liposome-encapsulated superoxide dismutase prevents liver necrosis induced by acetaminophen
TRANSCRIPT

AmericanJournal ofPathology, Vol. 136, No. 4, April 1990Copyrigbt X American Association ofPathologists
Liposome-encapsulated Superoxide DismutasePrevents Liver Necrosis Inducedby Acetaminophen
Dai Nakae,* Kazuhiko Yamamoto,* HitoshiYoshiji,* Tetsuo Kinugasa,* Hiroshi Maruyama,*John L. Farber,t and Yoichi Konishi*From the Department ofOncological Pathology, CancerCenter, Nara Medical University, Kashihara, Nara 634,Japan*; and the Department ofPathology, JeffersonMedical College, Philadelphia, Pennsylvaniat
Liposome-encapsulated human recombinant su-peroxide dismutase (LSOD) protected male ratsthat were pretreated with 3-methylcholanthrenefrom the liver necrosis produced by acetamino-phen. By contrast, SOD-free liposomes, free SOD, orheat-denaturedLSOD had noprotective effect. Lipo-some-encapsulated SOD did not simply delay theonset ofliver necrosis. A second dose ofLSOD at 12hoursprevented the necrosis ofthe liver as assessed24 hours after treatment with 500 mg/kg bodyweight of acetaminophen. Liposome-encapsulatedhuman recombinant superoxide dismutase did notalter the metabolism ofacetaminophen as assessedby either the rate or extent of the depletion of he-patic stores ofglutathione or by the extent of thecovalent binding of the metabolites of [3H]acet-aminophen to total liver cell proteins. Evidence ofthe peroxidation of lipids in the accumulation ofmalondialdebyde in the livers was detected within3 hours of the administration of acetaminophenand before the appearance of liver necrosis. Lipo-some-encapsulated human recombinant superox-ide dismutase prevented the accumulation of ma-londialdebyde in parallel with the prevention ofliver necrosis. Finally, LSOD also prevented thepo-tentiation by 1,3-bis(2-chloroethyl)-1-nitrosoureaofthe hepatotoxicity ofacetaminophen. These datadocument the participation of superoxide anionsin the hepatotoxicity of acetaminophen in intactrats. (AmJPathol 1990, 136:787-795)
The killing of rodent liver cells by acetaminophen, a widelyused analgesic, has been an important model in defining
the mechanisms by which drugs and other chemicals pro-duce lethal cell injury. Liver cell necrosis results from themetabolism of acetaminophen by cytochrome P450-de-pendent mixed-function oxidation. Whereas there is gen-eral agreement that N-acetyl-p-benzoquinone imine(NAPQI) is the reactive metabolite of the biotransforma-tion of acetaminophen," 2 the precise mechanism of itsformation is not defined, and the mechanism by which themetabolism of acetaminophen leads to the necrosis ofhepatocytes is a matter of continuing study.
Based on studies in the early 1970s,37 it has beenwidely held that the arylation of critical hepatic macromol-ecules by the reactive metabolite of acetaminophen,namely NAPQI, mediates the liver necrosis. By contrast,more recent studies with intact mice810 or cultured rathepatocytes"114 suggest a role for activated oxygen spe-cies in the hepatotoxicity of acetaminophen.
The sensitivity of cultured rat hepatocytes to acetamin-ophen is induced by pretreatment of the animals with 3-methylcholanthrene. Inhibition of glutathione reduc-tase by 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU), pre-viously shown to sensitize hepatocytes to an oxidativestress,15'17 potentiates the toxicity of acetaminophenwithout increasing the covalent binding of acetaminophenmetabolites." Superoxide dismutase and catalase pre-vent the cell killing.12 Pretreatment of the hepatocytes withthe ferric iron chelator deferoxamine, known to reduce thesensitivity of hepatocytes to an oxidative stress,16 pre-vents the cell killing without reducing covalent binding."These data document the participation of oxygen radicalsin the killing of cultured hepatocytes by acetaminophen inthis model. Furthermore, they suggest that hydroxyl radi-cals generated by an iron-catalyzed Haber Weiss reactionmediate the cell injury. Specifically, superoxide anions re-
Supported by grant 63010085 from the Ministry of Education, Science andCulture of Japan, Grants-in-Aid for Cancer Research and for the Compre-hensive 10-Year Strategy for Cancer Control from the Ministry of Healthand Welfare of Japan, and by grant DK 38309 from the National Institutesof Health.
Accepted for publication November 22,1989.Address reprint requests to Dai Nakae, MD, Departmerit of Oncologi-
cal Pathology, Cancer Center, Nara Medical University, 840 Shijo-cho,Kashihara, Nara 634, Japan.
787

788 Nakae et alAJPApril 1990, Vol 136, No. 4
duce an endogenous pool of ferric to ferrous iron. Thelatter reacts with hydrogen peroxide to generate the hy-droxyl radical. Thus, superoxide dismutase prevents theformation of hydroxyl radicals by inhibiting the formationof ferrous iron needed for the Fenton reaction. In turn,catalase prevents the formation of hydroxyl radicals byremoving hydrogen peroxide.
The present report was prompted by a need to confirmthat these recent insights into the action of acetamino-phen are relevant to the pathogenesis of liver necrosis inthe intact animal. The data show that the hepatotoxicityof acetaminophen can be prevented by treatment withliposome-encapsulated superoxide dismutase. Thus, acritical feature of the killing of cultured hepatocytes byacetaminophen has been reproduced in the intact ani-mals with the demonstration of the participation of super-oxide anions in the genesis of liver cell necrosis.
Materials And Methods
Animals
Male Sprague-Dawley rats (150 g) were obtained fromCharles River Japan Inc., (Atsugi, Kanagawa, Japan). Theanimals were fed ad libitum for 1 week before use (Orien-tal MF Pellet Diet; Oriental Yeast.Co. Ltd., Itabashi, Tokyo,Japan), and kept in stainless steel, wired cages in an air-conditioned room at 250C with a 12-hour light-dark cycle.All animals received 25 mg/kg of body weight of 3-methyl-cholanthrene (Sigma Chemical Co., St. Louis, MO) by in-traperitoneal injection of a 10 mg/ml solution in corn oil 18hours before use and then fasted overnight.
Reagents
Acetaminophen (APAP) (Sigma) was administered by in-traperitoneal injection of a 250 mg/ml solution in dimethylsulfoxide (Sigma). The BCNU (Bristol Laboratories, Syra-cuse, New York) was administered by intrapentoneal in-jection of 60 mg/kg of body weight as a 30-mg/ml solutionin dehydrated ethanol 2 hours before treatment of the ratswith APAP. Human recombinant Cu-Zn superoxide dis-mutase (SOD) (4000 U/mg of protein by the nitroblue tet-razolium assay) was a gift of the Toyo Jozo Co. Ltd.,Ohito, Shizuoka, Japan. The heat inactivation of SOD(confirmed by enzyme assay) was accomplished in an
autoclave at 1 1 0°C for 20 minutes. Superoxide dismutasewas encapsulated within liposomes by the method of Mi-chelson et al.19 In brief, phosphatidylcholine, cholesterol,and stearylamine (5:1:1) were dissolved in diethyl etherand evaporated to form a thin layer. Superoxide dismu-tase or heat-inactivated SOD was dissolved in 100 mmol/l
sodium phosphate buffer, pH 7.0, containing 5% glucoseand added to the layered lipids. The sample then washeated to 60°C, vortexed, and sonicated in the water bathfor 30 minutes. The resulting liposomes had an averagediameter of 500 nm and were collected by chromatogra-phy on Sephadex G-50 (Pharmacia LKB BiotechnologyGroup, Shimagawa, Tokyo, Japan) and centrifugation for1 hour at 20,000g. The resulting liposomes (LSOD) con-tained 571.4 U of SOD (0.143 mg of protein) per milligramof liposome lipid. Liposome-encapsulated heat-dena-tured SOD (LSODden) contained 0.143 mg of protein permilligram of liposome lipid. The SOD-free liposomes (56mg of lipid/ml), LSOD (32,000 U/mi), free SOD (FSOD,32,000 U/ml), and LSODden (8 mg of protein/ml) all wereadministered at the doses indicated in the text by intra-peritoneal injection as solutions or suspensions in a buffercontaining 100 mmol/i sodium phosphate, pH 7.0, and5% glucose.
Assessment of Liver Necrosis
Liver cell necrosis was quantitated by determination ofthe activities in the serum of aspartate aminotransferase(AST), alanine aminotransferase (ALT), and alkaline phos-phatase (ALP). The animals were anesthesized with ethylether. Blood was removed from the aorta at its bifurcation,allowed to clot at room temperature, and centrifuged for20 minutes at 3000g at 25°C. The serum was assayedfor AST and ALT as previously described2O and for ALPaccording to Bowers and McComb.21 After removal ofblood, the livers were excised, blotted, and used for histo-logic examination or as described below.
Covalent Binding
The covalent binding of the metabolites of [3H]acetamino-phen (p-[3H(G)]-hydroxyacetanilide, 9.9 Ci/mmol, NewEngland Nuclear Corp., Boston, MA, diluted with unla-beled APAP to 0.5 mCi/mmol and 250 mg/ml, adminis-tered by intraperitoneal injection at a dose of 500 mg/kgof body weight) was determined by modification of themethod of Jollow et al.22 A 1 -g sample of the liver or medialfemoral muscle was homogenized in 3 ml of ice-cold 50mmol/i sodium phosphate buffer, pH 7.4, using an Ultra-Turrax homogenizer (Junke and Kunkel KG, Staufen,West Germany). One milliliter of the homogenate wasmixed with 2 ml of ice-cold 10% trichloroacetic acid (TCA;100% solution from Wako Pure Chemical Industries,Osaka, Japan) and sonicated on ice by using a MicrosonUltrasonic Cell Disruptor (Heat Systems-Ultrasonics Inc.,Farmingdale, NY) at its maximal output for 15 minutes withintermittent interruptions. After centrifugation of the soni-

Acetaminophen Hepatotoxicity 789AJPApril 1990, Vol. 136, No. 4
cate for 10 minutes at 10,000g at 40C, the pellet was
taken and prepared according to Rao and Recknagel.23The dry protein residue was dissolved in 4.5 ml of 1 NNaOH at 500C. A 0.1-ml aliquot was mixed with 10 mlUltra-Gel II (Nacalai Tesque Inc., Kyoto, Japan) andcounted in a scintillation spectrometer. The protein con-
tent of the NaOH solution was measured using the BCAProtein Assay employing bicinchoninic acid (PierceChemical Co., Rockford, IL).
Measurement of Glutathione
Hepatic reduced glutathione was measured by modifica-tion of the method of Sedlak and Lindsay.24 Disodium eth-ylenediamine tetra-acetate (EDTA), tris(hydroxymethyl)-aminomethane (TRIZMA base), and reduced glutathione(GSH) were from Sigma. A 200-mg sample of liver washomogenized in 8 ml of ice-cold 20 mmol/l EDTA usingan Ultra-Turrax homogenizer. Five milliliters of the homog-enate or a standard solution was added to 4 ml of H20and 1 ml of 50% TCA and sonicated as above. After cen-trifugation of the sonicate for 15 minutes at 3000g, a 1 -mlaliquot of the supernatant was mixed with 2 ml of 400mmol/l Tris-HCI buffer, pH 8.9, containing 20 mmol/lEDTA and 0.05 ml of 10 mmol/l 5,5'-dithiobis-(2-nitro-ben-zoic acid) (Sigma). The resulting absorbance at 412 nmwas then measured. The protein content of the homoge-nate was measured with the BCA Protein Assay.
Measurement of Lipid Peroxidation
Hepatic lipid peroxidation was quantitated as the accumu-lation of malondialdehyde (MDA) by modification of themethod of Yagi.25 A 500-mg piece of liver was homoge-nized in 4.5 ml of ice-cold 50 mmol/l sodium phosphatebuffer, pH 7.4, using the Ultra-Turrax homogenizer. Afteradding 0.5 ml of ice-cold 50% TCA, the sample was soni-cated and centrifuged for 20 minutes at 20,000g at 40C.Two milliliters of the supernatant were mixed with 2 mlof 0.67% 2-thiobarbituric acid (Sigma) and boiled for 10minutes. After cooling on ice, 1.6 ml of n-butanol were
added and the solution was centrifuged for 15 minutes at3000g at 250C. The fluorescence of the supernatant wasdetermined at 515 nm excitation and 553 nm emission.Standards were prepared by diluting malondialdehydebis(dimethyl acetal) (Aldrich Chemical Co. Inc., Milwau-kee, WI) 1 :1000 with methanol. The resulting solution wasmixed 1:1 with 0.2 N HCI, left overnight at room tempera-ture, and 1.65 ml was added to 8.35 ml of methanol. Ap-propriate aliquots of this solution, containing 1.25 to 5nmol of MDA, were added to 4.5 ml of the phosphate
3.000 1-15.,00
2.000
1.-0
1,000
-10.0c
5,00c
450
C
150
3 6 3 6 3 6
HOURS
Figure 1. Time course ofinduction ofliver cell necrosis by 500mg/kg body weight of acetaminophen. The results are themean ± SD ofthe determinations onfour orfive animals. The3-hour valuesforAST, ALT, and ALP were significantly differ-entfrom the "O" time value at P < 0.05.
buffer used to homogenize the livers and then processedin a manner similar to these samples.
The statistical significance of the data was determinedby the paired Student's t-test.
Results
Acetaminophen-induced Liver Cell Necrosis
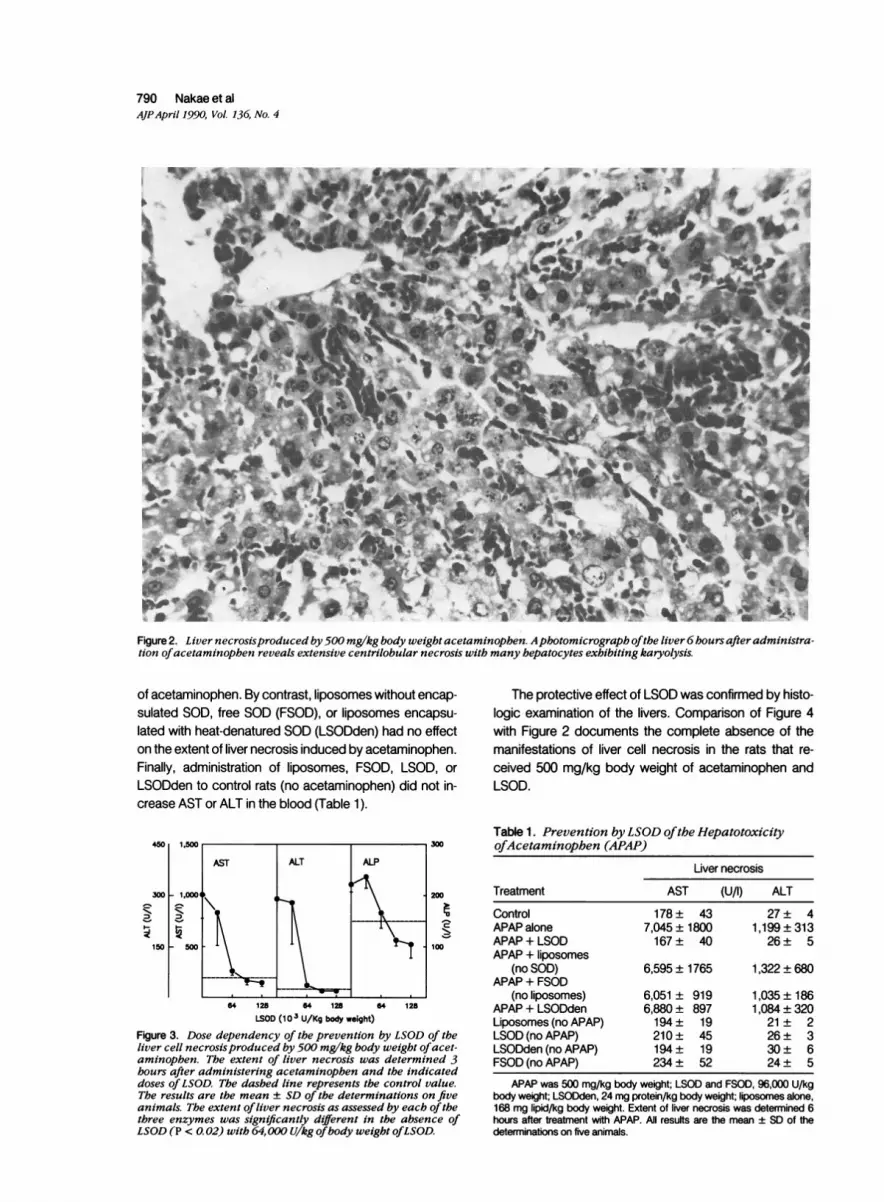
Fasted male rats induced with 3-methylcholanthrene weretreated with 500 mg/kg of body weight of acetamino-phen. Figure 1 illustrates the time course of the resultingliver cell necrosis as assessed by the content in the bloodof AST (left panel), ALT (middle panel), and ALP (rightpanel). The activities of each of these enzymes were ele-vated within 3 hours. By 6 hours, the activities of AST andALT had increased 100-fold, whereas that of ALP haddoubled. Histologic examination of the livers after 6 hoursconfirmed the presence of extensive centrilobular necro-
sis (Figure 2).
Prevention by LSOD of the Liver NecrosisInduced by Acetaminophen
Figure 3 details the dose-dependent prevention by LSODof the liver necrosis produced within 3 hours by 500 mg/kg body weight of acetaminophen. Doses of LSOD of64,000 U/kg of body weight or greater given immediatelyon treating the animals with APAP reduced the activity ofAST (left panel), ALT (middle panel), and ALP (right panel)in the blood to their control levels.
Table 1 details that this protection was still evident af-ter 6 hours. In addition, Table 1 gives the results of a num-ber of controls that documented the specificity of the pro-
tection by LSOD, which prevented the rise in both ASTand ALT produced at 6 hours by 500 mg/kg body weight
AST ALT ALP
/~~~~~~
790 Nakae et alAJPApril 1990, Vol. 136, No. 4
6 -
PJ v U e . ^ X _ i l~xf b
Figure2. Liver necrosisproduced by500 mg/kg body weightacetaminophen. Aphotomicrograph ofthe liver6hoursafteradministra-tion ofacetaminophen reveals extensive centrilobular necrosis with many hepatocytes exhibiting karyolysis.
of acetaminophen. By contrast, liposomes without encap-sulated SOD, free SOD (FSOD), or liposomes encapsu-lated with heat-denatured SOD (LSODden) had no effecton the extent of liver necrosis induced by acetaminophen.Finally, administration of liposomes, FSOD, LSOD, orLSODden to control rats (no acetaminophen) did not in-crease AST or ALT in the blood (Table 1).
450
300
150
1,000
- 500
200
a
100
64 128 64 128 64 128
LSOD (103 U/Kg body weight)
Figure 3. Dose dependency of the prevention by LSOD of theliver cell necrosisproduced by 500 mg/kg body weight ofacet-aminophen. The extent of liver necrosis was determined 3hours after administering acetaminophen and the indicateddoses of LSOD. The dashed line represents the control value.The results are the mean ± SD of the determinations on fiveanimals. The extent ofliver necrosis as assessed by each ofthethree enzymes was significantly different in the absence ofLSOD (P < 0. 02) with 64,000 U/kg ofbody weight ofLSOD.
The protective effect of LSOD was confirmed by histo-logic examination of the livers. Comparison of Figure 4with Figure 2 documents the complete absence of themanifestations of liver cell necrosis in the rats that re-ceived 500 mg/kg body weight of acetaminophen andLSOD.
Table 1. Prevention by LSOD ofthe HepatotoxicityofAcetaminophen (APAP)
Liver necrosis
Treatment AST (U/I) ALT
Control 178 ± 43 27 ± 4APAP alone 7,045 ± 1800 1,199 ± 313APAP + LSOD 167 ± 40 26 ± 5APAP + liposomes
(no SOD) 6,595 ± 1765 1,322 ± 680APAP + FSOD
(no liposomes) 6,051 ± 919 1,035 ± 186APAP + LSODden 6,880 ± 897 1,084 ± 320Liposomes (no APAP) 194 ± 19 21 ± 2LSOD (no APAP) 210 ± 45 26 ± 3LSODden (no APAP) 194 ± 19 30 ± 6FSOD (no APAP) 234 ± 52 24 ± 5
APAP was 500 mg/kg body weight; LSOD and FSOD, 96,000 U/kgbody weight; LSODden, 24 mg protein/kg body weight; liposomes alone,168 mg lipid/kg body weight. Extent of liver necrosis was determined 6hours after treatment with APAP. All resufts are the mean ± SD of thedeterminations on five animals.
AST ALT ALP
V~~~~~~~~~~~~~~~~~~~~~~~I ~_ _
I
I.00,W ,,,I

Acetaminophen Hepatotoxicity 791AJPApril 1990, Vol. 136, No. 4
Figure 4. Prevention by LSOD ofacetaminophen-induced liver cell necrosis. A photomicrograph 6 hours after the administration of500 mg/kg body weight ofacetaminophen and 96,000 U/kg body weight ofsuperoxide dismutase as LSOD reveals a normal liver.
Importantly, LSOD did not simply delay the develop-ment of liver cell necrosis in rats treated with 500 mg/kg body weight of acetaminophen. Rats given a secondinjection of LSOD at 12 hours were without liver necrosis24 hours after treatment with 500 mg/kg body weight ofacetaminophen (Table 2). Such protection at 24 hours de-pended on the dose of LSOD (Table 2) and was con-firmed by histologic examination of the livers (data notshown). In the absence of the second dose of LSOD, livernecrosis was now evident at 24 hours (Table 2). Finally,the data in Table 2 indicate that neither empty liposomes(no SOD), FSOD, nor LSODden given at both 'zero' timeand at 12 hours had a protective effect on the liver necro-sis produced by 500 mg/kg body weight of acetamino-phen after 24 hours.
LSOD Does Not Prevent the Depletionof Hepatic GSH
The hepatotoxicity of acetaminophen is dependent on itsmetabolism by the liver. Thus, an inhibition of the metabo-lism of acetaminophen by LSOD could be the basis of itsprevention of the liver necrosis. The effect of LSOD on the
metabolism of acetaminophen was examined in two ways:by the rate of depletion of GSH and by the extent of thecovalent binding of the metabolites of [3H]acetaminophen.
Table 2. LSOD Does Not Simply Delay the Onset ofAcetaminophen (APAP)-induced Liver Cell Necrosis
Liver necrosis
AST (U/I)Treatment
Control 183 ± 14APAP (24 hours) 15,525 ± 603APAP + LSOD (96,000 U/kg BW)
at 0 and 12 hours 190 ± 11APAP + LSOD (64,000 U/kg BW)
at 0 and 12 hours 398 ± 26APAP + LSOD (32,000 U/kg BW)
at 0 and 12 hours 12,645 ± 988APAP + LSOD (96,000 U/kg BW)
at 0 hour only 15,590 ± 537APAP + FSOD at 0 and 12 hours 15,569 ± 617APAP + liposomes (no SOD)
Oand 12 hours 15,550 ± 449APAP + LSODden at 0 and
12 hours 15,531 ± 609
APAP was 500 mg/kg of body weight (BW); FSOD, 96,000 U/kg (BW);LSODden, 24 mg denatured enzyme protein/kg (BW); liposomes alone,168 mg lipid/kg (BW). Extent of liver necrosis was determined 24 hoursafter treatment with APAP. All results are the mean ± SD of the determina-tions on five animals.
Jr V 111111111111,01"t..111WAMML- Iq
, .01e% IPT
:i.
b.d.
I
792 Nakae et alAJPApril 1990, Vol. 136, No. 4
6
a) 4002L.
1 3 6HOURS
Figure 5. Time course ofthe depletion ofGSH induced by 500mg/kg body weight of acetaminophen. The results are themean ± SD ofthe determinations on.five animals.
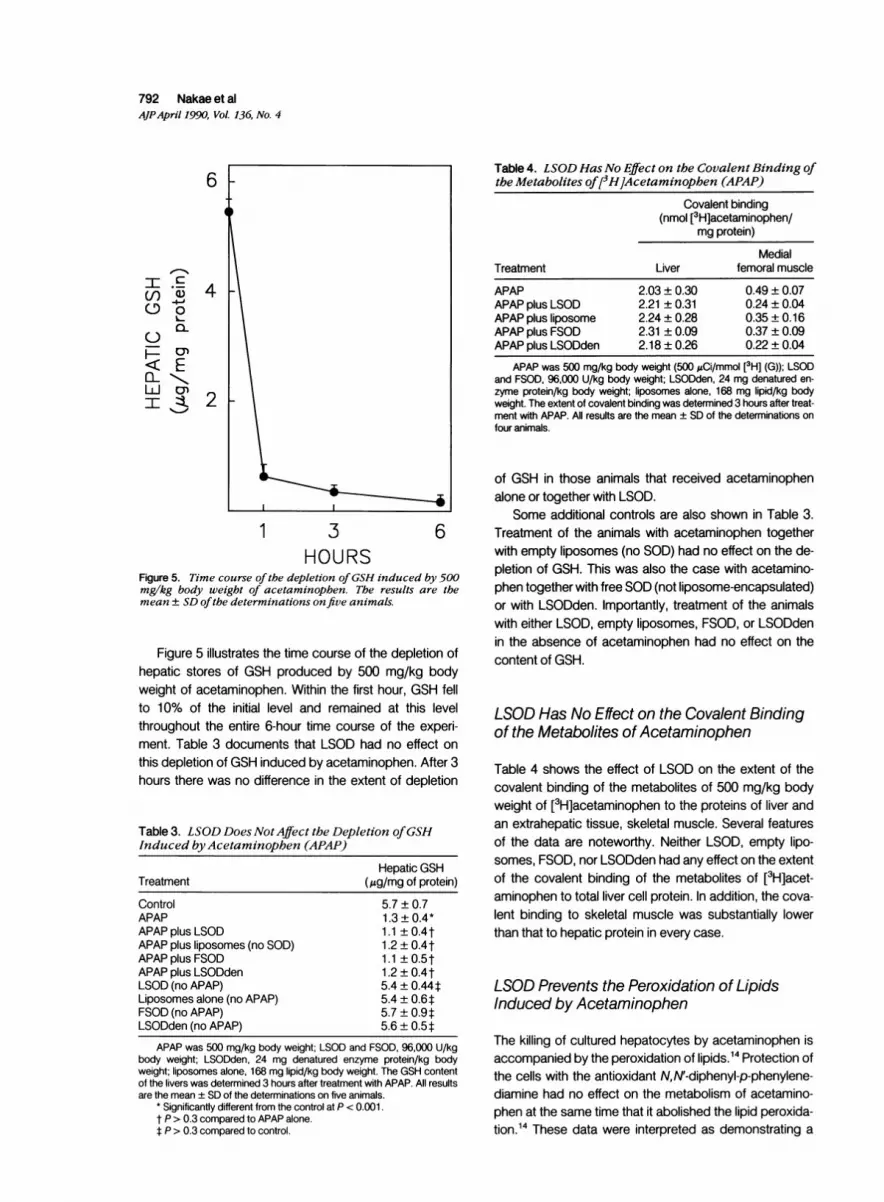
Figure 5 illustrates the time course of the depletion ofhepatic stores of GSH produced by 500 mg/kg bodyweight of acetaminophen. Within the first hour, GSH fellto 10% of the initial level and remained at this levelthroughout the entire 6-hour time course of the experi-ment. Table 3 documents that LSOD had no effect onthis depletion of GSH induced by acetaminophen. After 3hours there was no difference in the extent of depletion
Table 3. LSOD Does NotAffect the Depletion ofGSHInduced byAcetaminophen (APAP)
Treatment
ControlAPAPAPAP plus LSODAPAP plus liposomes (no SOD)APAP plus FSODAPAP plus LSODdenLSOD (no APAP)Liposomes alone (no APAP)FSOD (no APAP)LSODden (no APAP)
Hepatic GSH(,ug/mg of protein)
5.7 ± 0.71.3 ± 0.4*1.1 ±0.4t1.2±0.4t1.1 ±0.5t1.2±0.4t5.4 ± 0.44t5.4±0.6t5.7 ± 0.9t5.6±0.5t
APAP was 500 mg/kg body weight; LSOD and FSOD, 96,000 U/kgbody weight; LSODden, 24 mg denatured enzyme protein/kg bodyweight; liposomes alone, 168 mg lipid/kg body weight. The GSH contentof the livers was determined 3 hours after treatment with APAP. All resultsare the mean ± SD of the determinations on five animals.
* Significantly different from the control at P < 0.001.t P > 0.3 compared to APAP alone.t P > 0.3 compared to control.
Table 4. LSOD Has No Effect on the Covalent Binding ofthe Metabolites of13H]Acetaminophen (APAP)
Covalent binding(nmol [3H]acetaminophen/
mg protein)
MedialTreatment Liver femoral muscle
APAP 2.03 ± 0.30 0.49 ± 0.07APAP plus LSOD 2.21 ± 0.31 0.24 ± 0.04APAP plus liposome 2.24 ± 0.28 0.35 ± 0.16APAP plus FSOD 2.31 ± 0.09 0.37 ± 0.09APAP plus LSODden 2.18 ± 0.26 0.22 ± 0.04
APAP was 500 mg/kg body weight (500 1Ci/mmol [3H] (G)); LSODand FSOD, 96,000 U/kg body weight; LSODden, 24 mg denatured en-zyme protein/kg body weight; liposomes alone, 168 mg lipid/kg bodyweight. The extent of covalent binding was determined 3 hours after treat-ment with APAP. All results are the mean ± SD of the determinations onfour animals.
of GSH in those animals that received acetaminophenalone or together with LSOD.
Some additional controls are also shown in Table 3.Treatment of the animals with acetaminophen togetherwith empty liposomes (no SOD) had no effect on the de-pletion of GSH. This was also the case with acetamino-phen together with free SOD (not liposome-encapsulated)or with LSODden. Importantly, treatment of the animalswith either LSOD, empty liposomes, FSOD, or LSODdenin the absence of acetaminophen had no effect on thecontent of GSH.
LSOD Has No Effect on the Covalent Bindingof the Metabolites of Acetaminophen
Table 4 shows the effect of LSOD on the extent of thecovalent binding of the metabolites of 500 mg/kg bodyweight of [3H]acetaminophen to the proteins of liver andan extrahepatic tissue, skeletal muscle. Several featuresof the data are noteworthy. Neither LSOD, empty lipo-somes, FSOD, nor LSODden had any effect on the extentof the covalent binding of the metabolites of [3H]acet-aminophen to total liver cell protein. In addition, the cova-lent binding to skeletal muscle was substantially lowerthan that to hepatic protein in every case.
LSOD Prevents the Peroxidation of LipidsInduced by Acetaminophen
The killing of cultured hepatocytes by acetaminophen isaccompanied by the peroxidation of lipids.14 Protection ofthe cells with the antioxidant N,N'-diphenyl-p-phenylene-diamine had no effect on the metabolism of acetamino-phen at the same time that it abolished the lipid peroxida-tion.14 These data were interpreted as demonstrating a
V1)
CDa-5
LLJI

Acetaminophen Hepatotoxicity 793AJPApril 1990, Vol 136, No. 4
z
0
0
0~LLJ
0-
10.0
0
0
E
2.5
1 3 6
HOURSFigure 6. Prevention by LSOD oftheperoxidation oflipids in-duced by 500 mg/kg body weight ofacetaminopben. Animalstreated with acetaminophen alone (closed circles) or acet-aminophen plus LSOD (open circles) were given 96,000 U/kgbody weight ofsuperoxide dismutase. The results are the mean± SD ofthe determinations on three orfour animals.
role for lipid peroxidation in the hepatotoxicity of acetamin-ophen.14 We show here that LSOD prevented the peroxi-dation of lipids that occurs in the intact rat during thecourse of the intoxication with acetaminophen.
Figure 6 illustrates the effect of LSOD on the timecourse the peroxidation of lipids in rats that were intoxi-cated with 500 mg/kg body weight of acetaminophen.Lipid peroxidation was quantitated in the accumulation ofmalondialdehyde, which was first detected between 1and 3 hours after treating the rats with acetaminophen.Within 6 hours, the accumulation of MDA was more than10 nmol per gram of liver. Liposome-encapsulated humanrecombinant superoxide dismutase prevented the accu-mulation of malondialdehyde over the entire 6-hour timecourse of the experiment illustrated in Figure 6.
Table 5 confirms the specificity of the prevention oflipid peroxidation by LSOD. Empty liposomes, FSOD, or
LSODden did not prevent the peroxidation of lipids, a re-sult in agreement with their inability to prevent the liver cellnecrosis induced by acetaminophen. In addition, thesesame reagents had no effect in the absence of acetamin-ophen on the basal level of MDA.
LSOD Prevents the Potentiation by BCNUofAcetaminophen-induced Liver InjuryInhibition of glutathione reductase by BCNU potentiatesthe toxicity of acetaminophen both in the intact rat13 20 and
Table 5. Specifcity ofthe AntioxidantAction ofLSOD(APAP)
Treatment
ControlAPAPAPAP plus LSODAPAP plus liposomes (no SOD)APAP plus FSODAPAP plus LSODdenLSOD (no APAP)Liposomes (no APAP)FSOD (no APAP)LSODden (no APAP)
Lipid peroxidation(nmol MDA/g liver)
1.3±0.210.7 ± 0.71.1 ± 0.1
10.0 ± 0.110.9 ± 0.810.8 ± 0.41.1 ± 0.11.0±0.11.0 ± 0.01.2 ± 0.1
Acetaminophen was 500 mg/kg body weight; LSOD and FSOD,96,000 U/kg body weight; LSODden, 24 mg denatured enzyme protein/kg body weight; liposomes alone, 168mg lipid/kg body weight. The accu-mulation of MDA in the livers was determined 6 hours after treatment withAPAP. All results are the mean ± SD of the determinations on three ani-mals.
in primary cultures of hepatocytes.11-14 Table 6 showsthat LSOD also prevented the liver necrosis induced byacetaminophen after treatment of the rats with BCNU. Inthis experiment, the rats were injected with only 200 mg/kg body weight of acetaminophen, to reduce the extentof liver necrosis in the absence of BCNU.
Discussion
The data in the present report document that superoxideanions participate in the liver necrosis produced by acet-aminophen. In other words, the data document that thetoxicity of acetaminophen in the intact animal is related toan oxidative stress that is imposed by some conse-quence of the metabolism of this chemical. In addition,the data reproduce in the intact rat an important featureof the killing of cultured hepatocytes by acetaminophen,namely, the ability of SOD to prevent the toxicity of acet-aminophen.12 Thus the current report reinforces our previ-
Table 6. LSOD Prevents the Potentiation byBCNUofthe Toxicity ofAcetaminophen
Liver necrosis
Treatment AST (U/I) ALT
Control 120± 15 21 + 4APAP 209 ± 1* 38 ± 2*BCNU alone 327 ± 141 59 ± 29APAP+ BCNU 1,173± 43t 156 ± 6tAPAP + BCNU + LSOD 467± 25* 60± 1t
APAP was 200 mg/kg body weight; LSOD and FSOD, 96,000 U/kgbody weight; BCNU 60 mg/kg body weight. The extent of liver necrosiswas determined 3 hours after treatment with APAP. All results are themean ± SD of the determinations on three or four animals.
* Significantly different from control at P < 0.01.t Significantly different from BCNU alone or from APAP alone at P
<0.01.t Significantly different from APAP + BCNU at P < 0.001.

794 Nakae et alAJPApril 1990, Vol. 136, No. 4
ous conclusion2l that the essential features of the hepato-toxicity of acetaminophen in the intact rat are similar tothose determining the killing of cultured hepatocytes bythe same toxin.
The protection afforded by LSOD is fully attributable tothe action of SOD. Increasing protection was obtainedwith increasing doses of LSOD (Figure 3). Furthermore,empty liposomes or liposomes containing heat-inacti-vated SOD were ineffective (Table 1). Protection by SODdepended on the encapsulation of the superoxide dismu-tase. Free SOD did not protect, at least at doses that werefully protective when encapsulated within liposomes (Ta-ble 1). Interestingly, free SOD prevents the cell killing ofcultured hepatocytes by acetaminophen.12 This differ-ence between the action of SOD in the intact animal (Ta-ble 1) and in cell culture12 is most likely a dose-dependentphenomenon. Superoxide dismutase protects hepato-cytes from the toxicity of acetaminophen at much lowerdoses when it is encapsulated in a liposome rather thanfree in the culture medium.26 Similarly, it has been re-ported that liposome-encapsulated SOD penetrates cellsat a much faster rate than does free SOD.27
Prevention by LSOD of the liver necrosis produced byacetaminophen was achieved without inhibition of the me-tabolism of this toxin. Thus, LSOD did not prevent theacetaminophen-induced depletion of GSH (Table 3) or thecovalent binding of the metabolites of [3H]acetaminophen(Table 4). The latter result does not necessarily precludea role for covalent binding in the hepatotoxicity of acet-aminophen. However, it does argue that covalent bindingis not sufficient to cause liver necrosis. Furthermore, theconditions of the present study do not necessarily requirecovalent binding as a step in the genesis of liver necrosis.Covalent binding is most simply explained as an epiphe-nomenon of the metabolism of acetaminophen.
The data in the present report confirm the previousdemonstration that the toxicity of acetaminophen, both inthe intact animal8-10 and in cell culture,14 is accompaniedby the peroxidation of lipids. Importantly, LSOD preventedthis lipid peroxidation in parallel with the prevention of thenecrosis. Thus the lipid peroxidation must be a conse-quence of a mechanism that, on the one hand, superox-ide dismutase inhibits and that, on the other, is relatedto the lethal cell injury. The data presented here do notnecessarily indicate that the lipid peroxidation is a part ofthe same mechanism that kills the cells. However, previ-ous data suggested that lipid peroxidation is causally re-lated to killing of cultured hepatocytes by acetamino-phen.14 It then follows that the lipid peroxidation may sim-ilarly be related to the genesis of acetaminophen-inducedliver cell necrosis in the intact animal.
In summary, the demonstration that liposome-encap-sulated superoxide dismutase prevented the induction ofliver necrosis in the intact animal documents the participa-
tion of superoxide anions in the hepatotoxicity of acet-aminophen.
References
1. Hinson, JA: Biochemical toxicology of acetaminophen, Re-views in Biochemical Toxicology. Vol. 2. Edited by E Hodg-son, JR Bend, RM Philpot. New York, Elsevier, 1980, pp103-129
2. Dahlin DC, Miwa GT, Lu AYH, Nelson, SD: N-Acetyl-p-ben-zoquinone imine. A cytochrome P-450-mediated oxidationproduct of acetaminophen. Proc Natl Acad Sci USA 1984,81:1327-1331
3. Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Bro-die BB: Acetaminophen-induced hepatic necrosis: I. Role ofdrug metabolism. J Pharmacol Exp Ther 1973,187:185-194
4. Jollow DJ, Mitchell JR, Potter WZ, Davis DC, Gillette JR, Bro-die BB: Acetaminophen-induced hepatic necrosis: II. Role ofcovalent binding in vivo. J Pharmacol Exp Ther 1973, 187:195-202
5. Potter WZ, Davis DC, Mitchell JR, Jollow DJ, Gillette JR, Bro-die BB: Acetaminophen-induced hepatic necrosis: Ill. Cyto-chrome P-450-mediated covalent binding in vitro. J Pharma-col Exp Ther 1973, 187:203-210
6. Potter WZ, Thorgeirsson SS, Jollow DJ, Mitchell JR: Acet-aminophen-induced hepatic necrosis: V. Correlation of he-patic necrosis, covalent binding and glutathione depletion inhamsters. Pharmacology 1973,12:129-143
7. Jollow DJ, Thorgeirsson SS, Potter WZ, Hashimoto M, Mitch-ell JR: Acetaminophen-induced hepatic necrosis: VI. Meta-bolic disruption of toxic and non-toxic doses of acetamino-phen. Pharmacology 1974,12:251-271
8. Wendel A, Feuerstein S, Konz K-H: Acute paracetamol intox-ication of starved mice leads to lipid peroxidation in vivo.Biochem Pharmacol 1979,28:2051-2055
9. Wendel A, Feuerstein S: Drug-induced lipid peroxidation inmice: I. Modulation by monoxygenase activity, glutathioneand selenium status. Biochem Pharmacol 1981, 30:2513-2520
10. Wendel A, Jaeschke H, Gloger M: Drug-induced lipid peroxi-dation: II. Protection against paracetamol-induced liver ne-crosis by intravenous liposomally entrapped glutathione.Biochem Pharmacol 1982, 31:3601-3605
11. Gerson RJ, Casini A, Gilfor D, Serroni A, Farber JL: Oxygen-mediated cell injury in the killing of cultured hepatocytes byacetaminophen. Biochem Biophys Res Commun 1985,126:1129-1137
12. Kyle ME, Miccadei S, Nakae D, Farber JL: Superoxide dis-mutase and catalase protect cultured hepatocytes from thecytotoxicity of acetaminophen. Biochem Biophys Res Com-mun 1987, 149:889-896
13. Kyle ME, Nakae D, Serroni A, Farber JL: 1 ,3-(2-Chloroethyl)-1-nitrosourea potentiates the toxicity of acetaminophen bothin the phenobarbital-induced rat and in hepatocytes culturedfrom such animals. Mol Pharmacol 1988, 34:584-589
14. Farber JL, Leonard TB, Kyle ME, Nakae D, Serroni A, RogersSA: Peroxidation-dependent and peroxidation-independent

Acetaminophen Hepatotoxicity 795AJPApril 1990, Vol. 136, No. 4
mechanisms by which acetaminophen kills cultured rat he-patocytes. Arch Biochem Biophys 1988, 267:640-650
15. Babson JR, Abell NS, Reed DJ: Protective role of the gluta-thione redox cycle against adriamycin-mediated toxicity inisolated hepatocytes. Biochem Pharmacol 1981, 30:2299-2304
16. Bellomo S, Jewell SA, Thor H, Orrenius S: Regulation of theintracellular calcium compartmentation: Studies with iso-lated hepatocytes and t-butyl hydroperoxide. Proc NatlAcad Sci USA 1982, 79:6842-6846
17. Starke PE, Farber JL: Endogenous defenses against the tox-icity of hydrogen peroxide in cultured rat hepatocytes. J BiolChem 1985,260:86-92
18. Starke PE, Farber JL: Ferric iron and superoxide anions arerequired for the killing of cultured hepatocytes by hydrogenperoxide. Evidence for the participation of hydroxyl radicalsformed by an iron-catalyzed Haber-Weiss reaction. J BiolChem 1985, 260:10099-10104
19. Michelson AM, Puget K, Durosay P, Rousselet A: Penetra-tion of erythrocytes by superoxide dismutase, Biological andClinical Aspects of Superoxide and Superoxide Dismutase.Edited by WH Bannister, JV Bannister. New York, Elsevier1980, pp 348-366
20. Nakae D, Oakes JW, Farber JL: Potentiation in the intact ratof the hepatotoxicity of acetaminophen by 1,3-bis(2-chloro-ethyl)-1 -nitrosourea. Arch Biochem Biophys 1988, 267:651-659
21. Bowers GN Jr, McComb RB: Measurement of total alkalinephosphatase activity in human serum. Clin Chem 1975, 21:1988-1995
22. Jollow DJ, Mitchell JR, Zampaglione N, Gillette JR: Bromo-benzene-induced liver necrosis. Protective role of glutathi-one and evidence for 3,4-bromobenzene oxide as the he-patic metabolite. Pharmacology 1974,11:151-169
23. Rao KS, Recknagel RO: Early incorporation of carbon-la-beled carbon tetrachloride into rat liver particulate lipid andproteins. Exp Mol Pathol 1969,10:219-228
24. Sedlak J, Lindsay RH: Elimination of total, protein bound,and nonprotein sulfhydryl groups in tissue with Ellman's Re-agent. Anal Biochem 1968, 25:192-205
25. Yagi K: A simple flurometric assay for lipoperoxide in bloodplasma. Biochem Med 1976,15:212-216
26. Nakae D, Yoshiji H, Amanuma T, Kinugasa T, Farber JL,Konishi Y: Endocytosis-independent uptake of liposome-en-capsulated superoxide dismutase prevents the killing of cul-tured hepatocytes by tert-butyl hydroperoxide. Arch Bio-chem Biophys 1990 (In press)
27. Michelson AM, Puget K: Cell penetration by exogenous su-peroxide dismutase. Acta Physiol Scand 1980, 492(Suppl):67-80
Acknowledgments
The authors thank the staff of the Toyo Jozo Co., especially Drs.Kazumi Shiraiwa, Yuhji Nakano, and Yohshiroh Kobayashi fortheir cooperation, as well as for their generous supply of the re-agents needed for these studies.