photo-assisted electrochemical treatment of municipal wastewater reverse osmosis concentrate
TRANSCRIPT

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/authorsrights

Author's personal copy
Photo-assisted electrochemical treatment of municipal wastewaterreverse osmosis concentrate
Gil Hurwitz a,⇑, Eric M.V. Hoek a,b, Kai Liu c, Linhua Fan c, Felicity A. Roddick c
a Department of Civil & Environmental Engineering, California NanoSystems Institute and Institute of the Environment & Sustainability, University of California, Los Angeles, CA, USAb Department of Applied Chemistry, University of Johannesburg, Johannesburg, South Africac RMIT University, School of Civil, Environmental & Chemical Engineering, Melbourne, Victoria, Australia
h i g h l i g h t s
� Organics in RO concentrate were treated by a hybrid advanced oxidation process.� Hybrid photolytic and electrochemical process showed synergistic DOC degradation.� Electrochemical breaks down fluorescence and photolysis destroys core structure.� Hybrid treatment consumes less energy than photolysis alone.
a r t i c l e i n f o
Article history:Received 4 December 2013Received in revised form 19 March 2014Accepted 21 March 2014Available online 3 April 2014
Keywords:PhotolysisElectrochemical oxidationSynergyWastewaterReverse osmosis concentrate
a b s t r a c t
The combination of a photochemical (UV) and electrochemical (EL) process led to enhanced degradationof dissolved organic matter in reverse osmosis (RO) concentrate produced from municipal wastewater.Treatment by UV and EL alone resulted in 25% and 35% removal of dissolved organic carbon (DOC) after5 h, respectively. However, the hybrid process (UVEL) degraded more than 80% of DOC after the sametreatment time. Fluorescence excitation–emission matrix spectroscopy and size exclusion chromatogra-phy suggest simultaneous and cooperative degradation of backbone aliphatic bonds by UV and aromaticring cleavage by EL within the UVEL process. Overall, UVEL treatment led to efficient and non-selectivedegradation of dissolved organics over a wide range of molecular weights. Further, energy consumptionand halogenated by-product formation (typically a limiting factor for the application of oxidationtechnology) were reduced in the UVEL process relative to the UV and EL processes, respectively.
� 2014 Elsevier B.V. All rights reserved.
1. Introduction
Municipal wastewater effluent treated by RO produces highlypurified product water with many valuable uses (e.g., landscapeirrigation, industrial process water, and aquifer recharge).However, the RO process also produces a highly concentratedversion of the wastewater effluent, which may contain elevatedconcentrations of contaminants such as dissolved and suspendedsolids comprising BOD, COD, pathogens, and trace organics (e.g.,pharmaceuticals and personal care products, endocrine disruptingcompounds, and disinfection byproducts) not fully removed by thepreceding biological treatment process [1–3]. The potential humanand environmental health hazards associated with discharging
wastewater RO concentrate containing elevated levels ofpathogens, BOD/COD, and trace organics are not fully understood[4–10]. Therefore, it seems prudent to explore treatment technolo-gies that can ensure effluent discharge standards are met, particu-larly when dilution (i.e., mixing the RO concentrate with largevolumes of unconcentrated wastewater treatment plant effluent)is not possible. In particular, further disinfection and organicremoval may be necessary.
Both photochemical and electrochemical processes can be usedfor disinfection and organic removal. Direct UV irradiation ofwastewater gives rise to photo-activated chemical reactions thattranslate absorbed photonic energy into direct chemical bondbreakage or the production of free radicals [11,12]. Utilization ofphotolysis for advanced oxidation is typically achieved by theaddition of catalysts or UV reactive compounds (i.e., H2O2 andTiO2) to provide a source of the requisite hydroxyl radicals [12].Electrochemical oxidation targets mineralization of organiccompounds by electron abstraction at the anode surface or by the
http://dx.doi.org/10.1016/j.cej.2014.03.0841385-8947/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding author. Current address: Water Planet Engineering, 721 GlasgowAve, Unit D, Los Angeles, CA 90301, USA. Tel.: +1 (310) 215 8960; fax: +1 (310) 2158961.
E-mail address: [email protected] (G. Hurwitz).
Chemical Engineering Journal 249 (2014) 180–188
Contents lists available at ScienceDirect
Chemical Engineering Journal
journal homepage: www.elsevier .com/locate /ce j

Author's personal copy
production of highly reactive oxidants, such as �OH, produced in situelectrochemically [13]. In situ production of chemical reactantseliminates the need to purchase, transport, and store chemicalsproduced off-site, which can save money and energy as well asreduce carbon emissions and potential security vulnerabilities [14].
Electrochemical oxidation may be an attractive option for treat-ing wastewater RO concentrate because it becomes more efficientand effective with increased conductivity and chloride concentra-tion. High conductivity reduces the voltage needed to achieve atarget current density, which results in considerable energy sav-ings. However, electrochemical processes traditionally suffer fromhigh investment costs, especially for advanced anode materials,such as boron-doped diamond. For this reason, improving electro-chemical oxidation efficiency is paramount in order to reduce therequired active electrode area and, thus, the initial capital costfor a given electrochemical treatment system [15–17]. Advancedanode materials have garnered considerable attention recentlydue to their ability to improve oxidation efficiency by enhancingthe rate of direct oxidation at or near the electrode surface. Gener-ally, advanced anodes are characterized as ‘‘nonactive’’ materialswith high oxygen evolution overpotentials, such as antimony-doped tin oxide, lead dioxide, and boron-doped diamond, and favorthe complete oxidation of organics to CO2 due to their weak surfaceinteractions with hydroxyl radicals [18]. Boron-doped diamondalso shares the ability of some active materials, such as Ti/RuO2,to promote indirect bulk oxidation by the production of intermedi-ate oxidants, of which chlorine is commonly targeted [19,20]. Spe-cifically, chloride ions become oxidized at the anode; thus,introducing an entire family of potential secondary oxidants anddisinfectants. Indirect organic oxidation may occur via reactionwith chlorine radicals (�Cl) adsorbed on the anode surface or byreactive chlorine species such as Cl2, HOCl, or OCl� in the bulk solu-tion [13]. However, the formation of halogenated by-products is anobvious concern when treating waters containing both halide ionsand dissolved organic matter.
Significant research has been undertaken to determine theeffectiveness of hybrid photo-assisted electrochemical oxidationprocesses with particular emphasis on the UV excitation of photoa-nodes (e.g., thin-film TiO2, DSA�) [21–25]. However, due to its min-imal anodic photoactivity, we have not been able to find previousresearch studying the use of boron-doped diamond (a highly prom-ising material with superior electrochemical properties) electrodesin hybrid photo-assisted electrochemical oxidation processes [26].Herein, we tested a hybrid photo-assisted electrochemical process(UVEL) with the electrochemical and photochemical reactorsplaced in series within a batch recirculation system. This uniquereactor configuration can, therefore, take advantage of the electro-chemical benefits of boron-doped diamond electrodes (widepotential window, low background current, high chemical stability,high resistance to deactivation [27–30]) without the need to investenergy and resources to enhance its photoactivity. The aim of thisresearch is to study the practical limitations (extent of mineraliza-tion, energy consumption, by-product formation) of a photo-assisted electrochemical reactor, which integrates highly efficientboron-doped diamond electrodes to treat a target water of growingpublic and environmental concern.
2. Methodology
Biologically treated wastewater effluent was provided byWestern Treatment Plant (Victoria, Australia), which used asequential activated sludge-lagoon treatment (AS-lagoon) process.RO concentrate was prepared from the effluent using a laboratoryscale plate-and-frame unit (Sepa CF, GE-Osmonics, Minnetonka,MN) and a commercial interfacial composite RO membrane (AG;GE-Osmonics, Minnetonka, MN) operated at a constant pressure
and temperature of 17 bar and 20 �C and an average permeate fluxof 46.8 LMH. The effluent was concentrated to a recovery of 65%.Major water quality parameters are given in Table 1.
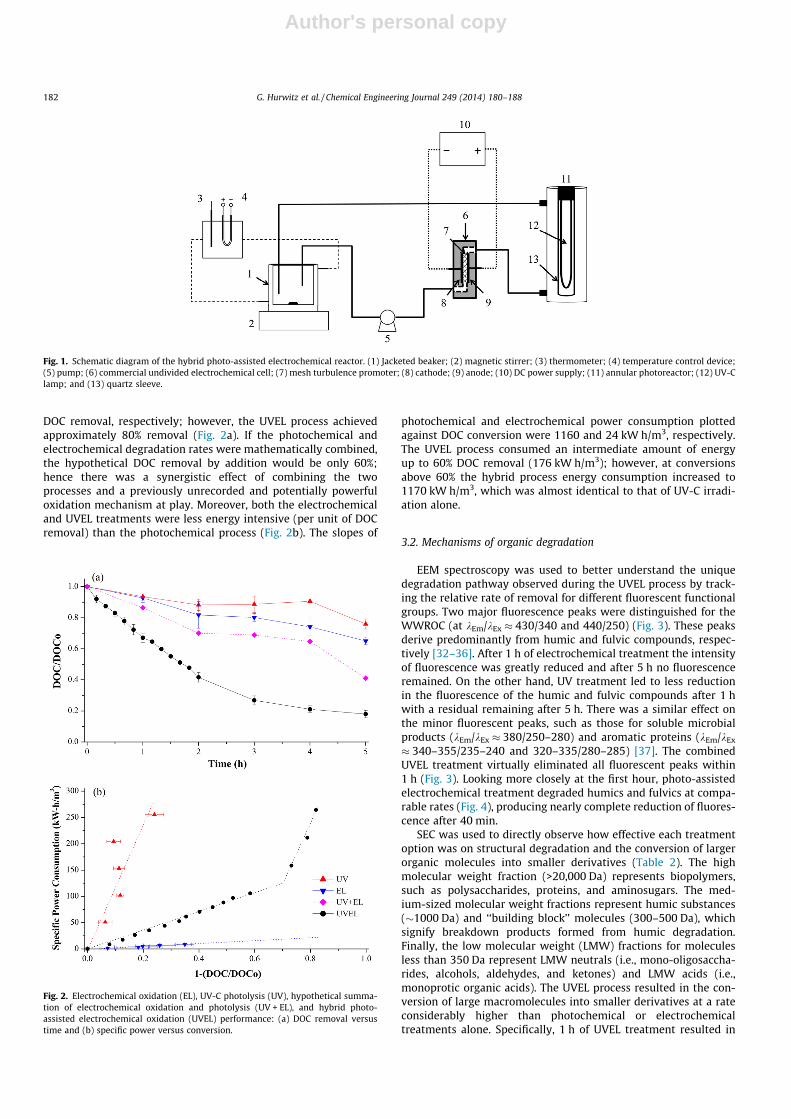
Electrochemical oxidation (EL) experiments were performed ina flow-through, undivided, spacer-filled electrochemical cell. ADiaChem� (Electrocell, Amherst, NY) boron-doped diamond anodewas used with a geometric active area of 10 cm2. Electrochemicaloxidation was performed at a constant current density of 20 mA/cm2 with a flow rate of 0.033 m3/h and Reynolds number of 360.Photolysis (UV) experiments were conducted using an annularreactor with a centrally mounted lamp. A low-pressure mercury-vapor lamp emitting at 254 nm with an intensity of 8500 lW/cm2 was used as the UV-C irradiation source (G36T15NU; Austra-lian Ultra Violet Services, Victoria, Australia). The average irradi-ated area was 464 cm2 with a path length of 1.94 cm; other UVreactor conditions were reported elsewhere [11]. A Titan 1500 chil-ler (Aqua-Medic, Bissendorf, Germany) was used to maintain aconstant solution temperature of 20 �C during all the experiments.The UVEL reactor subjected the wastewater RO concentrate(WWROC) to both electrochemical oxidation and UV photolysisin series, recirculation mode (Fig. 1). Specific power consumptionwas estimated from the power consumed over the operating timeand normalized by the total volume processed.
Dissolved organic carbon (DOC) was determined by measuringthe total organic carbon (TOC) content (Sievers 5310 C, GE,Boulder, Colorado, USA) of 0.45 lm filtered samples. The TOC ana-lyzer was equipped with an auto-sampler and inorganic carbonwas internally purged automatically (Sievers 900 ICR; GE, Boulder,Colorado, USA) prior to DOC analysis. The absorbance was mea-sured using a Unicam UV–vis spectrophotometer with a quartz cellof 1 cm pathlength. The excitation–emission matrix (EEM) spectraof the samples were obtained with a fluorescence spectrometer(LS55, Perkin Elmer, Waltham, Massachusetts, USA). The sizeexclusion chromatography (SEC) analyses were carried out at theWater Research Centre of the University of New South Wales,Australia. The SEC system (LC-OCD Model 8, DOC-Labor Dr. Hüber,Germany) consisted of a SEC column (Toyopearl TSK HW-50S,diameter 2 cm, length 25 cm) to separate organic molecules by size(i.e., 100–200,000 Da). Trihalomethane concentrations weredetermined by the Australian Water Quality Centre in Adelaide,Australia using previously described methods [31].
3. Results
3.1. DOC removal and energy consumption
After 5 h of treatment of the WWROC, the electrochemical andphotochemical processes resulted in approximately 25% and 35%
Table 1Water quality of secondary wastewater RO concentrate.
Parameter Unit Value
Conductivity mS/cm 3.8pH – 8.8DOC mg/L 22A254 /cm 0.42Na+ mg/L 600Ca2+ mg/L 47Mg2+ mg/L 53Cl� mg/L 954Br� mg/L 3.9SO4
2� mg/L 207TN, as N mg N/L 32TP, as P mg P/L 8Alkalinity, as CaCO3 mg CaCO3/L 242Color (Pt–Co) mg Pt–Co/L 55Turbidity NTU 0.5
G. Hurwitz et al. / Chemical Engineering Journal 249 (2014) 180–188 181
Author's personal copy
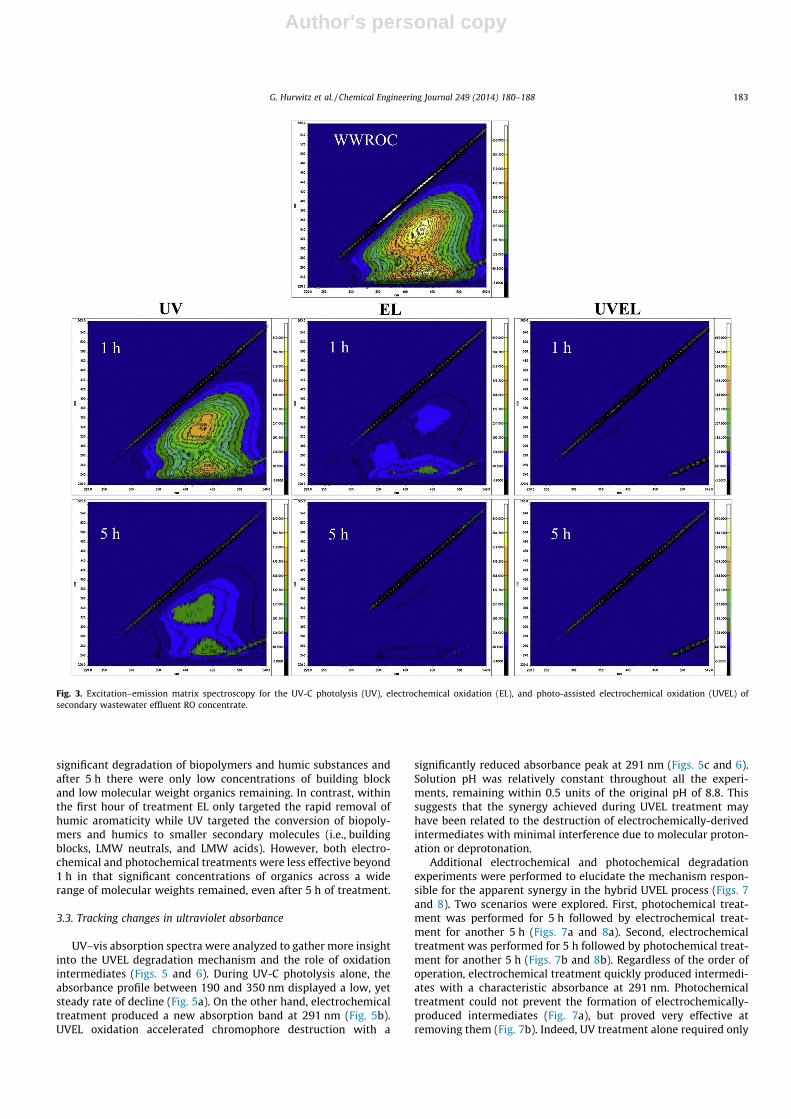
DOC removal, respectively; however, the UVEL process achievedapproximately 80% removal (Fig. 2a). If the photochemical andelectrochemical degradation rates were mathematically combined,the hypothetical DOC removal by addition would be only 60%;hence there was a synergistic effect of combining the twoprocesses and a previously unrecorded and potentially powerfuloxidation mechanism at play. Moreover, both the electrochemicaland UVEL treatments were less energy intensive (per unit of DOCremoval) than the photochemical process (Fig. 2b). The slopes of
photochemical and electrochemical power consumption plottedagainst DOC conversion were 1160 and 24 kW h/m3, respectively.The UVEL process consumed an intermediate amount of energyup to 60% DOC removal (176 kW h/m3); however, at conversionsabove 60% the hybrid process energy consumption increased to1170 kW h/m3, which was almost identical to that of UV-C irradi-ation alone.
3.2. Mechanisms of organic degradation
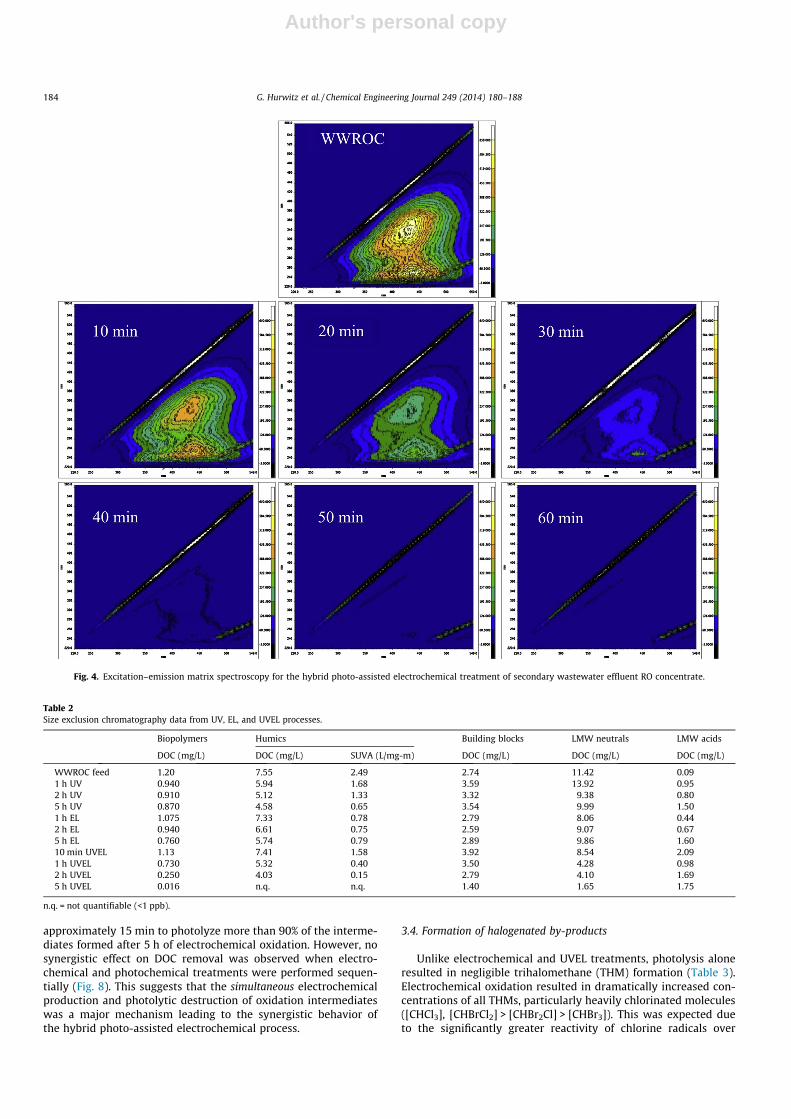
EEM spectroscopy was used to better understand the uniquedegradation pathway observed during the UVEL process by track-ing the relative rate of removal for different fluorescent functionalgroups. Two major fluorescence peaks were distinguished for theWWROC (at kEm/kEx � 430/340 and 440/250) (Fig. 3). These peaksderive predominantly from humic and fulvic compounds, respec-tively [32–36]. After 1 h of electrochemical treatment the intensityof fluorescence was greatly reduced and after 5 h no fluorescenceremained. On the other hand, UV treatment led to less reductionin the fluorescence of the humic and fulvic compounds after 1 hwith a residual remaining after 5 h. There was a similar effect onthe minor fluorescent peaks, such as those for soluble microbialproducts (kEm/kEx � 380/250–280) and aromatic proteins (kEm/kEx
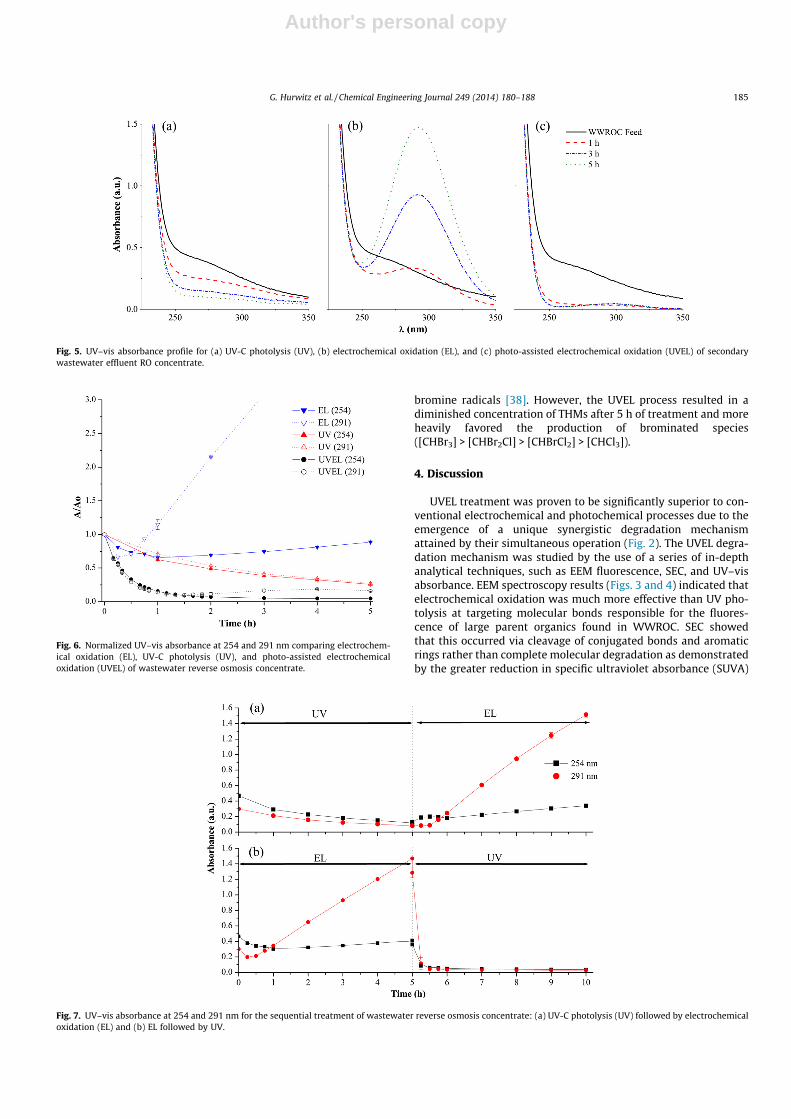
� 340–355/235–240 and 320–335/280–285) [37]. The combinedUVEL treatment virtually eliminated all fluorescent peaks within1 h (Fig. 3). Looking more closely at the first hour, photo-assistedelectrochemical treatment degraded humics and fulvics at compa-rable rates (Fig. 4), producing nearly complete reduction of fluores-cence after 40 min.
SEC was used to directly observe how effective each treatmentoption was on structural degradation and the conversion of largerorganic molecules into smaller derivatives (Table 2). The highmolecular weight fraction (>20,000 Da) represents biopolymers,such as polysaccharides, proteins, and aminosugars. The med-ium-sized molecular weight fractions represent humic substances(�1000 Da) and ‘‘building block’’ molecules (300–500 Da), whichsignify breakdown products formed from humic degradation.Finally, the low molecular weight (LMW) fractions for moleculesless than 350 Da represent LMW neutrals (i.e., mono-oligosaccha-rides, alcohols, aldehydes, and ketones) and LMW acids (i.e.,monoprotic organic acids). The UVEL process resulted in the con-version of large macromolecules into smaller derivatives at a rateconsiderably higher than photochemical or electrochemicaltreatments alone. Specifically, 1 h of UVEL treatment resulted in
Fig. 1. Schematic diagram of the hybrid photo-assisted electrochemical reactor. (1) Jacketed beaker; (2) magnetic stirrer; (3) thermometer; (4) temperature control device;(5) pump; (6) commercial undivided electrochemical cell; (7) mesh turbulence promoter; (8) cathode; (9) anode; (10) DC power supply; (11) annular photoreactor; (12) UV-Clamp; and (13) quartz sleeve.
Fig. 2. Electrochemical oxidation (EL), UV-C photolysis (UV), hypothetical summa-tion of electrochemical oxidation and photolysis (UV + EL), and hybrid photo-assisted electrochemical oxidation (UVEL) performance: (a) DOC removal versustime and (b) specific power versus conversion.
182 G. Hurwitz et al. / Chemical Engineering Journal 249 (2014) 180–188
Author's personal copy
significant degradation of biopolymers and humic substances andafter 5 h there were only low concentrations of building blockand low molecular weight organics remaining. In contrast, withinthe first hour of treatment EL only targeted the rapid removal ofhumic aromaticity while UV targeted the conversion of biopoly-mers and humics to smaller secondary molecules (i.e., buildingblocks, LMW neutrals, and LMW acids). However, both electro-chemical and photochemical treatments were less effective beyond1 h in that significant concentrations of organics across a widerange of molecular weights remained, even after 5 h of treatment.
3.3. Tracking changes in ultraviolet absorbance
UV–vis absorption spectra were analyzed to gather more insightinto the UVEL degradation mechanism and the role of oxidationintermediates (Figs. 5 and 6). During UV-C photolysis alone, theabsorbance profile between 190 and 350 nm displayed a low, yetsteady rate of decline (Fig. 5a). On the other hand, electrochemicaltreatment produced a new absorption band at 291 nm (Fig. 5b).UVEL oxidation accelerated chromophore destruction with a
significantly reduced absorbance peak at 291 nm (Figs. 5c and 6).Solution pH was relatively constant throughout all the experi-ments, remaining within 0.5 units of the original pH of 8.8. Thissuggests that the synergy achieved during UVEL treatment mayhave been related to the destruction of electrochemically-derivedintermediates with minimal interference due to molecular proton-ation or deprotonation.
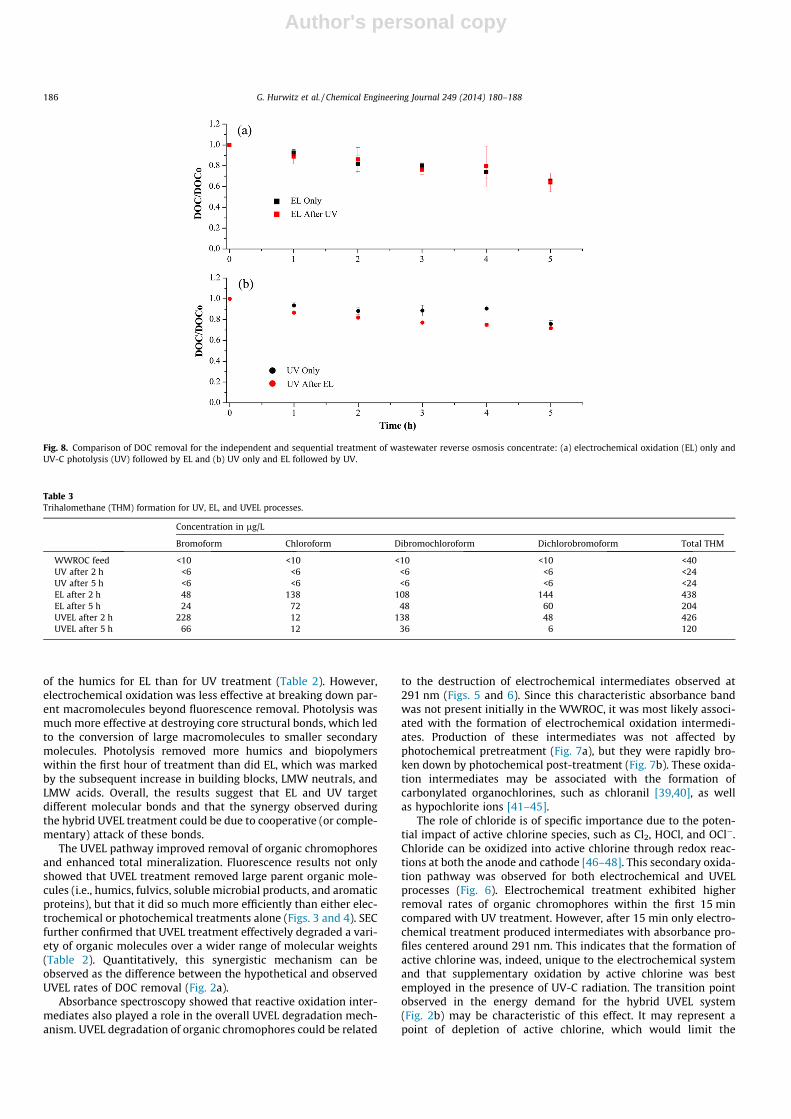
Additional electrochemical and photochemical degradationexperiments were performed to elucidate the mechanism respon-sible for the apparent synergy in the hybrid UVEL process (Figs. 7and 8). Two scenarios were explored. First, photochemical treat-ment was performed for 5 h followed by electrochemical treat-ment for another 5 h (Figs. 7a and 8a). Second, electrochemicaltreatment was performed for 5 h followed by photochemical treat-ment for another 5 h (Figs. 7b and 8b). Regardless of the order ofoperation, electrochemical treatment quickly produced intermedi-ates with a characteristic absorbance at 291 nm. Photochemicaltreatment could not prevent the formation of electrochemically-produced intermediates (Fig. 7a), but proved very effective atremoving them (Fig. 7b). Indeed, UV treatment alone required only
Fig. 3. Excitation–emission matrix spectroscopy for the UV-C photolysis (UV), electrochemical oxidation (EL), and photo-assisted electrochemical oxidation (UVEL) ofsecondary wastewater effluent RO concentrate.
G. Hurwitz et al. / Chemical Engineering Journal 249 (2014) 180–188 183
Author's personal copy
approximately 15 min to photolyze more than 90% of the interme-diates formed after 5 h of electrochemical oxidation. However, nosynergistic effect on DOC removal was observed when electro-chemical and photochemical treatments were performed sequen-tially (Fig. 8). This suggests that the simultaneous electrochemicalproduction and photolytic destruction of oxidation intermediateswas a major mechanism leading to the synergistic behavior ofthe hybrid photo-assisted electrochemical process.
3.4. Formation of halogenated by-products
Unlike electrochemical and UVEL treatments, photolysis aloneresulted in negligible trihalomethane (THM) formation (Table 3).Electrochemical oxidation resulted in dramatically increased con-centrations of all THMs, particularly heavily chlorinated molecules([CHCl3], [CHBrCl2] > [CHBr2Cl] > [CHBr3]). This was expected dueto the significantly greater reactivity of chlorine radicals over
Fig. 4. Excitation–emission matrix spectroscopy for the hybrid photo-assisted electrochemical treatment of secondary wastewater effluent RO concentrate.
Table 2Size exclusion chromatography data from UV, EL, and UVEL processes.
Biopolymers Humics Building blocks LMW neutrals LMW acids
DOC (mg/L) DOC (mg/L) SUVA (L/mg-m) DOC (mg/L) DOC (mg/L) DOC (mg/L)
WWROC feed 1.20 7.55 2.49 2.74 11.42 0.091 h UV 0.940 5.94 1.68 3.59 13.92 0.952 h UV 0.910 5.12 1.33 3.32 9.38 0.805 h UV 0.870 4.58 0.65 3.54 9.99 1.501 h EL 1.075 7.33 0.78 2.79 8.06 0.442 h EL 0.940 6.61 0.75 2.59 9.07 0.675 h EL 0.760 5.74 0.79 2.89 9.86 1.6010 min UVEL 1.13 7.41 1.58 3.92 8.54 2.091 h UVEL 0.730 5.32 0.40 3.50 4.28 0.982 h UVEL 0.250 4.03 0.15 2.79 4.10 1.695 h UVEL 0.016 n.q. n.q. 1.40 1.65 1.75
n.q. = not quantifiable (<1 ppb).
184 G. Hurwitz et al. / Chemical Engineering Journal 249 (2014) 180–188
Author's personal copy
bromine radicals [38]. However, the UVEL process resulted in adiminished concentration of THMs after 5 h of treatment and moreheavily favored the production of brominated species([CHBr3] > [CHBr2Cl] > [CHBrCl2] > [CHCl3]).
4. Discussion
UVEL treatment was proven to be significantly superior to con-ventional electrochemical and photochemical processes due to theemergence of a unique synergistic degradation mechanismattained by their simultaneous operation (Fig. 2). The UVEL degra-dation mechanism was studied by the use of a series of in-depthanalytical techniques, such as EEM fluorescence, SEC, and UV–visabsorbance. EEM spectroscopy results (Figs. 3 and 4) indicated thatelectrochemical oxidation was much more effective than UV pho-tolysis at targeting molecular bonds responsible for the fluores-cence of large parent organics found in WWROC. SEC showedthat this occurred via cleavage of conjugated bonds and aromaticrings rather than complete molecular degradation as demonstratedby the greater reduction in specific ultraviolet absorbance (SUVA)
Fig. 5. UV–vis absorbance profile for (a) UV-C photolysis (UV), (b) electrochemical oxidation (EL), and (c) photo-assisted electrochemical oxidation (UVEL) of secondarywastewater effluent RO concentrate.
Fig. 6. Normalized UV–vis absorbance at 254 and 291 nm comparing electrochem-ical oxidation (EL), UV-C photolysis (UV), and photo-assisted electrochemicaloxidation (UVEL) of wastewater reverse osmosis concentrate.
Fig. 7. UV–vis absorbance at 254 and 291 nm for the sequential treatment of wastewater reverse osmosis concentrate: (a) UV-C photolysis (UV) followed by electrochemicaloxidation (EL) and (b) EL followed by UV.
G. Hurwitz et al. / Chemical Engineering Journal 249 (2014) 180–188 185
Author's personal copy
of the humics for EL than for UV treatment (Table 2). However,electrochemical oxidation was less effective at breaking down par-ent macromolecules beyond fluorescence removal. Photolysis wasmuch more effective at destroying core structural bonds, which ledto the conversion of large macromolecules to smaller secondarymolecules. Photolysis removed more humics and biopolymerswithin the first hour of treatment than did EL, which was markedby the subsequent increase in building blocks, LMW neutrals, andLMW acids. Overall, the results suggest that EL and UV targetdifferent molecular bonds and that the synergy observed duringthe hybrid UVEL treatment could be due to cooperative (or comple-mentary) attack of these bonds.
The UVEL pathway improved removal of organic chromophoresand enhanced total mineralization. Fluorescence results not onlyshowed that UVEL treatment removed large parent organic mole-cules (i.e., humics, fulvics, soluble microbial products, and aromaticproteins), but that it did so much more efficiently than either elec-trochemical or photochemical treatments alone (Figs. 3 and 4). SECfurther confirmed that UVEL treatment effectively degraded a vari-ety of organic molecules over a wider range of molecular weights(Table 2). Quantitatively, this synergistic mechanism can beobserved as the difference between the hypothetical and observedUVEL rates of DOC removal (Fig. 2a).
Absorbance spectroscopy showed that reactive oxidation inter-mediates also played a role in the overall UVEL degradation mech-anism. UVEL degradation of organic chromophores could be related
to the destruction of electrochemical intermediates observed at291 nm (Figs. 5 and 6). Since this characteristic absorbance bandwas not present initially in the WWROC, it was most likely associ-ated with the formation of electrochemical oxidation intermedi-ates. Production of these intermediates was not affected byphotochemical pretreatment (Fig. 7a), but they were rapidly bro-ken down by photochemical post-treatment (Fig. 7b). These oxida-tion intermediates may be associated with the formation ofcarbonylated organochlorines, such as chloranil [39,40], as wellas hypochlorite ions [41–45].
The role of chloride is of specific importance due to the poten-tial impact of active chlorine species, such as Cl2, HOCl, and OCl�.Chloride can be oxidized into active chlorine through redox reac-tions at both the anode and cathode [46–48]. This secondary oxida-tion pathway was observed for both electrochemical and UVELprocesses (Fig. 6). Electrochemical treatment exhibited higherremoval rates of organic chromophores within the first 15 mincompared with UV treatment. However, after 15 min only electro-chemical treatment produced intermediates with absorbance pro-files centered around 291 nm. This indicates that the formation ofactive chlorine was, indeed, unique to the electrochemical systemand that supplementary oxidation by active chlorine was bestemployed in the presence of UV-C radiation. The transition pointobserved in the energy demand for the hybrid UVEL system(Fig. 2b) may be characteristic of this effect. It may represent apoint of depletion of active chlorine, which would limit the
Fig. 8. Comparison of DOC removal for the independent and sequential treatment of wastewater reverse osmosis concentrate: (a) electrochemical oxidation (EL) only andUV-C photolysis (UV) followed by EL and (b) UV only and EL followed by UV.
Table 3Trihalomethane (THM) formation for UV, EL, and UVEL processes.
Concentration in lg/L
Bromoform Chloroform Dibromochloroform Dichlorobromoform Total THM
WWROC feed <10 <10 <10 <10 <40UV after 2 h <6 <6 <6 <6 <24UV after 5 h <6 <6 <6 <6 <24EL after 2 h 48 138 108 144 438EL after 5 h 24 72 48 60 204UVEL after 2 h 228 12 138 48 426UVEL after 5 h 66 12 36 6 120
186 G. Hurwitz et al. / Chemical Engineering Journal 249 (2014) 180–188

Author's personal copy
effectiveness of electrochemically-driven secondary oxidation atwhich the slope of the specific energy consumption mirrored theUV process. Other synergistic oxidation mechanisms include theformation of highly reactive hydroxyl and chlorine radicals fromthe photolysis of electrogenerated active chlorine [24,49].
The UVEL process was seen to improve the energy efficiency ofUV photolysis and mitigate the electrochemical formation of disin-fection by-products (DBPs) at residence times exceeding 2 h. UVELresulted in a noticeable production of DBPs, specifically THMs, butat a rate lower than EL alone (Table 3). Although total THM concen-trations were comparable after the first hour of treatment for bothelectrochemical and UVEL treatments, the UVEL process showedmore efficient removal rates with time (120 and 204 ppb for UVELand electrochemical oxidation, respectively, after 5 h of treatment).Photo-irradiation reduced the production of heavily chlorinatedTHMs during UVEL treatment, but favored the formation of bromi-nated THMs. UV-C photolysis dramatically reduced the residencetimes of electrochemically-generated HOCl in the UVEL system,which allowed greater interaction between reactive bromine spe-cies and organic matter to selectively form brominated THMs.The increased steric obstruction of bromine compared with chlo-rine may have led to C–Br bonds that were less accessible to fur-ther radical or photolytic reactions [50]. Therefore, the overalleffect of UV irradiation within the UVEL system may have beenan increased formation rate and minimal destruction of bromi-nated organics. The mitigation of the formation of DBPs by electro-chemical oxidation is a promising result and should be furtherexploited in future UVEL studies, as should optimization of thehigh rate of mineralization and low selectivity for organic removal.
Although a significant majority of the UVEL energy demand wasdue to the power consumption of the UV lamp, which consumedover 95% of the total power for the process (Fig. 2), the electro-chemical cell will also be of focus for future optimization studies.The applied current density and resultant current efficiency playdominant roles in the overall kinetics and energy efficiency ofthe UVEL process. In this research, the target current density of20 mA/cm2 was chosen after a thorough literature review of com-parable EL applications [4,13,28,51,52]. However, future work willbe devoted to studying if the unique UVEL oxidation mechanismcan benefit with operation at current densities beyond what is rec-ommended by conventional electrochemical practice (i.e., upwardsof 100 mA/cm2). The role of current density will also be analyzedwith respect to its effect on the required electrode area in orderto drive down the UVEL capital investment costs. Further studiesshould also focus on reducing the power consumption of the pho-tochemical component of the UVEL process by exploring lowerenergy UV sources, such as UV-A and UV-B.
5. Conclusions
The combination of UV photolysis with anodic oxidation byboron-doped diamond led to non-selective and synergistic degra-dation of effluent organic matter in wastewater RO concentrate.The UVEL process effectively treated a wide array of organics foundin wastewater effluent at a rate that exceeded treatment by UV andEL individually or additively. The observed synergy was due in partto the cooperative destruction of select chemical bonds favored byeach process individually. Electrochemical oxidation was shown tobe less effective for the structural degradation of large macromol-ecules, but more effective for ring cleavage and the removal of aro-matic functionality, while photolysis preferentially targeted thedestruction of backbone aliphatic bonds and led to less aromaticdegradation. As a result, UVEL was able to effectively mineralizenot only large aromatic macromolecules, such as biopolymersand humics, but also smaller derivative compounds, such as low
molecular weight organic acids, without significant intermediatebottlenecking. Overall oxidation was further aided by the produc-tion of highly reactive hydroxyl and chlorine radicals. The chlorineradicals were produced by the photolysis of electrochemically gen-erated active chlorine and introduced a new class of reactants withboth high oxidation potentials and low selectivities.
The overall result of this research was the development of anadvanced oxidation process that utilized conventional plug-in pro-cesses with a boron-doped diamond electrode material to effec-tively degrade a wide range of dissolved organic matter found inwastewater RO concentrate. In doing so, energy efficiency wasenhanced and DBP formation mitigated compared with UV andEL alone, respectively. The authors find these results highly prom-ising and suggestive of future work focusing on the targetedremoval of recalcitrant emerging organic contaminants, such aspharmaceuticals and endocrine disrupting compounds, in waste-water RO concentrate prior to discharge into receiving bodies.
Acknowledgements
This research was supported in part by the UCLA Water Tech-nology Research Center through the California Department ofWater Resources Proposition 50 Grant Program (Agreement No.4600004120). We are also grateful to Dr. Sam Aroni from the UCLAInternational Institute’s Special Academic Cooperative Project aswell as the UCLA Department of Civil & Environmental EngineeringHorn fund and RMIT University for providing additional financialsupport.
References
[1] P.R. Gogate, A.B. Pandit, A review of imperative technologies for wastewatertreatment I: oxidation technologies at ambient conditions, Adv. Environ. Res. 8(2004) 501–551.
[2] G.-S. Wang, H.-W. Chen, S.-F. Kang, Catalyzed UV oxidation of organicpollutants in biologically treated wastewater effluents, Sci. Total Environ.277 (2001) 87–94.
[3] P. Westerhoff, H. Moon, D. Minakata, J. Crittenden, Oxidation of organics inretentates from reverse osmosis wastewater reuse facilities, Water Res. 43(2009) 3992–3998.
[4] K. Van Hege, M. Verhaege, W. Verstraete, Electro-oxidative abatement of low-salinity reverse osmosis membrane concentrates, Water Res. 38 (2004) 1550–1558.
[5] N. Bolong, A.F. Ismail, M.R. Salim, T. Matsuura, A review of the effects ofemerging contaminants in wastewater and options for their removal,Desalination 239 (2009) 229–246.
[6] G. Perez, A.R. Fernandez-Alba, A.M. Urtiaga, I. Ortiz, Electro-oxidation ofreverse osmosis concentrates generated in tertiary water treatment, WaterRes. 44 (2010) 2763–2772.
[7] J. Radjenovic, A. Bagastyo, R.A. Rozendal, Y. Mu, J. Keller, K. Rabaey,Electrochemical oxidation of trace organic contaminants in reverse osmosisconcentrate using RuO2/IrO2-coated titanium anodes, Water Res. 45 (2011)1579–1586.
[8] E. Sahar, I. David, Y. Gelman, H. Chikurel, A. Aharoni, R. Messalem, A. Brenner,The use of RO to remove emerging micropollutants following CAS/UF or MBRtreatment of municipal wastewater, Desalination 273 (2011) 142–147.
[9] V. Yangali-Quintanilla, A. Sadmani, M. McConville, M. Kennedy, G. Amy, AQSAR model for predicting rejection of emerging contaminants(pharmaceuticals, endocrine disruptors) by nanofiltration membranes, WaterRes. 44 (2010) 373–384.
[10] A.K. da Silva, M.J. Wells, A.N. Morse, M.-L. Pellegrin, S.M. Miller, J. Peccia, L.C.Sima, Emerging pollutants Part I: occurrence, fate and transport, WaterEnviron. Res. 84 (2012) 1878–1908.
[11] J. Thomson, F.A. Roddick, M. Drikas, Vacuum ultraviolet irradiation for naturalorganic matter removal, J. Water Supply Res. Technol. AQUA 53 (2004) 193–206.
[12] J.C. Crittenden, R.R. Trussell, D.W. Hand, K.J. Howe, G. Tchobanoglous, WaterTreatment: Principles and Design, second ed., John Wiley & Sons Inc., Hoboken,NJ, 2005.
[13] C.A. Martinez-Huitle, S. Ferro, Electrochemical oxidation of organic pollutantsfor the wastewater treatment: direct and indirect processes, Chem. Soc. Rev.35 (2006) 1324–1340.
[14] P. Canizares, R. Paz, C. Saez, M.A. Rodrigo, Costs of the electrochemicaloxidation of wastewaters: a comparison with ozonation and Fenton oxidationprocesses, J. Environ. Manage. 90 (2009) 410–420.
G. Hurwitz et al. / Chemical Engineering Journal 249 (2014) 180–188 187

Author's personal copy
[15] M. Panizza, P.A. Michaud, G. Cerisola, C. Comninellis, Electrochemicaltreatment of wastewaters containing organic pollutants on boron-dopeddiamond electrodes: prediction of specific energy consumption and requiredelectrode area, Electrochem. Commun. 3 (2001) 336–339.
[16] E. Alvarez-Guerra, A. Dominguez-Ramos, A. Irabien, Design of the PhotovoltaicSolar Electro-Oxidation (PSEO) process for wastewater treatment, Chem. Eng.Res. Des. 89 (2011) 2679–2685.
[17] C.G. Alfafara, T. Kawamori, N. Nomura, M. Kiuchi, M. Matsumura, Electrolyticremoval of ammonia from brine wastewater: scale-up, operation and pilot-scale evaluation, J. Chem. Technol. Biotechnol. 79 (2004) 291–298.
[18] C. Comninellis, Electrocatalysis in the electrochemical conversion/combustionof organic pollutants for waste-water treatment, Electrochim. Acta 39 (1994)1857–1862.
[19] M. Panizza, G. Cerisola, Electrochemical oxidation of 2-naphthol with in situelectrogenerated active chlorine, Electrochim. Acta 48 (2003) 1515–1519.
[20] J. Jeong, C. Kim, J. Yoon, The effect of electrode material on the generation ofoxidants and microbial inactivation in the electrochemical disinfectionprocesses, Water Res. 43 (2009) 895–901.
[21] M. Catanho, G.R.P. Malpass, A.J. Motheo, Photoelectrochemical treatment ofthe dye reactive red 198 using DSA� electrodes, Appl. Catal. B 62 (2006) 193–200.
[22] K. Shaw, P. Christensen, A. Hamnett, Photoelectrochemical oxidation oforganics on single-crystal TiO2: an in situ FTIR study, Electrochim. Acta 41(1996) 719–728.
[23] L. Pinhedo, R. Pelegrini, R. Bertazzoli, A.J. Motheo, Photoelectrochemicaldegradation of humic acid on a (TiO2)0.7(RuO2)0.3 dimensionally stableanode, Appl. Catal. B 57 (2005) 75–81.
[24] S. Xiao, J. Qu, X. Zhao, H. Liu, D. Wan, Electrochemical process combined withUV light irradiation for synergistic degradation of ammonia in chloride-containing solutions, Water Res. 43 (2009) 1432–1440.
[25] M.V.B. Zanoni, J.J. Sene, H. Selcuk, M.A. Anderson, Photoelectrocatalyticproduction of active chlorine on nanocrystalline titanium dioxide thin-filmelectrodes, Environ. Sci. Technol. 38 (2004) 3203–3208.
[26] K. Patel, K. Hashimoto, A. Fujishima, Photoelectrochemical investigations onboron-doped chemically vapour-deposited diamond electrodes, J. Photochem.Photobiol., A 65 (1992) 419–429.
[27] M. Panizza, A. Kapalka, C. Comninellis, Oxidation of organic pollutants on BDDanodes using modulated current electrolysis, Electrochim. Acta 53 (2008)2289–2295.
[28] M. Panizza, G. Cerisola, Application of diamond electrodes to electrochemicalprocesses, Electrochim. Acta 51 (2005) 191–199.
[29] P. Canizares, J. Lobato, R. Paz, M.A. Rodrigo, C. Saez, Electrochemical oxidationof phenolic wastes with boron-doped diamond anodes, Water Res. 39 (2005)2687–2703.
[30] G. Hurwitz, P. Pornwongthong, S. Mahendra, E.M.V. Hoek, Degradation ofphenol by synergistic chlorine-enhanced photo-assisted electrochemicaloxidation, Chem. Eng. J. 240 (2014) 235–243.
[31] W. Buchanan, F. Roddick, N. Porter, Formation of hazardous by-productsresulting from the irradiation of natural organic matter: comparison betweenUV and VUV irradiation, Chemosphere 63 (2006) 1130–1141.
[32] N. Her, G. Amy, D. McKnight, J. Sohn, Y. Yoon, Characterization of DOM as afunction of MW by fluorescence EEM and HPLC-SEC using UVA, DOC, andfluorescence detection, Water Res. 37 (2003) 4295–4303.
[33] L. Fan, T. Nguyen, F.A. Roddick, J.L. Harris, Low-pressure membrane filtration ofsecondary effluent in water reuse: pre-treatment for fouling reduction, J.Membr. Sci. 320 (2008) 135–142.
[34] A. Baker, Fluorescence excitation–emission matrix characterization of somesewage-impacted rivers, Environ. Sci. Technol. 35 (2001) 948–953.
[35] M.M.D. Sierra, M. Giovanela, E. Parlanti, E.J. Soriano-Sierra, Fluorescencefingerprint of fulvic and humic acids from varied origins as viewed by single-scan and excitation/emission matrix techniques, Chemosphere 58 (2005) 715–733.
[36] S.B. Abdelmelek, J. Greaves, K.P. Ishida, W.J. Cooper, W. Song, Removal ofpharmaceutical and personal care products from reverse osmosis retentateusing advanced oxidation processes, Environ. Sci. Technol. 45 (2011) 3665–3671.
[37] Z. Wang, Z. Wu, S. Tang, Characterization of dissolved organic matter in asubmerged membrane bioreactor by using three-dimensional excitation andemission matrix fluorescence spectroscopy, Water Res. 43 (2009) 1533–1540.
[38] U. von Gunten, Ozonation of drinking water: Part II. Disinfection and by-product formation in presence of bromide, iodide or chlorine, Water Res. 37(2003) 1469–1487.
[39] L. Codognoto, S.A.S. Machado, L.A. Avaca, Selective oxidation ofpentachlorophenol on diamond electrodes, J. Appl. Electrochem. 33 (2003)951–957.
[40] Y.J. Feng, X.Y. Li, Electro-catalytic oxidation of phenol on several metal-oxideelectrodes in aqueous solution, Water Res. 37 (2003) 2399–2407.
[41] C. Cunningham, K.F. Tipton, H.B.F. Dixon, Conversion of taurine into N-chlorotaurine (taurine chloramine) and sulphoacetaldehyde in response tooxidative stress, Biochem. J. 330 (1998) 939–945.
[42] R.M. Machado, T.W. Chapman, Kinetics and selectivity of aqueous propylenehalohydrination, Ind. Eng. Chem. Res. 28 (1989) 1789–1794.
[43] I.N. Najm, N.L. Patania, J.G. Jacangelo, S.W. Krasner, Evaluating surrogates fordisinfection by-products, AWWA 86 (1994) 98–106.
[44] Y.G. Feng, D.W. Smith, J.R. Bolton, Photolysis of aqueous free chlorine species(HOCl and OCl�) with 254 nm ultraviolet light, J. Environ. Eng. Sci. 6 (2007)277–284.
[45] M.J. Watts, K.G. Linden, Chlorine photolysis and subsequent OH radicalproduction during UV treatment of chlorinated water, Water Res. 41 (2007)2871–2878.
[46] C. Comninellis, A. Nerini, Anodic oxidation of phenol in the presence of NaClfor wastewater treatment, J. Appl. Electrochem. 25 (1995) 23–28.
[47] A.F. Adamson, B.G. Lever, W.F. Stones, The production of hypochlorite by directelectrolysis of sea water: electrode materials and design of cells for theprocess, J. Appl. Chem. 13 (1963) 483–495.
[48] D. Rajkumar, J.G. Kim, K. Palanivelu, Indirect electrochemical oxidation ofphenol in the presence of chloride for wastewater treatment, Chem. Eng.Technol. 28 (2005) 98–105.
[49] D. Gonenc, M. Bekbolet, Interactions of hypochlorite ion and humic acid:photolytic and photocatalytic pathways, Water Sci. Technol. 44 (2001) 205–210.
[50] D. Cassan, B. Mercier, F. Castex, A. Rambaud, Effects of medium-pressure UVlamps radiation on water quality in a chlorinated indoor swimming pool,Chemosphere 62 (2006) 1507–1513.
[51] M.A.Q. Alfaro, S. Ferro, C.A. Martínez-Huitle, Y.M. Vong, Boron doped diamondelectrode for the wastewater treatment, J. Braz. Chem. Soc. 17 (2006) 227–236.
[52] A. Kraft, Doped diamond: a compact review on a new, versatile electrodematerial, Int. J. Electrochem. Sci. 2 (2007) 355–385.
188 G. Hurwitz et al. / Chemical Engineering Journal 249 (2014) 180–188