reversible, allosteric small-molecule inhibitors of regulator of g protein signaling proteins
TRANSCRIPT

Reversible, Allosteric Small-Molecule Inhibitors of Regulator ofG Protein Signaling Proteins□S
Levi L. Blazer, David L. Roman, Alfred Chung, Martha J. Larsen, Benjamin M. Greedy,Stephen M. Husbands, and Richard R. NeubigDepartments of Pharmacology (L.L.B., A.C., R.R.N.) and Internal Medicine (Cardiovascular Medicine) (R.R.N.), and Center forChemical Genomics (M.J.L., R.R.N.), University of Michigan, Ann Arbor, Michigan; Division of Medical Chemistry and NaturalProducts, University of Iowa College of Pharmacy, Iowa City, Iowa (D.L.R.); and Department of Pharmacy and Pharmacology,University of Bath, Bath, United Kingdom (S.M.H.)
Received March 29, 2010; accepted June 15, 2010
ABSTRACTRegulators of G protein signaling (RGS) proteins are potent neg-ative modulators of G protein signaling and have been proposedas potential targets for small-molecule inhibitor development. Wereport a high-throughput time-resolved fluorescence resonanceenergy transfer screen to identify inhibitors of RGS4 and describethe first reversible small-molecule inhibitors of an RGS protein.Two closely related compounds, typified by CCG-63802 [((2E)-2-(1,3-benzothiazol-2-yl)-3-[9-methyl-2-(3-methylphenoxy)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl]prop-2-enenitrile)], inhibit the inter-action between RGS4 and G�o with an IC50 value in the lowmicromolar range. They show selectivity among RGS proteins
with a potency order of RGS 4 � 19 � 16 � 8 �� 7. Thecompounds inhibit the GTPase accelerating protein activity ofRGS4, and thermal stability studies demonstrate binding to theRGS but not to G�o. On RGS4, they depend on an interaction withone or more cysteines in a pocket that has previously been iden-tified as an allosteric site for RGS regulation by acidic phospho-lipids. Unlike previous small-molecule RGS inhibitors identified todate, these compounds retain substantial activity under reducingconditions and are fully reversible on the 10-min time scale. CCG-63802 and related analogs represent a useful step toward thedevelopment of chemical tools for the study of RGS physiology.
IntroductionNetworks of protein-protein interactions are crucial for
efficient cellular function. There has been significant interestin developing small-molecule protein-protein interaction in-hibitors (SMPPIIs) for use as research probes and potentialtherapeutic agents (Berg, 2003, 2008; Gadek and Nicholas,2003; Arkin and Wells, 2004; Blazer and Neubig, 2009). Thedevelopment of SMPPIIs has been difficult. One challenge
has been the lack of clearly identifiable small-molecule bind-ing sites on the relatively featureless protein-protein inter-action interface. A promising approach is the use of allostericpockets on the protein target to bypass this problem and,increasingly, there has been solid progress in SMPPII devel-opment (Berg, 2003, 2008; Arkin and Wells, 2004; Blazer andNeubig, 2009; Arkin and Whitty, 2009; Busschots et al., 2009;Niu and Chen, 2009).
RGS proteins are GTPase-accelerating proteins (GAPs) forheterotrimeric G protein � subunits (Berman et al., 1996).They increase the intrinsic rate of GTP hydrolysis by the G�,thus reconciling the paradox of the subsecond regulation of Gprotein signaling in vivo versus the relatively long half-life ofGTP bound to purified G� in vitro. In mammals, there aremore than 20 known RGS proteins that interact with limitedselectivity to most G� subtypes (Hollinger and Hepler, 2002;Neubig and Siderovski, 2002).
There is substantial interest in the therapeutic potential of
This work was supported by the Michigan Chemistry-Biology InterfaceTraining Program, which is funded through the National Institutes of HealthNational Institute of General Medical Sciences [Grant T32-GM00008597]; theNational Institutes of Health National Institute on Drug Abuse [Grant R01-DA023252]; and the National Institutes of Health National Institute of Gen-eral Medical Sciences [Grant F32-GM076821].
Article, publication date, and citation information can be found athttp://molpharm.aspetjournals.org.
doi:10.1124/mol.110.065128.□S The online version of this article (available at http://molpharm.
aspetjournals.org) contains supplemental material.
ABBREVIATIONS: RGS, regulator of G protein signaling; TR-FRET, time-resolved fluorescence resonance energy transfer; FCPIA, flow cytometryprotein interaction assay; SMPPII, small-molecule protein-protein interaction inhibitor; PPI, protein-protein interaction; GAP, GTPase-acceleratingprotein; GPCR, G protein-coupled receptor; DTT, dithiothreitol; MBP, maltose-binding protein; DMSO, dimethyl sulfoxide; PCR, polymerase chainreaction; BSA, bovine serum albumin; Tm, melting temperature; DRC, dose-response curve; CCG-63802, ((2E)-2-(1,3-benzothiazol-2-yl)-3-[9-methyl-2-(3-methylphenoxy)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl]prop-2-enenitrile); CCG-63808, ((2E)-2-(1,3-benzothiazol-2-yl)-3-[9-methyl-2-(4-fluorolphenoxy)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl]prop-2-enenitrile); CCG-4986, methyl-N-[(4-chlorophenyl)sulfonyl]-4-nitrobenzenesulfin-imidoate; CI-1033, N-[-4-[(3-chloro-4-fluorophenyl)amino]-7-[3-(4-morpholinyl)propoxy]-6-quinazolinyl]-2-propenamide.
0026-895X/10/7803-524–533$20.00MOLECULAR PHARMACOLOGY Vol. 78, No. 3Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics 65128/3619623Mol Pharmacol 78:524–533, 2010 Printed in U.S.A.
524
small-molecule modulators of RGS proteins (Zhong and Neu-big, 2001; Neubig and Siderovski, 2002; Riddle et al., 2005;Blazer and Neubig, 2009; Traynor et al., 2009). In brief, RGSinhibitors may potentiate signaling through GPCRs in a tis-sue-specific manner because of the localized expression pat-terns of many RGS proteins. This effect could be used toreduce side effects of clinically used GPCR agonists that stemfrom nontarget tissue receptor activation [e.g., �-opioid re-ceptor-dependent constipation during postoperative analge-sia (Bueno and Fioramonti, 1988)].
To understand the physiological ramifications of inhib-iting RGS protein GAP activity, we have developed twolines of mice that express mutant G�o or G�i2 and areinsensitive to RGS effects (G184S). These mice show dra-matic phenotypes, including resistance to diet-inducedobesity and antidepressant-like behavioral effects (Huanget al., 2006, 2008; Talbot et al., 2010). RGS4 is up-regulated in the dorsal horn of spinal cord during thedevelopment of neuropathic pain (Garnier et al., 2003),and RGS4 can inhibit several pain-modulating receptors(e.g., �-opioid receptor) (Garnier et al., 2003; Traynor andNeubig, 2005). Consequently, small-molecule modulatorsof RGS function should have utility as research tools andpotentially as therapeutics. Because of the wealth of infor-mation on the structure and function of RGS4, we chosethis protein as our primary target for validating the“drugability” of RGS proteins.
There have been several reported peptide inhibitors ofRGS4 and related family members (Roof et al., 2006, 2008;Wang et al., 2008) and one disclosed small-molecule inhibitor(Roman et al., 2007). Because of the physical properties of thepeptides, none of them function in a cellular environmentunless they are introduced intracellularly [e.g., by dialysisvia a patch pipette (Roof et al., 2006)]. The small-moleculecompound CCG-4986 [methyl-N-[(4-chlorophenyl)sulfonyl]-4-nitrobenzenesulfinimidoate] irreversibly inhibits RGS4 byreacting with one or more cysteine residues (Kimple et al.,2007; Roman et al., 2010), and its activity is lost in thepresence of free thiols. This mechanism of action makesCCG-4986 less desirable as a potential lead compound for
small-molecule probe development. Consequently, we under-took this study to identify novel RGS inhibitors that retainactivity under reducing conditions and ones that have areversible mechanism of action.
This article describes the identification and characteriza-tion of the first class of reversible small-molecule inhibitorsof an RGS protein. They were found in a biochemical high-throughput screen carried out in the presence of dithiothre-itol (DTT). They inhibit the binding and GAP activity ofRGS4 with G�o in a reversible manner through an interac-tion at an allosteric regulatory site on the RGS. These com-pounds represent an important step toward the developmentof tools for the study of RGS functions in physiological andpathophysiological situations.
Materials and MethodsReagents. Chemicals were purchased from Sigma-Aldrich (St.
Louis, MO) or Thermo Fisher Scientific (Waltham, MA) and werereagent grade or better. Alexa Fluor 488 succinimidyl ester andLanthaScreen Thiol-reactive Tb chelate were obtained from In-vitrogen (Carlsbad, CA). �[32P]GTP (10 mCi/ml) and [35S]GTP�S(12.5 mCi/ml) were obtained from PerkinElmer Life and Analyti-cal Sciences (Waltham, MA) and isotopically diluted with unla-beled nucleotide before use. Amylose resin was purchased fromNew England Biolabs (Ipswich, MA). Ni-NTA resin was purchasedfrom QIAGEN (Valencia, CA). Avidin-coated microspheres were pur-chased from Luminex (Austin, TX). The screening library was com-prised of a commercially available subset of compounds from ChemDiv(San Diego, CA) provided through a collaboration between the Univer-sity of Michigan Center for Chemical Genomics and the Novartis Insti-tute for Biomedical Research (East Hanover, NJ). CCG-63802 [((2E)-2-(1,3-benzothiazol-2-yl)-3-[9-methyl-2-(3-methylphenoxy)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl]prop-2-enenitrile)] and CCG-63808 [((2E)-2-(1,3-benzothiazol-2-yl)-3-[9-methyl-2-(4-fluorolphenoxy)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl]prop-2-enenitrile)] (see structures in Fig. 1)were purchased from ChemDiv, and compound identity was verified byNMR via ChemDiv and independent complete synthesis in the labora-tory of Dr. Stephen M. Husbands (University of Bath).
Compound Synthesis. In brief, 2-hydroxy-9-methyl-4H-pyrido[1,2-�]pyrimidin-4-one was prepared by the reaction of 2-amino-3-methylpyridine with diethyl malonate according to literature meth-
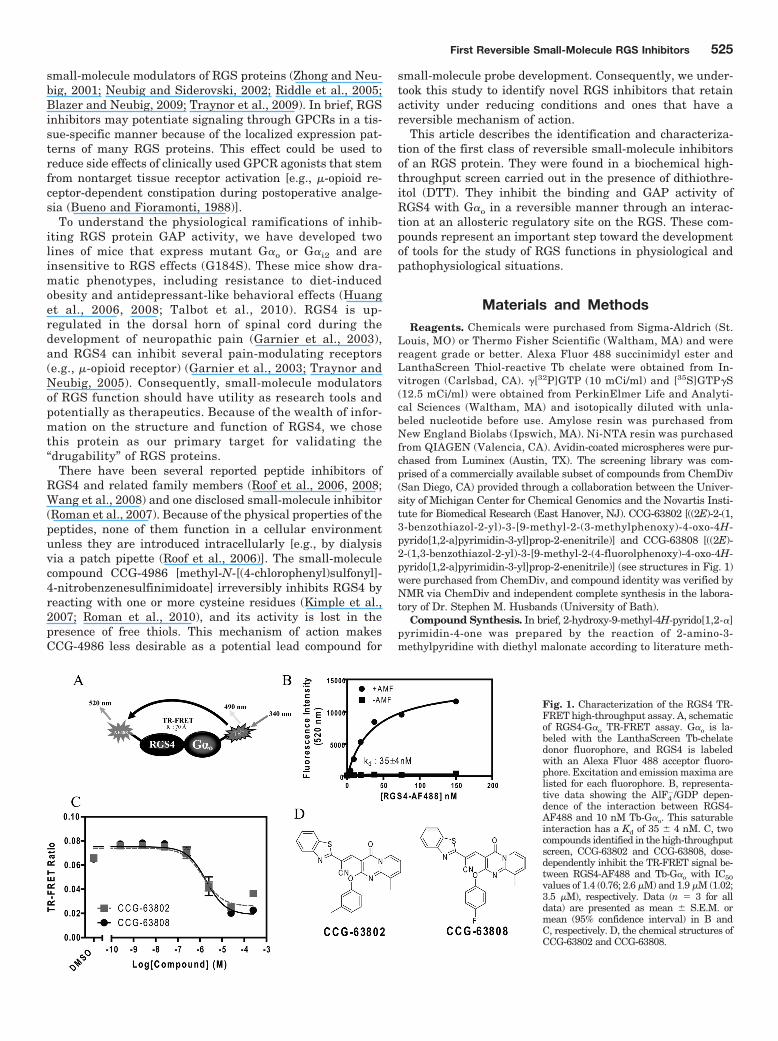
Fig. 1. Characterization of the RGS4 TR-FRET high-throughput assay. A, schematicof RGS4-G�o TR-FRET assay. G�o is la-beled with the LanthaScreen Tb-chelatedonor fluorophore, and RGS4 is labeledwith an Alexa Fluor 488 acceptor fluoro-phore. Excitation and emission maxima arelisted for each fluorophore. B, representa-tive data showing the AlF4
�/GDP depen-dence of the interaction between RGS4-AF488 and 10 nM Tb-G�o. This saturableinteraction has a Kd of 35 � 4 nM. C, twocompounds identified in the high-throughputscreen, CCG-63802 and CCG-63808, dose-dependently inhibit the TR-FRET signal be-tween RGS4-AF488 and Tb-G�o with IC50values of 1.4 (0.76; 2.6 �M) and 1.9 �M (1.02;3.5 �M), respectively. Data (n � 3 for alldata) are presented as mean � S.E.M. ormean (95% confidence interval) in B andC, respectively. D, the chemical structures ofCCG-63802 and CCG-63808.
First Reversible Small-Molecule RGS Inhibitors 525

ods (Ingalls and Popp, 1967). This material was first converted to2-chloro-9-methyl-4-oxo-4H-pyrido[1,2-�]pyrimidine-3-carbaldehydevia Vilsmeier formylation, and this product was then heated with4-fluorophenol to afford 2-(4-fluorophenoxy)-9-methyl-4-oxo-4H-pyrido[1,2-�]pyrimidine-3-carbaldehyde. Condensation of thiscompound with 2-benzothiazole acetonitrile using catalytic trieth-ylamine in dichloromethane provided CCG-63808 as an orangecrystalline solid (Supplemental Fig. 1). CCG-63802 was preparedin a similar manner, except 4-fluorophenol was replaced with3-methylphenol. Synthesized compounds were verified by 1H and13C NMR using a JEOL (Tokyo, Japan) �-270-MHz instrument: 1Hat 270 MHz, and Varian Inc. (Palo Alto, CA) Mercury-400-MHzinstrument: 1H at 400 MHz, 13C at 100 MHz; d in parts permillion, J in Hertz with tetramethylsilane as an internal stan-dard, by electrospray mass spectrometry using a micrOTOF(Bruker Daltonics, Billerica, MA) and microanalysis using aPerkinElmer Life and Analytical Sciences 240C analyzer.
Protein Expression and Purification. Human RGS4 was ex-pressed either from the pQE80RGS4 vector, which encodes 6� his-tidine-tagged and N-terminally truncated form of RGS4 that lacksthe first 18 residues (�N19RGS4), or the pKMRGS4 vector, whichencodes a maltose-binding protein (MBP)-�N19RGS4 fusion protein.The �N form of RGS4 was selected because it provides better proteinyield in prokaryotic expression systems. MBP-His6-RGS19�C11 (hu-man), MBP-His6-RGS7 (human), MBP-His6-RGS8 (human), andMBP-His6-RGS16 (human) were expressed from constructs madewith the pMALC2H10 vector as described previously (Roman et al.,2009). For the mutagenesis studies, �N51RGS4 (rat) wild type andcysteine 3 alanine mutants were expressed from the pMALC2H10vector. Mutagenesis was performed as described elsewhere (Romanet al., 2010) using the QuikChange multi site-directed mutagenesiskit (Stratagene, La Jolla, CA) where one or more of the cysteineresidues in the RGS domain of RGS4 were mutated to alanine.
All proteins were expressed in and harvested from BL21-DE3Escherichia coli via standard transformation, growth, and lysis pro-tocols (Lee et al., 1994; Lan et al., 1998, 2000; Roman et al., 2007;Roof et al., 2008). Histidine-tagged RGS4 was purified over a Ni-NTAaffinity column (QIAGEN) followed by cation exchange chromatog-raphy and size exclusion chromatography. MBP-tagged RGS pro-teins were purified with an amylose affinity column followed by sizeexclusion chromatography. Hexahistidine-tagged rat G�o was ex-pressed and purified as described previously (Lee et al., 1994). Gprotein activity was determined by [35S]GTP�S binding (Sternweisand Robishaw, 1984). In all cases, proteins were purified to �90%homogeneity before use.
Chemical Labeling of Purified G�o and RGS. For Alexa Fluor488 labeling of RGS4, �N19RGS4 was labeled with Alexa Fluor 488succinimidyl ester (Invitrogen) at a 5:1 (label/protein) stoichiometryin a total volume of 2.0 ml of 50 mM HEPES, pH 8.2 at 4°C, 100 mMNaCl, and 1 mM DTT. The reaction was performed while rotatingsamples in the dark for 1.5 h at 4°C. The reaction was quenched bythe addition of 1 mM glycine for 10 min at 4°C. Labeled RGS4 wasresolved from the reaction mixture by size exclusion chromatographyusing a 20-ml Sephadex G-25 desalting column (GE Healthcare,Little Chalfont, Buckinghamshire, UK). Degree of labeling was de-termined spectroscopically to be approximately 1:1.
Tb chelate labeling of G�o, G�o was labeled with the LanthaScreenTb thiol-reactive reagent (Invitrogen) at a 5:1 (label/protein) stoichi-ometry in a total volume of 1.0 ml of 50 mM HEPES, pH 7.25 at 4°C,100 mM NaCl, supplemented with 10 �M GDP and 0.8 mM Tris(2-carboxyethyl)phosphine. The reaction was allowed to proceed at 4°Cfor 1.5 h during rotation in the dark. The reaction was quenched bythe addition of 1 mM DTT for 20 min at 4°C. Labeled protein waspurified from the reaction mixture by size exclusion chromatographyusing a Sephadex G-25 desalting column (GE Healthcare). Degree oflabeling was determined spectroscopically to be approximately 1:1.The activity and effective concentration of the labeled G protein was
determined by [35S]GTP�S binding as described previously (Stern-weis and Robishaw, 1984).
For biotinylation of RGS proteins, RGS protein was mixed at a 3:1(label/protein) molar ratio with biotinamidohexanoic acid N-hy-droxysuccinimide ester (Sigma-Aldrich) in a buffer of 50 mMHEPES, pH 8.5 at 4°C, 100 mM NaCl, and 1 mM DTT. The reactionwas allowed to proceed at 4°C while rotating for 2 h and then wasquenched by the addition of a large molar excess of glycine for 10min. Labeled protein was purified from the reaction mixture by sizeexclusion chromatography using a Sephadex G-25 desalting column(GE Healthcare Biosciences).
Alexa Fluor 532 labeling was performed as described previously(Roman et al., 2007). Labeled protein was purified from the reactionmixture by size exclusion chromatography using a Sephadex G-25desalting column (GE Healthcare Biosciences).
Time-Resolved FRET. TR-FRET experiments were performedon a PHERAstar multipurpose microplate reader (BMG LabtechGmbH, Offenberg, Germany) using the LanthaScreen filter set.These experiments were based on the method of Leifert et al. (2006).For the saturation experiments, Tb-G�o was diluted to 20 nM in 50mM HEPES, pH 8.0, 100 mM NaCl, 0.1% Lubrol, 30 �M GDP, 5 mMNaF, 5 mM MgCl2, and 5 �M AlCl3 and allowed to activate for 10min on ice before use. RGS4-AF488 was serially diluted in 50 mMHEPES, pH 8.0 at room temperature, 100 mM NaCl, and 0.1%Lubrol (TR-FRET buffer). Ten microliters of the RGS4 dilution wasadded to a black nonstick, low-volume, 384-well plate (Corning LifeSciences, Lowell, MA) with a minimum of duplicate measurements.Ten microliters of Tb-G�o was added (10 nM final), and the mixturewas allowed to incubate at room temperature for 15 min in the dark.The nonspecific TR-FRET signal was determined by excluding AlCl3,MgCl2, and NaF from a set of samples. The fluorescence emission atboth 490 and 520 nm was measured from 50 flashes of 340-nmexcitation light per well. The data were collected in 10-�s bins, andthe delayed emission signal was integrated from 100 to 500 �s aftereach flash. TR-FRET data were analyzed as the ratio of emission at520 nm/490 nm.
High-Throughput Screening. High-throughput screening wasperformed at the University of Michigan Center for ChemicalGenomics. The approximately 40,000-compound screening collectionwas provided by the Novartis Institute for Biomedical Research andwas comprised of compounds selected from the ChemDiv screeninglibrary. Five microliters of 50 mM HEPES, pH 8.0 at room temper-ature, 100 mM NaCl, 0.1% Lubrol, and 1 mM DTT (TR-FRET buffer)was dispensed with a Multidrop (Thermo Fisher Scientific) intoevery well of a black nonstick, low-volume, 384-well plate. Twohundred nanoliters of each compound (2 mM stock, 20 �M final assayconcentration) or DMSO control was added to the plate with a pintool by using a Beckman BioMek FX liquid handler (BeckmanCoulter, Fullerton, CA). To this compound dilution, 5 �l of 200 nMAlexa Fluor 488-labeled RGS4 was added and incubated for 15 minat room temperature in the dark. Then, 10 �l of 20 nM Tb-labeledG�o was added to the mixture. For this assay, the positive inhibitioncontrol (i.e., no RGS4/G�o binding) was Tb-labeled G�o in the inac-tive GDP-bound state, and the negative control (i.e., full RGS4/G�o
binding) used G�o in the GDP/AlF4-bound state. This mixture wasincubated at room temperature in the dark for 15 min before analysiswith the PHERAstar plate reader. Data were compiled and analyzedby using the M-Screen database, a chemoinformatics suite developedby the Center for Chemical Genomics at the University of Michigan.Compounds that inhibited the TR-FRET signal �2 SD from thenegative control were considered “actives” and were chosen for dose-response follow-up experiments.
TR-FRET Dose-Response Experiments. Actives from the pri-mary screen were evaluated for concentration-dependent activity inthe TR-FRET assay. Compound dilutions were performed in DMSO,and 200 nl of diluted compound was spotted into the wells of a blacknonstick, low-volume, 384-well plate that contained 5 �l of TR-FRETbuffer. To the well, 5 �l of 200 nM Alexa Fluor 488-labeled RGS4 was
526 Blazer et al.
added and incubated at room temperature in the dark for 15 min.Then, 10 �l of 20 nM Tb-labeled G�o GDP/AlF4 was added to themixture and incubated at room temperature in the dark for 30 minbefore analysis on the PHERAstar plate reader. Compound dilutionscovered a final concentration range from 200 to 1.6 �M. Positive andnegative controls were performed as in the primary screening assay.Compounds whose dose-response curves (DRCs) were not fully de-fined by these concentrations were repeated by using a more appro-priate dilution scheme. Nonlinear least-squares regression fitting ofthe data were performed by using the data analysis component of theMScreen database.
Flow Cytometry Protein Interaction Assay ConcentrationDependence Experiments. Compounds that were confirmed in thefollow-up TR-FRET dose-response assay were tested as describedpreviously (Roman et al., 2007) in the flow cytometry protein inter-action assay (FCPIA). This was done in part to provide a comple-mentary set of biochemical data to filter out any compounds thatmight produce spectroscopic artifacts in the TR-FRET assay. Inbrief, biotinylated RGS proteins (5 nM, final assay concentration)were immobilized on Luminex LumAvidin beads and incubated withdiluted compound in 50 mM HEPES, pH 8.0 at room temperature,100 mM NaCl, 0.1% Lubrol, and 1 mM DTT, supplemented with 1%BSA. To each well of a 96-well PCR plate (Axygen, Union City, CA)Alexa Fluor 532-labeled G�o was added to a final concentration of 30nM. This mixture was incubated for 30 min at room temperature inthe dark, and then it was analyzed on a Luminex 200 flow cytometerfor the bead-associated fluorescence (median value). Nonlinear re-gression analysis of inhibition curves was performed with Prism 5.0(GraphPad Software Inc., San Diego CA).
FCPIA Reversibility Experiments. RGS-coated beads wereprepared as above and treated with 50 �M compound or vehicle(DMSO) for 15 min at room temperature. The RGS-containing beadswere then washed by resuspension in 1 ml of phosphate-bufferedsaline, pH 7.4 supplemented with 1% BSA, vortexing briefly, thenpelleting the beads by centrifugation. This procedure was repeated atotal of three times before 1000 beads were added to each quadru-plicate well of a 96-well PCR plate that contained Alexa Fluor 532-labeled G�o at a final concentration of 20 nM in the presence orabsence of 50 �M test compound. The mixture was incubated for 30min at room temperature and then analyzed on a Luminex 200 flowcytometer for bead-associated fluorescence. Data analysis was per-formed with Prism 5.0.
Single-Turnover GTPase Measurements. Compounds weretested for the ability to inhibit the RGS4-stimulated increase in GTPhydrolysis by G�o as described previously (Roof et al., 2006; Romanet al., 2007).
Thermal Stability Measurements. Untagged �N19RGS4 orHis6-G�o was added to the well of a 96-well ABI Prism opticalreaction plate (Applied Biosystems, Foster City CA) to a final con-centration of 5 or 2.5 �M, respectively in 50 to 60 �l of 50 mMHEPES, pH 8.0 with 150 mM NaCl. Test compounds were added tothe protein at the desired concentration and allowed to interact for15 min at room temperature. To each well, Sypro Orange dye (In-vitrogen) was added to a 5� final concentration (as described by thesupplier), and the plate was sealed with an optically clear adhesivefilm. Sypro Orange fluorescence was measured continuously in anABI HT7900 real-time PCR system during a stepwise gradient fromambient temperature to 90°C in 1°C steps lasting 30 s each. Datawere analyzed by fitting the obtained curves to a Boltzmann model(eq. 1).
I � L�U � L�
1 � e�Tm�T�
a
where I is fluorescence intensity (arbitrary units), L is the lower limitof the curve (°C), U is the upper limit of the curve (°C), T is temper-ature (°C), and a is a slope factor. Values obtained after the fluores-cence maximum occurred were excluded from the analysis.
ResultsDevelopment of a High-Throughput TR-FRET RGS4-
G�o Interaction Screen. We developed a biochemical TR-FRET assay by using purified human RGS4 labeled with theAlexa Fluor 488 acceptor fluorophore and purified G�o la-beled with the LanthaScreen Tb probe donor fluorophore(Fig. 1A). Using this system, we observed a saturable, alu-minum fluoride-dependent interaction between RGS4 andG� that has an affinity consistent with other reports of thisPPI in the literature (Fig. 1B) (Roman et al., 2007). In col-laboration with the Center for Chemical Genomics at theUniversity of Michigan, this assay was scaled to 384-wellformat and used to screen 44,000 small molecules for inhi-bition of RGS4/G�o binding in the presence of a thiol-reduc-ing agent (Table 1). Compounds from this screen were re-tested in the primary screening assay to confirm the initialresult and assess the concentration dependence of the inhi-bition using the original TR-FRET assay. Of the 162 com-pounds that met the 2-SD selection criteria for inhibition, 48were either unavailable or predicted to be chemically reactiveand were not followed up. The 114 selected compounds wereretested in TR-FRET DRC, and 11 were confirmed as inhib-itors with IC50 values 400 �M and Hill slopes 2.
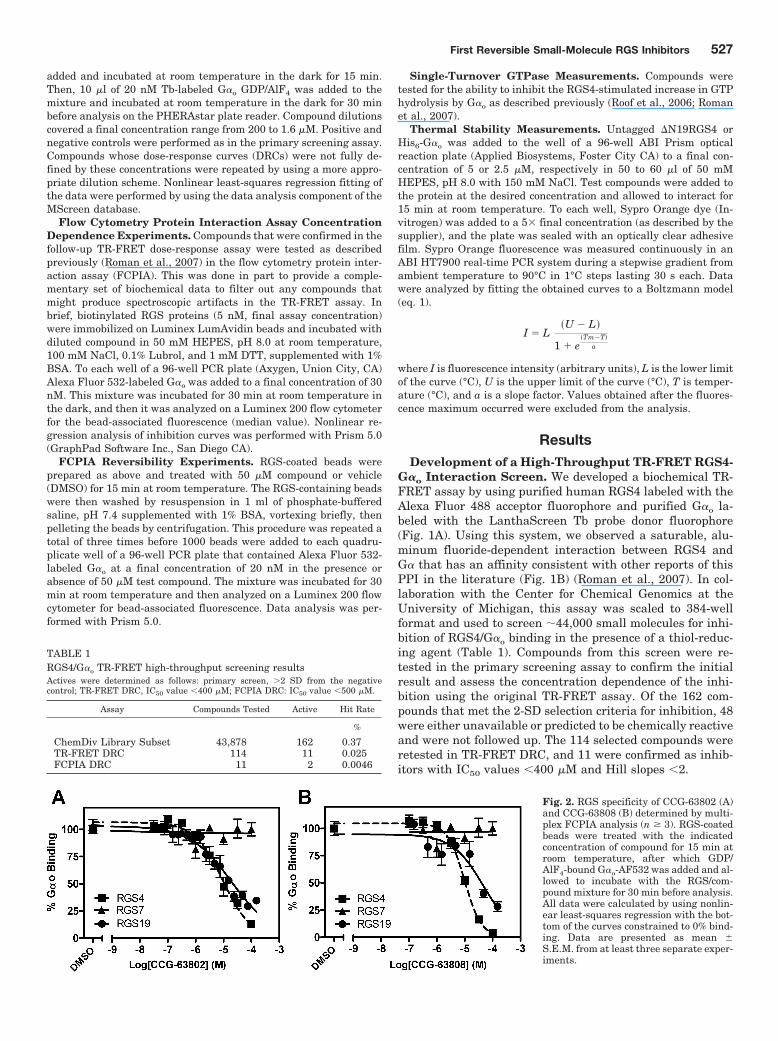
Fig. 2. RGS specificity of CCG-63802 (A)and CCG-63808 (B) determined by multi-plex FCPIA analysis (n � 3). RGS-coatedbeads were treated with the indicatedconcentration of compound for 15 min atroom temperature, after which GDP/AlF4-bound G�o-AF532 was added and al-lowed to incubate with the RGS/com-pound mixture for 30 min before analysis.All data were calculated by using nonlin-ear least-squares regression with the bot-tom of the curves constrained to 0% bind-ing. Data are presented as mean �S.E.M. from at least three separate exper-iments.
TABLE 1RGS4/G�o TR-FRET high-throughput screening resultsActives were determined as follows: primary screen, �2 SD from the negativecontrol; TR-FRET DRC, IC50 value 400 �M; FCPIA DRC: IC50 value 500 �M.
Assay Compounds Tested Active Hit Rate
%
ChemDiv Library Subset 43,878 162 0.37TR-FRET DRC 114 11 0.025FCPIA DRC 11 2 0.0046
First Reversible Small-Molecule RGS Inhibitors 527
The confirmed active compounds were obtained from thesupplier as fresh powders and tested by using the FCPIA, amethod that measures the binding of fluorescently taggedG�o to an RGS protein on beads (Roman et al., 2007). Of the11 compounds tested, 2 showed similar activity on RGS4 inboth the TR-FRET dose response and FCPIA experiments(Fig. 1C). The nine compounds that did not show activity inthis secondary assay are presumed to have been spectralartifacts or small-molecule aggregators that are likely to losefunction in the relatively stringent conditions of the FCPIAassay buffer (50 mM HEPES, 100 mM NaCl, 1% BSA, and0.1% Lubrol, pH 8.0).
The two active compounds that were identified from thisprimary screen were the closely related CCG-63808 andCCG-63802 (Fig. 1D). These compounds differ solely by thesubstituents on the phenyl moiety and have similar IC50
values in TR-FRET and FCPIA. The compounds also containa vinyl cyanide moiety that may function as a reversibleMichael acceptor.
CCG-63802 and CCG-63808 Selectively Inhibit G�o-RGS Interactions. Using TR-FRET to assess the RGS4-G�o
interaction, CCG-63802 and CCG-63808 had IC50 values of1.9 and 1.4 �M, respectively (Fig. 1C). To determine theselectivity of these compounds for different RGS proteins, theywere tested in an FCPIA competition experiment against apanel of five different RGS proteins (Fig. 2; Table 2). The com-pounds are 6- to 7-fold less potent in blocking G�o/RGS4interactions when tested with FCPIA (IC50 10 �M) thanwith the TR-FRET method. This is probably because of thehigh level of BSA (1%) in the FCPIA buffer sequesteringcompound and decreasing its apparent concentration in theassay. These compounds did not inhibit G� binding to RGS7,which is distantly related to RGS4, and they are 2- to 10-foldmore potent at RGS4 than on the other closely related R4
family members, RGS8 and RGS16 (Table 2). They are alsofairly active (IC50 20–50 �M) on the one RZ family membertested, RGS19.
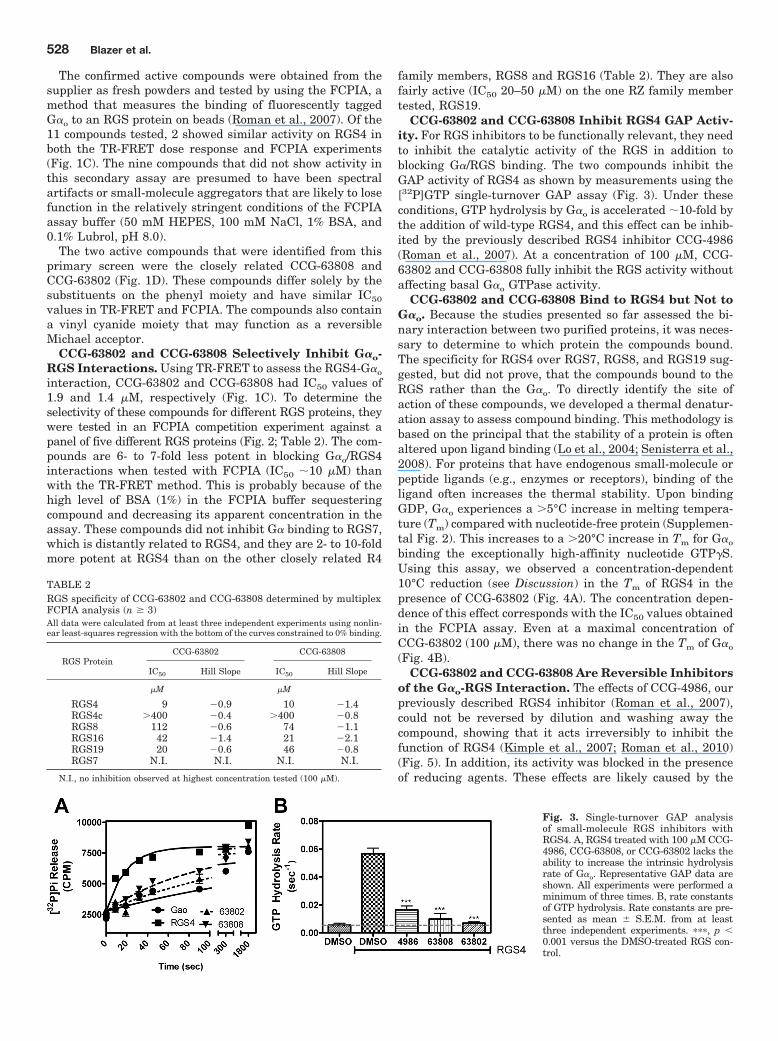
CCG-63802 and CCG-63808 Inhibit RGS4 GAP Activ-ity. For RGS inhibitors to be functionally relevant, they needto inhibit the catalytic activity of the RGS in addition toblocking G�/RGS binding. The two compounds inhibit theGAP activity of RGS4 as shown by measurements using the[32P]GTP single-turnover GAP assay (Fig. 3). Under theseconditions, GTP hydrolysis by G�o is accelerated 10-fold bythe addition of wild-type RGS4, and this effect can be inhib-ited by the previously described RGS4 inhibitor CCG-4986(Roman et al., 2007). At a concentration of 100 �M, CCG-63802 and CCG-63808 fully inhibit the RGS activity withoutaffecting basal G�o GTPase activity.
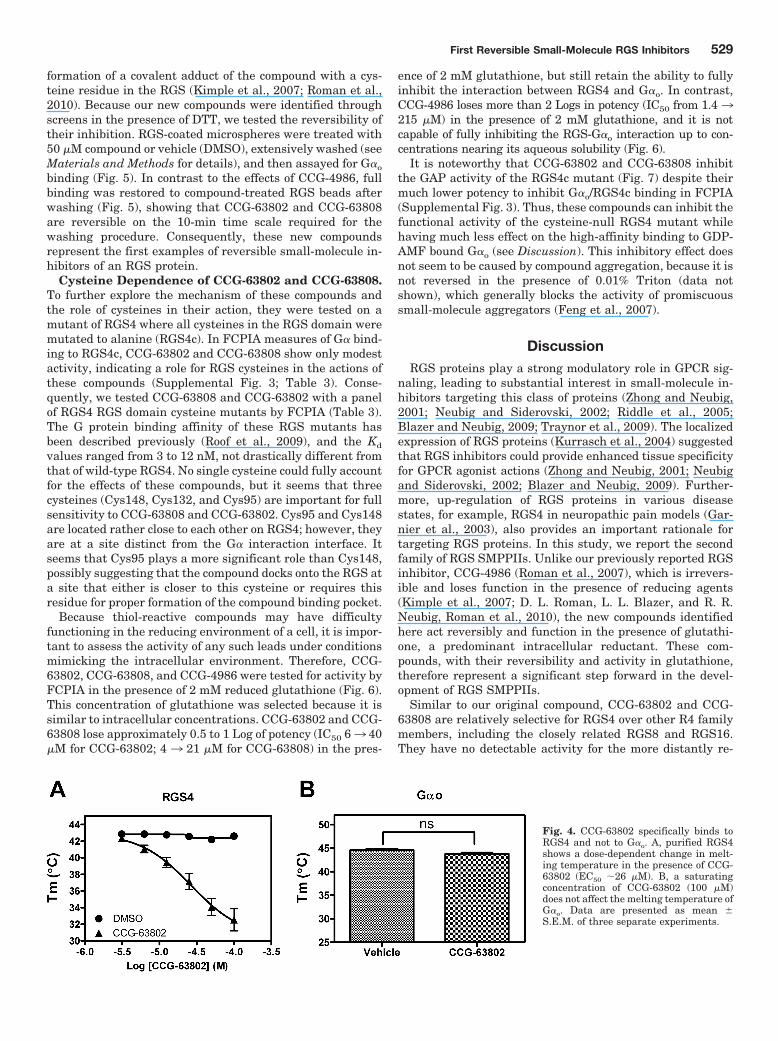
CCG-63802 and CCG-63808 Bind to RGS4 but Not toG�o. Because the studies presented so far assessed the bi-nary interaction between two purified proteins, it was neces-sary to determine to which protein the compounds bound.The specificity for RGS4 over RGS7, RGS8, and RGS19 sug-gested, but did not prove, that the compounds bound to theRGS rather than the G�o. To directly identify the site ofaction of these compounds, we developed a thermal denatur-ation assay to assess compound binding. This methodology isbased on the principal that the stability of a protein is oftenaltered upon ligand binding (Lo et al., 2004; Senisterra et al.,2008). For proteins that have endogenous small-molecule orpeptide ligands (e.g., enzymes or receptors), binding of theligand often increases the thermal stability. Upon bindingGDP, G�o experiences a �5°C increase in melting tempera-ture (Tm) compared with nucleotide-free protein (Supplemen-tal Fig. 2). This increases to a �20°C increase in Tm for G�o
binding the exceptionally high-affinity nucleotide GTP�S.Using this assay, we observed a concentration-dependent10°C reduction (see Discussion) in the Tm of RGS4 in thepresence of CCG-63802 (Fig. 4A). The concentration depen-dence of this effect corresponds with the IC50 values obtainedin the FCPIA assay. Even at a maximal concentration ofCCG-63802 (100 �M), there was no change in the Tm of G�o
(Fig. 4B).CCG-63802 and CCG-63808 Are Reversible Inhibitors
of the G�o-RGS Interaction. The effects of CCG-4986, ourpreviously described RGS4 inhibitor (Roman et al., 2007),could not be reversed by dilution and washing away thecompound, showing that it acts irreversibly to inhibit thefunction of RGS4 (Kimple et al., 2007; Roman et al., 2010)(Fig. 5). In addition, its activity was blocked in the presenceof reducing agents. These effects are likely caused by the
Fig. 3. Single-turnover GAP analysisof small-molecule RGS inhibitors withRGS4. A, RGS4 treated with 100 �M CCG-4986, CCG-63808, or CCG-63802 lacks theability to increase the intrinsic hydrolysisrate of G�o. Representative GAP data areshown. All experiments were performed aminimum of three times. B, rate constantsof GTP hydrolysis. Rate constants are pre-sented as mean � S.E.M. from at leastthree independent experiments. ���, p 0.001 versus the DMSO-treated RGS con-trol.
TABLE 2RGS specificity of CCG-63802 and CCG-63808 determined by multiplexFCPIA analysis (n � 3)All data were calculated from at least three independent experiments using nonlin-ear least-squares regression with the bottom of the curves constrained to 0% binding.
RGS ProteinCCG-63802 CCG-63808
IC50 Hill Slope IC50 Hill Slope
�M �M
RGS4 9 �0.9 10 �1.4RGS4c �400 �0.4 �400 �0.8RGS8 112 �0.6 74 �1.1RGS16 42 �1.4 21 �2.1RGS19 20 �0.6 46 �0.8RGS7 N.I. N.I. N.I. N.I.
N.I., no inhibition observed at highest concentration tested (100 �M).
528 Blazer et al.
formation of a covalent adduct of the compound with a cys-teine residue in the RGS (Kimple et al., 2007; Roman et al.,2010). Because our new compounds were identified throughscreens in the presence of DTT, we tested the reversibility oftheir inhibition. RGS-coated microspheres were treated with50 �M compound or vehicle (DMSO), extensively washed (seeMaterials and Methods for details), and then assayed for G�o
binding (Fig. 5). In contrast to the effects of CCG-4986, fullbinding was restored to compound-treated RGS beads afterwashing (Fig. 5), showing that CCG-63802 and CCG-63808are reversible on the 10-min time scale required for thewashing procedure. Consequently, these new compoundsrepresent the first examples of reversible small-molecule in-hibitors of an RGS protein.
Cysteine Dependence of CCG-63802 and CCG-63808.To further explore the mechanism of these compounds andthe role of cysteines in their action, they were tested on amutant of RGS4 where all cysteines in the RGS domain weremutated to alanine (RGS4c). In FCPIA measures of G� bind-ing to RGS4c, CCG-63802 and CCG-63808 show only modestactivity, indicating a role for RGS cysteines in the actions ofthese compounds (Supplemental Fig. 3; Table 3). Conse-quently, we tested CCG-63808 and CCG-63802 with a panelof RGS4 RGS domain cysteine mutants by FCPIA (Table 3).The G protein binding affinity of these RGS mutants hasbeen described previously (Roof et al., 2009), and the Kd
values ranged from 3 to 12 nM, not drastically different fromthat of wild-type RGS4. No single cysteine could fully accountfor the effects of these compounds, but it seems that threecysteines (Cys148, Cys132, and Cys95) are important for fullsensitivity to CCG-63808 and CCG-63802. Cys95 and Cys148are located rather close to each other on RGS4; however, theyare at a site distinct from the G� interaction interface. Itseems that Cys95 plays a more significant role than Cys148,possibly suggesting that the compound docks onto the RGS ata site that either is closer to this cysteine or requires thisresidue for proper formation of the compound binding pocket.
Because thiol-reactive compounds may have difficultyfunctioning in the reducing environment of a cell, it is impor-tant to assess the activity of any such leads under conditionsmimicking the intracellular environment. Therefore, CCG-63802, CCG-63808, and CCG-4986 were tested for activity byFCPIA in the presence of 2 mM reduced glutathione (Fig. 6).This concentration of glutathione was selected because it issimilar to intracellular concentrations. CCG-63802 and CCG-63808 lose approximately 0.5 to 1 Log of potency (IC50 63 40�M for CCG-63802; 4 3 21 �M for CCG-63808) in the pres-
ence of 2 mM glutathione, but still retain the ability to fullyinhibit the interaction between RGS4 and G�o. In contrast,CCG-4986 loses more than 2 Logs in potency (IC50 from 1.43215 �M) in the presence of 2 mM glutathione, and it is notcapable of fully inhibiting the RGS-G�o interaction up to con-centrations nearing its aqueous solubility (Fig. 6).
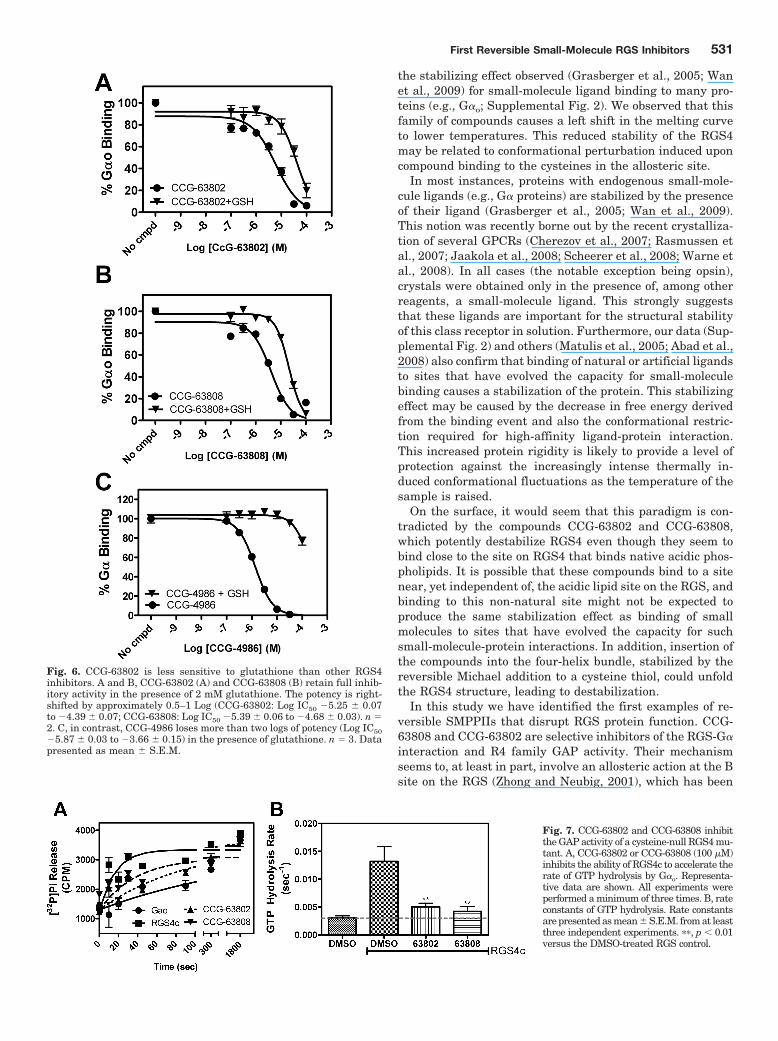
It is noteworthy that CCG-63802 and CCG-63808 inhibitthe GAP activity of the RGS4c mutant (Fig. 7) despite theirmuch lower potency to inhibit G�o/RGS4c binding in FCPIA(Supplemental Fig. 3). Thus, these compounds can inhibit thefunctional activity of the cysteine-null RGS4 mutant whilehaving much less effect on the high-affinity binding to GDP-AMF bound G�o (see Discussion). This inhibitory effect doesnot seem to be caused by compound aggregation, because it isnot reversed in the presence of 0.01% Triton (data notshown), which generally blocks the activity of promiscuoussmall-molecule aggregators (Feng et al., 2007).
DiscussionRGS proteins play a strong modulatory role in GPCR sig-
naling, leading to substantial interest in small-molecule in-hibitors targeting this class of proteins (Zhong and Neubig,2001; Neubig and Siderovski, 2002; Riddle et al., 2005;Blazer and Neubig, 2009; Traynor et al., 2009). The localizedexpression of RGS proteins (Kurrasch et al., 2004) suggestedthat RGS inhibitors could provide enhanced tissue specificityfor GPCR agonist actions (Zhong and Neubig, 2001; Neubigand Siderovski, 2002; Blazer and Neubig, 2009). Further-more, up-regulation of RGS proteins in various diseasestates, for example, RGS4 in neuropathic pain models (Gar-nier et al., 2003), also provides an important rationale fortargeting RGS proteins. In this study, we report the secondfamily of RGS SMPPIIs. Unlike our previously reported RGSinhibitor, CCG-4986 (Roman et al., 2007), which is irrevers-ible and loses function in the presence of reducing agents(Kimple et al., 2007; D. L. Roman, L. L. Blazer, and R. R.Neubig, Roman et al., 2010), the new compounds identifiedhere act reversibly and function in the presence of glutathi-one, a predominant intracellular reductant. These com-pounds, with their reversibility and activity in glutathione,therefore represent a significant step forward in the devel-opment of RGS SMPPIIs.
Similar to our original compound, CCG-63802 and CCG-63808 are relatively selective for RGS4 over other R4 familymembers, including the closely related RGS8 and RGS16.They have no detectable activity for the more distantly re-
Fig. 4. CCG-63802 specifically binds toRGS4 and not to G�o. A, purified RGS4shows a dose-dependent change in melt-ing temperature in the presence of CCG-63802 (EC50 26 �M). B, a saturatingconcentration of CCG-63802 (100 �M)does not affect the melting temperature ofG�o. Data are presented as mean �S.E.M. of three separate experiments.
First Reversible Small-Molecule RGS Inhibitors 529
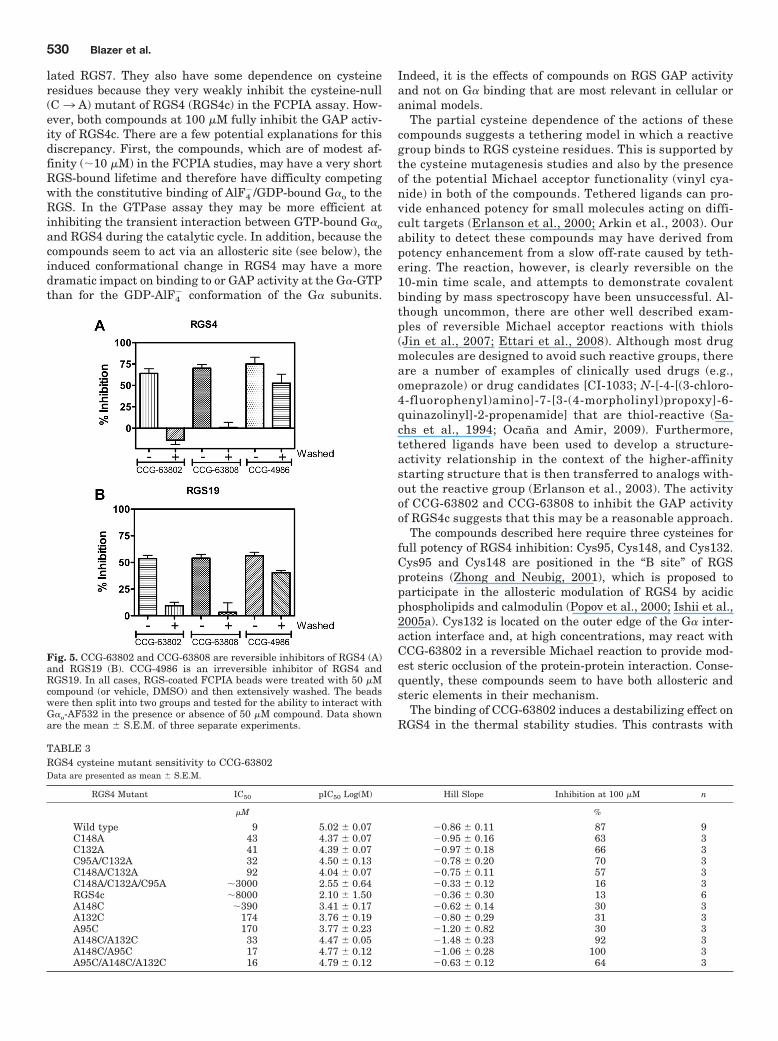
lated RGS7. They also have some dependence on cysteineresidues because they very weakly inhibit the cysteine-null(C3 A) mutant of RGS4 (RGS4c) in the FCPIA assay. How-ever, both compounds at 100 �M fully inhibit the GAP activ-ity of RGS4c. There are a few potential explanations for thisdiscrepancy. First, the compounds, which are of modest af-finity (10 �M) in the FCPIA studies, may have a very shortRGS-bound lifetime and therefore have difficulty competingwith the constitutive binding of AlF4
�/GDP-bound G�o to theRGS. In the GTPase assay they may be more efficient atinhibiting the transient interaction between GTP-bound G�o
and RGS4 during the catalytic cycle. In addition, because thecompounds seem to act via an allosteric site (see below), theinduced conformational change in RGS4 may have a moredramatic impact on binding to or GAP activity at the G�-GTPthan for the GDP-AlF4
� conformation of the G� subunits.
Indeed, it is the effects of compounds on RGS GAP activityand not on G� binding that are most relevant in cellular oranimal models.
The partial cysteine dependence of the actions of thesecompounds suggests a tethering model in which a reactivegroup binds to RGS cysteine residues. This is supported bythe cysteine mutagenesis studies and also by the presenceof the potential Michael acceptor functionality (vinyl cya-nide) in both of the compounds. Tethered ligands can pro-vide enhanced potency for small molecules acting on diffi-cult targets (Erlanson et al., 2000; Arkin et al., 2003). Ourability to detect these compounds may have derived frompotency enhancement from a slow off-rate caused by teth-ering. The reaction, however, is clearly reversible on the10-min time scale, and attempts to demonstrate covalentbinding by mass spectroscopy have been unsuccessful. Al-though uncommon, there are other well described exam-ples of reversible Michael acceptor reactions with thiols(Jin et al., 2007; Ettari et al., 2008). Although most drugmolecules are designed to avoid such reactive groups, thereare a number of examples of clinically used drugs (e.g.,omeprazole) or drug candidates [CI-1033; N-[-4-[(3-chloro-4-fluorophenyl)amino]-7-[3-(4-morpholinyl)propoxy]-6-quinazolinyl]-2-propenamide] that are thiol-reactive (Sa-chs et al., 1994; Ocana and Amir, 2009). Furthermore,tethered ligands have been used to develop a structure-activity relationship in the context of the higher-affinitystarting structure that is then transferred to analogs with-out the reactive group (Erlanson et al., 2003). The activityof CCG-63802 and CCG-63808 to inhibit the GAP activityof RGS4c suggests that this may be a reasonable approach.
The compounds described here require three cysteines forfull potency of RGS4 inhibition: Cys95, Cys148, and Cys132.Cys95 and Cys148 are positioned in the “B site” of RGSproteins (Zhong and Neubig, 2001), which is proposed toparticipate in the allosteric modulation of RGS4 by acidicphospholipids and calmodulin (Popov et al., 2000; Ishii et al.,2005a). Cys132 is located on the outer edge of the G� inter-action interface and, at high concentrations, may react withCCG-63802 in a reversible Michael reaction to provide mod-est steric occlusion of the protein-protein interaction. Conse-quently, these compounds seem to have both allosteric andsteric elements in their mechanism.
The binding of CCG-63802 induces a destabilizing effect onRGS4 in the thermal stability studies. This contrasts with
Fig. 5. CCG-63802 and CCG-63808 are reversible inhibitors of RGS4 (A)and RGS19 (B). CCG-4986 is an irreversible inhibitor of RGS4 andRGS19. In all cases, RGS-coated FCPIA beads were treated with 50 �Mcompound (or vehicle, DMSO) and then extensively washed. The beadswere then split into two groups and tested for the ability to interact withG�o-AF532 in the presence or absence of 50 �M compound. Data shownare the mean � S.E.M. of three separate experiments.
TABLE 3RGS4 cysteine mutant sensitivity to CCG-63802Data are presented as mean � S.E.M.
RGS4 Mutant IC50 pIC50 Log(M) Hill Slope Inhibition at 100 �M n
�M %
Wild type 9 5.02 � 0.07 �0.86 � 0.11 87 9C148A 43 4.37 � 0.07 �0.95 � 0.16 63 3C132A 41 4.39 � 0.07 �0.97 � 0.18 66 3C95A/C132A 32 4.50 � 0.13 �0.78 � 0.20 70 3C148A/C132A 92 4.04 � 0.07 �0.75 � 0.11 57 3C148A/C132A/C95A 3000 2.55 � 0.64 �0.33 � 0.12 16 3RGS4c 8000 2.10 � 1.50 �0.36 � 0.30 13 6A148C 390 3.41 � 0.17 �0.62 � 0.14 30 3A132C 174 3.76 � 0.19 �0.80 � 0.29 31 3A95C 170 3.77 � 0.23 �1.20 � 0.82 30 3A148C/A132C 33 4.47 � 0.05 �1.48 � 0.23 92 3A148C/A95C 17 4.77 � 0.12 �1.06 � 0.28 100 3A95C/A148C/A132C 16 4.79 � 0.12 �0.63 � 0.12 64 3
530 Blazer et al.
the stabilizing effect observed (Grasberger et al., 2005; Wanet al., 2009) for small-molecule ligand binding to many pro-teins (e.g., G�o; Supplemental Fig. 2). We observed that thisfamily of compounds causes a left shift in the melting curveto lower temperatures. This reduced stability of the RGS4may be related to conformational perturbation induced uponcompound binding to the cysteines in the allosteric site.
In most instances, proteins with endogenous small-mole-cule ligands (e.g., G� proteins) are stabilized by the presenceof their ligand (Grasberger et al., 2005; Wan et al., 2009).This notion was recently borne out by the recent crystalliza-tion of several GPCRs (Cherezov et al., 2007; Rasmussen etal., 2007; Jaakola et al., 2008; Scheerer et al., 2008; Warne etal., 2008). In all cases (the notable exception being opsin),crystals were obtained only in the presence of, among otherreagents, a small-molecule ligand. This strongly suggeststhat these ligands are important for the structural stabilityof this class receptor in solution. Furthermore, our data (Sup-plemental Fig. 2) and others (Matulis et al., 2005; Abad et al.,2008) also confirm that binding of natural or artificial ligandsto sites that have evolved the capacity for small-moleculebinding causes a stabilization of the protein. This stabilizingeffect may be caused by the decrease in free energy derivedfrom the binding event and also the conformational restric-tion required for high-affinity ligand-protein interaction.This increased protein rigidity is likely to provide a level ofprotection against the increasingly intense thermally in-duced conformational fluctuations as the temperature of thesample is raised.
On the surface, it would seem that this paradigm is con-tradicted by the compounds CCG-63802 and CCG-63808,which potently destabilize RGS4 even though they seem tobind close to the site on RGS4 that binds native acidic phos-pholipids. It is possible that these compounds bind to a sitenear, yet independent of, the acidic lipid site on the RGS, andbinding to this non-natural site might not be expected toproduce the same stabilization effect as binding of smallmolecules to sites that have evolved the capacity for suchsmall-molecule-protein interactions. In addition, insertion ofthe compounds into the four-helix bundle, stabilized by thereversible Michael addition to a cysteine thiol, could unfoldthe RGS4 structure, leading to destabilization.
In this study we have identified the first examples of re-versible SMPPIIs that disrupt RGS protein function. CCG-63808 and CCG-63802 are selective inhibitors of the RGS-G�
interaction and R4 family GAP activity. Their mechanismseems to, at least in part, involve an allosteric action at the Bsite on the RGS (Zhong and Neubig, 2001), which has been
Fig. 6. CCG-63802 is less sensitive to glutathione than other RGS4inhibitors. A and B, CCG-63802 (A) and CCG-63808 (B) retain full inhib-itory activity in the presence of 2 mM glutathione. The potency is right-shifted by approximately 0.5–1 Log (CCG-63802: Log IC50 �5.25 � 0.07to �4.39 � 0.07; CCG-63808: Log IC50 �5.39 � 0.06 to �4.68 � 0.03). n �2. C, in contrast, CCG-4986 loses more than two logs of potency (Log IC50�5.87 � 0.03 to �3.66 � 0.15) in the presence of glutathione. n � 3. Datapresented as mean � S.E.M.
Fig. 7. CCG-63802 and CCG-63808 inhibitthe GAP activity of a cysteine-null RGS4 mu-tant. A, CCG-63802 or CCG-63808 (100 �M)inhibits the ability of RGS4c to accelerate therate of GTP hydrolysis by G�o. Representa-tive data are shown. All experiments wereperformed a minimum of three times. B, rateconstants of GTP hydrolysis. Rate constantsare presented as mean � S.E.M. from at leastthree independent experiments. ��, p 0.01versus the DMSO-treated RGS control.
First Reversible Small-Molecule RGS Inhibitors 531

implicated in the physiological allosteric modulation of RGSproteins by acidic phospholipids and calmodulin (Ishii et al.,2005a,b). Further studies of the mechanism and structure-activity relationships for this compound class and translationto cellular and animal models of RGS function are currentlyunderway.
Acknowledgments
We thank Roger Sunahara (University of Michigan Pharmacology)and John Tesmer (University of Michigan Pharmacology and LifeSciences Institute) for helpful discussions with this project; the staffof the University of Michigan Center for Chemical Genomics forhigh-throughput screening and chemoinformatics assistance; theNovartis Institute for Biomedical Research for the gift of reagents,including the screening library; and the University of MichiganComprehensive Cancer Center for subsidizing the cost of DNA se-quencing. This work used the Cell and Molecular Biology Core of theMichigan Diabetes Research and Training Center, which is fundedby the National Institutes of Health National Institute of Diabetesand Digestive and Kidney Diseases (Grant DK020572).
ReferencesAbad MC, Askari H, O’Neill J, Klinger AL, Milligan C, Lewandowski F, Springer B,
Spurlino J, and Rentzeperis D (2008) Structural determination of estrogen-relatedreceptor � in the presence of phenol derivative compounds. J Steroid Biochem MolBiol 108:44–54.
Arkin MR and Wells JA (2004) Small-molecule inhibitors of protein-protein interac-tions: progressing towards the dream. Nat Rev Drug Discov 3:301–317.
Arkin MR and Whitty A (2009) The road less traveled: modulating signal transduc-tion enzymes by inhibiting their protein-protein interactions. Curr Opin ChemBiol 13:284–290.
Arkin MR, Randal M, DeLano WL, Hyde J, Luong TN, Oslob JD, Raphael DR, TaylorL, Wang J, McDowell RS, et al. (2003) Binding of small molecules to an adaptiveprotein-protein interface. Proc Natl Acad Sci USA 100:1603–1608.
Berg T (2003) Modulation of protein-protein interactions with small organic mole-cules. Angew Chem Int Ed Engl 42:2462–2481.
Berg T (2008) Small-molecule inhibitors of protein-protein interactions. Curr OpinDrug Discov Devel 11:666–674.
Berman DM, Kozasa T, and Gilman AG (1996) The GTPase-activating protein RGS4stabilizes the transition state for nucleotide hydrolysis. J Biol Chem 271:27209–27212.
Blazer LL and Neubig RR (2009) Small-molecule protein-protein interaction inhib-itors as CNS therapeutic agents: current progress and future hurdles. Neuropsy-chopharmacology 34:126–141.
Bueno L and Fioramonti J (1988) Action of opiates on gastrointestinal function.Baillieres Clin Gastroenterol 2:123–139.
Busschots K, De Rijck J, Christ F, and Debyser Z (2009) In search of small moleculesblocking interactions between HIV proteins and intracellular cofactors. Mol Bio-syst 5:21–31.
Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS,Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. (2007) High-resolution crystalstructure of an engineered human 2-adrenergic G protein-coupled receptor. Sci-ence 318:1258–1265.
Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, andWells JA (2000) Site-directed ligand discovery. Proc Natl Acad Sci USA 97:9367–9372.
Erlanson DA, Lam JW, Wiesmann C, Luong TN, Simmons RL, DeLano WL, ChoongIC, Burdett MT, Flanagan WM, Lee D, et al. (2003) In situ assembly of enzymeinhibitors using extended tethering. Nat Biotechnol 21:308–314.
Ettari R, Nizi E, Di Francesco ME, Dude MA, Pradel G, Vicík R, Schirmeister T,Micale N, Grasso S, and Zappala M (2008) Development of peptidomimetics witha vinyl sulfone warhead as irreversible falcipain-2 inhibitors. J Med Chem 51:988–996.
Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, and Austin CP(2007) A high-throughput screen for aggregation-based inhibition in a large com-pound library. J Med Chem 50:2385–2390.
Gadek TR and Nicholas JB (2003) Small molecule antagonists of proteins. BiochemPharmacol 65:1–8.
Garnier M, Zaratin PF, Ficalora G, Valente M, Fontanella L, Rhee MH, Blumer KJ,and Scheideler MA (2003) Up-regulation of regulator of G protein signaling 4expression in a model of neuropathic pain and insensitivity to morphine. J Phar-macol Exp Ther 304:1299–1306.
Grasberger BL, Lu T, Schubert C, Parks DJ, Carver TE, Koblish HK, CummingsMD, LaFrance LV, Milkiewicz KL, Calvo RR, et al. (2005) Discovery and cocrystalstructure of benzodiazepinedione HDM2 antagonists that activate p53 in cells.J Med Chem 48:909–912.
Hollinger S and Hepler JR (2002) Cellular regulation of RGS proteins: modulatorsand integrators of G protein signaling. Pharmacol Rev 54:527–559.
Huang X, Charbeneau RA, Fu Y, Kaur K, Gerin I, MacDougald OA, and Neubig RR(2008) Resistance to diet-induced obesity and improved insulin sensitivity in micewith a regulator of G protein signaling-insensitive G184S Gnai2 allele. Diabetes57:77–85.
Huang X, Fu Y, Charbeneau RA, Saunders TL, Taylor DK, Hankenson KD, RussellMW, D’Alecy LG, and Neubig RR (2006) Pleiotropic phenotype of a genomicknock-in of an RGS-insensitive G184S Gnai2 allele. Mol Cell Biol 26:6870–6879.
Ingalls EA and Popp FD (1967) The preparation, structure, and reactions of some“malonyl-�-aminopyridines.” J Heterocyclic Chem 4:523–526.
Ishii M, Fujita S, Yamada M, Hosaka Y, and Kurachi Y (2005a) Phosphatidylinositol3,4,5-trisphosphate and Ca2�/calmodulin competitively bind to the regulators ofG-protein-signalling (RGS) domain of RGS4 and reciprocally regulate its action.Biochem J 385:65–73.
Ishii M, Ikushima M, and Kurachi Y (2005b) In vivo interaction between RGS4 andcalmodulin visualized with FRET techniques: possible involvement of lipid raft.Biochem Biophys Res Commun 338:839–846.
Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP,and Stevens RC (2008) The 2.6 angstrom crystal structure of a human A2Aadenosine receptor bound to an antagonist. Science 322:1211–1217.
Jin F, Jin XY, Jin YL, Sohn DW, Kim SA, Sohn DH, Kim YC, and Kim HS (2007)Structural requirements of 2�,4�,6�-tris(methoxymethoxy) chalcone derivatives foranti-inflammatory activity: the importance of a 2�-hydroxy moiety. Arch PharmRes 30:1359–1367.
Kimple AJ, Willard FS, Giguere PM, Johnston CA, Mocanu V, and Siderovski DP(2007) The RGS protein inhibitor CCG-4986 is a covalent modifier of the RGS4G�-interaction face. Biochim Biophys Acta 1774:1213–1220.
Kurrasch DM, Huang J, Wilkie TM, and Repa JJ (2004) Quantitative real-timepolymerase chain reaction measurement of regulators of G-protein signalingmRNA levels in mouse tissues. Methods Enzymol 389:3–15.
Lan KL, Sarvazyan NA, Taussig R, Mackenzie RG, DiBello PR, Dohlman HG, andNeubig RR (1998) A point mutation in G�o and G�1 blocks interaction withregulator of G protein signaling proteins. J Biol Chem 273:12794–12797.
Lan KL, Zhong H, Nanamori M, and Neubig RR (2000) Rapid kinetics of regulator ofG-protein signaling (RGS)-mediated G�i and G�o deactivation. G� specificity ofRGS4 and RGS7. J Biol Chem 275:33497–33503.
Lee E, Linder ME, and Gilman AG (1994) Expression of G-protein � subunits inEscherichia coli. Methods Enzymol 237:146–164.
Leifert WR, Bailey K, Cooper TH, Aloia AL, Glatz RV, and McMurchie EJ (2006)Measurement of heterotrimeric G-protein and regulators of G-protein signalinginteractions by time-resolved fluorescence resonance energy transfer. Anal Bio-chem 355:201–212.
Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, and Ellestad G (2004)Evaluation of fluorescence-based thermal shift assays for hit identification in drugdiscovery. Anal Biochem 332:153–159.
Matulis D, Kranz JK, Salemme FR, and Todd MJ (2005) Thermodynamic stability ofcarbonic anhydrase: measurements of binding affinity and stoichiometry usingThermoFluor. Biochemistry 44:5258–5266.
Neubig RR and Siderovski DP (2002) Regulators of G-protein signalling as newcentral nervous system drug targets. Nat Rev Drug Discov 1:187–197.
Niu G and Chen X (2009) From protein-protein interaction to therapy response:molecular imaging of heat shock proteins. Eur J Radiol 70:294–304.
Ocana A and Amir E (2009) Irreversible pan-ErbB tyrosine kinase inhibitors andbreast cancer: current status and future directions. Cancer Treat Rev 35:685–691.
Popov SG, Krishna UM, Falck JR, and Wilkie TM (2000) Ca2�/calmodulin reversesphosphatidylinositol 3,4,5-trisphosphate-dependent inhibition of regulators of Gprotein-signaling GTPase-activating protein activity. J Biol Chem 275:18962–18968.
Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC,Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, et al. (2007) Crystalstructure of the human 2 adrenergic G-protein-coupled receptor. Nature 450:383–387.
Riddle EL, Schwartzman RA, Bond M, and Insel PA (2005) Multi-tasking RGSproteins in the heart: the next therapeutic target? Circ Res 96:401–411.
Roman D, Blazer LL, Monroy CA, and Neubig RR (2010) Allosteric inhibition of theregulator of G protein signaling-G protein-protein interaction by CCG-4986. MolPharmacol 78:360–365.
Roman DL, Ota S, and Neubig RR (2009) Polyplexed flow cytometry protein inter-action assay: a novel high-throughput screening paradigm for RGS protein inhib-itors. J Biomol Screen 14:610–619.
Roman DL, Talbot JN, Roof RA, Sunahara RK, Traynor JR, and Neubig RR (2007)Identification of small-molecule inhibitors of RGS4 using a high-throughput flowcytometry protein interaction assay. Mol Pharmacol 71:169–175.
Roof RA, Jin Y, Roman DL, Sunahara RK, Ishii M, Mosberg HI, and Neubig RR(2006) Mechanism of action and structural requirements of constrained peptideinhibitors of RGS proteins. Chem Biol Drug Des 67:266–274.
Roof RA, Roman DL, Clements ST, Sobczyk-Kojiro K, Blazer LL, Ota S, Mosberg HI,and Neubig RR (2009) A covalent peptide inhibitor of RGS4 identified in a focusedone-bead, one compound library screen. BMC Pharmacol 9:9.
Roof RA, Sobczyk-Kojiro K, Turbiak AJ, Roman DL, Pogozheva ID, Blazer LL,Neubig RR, and Mosberg HI (2008) Novel peptide ligands of RGS4 from a focusedone-bead, one-compound library. Chem Biol Drug Des 72:111–119.
Sachs G, Prinz C, Loo D, Bamberg K, Besancon M, and Shin JM (1994) Gastric acidsecretion: activation and inhibition. Yale J Biol Med 67:81–95.
Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP,and Ernst OP (2008) Crystal structure of opsin in its G-protein-interacting con-formation. Nature 455:497–502.
Senisterra GA, Soo Hong B, Park HW, and Vedadi M (2008) Application of high-throughput isothermal denaturation to assess protein stability and screen forligands. J Biomol Screen 13:337–342.
Sternweis PC and Robishaw JD (1984) Isolation of two proteins with high affinity forguanine nucleotides from membranes of bovine brain. J Biol Chem 259:13806–13813.
Talbot JN, Jutkiewicz EM, Graves SM, Clemans CF, Nicol MR, Mortensen RM,Huang X, Neubig RR, and Traynor JR (2010) RGS inhibition at G�i2 selectively
532 Blazer et al.

potentiates 5-HT1A-mediated antidepressant effects. Proc Natl Acad Sci USA107:11086–11091.
Traynor JR and Neubig RR (2005) Regulators of G protein signaling & drugs ofabuse. Mol Interv 5:30–41.
Traynor JR, Terzi D, Caldarone BJ, and Zachariou V (2009) RGS9–2: probing an intracel-lular modulator of behavior as a drug target. Trends Pharmacol Sci 30:105–111.
Wan KF, Wang S, Brown CJ, Yu VC, Entzeroth M, Lane DP, and Lee MA (2009)Differential scanning fluorimetry as secondary screening platform for small mol-ecule inhibitors of Bcl-XL. Cell Cycle 8:3943–3952.
Wang Y, Lee Y, Zhang J, and Young KH (2008) Identification of peptides that inhibitregulator of G protein signaling 4 function. Pharmacology 82:97–104.
Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Hender-son R, Leslie AG, Tate CG, and Schertler GF (2008) Structure of a 1-adrenergicG-protein-coupled receptor. Nature 454:486–491.
Zhong H and Neubig RR (2001) Regulator of G protein signaling proteins: novelmultifunctional drug targets. J Pharmacol Exp Ther 297:837–845.
Address correspondence to: Dr. Richard R. Neubig, Department of Phar-macology, University of Michigan Medical School, 1150 W. Medical CenterDrive, 1303 MSRB III, Ann Arbor, MI 48109. E-mail: [email protected]
First Reversible Small-Molecule RGS Inhibitors 533