposure of orbital implants.49,55
In extraocular and metastatic lesions, Rb cells arepoorly differentiated as compared to intraocular tu-mors.72 They appear as uniform masses of poorly differ-entiated small round cells with minimum necrosis andcalcification. No rosettes or florettes indicating retinaldifferentiation are found in metastatic Rb.
C. HISTOPATHOLOGY OFTRILATERAL RETINOBLASTOMA
Trilateral retinoblastomas are rare midline intra-cranial tumors. Also known as pinealoblastomas andparasellar neuroblastic tumors, they present in com-bination with bilateral Rb (Fig. 1).9,11 Like a patientwith bilateral Rb, trilateral patients typically have thegenetic form of the disease. Unlike most bilateral Rbpatients, these patients have developed midline cen-tral nervous system tumors that are typically fatal.
In these cases, tissue diagnosis would be helpful todifferentiate pinealoblastomas from neuroblastic tu-mors. Such differentiation, however, is not possiblebecause these lesions are identical both histopatho-logically and immunochemically.21,86 Midline tumorsare more readily diagnosed in today’s practice be-cause of the advent of new imaging techniques andit is quite likely that many of the previously de-scribed cases were misinterpreted as metastatic Rb tothe brain, as suggested by Shields and Shields.107
D. CYTOPATHOLOGY
1. Anterior Chamber Fine-Needle Aspiration Biopsy
Fine-needle aspiration biopsy (FNAB) and/or para-centesis of the anterior chamber are not typically

RISK FACTORS FOR Rb METASTASIS 13
performed by most ocular oncologists. This is due toconcerns about tumor cell seeding into the limbaland corneal needle tracts that have been histopatho-logically documented in humans.50,51 Although theviability of the spilled tumor cells is questionableand extraocular mass formation is rarely reported,the seriousness of this complication has unified theopinion of the ocular oncologists. It is our opinionthat diagnostic FNAB in questionable Rb casesshould be limited to extraordinarily unusual clinicalcircumstances. In a survey done among high-volumeocular oncologists, it was revealed that only six diag-nostic FNABs were done out of 3000 Rb patientsmanaged collectively during the last 15 years (Kar-cioglu, unpublished data).42,99 On the other hand,the extension of Rb outside of the globe through vit-rectomy ports and surgical incision may occur moreoften than one might think. Many of these cases donot find their way to the written literature.
2. Lumbar Puncture and Bone Marrow Biopsy Specimens
In today’s practice, metastasis of Rb is unlikely inthe absence of extraocular disease. Therefore, manyoncologists no longer perform bone marrow biopsy/aspirates and lumbar punctures in patients with in-traocular Rb.89 Although there is a possibility of he-matogenous (choroidal) spread of Rb, the detectionof tumor cells in blood, bone marrow aspirate, andbiopsy specimens has been low in patients with in-traocular disease.29,114
It can also be difficult to identify small numbers ofRb cells in blood and bone marrow specimens. This isbecause they are small, rounded cells that look like im-mature blood elements. Specimens can be misinter-preted and diagnoses of disseminated Rb have beenbased on misinterpretations of bone marrow speci-mens. In our experience, we have found electron mi-croscopy and immunoperoxidase staining help distin-guish Rb cells within bone marrow samples.87
Once the tumor extends into orbital soft tissues,however, tumor cells may disseminate into bonemarrow. Pratt et al reported two positive bone mar-row aspirates in 115 Rb cases; both cases were foundin Reese–Ellsworth stage III and IV eyes. In a series of101 patients reported by Karcioglu et al, 10 revealedpositive Rb diagnosis in bone marrow aspirates; all ofthese patients were in stage III and IV eyes.48
Cerebral spine fluid (CSF) cytology has been re-ported to be even less helpful than bone marrow as-pirates in screening for metastatic Rb. Pratt et alfound only one positive CSF cytology specimen outof 115 patients. The study by Karcioglu et al of cere-bral spinal fluid cytology specimens in 94 Rb pa-tients conversely showed four (4.2%) positive for Rb
cells. All patients with positive CSF cytology were inReese–Ellsworth stage III and IV eyes.48
The cases that are the most susceptible to developRb cell spillage into the subarachnoid space are theones with 8–10 mm optic nerve involvement. Pene-tration of the meningeal layers has been consideredan anatomic focus of weakness through which thetumor enters the subarachnoid space.
IV. SummaryRetinoblastoma is the most common primary in-
traocular tumor in children. Due to advances in di-agnosis and treatment, more than 90% of childrenare treated prior to extraocular extension or meta-static disease. Due to advances in chemotherapy, thepresence of retinoblastoma metastasis no longermeans patient death.
Risk factors for metastasis are related to patientage, sex, laterality, treatment, genetics, histopathol-ogy, and extraocular extension. Invasion of the uvea,orbit, and optic nerve continue to be the most im-portant current predictors of metastatic Rb. Bilater-ality and delays in diagnosis continue to be impor-tant factors. Genetic studies offer the potential ofpredicting which tumors are likely to be capable ofthe complex pathway called metastasis, which will re-cur within the eye and which will undergo apoptosisand senescence.
Findings that are currently helpful in clinical carecontinue to be available through clinical evalua-tions, genetic studies, and histopathologic evalua-tions of retrieved specimens. We hope this review ishelpful for those caring for patients with retinoblas-toma and for informed consent.
Methods of Literature SearchWe conducted a MEDLINE search covering the
years 1966 through 2000 (as available) using thesearch words retinoblastoma, race, gender, unilateral, bi-lateral, trilateral, chemotherapy, radiation, genetic, gene,chromosome, metastasis, metastases, pathology, angiogene-sis, orbit, optic nerve, choroid, pinealoma, and pinealoblas-toma. Additional references, particularly those be-fore 1966, were obtained from the bibliographies ofarticles obtained from the MEDLINE search. Due tothe availability of translation services, articles in En-glish, Spanish, French, and German received somepreference. When translations were not possible, En-glish abstracts were used as available.
References1. Abramson DH, Ellsworth RM: The surgical management of
retinoblastoma. Ophthalmic Surg 11:596–8, 19802. Abramson DH, Ellsworth RM, Grumbach N, Kitchin FD:
Retinoblastoma: survival, age at detection and comparison1914–1958, 1958–1983. J Pediatr Ophthalmol Strabismus22:246–50, 1985

14 Surv Ophthalmol 47 (1) January–February 2002 FINGER ET AL
3. Abramson DH, Ellsworth RM, Tretter P, et al: Treatment ofbilateral groups I through III retinoblastoma with bilateralradiation. Arch Ophthalmol 99:1761–2, 1981
4. Abramson DH, Greenfield DS, Ellsworth RM: Bilateral ret-inoblastoma. Correlations between age at diagnosis andtime course for new intraocular tumors. Ophthalmic Paedi-atr Genet 13:1–7, 1992
5. Abramson DH, Niksarli K, Ellsworth RM, Servodidio CA:Changing trends in the management of retinoblastoma:1951–1965 vs 1966–1980. J Pediatr Ophthalmol Strabismus31:32–7, 1994
6. Ainbinder DJ, Haik BG, Frei DF, et al: Gadolinium en-hancement: improved MRI detection of retinoblastoma ex-tension into the optic nerve. Neuroradiology 38:778–81,1996
7. Albert DM, Craft J, Sang DN: Ultrastructural retinoblas-toma: transmission and scanning electron microscopy, inJakobiec FA (ed): Ocular and Adnexal Tumors. Birming-ham, AL, Aesculapius, 1978, pp 157–71
8. Amati B, Alevizopoulos K, Vlach J: Myc and the cell cycle.Front Biosci 3:D250–68, 1998
9. Bader JL, Meadows AT, Zimmerman LE, et al: Bilateral ret-inoblastoma with ectopic intracranial retinoblastoma: tri-lateral retinoblastoma. Cancer Genet Cytogenet 5:203–13,1982
10. Bartek J, Bartkova J, Lukas J: The retinoblastoma proteinpathway in cell cycle control and cancer. Exp Cell Res 237:1–6, 1997
11. Blach LE, McCormick B, Abramson DH, Ellsworth RM: Tri-lateral retinoblastoma—incidence and outcome: a decadeof experience. Int J Radiat Oncol Biol Phys 29:729–33,1994
12. Burnier MN, McLean IW, Zimmerman LE, Rosenberg SH:Retinoblastoma. The relationship of proliferating cells toblood vessels. Invest Ophthalmol Vis Sci 31:2037–40, 1990
13. Cano J, Oliveros O, Yunis E: Phenotype variants, malig-nancy, and additional copies of 6p in retinoblastoma. Can-cer Genet Cytogenet 76:112–5, 1994
14. Carbajal UM: Metastasis in retinoblastoma. Am J Ophthal-mol 48:47–69, 1959
15. Christofori G, Semb H: The role of the cell-adhesion mole-cule E-cadherin as a tumour- suppressor gene. Trends Bio-chem Sci 24:73–6, 1999
16. Coupland SE, Bechrakis N, Schuler A, et al: Expression pat-terns of cyclin D1 and related proteins regulating G1-Sphase transition in uveal melanoma and retinoblastoma. BrJ Ophthalmol 82:961–70, 1998
17. de Sutter E, Havers W, Hopping W, et al: The prognosis ofretinoblastoma in terms of survival. A computer assistedstudy. Part II. Ophthalmic Paediatr Genet 8:85–8, 1987
18. DeBuen S: Retinoblastoma with spread by direct continuityto the contralateral optic nerve: report of a case. Am J Oph-thalmol 49:815–9, 1960
19. DerKinderen DJ, Koten JW, Van Romunde LK, et al: Earlydiagnosis of bilateral retinoblastoma reduces death andblindness. Int J Cancer 44:35–9, 1989
20. Devesa SS: The incidence of retinoblastoma. Am J Oph-thalmol 80:263–5, 1975
21. Donoso LA, Rorke LB, Shields JA, et al: S-antigen immu-noreactivity in trilateral retinoblastoma. Am J Ophthalmol103:57–62, 1987
22. Doz F, Khelfaoui F, Mosseri V, et al: The role of chemother-apy in orbital involvement of retinoblastoma. The experi-ence of a single institution with 33 patients. Cancer 74:722–32, 1994
23. Doz F, Peter M, Schleiermacher G, et al: N-MYC amplifica-tion, loss of heterozygosity on the short arm of chromo-some 1 and DNA ploidy in retinoblastoma. Eur J Cancer32A:645–9, 1996
24. Ellsworth RM: Orbital retinoblastoma. Trans Am Ophthal-mol Soc 72:79–88, 1974
25. Ellsworth RM: The treatment of retinoblastoma. Bibl Oph-thalmol 75:142–8, 1968
26. Erwenne CM, Franco EL: Age and lateness of referral as de-
terminants of extra-ocular retinoblastoma. OphthalmicPaediatr Genet 10:179–84, 1989
27. Finger PT, Czechonska G, Demirci H, Rausen A: Chemo-therapy for retinoblastoma: a current topic. Drugs 58:983–96, 1999
28. Finger, PT, Murphree AL: Ophthalmic brachytherapy:treatment of choroidal melanoma and retinoblastoma. In:Peyman G, Meffert S, Conway MD, Chou F (eds): Vitreoret-inal Surgical Techniques London, Martin Dunitz, 2001, pp.403–18.
29. Friedman HS, Kurtzberg J, Croker B, et al: Retinoblastomametastatic to bone marrow. A case with florid spread. ClinPediatr (Phila) 23:184–7, 1984
30. Friend SH, Bernards R, Rogelj S, et al: A human DNA seg-ment with properties of the gene that predisposes to retin-oblastoma and osteosarcoma. Nature 323:643–6, 1986
31. Fung YK, Murphree AL, Tang A, et al: Structural evidencefor the authenticity of the human retinoblastoma gene. Sci-ence 236:1657–61, 1987
32. Gallie BL, Budning A, DeBoer G, et al: Chemotherapy withfocal therapy can cure intraocular retinoblastoma withoutradiotherapy. Arch Ophthalmol 114:1321–8, 1996
33. Gallie BL, Campbell C, Devlin H, et al: Developmental ba-sis of retinal-specific induction of cancer by RB mutation.Cancer Res 59:1731s–35s, 1999
34. Gilbert F, Potluri VR, Short MP, et al: Retinoblastoma,chromosome abnormalities and oncogene expression.Ophthalmic Paediatr Genet 8:3–10, 1987
35. Gupta J, Han LP, Wang P, et al: Development of retinoblas-toma in the absence of telomerase activity. J Natl CancerInst 88:1152–7, 1996
36. Haik BG, Dunleavy SA, Cooke C, et al: Retinoblastoma withanterior chamber extension. Ophthalmology 94:367–70,1987
37. Haik BG, Saint Louis L, Smith ME, et al: Computed tomog-raphy of the nonrhegmatogenous retinal detachment inthe pediatric patient. Ophthalmology 92:1133–42, 1985
38. Haik BG, Saint Louis L, Smith ME, et al: Magnetic reso-nance imaging in the evaluation of leukocoria. Ophthal-mology 92:1143–52, 1985
39. Haik BG, Siedlecki A, Ellsworth RM, Sturgis-Buckhout L:Documented delays in the diagnosis of retinoblastoma.Ann Ophthalmol 17:731–2, 1985
40. Hanahan D, Weinberg RA: The hallmarks of cancer. Cell100:57–70, 2000
41. Harris CC, Hollstein M: Clinical implications of the p53 tu-mor-suppressor gene. N Engl J Med 329:1318–27, 1993
42. Herkes HE, Manschot WA: The danger of diagnostic bi-opsy in eyes suspected of an intraocular tumor. Ophthal-mologica 145:467–9, 1963
43. Hopping W, Waubke TN, Sack H: Orbital involvement inretinoblastoma. Mod Probl Ophthalmol 14:382–7, 1975
44. Hungerford J: Factors influencing metastasis in retinoblas-toma. Br J Ophthalmol 77:541, 1993
45. Hungerford J, Kingston J, Plowman N: Orbital recurrenceof retinoblastoma. Ophthalmic Paediatr Genet 8:63–8,1987
46. Ibarra MS, OBrien JM: Is screening for primitive neuroec-todermal tumors in patients with unilateral retinoblastomanecessary? J AAPOS 4:54–6, 2000
47. Jacquemin C, Karcioglu ZA: Detection of optic nerve in-volvement in retinoblastoma with enhanced computed to-mography. Eye 12:179–83, 1998
48. Karcioglu ZA, al-Mesfer SA, Abboud E, et al: Workup formetastatic retinoblastoma. A review of 261 patients. Oph-thalmology 104:307–12, 1997
49. Karcioglu ZA, al-Mesfer SA, Mullaney PB: Porous polyethyl-ene orbital implant in patients with retinoblastoma. Oph-thalmology 105:1311–6, 1998
50. Karcioglu ZA, Al-Rasheed W, Abboud E: Clinical and histo-pathological features of retinoblastoma presenting in chil-dren older than 5 years of age. Invest Ophthalmol Vis Sci39:S600, 1989
51. Karcioglu ZA, Gordon RA, Karcioglu GL: Tumor seeding

RISK FACTORS FOR Rb METASTASIS 15
in ocular fine needle aspiration biopsy. Ophthalmology 92:1763–7, 1985
52. Karcioglu ZA, Haik BG: Tissue Diagnosis: Intraocular Tu-mors. New York, McMillan, 1987
53. Karcioglu ZA, Haik BG, Gordon RA: Frozen section of theoptic nerve in retinoblastoma surgery. Ophthalmology 95:674–6, 1988
54. Karcioglu ZA, Huaman AM: Angiogenesis in retinoblas-toma. Middle East J Ophthalmol 4:17–25, 1996
55. Karcioglu ZA, Mullaney PB, Millar LC: Extrusion of porouspolyethylene orbital implant in recurrent retinoblastoma.Ophthal Plast Reconstr Surg 14:37–44, 1998
56. Karcioglu ZA, Parson, Bamgboye E: Retinoblastoma angio-genesis and prognosis. Exp Eye Res 63:S69, 1996
57. Khelfaoui F, Validire P, Auperin A, et al: Histopathologicrisk factors in retinoblastoma: a retrospective study of 172patients treated in a single institution. Cancer 77:1206–13,1996
58. Kiratli H, Bilgic S, Ozerdem U: Management of massive or-bital involvement of intraocular retinoblastoma. Ophthal-mology 105:322–6, 1988
59. Kivela T: Trilateral retinoblastoma: a meta-analysis of he-reditary retinoblastoma associated with primary ectopic in-tracranial retinoblastoma. J Clin Oncol 17:1829–37, 1999
60. Knudson AG Jr: Mutation and cancer: statistical study of re-tinoblastoma. Proc Natl Acad Sci USA 68:820–3, 1971
61. Knudson AG Jr, Hethcote HW, Brown BW: Mutation andchildhood cancer: a probabilistic model for the incidenceof retinoblastoma. Proc Natl Acad Sci USA 72:5116–20,1975
62. Kopelman JE, McLean IW, Rosenberg SH: Multivariateanalysis of risk factors for metastasis in retinoblastomatreated by enucleation. Ophthalmology 94:371–7, 1987
63. Kovanlikaya A, Nelson MD Jr, Murphree AL: Radiologicalcase of the month. Retinoblastoma presenting with orbitalcellulitis. Arch Pediatr Adolesc Med 150:873–4, 1996
64. Kvanta A, Steen B, Seregard S: Expression of vascular en-dothelial growth factor (VEGF) in retinoblastoma but notin posterior uveal melanoma. Exp Eye Res 63:511–8, 1996
65. Lee WH, Bookstein R, Hong F, et al: Human retinoblas-toma susceptibility gene: cloning, identification, and se-quence. Science 235:1394–9, 1987
66. Lee WH, Murphree AL, Benedict WF: Expression and am-plification of the N-myc gene in primary retinoblastoma.Nature 309:458–60, 1984
67. Magramm I, Abramson DH, Ellsworth RM: Optic nerve in-volvement in retinoblastoma. Ophthalmology 96:217–22,1989
68. Marcus DM, Brooks SE, Leff G, et al: Trilateral retinoblas-toma: insights into histogenesis and management. SurvOphthalmol 43:59–70, 1998
69. McLean I: Retinobastoma, retinocytomas, and pseudoret-inoblastomas. Philadelphia, Saunders, 1996
70. McLean IW, Rosenberg SH, Messmer EP, et al: Prognosticfactors in cases of retinoblastoma: analysis of 974 patientsfrom Germany and the United States treated by enucle-ation, in Bornfeld N, Gragoudas, ES, Hopping, W (eds):Tumors of the Eye. Amsterdam, Netherlands, Kugler, 1991,pp 69–72
71. Meli FJ, Boccaleri CA, Manzitti J, Lylyk P: Meningeal dis-semination of retinoblastoma: CT findings in eight pa-tients. AJNR Am J Neuroradiol 11:983–6, 1990
72. Merrium GR: Retinoblastoma analysis of 17 autopsies. ArchOphthalmol 44:71–108, 1950
73. Messmer EP, Heinrich T, Hopping W, et al: Risk factors formetastases in patients with retinoblastoma. Ophthalmology98:136–41, 1991
74. Moll AC, Kuik DJ, Bouter LM, et al: Incidence and survivalof retinoblastoma in The Netherlands: a register basedstudy 1862–1995. Br J Ophthalmol 81:559–62, 1997
75. Morgan G: Diffuse infiltrating retinoblastoma. Br J Oph-thalmol 55:600–6, 1971
76. Moshfeghi DM, Moshfeghi AA, Finger PT: Enucleation.Surv Ophthalmol 44:277–301, 2000
77. Muir KR, Smith H, Parkes SE, et al: Increasing incidence ofretinoblastoma? Arch Dis Child 65:915, 1990
78. Mullaney PB, Karcioglu ZA, al-Mesfer S, Abboud EB: Pre-sentation of retinoblastoma as phthisis bulbi. Eye 11:403–8,1997
79. Mullaney PB, Karcioglu ZA, Huaman AM, al-Mesfer S: Ret-inoblastoma associated orbital cellulitis. Br J Ophthalmol82:517–21, 1998
80. Murphree AL, Villablanca JG, Deegan WF 3rd, et al: Che-motherapy plus local treatment in the management of in-traocular retinoblastoma. Arch Ophthalmol 114:1348–56,1996
81. Nicholson DH, Norton EW: Diffuse infiltrating retinoblas-toma. Trans Am Ophthalmol Soc 78:265–89, 1980
82. Nork TM, Poulsen GL, Millecchia LL, et al: p53 regulatesapoptosis in human retinoblastoma. Arch Ophthalmol 115:213–9, 1997
83. Oliveros O, Yunis E: Chromosome evolution in retinoblas-toma. Cancer Genet Cytogenet 82:155–60, 1995
84. Peer J, Neufeld M, Baras M, et al: Rubeosis iridis in retino-blastoma. Histologic findings and the possible role of vas-cular endothelial growth factor in its induction. Ophthal-mology 104:1251–8, 1997
85. Pendergrass TW, Davis S: Incidence of retinoblastoma inthe United States. Arch Ophthalmol 98:1204–10, 1980
86. Pesin SR, Shields JA: Seven cases of trilateral retinoblas-toma. Am J Ophthalmol 107:121–6, 1989
87. Popoff N, Ellsworth RM: The fine structure of nuclear al-terations in retinoblastoma and in the developing humanretina: in vivo and in vitro observations. J Ultrastruct Res29:535–49, 1969
88. Pradhan DG, Sandridge AL, Mullaney P, et al: Radiationtherapy for retinoblastoma: a retrospective review of 120patients. Int J Radiat Oncol Biol Phys 39:3–13, 1997
89. Pratt CB, Meyer D, Chenaille P, Crom DB: The use of bonemarrow aspirations and lumbar punctures at the time of di-agnosis of retinoblastoma. J Clin Oncol 7:140–3, 1989
90. Redler LD, Ellsworth RM: Prognostic importance of chor-oidal invasion in retinoblastoma. Arch Ophthalmol 90:294–6, 1973
91. Reese AB, Ellsworth RM: The evaluation and current con-cept of retinoblastoma therapy. Trans Am Acad Ophthal-mol Otolaryngol 67:164–72, 1963
92. Robertson DM, Campbell RJ: Analysis of misdiagnosed ret-inoblastoma in a series of 726 enucleated eyes. Mod ProblOphthalmol 18:156–9, 1977
93. Romanella A, Abramson DH, Servodidio CA: Unusual pre-senting signs of retinoblastoma: a case study. J OphthalmicNurs Technol 10:98–102, 1991
94. Rootman J: Diseases of the Orbit. Philadelphia, Lippincott,1988
95. Rootman J, Ellsworth RM, Hofbauer J, Kitchen D: Orbitalextension of retinoblastoma: a clinicopathological study.Can J Ophthalmol 13:72–80, 1978
96. Rootman J, Hofbauer J, Ellsworth RM, Kitchen D: Invasionof the optic nerve by retinoblastoma: a clinicopathologicalstudy. Can J Ophthalmol 11:106–14, 1976
97. Rubenfeld M, Abramson DH, Ellsworth RM, Kitchin FD:Unilateral vs. bilateral retinoblastoma. Correlations be-tween age at diagnosis and stage of ocular disease. Oph-thalmology 93:1016–9, 1986
98. Rubin CM, Robison LL, Cameron JD, et al: Intraocular ret-inoblastoma group V: an analysis of prognostic factors. JClin Oncol 3:680–5, 1985
99. Sanders TE, Smith ME: Biopsy of intraocular tumors: a re-evaluation. Int Ophthalmol Clin 12:163–76, 1972
100. Schiffman JS, Grunwald GB: Differential cell adhesion andexpression of N-cadherin among retinoblastoma cell lines.Invest Ophthalmol Vis Sci 33:1568–74, 1992
101. Schilling H, Yang H, Havers W, et al: Changes of the RPEand choroidal invasion in 297 enucleated eyes with retino-blastoma, in IXth Symposium of the Retinoblastoma Soci-ety, Geneva, Switzerland, 1998
102. Schipper J: Retinoblastoma: a medical and experimental

16 Surv Ophthalmol 47 (1) January–February 2002 FINGER ET AL
study, in Radiation Oncology. Utrecht, Netherlands, Uni-versity of Utrecht, 1980, pp 144
103. Scott MH, Richard JM: Retinoblastoma in the state of Okla-homa: a clinicopathologic review. J Okla State Med Assoc86:111–8, 1993
104. Shields CL, Shields JA, Baez K, et al: Optic nerve invasionof retinoblastoma. Metastatic potential and clinical risk fac-tors. Cancer 73:692–8, 1994
105. Shields CL, Shields JA, Baez KA, et al: Choroidal invasionof retinoblastoma: metastatic potential and clinical risk fac-tors. Br J Ophthalmol 77:544–8, 1993
106. Shields CL, Shields JA, Shah P: Retinoblastoma in olderchildren. Ophthalmology 98:395–9, 1991
107. Shields JA, Shields CL: Intraocular Tumors: A Text and At-las. Philadelphia, Saunders, 1992
108. Shields JA, Shields CL, Donoso LA, Lieb WE: Changingconcepts in the management of retinoblastoma. Oph-thalmic Surg 21:72–6, 1990
109. Shields JA, Shields CL, Sivalingam V: Decreasing frequencyof enucleation in patients with retinoblastoma. Am J Oph-thalmol 108:185–8, 1989
110. Shields JA, Shields CL, Suvarnamani C, et al: Retinoblas-toma manifesting as orbital cellulitis. Am J Ophthalmol112:442–9, 1991
111. Skubitz AP, Grossman MD, McCarthy JB, et al: The de-creased adhesion of Y79 retinoblastoma cells to extracellu-lar matrix proteins is due to a deficit of integrin receptors.Invest Ophthalmol Vis Sci 35:2820–33, 1994
112. Squire J, Phillips RA, Boyce S, et al: Isochromosome 6p, aunique chromosomal abnormality in retinoblastoma: verifi-cation by standard staining techniques, new densitometricmethods, and somatic cell hybridization. Hum Genet 66:46–53, 1984
113. Stafford WR, Yanoff M, Parnell BL: Retinoblastomas ini-tially misdiagnosed as primary ocular inflammations. ArchOphthalmol 82:771–3, 1969
114. Stanford GB, Reese AB: Malignant cells in the blood of eyepatients. Trans Am Acad Ophthalmol Otolaryngol 75:102–9, 1971
115. Stannard C, Lipper S, Sealy R, Sevel D: Retinoblastoma: cor-relation of invasion of the optic nerve and choroid with prog-nosis and metastases. Br J Ophthalmol 63:560–70, 1979
116. Svedberg-Winholt H, Al-Moster SA, Riley FC: Survivaltrends of retinoblastoma patients with extraocular exten-sion. Congress of European Society of Ophthalmology Ab-stract: 502, 1999
117. Tamboli A, Podgor MJ, Horm JW: The incidence of retino-blastoma in the United States: 1974 through 1985. ArchOphthalmol 108:128–32, 1990
118. Tso MO, Fine BS, Zimmerman LE: The nature of retino-blastoma. II. Photoreceptor differentiation: an electron mi-croscopic study. Am J Ophthalmol 69:350–9, 1970
119. Tso MO, Zimmerman LE, Fine BS: The nature of retino-blastoma. I. Photoreceptor differentiation: a clinical andhistopathologic study. Am J Ophthalmol 69:339–49, 1970
120. Tso MO, Zimmerman LE, Fine BS, Ellsworth RM: A causeof radioresistance in retinoblastoma: photoreceptor differ-entiation. Trans Am Acad Ophthalmol Otolaryngol 74:959–69, 1970
121. Urquidi V, Tarin D, Goodison S: Role of telomerase in cellsenescence and oncogenesis. Annu Rev Med 51:65–79,2000
122. Weidner N, Semple JP, Welch WR, Folkman J: Tumor an-giogenesis and metastasis—correlation in invasive breastcarcinoma. N Engl J Med 324:1–8, 1991
123. Yanoff M, Fine BS: Ocular Pathology. New York, Harperand Row, 1984
The authors have no commercial or proprietary interest in anyproduct or concept discussed in this article. Supported in part by
The EyeCare Foundation, Inc., New York, (http://www.eyecarefoundation.org/) and K08 EY00 38201 Career DevelopmentAward of Research to Prevent Blindness (JWH).
Reprint address: Paul T. Finger, MD, The New York Eye CancerCenter, 115 East 61st Street, New York, New York, 10021.
OutlineI. Clinical risk factors
A. Demography1. Age at diagnosis2. Age at treatment3. Racial and gender distribution
B. Laterality1. Unilateral versus bilateral2. Trilateral disease
C. Local extension1. Optic nerve invasion2. Uveal invasion3. Orbital invasion
D. Timing of Diagnosis and Treatment1.Delay in diagnosis2. Eye conservation
II. Genetic and molecular pathophysiology of metastasis
A. Tumor invasion and metastasisB. Genetic events in the pathogenesis of Rb
1. Initiating event: Mutation of the Rb gene2. Early events
a. Increased copy number of chromosome 6pb. Increased copy number of chromosome 1q
3. Late eventsa. Telemerase activityb. Loss of chromosome 1pc. N-myc amplificationd. P53 inactivation
C. Pathophysiologic steps toward metastasis1. Angiogenesis2. Tissue invasion3. Deregulation of cell-to-cell adhesion
molecules4. Changes in integrins5. Other steps in metastasis
III. Histopathologic ParametersA. Enucleation specimens
1. Tumor morphologya. Endophytic Rbb. Exophytic Rbc. Plaque-like Rb
2. Local invasiona. Choroidal involvementb. Optic nerve involvementc. Orbital extensiond. Summary
3. Histopathologic evaluationa. Retinoblastoma histopathologyb. Vascular density
B. Orbital biopsy specimensC. Histopathology of trilateral retinoblastomaD. Cytopathology
1. Anterior chamber fine-needle aspiration biopsy
2. Lumbar puncture and bone marrow biopsy specimens
IV. Summary