replication-mediated instability of the gaa triplet repeat mutation in friedreich ataxia
TRANSCRIPT

Replication-mediated instability of the GAA tripletrepeat mutation in Friedreich ataxiaLaura M. Pollard1, Rajesh Sharma1, Mariluz Gomez1, Sonali Shah1, Martin B. Delatycki3,4,
Luigi Pianese5,7, Antonella Monticelli6,7, Bronya J.B. Keats8 and Sanjay I. Bidichandani1,2,*
1Department of Biochemistry and Molecular Biology and 2Department of Pediatrics, University of Oklahoma HealthSciences Center, Oklahoma City, OK 73104, USA, 3Bruce Leroy Centre for Genetic Health Research,Murdoch Children’s Research Institute, 4Department of Pediatrics, University of Melbourne, Australia,5BioGeM Consortium, 6IEOS-CNR and 7DBPCM, Federico II University, Naples, Italy and 8Department of Genetics,Louisiana State University Health Sciences Center, New Orleans, LA, USA
Received July 22, 2004; Revised and Accepted October 22, 2004
ABSTRACT
Friedreich ataxia is caused by the expansion of apolymorphic and unstable GAA triplet repeat in theFRDA gene, but the mechanisms for its instabilityare poorly understood. Replication of (GAA�TTC)n
sequences (9–105 triplets) in plasmids propagatedin Escherichia coli displayed length- and orienta-tion-dependent instability. There were small lengthvariations upon replication in both orientations, butlarge contractions were frequently observed whenGAA was the lagging strand template. DNA replicationwas also significantly slower in this orientation. Toevaluate the physiological relevance of our findings,we analyzed peripheral leukocytes from humansubjects carrying repeats of similar length (8–107triplets). Analysis of 9400 somatic FRDA moleculesusing small-pool PCR revealed a similar mutationalspectrum, including large contractions. The thresh-old length for the initiation of somatic instabilityin vivo was between 40 and 44 triplets, correspondingto the length of a eukaryotic Okazaki fragment.Consistent with the stabilization of premutationalleles during germline transmission, we also foundthat instability of somatic cells in vivo and repeatspropagated in E.coli were abrogated by (GAGGAA)n
hexanucleotide interruptions. Our data demonstratethat the GAA triplet repeat mutation in Friedreichataxia is destabilized, frequently undergoing largecontractions, during DNA replication.
INTRODUCTION
Friedreich ataxia, the most prevalent inherited ataxia, is causedby abnormal expansion of a GAA triplet repeat sequence inintron 1 of the FRDA gene (1,2) (http://www.geneclinics.org/).This sequence is both polymorphic and genetically unstable
(1,3,4). Normal alleles, which contain<32 triplets, are classifiedas being either short normal (SN; <12 triplets) or long normal(LN; >12 triplets). Disease-causing expanded (E) alleles, con-taining 66–1700 triplets, interfere with gene transcription atthe FRDA locus, thereby resulting in frataxin deficiency andFriedreich ataxia (5–9). SN and LN alleles are stably transmit-ted from parent to offspring (3,4). However, E alleles displayintergenerational instability, usually contracting by 20–30%upon paternal transmission, and showing equal tendenciesfor expansion and contraction during maternal transmission(10,11). Rare, intermediate-sized alleles, containing 33–65uninterrupted GAA triplets, are termed premutation (PM)alleles because they may undergo hyperexpansion upon inter-generational transmission, expanding by >5- to 10-fold to gen-erate E alleles in offspring [(3,4,12,13); unpublished data]. PMalleles and those at the long end of the LN spectrum maybe interrupted by (GAGGAA)n hexanucleotides, which arebelieved to result in their stabilization during germlinetransmission (3). By analyzing individual somatic genomesfrom peripheral blood leukocytes using small-pool PCR(SP-PCR), we previously showed that E alleles are extremelyunstable in vivo and show a marked predilection for largecontractions (14).
Unstable triplet repeats are known to cause at least 15inherited diseases (15,16). So far, only three of the ten possibletriplet repeat motifs are known to be associated with disease:(CGG�CCG)n, (CTG�CAG)n, and (GAA�TTC)n. Instabilityof (CGG�CCG)n and (CTG�CAG)n repeat sequences inbacteria and yeast is dependent upon both repeat lengthand the orientation of the repeat relative to the origin ofreplication (17–26). For example, the (CTG�CAG)n repeattract is more unstable, with a significant tendency for contrac-tions, when CTG is the template for lagging strand synthesis.Either strand of the (CGG�CCG)n or (CTG�CAG)n repeatsequence (i.e. the CNG triplet motif ) has a tendency toform hairpin structures. The relative thermodynamic stabilityof the hairpin formed by single-stranded CTG versus CAGon the lagging strand template is thought to be the basisfor the contraction bias in both bacterial and yeastsystems (17).
*To whom correspondence should be addressed at 975 NE, 10th, BRC458, Oklahoma City, OK 73104, USA. Tel: +1 405 271 1360; Fax: +1 405 271 3910;Email: [email protected]
Nucleic Acids Research, Vol. 32 No. 19 ª Oxford University Press 2004; all rights reserved
5962–5971 Nucleic Acids Research, 2004, Vol. 32, No. 19doi:10.1093/nar/gkh933
Published online November 8, 2004

Recent evidence suggests the possibility that GAA andTTC single-stranded sequences may form hairpin structures(27), which is a well-known property of both the CNG tri-plet repeats (28). However, the polypurine�polypyrimidine(GAA�TTC)n repeat differs from the two unstable CNG tripletrepeats in its ability to adopt triplex structures in supercoiledtemplates (5,7,29–32). Although a triplex structure formed bythe (GAA�TTC)n repeat is believed to be the molecular basisfor the transcriptional interference afforded by expandedFRDA alleles (5–7,9,30,33), its role in mediating geneticinstability is less clear. We proposed that errors during laggingstrand synthesis could be the basis for the genetic instability ofthe (GAA�TTC)n repeat based on our previous observationof large, spontaneous GAA contractions in somatic cellsin vivo (14), the known disparity in the relative affinities ofreplication protein A (RPA) for polypurine versus polypyri-midine sequences (34) and the propensity of single-stranded(GAA)n sequence to adopt altered structural conformations(35,36). Furthermore, Ohshima et al. (6) had previouslyreported orientation-dependent instability of (GAA�TTC)n
repeats, noting increased smearing of their cloned insertsafter restriction digest, indicative of a contraction bias in plas-mids when GAA was the template for lagging strand synthesis.
Therefore, in this study we conducted a more detailed invest-igation of the effect of orientation of DNA replication on theinstability of repeats, cloned from human carriers of severalFRDA alleles, in plasmids propagated in Escherichia coli. Wealso evaluated the instability of similar repeat lengths in per-ipheral blood leukocytes from human donors using SP-PCR.Here, we show that while small slippage-type events occurredin both orientations, large contractions of (GAA�TTC)n
sequences occurred primarily when GAA was the templatefor lagging strand synthesis. DNA replication was significantlyimpeded in this orientation, and the incremental accumulationof large contractions was coincident with the phase of maximalplasmid replication. We also observed the same spectrum ofmutations in peripheral leukocytes in vivo in human subjectscarrying FRDA alleles of similar length, including completereversions to the normal/premutation length. During the pre-paration of this manuscript, similar findings were reportedusing (GAA�TTC)n containing plasmids in yeast (37). Thisindicates that the mechanism of the strand-specific differential
instability of (GAA�TTC)n repeats appears to be conservedamong prokaryotes and eukaryotes, and the contraction bias isseen not only in prokaryotes and simple eukaryotes, but inhuman somatic cells as well. The threshold length for theinitiation of somatic instability in vivo coincided with thelength of a eukaryotic Okazaki fragment (38,39). (GAGGAA)n
hexanucleotide interruptions stabilized the (GAA�TTC)n
sequence in E.coli and human somatic cells, constitutingthe first direct evidence for a stabilizing role of interruptionsin the context of the human genome in vivo.
MATERIALS AND METHODS
Plasmid construction
Genomic DNA from whole blood of human subjects was usedto amplify 9, 21, 44, 81, 107, 119 and 163 pure (GAA�TTC)n
repeats and 114 (GAA�TTC)n repeats containing a (GAG-GAA)n hexanucleotide repeat interruption in intron 1 of theFRDA gene, using the following primers (40): GAA-104F(50-GGCTTAAACTTCCCACACGTGTT-30) and GAA-629R(50-AGGACCATCATGGCCACACTT-30), followed by nes-ted PCR using one of the following primer pairs: gaapst1-F(50-GCTCCGCTGCAGCCCAGTATCTACTAAAAAATAC-30)and gaaxba1-R (50-GATGCGTCTAGACGCGCGACACCA-CGCCCGGCTAAC-30), or ttcpst1-F (50-GCTCCGCTGCA-GCGCGCGACACCACGCCCGGCTAAC-30) and ttcxba1-R(50-GATGCGTCTAGACCCAGTATCTACTAAAAAATAC-30).
The PCR products were digested with PstI and XbaI, whichrecognize sequences located at the 50 end of the forward andreverse primers, respectively. The fragments containing(GAA�TTC)n repeats along with minimal flanking sequence(38 bp 50 and 35 bp 30 to the repeat) were ligated into the PstIand XbaI sites of pUC19. The pUC19 vector used containsthe ColE1 origin and a portion of the ROP gene making it alow copy number plasmid. The primer pairs determined theorientation of the repeat tract: ‘GAA’ orientation (gaapst1-F/gaaxba1-R) or ‘TTC’ orientation (ttcpst1-F/ttcxba1-R)(Figure 1). The following recombinant plasmids were selectedfor mutation analysis: 9, 21, 48, 82, 105 and 111 repeats in the‘GAA’ orientation, and 8, 21, 48, 79, 105 and 108 repeats in
Figure 1. (GAA�TTC)n constructs used to analyze the effect of differential orientation of replication on repeat instability. Several lengths of the (GAA�TTC)n
sequence were cloned into the PstI/XbaI sites of pUC19 in both orientations with respect to the unidirectional ColE1 origin of replication (arrow). Repeat-containingconstructs are depicted in the ‘GAA’ or ‘TTC’ orientations, with repeat lengths of n= 9, 21, 48, 82, 105 and 111 in the ‘GAA’ orientation, and n= 8, 21, 48, 79, 105 and108 in the ‘TTC’ orientation. Additionally, two interrupted tracts of 101* and 95* repeats were cloned in the ‘GAA’ and ‘TTC’ orientation, respectively. The blackboxes flanking the repeat represent minimal flanking sequence from intron 1 of the FRDA gene.
Nucleic Acids Research, 2004, Vol. 32, No. 19 5963

the ‘TTC’ orientation. Repeat lengths and orientation weredetermined by sequencing. The constructs containing(GAA)9, (GAA)48, (GAA)105, (GAA)111, (TTC)8, (TTC)48,(TTC)79, (TTC)105 and (TTC)108 were pure (GAA�TTC)n
repeats, and the other constructs contained either a single non-GAA interruption within the repeat tract as follows:(GAA)21 = (GAA)17(A)(GAA)4; (GAA)82 = (GAA)70(AA)(GAA)11; (TTC)21 = (TTC)4(TT)(TTC)17 or a (GAGGAA)5
hexanucleotide interruption along with additional non-GAAsequence as follows: (GAA)101* = (GAA)60GA(GAA)2GAGGA(GAA)18(GAGGAA)5(GAA)8; (TTC)95* = (TTC)8(TTC-CTC)5(TTC)18TCCTC(TTC)2TC(TTC)54. A control, non-repeat containing plasmid with a 289 bp insert of randomsequence (51% G/C content) was created by amplifyingexon 5 of the APTX (aprataxin) gene using primers:aptx5pst1-F (50-GCTCCGCTGCAGGTCTGTTTCCTTCTC-TTGT-30) and aptx5xba1-R (50-GATGCGTCTAGAGGAGC-CAGCAGCACTACC-30), which was similarly digested andinserted into the PstI and XbaI sites in pUC19.
Analysis of (GAA�TTC)n repeat instability
Individual bacterial (DH5a) colonies obtained by plating gly-cerol stocks of sequence verified plasmids were grown inliquid culture (Luria–Bertani + 100 mg/ml ampicillin) at30�C for 12–24 h. PCR of individual colonies was performedto confirm that the inserts were full-length repeat tracts imme-diately prior to initiation of cultures. Colonies representingeach repeat length and orientation of replication were culturedin triplicate, so that at each hour (between 12–24 h) threeseparate 5 ml cultures could be harvested for each construct.Detailed studies were carried out for GAA-82, TTC-79 and arandom sequence insert, in triplicate. Each culture was treatedas follows: 1 ml was used to estimate the OD600 as a measureof cell density (to plot bacterial growth curves), 1 ml was usedto set up a glycerol stock (for mutation studies, see below) andthe rest was used for plasmid DNA isolation (to measure theamount of plasmid replication). Plasmid DNA was applied toa Zeta-Probe1 membrane using a Bio-Dot1 microfiltrationapparatus (BioRad), probed with g-ATP32-labeled pUC19-Roligonucleotide and quantified by densitometry. Bacterialgrowth curves and plasmid replication results were used todetermine the time points for detailed mutation analysis: 12,16, 20 h (log phase), and 24 h (stationary phase) of culture. Wespecifically avoided multiple sub-culturing of E.coli and onlyanalyzed the mutational profile over a single log phase, whichresulted in low mutation frequencies (>80% of final insertswere full-length), thus minimizing the bias for contractionsand multiple mutations involving the same repeat tract.
Previous reports indicate that transformation of tripletrepeat-containing plasmids per se increases the instabilityof the repeat tract (41). Therefore, glycerol stocks forGAA-82 and TTC-79 at 12, 16, 20 and 24 h time pointswere plated and incubated at 37�C for 16 h. PCR of individualcolonies was performed to assess (GAA�TTC)n repeatinstability using primers: GS-F = (50-CCCAGTATCTAC-TAAAAAATAC-30) and GS-R (50-ACACCACGCCCGGC-TAACTTTTC-30). Relative sizes of PCR products weredetermined by electrophoresis on 3% agarose gels, coupledwith direct sequencing of selected products. Approximately100 colonies were analyzed for each orientation and time
point, in triplicate, and instability was defined as the percent-age of the total number of PCR products amplified whoselength was altered after replication in E.coli. Large contrac-tions were defined as products that lost >50% of their initialrepeat length.
To compare E.coli growth curves and instability of pureversus interrupted (GAA�TTC)n repeat tracts, 5 ml cultureswere inoculated with colonies containing plasmids with eitherthe pure or interrupted repeat tract inserted in both orienta-tions. Cultures were grown at 30�C for 10–24 h, in triplicate,such that three cultures for each construct were harvestedevery hour. Growth curves were generated as describedabove. Repeat tract instability was analyzed after 22 h ofgrowth, as described previously. Approximately 75 colonieswere analyzed for each of the four constructs, in triplicate.
SP-PCR analysis
The SP-PCR analysis was carried out as described previously(14,42). Briefly, serial dilutions of human genomic DNA,ranging from 6–600 pg, were prepared in siliconized micro-fuge tubes. PCR was performed using GAA-104F and GAA-629R, which allowed accurate sizing of alleles used in thepresent study. PCR products were resolved by electrophoresison 1–2% agarose gels (2% gels were used for the calculation ofthreshold length for the initiation of instability), and bandsdetected by Southern blotting using an end-labeled (TTC)9
oligonucleotide probe. The calculation of the average numberof individual FRDA molecules per reaction (the ‘gene equival-ent’) was performed by Poisson analysis as described pre-viously (42,43). The average quantity of genomic DNArequired for the amplification of one FRDA molecule was13.4 pg (95% CI 10.6–16.1 pg). For each genomic DNA sam-ple, multiple reactions were performed using ‘small pools’of 2.5–25 individual FRDA molecules per reaction todetect mutations. Mutation loads were calculated as the pro-portion of molecules that differed by >5% in size from theconstitutional (most common) allele determined by directsequencing.
Statistical methods
Comparison of medians was carried out using the Mann–Whitney U test, means were compared using the t-test, andfrequencies were compared by c2 analysis.
RESULTS
Large contractions of (GAA�TTC)n occur when ‘GAA’is the template for lagging strand synthesis
(GAA�TTC)n repeat tracts (n = 9, 21, 48, 80 and 105 triplets)were cloned in both orientations relative to the ColE1 origin ofreplication, such that either GAA or TTC would serve as thetemplate for lagging strand synthesis (Figure 1). Replication ofthese cloned (GAA�TTC)n triplet repeats, which were propag-ated in E.coli (DH5a), displayed length-dependent instabil-ity. Whereas repeat tracts containing <21 triplets werecompletely stable, repeat tracts containing 48 and �80 tripletswere moderately unstable and the construct containing >100triplets in the GAA orientation was highly unstable (data not
5964 Nucleic Acids Research, 2004, Vol. 32, No. 19

shown). We selected the moderately unstable repeat tractscontaining �80 triplets to perform a detailed analysis of theeffects of differential orientation of replication on GAA tripletrepeat instability, which was accomplished by analyzingmultiple individual replication events in bacterial colonies.The (GAA�TTC)n sequence was found to be significantlymore unstable when GAA was the template for lagging strandsynthesis (‘GAA’ orientation; GAA-82) compared withTTC (‘TTC’ orientation; TTC-79) (Figure 2A). The sameorientation-dependent instability was also noted when(GAA�TTC)n repeats were cloned in plasmids pBluescript II(Stratagene), pCR2.1 and pCR3.1 (Invitrogen) and replicatedin DH5a and TOP10 (Invitrogen) strains of E.coli (data notshown).
To further examine the role of replication in GAA tripletrepeat instability, we analyzed the mutational frequencies andspectra during the entire period of active replication of plas-mids in E.coli. Throughout the log phase of E.coli growth, bothbacterial growth and plasmid DNA replication were signifi-cantly slower when GAA was the template for lagging strandsynthesis (Figure 2B and C). A random sequence insert of thesame size showed the same profile as when TTC was thelagging strand template (Figure 2B and C). Even when
cultures were grown for 40 h, well into the stationary phase,bacterial density was significantly lower for GAA-82 than foreither TTC-79 or the random sequence control (data notshown). These data indicate that DNA replication is signifi-cantly slower when GAA is the template for lagging strandsynthesis.
Repeat instability was analyzed for GAA-82 and TTC-79after 12, 16, 20 (log phase), and 24 h (stationary phase) ofculture. Instability was significantly greater in the ‘GAA’orientation, and it increased throughout the exponentialphase of plasmid replication (Figure 2D). Contractions weremuch more prevalent than expansions in both orientations.However, analysis of all contraction events from 12–24 hrevealed that large contractions, involving >50% of theoriginal length, were observed primarily when GAA wasthe template for lagging strand synthesis (Figure 2A andE). Large contractions in the ‘TTC’ orientation were signifi-cantly less frequent (P < 0.001), and of lesser magnitude[median size of contractions = 25.5 versus 49.5 triplets(P < 0.001)]. Furthermore, despite the relatively blunted expo-nential replication phase, there was an incremental accumula-tion of large contractions throughout the log phase in the GAAorientation (Figure 2F), and indeed, most of the observed
Figure 2. Slowed replication and large contractions of the (GAA�TTC)n repeat when GAA is the template for lagging strand synthesis. Closed diamond = GAA-82,closed square=TTC-79, closed triangle= random sequence control and all error bars reflect–SD (A) Representative PCR products generated from colonies obtainedby plating the glycerol stock of a single culture (16 h) are shown. The repeat tract was significantly more unstable and prone to contractions when GAA was thetemplate for lagging strand synthesis. Arrows indicating the position of GAA-40 and TTC-40 represent the cutoff (�50%) used for defining small versus largecontractions. (B) Slower growth of E.coli and significantly blunted log phase when GAA was the template for lagging strand synthesis compared with TTC or randomsequence control. (C) Slower plasmid DNA replication when GAA was the template for lagging strand synthesis compared to TTC or random sequence control, asdetermined by dot blot analysis. RDU, relative densitometric units. (D) Percentage of colonies containing (GAA�TTC)n repeats of altered length (% instability) after12, 16, 20 and 24 h of culture indicates that there was a significant increase in instability over time for GAA-82 versus TTC-79 (P = 0.01 at 20 and 24 h). (E) Largecontractions (>50% loss of initial repeat length) were significantly more frequent with GAA-82 versus TTC-79 (P < 0.001). The median length of contractionproducts (indicated by horizontal lines) was significantly shorter for GAA-82 (25.5 repeats) than for TTC-79 (49.5 repeats) (P < 0.001). Only contraction events areshown in the graph, with the magnitude of change (in triplets) plotted on the y-axis. (F) Large contractions accumulated throughout the log phase when GAA was thetemplate for lagging strand synthesis.
Nucleic Acids Research, 2004, Vol. 32, No. 19 5965

instability was due to these large contractions (Figure 2Dand F).
We did not observe large expansions in either orientationof replication. Small contractions and expansions (involving<10% of the original tract length) were equally frequent in theGAA or TTC orientations (P = 0.41 and P = 0.37, respec-tively). Large contractions were far more prevalent than smallcontractions only in the ‘GAA’ orientation (P < 0.001)(Figure 2E). In summary, we noted large contractions predo-minantly when GAA is the lagging strand template, and thiswas coincident with plasmid DNA replication.
Similar mutational spectrum is observed at the FRDAlocus in human somatic cells in vivo
To test the physiological relevance of our observations, weperformed SP-PCR analysis of individual FRDA moleculescontaining repeat lengths similar to those tested in E.coli,derived from genomic DNA of peripheral blood leukocytesof human subjects. We have previously optimized this tech-nique for the analysis of GAA triplet repeat instability at theFRDA locus (14). Analysis of 2520 individual FRDA mole-cules from four heterozygous carriers of constitutional alleleswith 78 (n = 524 molecules), 81 (n = 700 molecules), 91(n = 328 molecules) and 105 (n = 968 molecules) uninterruptedtriplet repeats revealed the same spectrum of mutationsobserved in E.coli: small expansions and contractions, largecontractions and a paucity of large expansions or intermediate-sized contractions (Figure 3A and B). Of the 2520 moleculesanalyzed, we observed 283 variant bands (mutationload = 11.2%), with a significant contraction bias [55(2.2%) expansions versus 228 (9%) contractions, P < 0.001;Figure 3B]. In contrast to replication in E.coli, somaticinstability in leukocytes resulted in far more small versuslarge contractions. However, the extent of the large contrac-tions was similar in the two systems, with most large contrac-tions resulting in almost complete reversion to the normal size,and very few intermediate-sized contractions (Figure 3B). Itshould be noted that the frequency of large contractions ismost likely underestimated in our SP-PCR assay since allfour carriers used in this study also have a normal allele,which makes it nearly impossible to detect ‘complete’ rever-sion events of individual FRDA molecules (Figure 3A).
The threshold length for the initiation of somaticinstability at the FRDA locus coincides with thelength of a eukaryotic Okazaki fragment
Using SP-PCR, we previously showed that the thresholdlength for the initiation of somatic instability in vivo isbetween 26 and 44 uninterrupted GAA triplet repeats (14).Here, using the same technique, we analyzed a total of 5593individual FRDA molecules with constitutional allele sizesranging from 8 to 66 uninterrupted GAA triplet repeats tomore precisely define the threshold length. SP-PCR of(GAA)8–20 (n = 349 molecules), (GAA)30 (n = 560 molecules)and (GAA)39 (n = 1150 molecules) showed complete stability(Figure 4A and B) (data not shown). In contrast, SP-PCRanalysis of (GAA)44 (n = 2304 molecules) and (GAA)66
(n = 1230 molecules) showed somatic instability in vivo,with mutation loads of 6.3 and 30%, respectively (Figure 4Cand D). This indicates that the threshold length for the
initiation of somatic instability in peripheral leukocytesin vivo at the FRDA locus is between 40 and 44 uninterruptedtriplet repeats. The (GAA)39 allele, which showed no somaticinstability (Figure 3B), is known to have undergone hyperex-pansion in one of three germline transmissions, producing a(GAA)650 allele in the offspring. Therefore, it seems that theminimum length required for the initiation of somatic instabil-ity at the FRDA locus may be longer than that requiredfor germline instability.
(GAGGAA)n hexanucleotide interruptions stabilize the(GAA�TTC)n triplet repeat at the FRDA locus insomatic cells in vivo
To test whether (GAGGAA)n hexanucleotide interruptions in(GAA�TTC)n repeat tracts cause stabilization of tripletrepeats, as has been observed in E.coli (44) and predictedin human germline transmissions (3), SP-PCR analysis wasperformed using leukocytic DNA from carriers of two similar(GAA�TTC)n alleles. One carrier had a pure (GAA)107
allele, and another a (GAA)114* allele interrupted with a
Figure 3. SP-PCR analysis of (GAA�TTC)n alleles with 78–105 uninterruptedrepeats shows instability and a contraction bias in somatic cells in vivo. (A) Arepresentative Southern blot showing multiple ‘small pools’ of FRDAmolecules from human genomic DNA containing 9 and 78 GAA tripletrepeats. Somatic instability in vivo comprised frequent small contractions/expansions and some large contractions into the normal, non-disease sizerange. (B) Summary of mutations detected following SP-PCR analysis of2520 individual FRDA molecules containing (GAA�TTC)n with n = 78, 81,91, or 105 uninterrupted repeats. The x-axis represents the magnitude of change(%) from the constitutional (most common) GAA triplet repeat length,determined by sequencing, with negative and positive readings indicatingcontractions and expansions, respectively. Non-mutant bands are not plotted.
5966 Nucleic Acids Research, 2004, Vol. 32, No. 19
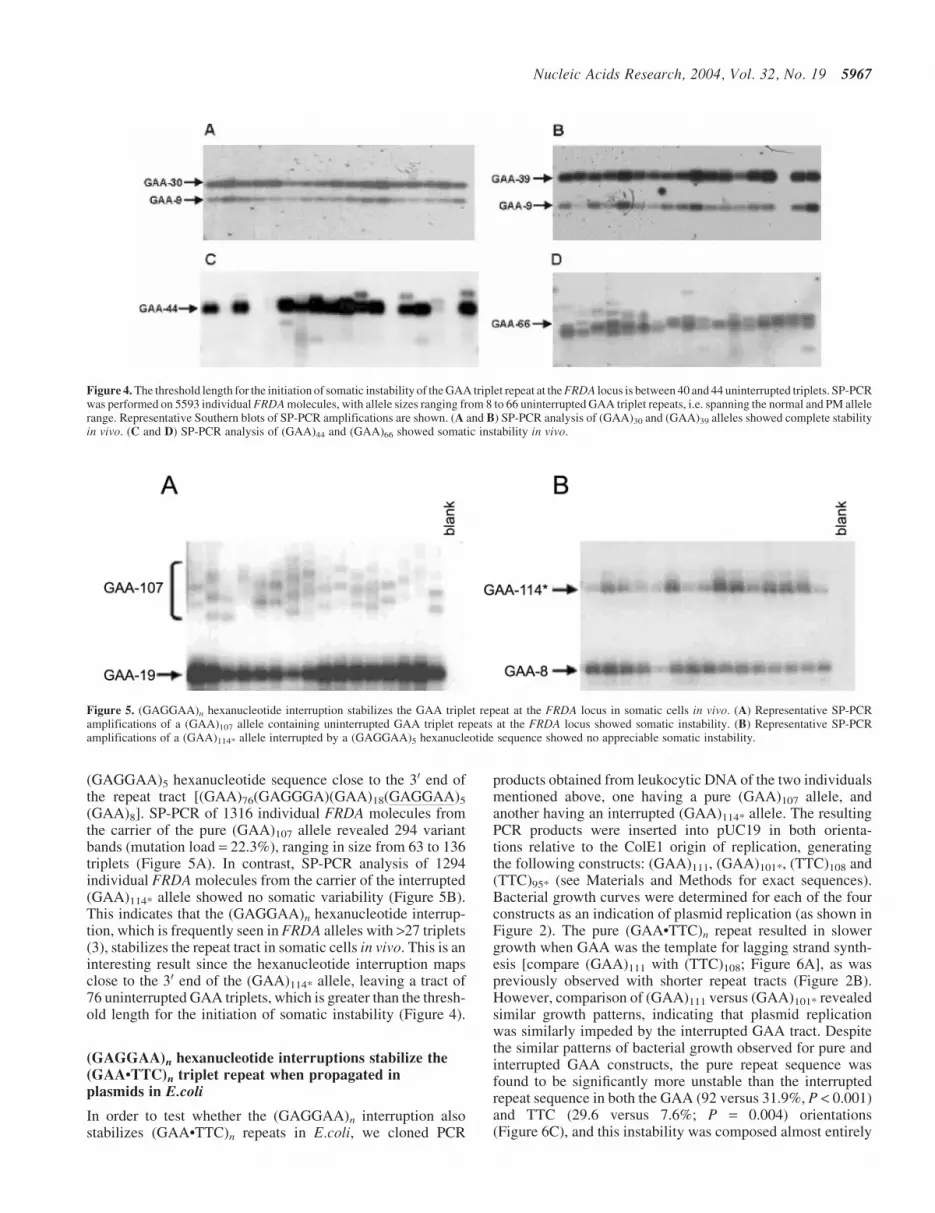
(GAGGAA)5 hexanucleotide sequence close to the 30 end ofthe repeat tract [(GAA)76(GAGGGA)(GAA)18(GAGGAA)5
(GAA)8]. SP-PCR of 1316 individual FRDA molecules fromthe carrier of the pure (GAA)107 allele revealed 294 variantbands (mutation load = 22.3%), ranging in size from 63 to 136triplets (Figure 5A). In contrast, SP-PCR analysis of 1294individual FRDA molecules from the carrier of the interrupted(GAA)114* allele showed no somatic variability (Figure 5B).This indicates that the (GAGGAA)n hexanucleotide interrup-tion, which is frequently seen in FRDA alleles with >27 triplets(3), stabilizes the repeat tract in somatic cells in vivo. This is aninteresting result since the hexanucleotide interruption mapsclose to the 30 end of the (GAA)114* allele, leaving a tract of76 uninterrupted GAA triplets, which is greater than the thresh-old length for the initiation of somatic instability (Figure 4).
(GAGGAA)n hexanucleotide interruptions stabilize the(GAA�TTC)n triplet repeat when propagated inplasmids in E.coli
In order to test whether the (GAGGAA)n interruption alsostabilizes (GAA�TTC)n repeats in E.coli, we cloned PCR
products obtained from leukocytic DNA of the two individualsmentioned above, one having a pure (GAA)107 allele, andanother having an interrupted (GAA)114* allele. The resultingPCR products were inserted into pUC19 in both orienta-tions relative to the ColE1 origin of replication, generatingthe following constructs: (GAA)111, (GAA)101*, (TTC)108 and(TTC)95* (see Materials and Methods for exact sequences).Bacterial growth curves were determined for each of the fourconstructs as an indication of plasmid replication (as shown inFigure 2). The pure (GAA�TTC)n repeat resulted in slowergrowth when GAA was the template for lagging strand synth-esis [compare (GAA)111 with (TTC)108; Figure 6A], as waspreviously observed with shorter repeat tracts (Figure 2B).However, comparison of (GAA)111 versus (GAA)101* revealedsimilar growth patterns, indicating that plasmid replicationwas similarly impeded by the interrupted GAA tract. Despitethe similar patterns of bacterial growth observed for pure andinterrupted GAA constructs, the pure repeat sequence wasfound to be significantly more unstable than the interruptedrepeat sequence in both the GAA (92 versus 31.9%, P < 0.001)and TTC (29.6 versus 7.6%; P = 0.004) orientations(Figure 6C), and this instability was composed almost entirely
Figure 4. The threshold length for the initiation of somatic instability of the GAA triplet repeat at the FRDA locus is between 40 and 44 uninterrupted triplets. SP-PCRwas performed on 5593 individual FRDA molecules, with allele sizes ranging from 8 to 66 uninterrupted GAA triplet repeats, i.e. spanning the normal and PM allelerange. Representative Southern blots of SP-PCR amplifications are shown. (A and B) SP-PCR analysis of (GAA)30 and (GAA)39 alleles showed complete stabilityin vivo. (C and D) SP-PCR analysis of (GAA)44 and (GAA)66 showed somatic instability in vivo.
Figure 5. (GAGGAA)n hexanucleotide interruption stabilizes the GAA triplet repeat at the FRDA locus in somatic cells in vivo. (A) Representative SP-PCRamplifications of a (GAA)107 allele containing uninterrupted GAA triplet repeats at the FRDA locus showed somatic instability. (B) Representative SP-PCRamplifications of a (GAA)114* allele interrupted by a (GAGGAA)5 hexanucleotide sequence showed no appreciable somatic instability.
Nucleic Acids Research, 2004, Vol. 32, No. 19 5967
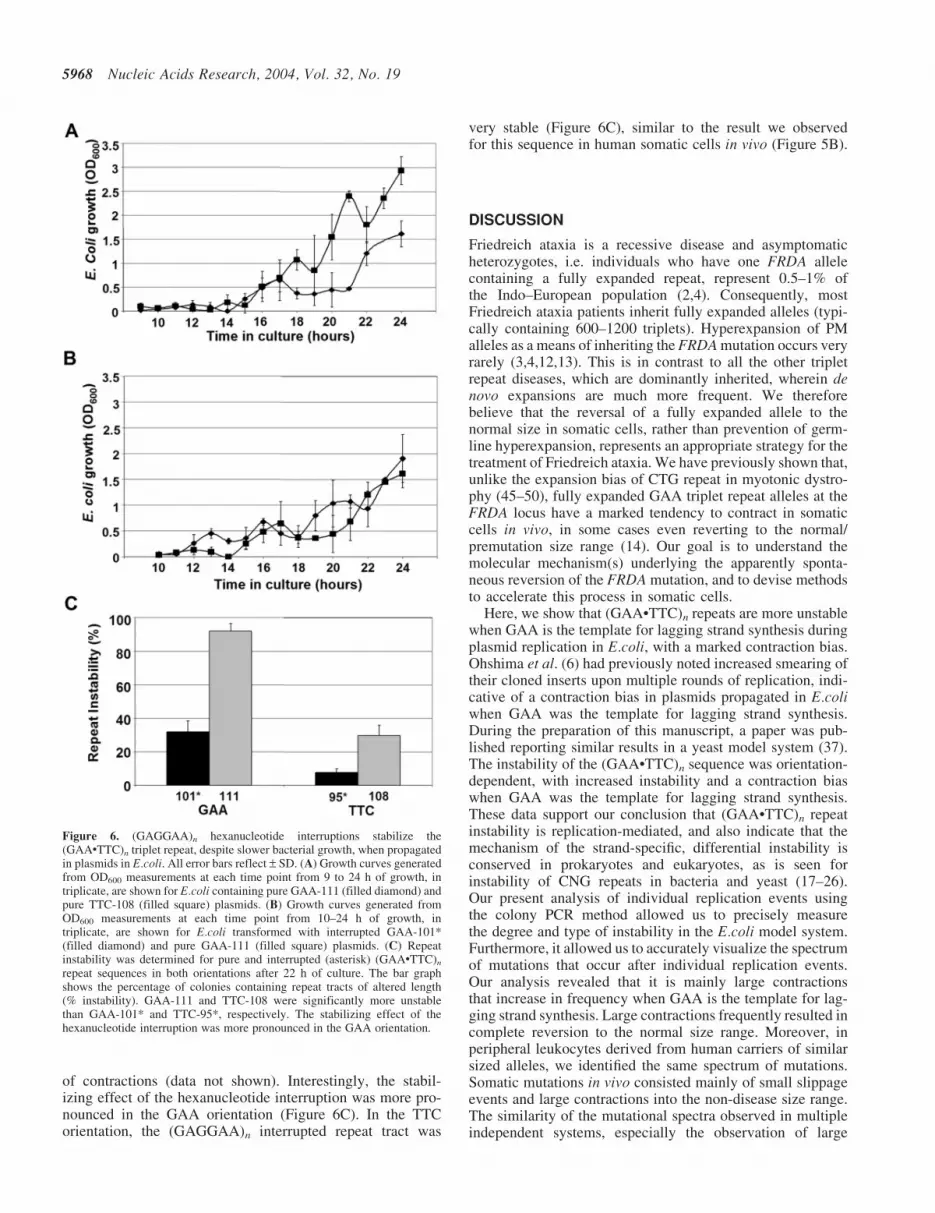
of contractions (data not shown). Interestingly, the stabil-izing effect of the hexanucleotide interruption was more pro-nounced in the GAA orientation (Figure 6C). In the TTCorientation, the (GAGGAA)n interrupted repeat tract was
very stable (Figure 6C), similar to the result we observedfor this sequence in human somatic cells in vivo (Figure 5B).
DISCUSSION
Friedreich ataxia is a recessive disease and asymptomaticheterozygotes, i.e. individuals who have one FRDA allelecontaining a fully expanded repeat, represent 0.5–1% ofthe Indo–European population (2,4). Consequently, mostFriedreich ataxia patients inherit fully expanded alleles (typi-cally containing 600–1200 triplets). Hyperexpansion of PMalleles as a means of inheriting the FRDA mutation occurs veryrarely (3,4,12,13). This is in contrast to all the other tripletrepeat diseases, which are dominantly inherited, wherein denovo expansions are much more frequent. We thereforebelieve that the reversal of a fully expanded allele to thenormal size in somatic cells, rather than prevention of germ-line hyperexpansion, represents an appropriate strategy for thetreatment of Friedreich ataxia. We have previously shown that,unlike the expansion bias of CTG repeat in myotonic dystro-phy (45–50), fully expanded GAA triplet repeat alleles at theFRDA locus have a marked tendency to contract in somaticcells in vivo, in some cases even reverting to the normal/premutation size range (14). Our goal is to understand themolecular mechanism(s) underlying the apparently sponta-neous reversion of the FRDA mutation, and to devise methodsto accelerate this process in somatic cells.
Here, we show that (GAA�TTC)n repeats are more unstablewhen GAA is the template for lagging strand synthesis duringplasmid replication in E.coli, with a marked contraction bias.Ohshima et al. (6) had previously noted increased smearing oftheir cloned inserts upon multiple rounds of replication, indi-cative of a contraction bias in plasmids propagated in E.coliwhen GAA was the template for lagging strand synthesis.During the preparation of this manuscript, a paper was pub-lished reporting similar results in a yeast model system (37).The instability of the (GAA�TTC)n sequence was orientation-dependent, with increased instability and a contraction biaswhen GAA was the template for lagging strand synthesis.These data support our conclusion that (GAA�TTC)n repeatinstability is replication-mediated, and also indicate that themechanism of the strand-specific, differential instability isconserved in prokaryotes and eukaryotes, as is seen forinstability of CNG repeats in bacteria and yeast (17–26).Our present analysis of individual replication events usingthe colony PCR method allowed us to precisely measurethe degree and type of instability in the E.coli model system.Furthermore, it allowed us to accurately visualize the spectrumof mutations that occur after individual replication events.Our analysis revealed that it is mainly large contractionsthat increase in frequency when GAA is the template for lag-ging strand synthesis. Large contractions frequently resulted incomplete reversion to the normal size range. Moreover, inperipheral leukocytes derived from human carriers of similarsized alleles, we identified the same spectrum of mutations.Somatic mutations in vivo consisted mainly of small slippageevents and large contractions into the non-disease size range.The similarity of the mutational spectra observed in multipleindependent systems, especially the observation of large
Figure 6. (GAGGAA)n hexanucleotide interruptions stabilize the(GAA�TTC)n triplet repeat, despite slower bacterial growth, when propagatedin plasmids in E.coli. All error bars reflect – SD. (A) Growth curves generatedfrom OD600 measurements at each time point from 9 to 24 h of growth, intriplicate, are shown for E.coli containing pure GAA-111 (filled diamond) andpure TTC-108 (filled square) plasmids. (B) Growth curves generated fromOD600 measurements at each time point from 10–24 h of growth, intriplicate, are shown for E.coli transformed with interrupted GAA-101*(filled diamond) and pure GAA-111 (filled square) plasmids. (C) Repeatinstability was determined for pure and interrupted (asterisk) (GAA�TTC)n
repeat sequences in both orientations after 22 h of culture. The bar graphshows the percentage of colonies containing repeat tracts of altered length(% instability). GAA-111 and TTC-108 were significantly more unstablethan GAA-101* and TTC-95*, respectively. The stabilizing effect of thehexanucleotide interruption was more pronounced in the GAA orientation.
5968 Nucleic Acids Research, 2004, Vol. 32, No. 19

contractions, suggests that a common mechanism may under-lie the instability of (GAA�TTC)n repeats.
The mechanism(s) responsible for GAA triplet repeatinstability are poorly understood. We propose that at leasttwo distinct mechanisms underlie the mutations we observed.Small length changes of the repeat tract (<10% variation) mayoccur as a result of slippage and mispairing during replicationof the triplet repeat. The equal frequency and magnitude ofsmall length changes in both ‘GAA’ and ‘TTC’ orientations isconsistent with the expectation that slippage is equally likelyto occur during leading or lagging strand synthesis, and withthe observation that GAA and TTC strands undergo approxi-mately the same degree of slippage during rolling circle repli-cation (51). However, our results implicate erroneous laggingstrand synthesis, when GAA is the template strand, as thelikely mechanism for the generation of large contractions ofthe (GAA�TTC)n repeat (Figure 7). The incremental accumu-lation of large contractions was associated with DNA replica-tion when GAA was the template for lagging strand synthesis.Our in vivo data show that the threshold length for the initia-tion of somatic instability is between 40 and 44 uninterruptedtriplets. Therefore, somatic instability in vivo may initiatewhen the length of the (GAA�TTC)n repeat exceeds the lengthof a eukaryotic Okazaki fragment. If the single-stranded lag-ging strand template were to adopt a stable or metastablesecondary structure(s), the replication machinery could bypassa variable number of repeats in the nascent strand, resulting incontraction of the repeat (Figure 7). The 50-fold lower affinityof RPA for polypurine sequences (34) and the selective abilityof GAA sequences to adopt intrastrand structures (17,35,36)lend further support to this mechanism. The frequent observa-tion of large contractions and paucity of intermediate-sizedcontractions could stem from a minimum length requirementfor GAA repeats to maintain a stable structure in order to bebypassed in the nascent strand. These data have importantimplications for understanding the molecular basis of somaticinstability at the FRDA locus.
It is likely that a structural transition may underlie theinitiation of somatic instability in vivo. The minimum lengthfor the formation of sticky DNA (59 triplets) (30) is greaterthan the 40–44 triplet repeat threshold for the initiation ofsomatic instability. However, Potaman et al. (32) showed thatthe (GAA�TTC)n repeat undergoes a structural transitionfrom a stable intramolecular triplex at 9–23 triplets to anintramolecular bi-triplex structure at 42 triplets, coincidingin length with the threshold for the initiation of somatic instab-ility in human cells. Interestingly, the (GAA)114* hexa-nucleotide interrupted allele was stable in somatic cells,despite containing a pure repeat tract of 76 triplets. Pure(GAA)111 and (TTC)108 sequences were also significantlymore unstable than interrupted (GAA)101* and (TTC)95*
sequences, respectively, when propagated in E.coli. How-ever, the interruption appeared to have no effect on theslowed bacterial growth. The mechanism by which thehexanucleotide interruption confers stability is unknown,but it may be due to the inability of the impure GAA tem-plate strand to adopt a stable secondary structure. Previouswork has shown that a repeat tract comprised entirely of(GAGGAA)n repetitive sequence (50% non-GAA) does notform sticky DNA, unlike pure (GAA)n repeat sequences (44).Interestingly, in the plasmid replication model the
hexanucleotide interruption resulted in significantly enhancedstability of the adjacent repeat when GAA was the laggingstrand template, despite interfering with efficient plasmidreplication. This dissociation of replication blockade andrepeat instability suggests that while bacterial growthand replication may be slowed by the homopurine�homopyrimidine nature of the sequence, purity of the tripletrepeat tract is required for GAA instability.
Figure 7. Model depicting the genesis of large contractions during laggingstrand synthesis of GAA triplet repeat sequences (in eukaryotic replication).‘H’, helicase; ‘PCNA’, proliferating cell nuclear antigen; RPA, replicationprotein A. Pold (large rectangle) replicates the leading strand at theadvancing fork. Pola/primase (gray filled circle) initiates Okazaki fragmentsynthesis, and pold elongates the nascent Okazaki fragment during laggingstrand synthesis. The single-stranded lagging strand template is bound by RPA).Non-repeat sequence and the complementary TTC repeat sequence are shownas solid black lines, and the GAA triplet repeat is shown as a dotted line. Whenthe GAA triplet repeat sequence expands beyond the length of an Okazakifragment (the threshold length of 40–44 triplets at the FRDA locus),individual fragments would have to be initiated, elongated and ligatedwithin the length of the repeat tract. We propose that the 50-fold reducedaffinity of RPA for purine-rich sequences [compared with pyrimidine-richsequences (34)] would allow the single-stranded GAA strand to adoptstable/metastable secondary structures (‘?’), which would result inbypassing of a variable number of GAA repeats in the nascent Okazakifragment, thus resulting in the experimentally observed contractions. Largecontractions (and the under-representation of intermediate sized contractions)in vivo involving alleles with 78–105 repeats could stem from the minimumlength required for the secondary structure(s) to be stable.
Nucleic Acids Research, 2004, Vol. 32, No. 19 5969

Furthermore, it seems that (GAA�TTC)n repeats are quan-titatively more unstable than (CTG�CAG)n and (CGG�CCG)n
repeats in somatic cells in vivo. SP-PCR analysis of similar-sized alleles of the other triplet repeats in leukocytes revealedvery low mutation frequencies compared with the mutationloads we observed with (GAA�TTC)n alleles (50,52). Thereason for these differences is not known, but it is interestingto note that during the evolution of the human genome,(GAA�TTC)n repeats have undergone significant instabilityas reflected by the development of a wide range of allelelengths in comparison with (CTG�CAG)n and (CGG�CCG)n
sequences (53).In conclusion, we have shown that the (GAA�TTC)n
repeat that causes Friedreich ataxia is destabilized duringDNA replication, undergoing large contractions when GAAserves as the lagging strand template. In somatic cells in vivo,the (GAA�TTC)n repeat at the FRDA locus initiates instabilitybetween 40 and 44 triplets, displays significant mutation load,and the mutational spectrum includes large contractions thatresult in reversion to non-disease alleles. These data indicatethat the FRDA mutation is reversible.
ACKNOWLEDGEMENTS
We are grateful to the patients and their families for participat-ing in this study. We would like to thank Dr Gillian Dalglieshfor critically reviewing this manuscript. This research was sup-ported in part by grants from the NIH/NINDS (NS047596),American Diabetes Association and OCAST to S.I.B. S.S.was supported by a fellowship from the Presbyterian HealthFoundation.
REFERENCES
1. Campuzano,V., Montermini,L., Molt�oo,M.D., Pianese,L., Coss�eee,M.,Cavalcanti,E., Monr�oos,F., Rodius,F., Duclos,F., Monticelli,A. et al.(1996) Friedreich’s ataxia: autosomal recessive disease caused by anintronic GAA triplet repeat expansion. Science, 271, 1423–1427.
2. Bidichandani,S.I. and Ashizawa,T. (2002) Friedreich ataxia,GeneReviews: Genetic Disease Online Reviews of GeneTests-GeneClinics. Copyright University of Washington, Seattle, WA, USA.
3. Montermini,L., Andermann,E., Labuda,M., Richter,A., Pandolfo,M.,Cavalcanti,F., Pianese,L., Iodice,L.,Farina,G., Monticelli,A. et al. (1997)The Friedreich ataxia GAA triplet repeat: premutation and normalalleles. Hum. Mol. Genet., 6, 1261–1266.
4. Coss�eee,M., Schmitt,M., Campuzano,V., Reutenauer,L., Moutou,C.,Mandel,J.L. and Koenig,M. (1997) Evolution of the Friedreich’s ataxiatrinucleotide repeat expansion: founder effect and premutations.Proc. Natl Acad. Sci., 94, 7452–7457.
5. Bidichandani,S.I., Ashizawa,T. and Patel,P.I. (1998) The GAAtriplet-repeat expansion in Friedreich ataxia interferes with transcriptionand may be associated with an unusual DNA structure. Am. J. Hum.Genet., 62, 111–121.
6. Ohshima,K., Montermini,L., Wells,R.D. and Pandolfo,M. (1998)Inhibitory effects of expanded GAA�TTC triplet repeats from intron 1of the Friedreich ataxia gene on transcription and replication in vivo.J. Biol. Chem., 273, 14588–14595.
7. Grabczyk,E. and Usdin, K (2000) The GAA�TTC triplet repeat expandedin Friedreich’s ataxia impedes transcription elongation by T7 RNApolymerase in a length and supercoil dependent manner. Nucleic AcidsRes., 28, 2815–2822.
8. Campuzano,V., Montermini,L., Lutz,Y., Cova,L., Hindelang,C.,Jiralespong,S., Trottier,Y., Kish,S.J., Faucheux,B., Trouillas,P. et al.(1997) Frataxin is reduced in Friedreich ataxia patients and is associatedwith mitochondrial membranes. Hum. Mol. Genet., 6, 1771–1780.
9. Sakamoto,N., Ohshima,K., Montermini,L., Pandolfo,M. and Wells,R.D.(2001) Sticky DNA, a self-associated complex formed at long GAA�TTCrepeats in intron 1 of the frataxin gene, inhibits transcription.J. Biol. Chem., 276, 27171–27177.
10. De Michele,G., Cavalcanti,F., Criscuolo,C., Pianese,L., Monticelli,A.,Filla,A. and Cocozza,S. (1998) Parental gender, age at birth andexpansion length influence GAA repeat intergenerational instability inthe X25 gene: pedigree studies and analysis of sperm from patientswith Friedreich’s ataxia. Hum. Mol. Genet., 7, 1901–1906.
11. Monr�oos,E., Molt�oo,M.D., Martinez,F., Ca~nnizares,J., Blanca,J.,Vilchez,J.J., Prieto,F., de Frutos,R. and Palau,F. (1997) Phenotypecorrelation and intergenerational dynamics of the Friedreich ataxiaGAA trinucleotide repeat. Am. J. Hum. Genet., 61, 101–110.
12. Epplen,C., Epplen,J.T., Frank,G., Miterski,B., Santos,E.J. and Schols,L.(1997) Differential stability of the (GAA)n tract in the Friedreich ataxia(STM7) gene. Hum. Genet., 99, 834–836.
13. Delatycki,M.B., Paris,D., Gardner,R.J., Forshaw,K., Nicholson,G.A.,Nassif,N., Williamson,R. and Forrest,S.M. (1998) Sperm DNA analysisin a Friedreich ataxia premutation carrier suggests both meioticand mitotic expansion in the FRDA gene. J. Med. Genet., 35,713–716.
14. Sharma,R., Bhatti,S., G�oomez,M., Clark,R.M., Murray,C., Ashizawa,T.and Bidichandani,S.I. (2002) The GAA triplet-repeat shows a high levelof somatic instability in vivo, with a significant predilection for largecontractions. Hum. Mol. Genet., 11, 2175–2187.
15. Cummings,C.J. and Zoghbi,H.Y. (2000) Fourteen and counting:unraveling trinucleotide repeat diseases. Hum. Mol. Genet. 9, 909–916.
16. Bowater,R.P. and Wells,R.D. (2001) The intrinsically unstable life ofDNA triplet repeats associated with human hereditary disorders.Prog. Nucleic Acid Res. Mol. Biol., 66, 159–202.
17. Kang,S., Jaworski,A., Ohshima,K. and Wells,R.D. (1995) Expansion anddeletion of CTG repeats from human disease genes are determined bythe direction of replication in E.coli. Nature Genet., 10, 213–218.
18. Samadashwily,S.M., Raca,G. and Mirkin,S.M. (1997) Trinucleotiderepeats affect DNA replication in vivo. Nature Genet., 17, 298–304.
19. Sarkar,P.S., Chang,H.C., Boudi,F.B. and Reddy,S. (1998) CTG repeatsshow bimodal amplification in E.coli. Cell, 95, 531–540.
20. Freudenreich,C.H., Stavenhagen,J.B. and Zakian,V.A. (1997) Stabilityof a CTG/CAG trinucleotide repeat in yeast is dependent on its orientationin the genome. Mol. Cell. Biol., 17, 2090–2098.
21. Miret,J.J., Pessoa-Brand~aao,L. and Lahue,R.S. (1998) Orientation-dependent and sequence-specific expansions of CTG/CAG trinucleotiderepeats in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 95,12438–12443.
22. Maurer,D.J., O’Callaghan,B.L. and Livingston,D.M. (1996) Orientationdependence of trinucleotide CAG repeat instability in Saccharomycescerevisiae. Mol. Cell. Biol., 16, 6617–6622.
23. Shimizu,M., Gellibolian,R., Oostra,B.A. and Wells,R.D. (1996) Cloning,characterization, and properties of plasmids containing CGG tripletrepeats from the FMR1 gene. J. Mol. Biol., 258, 614–626.
24. Hirst,M.C. and White,P.J. (1998) Cloned human FMR1 trinucleotiderepeats exhibit a length- and orientation-dependent instability suggestiveof in vivo lagging strand secondary structure. Nucleic Acids Res.,26, 2353–2358.
25. White,P.J., Borts,R.H. and Hirst,M.C. (1999) Stability of the humanfragile X (CGG)(n) triplet repeat array in Saccharomyces cerevisiaedeficient in aspects of DNA metabolism. Mol. Cell. Biol., 19, 5675–84.
26. Balakumaran,B.S., Freudenreich,C.H. and Zakian,V.A. (2000) CGG/CCG repeats exhibit orientation-dependent instability and orientation-independent fragility in Saccharomyces cerevisiae. Hum. Mol. Genet.,9, 93–100.
27. Heidenfelder,B.L., Makhov,A.M. and Topal,M.D. (2003) Hairpinformation in Friedreich’s ataxia triplet repeat expansion. J. Biol.Chem., 278, 2425–2431.
28. Gacy,A.M., Goellner,G., Juranic,N., Macura,S. and McMurray,C.T.(1995) Trinucleotide repeats that expand in human disease form hairpinstructures in vitro. Cell, 81, 533–540.
29. Gacy,A.M., Goellner,G.M., Spiro,C., Chen,X., Gupta,G.,Bradbury,E.M., Dyer,R.B., Mikesell,M.J., Yao,J.Z., Johnson,A.J. et al.(1998) GAA instability in Friedreich’s ataxia shares a common,DNA-directed and intraallelic mechanism with other trinucleotidediseases. Mol. Cell, 1, 583–593.
30. Sakamoto,N., Chastain,P.D., Parniewski,P., Ohshima,K., Pandolfo,M.,Griffith,J.D. and Wells,R.D. (1999) Sticky DNA: self-association
5970 Nucleic Acids Research, 2004, Vol. 32, No. 19

properties of long GAA�TTC repeats in R�R�Y triplex structures fromFriedreich’s ataxia. Mol. Cell., 3, 465–475.
31. Mariappan,S.V.S., Catasti,P., Silks,L.A.,III, Bradbury,E.M. andGupta,G. (1999) The high-resolution structure of the triplex formed bythe GAA/TTC triplet repeat associated with Friedreich’s ataxia.J. Mol. Biol., 285, 2035–2052.
32. Potaman,V.N., Oussatcheva,E.A., Lyubchenko,Y.L.,Shlyakhtenko,L.S., Bidichandani,S.I., Ashizawa,T. and Sinden,R.R.(2004) Length-dependent structure formation in Friedreich ataxia(GAA)n
�(TTC)n repeats. Nucleic Acids Res., 32, 1224–1231.33. Grabczyk,E. and Usdin,K. (2000) Alleviating transcript insufficiency
caused by Friedreich’s ataxia triplet repeats. Nucleic Acids Res.,28, 4930–4937.
34. Kim,C., Snyder,R.O. and Wold,M.S. (1992) Binding properties ofreplication protein A from human and yeast cells. Mol. Cell. Biol.,12, 3050–3059.
35. Suen,I., Rhodes,J.N., Christy,M., McEwen,B., Gray,D.M. and Mitas,M.(1999) Structural properties of Friedreich’s ataxia d(GAA) repeats.Biochim. Biophys. Acta, 1444, 14–24.
36. LeProust,E.M., Pearson,C.E., Sinden,R.R. and Gao,X. (2000)Unexpected formation of parallel duplex in GAA and TTC trinucleotiderepeats of Friedreich’s ataxia. J. Mol. Biol., 302, 1063–1080.
37. Krasilnikova,M.M. and Mirkin,S.M. (2004) Replication stalling atFriedreich’s ataxia (GAA)n repeats in vivo. Mol. Cell. Biol., 24,2286–2295.
38. Burhans,W.C., Vassilev,L.T., Caddle,M.S., Heintz,N.H. andDePamphilis,M.L. (1990) Identification of an origin of bidirectionalDNA replication in mammalian chromosomes. Cell, 62, 955–965.
39. Anderson,S. and DePamphilis,M.L. (1979) Metabolism of Okazakifragments during simian virus 40 DNA replication. J. Biol. Chem.,254, 11495–11504.
40. Filla,A., De Michele,G., Cavalcanti,F., Pianese,L., Monticelli,A.,Campanella,G. and Cocozza,S. (1996) The relationship betweentrinucleotide (GAA) repeat length and clinical features in Friedreichataxia. Am. J. Hum. Genet., 59, 554–560.
41. Hashem,V.I., Klysik,E.A., Rosche,W.A. and Sinden,R.R. (2002)Instability of repeated DNAs during transformation in Escherichia coli.Mutat. Res., 502, 39–46.
42. Gomes-Pereira,M., Bidichandani,S.I. and Monckton,D.G. (2004)Analysis of unstable triplet repeats using small pool polymerase chainreaction. In Kohwi,Y. (ed.), Methods in Molecular Biology, HumanaPress, Totowa, NJ, USA, Vol. 227, pp. 61–76.
43. Jeffreys,A.J., Tamaki,K., Mac Leon,A., Monckton,D.G., Neil,D.L. andArmour,J.A.L. (1994) Complex gene conversion events in germlinemutation at human minisatellites. Nature Genet., 6, 136–145.
44. Sakamoto,N., Larsson,J.E., Iyer,R.R., Montermini,L., Pandolfo,M. andWells,R.D. (2001) GGA�TCC-interrupted triplets in long GAA�TTCrepeats inhibit the formation of triplex and sticky DNA structures,alleviate transcription inhibition, and reduce genetic instabilities.J. Biol. Chem., 276, 27178–27187.
45. Monckton,D.G., Wong,L.C., Ashizawa,T. and Caskey,T. (1995) Somaticmosaicism, germline expansions, germline reversions andintergenerational reductions in myotonic dystrophy males: smallpool PCR analyses. Hum. Mol. Genet., 4, 1–8.
46. Ashizawa,T., Mockton,D.G., Vaishnav,S., Patel,B.J., Voskova,A. andCaskey,T. (1996) Instability of the expanded (CTG)n repeats in themyotonin protein kinase gene in cultured lymphoblastoid cell linesfrom patients with myotonic dystrophy. Genomics, 36, 47–53.
47. Martorell,L., Monckton,D.G., Gamez,J., Johnson,K.J., Gich,I., Lopez deMunain,A. and Baiget,M. (1998) Progression of somatic CTG repeatlength heterogeneity in the blood cells of myotonic dystrophy patients.Hum. Mol. Genet., 7, 307–312.
48. Fortune,M.T., Vassilopoulos,C., Coolbaugh,M.I., Siciliano,M.J. andMonckton,D.G. (2000) Dramatic, expansion-biased, age-dependent,tissue-specific somatic mosaicism in a transgenic mouse model oftriplet repeat instability. Hum. Mol. Genet., 9, 439–445.
49. Gomes-Pereira,M., Fortune,M.T. and Monckton,D.G. (2001) Mousetissue culture models of unstable triplet repeats: in vitro selection forlarger alleles, mutational expansion bias and tissue specific, but noassociation with cell division rates. Hum. Mol. Genet., 10, 845–854.
50. Martorell,L., Monckton,D.G., Sanchez,A., Lopez de Munain,A. andBaiget,M. (2001) Frequency and stability of the myotonic dystrophytype 1 premutation. Neurology., 56, 328–335.
51. Iyer,R.R. and Wells,R.D. (1999) Expansion and deletion of triplet repeatsequences in Escherichia coli on the leading strand of DNAreplication. J. Biol. Chem., 274, 3865–3877.
52. Mornet,E., Chateau,C., Hirst,M.C., Thepot,F., Taillandier,A., Cibois,O.and Serre,J.L. (1996) Analysis of germline variation at the FMR1CGG repeat shows variation in the normal-premutated borderline range.Hum. Mol. Genet., 5, 821–825.
53. Clark,R.M., Dalgliesh,G.L., Endres,D., G�oomez,M., Taylor,J. andBidichandani,S.I. (2004) Expansion of GAA triplet repeats in the humangenome: unique origin of the FRDA mutation at the center of anAlu. Genomics, 83, 373–383.
Nucleic Acids Research, 2004, Vol. 32, No. 19 5971