null mutation of connexin43 causes slow propagation of ventricular activation in the late stages of...
TRANSCRIPT

Gregory E. Morley and José JalifeDhananjay Vaidya, Houman S. Tamaddon, Cecilia W. Lo, Steven M. Taffet, Mario Delmar,
Late Stages of Mouse Embryonic DevelopmentNull Mutation of Connexin43 Causes Slow Propagation of Ventricular Activation in the
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2001 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/hh1101.0911072001;88:1196-1202; originally published online May 24, 2001;Circ Res.
http://circres.ahajournals.org/content/88/11/1196World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2001/05/17/hh1101.091107.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on October 8, 2014http://circres.ahajournals.org/Downloaded from by guest on October 8, 2014http://circres.ahajournals.org/Downloaded from

Null Mutation of Connexin43 Causes Slow Propagation ofVentricular Activation in the Late Stages of Mouse
Embryonic DevelopmentDhananjay Vaidya, Houman S. Tamaddon, Cecilia W. Lo, Steven M. Taffet, Mario Delmar,
Gregory E. Morley, José Jalife
Abstract—Connexin43 (Cx43) is the principal connexin isoform in the mouse ventricle, where it is thought to provideelectrical coupling between cells. Knocking out this gene results in anatomic malformations that nevertheless allow forsurvival through early neonatal life. We examined electrical wave propagation in the left (LV) and right (RV) ventriclesof isolated Cx43 null mutated (Cx432/2), heterozygous (Cx431/2), and wild-type (WT) embryos using high-resolutionmapping of voltage-sensitive dye fluorescence. Consistent with the compensating presence of the other connexins, noreduction in propagation velocity was seen in Cx432/2 ventricles at postcoital day (dpc) 12.5 compared with WT orCx431/2 ventricles. A gross reduction in conduction velocity was seen in the RV at 15.5 dpc (in cm/second, mean [1SE confidence interval], WT 9.9 [8.7 to 11.2], Cx431/2 9.9 [9.0 to 10.9], and Cx432/2 2.2 [1.8 to 2.7;P,0.005]) andin both ventricles at 17.5 dpc (in RV, WT 8.4 [7.6 to 9.3], Cx431/2 8.7 [8.1 to 9.3], and Cx432/2 1.1 [0.1 to 1.3;P,0.005]; in LV, WT 10.1 [9.4 to 10.7], Cx431/2 8.3 [7.8 to 8.9], and Cx432/2 1.7 [1.3 to 2.1;P,0.005]) correspondingwith the downregulation of Cx40. Cx40 and Cx45 mRNAs were detectable in ventricular homogenates even at 17.5 dpc,probably accounting for the residual conduction function. Neonatal knockout hearts were arrhythmic in vivo as well asex vivo. This study demonstrates the contribution of Cx43 to the electrical function of the developing mouse heart andthe essential role of this gene in maintaining heart rhythm in postnatal life.(Circ Res. 2001;88:1196-1202.)
Key Words: connexinn developmentn arrhythmian electrophysiologyn knockout
Gap junction channels (connexins) are specialized mem-brane structures that allow for direct communication
between neighboring cells.1 Although previous work docu-mented the spatial and temporal patterns of expression ofdifferent connexin isoforms,2–12 the functional correlates ofthis diversity of connexins are poorly understood. Analysis ofmice with null mutations has shed some light on the functionof specific connexin genes and their products.13–17 Con-nexin43 (Cx43) has widespread spatial distribution from the2-cell stage through postnatal life in the mouse.18 However,the null mutation of the Cx43 gene results in anatomicmalformations that are incompatible with postnatal life.13 Thecardiac phenotype of the Cx43 null mutation includes delayedlooping,19 intertrabecular pouching of the right ventricularcavity, and right ventricular outflow tract obstruction.13 Thepresence of other connexins, notably Cx40,19 may explainwhy the heart functions through gestation. However, it is notknown at what stage Cx43 becomes the predominant gapjunction involved in electrical impulse propagation.
Thus, the Cx432/2 embryo presents a unique system inwhich to study the role of this protein in electrical conductionin the developing heart as it replaces other connexins and
becomes the predominant cardiac isoform during late gesta-tion.20 Because of the small size and fragility of the embry-onic heart, traditional techniques to study electrical activitysuch as multiple extracellular electrodes or microelectrodemapping may present great technical difficulties. High-resolution optical mapping of voltage-dependent changes incardiac tissue21–23 has previously been used in the adultmouse,24,25 the embryonic chick,26,27 and other rodenthearts,26 as well as cellular preparations.28,29 This study usesoptical mapping to explore the role of Cx43 in cardiacimpulse propagation during the embryonic development andthe early postnatal life in the mouse.
Materials and MethodsHeart PreparationsAll animal care protocols conformed to institutional and NIHguidelines. The mouse colony was founded by a breeding pair(129Sv/C57BL6/CD1 strain) heterozygous for the Cx43 knockoutmutation. Pregnant dams were euthanized at 12.5, 15.5, and 17.5days postcoitum (dpc) to obtain embryonic hearts. While embryonicpreparations were superfused, hearts of 5 days postpartum (dpp)mice were also perfused via aortic cannulation. No pharmacologicalor mechanical motion-reduction techniques were used during any of
Original received January 24, 2001; revision received April 5, 2001; accepted April 5, 2001.From the Department of Pharmacology (D.V., H.S.T., M.D., G.E.M., J.J.) and Department of Microbiology and Immunology (S.M.T.), SUNY Upstate
Medical University, Syracuse, NY, and Department of Biology (C.W.L.), University of Pennsylvania, Philadelphia, Pa.Correspondence to José Jalife, MD, SUNY Upstate Medical University, 766 Irving Ave, Syracuse, NY 13210. E-mail [email protected]© 2001 American Heart Association, Inc.
Circulation Researchis available at http://www.circresaha.org
1196 by guest on October 8, 2014http://circres.ahajournals.org/Downloaded from

the experimental protocols. Voltage-sensitive dye fluorescence fromstained hearts was mapped optically on an upright microscopeequipped with a charge-coupled device camera (Dalsa Inc, modelCA-D1 128T) as discussed in detail previously.24,30
Pacing Protocol for Measurement ofConduction VelocityHearts at 12.5 and 15.5 dpc as well as those at 5 dpp were allowedto beat in sinus rhythm, and optical mapping records were obtainedto measure the heart rate. A suction glass electrode with an outerdiameter of 25 to 100mm and a fire-polished tip was used to deliverunipolar pacing stimuli to the ventricles. The left (LV) and right(RV) ventricles were separately paced at a basic cycle length (BCL)of 300 ms at 12.5 and 15.5 dpc, 200 ms at 17.5 dpc, and 100 ms at5 dpp using 4-ms stimuli at 1.5 times diastolic threshold. Activityduring pacing and during arrhythmias, if any, was recorded for 4seconds. Pacing was attempted in arrhythmic Cx432/2 hearts at 5 dppwith unipolar as well as bipolar silver/silver chloride electrodes withdiameters ranging from 0.2 to 1 mm. Optical movies of pacedactivity were signal-averaged as described previously.24,30,31 Theconduction velocity in the slowest direction was quantified for theRV and LV for all the time points studied.
RNase Protection AssayVentricles of hearts at 17.5 dpc used to extract total cellular RNAwere dissected in ice-cold Tyrode’s solution and immediately ho-mogenized in Triazol reagent (MRC Inc), and total tissue RNA wasextracted according to the manufacturer’s protocol. Antisense probeswere designed to recognize regions within the coding sequences ofCx40, Cx43, and Cx45. A probe for the mouse housekeeping genecyclophilin was used as a control for loading. RNase protection assay(RPA) was carried out using the Riboquant RPA kit (Pharmingen).At 17.5 days, the cyclophilin signal showed no genotype-dependentchanges (in million counts per 15 hours, wild type [WT] 3.5560.87[n54], Cx431/2 3.4760.38 [n58], and Cx432/2 3.6260.68 [n53]).Connexin RPA signals were quantified as a percentage of cyclophilinsignal.
Microelectrode RecordingsRecordings were obtained using 15- to 30-MVmicroelectrodes filledwith 3 mol/L KCl solution. The signals were appropriately condi-tioned and sampled at 15 to 20 kHz. The resting membrane potential,action potential amplitude, and maximum upstroke velocity (dV/dtmax) were analyzed in 15.5-dpc embryos.
Electrocardiography in Conscious MiceA custom-built chamber with four silver-silver chloride footpadsembedded in the floor was used to record ECGs in conscious mice at5 dpp. The pads were wetted with ECG electrode jelly, and the micewere placed in the chamber such that each foot of the mouse madecontact with a separate electrode. No physical restraint is required atthis age. Signals were amplified and low-pass filtered with adifferential amplifier (CyberAmp 380, Axon Instruments), digitized(Digidata 1200) at 5 kHz, and stored for offline analysis.
Statistical AnalysisAll values are reported as confidence intervals of mean6SE.Differences were considered significant at theP,0.05 level usingANOVA.
An expanded Materials and Methods section can be found in theonline data supplement available at http://www.circresaha.org.
ResultsViability of the PreparationHeart rates at sinus rhythm were recorded for superfusedembryonic hearts at 12.5 and 15.5 dpc, as well as perfused5-day postnatal hearts. The heart rates were comparable forall genotypes (in bpm, for 12.5 dpc, WT 117.6613.4 [n510],
Cx431/2 118.568.3 [n524], and Cx432/2 99.9610.7 [n510];for 15.5 dpc, WT 172.5622.1 [n515], Cx431/2 154.8617.6[n513], and Cx432/2 140.7619.3 [n514]; and for 5-daypostnatal, WT 444.5616.1 [n514], Cx431/2 436.5612.1[n515], and Cx432/2 all arrhythmic [n54]). The atria of all17.5-dpc embryonic hearts were crushed to make themasystolic. The above cycle lengths are comparable with thoserecorded previously for in vivo32,33and superfused embryonichearts34 as well as for perfused postnatal hearts.24,31,35
Optical Mapping of Paced Electrical ActivityBy 12.5 dpc, the muscular septum of the ventricle growsapace and is nearly fully formed.36 An anatomic deformity inthe angle of the bending of the heart tube is already apparentin the Cx432/2 heart by this time point.19 At this stage, bothCx43 and Cx40 are strongly expressed throughout the WTventricles.4 The velocities in the direction of slowest propa-gation during pacing of the RV and LV at a BCL of 300 msare tabulated in Table 1. The RV conduction velocities werereduced compared with the LV in all genotypes (P,0.005).However, no significant genotype-related change in velocitywas found in either ventricle.
At 16 dpc, the chamber formation in the heart is essentiallycomplete.36 Cx40 is no longer detected in the RV, although itis detected in the LV trabeculated myocardium.19 As seen inTable 1, no LV conduction velocity differences are seenbetween genotypes during ventricular pacing at the BCL of300 ms in 15.5-dpc hearts. However, RV conduction isgrossly reduced in the Cx432/2 hearts compared with theother genotypes (P,0.005). Thus, it appears that the presenceof Cx40 in the trabeculated myocardium of the LV isadequate to maintain propagation within the whole wall. Thevoltage-dependent fluorescence signal is expected to origi-nate throughout the depth of the ventricular wall37; the opticalmapping records may not reflect isolated slow conductionwithin the compact layers of the LV myocardium, if it occurs.
By day 18, Cx40 is downregulated throughout the left andright ventricular myocardium.19 The effect of this downregu-lation of all connexins other than Cx43 in the entire ventric-ular myocardium is seen as an extreme reduction in theconduction velocity in both ventricles of the Cx432/2 heart atthis stage (Table 1,P,0.005). Both the LV and the RV showsimilar conduction times in WT and Cx431/2 hearts. Videosof representative activation sequences in the LV and RV at allembryonic points can be found in an online data supplementavailable at http://www.circresaha.org.
RV and LV propagation was studied only in WT andCx431/2 mice at 5 dpp. As seen inTable 1, RV velocities aresignificantly different from those in the LV (P,0.05), al-though there is no significant genotype-dependent difference.At this stage, a small number of Cx432/2 knockout miceremain alive. However, none of the Cx432/2 hearts could bepaced for the measurement of conduction velocity.
Arrhythmias in Cx43 2/2 HeartsNo arrhythmias were observed in any of the embryonic heartsat 12.5 or 15.5 dpc. At 17.5 dpc, none of the 20 WT heartsand one of the 35 Cx431/2 hearts showed ventricular arrhyth-mia at some point during the protocol. Of the 20 Cx432/2
Vaidya et al Slow Conduction in Embryonic Cx432/2 Mice 1197
by guest on October 8, 2014http://circres.ahajournals.org/Downloaded from
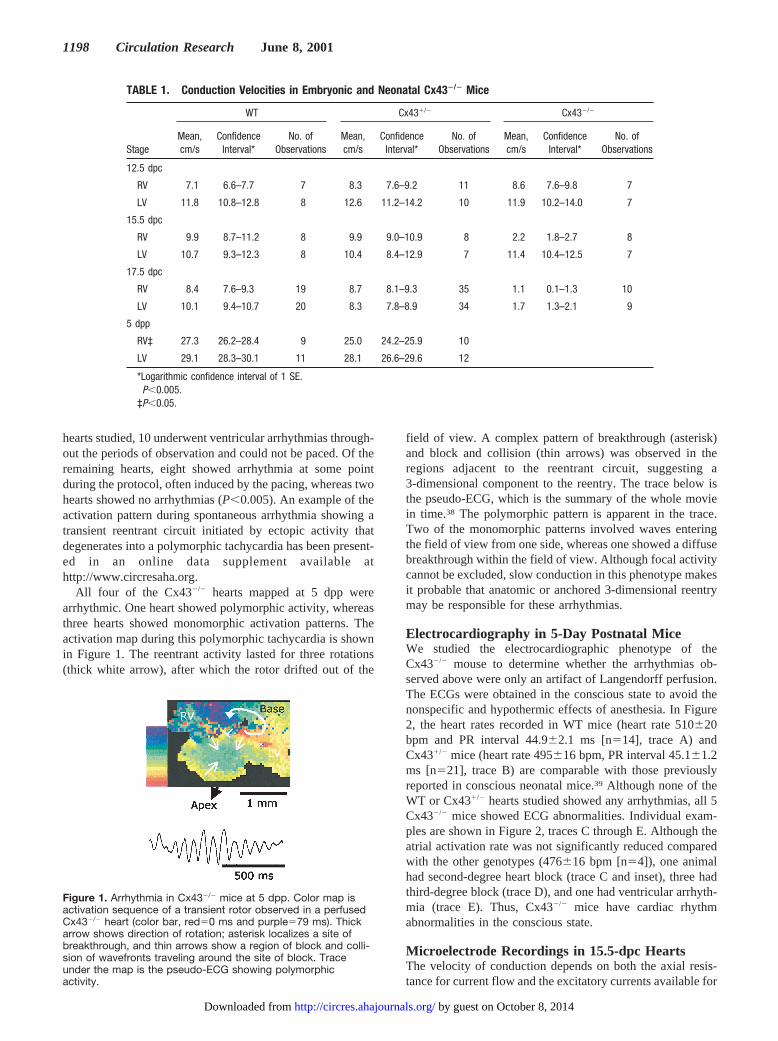
hearts studied, 10 underwent ventricular arrhythmias through-out the periods of observation and could not be paced. Of theremaining hearts, eight showed arrhythmia at some pointduring the protocol, often induced by the pacing, whereas twohearts showed no arrhythmias (P,0.005). An example of theactivation pattern during spontaneous arrhythmia showing atransient reentrant circuit initiated by ectopic activity thatdegenerates into a polymorphic tachycardia has been present-ed in an online data supplement available athttp://www.circresaha.org.
All four of the Cx432/2 hearts mapped at 5 dpp werearrhythmic. One heart showed polymorphic activity, whereasthree hearts showed monomorphic activation patterns. Theactivation map during this polymorphic tachycardia is shownin Figure 1. The reentrant activity lasted for three rotations(thick white arrow), after which the rotor drifted out of the
field of view. A complex pattern of breakthrough (asterisk)and block and collision (thin arrows) was observed in theregions adjacent to the reentrant circuit, suggesting a3-dimensional component to the reentry. The trace below isthe pseudo-ECG, which is the summary of the whole moviein time.38 The polymorphic pattern is apparent in the trace.Two of the monomorphic patterns involved waves enteringthe field of view from one side, whereas one showed a diffusebreakthrough within the field of view. Although focal activitycannot be excluded, slow conduction in this phenotype makesit probable that anatomic or anchored 3-dimensional reentrymay be responsible for these arrhythmias.
Electrocardiography in 5-Day Postnatal MiceWe studied the electrocardiographic phenotype of theCx432/2 mouse to determine whether the arrhythmias ob-served above were only an artifact of Langendorff perfusion.The ECGs were obtained in the conscious state to avoid thenonspecific and hypothermic effects of anesthesia. In Figure2, the heart rates recorded in WT mice (heart rate 510620bpm and PR interval 44.962.1 ms [n514], trace A) andCx431/2 mice (heart rate 495616 bpm, PR interval 45.161.2ms [n521], trace B) are comparable with those previouslyreported in conscious neonatal mice.39 Although none of theWT or Cx431/2 hearts studied showed any arrhythmias, all 5Cx432/2 mice showed ECG abnormalities. Individual exam-ples are shown in Figure 2, traces C through E. Although theatrial activation rate was not significantly reduced comparedwith the other genotypes (476616 bpm [n54]), one animalhad second-degree heart block (trace C and inset), three hadthird-degree block (trace D), and one had ventricular arrhyth-mia (trace E). Thus, Cx432/2 mice have cardiac rhythmabnormalities in the conscious state.
Microelectrode Recordings in 15.5-dpc HeartsThe velocity of conduction depends on both the axial resis-tance for current flow and the excitatory currents available for
TABLE 1. Conduction Velocities in Embryonic and Neonatal Cx432/2 Mice
Stage
WT Cx431/2 Cx432/2
Mean,cm/s
ConfidenceInterval*
No. ofObservations
Mean,cm/s
ConfidenceInterval*
No. ofObservations
Mean,cm/s
ConfidenceInterval*
No. ofObservations
12.5 dpc
RV† 7.1 6.6–7.7 7 8.3 7.6–9.2 11 8.6 7.6–9.8 7
LV 11.8 10.8–12.8 8 12.6 11.2–14.2 10 11.9 10.2–14.0 7
15.5 dpc
RV 9.9 8.7–11.2 8 9.9 9.0–10.9 8 2.2 1.8–2.7† 8
LV 10.7 9.3–12.3 8 10.4 8.4–12.9 7 11.4 10.4–12.5 7
17.5 dpc
RV 8.4 7.6–9.3 19 8.7 8.1–9.3 35 1.1 0.1–1.3† 10
LV 10.1 9.4–10.7 20 8.3 7.8–8.9 34 1.7 1.3–2.1† 9
5 dpp
RV‡ 27.3 26.2–28.4 9 25.0 24.2–25.9 10
LV 29.1 28.3–30.1 11 28.1 26.6–29.6 12
*Logarithmic confidence interval of 1 SE.†P,0.005.‡P,0.05.
Figure 1. Arrhythmia in Cx432/2 mice at 5 dpp. Color map isactivation sequence of a transient rotor observed in a perfusedCx432/2 heart (color bar, red50 ms and purple579 ms). Thickarrow shows direction of rotation; asterisk localizes a site ofbreakthrough, and thin arrows show a region of block and colli-sion of wavefronts traveling around the site of block. Traceunder the map is the pseudo-ECG showing polymorphicactivity.
1198 Circulation Research June 8, 2001
by guest on October 8, 2014http://circres.ahajournals.org/Downloaded from

depolarization. We excluded genotype-specific reduction inthe excitatory currents by recording the upstroke of the actionpotential in hearts at 15.5 dpc. Figure 3 shows representativeaction potential upstrokes and dV/dt signals from the RV andLV of the three genotypes. Faster upstroke velocities can beseen in Cx432/2 ventricles compared with WT and Cx431/2
ventricles. Table 2 summarizes the electrophysiologic prop-erties of three WT, 5 Cx431/2, and three Cx432/2 hearts.Although there is no significant difference between theresting membrane potential or the action potential amplitudebetween genotypes, the dV/dtmax is higher in the Cx432/2
mouse than in the WT and Cx431/2 hearts. The observationthat the dV/dtmax is not reduced in the Cx432/2 embryossuggests that the slow propagation seen at this time point isnot a result of a concurrent reduction in excitatory currents.This is consistent with the findings of Johnson et al,40 whostudied sodium currents in Cx432/2 animals at term. How-ever, that study found no increase in the upstroke velocity inisolated myocytes.40 The paradoxic increase in the upstrokevelocity gives credence to a computational prediction ofRudy and Quan41 that in the presence of unchanged excitatorycurrents, the higher input resistance of poorly coupled cellsmust result in higher upstroke velocities. Note that only RVand not LV conduction velocity was found to be reduced inthe Cx432/2 heart at 15.5 dpc. However, this conductionfunction may be sustained by the trabeculated myocardium,which expresses Cx40.19 The microelectrode recordings aresuperficial and likely to sample only the compact layer of the
ventricular myocardium, in which no Cx40 or Cx43 is foundin the Cx432/2 heart.19 This may explain the higher dV/dtmax
observed in microelectrode recordings from both ventricles atthis stage.
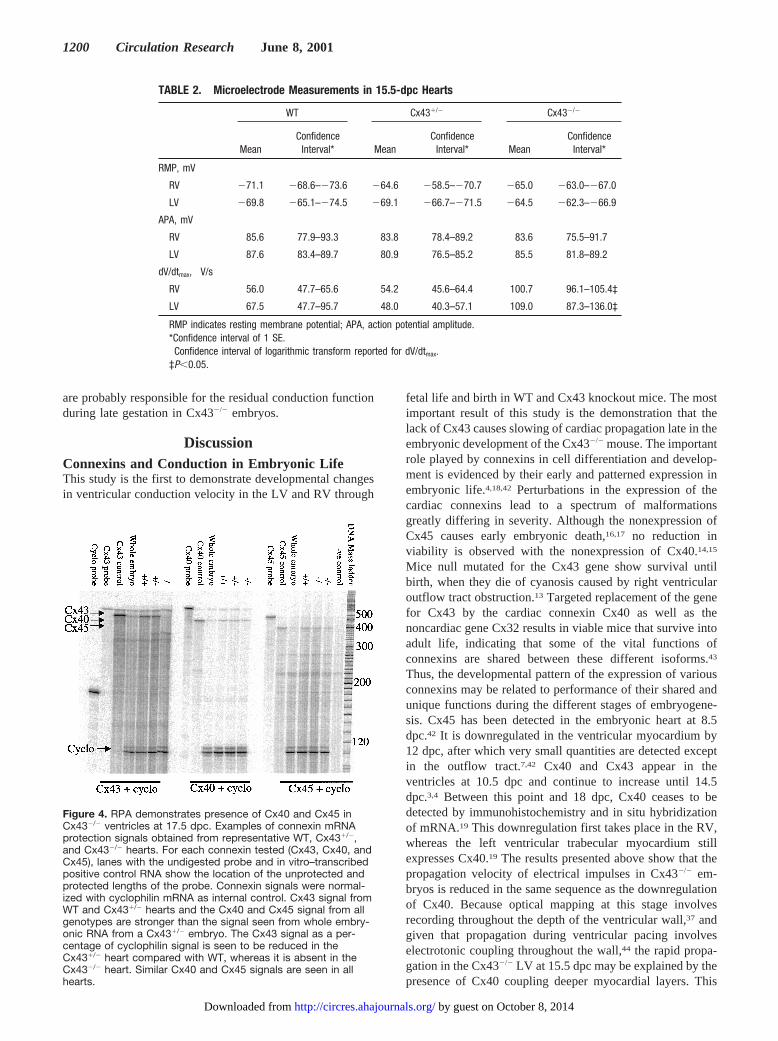
Presence of Other Connexins in the LateCx432/2 EmbryoBy day 18, little or no Cx40 has been demonstrated byimmunohistochemistry and in situ hybridization of mRNA,and little or no Cx45 has been demonstrated by in situhybridization.19 To determine what connexin is responsiblefor the residual slow conduction, we performed RPAs forCx40, Cx43, and Cx45 as a sensitive measure of RNAmessage in 17.5-dpc ventricles. As seen in Figure 4, signalsfor Cx43, Cx40 and Cx45 are all detected in WT and Cx431/2
hearts in quantities greater than that found in whole Cx431/2
embryonic RNA. Cx43 signal as a percentage of the cyclo-philin signal is reduced in the Cx431/2 heart (in percentage ofcyclophilin signal, WT 44.860.8 [n54] and Cx431/2
23.060.8 [n58],P51028). Cx40 and Cx45 bands are alsofound in Cx432/2 hearts. There is no genotype-specificdifference in the expression of Cx40 and Cx45 at this stage(WT n54, Cx431/2 n56, and Cx432/2 n53). Thus, Cx40 andCx45 are present in the embryonic ventricle at this stage and
Figure 2. ECGs in conscious 5-dpp mice. Trace A, WT mousehad an RR interval of 116 ms and a PR interval of 44 ms. TraceB, Cx431/2 mouse had an RR interval of 113 ms and a PR inter-val of 44 ms. No conscious WT or Cx431/2 mice studied werearrhythmic. Traces C through E, Examples of arrhythmias seenin Cx432/2 mice. Trace C, 2:1 atrioventricular propagation pat-tern, which is seen clearly in the 32.5-magnified, signal-averaged inset. Trace D, Atrioventricular dissociation. Trace E,Polymorphic ventricular arrhythmia with a few higher amplitudeventricular activations superimposed on an irregular baseline.Bar5500 ms for traces and 200 ms for inset.
Figure 3. Action potential upstrokes and dV/dt signals in 15.5-dpc mice. Shown are representative action potential upstrokes(upper traces) from the RV and LV of 15.5-dpc hearts, with thedV/dt signals (lower traces) plotted beneath each upstroke. Zerotransmembrane potential is shown by dotted lines near eachupstroke, whereas the baselines of the dV/dt signals represent 0V/s. Horizontal scale line (bottom right) represents 2 ms; verticalline represents 20 mV (transmembrane potential) and 50 V/s(dV/dt). Cx432/2 (2/2) hearts show steeper upstrokes andhigher peak dV/dt signals than either WT or Cx431/2 (1/2)hearts. Note that dV/dt signal traces have been shifted right forclarity of illustration.
Vaidya et al Slow Conduction in Embryonic Cx432/2 Mice 1199
by guest on October 8, 2014http://circres.ahajournals.org/Downloaded from
are probably responsible for the residual conduction functionduring late gestation in Cx432/2 embryos.
DiscussionConnexins and Conduction in Embryonic LifeThis study is the first to demonstrate developmental changesin ventricular conduction velocity in the LV and RV through
fetal life and birth in WT and Cx43 knockout mice. The mostimportant result of this study is the demonstration that thelack of Cx43 causes slowing of cardiac propagation late in theembryonic development of the Cx432/2 mouse. The importantrole played by connexins in cell differentiation and develop-ment is evidenced by their early and patterned expression inembryonic life.4,18,42 Perturbations in the expression of thecardiac connexins lead to a spectrum of malformationsgreatly differing in severity. Although the nonexpression ofCx45 causes early embryonic death,16,17 no reduction inviability is observed with the nonexpression of Cx40.14,15
Mice null mutated for the Cx43 gene show survival untilbirth, when they die of cyanosis caused by right ventricularoutflow tract obstruction.13 Targeted replacement of the genefor Cx43 by the cardiac connexin Cx40 as well as thenoncardiac gene Cx32 results in viable mice that survive intoadult life, indicating that some of the vital functions ofconnexins are shared between these different isoforms.43
Thus, the developmental pattern of the expression of variousconnexins may be related to performance of their shared andunique functions during the different stages of embryogene-sis. Cx45 has been detected in the embryonic heart at 8.5dpc.42 It is downregulated in the ventricular myocardium by12 dpc, after which very small quantities are detected exceptin the outflow tract.7,42 Cx40 and Cx43 appear in theventricles at 10.5 dpc and continue to increase until 14.5dpc.3,4 Between this point and 18 dpc, Cx40 ceases to bedetected by immunohistochemistry and in situ hybridizationof mRNA.19 This downregulation first takes place in the RV,whereas the left ventricular trabecular myocardium stillexpresses Cx40.19 The results presented above show that thepropagation velocity of electrical impulses in Cx432/2 em-bryos is reduced in the same sequence as the downregulationof Cx40. Because optical mapping at this stage involvesrecording throughout the depth of the ventricular wall,37 andgiven that propagation during ventricular pacing involveselectrotonic coupling throughout the wall,44 the rapid propa-gation in the Cx432/2 LV at 15.5 dpc may be explained by thepresence of Cx40 coupling deeper myocardial layers. This
TABLE 2. Microelectrode Measurements in 15.5-dpc Hearts
WT Cx431/2 Cx432/2
MeanConfidence
Interval* MeanConfidence
Interval* MeanConfidence
Interval*
RMP, mV
RV 271.1 268.6–273.6 264.6 258.5–270.7 265.0 263.0–267.0
LV 269.8 265.1–274.5 269.1 266.7–271.5 264.5 262.3–266.9
APA, mV
RV 85.6 77.9–93.3 83.8 78.4–89.2 83.6 75.5–91.7
LV 87.6 83.4–89.7 80.9 76.5–85.2 85.5 81.8–89.2
dV/dtmax,† V/s
RV 56.0 47.7–65.6 54.2 45.6–64.4 100.7 96.1–105.4‡
LV 67.5 47.7–95.7 48.0 40.3–57.1 109.0 87.3–136.0‡
RMP indicates resting membrane potential; APA, action potential amplitude.*Confidence interval of 1 SE.†Confidence interval of logarithmic transform reported for dV/dtmax.‡P,0.05.
Figure 4. RPA demonstrates presence of Cx40 and Cx45 inCx432/2 ventricles at 17.5 dpc. Examples of connexin mRNAprotection signals obtained from representative WT, Cx431/2,and Cx432/2 hearts. For each connexin tested (Cx43, Cx40, andCx45), lanes with the undigested probe and in vitro–transcribedpositive control RNA show the location of the unprotected andprotected lengths of the probe. Connexin signals were normal-ized with cyclophilin mRNA as internal control. Cx43 signal fromWT and Cx431/2 hearts and the Cx40 and Cx45 signal from allgenotypes are stronger than the signal seen from whole embry-onic RNA from a Cx431/2 embryo. The Cx43 signal as a per-centage of cyclophilin signal is seen to be reduced in theCx431/2 heart compared with WT, whereas it is absent in theCx432/2 heart. Similar Cx40 and Cx45 signals are seen in allhearts.
1200 Circulation Research June 8, 2001
by guest on October 8, 2014http://circres.ahajournals.org/Downloaded from

finding argues that conduction of electrical activation wavesin the ventricle is a shared function of Cx43 and Cx40. Thegross reduction in conduction velocity at 17.5 dpc in theCx432/2 mouse also highlights the fact that Cx43 is largelyresponsible for the conduction of electrical impulses in theventricles during late embryonic life. We may only speculatethat the 2.5- to 3-fold increase in conduction velocity seen inWT and Cx431/2 mice between embryonic life and 5 dpp mayoccur as a result of an increase in Cx43 expression,20 otherionic channels,45–47 or mechanical changes associated withcirculation48 that occur during this period.
Conduction in the Cx431/2 HeartThere have been conflicting reports about reductions inconduction velocity in the Cx431/2 mouse compared withWT.49,50 In this study no significant difference was found inthe conduction velocity in either ventricle. Indeed, previouslyreported differences in conduction velocities betweenCx431/2 and WT were less dramatic at birth than during adultlife.49 Computational studies51,52 and a recent experimentalstudy48 suggest that profound changes are required in theexpression of gap junction channels to produce relativelymodest changes in conduction velocity.
Residual Conduction in the Cx432/2 EmbryoCx40 has not been identified either by in situ hybridization ofmRNA or by immunohistochemistry in 18-dpc mice,19
whereas little if any Cx45 has been detected by in situhybridization at this stage either in WT, Cx431/2, or Cx432/2
mice.19 Our results in pooled whole-ventricular RNA at thisstage suggest that small quantities of Cx40 and Cx45 persistin the ventricles and may account for the residual conductionfunction seen in Cx432/2 embryos at this stage.
Relationship Between Excitatory Currentsand CouplingThe excitatory current that travels from cell to cell is providedby inward ionic flow through sodium and calcium channels.It has been shown that reduction in inward currents can leadto a slowing in propagation velocity before conduction blockensues.24,41,52However, this slowing of propagation is not asprofound as that which can be achieved by a reduction ofcoupling.41,52 A key difference between the two mechanismsis that the dV/dtmax is reduced with a primary deficit inexcitatory currents, whereas it has a biphasic relationshipwith the degree of coupling. The first phase of reduction incoupling is associated with an increase in the dV/dtmax causedby the higher-input resistance of the cell, whereas extremedegrees of uncoupling close to the onset of block areassociated with a decrease in the dV/dtmax. Our measurementsin the 15.5-dpc embryo show that the knockout heart has ahigh dV/dtmax, indicating that a reduction in excitatory cur-rents is not the primary cause of slow propagation. These dataare in agreement with previously published results showingthat the sodium current in isolated ventricular knockoutmyocytes is not reduced compared with WT.40 Indeed, thehigher dV/dtmax suggests that reduced coupling is the primarycause of the reduction in conduction velocity. It is possiblethat the higher dV/dtmax observed in the LV at this stage is due
to the poor coupling of epicardial cells, although well-coupled subendocardial cells maintain conduction function asdiscussed above.
Arrhythmias in Cx43 2/2 MiceArrhythmias were commonly observed in Cx432/2 mice at17.5 dpc, and as a rule after birth. The arrhythmias in 5-dppmice were observed in the conscious state as well as ex vivo.Although the triggers for spontaneous arrhythmias may befocal, slow conduction likely makes sustained reentrantarrhythmias possible. It is possible that the hypoxia sufferedby the mouse during postnatal life may be responsible for thearrhythmias. However, the embryonic circulation largelybypasses the pulmonary bed and receives oxygenation at theplacenta. It is not expected that the late embryonic heart ishypoxic. The observation of spontaneous as well as pacing-induced arrhythmias at 17.5 dpc suggests that reduction inconduction velocity makes the heart an arrhythmogenicsubstrate. This strengthens the conjecture that slow propaga-tion due to the absence of Cx43 and the hypoxia caused by theoutflow tract malformation are probable causes of death inthe Cx432/2 heart.
AcknowledgmentsThis work was supported in part by NIH Grants 2P01-HL3970711,HD36457 (to C.W.L.), and HL36059 (to C.W.L.); National ScienceFoundation Grant IBN-9905067 (to C.W.L.); and American HeartAssociation Grant 003056T (to G.E.M.).
References1. Yeager M. Structure of cardiac gap junction intercellular channels.J
Struct Biol. 1998;121:231–245.2. van Kempen MJ, Vermeulen JL, Moorman AF, Gros D, Paul DL, Lamers
WH. Developmental changes of connexin40 and connexin43 mRNAdistribution patterns in the rat heart.Cardiovasc Res. 1996;32:886–900.
3. Delorme B, Dahl E, Jarry-Guichard T, Marics I, Briand JP, Willecke K,Gros D, Theveniau-Ruissy M. Developmental regulation of connexin 40gene expression in mouse heart correlates with the differentiation of theconduction system.Dev Dyn. 1995;204:358–371.
4. Delorme B, Dahl E, Jarry-Guichard T, Briand JP, Willecke K, Gros D,Theveniau-Ruissy M. Expression pattern of connexin gene products at theearly developmental stages of the mouse cardiovascular system.Circ Res.1997;81:423–437.
5. Chen SC, Davis LM, Westphale EM, Beyer EC, Saffitz JE. Expression ofmultiple gap junction proteins in human fetal and infant hearts.PediatrRes. 1994;36:561–566.
6. Dahl E, Winterhager E, Traub O, Willecke K. Expression of gap junctiongenes, connexin40 and connexin43, during fetal mouse development.Anat Embryol. 1995;191:267–278.
7. Coppen SR, Severs NJ, Gourdie RG. Connexin45 (a6) expressiondelineates an extended conduction system in the embryonic and maturerodent heart.Dev Gen. 1999;24:82–90.
8. Anumonwo JM, Wang HZ, Trabka-Janik E, Dunham B, Veenstra RD,Delmar M, Jalife J. Gap junctional channels in adult mammalian sinusnodal cells: immunolocalization and electrophysiology.Circ Res. 1992;71:229–239.
9. Davis LM, Rodefeld ME, Green K, Beyer EC, Saffitz JE. Gap junctionprotein phenotypes of the human heart and conduction system.J Car-diovasc Electrophysiol. 1995;6:813–822.
10. Kanter HL, Laing JG, Beyer EC, Green KG, Saffitz JE. Multiple con-nexins colocalize in canine ventricular myocyte gap junctions.Circ Res.1993;73:344–350.
11. Davis LM, Kanter HL, Beyer EC, Saffitz JE. Distinct gap junction proteinphenotypes in cardiac tissues with disparate conduction properties.J AmColl Cardiol. 1994;24:1124–1132.
12. Gourdie RG, Severs NJ, Green CR, Rothery S, Germroth P, ThompsonRP. The spatial distribution and relative abundance of gap-junctionalconnexin40 and connexin43 correlate to functional properties of com-
Vaidya et al Slow Conduction in Embryonic Cx432/2 Mice 1201
by guest on October 8, 2014http://circres.ahajournals.org/Downloaded from

ponents of the cardiac atrioventricular conduction system.J Cell Sci.1993;105:985–991.
13. Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC,Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal micelacking connexin43.Science. 1995;267:1831–1834.
14. Simon AM, Goodenough DA, Paul DL. Mice lacking connexin40 havecardiac conduction abnormalities characteristic of atrioventricular blockand bundle branch block.Curr Biol. 1998;8:295–298.
15. Kirchhoff S, Nelles E, Hagendorff A, Kruger O, Traub O, Willecke K.Reduced cardiac conduction velocity and predisposition to arrhythmias inconnexin40-deficient mice.Curr Biol. 1998;8:299–302.
16. Kumai M, Nishii K, Nakamura K, Takeda N, Suzuki M, Shibata Y. Lossof connexin45 causes a cushion defect in early cardiogenesis.Devel-opment. 2000;127:3501–3512.
17. Kruger O, Plum A, Kim J, Winterhager E, Maxeiner S, Hallas G,Kirchhoff S, Traub O, Lamers WH, Willecke K. Defective vasculardevelopment in connexin 45-deficient mice.Development. 2000;127:4179–4193.
18. Davies TC, Barr KJ, Jones DH, Zhu D, Kidder GM. Multiple members ofthe connexin gene family participate in preimplantation development ofthe mouse.Dev Genet. 1996;18:234–243.
19. Ya J, Erdtsieck-Ernste EB, de Boer PA, van Kempen MJ, Jongsma H,Gros D, Moorman AF, Lamers WH. Heart defects in connexin43-deficient mice.Circ Res. 1998;82:360–366.
20. Fromaget C, el Aoumari A, Dupont E, Briand JP, Gros D. Changes in theexpression of connexin 43, a cardiac gap junctional protein, during mouseheart development.J Mol Cell Cardiol. 1990;22:1245–1258.
21. Davidenko JM, Pertsov AV, Salomonsz R, Baxter W, Jalife J. Stationaryand drifting spiral waves of excitation in isolated cardiac muscle.Nature.1992;355:349–351.
22. Salama G, Kanai AJ, Huang D, Efimov IR, Girouard SD, Rosenbaum DS.Hypoxia and hypothermia enhance spatial heterogeneities of repolar-ization in guinea pig hearts: analysis of spatial autocorrelation of opticallyrecorded action potential durations.J Cardiovasc Electrophysiol. 1998;9:164–183.
23. Kwaku KF, Dillon SM. Shock-induced depolarization of refractory myo-cardium prevents wave-front propagation in defibrillation.Circ Res.1996;79:957–973.
24. Morley GE, Vaidya D, Samie FH, Lo CW, Delmar M, Jalife J. Charac-terization of conduction in the ventricles of normal and heterozygousconnexin43 knockout mice using optical mapping.J Cardiovasc Electro-physiol. 1999;10:1361–1375.
25. Witkowski FX, Clark RB, Larsen TS, Melnikov A, Giles WR. Voltage-sensitive dye recordings of electrophysiological activation in aLangendorff-perfused mouse heart.Can J Cardiol. 1997;13:1077–1082.
26. Kamino K. Optical approaches to ontogeny of electrical activity andrelated functional organization during early heart development.PhysiolRev. 1991;71:53–91.
27. Chuck ET, Freeman DM, Watanabe M, Rosenbaum DS. Changing acti-vation sequence in the embryonic chick heart: implications for the devel-opment of the His-Purkinje system.Circ Res. 1997;81:470–476.
28. Fast VG, Darrow BJ, Saffitz JE, Kleber AG. Anisotropic activationspread in heart cell monolayers assessed by high-resolution opticalmapping: role of tissue discontinuities.Circ Res. 1996;79:115–127.
29. Windisch H, Ahammer H, Schaffer P, Muller W, Platzer D. Opticalmultisite monitoring of cell excitation phenomena in isolated cardiomyo-cytes.Pflügers Arch. 1995;430:508–518.
30. Tamaddon HS, Vaidya D, Simon AM, Paul DL, Jalife J, Morley GE.High-resolution optical mapping of the right bundle branch in connexin40knockout mice reveals slow conduction in the specialized conductionsystem.Circ Res. 2000;87:929–936.
31. Vaidya D, Morley GE, Samie FH, Jalife J. Reentry and fibrillation in themouse heart: a challenge to the critical mass hypothesis.Circ Res.1999;85:174–181.
32. Keller BB, MacLennan MJ, Tinney JP, Yoshigi M. In vivo assessment ofembryonic cardiovascular dimensions and function in day-10.5 to -14.5mouse embryos.Circ Res. 1996;79:247–255.
33. Srinivasan S, Baldwin HS, Aristizabal O, Kwee L, Labow M, Artman M,Turnbull DH. Noninvasive, in utero imaging of mouse embryonic heartdevelopment with 40-MHz echocardiography.Circulation. 1998;98:912–918.
34. Wildenthal K. Maturation of responsiveness to cardioactive drugs: dif-ferential effects of acetylcholine, norepinephrine, theophylline, tyramine,glucagon, and dibutyryl cyclic AMP on atrial rate in hearts of fetal mice.J Clin Invest. 1973;52:2250–2258.
35. Grupp IL, Subramaniam A, Hewett TE, Robbins J, Grupp G. Comparisonof normal, hypodynamic, and hyperdynamic mouse hearts using isolatedwork-performing heart preparations.Am J Physiol. 1993;265:H1401–H1410.
36. Kaufman MH. The Atlas of Mouse Development. San Diego, Calif:Academic Press; 1998.
37. Girouard SD, Laurita KR, Rosenbaum DS. Unique properties of cardiacaction potentials recorded with voltage-sensitive dyes.J Cardiovasc Elec-trophysiol. 1996;7:1024–1038.
38. Pertsov AM, Davidenko JM, Salomonsz R, Baxter WT, Jalife J. Spiralwaves of excitation underlie reentrant activity in isolated cardiac muscle.Circ Res. 1993;72:631–650.
39. Wang L, Swirp S, Duff H. Age-dependent response of the electrocardio-gram to K(1) channel blockers in mice.Am J Physiol. 2000;278:C73–C80.
40. Johnson CM, Green KG, Kanter EM, Bou-Abboud E, Saffitz JE, YamadaKA. Voltage-gated Na1 channel activity and connexin expression inCx43-deficient cardiac myocytes.J Cardiovasc Electrophysiol. 1999;10:1390–1401.
41. Rudy Y, Quan WL. A model study of the effects of the discrete cellularstructure on electrical propagation in cardiac tissue.Circ Res. 1987;61:815–823.
42. Alcolea S, Theveniau-Ruissy M, Jarry-Guichard T, Marics I, TzouanacouE, Chauvin JP, Briand JP, Moorman A, Lamers WH, Gros DB. Down-regulation of connexin 45 gene products during mouse heart devel-opment.Circ Res. 1999;84:1365–1379.
43. Plum A, Hallas G, Magin T, Dombrowski F, Hagendorff A, SchumacherB, Wolpert C, Kim J, Lamers WH, Evert M, Meda P, Traub O, WilleckeK. Unique and shared functions of different connexins in mice.Curr Biol.2000;10:1083–1091.
44. Keener JP, Panfilov AV. Three dimensional propagation in the heart: theeffects of geometry and fiber orientation on propagation in myocardium.In: Zipes DP, Jalife J, eds.Cardiac Physiology: From Cell to Bedside.Philadelphia, Pa: WB Saunders; 1995:335–347.
45. Davies MP, An RH, Doevendans P, Kubalak S, Chien KR, Kass RS.Developmental changes in ionic channel activity in the embryonic murineheart.Circ Res. 1996;78:15–25.
46. Wang L, Duff HJ. Developmental changes in transient outward current inmouse ventricle.Circ Res. 1997;81:120–127.
47. Wang L, Duff HJ. Identification and characteristics of delayed rectifierK1 current in fetal mouse ventricular myocytes.Am J Physiol. 1996;270:H2088–H2093.
48. Zhuang J, Yamada KA, Saffitz JE, Kleber AG. Pulsatile stretch remodelscell-to-cell communication in cultured myocytes.Circ Res. 2000;87:316–322.
49. Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM,Yamada KA, Saffitz JE. Slow ventricular conduction in mice het-erozygous for a connexin43 null mutation.J Clin Invest. 1997;99:1991–1998.
50. Thomas SA, Schuessler RB, Berul CI, Beardslee MA, Beyer EC,Mendelsohn ME, Saffitz JE. Disparate effects of deficient expression ofconnexin43 on atrial and ventricular conduction: evidence for chamber-specific molecular determinants of conduction.Circulation. 1998;97:686–691.
51. Jongsma HJ, Wilders R. Gap junctions in cardiovascular disease.CircRes. 2000;86:1193–1197.
52. Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue:roles of the sodium and L-type calcium currents during reduced excit-ability and decreased gap junction coupling.Circ Res. 1997;81:727–741.
1202 Circulation Research June 8, 2001
by guest on October 8, 2014http://circres.ahajournals.org/Downloaded from