journal of the iap karnataka state branch - citeseerx
TRANSCRIPT

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
1
Karnataka Paediatric Journal Vol. 25, No. 2 Quarterly ; Apr. - June 2011
Journal of the IAP
Karnataka State BranchCONTENTS
1. Minutes of the 2nd Executive Committee meeting of IAP-KSB 2011 held on 31-07-2011 3
2. APPROACH TO MANAGEMENT OF SECONDARY HYPERTENSION IN CHILDREN 4Vikram Singhal, Sucheta Rao, Nutan Kamath
3. OLIVOPONTOCEREBELLAR ATROPHY �A RARE NEUROLOGICAL DISORDER. 9Dr. Georgia, Dr.K.Shreedhara Avabratha, Dr.Habib Khan,Dr.B.Sanjeeva Rai, Dr.B Suresh
4. SMALL VESSEL VASCULITIS OF CENTRAL NERVOUS SYSTEM PRESENTING AS 12REFRACTORY FOCAL SEIZURES-A CASE REPORTPoppy Chadda, K Shreedhara Avabratha,Habib Khan,B.Sanjeev Rai and H B Suresh
5. RARE ASSOCIATION 0F POLAND�S SYNDROME WITH DEXTROCARDIA 15Dr. N.Rashmi; Dr. Narayanappa.D
6. A RARE BLISTERING DISEASE IN A NEWBORN 17James Daniel S, K Shreedhara Avabratha, Elizabeth Varkey Cherian, B Sanjeev Rai, Ramesh Bhat
7. STEVENS-JOHNSON SYNDROME���A CASE REPORT 20Dr. R.K. Jagadish Kumar, Dr. H.C. Krishna Kumar, Dr. Pawan Kumar, Dr. V.G. Manjunath, Dr. S. Mamatha
8. CASE REPORT - BIOTINIDASE DEFICIENCY IN INFANCY 24Dr.Rajashekar Murthy G.V , Dr Sanjay K.S. , Dr.Bharath Kumar Reddy K.R.
9. CASE REPORT: SCHIZENCEPHALY TYPE I � A CAUSE FOR STROKE IN CHILDREN 26Dr Sanjay K.S, Dr Rajashekar Murthy G, Dr Bharath Kumar Reddy K.R
10. NEONATAL POLYCYTHEMIA �A HOSPITAL BASED STUDY 28Dr.Narayanappa.D, Dr.N.Rashmi, Dr.Mohan B.K
11. CONGENITAL EMPHYSEMA: A CASE REPORT 34Roopa.Mangshetty. Sharangouda Patil. Shrikanth.S.W. Hosgouda K
12. HYPOTHYROIDISM & NEPHROTIC SYNDROME � A CAUSE OR EFFECT ? 36Nithya T, B Sanjeev Rai, Habib Ullah Khan, Aby Dany Varghese
13. COMMON PITFALLS IN DIAGNOSING RENAL PROBLEMS IN CHILDREN - Dr Arpana Iyengar 38
14. EVALUATION OF A CHILD WITH POLYURIA - Dr. Nagamani Agarwal 40
15. APPROACH TO ANTENATALLY DETECTED HYDRONEPHROSIS - Dr. R. Premalatha 42
EDITORIAL BOARD
EDITOR : EDITORIAL OFFICEDR. B. SANJEEV RAI Medicare Centre
Karangalpady, Mangalore - 575 003Ph : (0824) 2238399 (O),Fax : (0824) 2430361E-mail : [email protected]. : 94481-33494
MEMBERS :DR. HABEEB KHAN DR. PUSHPA KINI DR. SUDARSHAN SDR. RAMANATH MAHALE DR. SANTHOSH SOANS DR.KARUNAKAR BPDR. SUBRAMANYA NK
PAGE No.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
2
INDIAN ACADEMY OF PEDIATRICSKarnataka State Branch
Society Reg No: EKM – S460-2006-2007
OFFICE BEARERS FOR THE YEAR 2011
President Secretary Treasurer President ElectDr. R.T. Patil Dr. Ashok V. Badakali Dr. P. Subba Rao Dr.Suresh Babu
Bagalkot Mob: 9845365795 Bagalkot Mob: 9880227403 Magalore Mob: 9845872653 Davangere Mob: 9844096775
Historian Joint Secretary Editor K.P.J.Dr. Santosh Soans Dr. Pavan Hegde Dr. B. Sanjeev Rai.
Mangalore. Mob: 9343565558 Mangalore. Mob: 9845088116 Mangalore. Mob :9448133494
Executive Board MembersBagalkot : Dr. R. N. VanakiBangaluru : Dr. Basavaraj G. V.Belgaum : Dr. Shailesh PatilBellary : Dr. Kailash SoniBidar : Dr. Somashekhar BhalkeBijapur : Dr. M. M. PatilChikkamangalur : Dr. SundareshChitradurga : Dr. NatrajDakshina Kannada : Dr. Prasad NaikDavanagere : Dr. Naveen NadigDharwad : Dr. SudhindraGadag : Dr. Vijay NeelgundHassan : Dr. DineshKodagu : Dr. KrishnanandHaveri : Dr. Rajkumar MarolKolar : Dr. J. KrishnappaKollegal : Dr. Sridhar M.Koppal : Dr. Anand KumarMandya : Dr. NarendrababuMysore : Dr. Shrinivas MurthyRaichur : Dr. BalasubramanyaShimoga : Dr. Deepak ChirdoniTumkur : Dr. ShivaprakashUdupi : Dr. ShrikiranUttar Kannada : Dr. Dinesh HegdeCentral Council Executive Board MembersDr. Devraj Raichur. Mob : 9449864828Dr. Karunakar B.P. Mob : 9845263322Dr. Dinesh S.R. Mob : 9448006166Zonal CoordinatorsBangaluru : Dr. Subramanya N. KDharwad : Dr. Vijay KulkarniGulbarga : Dr. Arundathi PatilDavanagere : Dr. Deepak ChirdoniMysore : Dr. NarayanappaEx – Officio’sDr. K. DoddegowdaDr. S. R. Dinesh

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
3
Minutes of the 2nd Executive Committee meeting of
IAP-KSB 2011 held on 31-07-2011
The second EC meeting of IAP-KSB was held in Hotel Atriya, Bangaluru on 31th July
2011 during the 29th Annual CME programme of Lakeside Education Trust. Around 36
members were present.
The President Dr. R. T. Patil chaired the meeting and Dr. Ashok Badakali took the
charge of the proceeding of the meeting. Dr. R.T. Patil welcomed the office beares of IAP-KSB.
Dr. Ashok Badakali read the minutes of the last EC meeting held on 13.03.2011 at
Mangalore. Dr. T.U. Sukumaran National President of IAP was also present during the
meeting and seeked the members to conduct more academic activities and register under
family benefit scheme. Dr. (Smt) Mahantishetty N.S. state co-ordinator for NRP programme
briefed and requested zonal co-ordinator, district co-ordinator and trainer of trainers (TOT�s)
to conduct the Basic Neonatal Resuscitation Provider course in each districts as to fulfill the
IAP president�s action plan for the year 2011. Dr. Ashok Badakali read out various sub
committees. Dr. Gyanmurty and Dr. Basavaraj expressed tenure of sub committees should be
five years however all members expressed tenure should be decided at next G.B. meeting. Dr.
Deepak C.E. converner of IAP-Directory discussed how to make IAP-KSB directory and
financial assistance to IAP-KSB directory. Organizing Secretary Dr. Ramesh Pol informed all
the members that state conference will be held in Bagalkot on 14th to 16th October 2011. He
gave list of topics and the faculties, which was approved by the members.
Dr. Subramanya and Dr. Shrinath Mugali (Election Commissioner of IAP) expressed that
according to election code of conduct the person who is contesting for election are not
supposed become a faculty for IAP Conferences and other scientific activities. Dr. Subbra
Rao gave the quarterly account of IAP-KSB and showed positive balance of Rs. 29, 13,438/
-Dr. Sanjeev Rai told about the new design of KPJ and all the members were happy about
new design, he also suggested including sub-specialty series in KPJ and announced that
KPJ is now indexed journal but not indexed with pubmed and sought more articles from
medical colleges, practitioners and sub specialty chapters.
Dr. Subramanya has been nominated by central IAP as in charge for designing new
teaching slides for under graduates and post graduates.
Dr. Karunakar B.P. briefed on the minutes of EB meeting of central IAP held in June at
Cochin.
Dr. Ashok Badakali secretary IAP-KSB proposed the vote of Thanks.
Dr. R.T. Patil Dr. Ashok Badakali
President Secretary
IAP � KSB - 2011 IAP � KSB � 2011

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
4
APPROACH TO MANAGEMENT OF SECONDARYHYPERTENSION IN CHILDREN*Vikram Singhal, Sucheta Rao, Nutan Kamath
* Department of Pediatrics, Kasturba Medical College, Mangalore, Manipal University.
Hypertension is defined as
averagesystolic and/or diastolic pressure
>95th percentile for gender, age and height
on >3occasions.In developed countries, the
estimated prevalence in children is 1%-
2%.Surveys suggest a prevalence of 2%-5%
in Indian school children. Hypertension in
children can be essential (primary) or
secondary (identifiable underlying cause). A
secondary etiology may be suggested by
symptoms,examination findings or
laboratory abnormalities. Up to 85 percent of
children with hypertension have an
identifiable cause, most often renal
parenchymal disease. Anage-based approach
to the differential diagnosis is recommended.
The ABCDE mnemonic can be used to
help determine a secondary cause of
hypertension
TABLE I� Causes of Secondary Hypertension in Different Age Groups
Age Causes#
Newborns Renal artery thrombosis, renal artery stenosis, Congenital
malformation, coarctation of aorta,
Bronchopulmonary dysplasia
Infancy-6 yr Renal parenchymal disease(Chronic glomerulonephritis,
reflux nephropathy, obstructive uropathy, polycystic kidney
disease), renal artery stenosis , Coarctation of aorta
6-10 yr Renal artery stenosis, Renal parenchymal disease.
Adolescence Renal parenchymal disease
#__Others causes
Endocrine: Pheochromocytoma, cushing syndrome, congenital adrenal hyperplasia,
primary hyperaldosteronism, Liddle�s syndrome, neuroblastoma
Renal tumors: Wilms� tumor, nephroblastoma
Drugs: ibuprofen, naprosyn, peudoephedrine, carbamazepine, cyclosporine, tacrolimus,
methyl prednisolone, prednisolone, fludrocortisone, erythropoietin
A: ACCURACY, ALDOSTERONISM
Accuracy
The first step in diagnosing an
elevated blood pressure reading is to
investigate its accuracy. An inappropriate
blood pressure cuff for age or tight-fitting
sleeves that are not removed can give
falsely wrong readings. The cuffshould
encircle at least 80-100% of the armand the
bladder length should be >40% ofthe arm
circumference. Measurements should betaken
after 3 to 5 minutes of resting.White-coat
hypertension (blood pressure that is elevated
in the physician�s office but normal at other
times) accounts for about 20 percent of
patients with elevated readings.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
5
Appropriate charts with blood pressure
ranges based on gender, age, and height
percentilesfor children should be used.
Aldosteronism
Primary hyperaldosteronism is
defined as overproduction of aldosterone
independent of its usual regulator the renin-
angiotensin system.
B: BAD KIDNEYS, BRUITS
Bad Kidneys
Renal parenchymal disease can be a cause
or consequence of hypertension. The renal
damage decreases the kidneys� ability to
excrete salt and excess fluid (resulting in a
low renin state, as opposed to the high
renin state found in renovascular
hypertension).
Bruits
Renovascular hypertension results
from compromised arterial supply to the
kidneys and about 50% of patients have an
abdominal bruit identifiable on
examination.
C: COARCTATION, CATECHOLAMINES,
CUSHING�S SYNDROME
Coarctation of the Aorta
Coarctation of the aorta the second
most common cause of hypertension in
children, is more common in boys. In
neonates coarctation may present acutely as
congestive heart failure, but it is usually
diagnosed in children with the onset of
hypertension, difference between upper limb
and lower limb pulses or a cardiac
murmur.
Catecholamines
Excess catecholamine levels play a
role in white-coat hypertension and
pheochromocytoma. Acute stress induces
catecholamine release and often contributes
to hypertension.
Cushing�s Syndrome
Cushing�s syndrome can cause
hypertension via the mineralocorticoid effects
of excess glucocorticoids.
D: DRUGS, DIET
Drugs
Many prescription and nonprescription
drugs can cause or exacerbate hypertension
Eg. ibuprofen, naprosyn, peudoephedrine,
carbamazepine, cyclosporine, tacrolimus,
methyl prednisolone, prednisolone,
fludrocortisone, erythropoietin
Diet
Excess consumption of dietary sodium
is linked to chronic hypertension.Obesity
also can cause hypertension.
E: ENDOCRINE DISORDERS
Endocrine Disorders
Hypothyroidism induces decreased
cardiac output with a compensatory increase
in vascular tone, resulting in rise in diastolic
blood pressure whereas hyperthyroidism
induces increased cardiac output and
compensatory decreased vascular tone,
causing a greater increase in systolic blood
pressure.
Hyperparathyroidism (primary or
secondary to chronic renal insufficiency) is a
potentially reversible cause of hypertension.
However, only 30 to 40 percent of patients
with hyperparathyroidism have hypertension,
and parathyroidectomy does not reliably
resolve hypertension in patients with this
disorder.
In pheochromocytoma,the symptoms
can vary depending on the types of
catecholamines being produced, the amount
and frequency of their release into the
circulation.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
6
Figure 1: Algorithmic approach to evaluation of child with hypertension
Gradient between Upper and Lower limb
Abnormal urinalysis
Predominant WBC
Predominant RBC
Endocrine Renovascular lesion
Essential hypertension
Suspected Hypertension
Confirm Hypertension Check for • Proper cuff size • White Coat Hypertension
Detailed History Clinical Examination Full blood count ,Serum electrolytes, uric acid, renal function tests,Fasting lipid profile,Urinalysis,Renal ultrasound
Coarctation of Aorta- MRI Transthoracic Echocardiography
Reflux NephritisUrinary Tract Infection- Dimercaptosuccinicacid,DiethylenetriaminePentaacetic Acid,Micturatingcystourethrogram,Renal anomaly
Acute Glomerulonephritis
Lupus nephritisHenoch Schonlein Purpura
Renal Vein Thrombosis
Calculi,Infections
Thyroid- Thyroid stimulating hormone Computed tomography angiographyAldosteronism-Renin angiotensin activity Doppler ultrasonography of renalPheochromocytoma- 24-hour urinary arteries
fractionated MRI with gadolinium contrast mediametanephrines
Plasma free metanephrines Cushing syndrome- 24-hour urinary cortisol
Low-dose dexamethasoneSuppression
Congenital adrenal- 17-OH Progesteronehyperplasia

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
7
Treatment
It is imperative to differentiate
primary from secondary hypertension as
treatment of the underlying cause of
secondary hypertension canoften normalize
the blood pressure.
Principles of treatment
• The goal for treatment is reduction of
blood pressure to levels <95th
percentile, unless comorbid conditions
or target-organ damage is present,
when it should be lowered to
<90thpercentile of expected age, sex and
height of the child
• Therapy is initiated with one agent, at
an appropriate dose and the dose is
increased until the desired blood
pressure is achieved. If the highest dose
is not effective or if there are side
effects, a drug from a different class is
added or substituted
• Medications with a longer duration of
action (once, twice daily dosing) are
preferred forbetter compliance and
reduced side effects
• Dose adjustment of antihypertensive
medications can be made every 2-3
days.
Lifestyle modifications
• Dietary changes- Recommendations for
daily sodium intake range between 1-
1.5 g.
• Physical exercise- 30-60 minutes or
more of physical activity every day that
is developmentally appropriate,
enjoyable and involving a variety of
activities
• Weight loss- Reduction of BMI by 10%
is reported to lead to 8-12 mm Hg fall
in blood pressure.
The choice antihypertensive drugs
depend on the underlying cause.
Initial treatment with Calcium
channel blockers (CCB) or beta adrenergic
blockers (BB) or Angiotensin converting
enzyme inhibitor(ACEi)
If BP continues to be >95th centile:
Usecombination therapy - ACEi +
CCB or ACEi +Thiazides or CCB + BB.
(Watch for bradycardiawhen
combining BB and CCB)
If BP continues to be >95th centile:
Add thirdagent - ACEi + CCB + Diuretic/
BB.
Otheragents: prazosin, clonidine,
hydralazine.
Choice of drugs according to the cause
ofhypertension
• Acute glomerulonephritis :Loop diuretic
+ CCB or ACEi
• Renovascular hypertension: CCB+
diuretic
A BB instead of a CCB if ventricular
function is normal ormildly deranged
• Chronic kidney disease:CCB, ACEi or
BB
If two drugs are required, the ACEi
(or BB) should be combined with a CCB.
Drug step-down:It might be possible
in overweight children who have lost
sufficient weight and also in patients in
whom aspecific intervention has treated the
underlyingcause for hypertension.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
8
TABLE II� Choice of Antihypertensive Drugs
Drug Dose initial Maximum
Hypertensive emergencies
Nifedipine 0.25 mg/kg 0.5 mg/kg
Sodium nitroprusside 0.5 µg/kg/min IV 8 µg/kg/min IV
Labetalol 1 mg/kg/hr IV, can be given 3 mg/kg/hr IV
as bolus or steady infusion
Long-term therapy
Captopril Neonates 0.03 mg/kg/d 2 mg/kg/d
children 1.5 mg/kg/d 6 mg/kg/d
Enalapril 0.15 mg/kg/d 0.6 mg/kg/d
Losartan 0.7 mg/kg/d 1.4 mg/kg/d
Extended-release nifedipine 0.25 mg/kg/d 3 mg/kg/d
Amlodipine 0.1 mg/kg/dose 0.6 mg/kg/d
(maximum 20 mg/d)
Propranolol 1 mg/kg/d 8 mg/kg/d
Atenolol 1 mg/kg/d 8 mg/kg/d
Prazosin 0.05-0.1 mg/kg/d 0.5 mg/kg/d
Minoxidil 0.1-0.2 mg/kg/d 1 mg/kg/d
Hydrochlorothiazide 1 mg/kg/d 2-3 mg/kg/d
Furosemide 1 mg/kg/d 12 mg/kg/d
Further Reading:
1. Bagga A, Jain R, Vijayakumar M,
Kanitkar M, Ali U. Evaluation and
management of hypertension. Indian
Pediatr 2007; 44: 103-121.
2. Gulati S. Childhood Hypertension.
Indian Pediatr 2006; 43: 326-333.
3. Working Group on Management of
Congenital Heart Diseases in India.
Drug Therapy of Cardiac Diseases in
Children. Indian Pediatr 2009; 46: 310-
338.
4. Anthony J. V. , Dana M. N. Diagnosis
of Secondary Hypertension: An Age-
Based Approach. Am Fam
Physician.2010;82(12):1471-1478.
5. Edward O.Diagnosing Secondary
Hypertension.Am Fam Physician.2003;67(1):
67-74.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
9
Abstract
An 8 month old infant was broughtwith history of developmental delay and notfixing or following objects. Infant hadmicrocephaly and generalized hypotonia.Remaining general physical examinationand systemic examination was unremarkable.Developmental assessment showed globaldevelopmental delay with developmentalage corresponding to less than 2 months.Oto-acoustic emissions showed bilateralhearing loss. Ophthalmology evaluation torule out neurometabolic disorders revealednormal pupils, cornea and fundus. MRIscan features were suggestive ofolivopontocerebellar atrophy (OPCA).
Key words
Hypotonia, Hearing loss, Olivopontocerebellar
atrophy.
Introduction
Olivopontocerebellar atrophy (OPCA)
is a term coined by Dejerine and Thomas
which comprises a series of heterogenous
diseases whose only common factor is the
loss of neurons in the ventral portion of the
pons, inferior olives and cerebellar cortex1.
There may be neuronal loss to a variable
degree in the spinal cord, cerebral cortex
and basal ganglia. Clinically they manifest
as progressive cerebellar ataxia, tremor,
speech impairment, and in some instances,
marked extrapyramidal signs, cranial nerve
palsies, and peripheral neuropathy2. It is
rare in childhood and very few present in
the first year of life3. We report an 8 month
old baby with OPCA presenting with global
developmental delay for its rarity.
Case
An 8 month old male baby presentedwith history of developmental delay and notfixing or following objects. Baby was born tononconsanguineous parents as fulltermvacuum delivery. There was no history ofbirth asphyxia. Developmental historyrevealed global developmental delay withdevelopmental age corresponding to lessthan 2months. On examination babyweighed 5.75kg, Length-61cm, and Headcircumference-42cm (microcephaly). Babywas afebrile with normal heart rate andrespiratory rate. Anterior fontanelle wasopen (3x2 cm) and convergent squint waspresent. Facies was normal. Head lag waspresent and hypotonia was noticed in allfour limbs. Deep tendon reflexes were brisk.Cardiovascular, respiratory and abdominalexaminations were normal.
Blood counts, liver function testsand renal function test were within normallimits. ABG analysis, urine metabolicscreening, and ophthalmology evaluation
were done to rule out neurometabolic
disorders. ABG analysis was normal. Urine
for metabolic screening was negative.
Ophthalmology assessment revealed normal
pupils, cornea and fundus. Otoacoustic
emissions showed bilateral hearing loss.
MRI scan of brain showed prominence
of the cerebellar folia, fourth ventricle
and cerebellopontine angle cisterns
suggestive of cerebellar atrophy. The
prepontine and perimedullary cisternal
spaces were prominent with reduction
in the size of the pons. Features were
suggestive of olivopontocerebellar atrophy.
OLIVOPONTOCEREBELLAR ATROPHY �A RARENEUROLOGICAL DISORDER.
*Dr. Georgia, Dr.K.Shreedhara Avabratha, Dr.Habib Khan,Dr.B.Sanjeeva Rai, Dr.B Suresh
* Dept of Pediatrics and Radiology*, Fr.Muller Medical College Mangalore-575002.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
10
Discussion
In 1970, Konigsmarkand Weiner4
classified OPCA into five types, type II
being of recessive or sporadicinheritance, the other types beingautosomal dominant. It is now generallyaccepted that it is not a single disease, butis the result of a number of clinically andgenetically separate conditions. The firstreport of neonatal onset ofolivopontocerebellar atrophy with systemicfeatures was by Agamanolis et al4 in 1986.
They described a brother and sisterwith the condition and suggested that itmay have been caused by a primarylipoprotein disorder. Harding et al4, twoyears later, noticed low serumconcentrations of thyroid bindingglobulin and ceruloplasmin in the twocases that they reported, thus raisingthe possibility of an abnormality ofglycoproteins. A less severe disorder hasbeen described in recent years with manyfeatures in common with olivopontocerebellaratrophy of neonatal onset including failureto thrive, developmental delay, hypotonia,retinal abnormalities, liver disease, jointrestrictions, pericardial effusions, andcerebellar hypoplasia or atrophy. This hasbeen named disialotransferrin developmentaldeficiency (DDD) syndrome or carbohydratedeficient glycoprotein syndrome4.
Recently, a putative biochemicaldefect has been identified in some patientswith recessive or sporadic OPCA, which is
the deficiency of the enzyme glutamate
dehydrogenase which is involved in the
metabolism of the excitatory neurotransmitter
glutamate4. Other neurotransmitter
abnormalities have been described in
dominant OPCA3.
The diagnosis of olivopontocerebellar
atrophy rests primarily on morphological
evidence of degeneration of the cerebellar
cortex and its afferent pathways; it has been
well described in adults and to a lesserextent in older children. Notable features insporadic and the familial forms of OPCAare the extensive degeneration of the middlecerebral peduncles, the cerebellar whitematter, and the pontine, olivary, and arcuatenuclei. Loss of purkinje cells has beenvariable. Most likely this degenerationrepresents a terminal �dying back�� of axonsof the pontine and olivary nuclei withsecondary myelin degeneration. The extremeatrophy of the medullary olivary nucleivirtually identifies the process and isevident on MRIs5. Though we couldn�tdo all the biochemical investigations in ourcase, MRI features were suggestive ofOPCA.
Fig. A
Fig. B

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
11
Fig.A.MRI-Axial T2 weighted imaging
shows cerebellar and medullary atrophy
with prominent CSF spaces. Fig.B.MRI-
Sagital T1 weighted imaging shows atrophy
of pons,medulla and cerebellum and
prominent CSF spaces.
There is no specific treatment or cure for
this disease. Therapy is aimed at treating
symptoms and preventing complications.
This may include speech and physical
therapy, techniques to prevent choking,
walking aids to help with balance and
prevent falls.
In conclusion OPCA is a rare CNS
disorder with varied clinical manifestations
and characteristic MRI findings. This entity
should be considered whenever such
features are encountered.
REFERENCES
1. S Choi, M S Lee, W T Kim et al,
Olivopontocerebellar atrophy. Yonsei
Medical Journal 1988;29:233-237
2. Menkes J H, Heredodegenerative
diseases, Child Neurology, 7th edition,
Lippincot Williams and Wilkins,
Philadelphia, 2006:182-184
3. B N Harding, D B Dunger, D B Grant,
Familial olivopontocerebellar atrophy
with neonatal onset: a recessively
inherited syndrome with systemic and
biochemical abnormalities, Journal of
Neurology, Neurosurgery and
Psychiatry 1988;51:385-390
4. S P Horslen, P T Clayton, B N
Harding et al, Olivopontocerebellar
atrophy of neonatal onset and
disialotransferrin developmental
deficiency syndrome, Archives of
Disease in Childhood 1991;66:1027-
1032
Allan H.Ropper, Degenerative Diseases Of
The Newborn, Adams and Victors Principles
Of Neurology, Principles of Neurology,
Eighth edition, McGraw-Hill, New York
2005:935-936

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
12
Abstract:
Eight year old boy presented withrefractory focal seizures. Seizures persistedin spite of second line anticonvulsantdrugs.MRI revealed bilateral cerebralhemispheric multifocal non enhancinghyperintense lesions involving the graymatter and sub cortical white matter. Adiagnosis of small vessel vasculitis of thecentral nervous system was made. Childresponded to intravenous methylprednisolone followed by oral steroids.
Key words: Central nervous system,Refractory seizures, Vasculitis.
Introduction:
Childhood primary angiitis of thecentral nervous system is a recentlyrecognised rare inflammatory disease thatcauses severe neurological deficits andunexplained neurological symptomsincluding intractable seizures, hemiparesis,cranial nerve deficits,severe cognitivedeficits and decreased consciousness. Thereare two types of childhood primary angiitisof the CNS: medium-large vessel and smallvessel vasculitis.MRI is a sensitive but notspecific detector of vascular disease but iscertainly valuable in excluding otherconditions. In patients with small vesselchildhood primary angiitis of the CNS,angiography findings are typically negativeand thus diagnosis must be confirmed bybrain biopsy. Neurological outcome inpatients with small vessel childhoodprimary angiitis of the CNS can bedevastating and can result in death.However, some children with small vesseldisease have shown neurological recoveryafter immunosuppressive treatment,
SMALL VESSEL VASCULITIS OF CENTRAL NERVOUSSYSTEM PRESENTING AS REFRACTORY FOCALSEIZURES-A CASE REPORT*Poppy Chadda, K Shreedhara Avabratha,Habib Khan,B.Sanjeev Rai and H B Suresh
* Department of Paediatrics and *Radiology,Father Muller Medical College, Mangalore,Karnataka,India.
suggesting that the neurological deficitscaused by brain inflammation arereversible.We report one case who presentedwith refractory seizures and diagnosed tohave CNS small vessel vasculitis.
Case :
Eight year old boy presentedwith history of six episodes of focal seizureswith secondary generalisation and post ictaldrowsiness in the previous three days.Exceptfor headache there was no history offever,vomiting or trauma.There is no familyhistory of epilepsy. On examination, he wasafebrile, GCS was 15/15, HR-100/min,BP-100/60 mm of Hg,RR-26/min. Systemicexamination was normal and there was noneurological deficits. Initial blood counts,blood sugar and serum electrolytes werewithin normal limits.CSF analysis was alsonormal (CSF glucose-79, protein-16,cells -2lymphocytes)EEG showed leftcentrotemporal epileptiform discharges. CTand MRI of brain showed features of postictal oedema. Child was treated withloading dose of phenytoin followed bymaintainence dose. Seizures subsided for 2days.
Two days after admission childdeveloped recurrent focal seizures involvingthe left lower limb lasting for 1-2 minutesevery 30-60 minutes, which later becamepersistent. Child was put on valproate,leviteracetam and lamotrigine. Phenytoinwas tapered and stopped. But seizureactivity continued. Carbamazepine andphenobarbitone was also tried. Midazolaminfusion was also given. However seizureactivity did not subside. A course ofacyclovir was also administered.
Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
13
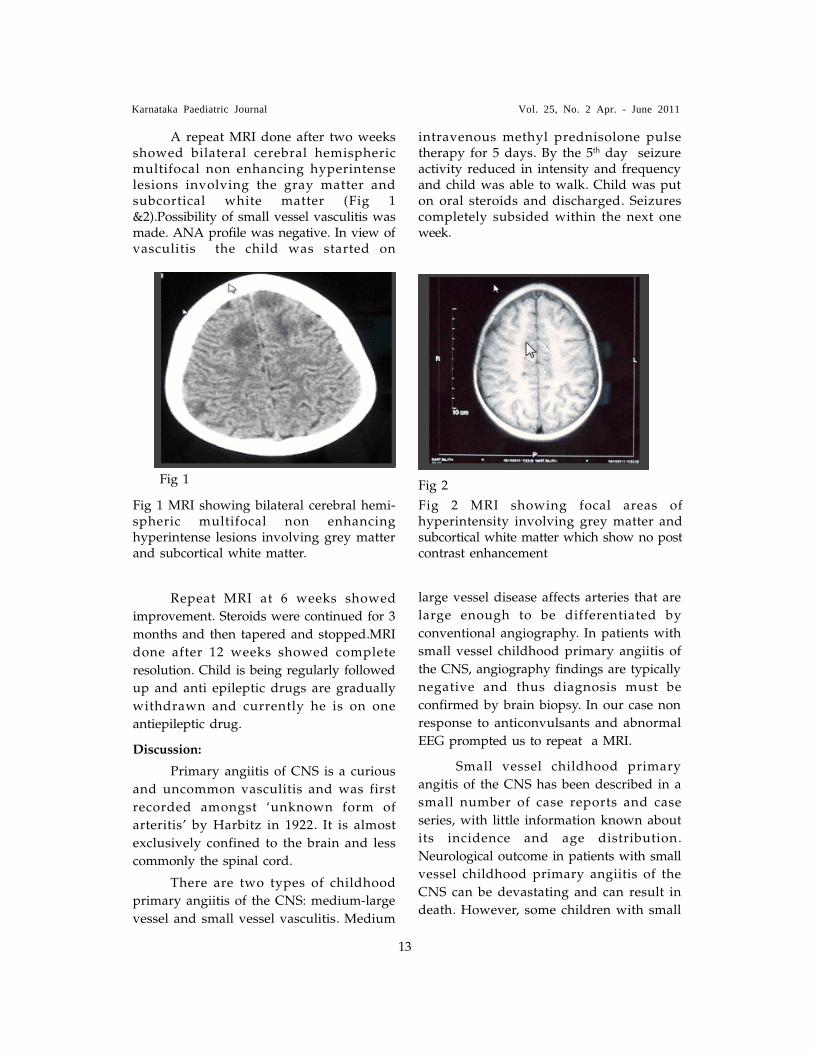
A repeat MRI done after two weeksshowed bilateral cerebral hemisphericmultifocal non enhancing hyperintenselesions involving the gray matter andsubcortical white matter (Fig 1&2).Possibility of small vessel vasculitis wasmade. ANA profile was negative. In view ofvasculitis the child was started on
Repeat MRI at 6 weeks showed
improvement. Steroids were continued for 3
months and then tapered and stopped.MRI
done after 12 weeks showed complete
resolution. Child is being regularly followed
up and anti epileptic drugs are gradually
withdrawn and currently he is on one
antiepileptic drug.
Discussion:
Primary angiitis of CNS is a curious
and uncommon vasculitis and was first
recorded amongst �unknown form of
arteritis� by Harbitz in 1922. It is almost
exclusively confined to the brain and less
commonly the spinal cord.
There are two types of childhood
primary angiitis of the CNS: medium-large
vessel and small vessel vasculitis. Medium
Fig 1 Fig 2
intravenous methyl prednisolone pulsetherapy for 5 days. By the 5th day seizureactivity reduced in intensity and frequencyand child was able to walk. Child was puton oral steroids and discharged. Seizurescompletely subsided within the next oneweek.
Fig 1 MRI showing bilateral cerebral hemi-spheric multifocal non enhancinghyperintense lesions involving grey matterand subcortical white matter.
Fig 2 MRI showing focal areas ofhyperintensity involving grey matter andsubcortical white matter which show no postcontrast enhancement
large vessel disease affects arteries that are
large enough to be differentiated by
conventional angiography. In patients with
small vessel childhood primary angiitis of
the CNS, angiography findings are typically
negative and thus diagnosis must be
confirmed by brain biopsy. In our case non
response to anticonvulsants and abnormal
EEG prompted us to repeat a MRI.
Small vessel childhood primary
angitis of the CNS has been described in a
small number of case reports and case
series, with little information known about
its incidence and age distribution.
Neurological outcome in patients with small
vessel childhood primary angiitis of the
CNS can be devastating and can result in
death. However, some children with small

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
14
vessel disease have shown neurological
recovery after immunosuppressive treatment,
suggesting that the neurological deficits
caused by brain inflammation are reversible.
This recovery contrasts with the irreversible
damage caused by acute ischaemia in
paediatric patients with medium-large vessel
disease.
A myriad of neurological symptoms,
signs or syndromes can occur in CNS
vasculitis, reflecting the potential for
infarction and ischaemia which may be
micro- or macroscopic, focal, multifocal or
diffuse and affect any part of the brain.
Most accounts of the disorder describe
headaches, focal or generalized seizures,
stroke-like episodes with hemispheric or
brainstem deficits, acute or subacute
encephalopathies, progressive cognitive
changes, chorea, myoclonus and other
movement disorders, and optic and other
cranial neuropathies. In short, there are few
neurological syndromes that are not
consistent with a vasculitic aetiology.
Systemic features such as fever, night
sweats, livedo reticularis, or
oligoarthropathy may also be present but
often are only revealed by direct questioning
of the patient. Our patient presented with
only seizures. The course is commonly acute
or subacute, but chronic progressive
presentations are also well described, as are
spontaneous relapses and remission.
Prospective randomised controlled
trials are understandably difficult because of
the rarity of the condition and the lack of
unifying diagnostic criteria. Retrospective
analyses done in various studies have
emerged significant support for the use of
steroids with or without cyclophosphamide
in confirmed cases.Our child responded
well to intravenous methyl prednisolone,
followed by oral prednisolone. Notwithstanding
the problems in recognition and diagnosis,
cerebral vasculitis is a highly treatable
condition for which prompt management
can radically improve the outcome, hence
every attempt should be made to diagnose
the same.
References
1) Hutchinson C, Elbers J,Halliday W etal.
Treatment of small vessel primary CNS
vasculitis in children-an open label
cohort study . Lancet neurology 2010; 9:
1078�84.
2) Yaari R, Anselm IA, Szer IS, Malicki
DM, Nespeca MP, Gleeson JG.
Childhood primary angiitis of the
central nervous system: two biopsy-
proven cases. J Pediatr 2004; 145: 693�
97.
3) Benseler SM, deVeber G, Hawkins C, et
al. Angiography-negative primary
central nervous system vasculitis in
children: a newly recognised
inflammatory central nervous system
disease. Arthritis Rheum 2005; 52:
2159�67.
4) Benseler SM, Silverman E, Aviv RI et al.
Primary central nervous system
vasculitis in children. Arthritis Rheum
2006; 54: 1291�97.
5) Lanthier S, Lortie A, Michaud J, Laxer
R, Jay V, deVeber G. Isolated angiitis of
the CNS in children. Neurology 2001;
56: 837�42.
6) Matsell DG, Keene DL, Jimenez C,
Humphreys P. Isolated angiitis of the
central nervous system in childhood.
Can J Neurol Sci 1990;17: 151�54.
7) F G. Joseph and N J. Scolding. Cerebral
vasculitis-a practical approach.Practical
Neurology 2002; 2, 80�93.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
15
A 7 year old boy presented to the
OPD with history of chest deformity noticed
since birth. There was no history of
recurrent chest infections, cyanosis or
breathing difficulty. He was born to non-
consanguinously married normal parents.
He was not investigated for the above
complaint anytime earlier. His general
physical examination revealed left sided
depressed hemithorax, with absent areola
and an inverted left nipple (fig 1).
There was no associated limb defect
or defective digits on the same side. No
other obvious external anomaly was made
out. Cardiovascular examination revealed a
right sided apex located in the 4th
intercostal space, with normal heart sounds
and no murmur. Respiratory system showed
decreased intensity of breath sounds over
the left lung fields. Per abdomen was
unremarkeable. Chest X-ray (fig 2) done
showed findings suggestive of dextrocardia
with defective left 2nd, 3rd and 4th ribs, with
normal diaphragms.
2D echo confirmed dextrocardia with
no structural cardiac abnormality.
Ultrasound abdomen showed normal
RARE ASSOCIATION 0F POLAND�S SYNDROME WITH
DEXTROCARDIA* Dr. N.Rashmi; Dr. Narayanappa.D
* Department of Pediatrics.JSS Medical College Hospital, JSS University, Mysore.
positions of all the organs, which ruled out
situs inversus.
Discussion:
Poland syndrome is a rare congenital
anomaly that was first described by Alfred
Poland in 1841. The incidence ranges from
1:10000 to 1:100000 as reported by different
authors.
The right side of the body is affected
three times more frequently than the left
and it is more common in boys than in
girls. It comprises of different anomalies
principally at musculo-skeletal system,
lungs, heart and kidneys.
Thorax deformity is the most
common feature of this syndrome. It
includes hypoplasia or absence of the
pectoralis minor and the sternal head of
pectoralis major muscles. The defect in the
chest wall is variable with the absence or
rudimentary development of the anterior
portion of 2, 3, 4, 5th ribs and their costal
cartilages. Breast together with nipple can
be absent or underdeveloped. Ipsilateral
hand anomalies can be seen as
brachydactyly, syndactyly or ectrodactyly

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
16
16
and are most important features of the
syndrome1,2,3. Different etiologic factors of
Poland syndrome are taken into account:
genetic, vascular compromise during early
stages of embryogenesis, but also teratogenic
effects of environmental xenobiotics.
More than 20 patients with
dextrocardia and left-sided Poland
syndrome have been previously described.
The association between these 2 rare
anomalies suggests a causal relationship,
but the etiopathogenetic mechanism has not
been clarified yet4.
Dextrocardia was reported in 5.6% of
a series of 144 patients with Poland
syndrome, and in 9.6% of those, the defect
was left-sided2,5,7.
In patients with isolated
dextrocardia, the incidence of congenital
heart disease has been estimated at 98%. In
dextrocardia with situs inversus this rate is
only 5%6. Congenital cardiovascular
anomalies have not been reported in Poland
syndrome with dextrocardia.
Dextrocardia in Poland syndrome is
associated with rib defects in all of cases,
whereas rib defects are reported in only
about 15% of patients with right-sided
Poland syndrome7.
Our case also supports the view that
the combination of Poland sequence and
dextrocardia is not coincidental and
dextrocardia may be part of the Poland
syndrome, especially left-sided. Probably,
mechanical factors during embryonic life
could explain the strong association between
left-sided Poland syndrome and
dextrocardia. Accordingly, partial agenesis
of 2 or more ribs is needed to displace the
heart toward the right side. The peculiar
features of dextrocardia when associated
with Poland syndrome (neither associated
with situs inversus nor complex intracardiac
anomalies) support this hypothesis.
References
1. Kevin P, David CS Jr. Disorders of
sternum and the thoracic wall. In :
Sabiston DC, Spencer FC (Eds.). Surgery
of the Chest, 6th ed. Philadelphia: WB
Saunders, 1995; 507-511.
2. Fokin AA, Robicsek F. Poland�s
syndrome revisited. Ann Thorac Surg
2002; 74: 2218- 2225.
3. Van Heest Ann E. Common orthopedic
problems II, Congenital disorders of
the hand and upper extremity.
Pediatr Clin North Am 1996; 43: 1113-
1133.
4. Michele Torre, Anwar Baban, Anna
Buluggiu, Sara Costanzo, et al. The
Journal of Thoracic and Cardiovascular
Surgery - 12 November 2009 (10.1016/
j.jtcvs.2009.08.024).
5. Bavinck JNB, Weaver DD. Subclavian
artery supply disruption sequence:
hypothesis of a vascular etiology for
Poland, Klippel- Feil, and Mobius
anomalies. Am J Med Genet. 1986;
23:903-18.
6. Chen JTT. The chest roentgenogram and
cardiac fluoroscopy. In: Alexander
RW, Schlant RC, Fuster V, editors:
Hurst�s the heart. Italian International
Edition. New York: McGraw-Hill; 1995.
p. 387-414.
7. Fraser FC, Teebi A, Walsh S, Pinsky L.
Poland sequence with dextrocardia:
which comes first? Am J Med Genet.
1997; 73: 194-6.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
17
Abstract
A term baby presented with multiple
bullae and erosions on lower limbs, back
and scalp. Minimal trauma resulted in
fresh lesions. There were a few oral and
perioral lesions. Skin biopsy from the lesion
confirmed the diagnosis of junctional
epidermolysis bullosa. Epidermolysis bullosa
is a rare congenital mechanobullous disease.
Key words: Epidermolysis bullosa,
blister, newborn
INTRODUCTION
Epidermolysis bullosa (EB) is a group
of congenital, hereditary blistering disorder
A RARE BLISTERING DISEASE IN A NEWBORN* James Daniel S, K Shreedhara Avabratha, Elizabeth Varkey Cherian, B Sanjeev Rai,
Ramesh Bhat
*Department of Pediatrics and # Department of Dermatology, Father Muller Medical College,Mangalore, Karnataka, India
that are characterized by blister formation
in response to little or no apparent trauma.
Hence the alternate term is mechanobullous
disease. There are three major types,
epidermolysis simplex, junctional
epidermolysis bullosa and dystrophic
epidermolysis bullosa which differs in
clinical and histologic features, inheritance
patterns and severity and prognosis1. It
usually presents either at birth or during
the neonatal period. The incidence and
prevalence of epidermolysis bullosa are
estimated to be 19.60 per million live births
and 8.22 per million population, respectively2.
We report a newborn with multiple blisters
Fig A. Showing blisteringin perioral regions
Fig B. Showingblistering anderosion onextremities
Fig C:Light microscopy(10X) of
biopsy specimen showing
subepidermal blistering

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
18
and erosions whose skin biopsy confirmed
the diagnosis junctional EB.
CASE
A term female baby born to gravida 3
mother, with a good apgar score was
referred on day1 with history of blistering
and erosion of skin since birth. There was
consanguinity( 2o relation) between the
parents, however there was no family
history of similar complaints. The birth
weight of the baby was 2200 grams. On
examination baby had erosion on the lower
limbs and bullae over the back and scalp
(Fig A). Minimal trauma resulted in fresh
lesions. There were few oral and perioral
lesions (Fig B). Systemic examination was
normal. A diagnosis of epidermolysis
bullosa was considered, with congenital
syphilis as a differential diagnosis. Mother
was tested negative for VDRL. Skin biopsy
confirmed the diagnosis of junctional
epidermolysis bullosa (Fig C). The baby was
treated with antibiotics � local and
systemic, and paraffin guaze dressing.
Inspite of the treatment the baby succumb to
illness.
DISCUSSION
Epidermolysis bullosa(EB) is a rare
group of inherited disorders that manifests
as blistering or erosion of the skin and in
some cases, the epithelial lining of other
organs, in response to little or no apparent
trauma. The following major types of
epidermolysis bullosa have been identified
Epidermolytic - Epidermolysis bullosa
simplex (EBS), Lucidolytic - Junctional
epidermolysis bullosa (JEB), Dermolytic -
Dystrophic epidermolysis bullosa (DEB)3.
Epidermolysis bullosa simplex is the
most common type characterized by blisters
in the palms and soles, while milia scarring
and nail dystrophy is uncommon in this
type of EB. The junctional EB is more
severe and is characterized by enamel
hypoplasia with moderate to severe
intraoral blistering and skin lesions where
as the dystrophic EB is the most severe
form of disease characterized by milia,
atrophy and nail dystrophy3.
Junctional epidermolysis bullosa(JEB)
is the rare form of epidermolysis bullosa
and has an incidence of 2.04 per million
live births and 0.44 per million population,
respectively2 which is characterized by
presence of enamel hypoplasia, manifested
as localized or more extensive thimble-like
pitting of some or all of the tooth surfaces.
It is therefore an extremely useful clinical
finding, although it cannot be used as a
diagnostic tool until after the primary teeth
have erupted3. There are two major JEB
subtypes JEB- Herlitz and JEB Non Herlitz.
The more severe one, JEB-Herlitz (JEB-
H), presents at birth and involves all skin
surfaces which is inherited as autosomal
recessive inheritance and is life threatening.
An affected usually presents with blisters
at birth or during early neonatal period with
blisters particularly in perioral area, scalp,
legs, diaper area and thorax with relative
sparing of feets and legs. Mucous membrane
involvement may be severe and presents as
ulcerations in respiratory, gastrointestinal
and genitourinary system.
An essentially pathognomonic finding
is exuberant granulation tissue which
usually arises within the first several
months to one to two years of life3. This
may involve not only the skin but also the
upper airway. Moderate to severe intraoral
blistering is invariably present, with some
eventual narrowing of the opening of the
mouth (�microstomia�) and reduced
extension of the tongue (�ankyloglossia�).

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
19
Most patients die within first three of
year as large moist erosive plaques may
provide a portal of entry for bacteria
causing septicemia which is the most
frequent cause of death1.
The second type is a less severe form,
JEB-non Herlitz which is a heterogenous
group which presents as severe blistering
in neonatal period which is difficult to
differentiate from Herlitz type as
conditions associated with it is seen
although in milder form. Generalized
atrophic benign epidermolysis bullosa and
JEB associated with pyloric stenosis are
variants of non-Herlitz JEB.
In all types of JEB, light microscopy
shows subepidermal blister and electron
microscopy shows cleavage plane in the
lamina lucida. Differentiation of the two
types of epidermolysis bullosa is also
dependent on electron microscopy findings
and antigenic staining 4 ,5. In our case,
lesions presented since birth and light
microscopy was suggestive of JEB, this fits
into JEB-H.
Differentiating EB from non-EB, or
one form of EB from another, can be very
difficult, especially in the neonatal period.
The following condition can be considered
in the differential diagnosis of EB: bullous
congenital ichthyosiform erythroderma;
staphylococcal scalded skin syndrome;
bullous impetigo; incontinentia pigmenti;
neonatal herpes simplex; autoimmune
bullous disease � pemphigus or herpes
gestationis; aplasia cutis; focal dermal
hypoplasia; congenital syphilis and
Gunther�s disease2.
The treatment of JEB is mainly
supportive with diet which gives adequate
calories, supplementation of iron and
prompt treatment of infections with
appropriate antibiotics. Transfusion of
packed cells may be required in patient not
responding to iron and erythropoietin.
Tissue engineered skin grafts may be
beneficiale.
In summary any baby presenting with
bullous skin lesions, EB should be
considered and every effort should be
made to confirm the diagnosis.
REFERENCES
1. Morelli JG. Vesicobullous disorders. In
Nelson Text book of Pediatrics.
Kleigman RM, Jenson HB, Behrman
RE and Stanton BF ed. Philadelphia,
Pennsylvania, Saunders 2007; 2685-
2693.
2. Fine JD and Burge SM. Genetic
Blistering Diseases. In Rooks Text book
of Dermatology. Burns T, Breathnach S,
Cox N and Griffiths C ed. Singapore,
Wiley Blacwel 2010; 39.1-39.32.
3. Fine JD. Epidermolysis Bullosa. In
Dermatology. Bolagnia JL, Jorizzo JL
and Rapini RP ed. Spain, Elsevier
2008; 457-466.
4. Fine JD. Inherited epidermolysis
bullosa. Orphanet Journal of Rare
Diseases 2010, 5:12.
5. Fine JD, Eady RAJ, Bauer JA, et al. The
classification of inherited
epidermolysis bullosa (EB): report of
the Third International Consensus
Meeting on Diagnosis and
Classification of EB. J Am Acad
Dermatol 2008, 58:931-950.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
20
ABSTRACT
Stevens-Johnson syndrome (SJS) and
toxic epidermal necrolysis (TEN) are
potentially fatal disorders, characterized by
high fever, wide-spread blistering exanthema
of macules, accompanied by mucosal and
oral involvement. The major causative drugs
were antibiotics, anticonvulsants, and
NSAIDs. The use of corticosteroids is based
on the idea that corticosteroids can
effectively suppress an excessive immune
response. We report 11 year old boy with
SJS caused by amoxicillin treated with
betamethasone, who recovered completely
without any sequele.
KEY WORDS: amoxicillin, SJS, betamethasone
INTRODUCTION
Stevens-Johnson syndrome is an
immune-complex�mediated hypersensitivity
disorder that may be caused by many drugs,
viral infections, and malignancies. Most
patients are in the second to fourth decade
of life; however, cases have been reported in
children as young as 3 months (1). SJS and
TEN are severe cutaneous reactions that
carry significant morbidity and mortality
risks for children who are affected (2). They
represent severity varients of the same
process with respect to mechanisms,
clinically, etiologically and histopathologically.
The incidence is 1 to 6 cases per million
person per years(3).Even though the exact
pathophysiology is unclear,drugs are the
important etiological factor. Supportive
therapy is the standard of care for SJS/TEN
STEVENS-JOHNSON SYNDROME���A CASE REPORT*DR.K JAGADISH KUMAR, DR.H.C.KRISHNA KUMAR, DR.PAWAN KUMAR,
DR.V.G.MANJUNATH, DR.S.MAMATHA
*Dept OF PEDIATRICS, JSS MEDICAL COLLEGE,JSS UNIVERSITY, MYSORE, KARNATAKA, INDIA.
and includes close monitoring of fluid and
electrolyte status, nutritional support,
meticulous wound care, and control of pain
and infection. We report 11 year old boy
with SJS caused by amoxicillin treated with
betamethasone who recovered completely
without any sequele.
CASE REPORT
A 11 year old boy presented with
fever since 5 days and skin lesions since 2
days. After 3 days of onset of fever he
developed red rashes over the trunk,which
gradually progressed to involve the whole
body over next three days.Later the rashes
became dark with blister formation .He
stopped taking orally also and mother
noticed red lips with oral ulcers.There was
no history of cough, difficuty in breathing
and pruritis.His urine out put was normal
and there was no history of dark coloured
urine and stools.He was treated with
amoxicillin and paracetamol for 2 days
before the onset of rashes.
On examination he was febrile, PR of
106/minute, BP of 90/60 mm of Hg,
Respiratory rate of 24/min oxygen
saturation was 98% in room air.Skin
examination revealed generalised red
maculopapular lesions all over the body
more on the face with few crusted lesions
exposing the red raw surface.There were
vesicles surrounded by erythematous
base.Nikolsky�s sign was absent.Oral cavity
appeared red,crusted with erosions over lips
,buccal mucosa,and genitalia.Detailed

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
21
ophthalmic examination revealed photophobia,
congested with white discharge bilaterally.
Other systemic examination was
unremarkable. Investigations revealed Hb
13gm/dl,TC 5,800cells/cu mm ,Differential
count of 80% Neutrophils,20% lymphocytes,
platelet count2.36 lakhs/cumm, normal
normocytic peripheral blood picture,CRP
positive,widal and peripheral smear for
malarial parasite were negative.Blood urea
22mg/dL,creatinine 0.6mg/dL,Blood sugar
136 mg/dL, ,Sodium 128mmol/l, potassium
4.2 m mol/L,chloride 100 mmol/l,chest X-
ray normal, blood culture was sterile. In
view of exposure to amoxicillin with typical
clinical features ,a diagnosis of SJS/TEN
was made .Boy was started with i.v
fluids,inj ranitidine, oral vitamin A 2 lakhs
per day and i.v.cefotaxim.Saline compresses
for the skin lesions,ofloxacin eye
drops,saline cleansing of oral mucosa with
application of glycerin with local
anaesthetic gel was given .I.V.betamethasone
4 mg once a day started with monitoring of
the vitals.He became afebrile by 4 days and
he started taking orally by 3 days and
became ambulent on the fifth day.Skin and
mucosal lesions started fading by 3 days.He
was completely off i.v fluids by fifth day.
Betamethasone was given for 7 days. By
10th day his skin and oral lesions healed
completely and discharged.
DISCUSSION
SJS and TEN are acute life threatening
mucocutaneous reactions characterised by
extensive necrosis and detachment of
epidermis.They start as erythematous
macules,evolve progressively to confluent
flaccid blisters with epithelial detachment.
They represent severity varients of the same
process with respect to mechanisms,
clinically, etiologically and histopathologically.
Pathologically, cell death results causing
separation of the epidermis from the dermis.
They differ only in the percentage of body
surface involvemet.The incidence is 1 to 6
cases per million people per years (3).Even
though the exact pathophysiology is unclear,
drugs are the important etiological factor.
Drugs particularly sulfonamides, NSAIDS,
anticonvulsants, antibiotics are the common
offenders (4).After the drug exposure SJS
clinically appears within 8 weeks (4 to 30
days).Fever, rhinitis may precede the
mucocutaneous lesions by 1 to 3
days.Burning eyes, pain on swallowing
progressively develops.The initial skin
lesions are dusky erythematous macules
which progressively coalese on
erythamatous base.The lesions evolve to
flaccid blisters and break easily. The typical
lesion has the appearance of a target. At
pressure points the necrotic epidermis gets
detachment exposing the red dermis.
Nikolsky�s sign will be positive. Usually
epithelial detachment occurs for 5-7 days
followed by re-epithelialisation.In our case
also; skin lesions were classical and
recovered by 7 days. If less than 10 % of
body surface area is involved it is SJS; more
than 30 % it is TEN; 10-30 % SJS /TEN(4).

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
22
In 90 % of the cases mucous membrane
involement occurs and atleast two or more
mucosal surfaces will be involved (3).It
begins as erythema followed by erosions of
buccal, ocular and genital mucosa. In SJS
pain from mucosal ulceration is severe and
skin tenderness is absent in contrast to toxic
epidermoal necrolysis (4). Individual lesions
typically should heal within 1-2 weeks,
unless secondary infection occurs. Most
patients recover without sequelae like our
case.85 % of cases will have conjunctival
lesions, manifested by hyperemia, chemosis,
photophobia, and lacrimation. Ocular
involvement can cause corneal scarring and
visual loss (1,3).Pulmonary involvement can
occur in 25 % of cases characterised by
bronchial hypersecretion and dyspnoea.
Raised blood urea is marker of
severity (3).Anemia, leucocytosis,
neutropenia, increased liver enzymes, raised
blood glucose can occur.The commonest
complication is sepsis due to epithelial
loss.Multiorgan failure, lung, ophthalmic
complications can also occur. The mortality
rate for TEN is 30 % and 5-12.5 % for SJS (1,
3,4). The single most important role for the
pediatrician is to detect Stevens-Johnson
syndrome/toxic epidermal necrolysis (SJS/
TEN) early and initiate the appropriate
emergency and inpatient management.
Treatment is primarily supportive and
symptomatic.Prompt withdrawal of the
offending drug and supportive care is very
important. Fluid and electrolyte balance is
the first priority, along with nutrition
support. Denuded skin lesions can be
cleaned with saline compresses.A daily
examination for infection is a must both
clinically and investigation wise. Eyes
shoud be taken care by ophthalmologist
with vitamin A, antibiotics and lubricants.
Some have advocated corticosteroids,
cyclophosphamide, plasmapheresis,
hemodialysis, and immunoglobulin.
The use of steroids is still
controversial (5,7,8, 9). The exact mode of
action of steroids in SJS/TEN is not known
(9). However, the prevailing consensus seems
to be that systemic glucocorticoids are
justified in the early and evolving disease
preferably within the first 72 hours of onset
to prevent widespread involvement or
during reappearance of erythema and/or
necrosis on newly regenerated skin. (6,9). In
a report of 52 cases of SJS and 65 cases
(2000-2006) of TEN from Japan, the authors
have used methyl prednisolone pulse (125-
1000 mg/day) for 3 days. They have
concluded that the mortality rates for
patients with SJS and TEN were 1.9% and
6.2% respectively which has decreased from
21.6% (58/269) during previous 17 years
(1981-1997) in which period steroids were
rarely used (5).Similar observations were
made by others also(9,10). Tripathi et al in
their report of 67 cases with SJS, 66 cases
recovered with steroid therapy (10). Steroid
pulse therapy at disease onset is of great
therapeutic importance in preventing ocular
complications also(11).Given the importance
of immune mechanisms in inflammatory
drug reactions, IVIG has emerged as a
potential immunomodulatory therapy for
SJS/TEN(2).IVIG seems to be a useful and
safe therapy for children with SJS/TEN. IVIG
doses of 0.5 to 1.0 g/kg administered over 3
days are most effective (2).
To conclude, SJS and TEN are
variations of the same disease expressed
with different severity .They generally begin
with a prodrome of high fever, sore throat,
and malaise, followed by the rapid onset of
cutaneous blistering, mucosal and eye

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
23
involvement. .SJS and TEN are rare but
serious disorders with significant morbidity
and mortality in children.Steroids seems to
be a useful therapy for SJS.
REFERENCES
1) Steven J Parrillo, Catherine V
Parrillo.Stevens-Johnson Syndrome.
eMedicine.Updated: May 25, 2010
2) Denise W. M, Peter J, Moise L. L. Use
of Intravenous Immunoglobulin in
Children with Stevens - Johnson
syndrome and Toxic Epidermal
Necrolysis: Seven Cases and Review of
the Literature. Pediatrics 2003;
112:1430-1436.
3) Valeyrie-Allanore L, Jaean-cLaude r,
Epidermal Necrolysis.In: Wolff K,
Goldsmith L A,,Katz S I ,Gilchrest B A
,Paller A S ,Leffell D J .editors.
Fitzpatrick�s Dermatology in General
Medicine., 7th Edition, vol-1, New-
york: Mc Graw Hill Medical;
2008.p.349-354
4) Joseph G.Moreli.Vesicobullous
disorders. In: Kliegman R M, Jenson H
B, Behrman R E,Stanton B F.editors.
Nelson Text Book of Pediatrics. 18th
Edition.vol-2, Philadelphia: W B
Saunders Company; 2008.p.2685-2688
5) Yamane Y, Aihara M, Ikezawa Z.
Analysis of Stevens-Johnson syndrome
and toxic epidermal necrolysis in
Japan from 2000 to 2006. Allergol Int
2007; 56:419-25.
6) Suresh Kumar.P.N, Biju Thomas,
Kishore Kumar, and Shibu Kumar
.Stevens�Johnson syndrome�toxic
epidermal necrolysis (SJS�TEN)
overlap associated with carbamazepine
use Indian J Psychiatry. 2005; 47: 121�
123.
7) Ginsburg CM.Stevens-Johnson
syndrome in children. Pediatr Infect
Dis. 1982; 1:155-8.
8) Cheriyan, Sarah, Patterson, Roy;
Greenberger, Paul A.; Grammer, Leslie
C.; Latall, John. The Outcome of
Stevens - Johnson syndrome Treated
with Corticosteroids.Allergy and
Asthma Proceedings.1995; 16:151-155.
9) Sharma VK, Sethuraman G, Minz A.
Stevens Johnson syndrome, toxic
epidermal necrolysis and SJS-TEN
overlap: A retrospective study of
causative drugs and clinical outcome.
Indian J Dermatol Venereol Leprol
2008; 74:238-40
10) Tripathi A, Ditto AM, Grammer LC,
Greenberger PA, McGrath KG, Zeiss
CR, et al. Corticosteroid therapy in an
additional 13 cases of Stevens-Johnson
syndrome: A total series of 67 cases.
Allergy Asthma Proc 2000; 21:101-5
11) Yamane Y, Aihara M, Ikezawa Z.
Analysis of Stevens-Johnson syndrome
and toxic epidermal necrolysis in
Japan from 2000 to 2006. Allergol Int
2007; 56:419-25.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
24
Abstract:
Biotinidase deficiency is described inpatients with neurological, dermatological,immunological and ophthalmologicalabnormalities, especially in profounddeficiency (<10%). We report a baby whopresented with refractory seizures and skinrashes, with abnormal tone. He had a highC5OH levels on TMS, suggestive ofholocarboxylase deficiency. Specific enzymeassay showed deficient biotinidase activityof 0.1 nmol/min/mL. On treatment thebaby showed response with absence ofseizures and disappearance of skin rash.
Introduction
Biotinidase is a mammalian cellenzyme that occurs at high levels in theliver, serum, and kidney. Multiplecarboxylase deficiency responsive to biotinadministration was first described in 1971.Wolf and colleagues further characterizedthe infantile form of multiple carboxylasedeficiency as biotinidase deficiency in the1980si.It can be profound (<10% enzymelevel) or partial (10-30% enzyme level).ii
Clinical manifestations include neurological,dermatological, immunological,
and ophthalmological abnormalities.We report a case of profound biotinidasedeficiency
Case Report
A 3-month old boy, born of nonconsanguineous marriage, presented withhistory of multiple seizures since 2 monthsof age. MRI brain at that time revealedischemic changes in the white matter andhe was on treatment with phenobarbitone.
On admission, he was found to have
jitteriness. Skin showed an erythematous
rash around neck and back. Alopecia and
seborrheic dermatitis present since birth.
Systemic examination revealed an increasedtone with exaggerated reflexes. Laboratoryinvestigations revealed normal hemogram,liver functions, and serum ammonia andserum electrolytes. Blood gas analysisshowed persistent severe metabolic acidosis,refractory to therapy. CSF was normal
After admission, baby was treated inintensive care for seizures which weredifficult to control. In view of poortherapeutic response and suspicious MRIreports, he was investigated further for ametabolic disorder. Plasma and urine aminoacidogram was normal. Tandem massspectrometry revealed increased C5OHlevels, suggestive of holocarboxylasedeficiency. Specific enzyme assay showeddeficient biotinidase activity of 0.1 nmol/
min/mL (normal >5nmol/min/mL).
Picture 1 � shows the face and scalp
to have an erythemaout rash along with
alopecia and seborrheic dermatitis.
He was started on oral biotin (10mg/day) along
with Carnitine (100mg/kg).The child improved
dramatically within few days with
normalization of sensorium and blood gas
reports, control of seizures, and disappearance
of skin lesions. He was discharged on biotin
CASE REPORT - BIOTINIDASE DEFICIENCY IN INFANCY*Dr.Rajashekar Murthy G.V , Dr Sanjay K.S. , Dr.Bharath Kumar Reddy K.R.
* Indira Gandhi Institute of Child Health, Bangalore

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
25
supplements and presently doing well after 3
months.
Discussion
The incidence of profound biotinidasedeficiency is estimated at 1 per 137,401population. The combined incidence ofpartial and profound deficiencies is 1 per61,067 populations1 However, onlyoccasional case reports are available inIndian literature.2 The gene that encodesbiotinidase is localized at 3p25. The mostcommon mutation, 98-104del7ins3 (which ispresent in approximately one half ofsymptomatic children), has been identified.A second, less common mutation, Arg538 R? C, has also been described.3
The appearance of symptoms seemsto be associated with metabolic stressors (eg,illness, fever, fasting), and children may notbe symptomatic until such time. The mostcommon symptom of presentation isseizures. Others include features ofdevelopmental delay, ataxia, neuropathy,auditory nerve dysfunction and spasticparaparesis. Dermatological manifestationsare particularly striking when they develop;these include alopecia and an eczematous,scaly perioral/facial rash. Although theymay be severe, the rash and alopeciatypically respond rapidly to biotinadministration over days to months. Respiratoryproblems in these children includehyperventilation, laryngeal stridor and apnea.
Most cases with Biotinidase deficiencyexhibit metabolic ketolactic acidosis, organicaciduria, and mild hyperammonemia.Children with biotinidase deficiency maydemonstrate cerebral edema, low attenuationof white matter signal, cerebral atrophy, andcompensatory ventricular enlargement.4 EEGfindings prior to treatment demonstrate poororganization of background and absence of
typical sleep morphology. Both thesefindings were seen in our child. Diagnosiscan be confirmed by Tandem Massspectrometry and serum enzyme analysis.
Therapy for biotinidase deficiency isoral biotin, typically administered at aninitial dose of 10 mg/d. Some patientsrequire higher dosages. If the enzymaticdefect is present but does not respond tolower dosages, consider a high-dose therapy(up to 40 mg/d). If children are left withresidual neurological disease, they mayrequire treatments for developmental delay,spasticity, and bulbar dysfunction inaddition to biotin. With treatment, patientshave an excellent prognosis and potential fora normal lifestylevii
1. Wolf B. Disorders of Biotin Metabolism.In: Scriver CR, Beaudet AL, Sly WS,Valle D (eds). The Metabolic andMolecular Bases of Inherited Disease.8th ed. New York: McGraw-Hill;2001.p.3935-3962.
2. McVoy, Julie R. Secor; Levy, Harvey L.;Lawler, Michael; Schmidt, Michael A.;Ebers, Douglas D.; Hart, Suzanne; Pettit,Denise Dove; Blitzer, Miriam G. et al.(1990). �Partial biotinidase deficiency:Clinical and biochemical features�. TheJournal of Pediatrics 116 (1): 78�83.
3. Wolf B. Worldwide survey of neonatalscreening for biotinidase deficiency. JInherit Metab Dis 1991; 14: 923-927.
4. Ramdas Dahiphale, Shreepal Jain,Mukesh Agrawal; Biotinidase deficiency;Indian Pediatrics 2008; 45:777-779
5. Hymes J, Stanley CM. Wolf B. Mutationsin BTD causing biotinidase deficiency.Hum Mutat 2001; 18: 375-381.
6. Lott IT, Lottenberg S, Nyhan WL,Buchsbaum MJ. Cerebral metabolicchanges after treatment in biotinidasedeficiency. J Inherit Metab Dis 1993: 16:399-400.
7. Weber P, Scholl S, Baumgartner ER.Outcome in patients with profoundbiotinidase deficiency: relevance ofnewborn screening. Dev Med ChildNeurol 2004; 46: 481-484.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
26
Abstract
Schizencephaly is a rare developmental
disorder of neuronal migration,
characterized by early focal destruction of
the germinal matrix and surrounding brain
before the cerebral hemispheres are fully
formed at 1-5 months of gestation .The
lesion is most likely related to multiple
aetiologies including genetic, toxic,
metabolic, vascular or infectious agents. This
case is reported due to its rarity. The
prevalence of schizencephaly is very
uncommon internationally.
Key Words: Schizencephaly, septum
pellucidum, septoptic dysplasia.
Introduction
Schizencephaly is an uncommon
disorder of neuronal migration characterized
by a cerebrospinal fluid�filled cleft, which is
lined by gray matter. The cleft extends
across the entire cerebral hemisphere, from
the ventricular surface (ependyma) to the
periphery (pial surface) of the brain1. This
disorder was originally described by
Wadsworth and Yakolev2. The clefts may be
unilateral or bilateral and may be closed
(fused lips), as in schizencephaly type I, or
separated (open lips), as in schizencephaly
type II. Schizencephaly can be distinguished
from porencephaly by the fact that in
schizencephaly the fluid-filled component, if
present, is entirely lined by heterotopic grey
matter while a porencephalic cyst is lined
mostly by white matter . The cardinal
neuropathological features are ³)Hemispheric
cleft ³³)communication of subarachnoid
space with lateral ventricle medially ³³³)
Infolding of grey matter along the cleft iv)
Multiple associated intracranial malformations
including polymicrigyria, absent septum
pellucidum, optic nerve hypoplasia.
Case Report:
A 15 month old male child presented
to us with complaints of inability to move
the left upper and lower limbs, noticed by
the parents since 5 months of age. The
development of the child was mildly delayed
in all domains. No history of convulsions
was present. The child was delivered full
term normally at a hospital with no
intrapartum or postpartum complications.
Child was the second born of a non
consanguineous parentage. On physical
examination child had normal head
circumference with no dysmorphic features.
Anthropometric measurements of weight and
length were within normal limits Vision and
hearing were normal. Muscle tone showed
spasticity in the left lower and upper limbs
with exaggerated reflexes. Babinski�s sign
was extensor on the left side. The child was
admitted for evaluation of the etiology of
stroke. Hematological and biochemical
parameters were within normal limits. CT
scan showed a Closes Lip Schizencephaly
on the right side with an absent septum
pellucidum.. MRI was suggested but the
patients were not willing for the same.
Counselling was given and physiotherapy
was advised.
CASE REPORT: SCHIZENCEPHALY TYPE I � A CAUSEFOR STROKE IN CHILDREN*Dr Sanjay K.S, Dr Rajashekar Murthy G, Dr Bharath Kumar Reddy K.R
*Indira Gandhi Institute of Child Health, Bangalore

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
27
Picture 1 � shows a closed lip
schizencephaly resulting in left sided
hemiparesis
Discussion
In schizencephaly, the neurons border
the edge of the cleft, implying a very early
disruption of the usual grey matter migration
during embryogenesis. The cause of the
disruption is not known, but likely the cause
may be either genetic or a physical insult, such
as infection, infarction, hemorrhage, toxin or
mutation. As genetic cause, it is found to have
a mutant gene,EMX21. The symptoms of
schizencephaly are variable. In closed type
(Unilateral case)- Mild hemisparesis and
seizure but normal development.In open type
there is mild to moderate developmental delay
with hemiparesis. In bilateral clefts there is
severe mental deficits, severe motor anomalies
including spastic quadreparesis2 MRI is the
imaging modality of choice3. Treatment
consists of treatment of seizures,
physiotherapy, and in cases that are
complicated by hydrocephalus, a
ventriculoperitoneal shunt is needed. In open
lip schizencephaly the patients die at early age.
Death is mainly due to failure to thrive, chronic
infections and respiratory problems. In closed
lip schizencephaly patients may not present
until later in infancy and they live upto
adulthood.
REFERENCES
1. Spalice A, Parisi P, Nicita F, Pizzardi G,
Del Balzo F, Iannetti P. Neuronal migration
disorders: clinical, neuroradiologic and
genetics aspects. Acta Paediatr. Mar
2009;98(3):421-33
2. Denis D, Chateil JF, Brun M, Brissaud O,
Lacombe D, Fontan D, Flurin V, Pedespan
J. Schizencephaly : Clinical imaging
features in 30 infantile cases. Brain Dev
2000 Dec ; 22(8)475-83
3. Tietjen, I.; Bodell, A.; Apse, K.; Mendonza,
A. M.; Chang, B. S.; Shaw, G. M.; Barkovich,
A. J.; Lammer, E. J.; Walsh, C. A. :
Comprehensive EMX2 genotyping of a
large schizencephaly case series. Am. J.
Med. Genet. 143A: 1313-1316, 2007
4. Gasparetto EL, Warszawiak D, de
Carvalho Neto A, Benites Filho PR, Bruck
I, Antoniuk S. Septo optic dysplasia plus
a case report. Arq Neuropsiquitar 2003
Sep; 671-6
5. Hayashi N, Tsutsumi Y, Barkovich AJ.
Morphological features and associated
anomalies of schizencephaly in the
clinical population: detailed analysis of
MR images. Neuroradiology 2002 May ;
44(5):418-27

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
28
Polycythemia is a silent clinical entity,
which if unrecognized can result in
significant morbidity. We present a
prospective study done on 1362 consecutive
inborn babies delivered in J.S.S Hospital,
Mysore during the period from 1st July 2004
to 30th June 2005. Babies were considered
polycythemic if the venous hematocrit was
65% or more. The incidence of polycythemia
was 2.28%. Majority of the polycythemic
babies had venous hematocrit between 65 to
66%. Most of the symptomatic and also
asymptomatic babies had borderline
polycythemia. Hence, the degree of
polycythemia did not have any relation
with the symptomatology.
Key words: Neonatal polycythemia,
Hematocrit, Symptomatology.
Introduction
Polycythemia and secondary
hyperviscosity are common problems in the
newborn period with reported incidence
ranging from 1% - 5% in total newborn
population(1,2).
The most widely accepted definition
is venous hematocrit 65% or greater(1,2,3).
Small for gestation age babies(3) and infant
of diabetic mother(4) are known to have an
increased incidence of polycythemia and
hyperviscosity.
In India, low birth weight babies
represent 30% of all live births each year.
More than half of these babies are born at
term(5). It is thus obvious that polycythemia
could be a real problem existing in our
country and babies need to be actively
screened for this condition.
Treatment of infants with polycythemia
is relatively simple but controversial. There
is no controversy with regard to treatment of
symptomatic newborn babies with
polycythemia. However, in a newborn with
asymptomatic polycythemia, the indication
of partial exchange transfusion is not as
universally accepted because of lack of
controlled data. Management in
asymptomatic infants should be individualized.
Clinical studies reveal some
measurable benefits following partial
exchange transfusion. Controversy exists
with respect to the long term benefits to
infants treated with partial exchange
transfusion(6).
Methods
All babies born at J.S.S Hospital,
Mysore from 1st July 2004 to 30th June 2005
were included in the study , irrespective of
the gestational age and birth weight.
Criteria for exclusion:
-delayed cord clamping.
-holding the baby below the level of
mother�s introitus.
-cord milking/stripping.
In all the cases, the umbilical cord
was clamped within 30 seconds after birth
of the baby and the babies were held at the
level of the mother�s introitus. Birth weight
was recorded to nearest 10 grams.
Gestational age was determined from
mother�s menstrual history and confirmed
by modified Ballard�s scoring. Intrauterine
growth retardation and macrosomia were
defined by birth weight according to
gestational age, less than 10th percentile and
more than 90th percentile, respectively.
NEONATAL POLYCYTHEMIA �A HOSPITAL BASED STUDY* Dr.Narayanappa.D, Dr.N.Rashmi, Dr.Mohan B.K
*Department of Pediatrics, J.S.S Medical College & Hospital, JSS University, Mysore, Karnataka, India

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
29
Capillary hematocrit was determined
on blood samples obtained by pricking a
neonate�s prewarmed heel (with medipoint
blood lancet) at 2- 4 hours of postnatal age.
Consent was obtained from the parent
present at the time of the procedure. All
capillary hematocrit were determined in
duplicate in 17 mm long and 1 mm wide
(internal diameter) capillary tubes spun at
10,000 RPM for 5 minutes, in a
microhematocrit centrifuge (REMI RM12C )
and the hematocrit read using a reading
device (REMI reading device) .
Steps of reading the haematocrit
using the reading device:
Step 1- position the capillary tube in
the slot so that the baseline of the reader
intersects base of the red cell column.
Step 2- move the tube holder so that
the top line intersects top of the plasma.
Step 3- adjust the knob so that the
middle line intersects top of the red cell
column.
Step 4- read hematocrit (as percent)
on the scale.
If the capillary hematocrit was 70%
or greater, a peripheral venous hematocrit
was determined immediately. The infants
were considered to be polycythemic if thevenous hematocrit was 65% or greater. Arepeat hematocrit was performed at 12 hours(or at any age if symptoms appeared) if theinitial hematocrit was high.
All neonates were examined by thesame investigator and the polycythemicbabies were categorized as symptomatic andasymptomatic. Particular attention was givento the presence of signs and symptomsattributable to polycythemia. Lethargy, poorfeeding, plethora, cyanosis, convulsions,icterus, tachypnea, etc., were looked for.
The following investigations were
sent for all polycythemic babies:
- Hb (gm/dl), platelet count (per/mm3),
- Blood glucose(mg/dl), serum calcium(mg/
dl), serum total bilirubin(mg/dl).
The laboratory abnormalities were defined
as follows-
• Thrombocytopenia defined as platelet
count 1,00,000/mm3.
• Hypoglycemia has been defined as
blood glucose less than 40 mg/dl
irrespective of birth weight and
gestational age.
• Hypocalcemia has been defined as
serum total calcium less than 7.0 mg/
dl.
• Hyperbilirubinemia has been defined as
serum bilirubin level of more than
12mg/dl.
In symptomatic babies, septicemia
was excluded by negative sepsis screen
(total leucocyte count, ESR, CRP) and blood
culture. Lumbar puncture was done in case
of convulsion to rule out meningitis and
cranial ultrasound was done to rule out any
structural anomalies of the brain.
Partial exchange transfusion was
performed for the following babies
- Those babies with venous hematocrit
≥65% with symptoms.
- Those babies with venous hematocrit
>70% without symptoms.
Asymptomatic babies with a venous
hematocrit of 65 to 70 % were only
observed.
The volume for partial exchange
transfusion was calculated using the
formula- (observed hematocrit- desired
hematocrit)×weight (kg)×blood volume (ml/
kg) observed hematocrit
Desired hematocrit was taken as 55%,
Blood volume taken as 80ml/kg.
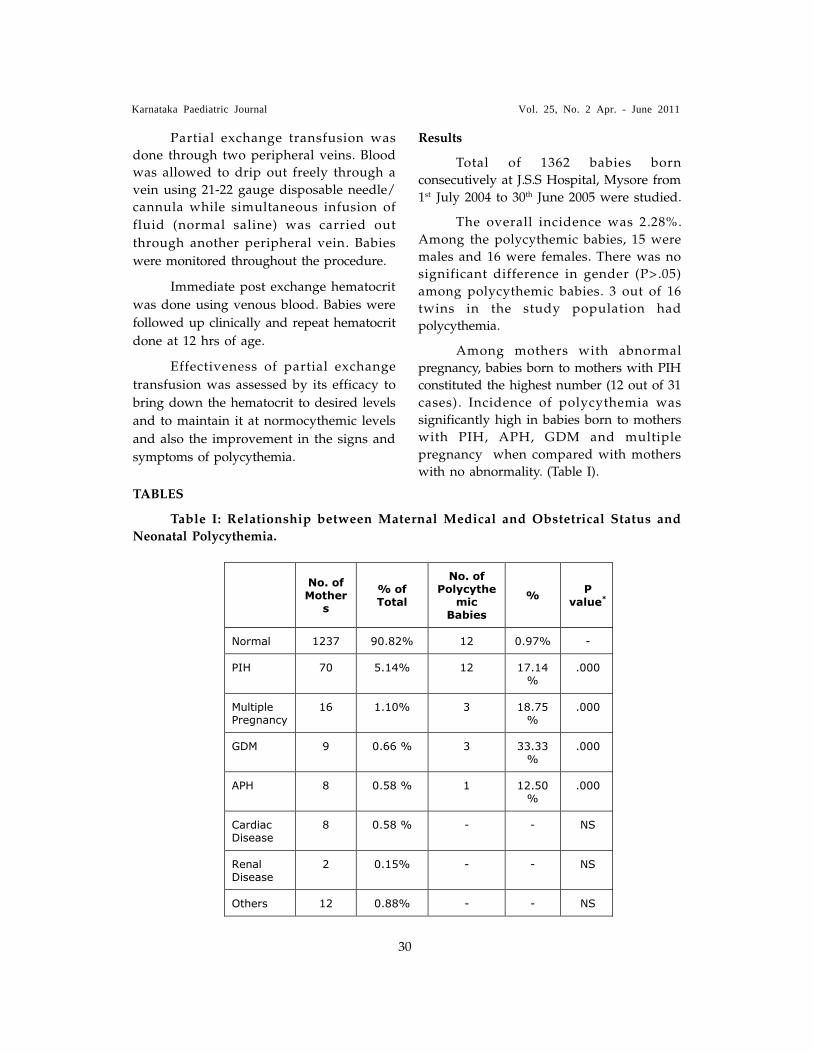
Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
30
Partial exchange transfusion was
done through two peripheral veins. Blood
was allowed to drip out freely through a
vein using 21-22 gauge disposable needle/
cannula while simultaneous infusion of
fluid (normal saline) was carried out
through another peripheral vein. Babies
were monitored throughout the procedure.
Immediate post exchange hematocrit
was done using venous blood. Babies were
followed up clinically and repeat hematocrit
done at 12 hrs of age.
Effectiveness of partial exchange
transfusion was assessed by its efficacy to
bring down the hematocrit to desired levels
and to maintain it at normocythemic levels
and also the improvement in the signs and
symptoms of polycythemia.
Results
Total of 1362 babies born
consecutively at J.S.S Hospital, Mysore from
1st July 2004 to 30th June 2005 were studied.
The overall incidence was 2.28%.
Among the polycythemic babies, 15 were
males and 16 were females. There was no
significant difference in gender (P>.05)
among polycythemic babies. 3 out of 16
twins in the study population had
polycythemia.
Among mothers with abnormal
pregnancy, babies born to mothers with PIH
constituted the highest number (12 out of 31
cases). Incidence of polycythemia was
significantly high in babies born to mothers
with PIH, APH, GDM and multiple
pregnancy when compared with mothers
with no abnormality. (Table I).
No. of Mother
s
% of Total
No. of Polycythe
mic Babies
% P
value*
Normal 1237 90.82% 12 0.97% -
PIH 70 5.14% 12 17.14
%
.000
Multiple
Pregnancy
16 1.10% 3 18.75
%
.000
GDM 9 0.66 % 3 33.33
%
.000
APH 8 0.58 % 1 12.50
%
.000
Cardiac
Disease
8 0.58 % - - NS
Renal
Disease
2 0.15% - - NS
Others 12 0.88% - - NS
TABLES
Table I: Relationship between Maternal Medical and Obstetrical Status and
Neonatal Polycythemia.
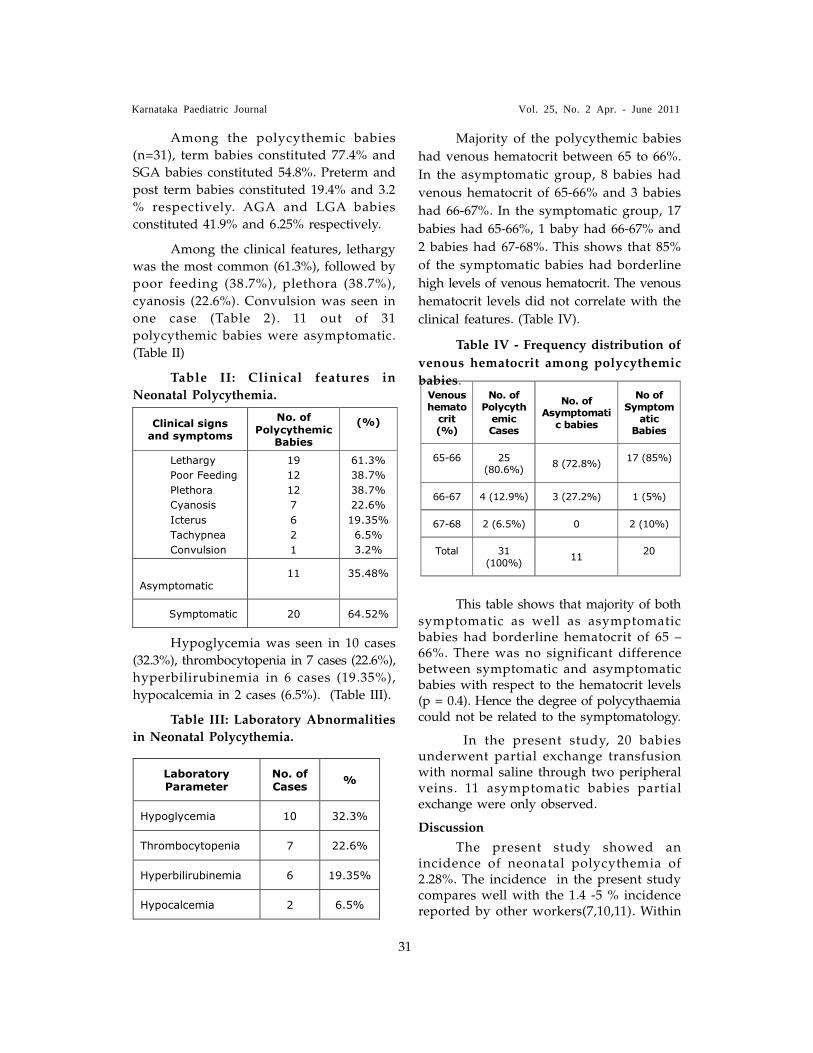
Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
31
Among the polycythemic babies
(n=31), term babies constituted 77.4% and
SGA babies constituted 54.8%. Preterm and
post term babies constituted 19.4% and 3.2
% respectively. AGA and LGA babies
constituted 41.9% and 6.25% respectively.
Among the clinical features, lethargy
was the most common (61.3%), followed by
poor feeding (38.7%), plethora (38.7%),
cyanosis (22.6%). Convulsion was seen in
one case (Table 2). 11 out of 31
polycythemic babies were asymptomatic.
(Table II)
Table II: Clinical features in
Neonatal Polycythemia.
Hypoglycemia was seen in 10 cases
(32.3%), thrombocytopenia in 7 cases (22.6%),
hyperbilirubinemia in 6 cases (19.35%),
hypocalcemia in 2 cases (6.5%). (Table III).
Table III: Laboratory Abnormalities
in Neonatal Polycythemia.
Majority of the polycythemic babies
had venous hematocrit between 65 to 66%.
In the asymptomatic group, 8 babies had
venous hematocrit of 65-66% and 3 babies
had 66-67%. In the symptomatic group, 17
babies had 65-66%, 1 baby had 66-67% and
2 babies had 67-68%. This shows that 85%
of the symptomatic babies had borderline
high levels of venous hematocrit. The venous
hematocrit levels did not correlate with the
clinical features. (Table IV).
Table IV - Frequency distribution of
venous hematocrit among polycythemic
babies.
This table shows that majority of bothsymptomatic as well as asymptomaticbabies had borderline hematocrit of 65 �66%. There was no significant differencebetween symptomatic and asymptomaticbabies with respect to the hematocrit levels(p = 0.4). Hence the degree of polycythaemiacould not be related to the symptomatology.
In the present study, 20 babiesunderwent partial exchange transfusionwith normal saline through two peripheralveins. 11 asymptomatic babies partialexchange were only observed.
Discussion
The present study showed anincidence of neonatal polycythemia of2.28%. The incidence in the present studycompares well with the 1.4 -5 % incidencereported by other workers(7,10,11). Within
Clinical signs
and symptoms
No. of
Polycythemic Babies
(%)
Lethargy
Poor Feeding
Plethora
Cyanosis
Icterus
Tachypnea
Convulsion
19
12
12
7
6
2
1
61.3%
38.7%
38.7%
22.6%
19.35%
6.5%
3.2%
Asymptomatic
11 35.48%
Symptomatic 20 64.52%
Laboratory Parameter
No. of Cases
%
Hypoglycemia 10 32.3%
Thrombocytopenia 7 22.6%
Hyperbilirubinemia 6 19.35%
Hypocalcemia 2 6.5%
Venous
hematocrit (%)
No. of
Polycythemic Cases
No. of Asymptomati
c babies
No of
Symptomatic
Babies
65-66 25
(80.6%) 8 (72.8%)
17 (85%)
66-67 4 (12.9%) 3 (27.2%) 1 (5%)
67-68 2 (6.5%) 0 2 (10%)
Total 31
(100%) 11
20

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
32
the polycythemic group, term babies andSGA babies constituted the highest. Thiswas similar to studies conducted byL.Krishnan(5), M.Singh(9), S.Singh(11). ButWiswell(10) showed that majority of thepolycythemic babies in the growth statusgroup were AGA.
Among the maternal risk factors forthe development of neonatal polycythemia,the present study showed that, PIH hadsignificantly (p=.000) high risk ofdevelopment of neonatal polycythemia. Thiswas similarly seen in studies conducted byVirginia (20.7%) and L.Krishnan (27%) (5,8).The study also showed that other maternalrisk factors like GDM, multiple pregnancy,APH significantly increased the incidence ofneonatal polycythemia.
The most common clinical featuresseen in this study included lethargy (61.3%),followed by poor feeding (38.7%), plethora(38.7%), cyanosis (22.6%), icterus (19.35%).Other studies showed similar result.
Other uncommon manifestations ofneonatal polycythemia, like necrotizingentrocolitis, intracranial hemorrhage,priapism were not encountered in thepresent study, unlike in other studies(12,13).
In the present study, 35.48 % of thepolycythemic babies were asymptomatic.This was similar to the study conducted byWiswell(10).
Most common laboratory abnormalityseen in neonatal polycythemia in this studywas hypoglycemia (32.3%), followed bythrombocytopenia (22.6%), hyperbilirubinemia(19.35%) and hypocalcemia (6.5%). Otherstudies also showed that hypoglycemia wasthe most common laboratory abnormalityassociated with polycythemia.
Majority of the babies had venoushematocrit in the range of 65-66% (25 out of31). In the asymptomatic group, 8 babieshad hematocrit of 65-66% and 3 babies had66-67%. In the symptomatic group, 17babies had hematocrit of 65�66%, i.e; 85% ofthe symptomatic babies had borderline high
levels of venous hematocrit. Hence, thevenous hematocrit levels did not have anyrelation with the clinical features.
Points to remember:
1) Polycythemia is a silent clinical entitywhich if unrecognized can result insignificant morbidity and mortality.
2) Close monitoring is necessary asclinical features in polycythemia may besubtle and babies may beasymptomatic.
3) Lethargy was the most commonsymptom and hypoglycaemia, the mostcommon laboratory finding.
4) The levels of venous hematocrit do nothave any relation with the symptomatology.
REFERENCES
1. Wirth FH, Goldberg KE, Lubchenco LO.Neonatal hyperviscosity: I Incidence.Pediatrics 1979; 63:833-836.
2. Stevens K, Wirth FH. Incidence ofNeonatal hyperviscosity at sea level. JPediatr 1980; 97: 118 � 119.
3. Humbert JR, Abelson WE, Battaglia FC.Polycythemia in small for gestationalage infants. J Pediatr 1969; 75: 812.
4. Letsky EA. Polycythemia in thenewborn. In: Text Book of Neonatology,2nd Edn. Eds. Roberton NRC.Edinburgh, Churchill Livingstone, 1992;p 719 � 723.
5. Krishnan L, Rahim A. NeonatalPolycythemia. Indian J Pediatr 1997;64:541-6
6. Goldberg K, Wirth FH, Hathaway WE etal. neonatal hyperviscosity: II. Effects ofpartial plasma exchange transfusion.Pediatrics 1982; 69: 419-425.
7. Ramamurthy RS, Brans TW. Neonatalpolycythemia: I. Criteria for diagnosis andtreatment. Pediatrics 1981; 68: 168 � 174.
8. Kurlat I, Sola. Neonatal polycythemia inappropriately grown infants ofhypertensive mothers. Acta Paediatr1992; 81(9):662-4

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
33
9. Singh M, Singhal PK, Paul VK et al.Polycythemia in the newborn � Doasymptomatic babies need exchangetransfusion? Indian pediatr 1990; 27:61-65.
10. Thomas E, Wiswell MC, Dern CornishMC. Neonatal polycythemia: Frequencyof clinical manifestations and otherassociated findings. Pediatrics 1986; 78: 26-30.
11. Singh S, Narang A, Bakoo O N.Polycythemia in Newborn. IndianPediatr 1990; 27: 349-353.
12. Hankanson DO, OH W. Necrotizingenterocolitis and hyperviscosity in thenewborn infant. J Pediatr 1977; 90: 458-461.
13. Black VD, Lubchenco LO. Neonatalpolycythemia and hyperviscosity. PediatrClin North Am 1982; 29: 1137-1148.
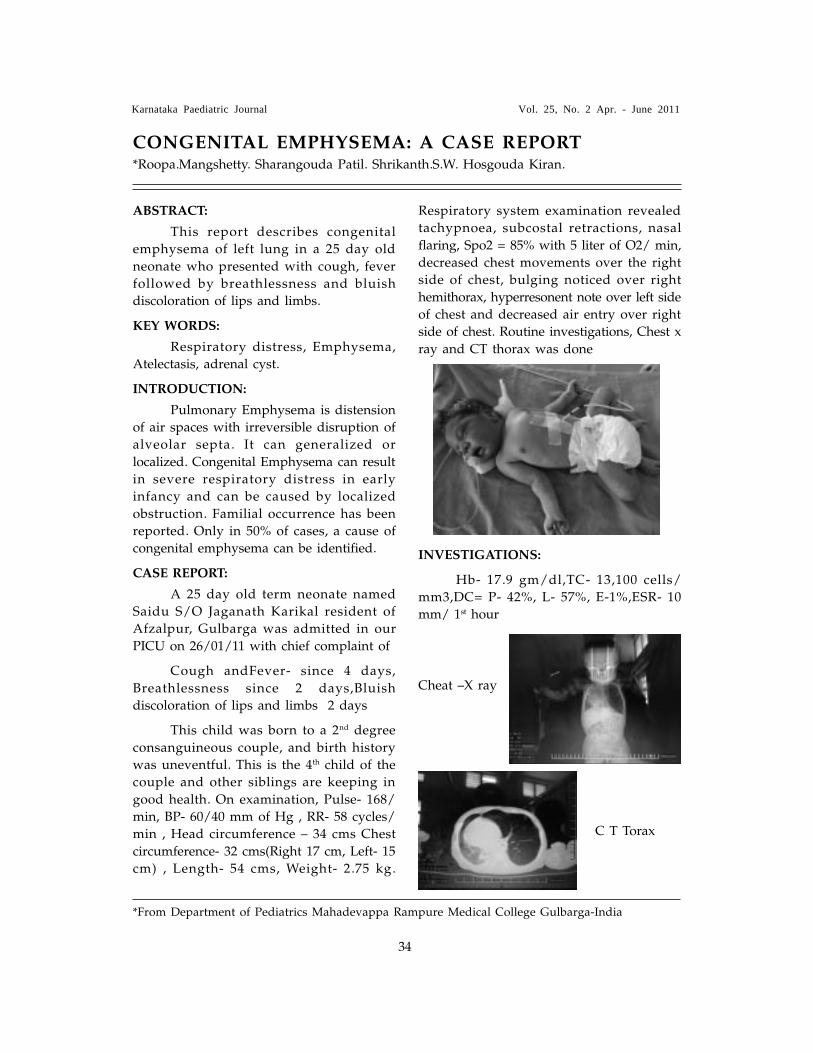
Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
34
ABSTRACT:
This report describes congenital
emphysema of left lung in a 25 day old
neonate who presented with cough, fever
followed by breathlessness and bluish
discoloration of lips and limbs.
KEY WORDS:
Respiratory distress, Emphysema,
Atelectasis, adrenal cyst.
INTRODUCTION:
Pulmonary Emphysema is distension
of air spaces with irreversible disruption of
alveolar septa. It can generalized or
localized. Congenital Emphysema can result
in severe respiratory distress in early
infancy and can be caused by localized
obstruction. Familial occurrence has been
reported. Only in 50% of cases, a cause of
congenital emphysema can be identified.
CASE REPORT:
A 25 day old term neonate named
Saidu S/O Jaganath Karikal resident of
Afzalpur, Gulbarga was admitted in our
PICU on 26/01/11 with chief complaint of
Cough andFever- since 4 days,
Breathlessness since 2 days,Bluish
discoloration of lips and limbs 2 days
This child was born to a 2nd degree
consanguineous couple, and birth history
was uneventful. This is the 4th child of the
couple and other siblings are keeping in
good health. On examination, Pulse- 168/
min, BP- 60/40 mm of Hg , RR- 58 cycles/
min , Head circumference � 34 cms Chest
circumference- 32 cms(Right 17 cm, Left- 15
cm) , Length- 54 cms, Weight- 2.75 kg.
Respiratory system examination revealed
tachypnoea, subcostal retractions, nasal
flaring, Spo2 = 85% with 5 liter of O2/ min,
decreased chest movements over the right
side of chest, bulging noticed over right
hemithorax, hyperresonent note over left side
of chest and decreased air entry over right
side of chest. Routine investigations, Chest x
ray and CT thorax was done
INVESTIGATIONS:
Hb- 17.9 gm/dl,TC- 13,100 cells/
mm3,DC= P- 42%, L- 57%, E-1%,ESR- 10
mm/ 1st hour
CONGENITAL EMPHYSEMA: A CASE REPORT*Roopa.Mangshetty. Sharangouda Patil. Shrikanth.S.W. Hosgouda Kiran.
*From Department of Pediatrics Mahadevappa Rampure Medical College Gulbarga-India
C T Torax
Cheat �X ray

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
35
Patchy areas of atelectasis noted in
posterior basal segment of right lower lobe
with loss of lung volume on right side
Hyperinflated left lung possibly
compensatory emphysema,Shift of
mediastinum and cardia to right, herniation
of left lung to right hemithorax, Wedge
shaped soft tissue attenuation in posterior
basal segment of right lower lobe of lung.
Treatment:
Injection Ceftriazone, Injection
Amikacin, Ambrodyl drops, Normal saline
nebulisation
REFERENCE:
1) Horak E, Bodner J, Gassener I et al:
congenital cystic lung disease:
diagnostic and therapeutic
considerations. Clin pediatr 2003; 42:
251-261
2) Karnak I, Senocak ME, Ciftci AO, et al:
congenital lobar emphysema:
diagnostic and therapeutic
considerations. J pediatr journal 1999;
34: 1347-1351
3) Chao MC, Karamzadeh AM, Ahuja G:
congenital lobar emphysema: an
otolaryngologic perspective. Int j
pediatr otorhinolaryngol 2005; 69: 549-
554
4) Mei- Zahar M, Konen O, Manson D,
Langer JC: is congenital lobar
emphysema a surgical desease? J
Pediatr surg 2006; 41: 1058-1061
5) Cumming JR, Macpherson RI, Chernick
V: unilateral hyperlucent lung
syndrome in children. J pediatr 1971:
78; 250-260.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
36
Abstract : A 3 year old girl with
clinical hypothyroidism with low T4 and
elevated serum TSH levels had
associated nephrotic range of proteinuria
with elevated lipid profile. Treatment
with steroids and thyroxine replacement
showed improvement in clinical and
laboratory parameters.
Keywords : Nephrotic syndrome,
hypothyroidism, proteinuria
Introduction
Nephrotic syndrome in children
is not an uncommon entity. Although
functional hypothyroidism is known to
occur in nephrotic syndrome, clinical
hypothyroidism is an unusual
association1.We report a case of nephrotic
syndrome with clinical hypothyroidism.
A 3 year old girl presented with
history of constipation, poor activity and
global developmental delay. She was a
term baby, appropriate for gestational age
, born of a non consanguineous marriage.
Physical examination findings included
coarse facies, periorbital puffiness, low set
ears, dry skin, umbilical hernia, mild
ascites and hypotonia. Developmental and
mental age was around 11/2 years .
Anthropometric measurements were
within normal limits. Developmental
delay was noticed at 8 months of age,
for which she was evaluated. Thyroid
function tests done then, were normal.
She had documented proteinuria for
Nephrology Section
HYPOTHYROIDISM & NEPHROTIC SYNDROME �A CAUSE OR EFFECT ?*Nithya T, B Sanjeev Rai, Habib Ullah Khan, Aby Dany Varghese
* Department of Pediatrics, Father Muller Medical College,Mangalore
past 4 months. Present investigations
revealed hypoalbuminemia (1.2 g/dl),
hypercholesterolemia (487mg/dl),
hypertriglyceridemia (602mg/dl) and
proteinuria (urine protein creatine ratio-
6.13). Renal function tests were within
normal limits. Serum C3 level was
normal. Abdominal ultrasound revealed
mild ascites with normal kidneys.
Radiological evaluation showed normal
bone age. The blood levels of total T3 and
T4 were 0.92 (0.8-2.0) and 4.58 (5.1- 12.0)
respectively. TSH was mildly elevated-
6.13 ( 0.27-4.2). Antithyroid antibodies were
negative. Urinary T3 and T4 was detectable
(1.09 and 1.17 respectively).
The child was started on oral
steroids (60mg/m2/day) and thyroxine
(10mcg/kg/day). Proteinuria resolved
within a week of treatment and showed
clinical signs of improvement � disappearance
of edema, better activity and apetite.
DiscussionNephrotic syndrome is characterized
by a marked increase in glomerularpermeability and presents with proteinuria,hypoproteinemia, edema and hypercholesterolemia.2

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
37
Proteinuria occurs due to changes incapillary endothelial cells, the glomerularbasement membrane, or podocytes, whichnormally filter serum protein selectivelyby size and charge. This results in urinaryloss of macromolecular proteins, primarilyalbumin but also opsonins, immunoglobulins,erythropoietin, transferrin, hormone-bindingproteins ( including thyroid-binding
globulin and vitamin D-binding protein),
and antithrombin III.
Urinary loss of thyroglobulin, free T4
,T3 with consequent fall in T3, T4, TBG
levels and rise in TSH are documented in
children with untreated nephrotic
syndrome and are reversible with disease
remission or following bilateral
nephrectomy and renal replacement
therapy .3,4 There is a positive correlation
between serum albumin and T4 levels ;
and degree of proteinuria and urinary loss
of T4. This is supported by normalization
of thyroid indices with onset of
remission of nephrotic syndrome .5
In our patient, hypothyroidism was
manifested with low T4 and elevated TSH
levels. Absence of thyroid antibodies and
goiter ruled out autoimmune etiology.
Normal thyroid function tests in the
first year of life and normal bone age
in Xray suggests that congenital
hypothyroidism is unlikely. Loss of
thyroid hormone in urine seems the
likely explanation for rising TSH levels
in this child. Thyroid hormone
replacement has resulted in clinical
improvement, which may be tapered
and stopped after attaining remission of
nephrotic syndrome5.
Glomerular disease giving rise to
protein loss with associated endocrine
dysfunction is not confined to thyroid
and clinicians should be alert to
alterations in other hormone systems-
notably hypothalami-pituitary-adrenal axis
and grwth hormone�IGF-1 system6.
In summary, there is an existence
of hypothyroid state in some infants
with nephrotic syndrome. Also, increasing
thyroxine requirements in a case of
hypothyroidism should be evaluated for
proteinuria. Routine thyroid screening
and early replacement therapy if
required, may be recommended for all
children with nephrotic syndrome.
References
1. Muranjan MN, Kher AS, Nadkarni UB,
Kamat JR. Congenital nephrotic
syndrome with clinical hypothyroidism.
Indian J Pediatr 1995;62:233-5.
2. Trouillier S,Delevaux I, Rance N et al.
Increasing thyroxine requirements in
primary hypothyroidism: don�t forget
the urinalysis! J Postgrad Med
2006;52:201-3.
3. Carpi A, Romano F, Massitelli M,
Ciardella F . Low protein supplemented
diet corrects altered serum thyroid
hormone and TSH concentrations in
patients with chronic nephrotic
syndrome. Thyroidology 1990;2:89-92.
4. Chadha V, Alon US. Bilateral
nephrectomy reverses hypothyroidism
in congenital nephrotic syndrome.
Pediatr Nephrol 1999;13:209-11.
5. Fonseca V, Thomas M, Katrak A, Sweny
P, Moorhead JF. Can urinary thyroid
hormone loss cause hypothyroidism?
Lancet 1991;338:475-6.
Haffner D, Tonshoff B, Blum WF, Vickers M,
Siebler T, Cronin MJ, et al. Insulin-like
growth factors (IGFs) and IGF binding
proteins, serum acid-labile subunit and
growth hormone binding protein in
nephrotic children. Kidney Int 1997;52:802-
10.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
38
Diagnosing renal disorders inchildren is challenging as manifestations ofrenal disease are vague and subtle. Inaddition, many of the signs and symptomsof renal disease mimic or simulate othersystemic diseases. It is therefore crucial forus pediatricians to have a high index ofsuspicion and bear in mind the clinicaldiversity of kidney disease.
The two important categories ofkidney diseases that present withcontrasting clinical scenarios are glomerulardisease and tubular disease. A typicalglomerular disease (nephrotic syndrome,glomerulonephritis, hemolytic uremicsyndrome) will present with any of thefollowing features of edema, oliguria andproteinuria with or without hematuria andhypertension. On the contrary, a tubulardisease (renal tubular acidosis, Barter�ssyndrome, and nephrogenic diabetesinsipidus) manifests as failure to thrive,growth retardation, polyuria and polydipsiawith or without bony deformities. Aglomerular or a tubular disease couldpresent as acute renal failure/ acute kidneyinjury or progress to chronic renal failure /chronic kidney disease.
Hematuria, oliguria and edema areobvious clinical markers of renal disease.However there are several other markers thatneed to be addressed which help us notmiss an underlying renal disease.
The �RED FLAGS�:
Growth: Failure to thrive, growthretardation or short stature are predominantmanifestations of renal diseases like renaltubular acidosis, Barter�s syndrome,nephrogenic diabetes insipidus, vitamin Ddependent and resistant rickets,vesicoureteral reflux, polycystic renal disease
COMMON PITFALLS IN DIAGNOSING RENALPROBLEMS IN CHILDREN*Dr Arpana Iyengar
*Associate Professor, Division of Pediatric Nephrology,Department of Pediatrics, St John�s MedicalCollege Hospital, Bangalore 560034
and renal dysplasia. In these conditionsthere is suboptimal growth inspite of normalrenal function. Growth retardation is ahallmark of chronic renal failure, childrenwith an underlying tubular disorder beingthe worst affected. Correcting metabolicacidosis helps to optimize growth.
Anemia: This could be the only clueto a probable diagnosis of a renal disease.Anemia is predominant in connective tissuediseases, hemolytic uremic syndrome, acuterenal failure secondary to malaria andintravascular hemolysis.Chronic anemia thatis refractory to iron therapy should beconsidered as a clue to exclude chronic renal failure.
Bony deformities: Every bonydeformity in childhood is not rickets.However, various forms of rickets in manychildren get missed and they end upreceiving orthopedic treatment. It is thereforeabsolutely necessary for us to screen for non-nutritional rickets in children withsignificant bony abnormalities. Rickets isalways diagnosed based on biochemicaland radiological findings. Growthretardation, polyuria and polydipsiaassociated with rickets could indicate renaltubular acidosis or Barter�s syndrome.Rickets predominantly affecting the lowerlimbs associated with teeth abnormalitiesand short stature without polyuria orpolydipsia is characteristic of hypophosphatemicrickets. Renal osteodystrophy presents asbony deformities and is a hallmark ofchronic renal failure.
Hypertension: Childhood hypertensioncan be asymptomatic. It is most often anincidental finding picked up during routineexamination. This emphasizes the need for aregular annual blood pressure recording inall children above 3 years of age. Childrenat risk for hypertension are growth restricted

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
39
neonates, obese children, children withrecurrent urinary tract infections, onmedications like steroids, cyclosporine andfollowing a glomerular disease.Hypertensive crisis could mimic meningitisor encephalitis with vomiting, headache,altered sensorium and seizures. A finding ofasymmetry in the size of the kidneys willgive a clue to diagnosis of renal arterystenosis. Reflux nephropathy with renalscars could present with hypertension.Hypertension is a manifestation of bothacute and chronic renal failure.
Urine:
Analysis : In day to day practice,transient urinary abnormalities accompanysystemic diseases. Febrile illness can presentwith transient proteinuria and microscopichematuria. It is important to closely followup these findings even after the childrecovers from the acute illness. Persistentproteinuria or microscopic hematuria formore than 2-3months needs a detailedevaluation inspite of the child beingasymptomatic to rule out Ig A nephropathy,Alport�s disease and connective tissuedisease. Microscopic hematuria may be apresentation of stone disease orhypercalciuria. The presence of white bloodcells in urine could indicate urinaryinfection or intake of nephrotoxic drugs likeNSAIDs causing interstitial nephritis.
Stream: Enquiring about the urinarystream, the flow, interruption or intermittentstream is important and could be the onlycomplaint in children with an underlyingposterior urethral valve or neurogenicbladder. Every enuresis is not night timebed wetting. Associated features of voidingdysfunction need to be excluded in childrenwith bedwetting.
Output: Decreased urine output isa straightforward symptom of renal disease.However it may be a symptom even whenrenal functions are normal like in childrenwith nephrotic syndrome and glomerulonephritis.In situations of acute renal failure, one canhave decreased, increased or even normalurine output. Therefore urine output is nota reliable indicator of renal functions. A
normal urine output in the presence ofmoderate to severe dehydration should raisethe possibility of an underlying tubulardisorder. Polyuria is most often missed as asignificant cause for failure to thrive andrecurrent dehydration.
Systemic manifestations: Prolongedfever, joint pains, skin rashes associatedwith hematuria, proteinuria or hypertensionusually indicates connective tissue diseaseor Henoch Scholein purpura nephritis. Feverwithout respiratory symptoms in infantsshould prompt us to rule out urinary infection.
Recurrent symptoms: Recurrence ofsymptoms is the key to diagnosing manyrenal disorders. Recurrent fever could be amanifestation of urinary tract infection,recurrent vomiting could due to metabolicacidosis in renal tubular acidosis, recurrentepisode of dehydration indicates polyurictubular diseases, recurrent hematuria goesagainst a post streptococcal glomerulonephritisand recurrent edema is characteristic ofnephrotic syndrome.
Syndromic associations: Markers for akidney disease at birth would includeabnormal antenatal renal scans (oligohydramnios,polyhydramnios, hydronephrosis, spinaldefects), a single umbilical artery, ear lobedefects, spinal and genital deformities, apalpable bladder or renal mass, hypoplasticlungs and Potter�s facies
Positive family history: A positivefamily history for renal disease could guideus make a diagnosis in children withvescicoureteral reflux, genetic forms ofnephrotic syndrome, tubular disorders andAlport�s syndrome.
Therapeutic indicators: Drugs needdose corrections in the presence of renalfailure and prolonged use of diuretics oraminoglycosides need regular monitoring ofserum creatinine. Children with nephroticsyndrome in shock should be treated withalbumin if they are unresponsive to fluid bolus.
To conclude detecting renal diseasesin children is a challenge which we canovercome with a detailed history taking andmeticulous examination. It would beessential to bear in mind the commonpitfalls in order to avoid missing adiagnosis of renal disease.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
40
Polyuria is defined as urine output
more than 4ml/kg/hour. Polyuric child
voids large amount of urine during day and
night as well. Polyuria is usually associated
with polydipsia which refers to excessive
thirst and intake of large volumes of liquid
to quench the thirst.
Accurate measurement of 24 hours
intake of fluids and urine output should be
done to establish the diagnosis of polyuria.
It is important to distinguish polyuria from
frequency of micturition where small volume
of urine passed frequently though the 24
hours urine output is within normal limits .
Polyuria may result from excessive
fluid intake (psychogenic polydipsia ),
increased osmotic load(solute diuresis),
failure to produce ADH(central diabetes
insipidus)or unresponsiveness to the action
of ADH in kidney(nephrogenic diabetes
insipidus). The final osmolality of the urine
is solely dependent on water permeability of
collecting ducts which is controlled by ADH.
Causes of polyuria in children
1.Solute diuresis(increased osmotic load)
Diabetes mellitus(due to glycosuria)
Hypercalcemia(due to hypercalciuria)
Bartter syndrome(due to sodium chloride)
Renal tubular acidosis(due to sodium
bicarbonate)
2. Failure of ADH production(central
diabetes insipidus-hypothalamic /pituitary
disorders)
Pituitary tumour
Post hypophysectomy
Infections-meningitis, encephalitis, tuberculosis
Basal fracture
Vascular aneurysm or thrombosis
EVALUATION OF A CHILD WITH POLYURIA*Dr. Nagamani Agarwal
* Associate Professor of Pediatrics, JJMMC, Davangere
3. Failure of renal response to ADH
Nephrogenic diabetes insipidus(x linked
recessive)
Acquired unresponsiveness to ADH
Obstructive uropathy
Chronic pyelonephritis
Reflux nephropathy
Hypokalemia
Hypercalcemia/nephrocalcinosis
Sickle cell disease
Chronic renal failure due to interstitial
nephritis
Drug induced-ampotericin,lithium,
tetracycline
4. Psychogenic or Primary polydipsia
Children with polyuria due to
pathological conditions may present with
failure to thrive, polydipsia, nocturnal
enuresis,febrile episodes due to dehydration
and convulsion. Failure to thrive is because
the constant increased intake of fluids limits
their appetite. Repeated episodes of
dehydration may result in mental retardation.
A thorough history and physical
examination may provide clues to the cause
of polyuria. Frequency of voiding and drinking
particularly at night is useful information
to assess the severity of the probem.
Physical examination should include
assessment of growth,anemia, hypertension,
bony deformities, renal or bladder mass,
evidence of dehydration and ophthalmic
evaluation.
Investigations and practical approach
First step is to confirm the presence of
polyuria by quantifying the 24 hours urine

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
41
output. Decide whether polyuria is
associated with polydipsia and failure tothrive Estimate blood sugar to excludediabetes mellitus and serum creatinine forrenal failure .Rule out hypokalemia andhypercalcemia .Arterial blood gas analysisis essential when renal tubular acidosis orBartter�s syndrome are suspected .Urineshould be tested for pH,specific gravity,sugar, protein and pyuria. Ultrasonographyof KUB region with MCU(micturitingcystourethrogram) or IVP(intravenouspyelography) to diagnose refluxnephropathy and obstructive uropathy .CTscan and MRI of brain may be needed whencentral diabetes insipidus is suspected.Urineand plasma osmolality should be assessedto decide if water diuresis is the cause ofpolyuria. Isoosmolar /hyperosmolar urine isfound in children with solute diuresis ornormal children. Ratio of Urine andplasma osmolality is more than 1.5. Inchildren with water diuresis (diabetesisipidus and psychogenic polydipsia) urineis hypoosmolar and ratio is less than 1.
Water deprivation test should bedone only in selected patients suspected tohave water diuresis. It helps to differentiatediabetes insipidus from psychogenicpolydipsia. The aim of water deprivation
test is to induce mild dehydration and thuschallenge the kidneys to preserve water. Ifchild presents with hypernatremia, this testis not necessary. The test carries the risk ofsevere hypernatremic dehydration especiallyin infants.
Water is withheld for 8 to 12 hours.Child is frequently weighed and closelyobserved. The test should be stopped ifweight loss exceeds 5%. The period of waterdeprivation should not exceed 4 hours ininfants and 8 hours in children. Urinespecific gravity and osmolality ,plasmaosmolality and serum sodium are testedevery 3 to 4 hours and at the end of the test. Following water deprivation normal childcan achieve a urine osmolality of over 900mosm/kg and ratio of urine to plasmaosmolality exceeds 1.5. Children withdiabetes insipidus(central or nephrogenic)fail to show a rise of urine osmolality whichremains below 300 mosm/kg water andratio is less than 1.Vasopressin test shouldbe done to differentiate central from nephrogenicdiabetes insipidus. Children with primarypolydipsia concentrate urine to varyingdegrees (>500mosm/kg) as prolongedpolyuria affects concentrating ability ofkidney due to washout of the medullary
counter-current concentration mechanism.Approach to a child with polyuria.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
42
Twenty two years old pregnant
woman was referred to pediatric OPD with
history of antenatal left renal
hydronephrosis detected at 30 weeks of
gestation, for advice regarding continuing
her pregnancy. We looked at the following
parameters. Fetal growth and liquor volume
was adequate. Hydronephrosis was
unilateral and there were no other major
congenital malformations. Hence she was
advised to continue her pregnancy and to
review soon after the delivery.
She delivered a normal weight male
baby at term. There was no abdominal
mass or palpable bladder. Urine stream was
good and the baby was feeding well. Baby
was started on antibiotic prophylaxis with
cephalexin and post natal ultra sound was
done on day 5, which showed left sided
moderate hydronephrosis. At 6 weeks of
age diuretic renogram showed left sided
pelviureteric junction obstruction with
differential renal function of 30% in left
kidney. Subsequently, micturating cystourethrogram
(MCUG) was done after administering
gentamycin 3mg/kg ½ hour before the
procedure, which revealed right sided grade
2 vesico-ureteric reflux (VUR).
Left PUJ obstruction with decreased
differential function was managed with
pyeloplasty and right grade 2 VUR was
managed conservatively with antibiotic
prophylaxis. The infant was advised close
APPROACH TO ANTENATALLY DETECTED HYDRONEPHROSIS*Dr. R. Premalatha
* Professor of Paediatrics, Paediatric Nephrologist .Bangalore Medical College & Research Institute(BMCRI) K.R. Road, Bangalore �560002
follow up to detect and treat urinary tract
infection.
This case illustrates that antenatally
detected hydronephrosis requires close
follow up, imaging and surgical or
conservative management. In fetus,
urological abnormalities are the most
common, with the incidence being 1 in 100.
50% of all abnormalities detected by prenatal
ultrasound are due to hydronephrosis.
Significant Hydronephrosis:
On antenatal scan pelvic diameter
>10mm at 24-26 weeks gestation, ratio of
anterio-posterior (AP) pelvic diameter to AP
renal diameter greater than 0.5 and
caliectasis indicate significant hydronephrosis.
Major causes:
}
Physiologic or non-obstructive dilatation of
upper urinary tract refers to mild dilatation,
where no cause is found and is transient.
}
Pelviureteric junction obstruction.
}
Vesico-ureteric reflux.
}
Posterior urethral valves.
}
Ureterocoele
}
Vesico ureteric junction obstruction.
}
Multi cystic dysplastic kidneys.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
43
Prenatal management:
Obstruction to urine flow occurring
before 24 weeks of gestation causes
dysplastic changes identifiable by an
increase in echogenecity of kidneys or
cortical cysts. If the obstruction is bilateral
and severe, amniotic fluid volume is
reduced and pulmonary hypoplasia occurs.
This may produce respiratory distress in the
new born and need for ventilation. Hence,
in presence of oligohydramnios, fetal growth
restriction and other major congenital
anomalies detected before 24 weeks,
pregnancy may be terminated. After 32
weeks of gestation, fetus is delivered
prematurely in a tertiary hospital, if lung
maturation is adequate and facilities are
available for management and obstruction is
managed postnatally. Fetal surgeries and
management of cases between 24 to 32
weeks is controversial. Bilateral hydronephrosis
without oligohydramnios and unilateral
hydronephrosis do not need intervention
Post natal management:
Neonates are assessed for abdominal
mass due to hydronephrosis or palpable
bladder. Urine stream is observed and urine
examined to detect urine infection. They are
started on prophylactic antibiotic like
cephlexin 10mg/kg, single daily dose for
first 3 months and thereafter septran 3mg/
kg or nitrofurantoin 1mg/kg is continued for
1year, to prevent urine infection. If imaging
detects obstruction, prophylactic antibiotics
are stopped, to prevent emergence of
resistant organisms.
First ultrasound is done 5 days after
birth, as physiologic oliguria during first
few days of life causes renal pelvis to
shrink transiently. If first ultrasound is
normal it is repeated after 3 months. If
hydronephrosis is detected on post natal
ultrasound, at 6 weeks of age MCU and
diuretic renogram are done. Diuretic
renogram done at 6 weeks is more accurate,
due to increased maturity of nephrons and
increased GFR. MCU is done to detect
vesico-ureteric reflux & posterior urethral
valves. Diuretic renogram is done to detect
pelvi-ureteric junction obstruction.
If hydronephrosis is severe or bilateral
or present in a solitary kidney or bladder is
palpable urgent evaluation and management
is required. As complete obstruction has a
potential to cause renal damage, urethral
catheter should be passed immediately to
drain the bladder and urgent U/S and
MCU are done. If first ultra sound is
normal, it is repeated after 5 days. After
release of obstruction, there will be a
diuretic phase requiring close monitoring
and replacement of fluid and electrolytes.
Uro-sepsis has to be detected early and
treated adequately. In bilateral cases renal
function and electrolytes may be abnormal.
Note that neonates take 1 week to
stabilize their s.creatinine to normal level of
0.4mg/dl from higher s.creatinine level at
birth which reflects maternal level.
Early detection of cases requiring
surgery and prompt referral to surgeons is
important. All children, whether managed
conservatively or surgically, require close
monitoring for UTI, hypertension, protinuria
and renal growth & function.
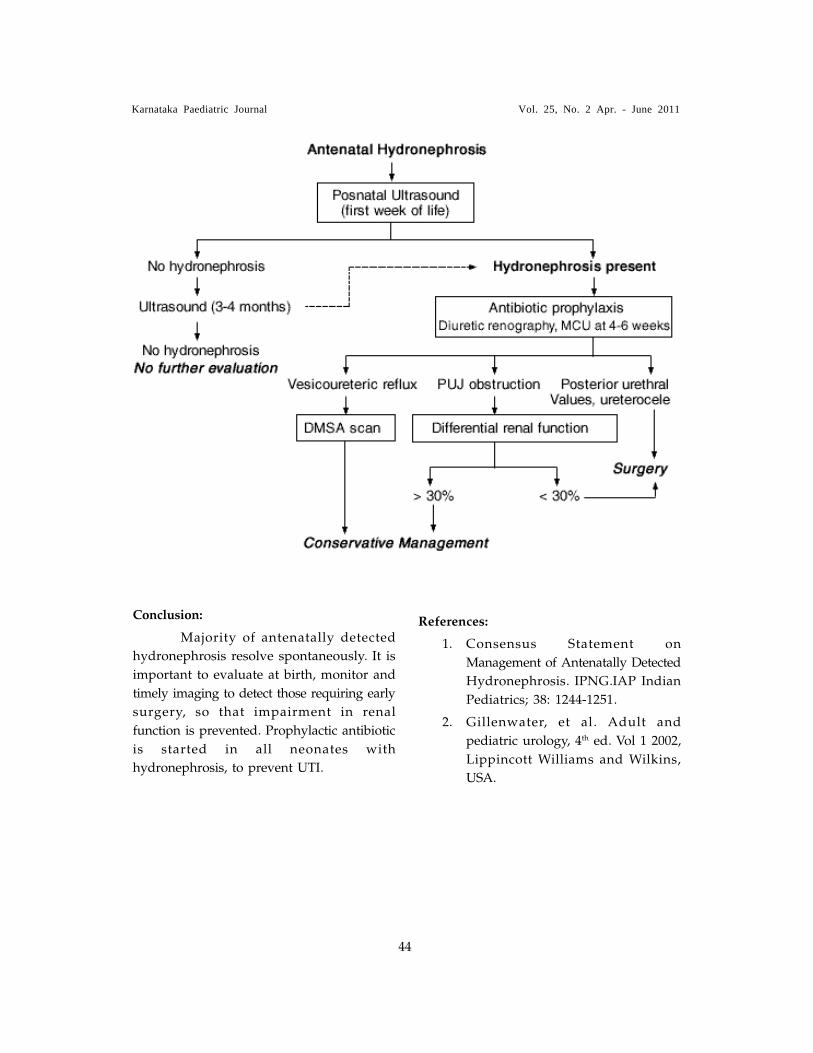
Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
44
Conclusion:
Majority of antenatally detected
hydronephrosis resolve spontaneously. It is
important to evaluate at birth, monitor and
timely imaging to detect those requiring early
surgery, so that impairment in renal
function is prevented. Prophylactic antibiotic
is started in all neonates with
hydronephrosis, to prevent UTI.
References:
1. Consensus Statement on
Management of Antenatally Detected
Hydronephrosis. IPNG.IAP Indian
Pediatrics; 38: 1244-1251.
2. Gillenwater, et al. Adult and
pediatric urology, 4th ed. Vol 1 2002,
Lippincott Williams and Wilkins,
USA.

Karnataka Paediatric Journal Vol. 25, No. 2 Apr. - June 2011
45
The Antimicrobial drugs therapy we think, say or do
Edited & Published by Dr.B Sanjeev Rai, Medicare center, Karangalpady, Mangalore 575003.
and Printed at Shrathi Printers & Publishers.Pvt. Ltd.Baikampady. Mangalore 575001
Editorial Office : Medicare Center, Karangalpady, Mangalore 575003,
email: [email protected]
Reg.no.RN/68641/23/AL/TC/92 ISSN 09755152
Antimicrobial Resistance:
No action today, No cure tomorrow
4 Way Test
Is It Economical
and beneficial
to the patient
Is the right Dose & Duration
Is the Best & First line
drug
Is It Essential