physics letters a2007
TRANSCRIPT

Physics Letters A 362 (2007) 73–83
www.elsevier.com/locate/pla
Investigation of structural stability and electronic propertiesof CuN, AgN and AuN by first principles calculations
M.B. Kanoun a, S. Goumri-Said b,∗
a Institut d’Electronique de Microélectronique et de Nanotechnologie, UMR CNRS 8520, Université de Lille I. Avenue Poincaré,BP 60069, 59652 Villeneuve d’Ascq Cedex, France
b Condensed Matter Theory Group, Department of Physics, Kaiserslautern University of Technology,Erwin-Schrödinger str. 46, Box 3049, D-67653 Kaiserslautern, Germany
Received 8 March 2006; received in revised form 26 September 2006; accepted 28 September 2006
Available online 16 October 2006
Communicated by R. Wu
Abstract
First-principles density-functional theory calculations using the full-potential (linearized) augmented plane waves plus local orbitals (FP-L/APW + lo) method within the local-density approximation (LDA) and the generalized gradient approximation (GGA) have been performed.We investigate the structural stability, bulk electronic and physical properties of a series of noble metal nitrides: CuN, AgN, and AuN. We analyzethe structural properties of these noble nitrides in order to extract the more stable structure. The analysis of electronic properties, is achievedby the calculation of the band structures, the densities (total and partial) of states, which show a metallic nature. The bonding is studied by thecalculation of the charge density contours in zincblende and rocksalt structures. It shows a covalent-like character because of the hybridizationbetween the nitrogen and noble metal states, added to a some ionic and metallic characters due to the electron transfer from the noble metal atomsto N atoms.© 2006 Elsevier B.V. All rights reserved.
PACS: 71.15.-m; 71.15.Mb; 71.20.Be
1. Introduction
Metal and semiconductor nitrides are an important class ofmaterials having properties of fundamental interest as well asthose used in a variety of applications [1–8]. Most of theseworks studied the nitrides of early transition metals, or the3d series of late transition metals [9–13]. The 4d and 5d se-ries of late transition metals Ru, Rh, Pd, Ag, Os, Ir, Pt, andAu, also known as noble metals are generally considered notto form nitrides [1], although Cu3N and Ag3N were reported[14,15]. Despite the wide interest in making ever better nitridesfor applications, the noble metal nitrides have evaded discov-ery until the recent synthesis of gold [16] and platinum nitrides.Recently, the highest pressure a new material, PtN, was synthe-
* Corresponding author.E-mail address: [email protected] (S. Goumri-Said).
0375-9601/$ – see front matter © 2006 Elsevier B.V. All rights reserved.doi:10.1016/j.physleta.2006.09.100
sized and recovered in zincblende structure back to atmosphericpressure, by Gregoryanz et al. [17]. This work have impor-tant implications for high-pressure experiments and technology,namely the possibility to synthesize other nitrides with transi-tion metals. The results also indicate that it might be possible toform nitrides with other noble metals such as Au, Ag, and Cu,which could have interesting electronic properties [17]. Therehave been several earlier first-principles studies into the bulkproperties and mechanical stability of the platinum nitrides indifferent phases [18–24].
It is interesting to know whether the other noble metals canform nitrides with the cubic structure. In the present work,we report first-principles all-electron density-functional full-potential (linearized) augmented plane waves plus local orbitals(FP-L/APW + lo) calculations for a series of noble metal ni-trides, namely, CuN, AgN and AuN. We are interested in thebulk properties, structural stability and electronic properties ofthese materials with zincblende (ZB), rocksalt (RS), caesium

74 M.B. Kanoun, S. Goumri-Said / Physics Letters A 362 (2007) 73–83
chloride (CsCl) and wurtzite (WZ) structures and in particu-lar report the lattice constants, and bulk modulus. We employthe local density approximation (LDA) and the generalized gra-dient approximation (GGA) for the exchange-correlation func-tional and investigate any improvements brought about by theGGA. Together with cubic PtN, they form a new noble metalnitrides, in which the metal atoms form a face-centered cubiclattice, while nitrogen atoms occupy all of the tetrahedral inter-stitial sites.
The Letter is organized as follows. In the next section, wewill briefly discuss the computational details. Section 3 willbe devoted to our own results of structural properties, bandstructures, density of states and bonding properties for CuN,AgN and AuN. The Letter ends with the main conclusionsabout the different properties determined for noble metal ni-trides.
2. Method of calculation
The calculations are performed in the framework of density-functional theory [25] within the LDA and GGA using thescheme of Perdew, Brouke and Ernzerhof [26]. We have em-ployed the scalar relativistic hybrid full potential (linearized)augmented plane waves plus local orbitals (FP-L/APW + lo)method as implemented in the WIEN2k code [27]. The ex-change and correlation effects are treated using the Ceperly–Alder [28] data as parameterized by Perdew and Wang [29].The core states were treated fully relativistically relaxed in aspherical approximation [30], while the valence states weretreated self-consistently within the semi-relativistic approxima-tion (no spin–orbit effects included). Wave functions, chargedensity and potential were expanded inside muffin-tin spheresof radius RMT by using spherical harmonics expansions, whilein the remaining space of the unit cell a plane wave basis setwas chosen. The plane wave cutoff of KMAX ∗RMT = 8 (whereKMAX is the maximum modulus for the reciprocal lattice vec-tor) was used for the expansion of the wave function in the inter-stitial region. Values of RMT were assumed to be 1.7, 1.83, 1.9and 1.6 (Bohr) for Cu, Ag, Au and N atoms, respectively. Thecharge density was Fourier expanded up to Gmax = 14
√Ry.
The maximum l value for the wave function expansions insidethe spheres was confined to lmax = 10. Accurate Brillouin inte-grations zone are performed using the standard special k pointsMonkhorst and Pack [31]. The corresponding integrating pointsover the irreductible Brillouin zone are 56 k points for CsCland RS phases, 55 k points for ZB phase and 48 k points forthe WZ phase. We investigated four different phases of CuN,AgN and AuN, (i) the ZB structure (space group F-43m), (ii) theRS structure (space group Fm-3m), (iii) CsCl structure (spacegroup Pm-3m), and (iv) WZ structure (space group P 63mc).The unit cells for the four phases are shown in Figs. 1(a)–(d) re-spectively. The first three phases have a = b = c, giving them acubic symmetry and the WZ phase is a hexagonal close-packedsublattices (with two formula units per unit cell), which can bedescribed by three structure parameters: a, c and an internal pa-rameter u. The self-consistent calculations are considered to be
(a) (b)
(c) (d)
Fig. 1. (a) Zincblende structure, (b) rocksalt structure, (c) CsCl structure and(d) wurtzite structure.
converged only when the calculated total energy of the crystalconverge to less than 1 mRyd.
3. Results and discussion
3.1. Structural properties
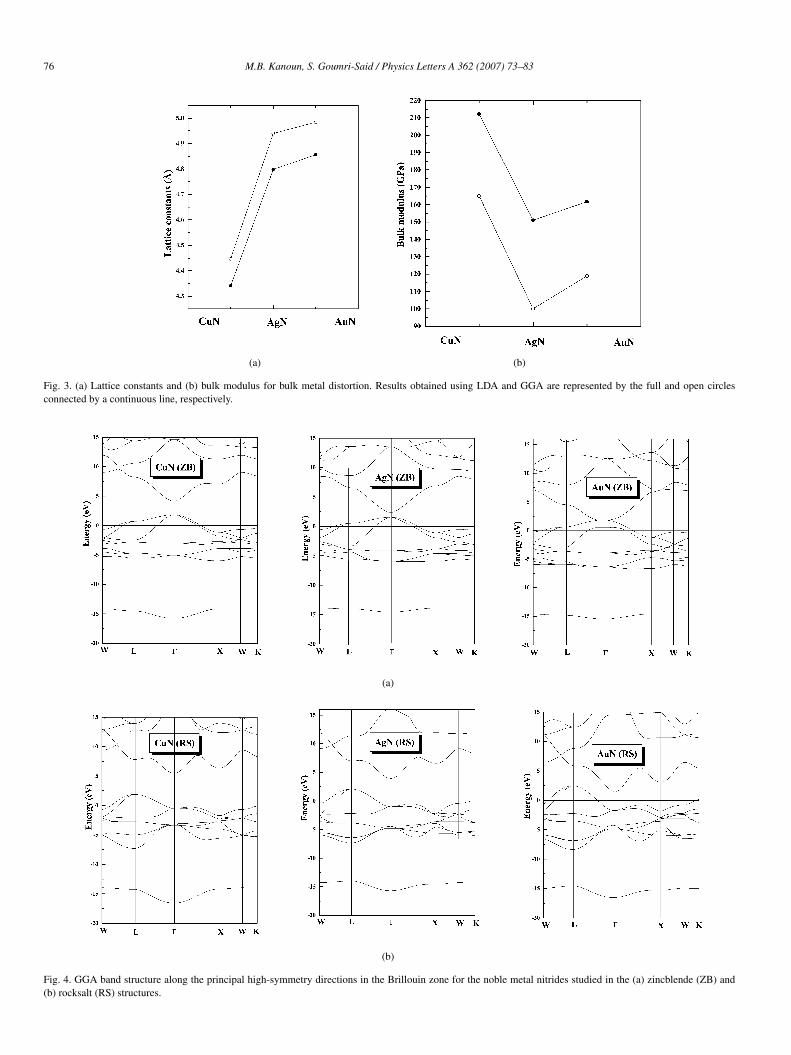
Total energy versus volume data for the rocksalt, CsCl,zincblende and wurtzite phases of CuN, AgN and AuN areshown in Fig. 2. Energies and volumes are per single copper-nitrides unit formula: there are two atom in wurtzite unit cell,and one in all the other cases. The data is fitted to the Mur-naghan equation of state [32] for each phase. In this way, we areable to obtain the equilibrium lattice constant, the bulk modulusand other structural parameters within LDA and GGA calcu-lations (see Table 1). Our calculated LDA and GGA latticeparameters for CuN, agree with the recent first principles lin-ear muffin-tin orbital (LMTO) calculation [33]. The zincblendephase is found to be the ground state of CuN structure. Inthe case of AgN and AuN, we found that the rocksalt phaseis the ground state structure. Therefore, we have presented inFig. 3, the lattice constants and bulk modulus for bulk metaldistortion. The trends can be seen more easily: on going fromCuN to AgN to AuN, it is seen that the lattice constant in-creases (Fig. 2(a)) and the bulk modulus decreases from CuNto AgN and increases from AgN to AuN (Fig. 3(b)). On go-ing down the columns of periodic table, the lattice constant

M.B. Kanoun, S. Goumri-Said / Physics Letters A 362 (2007) 73–83 75
(a) (b)
(c)
Fig. 2. Total energy (in eV) versus the atomic volume for different structures of CuN, AgN and AuN.
Table 1Lattice constants a, bulk modulus B , pressure derivative of bulk modulus B ′ of CuN, AgN and AuN in the different phases: ZB, RS, CsCl and WZ
Zincblende Rocksalt CsCl Wurtzite
LDA GGA LDA GGA LDA GGA LDA GGA
CuN a (Å) 4.341 4.447 4.074 4.185 2.54 2.61 3.077 3.17c (Å) – – – – – – 5.016 5.16B (GPa) 212.16 164.96 257.46 201.60 265.40 200.01 202.10 157.85B ′ 4.311 4.534 4.491 3.811 4.373 4.352 4.35 4.41
AgN a (Å) 4.79 4.94 4.476 4.606 2.78 2.87 3.41 3.54c (Å) – – – – – – 5.52 5.69B (GPa) 151.05 100.11 197.18 147.40 204.10 138.96 143.68 110.12B ′ 4.542 5.825 4.883 4.653 5.451 4.823 4.82 4.663
AuN a (Å) 4.856 4.984 4.541 4.662 2.831 2.915 3.45 3.600c (Å) – – – – – – 5.53 5.75B (GPa) 161.75 118.91 223.42 163.13 225.66 162.58 160.20 133.6B ′ 5.212 4.981 5.616 4.993 5.021 4.551 4.65 4.23
76 M.B. Kanoun, S. Goumri-Said / Physics Letters A 362 (2007) 73–83
(a) (b)
Fig. 3. (a) Lattice constants and (b) bulk modulus for bulk metal distortion. Results obtained using LDA and GGA are represented by the full and open circlesconnected by a continuous line, respectively.
(a)
(b)
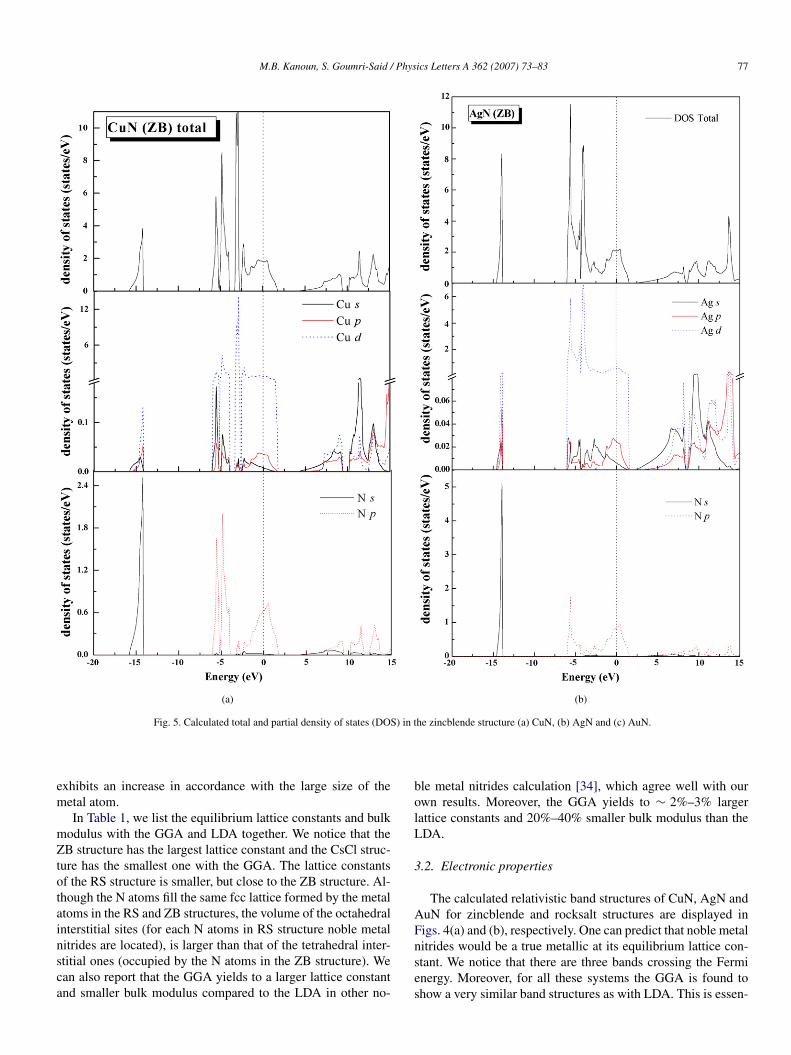
Fig. 4. GGA band structure along the principal high-symmetry directions in the Brillouin zone for the noble metal nitrides studied in the (a) zincblende (ZB) and(b) rocksalt (RS) structures.
M.B. Kanoun, S. Goumri-Said / Physics Letters A 362 (2007) 73–83 77
(a) (b)
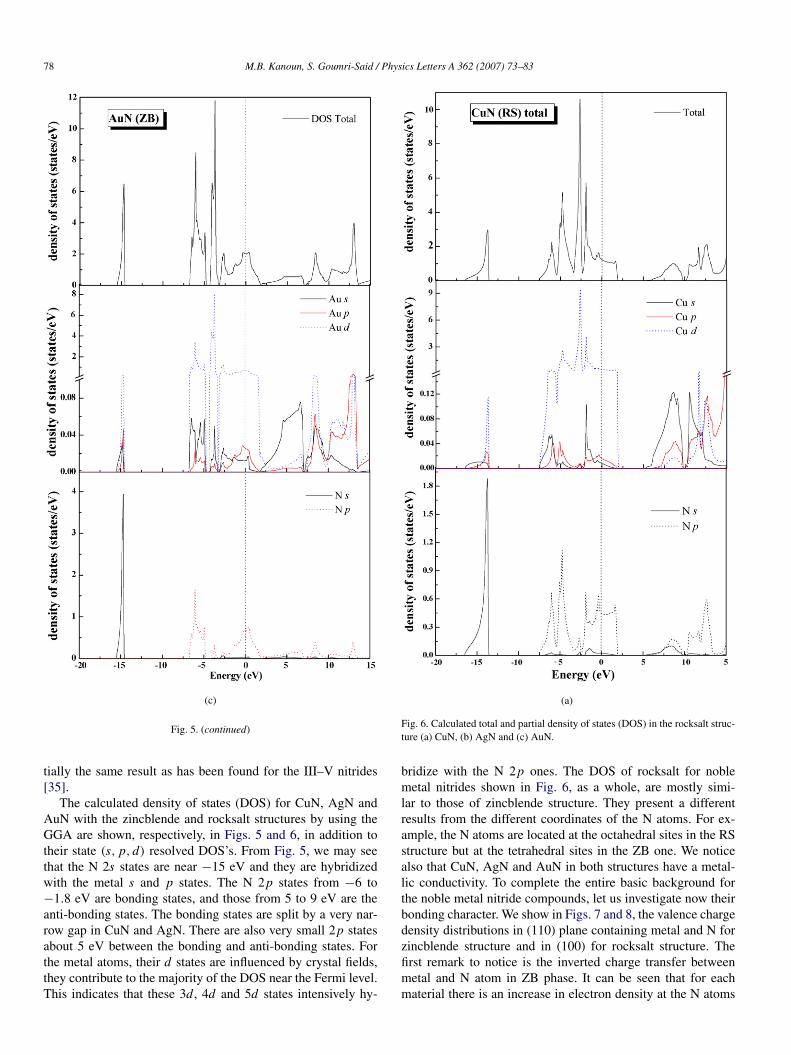
Fig. 5. Calculated total and partial density of states (DOS) in the zincblende structure (a) CuN, (b) AgN and (c) AuN.
exhibits an increase in accordance with the large size of themetal atom.
In Table 1, we list the equilibrium lattice constants and bulkmodulus with the GGA and LDA together. We notice that theZB structure has the largest lattice constant and the CsCl struc-ture has the smallest one with the GGA. The lattice constantsof the RS structure is smaller, but close to the ZB structure. Al-though the N atoms fill the same fcc lattice formed by the metalatoms in the RS and ZB structures, the volume of the octahedralinterstitial sites (for each N atoms in RS structure noble metalnitrides are located), is larger than that of the tetrahedral inter-stitial ones (occupied by the N atoms in the ZB structure). Wecan also report that the GGA yields to a larger lattice constantand smaller bulk modulus compared to the LDA in other no-
ble metal nitrides calculation [34], which agree well with ourown results. Moreover, the GGA yields to ∼ 2%–3% largerlattice constants and 20%–40% smaller bulk modulus than theLDA.
3.2. Electronic properties
The calculated relativistic band structures of CuN, AgN andAuN for zincblende and rocksalt structures are displayed inFigs. 4(a) and (b), respectively. One can predict that noble metalnitrides would be a true metallic at its equilibrium lattice con-stant. We notice that there are three bands crossing the Fermienergy. Moreover, for all these systems the GGA is found toshow a very similar band structures as with LDA. This is essen-
78 M.B. Kanoun, S. Goumri-Said / Physics Letters A 362 (2007) 73–83
(c)
Fig. 5. (continued)
tially the same result as has been found for the III–V nitrides[35].
The calculated density of states (DOS) for CuN, AgN andAuN with the zincblende and rocksalt structures by using theGGA are shown, respectively, in Figs. 5 and 6, in addition totheir state (s,p, d) resolved DOS’s. From Fig. 5, we may seethat the N 2s states are near −15 eV and they are hybridizedwith the metal s and p states. The N 2p states from −6 to−1.8 eV are bonding states, and those from 5 to 9 eV are theanti-bonding states. The bonding states are split by a very nar-row gap in CuN and AgN. There are also very small 2p statesabout 5 eV between the bonding and anti-bonding states. Forthe metal atoms, their d states are influenced by crystal fields,they contribute to the majority of the DOS near the Fermi level.This indicates that these 3d , 4d and 5d states intensively hy-
(a)
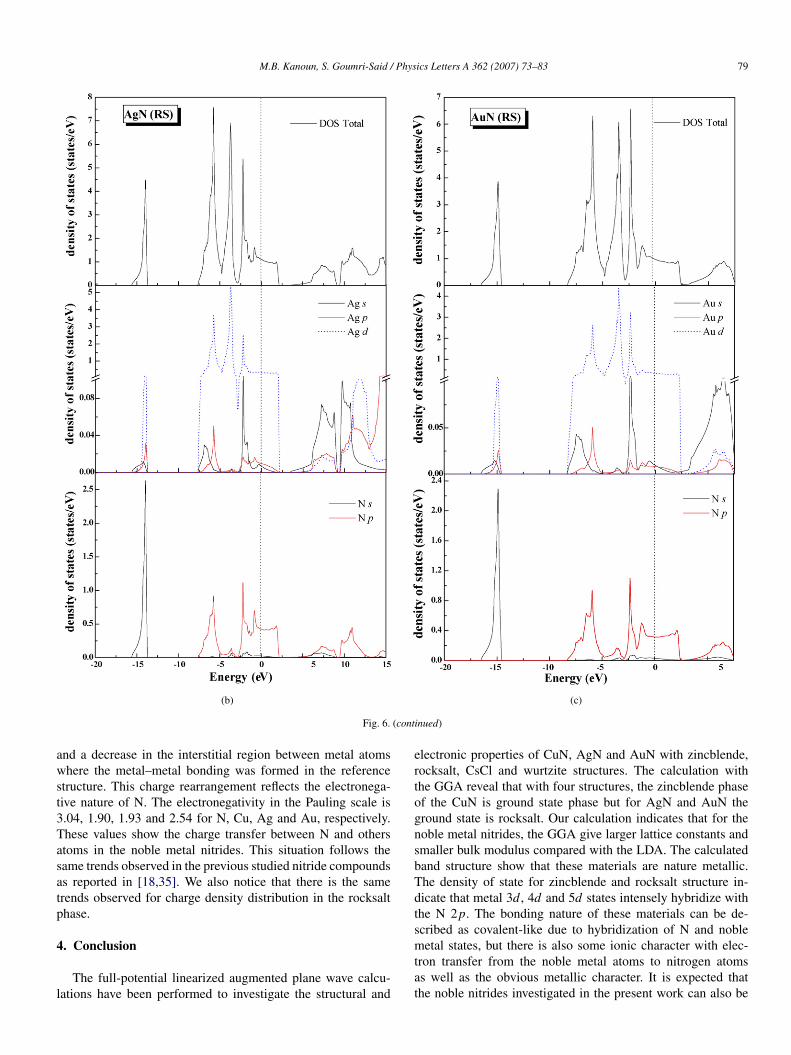
Fig. 6. Calculated total and partial density of states (DOS) in the rocksalt struc-ture (a) CuN, (b) AgN and (c) AuN.
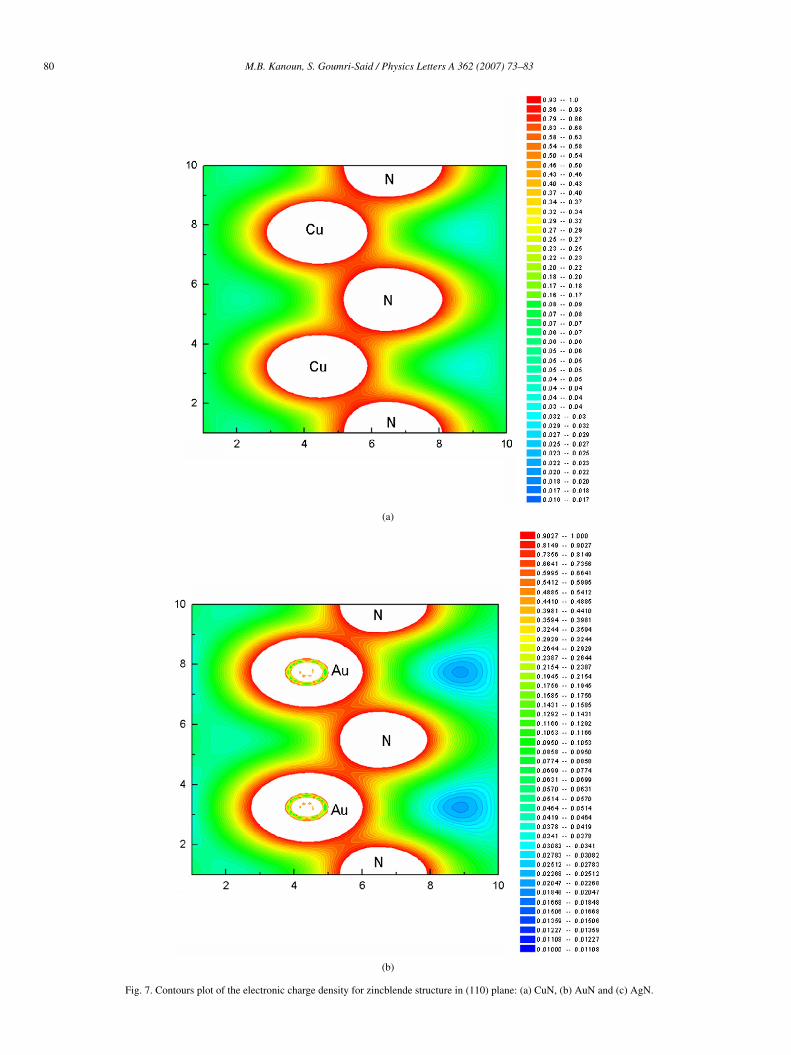
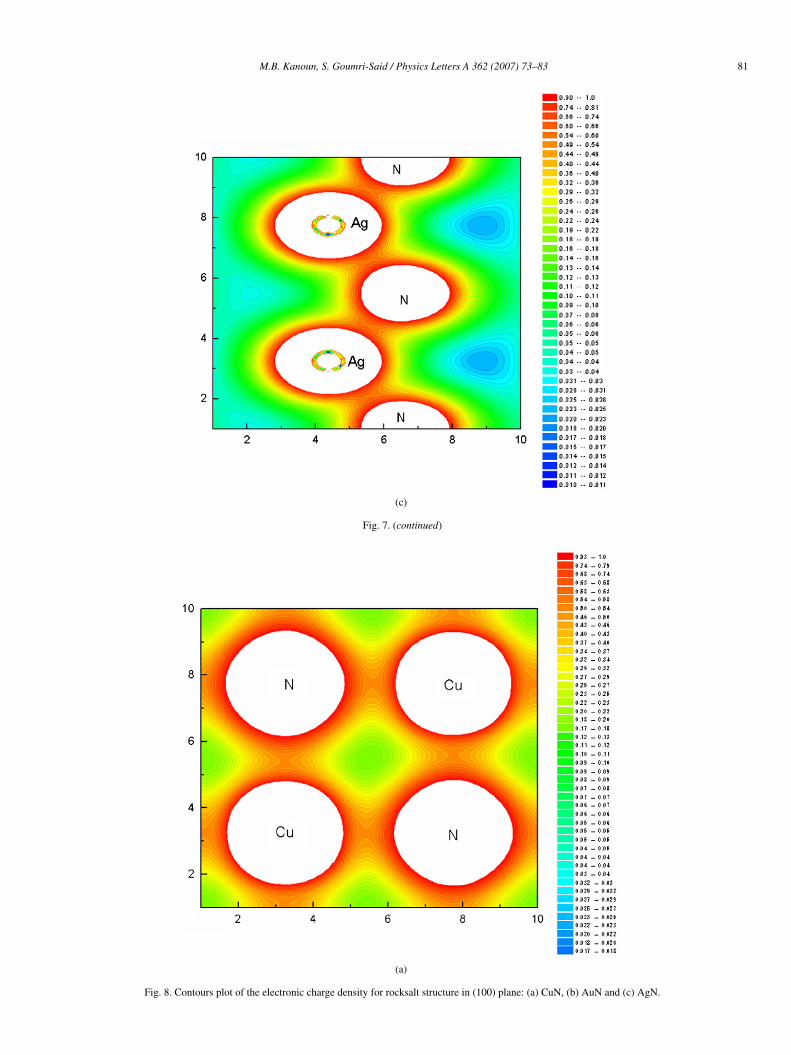
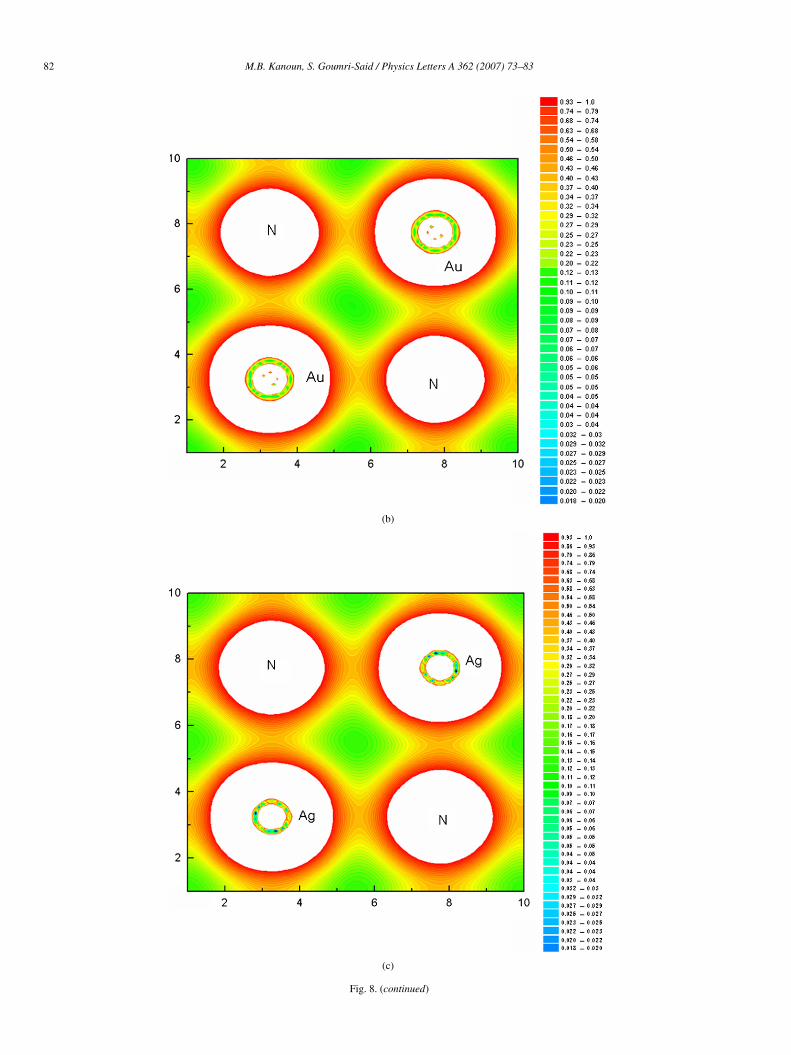
bridize with the N 2p ones. The DOS of rocksalt for noblemetal nitrides shown in Fig. 6, as a whole, are mostly simi-lar to those of zincblende structure. They present a differentresults from the different coordinates of the N atoms. For ex-ample, the N atoms are located at the octahedral sites in the RSstructure but at the tetrahedral sites in the ZB one. We noticealso that CuN, AgN and AuN in both structures have a metal-lic conductivity. To complete the entire basic background forthe noble metal nitride compounds, let us investigate now theirbonding character. We show in Figs. 7 and 8, the valence chargedensity distributions in (110) plane containing metal and N forzincblende structure and in (100) for rocksalt structure. Thefirst remark to notice is the inverted charge transfer betweenmetal and N atom in ZB phase. It can be seen that for eachmaterial there is an increase in electron density at the N atoms
M.B. Kanoun, S. Goumri-Said / Physics Letters A 362 (2007) 73–83 79
(b) (c)
Fig. 6. (continued)
and a decrease in the interstitial region between metal atomswhere the metal–metal bonding was formed in the referencestructure. This charge rearrangement reflects the electronega-tive nature of N. The electronegativity in the Pauling scale is3.04, 1.90, 1.93 and 2.54 for N, Cu, Ag and Au, respectively.These values show the charge transfer between N and othersatoms in the noble metal nitrides. This situation follows thesame trends observed in the previous studied nitride compoundsas reported in [18,35]. We also notice that there is the sametrends observed for charge density distribution in the rocksaltphase.
4. Conclusion
The full-potential linearized augmented plane wave calcu-lations have been performed to investigate the structural and
electronic properties of CuN, AgN and AuN with zincblende,rocksalt, CsCl and wurtzite structures. The calculation withthe GGA reveal that with four structures, the zincblende phaseof the CuN is ground state phase but for AgN and AuN theground state is rocksalt. Our calculation indicates that for thenoble metal nitrides, the GGA give larger lattice constants andsmaller bulk modulus compared with the LDA. The calculatedband structure show that these materials are nature metallic.The density of state for zincblende and rocksalt structure in-dicate that metal 3d , 4d and 5d states intensely hybridize withthe N 2p. The bonding nature of these materials can be de-scribed as covalent-like due to hybridization of N and noblemetal states, but there is also some ionic character with elec-tron transfer from the noble metal atoms to nitrogen atomsas well as the obvious metallic character. It is expected thatthe noble nitrides investigated in the present work can also be
80 M.B. Kanoun, S. Goumri-Said / Physics Letters A 362 (2007) 73–83
(a)
(b)
Fig. 7. Contours plot of the electronic charge density for zincblende structure in (110) plane: (a) CuN, (b) AuN and (c) AgN.
M.B. Kanoun, S. Goumri-Said / Physics Letters A 362 (2007) 73–83 81
(c)
Fig. 7. (continued)
(a)
Fig. 8. Contours plot of the electronic charge density for rocksalt structure in (100) plane: (a) CuN, (b) AuN and (c) AgN.
82 M.B. Kanoun, S. Goumri-Said / Physics Letters A 362 (2007) 73–83
(b)
(c)
Fig. 8. (continued)

M.B. Kanoun, S. Goumri-Said / Physics Letters A 362 (2007) 73–83 83
synthesized in forms of bulk or thin film under suitable condi-tions.
References
[1] L.E. Toth, Transition Metal Carbides and Nitrides, Academic Press, NewYork, 1971.
[2] H. Pierson, Handbook of Refractory Carbides and Nitrides: Properties,Characteristics and Applications, Noyes, Westwood, NJ, 1996.
[3] S.-H. Jhi, J. Ihm, S.G. Louie, M.L. Cohen, Nature (London) 399 (1999)132.
[4] P.F. McMillan, Nat. Mater. 1 (2003) 19.[5] S. Yamanaka, K. Hotehama, H. Kawaji, Nature (London) 392 (1998) 580.[6] A. Zerr, G. Miehe, R. Boehler, Nat. Mater. 2 (2003) 185.[7] P. Kroll, Phys. Rev. Lett. 90 (2003) 125501.[8] T. Maruyama, T. Morishita, Appl. Phys. Lett. 69 (1996) 890.[9] J.S. Chun, I. Petrov, J.E. Greene, J. Appl. Phys. 86 (1999) 3633.
[10] H. Al-Brithen, A.R. Smith, Appl. Phys. Lett. 77 (2000) 2485.[11] Q. Zhan, R. Yu, L.L. He, D.X. Li, Thin Solid Films 411 (2002) 225.[12] V. Ranjan, L. Bellaiche, E.J. Walter, Phys. Rev. Lett. 90 (2003) 257602.[13] Q. Zhan, R. Yu, L.L. He, D.X. Li, H.B. Nie, C.K. Ong, Mater. Lett. 57
(2003) 3904.[14] M.G. Moreno-Armenta, A. Martinez-Ruiz, N. Takeuchi, Solid State Sci. 6
(2004) 9.[15] E.S. Shanley, J.L. Ennis, Ind. Eng. Chem. Res. 30 (1991) 2503.[16] S. Krishnamurthy, M. Montalti, M.G. Wardle, M.J. Shaw, P.R. Briddon,
K. Svensson, M.R.C. Hunt, L. Šiller, Phys. Rev. B 70 (2004) 045414.[17] E. Gregoryanz, C. Sanloup, M. Somayazulu, J. Badro, G. Fiquet, H.K.
Mao, R.J. Hemley, Nat. Mater. 3 (2004) 294.[18] M.B. Kanoun, S. Goumri-Said, Phys. Rev. B 72 (2005) 113103.
[19] R. Yu, X.F. Zhang, Appl. Phys. Lett. 86 (2005) 121913.[20] B.R. Sahu, L. Kleinman, Phys. Rev. B 71 (2005) 041101R.[21] J. Uddin, G.E. Scuseria, Phys. Rev. B 72 (2005) 035101.[22] C.Z. Fan, L. Sun, J. Zhang, Y. Jia, Z. Wei, R. Liu, S. Zeng, W. Wang, Chin.
Sci. Bull. 50 (2005) 1079;C.Z. Fan, L. Sun, Y. Wang, Z. Wei, R. Liu, S. Zeng, W. Wang, Chin. Phys.Lett. 22 (2005) 2637.
[23] S. Patil, S. Khare, B. Tuttle, J. Boarding, S. Kodambakara, Phys. Rev. B 73(2006) 104118.
[24] A.F. Young, J.A. Montoya, C. Sanloup, M. Lazzeri, E. Gregoryanz, S.Scandolo, Phys. Rev. B 73 (2006) 153102.
[25] R. Dreizler, E.K.U. Gross, Density-Functional Theory, Springer-Verlag,New York, 1990.
[26] J.P. Perdew, S. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865.[27] P. Blaha, K. Schwarz, G.K.H. Madsen, D. Kvasnicka, J. Luitz, WIEN2k,
An Augmented-Plane-Wave + Local Orbitals Program for CalculatingCrystal Properties, Karlheinz Schwarz, Techn Wien, Austria, 2001, ISBN3-9501031-1-2.
[28] D.M. Ceperley, B.J. Alder, Phys. Rev. Lett. 45 (1980) 556.[29] J.P. Perdew, Y. Wang, Phys. Rev. B 45 (1992) 13244.[30] D.D. Koelling, B.N. Harmon, J. Phys. C 10 (1977) 3107.[31] H.J. Monkhorst, J.D. Pack, Phys. Rev. B 13 (1976) 5188.[32] F.D. Murnaghan, Proc. Natl. Acad. Sci. U.S.A. 30 (1944) 244.[33] H. Wang, D. Xue, Chin. Phys. Lett. 21 (2004) 1612.[34] C. Stampft, W. Mannstadt, R. Asahi, A.J. Freeman. Phys. Rev. B 63 (2001)
155106.[35] M.B. Kanoun, S. Goumri-Said, A.E. Merad, G. Merad, J. Cibert, H.
Aourag, Semicond. Sci. Technol. 19 (2004) 1220.