octane blending and oxidation chemistry of ethanol
TRANSCRIPT

Octane Blending and Oxidation Chemistry of
Ethanol-Hydrocarbon Mixtures
Hao Yuan
March 2018
Submitted in total fulfilment of the requirements of the degree of
Doctor of Philosophy
Supervised by
A/Prof. Yi Yang
Co-Supervised by
Prof. Michael Brear
Department of Mechanical Engineering
THE UNIVERSITY OF MELBOURNE
Produced on archival quality paper

Copyright © 2018 Hao Yuan
All rights reserved. No part of the publication may be reproduced in any form by print,
photoprint, microfilm or any other means without written permission from the author.

Abstract
The strong anti-knock property of ethanol makes it a preferred blending component for gasoline to
improve spark ignition (SI) engine performance. Despite its widespread use, understanding several,
particular aspects of ethanol’s interaction with different components of gasoline is still lacking.
This work therefore performs the following three studies to investigate the interactions among
ethanol and hydrocarbon fuels. First, a method for correlating octane numbers is developed for
toluene reference fuels (TRFs) blended with ethanol. This method combines linear regression and
exhaustive (or brute-force) searching for optimal correlations. The proposed correlations reproduce
the measured RON and MON with a maximum absolute error smaller than two octane numbers. De-
spite the empirical nature, the correlations demonstrate the significance of linear by mole blending
rules for TRF fuels and provide insights on the chemical interactions between ethanol and different
hydrocarbons. The work of the optimal octane number correlations has been published in Fuel [Yuan
et al., Fuel, 188 (2017), p.408].
Second, a five-component gasoline surrogate is developed to emulate the octane blending be-
haviours of gasoline and ethanol. The surrogate contains iso-pentane, n-pentane, cyclohexane, 1-
hexene, and 1,2,4-trimethylbenzene and is developed using extensive Cooperative Fuel Research (CFR)
engine testing. The formulated surrogate captures the synergistic RON blending behavior between the
target gasoline and ethanol over the entire blending range, with a hydrocarbon composition similar
to the target fuel.
Lastly, a Pressurised Flow Reactor (PFR) experimental study is carried out to study oxidation
chemistry of a fuel matrix including neat fuels, binaries, gasoline surrogates, and gasoline surro-
gates/ethanol mixtures. The measured species profiles are simulated with published kinetic models.
The result indicates that further investigations on toluene and its interaction chemistries with other
compounds are needed for understanding the oxidation of surrogate fuels.
iii


Declaration
This is to certify that:
1. the thesis comprises only my original work towards the PhD,
2. due acknowledgement has been made in the text to all other material used,
3. the thesis is fewer than 100,000 words in length, exclusive of tables, maps, bibliographies and
appendices.
Hao Yuan, March 2018
v


Acknowledgements
I would like to express my gratitude the following people for their supports during my PhD study.
This thesis would not have been possible without them.
• Yi Yang and Michael Brear (my academic supervisors)
Their constructive advice and insightful guidance have been of tremendous help to me during the
past four years of my PhD study.
• Zhongyuan Chen and Zhewen Lu
Zhongyuan helped me with the CFR engine experiments and we worked together for the past four
years. Zhewen helped to build the PFR and worked together with me on the PFR experiments.
• Tien Mun Foong and Al Knox
Tien Mun offered me great help in starting the CFR engine experiment and modelling at the beginning
of my PhD. Al provided technical supports in the CFR engine overhaul.
• James Anderson and Thomas Leone (research engineers at Ford)
James and Thomas provided the data for the engine modelling work and offered valuable suggestions
for my PhD research.
• Monica Pater
Thanks Monica for her help with purchasing of research equipment and chemicals.
• My friends within the Thermodynamics Group
Thank all of you for making the past four years such an enjoyable experience.
• My beloved family and girlfriend
Foremost, I wish to extend thanks to my family for the continuous and unquestioning support over the
last 30 years. I also would like to thank my girlfriend, Jie Jian, who shared all my sadness, happiness,
failure and success during my PhD study and wish her every success in her own PhD project.
vii


To my mum
ix


Contents
1 Introduction 1
1.1 Energy consumption and climate change . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.2 Biofuels for transportation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2.1 Increased biofuels production . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2.2 Ethanol as a fuel additive . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.3 Octane blending of ethanol and hydrocarbons . . . . . . . . . . . . . . . . . . . . . . . . 4
2 Literature Review 5
2.1 Overview of Knock . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
2.1.1 Essence of knock . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
2.1.2 Characteristics of knock . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
2.2 Anti-knock Characteristics of Ethanol/Hydrocarbon Blends . . . . . . . . . . . . . . . . 8
2.2.1 Octane numbers of ethanol/hydrocarbon blends . . . . . . . . . . . . . . . . . . . 8
2.2.2 Charge cooling effect . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2.3 Chemistries of Ethanol and Hydrocarbons . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
2.3.1 Experimental techniques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
2.3.1.1 Shock tube . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
2.3.1.2 Rapid compression machine . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.3.1.3 Well-stirred reactor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.3.1.4 Pressurised flow reactor . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2.3.2 Combustion chemistry of hydrocarbons and alcohol . . . . . . . . . . . . . . . . . 16
2.3.2.1 Combustion chemistry of alkanes . . . . . . . . . . . . . . . . . . . . . . 16
2.3.2.2 Combustion chemistry of aromatics . . . . . . . . . . . . . . . . . . . . . 19
2.3.2.3 Combustion chemistry of ethanol . . . . . . . . . . . . . . . . . . . . . . 21
2.4 Chemical Interactions of Fuel Mixtures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.4.1 Interactions between alkanes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.4.2 Interactions between PRF and toluene . . . . . . . . . . . . . . . . . . . . . . . . . 23
xi

2.4.2.1 Cross reactions via large radicals . . . . . . . . . . . . . . . . . . . . . . 23
2.4.2.2 Cross reactions via radical pool . . . . . . . . . . . . . . . . . . . . . . . 25
2.4.3 Interactions between ethanol and hydrocarbons . . . . . . . . . . . . . . . . . . . 27
2.5 Summary and research questions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
3 Experimental Methods 30
3.1 Overview . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
3.2 CFR engine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
3.2.1 Overview . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
3.2.2 The Structure of the CFR engine . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
3.2.3 Methods for standard octane number tests . . . . . . . . . . . . . . . . . . . . . . 33
3.3 Pressurised flow reactor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
3.3.1 Overview . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
3.3.2 Reactor structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
3.3.3 Mixer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
3.3.4 Sampling probe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
3.3.5 Experimental conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
3.4 Gas chromatography . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
3.4.1 Overview of the gas chromatography . . . . . . . . . . . . . . . . . . . . . . . . . 41
3.4.2 Identification and quantification of species . . . . . . . . . . . . . . . . . . . . . . 43
4 Optimal Octane Number Correlations for Toluene Reference Fuels (TRFs) Blended with
Ethanol 48
4.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
4.2 Algorithm for correlation development . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
4.2.1 The Scheffe polynomial based correlation . . . . . . . . . . . . . . . . . . . . . . . 49
4.2.2 Linear regression . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
4.2.3 Data for correlation development and validation . . . . . . . . . . . . . . . . . . . 51
4.2.4 Criterion for correlation development . . . . . . . . . . . . . . . . . . . . . . . . . 52
4.2.5 Procedures for optimal correlation development and validation . . . . . . . . . . 53
4.3 Optimal correlations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
4.3.1 Optimal RON correlation for TRF/ethanol mixtures . . . . . . . . . . . . . . . . . 55
4.3.1.1 Development of the optimal correlation . . . . . . . . . . . . . . . . . . 55
4.3.1.2 Validation of the optimal correlation . . . . . . . . . . . . . . . . . . . . 57
4.3.2 Optimal MON correlation for TRF/ethanol mixtures . . . . . . . . . . . . . . . . 58
xii

4.3.2.1 Development of the optimal correlation . . . . . . . . . . . . . . . . . . 58
4.3.2.2 Validation of the optimal correlation . . . . . . . . . . . . . . . . . . . . 58
4.4 Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
5 The Octane Numbers of Binary Mixtures and Gasoline Surrogates Blended with Ethanol 62
5.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
5.2 The RONs of binary mixtures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
5.2.1 Binary mixtures of hydrocarbons . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
5.2.2 Binary mixtures containing ethanol . . . . . . . . . . . . . . . . . . . . . . . . . . 72
5.3 The RONs of gasoline surrogates blended with ethanol . . . . . . . . . . . . . . . . . . . 73
5.3.1 Detailed hydrocarbon analysis for the Australian production gasoline . . . . . . 74
5.3.2 Strategy for emulating the octane number of the gasoline . . . . . . . . . . . . . . 76
5.3.3 Comparison between production gasoline and its surrogates when blended with
ethanol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
5.4 Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
6 Oxidation of Ethanol and Hydrocarbon Mixtures in a Flow Reactor 82
6.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
6.2 Kinetic modelling approach . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
6.3 Neat fuels . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
6.3.1 Isooctane . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
6.3.2 Ethanol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
6.3.3 Toluene . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
6.4 Test mechanism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
6.4.1 Sensitivity analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
6.4.2 Updated toluene sub-mechanism . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
6.5 Binary mixtures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
6.5.1 Ethanol and isooctane . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
6.5.2 Toluene and isooctane . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 99
6.5.3 Ethanol and toluene . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
6.6 Gasoline surrogates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102
6.6.1 PRF91 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102
6.6.2 TRF91-30 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 105
6.7 Gasoline surrogates/ethanol mixtures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 108
6.7.1 PRF91 and ethanol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 108
xiii

6.7.2 TRF91-30 and ethanol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 111
6.8 Comparison of fuel reactivities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 118
6.9 Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122
7 Conclusions and Recommendations for Future Research 123
7.1 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
7.2 Recommendations for future research . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
References 127
A Octane number data used for optimal correlation development 151
B Liquid volume based correlations 155
C Modelling of Trace Knock in a Modern SI Engine Fuelled by Ethanol and Gasoline Blends 157
C.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
C.2 Numerical methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158
C.3 Formulation of gasoline surrogates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 159
C.4 NO sub-model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 160
C.5 GT-Power modelling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162
C.5.1 Full flow model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162
C.5.2 Reverse run model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
C.6 Two-zone model of autoignition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167
C.7 Modelling of trace knock . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168
C.7.1 Raw pressure data . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168
C.7.2 Approach for modelling trace knock . . . . . . . . . . . . . . . . . . . . . . . . . . 170
C.7.3 Example of modelling approach . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
C.8 Modelling results and discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
C.8.1 UFI engine results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
C.8.1.1 Non-kinetic factors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
C.8.1.2 Effect of ethanol content . . . . . . . . . . . . . . . . . . . . . . . . . . . 173
C.8.2 DI engine results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 176
C.8.3 The effect of NO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 177
C.9 Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 179
D The kinetic model for the flow reactor 180
xiv

List of Figures
1.1 The outlook for (a) energy consumption and (b) oil demand before 2035 [1] . . . . . . . 2
1.2 Global biofuels production in the last ten years [5] . . . . . . . . . . . . . . . . . . . . . . 3
1.3 Annual U.S. average ethanol content of finished gasoline from 2010 to 2016 [17] . . . . . 3
2.1 The pressure trace of a knocking cycle and its corresponding non-knocking cycle sup-
pressed by tetraethyl lead [23] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
2.2 The Midgley and Boyd bouncing pin apparatus for knock detection [28] . . . . . . . . . 6
2.3 Image series for both non-knocking and knocking engine cycles [32] . . . . . . . . . . . 7
2.4 Measured (a) RONs and (b) MONs for the ethanol and gasoline blends . . . . . . . . . . 9
2.5 Measured (a) RONs and (b) MONs for ethanol blended with isooctane, n-heptane and
toluene . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2.6 Measured RON values for ethanol/gasoline blends under standard and modified con-
ditions [13] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
2.7 CAD model of the combustion chamber [45] . . . . . . . . . . . . . . . . . . . . . . . . . 12
2.8 The comparison between overall and effective octane numbers [14] . . . . . . . . . . . . 12
2.9 Separation of chemical octane and charge cooling effects on knock limit [11] . . . . . . . 13
2.10 Schematic of a shock tube/rapid compression machine . . . . . . . . . . . . . . . . . . . 14
2.11 Schematic of a well-stirred reactor [47] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2.12 Structure of a pressurised flow reactor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.13 Simplified scheme for the primary mechanism of oxidation of alkanes at low tempera-
tures [51] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
2.14 Simplified scheme for the oxidations of benzene and toluene [132] . . . . . . . . . . . . . 20
2.15 Measured MONs of toluene blended with isooctane [187] . . . . . . . . . . . . . . . . . . 24
2.16 Comparisons of cool flame (open symbols) and autoignition delay times (filled sym-
bols) of neat isooctane and isooctane/toluene mixture [190] . . . . . . . . . . . . . . . . . 26
3.1 The system of the CFR engine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
3.2 The structure of the CFR engine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
xv

3.3 The piston head (a) before and (b) after overhaul . . . . . . . . . . . . . . . . . . . . . . . 33
3.4 Schematic of the Pressurised Flow Reactor system . . . . . . . . . . . . . . . . . . . . . . 35
3.5 Structure of the Pressurised Flow Reactor . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
3.6 The mixer a) cutaway view and b) orifices distribution . . . . . . . . . . . . . . . . . . . 37
3.7 CO2 concentrations at 10 bar and 900 K in the flow reactor with air flow rate of 6.02 g/s
and CO2 flow rate of 0.71 g/s . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
3.8 The sampling probe a) cutaway view b) three thermocouples . . . . . . . . . . . . . . . 38
3.9 Reactor temperature profiles for isooctane oxidation at 10 bar and 900 K with equiva-
lence ratio of 0.058 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
3.10 Gas Chromatography-2010ATF plus from Shimadzu . . . . . . . . . . . . . . . . . . . . . 41
3.11 Flow chart of Gas Chromatography . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
3.12 The temperature program for GC analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
3.13 The spectrum of isooctane oxidation at 900 mm under 900 K and 10 bar . . . . . . . . . . 44
3.14 The spectrum of ethanol oxidation at 500 mm under 900 K and 10 bar . . . . . . . . . . . 45
3.15 The spectrum of toluene oxidation at 700 mm under 930 K and 10 bar . . . . . . . . . . . 45
3.16 The GC calibrations for (a) isooctane, (b) n-heptane, (c) toluene and (d) ethanol . . . . . 46
4.1 Data distribution on simplex lattices with filled circles representing development data
and open ones for validation data . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
4.2 Residual error between the development data and correlated RON from (a) linear by-
mole correlation, (b) five terms correlation, (c) six terms correlation and (d) seven terms
correlation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
4.3 Variation of a) R2 and b) MAE with optimal combination of terms in RON correlations
of increasing length . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
4.4 Residual error between the validation data and a) 7 and b) 8 term RON correlations on
a molar basis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
4.5 Residual error between the development data and correlated MON from (a) linear by-
mole correlation, and (b) seven terms correlation . . . . . . . . . . . . . . . . . . . . . . . 59
4.6 Variation of a) R2 and b) MAE with optimal combination of terms in MON correlations
of increasing length . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
4.7 Residual error between the validation data and a) 7 and b) 8 term MON correlations on
a molar basis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
5.1 Measured RONs for Australian production gasoline, PRF91, and TRF91s blended with
ethanol [9] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
xvi

5.2 RONs of isooctane blended with toluene on a a) volume basis and b) mole basis from
this study. RONs of isooctane blended with ethylbenzene on a c) volume basis and d)
mole basis from [208] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
5.3 RONs of n-heptane blended with toluene on a a) volume basis and b) mole basis from
this study. RONs of n-heptane blended with ethylbenzene on a c) volume basis and d)
mole basis from [208] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66
5.4 RONs of cyclohexane blended with toluene on a a) volume basis and b) mole basis from
this study. RONs of cyclopentane blended with ethylbenzene on a c) volume basis and
d) mole basis from [208] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
5.5 RONs of 1-hexene blended with toluene on a a) volume basis and b) mole basis from
this study. RONs of diisobutylene blended with ethylbenzene on a c) volume basis and
d) mole basis from [208] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
5.6 RONs of cyclohexane blended with isooctane on a a) volume basis and b) mole basis
from this study. RONs of methylcyclohexane blended with isooctane on a c) volume
basis and d) mole basis from [208] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
5.7 RONs of 1-hexene blended with isooctane on a a) volume basis and b) mole basis from
this study. RONs of 2-heptene blended with isooctane on a c) volume basis and d) mole
basis from [208] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
5.8 RONs of cyclohexane and 1-hexene blended with ethanol on a a) volume basis and b)
mole basis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72
5.9 The comparisons of the gasoline/ethanol mixture and different gasoline surrogates
blended with ethanol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
6.1 The measurements of (a) CO and CO2, and (b) isooctane from the neat isooctane oxida-
tion experiment at 900 K and 10 bar, and the modelling results from Mehl et al. (solid
lines), Andrae (dashed lines) and Atef et al. (dotted lines) using the corrected tempera-
ture profile from the three-thermocouple method (c) . . . . . . . . . . . . . . . . . . . . . 86
6.2 The measured intermediate species profiles from the neat isooctane oxidation exper-
iment at 900 K and 10 bar, and the modelling results from Mehl et al. (solid lines),
Andrae (dashed lines) and Atef et al. (dotted lines) . . . . . . . . . . . . . . . . . . . . . . 87
6.3 The reaction pathways for IC4H8, XC7H14, and YC7H14 from the isooctane experiment
at 900mm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88
6.4 The reaction pathway for IC3H5CHO from the isooctane experiment at 900mm . . . . . 88
xvii

6.5 The measurements of (a) CO and CO2, and (b) ethanol from the neat ethanol oxidation
at 900 K and 10 bar, and the modelling results from Mehl et al. (solid lines), Mittal et al.
(dotted lines), Marinov (dashdot lines) and Andrae (dashed lines) using the corrected
temperature profile from the three-thermocouple method (c) . . . . . . . . . . . . . . . . 90
6.6 The measured intermediate species profiles from the neat ethanol oxidation at 900 K
and 10 bar, and the modelling results from Mehl et al. (solid lines), Mittal et al. (dotted
lines), Marinov (dashdot lines) and Andrae (dashed lines) . . . . . . . . . . . . . . . . . 91
6.7 The reaction pathway for CH3CHO from the ethanol experiment at 500mm . . . . . . . 92
6.8 The measurements of CO and toluene (a) from the neat toluene oxidation at 930 K and
10 bar, and the modelling results from Mehl et al. (solid lines), Yuan et al. (dashdot
lines), Metcalfe et al. (dotted lines), Andrae (dashed lines), Zhang et al. (large dashed
lines), and Pelucchi et al. (large dashdot lines) using the corrected temperature profile
from the three-thermocouple method (b) . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
6.9 The measured benzene profile from the neat toluene oxidation at 930 K and 10 bar, and
the modelling results from Mehl et al. (solid line), Yuan et al. (dashdot line), Metcalfe et
al. (dotted line), Andrae (dashed line), Zhang et al. (large dashed lines), and Pelucchi
et al. (large dashdot lines) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
6.10 The brute-force sensitivity analysis of CO for the toluene oxidation at 930 K and 10 bar . 96
6.11 The measurements of CO and toluene from the neat toluene oxidation at 930 K and 10
bar, and the modelling results from Mehl et al. (solid lines) and TestMech (dashed lines) 98
6.12 The measured CO profiles of different binary mixtures (a-c) of isooctane and ethanol at
900 K and 10 bar, and the modelling results from Mehl et al. (solid lines) and TestMech
(dashed lines, overlapping with the solid lines) using the corrected temperature profile
from the three-thermocouple method (d) . . . . . . . . . . . . . . . . . . . . . . . . . . . 99
6.13 The measured CO profiles of different binary mixtures (a-c) of isooctane and toluene at
900 K and 10 bar, and the modelling results from Mehl et al. (solid lines) and TestMech
(dashed lines) using the corrected temperature profile from the three-thermocouple
method (d) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 100
6.14 The measured CO profiles of different binary mixtures (a-c) of ethanol and toluene at
900 K and 10 bar, and the modelling results from Mehl et al. (solid lines) and TestMech
(dashed lines) using the corrected temperature profile from the three-thermocouple
method (d) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
xviii

6.15 The measurements of (a) CO and CO2, and (b) isooctane and n-heptane from the PRF91
oxidation at 900 K and 10 bar, and the modelling results from Mehl et al. (solid lines),
TestMech (dashed lines) and TestMech without chemical interactions between parent
fuels or fuel-like species (dotted lines) using the corrected temperature profile from the
three-thermocouple method (c) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
6.16 The measured intermediate species profiles from the PRF91 oxidation experiment at
900 K and 10 bar, and the modelling results from Mehl et al. (solid lines) and TestMech
(dashed lines) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
6.17 The measured species profiles: (a) CO, CO2, and toluene, (b) isooctane and n-heptane
from the oxidation of TRF91-30 at 900 K and 10 bar, and the modelling results from Mehl
et al. (solid lines), TestMech (dashed lines) and TestMech without chemical interactions
between parent fuels or fuel-like species (dotted lines) using the corrected temperature
profile from the three-thermocouple method (c) . . . . . . . . . . . . . . . . . . . . . . . 106
6.18 The measured intermediate species profiles from the TRF91 oxidation experiment at
900 K and 10 bar, and the modelling results from Mehl et al. (solid lines) and Test-
Mech(dashed lines) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
6.19 The measured species profiles: (a) CO, CO2 and ethanol, (b) isooctane and n-heptane
from the oxidation of PRF91 blended with 73.7% ethanol by mole (50% by volume) at
900 K and 10 bar, and the modelling results from Mehl et al. (solid lines) and TestMech
(dashed lines) using the corrected temperature profile from the three-thermocouple
method (c) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
6.20 The measured intermediate species profiles from the oxidation of PRF91 blended with
73.7% ethanol by mole (50% by volume) at 900 K and 10 bar, and the modelling results
from Mehl et al. (solid lines) and TestMech (dotted lines) . . . . . . . . . . . . . . . . . . 110
6.21 The measured species profiles: (a) CO, isooctane, and n-heptane, (b) CO2, toluene, and
ethanol from the oxidation of TRF91-30 blended with 87.7% ethanol by mole (75% by
volume) at 900 K and 10 bar, and the modelling results from Mehl et al. (solid lines)
and TestMech (dashed lines) using the corrected temperature profile from the three-
thermocouple method (c) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
6.22 The measured intermediate species profiles the oxidation of TRF91-30 blended with
87.7% ethanol by mole (75% by volume) at 900 K and 10 bar, and the modelling results
from Mehl et al. (solid lines) and TestMech (dotted lines) . . . . . . . . . . . . . . . . . . 113
xix

6.23 The measured species profiles: (a) CO, isooctane, and n-heptane, (b) CO2, toluene, and
ethanol from the oxidation of TRF91-30 blended with 70.5% ethanol by mole (50% by
volume) at 900 K and 10 bar, and the modelling results from Mehl et al. (solid lines)
and TestMech (dashed lines) using the corrected temperature profile from the three-
thermocouple method (c) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 114
6.24 The measured intermediate species profiles the oxidation of TRF91-30 blended with
70.5% ethanol by mole (50% by volume) at 900 K and 10 bar, and the modelling results
from Mehl et al. (solid lines) and TestMech (dotted lines) . . . . . . . . . . . . . . . . . . 115
6.25 The measured species profiles: (a) CO, isooctane, and n-heptane, (b) CO2, ethanol, and
toluene from the oxidation of TRF91-30 blended with 44.3% ethanol by mole (25% by
volume) at 900 K and 10 bar, and the modelling results from Mehl et al. (solid lines)
and TestMech (dashed lines) using the corrected temperature profile from the three-
thermocouple method (c) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 116
6.26 The measured intermediate species profiles the oxidation of TRF91-30 blended with
44.3% ethanol by mole (25% by volume) at 900 K and 10 bar, and the modelling results
from Mehl et al. (solid lines) and TestMech (dotted lines) . . . . . . . . . . . . . . . . . . 117
6.27 The CO and corrected temperature comparisons among isooctane and ethanol . . . . . 118
6.28 The CO and corrected temperature comparisons for two binary mixtures: ethanol plus
isooctane and ethanol plus toluene . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
6.29 The measured CO profiles of PRF91 and TRF91-30: (a) without ethanol and (c) with
ethanol. The corrected temperature profiles of PRF91 and TRF91-30: (b) without ethanol
and (d) with ethanol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
B.1 Residual error between the development data and correlated RON from (a) linear by-
volume correlation, (b) seven terms correlation . . . . . . . . . . . . . . . . . . . . . . . . 155
B.2 Residual error between the development data and correlated MON from (a) linear by-
volume correlation, (b) seven terms correlation . . . . . . . . . . . . . . . . . . . . . . . . 156
C.1 Comparison of the simulated ignition delay of the formulated gasoline surrogate (Table
C.1) using the original LLNL model and the extended model containing NO in a con-
stant volume reactor without NO present initially. Equivalence ratio = 1, 30bar, 700-1200K161
C.2 Experimental CA50 vs. NMEP for ethanol/gasoline blends at 10:1 CR and 1500 rpm
with DI [11] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 163
C.3 The full flow GT-Power model for the single cylinder engine in [11] . . . . . . . . . . . . 163
xx

C.4 The sensitivity analysis for the convection multiplier, to the cylinder wall temperature,
Twall . Dashed lines represent the minimal RMSE at each wall temperature. RMSE values
(×104) are indicated by the numbers on the contours . . . . . . . . . . . . . . . . . . . . . 166
C.5 Unburned gas temperature profiles at different wall temperatures from the GT-Power
reverse run . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
C.6 Measured and simulated pressure traces from the GT-Power reverse run . . . . . . . . . 167
C.7 Raw and band pass filtered pressure traces (left), and power spectra from Fast Fourier
Transform (FFT) analysis (right) for the most advanced pressure traces under standard
knocking for isooctane in a CFR engine (a and b), and under trace knocking for E0, UFI,
NMEP=402kPa (c and d) and E50, UFI, NMEP=1324kPa (e and f) in a single-cylinder
engine from the experimental study [11] . . . . . . . . . . . . . . . . . . . . . . . . . . . . 169
C.8 Modelled results for E50 and UFI at NMEP=1324kPa with the MFB profile being swept 171
C.9 Comparison of measured and modelled spark timing for trace knock using different
representative traces for E0, E20 and E50. All cases are with UFI fueling . . . . . . . . . 174
C.10 Variation of modelled (using the 95th percentile advanced trace) and measured spark
timing for trace knock for E0, E20 and E50 . . . . . . . . . . . . . . . . . . . . . . . . . . . 175
C.11 MFB at autoignition for spark timings that are one degree earlier than the spark timing
for trace knock . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 175
C.12 Comparison of modelled and experimental spark timing for trace knock with DI and
UFI for E50. Modelling results are from 95th percentile most advanced pressure traces . 177
C.13 Modelled spark timings for trace knock without residual NO compared to equivalent
results with residual NO for different fuel mixtures and injection methods . . . . . . . . 178
D.1 The comparison between the modelled results of the neat isooctane oxidation at 900 K
and 10 bar in the PFR using Chemkin and the model developed in this study . . . . . . 181
xxi

List of Tables
3.1 Operating conditions for the RON and MON measurements [25, 26] . . . . . . . . . . . . 34
3.2 The composition of the dilute TEL [25, 26] . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
3.3 Experimental conditions for the PFR study . . . . . . . . . . . . . . . . . . . . . . . . . . 40
3.4 Response factors for gaseous fuels . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.5 Response factors for intermediate species in liquid phase . . . . . . . . . . . . . . . . . . 47
4.1 Terms of the Scheffe polynomial with four variables . . . . . . . . . . . . . . . . . . . . . 50
4.2 Coefficients of first order terms in the Scheffe polynomial . . . . . . . . . . . . . . . . . . 50
5.1 RONs of cyclohexane and 1-hexene from different studies [20, 207] . . . . . . . . . . . . 67
5.2 Interactions of binary mixtures on a mole basis . . . . . . . . . . . . . . . . . . . . . . . . 73
5.3 Volume fractions of hydrocarbon groups in the Australian production gasoline . . . . . 74
5.4 Top ten most abundant species in iso-, n- and cyclo-paraffins . . . . . . . . . . . . . . . . 75
5.5 Top ten most abundant species in aromatics and olefins . . . . . . . . . . . . . . . . . . . 75
5.6 Formulated gasoline surrogates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
5.7 Equivalence ratios of gasoline/ethanol and GS11/ethanol at standard knocking condi-
tions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
5.8 The physical properties of the gasoline and the gasoline surrogates . . . . . . . . . . . . 80
6.1 Test fuels and reaction mechanisms for modelling . . . . . . . . . . . . . . . . . . . . . . 83
6.2 Experimental conditions for the PFR study . . . . . . . . . . . . . . . . . . . . . . . . . . 84
6.3 Reaction changes to LLNL’s toluene sub-mechanism . . . . . . . . . . . . . . . . . . . . . 97
A.1 Octane number data used for developing the optimal correlations for TRF/ethanol mix-
tures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 151
A.2 Octane number data used for validating the optimal correlations for TRF/ethanol mix-
tures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153
C.1 Gasoline and surrogate fuel compositions (%vol) . . . . . . . . . . . . . . . . . . . . . . . 160
xxii

C.2 Gasoline and surrogate fuel properties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 160
C.3 Specifications for the single cylinder SI engine [11] . . . . . . . . . . . . . . . . . . . . . . 162
C.4 Experimental conditions for modelled trace knocking cases . . . . . . . . . . . . . . . . . 164
C.5 Vibration mode frequencies from [266] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 170
C.6 Comparison between peak frequencies from the FFT result and the prediction . . . . . . 170
C.7 Inputs to the two-zone modelling obtained from GT-Power for the UFI cases in Table
C.4 and their corresponding 95th percentile raw pressure traces . . . . . . . . . . . . . . 172
C.8 Inputs to the two-zone modelling obtained from GT-Power for the DI cases in Table C.4
and the 95th percentile raw pressure traces . . . . . . . . . . . . . . . . . . . . . . . . . . 176
xxiii

Chapter 1
Introduction
1.1 Energy consumption and climate change
With the growth of the world economy, more energy is required in the future. Based on the estimations
of the BP Energy Outlook in 2017 [1], the growth of the total energy consumption in the next 20
years is over 30% with the world economy to double in this period, as shown in Fig.1.1. Half of the
growth is expected to come from renewables, nuclear, and hydroelectric power, but the fossil energy
sources, such as coal, gas, and oil, still provide over three-quarters of total energy supplies. Among
all these conventional energies, the oil consumption is predicted to be the largest. Meanwhile, more
than half of the oil demands come from transportations, as shown in Fig.1.1(b). In the foreseeable
future, it is expected that Internal Combustion Engines (ICEs), which predominantly rely on oil, will
serve as the main propulsion systems for transportations [2] owing to low cost, high reliability, long
durability, and fast refuelling. The deep understanding and accurate control of combustion process
have enabled the emergence of novel engine technologies for higher efficiencies and lower emissions,
such as direct injection, turbocharging, and downsizing. New types of engine utilising advanced
combustion modes, such as spark assisted homogeneous charge compression ignition technology, are
emerging [3].
The predominant use of fossil fuels in ICEs produces a significant amount of carbon dioxide (CO2)
which accounts for approximately 25% of global greenhouse gas emissions responsible for global
warming [4] and other pollutants such as nitrogen oxides (NOx), carbon monoxide (CO), particulate
matter (PM), and soot. Compared with the fossil fuels, biofuels produced from biomass are renewable
and produce less CO2, soot, and unburned hydrocarbon (HC) emissions, which have been widely
used as alternative neat fuels or fuel additives.
1

0
2
4
6
8
10
12
14
16
18
1965 1975 1985 1995 2005 2015 2025 2035
Renewables Hydro Nuclear Coal
Gas
Oil
Billion toe
(a)
0
20
40
60
80
100
120
2000 2005 2010 2015 2020 2025 2030 2035
Mb/d
Non-combusted
Buildings
Industry
Ships, trains & planes
Trucks
Cars
Power
Transport
(b)
Figure 1.1: The outlook for (a) energy consumption and (b) oil demand before 2035 [1]
1.2 Biofuels for transportation
1.2.1 Increased biofuels production
To reduce the GHG emissions, biofuels have been used as, in most cases, fuel blending components
in nowadays transportations due to their cleaner emissions compared with the conventional fuels.
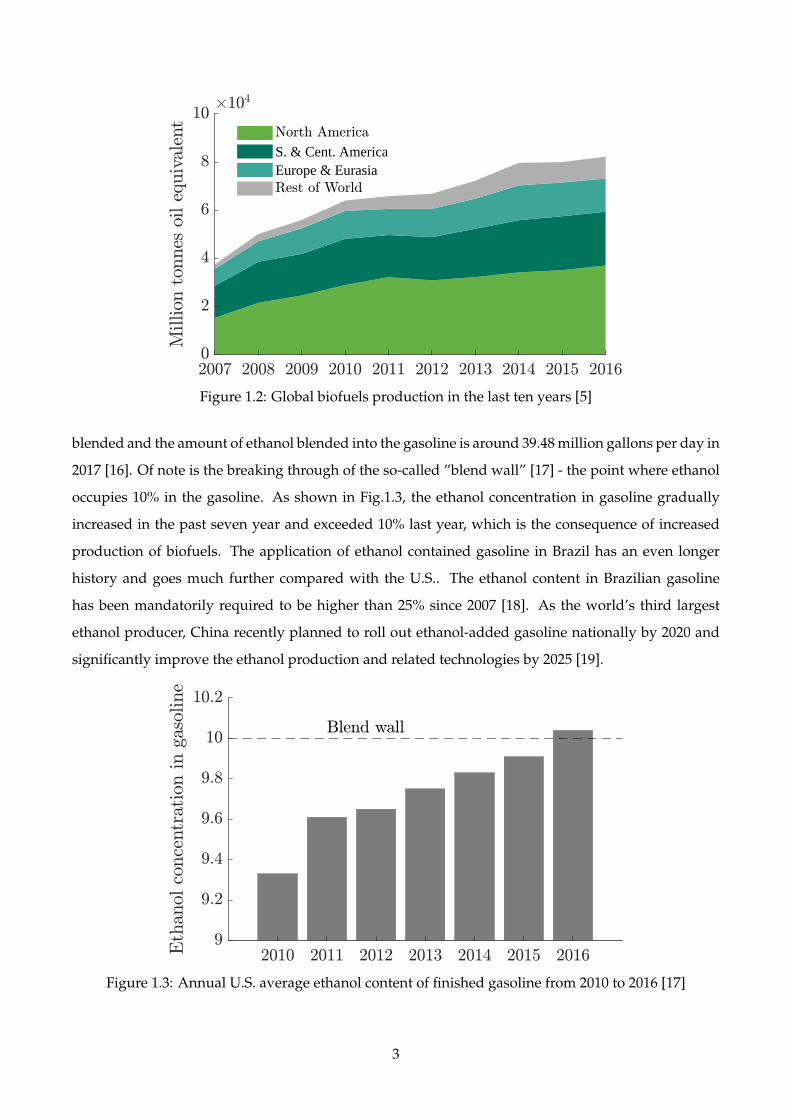
Fig.1.2 shows the global biofuels production in the past ten years [5]. The overall amount is twice of
the value from ten years ago, indicating increasing importance of biofuels. The increased production
enables higher levels of biofuels blending in the fossil fuels.
1.2.2 Ethanol as a fuel additive
Among all biofuels productions, ethanol is the predominant compound and has been extensively used
as a transportation fuel. Generally, renewable ethanol fuel can be sustainably produces in many coun-
tries [6, 7]. Besides, when blended with gasoline, ethanol reduces the emissions of CO and unburned
hydrocarbon in exhaust [8]. Finally, ethanol is known to have high octane numbers [9–12] and signifi-
cant charge cooling effect [9, 11–15], which suppress the knock in spark-ignition (SI) engines and thus
improves the engine efficiencies.
Ethanol has been widely used as a fuel additive in the gasoline with the blending ratios of 10%
or 85% by volume (known as E10 and E85) in most cases. As the largest ethanol production country,
U.S. blends ethanol extensively in the gasoline and nearly all their gasoline are sold with ethanol
2
S. & Cent. AmericaEurope & Eurasia
Figure 1.2: Global biofuels production in the last ten years [5]
blended and the amount of ethanol blended into the gasoline is around 39.48 million gallons per day in
2017 [16]. Of note is the breaking through of the so-called ”blend wall” [17] - the point where ethanol
occupies 10% in the gasoline. As shown in Fig.1.3, the ethanol concentration in gasoline gradually
increased in the past seven year and exceeded 10% last year, which is the consequence of increased
production of biofuels. The application of ethanol contained gasoline in Brazil has an even longer
history and goes much further compared with the U.S.. The ethanol content in Brazilian gasoline
has been mandatorily required to be higher than 25% since 2007 [18]. As the world’s third largest
ethanol producer, China recently planned to roll out ethanol-added gasoline nationally by 2020 and
significantly improve the ethanol production and related technologies by 2025 [19].
Figure 1.3: Annual U.S. average ethanol content of finished gasoline from 2010 to 2016 [17]
3

1.3 Octane blending of ethanol and hydrocarbons
With the increasing amount of ethanol blended into the gasoline, a number of experimental stud-
ies [9–11, 20] have been performed to investigate the blending behaviours between ethanol and hy-
drocarbon fuels. Among all these works, Foong et al. [9] shows that ethanol blends synergistically
with isooctane and n-heptane, but antagonistically with toluene. Besides, they also found that ethanol
blends synergistically with toluene reference fuels. Nevertheless, the causes for these non-linear oc-
tane blending behaviours are not well understood.
In practical applications, it is essential to understand and utilise these non-linear behaviours,
which helps to exploit the benefits from ethanol. More specifically, it is desirable to formulate base
gasoline with hydrocarbons blending synergistically with ethanol, which helps to further improve the
anti-knock performance of the fuel mixture. However, the gasoline is a complex fuel mixture con-
taining hundreds of different hydrocarbons which, in most cases, are expected to blend non-linearly
with ethanol. Besides, the interactions between these hydrocarbons should not be ignored either. It is
worthy noting that exploring all aforementioned octane blending behaviours is not realistic, and thus
fundamental experiments are required as well to understand the chemical origins of the non-linear
blending behaviours, which would provide insights into fuel design.
To sum up, despite the wide and increasing use of ethanol for gasoline blending, the optimal use
of ethanol with gasoline is not fully understood. To shed light on interactions between ethanol and
major components in the gasoline, this study investigates octane blending and oxidation chemistry of
ethanol and hydrocarbon mixtures.
4

Chapter 2
Literature Review
2.1 Overview of Knock
2.1.1 Essence of knock
Knock is the sound caused by the extremely rapid energy release in the unburned air-fuel mixture
(also known as ’end gas’) ahead of the propagating turbulent flame [21]. The abnormal combustion
in the end gas results in high local pressures whose non-uniform nature causes pressure waves to
propagate in the chamber. The oscillations of the pressure waves may cause the entire combustion
chamber to resonate at its natural frequency, which leads to a loud metallic pinging noise that defined
as knock [22].
A typical in-cylinder pressure trace of a knocking cycle of isooctane together with its correspond-
ing non-knocking trace suppressed by tetraethyl lead are shown in Figure 2.1 [23] where Pi and Pf rep-
resent the initial pressure rise and the later peak-peak pressure oscillation. Their responses for PRFs
were investigated by [24] under different compression ratios for both RON and MON conditions. It
was found that Pi correlates well with the knock intensity defined in the ASTM manual [25,26], while
Pf relates to the engine vibrations.
To quantify the knock intensity, a bouncing pin apparatus, shown in Fig.2.2, was developed by
Midgley and Boyd [27] back in 1922. When the engine knock occurs, the diaphragm vibrates due to
the in-cylinder pressure oscillations, which pushes the bouncing pin to close the electrical contacts.
The bouncing-pin fluctuations are measured by the gas evolution from an electrolytic cell filled by
sulphuric acid and distilled water. The electrical outputs, affected by the cycle to cycle variations, are
averaged to represent the knock intensity. The modern knock sensor used in the CFR engine is an
electronic emulation of the original bouncing pin apparatus but in a compact format.
The physical and chemical processes of engine knock have gained wide attention since the early
20th century [29–31]. Nowadays, it is generally accepted by the research community that the spon-
5
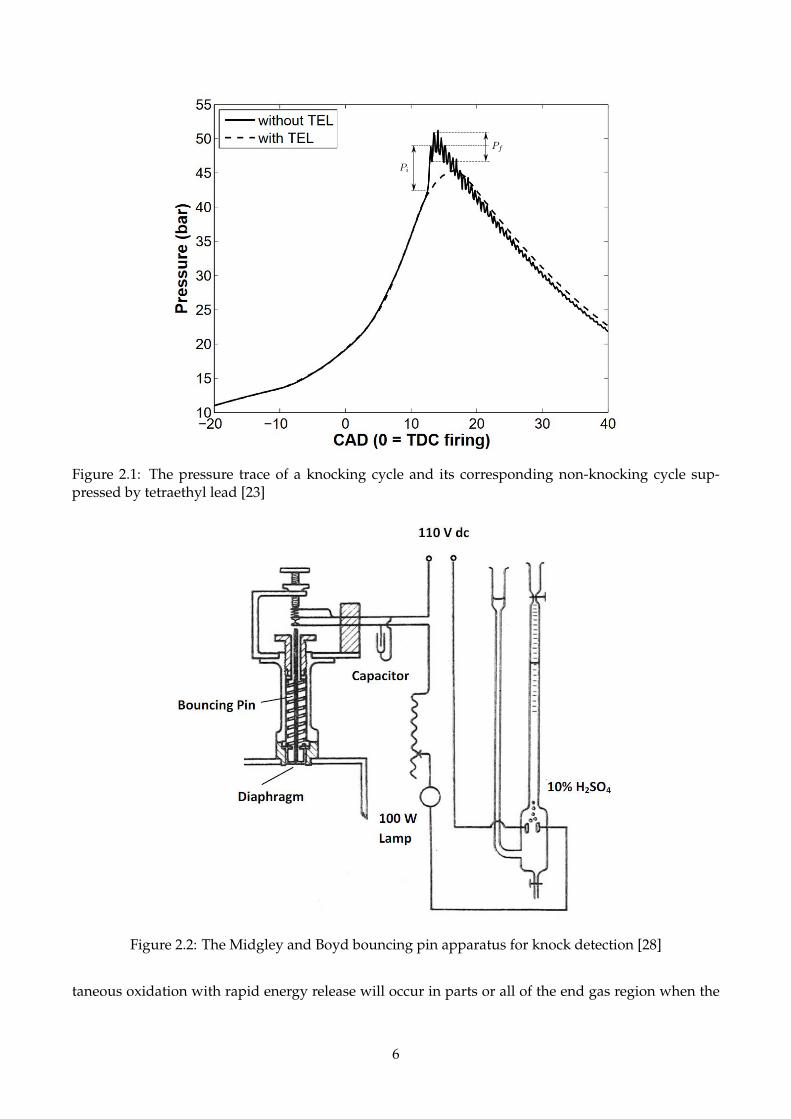
Figure 2.1: The pressure trace of a knocking cycle and its corresponding non-knocking cycle sup-pressed by tetraethyl lead [23]
Figure 2.2: The Midgley and Boyd bouncing pin apparatus for knock detection [28]
taneous oxidation with rapid energy release will occur in parts or all of the end gas region when the
6

pressure and temperatures of one or several gas pockets in the end gas are adequately high [21]. Dur-
ing the autoignition, the pressure trace first has a rapid increase and then oscillates with decaying
amplitude due to the pressure waves generated from the auto-ignited hot spots.
2.1.2 Characteristics of knock
With high-speed imaging, knocking combustion can be observed photographically. Figure 2.3 shows
the time series images for non-knocking and knocking cycles [32]. The normal flame front can be
clearly observed in the non-knocking cycle and the first image of the knocking cycle. The dark
crescent-shaped region ahead of the flame front is the unburned gas zone where autoignition occurs
(in frame G). Then the unburned gas zone becomes brighter and hotter with the propagation of the
autoignition region. Finally, the end gas gets burned completely in frame J.
Figure 2.3: Image series for both non-knocking and knocking engine cycles [32]
Autoignition occurs at places with the most favourable conditions for the low temperature oxi-
dations, and reaction propagation depends on the inhomogeneity of temperature and compositions
of the unburned gas. Based on the temperature gradients, the end gas autoignition could propagate
from the hot gas pocket in three modes [33]:
• With low temperature and steep temperature gradients, the end gas will produce a weak pres-
sure propagating from the centre and is attenuated. In this phase, combustion undergoes a
gradual transition to knock and is regarded as non-knocking combustion.
• With high temperature and small temperature gradients, the end gas will generate simultaneous
chemical reactions following the occurrence of the autoignition. The knock intensity is positively
correlated with the propagation speed of the reaction front. Moderate knock occurs under this
condition.
7

• With intermediate temperature and temperature gradients, the end gas will create strong shock
waves after the initiation of the chemical reactions. Strong pressure waves coupled with very
reactive end gas will generate an intensely illuminating flame. In this case, autoignition ends up
with a severe and damaging knock.
Knock detection techniques can generally be categorised into two types: direct measurements
based on in-cylinder parameters and indirect measurements such as sound, pressure or cylinder block
vibrations [34–36].
• The peak-peak value of the pressure oscillations after band pass filtering was applied to define
the knock intensity by [37].
• Fast Fourier transform (FFT) and power spectral density (PSD) of raw pressure trace were used
to characterise knock in [38, 39].
• The third derivative of the pressure trace, which generates a much higher absolute value when
knock happens, could also be applied to determine the knock onset point [40, 41].
• The occurrence of knock can be determined by engine vibration whose oscillation frequencies
depend on the size and shape of the chamber [42].
2.2 Anti-knock Characteristics of Ethanol/Hydrocarbon Blends
Ethanol, as an oxygenated gasoline blending component, has aroused worldwide interests in the last
two decades. Beneficial results have been reported in numerous studies related to ethanol fuelled SI
engine. Among all these studies, this review summarises ethanol’s two most important features: high
octane number and charge cooling effect.
2.2.1 Octane numbers of ethanol/hydrocarbon blends
In 1927, Graham Edgar [43] proposed an octane rating scale which defines the knock-limited com-
pression ratios for the blend fraction of the two Primary Reference Fuels (PRFs), namely iso-octane
and n-heptane. For instance, a PRF with an octane number of 80 is comprised of 80% iso-octane and
20% n-heptane by volume. Octane rating experiments are conducted in the Cooperative Fuel Re-
search Committee (CFR) engine with standard testing procedures [25, 26]. Research octane number
(RON) and motored octane number (MON) are two types of octane numbers associated with different
operating conditions.
8
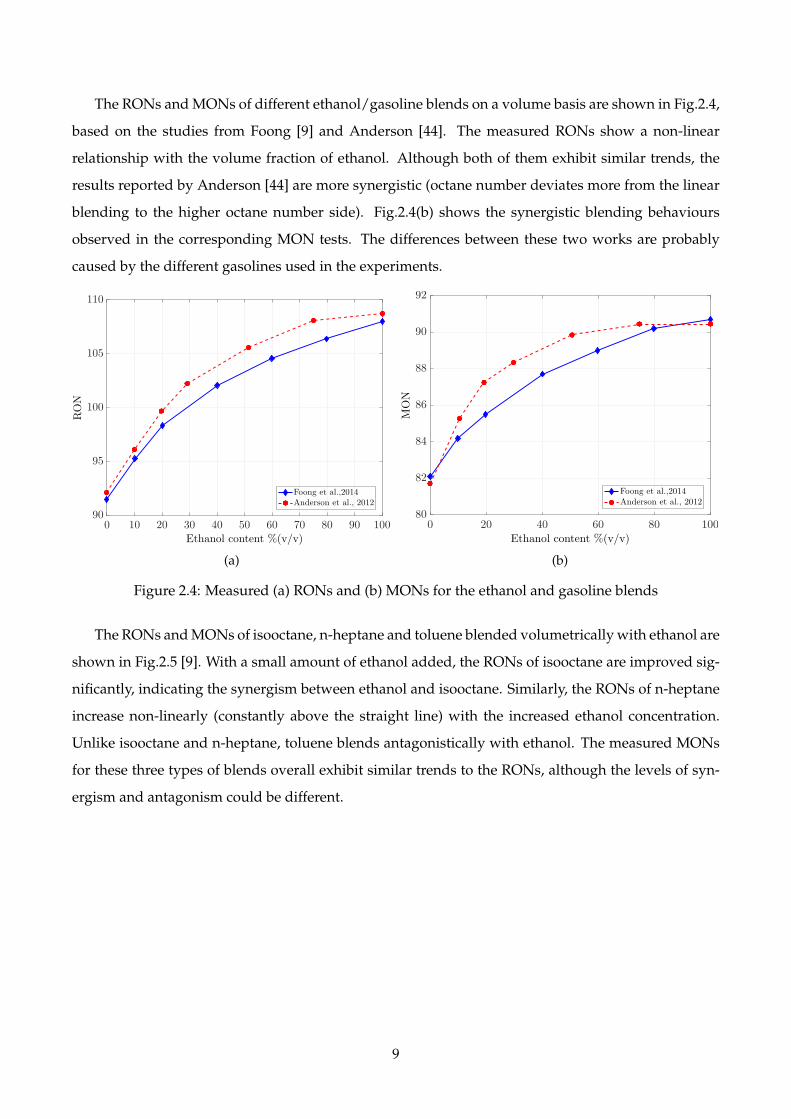
The RONs and MONs of different ethanol/gasoline blends on a volume basis are shown in Fig.2.4,
based on the studies from Foong [9] and Anderson [44]. The measured RONs show a non-linear
relationship with the volume fraction of ethanol. Although both of them exhibit similar trends, the
results reported by Anderson [44] are more synergistic (octane number deviates more from the linear
blending to the higher octane number side). Fig.2.4(b) shows the synergistic blending behaviours
observed in the corresponding MON tests. The differences between these two works are probably
caused by the different gasolines used in the experiments.
0 10 20 30 40 50 60 70 80 90 100
Ethanol content %(v/v)
90
95
100
105
110
RON
Foong et al.,2014Anderson et al., 2012
(a)
0 20 40 60 80 100
Ethanol content %(v/v)
80
82
84
86
88
90
92
MON
Foong et al.,2014Anderson et al., 2012
(b)
Figure 2.4: Measured (a) RONs and (b) MONs for the ethanol and gasoline blends
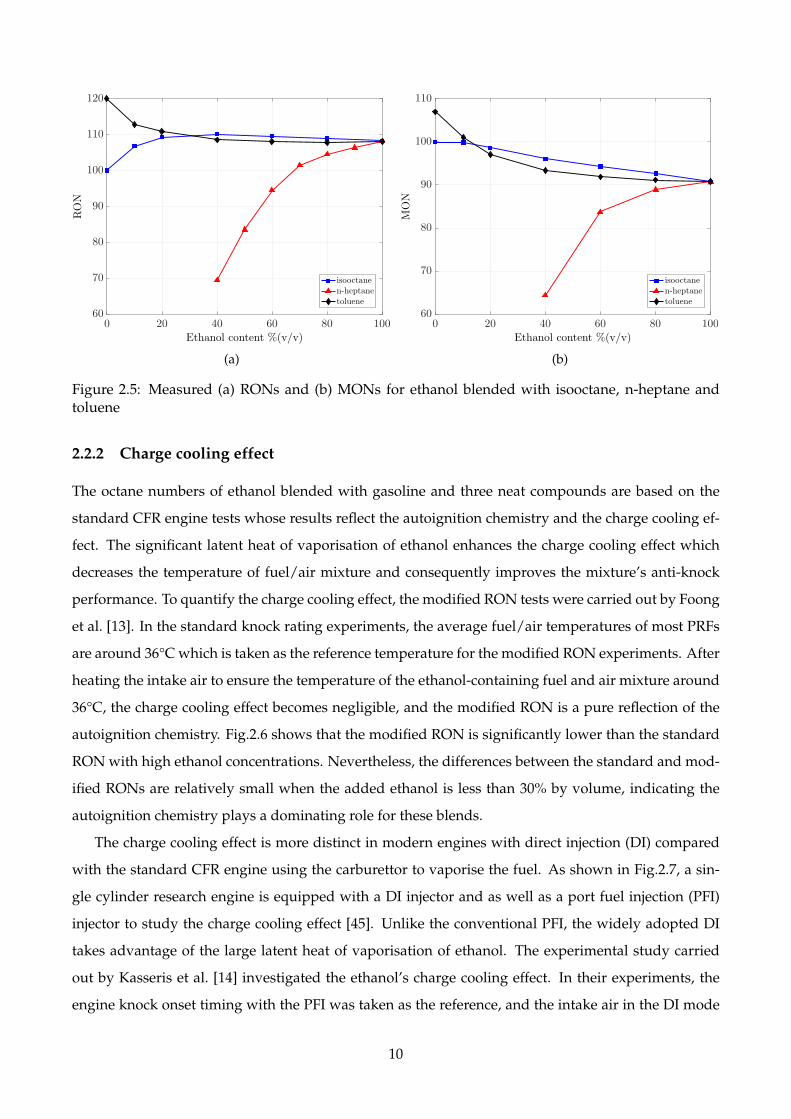
The RONs and MONs of isooctane, n-heptane and toluene blended volumetrically with ethanol are
shown in Fig.2.5 [9]. With a small amount of ethanol added, the RONs of isooctane are improved sig-
nificantly, indicating the synergism between ethanol and isooctane. Similarly, the RONs of n-heptane
increase non-linearly (constantly above the straight line) with the increased ethanol concentration.
Unlike isooctane and n-heptane, toluene blends antagonistically with ethanol. The measured MONs
for these three types of blends overall exhibit similar trends to the RONs, although the levels of syn-
ergism and antagonism could be different.
9
0 20 40 60 80 100
Ethanol content %(v/v)
60
70
80
90
100
110
120
RON
isooctanen-heptanetoluene
(a)
0 20 40 60 80 100
Ethanol content %(v/v)
60
70
80
90
100
110
MON
isooctanen-heptanetoluene
(b)
Figure 2.5: Measured (a) RONs and (b) MONs for ethanol blended with isooctane, n-heptane andtoluene
2.2.2 Charge cooling effect
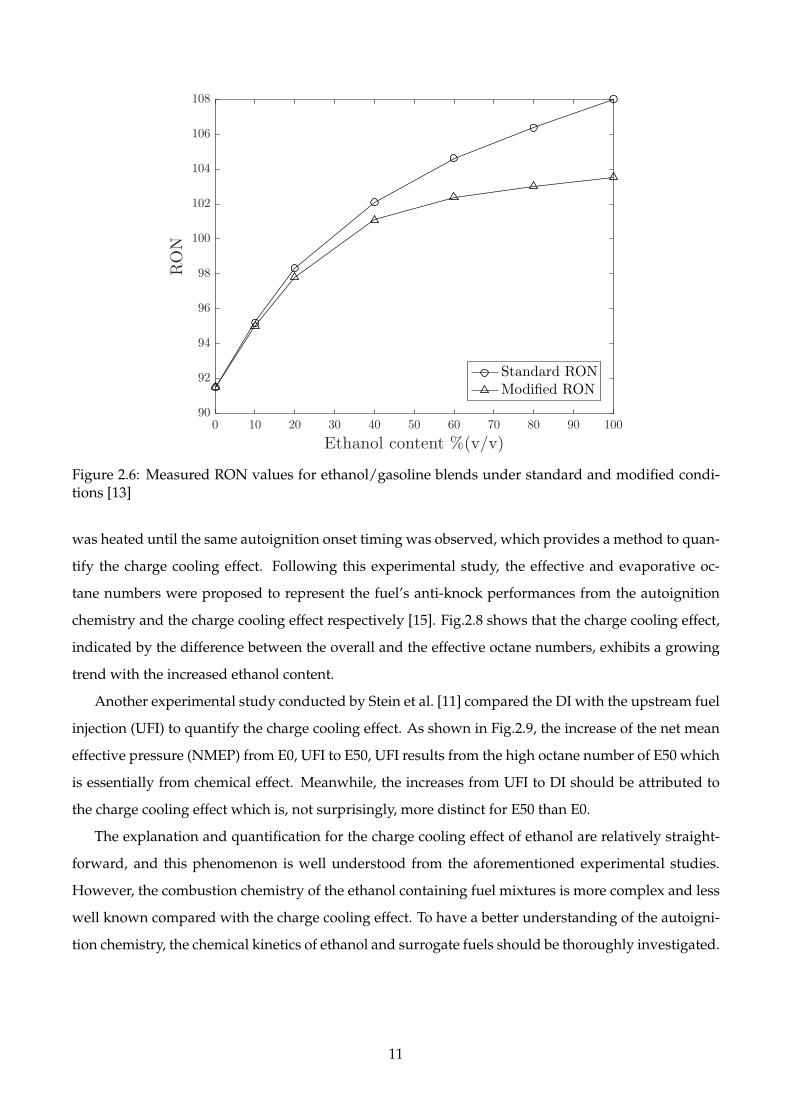
The octane numbers of ethanol blended with gasoline and three neat compounds are based on the
standard CFR engine tests whose results reflect the autoignition chemistry and the charge cooling ef-
fect. The significant latent heat of vaporisation of ethanol enhances the charge cooling effect which
decreases the temperature of fuel/air mixture and consequently improves the mixture’s anti-knock
performance. To quantify the charge cooling effect, the modified RON tests were carried out by Foong
et al. [13]. In the standard knock rating experiments, the average fuel/air temperatures of most PRFs
are around 36°C which is taken as the reference temperature for the modified RON experiments. After
heating the intake air to ensure the temperature of the ethanol-containing fuel and air mixture around
36°C, the charge cooling effect becomes negligible, and the modified RON is a pure reflection of the
autoignition chemistry. Fig.2.6 shows that the modified RON is significantly lower than the standard
RON with high ethanol concentrations. Nevertheless, the differences between the standard and mod-
ified RONs are relatively small when the added ethanol is less than 30% by volume, indicating the
autoignition chemistry plays a dominating role for these blends.
The charge cooling effect is more distinct in modern engines with direct injection (DI) compared
with the standard CFR engine using the carburettor to vaporise the fuel. As shown in Fig.2.7, a sin-
gle cylinder research engine is equipped with a DI injector and as well as a port fuel injection (PFI)
injector to study the charge cooling effect [45]. Unlike the conventional PFI, the widely adopted DI
takes advantage of the large latent heat of vaporisation of ethanol. The experimental study carried
out by Kasseris et al. [14] investigated the ethanol’s charge cooling effect. In their experiments, the
engine knock onset timing with the PFI was taken as the reference, and the intake air in the DI mode
10
0 10 20 30 40 50 60 70 80 90 100
Ethanol content %(v/v)
90
92
94
96
98
100
102
104
106
108
RON
Standard RONModified RON
Figure 2.6: Measured RON values for ethanol/gasoline blends under standard and modified condi-tions [13]
was heated until the same autoignition onset timing was observed, which provides a method to quan-
tify the charge cooling effect. Following this experimental study, the effective and evaporative oc-
tane numbers were proposed to represent the fuel’s anti-knock performances from the autoignition
chemistry and the charge cooling effect respectively [15]. Fig.2.8 shows that the charge cooling effect,
indicated by the difference between the overall and the effective octane numbers, exhibits a growing
trend with the increased ethanol content.
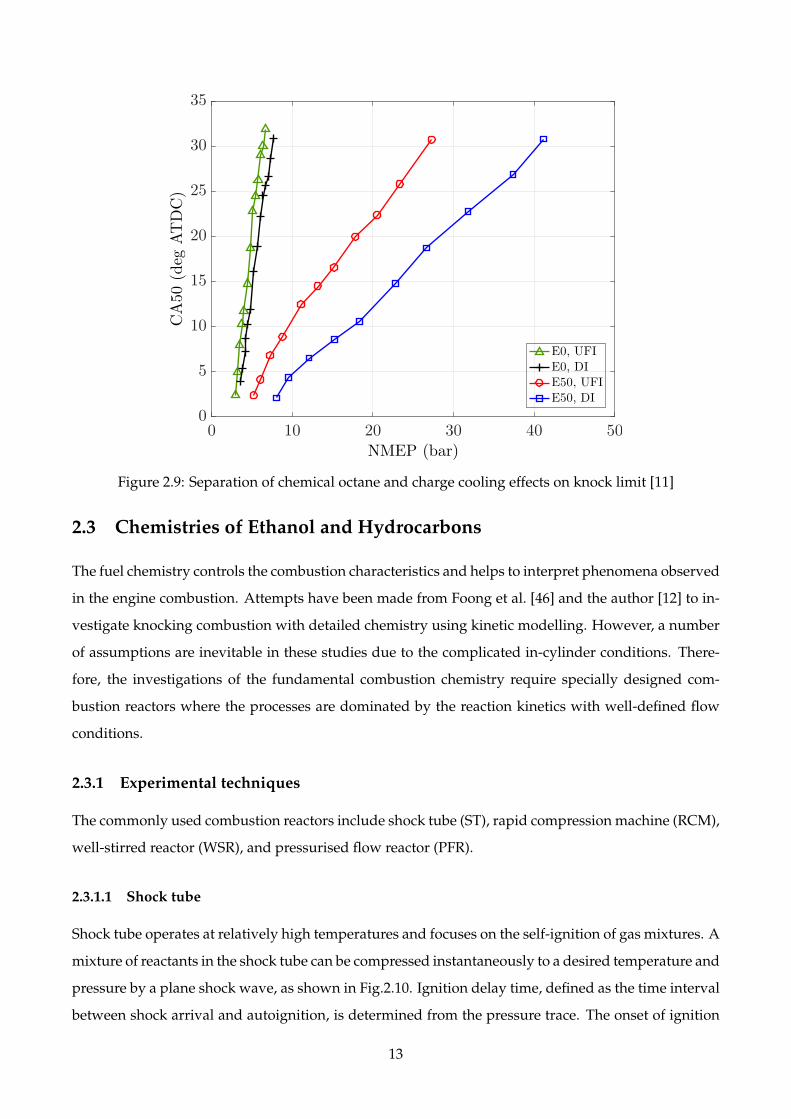
Another experimental study conducted by Stein et al. [11] compared the DI with the upstream fuel
injection (UFI) to quantify the charge cooling effect. As shown in Fig.2.9, the increase of the net mean
effective pressure (NMEP) from E0, UFI to E50, UFI results from the high octane number of E50 which
is essentially from chemical effect. Meanwhile, the increases from UFI to DI should be attributed to
the charge cooling effect which is, not surprisingly, more distinct for E50 than E0.
The explanation and quantification for the charge cooling effect of ethanol are relatively straight-
forward, and this phenomenon is well understood from the aforementioned experimental studies.
However, the combustion chemistry of the ethanol containing fuel mixtures is more complex and less
well known compared with the charge cooling effect. To have a better understanding of the autoigni-
tion chemistry, the chemical kinetics of ethanol and surrogate fuels should be thoroughly investigated.
11

Figure 2.7: CAD model of the combustion chamber [45]
0 20 40 60 80 100
Ethanol content %(v/v)
95
100
105
110
115
120
125
130
135
Octan
enumber
Effective octane numberOverall octane number
Figure 2.8: The comparison between overall and effective octane numbers [14]
12
0 10 20 30 40 50
NMEP (bar)
0
5
10
15
20
25
30
35
CA50
(deg
ATDC)
E0, UFIE0, DIE50, UFIE50, DI
Figure 2.9: Separation of chemical octane and charge cooling effects on knock limit [11]
2.3 Chemistries of Ethanol and Hydrocarbons
The fuel chemistry controls the combustion characteristics and helps to interpret phenomena observed
in the engine combustion. Attempts have been made from Foong et al. [46] and the author [12] to in-
vestigate knocking combustion with detailed chemistry using kinetic modelling. However, a number
of assumptions are inevitable in these studies due to the complicated in-cylinder conditions. There-
fore, the investigations of the fundamental combustion chemistry require specially designed com-
bustion reactors where the processes are dominated by the reaction kinetics with well-defined flow
conditions.
2.3.1 Experimental techniques
The commonly used combustion reactors include shock tube (ST), rapid compression machine (RCM),
well-stirred reactor (WSR), and pressurised flow reactor (PFR).
2.3.1.1 Shock tube
Shock tube operates at relatively high temperatures and focuses on the self-ignition of gas mixtures. A
mixture of reactants in the shock tube can be compressed instantaneously to a desired temperature and
pressure by a plane shock wave, as shown in Fig.2.10. Ignition delay time, defined as the time interval
between shock arrival and autoignition, is determined from the pressure trace. The onset of ignition
13

can be obtained from the emission/absorption spectra of intermediate combustion species. However,
the non-ideal conditions, i.e. the formation of boundary layers in reflected shock tube experiment,
limit the observation times to hundred of microseconds. Thus, experiment conditions are constrained
to pressure and temperature regimes with short chemical induction times.
Figure 2.10: Schematic of a shock tube/rapid compression machine
2.3.1.2 Rapid compression machine
With the similar schematic to the shock tube, the rapid compression machine is designed to emulate
the combustion process in reciprocating engines, which makes it a relatively complicated system. The
movement of a piston compresses premixed mixture in the combustion chamber to a small volume,
high pressure, and temperature, which initiates the ignition. The pressure and temperature histories
are controlled by the compression ratio, initial pressure and mixture composition. Similar to engines,
the physical phenomena inside the rapid compression machine are complicated. Unknown wall heat
transfer, blow-by due to piston crevices and large-scale disturbances of reacting mixture caused by
piston movement complicate the interpretation of the experimental results of the rapid compression
machine. Besides, it is challenging to use extractive sampling method to measure mixture composi-
tion. Normally, the data from rapid compression machine are modelled with a homogeneous reaction
condition and empirically determined heat loss function.
2.3.1.3 Well-stirred reactor
The well-stirred, or perfectly-stirred reactor is assumed to have entirely homogeneous mixing inside
the control volume, as shown in Fig.2.11 [47]. A common type of well stirred reactor is called jet-stirred
reactor which uses high velocity inlet jets to facilitate the mixing process.
An essential characteristics of the well-stirred reactor is the perfect mixing assumption which con-
siders the time required for the mixing is much shorter than the mean residence time of the fluid in the
reactor. However, the actual residence time in a WSR is less defined, which is supposed to follow a res-
idence time function, not a single value. This has to be assumed in the modelling. The WSR operates
with less dilution, short residence times, and higher temperatures. However, the non-ideal conditions
14

Figure 2.11: Schematic of a well-stirred reactor [47]
in the experiment, such as imperfect mixing and heterogeneous chemistry, make it complicated to
fully interpret the experimental results.
2.3.1.4 Pressurised flow reactor
The pressurised flow reactor is designed to provide a convective-reactive environment, where diffu-
sion along the flow direction is minor or can be neglected. The schematic of a flow reactor is shown in
Fig2.12. In the PFR experiment, the vaporised fuel and the heated oxidizer enter the reactor through
two separated lines and mix with each other via a mixer. The reaction starts to occur at the same time
due to the high temperature of the mixture.
When the mixture flows along a long insulated or heated reactor tube, a hot water cooled sampling
probe is used to extract the gas mixture continuously. The movement of the gas sampling probe is
driven by a program controlled motor, which could be applied to extract gas mixtures at different
positions. By measuring the gas temperature which determines the gas velocity, the tube distance can
be converted to the residence time. The time histories of the mole fractions of reactants, intermediates
and products can be measured by gas chromatography (GC) located downstream of the sampling
probe.
The ability of producing the species profiles of reactants, intermediates and products is the most
significant advantage of flow reactor compared with other reactors. A potential issue of the flow reac-
tor lies in the mixing process where inhomogeneous reactions are occurring. The negative effects from
15

Figure 2.12: Structure of a pressurised flow reactor
the inhomogeneity could be minimized by using a specially designed mixer providing fast mixing.
2.3.2 Combustion chemistry of hydrocarbons and alcohol
The experimental results from reactors mentioned above are applied to develop detailed combustion
chemistry which provide insights into understanding the autoignition phenomena in SI engines. The
reviews of comprehensive chemical mechanisms at high temperatures are available in [48–50], while
low temperature chemistry of hydrocarbons was reviewed by [51, 52]. The review mainly focuses on
the low temperature oxidations which are relevant to the autoignition in the SI engines. The state of
the art combustion chemistry of major hydrocarbon groups and ethanol will be introduced briefly.
2.3.2.1 Combustion chemistry of alkanes
Considering the significance of the low temperature chemistry in the engine knock, the general reac-
tion pathways for the oxidations of alkanes at low temperatures are first reviewed, whose scheme is
shown in Fig.2.13 [51]. The abstraction of a hydrogen atom (H-abstraction) from alkane by oxygen
or hydroxyl radical (•OH) initiates the reaction to produce alkyl (R•) and hydroperoxy (•OOH) radi-
cals. Since the rate constant of this type of reaction is sensitive to the radical structure, the branched
molecules (e.g., isooctane) have lower rate constants compared with those straight-chain ones (e.g.,
n-heptane) [53]. After H-abstraction, alkyl radicals react with oxygen molecules to generate a variety
of products [52]:
(2.1)R• + O2 ↔ ROO•
16

(2.2)R• + O2 → alkene + HO2•
The products from R• + O2 reaction change with pressure and temperature. Typically, at low tempera-
ture and moderate pressures, alkyl peroxy radical (•ROO) is the primary product, as shown in reaction
2.1.
•QOOH
HO2
• + alkeneRH
ROOH + O2
degeneratebranching
RO• + •OH
steps
RH •OH + cyclic ethers,aldehydes or ketones
R•
keto-hydroperoxides + • OH
R’• + H2 O2 ROO•degeneratebranching
steps
•OOQOOH
•U(OOH)2
O2
XO• + •OH
O2
R’• + alkene
(2)(1)
HO2•
RH + O2 or • OH
O2
R’• + H2O
initiation steps
H abstractions
(3)
Figure 2.13: Simplified scheme for the primary mechanism of oxidation of alkanes at low temperatures[51]
The thermally unstable alkyl peroxy radical may undergo different reaction paths, which results
in the varied progresses of autoignition. Firstly, the alkyl peroxy radical can dissociate back to the
alkyl radical and oxygen molecule. If at the same time, the temperature increases to favour reac-
tion 2.2, the overall reaction rate will be reduced, which leads to the so-called negative temperature
coefficient (NTC) regime. Secondly, with the low energy threshold, the alkyl peroxy radical decom-
poses to alkene and hydroperoxy radical even at room temperatures [54], as shown in reaction 2.3.
Both theoretical [55] and experimental [56] studies showed that this type of reaction is significant
for hydroperoxy radical production. However, hydroperoxy radical is not reactive, which slows or
effectively terminates the low temperature reactions.
(2.3)ROO• → alkene + HO2•
(2.4)ROO• ↔ •QOOH
17

The most important reaction path for alkyl peroxy radical, which leads to chain-propagating re-
actions, is the isomerization via internal H-atom abstraction to form hydroperoxy alkyl (•QOOH)
radical, as shown in reaction 2.4. This type of reaction undergoes a cyclic transition state, whose ac-
tivation energies for isomerization comprising the activation energy for H-abstraction and the strain
energy of the cyclic transition state. In this case, both the ring strain energy barriers and the type of
abstracted H atom affect the rate constants of these reactions. Then, the unstable hydroperoxy alkyl
radical decomposes to cyclic ether and highly reactive hydroxyl radical (•OH).
The unpaired electron of the carbon atom of hydroperoxy alkyl radical is vulnerable to the attack
from oxygen molecule, as shown in reaction 2.5, which is quite similar to the reaction between alkyl
radical and oxygen molecule.
(2.5)•QOOH + O2 ↔ •OOQOOH
Afterwards, the •OOQOOH radical goes through a second internal H-abstractions, similar to
ROO•, and forms the •Q(OOH)2 radical. The decomposition of this radical gives hydroxyl radical,
which is a chain-branching reaction:
(2.6)•OOQOOH ↔ •Q(OOH)2 → ketohydroperoxides + •OH→ •OQO + 2•OH
Another chain-branching reaction pathway in Fig.2.13 related to reaction 2.7 and 2.8. However, the
alkyl hydroperoxide is relatively stable, especially at low temperatures, which results in slow chain
branching reactions and thus contributes little to the production of hydroxyl radicals.
(2.7)ROO• + HO2• → ROOH + O2
(2.8)ROOH → RO• + •OH
The low temperature combustion chemistry of the alkanes provides fundamentals for the devel-
opment of the state of the art chemical mechanisms for various hydrocarbons of interest to practical
fuels. Among all these hydrocarbons, isooctane, n-heptane and pentanes are considered as important
in the production gasoline, and their chemical mechanisms are critical in terms of understanding the
engine knock.
As a representative branched alkane, isooctane is widely used as a surrogate gasoline fuel for
engine combustion research. The most well known detailed combustion model of isooctane at both
low and high temperatures was developed by Curran et al. [57] based on the experimental results
from a jet-stirred reactor [58], flow reactors [59–61], shock tubes [62–64] and motored engines [65, 66],
which cover the pressure range from 1 atm to 45 atm and temperature from 550 K to 1700 K with
equivalence ratio from 0.3 to 1.5. The model shows good agreements when compared with different
experimental results. Then, a gasoline surrogate mechanism developed by Mehl et al. [67] speeds up
18

the low temperature oxidation processes with updated rate constants and thermal properties, which
produces better agreements to experiments in various operating conditions. A very recent update for
isooctane mechanism comes from Atef et al. [68], which is motivated by matching the experimental
results [69–73] causing problems for previous mechanisms. With the implementations of the recent
results from computational studies in isooctane thermochemistry [74–77], low temperature oxidation
kinetics of normal and branched alkanes [78–81], and new alternative isomerization pathways, the
latest isooctane mechanism produces improved agreements to the existing experiments, especially
those at lower equivalence ratios.
N-heptane is a representative fuel for normal alkane, whose combustion chemistry is relatively
well understood. Numerous experimental studies were performed in shock tubes [62, 82–86], rapid
compression machines [87–90], jet-stirred reactors [58, 91–94], flow reactors [60, 95–97], flame exper-
iments [98–106], and engines [107–111] to study the oxidations of n-heptane over a wide range of
conditions. A detailed combustion mechanism of n-heptane was proposed by Curran et al. [112],
which not only perform well when matching experimental data but also provides a kinetic frame for
the mechanism development. This mechanism was modified by Mehl et al. [67] to incorporate the
updated decomposition rates of the alkyl and alkoxy radicals [113], the isomerization rates at low
temperature oxidation recommended by [114], and the new reaction pathways from [115, 116]. The
most recent update [117] for the mechanism includes AramcoMech 2.0 [118] for the C0 −C4 species,
the latest chemistry for three pentan isomers [119–121], and the base n-heptane sub-smechanism [112].
Although isooctane and n-heptane are normally considered as surrogate gasoline fuels, their frac-
tions in the Australia production gasoline are much less than iso-pentane and n-pentane. Several
experimental studies were conducted to investigate the oxidations of pentane isomers in rapid com-
pression machines [120, 122–125], shock tubes [120, 126, 127], a well-stirred reactor [128], an annular
flow reactor [129], and a CFR engine [130]. The state of the art chemical mechanism of pentane isomers
was developed by Bugler et al. [120] and very recently updated based on experimental results from
two jet-stirred reactors [131].
The chemical mechanisms for most alkanes have been renewed in recent two years with constantly
emerging experimental, theoretical and modelling studies. However, alkanes alone are not sufficient
to emulate the production gasoline with a significant amount of aromatics as octane boosters.
2.3.2.2 Combustion chemistry of aromatics
As important components in petroleum-derived fuels, aromatics typically show much slower oxida-
tion rates than alkanes, particularly at low temperatures. Understanding the detailed chemical kinet-
ics of aromatics is necessary to interpret the combustion characteristics of the production gasoline.
19

Toluene, a representative hydrocarbon in the aromatics family, has been used as a surrogate gaso-
line fuel along with n-heptane and isooctane to emulate the production gasoline. The major reaction
pathways and the state of the art chemical mechanisms for toluene will be reviewed.
Fig.2.14 presents the main reaction pathways of toluene and benzene oxidations proposed by
Brezinsky [132]. As a very important product of toluene oxidation, benzene may form phenyl rad-
ical, phenol, and phenoxy radical after being attacked by those small and reactive radicals. Besides,
the former two products, phenyl radical and phenol, react with small radicals to produce phenoxy
radical which decomposes to CO and cyclopentadienyl radical. The free electron of cyclopentadienyl
radical may combine an H atom to form cyclopentadiene or reacts with those oxygenated radicals to
generate cyclopentadionyl radical which produces butadienyl radical and CO.
Figure 2.14: Simplified scheme for the oxidations of benzene and toluene [132]
Toluene oxidation mechanism can be found on the right part of Fig.2.14. At the beginning, toluene
reacts with small radicals to form benzyl radical, cresol, cresoxy radical and benzene. Benzyl radical is
most abundant product from the first step oxidation. Although benzyl radical is very stable, especially
at low temperatures, it has several reaction pathways at relatively high temperatures. The benzyl rad-
ical may combine with itself to generate bibenzyl and react with small radicals to form other stable
molecules, such as benzyl alcohol, ethylbenzene and benzaldehyde. Apart from the reaction pathways
proposed in [132], a C7H6 molecule was observed from the decomposition of benzyl radical by the re-
20

cent experiment study [133]. The C7H6 was later found to be fulvenallene [134] and the corresponding
rate constant was theoretically computed by da Silva et al. [135]. Another important reaction is be-
tween benzyl and hydroperoxy radicals which mainly generate benzoxyl and hydroxyl radicals above
700K, but below this temperature, the primary product becomes benzylhydroperoxide. These benzyl
radical involved reactions all lead to chain-termination. Unlike alkanes above, aromatics, especially
those without long chain, generally don’t have low temperature chemistry as the cyclic transition state
cannot be formed.
The experimental studies for toluene oxidations have been performed using flow reactors [136–
138], jet-stirred reactors [139, 140], shock tubes [141–145], rapid compression machines [146] and lam-
inar premix flames measurements [147–149]. To model these experimental results, several detailed
chemical mechanisms [137, 139, 145, 150–155] have been developed previously. The recent modelling
study was conducted by Metcalfe et al. [138], which combines toluene sub-mechanism from [140]
and C0 −C4 sub-mechanism from [156–159]. This toluene mechanism [138] was incorporated into
the latest n-butylbenzene model by Nakamura et al. [160] which contains C0 −C4 sub-mechanism
from AramcoMech 1.3 [161] and alkyl-aromatics sub-mechanism from [162]. More recently, Yuan et
al. [163,164] proposed a kinetic model for toluene based on their experiments performed in flow reac-
tor and jet stirred reactor.
2.3.2.3 Combustion chemistry of ethanol
Ethanol, as a renewable fuel and an octane booster, has been added to the production gasoline world
widely. The oxidations of ethanol were investigated using shock tubes [165–170], flow reactors [171–
174], jet-stirred reactors [175, 176], rapid compression machine [170] and laminar flames [177–180].
The most well known detailed ethanol mechanism was developed by Marinov [181] which had
been validated against all available experimental data at that time. Marinov’s mechanism was first
improved by Li et al. [172, 173, 182] to model ethanol pyrolysis and oxidation in a pressurised flow
reactor by including modified rate parameters for the decomposition reactions. Later, Dagaut and
Togbe [183] updated Marinov’s mechanism with the kinetic parameters from quantum chemical cal-
culations for H atom abstraction from ethanol molecule. The existing ethanol mechanism continues to
be improved by experiments in different reactors [175, 184].
As critical reaction pathways, the rate constants for the four ethanol decomposition reactions (2.9
to 2.12) have aroused great interests in the research community. Based on the experimental and theo-
retical studies carried out by Li et al., the rate constants for reaction 2.11 and 2.12 are much lower than
those of reaction 2.9 and 2.10. Besides, Li et al. also presented that reaction 2.9 is strongly dependent
on temperature and is dominant over the temperature range of 300-2500 K at 1 atm.
21

(2.9)C2H5OH ↔ C2H4 + H2O
(2.10)C2H5OH ↔ •CH3 + •CH2OH
(2.11)C2H5OH ↔ •CH2CH3 + •OH
(2.12)C2H5OH ↔ •C•HCH3 + H2O
The reactions between ethanol and hydroxyl radical generate different products (2.13 to 2.15)
whose relative fractions are determined by the branching ratios. The ratios applied by both Mari-
nov [181] and Li et al. [172, 173, 182] are from empirical approaches. While a more recent study per-
formed by Mittal et al. [184] adopted the rate constants from Sivaramaskrishnan et al. [185] and tuned
the branching ratios to match the experimental results.
(2.13)C2H5OH + •OH ↔ CH3•CHOH + H2O
(2.14)C2H5OH + •OH ↔ •CH2CH2OH + H2O
(2.15)C2H5OH + •OH ↔ CH3CH2O• + H2O
Understanding the chemical mechanisms of the neat compounds is the prerequisite to explain the
behaviours of production gasoline and gasoline surrogates over a wide range of conditions. However,
the interactions among different components in these mixtures do exist and may play a significant role
concerning affecting the overall performances. In this regard, there has been an increasing awareness
of the necessity to investigate the chemical interactions.
2.4 Chemical Interactions of Fuel Mixtures
Although the oxidation kinetics for neat fuel compounds is relatively well understood, it is often chal-
lenging to predict the autoignition of fuel mixtures due to chemical interactions. These interactions
are typically divided into two types: the first is via large fuel-like radicals, and the second is via small
radicals.
2.4.1 Interactions between alkanes
The cross reactions for alkane mixtures have been studied by Andrae et al. [152, 186]. In their earlier
publication [186], the cross reactions between fuel-like radicals were incorporated in the mechanism to
explain the experimental results that PRF84 ignites much earlier than toluene/n-heptane mixture with
similar RON in HCCI engine at high intake pressure and low intake temperature, since the ignition
delays of these fuel mixtures are similar at low intake pressure and high intake temperature. They
22

argued that with the added cross reactions, the PRF mixture would be more reactive than toluene/n-
heptane mixture before the NTC regime. While at low intake pressure and high intake temperature,
which is within the NTC regime, the reactivity of toluene/n-heptane mixture was less affected by
the NTC effects compared with PRFs. It seems that the addition of the cross reactions increases the
reactivity of PRF before the NTC regime and thus improves the predictions of autoignition delays in
HCCI engine.
However, in their later experimental study [152], the rate constants of the cross reactions have
been re-evaluated. When validating the TRF mechanism against the shock tube autoignition delays,
the rate constants of the cross reactions were too high, which lead to significantly shorter predicted
ignition times than the measurements below 1000 K. Besides, excluding those cross reactions had little
influence to the modelling results, which suggests that the cross reactions may not be significant in the
PRF autoignition. The studies from Andrae et al. [152, 186] showed that the cross reactions between
fuel-like radicals are not significant at least when predicting the autoignition delays from the shock
tube. However, the cross reactions related to the small radicals are supposed to be important, which
are more likely to occur and even affect the overall reactivity.
2.4.2 Interactions between PRF and toluene
The measured MONs of isooctane and toluene mixtures are plotted in Fig.2.15, which indicate the fuel
interactions do occur as the MON of 75% isooctane and 25% toluene is lower than those of both neat
compounds [187]. It is necessary to understand the fuel interactions before interpreting the complex
behaviours of fuel mixtures.
2.4.2.1 Cross reactions via large radicals
The chemical interactions between PRF and toluene have been studied when developing the com-
prehensive kinetic mechanism for surrogate fuels [67, 152, 186]. At high temperatures, numerous in-
termediates, like alkenes and benzyl radical, coexist at the beginning of oxidation and tend to react
with each other. The cross reactions between large fuel-like radicals are divided into three groups, as
proposed by [188].
The first type is H-abstraction reaction, which was incorporated by Andrae et al. [152,186] in their
kinetic mechanisms. In reaction 2.16, RH represents toluene, benzene and benzaldehyde, while QH
denotes n-heptane, isooctane, C3H6 and iC4H8. The rate constants of this type of reactions are from
the studies by Bounaceur et al. [140] and Da Costa et al. [189].
(2.16)RH + Q ↔ R + QH
23
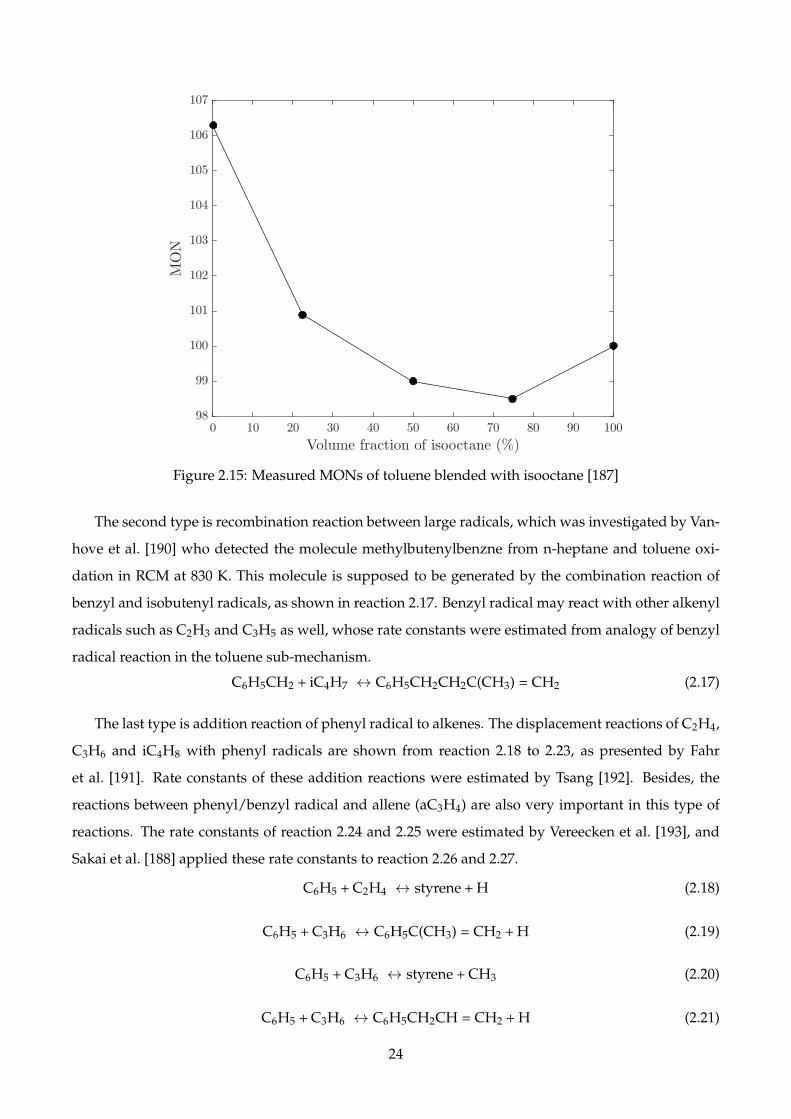
0 10 20 30 40 50 60 70 80 90 100
Volume fraction of isooctane (%)
98
99
100
101
102
103
104
105
106
107
MON
Figure 2.15: Measured MONs of toluene blended with isooctane [187]
The second type is recombination reaction between large radicals, which was investigated by Van-
hove et al. [190] who detected the molecule methylbutenylbenzne from n-heptane and toluene oxi-
dation in RCM at 830 K. This molecule is supposed to be generated by the combination reaction of
benzyl and isobutenyl radicals, as shown in reaction 2.17. Benzyl radical may react with other alkenyl
radicals such as C2H3 and C3H5 as well, whose rate constants were estimated from analogy of benzyl
radical reaction in the toluene sub-mechanism.
(2.17)C6H5CH2 + iC4H7 ↔ C6H5CH2CH2C(CH3) = CH2
The last type is addition reaction of phenyl radical to alkenes. The displacement reactions of C2H4,
C3H6 and iC4H8 with phenyl radicals are shown from reaction 2.18 to 2.23, as presented by Fahr
et al. [191]. Rate constants of these addition reactions were estimated by Tsang [192]. Besides, the
reactions between phenyl/benzyl radical and allene (aC3H4) are also very important in this type of
reactions. The rate constants of reaction 2.24 and 2.25 were estimated by Vereecken et al. [193], and
Sakai et al. [188] applied these rate constants to reaction 2.26 and 2.27.
(2.18)C6H5 + C2H4 ↔ styrene + H
(2.19)C6H5 + C3H6 ↔ C6H5C(CH3) = CH2 + H
(2.20)C6H5 + C3H6 ↔ styrene + CH3
(2.21)C6H5 + C3H6 ↔ C6H5CH2CH = CH2 + H
24

(2.22)C6H5 + iC4H8 ↔ C6H5C(CH3) = CH2 + CH3
(2.23)C6H5 + iC4H8 ↔ C6H5CH2C(CH3) = CH2 + H
(2.24)C6H5 + aC3H4 ↔ C6H5CH2 + C2H2
(2.25)C6H5 + aC3H4 ↔ C9H8 + H
(2.26)C6H5CH2 + aC3H4 ↔ C6H5CH2CH2 + C2H2
(2.27)C6H5CH2 + aC3H4 ↔ C10H10 + H
Although the fuel interactions on the large radical level have been observed in multiple studies,
their impacts on the fuel mixture performances could be limited as the associated elementary reactions
normally have very small rate constants. Note that the chemical interactions related to parent fuels
and parent fuel-like radicals were incorporated in the gasoline surrogate mechanisms developed by
Mehl et al. [67] and Andrae [187] respectively.
2.4.2.2 Cross reactions via radical pool
According to Vanhove et al. [193] and Andrae et al. [152], the cross reactions via radical pool may have
greater significance than those between large molecules and/or radicals. The ignition delays of neat
isooctane and isooctane/toluene mixtures are shown in Fig.2.16. Both the cool flame and the main
ignition delay times increase when toluene is added. Besides, the autoignition delay times of fuel
mixture decreases sharply above 830 K, suggesting the promoting effect of toluene on the reactivity
at high temperatures. The interactions between toluene and isooctane are of great significance com-
pared with those between toluene and n-heptane because both aromatics and iso-paraffins account
for significant fractions of gasoline. Based on the species analysis, Vanhove et al. [190] concluded that
toluene is unlikely to change the reaction pathways of isooctane oxidation, but may react with active
radicals from isooctane and produce stable benzyl radical to deactivate the reaction pool.
The investigation of fuel interactions via the radical pool is challenging, as it requires rigorous
species analysis to interpret how the elementary reactions related to the radical pool affect the mix-
ture’s performance. To predict the fuel mixture behaviours over a wide range of conditions, more
fundamental experimental and computational studies are required to provide accurate rate constants
and species profiles to better calibrate the existing chemical mechanisms.
25

isooctane/toluene
isooctane
isooctane/toluene
isooctane
Figure 2.16: Comparisons of cool flame (open symbols) and autoignition delay times (filled symbols)of neat isooctane and isooctane/toluene mixture [190]
26

2.4.3 Interactions between ethanol and hydrocarbons
The interactions between ethanol and hydrocarbons are known to be significant in the SI engines from
the experimental study performed by Foong et al. [9]. Meanwhile, several kinetic experimental studies
[86, 183, 194–198] were conducted to investigate the interactions between alcohols and hydrocarbons.
All these studies focused on the radical pool level competitions between alcohols and hydrocarbons.
Ethanol and n-heptane have very different reactivities, and their competition for small radicals
have been recently investigated by many groups. At low temperatures, both ethanol and n-heptane
undergo H abstraction to generate α-hydroxyethyl and heptyl respectively. The calculation from da
Silva et al. [199] suggested that, due to the influence of the OH group, the reaction of α-hydroxyethyl
and oxygen molecule proceeds almost exclusively to acetaldehyde and hydroperoxyl radical. As the
dominant product from H abstraction, α-hydroxyethyl prohibits ethanol’s chain-branching reaction,
which results in less OH radicals. At the beginning of the oxidation, n-heptane produces much more
OH radicals than ethanol. Later, the two fuels compete for the limited OH radicals at the same time.
Consequently, the consumption of n-heptane is decreased during this stage due to less OH radicals
available comparing with neat n-heptane oxidation; while ethanol gets relatively more OH radicals
and is consumed more rapidly than neat ethanol oxidation. After NTC regime, the decomposition of
hydrogen peroxide produces a significant amount of OH radicals consuming remaining fuels. Gener-
ally, for ethanol/hydrocarbon mixtures, ethanol acts as OH radical scavenger and therefore suppresses
the overall oxidation process. As two common alcohol compounds, ethanol and n-butanol have dif-
ferent lengths of the carbon chain. According to HCCI engine experiment by Saisirirat et al. [196],
both ethanol and n-butanol retard the start of combustion, but ethanol has a more pronounced effect
regarding suppressing combustion.
The oxidations of ethanol/gasoline surrogates were carried out by Dagaut and Togbe [183] and
Cancino et al. [200]. The kinetic models proposed by both groups can reproduce the experimental
results from the jet-stirred reactor and shock tube respectively. The chemical interactions involved in
these studies are still at the radical pool level.
Although the studies above all successfully reproduced their own experimental results by the
blended mechanisms, these kinetic models haven’t been validated for the complex fuel mixtures con-
taining practical gasoline surrogates and ethanol. Further experimental and modelling studies fo-
cusing on the oxidations of ethanol and hydrocarbon mixtures are necessary to understand the fuel
interactions.
27

2.5 Summary and research questions
As an abnormal combustion phenomenon in the engine, knock is a consequence of end gas autoigni-
tion. To suppress knock, ethanol is often added to production gasoline as an octane enhancer. Nu-
merous experimental studies show that ethanol increases both RON and MON of gasoline, and thus
improves anti-knock performance. However, the interactions between ethanol and production gaso-
line in the CFR engine are complicated. Foong et al . [9] carried out the initial experimental study
to understand the complex interactions. They formulated three TRF-based gasoline surrogates with
different amounts of toluene added to emulate the blending behaviours between ethanol and the
production gasoline. The results showed that the TRF-based gasoline surrogates blend more synergis-
tically with ethanol compared with the production gasoline at a similar RON and aromatic content..
Therefore, more engine tests are required to fully understand the interactions between ethanol and
the production gasoline.
Ethanol’s anti-knock behaviour has been extensively investigated mainly from two aspects: charge
cooling effect and chemical kinetics. The high latent heat of vaporisation of ethanol improves the
charge cooling effect which increases the knock onset limits and thus the engine efficiency. The chem-
ical effect of ethanol needs to be further clarified, especially when interacting with hydrocarbon fuels.
Although the interactions among larger species have been incorporated into the widely used gasoline
surrogate mechanisms developed by Mehl et al. [67] and Andrae [187], their impacts on the overall be-
haviours of fuel mixtures might not be significant due to the small rate constants of these elementary
reactions. Therefore, fuel interactions are expected to be more likely to occur with the involvement of
small reactive radicals. To predict the fuel interaction, the kinetic model should therefore have accu-
rate rate constants for these reactions involving small radicals, and more kinetic experiments for the
fuel mixtures are needed to calibrate the existing models.
This study, therefore, aims to investigate the fuel interactions in a CFR engine and combustion
chemistry of ethanol containing gasoline surrogates in a PFR. The following research questions are
proposed.
1. How should the non-linear octane blending of ethanol and toluene reference fuels (TRFs) be
represented?
Ethanol is known to blend non-linearly with surrogate fuels under standard knocking conditions
in the CFR engine [9]. This study proposes a statistical model to quantify these non-linearities
and predict the octane numbers of fuel mixtures containing ethanol and TRFs.
2. What is the gasoline surrogate that best emulates the anti-knock behaviours of production
gasoline when blended with ethanol?
28

As shown in the prior standard octane number test [9], ethanol blends more synergistically with
the TRF-based gasoline surrogates than with gasoline, suggesting that TRFs are not good enough
to emulate gasoline. Besides, the octane number blending behaviours between ethanol and the
hydrocarbon fuels other than TRF components are not known. Therefore, this study formu-
lates new gasoline surrogates to better emulate the knocking behaviours of the gasoline when
blended with ethanol.
3. How does ethanol interact with gasoline surrogates under engine representative conditions in
the PFR, and do existing mechanisms reproduce the measured species profiles?
Numerous kinetic experiments have been carried out by different groups to study the combus-
tion chemistry for neat fuels and fuel mixtures. However, no systematic experimental study has
been carried out to investigate how ethanol interacts with surrogate fuels and more importantly,
gasoline surrogates in flow reactors. Also, state of the art gasoline surrogate mechanisms haven’t
been fully calibrated to predict the behaviours of the fuel mixtures containing ethanol and gaso-
line surrogates. Therefore, this study performs PFR experiments to study the impacts of ethanol
on the reactivities of gasoline surrogates and validates the state of the art chemical mechanisms
using these measurements.
29

Chapter 3
Experimental Methods
3.1 Overview
This chapter first presents the experimental methods for the CFR engine and the PFR which are ap-
plied in this study to investigate the fuel interactions. Besides, the applications of the gas chromatog-
raphy (GC) in the PFR experiment for the identification and quantitative analysis of intermediate
species are elaborated.
3.2 CFR engine
3.2.1 Overview
The engine experiments in this study were carried out in a 1933 Waukesha CFR F1/F2 octane rating
engine, as shown in Fig.3.1. The CFR engine is driven by a dynamometer at a constant speed of 600
rpm for RON and 900 rpm for MON. In the experiment, liquid fuels, stored in the fuel bowls, are
vaporised by the hot air in the carburettor before entering the engine cylinder. Before the air is heated,
it goes through the dehumidifier to get rid of water vapours, as the introduction of water vapours
increases the overall reactivity of fuel and air mixture by providing hydroxyl radicals. In the standard
engine knock experiments, the compression ratio (CR) is estimated and adjusted based on the ASTM
manuals [25,26], while the fuel flow rate is tuned by raising or lowering the fuel bowl height to obtain
the standard knocking conditions. The lambda and knock meters, which are housed in a separate
electrical cabinet, show the fuel/air ratio and knock intensity respectively.
3.2.2 The Structure of the CFR engine
Fig.3.2 shows the detailed structure of the CFR engine. In Fig.3.2(a), the in-cylinder pressure oscilla-
tions during the standard knock rating tests are converted to voltages by the knock sensor mounted
30

Figure 3.1: The system of the CFR engine
on the top of the cylinder, and the dial indicator is applied to adjust the compression ratios specified
in [25, 26]. The condenser on the right upper part of the engine body uses the pressurised tap water
to dissipate heat away from the coolant flowing in the engine jacket. The CFR engine body is shown
in Fig.3.2(b), (c) and (d) from three different views with all auxiliary parts removed. The cylinder
inlet and outlet locate on the left and right side of the engine body respectively. The condenser inlet,
sitting on the right upper corner of the exhaust side, guides the engine coolant to the condenser and
get cooled.
Before starting the standard octane number measurements in this study, the CFR engine went
through a top overhaul to clean deposits on the cylinder wall and piston head. The comparison of
piston head before and after deposits cleaning is shown in Fig.3.3. The deposits act as an insulation
layer reducing the overall heat loss from the gas mixtures to the cylinder wall and piston head, which
makes test fuels more prone to knock and thus results in a lower octane number. In this study, all
knock rating tests are carried out right after the top overhaul when the CFR engine is in the good
condition.
31

(a) CFR engine (b) Left view
(c) Front view (d) Right view
Figure 3.2: The structure of the CFR engine
32

(a) (b)
Figure 3.3: The piston head (a) before and (b) after overhaul
3.2.3 Methods for standard octane number tests
The CFR engine test methods for the standard RON and MON have been specified in [25] and [26]
respectively, and their test conditions are listed in Table 3.1. Both methods determine the octane num-
bers of the sample fuel by comparing its standard knock intensity with those of two PRFs whose
octane numbers are known by definition. To obtain the standard knock intensity for the sample fuel
during the knock rating tests, the cylinder height representing the compression ratio needs to be first
estimated and gradually tuned based on the [25, 26]. Although it is desirable to measure both RON
and MON of the sample fuels to have comprehensive understandings of their knocking behaviours,
the measurements conducted in this study are mostly for RON, since MON is not as important as
RON in modern engines, especially under high loads.
To ensure the engine’s compliance with the ASTM standards [25, 26], the so-called ’Fit-for-Use’
tests were conducted using the toluene standardisation fuels with known octane numbers. If the dif-
ference between the known and measured octane numbers is within the allowed tolerance, the engine
is considered fit for knock ratings in a certain octane number range. Note that the PRFs are not capable
of rating any sample fuels with RON larger than 100, and different amounts of the dilute tetraethyl
lead (TEL) are blended into isooctane as bracket fuels for RON tests above 100. The compositions of
the dilute TEL are listed in Table 3.2.
33

Table 3.1: Operating conditions for the RON and MON measurements [25, 26]
Operating parameters RON MON
Engine speed 600±6 rpm 900±9 rpm
Intake air temperature 52±1.0 °Ca 38±2.8 °C
Mixture intake temperaturea N/A 149±1.0 °C
Intake air pressure Barometric Barometric
Intake air humidity 25-50g H2O/kg dry air 25-50g H2O/kg dry air
Coolant temperature 100±1.5 °C 100±1.5 °C
Oil pressure 172-207 kPa(g) 172-207 kPa(g)
Oil temperature 57±8.0 °C 57±8.0 °C
Spark timing 13 °BTDC 14-26 °BTDCc
a varied with barometric pressureb temperature measured right before engine inletc varied with the compression ratio
Table 3.2: The composition of the dilute TEL [25, 26]
Component TEL Ethylene dibromide Xylene N-heptane Other
Mass fraction (%) 18.2 10.6 52.5 17.8 0.9
3.3 Pressurised flow reactor
3.3.1 Overview
The schematic drawing of the PFR system is shown in Fig.3.4. The air comes from an oil-free com-
pressor and goes into the flow reactor after heated to a specified temperature. The air flow rate is
controlled by a needle valve and measured by a flow meter. A balanced air stream goes to the gap be-
tween reaction tube and wall heater to equalise pressures inside and outside of the reaction tube. The
nitrogen is divided into two streams: one is to pressurise liquid fuel out of the cylindrical tank, and
another one is used to vaporise the pressurised liquid fuel. These two lines, together with the subse-
quently merged line are all wrapped with tube heaters to ensure that the liquid fuel is fully vaporised
before entering the flow reactor. A strain gauge is used to measure the fuel tank weight to derive the
fuel flow rate which is confirmed by the Coriolis flow meter. The gas mixture in the flow reactor is
collected by a sampling probe and analysed by a Gas Chromatography, and the remaining mixture
is purged into the exhaust system. The reactor pressure is controlled by a back-pressure valve. More
detailed information are available in [201].
Although the reactor is designed to run at 50 bar and 1000 K, low pressure of 10 bar is used in this
34
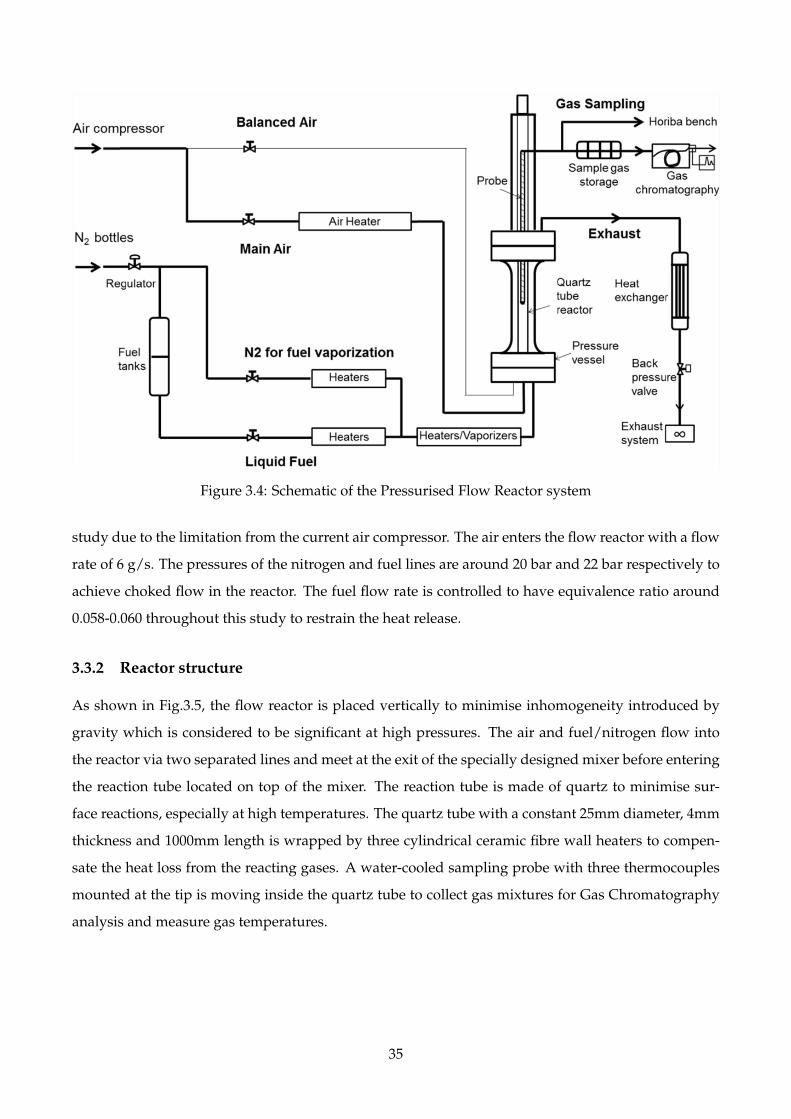
Figure 3.4: Schematic of the Pressurised Flow Reactor system
study due to the limitation from the current air compressor. The air enters the flow reactor with a flow
rate of 6 g/s. The pressures of the nitrogen and fuel lines are around 20 bar and 22 bar respectively to
achieve choked flow in the reactor. The fuel flow rate is controlled to have equivalence ratio around
0.058-0.060 throughout this study to restrain the heat release.
3.3.2 Reactor structure
As shown in Fig.3.5, the flow reactor is placed vertically to minimise inhomogeneity introduced by
gravity which is considered to be significant at high pressures. The air and fuel/nitrogen flow into
the reactor via two separated lines and meet at the exit of the specially designed mixer before entering
the reaction tube located on top of the mixer. The reaction tube is made of quartz to minimise sur-
face reactions, especially at high temperatures. The quartz tube with a constant 25mm diameter, 4mm
thickness and 1000mm length is wrapped by three cylindrical ceramic fibre wall heaters to compen-
sate the heat loss from the reacting gases. A water-cooled sampling probe with three thermocouples
mounted at the tip is moving inside the quartz tube to collect gas mixtures for Gas Chromatography
analysis and measure gas temperatures.
35
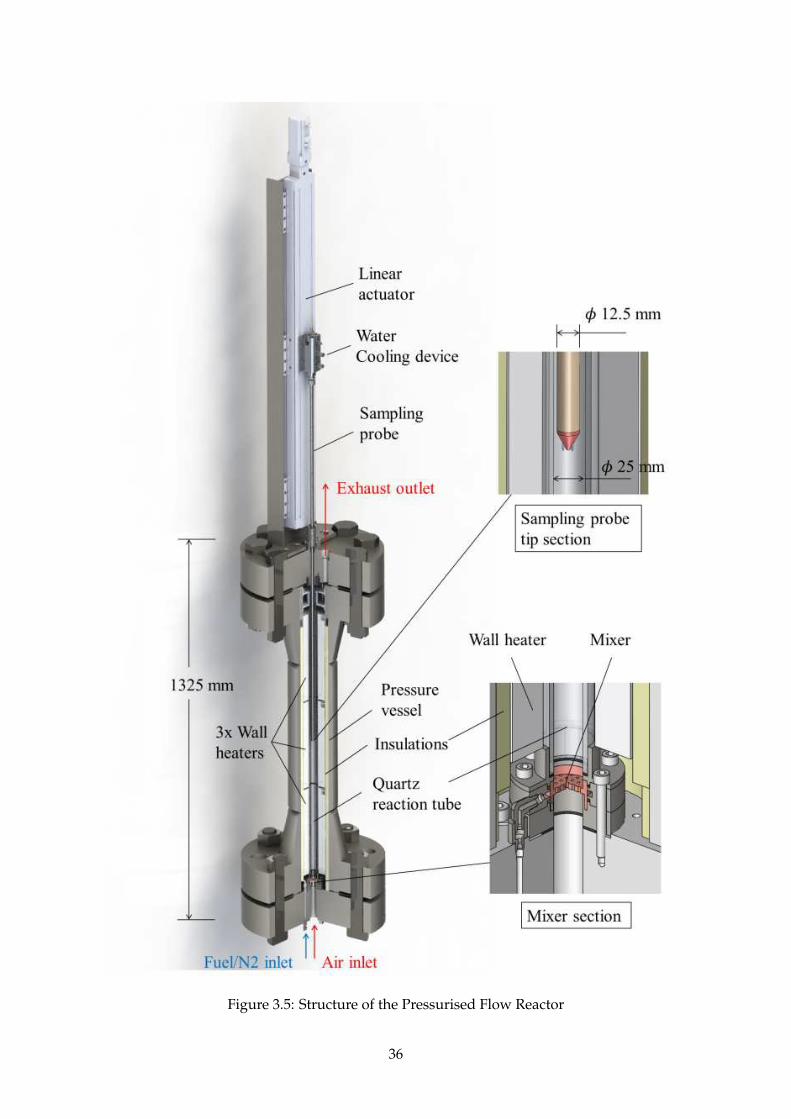
Figure 3.5: Structure of the Pressurised Flow Reactor
36

3.3.3 Mixer
In the reactor, the fuel, carried and heated by the nitrogen, starts to react with the preheated air once
they meet at the exit of the mixer, where both mixing and reaction occur at the same time. This compli-
cated process at the start of the reactions, which is also common for other kinetic experiments, includ-
ing stirred reactors, static reactors, leads to the so-called initiation problem in kinetic modelling, where
the compositions of gas mixtures are difficult to determine. Although this process is unavoidable, a
minimised mixing length does help to moderate the initiation problem. Fig.3.6 shows the specially
designed orifice-plate mixer which accelerates the mixing of fuel and air. The air flows into the reac-
tor through 21 evenly located orifices with the diameter of 1.75mm, while the fuel/nitrogen mixture
flows along four parallel through-channels which are in a direction perpendicular to the air flow path
and is injected via twelve small nozzles with the throat diameter of 0.18mm located in the centers of
the squares outlined by four large orifices. To further improve the mixing process, the fuel/nitrogen
flow is choked at small nozzles, which produces the same injected mass at each nozzle, regardless of
its position and pressure variations. In the experiments, the fuel/nitrogen pressure is always kept at
least twice of the reactor pressure to ensure the occurrence of the choked flow.
Figure 3.6: The mixer a) cutaway view and b) orifices distribution
To examine the mixing length of the specially designed mixer, a validation experiment of CO2/air
mixing was carried out [201]. In this experiment, the flow reactor was heated to 900 K and pressurised
to 10 bar with 6.02 g/s air flow coming from the large orifices, while CO2 was injected into the reactor
through small nozzles with the flow rate of 0.71 g/s. The mole fraction of CO2 was measured along
the centreline by a non-dispersive infrared (NDIR) analyser with a resolution of 20 PPM in Horiba
emission bench. As shown in Fig.3.7, the mole fraction of CO2 fluctuates at the start due to a huge
concentration difference, but rapidly reaches the equilibrium value around 100 mm downstream of
37
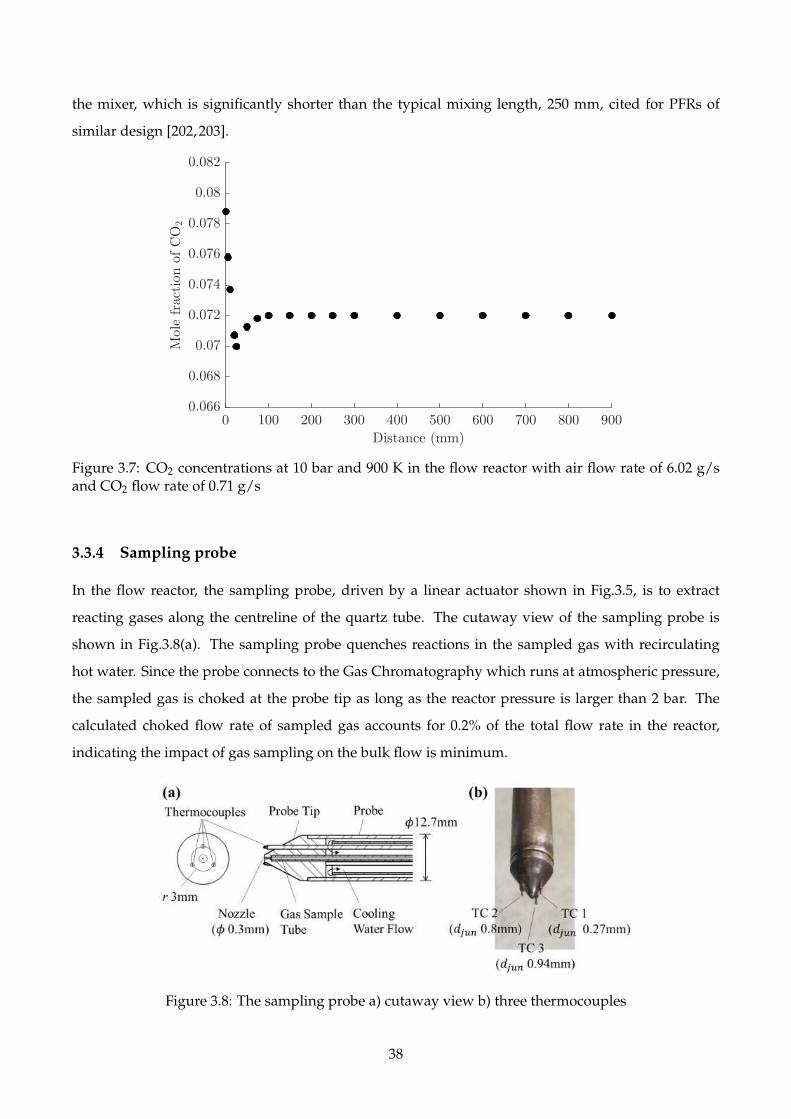
the mixer, which is significantly shorter than the typical mixing length, 250 mm, cited for PFRs of
similar design [202, 203].
0 100 200 300 400 500 600 700 800 900
Distance (mm)
0.066
0.068
0.07
0.072
0.074
0.076
0.078
0.08
0.082Mole
fractionofCO
2
Figure 3.7: CO2 concentrations at 10 bar and 900 K in the flow reactor with air flow rate of 6.02 g/sand CO2 flow rate of 0.71 g/s
3.3.4 Sampling probe
In the flow reactor, the sampling probe, driven by a linear actuator shown in Fig.3.5, is to extract
reacting gases along the centreline of the quartz tube. The cutaway view of the sampling probe is
shown in Fig.3.8(a). The sampling probe quenches reactions in the sampled gas with recirculating
hot water. Since the probe connects to the Gas Chromatography which runs at atmospheric pressure,
the sampled gas is choked at the probe tip as long as the reactor pressure is larger than 2 bar. The
calculated choked flow rate of sampled gas accounts for 0.2% of the total flow rate in the reactor,
indicating the impact of gas sampling on the bulk flow is minimum.
Figure 3.8: The sampling probe a) cutaway view b) three thermocouples
38

The sampling probe also measures gas temperatures which are critical for kinetic investigation.
To get accurate temperatures in combustion, heat radiation has to be handled carefully and rigor-
ously. Three K-type thermocouples with different junction sizes (0.27, 0.80 and 0.94 mm) and mea-
surement uncertainties of ±0.25% are mounted axis-symmetrically on the probe tip, as shown in
Fig.3.8(b). Based on the three-thermocouple method, the corrected gas temperature is calculated us-
ing the Eqn.3.1 and the uncertainty of the corrected temperature is estimated to be ±5 K at 900 K
(smaller at lower temperatures). The detailed description for the three-thermocouple method is avail-
able in [201]. The application of this method is illustrated with an example of isooctane oxidation at
10 bar and 900 K shown in Fig.3.9 where the corrected temperatures are compared with the measured
ones from three thermocouples. The thermocouple with smaller junction size is less affected by the
complicated heat radiations inside the reactor, and therefore has higher temperatures than those with
larger junction sizes, indicating the real gas temperatures should be measured using a thermocouple
with zero junction size. Although it is not practical and possible to have such a thermocouple, the
three thermocouple method provides a good estimation for the real gas temperatures which are, not
surprisingly, higher than measured temperatures. Note that this approach for the estimations of real
gas temperatures is proven to be theoretically rigorous, but the measured temperatures are known to
fluctuate in a certain range especially at lower temperatures, which might lead to obviously unrea-
sonable corrected temperatures in rare circumstances. To handle this issue, the problematic corrected
temperatures are interpolated using adjacent good results.
(3.1)Tgas =
T1 −
d1
d2
2/5
T2 −
T1 −
d1
d3
2/5
T3
T1
4 − T24
T14 − T3
4
1−
d1
d2
2/5
−
1−
d1
d3
2/5
T1
4 − T24
T14 − T3
4
3.3.5 Experimental conditions
The general operating conditions of PFR experiments in this work are listed in Table 3.3. To reach these
conditions, the PFR is first heated using hot air flow at 1100 K and 10 bar plus three wall heaters with
tunable power outputs. During the warming up, the metal temperatures of flanges are monitored and
used as an indication of the thermal equilibrium. When the metal temperatures are stable, nitrogen
flow at 500 K and 20 bar is introduced to the reactor, and the temperature of air flow and power
output of wall heater are tuned to reach a new equilibrium in the reactor tube. Once the temperatures
at the mixer exit, the outer surface of reactor wall, and the probe tip locating at the end of the tube
are all stabilised to the set value, e.g., 900 K, a new equilibrium is reached, and then the fuel will be
39

0 100 200 300 400 500 600 700 800 900 1000
Distance (mm)
820
840
860
880
900
920
940
960
Tem
perature
(K)
T1 with junction size of 0.27mmT2 with junction size of 0.80mmT3 with junction size of 0.94mmCorrected Temperature
Figure 3.9: Reactor temperature profiles for isooctane oxidation at 10 bar and 900 K with equivalenceratio of 0.058
injected into hot nitrogen flow at a slightly higher pressure around 23 bar. The fuel rate is measured
by the strain gauge and the Coriolis flow meter. The difference between these two measurements are
generally below 5%.
Table 3.3: Experimental conditions for the PFR study
PFR parameter Set value
Reactor pressure (bar) 10
Reactor nominal temperature (K) 900-930
Equivalence ratio 0.058-0.060
Air flow rate (g/s) 6
Nitrogen flow rate (g/s) 0.32
Reynolds number in the reactor tube 8000
Fuel/nitrogen pressure (bar) 20-21
Fuel/nitrogen temperature (K) 500
40

3.4 Gas chromatography
3.4.1 Overview of the gas chromatography
The reacting gases collected by the sampling probe go to the gas loops of the GC for quantitative
species analysis. Fig.3.10 shows the picture of GC used in this study, which contains GC itself and
an auxiliary sampling system installed on the left plate. The inlet and outlet of the sampling system
locate on top of the insulation container which accommodates rotator 94 and 93. On the upper left
of the container locates a manual controller for rotator 92 which connects ten sampling loops and is
placed in another insulation container mounted on the back of the plate (not shown in Fig.3.10). Each
rotator has a motor to drive it, and the motor for rotator 92 is installed on the lower middle part of
the front, while the other two motors for rotator 94 and 93 are attached on the back. The positions of
these rotators are shown on the indicators and controller. Besides the loop based sampling system,
two injectors are installed on the top of the GC for the typical manual injections.
Figure 3.10: Gas Chromatography-2010ATF plus from Shimadzu
The connection between the GC and the aforementioned auxiliary system is shown in Fig.3.11.
During the PFR experiments, gas samples are introduced into the GC via a sampling loop system,
which involves two working modes: sampling and analysis. In the sampling process, the rotator 94
stays in the current position, specified as position A, which guides the gas sample flow from point 1 to
6, and then to the inlet of the rotator 92. As shown in Fig.3.11, the rotator 92 has both inner and outer
41
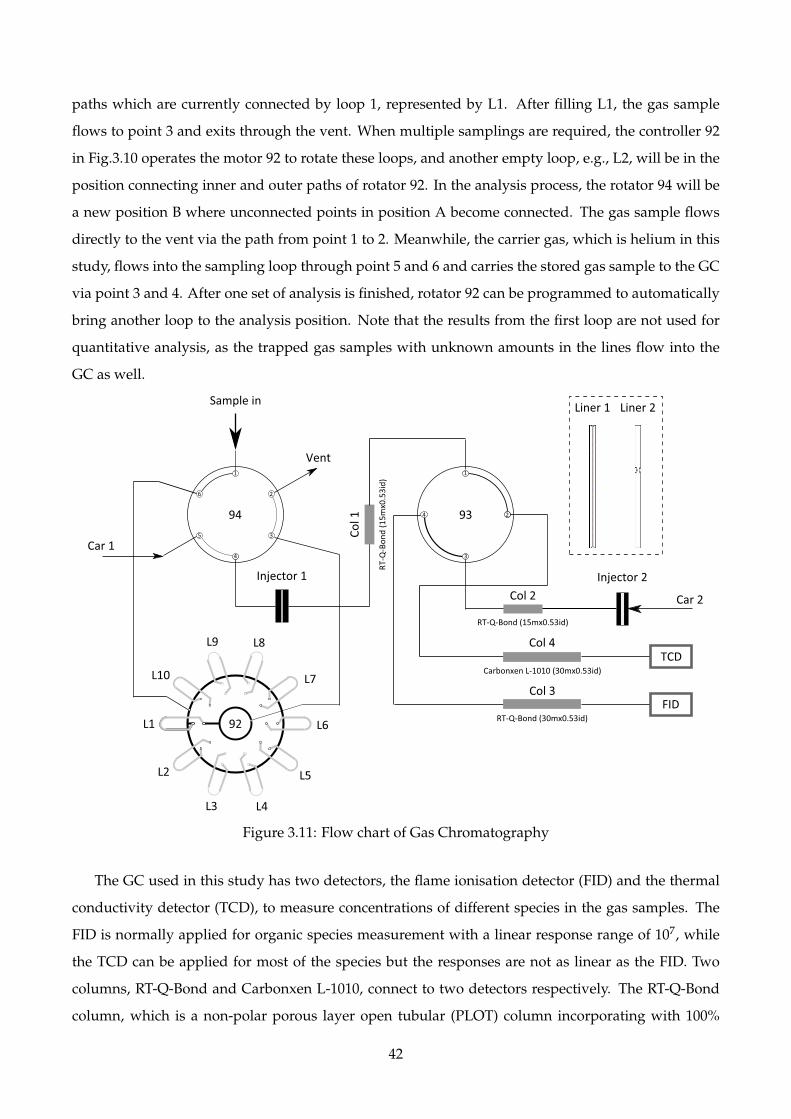
paths which are currently connected by loop 1, represented by L1. After filling L1, the gas sample
flows to point 3 and exits through the vent. When multiple samplings are required, the controller 92
in Fig.3.10 operates the motor 92 to rotate these loops, and another empty loop, e.g., L2, will be in the
position connecting inner and outer paths of rotator 92. In the analysis process, the rotator 94 will be
a new position B where unconnected points in position A become connected. The gas sample flows
directly to the vent via the path from point 1 to 2. Meanwhile, the carrier gas, which is helium in this
study, flows into the sampling loop through point 5 and 6 and carries the stored gas sample to the GC
via point 3 and 4. After one set of analysis is finished, rotator 92 can be programmed to automatically
bring another loop to the analysis position. Note that the results from the first loop are not used for
quantitative analysis, as the trapped gas samples with unknown amounts in the lines flow into the
GC as well.
Figure 3.11: Flow chart of Gas Chromatography
The GC used in this study has two detectors, the flame ionisation detector (FID) and the thermal
conductivity detector (TCD), to measure concentrations of different species in the gas samples. The
FID is normally applied for organic species measurement with a linear response range of 107, while
the TCD can be applied for most of the species but the responses are not as linear as the FID. Two
columns, RT-Q-Bond and Carbonxen L-1010, connect to two detectors respectively. The RT-Q-Bond
column, which is a non-polar porous layer open tubular (PLOT) column incorporating with 100%
42

divinylbenzene, performs excellently in term of separating hydrocarbons up to C12 and works fine
for oxygenated compounds. The other column, Carbonxen L-1010, is ideal for separating small com-
pounds, such as nitrogen, oxygen, carbon monoxide, carbon dioxide and light hydrocarbons up to C3.
In this study, only the FID was used for quantifying the compositions of the sampled reacting gases.
Although the FID cannot detect inorganic compounds, the NDIR analyser with a resolution of 20 PPM
in the Horiba emission bench was applied for CO and CO2 measurements. The reason for not using
the TCD was due to the excessive amounts oxygen and nitrogen in the sampled gases. Their large
concentrations produce huge peaks with long tails which affect the detection of other species with
small amounts.
Although the PFR experiments only necessitate the sampling loop system in quantitative analysis,
the GC has been equipped with two injectors containing different liners for calibration purpose, as
shown in Fig.3.11. The Injector 1, which contains Liner 1 with small volume, is essentially applied
as a pathway to the Col 1 for the gas samples coming from the sampling loop system, while the
Liner 2 with a large volume of 0.83 ml has been installed in Injector 2 is commonly used for manual
injections whose results are applied for the calibrations of loop volumes and concentrations of large
intermediates.
3.4.2 Identification and quantification of species
The most significant advantage of PFR is its capability of measuring the temporal species profiles
which provide insights into the fundamental fuel chemistry. Therefore, the species identification and
quantification are critical to obtain accurate profiles for combustion chemistry study.
In the PFR experiment, the sampled reacting gas was transferred in a heated sampling pipe to the
sampling loop system of GC. The temperatures of both pipe and loop system were kept at 200 °C to
prevent condensation. Besides, a specially designed temperature program, as shown in Fig.3.12, was
used for GC analysis to achieve efficient separations. Finally, the loop injection volumes were used to
calculate the total moles of reacting gases which quantify the mole fractions of detected species. Note
that no split was applied to the GC analysis performed in this study.
The detected peaks on the spectrum need to be identified before the quantitative analysis. The
identification of the parent fuels and small gaseous species are relatively straightforward. The react-
ing gas collected at the start of the reactor contains mainly parent fuels whose peaks stand out on the
spectrum. A gas standard containing 27 species from C1 to C6 was applied for the identification of
the small gaseous species by comparing their retention times. Nevertheless, it is difficult to identify
the large and uncommon intermediates which are not likely to be included in the gas standards. In
this case, the literature results under similar conditions, together with the predictions from kinetic
43

Figure 3.12: The temperature program for GC analysis
modellings were combined to speculate those unknown intermediates. Then, the vapours of the spec-
ulated compounds were introduced to the GC whose retention times can be compared with those from
the PFR experiments. If the retention times agree well, the identifications are considered to be success-
ful. Note that the vapours of the guessed compounds were carried by a gas standard containing 1000
PPM propane balanced by nitrogen. The introduction of the propane standard provides an internal
standard for identification to take care of the retention time drifts. The identified intermediate species
for isooctane, ethanol, and toluene oxidations are shown in Fig.3.13, 3.14, and 3.15 respectively.
CH4C2H4
C3H6
CH3CHO
IC4H8
CH3COCH3
IC3H5CHO
XC7H14YC7H14
IC8H18
5.0 10.0 15.0 20.0 25.0 30.0 35.0 40.0 45.0 50.0 min0
25
50
75
100
125
150
175
200mV
Figure 3.13: The spectrum of isooctane oxidation at 900 mm under 900 K and 10 bar
The intermediate species can be divided into three groups: the parent fuels, the large intermedi-
ates in the liquid phase and the small molecules in the gaseous phase. Each group has its unique
quantification method.
44

5.0 10.0 15.0 20.0 25.0 30.0 35.0 40.0 45.0 50.0 min
0
250
500
750
1000
1250mV
CH4C2H4
CH3CHO
C2H5OH
Figure 3.14: The spectrum of ethanol oxidation at 500 mm under 900 K and 10 bar
5.0 10.0 15.0 20.0 25.0 30.0 35.0 40.0 45.0 50.0 55.0 60.0 65.0 min0
250
500
750
1000
1250
1500
1750
2000mV
C6H5CH3
C6H6
Figure 3.15: The spectrum of toluene oxidation at 700 mm under 930 K and 10 bar
45
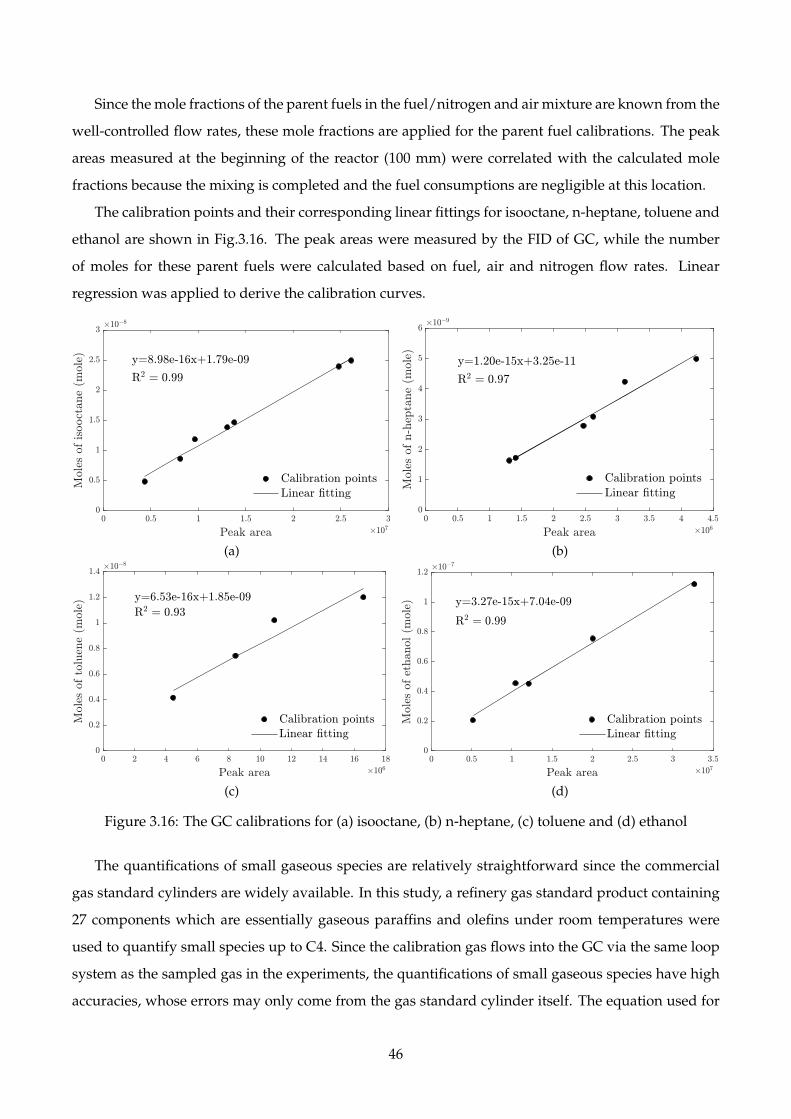
Since the mole fractions of the parent fuels in the fuel/nitrogen and air mixture are known from the
well-controlled flow rates, these mole fractions are applied for the parent fuel calibrations. The peak
areas measured at the beginning of the reactor (100 mm) were correlated with the calculated mole
fractions because the mixing is completed and the fuel consumptions are negligible at this location.
The calibration points and their corresponding linear fittings for isooctane, n-heptane, toluene and
ethanol are shown in Fig.3.16. The peak areas were measured by the FID of GC, while the number
of moles for these parent fuels were calculated based on fuel, air and nitrogen flow rates. Linear
regression was applied to derive the calibration curves.
(a) (b)
(c) (d)
Figure 3.16: The GC calibrations for (a) isooctane, (b) n-heptane, (c) toluene and (d) ethanol
The quantifications of small gaseous species are relatively straightforward since the commercial
gas standard cylinders are widely available. In this study, a refinery gas standard product containing
27 components which are essentially gaseous paraffins and olefins under room temperatures were
used to quantify small species up to C4. Since the calibration gas flows into the GC via the same loop
system as the sampled gas in the experiments, the quantifications of small gaseous species have high
accuracies, whose errors may only come from the gas standard cylinder itself. The equation used for
46

response factor calculation in this study is given by
(3.2)RFi =ni
Ai
where RFi is the response factor of species i, ni represents the number of moles for species i, and Ai
denotes the corresponding peak area. The response factors for gaseous fuels of interest in this study
are listed in Table 3.4. Note that the calibration curves in Fig.3.16 and the response factors in Table 3.4
convert the measured peak areas to the number of moles. Since the total moles of the reacting gas in
the loop are known, it is straightforward to calculate the mole factions of the measured species.
Table 3.4: Response factors for gaseous fuels
Fuels CH4 C2H4 C3H6 IC4H8
Response factor 4.90e-15 2.50e-15 2.77e-15 2.06e-15
Compared with the parent fuels, the large intermediates in liquid phase at room temperatures
are difficult to be quantified. Most of these intermediates are expensive and are not as common as
the parent fuels. Therefore, it is not practical to quantify these intermediate species using the same
method as the parent fuels as it requires large quantities. In this case, the response factors of these large
intermediates are estimated from the well quantified species based on the effective carbon numbers
(ECN) [204, 205] using Eqn.3.3. The resulting response factors are listed in Table 3.5.
(3.3)RFi =RFre f ECNre f
ECNi
Table 3.5: Response factors for intermediate species in liquid phase
Fuels CH3CHO CH3COCH3 IC3H5CHO XC7H14 YC7H14 C6H6
ECN 1.06 2.10 3.42 6.77 6.77 5.81
Response factor 4.89e-15a 3.73e-15b 2.29e-15b 1.15e-15b 1.15e-15b 6.17e-16c
a Taking ethanol as reference with an ECN of 1.48b Taking IC4H8 as reference with an ECN of 3.80c Taking toluene as reference with an ECN of 6.78
47

Chapter 4
Optimal Octane Number Correlations for
Toluene Reference Fuels (TRFs) Blended
with Ethanol
4.1 Introduction
Ethanol is known to interact non-linearly with isooctane, n-heptane, toluene, and PRF-, TRF-based
gasoline surrogates in the CFR engine under the standard knocking conditions [9], but the quantita-
tive understanding of these interaction behaviours is not complete. Therefore, this chapter proposes
optimal octane number correlations to quantify these interactions based on a systematically and rig-
orously statistical approach, which provides basis for ethanol blending into the gasoline and octane
number estimations for TRF-based gasoline surrogates. Note that this chapter is revised from a re-
cently published paper by the author [206].
The octane numbers of neat compounds were extensively measured in the 1950s by the American
Petroleum Institute [207]. Publications that present the octane numbers of mixtures are relatively
sparse. The octane blending of mixtures can be quite complicated [9, 208], particularly when ethanol
is involved. The use of ethanol in SI engines has received significant attention in recent years [9,
10, 12, 44, 209, 210], and it is commonly blended with gasoline for its low autoignition reactivity and
high heat of vaporisation, both of which result in ethanol’s relatively high octane number. Octane
number measurements have been conducted for ethanol blended with different commercial gasoline
[10,44], primary reference fuels (PRFs) (mixtures of n-heptane and isooctane) [9,211–213], and toluene
reference fuels (TRFs) (mixtures of PRF and toluene) [9, 211–213]. Significant non-linear blending
has been reported in some cases. Proper account of such non-linear and sometimes non-monotonic
behaviours is essential for optimal blending of ethanol into gasoline of different compositions, as well
48

as, for reliable estimation of octane numbers in developing gasoline surrogates.
In contrast to equivalent correlation studies of TRFs [214–216] and gasoline distillate blends [217–
220], few correlations have been proposed for TRF/ethanol mixtures. Two studies are recently re-
ported on this topic [213, 221]. While these studies reported good agreement between measured and
correlated octane numbers, the correlations often do not necessarily return the octane numbers of the
neat TRF/ethanol components. Such a function therefore likely results in a limited accuracy, par-
ticularly when the mixture approaches one of these neat components. Besides, complex forms of
correlation are often proposed where the physical meaning of the terms involved are not clear.
An alternative method for octane numbers correlation is raised in this study. While this method
can be generalised to any mixture, it is applied to the study of TRF/ethanol fuels. This method makes
optimal use of Scheffe polynomials [222], which provide a composition-based model for mixture prop-
erties. It involves the use of linear regression and exhaustive (or brute-force) searching of polynomials
with the fewest terms to correlate measured, correlation development data and meet specified ac-
curacy requirements. Correlations based on the mole fractions are developed and validated for the
RONs and MONs of TRF/ethanol mixtures, using data available in the literature and data obtained
from our group. Correlations based on the mole fractions are presented for the RONs and MONs of
TRF/ethanol mixtures; volume fraction is not used for being a non-conserved property in fuel blend-
ing [223], which has also been reported poorly correlated with ON [215].
4.2 Algorithm for correlation development
4.2.1 The Scheffe polynomial based correlation
Non-linear interactions are often observed in octane number measurements for fuel mixtures. Such
effects are sometimes described as synergistic (i.e. super-linear) or antagonistic (i.e. sub-linear) blend-
ing. The Scheffe polynomial [222] has been used to characterise such behaviours since it is designed
to deal with mixtures [224–226]. The complete Scheffe polynomial octane number correlation of four
components mixtures contains a total of 35 terms, as shown in Table 4.1, and can be written as
(4.1)
ON =4
∑i=1
βixi +3
∑i<j
4
∑j
βijxixj +3
∑i<j
4
∑j
δijxixj(xi − xj) +3
∑i<j
4
∑j
γijxixj(xi − xj)2 +2
∑i<j
3
∑j<k
4
∑k
βiijkx2i xjxk
+2
∑i<j
3
∑j<k
4
∑k
βijjkxix2j xk +
2
∑i<j
3
∑j<k
4
∑k
βijkkxixjx2k +
1
∑i<j
2
∑j<k
3
∑k<l
4
∑l
βijklxixjxkxl
where xi denotes mole fraction for each fuel compound in the mixture. x1, x2, x3 and x4 are defined
to be isooctane, n-heptane, toluene and ethanol, respectively. The coefficient βi of the first-order (lin-
ear) terms is the octane number of neat component, which for isooctane and n-heptane are set to be
49

100 and 0 by the octane number definition (Table 4.2). Although those for toluene and ethanol could
also be fixed, there is disagreement in the literature as to the RON and MON of the two compounds,
particularly for toluene, whose reported values from different studies [207, 215, 227] including mea-
surements from this work vary by 4-5 ON. To avoid over-complicating the problem, the RON and
MON of ethanol are fixed to the measured values from the previous study from our group [9], while
allowing the βi coefficients quantifying toluene’s RON and MON to be part of the correlation method.
With setting βi of the first order terms as the components octane number, Scheffe polynomial
allows the correlations return to the octane numbers of neat compounds when they are used, which is
a distinct difference from similar correlations in the literature.
Table 4.1: Terms of the Scheffe polynomial with four variables
NO. Term NO. Term NO. Term NO. Term NO. Term
1 x1 8 x1x2 15 x2x4(x2 − x4) 22 x3x4(x3 − x4)2 29 x1x23x4
2 x2 9 x1x3 16 x3x4(x3 − x4) 23 x21x2x3 30 x2x2
3x4
3 x3 10 x2x3 17 x1x2(x1 − x2)2 24 x21x2x4 31 x1x2x2
3
4 x4 11 x1x2(x1 − x2) 18 x1x3(x1 − x3)2 25 x21x3x4 32 x1x2x2
4
5 x1x4 12 x1x3(x1 − x3) 19 x1x4(x1 − x4)2 26 x22x3x4 33 x1x3x2
4
6 x2x4 13 x1x4(x1 − x4) 20 x2x3(x2 − x3)2 27 x1x22x3 34 x2x3x2
4
7 x3x4 14 x2x3(x2 − x3) 21 x2x4(x2 − x4)2 28 x1x22x4 35 x1x2x3x4
Table 4.2: Coefficients of first order terms in the Scheffe polynomial
Variable Compound βi/RON βi/MON
x1 isooctane 100 100
x2 n-heptane 0 0
x3 toluenea var var
x4 ethanol 108 90.7a RON=120, MON=103.5 from [207]RON=116, MON=101.8 from [215]RON=117.2, MON=110.5 from [227]RON=117.4, MON=106.9 from our work
4.2.2 Linear regression
All candidate polynomials are evaluated by linear regression. With m different fuels of known (i.e.
measured) ON and a polynomial with n terms, this linear regression can be expressed as
50

y = Xβ (4.2)
where y is a vector representing the measured ONs for the m fuels, X is an m× n matrix containing
the value of the indeterminate of each of the n polynomial terms for these m fuels (i.e. x1, x2,..., x1x2,...,
x1x2(x1− x2),...), and β is a vector denoting n coefficients of these polynomial terms. To solve for vector
β, the rank of X must be no less than n, i.e. the number of fuels of linearly independent composition
must be at least equal to the number of polynomial terms. Otherwise, the rank deficiency problem will
appear. Given the number of fuels (m > 50) is considerably larger than the number of terms (n < 10),
rank deficiency is not an issue for correlations reported here.
4.2.3 Data for correlation development and validation
This section used the following, independent data sets, which are also presented in full tabular form
in Appendix A.
• The data for developing the correlations
TRF - ASTM standards [25, 26], Morgan et al. [214] and Knop et al. [215]
TRF/ethanol - Foong et al. [9]
• The data for validating the correlations
TRF - this work, and Lund [211–213]
TRF/ethanol - Lund [211–213]
It is noted that the development data used in this study is restricted to a range of 80 to 120 regarding
RON, since this spans a plausible range of production gasoline, while avoiding excessive polynomial
complexity that arises from fitting data at low ONs and which is of limited, practical interest. These
limits could, of course, be relaxed, with longer, optimal polynomials then inevitably resulting. The
distributions of development and validation data are plotted in four simplex lattices as shown in
Fig.4.1, each with one component omitted. Note that quaternary mixtures are not shown in Fig.4.1,
but are listed in Appendix A. The contour line representing RON = 80 is drawn in Fig.4.1(a), (c)
and (d), and no contour line is plotted in Fig.4.1(b), as the entire surface has octane number higher
than 100. It is clear that both the development and validation data are located in the regions with
octane number above 80. Considering that octane number tests are normally carried out in a range of
application interests, and coordinated efforts have not been made to map these surfaces systematically,
it is not surprising to find unevenly distributed data points on these simplex lattices, which might
make developed correlations perform poorly in the regions where data points are scarce. In this case,
51

RON=80
0.2 0.4 0.6 0.8
0.8
0.6
0.4
0.2 0.8
0.6
0.4
0.2
n−heptane
toluene ethanol
(a) toluene/n-heptane/ethanol
0.2 0.4 0.6 0.8
0.8
0.6
0.4
0.2 0.8
0.6
0.4
0.2
ethanol
isooctane toluene
(b) isooctane/toluene/ethanol
RON=80
0.2 0.4 0.6 0.8
0.8
0.6
0.4
0.2 0.8
0.6
0.4
0.2
n−heptane
isooctane ethanol
(c) isooctane/n-heptane/ethanol
RON=80
0.2 0.4 0.6 0.8
0.8
0.6
0.4
0.2 0.8
0.6
0.4
0.2
n−heptane
isooctane toluene
(d) isooctane/n-heptane/toluene
Figure 4.1: Data distribution on simplex lattices with filled circles representing development data andopen ones for validation data
the selection of the criterion for correlation development has to be careful to avoid over-constraining
the correlation, as discussed in the following.
4.2.4 Criterion for correlation development
The optimal correlations should be accurate enough to constrain the residual errors within an accept-
able range, but should not over-constrain the data, because measurements from different sources have
different uncertainties, and correlations may get involved with these experimental errors if containing
52

more terms than necessary. That is, an over-constrained correlation may correlate the development
data very well but perform poorly with independent, validation data, due to excessive terms pro-
ducing erratic results. Therefore, a balance needs to be maintained between accurate correlation and
over-fitting. In this work, the Scheffe polynomial starts from the four linear first-order terms and is
increased by adding one term a time. The coefficient of determination (R2) and the maximum abso-
lute error (MAE) between the measured and correlated values are examined in parallel for all cases.
Both R2 and MAE are used with the consideration that R2 reflects the statistical performance of the
correlations, and MAE indicates how individual datasets are correlated. The latter apparently rep-
resents a more practical measure of accuracy but is subject to the experimental uncertainties specific
to the dataset used. During the correlation development, if R2 and MAE are improved significantly
with adding one term, more term(s) is added. If the improvement is minor and the highest R2 and the
lowest MAE don’t yield the same correlation, the process then stops, with the consideration that the
statistical measure and dataset specific measure of the correlation quality start to deviate, suggesting
that over-constraint may occur. In this case, the last polynomial is called the optimal correlation which
should contain the fewest terms that can correlate the development data reasonably well.
4.2.5 Procedures for optimal correlation development and validation
The method for determining the optimal octane number correlation of any fuel mixture places three
requirements.
1. The candidate polynomials must only depend on some measure of the fuel composition and
must return the octane numbers of the neat mixture components.
2. The polynomial selected must belong to the sub-set of candidate polynomials with the fewest
terms that meet the criterion mentioned above.
3. The polynomial selected must have the highest R2 or smallest MAE with the development data
amongst all of those belonging to the sub-set of candidate polynomial.
The first requirement ensures that correlation can be determined from physically meaningful quan-
tities and can be expected to be accurate over a wide range of mixtures, e.g. from neat compounds
to quaternary TRF/ethanol mixtures. Scheffe polynomials [222], which provide a composition-based
model for mixture properties, meet this requirement. The second requirement recognises that differ-
ences with data generally decrease with more polynomial terms, and that we should, therefore, seek
the polynomial with the fewest terms that is acceptably accurate. The third requirement then sim-
ply states that the final, selected polynomial should be most accurate of those simplest polynomials
considered.
53

Importantly, this method can be generalised to any mixture of m components by varying the num-
ber of terms in the Scheffe polynomial [222]. Requirements 2 and 3 above extend our previous work
that applied Scheffe polynomials to study the octane rating of four components, liquefied petroleum
gases (LPG) [228], and are required to deal with over-constraining of candidate polynomials and ex-
cessive polynomial complexity. Of course, these two requirements can be replaced by alternatives
should that be considered more appropriate.
The method commences by considering a linear Scheffe polynomial for the mixture. This is simply
the sum of the RONs or MONs of the neat components weighted by their mole fractions, i.e. n initially
equals 4 for TRF/ethanol mixtures. Linear regression was carried out for all polynomials formed from
all possible combinations of all terms in the n term Scheffe polynomial, with only the linear terms
always required to be present.
Once the optimal correlation has been identified using the development data and the criterion
above, it is then tested using the independent, validation data. Because the development and valida-
tion datasets are independent, the R2 and the MAE from all development data do not infer the same
results when undertaking the validation. This is only guaranteed to occur if both the development
and validation datasets are free from error and the distribution of the development data is optimal
across the parameter space. However, the optimal distribution of the development data is not known
a priori, and the errors in the development and validation datasets are unknown as well. As such,
it is hoped to achieve a comparable R2 and MAE with the validation data as a check on the overall
effectiveness of this approach; this is not a definitive test of the correctness of this method.
A sense of the scale of this exhaustive or brute-force procedure can be obtained by considering can-
didate polynomials with different numbers of terms. A n = 7 term polynomial describing a quaternary
mixture (e.g. a TRF/ethanol mixture) has 4495 different combinations, with the four first-order terms
being fixed and only 3 terms picked out of 31. A n = 10 term polynomial has 736281 such polynomial
combinations. This method identified all optimal polynomials presented in this study in at most a few
hours using a normal, desktop workstation running Matlab. Since this is far less time and cost that
involved in measuring the development and validation data in the first place, an exhaustive searching
approach is justified.
4.3 Optimal correlations
Although liquid volume fraction is used to define octane numbers of PRFs, it is found to be not suitable
for the optimal correlation development [10, 44, 214, 215], as also demonstrated in Appendix B. In this
work, the optimal correlations are developed on a mole basis.
54

4.3.1 Optimal RON correlation for TRF/ethanol mixtures
4.3.1.1 Development of the optimal correlation
The linear-by-mole correlation (Eq.4.3), which contains four first order terms for isooctane, n-heptane,
toluene and ethanol, was first demonstrated with the octane number of toluene allowed to vary (as
discussed in Section 4.2.1). The residual errors between the development data and linear blending are
shown in Fig.4.2(a), indicating an overall poor correlation. This is in sharp contrast to the neat TRF
cases for which the linear-by-mole rule was reported working satisfactorily [215]. It is noted that in
Fig.4.2(a) that a significant proportion of residual errors are above 0, because the fuels used here are
mostly synergistic blending mixtures, causing the linear-by-mole rule underestimating RON.
(4.3)RON4,terms = 100x1 + 0x2 + 116.4x3 + 108x4
Non-linear terms are then added one at a time to improve the correlation through the exhaustive
searching described in Section 4.2.5. The first term identified, i.e. that produces the highest R2 and
smallest MAE for 5-term Scheffe polynomials, is 63.2x2x4, as shown in Eq.4.4. The positive coefficient
for this term is consistent with the synergistic octane blending between n-heptane (x2) and ethanol (x4)
observed in the recent study from our group [9]. With this term added, distribution of the residual
errors shifts closer to 0 as a whole, as shown in Figs.4.2(b), confirming that the added term accounts
for synergisms in the fuel mixtures.
(4.4)RON5,terms = 100x1 + 0x2 + 115.9x3 + 108x4 + 63.2x2x4
To further improve the correlation, a second non-linear term, 26.1x1x4, is identified and added
to the polynomial (Eq.4.5). This positive coefficient is also consistent with the synergism observed
between isooctane and ethanol in octane blending [9]. Meanwhile, the 63.2x2x4 term previously found
in Eq.4.4 is replaced by a term composed of the same molar fractions but a negative coefficient, -
97.8x2x4(x2 − x4). Since x2 is smaller than x4 for all development data used here, this term is always
positive and thus still captures the synergism between n-heptane and ethanol. With the two terms
added, the positive residual errors, resulted from synergistic blending, are constrained to 2 ON as in
Fig.4.2(c).
(4.5)RON6,terms = 100x1 + 0x2 + 115.7x3 + 108x4 + 26.1x1x4 − 97.8x2x4(x2 − x4)
A third term is further identified, −9.1x3x4, and added to the polynomial (Eq.4.6). The negative
contribution to the mixture octane numbers by this term is also consistent with the antagonistic blend-
ing between toluene and ethanol as reported in [9]. With this term, residual errors below 0 are mostly
corrected, as shown in Fig.4.2(d).
(4.6)RON7,terms = 100x1 + 0x2 + 116.2x3 + 108x4 + 27.0x1x4 − 9.1x3x4 − 98.4x2x4(x2 − x4)
55

RON
80 85 90 95 100 105 110 115 120
Residual
-10
-8
-6
-4
-2
0
2
4
6
8
10
(a)
RON
80 85 90 95 100 105 110 115 120
Residual
-10
-8
-6
-4
-2
0
2
4
6
8
10
(b)
RON
80 85 90 95 100 105 110 115 120
Residual
-6
-4
-2
0
2
4
6
(c)
RON
80 85 90 95 100 105 110 115 120
Residual
-6
-4
-2
0
2
4
6
(d)
Figure 4.2: Residual error between the development data and correlated RON from (a) linear by-molecorrelation, (b) five terms correlation, (c) six terms correlation and (d) seven terms correlation
Apparently, as more non-linear terms are added, the development data can be better correlated
with significantly improved R2 and MAE. However, the improvement starts to diminish at some point
and the risk of over-constraining the correlation to measurement uncertainties increases, as discussed
in Section 4.2.4. Fig.4.3(a) and (b) show the variations of the R2 and MAE with the number of terms. In
each figure, results from searching by the highest R2 and by the smallest MAE are plotted. It is noted
that after 7 terms, R2 only increases slightly for both searching methods. Besides, from 7-term to 8-
term, the two searching methods result in different polynomials, as shown in the locally magnified
plots in Fig.4.3. In particular, the MAE produced by searching the highest R2 is larger with the 8-term
correlation than with the 7-term correlation (Fig.4.3(b)). These observations indicate that the statistics-
based measure (R2) and dataset specific measure (MAE) for correlation quality start to deviate and
thus suggest over-constraining of the correlation. The correlation development thus stops at 7 terms,
and Eq.4.6 is called the optimal correlation for RON of TRF/ethanol mixtures, with respect to the
56
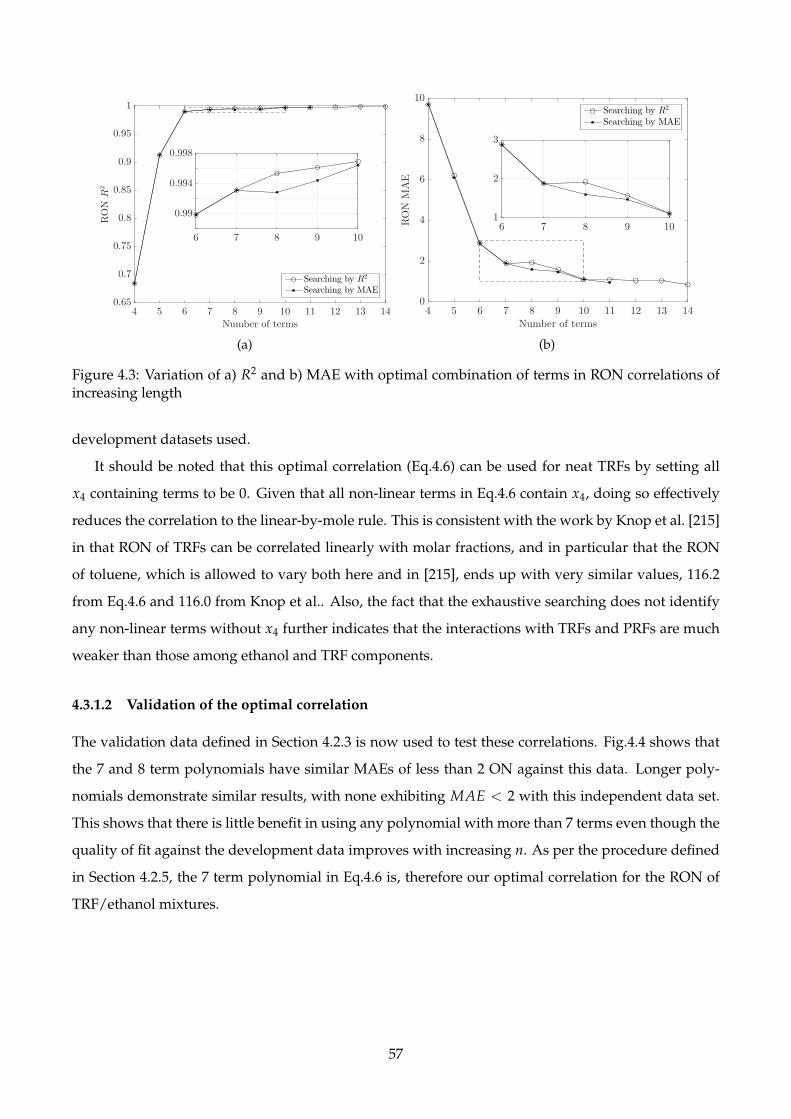
Number of terms
4 5 6 7 8 9 10 11 12 13 14
RON
R2
0.65
0.7
0.75
0.8
0.85
0.9
0.95
1
Searching by R2
Searching by MAE
6 7 8 9 10
0.99
0.994
0.998
(a)
Number of terms
4 5 6 7 8 9 10 11 12 13 14
RON
MAE
0
2
4
6
8
10
Searching by R2
Searching by MAE
6 7 8 9 101
2
3
(b)
Figure 4.3: Variation of a) R2 and b) MAE with optimal combination of terms in RON correlations ofincreasing length
development datasets used.
It should be noted that this optimal correlation (Eq.4.6) can be used for neat TRFs by setting all
x4 containing terms to be 0. Given that all non-linear terms in Eq.4.6 contain x4, doing so effectively
reduces the correlation to the linear-by-mole rule. This is consistent with the work by Knop et al. [215]
in that RON of TRFs can be correlated linearly with molar fractions, and in particular that the RON
of toluene, which is allowed to vary both here and in [215], ends up with very similar values, 116.2
from Eq.4.6 and 116.0 from Knop et al.. Also, the fact that the exhaustive searching does not identify
any non-linear terms without x4 further indicates that the interactions with TRFs and PRFs are much
weaker than those among ethanol and TRF components.
4.3.1.2 Validation of the optimal correlation
The validation data defined in Section 4.2.3 is now used to test these correlations. Fig.4.4 shows that
the 7 and 8 term polynomials have similar MAEs of less than 2 ON against this data. Longer poly-
nomials demonstrate similar results, with none exhibiting MAE < 2 with this independent data set.
This shows that there is little benefit in using any polynomial with more than 7 terms even though the
quality of fit against the development data improves with increasing n. As per the procedure defined
in Section 4.2.5, the 7 term polynomial in Eq.4.6 is, therefore our optimal correlation for the RON of
TRF/ethanol mixtures.
57

RON
80 90 100 110 120
Residual
-6
-4
-2
0
2
4
6
(a)
RON
80 90 100 110 120
Residual
-6
-4
-2
0
2
4
6
(b)
Figure 4.4: Residual error between the validation data and a) 7 and b) 8 term RON correlations on amolar basis
4.3.2 Optimal MON correlation for TRF/ethanol mixtures
4.3.2.1 Development of the optimal correlation
The optimal MON correlation was developed using the same method as for RON. Here the MON
range extends to lower than 80 because of the non-zero octane sensitivity of the fuels used in the RON
correlation. The linear-by-mole blending rule was first attempted, and the correlation and residual er-
rors are shown in Eq.4.7 and Fig.4.5(a) respectively. Three higher order terms: 76.7x2x4, 12.8x1x4, and
−6.4x3x4 were added consecutively from exhaustive searching to account for the non-linear blending
behaviours, as shown in Eq.4.8 and Fig.4.5(b). Coefficients of these terms again suggest the synergis-
tic blending of ethanol/n-heptane and ethanol/isooctane, and antagonistic blending between ethanol
and toluene, which are all similar to the RON case. As shown in Fig.4.6, adding another higher or-
der term to Eq.4.8 doesn’t increase R2 significantly, whereas the correlation with the maximum R2
becomes different from the one with the minimum MAE, suggesting that the seven terms polynomial
in Eq.4.8 is the optimal correlation.
(4.7)MON4,terms = 100x1 + 0x2 + 102.4x3 + 90.7x4
(4.8)MON7,terms = 100x1 + 0x2 + 102.0x3 + 90.7x4 + 12.8x1x4 + 76.7x2x4 − 6.4x3x4
4.3.2.2 Validation of the optimal correlation
The validation data defined in Section 4.2.3 is again used to test these correlations. As with the RON,
Fig.4.7 shows that the 7 and 8 term polynomials have similar MAEs of less than 2 ON against the
validation data. Longer polynomials again demonstrate similar results, with none exhibiting MAE <
58

MON
70 75 80 85 90 95 100 105
Residual
-15
-10
-5
0
5
10
15
(a)
MON
70 75 80 85 90 95 100 105
Residual
-6
-4
-2
0
2
4
6
(b)
Figure 4.5: Residual error between the development data and correlated MON from (a) linear by-molecorrelation, and (b) seven terms correlation
Number of terms4 5 6 7 8 9 10 11
MON
R2
0.7
0.75
0.8
0.85
0.9
0.95
1
Searching by R2
Searching by MAE
6 7 8 90.98
0.985
0.99
0.995
(a)
Number of terms
4 5 6 7 8 9 10 11
MON
MAE
0
2
4
6
8
10
12
14
Searching by R2
Searching by MAE
6 7 8 91
1.5
2
2.5
(b)
Figure 4.6: Variation of a) R2 and b) MAE with optimal combination of terms in MON correlations ofincreasing length
2 with this independent data set. The procedure defined in Section 4.2.5 therefore now infers that the
7 term polynomial in Eq.4.8 is the optimal correlation for the MON of TRF/ethanol mixtures.
59
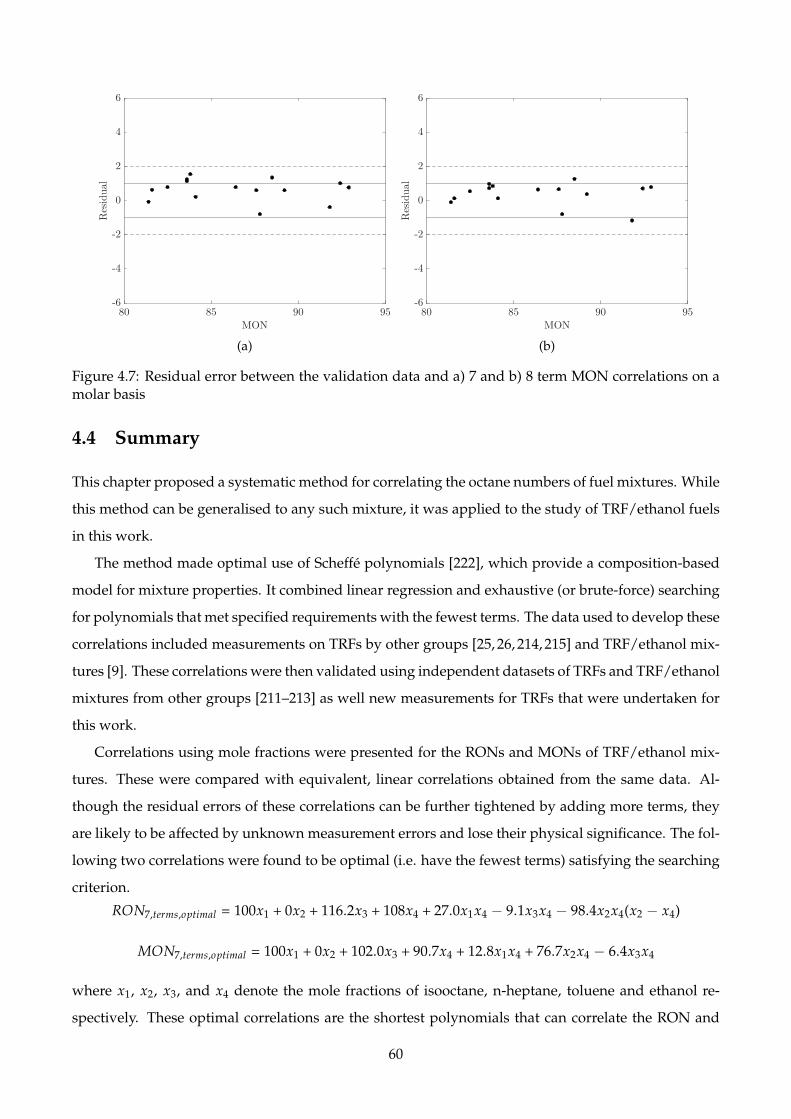
MON
80 85 90 95
Residual
-6
-4
-2
0
2
4
6
(a)
MON
80 85 90 95
Residual
-6
-4
-2
0
2
4
6
(b)
Figure 4.7: Residual error between the validation data and a) 7 and b) 8 term MON correlations on amolar basis
4.4 Summary
This chapter proposed a systematic method for correlating the octane numbers of fuel mixtures. While
this method can be generalised to any such mixture, it was applied to the study of TRF/ethanol fuels
in this work.
The method made optimal use of Scheffe polynomials [222], which provide a composition-based
model for mixture properties. It combined linear regression and exhaustive (or brute-force) searching
for polynomials that met specified requirements with the fewest terms. The data used to develop these
correlations included measurements on TRFs by other groups [25, 26, 214, 215] and TRF/ethanol mix-
tures [9]. These correlations were then validated using independent datasets of TRFs and TRF/ethanol
mixtures from other groups [211–213] as well new measurements for TRFs that were undertaken for
this work.
Correlations using mole fractions were presented for the RONs and MONs of TRF/ethanol mix-
tures. These were compared with equivalent, linear correlations obtained from the same data. Al-
though the residual errors of these correlations can be further tightened by adding more terms, they
are likely to be affected by unknown measurement errors and lose their physical significance. The fol-
lowing two correlations were found to be optimal (i.e. have the fewest terms) satisfying the searching
criterion.
RON7,terms,optimal = 100x1 + 0x2 + 116.2x3 + 108x4 + 27.0x1x4 − 9.1x3x4 − 98.4x2x4(x2 − x4)
MON7,terms,optimal = 100x1 + 0x2 + 102.0x3 + 90.7x4 + 12.8x1x4 + 76.7x2x4 − 6.4x3x4
where x1, x2, x3, and x4 denote the mole fractions of isooctane, n-heptane, toluene and ethanol re-
spectively. These optimal correlations are the shortest polynomials that can correlate the RON and
60

MON of both the development data and validation data with MAE < 2ON, obtained by employing
two goodness-of-fit indicators, R2 and MAE. The non-linear terms identified from the correlation de-
velopment are well represented by the interactions observed in binary mixtures of ethanol and TRF
components, imparting physical significance for these terms selected from exhaustive searching. Nev-
ertheless, these correlations can return the linear-by-mole blending rules for neat TRF mixtures with
the fitted RON and MON of toluene agreeing excellently with the literature values. This demonstrates
the consistency of these correlations over a wide range of fuel compositions, an attribute lacking for
most fuel properties correlations previously reported.
61

Chapter 5
The Octane Numbers of Binary Mixtures
and Gasoline Surrogates Blended with
Ethanol
5.1 Introduction
The work from Chapter 4, together with the prior experimental study [9] explore the blending be-
haviours between ethanol and the TRF-based gasoline surrogates. Nevertheless, the interactions be-
tween ethanol and gasoline should be more complex due to the composition of practical fuels. In this
regard, it is necessary to add more hydrocarbons to the test matrix, which extends the understand-
ings of ethanol/hydrocarbon and hydrocarbon/hydrocarbon interactions and provides fundamental
information to formulate more practical gasoline surrogates.
Australian production gasoline with approximately 30% aromatics by volume blends slightly syn-
ergistically with ethanol, as shown in Fig.5.1. In comparison, the TRF-based gasoline surrogates,
which have a fixed RON of 91 and varied toluene fractions, exhibit significant synergistic behaviours
when blended with ethanol as shown in Fig.5.1, indicating that the TRFs are not sufficient to emulate
the production gasoline.
To develop the gasoline surrogates which better emulate the gasoline’s blending behaviours with
ethanol, systematic studies of the octane numbers for binary fuel mixtures and multi-component gaso-
line surrogates were carried out in the CFR engine. These mixtures were formulated by hydrocarbons
which were selected based on the detailed hydrocarbon analysis (DHA) results of the Australian regu-
lar grade unleaded gasoline with RON of 91. The study has two steps. In the first step, binary mixtures
containing the selected hydrocarbons and ethanol were tested to investigate how neat fuels interact
with each other, which provides the foundation for the following complicated gasoline surrogates for-
62

Volume fraction of ethanol (%)0 20 40 60 80 100
RON
90
95
100
105
110
Gasoline
PRF91
TRF91-15
TRF91-30
TRF91-45
Figure 5.1: Measured RONs for Australian production gasoline, PRF91, and TRF91s blended withethanol [9]
mulation. In the second step, various gasoline surrogates were formulated and tested to develop a
surrogate that can reproduce the interactions between production gasoline and ethanol. Note that this
surrogate only focuses on reproducing the octane number blending with ethanol and other properties
are not used as targets or constraints, although it turns out that some properties of the surrogate match
reasonably well with the gasoline.
5.2 The RONs of binary mixtures
Before formulating new gasoline surrogates, it is necessary to understand the blending behaviours
of binary mixtures, which provides the required basis for more complex gasoline surrogate formula-
tion. Considering that the gasoline contains hundreds of hydrocarbons, the interactions among these
hydrocarbons are critical to formulate gasoline surrogates. Therefore, this section first investigates
how isooctane, n-heptane, cyclohexane, and 1-hexene interact with toluene, and then the blending
behaviours of isooctane/cyclohexane and isooctane/1-hexene mixtures. Lastly, the octane number
measurements of ethanol blended with cyclohexane and 1-hexene were carried out in this study to
supplement the prior experimental study focusing on blending ethanol with isooctane, n-heptane,
and toluene [9].
5.2.1 Binary mixtures of hydrocarbons
Although the octane numbers of neat hydrocarbons have been measured extensively in the API Re-
search Project 45 [207], practical fuels are complex mixtures in which the interactions among hydro-
63

carbons can be significant and thus their octane numbers cannot be determined by the measurements
of these neat hydrocarbons. In this case, a special committee of Research Project 45 measured the
octane numbers of fuel mixtures of interest at that time, and the measurements were summarised
by Scott in 1958 [208]. Since then, few systematic studies have been conducted on octane numbers
of hydrocarbon mixtures. Until recently, Kalghatgi et al. [227, 229] measured the octane numbers of
toluene reference fuels for knock predictions. However, the octane numbers of the other hydrocarbon
mixtures which could potentially be the surrogate compounds, are not well known.
This study reports the octane numbers of binary hydrocarbon mixtures which will be used in
the later gasoline surrogate formulation. In comparison with the measurements from Scott [208],
some of the hydrocarbons tested in this study are different from those reported in Scott’s results, but
compounds from the same hydrocarbon group have been used. In this regard, the binary mixtures
from [208] which are similar to those tested in this study are selected for comparison.
The RONs of isooctane and toluene mixtures are shown in Fig.5.2 (a) and (b) on a volume and
mole basis respectively. In comparison, the measurements of isooctane and ethylbenzene mixtures
from Scott [208] are plotted in Fig.5.2 as well. Unlike isooctane and toluene mixtures, which blend
linearly on a volume basis and antagonistically in mole fractions, significant synergism was observed
when isooctane mixed with ethylbenzene, as the RON of 50% isooctane/50% ethylbenzene exceeds
both neat compounds. It is noticed that the mixture reactivity relative to the neat hydrocarbons is
reduced significantly when methyl group is replaced by ethyl group on the benzene ring.
N-heptane blends synergistically with toluene on a volume basis, and linearly on a mole basis, as
shown in Fig.5.3 (a) and (b) respectively. In comparison, synergisms between n-heptane and ethylben-
zene on both volume and mole bases were reported by Scott [208] and shown in Fig.5.3 (c) and (d),
which, again, suggests that the ethyl group on the benzene ring decreases reactivity compared with
the methyl group when interacting with n-heptane.
Cyclo-paraffins and olefins are important constituents of the gasoline. Therefore, cyclohexane and
1-hexene are chosen as representative compounds for cyclo-paraffins and olefins respectively in this
study. It is found that cyclohexane blends antagonistically with toluene on both volume and mole
bases, as shown in Fig.5.4(a) and (b). Similar behaviours were found in 1-hexene and toluene mixtures
as well, as shown in Fig.5.5(a) and (b). The measured RONs of cyclohexane and 1-hexene are listed in
Table 5.1. Note that the measurements from this study are lower than the values reported by the API
Research Project 45 [207], especially for 1-hexene. However, the RON of 1-hexene recently reported
by Badra et al. [20] is close to the measurement from this study. Since these two representative fuels
were not included in Scott’s test matrix [208], cyclopentane and diisobutylene were used instead. Sim-
ilar antagonistic blending behaviours were observed when cyclopentane and diisobutylene blended
64
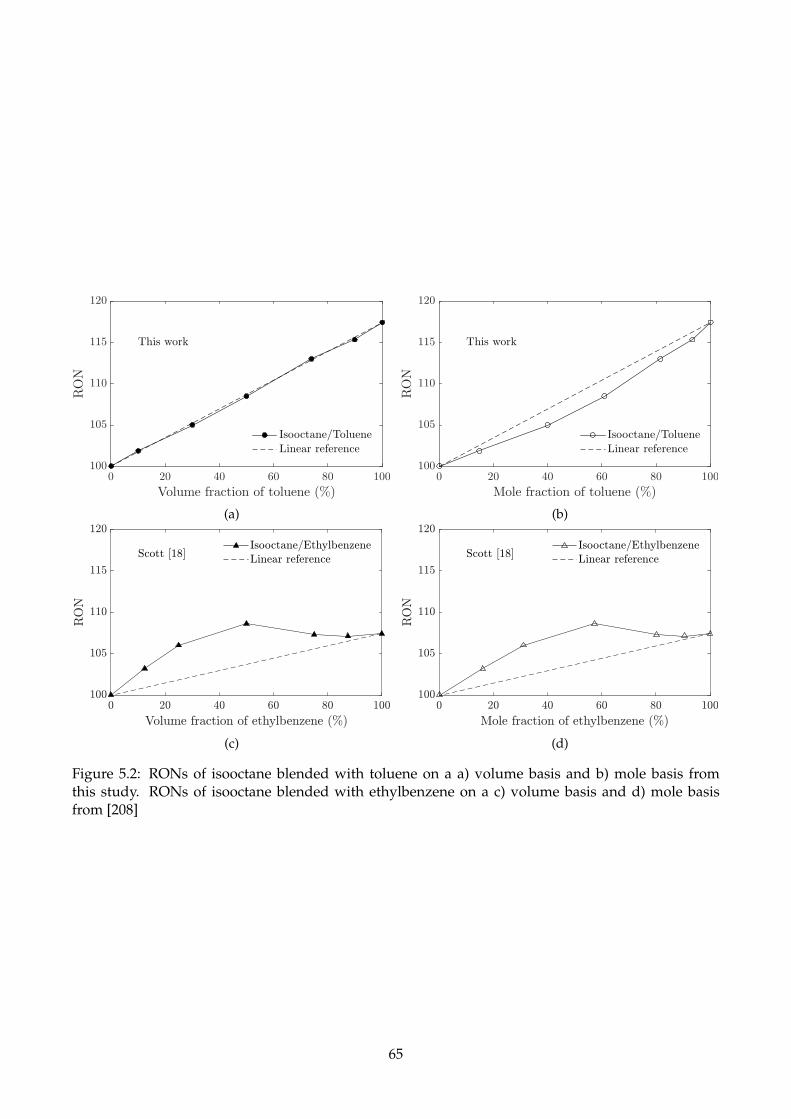
Volume fraction of toluene (%)0 20 40 60 80 100
RON
100
105
110
115
120
This work
Isooctane/Toluene
Linear reference
(a)
Mole fraction of toluene (%)0 20 40 60 80 100
RON
100
105
110
115
120
This work
Isooctane/Toluene
Linear reference
(b)
(c) (d)
Figure 5.2: RONs of isooctane blended with toluene on a a) volume basis and b) mole basis fromthis study. RONs of isooctane blended with ethylbenzene on a c) volume basis and d) mole basisfrom [208]
65
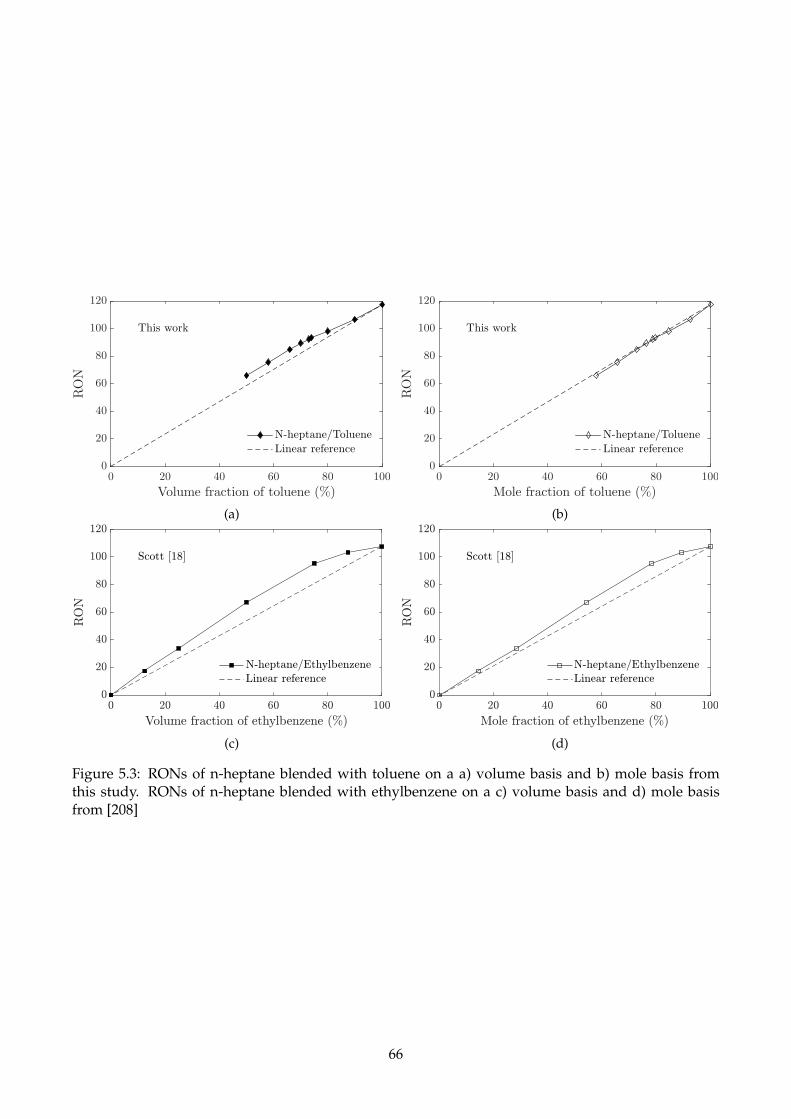
Volume fraction of toluene (%)0 20 40 60 80 100
RON
0
20
40
60
80
100
120
This work
N-heptane/Toluene
Linear reference
(a)
Mole fraction of toluene (%)0 20 40 60 80 100
RON
0
20
40
60
80
100
120
This work
N-heptane/Toluene
Linear reference
(b)
(c) (d)
Figure 5.3: RONs of n-heptane blended with toluene on a a) volume basis and b) mole basis fromthis study. RONs of n-heptane blended with ethylbenzene on a c) volume basis and d) mole basisfrom [208]
66

with ethylbenzene, as shown in Fig.5.4 and 5.5 respectively. Although ethylbenzene exhibits more
complex blending behaviours with other hydrocarbons, it is of interest to find that toluene blends
antagonistically with most representative fuels (except n-heptane) on a mole basis in this study.
Table 5.1: RONs of cyclohexane and 1-hexene from different studies [20, 207]
Fuel This study API Project 45 [207] Badra et al. [20]
1-hexene 72.7 76.4 73.6
Cyclohexane 82.2 83.0 -
The RONs of cyclohexane and 1-hexene blended with isooctane were measured in this study as
well. As shown in Fig.5.6 (a) and (b), cyclohexane blends linearly with isooctane on a volume basis
and synergistically on a mole basis. In comparison, similar trends were found when blending methyl-
cyclohexane with isooctane [208], as shown in Fig.5.6(c) and (d). Synergistic blending behaviours were
observed for 1-hexene/isooctane and 2-heptene/isooctane [208] mixtures, as shown in Fig.5.7.
67
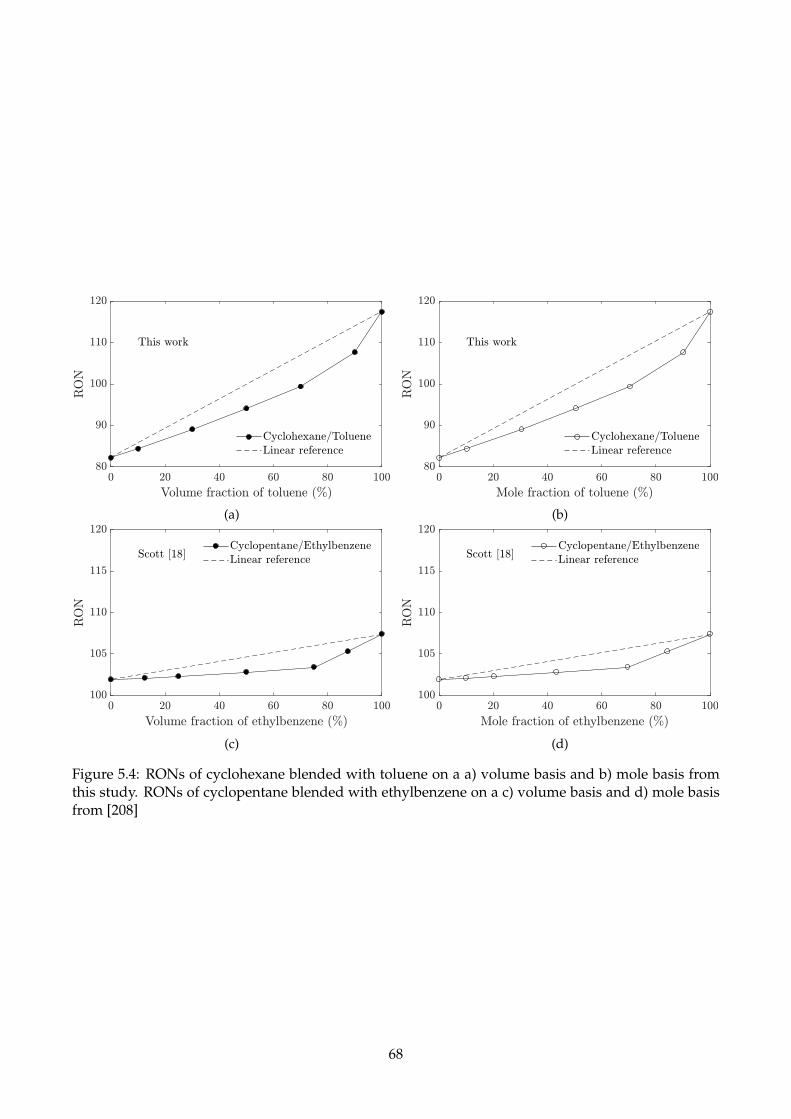
(a) (b)
(c) (d)
Figure 5.4: RONs of cyclohexane blended with toluene on a a) volume basis and b) mole basis fromthis study. RONs of cyclopentane blended with ethylbenzene on a c) volume basis and d) mole basisfrom [208]
68

(a) (b)
(c) (d)
Figure 5.5: RONs of 1-hexene blended with toluene on a a) volume basis and b) mole basis from thisstudy. RONs of diisobutylene blended with ethylbenzene on a c) volume basis and d) mole basisfrom [208]
69

(a) (b)
(c) (d)
Figure 5.6: RONs of cyclohexane blended with isooctane on a a) volume basis and b) mole basis fromthis study. RONs of methylcyclohexane blended with isooctane on a c) volume basis and d) mole basisfrom [208]
70
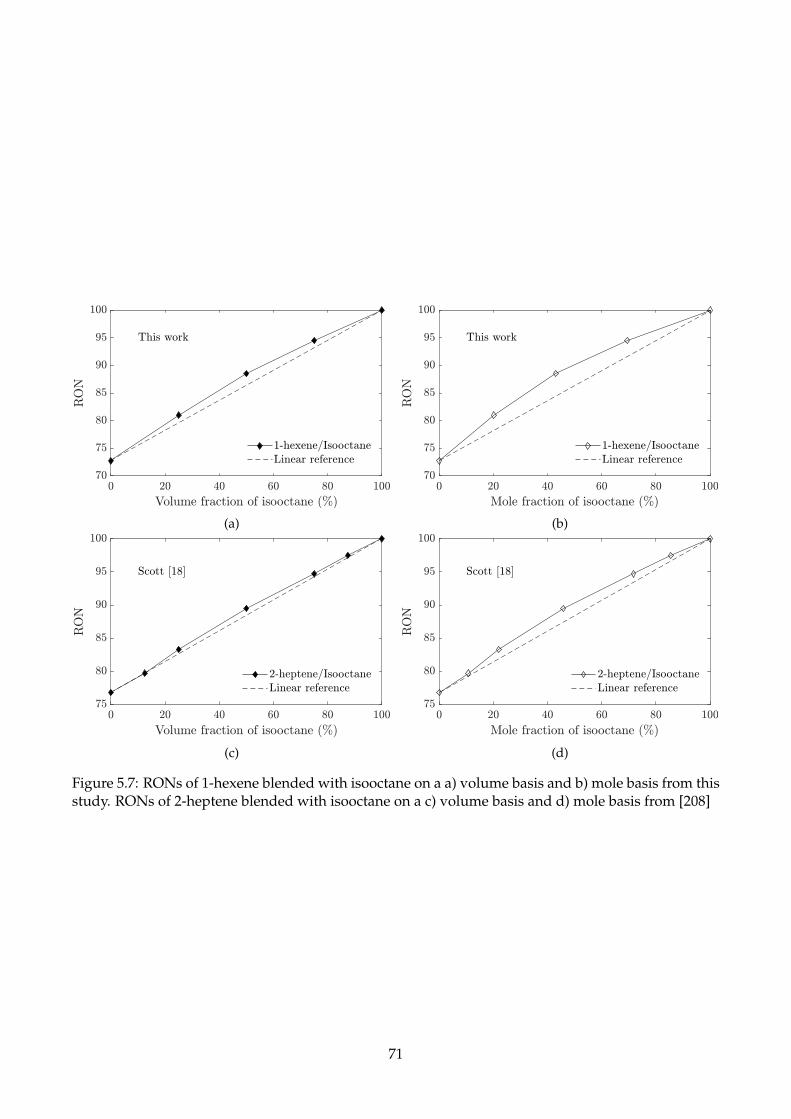
(a) (b)
(c) (d)
Figure 5.7: RONs of 1-hexene blended with isooctane on a a) volume basis and b) mole basis from thisstudy. RONs of 2-heptene blended with isooctane on a c) volume basis and d) mole basis from [208]
71

5.2.2 Binary mixtures containing ethanol
The blending behaviours of isooctane, n-heptane, and toluene with ethanol have been systematically
studied in Foong’s work [9]. However, the interactions of other hydrocarbon groups, such as cyclo-
paraffins and olefins, with ethanol are not known. Fig.5.8 shows the RONs of cyclohexane and 1-
hexene blended with ethanol. The synergistic blending behaviours were observed from these two
binary mixtures, which are more evident on a volume basis than on a mole basis.
Volume fraction of ethanol (%)0 20 40 60 80 100
RON
70
80
90
100
110
This work
Cyclohexane1-hexeneLinear reference
(a)
Mole fraction of ethanol (%)0 20 40 60 80 100
RON
70
80
90
100
110
This work
Cyclohexane1-hexeneLinear reference
(b)
Figure 5.8: RONs of cyclohexane and 1-hexene blended with ethanol on a a) volume basis and b) molebasis
Combining Foong’s work [9] and the results of this study, it is found that the commonly used rep-
resentative fuels of different hydrocarbon groups (except toluene) blend synergistically with ethanol.
On the other hand, toluene blends antagonistically with all studied components (except n-heptane)
on a mole basis.
The octane number interactions among hydrocarbons and ethanol are known to be complicated
and, often non-linear, which are summarised in Table 5.2. It is difficult to draw definitive conclusions
for octane number predictions of complex fuel mixtures, especially when the uncommon components
get involved. The RON tests for the binary mixtures here obtained thus provide the foundations for
the more complex gasoline surrogate formulation.
72
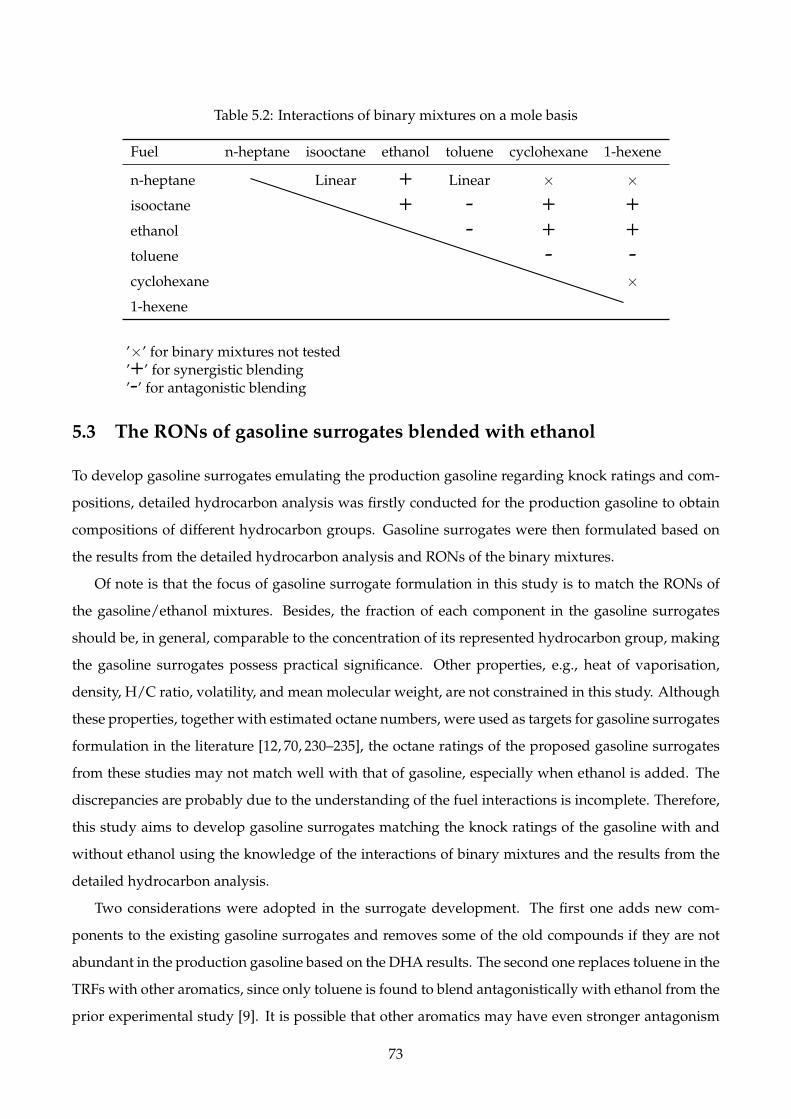
Table 5.2: Interactions of binary mixtures on a mole basis
Fuel n-heptane isooctane ethanol toluene cyclohexane 1-hexene
n-heptane Linear + Linear × ×isooctane + - + +ethanol - + +toluene - -cyclohexane ×1-hexene
’×’ for binary mixtures not tested’+’ for synergistic blending’-’ for antagonistic blending
5.3 The RONs of gasoline surrogates blended with ethanol
To develop gasoline surrogates emulating the production gasoline regarding knock ratings and com-
positions, detailed hydrocarbon analysis was firstly conducted for the production gasoline to obtain
compositions of different hydrocarbon groups. Gasoline surrogates were then formulated based on
the results from the detailed hydrocarbon analysis and RONs of the binary mixtures.
Of note is that the focus of gasoline surrogate formulation in this study is to match the RONs of
the gasoline/ethanol mixtures. Besides, the fraction of each component in the gasoline surrogates
should be, in general, comparable to the concentration of its represented hydrocarbon group, making
the gasoline surrogates possess practical significance. Other properties, e.g., heat of vaporisation,
density, H/C ratio, volatility, and mean molecular weight, are not constrained in this study. Although
these properties, together with estimated octane numbers, were used as targets for gasoline surrogates
formulation in the literature [12, 70, 230–235], the octane ratings of the proposed gasoline surrogates
from these studies may not match well with that of gasoline, especially when ethanol is added. The
discrepancies are probably due to the understanding of the fuel interactions is incomplete. Therefore,
this study aims to develop gasoline surrogates matching the knock ratings of the gasoline with and
without ethanol using the knowledge of the interactions of binary mixtures and the results from the
detailed hydrocarbon analysis.
Two considerations were adopted in the surrogate development. The first one adds new com-
ponents to the existing gasoline surrogates and removes some of the old compounds if they are not
abundant in the production gasoline based on the DHA results. The second one replaces toluene in the
TRFs with other aromatics, since only toluene is found to blend antagonistically with ethanol from the
prior experimental study [9]. It is possible that other aromatics may have even stronger antagonism
73

than toluene when blended with ethanol, which may help to decrease the octane number difference.
The gasoline surrogates proposed in this study all perform better than the simple TRFs, which sug-
gests the added surrogate compounds are critical in terms of emulating the blending behaviours of
the gasoline and ethanol. The experimental results reported in this chapter together with the optimal
correlations developed in Chapter 4 provide implications for fuel surrogate design.
5.3.1 Detailed hydrocarbon analysis for the Australian production gasoline
The Australian gasoline with RON of 91 was analysed by Independent Petroleum Laboratory (IPL)
in New Zealand and categorized into five groups: iso-, n-, cyclo- paraffins, aromatics and olefins
using the PIANO method which considers cyclo-olefins as olefins and is thus different from the more
commonly used PIONA method. The volume fraction of each group is summarised in Table 5.3 which
shows that iso-paraffins, n-paraffins and aromatics are major constituents in the gasoline, while cyclo-
paraffins and olefins, whose volume fractions are slightly less than n-paraffins, should be considered
as important constituents as well.
Table 5.3: Volume fractions of hydrocarbon groups in the Australian production gasoline
Group iso-paraffins n-paraffins cyclo-paraffins aromatics olefins others
vol (%) 39.9 10.7 9.1 24.8 9.9 5.6
The result of DHA test contains more than 300 species which have been sorted into different hy-
drocarbon groups. Table 5.4 and 5.5 list top ten most abundant species in these five groups and the
species with the largest fraction in each group is marked in bold. Although isooctane and n-heptane
are commonly used as the representative compounds of iso-paraffins and n-paraffins respectively,
their contents in the gasoline are much lower than iso-pentane and n-pentane. Besides, cyclohexane
and toluene are important surrogate compounds as well owing to their absolute concentrations. Un-
like other hydrocarbon groups, olefins don’t have a single species which has a significantly higher
volume fraction than the rest. Since most olefins in the gasoline are C6, the simplest C6 olefin, 1-
hexene, is a reasonable choice to represent olefins.
74

Table 5.4: Top ten most abundant species in iso-, n- and cyclo-paraffins
iso-paraffins n-paraffins cyclo-paraffinsSpecies vol (%) Species vol (%) Species vol (%)
iso-pentane 11.1 n-butane 1.3 cyclopentane 0.4
2-methylpentane 5.0 n-pentane 4.0 cyclohexane 2.7
3-methylpentane 3.3 n-hexane 3.3 methylcyclohexane 0.5
2,3-dimethylbutane 1.2 n-heptane 1.0 1c,3-dimethylcyclopentane 0.2
2,2-dimethylbutane 0.8 n-octane 0.5 1t,3-dimethylcyclopentane 0.2
2,2-dimethylpentane 1.9 n-nonane 0.3 1t,2-dimethylcyclopentane 0.2
2-methylhexane 1.7 n-decane 0.2 1c,3-dimethylcyclohexane 0.7
3-methylhexane 1.7 n-undecane 0.1 1t,4-dimethylcyclohexane 0.2
isooctane 0.6 n-dodecane 0.1 1c,2c,4-trimethylcyclopentane 0.2
2-methylhexane 0.7 n-tridecane 0.1 1c,4-dimethylcyclohexane 0.1
Table 5.5: Top ten most abundant species in aromatics and olefins
aromatics olefinsSpecies vol (%) Species vol (%)
benzene 1.0 isoprene 1.1
toluene 8.7 2-methylbutene-1 0.9
1,3-dimethylbenzene 3.7 1,4-pentadiene 0.4
1,2-dimethylbenzene 1.9 3,3-dimethylbutene-1 1.7
1,4-dimethylbenzene 1.5 t-hexene-2 0.5
ethylbenzene 1.1 4-methylcyclopentene 0.3
1,3-methylethylbenzene 1.3 3-methylcyclopentene 0.3
1,4-methylethylbenzene 0.6 2-methyl-2-hexene 0.4
1,2,3-trimethylbenzene 0.5 t-heptene-3 0.3
1,2-dimethyl-4-ethylbenzene 0.6 3-methyl-t-hexene-2 0.3
75

5.3.2 Strategy for emulating the octane number of the gasoline
A good gasoline should match RON, MON, heat of vaporisation, etc, which are important for SI engine
combustion. In the prior study, Foong et al. [9] formulated the three TRF-based gasoline surrogates
with 15%, 30% and 45% toluene by volume. To make these mixtures have RONs around 91, the
base PRF composition was adjusted. The same method is applied in this study to formulate the new
gasoline surrogates.
As shown in Fig.5.1, the TRF-based gasoline surrogates have higher octane numbers than the pro-
duction gasoline when blended with ethanol. Although this difference can be reduced by increasing
the fraction of toluene and decreasing the isooctane in the mixture using the optimal correlations in
Chapter 4 to estimate the octane number, the toluene content becomes too high relative to the aromat-
ics content typically found in production gasoline (25% in this study and 32% from Foong [23]). The
purpose of this study is to find the gasoline surrogates similar to the production gasoline regarding
both compositions and octane numbers when blended with ethanol.
Several strategies have been applied to formulate better gasoline surrogates.
• Cyclohexane and 1-hexene were added into TRFs, and three different gasoline surrogates named
GS1, GS2, and GS3 were developed, as shown in Table 5.6. The volume fractions of toluene in
these three mixtures are fixed to be 30%, which is similar to the concentration of aromatics in the
gasoline. Since the volume fractions of both cyclo-paraffins and olefins are around 10%, the GS3
is developed to meet these compositional requirements. In comparison, GS1 and GS2 have 20%
cyclohexane and 1-hexene respectively.
• Since toluene is known to have antagonistic blending behaviours with most hydrocarbons from
the previous binary mixtures experiments, other aromatics, including p-, m-, o-xylene, ethyl-
benzene, and 1,2,4-trimethylbenzene, were used to replace toluene to test whether they exhibit
stronger antagonism when blended with ethanol, and these mixtures are denoted by GS6 to
GS10.
• Based on the study of aromatics, 1,2,4-trimethylbenzene was selected to represent aromatics.
Besides, iso-pentane and n-pentane were also chosen for being the most abundant compound in
their respective class. Note that the additions of C5 components with low boiling points could
help to emulate the gasoline’s volatility (quantified by reid vapour pressure).
Note that all surrogate fuel were formulated to have the RON of 91. This was achieved by adjusting
the ratio of iso-paraffins and n-paraffins via trial-and-error method.
76

Table 5.6: Formulated gasoline surrogates
Surrogate Detailed compositions (vol %)
GS1 30% toluene+20% cyclohexane+50% PRF77
GS2 30% toluene+20% 1-hexene+50% PRF78
GS3 30% toluene+10% cyclohexane+10% 1-hexene+50% PRF77.5
GS4 30% toluene+10% cyclohexane+10% 1-hexene+32% i-C5+18% n-C5
GS5 30% 1,2,4-trimethylbenzene+10% cyclohexane+10% 1-hexene+50% PRF78
GS6 30% p-xylene+70% PRF70
GS7 30% m-xylene+70% PRF72
GS8 30% o-xylene+70% PRF81
GS9 30% 1,2,4-trimethylbenzene+70% PRF76
GS10 30% ethylbenzene+70% PRF74
GS11 30% 1,2,4-trimethylbenzene+10% cyclohexane+10% 1-hexene+38% i-C5+12% n-C5
5.3.3 Comparison between production gasoline and its surrogates when blended with
ethanol
Since the largest octane number difference between gasoline and its TRF-based surrogates appears
when approximately 40% (by volume) ethanol is added, the formulated eleven gasoline surrogates
listed in Table 5.6 were blended with the same amount of ethanol to compare their knock propensities
with the gasoline.
The effects of adding cyclohexane and 1-hexene into TRFs with 30% toluene by volume are shown
in Fig.5.9 (a). With 20% cyclohexane added, the mixture of GS1 and 40% ethanol has RON of 104.0
which is lower than the result of 105.1 from TRF91-30 reported in the prior experimental study [9].
The octane number is further decreased by 0.6 with GS2 which has 20% 1-hexene instead of cyclohex-
ane. However, production gasolines generally don’t contain such a large amount of cyclo-paraffins or
olefins, and the more reasonable surrogate is GS3 which has 10% cyclohexane and 10% 1-hexene re-
spectively. The octane number from GS3 is right in the middle of GS1 and GS2, indicating apparently
a linear trend when varying the concentrations of 1-hexene and cyclohexane.
Although GS1, GS2, and GS3 reduce the octane number difference, it is still not close to the target
value 101.6 which is the octane number of the production gasoline blended with 40% ethanol. In this
case, other aromatic hydrocarbons, including p-xylene, m-xylene, o-xylene, 1,2,4-trimethylbenzene,
and ethylbenzene, were used to replace toluene in TRF91-30. The results are plotted in Fig.5.9 (b). Of
interest is GS9 containing 30% 1,2,4-trimethylbenzene, whose octane number is 103.6 with 40% ethanol
added, while the measurements for the other four surrogates range from 104.2 to 104.7, suggesting
77

1,2,4-trimethylbenzene produces a stronger antagonistic effect with ethanol than other aromatics re-
garding decreasing the octane number gap.
Considering the strong antagonistic effect of 1,2,4-trimethylbenzene, this aromatic compound is
applied to replace toluene in GS3 to formulate GS5 which has RON of 103.0 with 40% ethanol added,
as shown in Fig.5.9(c). Also, another attempt is to use iso-pentane and n-pentane instead of isooc-
tane and n-heptane in the gasoline surrogates, as the former two have much higher concentrations in
the gasoline. Therefore, another five-component gasoline surrogate GS4 is formed, and the resulting
octane number is 103.1. GS4 and GS5 are thus two gasoline surrogates having the lowest octane num-
bers than the other mixtures with 40% ethanol added. Considering that 1,2,4-trimethylbenzene and
C5 paraffins both reduce the octane number difference effectively, another gasoline surrogate, GS11,
containing all aforementioned favourable compounds, is formulated, which has RON of 102.1 with
40% ethanol blended. With GS11, the RON difference has been reduced to 0.5 which is smaller than
the tolerance of 0.9 for RON measurements of this octane number range. To have a comprehensive
comparison between GS11 and the gasoline, different amounts of ethanol were added into GS11 and
the resulting RONs are plotted in Fig.5.9(c). Although the largest RON difference across the entire
range is 0.7 with 60% ethanol addition, it is found that the differences are very small at lower ethanol
concentrations which are practical range of current ethanol blending.
Due to the nature of octane number tests, the equivalence ratios had to be varied to achieve
the standard knocking conditions. The equivalence ratios of gasoline/ethanol and GS11/ethanol at
standard knocking conditions are listed in Table 5.7 which includes previous measurements from
Foong [23] as well. It is apparent that the equivalence ratios of gasoline/ethanol mixtures reported in
this study are similar to Foong’s results [23] and comparable to those of GS11 and ethanol mixtures.
This study also compares other physical properties between the gasoline and the formulated gasoline
surrogates, as shown in Table 5.8. Note that the heat of vaporisation is quantified by the mixture tem-
perature which is measured downstream of the carburettor and right before the intake port. Table 5.8
shows that the mixture temperatures of both the gasoline and GS11 are 291 K, indicating the similar
level of heat of vaporisation. Besides, the contents of iso-pentane and n-pentane in GS11 are very sim-
ilar to the volume fractions of iso-paraffins and n-paraffins which are 39.9% and 10.7% respectively in
the gasoline. In the following MON tests, the MONs of GS11 with and without 40% ethanol are 88.0
and 81.6 respectively, which are higher than the values of 85.4 and 80.3 from the gasoline. This is not
totally unexpected, as the formulation is focused on RON instead of MON.
The above results indicate that although isooctane, n-heptane, and toluene are commonly regarded
as useful surrogate compounds, they cannot match the anti-knock behaviours of the production gaso-
line when blended with ethanol if their concentrations are constrained by the hydrocarbon class dis-
78
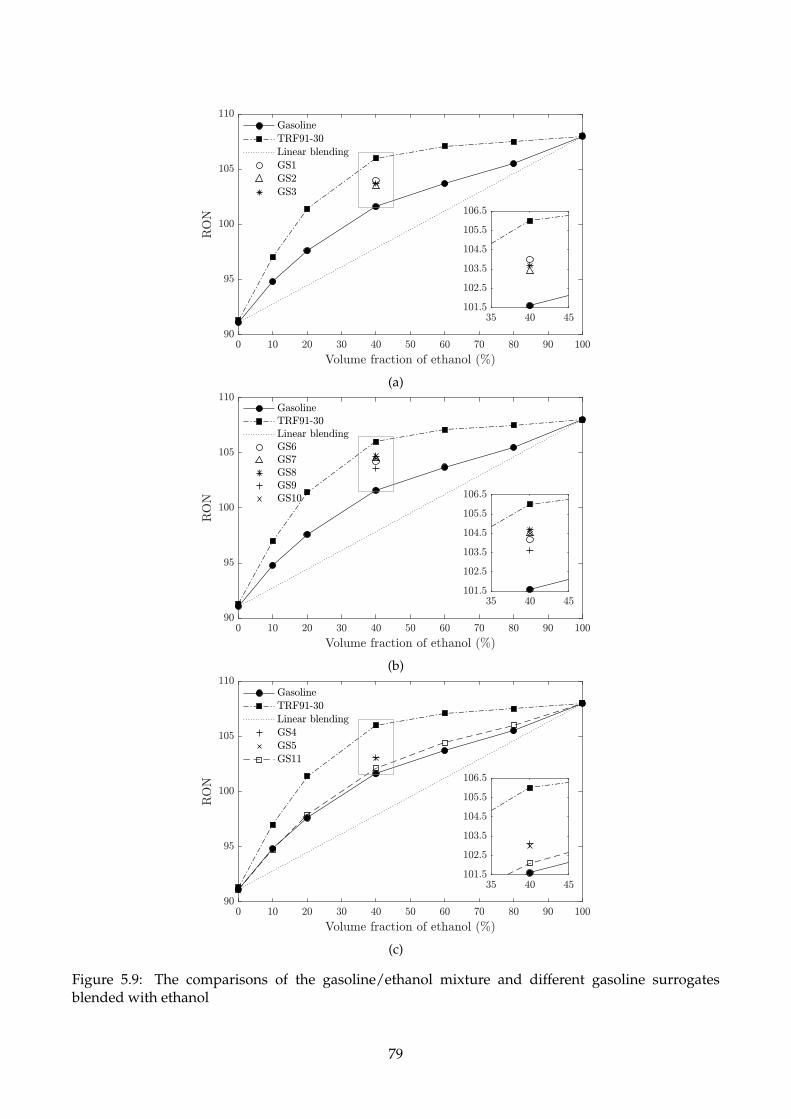
(a)
(b)
(c)
Figure 5.9: The comparisons of the gasoline/ethanol mixture and different gasoline surrogatesblended with ethanol
79

Table 5.7: Equivalence ratios of gasoline/ethanol and GS11/ethanol at standard knocking conditions
Mixtures E0 E10 E20 E40 E60 E80 E100
Gasoline/ethanol a 1.05 1.03 1.03 0.98 0.99 1.02 1.03
Gasoline/ethanol b 1.06 1.04 1.04 1.02 1.01 1.01 1.03
GS11/ethanol a 1.14 1.07 1.03 0.99 1.00 1.05 1.03a Data from this studyb Data from Foong’s experiments [23]
Table 5.8: The physical properties of the gasoline and the gasoline surrogates
Fueldensity(g/ml)
H/C ratioMW(g/mol)
RON(+40% EtOH)
Mixture T atRON test (K)
Gasoline 0.73 1.87 92.06 101.6 291
GS1 0.61 1.81 97.48 104 -
GS2 0.61 1.80 97.98 103.4 -
GS3 0.75 1.80 97.73 103.7 289
GS4 0.72 1.84 80.71 103.1 291
GS5 0.75 1.87 107.00 103.0 -
GS6 1.16 1.89 108.21 104.2 -
GS7 1.17 1.89 108.40 104.5 -
GS8 1.25 1.88 109.22 104.7 -
GS9 1.21 1.92 113.55 103.6 -
GS10 1.20 1.89 108.58 104.5 -
GS11 0.72 1.91 87.28 102.1 291
80

tributions of the production gasoline. The octane number measurements performed in this study
suggests that iso-pentane, n-pentane and 1,2,4-trimethylbenzene should be applied as the surrogate
compounds to emulate the chemical interactions between the production gasoline and ethanol. Simi-
lar compounds have been used by other study [20] to formulate gasoline surrogates as well.
5.4 Summary
To emulate the knocking behaviours of gasoline/ethanol mixture, this chapter first studied the RONs
of binary mixtures which provide fundamental knowledge for the interactions between different hy-
drocarbons and between hydrocarbon and ethanol. It is found that most hydrocarbons selected blend
synergistically with ethanol, except aromatic fuels showing antagonistic trend.
Based on the results of the detailed hydrocarbon analysis of an Australian market gasoline, cy-
clohexane and 1-hexene, each at 10% by volume, were used as the representative compounds for
cycloparaffins and olefins. Further, 1,2,4-trimethylbenzene with a 30% volume fraction was used to
represent aromatics for its strongest antagonism when blended with ethanol, which is required to com-
pensate the synergism exhibited by ethanol blending with other hydrocarbons. Finally, iso-pentane
and n-pentane were selected as the representative compounds of paraffins due to their significant con-
centrations in the production gasoline, and their relative volume fractions were the balancing factor
to make the mixture have a RON of 91. It is found that the proposed gasoline surrogate closely repro-
duced the octane blending behaviours with ethanol over the entire blending range, with a maximum
ON difference of 0.7 unit. Compared with commonly used TRF mixtures, this result indicated that
including additional components from other hydrocarbon classes and using different paraffin and
aromatic compounds than those in TRFs are necessary to emulate the interaction between gasoline
and ethanol.
81

Chapter 6
Oxidation of Ethanol and Hydrocarbon
Mixtures in a Flow Reactor
6.1 Introduction
Octane rating is closely related to autoignition chemistry of the unburned fuel/air mixtures in SI
engines. Kinetic modelling of SI engines autoignition has been attempted for gasoline and ethanol
mixtures by Foong et al. [46] and the author ([Yuan et al., SAE Paper 2015-01-1242], reported in Ap-
pendix C). These efforts suggested that the existing chemistry models are unable to reproduce the
octane behaviours observed in the CFR engine experiments. Therefore, this chapter uses a recently
built PFR which is designed to operate at up to 1000 K and 50 bar in Thermodynamics Laboratory,
the University of Melbourne, to investigate the autoignition chemistry of hydrocarbon and ethanol
mixtures. The PFR experiments in this study were carried out at a nominal temperature around 900 K
which is related to the autoignition temperature in the CFR engine [46]. Note that the knock related
chemistry may occur at lower temperatures. However, not all fuels tested in this study have low tem-
perature chemistry, and the purpose of this study is to systematically check the existing models with
a comprehensive fuel matrix. Therefore, the intermediate temperature of 900 K is chosen in this study.
This PFR study focuses on evaluating the existing chemical mechanisms that can be used to reproduce
the engine measurements.
Neat fuels, binary mixtures, gasoline surrogates and their mixtures with ethanol were tested in
the PFR at 900 K (except 930 K for neat toluene) and 10 bar. The fuel matrix and the PFR operating
conditions in this work are listed in Table 6.1 and 6.2 respectively. The Horiba emission bench was
used to measure the CO concentration with a non-dispersive infrared (NDIR) analyser with a resolu-
tion of 20 PPM. Other intermediate species including the parent fuels were measured using the gas
chromatography (GC) discussed in Chapter 3, whose measurement uncertainties are around 10%. As
82

mentioned in Chapter 3, the reacting gas temperatures are calculated based on measurements from
three K-type thermocouples using the so called three-thermocouple method [201].
Table 6.1: Test fuels and reaction mechanisms for modelling
Fuel Mechanism Composition
IsooctaneAtef et al. [68]Mehl et al. [67]Andrae [187]
isooctanegasoline surrogate and ethanolgasoline surrogate and ethanol
Ethanol
Marinov [181]Mittal et al. [184]Mehl et al . [67]Andrae [187]
ethanolethanolgasoline surrogate and ethanolgasoline surrogate and ethanol
Toluene
Metcalfe et al. [138]Yuan et al. [163]Mehl et al . [67]Andrae [187]
toluenetoluenegasoline surrogate and ethanolgasoline surrogate and ethanol
Isooctane/ethanolIsooctane/tolueneEthanol/toluene
Mehl et al. [67]TestMecha
gasoline surrogate and ethanolgasoline surrogate and ethanol
PRF91b
TRF91-30cMehl et al. [67]TestMech
gasoline surrogate and ethanolgasoline surrogate and ethanol
PRF91/ethanolTRF91-30/ethanol
Mehl et al. [67]TestMech
gasoline surrogate and ethanolgasoline surrogate and ethanol
a TestMech: the mechanism from this studyb PRF91: 91% isooctane and 9% n-heptanec TRF91-30: 53.2% isooctane, 17.0% n-heptane, and 29.8% toluene [9]
83

Table 6.2: Experimental conditions for the PFR study
PFR parameter Set value
Reactor pressure (bar) 10
Reactor nominal temperature (K) 900-930
Equivalence ratio 0.058-0.060
Air flow rate (g/s) 6
Nitrogen flow rate (g/s) 0.32
Reynolds number in the reactor tube 8000
Fuel/nitrogen pressure (bar) 20-21
Fuel/nitrogen temperature (K) 500
6.2 Kinetic modelling approach
To have good understandings of the fuel chemistry, the measured species profiles are modelled with
state of the art detailed chemical mechanisms in CHEMKIN-Pro [236]. The kinetic models chosen in
this study are either developed very recently or widely used in the research community and are listed
in Table 6.1.
The kinetic modelling of the PFR was elaborated in [237] and later incorporated in the commercial
software CHEMKIN-Pro [236]. Since the reacting gas temperatures are measured and corrected in
this study, the temperature changes along the reactor length are known and the temperature profile is
taken as an input for the kinetic modelling, which reflects both energy generation from the chemical
reactions and heat loss to the surroundings. At each integration step, the gas temperature is interpo-
lated from the imported temperature profile. Although CHEMKIN-Pro uses a differential/algebraic
system solver called DASSL [238] which applies adaptive step size to handle fast-changing variations
in the solution, it may encounter numerical errors with the default maximum step size. To solve this
issue, a smaller maximum time step is required.
6.3 Neat fuels
6.3.1 Isooctane
As one of the primary reference fuels, isooctane was first tested in this study. The measured CO,
CO2, and isooctane profiles are plotted in Fig.6.1 as a function of the flow distance. The CO forma-
tion remains at zero until 400 mm whereas CO2 starts to form around 750 mm. The consumption of
isooctane becomes significant after 200 mm. These results were modelled using the latest isooctane
84

mechanisms from Atef et al. [68] and two gasoline surrogate mechanisms from Mehl et al. [67] and
Andrae [187]. The modelling results are also shown in Fig.6.1. Among these mechanisms, the one
from Mehl et al. [67] best reproduces the measured species profiles, while Andrae’s mechanism [187]
and Atef et al.’s mechanism [68] under-predicts and over-predicts the isooctane oxidation reactivity
respectively in the PFR.
The profiles of intermediate species are plotted separately with the corresponding modelling re-
sults in Fig.6.2. Note that the flame ionization detector used in this study cannot detect inorganic
substances and highly oxygenated species. This is why some common intermediates from isooc-
tane oxidation, like hydrogen and formaldehyde, are not included in Fig.6.2. The most significant
and abundant intermediate species is isobutylene (IC4H8) whose profile is well predicted by Mehl et
al. [67], slightly over-predicted by Andrae [187], and significantly under-predicted by the most recent
mechanism from Atef et al. [68]. The reaction pathways for IC4H8 are shown in Fig.6.3. As for the
predictions of the small species, such as methane, ethylene, and propylene, the model from Mehl et
al. [67], again, performs the best, while the other two models [68, 187] reasonably reproduce the pro-
files. It is noticed that these models all under-predict the profiles for the oxygenated compounds, but
Mehl et al.’s model [67] exhibits good fidelity to the measurements in terms of matching the posi-
tions where the peak values appear. The reaction pathway for one of these oxygenated compounds,
IC3H5CHO, is show in Fig.6.4. The largest intermediate species detected from the isooctane oxidation
are XC7H14 (2,4-dimethyl-1-pentene) and YC7H14 (2,4-dimethyl-2-pentene) whose concentrations are
much smaller than the others’. The mechanism developed by Mehl et al. [67] over-estimates the con-
centration of XC7H14 and under-estimates YC7H14. The reaction pathways for XC7H14 and YC7H14
are shown in Fig.6.3.
The intermediate species shown in Fig.6.2 have been reported by previous experimental studies
[58, 59, 61, 239] as well. Among all these works, the flow reactor experiments from Chen et al. [61]
have the closest experimental conditions to this study. The peak values of different hydrocarbon
species from these two experiments are similar, except for the oxygenated compounds. The measured
oxygenated compounds from this study have higher concentrations than those from Chen et al. [61].
Since the isooctane sub-model from the gasoline surrogate mechanism [67] was developed based on
the measurements from Chen et al. [61], this is why the existing model under-estimates the profiles of
the oxygenated compounds from this study. Currently, the causes for the discrepancies between the
measured oxygenated compounds from this study and Chen et al. [61] are not known, and thus more
kinetic experiments are required to explain the differences, which could be a future work of this study.
85
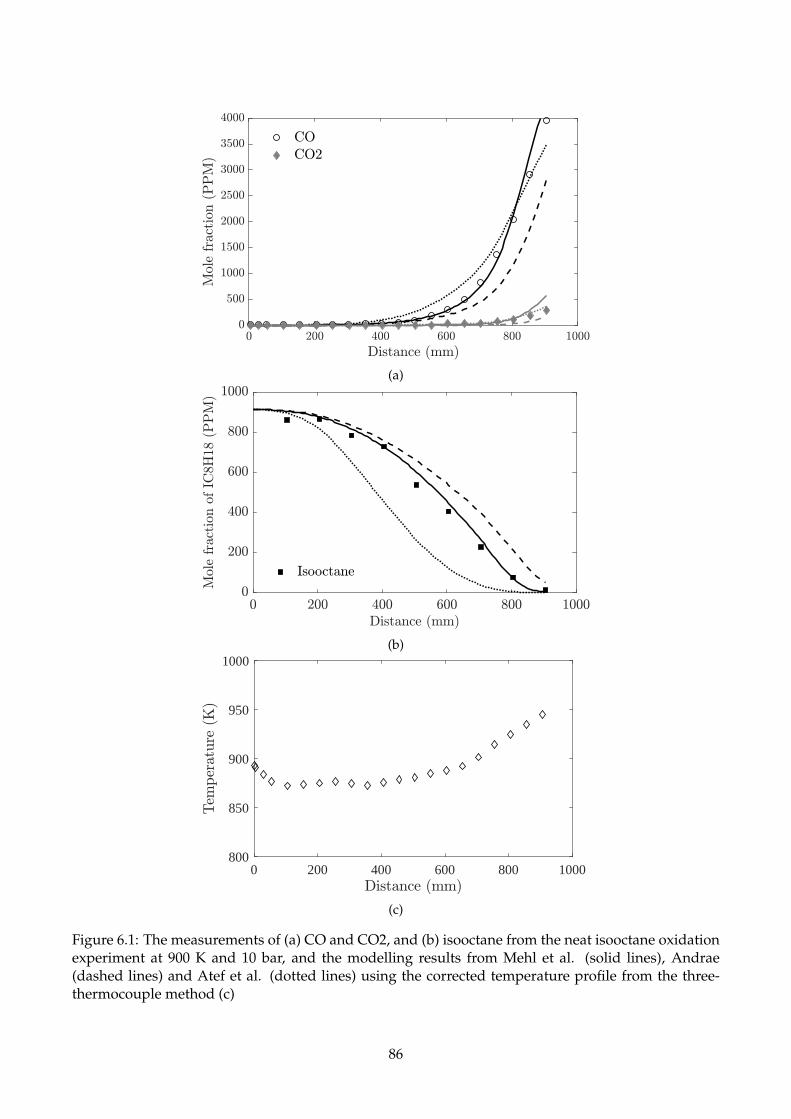
(a)
(b)
0 200 400 600 800 1000800
850
900
950
1000
(c)
Figure 6.1: The measurements of (a) CO and CO2, and (b) isooctane from the neat isooctane oxidationexperiment at 900 K and 10 bar, and the modelling results from Mehl et al. (solid lines), Andrae(dashed lines) and Atef et al. (dotted lines) using the corrected temperature profile from the three-thermocouple method (c)
86
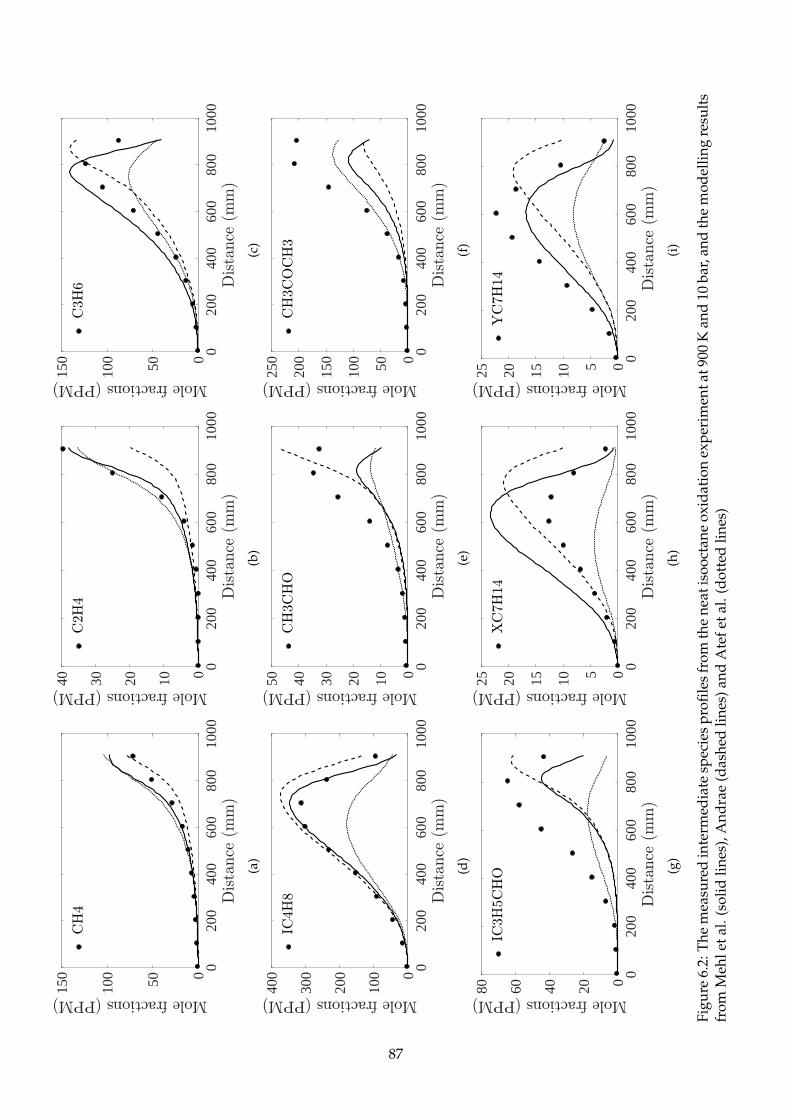
020
040
060
080
010
00050100
150
(a)
020
040
060
080
010
00010203040
(b)
020
040
060
080
010
00050100
150
(c)
020
040
060
080
010
000
100
200
300
400
(d)
020
040
060
080
010
0001020304050
(e)
020
040
060
080
010
00050100
150
200
250
(f)
020
040
060
080
010
00020406080
(g)
020
040
060
080
010
000510152025
(h)
020
040
060
080
010
000510152025
(i)
Figu
re6.
2:Th
em
easu
red
inte
rmed
iate
spec
ies
profi
les
from
the
neat
isoo
ctan
eox
idat
ion
expe
rim
enta
t900
Kan
d10
bar,
and
the
mod
ellin
gre
sult
sfr
omM
ehle
tal.
(sol
idlin
es),
And
rae
(das
hed
lines
)and
Ate
feta
l.(d
otte
dlin
es)
87
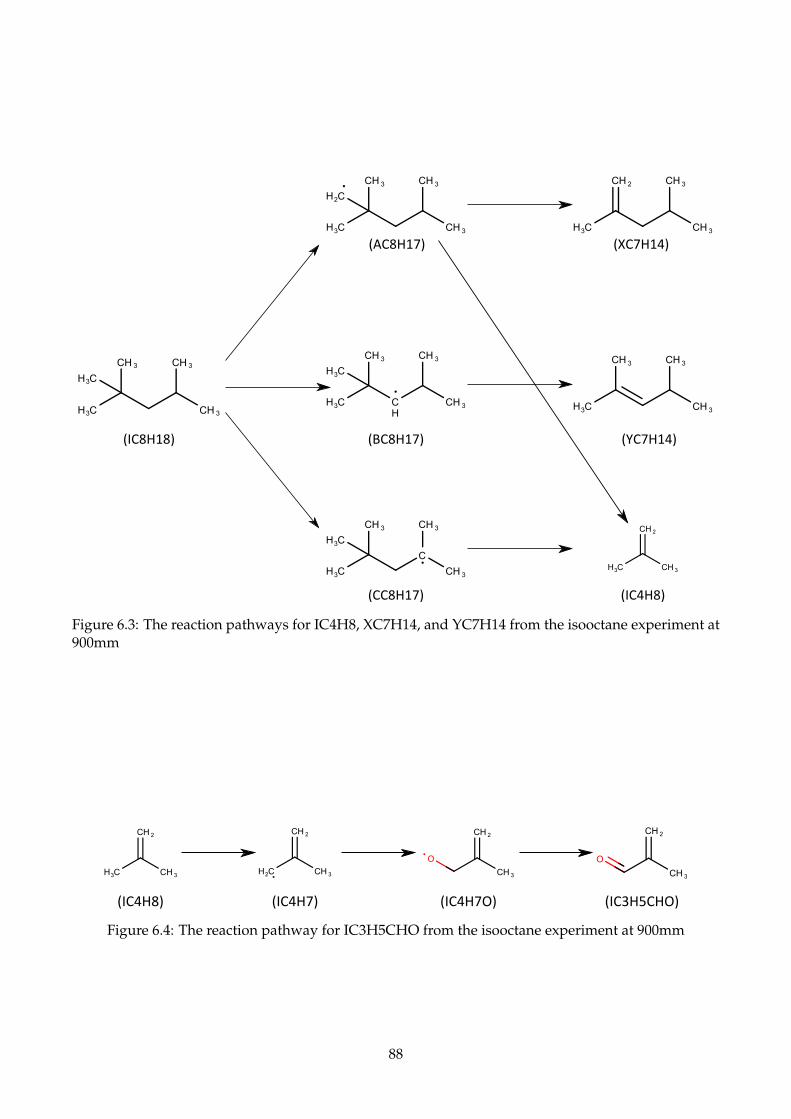
Figure 6.3: The reaction pathways for IC4H8, XC7H14, and YC7H14 from the isooctane experiment at900mm
(IC4H8) (IC4H7) (IC4H7O) (IC3H5CHO)
Figure 6.4: The reaction pathway for IC3H5CHO from the isooctane experiment at 900mm
88

6.3.2 Ethanol
The CO, CO2, and fuel profiles for ethanol, together with the modelling results are shown in Fig.6.5.
Marinov’s mechanism [181] is one of the earliest comprehensive mechanisms for ethanol, which under-
predicts the ethanol’s reactivity in this study. Three more recent mechanisms from Mehl et al. [67],
Andrae [187], and Mittal et al. [184] perform similarly, which produce excellent agreements for CO
and ethanol profiles but fail to match the CO plateau. The agreements suggest that the models accu-
rately capture the chemistry where ethanol converts to CO. The discrepancies in the CO plateau are
significant, which are probably caused by the problematic kinetics of CO to CO2 conversion as the
CO2 profile is over-predicted by all these three mechanisms.
The measured intermediate species from the ethanol oxidation are methane, ethylene, and ac-
etaldehyde, as shown in Fig.6.6. The most abundant intermediate species is acetaldehyde whose pro-
file is overall well-predicted by Mehl et al. [67] and the reaction pathways for acetaldehyde are shown
in Fig.6.7. Besides, the methane profile is well captured with the same mechanism as well. However,
this mechanism has difficulty to reproduce the ethylene profile and the level of over-prediction is sig-
nificant. In comparison, the predictions from Andrae’s model [187] match well with the measured
ethylene profile but not with the other species. The discrepancies between the measurements and
modellings are likely due to the super lean experimental conditions, against which the existing kinetic
models may not be fully validated.
The intermediates shown in Fig.6.6 were also reported in a flow reactor [202], a jet-stirred reac-
tor [175], and a rapid compression machine [240]. Although it is difficult to compare the measure-
ments from the different reactors due to different experimental conditions, the relative concentrations
of the major intermediate, acetaldehyde, from these experiments are overall comparable with the mea-
surement in this study, especially when taking their initial contents of ethanol into account. Note that
in the most recent rapid compression machine study [240], the initial mole fraction of ethanol is one
order of magnitude higher than those of the aforementioned experiments, making its results not com-
parable with others.
89
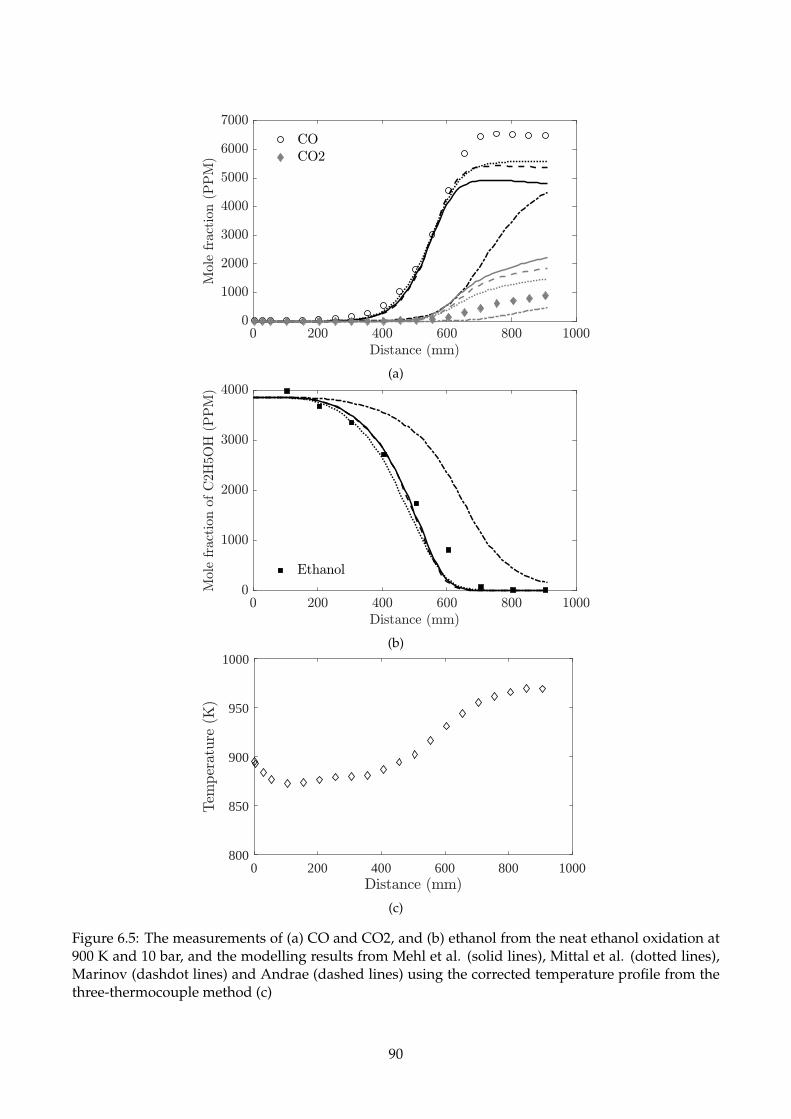
(a)
(b)
0 200 400 600 800 1000800
850
900
950
1000
(c)
Figure 6.5: The measurements of (a) CO and CO2, and (b) ethanol from the neat ethanol oxidation at900 K and 10 bar, and the modelling results from Mehl et al. (solid lines), Mittal et al. (dotted lines),Marinov (dashdot lines) and Andrae (dashed lines) using the corrected temperature profile from thethree-thermocouple method (c)
90

0 200 400 600 800 10000
100
200
300
400
500
600
700
(a)
0 200 400 600 800 10000
200
400
600
800
(b)
0 200 400 600 800 10000
200
400
600
800
(c)
Figure 6.6: The measured intermediate species profiles from the neat ethanol oxidation at 900 K and10 bar, and the modelling results from Mehl et al. (solid lines), Mittal et al. (dotted lines), Marinov(dashdot lines) and Andrae (dashed lines)
91

Figure 6.7: The reaction pathway for CH3CHO from the ethanol experiment at 500mm
6.3.3 Toluene
Comparing with isooctane and ethanol, toluene has much lower reactivity and barely reacts at 900 K.
Therefore, the experiment was conducted at a higher temperature, 930 K. Even at this temperature,
the measured CO concentrations are significantly lower than those of isooctane and ethanol, as shown
in Fig.6.8. The modelling results show that only the predictions from the mechanisms of Metcalfe
et al. [138] and Zhang et al. [241] agree reasonably well with the measurements. The other three
mechanisms from Mehl et al. [67], Yuan et al. [163] and, in particular, Andrae [187] and Pelucchi et
al. [242] substantially over-predict the toluene’s reactivity. Therefore, it is necessary to examine the
existing toluene sub-model to reproduce the measured species profiles in this study.
Since the reactivity of toluene at 930 K is low, benzene is the only intermediate detected by the
GC and has a much smaller fraction compared with toluene. The comparison between the measured
benzene profile and the modelling results from the aforementioned mechanisms is shown in Fig.6.9.
Although the kinetic models from Mehl et al. [67] and Yuan et al. [163] over-predict the overall reac-
tivity, their predictions on the benzene formation are lower than the measurement. This inconsistency
may come from the mechanisms themselves or the measurement uncertainties of species with small
amounts.
As one of the most important intermediate species, benzene was detected in two recent experi-
mental studies performed in the flow reactor [138] and the jet-stirred reactor [163] respectively. Under
comparable conditions to our experiments, similarly small levels of benzene concentrations were ob-
served in these experiments.
92
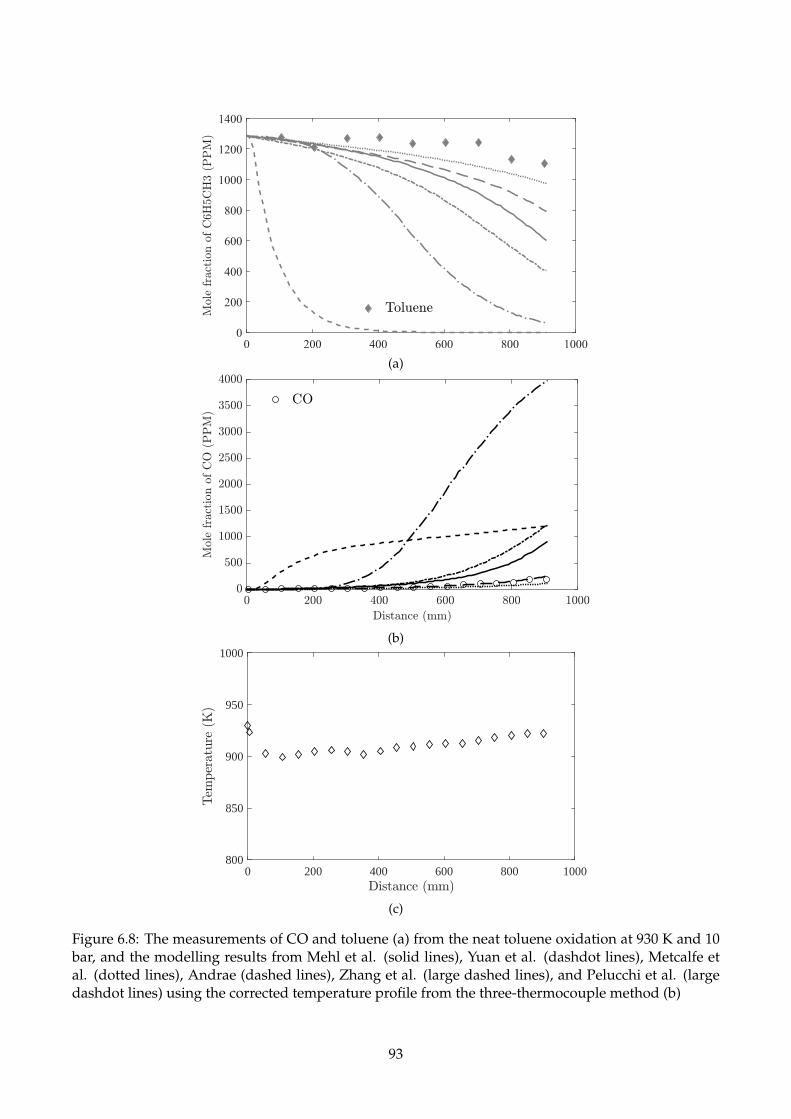
(a)
(b)
0 200 400 600 800 1000800
850
900
950
1000
(c)
Figure 6.8: The measurements of CO and toluene (a) from the neat toluene oxidation at 930 K and 10bar, and the modelling results from Mehl et al. (solid lines), Yuan et al. (dashdot lines), Metcalfe etal. (dotted lines), Andrae (dashed lines), Zhang et al. (large dashed lines), and Pelucchi et al. (largedashdot lines) using the corrected temperature profile from the three-thermocouple method (b)
93

Figure 6.9: The measured benzene profile from the neat toluene oxidation at 930 K and 10 bar, and themodelling results from Mehl et al. (solid line), Yuan et al. (dashdot line), Metcalfe et al. (dotted line),Andrae (dashed line), Zhang et al. (large dashed lines), and Pelucchi et al. (large dashdot lines)
6.4 Test mechanism
In order to improve the modelling of species profiles from the neat toluene oxidation, a test mechanism
is proposed based on the toluene sub-model from Mehl et al.’s gasoline surrogate mechanism [67].
6.4.1 Sensitivity analysis
In this study, an in-house PFR model was developed in Python with the VODE solver and designed
to run in parallel on a high performance computing (HPC) system for the sensitivity analysis. The
details of this in-house model can be found in Appendix D.
A sensitivity analysis was carried out for the toluene oxidation at 930 K and 10 bar using the gaso-
line surrogate mechanism from Mehl et al. [67]. When conducting the analysis, each pre-exponential
factor, A, was increased by two times to produce a temporary chemical mechanism, and the total
number of the temporary mechanisms is over 10,000. All these generated mechanisms were applied
to model the neat toluene oxidation, and their resulting maximum CO concentrations, denoted by
COi, were compared with the CO concentration from the original gasoline surrogate mechanism, rep-
resented by COref. Therefore, the sensitivity coefficient (SC) is given by:
(6.1)SC =COi − COre f
COre f
The elementary reactions having the top 30 largest absolute sensitivity coefficients for the toluene
oxidation at 930 K and 10 bar are shown in Fig.6.10 where positive sensitivity coefficients indicate that
increasing the rate constants of the corresponding elementary reactions enhances the CO formation
94

and negative coefficients suggest the opposite trend. It is noticed that the high-sensitivity elementary
reactions mostly contain toluene or toluene-like species.
6.4.2 Updated toluene sub-mechanism
To update the existing toluene sub-mechanism, the rate constants of those most sensitive elementary
reactions require careful investigations. Considering the sparsity of the accurate rate constants for
these high-sensitivity reactions, either from experimental measurements or theoretical computations,
only the top eight most sensitive elementary reactions are analysed for updates in their rate constants.
Since Metcalfe et al.’s mechanism [138] performs the best as shown in Fig.6.8, the rate constants from
Mehl et al’s model [67] were replaced by those from [138], if related fundamental experiments and
theoretical computations are not available.
Reaction 6.2 is found to be the most sensitive elementary reaction from the sensitivity analysis
as shown in Fig.6.10. Both Mehl et al. [67] and Metcalfe et al. [138] only assigned pre-exponential
factors as the rate constants for this elementary reaction, indicating the reaction is independent of
temperature and pressure in their mechanisms. However, the quantum chemical study of da Silva
and Bozzelli [243] proposed a more complex and lower rate constant for Reaction 6.2 compared with
the values used by those two mechanisms. Note that the rate constants from da Silva and Bozzelli [243]
are at atmospheric pressure. Zhang et al. [241] extended the rate constants to different pressures with
Quantum-Rice˙Ramsperger-Kassel theory. Therefore, this study uses the updated rate constants from
Zhang et al. [241] for Reaction 6.2.
(6.2)C6H5CH2J + HO2 = C6H5CH2OJ + OH
The rate constants of Reaction 6.3 and 6.4 from Mehl et al. [67] were updated by the values from
Baulch et al. [244] and Metcalfe et al. [138]. The updated rate constants were used in Metcalfe et al.’s
toluene mechanism [138] which produce good agreements with the measurements from this study, as
shown in Fig.6.8. The changes made to the toluene sub-model in the gasoline surrogate model [67] are
listed in Table 6.3.
(6.3)C6H5CH3 + HO2 = C6H5CH2J + H2O2
(6.4)C6H5OJ + HO2 = RODC6JDO + OH
Although Reaction 6.5 to 6.9 have high sensitivities, this study keeps the original rate constants
from Mehl et al. [67] for these reactions due to the following reasons. First, the rate constants of these
five reactions haven’t been investigated by fundamental experiments or theoretical computations. It
is difficult to figure out whether the original rate constants from Mehl et al. [67] need to be updated.
Second, Reaction 6.5 and 6.6 don’t appear in Metcalfe et al.’s model [138], while the rate constants of
95
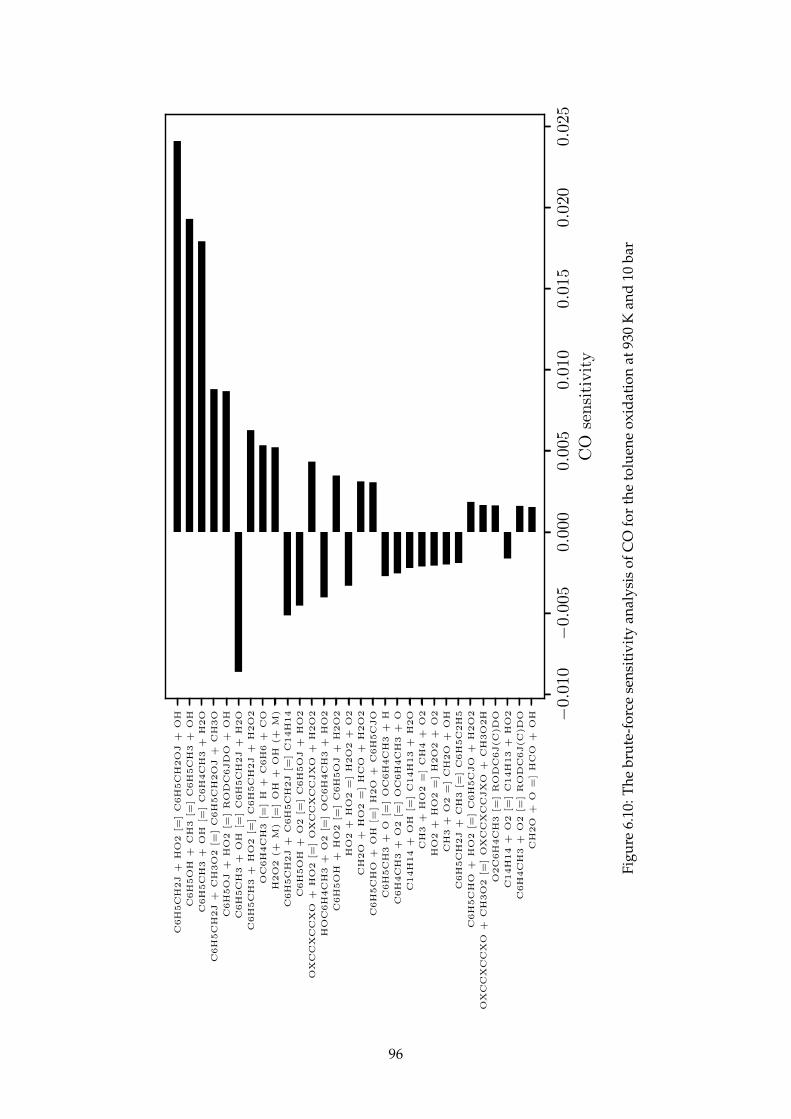
−0.0
10
−0.0
050.
000
0.00
50.
010
0.01
50.
020
0.02
5
CO
sen
siti
vit
y
C6H
5C
H2J
+H
O2
[=]
C6H
5C
H2O
J+
OH
C6H
5O
H+
CH
3[=
]C
6H
5C
H3
+O
H
C6H
5C
H3
+O
H[=
]C
6H
4C
H3
+H
2O
C6H
5C
H2J
+C
H3O
2[=
]C
6H
5C
H2O
J+
CH
3O
C6H
5O
J+
HO
2[=
]R
OD
C6JD
O+
OH
C6H
5C
H3
+O
H[=
]C
6H
5C
H2J
+H
2O
C6H
5C
H3
+H
O2
[=]
C6H
5C
H2J
+H
2O
2
OC
6H
4C
H3
[=]
H+
C6H
6+
CO
H2O
2(+
M)
[=]
OH
+O
H(+
M)
C6H
5C
H2J
+C
6H
5C
H2J
[=]
C14H
14
C6H
5O
H+
O2
[=]
C6H
5O
J+
HO
2
OX
CC
XC
CX
O+
HO
2[=
]O
XC
CX
CC
JX
O+
H2O
2
HO
C6H
4C
H3
+O
2[=
]O
C6H
4C
H3
+H
O2
C6H
5O
H+
HO
2[=
]C
6H
5O
J+
H2O
2
HO
2+
HO
2=
]H
2O
2+
O2
CH
2O
+H
O2
=]
HC
O+
H2O
2
C6H
5C
HO
+O
H[=
]H
2O
+C
6H
5C
JO
C6H
5C
H3
+O
[=]
OC
6H
4C
H3
+H
C6H
4C
H3
+O
2[=
]O
C6H
4C
H3
+O
C14H
14
+O
H[=
]C
14H
13
+H
2O
CH
3+
HO
2=
]C
H4
+O
2
HO
2+
HO
2=
]H
2O
2+
O2
CH
3+
O2
=]
CH
2O
+O
H
C6H
5C
H2J
+C
H3
[=]
C6H
5C
2H
5
C6H
5C
HO
+H
O2
[=]
C6H
5C
JO
+H
2O
2
OX
CC
XC
CX
O+
CH
3O
2[=
]O
XC
CX
CC
JX
O+
CH
3O
2H
O2C
6H
4C
H3
[=]
RO
DC
6J(C
)D
O
C14H
14
+O
2[=
]C
14H
13
+H
O2
C6H
4C
H3
+O
2[=
]R
OD
C6J(C
)D
O
CH
2O
+O
=]
HC
O+
OH
Figu
re6.
10:T
hebr
ute-
forc
ese
nsit
ivit
yan
alys
isof
CO
for
the
tolu
ene
oxid
atio
nat
930
Kan
d10
bar
96

Table 6.3: Reaction changes to LLNL’s toluene sub-mechanism
Reaction A n EA Reference
C6H5CH2+HO2=C6H5CH2OJ+OH 1.19e9 1.03 -2250 [243]a
C6H5CH3+HO2=C6H5CH2J+H2O2 3.97e11 0 14069 [244]
C6H5OJ+HO2=RODC6JDO+OH 2.00e12 0 0 [138]a This rate constant from da Silva and Bozzelli [243] is at atmospheric pres-sure and thus not directly used in the TestMech
Reaction 6.7 to 6.9 from both mechanisms are very similar or the same. This study uses the original
rate constants from Mehl et al. [67] for Reaction 6.5 to 6.9.
(6.5)C6H5OH + CH3 = C6H5CH3 + OH
(6.6)C6H5CH2J + CH3O2 = C6H5CH2OJ + CH3O
(6.7)C6H5CH3 + OH = C6H4CH3 + H2O
(6.8)OC6H4CH3 = H + C6H6 + CO
(6.9)C6H5CH3 + OH = C6H5CH2J + H2O
Based on the sensitivity analysis, the rate constants of Reaction 6.2, 6.3, and 6.4 were updated
to formulate a TestMech, which significantly reduce the discrepancies between the measured and
modelled CO profiles, as show in Fig.6.11. Given the good agreements of the toluene oxidation at 930
K and 10 bar using the TestMech, it is of great interest to investigate whether the behaviours of fuel
mixtures can be reasonably reproduced. Note that the changes only affect toluene-related reactions,
which means the TestMech performs identically to the original gasoline surrogate mechanism [67]
when applied to model fuel mixture without toluene-like components.
97

Figure 6.11: The measurements of CO and toluene from the neat toluene oxidation at 930 K and 10bar, and the modelling results from Mehl et al. (solid lines) and TestMech (dashed lines)
6.5 Binary mixtures
The proposed TestMech is capable of predicting the measurements of all neat fuels tested in this study.
However, practical fuels are all mixtures. In this regard, it is critical to understand the behaviours
of fuel mixtures in the PFR, which are good tests for the state of the art surrogate mechanisms as
well. Therefore, binary mixtures were studied in this subsection, which includes ethanol/isooctane,
toluene/isooctane, and ethanol/toluene. Apart from the TestMech, the gasoline surrogate mechanism
from Mehl et al. [67] is also applied in the following kinetic modellings for fuel mixtures. This mecha-
nism [67] was used by several recent experimental studies [245–248] to model ignition delays in shock
tubes and rapid compression machines. It was found that the mechanism [67] performs reasonably
well in terms of predicting experimental results but requires improvements for TRF mixtures with
high toluene fraction [246], which is consistent with the results from the experiments of the neat com-
pounds in this study. To the knowledge of the author, the mechanism [67] has not been systematically
validated with the species profiles from fuel mixtures in either flow reactors or jet-stirred reactors.
6.5.1 Ethanol and isooctane
Ethanol and isooctane mixtures were first tested in this study at 900 K and 10 bar. The mole frac-
tions of ethanol in these mixtures are 25%, 50%, and 74%. The measured CO profiles and corrected
temperature profiles using the three-thermocouple method are shown in Fig.6.12. Both Mehl et al.’s
gasoline surrogate model [67] and the TestMech proposed by this study were applied to model the CO
profiles. The modelling results well reproduced the measured CO profiles as shown in Fig.6.12, indi-
cating the interactions between isooctane and ethanol are well taken care of by the existing chemical
98
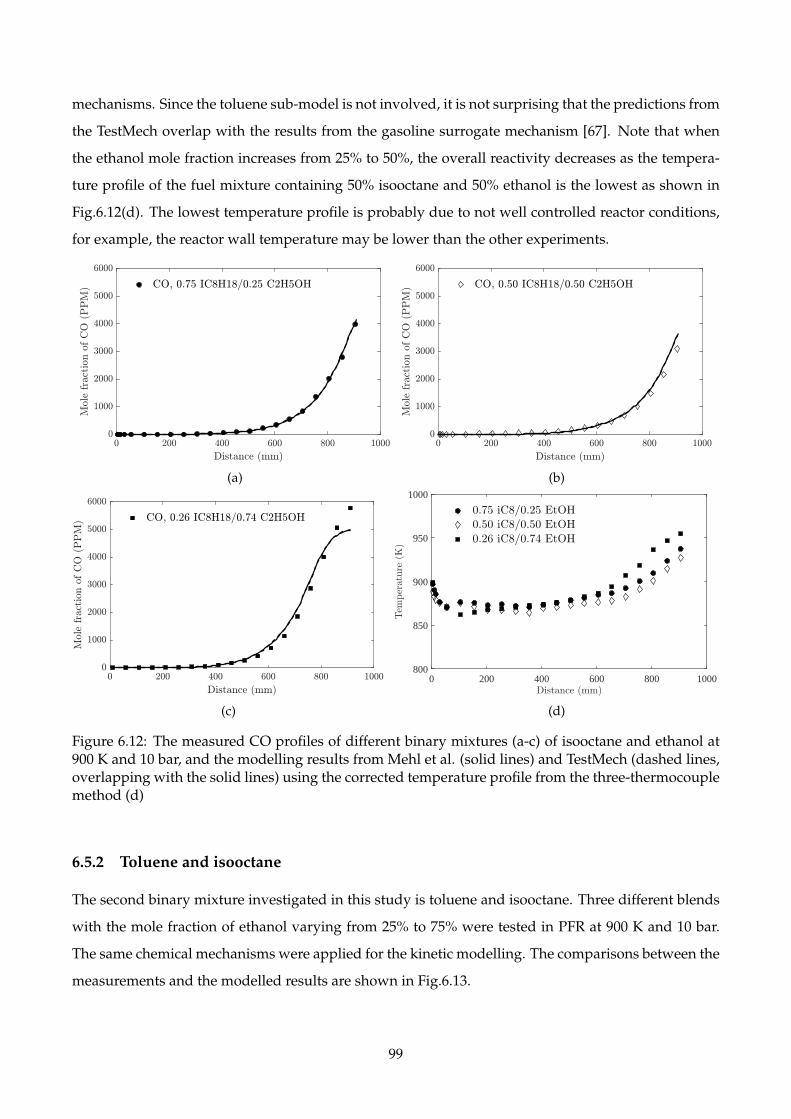
mechanisms. Since the toluene sub-model is not involved, it is not surprising that the predictions from
the TestMech overlap with the results from the gasoline surrogate mechanism [67]. Note that when
the ethanol mole fraction increases from 25% to 50%, the overall reactivity decreases as the tempera-
ture profile of the fuel mixture containing 50% isooctane and 50% ethanol is the lowest as shown in
Fig.6.12(d). The lowest temperature profile is probably due to not well controlled reactor conditions,
for example, the reactor wall temperature may be lower than the other experiments.
(a) (b)
(c)
Distance (mm)0 200 400 600 800 1000
Tem
perature
(K)
800
850
900
950
1000
0.75 iC8/0.25 EtOH0.50 iC8/0.50 EtOH0.26 iC8/0.74 EtOH
(d)
Figure 6.12: The measured CO profiles of different binary mixtures (a-c) of isooctane and ethanol at900 K and 10 bar, and the modelling results from Mehl et al. (solid lines) and TestMech (dashed lines,overlapping with the solid lines) using the corrected temperature profile from the three-thermocouplemethod (d)
6.5.2 Toluene and isooctane
The second binary mixture investigated in this study is toluene and isooctane. Three different blends
with the mole fraction of ethanol varying from 25% to 75% were tested in PFR at 900 K and 10 bar.
The same chemical mechanisms were applied for the kinetic modelling. The comparisons between the
measurements and the modelled results are shown in Fig.6.13.
99
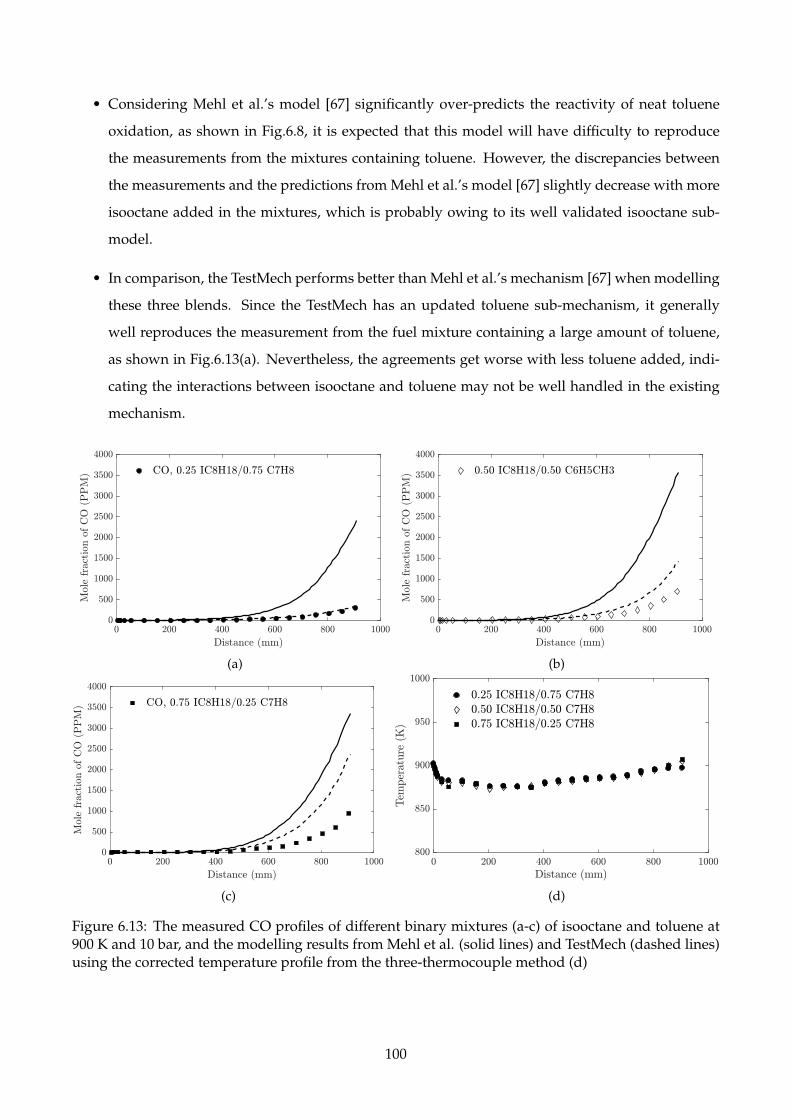
• Considering Mehl et al.’s model [67] significantly over-predicts the reactivity of neat toluene
oxidation, as shown in Fig.6.8, it is expected that this model will have difficulty to reproduce
the measurements from the mixtures containing toluene. However, the discrepancies between
the measurements and the predictions from Mehl et al.’s model [67] slightly decrease with more
isooctane added in the mixtures, which is probably owing to its well validated isooctane sub-
model.
• In comparison, the TestMech performs better than Mehl et al.’s mechanism [67] when modelling
these three blends. Since the TestMech has an updated toluene sub-mechanism, it generally
well reproduces the measurement from the fuel mixture containing a large amount of toluene,
as shown in Fig.6.13(a). Nevertheless, the agreements get worse with less toluene added, indi-
cating the interactions between isooctane and toluene may not be well handled in the existing
mechanism.
(a) (b)
(c) (d)
Figure 6.13: The measured CO profiles of different binary mixtures (a-c) of isooctane and toluene at900 K and 10 bar, and the modelling results from Mehl et al. (solid lines) and TestMech (dashed lines)using the corrected temperature profile from the three-thermocouple method (d)
100
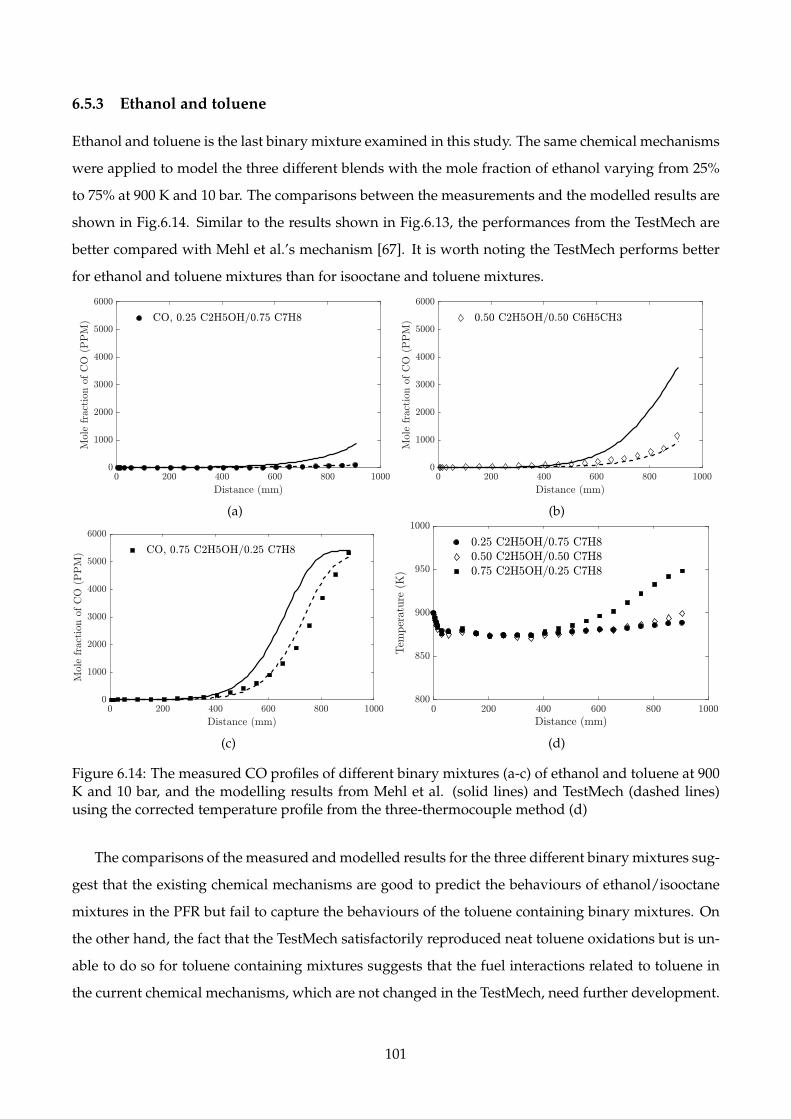
6.5.3 Ethanol and toluene
Ethanol and toluene is the last binary mixture examined in this study. The same chemical mechanisms
were applied to model the three different blends with the mole fraction of ethanol varying from 25%
to 75% at 900 K and 10 bar. The comparisons between the measurements and the modelled results are
shown in Fig.6.14. Similar to the results shown in Fig.6.13, the performances from the TestMech are
better compared with Mehl et al.’s mechanism [67]. It is worth noting the TestMech performs better
for ethanol and toluene mixtures than for isooctane and toluene mixtures.
(a) (b)
(c) (d)
Figure 6.14: The measured CO profiles of different binary mixtures (a-c) of ethanol and toluene at 900K and 10 bar, and the modelling results from Mehl et al. (solid lines) and TestMech (dashed lines)using the corrected temperature profile from the three-thermocouple method (d)
The comparisons of the measured and modelled results for the three different binary mixtures sug-
gest that the existing chemical mechanisms are good to predict the behaviours of ethanol/isooctane
mixtures in the PFR but fail to capture the behaviours of the toluene containing binary mixtures. On
the other hand, the fact that the TestMech satisfactorily reproduced neat toluene oxidations but is un-
able to do so for toluene containing mixtures suggests that the fuel interactions related to toluene in
the current chemical mechanisms, which are not changed in the TestMech, need further development.
101

6.6 Gasoline surrogates
Comparing with the binary mixtures, the gasoline surrogates have more implications for the practi-
cal applications. This study chooses PRF91 and TRF91-30 with 30% toluene proposed by [9] as the
gasoline surrogates, which were tested in the PFR under 900 K and 10 bar.
6.6.1 PRF91
PRFs are known as the simplest gasoline surrogates which can match the RON of the production
gasoline. In this study, PRF91 is blended to emulate the gasoline with RON of 91. The compar-
isons between the measured profiles from the PRF91 oxidation and the modelling results are shown
in Fig.6.15. Both the gasoline surrogate mechanism [67] and the TestMech well predict the profiles
of CO, CO2, and the parent fuels. Since toluene is not included in PRF91, it is not surprising that
the TestMech behaves exactly the same as the original mechanism from Mehl et al. [67]. In order to
quantify the impact of the chemical interactions between the parent fuels or fuel-like species to the
reactivity, the related elementary reactions (Reaction 5724 to 5935 from the original gasoline surrogate
model [67]) were deleted from the TestMech. The predictions from the mechanism excluding these
chemical interactions are also shown in Fig.6.15. It is found that removing chemical interactions has
negligible impact on the reactivity.
The profiles of the intermediate species from the PRF91 oxidation and the corresponding mod-
elling results are compared in Fig.6.16. The major species measured from the PRF91 oxidation are
similar to those from isooctane since isooctane is the major component of PRF91. Generally, the pro-
files of the hydrocarbon intermediates are well captured by these two mechanisms. However, the
maximum concentrations of the two oxygenated compounds, acetone and methacrolein, are under-
predicted. It is noticed that the mechanisms exhibit good fidelity in terms of reproducing the overall
trends for these two oxygenated compounds.
102
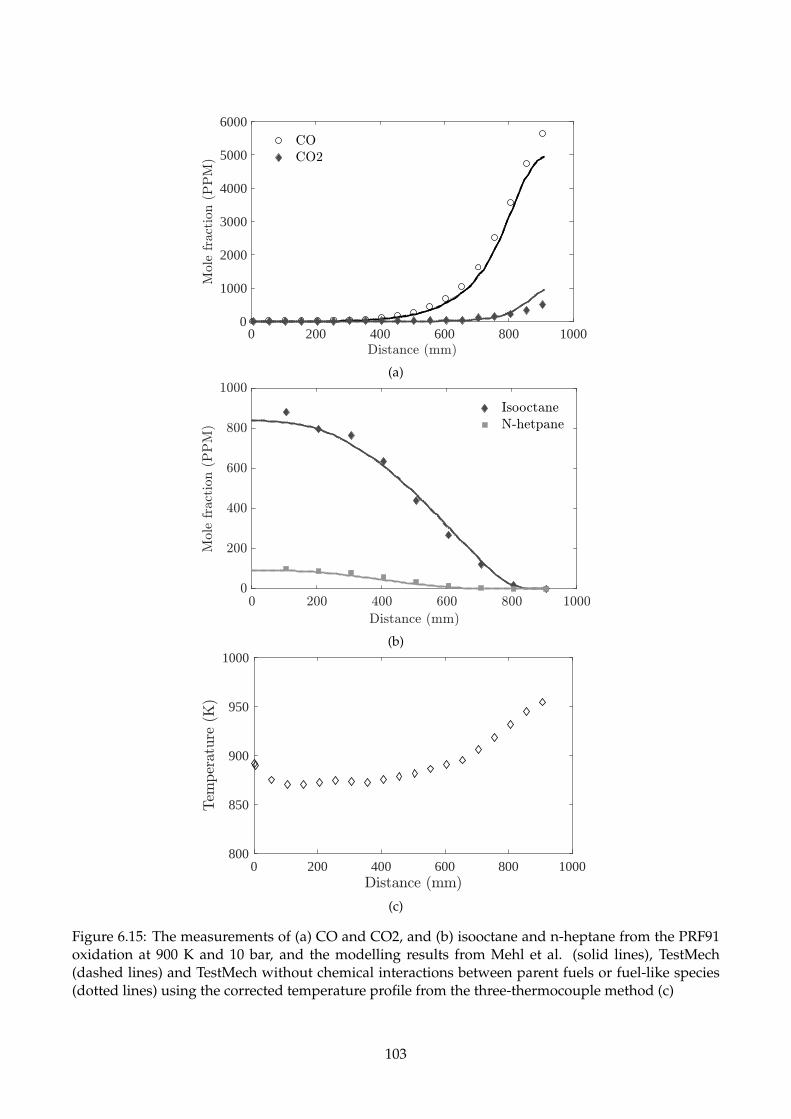
0 200 400 600 800 10000
1000
2000
3000
4000
5000
6000
(a)
(b)
0 200 400 600 800 1000800
850
900
950
1000
(c)
Figure 6.15: The measurements of (a) CO and CO2, and (b) isooctane and n-heptane from the PRF91oxidation at 900 K and 10 bar, and the modelling results from Mehl et al. (solid lines), TestMech(dashed lines) and TestMech without chemical interactions between parent fuels or fuel-like species(dotted lines) using the corrected temperature profile from the three-thermocouple method (c)
103

020
040
060
080
010
00020406080100
(a)
020
040
060
080
010
00020406080100
(b)
020
040
060
080
010
00050100
150
(c)
020
040
060
080
010
00050100
150
200
250
300
350
(d)
020
040
060
080
010
00050100
150
200
250
(e)
020
040
060
080
010
00010203040506070
(f)
020
040
060
080
010
000510152025
(g)
020
040
060
080
010
000510152025
(h)
Figu
re6.
16:T
hem
easu
red
inte
rmed
iate
spec
ies
profi
les
from
the
PRF9
1ox
idat
ion
expe
rim
enta
t900
Kan
d10
bar,
and
the
mod
ellin
gre
sult
sfr
omM
ehle
tal.
(sol
idlin
es)a
ndTe
stM
ech
(das
hed
lines
)
104

6.6.2 TRF91-30
Although the existing chemical mechanisms well reproduce the species profiles from the PRF91 ox-
idation, PRF91 is not an ideal gasoline surrogate since it has no octane sensitivity. In comparison,
TRF91-30 not only has RON of 91 but also has 30% toluene by volume which is similar to Australian
gasoline. Besides, TRF91-30 is a sensitive fuel, making it a good emulation for the gasoline. The
measured profiles of CO and the parent fuels from the TRF91-30 oxidation at 900 K and 10 bar, to-
gether with the modelling results are shown in Fig.6.17. Although the TestMech performs better than
the Mehl et al.’s model [67], both of them over-predict the mixture’s reactivity, which is most likely
due to the significantly over-predicted toluene reactivity. Besides, the predictions for the profiles of
isooctane and n-heptane are clearly worse compared with the results shown in Fig.6.15, indicating the
over-estimated toluene reactivity may affect the predictions of the other fuels. Alternatively, the poor
agreements could also come from the fuel interactions, given the TestMech is capable of capturing
the reactivity of each neat compound, as shown in section 6.3. To figure out how the fuel interac-
tions affect the reactivity of TRF91-30, the related elementary reactions (Reaction 5724 to 5935 from
the original gasoline surrogate model [67]) were removed and the resulting predictions are shown in
Fig.6.17 as well. The overall reactivity is slightly decreased without chemical interactions, suggesting
that these interactions may not be significant under the experimental conditions in this study. Note
that this is not saying the fuel interactions are not important. It is possible that the rate constants of
these elementary reactions cannot accurately capture the chemical interactions in this study and thus
require improvements.
The major intermediate species from TRF91-30 and the modelling results are shown in Fig.6.18.
With the presence of toluene, the overall agreements from both mechanisms are getting worse, espe-
cially when compared with the results shown in Fig.6.16. With 30% toluene added, the predictions of
the major products from the isooctane oxidation, such as isobutylene and acetone, reach peak values
earlier than the measurements, which is consistent with the over-predicted CO profile.
From the PFR experiments of the gasoline surrogates, both Mehl et al.’s mechanism [67] and the
TestMech well reproduce the species profiles from PRF91 but significantly over-predicted the reactiv-
ity of TRF91-30 due to the existence of toluene. This finding is consistent with the results from the
binary mixtures. The next step of this study is to explore the behaviours of fuel mixtures containing
gasoline surrogates and ethanol.
105
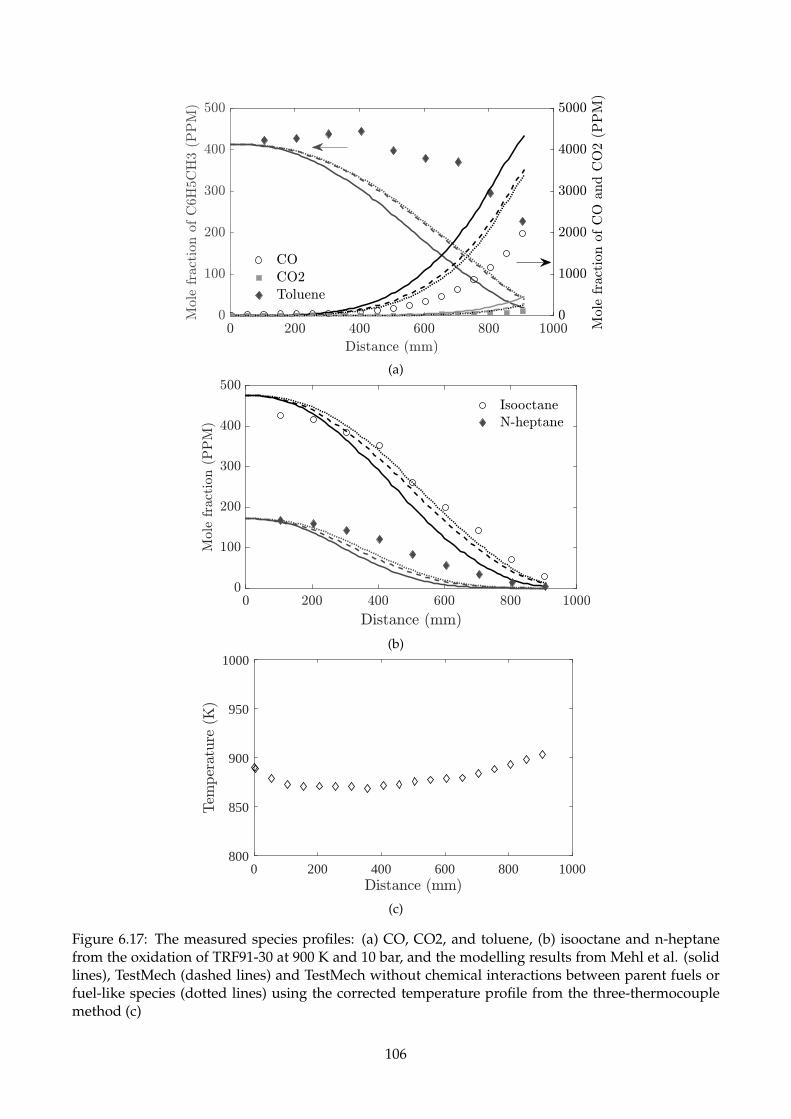
(a)
(b)
0 200 400 600 800 1000800
850
900
950
1000
(c)
Figure 6.17: The measured species profiles: (a) CO, CO2, and toluene, (b) isooctane and n-heptanefrom the oxidation of TRF91-30 at 900 K and 10 bar, and the modelling results from Mehl et al. (solidlines), TestMech (dashed lines) and TestMech without chemical interactions between parent fuels orfuel-like species (dotted lines) using the corrected temperature profile from the three-thermocouplemethod (c)
106

020
040
060
080
010
000102030405060
(a)
020
040
060
080
010
00020406080100
120
(b)
020
040
060
080
010
00020406080100
(c)
020
040
060
080
010
00050100
150
200
(d)
020
040
060
080
010
00050100
150
200
(e)
020
040
060
080
010
0001020304050
(f)
020
040
060
080
010
00024681012
(g)
020
040
060
080
010
0002468101214
(h)
Figu
re6.
18:T
hem
easu
red
inte
rmed
iate
spec
ies
profi
les
from
the
TRF9
1ox
idat
ion
expe
rim
enta
t900
Kan
d10
bar,
and
the
mod
ellin
gre
sult
sfr
omM
ehle
tal.
(sol
idlin
es)a
ndTe
stM
ech(
dash
edlin
es)
107

6.7 Gasoline surrogates/ethanol mixtures
6.7.1 PRF91 and ethanol
Ethanol was first blended into the simplest gasoline surrogate PRF91 with a mole fraction of 73.7%
(50% by volume). The fuel mixture was tested in the PFR at 900 K and 10 bar. The species profiles of
CO and the parent fuels from the oxidation of PRF91 blended with 73.7% ethanol by mole (50% by
volume), together with the modelling results using Mehl et al.’s mechanism [67] and the TestMech are
shown in Fig.6.19. Without the presence of toluene, both mechanisms well predict the profiles of CO
and three parent fuels and the TestMech performs exactly the same as Mehl et al.’s mechanism [67].
More importantly, the good agreements suggest that the interactions among isooctane, n-heptane, and
ethanol are well modelled, although the predicted ethanol profile has an earlier plateau compared
with the measurement. The mismatched plateau is less likely due to the fuel interactions since the
modelling of neat ethanol has the same issue, as shown in Fig.6.5.
With 73.7% (50% by volume) ethanol added, acetaldehyde becomes the most abundant interme-
diate species whose profile is well captured by both Mehl et al.’s mechanism [67] and the TestMech,
as shown in Fig.6.20. Note that acetaldehyde is the common product from both isooctane and ethanol
oxidations. The gasoline surrogate mechanism [67] well captures the acetaldehyde profile from the
ethanol oxidation, as shown in Fig.6.6(c), but has difficulty to match the peak value of acetaldehyde
profile from the isooctane oxidation which is shown in Fig.6.2(e). Since ethanol is much more abun-
dant than isooctane in the mixture, the formation of acetaldehyde mainly comes from the ethanol
oxidation, which is known to be well predicted by the mechanism [67]. For the same reason, most
of ethylene comes from ethanol as well and the over-predicted ethylene profile is consistent with
the result shown in Fig.6.6(b). Apart from acetaldehyde, the other two oxygenated compounds from
isooctane oxidation are under-predicted, which were also observed in the neat isooctane experiment.
Other than predicting these two oxygenates and ethylene, the existing mechanisms perform overall
satisfactorily.
108

(a)
(b)
0 200 400 600 800 1000800
850
900
950
1000
(c)
Figure 6.19: The measured species profiles: (a) CO, CO2 and ethanol, (b) isooctane and n-heptanefrom the oxidation of PRF91 blended with 73.7% ethanol by mole (50% by volume) at 900 K and 10bar, and the modelling results from Mehl et al. (solid lines) and TestMech (dashed lines) using thecorrected temperature profile from the three-thermocouple method (c)
109
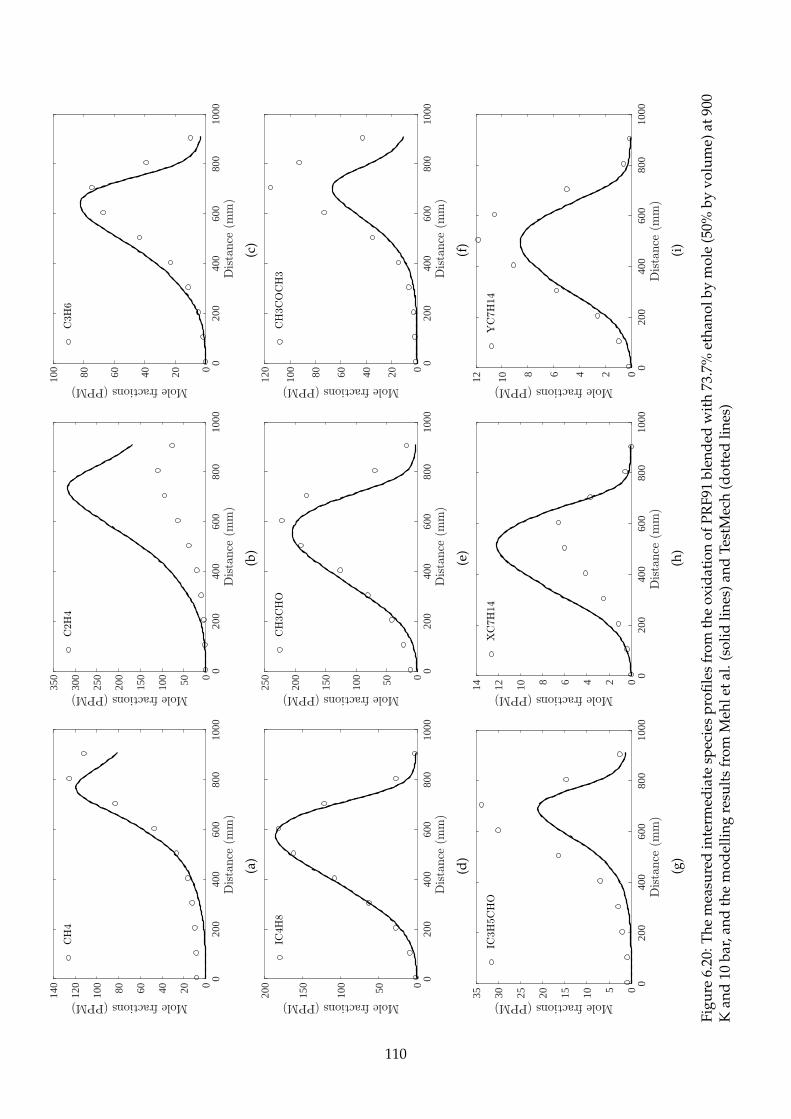
020
040
060
080
010
00020406080100
120
140
(a)
020
040
060
080
010
00050100
150
200
250
300
350
(b)
020
040
060
080
010
00020406080100
(c)
020
040
060
080
010
00050100
150
200
(d)
020
040
060
080
010
00050100
150
200
250
(e)
020
040
060
080
010
00020406080100
120
(f)
020
040
060
080
010
0005101520253035
(g)
020
040
060
080
010
0002468101214
(h)
020
040
060
080
010
00024681012
(i)
Figu
re6.
20:T
hem
easu
red
inte
rmed
iate
spec
ies
profi
les
from
the
oxid
atio
nof
PRF9
1bl
ende
dw
ith
73.7
%et
hano
lby
mol
e(5
0%by
volu
me)
at90
0K
and
10ba
r,an
dth
em
odel
ling
resu
lts
from
Meh
leta
l.(s
olid
lines
)and
Test
Mec
h(d
otte
dlin
es)
110

6.7.2 TRF91-30 and ethanol
To study the ethanol’s effects on the reactivity of TRF91-30, different amounts of ethanol were added
into the gasoline surrogates, and these mixtures were tested in PFR at 900 K and 10 bar. The same
chemical mechanisms were applied for the kinetic modellings.
The species profiles of CO and the parent fuels from the oxidation of TRF91-30 blended with 87.7%
ethanol by mole (75% by volume), together with the modelling results are shown in Fig.6.21. The
plateau from the measured CO profile indicates that the overall reactivity has been significantly im-
proved by the ethanol addition. With such a large ethanol content, the toluene sub-models in both
chemical mechanisms play a much less significant role, as the mole fraction of toluene in the fuel
mixture is less than 5%. Of interest is that the modelled isooctane and n-heptane profiles agree much
better with the measurements than in the neat TRF91 case, as shown in Fig.6.17.
Consistent with the reactivity measurements, the addition of 87.7% ethanol makes the mixture be-
have more like ethanol, which is embodied by the over-predicted ethylene profiles from both Mehl et
al.’s model [67] and the TestMech, as shown in Fig.6.22. Meanwhile, the profiles of the oxygenated
compounds are still under-estimated. Note that the absolute amounts of the C7 olefins become negli-
gible due to the small content of isooctane in the mixture, and thus these species are not included in
Fig.6.22.
With the addition of 70.5% ethanol by mole (50% by volume), the overall performances of the
gasoline surrogate mechanisms become worse, as shown in Fig.6.23. Compared with the predictions
of isooctane and n-heptane profiles, the consumption rates of toluene and ethanol are significantly
over-estimated, resulting in an over-predicted CO profile. The profiles of intermediate species and
the modelling results of the TRF mixture with 70.5% ethanol by mole (50% by volume) are shown
in Fig.6.24. The overall agreements are consistent with the results shown in Fig.6.22, suggesting that
ethanol still plays a significant role during the oxidation process. With the decreased ethanol content,
the fraction of isooctane increases accordingly, which produces detectable amounts of C7 olefins.
When the ethanol concentration drops to 44.3% by mole (25% by volume), the predictions become
the worst among all ethanol-containing TRF91-30 mixtures. The consumption rates of all parent fuels
are over-estimated, as shown in Fig.6.25, which are probably due to the problematic toluene sub-
mechanism. The intermediates’ profiles and the modelling results are shown in Fig.6.26. The predicted
profiles reach peaks earlier than the measurements, which is similar to the results shown in Fig.6.18.
It is probably because both cases have significant amounts of toluene in the fuel mixtures, and the
problematic toluene sub-mechanisms over-predict the reactivity, making the modelled profiles reach
peaks values earlier.
In summary, the gasoline surrogate mechanism from Mehl et al. [67] performs satisfactorily in
111

(a)
(b)
0 200 400 600 800 1000800
850
900
950
1000
(c)
Figure 6.21: The measured species profiles: (a) CO, isooctane, and n-heptane, (b) CO2, toluene, andethanol from the oxidation of TRF91-30 blended with 87.7% ethanol by mole (75% by volume) at 900 Kand 10 bar, and the modelling results from Mehl et al. (solid lines) and TestMech (dashed lines) usingthe corrected temperature profile from the three-thermocouple method (c)
112

020
040
060
080
010
00050100
150
(a)
020
040
060
080
010
000
100
200
300
400
500
600
(b)
020
040
060
080
010
00010203040
(c)
020
040
060
080
010
00010203040506070
(d)
020
040
060
080
010
000
100
200
300
400
(e)
020
040
060
080
010
00020406080100
(f)
020
040
060
080
010
0002468101214
(g)
Figu
re6.
22:T
hem
easu
red
inte
rmed
iate
spec
ies
profi
les
the
oxid
atio
nof
TRF9
1-30
blen
ded
wit
h87
.7%
etha
nolb
ym
ole
(75%
byvo
lum
e)at
900
Kan
d10
bar,
and
the
mod
ellin
gre
sult
sfr
omM
ehle
tal.
(sol
idlin
es)a
ndTe
stM
ech
(dot
ted
lines
)
113

(a)
(b)
0 200 400 600 800 1000800
850
900
950
1000
(c)
Figure 6.23: The measured species profiles: (a) CO, isooctane, and n-heptane, (b) CO2, toluene, andethanol from the oxidation of TRF91-30 blended with 70.5% ethanol by mole (50% by volume) at 900 Kand 10 bar, and the modelling results from Mehl et al. (solid lines) and TestMech (dashed lines) usingthe corrected temperature profile from the three-thermocouple method (c)
114
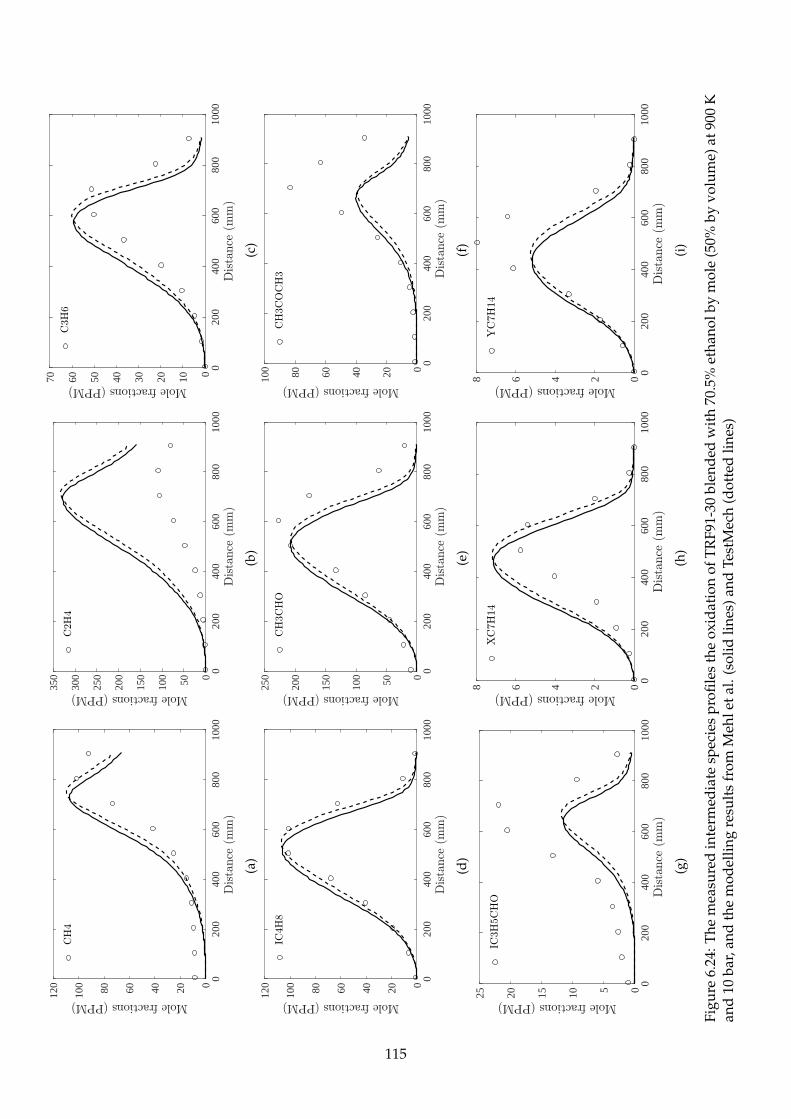
020
040
060
080
010
00020406080100
120
(a)
020
040
060
080
010
00050100
150
200
250
300
350
(b)
020
040
060
080
010
00010203040506070
(c)
020
040
060
080
010
00020406080100
120
(d)
020
040
060
080
010
00050100
150
200
250
(e)
020
040
060
080
010
00020406080100
(f)
020
040
060
080
010
000510152025
(g)
020
040
060
080
010
0002468
(h)
020
040
060
080
010
0002468
(i)
Figu
re6.
24:T
hem
easu
red
inte
rmed
iate
spec
ies
profi
les
the
oxid
atio
nof
TRF9
1-30
blen
ded
wit
h70
.5%
etha
nolb
ym
ole
(50%
byvo
lum
e)at
900
Kan
d10
bar,
and
the
mod
ellin
gre
sult
sfr
omM
ehle
tal.
(sol
idlin
es)a
ndTe
stM
ech
(dot
ted
lines
)
115
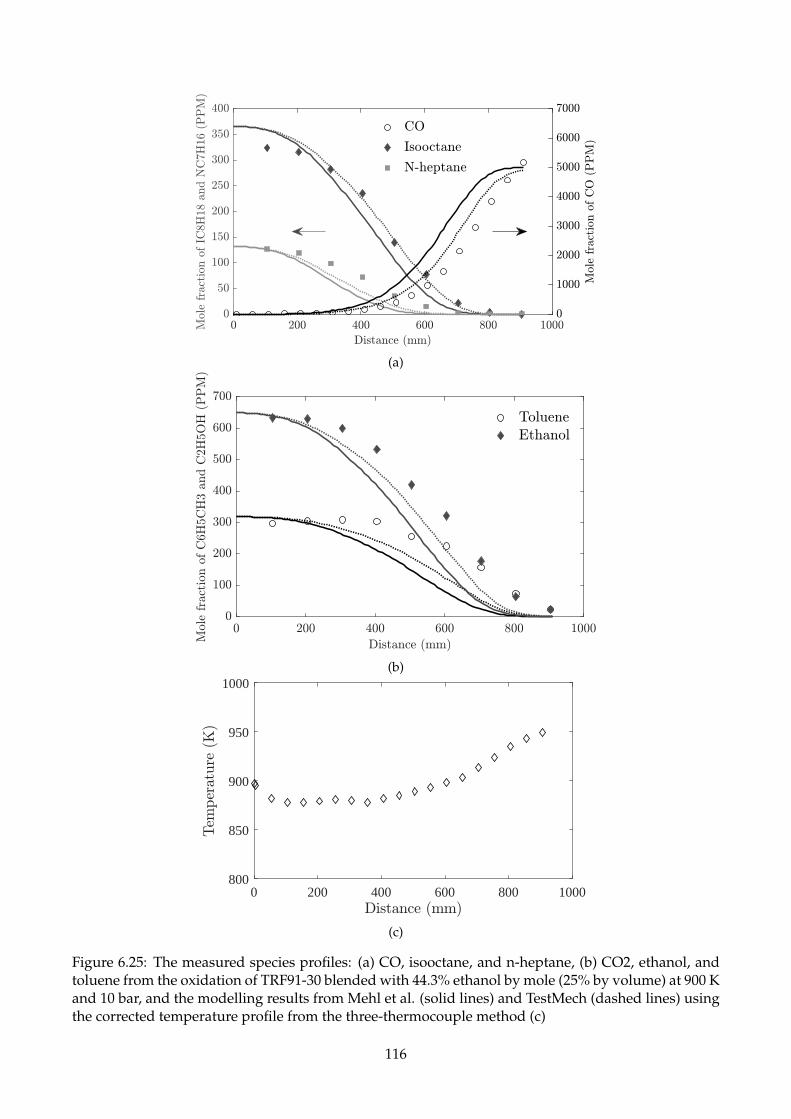
(a)
(b)
0 200 400 600 800 1000800
850
900
950
1000
(c)
Figure 6.25: The measured species profiles: (a) CO, isooctane, and n-heptane, (b) CO2, ethanol, andtoluene from the oxidation of TRF91-30 blended with 44.3% ethanol by mole (25% by volume) at 900 Kand 10 bar, and the modelling results from Mehl et al. (solid lines) and TestMech (dashed lines) usingthe corrected temperature profile from the three-thermocouple method (c)
116
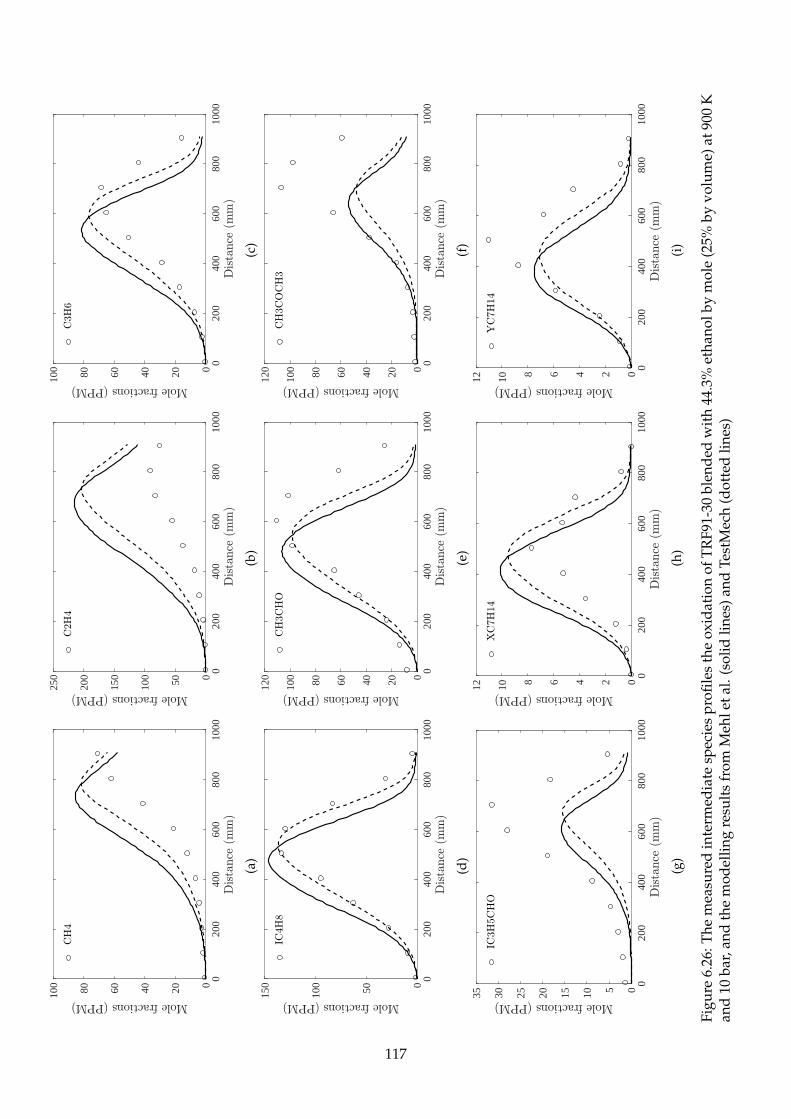
020
040
060
080
010
00020406080100
(a)
020
040
060
080
010
00050100
150
200
250
(b)
020
040
060
080
010
00020406080100
(c)
020
040
060
080
010
00050100
150
(d)
020
040
060
080
010
00020406080100
120
(e)
020
040
060
080
010
00020406080100
120
(f)
020
040
060
080
010
0005101520253035
(g)
020
040
060
080
010
00024681012
(h)
020
040
060
080
010
00024681012
(i)
Figu
re6.
26:T
hem
easu
red
inte
rmed
iate
spec
ies
profi
les
the
oxid
atio
nof
TRF9
1-30
blen
ded
wit
h44
.3%
etha
nolb
ym
ole
(25%
byvo
lum
e)at
900
Kan
d10
bar,
and
the
mod
ellin
gre
sult
sfr
omM
ehle
tal.
(sol
idlin
es)a
ndTe
stM
ech
(dot
ted
lines
)
117
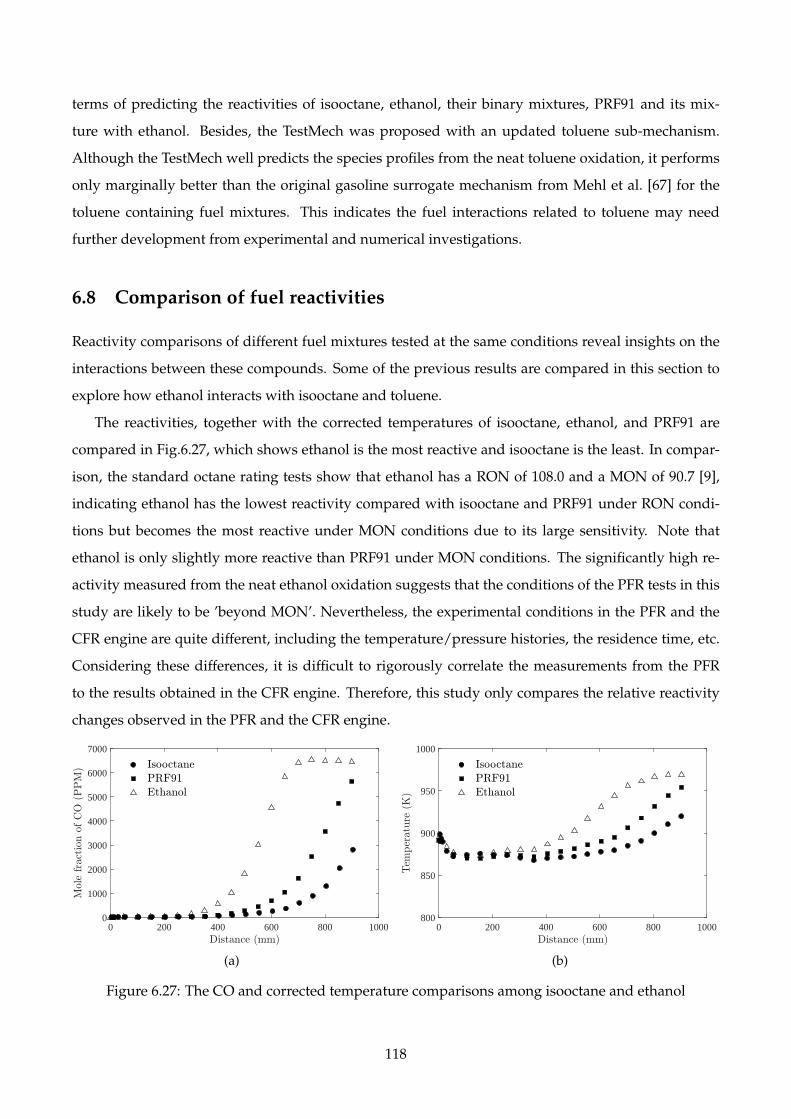
terms of predicting the reactivities of isooctane, ethanol, their binary mixtures, PRF91 and its mix-
ture with ethanol. Besides, the TestMech was proposed with an updated toluene sub-mechanism.
Although the TestMech well predicts the species profiles from the neat toluene oxidation, it performs
only marginally better than the original gasoline surrogate mechanism from Mehl et al. [67] for the
toluene containing fuel mixtures. This indicates the fuel interactions related to toluene may need
further development from experimental and numerical investigations.
6.8 Comparison of fuel reactivities
Reactivity comparisons of different fuel mixtures tested at the same conditions reveal insights on the
interactions between these compounds. Some of the previous results are compared in this section to
explore how ethanol interacts with isooctane and toluene.
The reactivities, together with the corrected temperatures of isooctane, ethanol, and PRF91 are
compared in Fig.6.27, which shows ethanol is the most reactive and isooctane is the least. In compar-
ison, the standard octane rating tests show that ethanol has a RON of 108.0 and a MON of 90.7 [9],
indicating ethanol has the lowest reactivity compared with isooctane and PRF91 under RON condi-
tions but becomes the most reactive under MON conditions due to its large sensitivity. Note that
ethanol is only slightly more reactive than PRF91 under MON conditions. The significantly high re-
activity measured from the neat ethanol oxidation suggests that the conditions of the PFR tests in this
study are likely to be ’beyond MON’. Nevertheless, the experimental conditions in the PFR and the
CFR engine are quite different, including the temperature/pressure histories, the residence time, etc.
Considering these differences, it is difficult to rigorously correlate the measurements from the PFR
to the results obtained in the CFR engine. Therefore, this study only compares the relative reactivity
changes observed in the PFR and the CFR engine.
0 200 400 600 800 10000
1000
2000
3000
4000
5000
6000
7000
(a)
0 200 400 600 800 1000800
850
900
950
1000
(b)
Figure 6.27: The CO and corrected temperature comparisons among isooctane and ethanol
118

The reactivity comparisons between ethanol/isooctane and ethanol/toluene are shown in Fig.6.28.
Although the measured CO profiles from the PFR are not directly comparable with the octane numbers
from the CFR engine, they do reflect the reactivities of different fuels and their mixtures. As shown in
Fig.6.28(a), isooctane is much more reactive than toluene which barely reacts at 900 K. With ethanol
added, the reactivity difference between these two fuels are diminishing, as shown in Fig.6.28(c) and
(e). When the ethanol concentration reaches approximately 75%, the reactivity of ethanol and isooc-
tane mixture is only slightly higher than that of the mixture containing ethanol and toluene, suggesting
that the impact of ethanol is more significant on toluene than on isooctane in terms of improving the
mixture’s reactivity, especially when considering isooctane is much more reactive than toluene.
Similar impact was also found with gasoline surrogates, as shown in Fig.6.29. Although both
PRF91 and TRF91-30 have RON of 91, PRF91 has a higher CO concentration compared with TRF91-30
in the PFR experiment. After adding ethanol with a volume fraction of 50% (73.7% by mole in PRF91
and 70.5% by mole in TRF91-30) to these two gasoline surrogates, their reactivities become very similar
and the reactivity increment of PRF91 is not significant, indicating that the interaction between ethanol
and isooctane (the major component in PRF91) does not contribute much to improve the mixture’s
reactivity, but the interaction between ethanol and toluene does. The results from both Fig.6.28 and
6.29 are consistent with the conclusion from the prior study [9] that ethanol blends synergistically
with isooctane but antagonistically with toluene in terms of the octane rating, as shown in Fig.2.5(a).
Although it is not intended to use the PFR results to explain the octane number blending behaviours
reported in [9] due to the different conditions in the two experiments, it is interesting to find the
relative reactivity changes in the PFR and the CFR engine are consistent, suggesting the combustion
chemistry behind these experiments may be similar.
The motivation of the reactivity comparisons for different fuels and fuel mixtures is trying to fig-
ure out how hydrocarbons interact with ethanol under simple and well-controlled thermodynamic
conditions in the PFR.
119
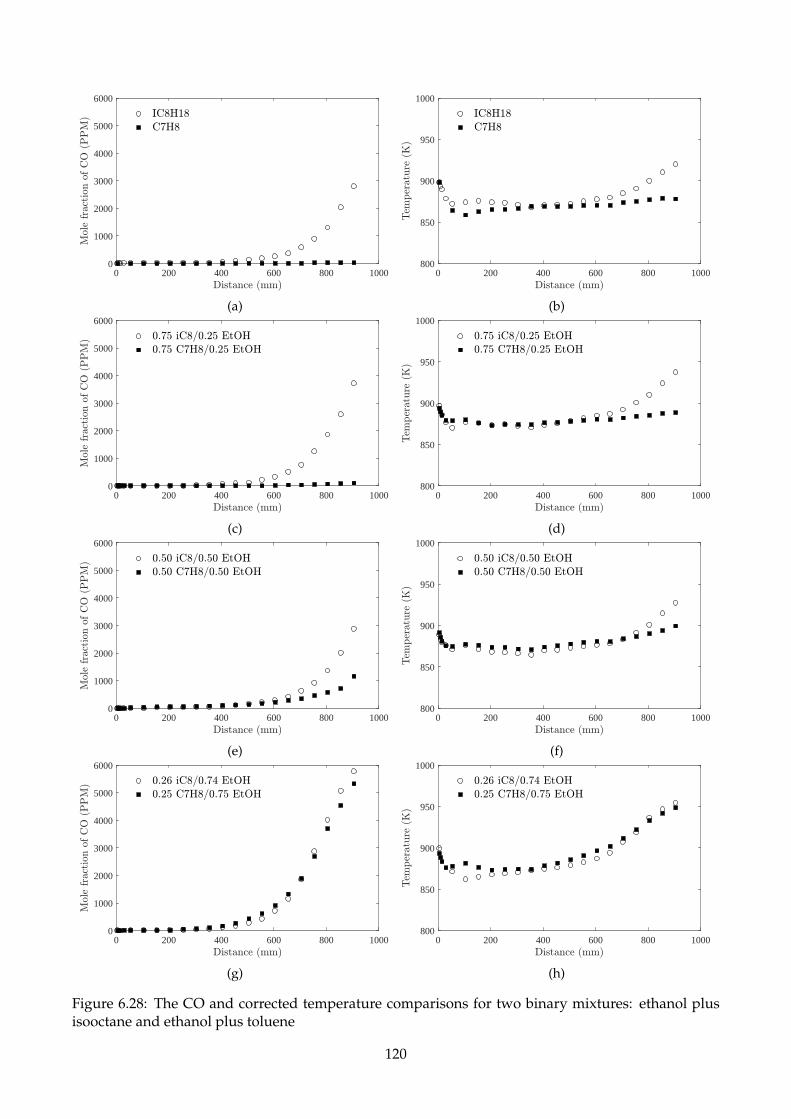
0 200 400 600 800 10000
1000
2000
3000
4000
5000
6000
(a)
0 200 400 600 800 1000800
850
900
950
1000
(b)
0 200 400 600 800 10000
1000
2000
3000
4000
5000
6000
(c)
0 200 400 600 800 1000800
850
900
950
1000
(d)
0 200 400 600 800 10000
1000
2000
3000
4000
5000
6000
(e)
0 200 400 600 800 1000800
850
900
950
1000
(f)
0 200 400 600 800 10000
1000
2000
3000
4000
5000
6000
(g)
0 200 400 600 800 1000800
850
900
950
1000
(h)
Figure 6.28: The CO and corrected temperature comparisons for two binary mixtures: ethanol plusisooctane and ethanol plus toluene
120
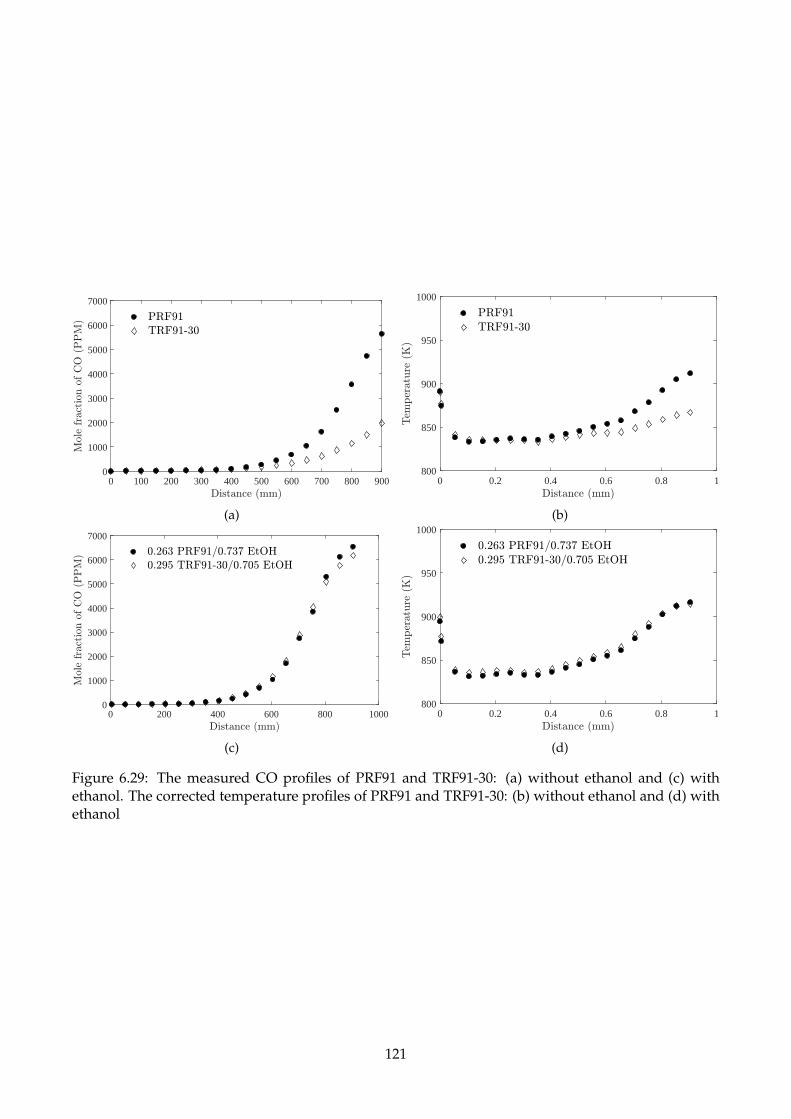
0 100 200 300 400 500 600 700 800 9000
1000
2000
3000
4000
5000
6000
7000
(a)
0 0.2 0.4 0.6 0.8 1800
850
900
950
1000
(b)
0 200 400 600 800 10000
1000
2000
3000
4000
5000
6000
7000
(c)
0 0.2 0.4 0.6 0.8 1800
850
900
950
1000
(d)
Figure 6.29: The measured CO profiles of PRF91 and TRF91-30: (a) without ethanol and (c) withethanol. The corrected temperature profiles of PRF91 and TRF91-30: (b) without ethanol and (d) withethanol
121

6.9 Summary
The PFR experiments for isooctane, ethanol, toluene, and their mixtures were performed at 900 K
(except toluene at 930 K) and 10 bar. CO, the parent fuels, and the intermediate species were measured
along the reactor length and modelled with the state of the art chemical mechanisms.
First, these mechanisms, in general, well reproduce the reactivities of isooctane and ethanol (except
CO plateau), their binary mixtures, PRF and PRF/ethanol mixture. Overall, the profiles of the major
intermediate species are well matched, but some improvements are needed for certain intermediate
species, e.g., oxygenates for isooctane and ethylene for ethanol. However, the existing chemical mech-
anisms become problematic when applied to model the neat toluene oxidation. Except the Princeton’s
mechanism [138], the other mechanisms significantly over-estimate the reactivity of toluene. As for
the toluene containing fuel mixtures, the problematic toluene sub-model in the gasoline surrogate
mechanism [67] results in larger discrepancies with a higher concentration of toluene in the mixture.
Second, the TestMech with an updated toluene sub-model was proposed to have close agreements
with the species profiles from the neat toluene experiment. However, the TestMech only performs
marginally better than the gasoline surrogate mechanism [67] for fuel mixtures, suggesting the inter-
action chemistry needs further development.
Lastly, the reactivity comparisons show that the impact of ethanol is more evident on toluene than
on isooctane in terms of improving the mixture’s reactivity, which is consistent with the conclusions
from the prior octane number study [9] despite the major differences between the two experiments.
122

Chapter 7
Conclusions and Recommendations for
Future Research
7.1 Conclusions
This thesis studied the octane blending and oxidation chemistry of ethanol-hydrocarbon mixtures. It
makes three contributions to our understanding of this problem.
1. This study quantifies the non-linear octane blending between ethanol and the surrogate fuels.
A systematic method for quantifying non-linear octane blending behaviours observed in fuel
mixtures was proposed. The method made use of Scheffe polynomials, which provide a com-
position based model for mixture properties, including octane numbers. It combined linear
regression and exhaustive (or brute-force) searching for polynomials that met specified require-
ments with the fewest terms. The following optimal correlations for the RON and MON of
TRF/ethanol mixtures were found, which achieve maximum absolute error of less than two
octane numbers across TRF/ethanol mixtures with a RON between 80 and 120.
RON = 100x1 + 0x2 + 116.2x3 + 108x4 + 27.0x1x4 − 98.4x2x4(x2 − x4)− 9.1x3x4MON = 100x1 + 0x2 + 102.0x3 + 90.7x4 + 12.8x1x4 + 76.7x2x4 − 6.4x3x4
where x1, x2, x3 and x4 denote the mole fractions of isooctane, n-heptane, toluene and ethanol
respectively. These correlations were obtained from a systematic method and reveal some signif-
icant results. For example, the significance of the linear by mole blending rule for TRF mixtures
is reconfirmed by correlations, since the non-linear terms are all binary and all contain ethanol
(x4). Furthermore, the coefficients of these non-linear terms correspond to the levels of synergis-
tic or antagonistic blending of different binary mixtures, as previously reported in the literature.
123

2. This work develops a gasoline surrogate that more accurately emulates the octane blending be-
haviours of production gasoline with ethanol.
To emulate the knocking behaviours of gasoline/ethanol mixtures, this work first measured the
RONs of binary mixtures to gain fundamental understanding for the interactions between dif-
ferent hydrocarbons and between hydrocarbon and ethanol. It is found that most hydrocarbons
selected blend synergistically with ethanol, except aromatic fuels showing antagonistic trend.
Based on the results of the detailed hydrocarbon analysis of an Australian market gasoline, cy-
clohexane and 1-hexene, each at 10% by volume, were used as the representative compounds
for cycloparaffins and olefins. Further, 1,2,4-trimethylbenzene with a 30% volume fraction was
used to represent aromatics for its strongest antagonism when blended with ethanol, which is re-
quired to compensate the synergism exhibited by ethanol blending with other hydrocarbons. Fi-
nally, iso-pentane and n-pentane were selected as the representative compounds of paraffins due
to their significant concentrations in the production gasoline, and their relative volume fractions
were the balancing factor to make the mixture have a RON of 91. It is found that the proposed
gasoline surrogate closely reproduced the octane blending behaviours with ethanol over the en-
tire blending range, with a maximum ON difference of 0.7 unit. Compared with commonly used
TRF mixtures, this surrogate formulation indicates that including additional components from
other hydrocarbon classes and using different paraffin and aromatic compounds than those in
TRFs are necessary to emulate the interaction between gasoline and ethanol. The developed
gasoline surrogate is the first of its kind in attempt to emulate the octane blending behaviour of
the gasoline and ethanol. In addition to capture the blending behaviours, the compositions of
the gasoline surrogate match with the hydrocarbon class distributions in the gasoline.
3. This work studied ethanol interaction with different hydrocarbons in the pressurised flow re-
actor, finding that whilst current mechanisms often captured the measured trends, those for
toluene and toluene-rich mixtures are lacking.
A systematic PFR experimental study was carried out at 900 K (except toluene at 930 K) and
10 bar to study oxidation chemistry of a comprehensive fuel matrix including neat fuels, bi-
nary mixtures, gasoline surrogates, and gasoline surrogates/ethanol mixtures. GC with an FID
analyser was applied for species measurements. The measurements are consistent with those
reported previously from jet-stirred reactors and rapid compression machines, and other flow
reactors on the relevant fuels.
The measured detailed species profiles are used to systematically validate the existing chemi-
cal mechanisms. The gasoline surrogate mechanism from Mehl et al. [67] reproduces most of
the species profiles from the oxidations of ethanol, isooctane, their binary mixtures, PRF, and
124

PRF/ethanol mixtures. Large discrepancies were observed between the measured profiles and
the modelled results for neat toluene and toluene-containing fuel mixtures. Revising some mis-
used elementary reactions rate constants (TestMech) improved the modelling of the neat toluene
but was still unable to capture the behaviours of the mixtures, indicating that the toluene mech-
anism, and probably its interactions with other compounds need major improvements, which
are required to develop a valid gasoline surrogate mechanism.
Under the experimental conditions in this study, it is found that ethanol promotes toluene oxi-
dation more than it does to isooctane, which is consistent with the results from the prior octane
blending behaviours in the CFR engine [9].
7.2 Recommendations for future research
1. Experimental studies for toluene oxidation under various conditions and development of a de-
tailed toluene mechanism
This study shows that the existing toluene sub-model significantly over-estimates the reactiv-
ity of toluene and toluene-containing fuel mixtures, which indicates that the understanding of
the toluene oxidation is not complete. Therefore, more kinetic experiments are requires to in-
vestigate the oxidation of toluene at different temperatures, pressures, and equivalence ratios.
With more experimental results, a new detailed toluene mechanism can be proposed, which is
required for further gasoline surrogate mechanism development.
2. Experimental studies for binary mixtures to understand fuel interactions
It has been shown in this work that the TestMech can reproduce the species profiles from neat
toluene but didn’t perform well for toluene containing binary mixtures, indicating the chemical
interactions related to toluene need improvement. Besides, it is also found ethanol promotes
toluene oxidation more than does to isooctane, but the combustion chemistry behind this ob-
servation is not clear. Hence, more kinetic experiments are required to understand the fuel
interactions of these binary mixtures, which is essential to develop a detailed gasoline surrogate
mechanism.
3. Experimental studies for neat fuels and their mixtures at lower temperatures
The reactivity comparison between ethanol and PRF91 suggests that the experimental condi-
tion in this study is ’beyond MON’. To have comprehensive understandings of the combustion
chemistry, it is necessary to carry out these experiments at lower temperatures where the low
temperature chemistry is more significant.
125

4. Experimental studies for the oxidations of isooctane and ethanol
Although the existing chemical mechanisms capture most of the species profiles from isooctane
and ethanol oxidation, the oxygenated compounds from isooctane and ethylene from ethanol
are not well reproduced. Also, the CO plateau issue near the end of the reactor at high reactivity
conditions should be resolved. Thus, it is necessary to carry out more kinetic experiments for
isooctane and ethanol under different conditions in the PFR to improve the existing mechanisms.
126

References
[1] BP Global. BP Energy Outlook, 2017 edition, 2017.
[2] Jose Ramon Serrano. Imagining the Future of the Internal Combustion Engine for Ground Trans-
port in the Current Context. Applied Sciences, 7:1001, 2017.
[3] Mazda announces breakthrough in long-coveted engine technology. Yahoo finance, 2017.
[4] J. Hansen, D. Johnson, A. Lacis, S. Lebedeff, P. Lee, D. Rind, and G. Russell. Climate Impact of
Increasing Atmospheric Carbon Dioxide. Science, 213(4511):957–966, 1981.
[5] BP Global. BP Statistical Review of World Energy, 2017.
[6] Alexander E. Farrell, Richard J. Plevin, Brian T. Turner, Andrew D. Jones, Michael O’Hare, and
Daniel M. Kammen. Ethanol Can Contribute to Energy and Environmental Goals. Science,
311(5760):506–508, 2006.
[7] Jose Goldemberg. Ethanol for a Sustainable Energy Future. Science, 315(5813):808–810, 2007.
[8] Avinash Kumar Agarwal. Biofuels (alcohols and biodiesel) applications as fuels for internal
combustion engines. Progress in Energy and Combustion Science, 33(3):233–271, 2007.
[9] Tien Mun Foong, Kai J. Morganti, Michael J. Brear, Gabriel da Silva, Yi Yang, and Frederick L.
Dryer. The octane numbers of ethanol blended with gasoline and its surrogates. Fuel, 115:727–
739, 2014.
[10] J. E. Anderson, U. Kramer, S. A. Mueller, and T. J. Wallington. Octane numbers of ethanol- and
methanol- gasoline blends estimated from molar concentrations. Energy Fuels, 24(12):6576–6585,
2010-12-16.
[11] Robert A. Stein, Dusan Polovina, Kevin Roth, Michael Foster, Michael Lynskey, Todd Whiting,
James E. Anderson, Michael H. Shelby, Thomas G. Leone, and Steven VanderGriend. Effect of
Heat of Vaporization, Chemical Octane, and Sensitivity on Knock Limit for Ethanol - Gasoline
Blends. SAE Int. J. Fuels Lubr., 5(2):823–843, 2012.
127

[12] Hao Yuan, Tien Mun Foong, Zhongyuan Chen, Yi Yang, Michael Brear, Thomas Leone, and
James E. Anderson. Modeling of trace knock in a modern SI engine fuelled by ethanol/gasoline
blends. SAE Technical Paper, 2015-01-1242, 2015.
[13] Tien Mun Foong, Kai J. Morganti, Michael J. Brear, Gabriel da Silva, Yi Yang, and Frederick L.
Dryer. The effect of charge cooling on the RON of ethanol/gasoline blends. SAE Int. J. Fuels
Lubr., 6(1):31–43, 2013.
[14] Emmanuel Kasseris and John Heywood. Charge cooling effects on knock limits in SI DI engines
using Gasoline/Ethanol blends: Part 1-quantifying charge cooling. SAE Technical Paper, 2012-
01-1275, 2012.
[15] Emmanuel Kasseris and John Heywood. Charge cooling effects on knock limits in SI DI engines
using Gasoline/Ethanol blends: Part 2-effective octane numbers. SAE Int. J. Fuels Lubr., 5(2):844–
854, 2012.
[16] Renewable Fuel Association. 2017 Ethanol Industry Outlook, 2017.
[17] Renewable Fuel Association. Ethanol Consumption Breaks Through the ”Blend Wall” in 2016,
2017.
[18] Livestock Ministry of Agriculture and Food Supply. This decree fixed the mandatory blend at
25% starting july 1st, 2007, 2007.
[19] Chinese Government. The Implementation of the Expansion of Ethanol Production and the
Promotion of Ethanol-added Gasoline, 2017.
[20] Jihad Badra, Abdullah S. AlRamadan, and S. Mani Sarathy. Optimization of the octane response
of gasoline/ethanol blends. Applied Energy, 203:778–793, 2017.
[21] John B. Heywood. Internal combustion engine fundamentals. McGraw-Hill, 1988.
[22] J. M. Towers and R. L. Hoekstra. Engine knock, a renewed concern in motorsports - a literature
review. SAE Technical Paper, 983026, 1998.
[23] Tien Mun Foong. On the Autoignition of Ethanol/Gasoline Blends in Spark-Ignition Engines. Ph.D.
thesis, The University of Melbourne, 2013.
[24] V. Arrigoni, G. M. Cornetti, Spallanzani G., Calvi F., and A. Tontodonati. High speed knock in
s.i. engines. SAE Technical Paper, 741056, 1974.
128

[25] ASTM International. Standard test method for research octane number of spark-ignition engine
fuel, 2011. ASTM D2699-11.
[26] ASTM International. Standard test method for motor octane number of spark-ignition engine
fuel, 2011. ASTM D2700-11.
[27] T. Midgley and T. Boyd. Methods of measuring detonation in engines. SAE Technical Paper,
220004, 1922.
[28] H.R. Ricardo. The influence of various fuels on the performance of internal combustion engines.
Report of the Empire Motor Fuels Committee, The Institution of Automobile Engineers vol XVIII Pt
I, 1924.
[29] Dugald Clerk. Investigations on gaseous explosions. part II. explosive reactions considered in
reference to internal combustion engines. introductory survey. Trans. Faraday Soc., 22(0):338–340,
1926.
[30] D. Downs, A. D. Walsh, and R. W. Wheeler. A study of the reactions that lead to ’Knock’ in the
spark-ignition engine. Phil. Trans. R. Soc. Lond. A, 243(870):463–524, 1951.
[31] Charles K. Westbrook, Yasuhiro Mizobuchi, Thierry J. Poinsot, Phillip J. Smith, and Jurgen War-
natz. Computational combustion. Proceedings of the Combustion Institute, 30(1):125–157, 2005.
[32] Nobuyuki Kawahara, Eiji Tomita, and Yoshitomo Sakata. Auto-ignited kernels during knocking
combustion in a spark-ignition engine. Proceedings of the Combustion Institute, 31(2):2999–3006,
2007.
[33] M. Poschl and T. Sattelmayer. Influence of temperature inhomogeneities on knocking combus-
tion. Combustion and Flame, 153(4):562–573, 2008.
[34] R. Worret, S. Bernhardt, F. Schwarz, and U. Spicher. Application of different cylinder pressure
based knock detection methods in spark ignition engines. SAE Technical Paper, 2002-01-1668,
2002.
[35] Enrico Corti and Claudio Forte. Statistical analysis of indicating parameters for knock detection
purposes. SAE Technical Paper, 2009-01-0237, 2009.
[36] F. Millo and C. V. Ferraro. Knock in S.I. engines: A comparison between different techniques for
detection and control. SAE Technical Paper, 982477, 1998.
[37] Kwang Min Chun and John B. Heywood. Characterization of knock in a spark-ignition engine.
SAE Technical Paper, 890156, 1989.
129

[38] Gao Xiaofeng, Richard Stone, Chris Hudson, and Ian Bradbury. The detection and quantification
of knock in spark ignition engines. SAE Technical Paper, 932759, 1993.
[39] Michael F. J. Brunt, Christopher R. Pond, and John Biundo. Gasoline engine knock analysis
using cylinder pressure data. SAE Technical Paper, 980896, 1998.
[40] M. D. Checkel and J. D. Dale. Computerized knock detection from engine pressure records. SAE
Technical Paper, 860028, 1986.
[41] Tassos H. Valtadoros, Victor W. Wong, and John B. Heywood. Engine knock characteristics at
the audible level. SAE Technical Paper, 910567, 1991.
[42] Kwang Min Chun and Kyung Woon Kim. Measurement and analysis of knock in a SI engine
using the cylinder pressure and block vibration signals. SAE Technical Paper, 940146, 1994.
[43] G. Edgar. Measurement of knock characteristics of gasoline in terms of a standard fuel. Ind.
Engg. Chem., pages 145–146, 1927.
[44] James E. Anderson, Thomas G. Leone, Michael H. Shelby, Timothy J. Wallington, Jeffrey J. Bizub,
Michael Foster, Michael G. Lynskey, and Dusan Polovina. Octane numbers of ethanol-gasoline
blends: Measurements and novel estimation method from molar composition. SAE Technical
Paper, 2012-01-1274, 2012.
[45] Paul Whitaker, Yuan Shen, Christian Spanner, Heribert Fuchs, Apoorv Agarwal, and Kevin
Byrd. Development of the combustion system for a flexible fuel turbocharged direct injection
engine. SAE Int. J. Engines, 3(1)(2010-01-0585):326–354, 2010.
[46] Tien Mun Foong, Michael J. Brear, Kai J. Morganti, Gabriel da Silva, Yi Yang, and Frederick L.
Dryer. Modeling End-Gas Autoignition of Ethanol/Gasoline Surrogate Blends in the Coopera-
tive Fuel Research Engine. Energy & Fuels, 31(3):2378–2389, 2017.
[47] Stephen Turns. An Introduction to Combustion: Concepts and Applications. McGraw-Hill Educa-
tion, 2011.
[48] Charles K. Westbrook and Frederick L. Dryer. Chemical kinetic modeling of hydrocarbon com-
bustion. Progress in Energy and Combustion Science, 10(1):1–57, 1984.
[49] Charles K. Westbrook. Chemical kinetics of hydrocarbon ignition in practical combustion sys-
tems. Proceedings of the Combustion Institute, 28(2):1563–1577, 2000.
[50] John M. Simmie. Detailed chemical kinetic models for the combustion of hydrocarbon fuels.
Progress in Energy and Combustion Science, 29(6):599–634, 2003.
130

[51] F. Battin-Leclerc. Detailed chemical kinetic models for the low-temperature combustion of hy-
drocarbons with application to gasoline and diesel fuel surrogates. Progress in Energy and Com-
bustion Science, 34(4):440–498, August 2008.
[52] Judit Zador, Craig A. Taatjes, and Ravi X. Fernandes. Kinetics of elementary reactions in low-
temperature autoignition chemistry. Progress in Energy and Combustion Science, 37(4):371–421,
August 2011.
[53] Zhengkai Xi, Wha Jin Han, and Kyle D. Bayes. Temperature dependence of the rate constant for
the reaction of neopentyl radicals with oxygen. J. Phys. Chem., 92(12):3450–3453, 1988.
[54] E. W. Kaiser. Formation of C3H6 from the reaction C3H7 + o2 between 450 and 550 k. J. Phys.
Chem. A, 102(29):5903–5906, 1998.
[55] James A. Miller, Stephen J. Klippenstein, and Struan H. Robertson. A theoretical analysis of the
reaction between ethyl and molecular oxygen. Proceedings of the Combustion Institute, 28(2):1479–
1486, 2000.
[56] Edgar G. Estupinan, Stephen J. Klippenstein, and Craig A. Taatjes. Measurements and modeling
of HO2 formation in the reactions of n-C3H7 and i-C3H7 radicals with o2†. J. Phys. Chem. B,
109(17):8374–8387, 2005.
[57] H. J. Curran, P. Gaffuri, W. J. Pitz, and C. K. Westbrook. A comprehensive modeling study of
iso-octane oxidation. Combustion and Flame, 129(3):253–280, 2002.
[58] Philippe Dagaut, Marcelline Reuillon, and Michel Cathonnet. High Pressure Oxidation of Liq-
uid Fuels From Low to High Temperature. 1. n-Heptane and iso-Octane. Combustion Science and
Technology, 95(1-6):233–260, 1993.
[59] F. L. DRYER and K. BREZINSKY. A Flow Reactor Study of the Oxidation of n-Octane and Iso-
Octane. Combustion Science and Technology, 45(3-4):199–212, 1986.
[60] C. V. Callahan, T. J. Held, F. L. Dryer, R. Minetti, M. Ribaucour, L. R. Sochet, T. Faravelli,
P. Gaffuri, and E. Rani. Experimental data and kinetic modeling of primary reference fuel mix-
tures. Symposium (International) on Combustion, 26(1):739–746, 1996.
[61] J.-S. CHEN, T. A. LITZINGER, and H. J. CURRAN. The Lean Oxidation of Iso-Octane in the
Intermediate Temperature Regime at Elevated Pressures. Combustion Science and Technology,
156(1):49–79, 2000.
131

[62] D. J. Vermeer, J. W. Meyer, and A. K. Oppenheim. Auto-ignition of hydrocarbons behind re-
flected shock waves. Combustion and Flame, 18(3):327–336, 1972.
[63] K. Fieweger, R. Blumenthal, and G. Adomeit. Shock-tube investigations on the self-ignition
of hydrocarbon-air mixtures at high pressures. Symposium (International) on Combustion,
25(1):1579–1585, 1994.
[64] K. Fieweger, R. Blumenthal, and G. Adomeit. Self-ignition of S.I. engine model fuels: A shock
tube investigation at high pressure. Combustion and Flame, 109(4):599–619, 1997.
[65] Willaim R. Leppard. The autoignition chemistries of primary reference fuels, olefin/paraffin
binary mixtures, and non-linear octane blending. SAE Technical Paper, 922325, 1992.
[66] H. Li, S. Prabhu, D. Miller, and N. Cernansky. Autoignition chemistry studies on primary refer-
ence fuels in a motored engine. SAE Technical Paper, 942062, 1994.
[67] Marco Mehl, William J. Pitz, Charles K. Westbrook, and Henry J. Curran. Kinetic modeling of
gasoline surrogate components and mixtures under engine conditions. Proceedings of the Com-
bustion Institute, 33(1):193–200, 2011.
[68] Nour Atef, Goutham Kukkadapu, Samah Y. Mohamed, Mariam Al Rashidi, Colin Banyon,
Marco Mehl, Karl Alexander Heufer, Ehson F. Nasir, A. Alfazazi, Apurba K. Das, Charles K.
Westbrook, William J. Pitz, Tianfeng Lu, Aamir Farooq, Chih-Jen Sung, Henry J. Curran, and
S. Mani Sarathy. A comprehensive iso-octane combustion model with improved thermochem-
istry and chemical kinetics. Combustion and Flame, 178:111–134, April 2017.
[69] M. Hartmann, I. Gushterova, M. Fikri, C. Schulz, R. Schießl, and U. Maas. Auto-ignition of
toluene-doped n-heptane and iso-octane/air mixtures: High-pressure shock-tube experiments
and kinetics modeling. Combustion and Flame, 158(1):172–178, 2011.
[70] S. Mani Sarathy, Goutham Kukkadapu, Marco Mehl, Weijing Wang, Tamour Javed, Sungwoo
Park, Matthew A. Oehlschlaeger, Aamir Farooq, William J. Pitz, and Chih-Jen Sung. Ignition
of alkane-rich FACE gasoline fuels and their surrogate mixtures. Proceedings of the Combustion
Institute, 35(1):249–257, 2015.
[71] M. Mehl, W. Pitz, M. Sjoberg, , and J. Dec. Autoignition chemistry studies on primary reference
fuels in a motored engine. SAE Technical Paper, 2009-01-1806, 2009.
[72] Wontae Hwang, John Dec, and Magnus Sjoberg. Spectroscopic and chemical-kinetic analysis
of the phases of HCCI autoignition and combustion for single- and two-stage ignition fuels.
Combustion and Flame, 154(3):387–409, 2008.
132

[73] D. Vuilleumier, H. Selim, R. Dibble, and M. Sarathy. Exploration of heat release in a homoge-
neous charge compression ignition engine with primary reference fuels. SAE Technical Paper,
2013-01-2622, 2013.
[74] Suarwee Snitsiriwat and Joseph W. Bozzelli. Thermochemical Properties for Isooctane and Car-
bon Radicals: Computational Study. The Journal of Physical Chemistry A, 117(2):421–429, 2013.
[75] Itsaso Auzmendi-Murua and Joseph W. Bozzelli. Thermochemistry, Reaction Paths, and Ki-
netics on the Secondary Isooctane Radical Reaction with 3O2. International Journal of Chemical
Kinetics, 46(2):71–103, 2014.
[76] Suarwee Snitsiriwat and Joseph W. Bozzelli. Thermochemistry, Reaction Paths, and Kinetics on
the tert-Isooctane Radical Reaction with O2. The Journal of Physical Chemistry A, 118(26):4631–
4646, 2014.
[77] HongBo Ning, ChunMing Gong, ZeRong Li, and XiangYuan Li. Pressure-Dependent Kinetics of
Initial Reactions in Iso-octane Pyrolysis. The Journal of Physical Chemistry A, 119(18):4093–4107,
2015.
[78] Stephanie M. Villano, Lam K. Huynh, Hans-Heinrich Carstensen, and Anthony M. Dean. High-
Pressure Rate Rules for Alkyl + O2 Reactions. 1. The Dissociation, Concerted Elimination,
and Isomerization Channels of the Alkyl Peroxy Radical. The Journal of Physical Chemistry A,
115(46):13425–13442, 2011.
[79] Stephanie M. Villano, Lam K. Huynh, Hans-Heinrich Carstensen, and Anthony M. Dean. High-
Pressure Rate Rules for Alkyl + O2 Reactions. 2. The Isomerization, Cyclic Ether Formation,
and β-Scission Reactions of Hydroperoxy Alkyl Radicals. The Journal of Physical Chemistry A,
116(21):5068–5089, 2012.
[80] Akira Miyoshi. Systematic Computational Study on the Unimolecular Reactions of Alkylper-
oxy (RO2), Hydroperoxyalkyl (QOOH), and Hydroperoxyalkylperoxy (O2qooh) Radicals. The
Journal of Physical Chemistry A, 115(15):3301–3325, 2011.
[81] Sandeep Sharma, Sumathy Raman, and William H. Green. Intramolecular Hydrogen Migration
in Alkylperoxy and Hydroperoxyalkylperoxy Radicals: Accurate Treatment of Hindered Rotors.
The Journal of Physical Chemistry A, 114(18):5689–5701, 2010.
[82] C. M. Coats and Alan Williams. Investigation of the ignition and combustion of n-heptane-
oxygen mixtures. Symposium (International) on Combustion, 17(1):611–621, 1979.
133

[83] H. K. Ciezki and G. Adomeit. Shock-tube investigation of self-ignition of n-heptane-air mixtures
under engine relevant conditions. Combustion and Flame, 93(4):421–433, 1993.
[84] J. Herzler, L. Jerig, and P. Roth. Shock tube study of the ignition of lean n-heptane/air mixtures at
intermediate temperatures and high pressures. Proceedings of the Combustion Institute, 30(1):1147–
1153, 2005.
[85] D. F. Davidson, Z. Hong, G. L. Pilla, A. Farooq, R. D. Cook, and R. K. Hanson. Multi-species
time-history measurements during n-heptane oxidation behind reflected shock waves. Combus-
tion and Flame, 157(10):1899–1905, 2010.
[86] Jiaxiang Zhang, Shaodong Niu, Yingjia Zhang, Chenglong Tang, Xue Jiang, Erjiang Hu, and
Zuohua Huang. Experimental and modeling study of the auto-ignition of n-heptane/n-butanol
mixtures. Combustion and Flame, 160(1):31–39, 2013.
[87] R. Minetti, M. Carlier, M. Ribaucour, E. Therssen, and L. R. Sochet. A rapid compression ma-
chine investigation of oxidation and auto-ignition of n-Heptane: Measurements and modeling.
Combustion and Flame, 102(3):298–309, 1995.
[88] A. Cox, J. F. Griffiths, C. Mohamed, H. J. Curran, W. J. Pitz, and C. K. Westbrook. Extents of
alkane combustion during rapid compression leading to single-and two-stage ignition. Sympo-
sium (International) on Combustion, 26(2):2685–2692, 1996.
[89] Emma J. Silke, Henry J. Curran, and John M. Simmie. The influence of fuel structure on combus-
tion as demonstrated by the isomers of heptane: a rapid compression machine study. Proceedings
of the Combustion Institute, 30(2):2639–2647, 2005.
[90] J. F. Griffiths, K. J. Hughes, M. Schreiber, and C. Poppe. A unified approach to the reduced
kinetic modeling of alkane combustion. Combustion and Flame, 99(3):533–540, 1994.
[91] A. Chakir, M. Bellimam, J. C. Boettner, and M. Cathonnet. Kinetic study of n-heptane oxidation.
International Journal of Chemical Kinetics, 24(4):385–410, 1992.
[92] Philippe Dagaut, Marcelline Reuillon, and Michel Cathonnet. Experimental study of the oxida-
tion of n-heptane in a jet stirred reactor from low to high temperature and pressures up to 40
atm. Combustion and Flame, 101(1):132–140, 1995.
[93] Olivier Herbinet, Benoit Husson, Zeynep Serinyel, Maximilien Cord, Valerie Warth, Rene Four-
net, Pierre-Alexandre Glaude, Baptiste Sirjean, Frederique Battin-Leclerc, Zhandong Wang,
Mingfeng Xie, Zhanjun Cheng, and Fei Qi. Experimental and modeling investigation of the
low-temperature oxidation of n-heptane. Combustion and Flame, 159(12):3455–3471, 2012.
134

[94] H. M. Hakka, R. F. Cracknell, A. Pekalski, P. A. Glaude, and F. Battin-Leclerc. Experimental and
modeling study of ultra-rich oxidation of n-heptane. Fuel, 144:358–368, 2015.
[95] T. J. Held, A. J. Marchese, and F. L. Dryer. A Semi-Empirical Reaction Mechanism for n-Heptane
Oxidation and Pyrolysis. Combustion Science and Technology, 123(1-6):107–146, 1997.
[96] David B. Lenhert, David L. Miller, Nicholas P. Cernansky, and Kevin G. Owens. The oxidation
of a gasoline surrogate in the negative temperature coefficient region. Combustion and Flame,
156(3):549–564, 2009.
[97] P.S. Veloo, S. Jahangirian, and F.L. Dryer. An experimental and kinetic modeling study of the
two stage autoignition kinetic behavior of c7, c10, c12, and c14 n-alkanes. Proceedings of the
Spring Technical MEtting of the Central States Section of the Combustion Institute, Dayton, OH, 2012.
[98] A. J. Smallbone, W. Liu, C. K. Law, X. Q. You, and H. Wang. Experimental and modeling study
of laminar flame speed and non-premixed counterflow ignition of n-heptane. Proceedings of the
Combustion Institute, 32(1):1245–1252, 2009.
[99] Chunsheng Ji, Enoch Dames, Yang L. Wang, Hai Wang, and Fokion N. Egolfopoulos. Propaga-
tion and extinction of premixed C5–C12 n-alkane flames. Combustion and Flame, 157(2):277–287,
2010.
[100] A. P. Kelley, A. J. Smallbone, D. L. Zhu, and C. K. Law. Laminar flame speeds of C5 to C8
n-alkanes at elevated pressures: Experimental determination, fuel similarity, and stretch sensi-
tivity. Proceedings of the Combustion Institute, 33(1):963–970, 2011.
[101] L. Sileghem, V. A. Alekseev, J. Vancoillie, K. M. Van Geem, E. J. K. Nilsson, S. Verhelst, and
A. A. Konnov. Laminar burning velocity of gasoline and the gasoline surrogate components
iso-octane, n-heptane and toluene. Fuel, 112:355–365, 2013.
[102] P. Dirrenberger, P. A. Glaude, R. Bounaceur, H. Le Gall, A. Pires da Cruz, A. A. Konnov, and
F. Battin-Leclerc. Laminar burning velocity of gasolines with addition of ethanol. Fuel, 115:162–
169, 2014.
[103] Chunde Yao, Chuanhui Cheng, Shiyu Liu, Zhenyu Tian, and Jing Wang. Identification of in-
termediates in an n-heptane/oxygen/argon low-pressure premixed laminar flame using syn-
chrotron radiation. Fuel, 88(9):1752–1757, 2009.
[104] Jinou Song, Chunde Yao, Shiyu Liu, Zhenyu Tian, and Jing Wang. Experiment study of oxy-
genates impact on n-heptane flames with tunable synchrotron vacuum UV photoionization.
Fuel, 88(11):2297–2302, 2009.
135

[105] Gen Chen, Wu Yu, Jin Fu, Jun Mo, Zuohua Huang, Jiuzhong Yang, Zhandong Wang, Hanfeng
Jin, and Fei Qi. Experimental and modeling study of the effects of adding oxygenated fuels to
premixed n-heptane flames. Combustion and Flame, 159(7):2324–2335, 2012.
[106] Lars Seidel, Kai Moshammer, Xiaoxiao Wang, Thomas Zeuch, Katharina Kohse-Hoinghaus, and
Fabian Mauss. Comprehensive kinetic modeling and experimental study of a fuel-rich, pre-
mixed n-heptane flame. Combustion and Flame, 162(5):2045–2058, 2015.
[107] A. Cavaliere, A. Ciajolo, A. D’Anna, R. Mercogliano, and R. Ragucci. Autoignition of n-heptane
and n-tetradecane in engine-like conditions. Combustion and Flame, 93(3):279–286, 1993.
[108] Dae Sik Kim and Chang Sik Lee. Improved emission characteristics of HCCI engine by various
premixed fuels and cooled EGR. Fuel, 85(5):695–704, 2006.
[109] Xing-Cai Lu, Wei Chen, and Zhen Huang. A fundamental study on the control of the HCCI
combustion and emissions by fuel design concept combined with controllable EGR. Part 1. The
basic characteristics of HCCI combustion. Fuel, 84(9):1074–1083, 2005.
[110] Xing-Cai Lu, Wei Chen, and Zhen Huang. A fundamental study on the control of the HCCI
combustion and emissions by fuel design concept combined with controllable EGR. Part 2. Effect
of operating conditions and EGR on HCCI combustion. Fuel, 84(9):1084–1092, 2005.
[111] James P. Szybist, Andre L. Boehman, Daniel C. Haworth, and Hibiki Koga. Premixed ignition
behavior of alternative diesel fuel-relevant compounds in a motored engine experiment. Com-
bustion and Flame, 149(1):112–128, 2007.
[112] H. J. Curran, P. Gaffuri, W. J. Pitz, and C. K. Westbrook. A Comprehensive Modeling Study of
n-Heptane Oxidation. Combustion and Flame, 114(1):149–177, 1998.
[113] H. J. Curran. Rate constant estimation for C1 to C4 alkyl and alkoxyl radical decomposition.
International Journal of Chemical Kinetics, 38(4):250–275, 2006.
[114] Li Zhu, Joseph W. Bozzelli, and Lisa M. Kardos. Thermochemical Properties, δ f h°(298), S°(298),
and Cp°(T), for n-Butyl and n-Pentyl Hydroperoxides and the Alkyl and Peroxy Radicals, Tran-
sition States, and Kinetics for Intramolecular Hydrogen Shift Reactions of the Peroxy Radicals.
The Journal of Physical Chemistry A, 111(28):6361–6377, 2007.
[115] Marco Mehl, Guillaume Vanhove, William J. Pitz, and Eliseo Ranzi. Oxidation and combus-
tion of the n-hexene isomers: A wide range kinetic modeling study. Combustion and Flame,
155(4):756–772, 2008.
136

[116] Marco Mehl, William J. Pitz, Charles K. Westbrook, Kenji Yasunaga, Christine Conroy, and
Henry J. Curran. Autoignition behavior of unsaturated hydrocarbons in the low and high tem-
perature regions. Proceedings of the Combustion Institute, 33(1):201–208, 2011.
[117] Kuiwen Zhang, Colin Banyon, John Bugler, Henry J. Curran, Anne Rodriguez, Olivier Herbinet,
Frederique Battin-Leclerc, Christine B’Chir, and Karl Alexander Heufer. An updated experi-
mental and kinetic modeling study of n-heptane oxidation. Combustion and Flame, 172:116–135,
2016.
[118] Chong-Wen Zhou, Yang Li, Eoin O’Connor, Kieran P. Somers, Sebastien Thion, Charles Keesee,
Olivier Mathieu, Eric L. Petersen, Trent A. DeVerter, Matthew A. Oehlschlaeger, Goutham
Kukkadapu, Chih-Jen Sung, Majed Alrefae, Fathi Khaled, Aamir Farooq, Patricia Dirrenberger,
Pierre-Alexandre Glaude, Frederique Battin-Leclerc, Jeffrey Santner, Yiguang Ju, Timothy Held,
Francis M. Haas, Frederick L. Dryer, and Henry J. Curran. A comprehensive experimental and
modeling study of isobutene oxidation. Combustion and Flame, 167:353–379, 2016.
[119] John Bugler, Kieran P. Somers, Emma J. Silke, and Henry J. Curran. Revisiting the Kinetics and
Thermodynamics of the Low-Temperature Oxidation Pathways of Alkanes: A Case Study of the
Three Pentane Isomers. The Journal of Physical Chemistry A, 119(28):7510–7527, 2015.
[120] John Bugler, Brandon Marks, Olivier Mathieu, Rachel Archuleta, Alejandro Camou, Claire
Gregoire, Karl A. Heufer, Eric L. Petersen, and Henry J. Curran. An ignition delay time and
chemical kinetic modeling study of the pentane isomers. Combustion and Flame, 163:138–156,
2016.
[121] Kuiwen Zhang, Colin Banyon, Casimir Togbe, Philippe Dagaut, John Bugler, and Henry J. Cur-
ran. An experimental and kinetic modeling study of n-hexane oxidation. Combustion and Flame,
162(11):4194–4207, 2015.
[122] C. K. Westbrook, H. J. Curran, W. J. Pitz, J. F. Griffiths, C. Mohamed, and S. K. Wo. The effects
of pressure, temperature, and concentration on the reactivity of alkanes: Experiments and mod-
eling in a rapid compression machine. Symposium (International) on Combustion, 27(1):371–378,
1998.
[123] M. Ribaucour, R. Minetti, L. R. Sochet, H. J. Curran, W. J. Pitz, and C. K. Westbrook. Ignition of
isomers of pentane: An experimental and kinetic modeling study. Proceedings of the Combustion
Institute, 28(2):1671–1678, 2000.
[124] R Minetti, A Roubaud, E Therssen, M Ribaucour, and L. R Sochet. The chemistry of pre-ignition
of n-pentane and 1-pentene. Combustion and Flame, 118(1):213–220, 1999.
137

[125] R. MINETTI, M. RIBAUCOUR, M. CARLIER, and L. R. SOCHET. Autoignition Delays of a
Series of Linear and Branched Chain Alkanes in the Intermediate Range of Temperature. Com-
bustion Science and Technology, 113(1):179–192, 1996.
[126] Alexander Burcat, Karl Scheller, and Assa Lifshitz. Shock-tube investigation of comparative
ignition delay times for C1-C5 alkanes. Combustion and Flame, 16(1):29–33, 1971.
[127] V. P. Zhukov, V. A. Sechenov, and A. Yu. Starikovskii. Self-ignition of a lean mixture of n-pentane
and air over a wide range of pressures. Combustion and Flame, 140(3):196–203, 2005.
[128] Charles K. Westbrook, William J. Pitz, Mark M. Thornton, and Philip C. Malte. A kinetic mod-
eling study of n-pentane oxidation in a well-stirred reactor. Combustion and Flame, 72(1):45–62,
1988.
[129] F. S. Gonzalez and S. Sandler. An experimental study of the oxidation of n-pentane in the high
temperature pre-ignition region. Combustion and Flame, 26:35–44, 1976.
[130] Song Cheng, Yi Yang, Michael J. Brear, Dongil Kang, Stanislav Bohac, and Andre L. Boehman.
Autoignition of pentane isomers in a spark-ignition engine. Proceedings of the Combustion Insti-
tute, 36(3):3499–3506, 2017.
[131] John Bugler, Anne Rodriguez, Olivier Herbinet, Frederique Battin-Leclerc, Casimir Togbe, Guil-
laume Dayma, Philippe Dagaut, and Henry J. Curran. An experimental and modelling study of
n-pentane oxidation in two jet-stirred reactors: The importance of pressure-dependent kinetics
and new reaction pathways. Proceedings of the Combustion Institute, 36(1):441–448, 2017.
[132] Kenneth Brezinsky. The high-temperature oxidation of aromatic hydrocarbons. Progress in En-
ergy and Combustion Science, 12(1):1–24, 1986.
[133] Matthew A. Oehlschlaeger, David F. Davidson, and Ronald K. Hanson. Investigation of the reac-
tion of toluene with molecular oxygen in shock-heated gases. Combustion and Flame, 147(3):195–
208, 2006.
[134] Carlo Cavallotti, Marco Derudi, and Renato Rota. On the mechanism of decomposition of the
benzyl radical. Proceedings of the Combustion Institute, 32(1):115–121, 2009.
[135] Gabriel da Silva, John A. Cole, and Joseph W. Bozzelli. Thermal decomposition of the benzyl
radical to fulvenallene (C7H6) + h. J. Phys. Chem. A, 113(21):6111–6120, 2009.
[136] K. Brezinsky, T. A. Litzinger, and I. Glassman. The high temperature oxidation of the methyl
side chain of toluene. International Journal of Chemical Kinetics, 16(9):1053–1074, 1984.
138

[137] Stephen D. Klotz, Kenneth Brezinsky, and Irvin Glassman. Modeling the combustion of toluene-
butane blends. Symposium (International) on Combustion, 27(1):337–344, 1998.
[138] W. K. Metcalfe, S. Dooley, and F. L. Dryer. Comprehensive Detailed Chemical Kinetic Modeling
Study of Toluene Oxidation. Energy & Fuels, 25(11):4915–4936, 2011.
[139] P. Dagaut, G. Pengloan, and A. Ristori. Oxidation, ignition and combustion of toluene: Experi-
mental and detailed chemical kinetic modeling. Physical Chemistry Chemical Physics, 4(10):1846–
1854, 2002.
[140] Experimental and modeling study of the oxidation of toluene. International Journal of Chemical
Kinetics, 37(1).
[141] D. F. Davidson, B. M. Gauthier, and R. K. Hanson. Shock tube ignition measurements of iso-
octane/air and toluene/air at high pressures. Proceedings of the Combustion Institute, 30(1):1175–
1182, 2005.
[142] Subith S. Vasu, David F. Davidson, and Ronald K. Hanson. Shock-Tube Experiments and Kinetic
Modeling of Toluene Ignition. Journal of Propulsion and Power, 26(4):776–783, 2010.
[143] Y. Sakai, T. Inamura, T. Ogura, M. Koshi, and W. Pitz. Detailed kinetic modeling of toluene
combustion over a wide range of temperature and pressure. SAE Technical Paper, 2007-01-1885,
2007.
[144] Hsi-Ping S. Shen, Jeremy Vanderover, and Matthew A. Oehlschlaeger. A shock tube study of the
auto-ignition of toluene/air mixtures at high pressures. Proceedings of the Combustion Institute,
32(1):165–172, 2009.
[145] R. Sivaramakrishnan, R. S. Tranter, and K. Brezinsky. High-pressure, high-temperature oxida-
tion of toluene. Combustion and Flame, 139(4):340–350, 2004.
[146] A. Roubaud, R. Minetti, and L. R. Sochet. Oxidation and combustion of low alkylbenzenes at
high pressure: comparative reactivity and auto-ignition. Combustion and Flame, 121(3):535–541,
2000.
[147] S. G. Davis, H. Wang, K. Breinsky, and C. K. Law. Laminar flame speeds and oxidation kinetics
of benene-air and toluene-air flames. Symposium (International) on Combustion, 26(1):1025–1033,
1996.
[148] Taichang Zhang, Lidong Zhang, Xin Hong, Kuiwen Zhang, Fei Qi, Chung K. Law, Taohong Ye,
Pinghui Zhao, and Yiliang Chen. An experimental and theoretical study of toluene pyrolysis
139

with tunable synchrotron VUV photoionization and molecular-beam mass spectrometry. Com-
bustion and Flame, 156(11):2071–2083, 2009.
[149] Yuyang Li, Lidong Zhang, Zhenyu Tian, Tao Yuan, Jing Wang, Bin Yang, and Fei Qi. Experimen-
tal Study of a Fuel-Rich Premixed Toluene Flame at Low Pressure. Energy & Fuels, 23(3):1473–
1485, 2009.
[150] J. L. Emdee, K. Brezinsky, and I. Glassman. A kinetic model for the oxidation of toluene near
1200 K. The Journal of Physical Chemistry, 96(5):2151–2161, 1992.
[151] R. P. LINDSTEDT and L. Q. MAURICE. Detailed Kinetic Modelling of Toluene Combustion.
Combustion Science and Technology, 120(1-6):119–167, 1996.
[152] J. C. G. Andrae, P. Bjornbom, R. F. Cracknell, and G. T. Kalghatgi. Autoignition of toluene
reference fuels at high pressures modeled with detailed chemical kinetics. Combustion and Flame,
149(1):2–24, 2007.
[153] Y. Sakai, H. Ozawa, T. Ogura, A. Miyoshi, M. Koshi, and W. Pitz. Effects of toluene addition to
primary reference fuel at high temperature. SAE Technical Paper, 2007-01-4104, 2007.
[154] K. Narayanaswamy, G. Blanquart, and H. Pitsch. A consistent chemical mechanism for oxida-
tion of substituted aromatic species. Combustion and Flame, 157(10):1879–1898, 2010.
[155] Zhenyu Tian, William J. Pitz, Rene Fournet, Pierre-Alexander Glaude, and Frederique Battin-
Leclerc. A detailed kinetic modeling study of toluene oxidation in a premixed laminar flame.
Proceedings of the Combustion Institute, 33(1):233–241, 2011.
[156] Juan Li, Zhenwei Zhao, Andrei Kazakov, Marcos Chaos, Frederick L. Dryer, and James J. Scire. A
comprehensive kinetic mechanism for CO, CH2o, and CH3oh combustion. International Journal
of Chemical Kinetics, 39(3):109–136, 2007.
[157] D. Healy, H. J. Curran, J. M. Simmie, D. M. Kalitan, C. M. Zinner, A. B. Barrett, E. L. Petersen,
and G. Bourque. Methane/ethane/propane mixture oxidation at high pressures and at high,
intermediate and low temperatures. Combustion and Flame, 155(3):441–448, 2008.
[158] S. G. Davis, C. K. Law, and H. Wang. Propene pyrolysis and oxidation kinetics in a flow reactor
and laminar flames. Combustion and Flame, 119(4):375–399, 1999.
[159] Alexander Laskin, Hai Wang, and Chung K. Law. Detailed kinetic modeling of 1,3-butadiene
oxidation at high temperatures. International Journal of Chemical Kinetics, 32(10):589–614, 2000.
140

[160] Hisashi Nakamura, Daniel Darcy, Marco Mehl, Colin J. Tobin, Wayne K. Metcalfe, William J.
Pitz, Charles K. Westbrook, and Henry J. Curran. An experimental and modeling study of shock
tube and rapid compression machine ignition of n-butylbenzene/air mixtures. Combustion and
Flame, 161(1):49–64, 2014.
[161] Wayne K. Metcalfe, Sinead M. Burke, Syed S. Ahmed, and Henry J. Curran. A hierarchical and
comparative kinetic modeling study of c1-c2 hydrocarbon and oxygenated fuels. International
Journal of Chemical Kinetics, 45(10):638–675, 2013.
[162] D. Darcy, M. Mehl, J. M. Simmie, J. Wurmel, W. K. Metcalfe, C. K. Westbrook, W. J. Pitz, and
H. J. Curran. An experimental and modeling study of the shock tube ignition of a mixture
of n-heptane and n-propylbenzene as a surrogate for a large alkyl benzene. Proceedings of the
Combustion Institute, 34(1):411–418, 2013.
[163] Wenhao Yuan, Yuyang Li, Philippe Dagaut, Jiuzhong Yang, and Fei Qi. Investigation on the
pyrolysis and oxidation of toluene over a wide range conditions. I. Flow reactor pyrolysis and
jet stirred reactor oxidation. Combustion and Flame, 162(1):3–21, 2015.
[164] Wenhao Yuan, Yuyang Li, Philippe Dagaut, Jiuzhong Yang, and Fei Qi. Investigation on the
pyrolysis and oxidation of toluene over a wide range conditions. II. A comprehensive kinetic
modeling study. Combustion and Flame, 162(1):22–40, 2015.
[165] K. Natarajan and K. A. Bhaskaran. Experimental and analytical investigation of high tempera-
ture ignition of ethanol. Int. Symp. Shock Waves, 13:834, 1981.
[166] Mary P. Dunphy and John M. Simmie. High-temperature oxidation of ethanol. Part 1.—Ignition
delays in shock waves. Journal of the Chemical Society, Faraday Transactions, 87(11):1691–1696,
1991.
[167] Henry J. Curran, Mary P. Dunphy, John M. Simmie, Charles K. Westbrook, and William J. Pitz.
Shock tube ignition of ethanol, isobutene and MTBE: Experiments and modeling. Symposium
(International) on Combustion, 24(1):769–776, 1992.
[168] K. A. Heufer and H. Olivier. Determination of ignition delay times of different hydrocarbons in
a new high pressure shock tube. Shock Waves, 20(4):307–316, 2010.
[169] L. R. Cancino, M. Fikri, A. A. M. Oliveira, and C. Schulz. Measurement and Chemical Kinetics
Modeling of Shock-Induced Ignition of Ethanol-Air Mixtures. Energy & Fuels, 24(5):2830–2840,
2010.
141

[170] Changyoul Lee, Stijn Vranckx, Karl A. Heufer, Sergey V. Khomik, Yasar Uygun, Herbert
Olivier, and Ravi X. Fernandez. On the Chemical Kinetics of Ethanol Oxidation: Shock Tube,
Rapid Compression Machine and Detailed Modeling Study. Zeitschrift fur Physikalische Chemie,
226(1):1–28, 2011.
[171] T. S. Norton and F. L. Dryer. An experimental and modeling study of ethanol oxidation kinetics
in an atmospheric pressure flow reactor. Int. J. Chem. Kinet., 24(4):319–344, 1992.
[172] Juan Li, Andrei Kazakov, and Frederick L. Dryer. Ethanol pyrolysis experiments in a variable
pressure flow reactor. International Journal of Chemical Kinetics, 33(12):859–867, 2001.
[173] Juan Li, Andrei Kazakov, and Frederick L. Dryer. Experimental and Numerical Studies of
Ethanol Decomposition Reactions. The Journal of Physical Chemistry A, 108(38):7671–7680, 2004.
[174] Francis M. Haas, Marcos Chaos, and Frederick L. Dryer. Low and intermediate temperature
oxidation of ethanol and ethanol–PRF blends: An experimental and modeling study. Combustion
and Flame, 156(12):2346–2350, 2009.
[175] N. Leplat, P. Dagaut, C. Togbe, and J. Vandooren. Numerical and experimental study of ethanol
combustion and oxidation in laminar premixed flames and in jet-stirred reactor. Combustion and
Flame, 158(4):705–725, 2011.
[176] A. Frassoldati, A. Cuoci, T. Faravelli, and E. Ranzi. Kinetic Modeling of the Oxidation of Ethanol
and Gasoline Surrogate Mixtures. Combustion Science and Technology, 182(4-6):653–667, 2010.
[177] Omer L. Gulder. Laminar burning velocities of methanol, ethanol and isooctane-air mixtures.
Symposium (International) on Combustion, 19(1):275–281, 1982.
[178] F. N. Egolfopoulos, D. X. Du, and C. K. Law. A study on ethanol oxidation kinetics in lami-
nar premixed flames, flow reactors, and shock tubes. Symposium (International) on Combustion,
24(1):833–841, 1992.
[179] T. S. Kasper, P. Oßwald, M. Kamphus, and K. Kohse-Hoinghaus. Ethanol flame structure inves-
tigated by molecular beam mass spectrometry. Combustion and Flame, 150(3):220–231, 2007.
[180] Priyank Saxena and Forman A. Williams. Numerical and experimental studies of ethanol flames.
Proceedings of the Combustion Institute, 31(1):1149–1156, 2007.
[181] Nick M. Marinov. A detailed chemical kinetic model for high temperature ethanol oxidation.
Int. J. Chem. Kinet., 31(3):183–220, 1999.
142

[182] J. Li, A. Kazakov, M. Chaos, and F.L. Dryer. Chemical kinetics of ethanol oxidation. 5th US
combustion meeting, 2007.
[183] Philippe Dagaut and Casimir Togbe. Experimental and modeling study of the kinetics of oxida-
tion of ethanol−gasoline surrogate mixtures (e85 surrogate) in a jet-stirred reactor. Energy Fuels,
22(5):3499–3505, 2008.
[184] Gaurav Mittal, Sinead M. Burke, Varun A. Davies, Bikash Parajuli, Wayne K. Metcalfe, and
Henry J. Curran. Autoignition of ethanol in a rapid compression machine. Combustion and
Flame, pages 1164–1171, 2013.
[185] R. Sivaramakrishnan, M.-C. Su, J. V. Michael, S. J. Klippenstein, L. B. Harding, and B. Ruscic.
Rate constants for the thermal decomposition of ethanol and its bimolecular reactions with OH
and d: Reflected shock tube and theoretical studies. J. Phys. Chem. A, 114(35):9425–9439, 2010.
[186] Johan Andrae, David Johansson, Pehr Bjornbom, Per Risberg, and Gautam Kalghatgi. Co-
oxidation in the auto-ignition of primary reference fuels and n-heptane/toluene blends. Com-
bustion and Flame, 140(4):267–286, 2005.
[187] J. C. G. Andrae. Development of a detailed kinetic model for gasoline surrogate fuels. Fuel,
87(10–11):2013–2022, 2008.
[188] Yasuyuki Sakai, Akira Miyoshi, Mitsuo Koshi, and William J. Pitz. A kinetic modeling study
on the oxidation of primary reference fuel–toluene mixtures including cross reactions between
aromatics and aliphatics. Proceedings of the Combustion Institute, 32(1):411–418, 2009.
[189] I. Da Costa, R. Fournet, F. Billaud, and F. Battin-Leclerc. Experimental and modeling study of
the oxidation of benzene. Int. J. Chem. Kinet., 35(10):503–524, 2003.
[190] G. Vanhove, G. Petit, and R. Minetti. Experimental study of the kinetic interactions in the low-
temperature autoignition of hydrocarbon binary mixtures and a surrogate fuel. Combustion and
Flame, 145(3):521–532, 2006.
[191] Askar Fahr and Stephen E. Stein. Gas-phase reactions of phenyl radicals with aromatic
molecules. J. Phys. Chem., 92(17):4951–4955, 1988.
[192] Wing Tsang. Chemical kinetic data base for combustion chemistry part v. propene. Journal of
Physical and Chemical Reference Data, 20(2):221–273, 1991.
[193] Luc Vereecken and Jozef Peeters. Reactions of chemically activated C9H9 species II: the reaction
of phenyl radicals with allene and cyclopropene, and of benzyl radicals with acetylene. Phys.
Chem. Chem. Phys., 5(13):2807–2817, 2003.
143

[194] Philippe Dagaut and Casimir Togbe. Experimental and modeling study of the kinetics of ox-
idation of butanol−n-heptane mixtures in a jet-stirred reactor. Energy Fuels, 23(7):3527–3535,
2009.
[195] Philippe Dagaut and Casimir Togbe. Experimental and modeling study of the kinetics of oxida-
tion of ethanol-n-heptane mixtures in a jet-stirred reactor. Fuel, 89(2):280–286, 2010.
[196] P. Saisirirat, C. Togbe, S. Chanchaona, F. Foucher, C. Mounaim-Rousselle, and P. Dagaut.
Auto-ignition and combustion characteristics in HCCI and JSR using 1-butanol/n-heptane and
ethanol/n-heptane blends. Proceedings of the Combustion Institute, 33(2):3007–3014, 2011.
[197] Darshan M. A. Karwat, Scott W. Wagnon, Margaret S. Wooldridge, and Charles K. West-
brook. On the combustion chemistry of n-heptane and n-butanol blends. J. Phys. Chem. A,
116(51):12406–12421, 2012.
[198] Zheng Yang, Yong Qian, Xin Yang, Yue Wang, Ya Wang, Zhen Huang, and Xingcai Lu. Autoigni-
tion of n-butanol/n-heptane blend fuels in a rapid compression machine under low-to-medium
temperature ranges. Energy Fuels, 27(12):7800–7808, 2013.
[199] Gabriel da Silva, Joseph W. Bozzelli, Long Liang, and John T. Farrell. Ethanol Oxidation: Ki-
netics of the alpha-Hydroxyethyl Radical + O2 Reaction. The Journal of Physical Chemistry A,
113(31):8923–8933, 2009.
[200] L. R. Cancino, M. Fikri, A. A. M. Oliveira, and C. Schulz. Ignition delay times of ethanol-
containing multi-component gasoline surrogates: Shock-tube experiments and detailed model-
ing. Fuel, 90(3):1238–1244, 2011.
[201] Zhewen Lu, Julien Cochet, Nicolas Leplat, Yi Yang, and Michael J. Brear. A high-pressure plug
flow reactor for combustion chemistry investigations. Measurement Science and Technology, 2017.
[202] Juan Li. Experimental and numerical studies of ethanol chemical kinetics. Ph.D. thesis, Princeton
University, 2004.
[203] T.J. Held. The oxidation of methanol, isobutene and methyl tertiary-butyl ether. Ph.D. thesis, Princeton
University, 1993.
[204] James T. Scanlon and Donald E. Willis. Calculation of Flame Ionization Detector Relative Re-
sponse Factors Using the Effective Carbon Number Concept. Journal of Chromatographic Science,
23(8):333–340, 1985.
144

[205] Andrew D. Jorgensen, Kurt C. Picel, and Vassilis C. Stamoudis. Prediction of gas chromatogra-
phy flame ionization detector response factors from molecular structures. Analytical Chemistry,
62(7):683–689, 1990.
[206] Hao Yuan, Yi Yang, Michael J. Brear, Tien Mun Foong, and James E. Anderson. Optimal octane
number correlations for mixtures of toluene reference fuels (TRFs) and ethanol. Fuel, 188:408–
417, 2017.
[207] American Petroleum Institute. A.P.I. Research Project 45. ASTM Special Technical Publication
No. 225, 1941.
[208] ESY Scott. Knock characteristics of hydrocarbon mixtures. Proc. API Div. refin., 38(III):90–111,
1958.
[209] Raymond L. Speth, Eric W. Chow, Robert Malina, Steven R. H. Barrett, John B. Heywood, and
William H. Green. Economic and environmental benefits of higher-octane gasoline. Environ. Sci.
Technol., 48(12):6561–6568, 2014.
[210] Thomas G. Leone, Edward D. Olin, James E. Anderson, Hosuk H. Jung, Michael H. Shelby, and
Robert A. Stein. Effects of fuel octane rating and ethanol content on knock, fuel economy, and
CO2 for a turbocharged DI engine. SAE Int. J. Fuels Lubr, 7(1):9–28, 2014.
[211] Hadeel Solaka, Martin Tuner, and Bengt Johansson. Analysis of surrogate fuels effect on ignition
delay and low temperature reaction during partially premixed combustion. SAE Technical Paper,
2013-01-0903, 2013.
[212] Ida Truedsson, Martin Tuner, Bengt Johansson, and William Cannella. Pressure sensitivity of
hcci auto-ignition temperature for gasoline surrogate fuels. SAE Technical Paper, 2013-01-1669,
2013.
[213] Solaka Aronsson, Martin Tuner, and Bengt Johansson. Using oxygenated gasoline surrogates
compositions to map ron and mon. SAE Technical Paper, 2014-01-1303, 2014.
[214] Neal Morgan, Andrew Smallbone, Amit Bhave, Markus Kraft, Roger Cracknell, and Gautam
Kalghatgi. Mapping surrogate gasoline compositions into RON/MON space. Combustion and
Flame, 157(6):1122–1131, 2010.
[215] Vincent Knop, Melanie Loos, Cecile Pera, and Nicolas Jeuland. A linear-by-mole blending rule
for octane numbers of n-heptane/iso-octane/toluene mixtures. Fuel, 115:666–673, 2014.
145

[216] Jihad A. Badra, Nehal Bokhumseen, Najood Mulla, S. Mani Sarathy, Aamir Farooq, Gautam
Kalghatgi, and Patrick Gaillard. A methodology to relate octane numbers of binary and ternary
n-heptane, iso-octane and toluene mixtures with simulated ignition delay times. Fuel, 160:458–
469, 2015.
[217] Prasenjeet Ghosh, Karlton J. Hickey, and Stephen B. Jaffe. Development of a detailed gasoline
composition-based octane model. Ind. Eng. Chem. Res., 45(1):337–345, 2005.
[218] N. Nikolaou, C. E. Papadopoulos, I. A. Gaglias, and K. G. Pitarakis. A new non-linear calculation
method of isomerisation gasoline research octane number based on gas chromatographic data.
Fuel, 83(4):517–523, 2004.
[219] C. MORLEY. A Fundamentally Based Correlation Between Alkane Structure and Octane Num-
ber. Combustion Science and Technology, 55(4-6):115–123, October 1987.
[220] Gorana Protic-Lovasic, Nada Jambrec, Djurdja Deur-Siftar, and Mladen V. Prostenik. Determi-
nation of catalytic reformed gasoline octane number by high resolution gas chromatography.
Fuel, 69(4):525–528, April 1990.
[221] Abdullah S. AlRamadan, S. Mani Sarathy, Muneeb Khurshid, and Jihad Badra. A blending rule
for octane numbers of PRFs and TPRFs with ethanol. Fuel, 180:175–186, September 2016.
[222] Henry Scheffe. Experiments with mixtures. Journal of the Royal Statistical Society. Series B (Method-
ological), 20(2):344–360, 1958.
[223] Francis M. Haas and Frederick L. Dryer. Application of blending rules for ignition quality
metrics: A comment on “a linear-by-mole blending rule for octane numbers of n-heptane/iso-
octane/toluene mixtures”. Fuel, 120:240–242, 2014.
[224] N.G. Becker. Models for the response of a mixture. Journal of the Royal Statistical Society. Series B
(Methodological), 30(2):349–358, 1968.
[225] S.D. Dimitrov and D.I. Kamenski. Binary mixture properties: fitting with canonical rational
functions. Computer and Chemical Engineering, 23(8):1011–1019, 1999.
[226] Norman R. Draper and Friedrich Pukelsheim. Mixture models based on homogeneous polyno-
mials. Journal of Statistical Planning and Inference, 71(1-2):303–311, 1998.
[227] Gautam Kalghatgi, Hassan Babiker, and Jihad Badra. A simple method to predict knock using
toluene, n-heptane and iso-octane blends (TPRF) as gasoline surrogates. SAE Int. J. Engines,
8(2):505–519, 2015.
146

[228] Kai J. Morganti, Tien Mun Foong, Michael J. Brear, Gabriel da Silva, Yi Yang, and Frederick L.
Dryer. The research and motor octane numbers of liquefied petroleum gas (LPG). Fuel, 108:797–
811, 2013.
[229] Gautam Kalghatgi, Robert Head, Junseok Chang, Yoann Viollet, Hassan Babiker, and Amer
Amer. An alternative method based on toluene/n-heptane surrogate fuels for rating the anti-
knock quality of practical gasolines. SAE Int. J. Fuels Lubr., 7(3):663–672, 2014.
[230] M. Mehl, J. Y. Chen, W. J. Pitz, S. M. Sarathy, and C. K. Westbrook. An Approach for Formulat-
ing Surrogates for Gasoline with Application toward a Reduced Surrogate Mechanism for CFD
Engine Modeling. Energy & Fuels, 25(11):5215–5223, 2011.
[231] Goutham Kukkadapu, Kamal Kumar, Chih-Jen Sung, Marco Mehl, and William J. Pitz. Au-
toignition of gasoline and its surrogates in a rapid compression machine. Proceedings of the
Combustion Institute, 34(1):345–352, 2013.
[232] Combustion and emissions modeling of a gasoline hcci engine using model fuels. 2009-01-0669,
pages = , journal = SAE Technical Paper, author = Puduppakkam, Karthick V. and Liang, Long
and Naik, Chitralkumar V. and Meeks, Ellen and Bunting, Bruce G., year = 2009.
[233] Vincent Knop, Cecile Pera, and Florence Duffour. Validation of a ternary gasoline surrogate in
a CAI engine. Combustion and Flame, 160(10):2067–2082, 2013.
[234] Cecile Pera and Vincent Knop. Methodology to define gasoline surrogates dedicated to auto-
ignition in engines. Fuel, 96:59–69, 2012.
[235] Ahfaz Ahmed, Gokop Goteng, Vijai S. B. Shankar, Khalid Al-Qurashi, William L. Roberts, and
S. Mani Sarathy. A computational methodology for formulating gasoline surrogate fuels with
accurate physical and chemical kinetic properties. Fuel, 143:290–300, 2015.
[236] CHEMKIN-PRO. CHEMKIN-PRO 15131, Reaction Design: San Diego, 2013.
[237] Richard S. Larson. Plug: a fortran program for the analysis of plug flow reactors with gas-phase
and surface chemistry, January 1996.
[238] Linda R. Petzold. A description of DASSL: a differential/algebraic system solver, August 1982.
[239] X. He, S. M. Walton, B. T. Zigler, M. S. Wooldridge, and A. Atreya. Experimental investiga-
tion of the intermediates of isooctane during ignition. International Journal of Chemical Kinetics,
39(9):498–517, 2007.
147

[240] Cesar L. Barraza-Botet, Scott W. Wagnon, and Margaret S. Wooldridge. Combustion Chemistry
of Ethanol: Ignition and Speciation Studies in a Rapid Compression Facility. The Journal of
Physical Chemistry A, 120(38):7408–7418, 2016.
[241] Yingjia Zhang, Kieran P. Somers, Marco Mehl, William J. Pitz, Roger F. Cracknell, and Henry J.
Curran. Probing the antagonistic effect of toluene as a component in surrogate fuel models at
low temperatures and high pressures. A case study of toluene/dimethyl ether mixtures. Pro-
ceedings of the Combustion Institute, 36(1):413–421, January 2017.
[242] M. Pelucchi, C. Cavallotti, T. Faravelli, and S. J. Klippenstein. H-Abstraction reactions by OH,
HO2, O, O2 and benzyl radical addition to O2 and their implications for kinetic modelling of
toluene oxidation. Physical Chemistry Chemical Physics, 2018.
[243] Gabriel da Silva and Joseph W. Bozzelli. Kinetic modeling of the benzyl+HO2 reaction. Proceed-
ings of the Combustion Institute, 32(1):287–294, 2009.
[244] D. L. Baulch, C. J. Cobos, R. A. Cox, P. Frank, G. Hayman, Th. Just, J. A. Kerr, T. Murrells, M. J.
Pilling, J. Troe, R. W. Walker, and J. Warnatz. Evaluated Kinetic Data for Combustion Modeling.
Supplement I. Journal of Physical and Chemical Reference Data, 23(6):847–848, 1994.
[245] Goutham Kukkadapu, Kamal Kumar, Chih-Jen Sung, Marco Mehl, and William J. Pitz. Au-
toignition of gasoline surrogates at low temperature combustion conditions. Combustion and
Flame, 162(5):2272–2285, 2015.
[246] Tamour Javed, Changyoul Lee, Mohammed AlAbbad, Khalil Djebbi, Mohamed Beshir, Jihad
Badra, Henry Curran, and Aamir Farooq. Ignition studies of n-heptane/iso-octane/toluene
blends. Combustion and Flame, 171:223–233, September 2016.
[247] Abdullah S. AlRamadan, Jihad Badra, Tamour Javed, Mohammed Al-Abbad, Nehal Bokhum-
seen, Patrick Gaillard, Hassan Babiker, Aamir Farooq, and S. Mani Sarathy. Mixed butanols
addition to gasoline surrogates: Shock tube ignition delay time measurements and chemical
kinetic modeling. Combustion and Flame, 162(10):3971–3979, 2015.
[248] Edirin Agbro, Alison S. Tomlin, Malcolm Lawes, Sungwoo Park, and S. Mani Sarathy. The
influence of n-butanol blending on the ignition delay times of gasoline and its surrogate at high
pressures. Fuel, 187(Supplement C):211–219, 2017.
[249] O. Stenlaas, P. Einewall, R. Egnell, and B. Johansson. Measurement of Knock and Ion Current
in a Spark Ignition Engine with and without NO Addition to the Intake Air. Technical report,
2003.
148

[250] P.J. Roberts and C.G.W. Sheppard. The Influence of Residual Gas NO Content on Knock Onset
of Iso-Octane, PRF, TRF and ULG Mixtures in SI Engines. SAE Int. J. Engines, 6(4):2028–2043,
2013.
[251] Kai J. Morganti, Michael J. Brear, Gabriel da Silva, Yi Yang, and Frederick L. Dryer. The autoigni-
tion of Liquefied Petroleum Gas (LPG) in spark-ignition engines. Proceedings of the Combustion
Institute, 2014.
[252] J. S. Cowart, J. C. Keck, J. B. Heywood, C. K. Westbrook, and W. J. Pitz. Engine knock predictions
using a fully-detailed and a reduced chemical kinetic mechanism. Symposium (International) on
Combustion, 23(1):1055–1062, 1991.
[253] Henry J. Curran, Paolo Gaffuri, William J. Pitz, Charles K. Westbrook, and William R. Leppard.
Autoignition chemistry in a motored engine: An experimental and kinetic modeling study. Sym-
posium (International) on Combustion, 26(2):2669–2677, 1996.
[254] Iltesham Zameer Syed, Abhijit Mukherjee, and Jeffrey D. Naber. Numerical Simulation of Au-
toignition of Gasoline-Ethanol/Air Mixtures under Different Conditions of Pressure, Tempera-
ture, Dilution, and Equivalence Ratio. Technical report, 2011.
[255] Marlan Perumal and Gareth Floweday. An Investigation of Cascading Autoignition and Octane
Number using a Multi-zone Model of the CFR Engine. Technical Report 2011-01-0850, 2011.
[256] L. Sileghem, T. Wallner, and S. Verhelst. A quasi-dimensional model for SI engines fueled with
gasoline–alcohol blends: Knock modeling. Fuel, 140:217–226, 2015.
[257] Shahrokh Hajireza, Bengt Sunden, and Fabian Mauss. A Three-Zone Model for Investigation
of Gas Behavior in the Combustion Chamber of SI Engines in Relation to Knock. SAE Technical
Paper, 1999-01-0219, 1999.
[258] G. D’Errico, T. Lucchini, A. Onorati, M. Mehl, T. Faravelli, E. Ranzi, S. Merola, and B. M.
Vaglieco. Development and Experimental Validation of a Combustion Model with Detailed
Chemistry for Knock Predictions. SAE Technical Paper, 2007-01-0938, 2007.
[259] Shahrokh Hajireza, Fabian Mauss, and Bengt Sunden. Two-zone model of gas thermodynamic
state in SI engines with relevance for knock. COMODIA, pages 203–208, 1998.
[260] A. Gogan, B. Sunden, L. Montorsi, S. S. Ahmedand, and F. Mauss. Knock Modeling: an Inte-
grated Tool for Detailed Chemistry and Engine Cycle Simulation. SAE Technical Paper, 2003-01-
3122, 2003.
149

[261] Gamma Technologies. GT-Power user’s manual, version 7.3, 2012.
[262] Philippe Dagaut and Andre Nicolle. Experimental study and detailed kinetic modeling of the
effect of exhaust gas on fuel combustion: mutual sensitization of the oxidation of nitric oxide
and methane over extended temperature and pressure ranges. Combustion and Flame, 140(3):161–
171, 2005.
[263] David G. Goodwin, Harry K. Moffat, and Raymond L. Speth. Cantera: An object-oriented soft-
ware toolkit for chemical kinetics, thermodynamics, and transport processes, 2016.
[264] G. Woschni. A Universally Applicable Equation for the Instantaneous Heat Transfer Coefficient
in the Internal Combustion Engine. SAE Technical Paper, 670931, 1967.
[265] D. G. Goodwin. An open source, extensible software suite for CVD process simulation. Technical
report, 2003.
[266] C. Draper. Pressure waves accompanying detonation in the internal combustion engine. J.
Aeronaut. Sci., 5(6):219–226, 1938.
[267] Zhongyuan Chen, Hao Yuan, Yi Yang, and Michael Brear. The Effect of Nitric Oxide on Knock
Onset of iso-Octane in a CFR Spark-Ignition Engine. The 10th Asia-Pacific Conference on Com-
bustion, Beijing, July 2015.
[268] P. Brown, G. Byrne, and A. Hindmarsh. VODE: A Variable-Coefficient ODE Solver. SIAM Journal
on Scientific and Statistical Computing, 10(5):1038–1051, 1989.
150

Appendix A
Octane number data used for optimal
correlation development
Table A.1: Octane number data used for developing the optimal correlations for TRF/ethanol mixtures
NO.iC8H18
(mol%)
iC8H18
(vol%)
nC7H16
(mol%)
nC7H16
(vol%)
C7H8
(mol%)
C7H8
(vol%)
C2H6O
(mol%)
C2H6O
(vol%)RON MON Ref.
1 0.0 0.0 27.2 34.0 72.8 66.0 0.0 0.0 85.2 74.8 [25, 26]
2 0.0 0.0 23.7 30.0 76.3 70.0 0.0 0.0 89.3 78.2 [25, 26]
3 0.0 0.0 20.3 26.0 79.7 74.0 0.0 0.0 93.4 81.5 [25, 26]
4 3.5 5.0 16.5 21.0 80.0 74.0 0.0 0.0 96.9 85.2 [25, 26]
5 7.0 10.0 12.6 16.0 80.4 74.0 0.0 0.0 99.8 88.7 [25, 26]
6 10.5 15.0 8.7 11.0 80.8 74.0 0.0 0.0 103.3 92.6 [25, 26]
7 14.1 20.0 4.8 6.0 81.1 74.0 0.0 0.0 107.6 96.6 [25, 26]
8 18.4 26.0 0.0 0.0 81.6 74.0 0.0 0.0 113.0 100.8 [25, 26]
9 66.0 73.0 13.0 12.0 21.0 15.0 0.0 0.0 91.0 88.4 [9]
10 45.0 53.0 16.0 17.0 39.0 30.0 0.0 0.0 91.4 86.1 [9]
11 27.0 35.0 18.0 20.0 55.0 45.0 0.0 0.0 91.0 83.5 [9]
12 12.0 16.7 13.5 16.7 74.5 66.7 0.0 0.0 98.0 87.4 [214]
13 59.9 66.7 16.9 16.7 23.2 16.7 0.0 0.0 87.0 84.0 [214]
14 39.2 50.0 0.0 0.0 60.8 50.0 0.0 0.0 110.0 99.3 [214]
15 0.0 0.0 20.8 26.6 79.2 73.4 0.0 0.0 92.3 80.7 [215]
16 65.0 72.2 10.0 9.9 25.0 17.9 0.0 0.0 93.7 90.3 [215]
17 35.0 43.5 15.0 16.5 50.0 40.0 0.0 0.0 93.0 85.8 [215]
18 42.8 51.5 13.7 14.7 43.5 33.8 0.0 0.0 93.0 86.7 [215]
19 0.0 0.0 16.0 20.8 84.0 79.2 0.0 0.0 97.7 86.2 [215]
151

20 56.5 65.0 9.8 10.0 33.7 25.0 0.0 0.0 95.2 90.5 [215]
21 27.0 35.0 13.0 15.0 60.0 50.0 0.0 0.0 96.3 87.3 [215]
22 63 69.2 17 16.6 20.0 14.2 0.0 0.0 86.6 84.2 [215]
23 69.0 74.1 17.0 16.2 14 9.7 0.0 0.0 85.7 84.6 [215]
24 34.0 42.8 12.3 13.7 53.7 43.5 0.0 0.0 96.3 88.3 [215]
25 76.2 90.0 0.0 0.0 0.0 0.0 23.8 10.0 106.8 99.9 [9]
26 58.7 80.0 0.0 0.0 0.0 0.0 41.3 20.0 109.4 99.1 [9]
27 34.8 60.0 0.0 0.0 0.0 0.0 65.2 40.0 110.2 95.9 [9]
28 19.2 40.0 0.0 0.0 0.0 0.0 80.8 60.0 109.6 94.2 [9]
29 8.2 20.0 0.0 0.0 0.0 0.0 91.8 80.0 109.0 92.6 [9]
30 0.0 0.0 0.0 0.0 0.0 0.0 100.0 100.0 108.0 90.7 [9]
31 0.0 0.0 28.6 50.0 0.0 0.0 71.4 50.0 83.8 - [9]
32 0.0 0.0 21.1 40.0 0.0 0.0 78.9 60.0 94.7 83.8 [9]
33 0.0 0.0 14.7 30.0 0.0 0.0 85.3 70.0 101.6 - [9]
34 0.0 0.0 9.1 20.0 0.0 0.0 90.9 80.0 104.7 88.9 [9]
35 0.0 0.0 4.3 10.0 0.0 0.0 95.7 90.0 106.5 - [9]
36 0.0 0.0 0.0 0.0 83.3 90.0 16.7 10.0 112.8 101 [9]
37 0.0 0.0 0.0 0.0 68.8 80.0 31.2 20.0 110.9 97 [9]
38 0.0 0.0 0.0 0.0 45.3 60.0 54.7 40.0 108.6 93.3 [9]
39 0.0 0.0 0.0 0.0 26.9 40.0 73.1 60.0 108.1 91.9 [9]
40 0.0 0.0 0.0 0.0 12.1 20.0 87.9 80.0 107.9 91.1 [9]
41 6.7 12.0 30.3 48.0 0.0 0.0 63.0 40.0 80.7 - [9]
42 5.1 10.0 23.0 40.0 0.0 0.0 71.8 50.0 91.5 - [9]
43 3.8 8.0 17.0 32.0 0.0 0.0 79.3 60.0 99.1 - [9]
44 1.6 4.0 7.3 16.0 0.0 0.0 91.1 80.0 105.8 - [9]
45 17.5 28.0 29.6 42.0 0.0 0.0 52.8 30.0 80.6 - [9]
46 13.6 24.0 22.9 36.0 0.0 0.0 63.5 40.0 90.4 - [9]
47 10.3 20.0 17.4 30.0 0.0 0.0 72.3 50.0 97.9 - [9]
48 7.6 16.0 12.8 24.0 0.0 0.0 79.7 60.0 102.7 - [9]
49 3.2 8.0 5.5 12.0 0.0 0.0 91.3 80.0 106.6 - [9]
50 34.2 48.0 25.7 32.0 0.0 0.0 40.1 20.0 83.5 - [9]
51 26.6 42.0 20.0 28.0 0.0 0.0 53.4 30.0 92.0 - [9]
52 20.5 36.0 15.4 24.0 0.0 0.0 64.1 40.0 98.9 - [9]
53 11.4 24.0 8.6 16.0 0.0 0.0 80.1 60.0 105.5 - [9]
54 4.9 12.0 3.7 8.0 0.0 0.0 91.5 80.0 107.6 - [9]
55 59.8 72.0 16.9 18.0 0.0 0.0 23.4 10.0 89.5 - [9]
152
56 46.3 64.0 13.0 16.0 0.0 0.0 40.7 20.0 97.0 - [9]
57 27.6 48.0 7.8 12.0 0.0 0.0 64.6 40.0 105.7 - [9]
58 15.3 32.0 4.3 8.0 0.0 0.0 80.4 60.0 107.7 - [9]
59 6.5 16.0 1.8 4.0 0.0 0.0 91.6 80.0 108.3 - [9]
60 77.5 85.5 9.7 9.5 0.0 0.0 12.7 5.0 94.1 - [9]
61 67.9 81.0 8.5 9.0 0.0 0.0 23.6 10.0 97.6 - [9]
62 52.5 72.0 6.6 8.0 0.0 0.0 41.0 20.0 103.6 - [9]
63 68.7 81.9 7.7 8.1 0.0 0.0 23.6 10.0 98.7 94.3 [9]
64 53.1 72.8 5.9 7.2 0.0 0.0 41.0 20.0 103.8 95.3 [9]
65 31.5 54.6 3.5 5.4 0.0 0.0 65.0 40.0 108.0 94.5 [9]
66 17.4 36.4 1.9 3.6 0.0 0.0 80.7 60.0 108.4 93.4 [9]
67 7.4 18.2 0.8 1.8 0.0 0.0 91.7 80.0 108.4 92.2 [9]
68 51.4 65.3 9.9 11.2 16.5 13.5 22.1 10.0 97.8 91.7 [9]
69 40.3 58.1 7.8 9.9 12.9 12.0 39.0 20.0 102.6 93.2 [9]
70 24.4 43.6 4.7 7.4 7.8 9.0 63.0 40.0 107.1 93.6 [9]
71 13.7 29.0 2.6 5.0 4.4 6.0 79.3 60.0 107.7 92.6 [9]
72 5.9 14.5 1.1 2.5 1.9 3.0 91.1 80.0 107.8 91.7 [9]
73 35.5 47.9 12.8 15.3 30.9 26.8 20.8 10.0 97.0 89.4 [9]
74 28.2 42.6 10.1 13.6 24.5 23.8 37.2 20.0 101.4 91.1 [9]
75 17.4 31.9 6.3 10.2 15.1 17.9 61.2 40.0 106.0 92.1 [9]
76 9.8 21.3 3.5 6.8 8.6 11.9 78.0 60.0 107.1 92 [9]
77 4.3 10.6 1.5 3.4 3.7 6.0 90.5 80.0 107.5 91.4 [9]
78 21.9 31.2 14.4 18.3 44.0 40.5 19.7 10.0 96.0 87.2 [9]
79 17.5 27.8 11.6 16.2 35.3 36.0 35.5 20.0 100.2 89.1 [9]
80 11.0 20.8 7.3 12.2 22.2 27.0 59.5 40.0 104.6 90.9 [9]
81 6.3 13.9 4.2 8.1 12.7 18.0 76.8 60.0 106.3 91.2 [9]
82 2.8 6.9 1.8 4.1 5.6 9.0 89.8 80.0 107.1 91.1 [9]
Table A.2: Octane number data used for validating the optimal correlations for TRF/ethanol mixtures
NO.iC8H18
(mol%)
iC8H18
(vol%)
nC7H16
(mol%)
nC7H16
(vol%)
C7H8
(mol%)
C7H8
(vol%)
C2H6O
(mol%)
C2H6O
(vol%)RON MON Ref.
1 61.3 69.4 8.5 8.5 30.3 22.1 0.0 0.0 96.1 91.8 [213]
2 64.7 70.5 18.1 17.5 17.2 12.0 0.0 0.0 85.1 83.8 [213]
3 39.5 48.4 12.4 13.5 48.2 38.1 0.0 0.0 94.8 87.8 [213]
4 50.5 58.0 19.6 20.0 29.9 22.1 0.0 0.0 85.1 81.6 [213]
5 85.2 89.9 0.0 0.0 14.8 10.1 0.0 0.0 101.9 - This study
153
6 60.0 70.0 0.0 0.0 40.0 30.0 0.0 0.0 105.0 - This study
7 39.1 49.9 0.0 0.0 60.9 50.1 0.0 0.0 108.5 - This study
8 18.4 25.9 0.0 0.0 81.6 74.1 0.0 0.0 113.0 - This study
9 6.7 10.0 0.0 0.0 93.4 90.0 0.0 0.0 115.3 - This study
10 34.5 45.0 25.9 30.0 17.9 15.0 21.7 10.0 81.6 77.2 [211]
11 34.6 49.9 15.6 20.0 10.8 10.0 39.1 20.1 94.7 88.5 [212]
12 19.3 29.9 14.5 19.9 30.0 30.0 36.3 20.1 97.0 87.6 [212]
13 6.3 10.0 28.5 39.9 29.5 30.1 35.7 20.1 80.8 73.0 [212]
14 19.2 27.5 23.6 29.9 32.6 30.1 24.6 12.5 85.3 78.6 [212]
15 27.2 37.5 24.5 29.9 22.6 20.0 25.6 12.6 83.8 78.2 [212]
16 35.9 47.5 25.6 30.0 11.8 10.0 26.7 12.6 81.6 77.9 [212]
17 35.0 44.9 17.5 20.0 36.4 30.1 11.0 5.0 90.2 84.1 [212]
18 55.4 65.0 19.2 20.0 13.3 10.0 12.1 5.0 86.1 83.6 [213]
19 34.8 47.4 16.5 20.0 22.8 20.0 25.9 12.6 92.3 86.4 [212]
20 19.8 29.9 22.3 29.9 20.6 20.0 37.3 20.1 87.9 81.4 [212]
21 66.9 80.0 9.4 10.0 0.0 0.0 23.7 10.0 96.4 92.9 [213]
22 10.9 19.0 27.7 42.9 0.0 0.0 61.4 38.1 84.4 78.7 [213]
23 40.9 53.9 12.8 15.0 24.8 21.1 21.4 10.0 95.5 89.2 [213]
24 51.0 62.0 20.8 22.5 9.6 7.5 18.6 8.0 85.1 82.5 [213]
25 63.3 75.5 9.9 10.5 7.8 6.0 19.0 8.0 96.3 92.4 [213]
26 65.2 73.0 19.1 19.0 5.6 4.0 10.1 4.0 84.9 83.6 [213]
154
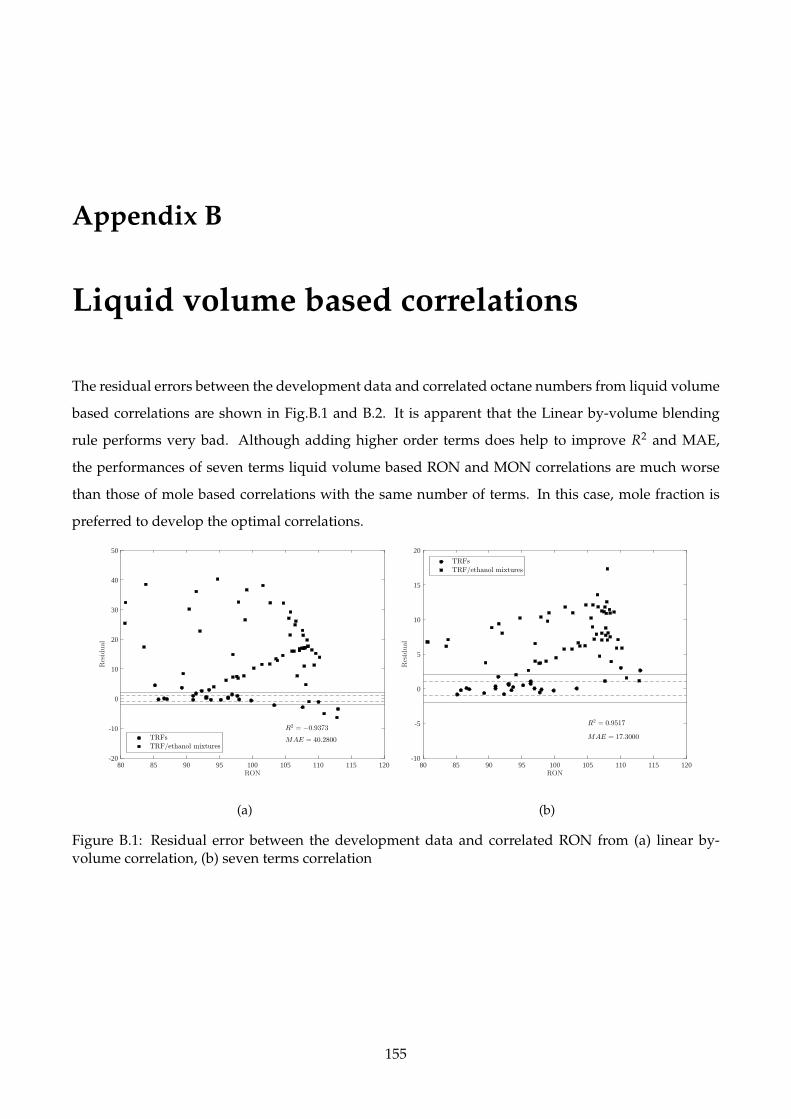
Appendix B
Liquid volume based correlations
The residual errors between the development data and correlated octane numbers from liquid volume
based correlations are shown in Fig.B.1 and B.2. It is apparent that the Linear by-volume blending
rule performs very bad. Although adding higher order terms does help to improve R2 and MAE,
the performances of seven terms liquid volume based RON and MON correlations are much worse
than those of mole based correlations with the same number of terms. In this case, mole fraction is
preferred to develop the optimal correlations.
RON80 85 90 95 100 105 110 115 120
Residual
-20
-10
0
10
20
30
40
50
R2 = −0.9373
MAE = 40.2800TRFsTRF/ethanol mixtures
(a)
RON80 85 90 95 100 105 110 115 120
Residual
-10
-5
0
5
10
15
20
R2 = 0.9517
MAE = 17.3000
TRFsTRF/ethanol mixtures
(b)
Figure B.1: Residual error between the development data and correlated RON from (a) linear by-volume correlation, (b) seven terms correlation
155
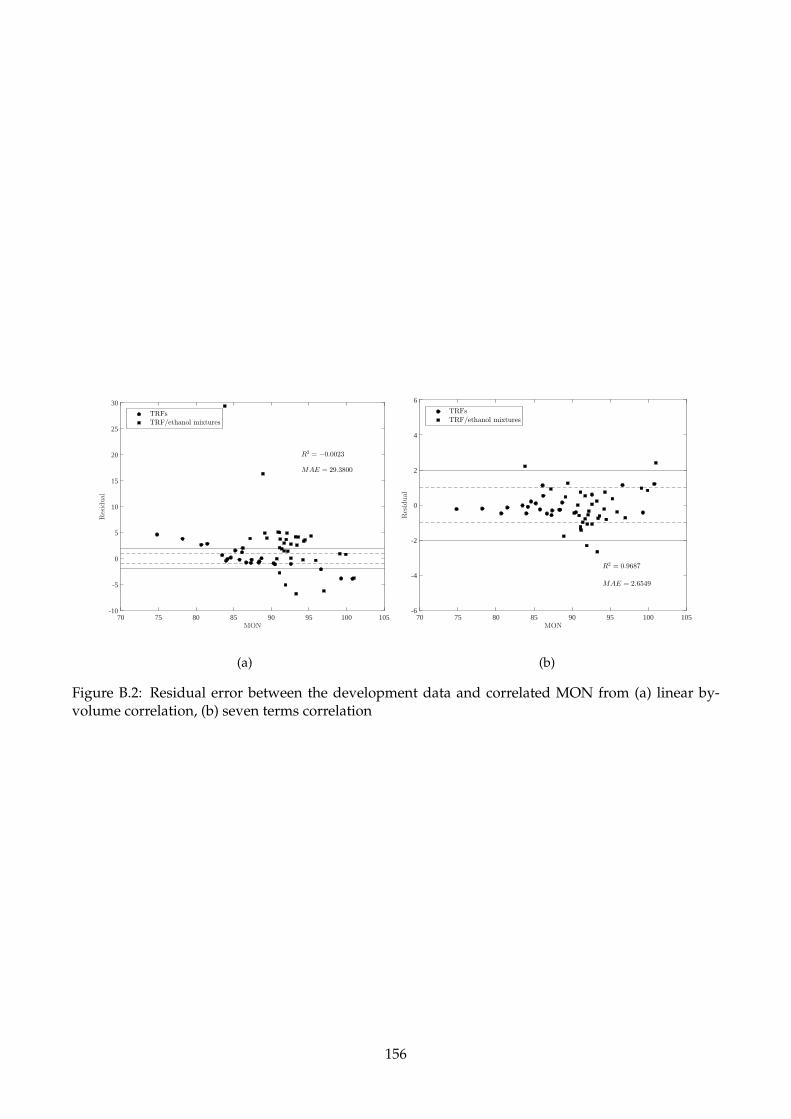
MON70 75 80 85 90 95 100 105
Residual
-10
-5
0
5
10
15
20
25
30
R2 = −0.0023
MAE = 29.3800
TRFsTRF/ethanol mixtures
(a)
MON70 75 80 85 90 95 100 105
Residual
-6
-4
-2
0
2
4
6
R2 = 0.9687
MAE = 2.6549
TRFsTRF/ethanol mixtures
(b)
Figure B.2: Residual error between the development data and correlated MON from (a) linear by-volume correlation, (b) seven terms correlation
156

Appendix C
Modelling of Trace Knock in a Modern SI
Engine Fuelled by Ethanol and Gasoline
Blends
C.1 Introduction
A systematic method has been proposed in [23] to model the knocking behaviours in spark ignition
engines using two-zone kinetic model coupled with detailed chemistry. However, in most scenarios,
engine knocks are not as strong as reported in [23] and, most likely, occur intermittently in real pro-
duction engines, which is commonly known as trace knock. The trace knock is the borderline between
non-knocking and knocking combustions. If the spark timing is advanced, there will be a larger heat
release inside the engine cylinder, resulting in knocking combustion, and vice versa. Unlike those
strong knocking behaviours, no distinct incipient jump, indicating autoignition, can be seen on the
pressure traces from trace knock. This complicates the numerical modelling since the autoignition
is insignificant and no clear clue is available from the pressure traces suggesting the occurrence of
autoignition.
Hence, this study is aiming to model the trace knocks from a single-cylinder research engine [11].
The objectives of this study are as follows.
• To propose a systematic method for trace knock modelling in SI engines
• To model the effect of ethanol on trace knock with the proposed method and an existing kinetic
model for gasoline surrogates
• To model the effect of DI on trace knock
157

• To investigate the effect of residual NO on trace knock.
The last objective relates to the potentially significant impact of NO on autoignition [249–251],
which has been ignored in almost all prior kinetic modelling of gasoline combustion. Note that this
appendix is revised from a previously published paper by the author [12].
C.2 Numerical methods
To apply detailed kinetic models in studying fuel effects on fundamental engine combustion, zero-
dimensional thermodynamic models are commonly used, since they are substantially less compu-
tationally expensive than multidimensional models, without solving for the fluid dynamics in the
engine cylinder.
Zero-dimensional models may consider single-zone [187, 252–254] or multiple zones [255–258] in
the combustion chamber. Chemical kinetics coupled with thermodynamic governing equations are
solved in each zone where the gas properties are assumed to be spatially uniform. Single-zone mod-
els typically apply closed homogeneous reactors with time-varying volume to model the piston move-
ment. The simulated autoignition timing can be compared with the measured knock onset timings.
Since the single-zone does not consider the flame propagation and its compression on the end gases,
higher compression ratios have to be used to compensate this effect, which makes it hard to com-
pare the modelling with an actual engine experiment. On the other hand, multi-zone models take
flame propagation and its additional compression of the end gas into consideration. The mostly used
two-zone models separate the combustion chamber into a burned zone and an unburned zone. Ig-
nition delay correlations have been applied in some previous two-zone models to simulate knock
onset [255, 256], which apparently could not provide any insight into the thorough understanding
of observed fuel effects. Two-zone models coupled with detailed combustion chemistry have been
reported by [257–260], and this approach is used in this study.
Common to all kinetic modelling of engine combustion, the initial and boundary conditions, in
particular, the temperature and compositions of initial mixture and the heat transfer during compres-
sion and combustion, are critical to autoignition onset timing. However, it is hard to measure these
parameters experimentally and thus are usually only estimated or even ignored sometimes. This
significantly reduces the accuracy of these models and results in a major defect of many existing ap-
proaches.
The uncertainties of the initial and boundary conditions, such as residual gas fraction, the heat
transfer, the temperature at intake valve closure (TIVC) and the cylinder wall temperature, are ad-
dressed by a commercial software package (GT-Power [261]). Besides, GT-Power derives the mass
158

fraction burned (MFB) profiles from the measured pressure traces, which determine the flame propa-
gation and following conditions in the engine chamber. Afterwards, an in-house two-zone model [23]
with the detailed combustion kinetics is used to estimate the critical in-cylinder conditions that affect
autoignition.
It should be noted that since the in-cylinder gas flow is not modelled, zero-dimensional models, no
matter how many zones they have, are unable to locate local hot spots in the end gas which most likely
trigger autoignition. Nevertheless, the homogeneous end gas assumption is considered acceptable
regarding reproducing the overall trend of autoignition.
C.3 Formulation of gasoline surrogates
Gasoline is a mixture of thousands of different fuel components, which can be categorised into four
major groups: paraffins (straight-chain and branched), aromatics, olefins, cycloparaffins. It is not
realistic to model everything contained in gasoline, and this very complicated mixture can be ap-
proximated by surrogate fuels comprised of representative components from each of hydrocarbon
group. In this study, a four-component surrogate fuel consisting of iso-octane, n-heptane, toluene and
2-pentene, proposed by Lawrence Livermore National Laboratory (LLNL) [67], is applied to emulate
the gasoline used in the experiments [11]. The kinetic model of the surrogate mixture, which is from
LLNL [67] as well, is then utilised for the autoignition modelling. Note that this chemical mechanism
contains a sub-model for ethanol, and therefore no additional model is required for investigating the
ethanol/gasoline blends.
Four fuel properties: RON, MON, H/C ratio and lower heating value (LHV) are constrained to
formulate the surrogate gasoline composition. Simple mixing rules based on molar fractions [234] are
used to calculate mixture properties for the gasoline as well as the ethanol/gasoline blends [10], i.e.
n
∑i=1
xi = 1 (C.1)
n
∑i=1
xiRONi = RON (C.2)
n
∑i=1
xi MONi = MON (C.3)
∑ xi Hi
∑ xiCi=
HC
(C.4)
159

n
∑i=1
xiLHVi = LHV (C.5)
where n is the number of representative components, xi is the molar fraction, Hi and Ci are the number
of hydrogen and carbon atoms respectively, RONi and MONi are research and motor octane numbers,
and LHVi is the lower heating value of the ith component in the surrogate mixture.
Since autoignition is of primary concern, priority was given to matching the RON MON, when
considering these constraints. In addition, the gasoline compositions reported in the original single-
cylinder, research engine experiments [11, 44] were considered to make the surrogate fuels have a
similar hydrocarbon type distribution to the actual test fuels. Table C.1 shows the compositions of
the original and resulting gasoline surrogates. The properties of the two fuels are shown in Table
C.2. It is clear that the RON and MON are matched closely, and the H/C ratio and LHV are matched
reasonably well. The naphthenes, whose composition is less than 5 vol% in the test gasoline, are not
considered in the surrogates.
Table C.1: Gasoline and surrogate fuel compositions (%vol)
Fuel iso-paraffins n-paraffins naphthenes olefins aromatics
Gasoline 42.4 21.2 4.9 5.8 25.7
surrogate 44.6 21.0 0.0 6.5 27.9
Table C.2: Gasoline and surrogate fuel properties
Parameters RON MON H/C ratio LHV (MJ/kg)
Gasoline 87.7 81.5 1.94 42.4
surrogate 87.7 81.5 1.85 43.4
C.4 NO sub-model
NO has been identified as a particularly significant trace species that affect autoignition in spark igni-
tion engines [249–251]. The NO sub-model, however, is not available in most chemical mechanisms,
including the one used in this study [67]. In this case, the NO mechanism proposed by [262] was incor-
porated into the four-component LLNL gasoline mechanism for this study. One repeated elementary
reaction (appearing in both mechanisms) was removed from the NO sub-model, which means the
original gasoline surrogate model remained intact. To confirm this, the ignition delay of the new
160

NO-containing gasoline mechanism and the original LLNL mechanism was modelled in a constant
volume reactor.
The governing equations for a constant volume reactor [47] are expressed as
dTdt
=(Q/V) + RuT ∑ wi −∑ (hiwi)
∑ [[Xi](cp,i − Ru)](C.6)
dPdt
= RuT ∑ wi + Ru ∑ [Xi]dTdt
(C.7)
where T, P, Q, V, and Ru are temperature, pressure, rate of heat transferred to the system, reactor
volume and gas constant, [Xi] represents mole concentration for species i, cp,i and hi denote molar
constant-pressure specific heat and molar enthalpy of species i, and wi is the net production rate for
species i, which can be derived from chemical kinetics. These equations are solved in Matlab using
ode15s solver with kinetic parameters provided by Cantera [263]. Whilst the ignition delay timing
is indicated by the rapid temperature increase, e.g. 400K increase in one time step. The modelled
ignition delays with the original and blended mechanisms are compared in Fig.C.1, which confirms
the integrity of the original mechanism.
1000K/T
0.8 0.9 1 1.1 1.2 1.3 1.4 1.5
Ignitiondelay
time(m
s)
10−1
100
101
102
Blended mechanismOriginal mechanism
Figure C.1: Comparison of the simulated ignition delay of the formulated gasoline surrogate (TableC.1) using the original LLNL model and the extended model containing NO in a constant volumereactor without NO present initially. Equivalence ratio = 1, 30bar, 700-1200K
161

C.5 GT-Power modelling
The single-cylinder, research engine used in the prior, experimental study [11] was first modelled
using GT-Power. The engine model contains the combustion chamber geometry and sub-models for
the intake and exhaust systems. The engine specifications are shown in Table C.3.
Table C.3: Specifications for the single cylinder SI engine [11]
Engine parameter Value
Displaced volume 618.8 cc
Bore 88.5 mm
Stroke 100.6 mm
Connecting rod 166.5 mm
Compression ratio 10:1
Number of valves 4
Inlet valve open 10◦ BTDC at 0.04 mm lift
Inlet valve close 52◦ ABDC at 0.08 mm lift
Exhaust valve open 63.5◦ BBDC at 0.04 mm lift
Exhaust valve close 9.5◦ ATDC at 0.08 mm lift
The experiments modelled in this study were carried out at a constant engine speed (1500 rpm)
and a compression ratio of 10:1. In all experiments, the equivalence ratio was set to unity and the
engine load, represented by the net effective mean pressure (NMEP), was increased by increasing the
intake pressure (Pin). With higher Pin, the spark timing was retarded to shift the combustion phasing,
which is indicated by 50% mass fraction burned or CA50, until the occurrence of trace knock. Fig.C.2
shows example results from these earlier experiments.
The operating parameters (obtained from [11]) for modelled cases in this study are summarised in
Table C.4. Three types of fuels are modelled, including neat gasoline (E0) and two ethanol/gasoline
blends (E20, E50). Besides, two injection methods: DI and UFI are modelled, the latter indicates
fuel injection far upstream of the intake port [11]. Since the intake temperature downstream of UFI
was maintained constant at 52◦C for all experiments, the UFI cases had no charge cooling from fuel
vaporisation.
C.5.1 Full flow model
A full flow model shown in Fig.C.3 was built in GT-Power for the single cylinder engine in [11].
This model simulates the state of the air/fuel mixture at intake valve closure (IVC), particularly the
162
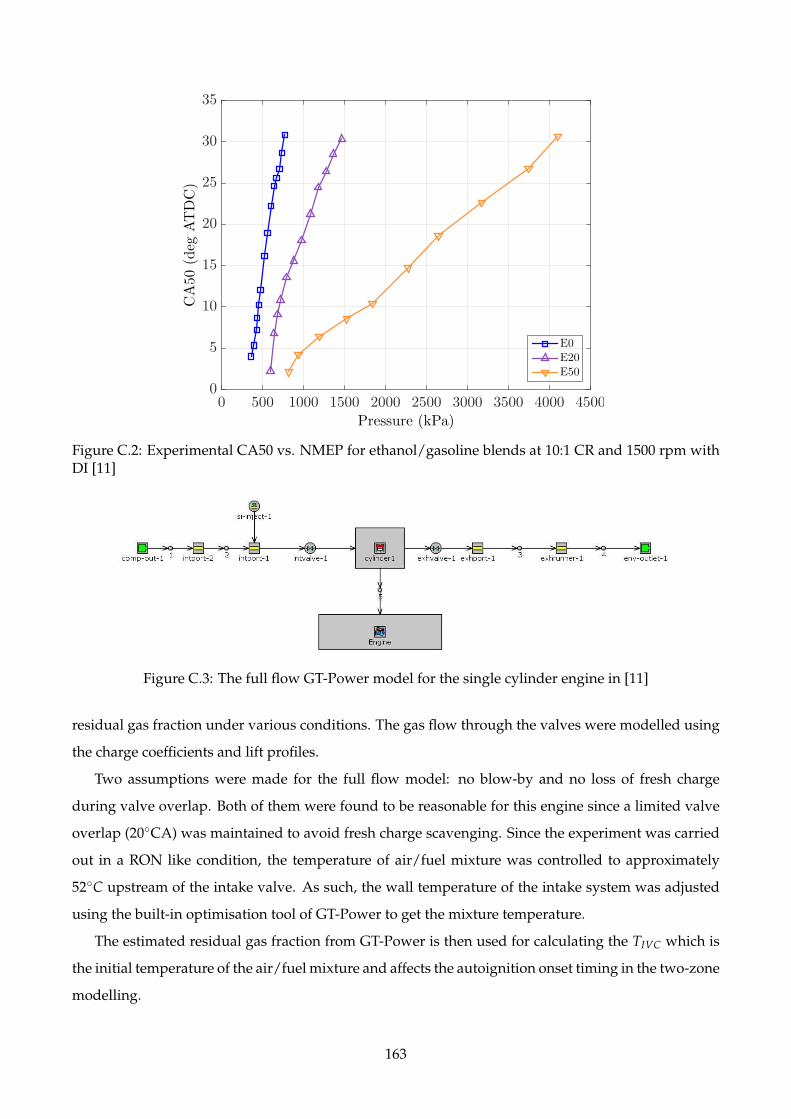
Pressure (kPa)
0 500 1000 1500 2000 2500 3000 3500 4000 4500
CA50(deg
ATDC)
0
5
10
15
20
25
30
35
E0E20E50
Figure C.2: Experimental CA50 vs. NMEP for ethanol/gasoline blends at 10:1 CR and 1500 rpm withDI [11]
Figure C.3: The full flow GT-Power model for the single cylinder engine in [11]
residual gas fraction under various conditions. The gas flow through the valves were modelled using
the charge coefficients and lift profiles.
Two assumptions were made for the full flow model: no blow-by and no loss of fresh charge
during valve overlap. Both of them were found to be reasonable for this engine since a limited valve
overlap (20◦CA) was maintained to avoid fresh charge scavenging. Since the experiment was carried
out in a RON like condition, the temperature of air/fuel mixture was controlled to approximately
52◦C upstream of the intake valve. As such, the wall temperature of the intake system was adjusted
using the built-in optimisation tool of GT-Power to get the mixture temperature.
The estimated residual gas fraction from GT-Power is then used for calculating the TIVC which is
the initial temperature of the air/fuel mixture and affects the autoignition onset timing in the two-zone
modelling.
163
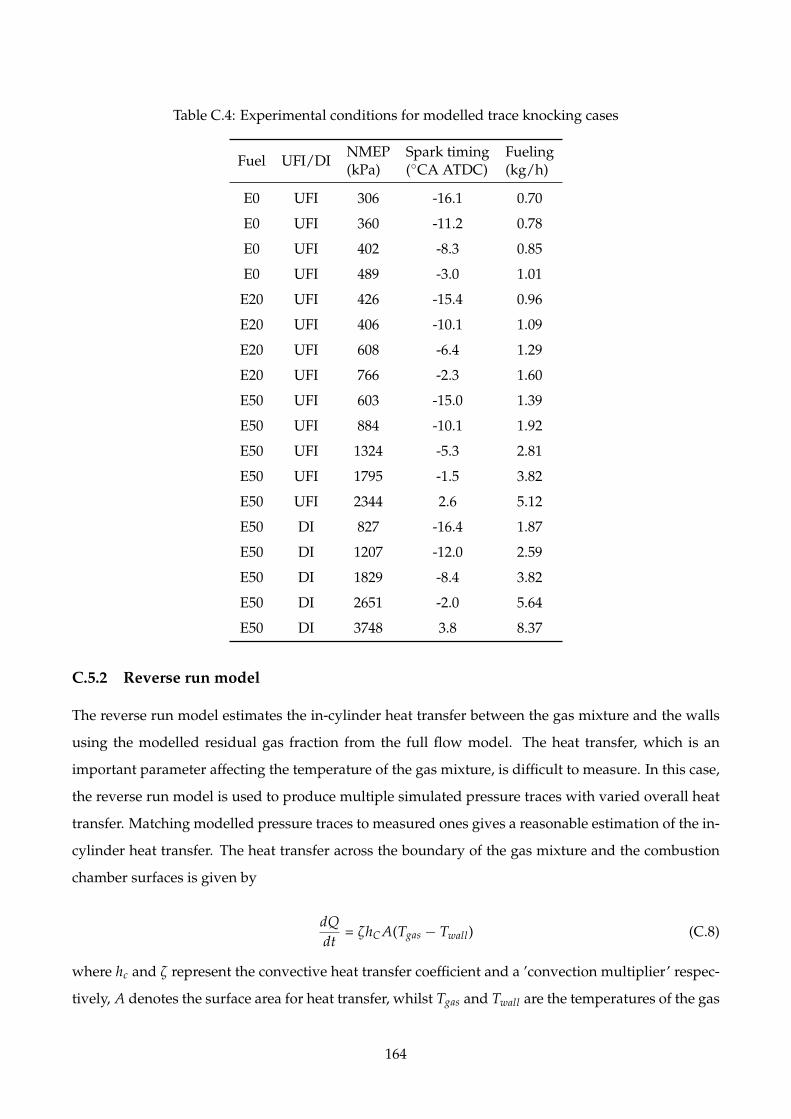
Table C.4: Experimental conditions for modelled trace knocking cases
Fuel UFI/DINMEP(kPa)
Spark timing(◦CA ATDC)
Fueling(kg/h)
E0 UFI 306 -16.1 0.70
E0 UFI 360 -11.2 0.78
E0 UFI 402 -8.3 0.85
E0 UFI 489 -3.0 1.01
E20 UFI 426 -15.4 0.96
E20 UFI 406 -10.1 1.09
E20 UFI 608 -6.4 1.29
E20 UFI 766 -2.3 1.60
E50 UFI 603 -15.0 1.39
E50 UFI 884 -10.1 1.92
E50 UFI 1324 -5.3 2.81
E50 UFI 1795 -1.5 3.82
E50 UFI 2344 2.6 5.12
E50 DI 827 -16.4 1.87
E50 DI 1207 -12.0 2.59
E50 DI 1829 -8.4 3.82
E50 DI 2651 -2.0 5.64
E50 DI 3748 3.8 8.37
C.5.2 Reverse run model
The reverse run model estimates the in-cylinder heat transfer between the gas mixture and the walls
using the modelled residual gas fraction from the full flow model. The heat transfer, which is an
important parameter affecting the temperature of the gas mixture, is difficult to measure. In this case,
the reverse run model is used to produce multiple simulated pressure traces with varied overall heat
transfer. Matching modelled pressure traces to measured ones gives a reasonable estimation of the in-
cylinder heat transfer. The heat transfer across the boundary of the gas mixture and the combustion
chamber surfaces is given by
dQdt
= ζhC A(Tgas − Twall) (C.8)
where hc and ζ represent the convective heat transfer coefficient and a ’convection multiplier’ respec-
tively, A denotes the surface area for heat transfer, whilst Tgas and Twall are the temperatures of the gas
164

mixture and surfaces respectively. In the reverse run model, Woschni’s correlation [264] was used to
calculate the convective heat transfer coefficient, with the convection multiplier to allow calibration of
the Woschni heat transfer model. The Woschni correlation is given by
h = 3.26B−0.2 p0.8T−0.55w0.8 (C.9)
where B is the bore diameter, p denotes the in-cylinder pressure, T represents the temperature, and w
is the average cylinder gas velocity, which is given by
w = C1Sp + C2VdTIVC
pIVCVIVC(p− pm) (C.10)
where Sp is the mean piston speed, Vd is the displaced volumes, pIVC, TIVC and VIVC represent in-
cylinder pressure, temperature and volume at IVC timing respectively, and pm is the motored pressure
with the same crank angle as p. C1 and C2 are 2.28 and 0 during compression, and these values change
to 2.28 and 0.00324 in both combustion and expansion strokes.
In order to calibrate the Woschni’s model, the in-cylinder wall temperatures, which are usually
very difficult to measure, need to be estimated. Therefore, a range of values for Twall and the con-
vection multiplier are examined to test their impact on the agreement between modelling and mea-
surement. In this sensitivity analysis, a single wall temperature is assumed for all contact surfaces for
simplicity, which varied from 383 K to 478 K; while the convection multiplier was set to vary from 0.50
to 1.48. The difference between the modelled pressure traces and measurements from [11] is indicated
by the normalised root mean squared error (RMSE), which is calculated by
RMSE =
√∑(pmeas − psim)2/n
IMEP(C.11)
where pmeas is the measured in-cylinder pressure of the median pressure trace (out of 300 cycles in
the experimental study [11]), psim is the modelled in-cylinder pressure and n is the number of crank
angles.
Fig.C.4 shows examples of the surfaces of RMSE for E0 and E50 with varying convection multiplier
and wall temperature. The dashed line represents the locus of minimum RMSE, i.e., the best agree-
ment between the modelled and measured pressure traces at each wall temperature. It is clear from
Fig.C.4 that convection multiplier only varied slightly in the tested wall temperature range, indicating
the in-cylinder heat transfer is relatively insensitive to wall temperature.
The insensitivity is further confirmed by the unburned gas temperatures from the reverse runs, as
shown in Fig.C.5. The three sets of gas temperature profiles, with Twall varying from 383K to 463K,
look identical before reaching the peak. Although they start to deviate after the peak temperature, the
165

1.5
1.5
2
2
2.5
2.5
3
3
3.5
3.54
4.55
5.5
6 6
Temperature (K)
383 393 403 413 423 433 443 453 463 473
Convectionmultiplier
0.5
0.6
0.7
0.8
0.9
1
1.1
1.2
1.3
1.4
(a) E0.UFI, NMEP=402kPa
1
1
1.5
1.5
2
2
2.5
2.5
3
3
3.5
3.5
4
4
4.5
4.5
5
5
Temperature (K)
383 393 403 413 423 433 443 453 463 473
Convectionmultiplier
0.5
0.6
0.7
0.8
0.9
1
1.1
1.2
1.3
1.4
(b) E50.UFI, NMEP=1324kPa
Figure C.4: The sensitivity analysis for the convection multiplier, to the cylinder wall temperature,Twall . Dashed lines represent the minimal RMSE at each wall temperature. RMSE values (×104) areindicated by the numbers on the contours
absolute differences are still quite small at the end of combustion.
Crank Angle (deg)
-20 0 20 40 60 80
Tem
perature
(K)
550
600
650
700
750
800
850
900
950
1000383K423K463K
(a) E0.UFI, NMEP=402kPa
Crank Angle (deg)
-20 0 20 40 60 80 100
Tem
perature
(K)
550
600
650
700
750
800
850
900
950383K423K463K
(b) E50.UFI, NMEP=1324kPa
Figure C.5: Unburned gas temperature profiles at different wall temperatures from the GT-Powerreverse run
Based on the results from Fig.C.4 and C.5, it is reasonable to assume a constant wall temperature
of 423K for all modelling in this work, with the convection multiplier optimised for a given measured
pressure trace. This assumption significantly simplifies the reverse run modelling, since the only pa-
rameter needs to be varied is the convection multiplier when estimating the in-cylinder heat transfer.
Fig.C.6 then shows the close agreement between the resulting modelled pressure traces and those
measured over the range of fuels in this study. Besides, the mass fraction burned (MFB) profiles were
derived from the matched modelled pressure trace. Therefore, a systematic approach estimating the
166
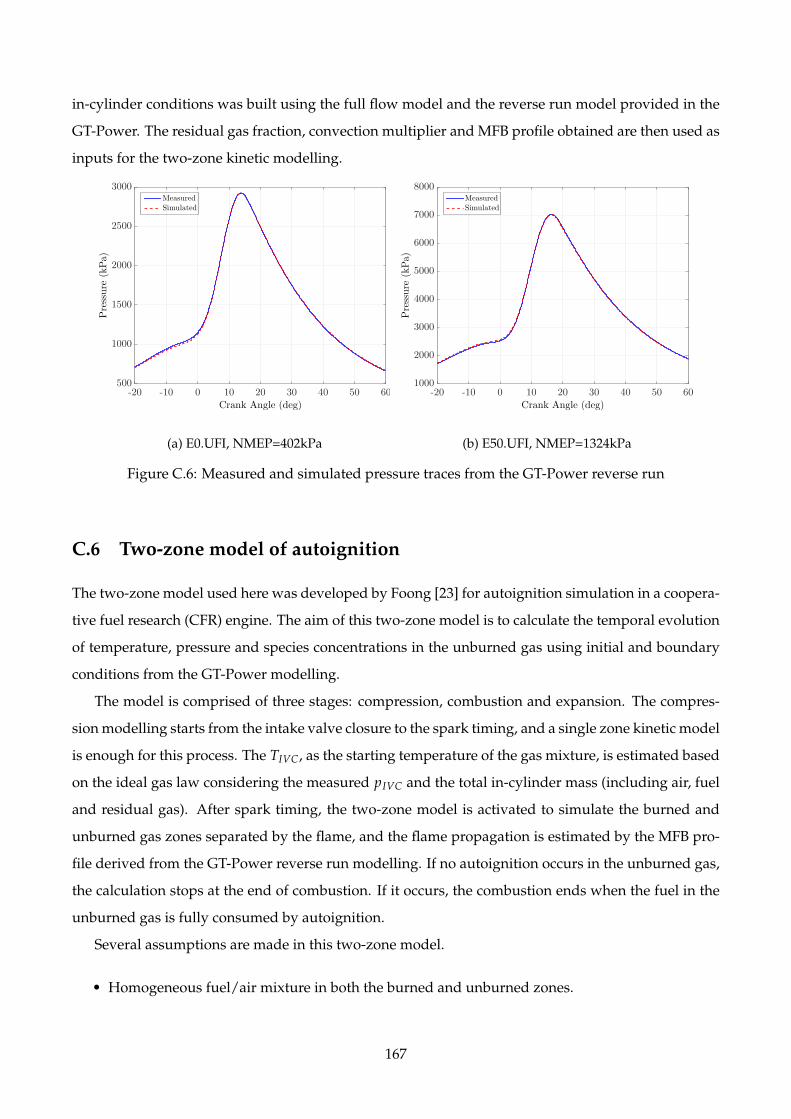
in-cylinder conditions was built using the full flow model and the reverse run model provided in the
GT-Power. The residual gas fraction, convection multiplier and MFB profile obtained are then used as
inputs for the two-zone kinetic modelling.
Crank Angle (deg)
-20 -10 0 10 20 30 40 50 60
Pressure
(kPa)
500
1000
1500
2000
2500
3000MeasuredSimulated
(a) E0.UFI, NMEP=402kPa
Crank Angle (deg)
-20 -10 0 10 20 30 40 50 60Pressure
(kPa)
1000
2000
3000
4000
5000
6000
7000
8000MeasuredSimulated
(b) E50.UFI, NMEP=1324kPa
Figure C.6: Measured and simulated pressure traces from the GT-Power reverse run
C.6 Two-zone model of autoignition
The two-zone model used here was developed by Foong [23] for autoignition simulation in a coopera-
tive fuel research (CFR) engine. The aim of this two-zone model is to calculate the temporal evolution
of temperature, pressure and species concentrations in the unburned gas using initial and boundary
conditions from the GT-Power modelling.
The model is comprised of three stages: compression, combustion and expansion. The compres-
sion modelling starts from the intake valve closure to the spark timing, and a single zone kinetic model
is enough for this process. The TIVC, as the starting temperature of the gas mixture, is estimated based
on the ideal gas law considering the measured pIVC and the total in-cylinder mass (including air, fuel
and residual gas). After spark timing, the two-zone model is activated to simulate the burned and
unburned gas zones separated by the flame, and the flame propagation is estimated by the MFB pro-
file derived from the GT-Power reverse run modelling. If no autoignition occurs in the unburned gas,
the calculation stops at the end of combustion. If it occurs, the combustion ends when the fuel in the
unburned gas is fully consumed by autoignition.
Several assumptions are made in this two-zone model.
• Homogeneous fuel/air mixture in both the burned and unburned zones.
167

• The volume of the flame is neglected, and therefore mass transfer and enthalpy exchange be-
tween the two zones occur instantaneously.
• The flame is at chemical equilibrium.
• No heat transfer between the two zones.
The two-zone model was implemented in MATLAB, with kinetic information derived from Can-
tera 1.8 [265].
C.7 Modelling of trace knock
C.7.1 Raw pressure data
Trace knock, which has no distinct pressure jump, is the borderline between knocking and non-
knocking combustion. If the spark timing is advanced slightly, e.g. 1 ◦CA, the engine combustion
may transfer from non-knocking to knocking and vice versa. To have a good understanding on trace
knock, the raw pressure data from trace knocking conditions are compared a ’typical’ knocking pres-
sure trace. Fig.C.7(a) shows a typical knocking raw pressure trace for isooctane at so-called ’standard
knock intensity’ condition in our CFR engine, which is from [23] and represents a condition in which
autoignition and knock occur in all cycles. The trace knock results in Fig.C.7(c) and (e) are for the
E0 and E50 cases as shown in Fig.C.6. The pressure traces selected for trace knock conditions are
the most advanced ones, which have the most intensive autoignition comparing to other relatively
retarded traces. It would be interesting to investigate whether the characteristics of typical knock-
ing pressure trace repeat themselves on trace knocks. Band-pass filtering and Fast Fourier Transform
(FFT) are two common techniques used to characterise knocking pressure traces, and their results are
shown in Fig.C.7 as well. The band-pass filter has high-pass and cut-off frequencies of 4kHz and
25kHz respectively.
Fig.C.7(a) shows the commonly observed rapid change in the pressure rise rate and the resulting
pressure oscillations for the knocking of isooctane in the CFR engine. Nevertheless, such oscillations
cannot be seen for the trace knocking cycles for both E0, UFI, NMEP=402kPa (Fig.C.7(c)) and E50,
UFI, NMEP=1324kPa (Fig.C.7(e)) in the single-cylinder Ford engine, most likely because trace knock is
intermittent. Meanwhile, the FFT result of the typical knocking pressure trace of isooctane has several
clear peaks at different frequencies, as shown in Fig.C.7(b), which correspond to various vibration
modes reported by [266].The equation used to predict these peak frequencies is given by
fm,n =Cρm,n
πB(C.12)
168

(a)
Frequency (kHz)
4 6 8 10 12 14 16 18
Magnitude
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
(b)
(c)
Frequency (kHz)
4 6 8 10 12 14 16 18 20 22 24
Magnitude
0
0.1
0.2
0.3
0.4
0.5
(d)
(e)
Frequency (kHz)
4 6 8 10 12 14 16 18 20 22 24
Magnitude
0
0.1
0.2
0.3
0.4
0.5
(f)
Figure C.7: Raw and band pass filtered pressure traces (left), and power spectra from Fast FourierTransform (FFT) analysis (right) for the most advanced pressure traces under standard knocking forisooctane in a CFR engine (a and b), and under trace knocking for E0, UFI, NMEP=402kPa (c and d)and E50, UFI, NMEP=1324kPa (e and f) in a single-cylinder engine from the experimental study [11]
169
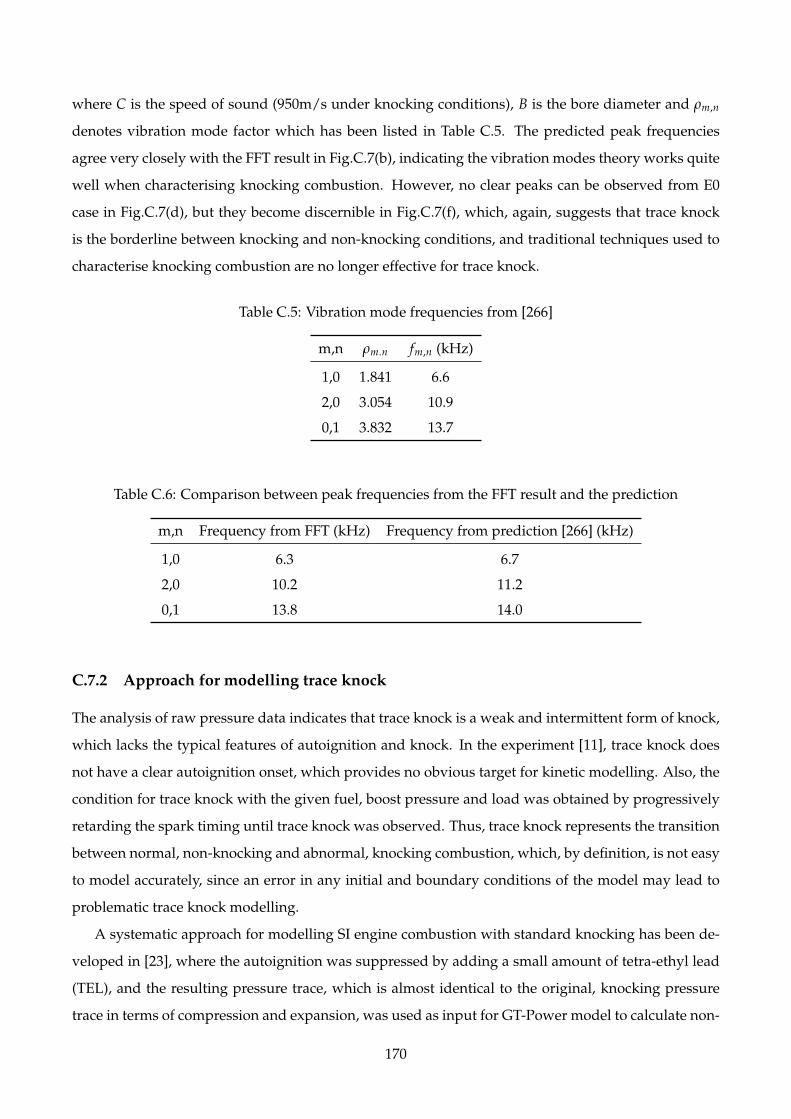
where C is the speed of sound (950m/s under knocking conditions), B is the bore diameter and ρm,n
denotes vibration mode factor which has been listed in Table C.5. The predicted peak frequencies
agree very closely with the FFT result in Fig.C.7(b), indicating the vibration modes theory works quite
well when characterising knocking combustion. However, no clear peaks can be observed from E0
case in Fig.C.7(d), but they become discernible in Fig.C.7(f), which, again, suggests that trace knock
is the borderline between knocking and non-knocking conditions, and traditional techniques used to
characterise knocking combustion are no longer effective for trace knock.
Table C.5: Vibration mode frequencies from [266]
m,n ρm.n fm,n (kHz)
1,0 1.841 6.6
2,0 3.054 10.9
0,1 3.832 13.7
Table C.6: Comparison between peak frequencies from the FFT result and the prediction
m,n Frequency from FFT (kHz) Frequency from prediction [266] (kHz)
1,0 6.3 6.7
2,0 10.2 11.2
0,1 13.8 14.0
C.7.2 Approach for modelling trace knock
The analysis of raw pressure data indicates that trace knock is a weak and intermittent form of knock,
which lacks the typical features of autoignition and knock. In the experiment [11], trace knock does
not have a clear autoignition onset, which provides no obvious target for kinetic modelling. Also, the
condition for trace knock with the given fuel, boost pressure and load was obtained by progressively
retarding the spark timing until trace knock was observed. Thus, trace knock represents the transition
between normal, non-knocking and abnormal, knocking combustion, which, by definition, is not easy
to model accurately, since an error in any initial and boundary conditions of the model may lead to
problematic trace knock modelling.
A systematic approach for modelling SI engine combustion with standard knocking has been de-
veloped in [23], where the autoignition was suppressed by adding a small amount of tetra-ethyl lead
(TEL), and the resulting pressure trace, which is almost identical to the original, knocking pressure
trace in terms of compression and expansion, was used as input for GT-Power model to calculate non-
170
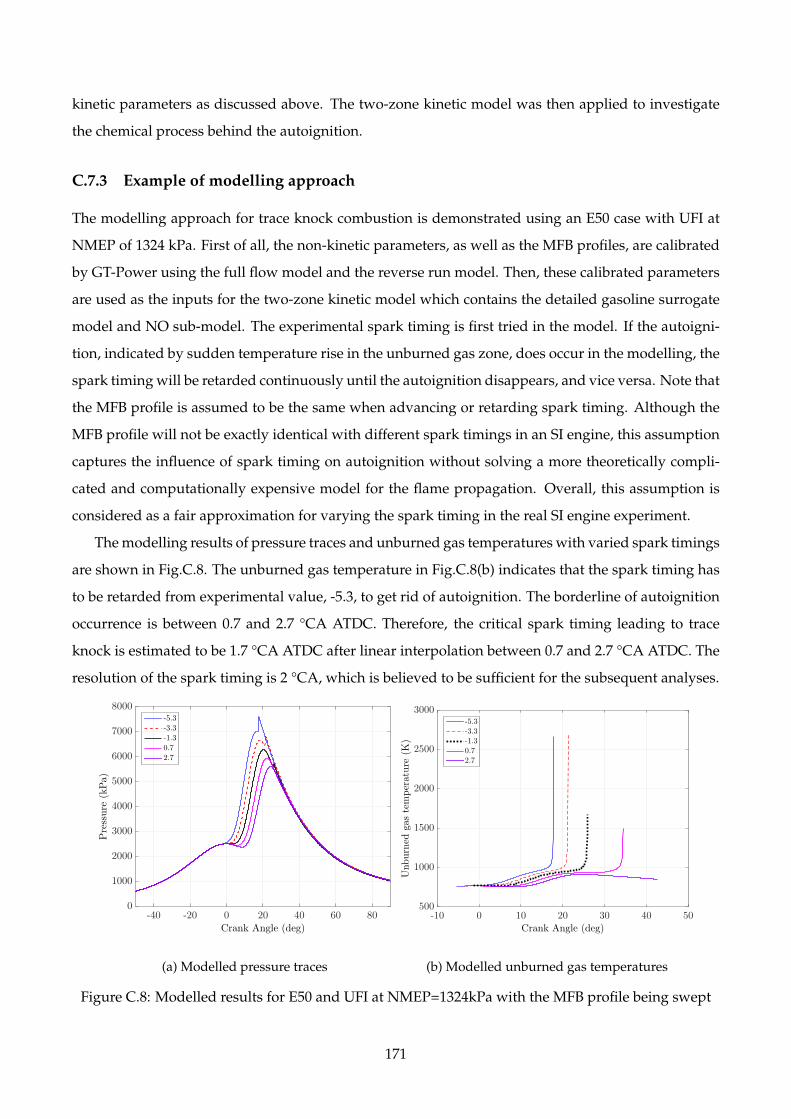
kinetic parameters as discussed above. The two-zone kinetic model was then applied to investigate
the chemical process behind the autoignition.
C.7.3 Example of modelling approach
The modelling approach for trace knock combustion is demonstrated using an E50 case with UFI at
NMEP of 1324 kPa. First of all, the non-kinetic parameters, as well as the MFB profiles, are calibrated
by GT-Power using the full flow model and the reverse run model. Then, these calibrated parameters
are used as the inputs for the two-zone kinetic model which contains the detailed gasoline surrogate
model and NO sub-model. The experimental spark timing is first tried in the model. If the autoigni-
tion, indicated by sudden temperature rise in the unburned gas zone, does occur in the modelling, the
spark timing will be retarded continuously until the autoignition disappears, and vice versa. Note that
the MFB profile is assumed to be the same when advancing or retarding spark timing. Although the
MFB profile will not be exactly identical with different spark timings in an SI engine, this assumption
captures the influence of spark timing on autoignition without solving a more theoretically compli-
cated and computationally expensive model for the flame propagation. Overall, this assumption is
considered as a fair approximation for varying the spark timing in the real SI engine experiment.
The modelling results of pressure traces and unburned gas temperatures with varied spark timings
are shown in Fig.C.8. The unburned gas temperature in Fig.C.8(b) indicates that the spark timing has
to be retarded from experimental value, -5.3, to get rid of autoignition. The borderline of autoignition
occurrence is between 0.7 and 2.7 °CA ATDC. Therefore, the critical spark timing leading to trace
knock is estimated to be 1.7 °CA ATDC after linear interpolation between 0.7 and 2.7 °CA ATDC. The
resolution of the spark timing is 2 °CA, which is believed to be sufficient for the subsequent analyses.
Crank Angle (deg)
-40 -20 0 20 40 60 80
Pressure
(kPa)
0
1000
2000
3000
4000
5000
6000
7000
8000-5.3
-3.3
-1.3
0.7
2.7
(a) Modelled pressure traces
Crank Angle (deg)
-10 0 10 20 30 40 50
Unburned
gastemperature
(K)
500
1000
1500
2000
2500
3000-5.3
-3.3
-1.3
0.7
2.7
(b) Modelled unburned gas temperatures
Figure C.8: Modelled results for E50 and UFI at NMEP=1324kPa with the MFB profile being swept
171
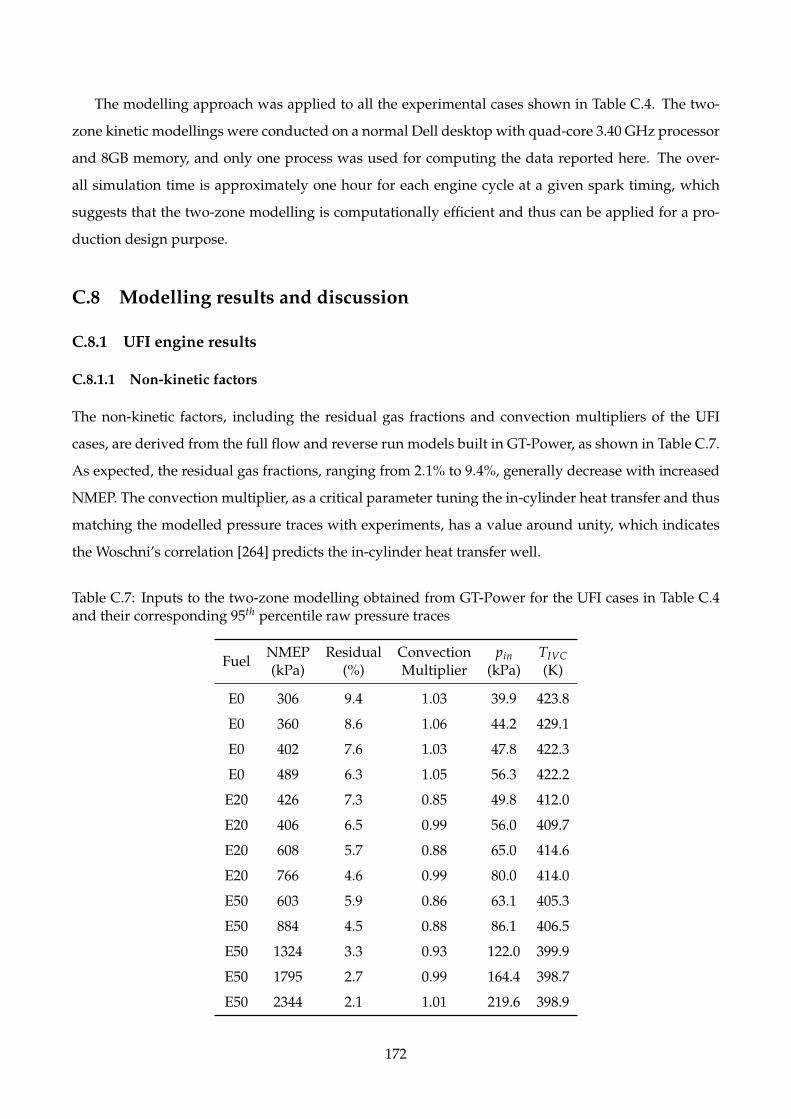
The modelling approach was applied to all the experimental cases shown in Table C.4. The two-
zone kinetic modellings were conducted on a normal Dell desktop with quad-core 3.40 GHz processor
and 8GB memory, and only one process was used for computing the data reported here. The over-
all simulation time is approximately one hour for each engine cycle at a given spark timing, which
suggests that the two-zone modelling is computationally efficient and thus can be applied for a pro-
duction design purpose.
C.8 Modelling results and discussion
C.8.1 UFI engine results
C.8.1.1 Non-kinetic factors
The non-kinetic factors, including the residual gas fractions and convection multipliers of the UFI
cases, are derived from the full flow and reverse run models built in GT-Power, as shown in Table C.7.
As expected, the residual gas fractions, ranging from 2.1% to 9.4%, generally decrease with increased
NMEP. The convection multiplier, as a critical parameter tuning the in-cylinder heat transfer and thus
matching the modelled pressure traces with experiments, has a value around unity, which indicates
the Woschni’s correlation [264] predicts the in-cylinder heat transfer well.
Table C.7: Inputs to the two-zone modelling obtained from GT-Power for the UFI cases in Table C.4and their corresponding 95th percentile raw pressure traces
FuelNMEP(kPa)
Residual(%)
ConvectionMultiplier
pin(kPa)
TIVC(K)
E0 306 9.4 1.03 39.9 423.8
E0 360 8.6 1.06 44.2 429.1
E0 402 7.6 1.03 47.8 422.3
E0 489 6.3 1.05 56.3 422.2
E20 426 7.3 0.85 49.8 412.0
E20 406 6.5 0.99 56.0 409.7
E20 608 5.7 0.88 65.0 414.6
E20 766 4.6 0.99 80.0 414.0
E50 603 5.9 0.86 63.1 405.3
E50 884 4.5 0.88 86.1 406.5
E50 1324 3.3 0.93 122.0 399.9
E50 1795 2.7 0.99 164.4 398.7
E50 2344 2.1 1.01 219.6 398.9
172

C.8.1.2 Effect of ethanol content
The critical spark timings for trace knocks of all UFI cases listed in Table C.7 were modelled by the
two-zone kinetic model, and the results were plotted against NMEP for E0, E20 and E50, as shown in
Fig.C.9. The most advanced, the 95th percentile and the median pressure traces were modelled for all
cases in Table C.7. In general, similar trends are observed from the three different raw pressure traces
regarding the variation of spark timing for knock limited combustion with NMEP. However, absolute,
systematic differences between experimental and modelling exist in all cases.
The largest systematic differences between the modelled and measured spark timings come from
the most advanced traces, which is probably due to two factors: the burning rates are faster with the
most advanced traces, leading to more rapid compression of the unburned gas, and the convection
multipliers for more advanced cycles are smaller, resulting in less heat loss, both of which increase
the unburned gas temperature and thus more retarded critical spark timing is required to get rid of
autoignition. Although the autoignition in the unburned gas zone is used to infer the critical spark
timing for the trace knock, it may not necessarily lead to knocking combustion, since the mass in the
unburned gas zone may not be sufficient to produce knock if the autoignition event occurs too late.
In this regard, some additional constrains should be imposed to guarantee the occurrence of knock,
which helps to reduce the gap between the modelling and experiment. An example given by [256]
suggests that the autoignition should occur early enough and the mass left in the end gas should be
sufficient to produce engine knock. However, it can be arbitrary and subjective to quantify ’early
timing’ and ’sufficient mass’, which will not be explored in this work.
Note that the differences of critical spark timings between the three types of raw pressure traces
are overall consistent across the NMEP range. However, larger discrepancies are found at relatively
low loads, which might result from greater randomness in those most advanced pressure traces under
such conditions. Considering the similar and systematic differences in the modelled critical spark
timing for trace knock using the most advanced, 95th percentile and the median pressure traces, the
95th percentile trace, unaffected by the randomness and representative of the advanced cycles having
trace knock, is applied in the rest modelling of this study.
The modelled critical spark timings using the 95th percentile advanced pressure traces are com-
pared with measurements in Fig.C.10. It is evident that the modelled spark timings are consistently
later than measured ones. Besides the choice of the representative trace and aforementioned non-
kinetic modelling assumptions, two possible causes will be elaborated here.
First, the experiment [11] used a pressure-based criterion to determine trace knock, which might be
less stringent than the one used in the two-zone kinetic modelling. As mentioned before and shown
in Fig.C.8(b), the critical spark timing in the modelling was obtained by analysing the temperature
173
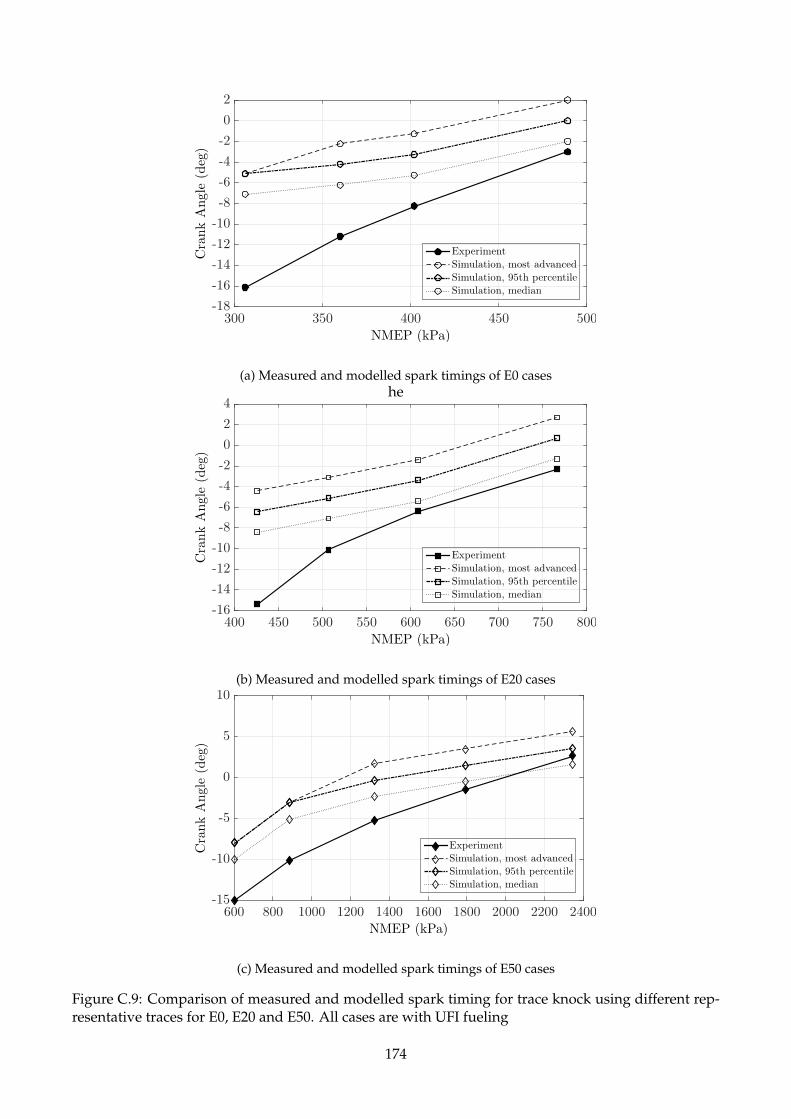
NMEP (kPa)
300 350 400 450 500
CrankAngle
(deg)
-18
-16
-14
-12
-10
-8
-6
-4
-2
0
2
Experiment
Simulation, most advanced
Simulation, 95th percentile
Simulation, median
(a) Measured and modelled spark timings of E0 caseshe
NMEP (kPa)
400 450 500 550 600 650 700 750 800
CrankAngle
(deg)
-16
-14
-12
-10
-8
-6
-4
-2
0
2
4
Experiment
Simulation, most advanced
Simulation, 95th percentile
Simulation, median
(b) Measured and modelled spark timings of E20 cases
NMEP (kPa)
600 800 1000 1200 1400 1600 1800 2000 2200 2400
CrankAngle
(deg)
-15
-10
-5
0
5
10
Experiment
Simulation, most advanced
Simulation, 95th percentile
Simulation, median
(c) Measured and modelled spark timings of E50 cases
Figure C.9: Comparison of measured and modelled spark timing for trace knock using different rep-resentative traces for E0, E20 and E50. All cases are with UFI fueling
174

NMEP (kPa)
0 500 1000 1500 2000 2500
CrankAngle
(deg)
-20
-15
-10
-5
0
5
E0, experiment
E0, simulation
E20, experiment
E20, simulation
E50, experiment
E50, simulation
Figure C.10: Variation of modelled (using the 95th percentile advanced trace) and measured sparktiming for trace knock for E0, E20 and E50
rise in the unburned gas zone. However, the comparison between Fig.C.8(a) and (b) suggests that a
temperature rise in the unburned gas zone may not always lead to a discernible pressure jump when
the mass left in the unburned gas zone at autoignition is not enough. Fig.C.11 shows that the MFB at
autoignition timing is approximately 90% for all cases modelled in this work.
NMEP (kPa)
0 500 1000 1500 2000 2500 3000 3500 4000
MFB
atautoignitiontiming
0.5
0.55
0.6
0.65
0.7
0.75
0.8
0.85
0.9
0.95
1
E0, UFI
E20, UFI
E50, UFI
E50, DI
Figure C.11: MFB at autoignition for spark timings that are one degree earlier than the spark timingfor trace knock
175

Second, although the gasoline surrogate model from LLNL captured the effect of increasing NMEP
and ethanol addition on trace knock very well, it might be too reactive and thus results in retarded
critical spark timing. Further investigations on this kinetic model are necessary to address this possi-
bility, which is not within the scope of this study. Since the variations in the modelled spark timing
with the different choices of raw pressure traces are comparable to the discrepancies between any
of the modelling results and corresponding experimental measurement, it is difficult to categorically
evaluate the accuracy of the kinetic model due to other modelling uncertainties.
C.8.2 DI engine results
Direct injection (DI) reduces the temperature of the fresh charge by taking advantage of the fuel’s
charge cooling effect and thus helps to suppress engine knock. Since the charge cooling effect of
ethanol is more significant than other hydrocarbons in gasoline, it is of great interest to introduce
ethanol containing mixtures to the SI engine using DI.
In the experimental study [11], fuel was injected during the intake stroke which was well before
IVC (52 °CA ABDC). Thus, it is reasonable to assume the fuel is fully vaporised before IVC. This
indicates that the temperature difference at IVC between UFI and DI with similar NMEP is reflected
by the measured in-cylinder pressure based on the ideal gas law, and the same overall modelling
approach for UFI cases can be applied directly to DI cases. To be consistent, the 95th percentile most
advanced pressure traces were chosen for DI modelling, and the non-kinetic parameters derived from
GT-Power are listed in Table C.8.
Table C.8: Inputs to the two-zone modelling obtained from GT-Power for the DI cases in Table C.4 andthe 95th percentile raw pressure traces
NMEP(kPa)
Residual(%)
ConvectionMultiplier
pin(kPa)
TIVC(K)
827 5.0 0.89 76.5 380.1
1207 3.71 1.06 104.5 376.8
1829 2.61 0.99 152.2 369.7
2651 1.80 1.12 219.6 366.7
3748 1.24 1.13 319.1 365.5
The modelled and measured spark timings for trace knock of UFI and DI with E50 are shown in
Fig.C.12. Similar to the experimental observations, the modelling results show that the DI improves
knock-limited performance for SI engine. The differences in TIVC, which are approximately 25-30 K
lower with DI due to charge cooling effect, cause the differences between the modelled UFI and DI
176
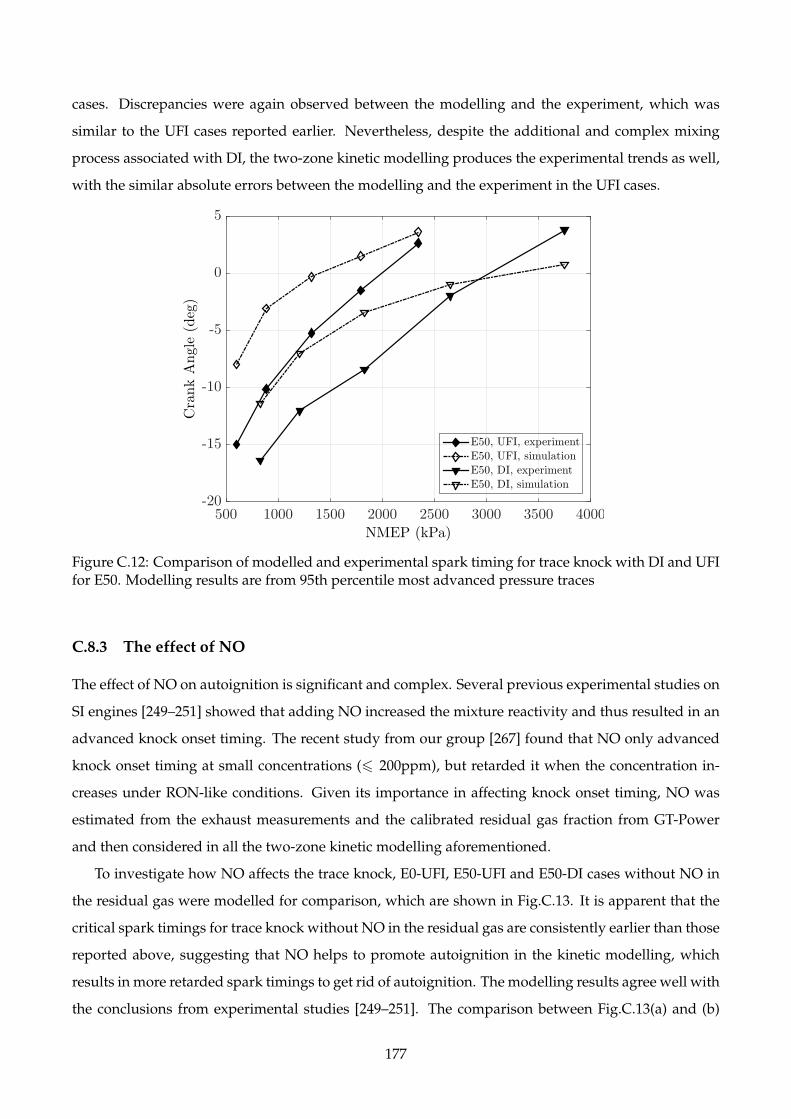
cases. Discrepancies were again observed between the modelling and the experiment, which was
similar to the UFI cases reported earlier. Nevertheless, despite the additional and complex mixing
process associated with DI, the two-zone kinetic modelling produces the experimental trends as well,
with the similar absolute errors between the modelling and the experiment in the UFI cases.
NMEP (kPa)
500 1000 1500 2000 2500 3000 3500 4000
CrankAngle
(deg)
-20
-15
-10
-5
0
5
E50, UFI, experiment
E50, UFI, simulation
E50, DI, experiment
E50, DI, simulation
Figure C.12: Comparison of modelled and experimental spark timing for trace knock with DI and UFIfor E50. Modelling results are from 95th percentile most advanced pressure traces
C.8.3 The effect of NO
The effect of NO on autoignition is significant and complex. Several previous experimental studies on
SI engines [249–251] showed that adding NO increased the mixture reactivity and thus resulted in an
advanced knock onset timing. The recent study from our group [267] found that NO only advanced
knock onset timing at small concentrations (6 200ppm), but retarded it when the concentration in-
creases under RON-like conditions. Given its importance in affecting knock onset timing, NO was
estimated from the exhaust measurements and the calibrated residual gas fraction from GT-Power
and then considered in all the two-zone kinetic modelling aforementioned.
To investigate how NO affects the trace knock, E0-UFI, E50-UFI and E50-DI cases without NO in
the residual gas were modelled for comparison, which are shown in Fig.C.13. It is apparent that the
critical spark timings for trace knock without NO in the residual gas are consistently earlier than those
reported above, suggesting that NO helps to promote autoignition in the kinetic modelling, which
results in more retarded spark timings to get rid of autoignition. The modelling results agree well with
the conclusions from experimental studies [249–251]. The comparison between Fig.C.13(a) and (b)
177
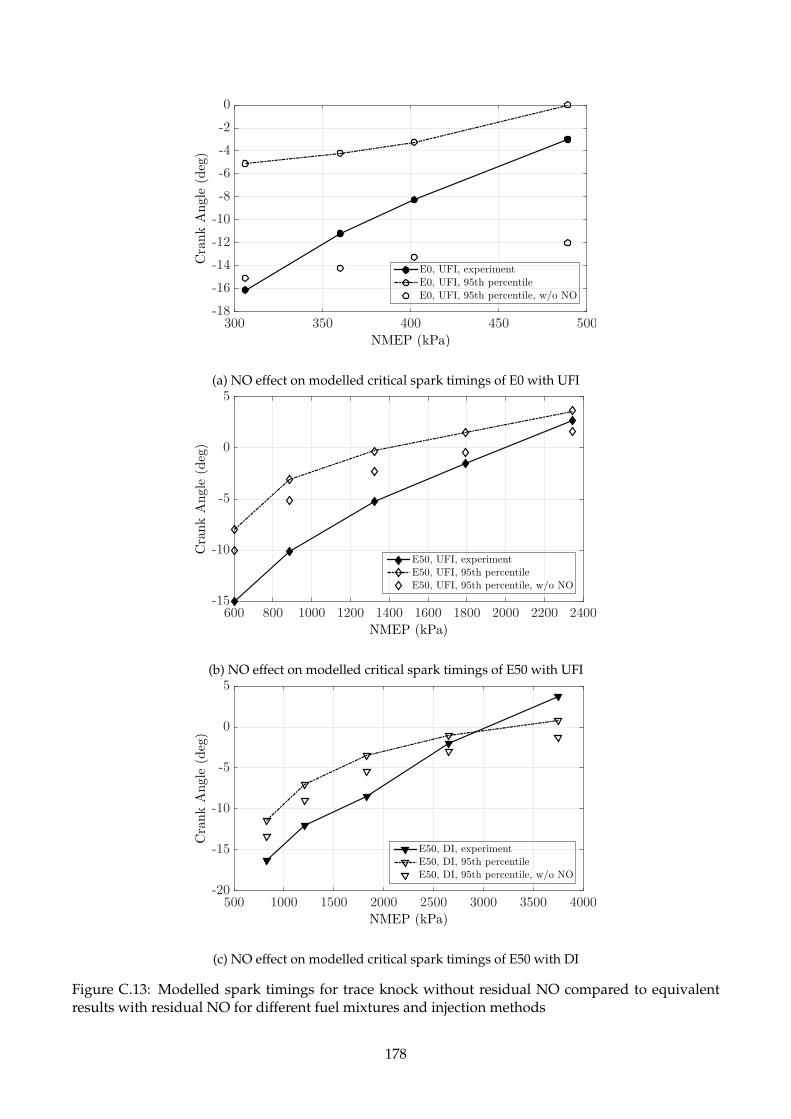
NMEP (kPa)
300 350 400 450 500
CrankAngle
(deg)
-18
-16
-14
-12
-10
-8
-6
-4
-2
0
E0, UFI, experimentE0, UFI, 95th percentileE0, UFI, 95th percentile, w/o NO
(a) NO effect on modelled critical spark timings of E0 with UFI
NMEP (kPa)
600 800 1000 1200 1400 1600 1800 2000 2200 2400
CrankAngle
(deg)
-15
-10
-5
0
5
E50, UFI, experimentE50, UFI, 95th percentileE50, UFI, 95th percentile, w/o NO
(b) NO effect on modelled critical spark timings of E50 with UFI
NMEP (kPa)
500 1000 1500 2000 2500 3000 3500 4000
CrankAngle
(deg)
-20
-15
-10
-5
0
5
E50, DI, experimentE50, DI, 95th percentileE50, DI, 95th percentile, w/o NO
(c) NO effect on modelled critical spark timings of E50 with DI
Figure C.13: Modelled spark timings for trace knock without residual NO compared to equivalentresults with residual NO for different fuel mixtures and injection methods
178

shows that the effect of NO is more significant for E0 than E50, as the spark advance is approximately
11-12 °CA for E0 but only 2 °CA for E50, which indicates that the interactions between NO and the
hydrocarbon fuels may be weakened by ethanol at low temperatures. In general, it is necessary to
include the NO sub-model for accurate autoignition modelling.
C.9 Summary
This study proposed a systematic numerical approach for trace knock modelling in a modern, single-
cylinder SI engine fuelled by ethanol/gasoline mixture. GT-Power and two-zone model were used
for the calibration of non-kinetic parameters and the prediction of autoignition onset timing respec-
tively. The modelling results agreed well with the experimental data [11] in terms of the trend of the
critical spark timings for trace knock under various conditions. The two-zone kinetic model used an
ethanol-containing gasoline surrogate mechanism [67] coupled with a NO sub-model [262] to simulate
autoignition in the unburned gas zone. For the purpose of modelling trace knock, the mass fraction
burned (MFB) profile from GT-Power was shifted accordingly with the change of the spark timing
until no autoignition occurred, and the critical spark timing was then obtained for trace knock.
The influence of ethanol on the autoignition, including its low autoignition reactivity and high
charge cooling, was first investigated by modelling upstream, pre-vaporized fuel injection (UFI) and
direct injection (DI) cases. As expected, the charge cooling effects of ethanol significantly improve
knock-limited performance with DI. Although some systematic, absolute differences do exist when
comparing modelled critical spark timings with those from the measurements, the relative trends
with engine load and ethanol content were well captured. Also, the effect of NO on knock-limited
combustion was investigated. The results are consistent with the experimental findings from the liter-
ature, but ethanol appears to weaken the interactions between NO and the hydrocarbon fuels at low
temperatures.
179

Appendix D
The kinetic model for the flow reactor
The governing equations for the PFR, ignoring heat loss and surface chemistry for simplicity, are
expressed in the format of the ODE system [47], as shown from Eqn.D.1 to D.3.
(D.1)dρ
dx=
1−Ru
cpMWmix
ρ2v2x
1
A
dA
dx
+ρRu
vxcp MWmix∑N
i=1 MWiωi
hi −MWmix
MWicpT
P
1 +v2
x
cpT
− ρv2x
(D.2)dT
dx=
v2x
ρcp
dρ
dx+
v2x
cp
1
A
dA
dx
− 1
vxρcp
N
∑i=1
hiωi MWi −Q′′C
mcp
(D.3)dYi
dx=
ωi MWi
ρvx
where ρ, T, Yi and x indicate the mixture density, temperature, mass fraction of species i and reactor
length, P is the reactor pressure, cp is the constant-pressure specific heat, MWmix is molecular weight
of the mixture, Ru is the universal gas constant, A is the cross section area of the reactor tube (constant
in our PFR), vx is the axial gas velocity, hi is the specific enthalpy of species i, Q′′
is the heat loss to
the surrounding, C is the circumference of the reactor tube, m is the total mass flow rate, and ωi is the
molar net production rate.
In this study, Eqn.D.1 and D.3 are solved with the VODE solver [268] in Python when carrying out
the global sensitivity analysis on a high performance computing (HPC) system named FIREBOX in our
group. Note that the temperature change with reactor length is provided from the experiments and
thus Eqn.D.2 is no longer needed. It generally takes twelve hours to analyse a chemical mechanism
containing approximately 10,000 elementary reactions using 30 cores and 100 GB memory. To validate
180

this in-house model, the modelled results for isooctane oxidation in the PFR using Chemkin and the
developed model are compared in Fig.D.1. The almost identical results suggest that the in-house
model is good enough to replace Chemkin in the PFR modelling.
0 200 400 600 800 10000
1
2
3
4
510-3
Figure D.1: The comparison between the modelled results of the neat isooctane oxidation at 900 K and10 bar in the PFR using Chemkin and the model developed in this study
181