novel peptide ligands with dual acting pharmacophores designed for the pathophysiology of...
TRANSCRIPT

Research Report
Novel peptide ligands with dual acting pharmacophoresdesigned for the pathophysiology of neuropathic pain
Katherine E. Hanlona, Dave S. Hermana, Richard S. Agnesb, Tally M. Largent-Milnesa,Isuru R. Kumarasingheb, Sho W. Maa, Wenhong Guoa, Yeon-Sun Leea,b,Michael H. Ossipova, Victor J. Hrubyb, Josephine Laia, Frank Porrecaa, Todd W. Vanderaha,⁎aDepartment of Pharmacology, University of Arizona Health Sciences Center, P.O. Box 245050, 1501 N Campbell Ave., Tucson, AZ 85724, USAbDepartment of Chemistry, University of Arizona, P.O. Box 210041, 1306 E University Blvd, Tucson, AZ 85721, USA
A R T I C L E I N F O A B S T R A C T
Article history:Accepted 14 April 2011Available online 20 April 2011
The conventional design of high affinity drugs targeted to a single molecule has not resultedin clinically useful therapies for pain relief. Recent reviews have suggested that newlydesigned analgesic drugs should incorporate multiple targets. The distributions ofcholecystokinin (CCK) and CCK receptors in the central nervous system (CNS) overlapsignificantly with endogenous opioid systems and can be dually targeted. CCK has beenshown to act as an endogenous “anti-analgesic” peptide and neuropathic pain conditionspromote endogenous CCK release in CNS regions of painmodulation. Administration of CCKinto nuclei of the rostral ventromedial medulla induces pronociceptive behaviors in rats.RSA 504 and RSA 601 are novel bifunctional compounds developed to target neuropathicpain by simultaneously acting as agonists at two distinct opioid receptors and antagonizingCCK receptors in the CNS. RSA 504 and RSA 601 demonstrate agonist activity in vitro andantihypersensitivity to mechanical and thermal stimuli in vivo using the spinal nerveligation model of neuropathic pain. Intrathecal administration of RSA 504 and RSA 601 didnot demonstrate antinociceptive tolerance over 7 days of administration and did not displaymotor impairment or sedation using a rotarod. These are the first behavioral studies thatdemonstrate howmulti-targetedmolecule design can address the pathology of neuropathicpain. These compounds with δ and μ opioid agonist activity and CCK antagonist activitywithin onemolecule offer a novel approach with efficacy for neuropathic pain while lackingthe side effects typically caused by conventional opioid therapies.
© 2011 Elsevier B.V. All rights reserved.
Keywords:Neuropathic painSpinal nerve ligationCholecystokininOpioids
B R A I N R E S E A R C H 1 3 9 5 ( 2 0 1 1 ) 1 – 1 1
⁎ Corresponding author at: University of Arizona, Department of Pharmacology, 1501 N. Campbell Ave., Tucson, AZ 85724, USA. Fax: +1 520626 4779.
E-mail address: [email protected] (T.W. Vanderah).Abbreviations: AMPA, α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate; AUC, area under curve; CCK, cholecystokinin; CGRP,
calcitonin gene related peptide; CNS, central nervous system; hDOR, human δ opioid receptor; GABA, γ-aminobutyric acid; HEK, humanembryonic kidney cells; IMDM, Iscove's modified Dulbecco's medium; MOR, μ opioid receptor; MS, morphine sulfate; NOS, nitric oxidesynthase; NRI, norepinephrine reuptake inhibitor; PAG, periaqueductal gray; PKC, protein kinase C; RVM, rostral ventromedial medulla;SNL, spinal nerve ligation; SNRI, serotonin-norepinephrine reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor
0006-8993/$ – see front matter © 2011 Elsevier B.V. All rights reserved.doi:10.1016/j.brainres.2011.04.024
ava i l ab l e a t www.sc i enced i r ec t . com
www.e l sev i e r . com/ loca te /b ra i n res

1. Introduction
Chronic pain affects 78million people in the United States andat least 75 million Europeans (Breivik et al., 2006; Loeser andBonica, 2001). For many patients chronic pain is untreated orundermanaged due to a lack of novel medications that aretolerable. Recently, the National Institutes for Health de-scribed the need for designing medications that may havemultiple sites of action to better address the pathology ofchronic and/or neuropathic pain (Woodcock et al., 2007),stimulating the development of mechanism-based, individu-alized pain therapies.
Neuropathic pain, demonstrated by hypersensitivity tonoxious and innocuous stimuli, is defined in broad terms aspaincausedbydamageordysfunction in theperipheral nervoussystem (Merskey, 2002) leading to dysfunction of pain fibers ofthe CNS (Ossipov et al., 2000). This type of pain is generallydiversewith unpredictable etiology; additionally, somepatientspresent with genetic conditions, making treatment outcomeevenmoredifficult to predict (Ossipov andPorreca, 2005).Opiatetherapy is one of themost commonly prescribed treatments forchronic neuropathic pain, however opioids do not address themechanisms of neuropathic pain and often have limitedefficacy against this type of pain. Opioids are plagued byanalgesic tolerance, addiction, medication overuse hypersensi-tivities and other physical side effects (Jensen et al., 2001).Regardless of whether it is caused by a disease state or a nervetrauma, neuropathic pain generates unique challenges for thedesign of novel analgesic treatments due to the multiplemechanisms driving pain sequela.
Of particular importance are the descending facilitatorypathways of the brainstem, specifically CCK, which is co-localized in the CNS with endogenous opioids and opioidreceptors (Ghilardi et al., 1992; Zhang et al., 2009). CCK releaseis increased in multiple models of injury in the descendingpain facilitatory pathways (Kovelowski et al., 2000; Friedrichand Gebhart, 2003). Together, these data suggest that CCKplays a significant role in pain processing and endogenousopioid activity. In addition to activating nociceptive descend-ing pain facilitatory neurons (Heinricher and Neubert, 2004),CCK administered into the RVM of normal rats produces timedependent behavioral signs of mechanical and thermalhypersensitivity of the viscera and hindpaw (Friedrich andGebhart, 2003; Xie et al., 2005). Studies have demonstrated thatexogenous opioid therapy also induces the release of CCK inthe spinal cord, frontal cortex and brainstem (Becker et al.,2000; Gustafsson et al., 2001; Xie et al., 2005). Conversely,administration of a CCK antagonist into the RVM produces atime dependent attenuation of thermal and mechanicalhypersensitivity in the rat L5/L6 SNL model (Kovelowski etal., 2000); the opioid-induced hyperalgesia model (Xie et al.,2005); as well as a model of visceral hypersensitivity (Friedrichand Gebhart, 2003). Lastly, systemic CCK antagonists signifi-cantly enhance the analgesic effects of opioids for thetreatment of neuropathic pain and attenuate the developmentof opioid-induced antinociceptive tolerance in animals andhumans (Dourish et al., 1990; McCleane, 1998; McCleane, 2002;McCleane, 2003; Nichols et al., 1996; Wiesenfeld-Hallin et al.,2002; Xie et al., 2005).
An emerging trend encompasses the use of hetero-bivalent compounds in the development of novel therapeu-tics. Licofelone, a pyrrolizine analogue, simultaneouslyinhibits both cyclooxygenases and 5-lipoxygenase; currentlyin clinical trials for osteoarthritis, it is the first NSAID toinhibit leukotriene and prostaglandin formation simulta-neously (Raynauld et al., 2008). The only centrally actinganalgesic approved in the United States since the 1970s isitself a dual pharmacophore: tapentadol (Nucynta©) is a μopioid agonist and norepinephrine reuptake inhibitor in asingle molecule (Frampton, 2010). Tapentadol sets itselfapart from traditional opiate therapy with reduced sideeffects—most notably fewer patients experienced constipa-tion, nausea, vomiting, pruritis and dizziness when com-pared to oxycodone in a long term safety and tolerabilitytrial (Wild et al., 2010). Dual target non analgesics have alsoshown efficacy in chronic pain states: duloxetine (Cym-balta©), an SNRI, has been approved in the treatment offibromyalgia and diabetic neuropathy in addition to majordepressive disorder and generalized anxiety disorder in theUnited States (Wright et al., 2011). SNRIs alone have notresulted in the treatment of all types of chronic, neuropathicpain suggesting alternative underlying disease states suchas increased levels of CCK. The increasing occurrence ofbifunctional compounds in pain therapies is not accidental:it has been suggested that multivalent therapies offer agreater disease specificity and greater affinity than existingmonovalent therapies (Lezoualc'h et al., 2009).
The bi-functional compounds examined here, RSA 504 (H-Tyr1-DNle2-Gly3-Trp4-Nle5-Asp6-Phe7-NH2) (Agnes et al., 2008)and RSA 601 (H-Tyr1-DPhe2-Gly3-DTrp4-NMeNle5-Asp6-Phe7-NH2) (Hruby et al., 2003) were based on the structure ofSNF9007, a novel peptide analogue of CCK. SNF9007was foundto act simultaneously at the δ and μ opioid receptors toproduce antinociception (Williams et al., 1994) but lackedefficacy due to some CCK agonist activity. The RSA com-pounds, designed to act as both opioid agonists and CCKantagonists, have nanomolar binding affinity at both receptorclasses with no CCK agonist activity and clear in vitro and invivo antagonist activity at the CCK receptors. RSA 504(913.02 g/mol) possesses Ki values of 23 nM and 27 nM at theδ and μ opioid receptors, respectively with Ki's of 11.2 nM and16 nM at the CCK-1 and CCK-2 receptors, respectively (Agneset al., 2008). RSA 601 (m.w. 962.04 g/mol) displays 0.55 nM and5.7 nM affinity at the δ and μ opioid receptors, respectively, aswell as 1100 and 1.6 nM affinity for the CCK-1 and CCK-2receptors, respectively (Hruby et al., 2003).
Overall, many compounds for neuropathic pain have beendesigned to act at a single selective target in a particularanimal model of pain; however, they have not resulted inadvancing clinical therapies. The RSA 504 and 601 compoundswere designed toward the pathology of neuropathic pain inwhich two pharmacophores with different mechanisms ofaction result in better, longer lasting efficacy over the currentmonotherapy. These single entity compounds with agonistactivity at the δ/μ opioid receptors in addition to CCKantagonist activity demonstrate reduced adverse side effectsversus multiple compounds with pharmacokinetic and/ordrug interactions. We advance the idea that these dual actingcompounds address the pathology seen in many chronic pain
2 B R A I N R E S E A R C H 1 3 9 5 ( 2 0 1 1 ) 1 – 1 1
syndromes, offering amulti-targeted approach to significantlyimprove antinociceptive efficacy.
2. Results
2.1. Biological activities in vitro
The RSA compounds were designed toward the pathology ofneuropathic pain containing both CCK antagonist and opioidagonist pharmacophores. To expand upon the in vitro activi-ties published by the Hruby and colleagues (Agnes et al., 2008;Hruby et al., 2003) demonstrating 0.45 or 23 nM in the MVD fordelta opioid receptor activity and 63 or 210 nM in GPI for muopioid receptor activity for RSA 601 or 504 respectively. Thebiological activity of RSA 601 was assessed using GTPγSbinding assay for opioid receptors. At the hDOR in the GTP-γ-S binding assay RSA 601 displayed an EC50 of 20.9 nM andEmax of 33.8 nM; at MOR the EC50 was 520 nM and demonstrat-ed an Emax of 52.7 nM. RSA 504 shows similar activity in theGTP-γ-S binding assay with an EC50 of 5.5 nM and an Emax of81 nM at the hDOR and an EC50 of 0.47 nM and Emax of 110 nMat the MOR (Agnes et al., 2008).
2.2. RSA 504 and 601 attenuate tactile allodynia whenadministered intrathecally
The L5/L6 SNL model was used to evaluate efficacy againstmechanical hypersensitivity. Prior to and after injury, allanimals were evaluated for mechanical response to non-noxious probing of the left hind paw with calibrated von Freyfilaments. The mean paw withdrawal threshold before SNLwas 15.0±0.0 g. Seven days after nerve ligation, the meanwithdrawal threshold was 4.7±1.02 g, indicating the develop-ment of tactile allodynia (n=74). The animals were separatedat random into i.t. treatment groups: 33% EtOH vehicle,morphine (10 μg), gabapentin (30 μg), RSA 504 (3, 10, or 30 μg),
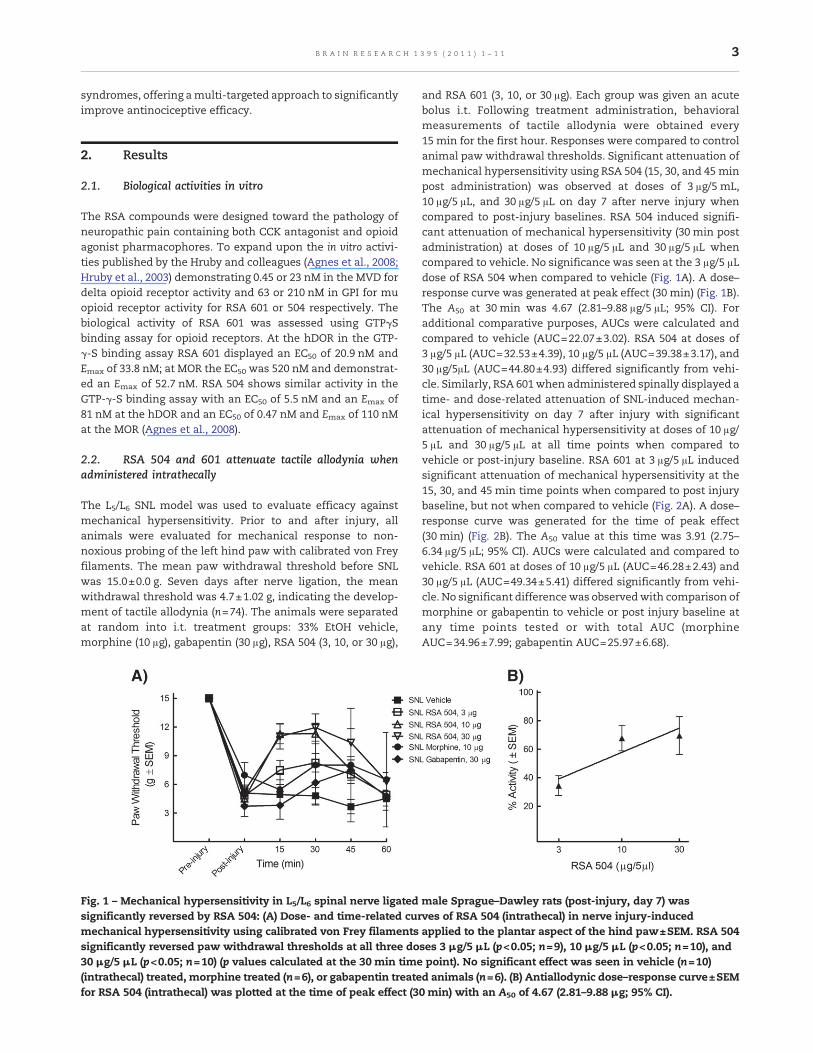
and RSA 601 (3, 10, or 30 μg). Each group was given an acutebolus i.t. Following treatment administration, behavioralmeasurements of tactile allodynia were obtained every15 min for the first hour. Responses were compared to controlanimal paw withdrawal thresholds. Significant attenuation ofmechanical hypersensitivity using RSA 504 (15, 30, and 45 minpost administration) was observed at doses of 3 μg/5 mL,10 μg/5 μL, and 30 μg/5 μL on day 7 after nerve injury whencompared to post-injury baselines. RSA 504 induced signifi-cant attenuation of mechanical hypersensitivity (30 min postadministration) at doses of 10 μg/5 μL and 30 μg/5 μL whencompared to vehicle. No significance was seen at the 3 μg/5 μLdose of RSA 504 when compared to vehicle (Fig. 1A). A dose–response curve was generated at peak effect (30 min) (Fig. 1B).The A50 at 30 min was 4.67 (2.81–9.88 μg/5 μL; 95% CI). Foradditional comparative purposes, AUCs were calculated andcompared to vehicle (AUC=22.07±3.02). RSA 504 at doses of3 μg/5 μL (AUC=32.53±4.39), 10 μg/5 μL (AUC=39.38±3.17), and30 μg/5μL (AUC=44.80±4.93) differed significantly from vehi-cle. Similarly, RSA 601when administered spinally displayed atime- and dose-related attenuation of SNL-induced mechan-ical hypersensitivity on day 7 after injury with significantattenuation of mechanical hypersensitivity at doses of 10 μg/5 μL and 30 μg/5 μL at all time points when compared tovehicle or post-injury baseline. RSA 601 at 3 μg/5 μL inducedsignificant attenuation of mechanical hypersensitivity at the15, 30, and 45 min time points when compared to post injurybaseline, but not when compared to vehicle (Fig. 2A). A dose–response curve was generated for the time of peak effect(30 min) (Fig. 2B). The A50 value at this time was 3.91 (2.75–6.34 μg/5 μL; 95% CI). AUCs were calculated and compared tovehicle. RSA 601 at doses of 10 μg/5 μL (AUC=46.28±2.43) and30 μg/5 μL (AUC=49.34±5.41) differed significantly from vehi-cle. No significant differencewas observedwith comparison ofmorphine or gabapentin to vehicle or post injury baseline atany time points tested or with total AUC (morphineAUC=34.96±7.99; gabapentin AUC=25.97±6.68).
Fig. 1 – Mechanical hypersensitivity in L5/L6 spinal nerve ligated male Sprague–Dawley rats (post-injury, day 7) wassignificantly reversed by RSA 504: (A) Dose- and time-related curves of RSA 504 (intrathecal) in nerve injury-inducedmechanical hypersensitivity using calibrated von Frey filaments applied to the plantar aspect of the hind paw±SEM. RSA 504significantly reversed paw withdrawal thresholds at all three doses 3 μg/5 μL (p<0.05; n=9), 10 μg/5 μL (p<0.05; n=10), and30 μg/5 μL (p<0.05; n=10) (p values calculated at the 30 min time point). No significant effect was seen in vehicle (n=10)(intrathecal) treated, morphine treated (n=6), or gabapentin treated animals (n=6). (B) Antiallodynic dose–response curve±SEMfor RSA 504 (intrathecal) was plotted at the time of peak effect (30 min) with an A50 of 4.67 (2.81–9.88 μg; 95% CI).
3B R A I N R E S E A R C H 1 3 9 5 ( 2 0 1 1 ) 1 – 1 1
2.3. RSA 504 and 601 attenuate thermal hyperalgesiawhen administered intrathecally
The L5/L6 SNLmodel was also used to evaluate efficacy againstthermal hypersensitivity. Prior to and after injury, all animalswere evaluated for hyperalgesic response to noxious probingof the left hind paw with an infrared cylinder. The mean pawwithdrawal latency before SNL was 22.65±1.6 s (n=80). Sevendays after nerve ligation, the mean withdrawal latency was12.21±0.26 s (n=80), indicating the development of thermal
hyperalgesia. The animals were separated at random intoi.t. treatment groups: 33% EtOH vehicle, morphine (10 mg),gabapentin (30 mg), RSA 504 (1, 10, or 30 μg), and RSA 601(1, 6, or 30 μg). An acute bolus was given i.t. to rats that hadundergone L5/L6 SNL 7 days earlier. Additionally, shamcontrol groups were included in the studies in which theanimals received the same surgery as SNL animals minusthe ligation of the L5 and L6 spinal nerves. No change inpaw withdrawal latency was observed in sham-operatedanimals from pre-surgical baseline (22.09±0.76 s, n=18) to
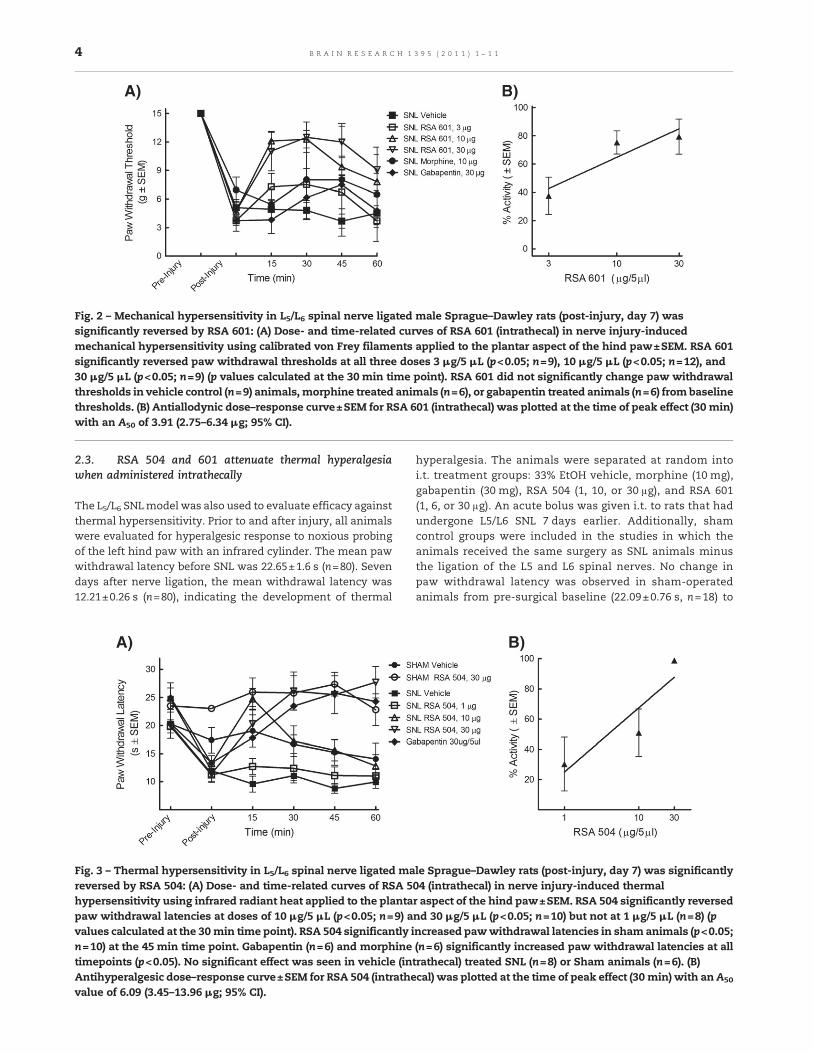
Fig. 2 – Mechanical hypersensitivity in L5/L6 spinal nerve ligated male Sprague–Dawley rats (post-injury, day 7) wassignificantly reversed by RSA 601: (A) Dose- and time-related curves of RSA 601 (intrathecal) in nerve injury-inducedmechanical hypersensitivity using calibrated von Frey filaments applied to the plantar aspect of the hind paw±SEM. RSA 601significantly reversed paw withdrawal thresholds at all three doses 3 μg/5 μL (p<0.05; n=9), 10 μg/5 μL (p<0.05; n=12), and30 μg/5 μL (p<0.05; n=9) (p values calculated at the 30 min time point). RSA 601 did not significantly change paw withdrawalthresholds in vehicle control (n=9) animals, morphine treated animals (n=6), or gabapentin treated animals (n=6) from baselinethresholds. (B) Antiallodynic dose–response curve±SEM for RSA 601 (intrathecal) was plotted at the time of peak effect (30 min)with an A50 of 3.91 (2.75–6.34 μg; 95% CI).
Fig. 3 – Thermal hypersensitivity in L5/L6 spinal nerve ligated male Sprague–Dawley rats (post-injury, day 7) was significantlyreversed by RSA 504: (A) Dose- and time-related curves of RSA 504 (intrathecal) in nerve injury-induced thermalhypersensitivity using infrared radiant heat applied to the plantar aspect of the hind paw±SEM. RSA 504 significantly reversedpaw withdrawal latencies at doses of 10 μg/5 μL (p<0.05; n=9) and 30 μg/5 μL (p<0.05; n=10) but not at 1 μg/5 μL (n=8) (pvalues calculated at the 30 min time point). RSA 504 significantly increased pawwithdrawal latencies in shamanimals (p<0.05;n=10) at the 45 min time point. Gabapentin (n=6) and morphine (n=6) significantly increased paw withdrawal latencies at alltimepoints (p<0.05). No significant effect was seen in vehicle (intrathecal) treated SNL (n=8) or Sham animals (n=6). (B)Antihyperalgesic dose–response curve±SEM for RSA 504 (intrathecal) was plotted at the time of peak effect (30 min) with an A50
value of 6.09 (3.45–13.96 μg; 95% CI).
4 B R A I N R E S E A R C H 1 3 9 5 ( 2 0 1 1 ) 1 – 1 1
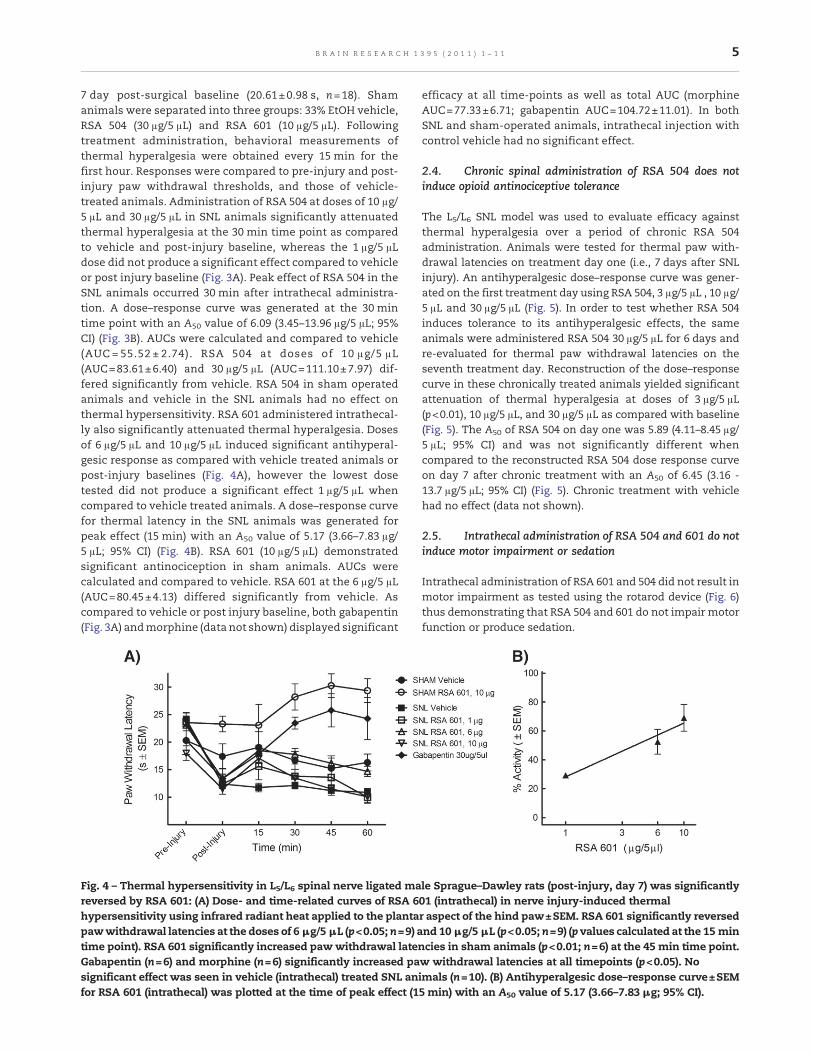
7 day post-surgical baseline (20.61±0.98 s, n=18). Shamanimals were separated into three groups: 33% EtOH vehicle,RSA 504 (30 μg/5 μL) and RSA 601 (10 μg/5 μL). Followingtreatment administration, behavioral measurements ofthermal hyperalgesia were obtained every 15 min for thefirst hour. Responses were compared to pre-injury and post-injury paw withdrawal thresholds, and those of vehicle-treated animals. Administration of RSA 504 at doses of 10 μg/5 μL and 30 μg/5 μL in SNL animals significantly attenuatedthermal hyperalgesia at the 30 min time point as comparedto vehicle and post-injury baseline, whereas the 1 μg/5 μLdose did not produce a significant effect compared to vehicleor post injury baseline (Fig. 3A). Peak effect of RSA 504 in theSNL animals occurred 30 min after intrathecal administra-tion. A dose–response curve was generated at the 30 mintime point with an A50 value of 6.09 (3.45–13.96 μg/5 μL; 95%CI) (Fig. 3B). AUCs were calculated and compared to vehicle(AUC = 55.52 ± 2.74). RSA 504 at doses of 10 μg/5 μL(AUC=83.61±6.40) and 30 μg/5 μL (AUC=111.10±7.97) dif-fered significantly from vehicle. RSA 504 in sham operatedanimals and vehicle in the SNL animals had no effect onthermal hypersensitivity. RSA 601 administered intrathecal-ly also significantly attenuated thermal hyperalgesia. Dosesof 6 μg/5 μL and 10 μg/5 μL induced significant antihyperal-gesic response as compared with vehicle treated animals orpost-injury baselines (Fig. 4A), however the lowest dosetested did not produce a significant effect 1 μg/5 μL whencompared to vehicle treated animals. A dose–response curvefor thermal latency in the SNL animals was generated forpeak effect (15 min) with an A50 value of 5.17 (3.66–7.83 μg/5 μL; 95% CI) (Fig. 4B). RSA 601 (10 μg/5 μL) demonstratedsignificant antinociception in sham animals. AUCs werecalculated and compared to vehicle. RSA 601 at the 6 μg/5 μL(AUC=80.45±4.13) differed significantly from vehicle. Ascompared to vehicle or post injury baseline, both gabapentin(Fig. 3A) andmorphine (data not shown) displayed significant
efficacy at all time-points as well as total AUC (morphineAUC=77.33±6.71; gabapentin AUC=104.72±11.01). In bothSNL and sham-operated animals, intrathecal injection withcontrol vehicle had no significant effect.
2.4. Chronic spinal administration of RSA 504 does notinduce opioid antinociceptive tolerance
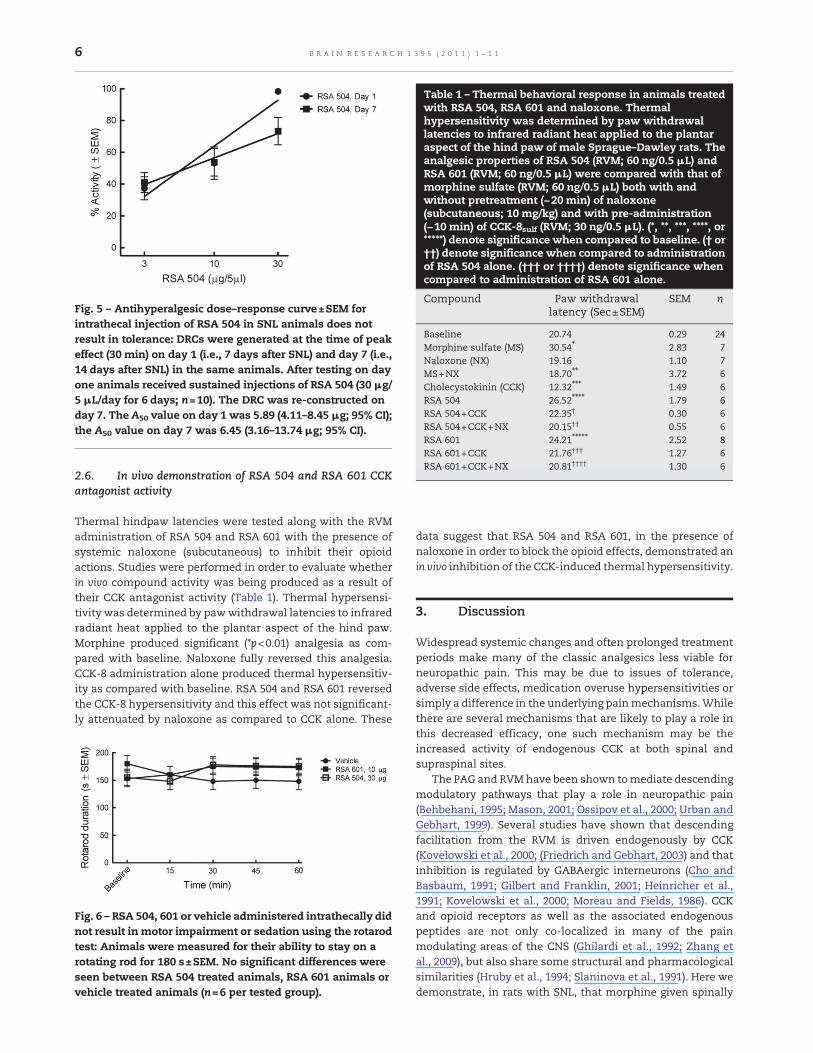
The L5/L6 SNL model was used to evaluate efficacy againstthermal hyperalgesia over a period of chronic RSA 504administration. Animals were tested for thermal paw with-drawal latencies on treatment day one (i.e., 7 days after SNLinjury). An antihyperalgesic dose–response curve was gener-ated on the first treatment day using RSA 504, 3 μg/5 μL , 10 μg/5 μL and 30 μg/5 μL (Fig. 5). In order to test whether RSA 504induces tolerance to its antihyperalgesic effects, the sameanimals were administered RSA 504 30 μg/5 μL for 6 days andre-evaluated for thermal paw withdrawal latencies on theseventh treatment day. Reconstruction of the dose–responsecurve in these chronically treated animals yielded significantattenuation of thermal hyperalgesia at doses of 3 μg/5 μL(p<0.01), 10 μg/5 μL, and 30 μg/5 μL as compared with baseline(Fig. 5). The A50 of RSA 504 on day one was 5.89 (4.11–8.45 μg/5 μL; 95% CI) and was not significantly different whencompared to the reconstructed RSA 504 dose response curveon day 7 after chronic treatment with an A50 of 6.45 (3.16 -13.7 μg/5 μL; 95% CI) (Fig. 5). Chronic treatment with vehiclehad no effect (data not shown).
2.5. Intrathecal administration of RSA 504 and 601 do notinduce motor impairment or sedation
Intrathecal administration of RSA 601 and 504 did not result inmotor impairment as tested using the rotarod device (Fig. 6)thus demonstrating that RSA 504 and 601 do not impair motorfunction or produce sedation.
Fig. 4 – Thermal hypersensitivity in L5/L6 spinal nerve ligated male Sprague–Dawley rats (post-injury, day 7) was significantlyreversed by RSA 601: (A) Dose- and time-related curves of RSA 601 (intrathecal) in nerve injury-induced thermalhypersensitivity using infrared radiant heat applied to the plantar aspect of the hind paw±SEM. RSA 601 significantly reversedpawwithdrawal latencies at the doses of 6 μg/5 μL (p<0.05; n=9) and 10 μg/5 μL (p<0.05; n=9) (p values calculated at the 15 mintime point). RSA 601 significantly increased pawwithdrawal latencies in sham animals (p<0.01; n=6) at the 45 min time point.Gabapentin (n=6) and morphine (n=6) significantly increased paw withdrawal latencies at all timepoints (p<0.05). Nosignificant effect was seen in vehicle (intrathecal) treated SNL animals (n=10). (B) Antihyperalgesic dose–response curve±SEMfor RSA 601 (intrathecal) was plotted at the time of peak effect (15 min) with an A50 value of 5.17 (3.66–7.83 μg; 95% CI).
5B R A I N R E S E A R C H 1 3 9 5 ( 2 0 1 1 ) 1 – 1 1
2.6. In vivo demonstration of RSA 504 and RSA 601 CCKantagonist activity
Thermal hindpaw latencies were tested along with the RVMadministration of RSA 504 and RSA 601 with the presence ofsystemic naloxone (subcutaneous) to inhibit their opioidactions. Studies were performed in order to evaluate whetherin vivo compound activity was being produced as a result oftheir CCK antagonist activity (Table 1). Thermal hypersensi-tivity was determined by pawwithdrawal latencies to infraredradiant heat applied to the plantar aspect of the hind paw.Morphine produced significant (*p<0.01) analgesia as com-pared with baseline. Naloxone fully reversed this analgesia.CCK-8 administration alone produced thermal hypersensitiv-ity as compared with baseline. RSA 504 and RSA 601 reversedthe CCK-8 hypersensitivity and this effect was not significant-ly attenuated by naloxone as compared to CCK alone. These
data suggest that RSA 504 and RSA 601, in the presence ofnaloxone in order to block the opioid effects, demonstrated anin vivo inhibition of the CCK-induced thermal hypersensitivity.
3. Discussion
Widespread systemic changes and often prolonged treatmentperiods make many of the classic analgesics less viable forneuropathic pain. This may be due to issues of tolerance,adverse side effects, medication overuse hypersensitivities orsimply a difference in the underlying painmechanisms.Whilethere are several mechanisms that are likely to play a role inthis decreased efficacy, one such mechanism may be theincreased activity of endogenous CCK at both spinal andsupraspinal sites.
The PAG and RVMhave been shown tomediate descendingmodulatory pathways that play a role in neuropathic pain(Behbehani, 1995; Mason, 2001; Ossipov et al., 2000; Urban andGebhart, 1999). Several studies have shown that descendingfacilitation from the RVM is driven endogenously by CCK(Kovelowski et al., 2000; (Friedrich and Gebhart, 2003) and thatinhibition is regulated by GABAergic interneurons (Cho andBasbaum, 1991; Gilbert and Franklin, 2001; Heinricher et al.,1991; Kovelowski et al., 2000; Moreau and Fields, 1986). CCKand opioid receptors as well as the associated endogenouspeptides are not only co-localized in many of the painmodulating areas of the CNS (Ghilardi et al., 1992; Zhang etal., 2009), but also share some structural and pharmacologicalsimilarities (Hruby et al., 1994; Slaninova et al., 1991). Here wedemonstrate, in rats with SNL, that morphine given spinally
Fig. 5 – Antihyperalgesic dose–response curve±SEM forintrathecal injection of RSA 504 in SNL animals does notresult in tolerance: DRCs were generated at the time of peakeffect (30 min) on day 1 (i.e., 7 days after SNL) and day 7 (i.e.,14 days after SNL) in the same animals. After testing on dayone animals received sustained injections of RSA 504 (30 μg/5 μL/day for 6 days; n=10). The DRC was re-constructed onday 7. The A50 value on day 1 was 5.89 (4.11–8.45 μg; 95% CI);the A50 value on day 7 was 6.45 (3.16–13.74 μg; 95% CI).
Fig. 6 – RSA 504, 601 or vehicle administered intrathecally didnot result in motor impairment or sedation using the rotarodtest: Animals were measured for their ability to stay on arotating rod for 180 s±SEM. No significant differences wereseen between RSA 504 treated animals, RSA 601 animals orvehicle treated animals (n=6 per tested group).
Table 1 – Thermal behavioral response in animals treatedwith RSA 504, RSA 601 and naloxone. Thermalhypersensitivity was determined by paw withdrawallatencies to infrared radiant heat applied to the plantaraspect of the hind paw of male Sprague–Dawley rats. Theanalgesic properties of RSA 504 (RVM; 60 ng/0.5 μL) andRSA 601 (RVM; 60 ng/0.5 μL) were compared with that ofmorphine sulfate (RVM; 60 ng/0.5 μL) both with andwithout pretreatment (−20 min) of naloxone(subcutaneous; 10 mg/kg) and with pre-administration(−10 min) of CCK-8sulf (RVM; 30 ng/0.5 μL). (*, **, ***, ****, or*****) denote significance when compared to baseline. († or††) denote significance when compared to administrationof RSA 504 alone. (††† or ††††) denote significance whencompared to administration of RSA 601 alone.
Compound Paw withdrawallatency (Sec±SEM)
SEM n
Baseline 20.74 0.29 24Morphine sulfate (MS) 30.54⁎ 2.83 7Naloxone (NX) 19.16 1.10 7MS+NX 18.70⁎⁎ 3.72 6Cholecystokinin (CCK) 12.32⁎⁎⁎ 1.49 6RSA 504 26.52⁎⁎⁎⁎ 1.79 6RSA 504+CCK 22.35† 0.30 6RSA 504+CCK+NX 20.15†† 0.55 6RSA 601 24.21⁎⁎⁎⁎⁎ 2.52 8RSA 601+CCK 21.76††† 1.27 6RSA 601+CCK+NX 20.81†††† 1.30 6
6 B R A I N R E S E A R C H 1 3 9 5 ( 2 0 1 1 ) 1 – 1 1

lacks efficacy for mechanical hypersensitivity after nerveligation. However, when administered spinally, both RSA 504and RSA 601 effectively inhibited tactile and thermal hyper-sensitivity caused by L5/L6 SNL at doses that do not producesedation. By having dual pharmacophores, opioid agonist/CCKantagonist activity, the RSA compounds are more likely toreverse nerve injury-induced hypersensitivities.
Previous studies have demonstrated that a single injectionof a CCK-2 antagonist into the RVM reversed L5/L6 SNL-induced mechanical and thermal hypersensitivity in a timedependent manner with peak effect occurring 10 min postinjection (Kovelowski et al., 2000). Similarly, mechanicalhypersensitivity from nerve injury was reversed by spinalco-administration of a CCK-2 antagonist and morphine(Dourish et al., 1990) or spinal CCK-2 antagonist and inhibitorsof endogenous enkephalin (Nichols et al., 1996). Studies byFriedrich and Gebhart (Friedrich and Gebhart, 2003) demon-strated that a CCK-2 antagonist administered into the RVMdose- and time-dependently reversed a visceral hypersensi-tive response induced by chronic inflammation of the GI tract.These data suggest that peripheral nerve injury to the lumbarregion or chronic inflammation of visceral afferents results inthe increase in endogenous CCK in the spinal cord and RVMpromoting behavioral signs of pain and, therefore, a com-pound that acts to inhibit CCK receptors and activate opioidreceptors is more likely to inhibit neuropathic and inflamma-tory pain.
Although in rodent studies the CCK-2 receptor does seemto be the primary arbiter of CCK descending modulation of theendogenous opioid system, this may not be the case inhumans. Human trials conducted by McCleane evaluated theeffects of the CCK-2 specific antagonist L365,260 in conjunc-tionwithmorphine and did not show any augmentation to theanalgesic effects of morphine (McCleane, 2003). Conversely,proglumide, a non-specific CCK inhibitor, did augment theanalgesic efficacy of morphine in human trials (McCleane,1998), suggesting that in humans both CCK-1 and CCK-2 mayplay a role in pain modulation. Therefore, a non-selective CCKantagonist may be the most appropriate for clinical develop-ment. The compounds tested here have binding affinity in thenanomolar range at both CCK receptors, and simultaneouslyagonize δ and μ opioid receptors; a characteristic that mayfurther increase utility of these compounds. Along withincreases in affinity and specificity, patient care is alsoimproved by simultaneously counteracting multiple facets ofone disease state, lowering required doses, and reducing therisk of side effects and drug–drug interactions.
Our studies demonstrate that RSA 601, and to a lesserextent RSA 504, resulted in a significant increase in thermalthresholds above baseline in sham animals. By measuringtheir ability to attenuate CCK-8sulf induced thermal hyper-algesia using naïve animals in the presence of naloxone, theRSA compoundswere shown to act as CCK antagonists in vivo.Since these compounds have opioid agonist activity, weblocked their ability to act at opioid receptors using naloxoneand found that the CCK antagonist pharmacophore wasactive against CCK-8-induced thermal hyperalgesia confirm-ing their dual acting biological activity. Preclinical studieshave shown that long-term opioids can elicit abnormal painstates manifested as paradoxical algesia and hyperesthesias
(Mao et al., 1995; Trujillo and Akil, 1991; Woolf, 1981; YakshandHarty, 1988). The development of thermal hyperalgesia inresponse to either repeated injections or constant infusion ofmorphine has also been reported (Mao et al., 1994, 1995).Thus, opioids given over time may maintain their level ofefficacy, but the concurrent development of hyperalgesiacould serve to counteract the antinociceptive effect ofopioids, producing an impression of tolerance (Colpaert,1996; Laulin et al., 1998; Laulin et al., 1999). Systems thatmay be activated upon sustained morphine administrationinclude the descending pain facilitatory pathways from theRVM up-regulating the endogenous pronociceptive transmit-ter CCK.
Using in vivomicrodialysis, Afrahand colleagues (Afrah et al.,2001) demonstrated that peripheral nerve injury results in a 3fold increase in spinal endogenous CCK levels 7 days afterinjury. Likewise, using in vivo microdialysis we have demon-strated a 2- to 3-fold increase in endogenous CCK in the RVM,7 days after L5/L6 SNL (abstract SfN, Vanderah Lab, 2008). Theexogenous administration of CCK into the RVM results in a timedependent mechanical and thermal hypersensitivity as well asvisceral hyperalgesia in naïve animals (Friedrich and Gebhart,2003; Xie et al., 2005). CCK is thought to promote pain byactivating a pain facilitatory pathway in the RVM. This pathwayhas been described as neurons that fire when pain is present,termed “ON cells” and are directly activated by the administra-tion of CCK (Heinricher and Neubert, 2004). Collectively, thesedata strongly suggest that nerve injury or chronic inflammationresults in the increase of endogenousCCK in the spinal cord andRVM resulting in pain facilitation, strongly supporting theconcept of having a compound such as RSA 504 or 601 that notonly activates opioid receptors but also blocks CCK receptors toinhibit neuropathic pain and antinociceptive tolerance.
Here we demonstrate that chronic administration of RSA504 does not induce the onset of opioid antinociceptivetolerance: a promising finding consistent with previousfindings that a CCK antagonist can reverse morphine-inducedantinociceptive tolerance (Dourish et al., 1990). Likewise,several studies in humans with chronic pain have demon-strated significant opioid sparing effects by co-administering aCCK antagonist with an opioid and/or pain relief (McCleane,1998; McCleane, 2002; McCleane, 2003). Unfortunately, suchhuman studies cannot proceed since CCK antagonists, ap-proved for human use, are no longer being manufacturedmost likely due to expired patents.
Over the past 20 years companies have continued to designhighly selective compounds for a single target in order toattenuate neuropathic pain, however, such strategies have notresulted in a plethora of highly efficacious compounds thatlack unwanted side effects. By designing compounds thattarget μ and δ opioid receptors, as well as the CCK receptors,we have produced novel compounds that demonstrate better,longer lasting antinociceptive efficacy in a neuropathic painmodel. Novel compounds are being designed to targetmultiple sites that may offer better long-term efficacy and/orlack side effects due to over stimulation of one moleculartarget. Here we report in vivo behavioral studies withcompounds designed to have agonist and antagonist activityat two distinctly different receptors for neuropathic pain. CCKis upregulated under conditions of neuropathic pain and
7B R A I N R E S E A R C H 1 3 9 5 ( 2 0 1 1 ) 1 – 1 1

therefore a compound is needed to block its pro-algesiceffects. Unlike morphine, RSA 504 with CCK antagonistactivity and opioid agonist activity did not result in anti-hyperalgesic tolerance over a 7 day period in nerve injuredanimals suggesting that the sustained administration of suchdual acting pharmacophores may be effective over a longperiod of time in chronic pain patients. The results of thisstudy show that the design of single compounds with dualpharmacophores leads to promising therapeutic agents for thetreatment of neuropathic pain lacking unwanted side effectssuch as tolerance, sedation and opioid-induced hyperalgesia.
4. Experimental procedures
4.1. Peptides
The peptides reported here were synthesized by standardsolid phase peptide synthesis methods (Agnes et al., 2008;Hruby et al., 2003) by the Hruby lab in the University of Arizonadepartment of Chemistry.
4.2. In vitro assays
4.2.1. GTPγS bindingGTPγS binding was done using membranes from cells thatexpress the human δ or rat μ opioid receptors. The procedurefor this analysis was based on that of Lorenzen et al.(1993).Reactions were initiated by the addition of an aliquot ofmembrane preparation (15 μg) to a final volume of 300 μL ofincubation mix [50 mM HEPES, pH.7.4, 1 mM EDTA, 5 mMMgCl2, 30 μM GDP, 1 mM dithiothreitol, 100 mM NaCl, 100 μMphenylmethylsulfonyl fluoride, 0.1% bovine serum albumin,0.1 nM [35S]GTPγS (1250 Ci/mmol)] and indicated concentra-tion range of agonist and incubated for 60 min at 30 °C. Basallevel of [35S]GTPγS binding was defined as the amount boundin the absence of agonist. Nonspecific binding was deter-mined in the presence of 10 μM unlabeled GTPγS. Reactionswere performed in triplicate and terminated by rapidfiltration through Whatman GF/B filters presoaked in waterfollowed by four washes with ice-cold wash buffer (50 mMTris, 5 mMMgCl2, and 100 mMNaCl, pH 7.4). The radioactivitywas determined by liquid scintillation counting. Data werefitted by nonlinear least-squares analysis using GraphPadPrism.
4.3. In vivo assays
4.3.1. AnimalsThe experiments contained herein were carried out usingmale Sprague–Dawley rats (250–350 g; Harlan; Indianapolis,IN). All animals weremaintained on a 12/12 hr light/dark cycle(lights on at 07:00 am) and provided food and water ad libitumexcept as noted during the experimental procedures. Allexperiments were performed under an approved protocol bythe Institutional Animal Care and Use Committee of theUniversity of Arizona, and in accordance with policies andguidelines for the care and use of laboratory animals asadopted by International Association for the Study of Pain andthe National Institutes of Health.
4.3.2. L5/L6 SNL surgerySNL injury was induced in male Sprague–Dawley rats asdescribed by Chung and colleagues (Kim and Chung, 1992).Anesthesia was induced with 2% isoflurane in O2 at 5 L/minand maintained with 2.5% isoflurane in O2. The dorsalvertebral column from L4 to S2 was exposed; the L5 and L6spinal nerves were tightly ligated distal to the dorsal rootganglion using 4-0 silk suture. The incision was closed and theanimals were allowed to recover for 5–7 days. Rats thatexhibited motor deficiency (such as paw dragging) or failureto exhibit subsequent tactile allodynia were excluded fromfurther testing (<5%). Sham control rats underwent the sameoperation and handling as the experimental animals, butwithout ligation of the L5/L6 spinal nerves.
4.3.3. Intrathecal catheter surgeryMale Sprague–Dawley rats were prepared for intrathecal drugadministration as described by Yaksh and Rudy (Yaksh andRudy, 1976) by placing anesthetized (ketamine/xylazine100 mg/kg, intraperitoneal) animals in a stereotaxic headholder. The cisterna magna was exposed, an incision wasmade and animals were implanted with a catheter (PE: 10,8 mm) that terminated in the lumbar region of the spinal cord.The animals were allowed to recover 5–7 days post-surgerybefore any pharmacological manipulations were made. Anyrats showing impaired motor skills (e.g. paralysis) or greaterthan 10% weight loss were excluded from experiments (< 5%).
4.3.4. RVM cannulationMale Sprague–Dawley rats were cannulated as described bythe Porreca group (Roberts et al., 2009). Rats were anesthetized(ketamine/xylazine 100 mg/kg, intraperitoneal) and placedinto a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA).A 2 cm incision was made in the scalp, and the underlyingconnective tissue was retracted with hemostats to expose theskull. Paired guide cannulae 1.2 mm apart (26GA, #C235G-1.2 mm; Plastics One Inc., Roanoke, VA, USA) were directedtoward the RVM (10.8 mm caudal to bregma, 0.6 mm to eachside of the sagittal suture, and 7.0 mm ventral to the duramater surface). Bone wax was used to seal the hole around thecannula. The paired guide cannula was secured to the skullusing stainless steel screws and dental acrylic. A dummycannula (#C235DC; Plastics One Inc.) was inserted to preventcontaminants from entering the RVM guide cannula. Ratswere given an injection of antibiotic (amikacin C, 5 mg/kg,intraperitoneal) and allowed to recover for 7 days.
4.3.5. Drug administrationRSA 504 and RSA 601 were dissolved in 33% EtOH and broughtto volume with MilliPore H2O. CCK-8sulf was dissolved inMillipore H2O. Morphine sulfate was dissolved in 0.9% saline.Deltorphin (Bachem) was dissolved in 30% DMSO and broughtto volume with 0.9% saline. Naloxone (RBI) was dissolved in0.9% saline. For intrathecal compound administration, allcompounds were injected in a 5 μL volume followed by a 9 μLsaline flush. Blinded testing took place 15, 30, 45 and 60 minafter drug injection, and dose–response curveswere generatedfrom data gathered at the time of peak effect. RVM drugadministrations were performed by slowly expelling 0.5 μLbilaterally through an injection cannula protruding 1 mm
8 B R A I N R E S E A R C H 1 3 9 5 ( 2 0 1 1 ) 1 – 1 1

beyond the tip of the guide to prevent backflow of drug into theguide cannula. Naloxone (10 mg/kg), when used, was admin-istered subcutaneously 20 min before behavioral testing.
4.3.6. Behavioral assessment
4.3.6.1. Thermal hypersensitivity. Thermal hypersensitivitywas assessed using the rat plantar test following a modifiedmethod of Hargreaves and colleagues (Hargreaves et al., 1988).Male Sprague–Dawley rats were allowed to acclimate withinPlexiglas enclosures on a clear glass plate. A mobile radiantheat source (Ugo Basile, Italy) was located under the glassplate and focused onto the hind paw. Paw withdrawallatencies were recorded in seconds at 15 min intervals for60 min. An automatic cut off point of 33.0 s was set to preventtissue damage. The apparatus was calibrated to give a pawwithdrawal latency of approximately 20 s on the uninjuredpaw. The radiant heat source was activated with a timer andfocused onto the plantar surface of the hindpaw. A motiondetector which halted both heat source and timer when thepaw was withdrawn determined paw-withdrawal latencies.The ipsilateral paws of SNL and sham animals were testedusing the radiant heat source. The contralateral pawswere nottested so that the injured paw would not be forced to bearweight unnecessarily. No thermal hypersensitivity was seenin sham animals. Crossover studies were performed whenpossible to reduce the total number of animals used: SNL orsham animals receiving vehicle during acute phase testingreceived a dose of either RSA 504 or 601 aminimumof 5 h laterfor additional acute testing after repeating post-injury base-line testing.
Injury % Activity Calculation (% Antihyperalgesia): %Activity=100×(withdrawal latency post drug−post surgerybaseline latency)/ (pre surgery baseline latency−post surgerybaseline latency)
Sham operated percent activities (% analgesia): % Activi-ty=100×(withdrawal latency post drug−post sham baselinelatency) / (pre sham baseline latency−post sham baselinelatency)
4.3.6.2. Mechanical hypersensitivity. The assessment of me-chanical hypersensitivity consisted of measuring the with-drawal threshold of the paw ipsilateral to the site of nerveinjury in response to probing with a series of calibrated vonFrey filaments. Prior to the SNL surgery, male Sprague–Dawleyrats were tested for pre-injury baselinemechanical sensitivity.Each filament was applied perpendicularly to the plantarsurface of the left hind paw of rats kept in suspended wire-mesh cages. 7 days post SNL surgery on the left hind limb,measurements were taken both before (post-injury baseline)and after administration of RSA 504, RSA 601, or vehicle at 15-min intervals for 60 min. The withdrawal threshold wasdetermined by sequentially increasing and decreasing thestimulus strength (“up–down” method) analyzed using aDixon non-parametric test (Chaplan et al., 1994) andexpressed as the mean withdrawal threshold. SNL animalswere tested with the von Frey filaments while sham animalswere not since maximum threshold was set to 15 g. Highercalibrated filaments (>15 g) result in the actual lifting of thepaw that may be misinterpreted as a mechanical withdrawal
(the higher calibrated filaments support the weight of thepaw). Crossover studies were performed when possible toreduce the total number of animals used: SNL or shamanimals receiving vehicle during acute phase testing receiveda dose of either RSA 504 or 601 a minimum of 5 hours later foradditional acute testing after repeating post-injury baselinetesting.
Injury % Activity Calculation (% Antiallodynia): % Activi-ty=100×(withdrawal threshold post drug−post surgery base-line threshold)/ (pre surgery baseline threshold−post surgerybaseline threshold)
4.3.6.3. Motor function. Male Sprague–Dawley rats weretrained to ambulate on a rotarod device (Columbus Instru-ments International, Columbus, OH) as previously described(Vanderah et al., 2008) until all could remain on the device for aduration of 180 s at a speed of 10 revolutions per minute. Theratswere tested again after administration of RSA 504, RSA 601or vehicle, and the time the rats were able to remain on thedevice without falling was recorded at 15-min intervals for60 min. The κ opioid agonist U50,488 was used as a positivecontrol (not shown). A maximum cutoff time of 180 s wasused.
4.4. Statistical analysis
[35S]GTPγS binding data were analyzed by non-linear regres-sion analysis using GraphPad Inplot. For binding affinity, theKi value(s) for each ligandwas calculated from the IC50 value(s)based on the Cheng and Prusoff equation from at least threeindependent experiments. For GTPγS binding, potency wasexpressed as log EC50±S.E.M. Maximal effect was expressed asEmax±S.E.M.
Thermal and tactile hypersensitivity data were analyzed aspreviously published using one-way analysis of variancefollowed by students Neuman–Kuels testing for multiplecomparisons in FlashCalc (Vanderah et al., 2000; Xie et al.,2005; Zhang et al., 2009). Differences were considered to besignificant if p≤0.05. When possible, potencies (or A50) weredetermined by regression analysis of dose–response curve (logdose [x] vs response [y]) using a 95% confidence intervalaccording to the method of analysis of the Graded Dose–Response (Tallarida and Murray, 1987). For the calculations ofA50's, the minimal possible response was set to 0%.
Acknowledgments
1. These studieswere supportedbyNIDAgrant 2PO1DA006284.1.2. All peptides in this study were generously provided by the
Hruby lab in the Department of Chemistry at the Universityof Arizona, Tucson, AZ.
R E F E R E N C E S
Afrah, A.W., Gustafsson, H., Olgart, L., Brodin, E., Stiller, C.O., 2001.Changes in spinal cholecystokinin release after peripheralaxotomy. Neuroreport. 12, 49–52.
9B R A I N R E S E A R C H 1 3 9 5 ( 2 0 1 1 ) 1 – 1 1

Agnes, R.S., Ying, J., Kover, K.E., Lee, Y.S., Davis, P., Ma, S.W.,Badghisi, H., Porreca, F., Lai, J., Hruby, V.J., 2008. Structure–activity relationships of bifunctional cyclic disulfide peptidesbased on overlapping pharmacophores at opioid and chole-cystokinin receptors. Peptides. 29, 1413–1423.
Becker, C., Pohl, M., Thiebot, M.-H., Collin, E., Hamon, M., Cesslin,F., Benoliel, J.-J., 2000. δ-opioid receptor-mediated increase incortical extracellular levels of cholecystokinin-like materialby subchronic morphine in rats. Neuropharmacology. 39,161–171.
Behbehani, M.M., 1995. Functional characteristics of the midbrainperiaqueductal gray. Prog Neurobiol. 46, 575–605.
Breivik, H., Collett, B., Ventafridda, V., Cohen, R., Gallacher, D.,2006. Survey of chronic pain in Europe: prevalence, impact ondaily life, and treatment. Eur J Pain. 10, 287–333.
Chaplan, S.R., Bach, F.W., Pogrel, J.W., Chung, J.M., Yaksh, T.L.,1994. Quantitative assessment of tactile allodynia in the ratpaw. J Neurosci Methods. 53, 55–63.
Cho, H.J., Basbaum, A.I., 1991. GABAergic circuitry in the rostralventral medulla of the rat and its relationship to descendingantinociceptive controls. Journal of Comparative Neurology.303, 316–328.
Colpaert, F.C., 1996. System theory of pain and of opiate analgesia:no tolerance to opiates. Pharmacol Rev. 48, 355–402.
Dourish, C.T., O'Neill, M.F., Coughlan, J., Kitchener, S.J., Hawley, D.,Iversen, S.D., 1990. The selective CCK-B receptor antagonist L-365,260 enhances morphine analgesia and prevents morphinetolerance in the rat. Eur J Pharmacol. 176, 35–44.
Frampton, J.E., 2010. Tapentadol immediate release: a review of itsuse in the treatment of moderate to severe acute pain. Drugs.70, 1719–1743.
Friedrich, A.E., Gebhart, G.F., 2003. Modulation of visceral hyper-algesia by morphine and cholecystokinin from the rat rostro-ventral medial medulla. Pain. 104, 93–101.
Ghilardi, J.R., Allen, C.J., Vigna, S.R., McVey, D.C., Mantyh, P.W.,1992. Trigeminal and dorsal root ganglion neurons expressCCK receptor binding sites in the rat, rabbit, and monkey:possible site of opiate-CCK analgesic interactions. J Neurosci.12, 4854–4866.
Gilbert, A.K., Franklin, K.B., 2001. GABAergic modulation ofdescending inhibitory systems from the rostral ventromedialmedulla (RVM). Dose–response analysis of nociception andneurological deficits. Pain 90, 25–36.
Gustafsson, H., Afrah, A.W., Stiller, C.O., 2001. Morphine-inducedin vivo release of spinal cholecystokinin is mediated by delta-opioid receptors—effect of peripheral axotomy. J Neurochem.78, 55–63.
Hargreaves, K., Dubner, R., Brown, F., Flores, C., Joris, J., 1988. Anew and sensitive method for measuring thermal nociceptionin cutaneous hyperalgesia. Pain. 32, 77–88.
Heinricher, M.M., Haws, C.M., Fields, H.L., 1991. Evidence for GABA-mediated control of putative nociceptivemodulating neurons inthe rostral ventromedial medulla: iontophoresis of bicucullineeliminates the off-cell pause. Somatosens Mot Res. 8, 215–225.
Heinricher, M.M., Neubert, M.J., 2004. Neural basis for thehyperalgesic action of cholecystokinin in the rostral ventro-medial medulla. J Neurophysiol. 92, 1982–1989.
Hruby, V., Fang, S.-N., Kramer, T., Davis, P., Parkhurst, D.,Nikiforovish, G., Boteju, L., Slaminova, J., Yamamura, H., Burks,T., 1994. Analogues of cholecystokinin26-33 selective for B-typeCCK receptors possess delta opioid receptor agonist activity invitro and in vivo: evidence for similarities in CCK-B and deltaopioid receptor requirements. In peptides: Chemistry, Struc-ture and Biology. Vol., R. Hodges, J. Smith, ed.^eds. ESCOMPublishers, Leiden, pp. 669–671.
Hruby, V.J., Agnes, R.S., Davis, P., Ma, S.W., Lee, Y.S., Vanderah,T.W., Lai, J., Porreca, F., 2003. Design of novel peptide ligandswhich have opioid agonist activity and CCK antagonist activityfor the treatment of pain. Life Sci. 73, 699–704.
Jensen, T.S., Gottrup, H., Sindrup, S.H., Bach, F.W., 2001. Theclinical picture of neuropathic pain. Eur J Pharmacol. 429,1–11.
Kim, S.H., Chung, J.M., 1992. An experimental model for peripheralneuropathy produced by segmental spinal nerve ligation in therat. Pain. 50, 355–363.
Kovelowski, C.J., Ossipov, M.H., Sun, H., Lai, J., Malan, T.P., Porreca,F., 2000. Supraspinal cholecystokinin may drive tonic des-cending facilitationmechanisms tomaintain neuropathic painin the rat. Pain. 87, 265–273.
Laulin, J.P., Larcher, A., Celerier, E., Le Moal, M., Simonnet, G.,1998. Long-lasting increased pain sensitivity in rat followingexposure to heroin for the first time. Eur J Neurosci. 10,782–785.
Laulin, J.P., Celerier, E., Larcher, A., Le Moal, M., Simonnet, G., 1999.Opiate tolerance to daily heroin administration: an apparentphenomenon associated with enhanced pain sensitivity.Neuroscience. 89, 631–636.
Lezoualc'h, F., Jockers, R., Berque-Bestel, I., 2009. Multivalent-based drug design applied to serotonin 5-HT(4) receptoroligomers. Curr Pharm Des. 15, 719–729.
Loeser, J.D., Bonica, J.J., 2001. Bonica's management of pain. Vol.,Lippincott Williams & Wilkins, Philadelphia, Penn.
Lorenzen, A., Fuss, M., Vogt, H., Schwabe, U., 1993. Measurement ofguanine nucleotide-binding protein activation by A1adenosine receptor agonists in bovine brain membranes:stimulation of guanosine-5'-O-(3-[35S]thio)triphosphate bind-ing. Mol Pharmacol. 44, 115–123.
Mao, J., Price, D.D., Mayer, D.J., 1994. Thermal hyperalgesia inassociation with the development of morphine tolerance inrats: roles of excitatory amino acid receptors and proteinkinase C. Journal of Neuroscience. 14, 2301–2312.
Mao, J., Price, D.D., Mayer, D.J., 1995. Mechanisms of hyperalgesiaand morphine tolerance: a current view of their possibleinteractions. Pain. 62, 259–274.
Mason, P., 2001. Contributions of the medullary raphe andventromedial reticular region to pain modulation and otherhomeostatic functions. Annu Rev Neurosci. 24, 737–777.
McCleane, G.J., 1998. The cholecystokinin antagonist proglumideenhances the analgesic efficacy of morphine in humans withchronic benign pain. Anesth Analg. 87, 1117–1120.
McCleane, G.J., 2002. A phase 1 study of the cholecystokinin (CCK)B antagonist L-365,260 in human subjects taking morphine forintractable non-cancer pain. Neurosci Lett. 332, 210–212.
McCleane, G.J., 2003. A randomised, double blind, placebo con-trolled crossover study of the cholecystokinin 2 antagonist L-365,260 as an adjunct to strong opioids in chronic humanneuropathic pain. Neurosci Lett. 338, 151–154.
Merskey, H., 2002. Clarifying definition of neuropathic pain. Pain.96, 408–409.
Moreau, J.L., Fields, H.L., 1986. Evidence for GABA involvement inmidbrain control of medullary neurons that modulate noci-ceptive transmission. Brain Research. 397, 37–46.
Nichols, M.L., Bian, D., Ossipov, M.H., Malan Jr., T.P., Porreca, F.,1996. Antiallodynic effects of a CCKB antagonist in rats withnerve ligation injury: role of endogenous enkephalins. Neu-rosci Lett. 215, 161–164.
Ossipov, M.H., Lai, J., Malan, T.P.J., Porreca, F., 2000. Spinal andsupraspinal mechanisms of neuropathic pain. Annals of theNew York Academy of Sciences. 909, 12–24.
Ossipov, M.H., Porreca, F., 2005. Challenges in the development ofnovel treatment strategies for neuropathic pain. NeuroRx. 2,650–661.
Raynauld, J.P., Martel-Pelletier, J., Abram, F., Dorais, M., Haraoui, B.,Choquette, D., Bias, P., Emmert, K.H., Laufer, S., Pelletier, J.P.,2008. Analysis of the precision and sensitivity to change ofdifferent approaches to assess cartilage loss by quantitativeMRI in a longitudinal multicentre clinical trial in patients withknee osteoarthritis. Arthritis Res Ther. 10, R129.
10 B R A I N R E S E A R C H 1 3 9 5 ( 2 0 1 1 ) 1 – 1 1

Roberts, J., Ossipov, M.H., Porreca, F., 2009. Glial activation in therostroventromedial medulla promotes descending facilitationto mediate inflammatory hypersensitivity. Eur J Neurosci. 30,229–241.
Slaninova, J., Knapp, R.J., Wu, J.J., Fang, S.N., Kramer, T., Burks, T.F.,Hruby, V.J., Yamamura, H.I., 1991. Opioid receptor bindingproperties of analgesic analogues of cholecystokinin octapep-tide. Eur J Pharmacol. 200, 195–198.
Tallarida, R.J., Murray, R.B., 1987. Manual of PharmacologicalCalculations with Computer Programs. Springer-Verlag Pub-lishers, New York, NY, Vol.
Trujillo, K.A., Akil, H., 1991. Opiate tolerance and dependence:recent findings and synthesis. New Biologist. 3, 915–923.
Urban, M.O., Gebhart, G.F., 1999. Supraspinal contributions tohyperalgesia. Proc Natl Acad Sci U S A. 96, 7687–7692.
Vanderah, T.W., Gardell, L.R., Burgess, S.E., Ibrahim, M., Dogrul, A.,Zhong, C.M., Zhang, E.T., Malan Jr., T.P., Ossipov, M.H., Lai, J.,Porreca, F., 2000. Dynorphin promotes abnormal pain andspinal opioid antinociceptive tolerance. J Neurosci. 20,7074–7079.
Vanderah, T.W., Largent-Milnes, T., Lai, J., Porreca, F., Houghten, R.A., Menzaghi, F., Wisniewski, K., Stalewski, J., Sueiras-Diaz, J.,Galyean, R., Schteingart, C., Junien, J.L., Trojnar, J., Riviere, P.J.,2008. Novel D-amino acid tetrapeptides produce potent anti-nociception by selectively acting at peripheral kappa-opioidreceptors. Eur J Pharmacol. 583, 62–72.
Wiesenfeld-Hallin, Z., Xu, X.J., Hokfelt, T., 2002. The role of spinalcholecystokinin in chronic pain states. Pharmacol Toxicol. 91,398–403.
Wild, J.E., Grond, S., Kuperwasser, B., Gilbert, J., McCann, B., Lange,B., Steup, A., Haufel, T., Etropolski, M.S., Rauschkolb, C., Lange,
R., 2010. Long-term safety and tolerability of tapentadolextended release for themanagement of chronic low back painor osteoarthritis pain. Pain Pract. 10, 416–427.
Williams, C., Rosenfeld, G., Dafney, N., Fang, S.-N., Hruby, V.,Bowden, G., Cullinan, C., Burks, T., 1994. SNF9007: a novelanalgesic that acts simultaneously at delta-1, delta-2, and muopioid receptors. Journal of Pharmacology And ExperimentalTherapeutics. 269, 750–755.
Woodcock, J., Witter, J., Dionne, R.A., 2007. Stimulating thedevelopment of mechanism-based, individualized pain thera-pies. Nat Rev Drug Discov. 6, 703–710.
Woolf, C.J., 1981. Intrathecal high dose morphine produceshyperalgesia in the rat. Brain Research. 209, 491–495.
Wright, A., Luedtke, K.E., Vandenberg, C., 2011. Duloxetine in thetreatment of chronic pain due to fibromyalgia and diabeticneuropathy. J Pain Res. 4, 1–10.
Xie, J.Y., Herman, D.S., Stiller, C.O., Gardell, L.R., Ossipov, M.H., Lai,J., Porreca, F., Vanderah, T.W., 2005. Cholecystokinin in therostral ventromedial medulla mediates opioid-inducedhyperalgesia and antinociceptive tolerance. J Neurosci. 25,409–416.
Yaksh, T.L., Rudy, T.A., 1976. Chronic catheterization of the spinalsubarachnoid space. Physiol Behav. 17, 1031–1036.
Yaksh, T.L., Harty, G.J., 1988. Pharmacology of the allodynia in ratsevoked by high dose intrathecal morphine. Journal of Phar-macology & Experimental Therapeutics. 244, 501–507.
Zhang, W., Gardell, S., Zhang, D., Xie, J.Y., Agnes, R.S., Badghisi, H.,Hruby, V.J., Rance, N., Ossipov, M.H., Vanderah, T.W., Porreca,F., Lai, J., 2009. Neuropathic pain is maintained by brainstemneurons co-expressing opioid and cholecystokinin receptors.Brain. 132, 778–787.
11B R A I N R E S E A R C H 1 3 9 5 ( 2 0 1 1 ) 1 – 1 1