microbial diversity in a thermophilic aerobic biofilm process: analysis by length heterogeneity pcr...
TRANSCRIPT

Water Research 37 (2003) 2259–2268
Microbial diversity in a thermophilic aerobic biofilm process:analysis by length heterogeneity PCR (LH-PCR)
Marja A. Tiirola1, Juhani E. Suvilampi, Markku S. Kulomaa, Jukka A. Rintala*
Department of Biological and Environmental Science, P.O. Box 35, Jyv .askyl .a FIN-40351, University of Jyv .askyl .a, Finland
Received 15 March 2001; received in revised form 4 November 2002; accepted 29 November 2002
Abstract
A two-stage pilot-scale thermophilic aerobic suspended carrier biofilm process (SCBP) was set up for the on-site
treatment of pulp and paper mill whitewater lining. The microbial diversity in this process was analyzed by length
heterogeneity analysis of PCR-amplified 16S ribosomal DNA. The primer pair selected for PCR amplification was first
evaluated by a computational analysis of fragment lengths in ten main phylogenetical eubacterial groups. The fragment
contained the first third of the 16S rRNA gene, which was shown to vary naturally between 465 and 563 bp in length.
The length heterogeneity analysis of polymerase chain reaction (LH-PCR) profile of the biomass attached to carrier
elements was found to be diverse in both stages of the SCBP. During normal operating conditions, sequences belonging
to beta-Proteobacteria, Cytophaga/Flexibacter/Bacteroides group and gamma-Proteobacteria were assigned to the most
prominent LH-PCR peak. Samples from the suspended biomass consisted of completely different bacterial populations,
which were, however, similar in the serial reactors. The pilot process experienced alkaline shocks, after which Bacillus-
like sequences were detected in both the biofilm and suspended biomass. However, when the conditions were reversed,
the normal microbial population in the biofilm recovered rapidly without further biomass inoculations. This study
shows that LH-PCR is a valuable method for profiling microbial diversity and dynamics in industrial wastewater
processes.
r 2003 Elsevier Science Ltd. All rights reserved.
Keywords: Microbial diversity; Thermophilic; Aerobic; Biofilm process; Pulp and paper mill; LH-PCR
1. Introduction
Biological processes, both aerobic and anaerobic, are
widely applied in the treatment of pulp and paper
industry wastewaters. Although wastewaters are often
hot (about 50–60�C), biological processes are currently
run under mesophilic conditions, at temperatures below
40�C. Thus, many pulp and paper mill wastewaters are
cooled before being subjected to mesophilic treatment.
Biological treatment at high temperatures could be a
preferred option in the treatment of hot wastewaters and
in the internal treatment of circulating process waters,
since it would reduce operating and investment costs as
heat exchangers would not be needed and simpler
process configurations could be applied. However, it
has been reported that thermophilic bacteria may fail to
aggregate, making biomass separation from the treated
effluent a key design criterion. Currently, little is known
about the microbiological aspects of thermophilic
aerobic wastewater treatment processes. The weak floc
formation ability of these processes may be due to lack
of floc-forming species [1] or the porous nature and
pinpoint size of the flocs [2]. In previous microbial
cultivation studies, only Bacillus and Bacillus-like
organisms have been isolated from thermophilic aerobic
wastewater treatment reactors (for a review, see [1]). In
biofilm processes, such as the suspended carrier biofilm
process (SCBP), a biofilm is formed on the carrier
*Corresponding author.
E-mail address: [email protected] (J.A. Rintala).1Also for correspondence.
0043-1354/03/$ - see front matter r 2003 Elsevier Science Ltd. All rights reserved.
doi:10.1016/S0043-1354(02)00631-0

surface preventing the degrading population from
washing out. Thermophilic aerobic biofilm processes
have so far been studied only in a few pilot or laboratory
studies. These studies have suggested stable perfor-
mance, high loading rates and short hydraulic retention
times (HRTs) [3,4,5,27].
Molecular techniques offer new possibilities for
profiling microbial dynamics which complement tradi-
tional cultivation studies. Most of these techniques are
based on the genes encoding ribosomal RNA (rDNA
genes). One of the recently introduced molecular
methods is length heterogeneity analysis of polymerase
chain reaction-amplified DNA (LH-PCR) of 16S rDNA
[6]. The analysis is based on the natural length variation
within 16S rDNA genes. The variable region is amplified
by PCR with fluorescently labeled universal primers that
recognize the region in all eubacteria. The peak
intensities within each LH-PCR size class are assumed
to be proportional to the original template concentra-
tions. The LH-PCR-method has been applied in screen-
ing microbial diversity in bacterioplankton [6] and, more
recently, in the examination of soil samples as well as the
origin of microbial faecal pollution in coastal waters
[7,8]. According to Ritchie et al. [8], LH-PCR is an easy,
rapid and reproducible method, but the authors
encountered several problem areas that require further
examination. First, the use of commercial fluorescent
size standards yielded fragment lengths that did not
correspond to the lengths of the sequenced 16S rDNA
fragments. Second, although more than 30 000 partial or
complete 16S rDNA sequences from cultivated or
uncultivated bacterial species have been submitted to
the public databases, there exists no exhaustive fragment
length database to directly compare and associate LH-
PCR lengths with native microorganisms. Comparison
of fragment lengths of native bacteria in different
phylogenetic groups is of especial importance, because
the usefulness of length heterogeneity analysis relies on
the evolutionary rate (deletions and insertions) of the
variable region of 16S rDNA that is selected for
analysis.
The objective of our study was to apply LH-PCR to
characterize bacterial populations in an on-site pilot
study using an SCBP to treat groundwood mill (GWM)
circulation water at temperatures between 44�C and
59�C. The process performance is described in greater
detail by Suvilampi et al. [9]. The microbial community
in both the suspended biomass and the biofilm attached
to carrier elements was analyzed from several samples
obtained during stable operation and during operational
disturbances. The population profiles were also com-
pared with those present in the feed and inoculum. A
computer-assisted analysis of the length variation of the
fragment sizes in different phylogenetic groups was
conducted to reveal the usefulness and limitations of the
LH-PCR method, and to assist in the interpretation of
the resulting LH-PCR patterns. For comparison, a
second dimension, GC-%, was also analyzed. GC-% is a
parameter that affects the mobility of DNA fragments in
denaturing gradient gel electrophoresis (DGGE) [10], a
commonly used molecular method for analyzing com-
plex microbial communities.
2. Materials and methods
2.1. Process water
The GWM circulation water used in all the experi-
ments was obtained from the grinder whitewater lining,
a stream also containing varying amounts of whitewater
from the paper machine. The characteristics of the
influent process water after 1mm pre-screening are
shown in Table 1.
2.2. SCBP pilot process and experimental set-up
Two serial aerobic reactors (referred to as R1 and R2
as single and R12 as serial reactors) with total volume of
2m3 were run in a 37-day trial. Half of the reactor
volume was filled with carrier elements (Flootek RF 438
black in R1 and RF 438 gray in R2, densities 0.95 and
1.05 gml�3, respectively), which provided an effective
surface area of 190m2m�3 in both reactors. A 250 l
lamell settling unit with sludge recirculation was
installed after the reactor stage. The technical operation
of the process was first tested by continuous feeding with
the whitewater for 4 days. Subsequently, on experi-
mental day 0, the reactors were inoculated with 60 l
(volatile suspended solids (VSS) 8.6 g l�1) of mixed
liquor from the mesophilic full-scale activated sludge
plant treating pulp and paper mill wastewater. The
inoculation temperature was 50�C.
2.3. Sampling and analyses
An automatic sampling device collected 24 h compo-
site samples from the feed line after screening, effluent
Table 1
Characteristics of groundwood mill circulation water used as
feed
N Average7SD
Temperature (�C) 81 6075
SS (mg l�1) 18 4607270
VSS (mg l�1) 18 4407260
DOC (mg l�1) 32 430790
TCOD (mg l�1) 9 13007260
SCOD (mg l�1) 9 11007250
SBOD7 (mg l�1) 9 6507140
N=number of samples, SD=standard deviation.
M.A. Tiirola et al. / Water Research 37 (2003) 2259–22682260

from R1, and final effluent after R2. Samples were
stored at �20�C if not immediately analyzed. VSS were
analyzed according to Standard Methods [11]. Scraped
biomass of the carriers was used to determine VS-fix.
Total VSS in the reactors was the sum of VSS and VS-
fix. Dissolved organic carbon (DOC) was measured with
a Shimadzu TOC-5050A total organic carbon analyzer.
Total and soluble chemical oxygen demand (TCOD and
SCOD) and soluble biological oxygen demand (SBOD7)
were analyzed according to Finnish standards [12,13],
respectively. All the samples were S&S GFA-filtered
except DOC and SCOD-samples were S&S GF50
filtered and TCOD samples were non-filtered.
2.4. Database examination
The 16S rDNA sequences, bases from 8 to 534
according to E. coli numbering [14], of 10 major lineages
of the bacteria domain were retrieved from the EMBL
database release 55 using the sequence retrieval system
(SRS) (EBI, Hinxton, UK). All the classified sequences
that were long enough were collected from the
phylogenetical groups of Cytophaga/Flexibacter/Bacter-
oides, epsilon-Proteobacteria, cyanobacteria, and Ther-
mus/Deinococcus. From the other groups the sequences
were limited to 150–200 per group by systematic
sampling to avoid overrepresentation of some groups.
For the Gram-positive group with low G+C content,
sequences were collected by the search words Clostri-
diaceae, Bacillaceae, Lactobacillaceae and Streptococca-
ceae. Sequences from uncultured species were generally
not classified and therefore could not be included. The
final data set comprised 1263 sequences. The GCG
program package (Genetics Computer Group, Madison,
WI) was used for the computational analysis of
fragment lengths and guanidine and cytosine content
(GC-%). The LH-PCR length data can be obtained
from M.T. on request.
2.5. DNA extraction
The biofilm of a plastic carrier was scraped mechani-
cally into 100ml of sterile water. 1ml sample of the
carrier biomass or suspended biomass was centrifuged in
a microcentrifuge tube at 14 000g 10min, and the pellet
was used for the analysis. The tube was filled with 0.4ml
of extraction buffer (10mM Tris–HCl pH 8.0, 1mM
EDTA, 0.2mgml�1 proteinase K, 1% sodium dodecyl
sulfate), and incubated at 37�C for 1 h. Cell lysis was
ensured by bead-millig: 0.6 g of glass beads (diameter
0.1mm) and 0.4ml of chloroform-isoamyl alcohol (24:1)
were added to the samples and the tubes were shaken at
1600 rpm for 3min using a cell homogenizer (Mikro-U
Dismembrator, B. Braun Biotech International, Mes-
slungen, Germany). The tubes were then centrifuged at
14 000g for 10min. The upper phase was re-extracted
with chloroform-isoamyl alcohol (25:24:1), purified with
ethanol precipitation (in the presence of 0.2M NaCl),
and finally dissolved into 100ml of molecular biology
grade water (50->30, Boulder, CO).
2.6. LH-PCR analysis
The primers for PCR were synthesized in the Institute
of Biotechnology at the University of Helsinki (Helsinki,
Finland) or in T–A–G–Copenhagen ApS (Copenhagen,
Denmark). Specific amplification of eubacterial
sequences was performed with primers fD1 (50-
AGAGTTTGATCCTGGCTCAG-30) [15] and an
IRD800 labeled PRUN518r (50-ATTACCGCGGCTG
CTGG-30) [10] for the LH-PCR analysis corresponding
to the fragment size of the computational analysis. For
the cloning and sequencing experiments eubacterial
specific primers fD1 and Com2-Ph (50-CCGTCAATT
CCTTTGAGTTT-30) [16] were also used to amplify a
longer sequence (B900 bp) of 16S rDNA gene. In the
PCR reactions, 1 ml of purified DNA was used as a
template in 50 ml PCR mixture containing 0.2mM of
dNTPs, 0.3mM of each primer, 1�DynaZyme reaction
buffer, 0.2mgml�1 BSA and 2 U DynaZyme F501-KL
polymerase (FinnZymes, Espoo, Finland). The PCR
procedure included an initial denaturation step at 95�C
for 5min and 30 cycles of amplification (94�C for 30 s,
55�C for 1min and 72�C for 3min). Gel electrophoresis
was performed with an automated LI-COR 4200
sequencer (LI-COR BioTech, Lincoln, NE) for six hours
or overnight using 6% Long Ranger denaturing poly-
acrylamide gel (FMC Bioproducts, Rockland, ME).
Data were analyzed using Quantity One software (Bio-
Rad Laboratories, Hercules, CA). For the development
of DNA length standards, templates from selected
strains of Sphingomonas sp., Yersinia sp. and Lactoba-
cillus sp. were amplified with the same 16S rDNA-
specific PCR primers as the examined samples. The
products were cloned into the pGEM-T vector (Prome-
ga, Madison, WI), and analyzed by bidirectional
sequencing. Size standards of 470, 527 and 553 bp were
prepared by amplifying these clones by the same method
and primers as used for the reactor samples.
2.7. Cloning and sequencing
The eubacterial 16S rDNA products obtained from
the reactor samples were excised from 0.8% agarose gel
and purified with Ultrafree-DA filter units (Millipore,
Bedford, MA, USA). The products were cloned into the
pGEM-T vector and sequenced using simultaneous
bidirectional cycle sequencing with a SequiTherm Excel
II DNA Sequencing Kit (Epicentre Technologies,
Madison, WI), sequencing primer pairs T7/SP6 or
PRUN518r/fD1 labeled with IRD800 and IRD700 dyes,
and the automated LI-COR 4200 sequencer. The
M.A. Tiirola et al. / Water Research 37 (2003) 2259–2268 2261

sequences (in average 400–600 bp long) have been
deposited in the EMBL database under accession
numbers AJ303332–AJ303341 and AJ298266–
AJ298277. The SCBP sequences are marked in the
database with A, S, D or F depending on the DNA
source (attached biomass, suspended biomass, attached
biomass in disturbed conditions and in feed water,
respectively). Sequences were compared with the data-
base with the BLAST program [17].
3. Results
3.1. Thermophilic process performance and biomass
retention
The temperature in the two-stage process ranged from
44�C to 59�C according to the feed water temperature
(range 48–68�C). In the two-stage process (R12) and
single reactor (R1), loading rates were gradually
increased by decreasing the HRT (Table 2). During the
run, R1 removed generally 47–55% and R12 55–70% of
DOC. The processes experienced four alkaline pH
shocks, which decreased only temporarily (generally
for 1 day) the DOC removal, especially in R1. On day 8
the DOC removal in R12 declined from 60% to 41%
after two pH shocks lasting about 10 h on days 7 and 8
(pH up to 12 in R1 and pH 9 in R2). DOC removal
recovered to 55% within 6 h following the last pH shock.
During the disturbances, the temperature was lower (40–
45�C) than under normal operation (52–55�) owing to
the recovery period of 6–12 h during which the flow of
feed to the reactor was suspended.
The concentration of attached biomass increased in
both reactors attained a maximum of 2.6 kg VS-fixm�3
in R1 on day 24. On day 30, the concentration of
attached biomass decreased to 1.4 kg VS-fixm�3 owing
to washout. This was apparently caused by two pH
shocks; pH rose to 11 on days 24 and 29 (for 12 and 6 h,
respectively). In R2 the concentration of attached
biomass increased to 2.9 kg VS-fixm�3 during the run
despite the disturbances. Suspended biomass concentra-
tions in both reactors ranged typically from 0.6 to 1.2 kg
VSSm�3. These figures mean that 60–80% of the total
biomass (VS-fix+VSS) was attached on the carriers.
3.2. Computational analysis of length variation in
LH-PCR
Ribosomal sequences from ten major lineages of the
bacteria domain were examined to assist the general
interpretation of LH-PCR profiles and to reveal the
usefulness of the method. The fragment chosen for
Table 2
Temperature, pH, HRT, loading rate, biomass and DOC removal in the two-stage SCBP treating groundwood mill circulation water in
the first stage reactor (R1) and second stage reactor (R2) on the days when microbial samples were taken for molecular analyses
Day Reactor unit T (�C) pH HRTa
(h)
Loading rate (kg
SCODm�3 d�1)
Suspended and
attached biomass
(kg VSSm�3/kg
VS-fixm�3)
VS-fix/total VSS
ratio (%)
DOC
removala
(%)
3 R1 50 7.0 4.1 8.5 1.0/1.4 58 55
R2 49 7.5 8.3 4.2 1.0/1.0 50 60
8 R1 48 12.0 4.0 6.1 0.7/1.6 70 nd.
R2 46 9.0 8.0 3.1 0.6/1.2 67 40
13 R1 50 6.5 2.5 9.8 1.0/2.2 69 49
R2 50 7.2 5.0 4.9 0.8/1.2 60 69
17 R1 52 7.0 3.3 6.5 0.6/2.6 81 60
R2 51 7.5 6.7 3.3 0.4/2.1 84 73
21 R1 53 7.0 3.3 8.8 0.9/2.6 75 51
R2 52 7.5 6.7 4.4 0.5/2.1 81 65
30 R1 50 11.0 1.9 9.0 1.2/1.4 54 26
R2 50 9.0 3.8 4.5 1.0/2.1 68 50
35 R1 55 7.0 1.6 19.0 0.5/1.2 71 43
R2 55 7.5 3.2 9.5 0.5/2.5 83 55
aR2 figures HRT and DOC removal after two-stage process.
M.A. Tiirola et al. / Water Research 37 (2003) 2259–22682262
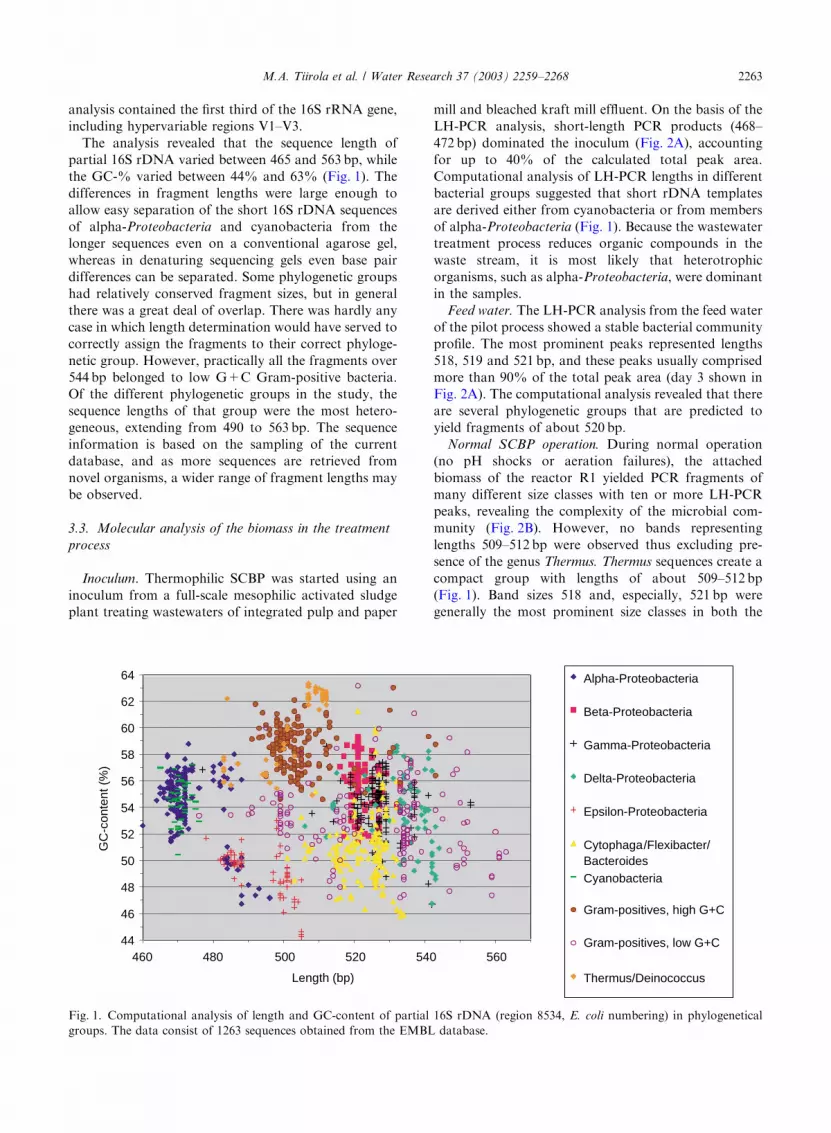
analysis contained the first third of the 16S rRNA gene,
including hypervariable regions V1–V3.
The analysis revealed that the sequence length of
partial 16S rDNA varied between 465 and 563 bp, while
the GC-% varied between 44% and 63% (Fig. 1). The
differences in fragment lengths were large enough to
allow easy separation of the short 16S rDNA sequences
of alpha-Proteobacteria and cyanobacteria from the
longer sequences even on a conventional agarose gel,
whereas in denaturing sequencing gels even base pair
differences can be separated. Some phylogenetic groups
had relatively conserved fragment sizes, but in general
there was a great deal of overlap. There was hardly any
case in which length determination would have served to
correctly assign the fragments to their correct phyloge-
netic group. However, practically all the fragments over
544 bp belonged to low G+C Gram-positive bacteria.
Of the different phylogenetic groups in the study, the
sequence lengths of that group were the most hetero-
geneous, extending from 490 to 563 bp. The sequence
information is based on the sampling of the current
database, and as more sequences are retrieved from
novel organisms, a wider range of fragment lengths may
be observed.
3.3. Molecular analysis of the biomass in the treatment
process
Inoculum. Thermophilic SCBP was started using an
inoculum from a full-scale mesophilic activated sludge
plant treating wastewaters of integrated pulp and paper
mill and bleached kraft mill effluent. On the basis of the
LH-PCR analysis, short-length PCR products (468–
472 bp) dominated the inoculum (Fig. 2A), accounting
for up to 40% of the calculated total peak area.
Computational analysis of LH-PCR lengths in different
bacterial groups suggested that short rDNA templates
are derived either from cyanobacteria or from members
of alpha-Proteobacteria (Fig. 1). Because the wastewater
treatment process reduces organic compounds in the
waste stream, it is most likely that heterotrophic
organisms, such as alpha-Proteobacteria, were dominant
in the samples.
Feed water. The LH-PCR analysis from the feed water
of the pilot process showed a stable bacterial community
profile. The most prominent peaks represented lengths
518, 519 and 521 bp, and these peaks usually comprised
more than 90% of the total peak area (day 3 shown in
Fig. 2A). The computational analysis revealed that there
are several phylogenetic groups that are predicted to
yield fragments of about 520 bp.
Normal SCBP operation. During normal operation
(no pH shocks or aeration failures), the attached
biomass of the reactor R1 yielded PCR fragments of
many different size classes with ten or more LH-PCR
peaks, revealing the complexity of the microbial com-
munity (Fig. 2B). However, no bands representing
lengths 509–512 bp were observed thus excluding pre-
sence of the genus Thermus. Thermus sequences create a
compact group with lengths of about 509–512 bp
(Fig. 1). Band sizes 518 and, especially, 521 bp were
generally the most prominent size classes in both the
44
46
48
50
52
54
56
58
60
62
64
460 480 500 520 540 560
Length (bp)
GC
-con
tent
(%
)
Alpha-Proteobacteria
Beta-Proteobacteria
Gamma-Proteobacteria
Delta-Proteobacteria
Epsilon-Proteobacteria
Cytophaga/Flexibacter/BacteroidesCyanobacteria
Thermus/Deinococcus
Gram-positives, low G+C
Gram-positives, high G+C
Fig. 1. Computational analysis of length and GC-content of partial 16S rDNA (region 8534, E. coli numbering) in phylogenetical
groups. The data consist of 1263 sequences obtained from the EMBL database.
M.A. Tiirola et al. / Water Research 37 (2003) 2259–2268 2263

serial reactors. However, reactor R2 had a slightly
different bacterial population compared to reactor R1
(Fig. 2D).
Suspended biomass samples showed clearly different
LH-PCR patterns as compared to the attached biomass,
indicating a different bacterial composition. Suspended
biomass produced high amounts of relatively long LH-
PCR fragments, especially 536 and 538 bp (Fig. 2C),
suggesting that Bacillus-related species may be abun-
dant. We made a complete analysis of all the Bacillus
and Paenibacillus sequences in the database (368
sequences), and found that most of the sequences
belonging to these genera (over 90%) yield lengths
between 531 and 538 bp.
Disturbed SCBP operation. The LH-PCR analysis
showed that after the pH shocks (days 8 and 30) the
attached biomass also experienced a major shift towards
microbial populations that produced higher fragment
Fig. 2. Fragment analysis of partial 16S rDNA amplified from samples of inoculated mesophilic sludge and feed water (day 3) (A),
attached biomass (B) and suspended biomass (C) of the reactor R1 of the pilot-scale aerobic thermophilic SCBP during normal
operating conditions. Comparisons of samples from the serial reactors R1 and R2 are shown in lower figures, (D) attached biomass
samples, day 17, during normal operating conditions, (E) attached biomass samples, day 8, during disturbed operating conditions.
M.A. Tiirola et al. / Water Research 37 (2003) 2259–22682264

lengths in LH-PCR analysis. This was observed
especially in reactor R1, which was more severely
disturbed (Figs. 2D and E).
Some of the PCR products were cloned and a total of
22 clones were selected to reveal the bacterial diversity in
the thermophilic process (Table 3). Although the
number of sequenced clones was small, the results show
that several Gram-negative genera from beta-Proteo-
bacteria, gamma-Proteobacteria and Cytophaga–Flexi-
bacter–Bacteroides group can thrive in the thermophilic
process as a part of the attached biomass (biofilm). All
these clones shared the LH-PCR length of 521 bp.
Sequences of species belonging to Bacillaceae (Anox-
ybacillus, Thermobacillus and Paenibacillus having LH-
PCR lengths 535, 550 and 538 bp, respectively) were
sequenced from the suspended biomass but also from
the biofilm sample collected during disturbed conditions
6 h after alkaline shock.
4. Discussion
In this study, the microbial diversity in an aerobic
thermophilic SCBP was analyzed by LH-PCR. This
method could offer a rapid tool for characterization of
microbial communities in many kinds of process
industry. Thermophilic aerobic processes have pre-
viously been studied using laboratory cultivation proce-
dures. Recent studies by Konopka et al. [28] and LaPara
et al. [18–21] have shown the advantages of the modern
culture-independent DNA methods, including DGGE
and sequence analysis of 16S rDNA clones.
According to our findings the microbial community of
the thermophilic SCBP exhibited wide bacterial diversity
in temperatures ranging from 44�C to 59�C. Archae-
abacteria-specific PCR primers were also tested, but no
amplification product was obtained from inoculum or
subsequently from feed water or reactor samples (data
not shown), which suggests that eubacteria thrive in this
thermophilic aerobic process. Noteworthy changes in
the microbial diversity of the biofilm were connected to
the occurrences of alkaline shock conditions, which may
also occur in full-scale mill conditions. During normal
operation, several different LH-PCR band patterns were
obtained from attached biomass, but a single dominant
band was observed most of the time. However, the
sequencing studies showed that even this main band was
a mixture of sequences from members of several
bacterial groups (at least beta- and gamma-Proteobac-
teria, and Cytophaga–Flexibacter–Bacteroides group).
Suspended biomass samples showed a great variance
suggesting that different microbial populations grew in
bursts. This was most likely due to repetitive adjust-
ments in the HRT. In a previous study [18], it has been
shown that the dominant phylotypes of the thermophilic
bioreactors change as a function of HRT. Therefore the
shifts in the community structure in the suspended
biomass samples may be due to decreased HRT during
operation of the pilot system, which increased the
selective pressure for the high growth rate. The
Table 3
Phylogenetic affiliation and closest cultured organisms of the cloned 16S rDNA sequences collected from the thermophilic wastewater
treatment system
Group Accession no. Source Best match in EMBL database Identity (%)
Alpha-Proteobacteria AJ303339 S Erythrobacter sp. AS–45 95
AJ303341 F Rhodobacter gluconicum acc. AB077986 92
Beta-Proteobacteria AJ303332 A Hydrogenophaga intermedia S1 95
AJ303334 A Hydrogenophaga palleronii DSM 63 96
AJ303337 A Hydrogenophilus thermoluteolus TH–1 98
AJ298276 D Tepidimonas ignava SPS-1037T 98
Gamma-Proteobacteria AJ303333 A Sulfur-oxidizing bacterium OAII2 91
Cytophaga/Flexibacter/Bacteroides AJ303335, AJ303336 A Cytophaga sp. BD2–2 94
Gram-positives, low G+C AJ303338, AJ303340, AJ298277 S, D Paenibacillus granivorans A30 94–95
AJ298267, AJ298268 F Alicyclobacillus acidocaldarius DSM 446 97–98
AJ298269-AJ298272 D Anoxybacillus gonensis G2T 97–99
AJ298273-AJ298275 D Thermobacillus xylanilyticus XE 95–98
Thermus/Deinococcus AJ298266 F Fervidobacterium islandicum AW–1 94
F, feed water (day 13); A, attached biomass (reactor R1, day 21); S, suspended biomass (reactor R1, day 21); D, attached biomass
during disturbed conditions (reactor R1, day 8).
M.A. Tiirola et al. / Water Research 37 (2003) 2259–2268 2265

differences between the attached and suspended biomass
can be explained by the different growth rates and/or
adhesion behaviors of bacterial groups. It is known that
early colonizing organisms are usually gram-negative
bacteria, whereas gram-positive bacteria have only
rarely been recorded on surfaces in aquatic habitats
[22]. However, after disturbed operation (alkaline shock)
Bacillus–related sequences were obtained also from the
attached biofilm. This result suggests that fast-growing
Gram-positive bacteria can colonize carrier surfaces, but
other bacteria with higher affinity for the substrate may
outcompete them from the biofilm environment under
stable conditions. Many of the most alkaliphilic micro-
organisms known are mesophilic bacteria of the genus
Bacillus [23]. Highly resistant cell walls, the tendency to
sporulate and, most importantly, short generation time
may favor these bacteria over others during or following
extreme conditions. Their short generation time may
also work in their favor in suspended biomass, since
slow-growing bacteria may be washed out during short
HRTs (down to 1.6 h in the single reactor).
The SCBP pilot study was established with two serial
aerated reactors. Most of the DOC removal occurred in
the first reactor R1 from which suspended biomass was
continuously washed out to the reactor R2. However,
suspended biomass content was higher in R1 than in R2
(effluent samples) which might indicate rapid reduction
in biomass weight by maintenance and decay processes
in the latter reactor. During normal operating condi-
tions, both the serial reactors showed very similar LH-
PCR profiles of bacterial diversity in the suspended
biomass samples, which is logical considering the
continuous inoculation from R1 Following pH-shock,
bacterial composition shifted more in the first reactor
(R1), in which the pH change was more severe.
However, the bacterial community of the process
recovered quickly, as revealed by process parameters
and molecular markers. Fast recovery is in accordance
with the high growth rate of thermophiles [24].
The mesophilic sludge used as an inoculum consisted
of a different microbial composition, as concluded from
its different LH-PCR patterns when compared to either
suspended or attached biomass in the thermophilic
SCBP. On the other hand, some of the prominent peaks
obtained from the attached biomass were similar to
those from the feed water from the pulp and paper mill
whitewater lining. However, computational calculations
revealed that many genera, even different subdivisions,
produce similar LH-PCR lengths. Nevertheless, LH-
PCR was shown to be convenient for preliminary
screening and selecting the samples that were of interest
for further experiments. Compared to DGGE, which is a
commonly used molecular tool for profiling bacterial
diversity, LH-PCR gives more reproducible results and
numerical values. In LH-PCR, fragment sizes can be
compared to exact lengths derived from the database. In
DGGE, the predictions of the mobility of the DNA
fragments in the gel (melting behavior) are only a guide,
as shown in the work of van Orsouw et al. [25]. Some of
the previous studies have also reported a discrepancy in
the LH-PCR analysis when using commercial fluores-
cently labeled size standards [7,8]. We also encountered
the same problem when using the capillary electrophor-
esis system (ABI310) and GeneScan-500 TAMRA size
standard (Applied Biosystems, Foster City, CA, USA).
The problem was solved easily and cost-efficiently by
using ‘home-made’ size standards that originated from
previously cloned ribosomal fragments. Using these
standards, correct band sizes could be obtained in the
LH-PCR analysis, as confirmed by sequencing.
The basic idea of LH-PCR is to use fluorescently
labeled primers to allow fast and accurate detection with
an automated sequencer. This approach does not allow
direct sequencing analysis of the bands of interest,
because the bands can not be excised from the gel. Other
electrophoresis systems can also be used. We have tested
a manual sequencing system and capillary electrophor-
esis system for LH-PCR analysis and found that these
systems have some notable advantages. Capillary
electrophoresis allowed faster (analysis time less than
1 h) and simpler operation, and might thus be more
practical in the daily control of industrial bioprocesses.
With a manual sequencing system, the whole analysis
including silver staining may take as long as 1–2 days,
but the manual sequencing apparatus is relatively
inexpensive and the method allows the extraction of
the band of interest for sequencing.
Currently, LH-PCR may be one of the most attractive
methods available, if one is seeking a fast, reproducible
and predictable method for the preliminary screening of
microbial systems over time and space. It could become
a valuable tool in screening microbial dynamics in
environmental and biotechnological fields. For future
development, a two-dimensional analysis of both
sequence length and G+C—content would be interest-
ing for the analysis of complex microbial communities.
As shown in the computational analysis (Fig. 1), the
phylogenetic division of many bacteria could be better
estimated by their two-dimensional coordinates. Two-
dimensional electrophoresis has been used in genetic
mutation analyses [26] using non-denaturing conditions
in length analysis and DGGE for the analysis of the
second dimension. However, as it is technically not yet
possible to analyze the first dimension in two-dimen-
sional electrophoresis using denaturing conditions,
measurements may be too inaccurate to yield relevant
numerical data, new technical solutions are needed. The
number of bacterial sequences available for comparison
are also constantly increasing; all that is lacking is the
availability of user-friendly biocomputing programs to
allow real-time simulations of sequence-based biomar-
kers of the database.
M.A. Tiirola et al. / Water Research 37 (2003) 2259–22682266

5. Conclusions
1. Database analysis of 16S rDNA sequences revealed
that the length heterogeneity of the first third of the
16S rDNA spans at least 465–563 bp.
2. Some qualitative predictions of the microbial com-
munity in bioprocesses can be made directly from the
LH-PCR length profiles, but for more detailed
analysis, sequencing or other DNA analyses are
needed.
3. The aerobic thermophilic biofilm process showed
stable performance on mill premises with generally
70–55% DOC removal in the two-stage process
with HRTs of 8–3 h. The community structure
differed from that of the seed sludge, and recovered
rapidly from disturbances caused by extreme pH
variations.
4. Different LH-PCR profiles for suspended and
attached biomass show that different microbial
populations follow as a function of the applied
selection criterion.
5. Alkaline pH shock led to a microbial community
where thermophilic Bacillus-associated microbes
were present, whereas the same bacteria did not play
significant role in the attached biofilm during non-
disturbed operation.
Acknowledgements
This study was supported by the funding of the
Graduate School for Environmental Ecology,
Ecotoxicology and Ecotechnology (EEEE) at the Uni-
versity of Jyv.askyl.a. We thank Maarit Kivim.aki for
assistance in the computational analysis, Irene Helkala
for technical assistance in laboratory, and Mervi
Ahlroth for helpful comments during the writing
process.
References
[1] LaPara T, Alleman J. Thermophilic aerobic biological
wastewater treatment. Water Res 1999;33:895–908.
[2] Tripathi CS, Allen DG. Comparison of mesophilic and
thermophilic aerobic biological treatment in sequencing
batch reactors treating bleached kraft pulp mill effluents.
Water Res 1999;33:836–46.
[3] Malmqvist (A, Welander T, Gunnarson L. Suspended-
carrier biofilm technology for treatment of pulp and paper
industry effluents. Symposium pre-print, the Fifth IAWQ
Symposium on Forest Industry Wastewaters, Vancouver,
Canada, 1996.
[4] Ragona CSF, Hall ER. Parallel operation of ultrafiltration
and aerobic membrane bioreactor treatment systems for
mechanical newsprint whitewater at 55�C. Water Sci
Technol 1998;38(4–5):307–14.
[5] Jahren SJ. Thermophilic treatment of concentrated waste-
water using biofilm carriers. Doctoral thesis. Norwegian
University of Science and Technology, 1999.
[6] Suzuki M, Rapp!e MS, Giovannoni SJ. Kinetic bias in
estimates of coastal picoplankton community structure
obtained by measurements of small-subunit rRNA gene
PCR amplicon length heterogeneity. Appl Environ Micro-
biol 1998;64:4522–9.
[7] Bernhard AE, Field KG. Identification of nonpoint
sources of faecal pollution in coastal waters by using
host-specific 16S ribosomal DNA genetic markers from
faecal anaerobes. Appl Environ Microbiol 2000;66:
1587–94.
[8] Ritchie NJ, Schutter ME, Dick RP, Myrold DD. Use of
length heterogeneity PCR and fatty acid methyl ester
profiles to characterize microbial communities in soil. Appl
Environ Microbiol 2000;66:1668–75.
[9] Suvilampi J, Rintala J, Nuortila-Jokinen J. On-site aerobic
suspended carrier biofilm treatment for pulp and paper
mill process water under thermophilic conditions, sub-
mitted for publication.
[10] Muyzer G, De Waal EC, Uitterlinden AG. Profiling
complex microbial populations by denaturing gradient
gel electrophoresis analysis of polymerase chain reaction-
amplified genes coding for 16S rRNA. Appl Environ
Microbiol 1993;62:2156–62.
[11] APHA. Standard methods for estimation of water and
wastewater, 20th ed. Washington, DC: American Public
Health Association, 1998.
[12] SFS 5504. Determination of chemical oxygen demand
(CODCr) in water with closed tube method, oxidation with
dichromate. Helsinki, Finland: Finnish Standard Associa-
tion, 1988.
[13] SFS 3019. Determination of biochemical oxygen demand
(BOD) of water. Dilution method. Helsinki, Finland:
Finnish Standards Association, 1979.
[14] Brosius J, Palmer ML, Kennedy JP, Noller HF. Complete
nucleotide sequence of the ribosomal RNA gene from
Escherichia coli. Proc Natl Acad Sci 1978;75:4801–5.
[15] Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 16S
ribosomal DNA amplification for phylogenetic study. J
Bacteriol 1991;173:697–703.
[16] Schwieger F, Tebbe CC. A new approach to utilize PCR-
single-strand-conformation polymorphism for 16S rRNA
gene-based microbial community analysis. Appl Environ
Microbiol 1998;64:4870–6.
[17] Altschul SF, Madden TJ, Sch.affer AA, Zhang J, Zhang Z,
Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a
new generation of protein database search programs.
Nucleic Acids Res 1997;25:3389–402.
[18] LaPara TM, Konopka A, Nakatsu CH, Alleman JE.
Thermophilic aerobic wastewater treatment in continuous-
flow bioreactors. J Environ Eng 2000;126:739–44.
[19] LaPara TM, Nakatsu CH, Pantea L, Alleman JE.
Phylogenetic analysis of bacterial communities in meso-
philic and thermophilic bioreactors treating pharmaceu-
tical wastewater. Appl Environ Microbiol 2000;66:3951–9.
[20] LaPara TM, Konopka A, Nakatsu CH, Alleman JE.
Thermophilic aerobic treatment of synthetic wastewater in
a membrane-coupled bioreactor. J Ind Microbiol Biotech-
nol 2001;26:203–9.
M.A. Tiirola et al. / Water Research 37 (2003) 2259–2268 2267

[21] LaPara TM, Nakatsu CH, Pantea L, Alleman JE. Aerobic
thermophilic treatment of a pharmaceutical wastewater:
effect of temperature on cod removal and bacterial
community development. Water Res 2001;35:4417–25.
[22] Marshall K. Planktonic versus sessile life of prokaryotes.
In: Balows A, Tr .uper HG, Dworkin, Harder W, Schleifer
K-H, editors. The prokaryotes: a handbook on the biology
of bacteria. New York: Springer, 1991. p. 262–75.
[23] Wiegel J. Anaerobic alkalithermophiles, a novel group of
extremophiles. Extremophiles 1998;2:257–67.
[24] Sundaram T. Physiology and growth of thermophilic
bacteria. In: Brock TD, editor. Thermophiles: general,
molecular, and applied microbiology. Wiley Series in
Ecological and Applied Microbiology. New York: Wiley/
Interscience, Wiley, 1986. p. 75–106.
[25] van Orsow NJ, Dhanda RK, Rines RD, Smith WM,
Sigalas I, Eng C, Vijg J. Rapid design of denaturing
gradient-based two-dimensional electrophoretic gene mu-
tational scanning tests. Nucleic Acids Res 1998;26:
2398–406.
[26] Uitterlinden AG. Gene and genome scanning by two-
dimensional DNA typing. Electrophoresis 1995;16:182–96.
[27] Malmqvist (A, Ternstr .om A, Welander T. In-mill biological
treatment for paper mill closure. Water Sci Tech
1999;40(11–12):43–50.
[28] Konopka A, Zakhorova T, LaPara T. Bacterial function
and community structure in reactors treating bio-
polymers and surfactants at mesophilic and thermo-
philic temperatures. J Indus Microbiol Biotech 1999;23:
127–32.
M.A. Tiirola et al. / Water Research 37 (2003) 2259–22682268