preserving mitochondrial function prevents the proteasomal degradation of gtp cyclohydrolase i
TRANSCRIPT

Free Radical Biology and Medicine 53 (2012) 216–229
Contents lists available at SciVerse ScienceDirect
Free Radical Biology and Medicine
0891-58
http://d
n Corr
E-m1 Th
journal homepage: www.elsevier.com/locate/freeradbiomed
Original Contribution
Preserving mitochondrial function prevents the proteasomal degradationof GTP cyclohydrolase I
Shruti Sharma a,1, Xutong Sun a,1, Sanjiv Kumar a, Ruslan Rafikov a, Angela Aramburo b, Gokhan Kalkan b,Jing Tian a, Imran Rehmani a, Suphin Kallarackal a, Jeffrey R. Fineman b,c, Stephen M. Black a,n
a Program in Pulmonary Vascular Disease, Vascular Biology Center, Georgia Health Sciences University, Augusta, GA 30912, USAb Department of Pediatrics, University of California at San Francisco, San Francisco, CA 94143, USAc Cardiovascular Research Institute, University of California at San Francisco, San Francisco, CA 94143, USA
a r t i c l e i n f o
Article history:
Received 12 January 2011
Received in revised form
18 March 2012
Accepted 24 March 2012Available online 16 April 2012
Keywords:
Mitochondrial dysfunction
BH4
Hsp90
Hsp70
CHIP
Ubiquitination
Free radicals
49/$ - see front matter & 2012 Elsevier Inc. A
x.doi.org/10.1016/j.freeradbiomed.2012.03.01
esponding author. Fax: þ1 706 721 9799.
ail address: [email protected] (S.M. B
ese authors contributed equally to this work
a b s t r a c t
The development of pulmonary hypertension is a common accompaniment of congenital heart disease
(CHD) with increased pulmonary blood flow. Our recent evidence suggests that asymmetric dimethy-
larginine (ADMA)-induced mitochondrial dysfunction causes endothelial nitric oxide synthase (eNOS)
uncoupling secondary to a proteasome-dependent degradation of GTP cyclohydrolase I (GCH1) that
results in a decrease in the NOS cofactor tetrahydrobiopterin (BH4). Decreases in NO signaling are
thought to be an early hallmark of endothelial dysfunction. As L-carnitine plays an important role in
maintaining mitochondrial function, in this study we examined the protective mechanisms and the
therapeutic potential of L-carnitine on NO signaling in pulmonary arterial endothelial cells and in a
lamb model of CHD and increased pulmonary blood flow (Shunt). Acetyl-L-carnitine attenuated the
ADMA-mediated proteasomal degradation of GCH1. This preservation was associated with a decrease
in the association of GCH1 with Hsp70 and the C-terminus of Hsp70-interacting protein (CHIP) and a
decrease in its ubiquitination. This in turn prevented the decrease in BH4 levels induced by ADMA and
preserved NO signaling. Treatment of Shunt lambs with L-carnitine also reduced GCH1/CHIP interac-
tions, attenuated the ubiquitination and degradation of GCH1, and increased BH4 levels compared to
vehicle-treated Shunt lambs. The increases in BH4 were associated with decreased NOS uncoupling and
enhanced NO generation. Thus, we conclude that L-carnitine may have a therapeutic potential in the
treatment of pulmonary hypertension in children with CHD with increased pulmonary blood flow.
& 2012 Elsevier Inc. All rights reserved.
Introduction
Our recent studies have shown that mitochondrial dysfunction isan important contributing factor to the decrease in NO signaling inour lamb model of pulmonary hypertension with increased pul-monary blood flow (Shunt) [1]. Furthermore, we have also pre-viously shown that asymmetric dimethylarginine (ADMA), anendogenous competitive inhibitor of NO synthase (NOS), inducesmitochondrial dysfunction and decreases heat shock protein 90(Hsp90) chaperone activity through a reduction in cellular ATPlevels in cultured ovine pulmonary arterial endothelial cells(PAECs) [2]. Thus, preventing mitochondrial dysfunction may havepotential for therapeutic interventions to preserve NO signaling andprevent the development of the endothelial dysfunction associatedwith the development of a number of cardiovascular diseases.
ll rights reserved.
6
lack).
.
The mitochondrial metabolic pathways acyl-CoA and carnitineacetyltransferase have been shown to be of critical importance formaintaining normal mitochondrial function. Previous studies impli-cate compromised carnitine metabolism as an important mediatorin the development of mitochondrial injury [3]. Studies also showthat under conditions of metabolic stress, mitochondria accumulateacyl-CoA, which is normally maintained in homeostasis with carni-tine (reviewed in [4]). Recently, we also demonstrated a disruptionin carnitine homeostasis in these shunted lambs, in association withmitochondrial dysfunction and decreased endothelial (e) NOS/Hsp90 interactions, which contributed to eNOS uncoupling anddecreased NO signaling [1]. Although the mechanism underlyingthe increase in eNOS uncoupling in Shunt lambs is unclear, itappears to involve decreases in the NOS cofactor tetrahydrobiop-terin (BH4) [5]. Further, recent data suggest that GTP cyclohydrolaseI (GCH1), the first and rate-limiting enzyme in the de novo pathwayof BH4 biosynthesis, is a client protein for Hsp90 [6] and we haveshown that ADMA decreases Hsp90 activity in PAECs [2].
As carnitine plays an important role in maintaining mitochon-drial function and has been shown to have protective effects in

S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229 217
various pathologic conditions such as chronic renal failure [7], wehypothesized that supplementation with L-carnitine could pre-vent the mitochondrial dysfunction and loss of Hsp90 activityassociated with increased levels of ADMA, preserving BH4 levelsand NO signaling. Thus in this study, we examined the effects ofL-carnitine supplementation on ADMA-induced mitochondrialdysfunction in cultured PAECs and in our Shunt model. Our datademonstrate that acetyl-L-carnitine (ALC) abolishes the ADMA-induced decrease in GCH1 protein levels by attenuating theC-terminus of Hsp70-interacting protein (CHIP)-dependent ubi-quitination and proteasomal degradation of GCH1. This preventsthe ADMA-mediated decrease in BH4 levels, which, in turn,preserves NO signaling and attenuates eNOS uncoupling. We alsofound that the oral administration of carnitine effectivelyreversed the decrease in BH4 levels in Shunt lambs and thisresulted in increased NO bioavailability and decreased eNOSuncoupling. Taken together, our data indicate that L-carnitineabolishes ADMA-induced GCH1 degradation both in vitro andin vivo. Our studies suggest that L-carnitine may have therapeuticpotential in the treatment of pulmonary hypertension in childrenwith congenital heart disease (CHD) with increased pulmonaryblood flow.
Material and methods
Surgical preparations and experimental protocol
Twelve mixed-breed Western pregnant ewes (137–141 daygestation, term¼145 day) were operated on as previouslydescribed in detail [8,9]. Lambs received daily treatment withoral L-carnitine (n¼6; 100 mg/kg/day) or its vehicle (controllambs, n¼6) beginning no later than 12 h after delivery. Fourweeks after spontaneous delivery, just before sacrifice, thepatency of the vascular graft was confirmed by inspection andchanges in oxygen saturation. At the end of the protocol, all lambswere killed with a lethal injection of sodium pentobarbitalfollowed by bilateral thoracotomy as described in the NIH Guide-
lines for the Care and Use of Laboratory Animals. All protocols andprocedures were approved by the Committees on AnimalResearch at the University of California at San Francisco andGeorgia Health Sciences University.
Cell culture and treatment
Primary cultures of ovine PAECs were isolated and cultured asdescribed previously [10]. Before any treatments, PAECs wereserum-starved for 2 h and then pretreated with 100 mM ALC(Sigma, St. Louis, MO, USA) for 2 h, followed by incubation with5 mM ADMA (Sigma) for an additional 2 h. For some studies, cellswere treated with the proteasome inhibitor lactacystin (20 mM;Calbiochem, La Jolla, CA, USA), vehicle (0.1% dimethyl sulfoxide(DMSO)), PEGylated superoxide dismutase (PEG-SOD; 100 U/ml),or PEG-catalase (100 U/ml).
Determination of mitochondrial reactive oxygen species (ROS) levels
MitoSOX red (Molecular Probes), a fluorogenic dye for selec-tive detection of ROS levels in the mitochondria of live cells, wasused. Briefly, cells were washed with fresh medium and thenincubated in medium containing MitoSOX red (2 mM), for 30 minat 37 1C in the dark, and then subjected to fluorescence micro-scopy at an excitation of 510 nm and an emission at 580 nm. AnOlympus IX51 microscope equipped with a CCD camera (HamamatsuPhotonics) was used for acquisition of fluorescence images. Theaverage fluorescence intensities (to correct for differences in cell
number) were quantified using ImagePro Plus version 5.0 imagingsoftware (Media Cybernetics).
Analysis of mitochondrial membrane potential
Mitochondrial membrane potential was analyzed as previouslydescribed using the lipophilic cation 5,50,6,60-tetrachloro-1,10,3,30-tetraethylbenzimidazolylcarbocyanine iodide [11,12], which fluor-esces red in its multimeric form in healthy mitochondria and is theactive reagent in the DePsipher mitochondrial potential assay kit(Trevigen, Gaithersburg, MD, USA). PAECs were seeded onto 24-well plates and treated in the presence or absence of ALC (100 mM,2 h), followed ADMA exposure (5 mM, 2 h). DePsipher reagent(10 mg/ml) was then added and the samples were incubated fora further 20 min. After an additional wash with Dulbecco’sphosphate-buffered saline (DPBS), the cytosolic monomer (green)form was observed and quantified by fluorescence microscopy at530 nm.
Proteasome activity assay
Proteasome activity in PAECs was estimated using a protea-some activity assay kit (Chemicon, Billerica, MA, USA) accordingto the manufacturer’s guidelines. The assay is based on detectionof the fluorophore 7-amino-4-methylcoumarin (AMC) after clea-vage from the labeled substrate, LLVY–AMC. The free AMCfluorescence was quantified using an excitation at 380 nm andan emission at 460 nm using a fluorescence plate reader (ThermoFisher).
Determination of cellular ATP levels
ATP levels were estimated using a rapid, quantitative, biolu-minescence determination, Enzylight ATP Assay Kit (BioAssaySystems, Hayward, CA, USA), according to the manufacturer’sguidelines. ATP is consumed and light is emitted when fireflyluciferase catalyzes the oxidation of luciferin. The amount of lightemitted during the reaction is proportional to the availability ofATP. Luminescence was determined using a Fluoroscan Ascent FLplate luminometer (ThermoElectron Corp.).
Expression of a dominant negative mutant of Hsp90
The day before transfection, 1.5�105 cells were seeded ineach well of a six-well plate and fresh Dulbecco’s modified Eagle’smedium (DMEM) containing serum and antibiotics was added. Onthe day of transfection, the medium was changed to one withoutantibiotics. The cells were transiently transfected with a domi-nant negative mutant of Hsp90 or the parental vector (pcDNA3)using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), accord-ing to the manufacturer’s instructions. The Hsp90 dominantnegative mutant exerts its affect through an inability to bindATP [13]. The plasmid was obtained commercially (AddgeneCambridge, MA, USA; plasmid 22480).
Hsp70 and CHIP RNA interference assays
PAECs were transfected with the appropriate small interferingRNA (siRNA) using HiPerFect transfection reagent (Qiagen) asdescribed previously [14]. Briefly, the day before transfection,1.5�105 cells were seeded in each well of a six-well plate andfresh DMEM containing serum and antibiotics was added. On theday of transfection, the medium was changed to one withoutantibiotics. For each well, 6 ml of a 10 mM siRNA stock of Hsp70(Santa Cruz Biotechnology), CHIP (Santa Cruz Biotechnology), orthe control (a scrambled siRNA with no known homology to any

S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229218
human gene) was diluted into 100 ml of DMEM without serum(to give a final siRNA concentration of 30 nM). To this was added12 ml of HiPerFect transfection reagent. The solution was vor-texed, incubated for 10 min at room temperature, and addeddropwise to the cells. As the siRNA’s utilized were designedagainst human mRNA sequences their ability to silence Hsp70and CHIP was validated by Western blot 48 h after transfection(Figs. 5A and 7A, respectively).
Measurement of GCH1 activity
Cells were washed in PBS and lysed using 50 mM Tris–HClbuffer (pH 7.4) containing 1 mM MgCl2, 100 mM KCl, 1 mM EDTA,1 mM dithiothreitol (DTT), 1 mM phenylmethanesulfonyl fluor-ide, and protease inhibitor cocktail (Sigma). Aliquots (100 ml)were assayed for activity as described previously [15,16]. Briefly,cell lysates were incubated at 37 1C with 1 mM GTP and 50 mg/mlbovine serum albumin, in a total volume of 200 ml, for either 0 or60 min. Reactions were terminated by rapid cooling on water icefollowed by the addition of 8 ml 5 M HCl. The reaction mixtureswere then oxidized with 2% KI/1% I2 for 60 min in the darkfollowed by the addition of 2% ascorbic acid. Samples were thenincubated with 5 U of alkaline phosphatase at pH 8.0 for 60 min at37 1C and the reaction was stopped by acidification using 5 M HCl.The neopterin content of each sample was then analyzed by HPLC(Shimadzu, Japan) using a C18 reverse-phase column elutedwith aqueous 0.05% trifluoroacetic acid at a flow of 0.5 ml/min.Neopterin was detected by fluorescence with excitation at350 nm and emission at 440 nm; authentic neopterin was usedas standard. The protein content of samples was determinedusing the BCA assay and activity was expressed as the amountof neopterin formed (pmol/mg protein).
Quantification of tetrahydrobiopterin levels by high-performance
liquid chromatography
BH4 levels were determined using the differential iodine oxida-tion method as we have previously published [17,18]. Briefly, PAECswere homogenized in an extraction buffer (50 mM Tris–HCl, 1 mMEDTA, 1 mM DTT; pH 7.4) and divided into equal volumes betweentwo centrifuge tubes containing either 1 M NaOH (base) or 1 MH3PO4 (acid). A solution of 1% iodine in 2% potassium iodide wasadded to each tube and the samples were incubated in the dark atroom temperature for 90 min. H3PO4 (1 M) was then added to thetubes containing NaOH. In both the acidic and the basic samples theexcess iodine was removed by the addition of 2% ascorbic acid andthen centrifuged at 15,000g for 10 min to remove the precipitatedprotein. Each sample was then analyzed by HPLC using a SpherisorbODS-1 column (Waters, Franklin, MA, USA) and fluorescencedetection (350 nm excitation, 450 nm emission). The area underthe curve obtained by oxidation under acidic conditions representstotal biopterins, i.e., the sum of BH4, BH2, and free biopterin(BH4þBH2þB). In contrast, alkaline oxidation measures BH2 andfree biopterin (BH2þB). Thus, BH4 levels were calculated as thedifference between the values obtained under acidic and alkalineconditions. A standard curve of freshly made tetrahydrobiopterin(Sigma) was also included to allow the area under each curve to beconverted into a concentration. BH4 levels were then normalized tototal protein using the Bradford assay and expressed as femtomolesper microgram of protein.
Western blot analysis
For the Western blot analyses, 20 mg of protein prepared fromtotal cell lysates was separated on 4%–20% SDS-polyacrylamidegels and transferred to polyvinylidene difluoride (PVDF)
membranes. Immunoblotting was carried out using the appro-priate antibodies in Tris-base buffered saline with 0.1% Tween 20and 5% nonfat dried milk. After being washed, the membraneswere probed with horseradish peroxidase-conjugated goatantiserum to rabbit or mouse. Reactive bands were visualizedand quantified as mentioned above. The generation of theanti-GCH1 antibody has been previously described [14]. Theanti-SOD2 antibody and the anti-UCP2 antibody were purchasedfrom Lifespan Biosciences (Seattle, WA, USA). Loading was nor-malized by reprobing the membranes with an antibody specific tob-actin.
Immunoprecipitation analysis
For the in vitro studies, PAECs were harvested in lysis buffersupplemented with protease inhibitor cocktail as we havedescribed [1]. Cell and tissue lysates were then clarified bycentrifugation at 20,000g (20 min at 4 1C), the protein concentra-tions were determined, and 1 mg of each lysate was incubatedovernight at 4 1C with anti-Hsp90 (BD Transduction Laboratories,San Jose, CA, USA), anti-Hsp70 (Stressgen, Ann Arbor, MI, USA), oranti-CHIP (ABR Affinity BioReagents, Golden, CO, USA) antibodies(2 mg of each antibody). Immunocomplexes were adsorbed to40 ml of protein G plus protein A agarose (Calbiochem) and thenincubated for 2 h at 4 1C. The immune complexes were precipi-tated by centrifugation, washed three times with lysis buffer,boiled in SDS sample buffer, and subjected to SDS-PAGE and thentransferred to PVDF membranes. The membranes were blockedwith 5% nonfat dry milk in Tris-buffered saline containing 0.1%Tween. A specific antiserum we have raised against GCH1 protein[5] was then added and incubated overnight at 4 1C. After beingwashed, the membranes were probed with horseradish perox-idase-conjugated goat antiserum to rabbit. Reactive bands werevisualized using chemiluminescence (Super Signal West Femto;Pierce) on a Kodak 440CF image station (New Haven, CT, USA).Bands were quantified using Kodak Image Station software(Kodak 1D 3.6). The efficiency of each immunoprecipitation wasnormalized by reprobing the membranes with the appropriateimmunoprecipitation antibody (Hsp90, Hsp70, or CHIP). In somecases, immunoprecipitated lysates were run on duplicate blots,with one blot probed for GCH1 and the other probed for CHIP tonormalize the immunoprecipitation (Fig. 6A, Hsp70 siRNA, GCH1/CHIP interaction).
Real-time quantitative (q) RT-PCR
For the in vivo studies using peripheral lung tissue, qRT-PCR usingSYBR Green I dye for specific detection of double-stranded DNA wasemployed. Briefly, total RNA was extracted using the RNeasy kit(Qiagen), and 1 mg of total RNA was reverse-transcribed using theQuantiTect reverse transcription kit (Qiagen). Primers for GCH1 andb-actin were designed by Primer 3. The sequences were GCH1forward, 50-TCTTCACCAAGGGCTACCAG-30, reverse, 50-GGACCTTTC-CAACAAATGGA-30; b-actin forward, 50-CTCTTCCAGCCTTCCTTCCT-30,reverse, 50-GGGCAGTGATCTCTTTCTGC-30. Real-time quantitative PCRwas conducted using an Mx4000 Multiplex Quantitative PCR System(Stratagene), using 2 ml of RT product, 12.5 ml of QuantiTect SYBRGreen PCR master mix (Qiagen), and primers (400 nM) in a totalvolume of 25 ml. The following thermocycling conditions wereemployed: 95 1C for 10 min, followed by 95 1C for 30 s, 55 1C for60 s, and 72 1C for 30 s for 45 cycles. The threshold cycles (Ct) of aserially diluted control sample were plotted to generate a standardcurve. Concentration of each sample was calculated by plotting its Ct
on the standard curve and then normalized using the mRNA levels ofb-actin as an internal control.

S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229 219
Detection of ubiquitinated GTP cyclohydrolase I
Ubiquitinated GCH1 from both PAECs and peripheral lung wasenriched using affinity beads comprising a GST-fusion proteincontaining a ubiquitin-associated sequence conjugated toglutathione–agarose using a ubiquitinated protein enrichmentkit (Calbiochem) according to the manufacturer’s guidelines. Theresulting samples were then separated by SDS-PAGE and analyzedby Western blotting using our specific antiserum raised againstGCH1 [17].
Exposure of pulmonary endothelial cells to shear stress
Laminar shear stress was applied using a cone-plate visc-ometer that accepts six-well tissue culture plates, as describedpreviously [19,20]. Using this apparatus we exposed PAECs to alaminar flow rate of 20 dyn/cm2 to represent physiologicallevels of laminar shear stress in the major human arteries, whichis in the range of 5–20 dyn/cm2 with localized increases to30–100 dyn/cm2.
Measurement of NOx levels
NO generated by PAECs was measured using an NO-sensitiveelectrode with a 2-mm-diameter tip (ISO-NOP sensor; WPI)connected to an NO meter (ISO-NO Mark II; WPI) as describedpreviously [21]. To measure NO levels in peripheral lung tissue,lysates were initially treated with cold ethanol to remove proteinsand then we utilized a Sievers 280i nitric oxide analyzer (GEHealthcare) to determine NOx levels as we have described [1].Results were analyzed by measuring the area under the curve ofthe chemiluminescence signal using the Liquid software (GEHealthcare).
Measurement of superoxide levels
To detect superoxide generation in intact cells and peripherallung tissue, electron paramagnetic resonance (EPR) was carriedout using the spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine �HCl (CMH HCl; Alexis Biochemicals, SanDiego, CA, USA) as we have described previously [22,23]. CMH HClwas chosen based on a prior publication in which it was shown toreact more rapidly with superoxide and produce a more stableadduct than spin traps [24]. For peripheral lungs, approximately0.1 g of tissue was sectioned and powdered from fresh-frozenbiopsies of lung tissue and immediately immersed, while stillfrozen, in 100 ml of EPR buffer (PBS supplemented with 5 mMdiethyldithiocarbamate (Sigma–Aldrich) and 25 mM desferrioxa-mine (Sigma–Aldrich)). In addition, to determine the relativecontribution of uncoupled NOS activity to superoxide production,equivalent samples were preincubated in 100 mM S-ethyl-N-[4-(trifluoromethyl)phenyl]isothiourea (ETU; Sigma), a nonspecificinhibitor of NOS isoforms. All samples were then incubated for30 min on ice. Sample volumes were then adjusted with EPRbuffer and 20 mg/ml CMH HCl to achieve a final CMH HClconcentration of 5 mg/ml. Samples were further incubated for30 min on ice and then centrifuged at 14,000g for 15 min at 4 1C.For cell culture EPR, cells were plated in six-well plates and 24 hbefore experiment, standard medium was replaced with L-argi-nine and phenol red-free DMEM (Athena Enzyme System, Balti-more, MD, USA). PAECs were serum-starved for 2 h, followed bypretreatment with or without ALC (100 mM, 2 h), in the presenceor absence of the isothiourea ETU (100 mM, 30 min, to estimatedNOS-derived superoxide). Ethyl-substituted isothiourea is apotent inhibitor of NOS with little isoform selectivity, as pre-viously described [25]. The cells were then treated with ADMA
(5 mM, 2 h) followed by exposure or not to shear stress (20 dyn/cm2,15 min). In the final hour of incubation with ADMA, 20 ml of spintrap stock solution consisting of CMH (20 mM in DPBS þ25 mMdesferrioxamine; Calbiochem) and 5 mM diethyldithiocarbamate(Alexis Biochemicals, Lausen, Switzerland) plus 2 ml DMSO wasadded to each well. Upon completion of incubation, adherent cellswere trypsinized and pelleted at 500g. The cell pellet was washedand suspended in a final volume of 40 ml DPBS (þdesferrioxamine,diethyldithiocarbamate). Thirty-five microliters of tissue super-natant or cell suspension was loaded into a 50-ml capillary tubeand analyzed with a MiniScope MS200 ESR (Magnettech, Berlin,Germany) at a microwave power of 10 mW, modulation ampli-tude of 2000 mG. The rest of the sample was analyzed for proteincontent using the Bradford assay (Bio-Rad). EPR spectra wereanalyzed using Analysis version 2.02 software (Magnettech),whereby the EPR maximum and minimum spectral amplitudesfor the CM � superoxide spin trap product waveform werequantified. NOS-derived superoxide levels were determined bysubtracting the superoxide values in the presence of ETU from thesuperoxide values in the absence of ETU. To convert cell andtissue EPR waveforms into units of superoxide we used 1 mUof xanthine oxidase to generate 1 nM/min of superoxide over a60-min period. Using this we found a linear correlation betweenthe waveform amplitudes generated using CMH in cells andtissues and the generation of superoxide by xanthine oxidase.Using this standard curve we were able to convert waveformamplitudes into nmol of superoxide produced/min/mg protein ineach reaction condition.
Mitochondrial isolation for superoxide estimation
Mitochondria were isolated from 2�107 cells using the Piercemitochondria isolation kit, as described previously [2]. Then, 2 mgof isolated mitochondrial protein was adjusted with EPR bufferplus 50 ml of spin trap stock solution consisting of CMH (20 mM inDPBSþ25 mM desferrioxamine; Calbiochem) to achieve equalprotein content and CMH concentration. The samples were thenincubated for 30 min on ice and then pelleted. The supernatant(35 ml) was then loaded into a 50-ml capillary tube and analyzedfor superoxide generation as described above.
Statistical analysis
Statistical analysis was performed using GraphPad Prismversion 4.01 for Windows (GraphPad Software). The means7SEMwere calculated for all samples. Values lying 42 SD from themean were not used for further analysis. Statistical significancewas determined either by the unpaired t test (for two groups) orby ANOVA (for three or more groups) with Newman–Keuls posthoc testing. A value of Po0.05 was considered significant.
Results
ALC prevents ADMA-mediated mitochondrial dysfunction and GCH1
degradation in pulmonary arterial endothelial cells
To examine the effect of ALC on mitochondrial function, weinitially carried out a dose–response analysis by pretreatingPAECs with ALC (0–1 mM, 2 h) followed by exposure to ADMA(5 mM, 2 h). Our data indicate that ADMA exposure produces asignificant increase in mitochondrial ROS generation (Fig. 1A),indicating an increase in oxidative stress within the mitochondria,and this is dose-dependently attenuated by ALC supplementationwith a maximum effect observed at 100 mM ALC. The mitochon-drial ROS is probably superoxide as the addition of PEG-SOD

Fig. 1. Acetyl-L-carnitine prevents the ADMA-mediated mitochondrial dysfunction and decreases in GTP cyclohydrolase I in pulmonary arterial endothelial cells. PAECs
were pretreated with ALC (0–1 mM) for 2 h and then exposed to ADMA (5 mM, 2 h). The effects on mitochondrial function were then determined by examining the effect
on mitochondrial ROS levels, using MitoSOX red fluorescence. Representative images are shown. (A) ADMA caused a significant increase in MitoSOX fluorescence that was
dose-dependently decreased in the presence of ALC or when PAECs were preincubated with PEG-SOD (100 U/ml). (B) To confirm the MitoSOX data, mitochondrial fractions
were prepared and superoxide levels were determined by EPR. ADMA increased mitochondrial superoxide levels in PAECs and ALC abolished this increase. (C) ADMA also
caused a disruption of the mitochondrial membrane potential as estimated using the DePsipher compound and fluorescence microscopy. ALC pretreatment preserved the
mitochondrial membrane potential. Western blot analysis also revealed that ADMA (D) decreased the levels of SOD2 and (E) increased the levels of UCP2. Again ALC
pretreatment prevented these changes. (F) Cumulatively, the mitochondrial dysfunction induced by ADMA led to a significant decrease in cellular ATP levels. The decrease
in ATP levels was prevented by ALC. In addition (G) protein extracts and (H) mRNA were prepared from cells were treated with ALC (100 mM) and ADMA and subjected to
Western blot analysis and qRT-PCR, respectively. A representative image is shown of the Western blot using an antibody specific for GCH1 and loading normalized by
reprobing the membranes with an antibody specific to b-actin. ALC prevented the ADMA-mediated decrease in GCH1 protein levels. ADMA either alone or in combination
with ALC had no effect on GCH1 mRNA levels, although ALC alone significantly increased GCH1 mRNA levels. (I) Cells were incubated with ADMA for 2 h and then ALC was
added (100 mM, 0–2 h post-ADMA exposure). Postexposure with ALC preserved GCH1 protein levels. (J) Further, increasing the redox scavenging potential of the cells by
adding PEG-SOD (100 U/ml) or PEG-catalase (100 U/ml) prevented the ADMA-mediated decrease in GCH1 protein levels. Values are means7SEM, n¼8–10; *Po0.05 vs
untreated, yPo0.05 vs ADMA alone, zPo0.05 vs previous dose.
S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229220
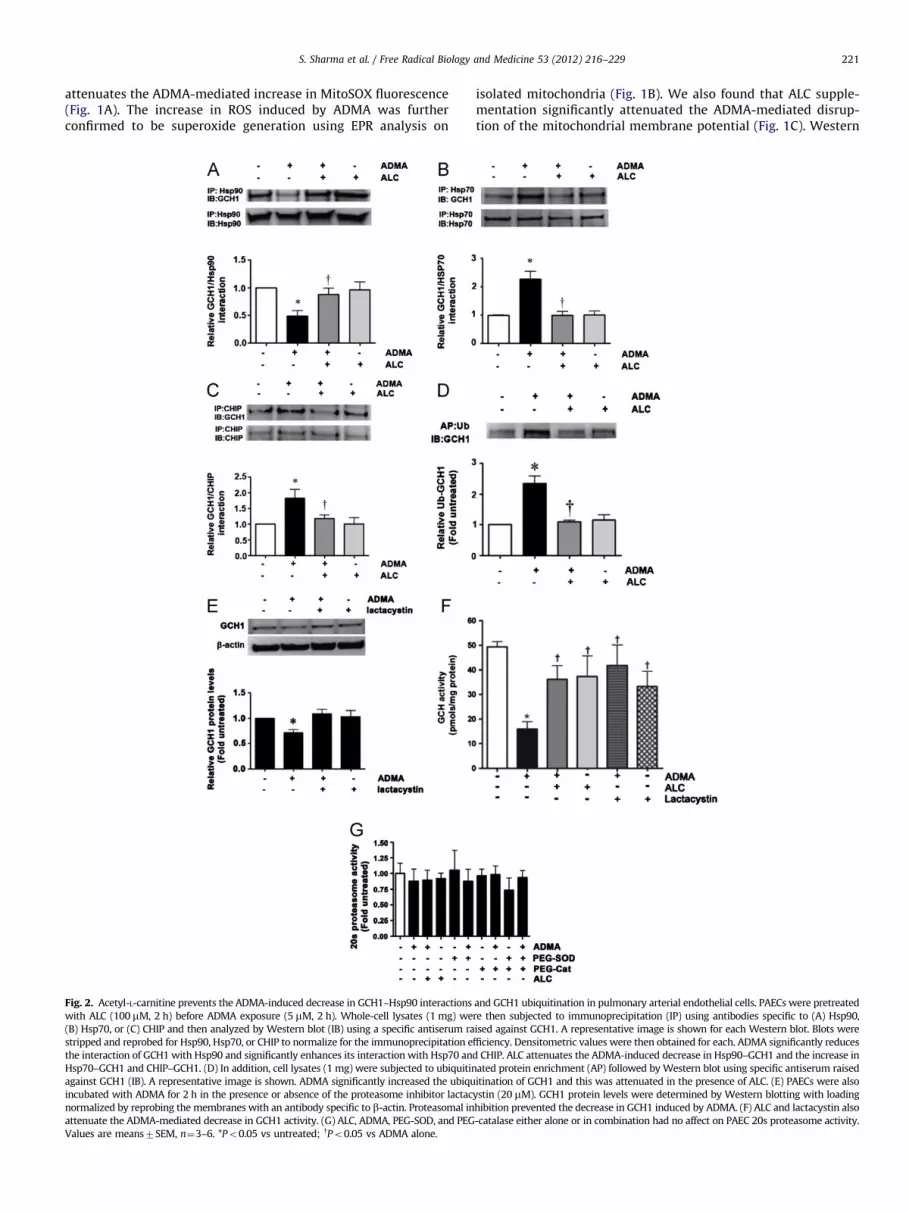
S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229 221
attenuates the ADMA-mediated increase in MitoSOX fluorescence(Fig. 1A). The increase in ROS induced by ADMA was furtherconfirmed to be superoxide generation using EPR analysis on
Fig. 2. Acetyl-L-carnitine prevents the ADMA-induced decrease in GCH1–Hsp90 interactions
with ALC (100 mM, 2 h) before ADMA exposure (5 mM, 2 h). Whole-cell lysates (1 mg) we
(B) Hsp70, or (C) CHIP and then analyzed by Western blot (IB) using a specific antiserum ra
stripped and reprobed for Hsp90, Hsp70, or CHIP to normalize for the immunoprecipitation e
the interaction of GCH1 with Hsp90 and significantly enhances its interaction with Hsp70 an
Hsp70–GCH1 and CHIP–GCH1. (D) In addition, cell lysates (1 mg) were subjected to ubiquitin
against GCH1 (IB). A representative image is shown. ADMA significantly increased the ubiqu
incubated with ADMA for 2 h in the presence or absence of the proteasome inhibitor lactac
normalized by reprobing the membranes with an antibody specific to b-actin. Proteasomal in
attenuate the ADMA-mediated decrease in GCH1 activity. (G) ALC, ADMA, PEG-SOD, and PEG
Values are means7SEM, n¼3–6. *Po0.05 vs untreated; yPo0.05 vs ADMA alone.
isolated mitochondria (Fig. 1B). We also found that ALC supple-mentation significantly attenuated the ADMA-mediated disrup-tion of the mitochondrial membrane potential (Fig. 1C). Western
and GCH1 ubiquitination in pulmonary arterial endothelial cells. PAECs were pretreated
re then subjected to immunoprecipitation (IP) using antibodies specific to (A) Hsp90,
ised against GCH1. A representative image is shown for each Western blot. Blots were
fficiency. Densitometric values were then obtained for each. ADMA significantly reduces
d CHIP. ALC attenuates the ADMA-induced decrease in Hsp90–GCH1 and the increase in
ated protein enrichment (AP) followed by Western blot using specific antiserum raised
itination of GCH1 and this was attenuated in the presence of ALC. (E) PAECs were also
ystin (20 mM). GCH1 protein levels were determined by Western blotting with loading
hibition prevented the decrease in GCH1 induced by ADMA. (F) ALC and lactacystin also
-catalase either alone or in combination had no affect on PAEC 20s proteasome activity.
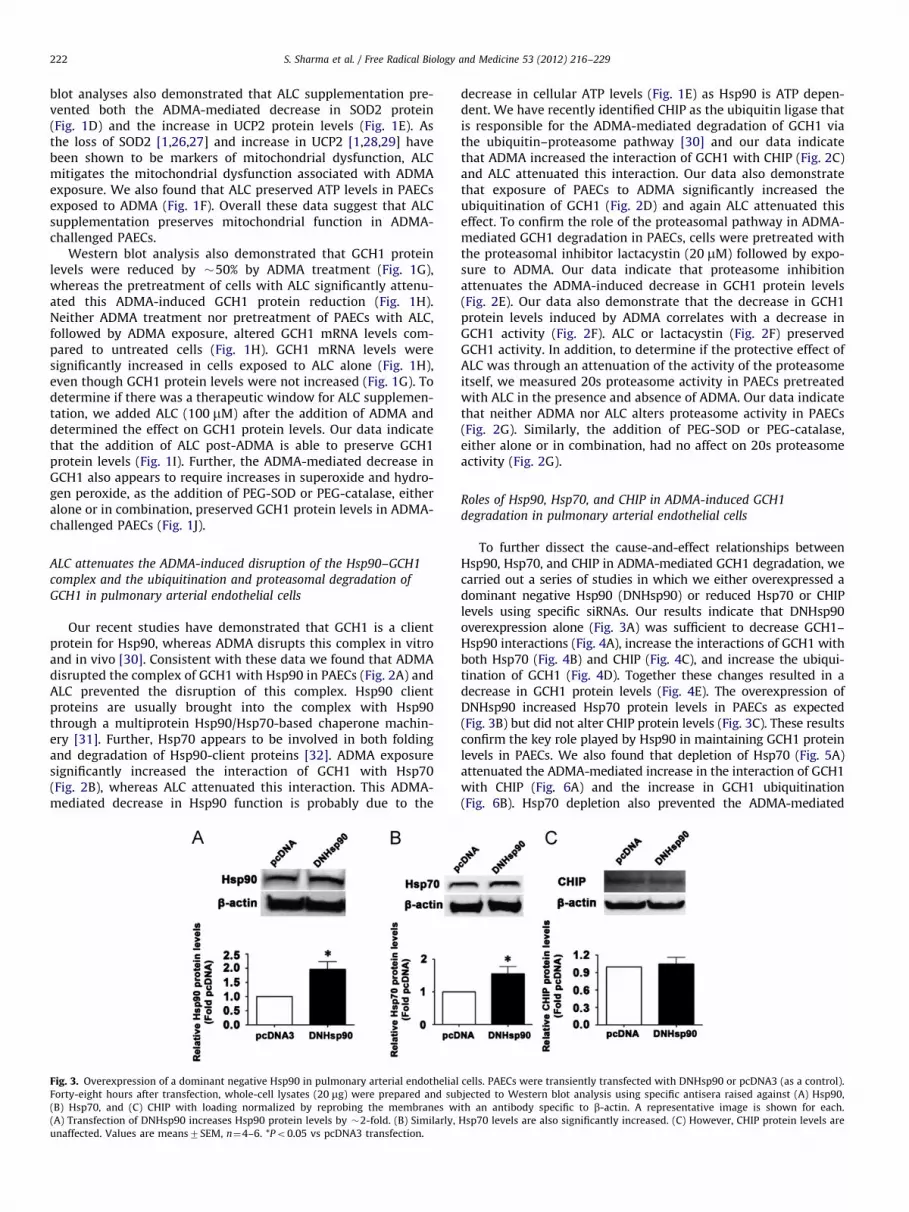
S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229222
blot analyses also demonstrated that ALC supplementation pre-vented both the ADMA-mediated decrease in SOD2 protein(Fig. 1D) and the increase in UCP2 protein levels (Fig. 1E). Asthe loss of SOD2 [1,26,27] and increase in UCP2 [1,28,29] havebeen shown to be markers of mitochondrial dysfunction, ALCmitigates the mitochondrial dysfunction associated with ADMAexposure. We also found that ALC preserved ATP levels in PAECsexposed to ADMA (Fig. 1F). Overall these data suggest that ALCsupplementation preserves mitochondrial function in ADMA-challenged PAECs.
Western blot analysis also demonstrated that GCH1 proteinlevels were reduced by �50% by ADMA treatment (Fig. 1G),whereas the pretreatment of cells with ALC significantly attenu-ated this ADMA-induced GCH1 protein reduction (Fig. 1H).Neither ADMA treatment nor pretreatment of PAECs with ALC,followed by ADMA exposure, altered GCH1 mRNA levels com-pared to untreated cells (Fig. 1H). GCH1 mRNA levels weresignificantly increased in cells exposed to ALC alone (Fig. 1H),even though GCH1 protein levels were not increased (Fig. 1G). Todetermine if there was a therapeutic window for ALC supplemen-tation, we added ALC (100 mM) after the addition of ADMA anddetermined the effect on GCH1 protein levels. Our data indicatethat the addition of ALC post-ADMA is able to preserve GCH1protein levels (Fig. 1I). Further, the ADMA-mediated decrease inGCH1 also appears to require increases in superoxide and hydro-gen peroxide, as the addition of PEG-SOD or PEG-catalase, eitheralone or in combination, preserved GCH1 protein levels in ADMA-challenged PAECs (Fig. 1J).
ALC attenuates the ADMA-induced disruption of the Hsp90–GCH1
complex and the ubiquitination and proteasomal degradation of
GCH1 in pulmonary arterial endothelial cells
Our recent studies have demonstrated that GCH1 is a clientprotein for Hsp90, whereas ADMA disrupts this complex in vitroand in vivo [30]. Consistent with these data we found that ADMAdisrupted the complex of GCH1 with Hsp90 in PAECs (Fig. 2A) andALC prevented the disruption of this complex. Hsp90 clientproteins are usually brought into the complex with Hsp90through a multiprotein Hsp90/Hsp70-based chaperone machin-ery [31]. Further, Hsp70 appears to be involved in both foldingand degradation of Hsp90-client proteins [32]. ADMA exposuresignificantly increased the interaction of GCH1 with Hsp70(Fig. 2B), whereas ALC attenuated this interaction. This ADMA-mediated decrease in Hsp90 function is probably due to the
Fig. 3. Overexpression of a dominant negative Hsp90 in pulmonary arterial endothelial
Forty-eight hours after transfection, whole-cell lysates (20 mg) were prepared and sub
(B) Hsp70, and (C) CHIP with loading normalized by reprobing the membranes w
(A) Transfection of DNHsp90 increases Hsp90 protein levels by �2-fold. (B) Similarly,
unaffected. Values are means7SEM, n¼4–6. *Po0.05 vs pcDNA3 transfection.
decrease in cellular ATP levels (Fig. 1E) as Hsp90 is ATP depen-dent. We have recently identified CHIP as the ubiquitin ligase thatis responsible for the ADMA-mediated degradation of GCH1 viathe ubiquitin–proteasome pathway [30] and our data indicatethat ADMA increased the interaction of GCH1 with CHIP (Fig. 2C)and ALC attenuated this interaction. Our data also demonstratethat exposure of PAECs to ADMA significantly increased theubiquitination of GCH1 (Fig. 2D) and again ALC attenuated thiseffect. To confirm the role of the proteasomal pathway in ADMA-mediated GCH1 degradation in PAECs, cells were pretreated withthe proteasomal inhibitor lactacystin (20 mM) followed by expo-sure to ADMA. Our data indicate that proteasome inhibitionattenuates the ADMA-induced decrease in GCH1 protein levels(Fig. 2E). Our data also demonstrate that the decrease in GCH1protein levels induced by ADMA correlates with a decrease inGCH1 activity (Fig. 2F). ALC or lactacystin (Fig. 2F) preservedGCH1 activity. In addition, to determine if the protective effect ofALC was through an attenuation of the activity of the proteasomeitself, we measured 20s proteasome activity in PAECs pretreatedwith ALC in the presence and absence of ADMA. Our data indicatethat neither ADMA nor ALC alters proteasome activity in PAECs(Fig. 2G). Similarly, the addition of PEG-SOD or PEG-catalase,either alone or in combination, had no affect on 20s proteasomeactivity (Fig. 2G).
Roles of Hsp90, Hsp70, and CHIP in ADMA-induced GCH1
degradation in pulmonary arterial endothelial cells
To further dissect the cause-and-effect relationships betweenHsp90, Hsp70, and CHIP in ADMA-mediated GCH1 degradation, wecarried out a series of studies in which we either overexpressed adominant negative Hsp90 (DNHsp90) or reduced Hsp70 or CHIPlevels using specific siRNAs. Our results indicate that DNHsp90overexpression alone (Fig. 3A) was sufficient to decrease GCH1–Hsp90 interactions (Fig. 4A), increase the interactions of GCH1 withboth Hsp70 (Fig. 4B) and CHIP (Fig. 4C), and increase the ubiqui-tination of GCH1 (Fig. 4D). Together these changes resulted in adecrease in GCH1 protein levels (Fig. 4E). The overexpression ofDNHsp90 increased Hsp70 protein levels in PAECs as expected(Fig. 3B) but did not alter CHIP protein levels (Fig. 3C). These resultsconfirm the key role played by Hsp90 in maintaining GCH1 proteinlevels in PAECs. We also found that depletion of Hsp70 (Fig. 5A)attenuated the ADMA-mediated increase in the interaction of GCH1with CHIP (Fig. 6A) and the increase in GCH1 ubiquitination(Fig. 6B). Hsp70 depletion also prevented the ADMA-mediated
cells. PAECs were transiently transfected with DNHsp90 or pcDNA3 (as a control).
jected to Western blot analysis using specific antisera raised against (A) Hsp90,
ith an antibody specific to b-actin. A representative image is shown for each.
Hsp70 levels are also significantly increased. (C) However, CHIP protein levels are

Fig. 4. Inhibition of Hsp90 alone is sufficient to stimulate GCH1 ubiquitination and degradation in pulmonary arterial endothelial cells. PAECs were transiently transfected
with DNHsp90 or pcDNA3 (as a control). Forty-eight hours after transfection, whole-cell lysates (1 mg) were subjected to immunoprecipitation (IP) using antibodies
specific to (A) Hsp90, (B) Hsp70, or (C) CHIP and then analyzed by Western blot (IB) using an anti-GCH1 antibody. Blots were stripped and reprobed with the appropriate IP
antibody to normalize for immunoprecipitation efficiency. Representative images are shown for each. The overexpression of DNHsp90 (A) decreases GCH1–Hsp90
interactions but increases both (B) GCH1–Hsp70 and (C) GCH1–CHIP interactions. (D) Ubiquitinated protein enrichment (AP) followed by Western blot with an anti-GCH1
antibody was also used to measure GCH1 ubiquitination (IB). A representative image is shown. There is a significant increase in ubiquitinated GCH1. (E) Whole-cell lysates
(20 mg) were also subjected to Western blot analysis using a specific antiserum raised against GCH1 with loading normalized by reprobing the membranes with an
antibody specific to b-actin. A representative image is shown along with the densitometric analysis indicating that DNHsp90 overexpression significantly decreased GCH1
protein levels. Values are means7SEM, n¼4–6. *Po0.05 vs pcDNA3 transfection.
Fig. 5. Depletion of Hsp70 in pulmonary arterial endothelial cells. PAECs were transiently transfected with a siRNA for Hsp70 or a control siRNA. Forty-eight hours after
transfection, whole-cell lysates (20 mg) were prepared and subjected to Western blot analysis using specific antisera raised against (A) Hsp70, (B) Hsp90, and (C) CHIP with
loading normalized by reprobing the membranes with an antibody specific to b-actin. A representative image is shown for each. (A) Transfection with the Hsp70 siRNA
reduces Hsp70 protein levels by �50% without altering (B) Hsp90 or (C) CHIP protein levels. Values are means7SEM, n¼6. *Po0.05 vs control siRNA.
S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229 223
decrease in GCH1 protein levels (Fig. 6C). Depletion of Hsp70 didnot alter Hsp90 (Fig. 5B) or CHIP protein levels (Fig. 5C). Further, ourdata indicate that the depletion of CHIP (Fig. 7A) does not preventthe ADMA-mediated increase in Hsp70–GCH1 interactions (Fig. 8A).However, depletion of CHIP attenuated both the increase in GCH1ubiquitination (Fig. 8B) and the ADMA-mediated decrease in GCH1protein levels (Fig. 8C). Depletion of CHIP did not alter Hsp90
(Fig. 7B) or Hsp70 protein levels (Fig. 7C). Collectively, thesefindings delineate the temporal series of events induced by ADMAthat results in GCH1 ubiquitination and degradation: ADMA initi-ally causes a reduction in Hsp90 activity that reduces its interactionwith GCH1; Hsp70 is then recruited and this allows CHIP to interactwith the complex. GCH1 is then ubiquitinated via CHIP and targetedfor proteasomal degradation.
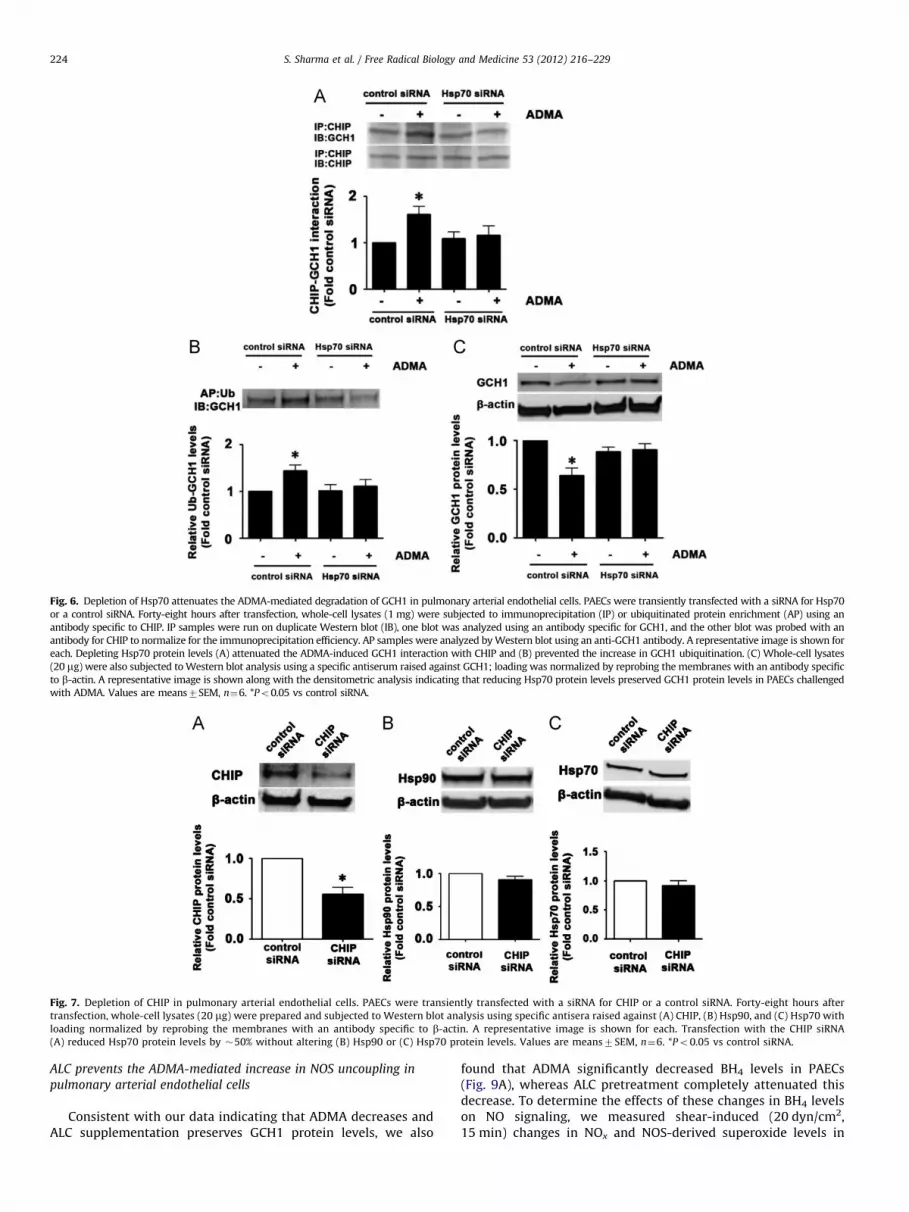
Fig. 6. Depletion of Hsp70 attenuates the ADMA-mediated degradation of GCH1 in pulmonary arterial endothelial cells. PAECs were transiently transfected with a siRNA for Hsp70
or a control siRNA. Forty-eight hours after transfection, whole-cell lysates (1 mg) were subjected to immunoprecipitation (IP) or ubiquitinated protein enrichment (AP) using an
antibody specific to CHIP. IP samples were run on duplicate Western blot (IB), one blot was analyzed using an antibody specific for GCH1, and the other blot was probed with an
antibody for CHIP to normalize for the immunoprecipitation efficiency. AP samples were analyzed by Western blot using an anti-GCH1 antibody. A representative image is shown for
each. Depleting Hsp70 protein levels (A) attenuated the ADMA-induced GCH1 interaction with CHIP and (B) prevented the increase in GCH1 ubiquitination. (C) Whole-cell lysates
(20 mg) were also subjected to Western blot analysis using a specific antiserum raised against GCH1; loading was normalized by reprobing the membranes with an antibody specific
to b-actin. A representative image is shown along with the densitometric analysis indicating that reducing Hsp70 protein levels preserved GCH1 protein levels in PAECs challenged
with ADMA. Values are means7SEM, n¼6. *Po0.05 vs control siRNA.
Fig. 7. Depletion of CHIP in pulmonary arterial endothelial cells. PAECs were transiently transfected with a siRNA for CHIP or a control siRNA. Forty-eight hours after
transfection, whole-cell lysates (20 mg) were prepared and subjected to Western blot analysis using specific antisera raised against (A) CHIP, (B) Hsp90, and (C) Hsp70 with
loading normalized by reprobing the membranes with an antibody specific to b-actin. A representative image is shown for each. Transfection with the CHIP siRNA
(A) reduced Hsp70 protein levels by �50% without altering (B) Hsp90 or (C) Hsp70 protein levels. Values are means7SEM, n¼6. *Po0.05 vs control siRNA.
S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229224
ALC prevents the ADMA-mediated increase in NOS uncoupling in
pulmonary arterial endothelial cells
Consistent with our data indicating that ADMA decreases andALC supplementation preserves GCH1 protein levels, we also
found that ADMA significantly decreased BH4 levels in PAECs(Fig. 9A), whereas ALC pretreatment completely attenuated thisdecrease. To determine the effects of these changes in BH4 levelson NO signaling, we measured shear-induced (20 dyn/cm2,15 min) changes in NOx and NOS-derived superoxide levels in
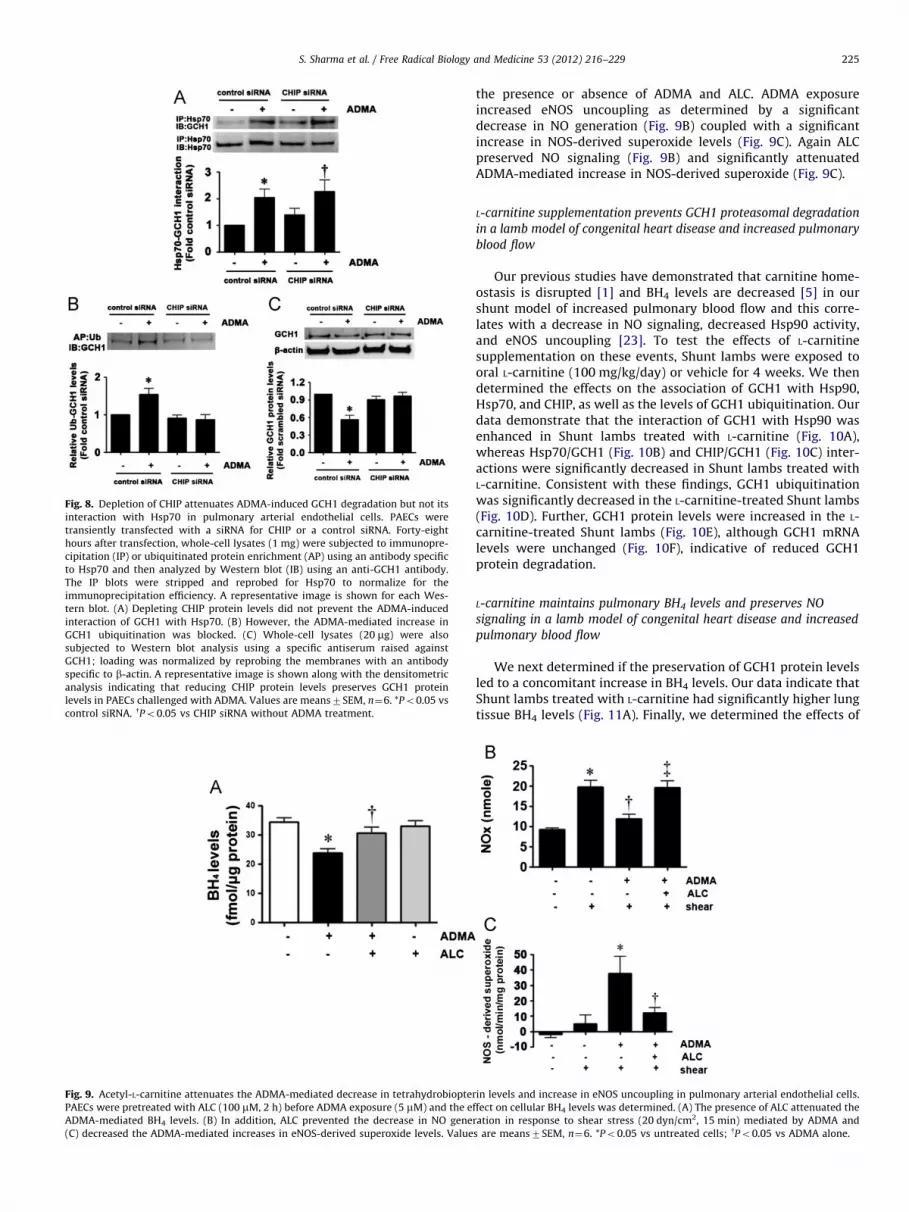
Fig. 8. Depletion of CHIP attenuates ADMA-induced GCH1 degradation but not its
interaction with Hsp70 in pulmonary arterial endothelial cells. PAECs were
transiently transfected with a siRNA for CHIP or a control siRNA. Forty-eight
hours after transfection, whole-cell lysates (1 mg) were subjected to immunopre-
cipitation (IP) or ubiquitinated protein enrichment (AP) using an antibody specific
to Hsp70 and then analyzed by Western blot (IB) using an anti-GCH1 antibody.
The IP blots were stripped and reprobed for Hsp70 to normalize for the
immunoprecipitation efficiency. A representative image is shown for each Wes-
tern blot. (A) Depleting CHIP protein levels did not prevent the ADMA-induced
interaction of GCH1 with Hsp70. (B) However, the ADMA-mediated increase in
GCH1 ubiquitination was blocked. (C) Whole-cell lysates (20 mg) were also
subjected to Western blot analysis using a specific antiserum raised against
GCH1; loading was normalized by reprobing the membranes with an antibody
specific to b-actin. A representative image is shown along with the densitometric
analysis indicating that reducing CHIP protein levels preserves GCH1 protein
levels in PAECs challenged with ADMA. Values are means7SEM, n¼6. *Po0.05 vs
control siRNA. yPo0.05 vs CHIP siRNA without ADMA treatment.
Fig. 9. Acetyl-L-carnitine attenuates the ADMA-mediated decrease in tetrahydrobiopter
PAECs were pretreated with ALC (100 mM, 2 h) before ADMA exposure (5 mM) and the e
ADMA-mediated BH4 levels. (B) In addition, ALC prevented the decrease in NO gene
(C) decreased the ADMA-mediated increases in eNOS-derived superoxide levels. Value
S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229 225
the presence or absence of ADMA and ALC. ADMA exposureincreased eNOS uncoupling as determined by a significantdecrease in NO generation (Fig. 9B) coupled with a significantincrease in NOS-derived superoxide levels (Fig. 9C). Again ALCpreserved NO signaling (Fig. 9B) and significantly attenuatedADMA-mediated increase in NOS-derived superoxide (Fig. 9C).
L-carnitine supplementation prevents GCH1 proteasomal degradation
in a lamb model of congenital heart disease and increased pulmonary
blood flow
Our previous studies have demonstrated that carnitine home-ostasis is disrupted [1] and BH4 levels are decreased [5] in ourshunt model of increased pulmonary blood flow and this corre-lates with a decrease in NO signaling, decreased Hsp90 activity,and eNOS uncoupling [23]. To test the effects of L-carnitinesupplementation on these events, Shunt lambs were exposed tooral L-carnitine (100 mg/kg/day) or vehicle for 4 weeks. We thendetermined the effects on the association of GCH1 with Hsp90,Hsp70, and CHIP, as well as the levels of GCH1 ubiquitination. Ourdata demonstrate that the interaction of GCH1 with Hsp90 wasenhanced in Shunt lambs treated with L-carnitine (Fig. 10A),whereas Hsp70/GCH1 (Fig. 10B) and CHIP/GCH1 (Fig. 10C) inter-actions were significantly decreased in Shunt lambs treated withL-carnitine. Consistent with these findings, GCH1 ubiquitinationwas significantly decreased in the L-carnitine-treated Shunt lambs(Fig. 10D). Further, GCH1 protein levels were increased in the L-carnitine-treated Shunt lambs (Fig. 10E), although GCH1 mRNAlevels were unchanged (Fig. 10F), indicative of reduced GCH1protein degradation.
L-carnitine maintains pulmonary BH4 levels and preserves NO
signaling in a lamb model of congenital heart disease and increased
pulmonary blood flow
We next determined if the preservation of GCH1 protein levelsled to a concomitant increase in BH4 levels. Our data indicate thatShunt lambs treated with L-carnitine had significantly higher lungtissue BH4 levels (Fig. 11A). Finally, we determined the effects of
in levels and increase in eNOS uncoupling in pulmonary arterial endothelial cells.
ffect on cellular BH4 levels was determined. (A) The presence of ALC attenuated the
ration in response to shear stress (20 dyn/cm2, 15 min) mediated by ADMA and
s are means7SEM, n¼6. *Po0.05 vs untreated cells; yPo0.05 vs ADMA alone.
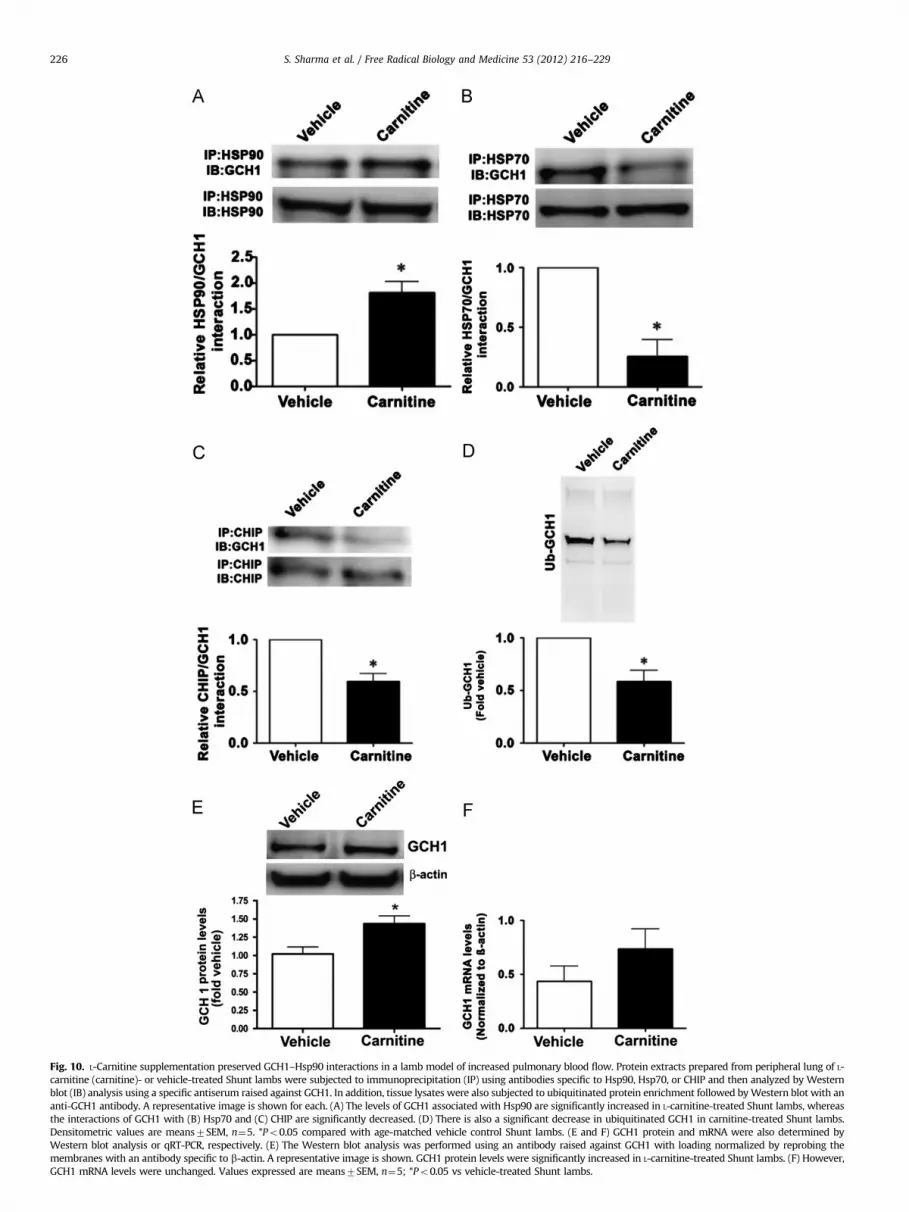
Fig. 10. L-Carnitine supplementation preserved GCH1–Hsp90 interactions in a lamb model of increased pulmonary blood flow. Protein extracts prepared from peripheral lung of L-
carnitine (carnitine)- or vehicle-treated Shunt lambs were subjected to immunoprecipitation (IP) using antibodies specific to Hsp90, Hsp70, or CHIP and then analyzed by Western
blot (IB) analysis using a specific antiserum raised against GCH1. In addition, tissue lysates were also subjected to ubiquitinated protein enrichment followed by Western blot with an
anti-GCH1 antibody. A representative image is shown for each. (A) The levels of GCH1 associated with Hsp90 are significantly increased in L-carnitine-treated Shunt lambs, whereas
the interactions of GCH1 with (B) Hsp70 and (C) CHIP are significantly decreased. (D) There is also a significant decrease in ubiquitinated GCH1 in carnitine-treated Shunt lambs.
Densitometric values are means7SEM, n¼5. *Po0.05 compared with age-matched vehicle control Shunt lambs. (E and F) GCH1 protein and mRNA were also determined by
Western blot analysis or qRT-PCR, respectively. (E) The Western blot analysis was performed using an antibody raised against GCH1 with loading normalized by reprobing the
membranes with an antibody specific to b-actin. A representative image is shown. GCH1 protein levels were significantly increased in L-carnitine-treated Shunt lambs. (F) However,
GCH1 mRNA levels were unchanged. Values expressed are means7SEM, n¼5; *Po0.05 vs vehicle-treated Shunt lambs.
S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229226

Fig. 11. L-Carnitine supplementation protects tetrahydrobiopterin levels and enhances NOS coupling in lambs with increased pulmonary blood flow. BH4 levels in the
peripheral lung of L-carnitine- or vehicle-treated Shunt lambs were measured by HPLC. (A) BH4 levels were significantly increased after carnitine treatment. In addition,
L-carnitine (B) enhanced tissue NOx levels and (C) decreased NOS-derived superoxide, indicative of enhanced eNOS coupling. Values expressed are means7SEM, n¼5;
*Po0.05 vs vehicle-treated Shunt lambs.
S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229 227
L-carnitine treatment on NO signaling. We identified a significantincrease in NO signaling, as determined by enhanced tissue NOx
levels (Fig. 11B), and this correlated with a significant decrease inNOS-derived superoxide levels (Fig. 11C), indicating that L-carni-tine increases eNOS coupling.
Discussion
Mitochondria are the source of superoxide anion radicals andhydrogen peroxide under physiologic and pathologic conditions. Theincreased production of reactive oxygen species from the mitochon-dria can be deleterious to the cell because of their ability to inducelipid peroxidation, protein oxidation, and DNA damage [33,34].Accumulating evidence suggests that mitochondrial dysfunction isassociated with pulmonary hypertension [35,36]. L-carnitine playsan important role in maintaining the balance of acyl-CoA-relatedfunctions in mitochondria and can be used pharmacologically tomodulate mitochondrial dysfunction (reviewed in [4]). In this study,we found that ALC significantly attenuated the decrease in GCH1protein levels caused by exposure to ADMA. Our data indicate thatALC preserves GCH1 protein levels by attenuating ADMA-mediatedGCH1 degradation. Interestingly, we also found that ALC treatmentalone significantly increased GCH1 mRNA levels although GCH1protein levels were not significantly increased. One possible expla-nation is that ALC itself may lead to some level of cellular stress [37].Indeed intraperitoneal injection of L-carnitine is associated with alocalized inflammatory macrophage activation in the peritonealcavity [38], and GCH1 mRNA levels can be strongly elevated in cellstreated with inflammatory stimuli [39]. The fact that we did not seechanges in GCH1 protein levels that mirrored the changes in mRNAmay be due to the fact that our 2-h study did not allow sufficienttime for significant increases in GCH1 translation to occur. However,further investigations will be needed to clarify the possible regula-tory mechanisms of L-carnitine on GCH1 gene and protein expres-sion. Further, from our studies it seems that ALC had multiple effectson the cell, including both ubiquitination and transcription of GCH1,
as well as modifying both mitochondrial function and mitochon-drion-derived superoxide. However, we speculate that the cardinalevent of ALC is the preservation of mitochondrial function and thatall downstream protective measures stem from this. Based on thework of Ames’ group [40–42] it is likely that ALC preserves theacetyl-CoA:acyl-CoA balance in the mitochondria that is disruptedby ADMA. As increases in acyl-CoA are known to decrease ATPgeneration the preservation of acetyl-CoA levels will preserve ATPlevels. This in turn will maintain Hsp90 in an active form andpreserve the Hsp90–GCH1 complex. This in turn will prevent Hsp70and CHIP recruitment and GCH1 ubiquitination and degradation.
Hsp90 has been shown to interact with a number of proteinsthat are required for efficient NO biosynthesis, including eNOS[43] and soluble guanylate cyclase [43]. Further, the chaperoneactivity of Hsp90 is ATP dependent. Our previous studies demon-strate that ADMA can have an indirect effect on eNOS couplingthrough its ability to cause mitochondrial dysfunction and adecrease in Hsp90 chaperone activity [2]. Our recent data haveshown that GCH1 is a client protein of Hsp90 and that Hsp90–GCH1 interactions are disrupted in ADMA-exposed cells [30], andhere we found that ALC administration during ADMA exposurepreserved mitochondrial function and cellular ATP levels andprevented the ADMA-induced disruption of the Hsp90–GCH1complex. These findings suggest that, under the oxidative stressconditions induced by high levels of ADMA, L-carnitine canregulate Hsp90 chaperone activity by maintaining mitochondrialfunction and so favor the production of NO signaling molecules.Increasing evidence suggests that Hsp90-client proteins bind toHsp70 before degradation and our recent study demonstratedthat CHIP targets the proteasomal degradation of GCH1 [30].Here, we expand on this finding by demonstrating that GCH1 isdegraded via a signaling pathway that initially involves Hsp90inhibition followed by the sequential recruitment of Hsp70 andCHIP, which leads to GCH1 ubiquitination and protein degrada-tion. ALC was able to significantly prevent these events andpreserve the interaction of GCH1 with Hsp90. Recent studiesfrom the laboratories of Zou [44,45] and Vasquez-Vivar [16,46]

Fig. 12. A possible mechanism by which ADMA disrupts mitochondrial function and attenuates NO signaling in lambs with increased pulmonary blood flow. ADMA
induces mitochondrial dysfunction, reducing cellular ATP levels. As the activity of Hsp90 is ATP dependent, GCH1–Hsp90 interactions are disrupted. This leads to increased
binding of Hsp70 to GCH1, leading to the recruitment of CHIP. CHIP then ubiquitinates GCH1, targeting it for proteasomal degradation. The resulting BH4 deficiency
induces eNOS uncoupling. L-Carnitine supplementation short-circuits this degradation pathway by maintaining mitochondrial function.
S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229228
demonstrated that ubiquitinated GCH1 levels in endothelial cellswere significantly elevated under conditions of oxidative stress,which led to GCH1 degradation. Our data add to these priorstudies by demonstrating that maintaining mitochondrial func-tion through the administration of ALC significantly attenuatesthe GCH1 ubiquitination induced by ADMA. Further, enhancingthe removal of superoxide (by the addition of PEG-SOD) orhydrogen peroxide (by the addition of PEG-catalase) also pre-serves GCH1 protein levels in ADMA-challenged PAECs, suggest-ing that oxidative stress is also involved in the degradationprocess.
Increasing evidence indicates that optimal concentrations of BH4
are of fundamental importance for the normal function of eNOS invascular endothelial cells, and endothelial BH4 bioavailability hasemerged as a rationale therapeutic target in a number of vasculardisease states [47]. Studies indicate that supplementation with BH4 orincreasing BH4 synthesis using adenoviral gene delivery of GCH1 canrestore BH4 levels and normalize eNOS function [47]. Our resultsindicate that ALC preserved BH4 levels in PAECs exposed to ADMA.Furthermore, the preservation of BH4 levels was sufficient to preventthe increase in eNOS uncoupling that we have previously demon-strated in PAECs exposed to ADMA. Together, these data suggest thatincreasing and/or sustaining BH4 levels may be dominant over theuncoupling of eNOS associated with increases in ADMA [2]. Given theincreasing number of studies that have shown an association ofincreases in ADMA with cardiovascular risk, enhancing BH4 levelsmay have therapeutic utility and L-carnitine supplementation is apotentially useful agent.
We also extended our cell culture studies to a clinically relevantlamb model that mimics a CHD that results in increased pulmonaryblood flow (Shunt). Children with these types of CHD develop earlyand progressive alterations in pulmonary vascular function thatcause significant morbidity [8]. The mechanisms involved in thispulmonary vascular disease are not fully understood. However, ourprevious studies using our Shunt model have shown that there is aprogressive decline in NO signaling that leads to the development of
endothelial dysfunction [23]. This decline in bioavailable NO corre-sponds to a complex series of temporal events that involves thedisruption of carnitine homeostasis and the development of mito-chondrial dysfunction [1], loss of Hsp90 chaperone activity [1], andincreased eNOS uncoupling [23] related, at least in part, to areduction in BH4 levels [5]. Here, we show that ALC supplementa-tion preserves Hsp90 chaperone activity and its interaction withGCH1 interaction, which suppresses the Hsp70- and CHIP-depen-dent GCH1 ubiquitination and degradation of GCH1 and, as a result,these alterations led to an increase in BH4 levels and, subsequently,preserved NO signaling and reduced eNOS uncoupling (Fig. 12). Asone of the most important consequences of endothelial injury is adecrease in bioavailable NO, with subsequent endothelial dysfunc-tion, our findings that ALC preserves NO signaling could lead to anew effective therapy for children born with CHD and increasedpulmonary blood flow.
In summary, our data show the effects of L-carnitine on ADMA-induced BH4 deficiency both in vitro and in vivo and suggest thatthe protective effects of L-carnitine on mitochondrial functioncould be an effective preventive therapy against the loss of NOsignaling during the development of pulmonary hypertension.Our study provides a novel insight regarding the molecular basisfor the beneficial potential of L-carnitine through its ability topreserve BH4 biosynthesis in cardiovascular disease states. Ourresults also reinforce the concept that preventing or attenuatingthe development of mitochondrial dysfunction may be a ther-apeutic approach for the treatment of a number of cardiovasculardisease states including pulmonary hypertension.
Acknowledgments
This research was supported in part by Grants HL60190 (toS.M.B.), HL67841 (to S.M.B.), HL084739 (to S.M.B.), R21HD057406(to S.M.B.), and HL61284 (to J.R.F.), all from the National Institutesof Health; by a grant from the Fondation Leducq (to S.M.B. and

S. Sharma et al. / Free Radical Biology and Medicine 53 (2012) 216–229 229
J.R.F.); 09BGIA2310050 from the Southeast Affiliates of the Amer-ican Heart Association (to S.S.); and Cardiovascular DiscoveryInstitute Seed Awards (to S.S. and S.K.).
References
[1] Sharma, S.; Sud, N.; Wiseman, D. A.; Carter, A. L.; Kumar, S.; Hou, Y.; Rau, T.;Wilham, J.; Harmon, C.; Oishi, P.; Fineman, J. R.; Black, S. M. Altered carnitinehomeostasis is associated with decreased mitochondrial function and alterednitric oxide signaling in lambs with pulmonary hypertension. Am. J. Physiol.Lung Cell Mol. Physiol. 294:L46–56; 2008.
[2] Sud, N.; Wells, S. M.; Sharma, S.; Wiseman, D. A.; Wilham, J.; Black, S. M.Asymmetric dimethylarginine inhibits HSP90 activity in pulmonary arterialendothelial cells: role of mitochondrial dysfunction. Am. J. Physiol. Lung CellMol. Physiol. 294:C1407–1418; 2008.
[3] Liu, J.; Head, E.; Gharib, A. M.; Yuan, W.; Ingersoll, R. T.; Hagen, T. M.; Cotman, C.W.; Ames, B. N. Memory loss in old rats is associated with brain mitochondrialdecay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-a-lipoic acid. Proc. Natl. Acad. Sci. USA 99:2356–2361; 2002.
[4] Zammit, V. A.; Ramsay, R. R.; Bonomini, M.; Arduini, A. Carnitine, mitochon-drial function and therapy. Adv. Drug Delivery Rev. 61:1353–1362; 2009.
[5] Grobe, A. C.; Wells, S. M.; Benavidez, E.; Oishi, P.; Azakie, A.; Fineman, J. R.;Black, S. M. Increased oxidative stress in lambs with increased pulmonaryblood flow and pulmonary hypertension: role of NADPH oxidase and endothelialNO synthase. Am. J. Physiol. Lung Cell Mol. Physiol. 290:L1069–1077; 2006.
[6] Swick, L.; Kapatos, G. A yeast 2-hybrid analysis of human GTP cyclohydrolaseI protein interactions. J. Neurochem. 97:1447–1455; 2006.
[7] Calo, L. A.; Semplicini, A.; Davis, P. A. Antioxidant and antiinflammatory effectof carvedilol in mononuclear cells of hypertensive patients. Am. J. Med.118:201–202; 2005.
[8] Reddy, V. M.; Meyrick, B.; Wong, J.; Khoor, A.; Liddicoat, J. R.; Hanley, F. L.;Fineman, J. R. In utero placement of aortopulmonary shunts: a model ofpostnatal pulmonary hypertension with increased pulmonary blood flow inlambs. Circulation 92:606–613; 1995.
[9] Reddy, V. M.; Meyrick, B.; Wong, J.; Khoor, A.; Liddicoat, J. R.; Hanley, F. L.;Fineman, J. R. In utero placement of aortopulmonary shunts: a model ofpostnatal pulmonary hypertension with increased pulmonary blood flow inlambs. Circulation 92:1–8; 1995.
[10] Kelly, L. K.; Wedgwood, S.; Steinhorn, R. H.; Black, S. M. Nitric oxide decreasesendothelin-1 secretion through the activation of soluble guanylate cyclase.Am. J. Physiol. Lung Cell Mol. Physiol. 286:L984–991; 2004.
[11] Cossarizza, A.; Baccarani-Contri, M.; Kalashnikova, G.; Franceschi, C. A newmethod for the cytofluorimetric analysis of mitochondrial membrane poten-tial using the J-aggregate forming lipophilic cation 5,50 ,6,60-tetrachloro-1,10 ,3,30-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem. Biophys.Res. Commun. 197:40–45; 1993.
[12] Wiseman, D. A.; Wells, S. M.; Hubbard, M.; Welker, J. E.; Black, S. M.Alterations in zinc homeostasis underlie endothelial cell death induced byoxidative stress from acute exposure to hydrogen peroxide. Am. J. Physiol.Lung Cell Mol. Physiol. 292:L165–177; 2007.
[13] Miao, R. Q.; Fontana, J.; Fulton, D.; Lin, M. I.; Harrison, K. D.; Sessa, W. C.Dominant-negative Hsp90 reduces VEGF-stimulated nitric oxide release andmigration in endothelial cells. Arterioscler Thromb. Vasc. Biol. 28:105–111; 2008.
[14] Sun, X.; Kumar, S.; Tian, J.; Black, S. M. Estradiol increases guanosine 50-triphosphate cyclohydrolase expression via the nitric oxide-mediated activa-tion of cyclic adenosine 50-monophosphate response element binding pro-tein. Endocrinology 150:3742–3752; 2009.
[15] Milstien, S.; Jaffe, H.; Kowlessur, D.; Bonner, T. I. Purification and cloning ofthe GTP cyclohydrolase I feedback regulatory protein, GFRP. J. Biol. Chem.271:19743–19751; 1996.
[16] Kalivendi, S.; Hatakeyama, K.; Whitsett, J.; Konorev, E.; Kalyanaraman, B.;Vasquez-Vivar, J. Changes in tetrahydrobiopterin levels in endothelial cellsand adult cardiomyocytes induced by LPS and hydrogen peroxide—a role forGFRP? Free Radic. Biol. Med. 38:481–491; 2005.
[17] Kumar, S.; Sun, X.; Sharma, S.; Aggarwal, S.; Ravi, K.; Fineman, J. R.; Black, S.M. GTP cyclohydrolase I expression is regulated by nitric oxide: role of cyclicAMP. Am. J. Physiol. Lung Cell Mol. Physiol. 297:L309–317; 2009.
[18] Wainwright, M. S.; Arteaga, E.; Fink, R.; Ravi, K.; Chace, D. H.; Black, S. M.Tetrahydrobiopterin and nitric oxide synthase dimer levels are not changedfollowing hypoxia–ischemia in the newborn rat. Brain Res. Dev. Brain Res.156:183–192; 2005.
[19] Dewey Jr C. F.; Bussolari, S. R.; Gimbrone Jr M. A.; Davies, P. F. The dynamicresponse of vascular endothelial cells to fluid shear stress. J. Biomech. Eng.103:177–185; 1981.
[20] Wedgwood, S.; Bekker, J. M.; Black, S. M. Shear stress regulation ofendothelial NOS in fetal pulmonary arterial endothelial cells involves PKC.Am. J. Physiol. Lung Cell Mol. Physiol. 281:L490–498; 2001.
[21] Wiseman, D. A.; Wells, S. M.; Wilham, J.; Hubbard, M.; Welker, J. E.; Black, S.M. Endothelial response to stress from exogenous Zn2þ resembles that ofNO-mediated nitrosative stress, and is protected by MT-1 overexpression.Am. J. Physiol. Lung Cell Mol. Physiol. 291:C555–568; 2006.
[22] Sud, N.; Sharma, S.; Wiseman, D. A.; Harmon, C.; Kumar, S.; Venema, R. C.;Fineman, J. R.; Black, S. M. Nitric oxide and superoxide generation from
endothelial NOS: modulation by HSP90. Am. J. Physiol. Lung Cell Mol. Physiol.293:L1444–1453; 2007.
[23] Oishi, P. E.; Wiseman, D. A.; Sharma, S.; Kumar, S.; Hou, Y.; Datar, S. A.;Azakie, A.; Johengen, M. J.; Harmon, C.; Fratz, S.; Fineman, J. R.; Black, S. M.Progressive dysfunction of nitric oxide synthase in a lamb model ofchronically increased pulmonary blood flow: a role for oxidative stress. Am.J. Physiol. Lung Cell Mol. Physiol. 295:L756–766; 2008.
[24] Dikalov, S. I.; Li, W.; Mehranpour, P.; Wang, S. S.; Zafari, A. M. Production ofextracellular superoxide by human lymphoblast cell lines: comparison ofelectron spin resonance techniques and cytochrome C reduction assay.Biochem. Pharmacol. 73:972–980; 2007.
[25] Southan, G. J.; Szabo, C.; Thiemermann, C. Isothioureas: potent inhibitors ofnitric oxide synthases with variable isoform selectivity. Br. J. Pharmacol.114:510–516; 1995.
[26] Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G. Q.; Bien-Ly, N.; Puolivali, J.;Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in mitochondrial super-oxide dismutase modulates Alzheimer’s disease-like pathology and acceler-ates the onset of behavioral changes in human amyloid precursor proteintransgenic mice. J. Neurosci. 26:5167–5179; 2006.
[27] Liang, L. P.; Patel, M. Mitochondrial oxidative stress and increased seizuresusceptibility in Sod2(�/þ) mice. Free Radic. Biol. Med. 36:542–554; 2004.
[28] Andrews, Z. B.; Diano, S.; Horvath, T. L. Mitochondrial uncoupling proteins in theCNS: in support of function and survival. Nat. Rev. Neurosci. 6:829–840; 2005.
[29] Hoffstedt, J.; Folkesson, R.; Wahrenberg, H.; Wennlund, A.; van Harmelen, V.;Arner, P. A marked upregulation of uncoupling protein 2 gene expression inadipose tissue of hyperthyroid subjects. Horm. Metab. Res. 32:475–479; 2000.
[30] Sun, X.; Fratz, S.; Sharma, S.; Hou, Y.; Rafikov, R.; Kumar, S.; Rehmani, I.; Tian, J.;Smith, A.; Schreiber, C.; Reiser, J.; Naumann, S.; Haag, S.; Hess, J.; Catravas, J. D.;Patterson, C.; Fineman, J. R.; Black, S. M. C-terminus of heat shock protein 70-interacting protein-dependent GTP cyclohydrolase I degradation in lambs withincreased pulmonary blood flow. Am. J. Respir. Cell Mol. Biol. 45:163–171; 2011.
[31] Pratt, W. B.; Toft, D. O. Regulation of signaling protein function andtrafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med.(Maywood) 228:111–133; 2003.
[32] Bercovich, B.; Stancovski, I.; Mayer, A.; Blumenfeld, N.; Laszlo, A.; Schwartz,A. L.; Ciechanover, A. Ubiquitin-dependent degradation of certain proteinsubstrates in vitro requires the molecular chaperone Hsc70. J. Biol. Chem.272:9002–9010; 1997.
[33] Tribe, R. M.; Poston, L. Oxidative stress and lipids in diabetes: a role inendothelium vasodilator dysfunction? Vasc. Med. 1:195–206; 1996.
[34] Ronson, R. S.; Nakamura, M.; Vinten-Johansen, J. The cardiovascular effectsand implications of peroxynitrite. Cardiovasc. Res. 44:47–59; 1999.
[35] Daicho, T.; Yagi, T.; Abe, Y.; Ohara, M.; Marunouchi, T.; Takeo, S.; Tanonaka, K.Possible involvement of mitochondrial energy-producing ability in thedevelopment of right ventricular failure in monocrotaline-induced pulmon-ary hypertensive rats. J. Pharmacol. Sci. 111:33–43; 2009.
[36] Ward, J. P.; McMurtry, I. F. Mechanisms of hypoxic pulmonary vasoconstric-tion and their roles in pulmonary hypertension: new findings for an oldproblem. Curr. Opin. Pharmacol. 9:287–296; 2009.
[37] Alves, E.; Binienda, Z.; Carvalho, F.; Alves, C. J.; Fernandes, E.; de LourdesBastos, M.; Tavares, M. A.; Summavielle, T. Acetyl-L-carnitine provideseffective in vivo neuroprotection over 3,4-methylenedioximethampheta-mine-induced mitochondrial neurotoxicity in the adolescent rat brain.Neuroscience 158:514–523; 2009.
[38] Dionyssopoulou, E.; Vassiliadis, S.; Evangeliou, A.; Koumantakis, E. E.;Athanassakis, I. Constitutive or induced elevated levels of L-carnitine corre-late with the cytokine and cellular profile of endometriosis. J. Reprod.Immunol. 65:159–170; 2005.
[39] Linscheid, P.; Schaffner, A.; Blau, N.; Schoedon, G. Regulation of 6-pyruvoyl-tetrahydropterin synthase activity and messenger RNA abundance in humanvascular endothelial cells. Circulation 98:1703–1706; 1998.
[40] Shenk, J. C.; Liu, J.; Fischbach, K.; Xu, K.; Puchowicz, M.; Obrenovich, M. E.;Gasimov, E.; Alvarez, L. M.; Ames, B. N.; Lamanna, J. C.; Aliev, G. The effect ofacetyl-L-carnitine and R-a-lipoic acid treatment in ApoE4 mouse as a modelof human Alzheimer’s disease. J. Neurol. Sci. 283:199–206; 2009.
[41] Long, J.; Gao, F.; Tong, L.; Cotman, C. W.; Ames, B. N.; Liu, J. Mitochondrialdecay in the brains of old rats: ameliorating effect of a-lipoic acid and acetyl-L-carnitine. Neurochem. Res. 34:755–763; 2009.
[42] Aliev, G.; Liu, J.; Shenk, J. C.; Fischbach, K.; Pacheco, G. J.; Chen, S. G.;Obrenovich, M. E.; Ward, W. F.; Richardson, A. G.; Smith, M. A.; Gasimov, E.;Perry, G.; Ames, B. N. Neuronal mitochondrial amelioration by feeding acetyl-L-carnitine and lipoic acid to aged rats. J. Cell Mol. Med. 13:320–333; 2009.
[43] Zhang, G. G.; Shi, R. Z.; Jiang, D. J.; Chen, Y. R.; Jia, C.; Tang, Z. Y.; Bai, Y. P.;Xiao, H. B.; Li, Y. J. Involvement of the endothelial DDAH/ADMA pathway innitroglycerin tolerance: the role of ALDH-2. Life Sci. 82:699–707; 2008.
[44] Wang, S.; Xu, J.; Song, P.; Viollet, B.; Zou, M. H. In vivo activation of AMP-activated protein kinase attenuates diabetes-enhanced degradation of GTPcyclohydrolase I. Diabetes 58:1893–1901; 2009.
[45] Xu, J.; Wang, S.; Wu, Y.; Song, P.; Zou, M. H. Tyrosine nitration of PA700activates the 26S proteasome to induce endothelial dysfunction in mice withangiotensin II-induced hypertension. Hypertension 54:625–632; 2009.
[46] Whitsett, J.; Picklo Sr M. J.; Vasquez-Vivar, J. 4-Hydroxy-2-nonenal increasessuperoxide anion radical in endothelial cells via stimulated GTP cyclohydrolaseproteasomal degradation. Arterioscler. Thromb. Vasc. Biol. 27:2340–2347; 2007.
[47] Schmidt, T. S.; Alp, N. J. Mechanisms for the role of tetrahydrobiopterin inendothelial function and vascular disease. Clin. Sci. (London) 113:47–63; 2007.