int131: a selective modulator of pparγ
TRANSCRIPT

doi:10.1016/j.jmb.2009.01.025 J. Mol. Biol. (2009) 386, 1301–1311
Available online at www.sciencedirect.com
INT131: A Selective Modulator of PPARγ
Alykhan Motani†, Zhulun Wang†, Jennifer Weiszmann†,Lawrence R. McGee, Gary Lee, Qingxiang Liu, Jocelyn Staunton,Zexu Fang, Helen Fuentes, Michelle Lindstrom, Jinsong Liu,Donna H. T. Biermann, Juan Jaen, Nigel P. C. Walker,R. Marc Learned, Jin-Long Chen and Yang Li⁎
Amgen, Inc., 1120 VeteransBoulevard, South San Francisco,CA 94080, USA
Received 29 September 2008;received in revised form15 January 2009;accepted 16 January 2009Available online22 January 2009
*Corresponding author. E-mail addr† A.M., Z.W., and J.W. contributedPresent addresses: L.J., Guangzho
China; J.J., ChemoCentryx, Inc., 850Abbreviations used: PPARγ, pero
ligand-binding domain; Gal4-UAS,time-resolved fluorescence; qPCR, q
0022-2836/$ - see front matter © 2009 E
The nuclear hormone receptor peroxisome proliferator-activated receptor γ(PPARγ; NR1C3) plays a central role in adipogenesis and is the moleculartarget of the thiazolidinedione class of antidiabetic drugs. To overcome thewell-known shortcomings of thiazolidinediones, we have identified INT131(formerly T131 and AMG131) as a potent selective ligand for PPARγ that isstructurally and pharmacologically distinct from glitazone agonists. In vitrobiochemical and cell-based functional assays showed that INT131 mediatesa distinct pattern of coregulator recruitment to PPARγ. In adipocytes,INT131 showed minimal stimulation of adipocyte differentiation andpartially activated PPARγ target genes involved in adipogenesis and, atthe same time, showedmore agonistic activity on another set of target genesthat may influence insulin sensitivity directly. These unique properties ofINT131 may provide a mechanistic basis for its distinct pharmacologicalprofile. In vivo, increases in glucose tolerance were observed in Zucker (fa/fa)rats following a 14-day oral treatment with INT131. Although the maximalefficacies of INT131 and rosiglitazone were similar with respect toimprovements in glucose tolerance, INT131 had less effect on heart andlungweights, weight gain, hemodilution, and plasma volume. Thus, INT131appears to selectively modulate PPARγ responses in an in vivo preclinicalmodel, showing antidiabetic efficacy while exhibiting an improved hemo-dynamic and cardiovascular adverse effect profile compared to the fullagonist rosiglitazone. X-ray crystallography revealed that INT131 interactswith PPARγ through a distinct binding mode, forming primarily hydro-phobic contacts with the ligand-binding pocket without direct hydrogen-bonding interactions to key residues in helix 12 that are characteristic of fullagonists. Mutagenesis studies on Tyr473 in helix 12 demonstrated thisresidue as essential for rosiglitazone-induced receptor activation, but non-essential for INT131 function in vitro, providing one possible moleculardeterminant for INT131's distinct pharmacology. INT131 is currently beingevaluated in a clinical setting as a therapeutic agent for the treatment of type2 diabetes.
© 2009 Elsevier Ltd. All rights reserved.
Edited by K. Morikawa
Keywords: peroxisome proliferator-activated receptor γ; T131; INT131; TZD;diabetesess: [email protected] to this work.
u Institute of Biomedicine and Health, Guangzhou Science Park, Guangzhou 510663,Maude Avenue, Mountain View, CA 94043, USA.xisome proliferator-activated receptor γ; TZD, thiazolidinedione; LBD,Gal4 upstream-activating sequence; LXR, liver X receptor; HTRF, homogeneousuantitative PCR.
lsevier Ltd. All rights reserved.

1302 PPARγ Modulator
Introduction
Peroxisome proliferator-activated receptor γ(PPARγ; NR1C3) is amember of the nuclear hormonereceptor superfamily of ligand-activated transcrip-tion factors.1,2 Together with peroxisome proliferator-activated receptor α (NR1C1) and peroxisome pro-liferator-activated receptor δ (NR1C2), they form asubfamily of “lipid-sensing” receptors that regulateimportant aspects of lipid and glucose homeostasis.The important roles of PPARγ in the functions ofadipocytes, liver, muscle, and macrophages have adirect impact on type 2 diabetes, dyslipidemia,atherosclerosis, and cardiovascular diseases.3,4Two drugs from the thiazolidinedione (TZD) class
of compounds—rosiglitazone (Avandia™; GlaxoS-mithKline) and pioglitazone (Actos™; Takeda Phar-maceuticals)—have been prescribed either asmonotherapy or in combination with sulfonylurea,metformin, or insulin to control hyperglycemia indiabetic patients through the activation of PPARγ.5
Although TZDs lower glucose and improve lipid andadipokine profiles in type 2 diabetics, they have alsobeen associated with edema, cardiomegaly, anemia,and hemodilution.6 Edema is one of the most signifi-cant dose-limiting undesirable effects for PPARγagonists and has been suggested to perhaps negatethe beneficial effects of TZDs on the promotion ofcardiovascular health, leading to increased incidenceof congestive heart failure particularly in patientswith preexisting cardiovascular disease.7–12 OtherPPARγ full agonists with unrelated chemical struc-tures show similar profiles.13 Hence, it has been pro-posed that new drugs thatminimize edema and otherrelated side effects may reduce cardiovascular risksassociated with the treatment.10
There appears to be a dichotomy for PPARγ func-tion in relation to insulin sensitivity, since accumu-lating genetic and pharmacological evidences alsosuggest that reduction in PPARγ activity and/orselectivemodulationof receptor activitymay improveinsulin sensitivity. For example, a common human
Fig. 1. Chemical structure and receptor affinity of INT131.and rosiglitazone for PPARγ in filtration binding assay.
PPARγ polymorphism, Pro12Ala (which causespartial loss of PPARγ function), and heterozygosityin rodents have both been associated with improve-ments in insulin sensitivity and control of bodyweight gain.14,15 Several compounds (e.g., T2384,SR202, FMOC, nTZDpa, GW0072, and FK614) haveunique modulator activities on the receptor and havebeen shown to improve insulin resistance in rodentmodels without certain undesired adverse effects.10,16
Although the underlying mechanism for these obser-vations is not clear, it may involve the production ofsmaller, more insulin-sensitive fat cells;modulation ofadipokine secretion; and/or increases in fatty acidoxidation in muscle—factors that, in combination,ultimately resulted in the improvement in plasmalipid parameters and insulin sensitivity.17 Theseobservations, in addition to the analysis of otherPPARγ polymorphisms, have led to the suggestionthat the undesirable side effects associated with TZDtreatment could potentially be decoupled from theirinsulin-sensitizing effects.14,18 Presumably, a PPARγligand that retains insulin-sensitizing effects and exhi-bits improvement in fluid retention may result inimproved cardiovascular safety outcome.Here we describe a novel, potent, and selective
PPARγ ligand, INT131, which may represent such amolecule. Using a variety of biochemical and cell-based assays and X-ray crystallographic studies, wedemonstrate that INT131 is a selective PPARγmodulator. In a rodent model of diabetes, INT131effectively improved glucose tolerance withoutinducing cardiomegaly, hemodilution, and plasmavolume expansion.
Results
INT131 is a potent and highly selective PPARγpartial agonist
In a search for novel PPARγ ligands with activitiesdistinct from those of TZDs, a chemical compound
(a) Chemical structure of INT131. (b) Affinities of INT131

1303PPARγ Modulator
library was screened for molecules that can bind tothe PPARγ ligand-binding domain (LBD), and novelligands with partial or antagonistic activity onreceptor function were then followed up. INT131was derived from subsequent chemistry effort on alead chemical series from this screening effort and isa novel PPARγ ligand that binds to the receptorwith high affinity,19 capable of displacing [3H]rosiglitazone with a Ki of 10 nM (Fig. 1). INT131shows a high selectivity for PPARγ against othernuclear receptors. In cell-based reporter gene assayswhere HEK293 cells were transiently transfectedwith a Gal4 upstream-activating sequence (Gal4-UAS) reporter and expression constructs encodingthe LBDs of various receptors fused to the Gal4-DNA binding domain, no activation of peroxisomeproliferator-activated receptor α, peroxisome pro-liferator-activated receptor δ, farnesoid X receptor,liver X receptor (LXR), pregnane X receptor, orretinoid X receptor α at concentrations below 10 μMwas observed (data not shown).INT131 can partially activate PPARγ LBD function
in biochemical and cell-based assays. Using a homo-geneous time-resolved fluorescence (HTRF) assay tomonitor the interaction of the receptor LBD withshort peptides (∼20 amino acids) derived fromtranscription coregulatory proteins, INT131 partiallyrecruited a peptide (with the sequence GNTKNH-PMLMNLLKDNPADQF) derived from the DRIP205coactivator protein. In a dose-dependent fashion,INT131 promoted the binding of the DRIP205 pep-tide to a maximal level of approximately 30%relative to that conferred by rosiglitazone (Fig. 2a),with an EC50 value of 4 nM. INT131 also effectivelyantagonized the effects of rosiglitazone in a dose-dependent manner (Fig. 2a).
Fig. 2. INT131 can partially activate PPARγ LBD functionrosiglitazone and INT131 in the presence and in the absence oftransferase PPARγ LBD and peptide derived from coactivatoINT131 in the presence and in the absence of 0.3 μM rosiglitaztransiently transfected with an expression construct that condomain, together with a luciferase reporter gene under the tra
The effect of INT131 on the transcriptional activityof PPARγ in a cell-based reporter gene assay wasalso examined. HEK293 cells were transientlytransfected with an expression construct containingthe PPARγ LBD fused to the Gal4-DNA bindingdomain, together with a luciferase reporter geneunder the transcriptional control of the Gal4-UAS.As shown in Fig. 2b, rosiglitazone activated thetranscription of the reporter gene by approximately12-fold, whereas activation of receptor-mediatedgene expression by INT131 in this context was only3-fold. In addition, INT131 is a potent inhibitor ofPPARγ transactivation in the presence of rosiglita-zone (Fig. 2b).
INT131 does not have a direct contact withhelix 12
To facilitate the understanding of the unique phar-macological profile of INT131, we determined thethree-dimensional cocrystal structure of INT131withPPARγLBD in the presence of a peptide derived fromSRC-1 coactivator protein using X-ray crystallogra-phy up to a resolution of 2.4 Å. Resembling otherPPARγ ligands, INT131 binds in the ligand-bindingpocket of the PPARγ LBD (Fig. 3a) behind helix 3 andwraps around C285 from helix 3 in a U-shaped con-formation. The overall structure of the PPARγ LBDshowed no significant differences from other pre-viously reported PPARγ LBD structures.20–23
The binding of INT131 partially overlaps with thatof TZDs (Fig. 3a and b), with the central 2,6-dichlorophenyl ring (B-ring) occupying the narrowchannel between helices 3 and 7, similar to the posi-tioning of the central benzene ring of rosiglitazone.23
In addition, the quinoline ring of INT131 and the
in biochemical and cell-based assays. (a) Dose response of0.3 μM rosiglitazone in an HTRF assay with glutathione S-r DRIP205 protein. (b) Dose response of rosiglitazone andone in a cell-based reporter gene assay. HEK293 cells weretained the PPARγ LBD fused to the Gal4-DNA bindingnscriptional control of the Gal4-UAS.

Fig. 3. Crystal structures of INT131-bound PPARγ. (a and b) The overall cocrystal structure of INT131 (in cyan) in thePPARγ LBD domain (in gray/red ribbon) in the presence of SRC-1 coactivator peptide (in yellow ribbon). Rosiglitazone(in pink) is superimposed on the structure. (c) The ligand-binding pocket of PPARγ in the presence of INT131. Importantresidues, as well as two water molecules involved in the ligand–protein interactions, are indicated. The molecular surfacewas prepared without helix 3. (d) Dose response of rosiglitazone and INT131 on either wild-type or Y473A mutantreceptors in a cell-based reporter gene assay. HEK293 cells were transiently transfected with an expression construct thatcontained either the wild-type or the mutant Y473A PPARγ LBD fused to the Gal4-DNA binding domain, together with aluciferase reporter gene under the transcriptional control of the Gal4-UAS.
1304 PPARγ Modulator
TZD pyridyl ring are both positioned in the ligandentry area between helix 3 and the adjacent β-sheet.However, the sulfonamide linker and the 2,4-dichlo-rophenyl ring (A-ring) of INT131 occupy a distinctspace in the ligand-binding pocket compared withthe TZDhead group. Instead of pointing into the areaclusteredwith residues Y473 (AF2 helix), H449 (helix
11), and H323 (helix 5), the 2,4-dichlorophenyl group(A-ring) of INT131 resides between helices 7 and 3(Fig. 3b). This volume is not accessed by the TZDs;instead, F363 from helix 7 occupies this area in theTZD PPARγ structure, as well as in the PPARγ apostructure. In the presence of INT131, F363 adopts adifferent side-chain rotamer conformation and opens

1305PPARγ Modulator
up the space between helices 7 and 3 to accommodatethe A-ring in INT131. Furthermore, F363 forms aπ–π stacking interaction with the A-ring of INT131(Fig. 3c).Interestingly, two water molecules were also
detected in the cocrystal immediately adjacent tothe INT131 head group (Fig. 3c), one of which islocated near the N-atom in the INT131 sulfonamidegroup. These two water molecules participate in ahydrogen-bond network that involves residuesY473, H323, H449, and Y327 and the sulfonamidegroup of INT131. The sulfonamide nitrogen atommakes a direct hydrogen bond with the hydroxylmoiety of Y327, with a distance of 2.7 Å. Y327 makesa hydrogen bond with one water molecule (2.5 Å),which is in hydrogen bond with the second watermolecule (2.6 Å). The secondwater molecule serves ahydrogen-bonding network center that forms hydro-gen bonds with Y473 (2.5 Å), H449 (2.9 Å), and H323(3.2 Å). In addition, one of the oxygen atoms of thesulfonamide group picks up a weak hydrogen bond(3.5 Å) with K367, which is also in hydrogen bondwithH449 (3.1 Å). It is generally believed that PPARγagonists activate the receptor through direct inter-actions with AF2 helix.23 This interaction places theAF2 helix in a conformation that facilitates therecruitment of coactivator proteins and the activa-tion of the transcriptional machinery. In contrast,INT131 makes no direct interaction with the AF2helix, rather the high-affinity interactions betweenINT131 and receptor result from the numeroushydrophobic van der Waals contacts and water-mediated interactions in the large ligand-bindingpocket of PPARγ LBD. This lack of direct specifichydrogen-bonding interactions between INT131 andthe AF2 helix differentiates INT131 from typicalagonists such as TZDs.23 We believe that the indirect
Fig. 4. The effects of INT131 on PPARγ activity in HTRdifferences between the binding of INT131 and the bindingdescribed previously,25 consist of 20 amino acids derived from
water-mediated interactions between INT131 andthe AF2 helix through Y473 only partially stabilizethe AF2 helix in its active conformation, possibleexplaining the partial agonistic properties observedfor INT131.To further differentiate INT131 and rosiglitazone
interactions with the receptor and to directly test theimportance of Y473 in helix 12 in INT131 androsiglitazone-induced receptor activation, we cons-tructed amutant PPARγ LBDwhere Y473 is replacedwith alanine. An expression construct containingeither the wild-type PPARγ LBD or the mutantY473A PPARγ LBD fused to the Gal4-DNA bindingdomain was cotransfected with a luciferase reportergene into HEK293 cells, as in Fig. 2b. As shown inFig. 3d, while rosiglitazone activated the transcrip-tion of the reporter gene on wild-type receptor asbefore, the Y473A mutation completely abolishedthe activation by rosiglitazone. This observation issimilar to a previous report, which also suggestedthat the affinity of rosiglitazone for the mutantreceptor has been greatly reduced.24 In contrast,Y473A mutation had no significant effect on neitherthe efficacy nor the potency of INT131-inducedreceptor activation (Fig. 3d). These results confirmedthe differences observed in the cocrystal structureshown in Fig. 3a–c and suggest that this may indeeddirectly contribute to the functional differencesobserved between INT131 and rosiglitazone.
INT131 is a context-dependent PPARγmodulator
To further characterize and understand the uniquepharmacology of INT131, we examined its effectswith a panel of biochemical and cellular assays.PPARγ has been demonstrated to form protein–
F assay. Peptide fingerprinting in HTRF assay revealsof rosiglitazone to the receptor. The peptides used, asvarious coactivator and corepressor proteins.

1306 PPARγ Modulator
protein interactions with a variety of transcriptionalcoactivator and corepressor proteins to regulatetarget gene expression. To monitor the uniquereceptor conformation induced upon INT131 bind-ing and the consequences of the recruitment ofvarious transcriptional regulatory proteins, we exa-mined the interactions between PPARγ LBD andpeptides derived from a number of different coacti-vator or corepressor proteins in the HTRF assay, asdescribed previously.25 As shown in Fig. 4, distinctpatterns of coregulator recruitment to PPARγ wereobserved in response to INT131 and rosiglitazone.First, several peptides, including TIF1, RIP140-2,RIP140-4, and RIP140-8, exhibited a similar patternof interaction with the receptor upon binding ofthese two ligands. A second group of peptidesexhibited partial recruitment by INT131 compared torosiglitazone, including DRIP205 and SRC2-3 (asindicated by arrows in Fig. 4). A third group ofpeptides, including SRC3-3-derived, CBP-derived,and p300-derived peptides, was recruited to thereceptor upon rosiglitazone binding, but not uponINT131 binding. Finally, the peptides derived fromNCoR and SMRT corepressors were displaced byrosiglitazone—but only partially (NCoR)—or notdisplaced (SMRT) by INT131 at the concentrationstested. These results suggest that INT131 can inducea distinct receptor conformation upon binding thatcould result in a unique pattern of coactivator andcorepressor protein recruitment.
INT131 weakly promoted in vitro differentiationof mouse preadipocytes and differentiallyinduced PPARγ target genes
The effects of INT131 on adipocyte differentiationin vitro were examined (Fig. 5). PPARγ agonists areknown to promote the conversion of a variety ofpreadipocyte cell lines and primary cells into matureadipocytes. Incubation of 3T3L1 cells, a mouse pre-adipocyte cell line, with rosiglitazone resulted in effi-cient conversion into adipocytes, as indicated byincreased lipid accumulation (Fig. 5a) and inductionof a panel of adipogenic genes and other PPARγtarget genes (Fig. 5b and c). In contrast, when 3T3L1cells were treated with INT131, very little lipid accu-mulation and only modest induction of adipogenicgenes and a group of PPARγ target genes wereobserved (Fig. 5a and b) (e.g., aP2, adiponectin,PEPCK, LXR, Cox7A, and Elvol3 were induced onlyto b15% of the level observed for rosiglitazone) (Fig.5b). Furthermore, INT131was able to efficiently anta-gonize the effects of rosiglitazone on lipid accumula-tion in these cells (Fig. 5a). These results are consistentwith the partial agonist activities of INT131 observedin other assays. Interestingly, several PPARγ targetgenes appear to be differentially regulated by INT131(e.g., INT131 induced the expression of 11β-HSD1,and FGF21 reached N60% of the level observed forrosiglitazone) (Fig. 5c). These data further supportedcontext-dependent selective modulator activities thatare unique to INT131 and differentiate them fromthose of the rosiglitazone class of PPARγ agonists.
In vivo profile of INT131 versus rosiglitazone
The in vivo effects of INT131 were evaluated in arat model of insulin resistance, the Zucker (fa/fa) rat.Compounds were administered once daily by oralgavage for 14 days. INT131 exhibited maximalefficacy (Fig. 6a) comparable to that of rosiglitazonewith respect to plasma glucose clearance in an oralglucose tolerance test. INT131 also reduced baselineinsulin levels, suggesting that INT131, similar torosiglitazone, could improve insulin sensitivity intreated animals. Despite the comparable efficacies ofthese agents with respect to glucose clearance,rosiglitazone exhibited additional reductions inlipid parameters and insulin levels at the highestdose (Fig. 6a). However, these were also accompa-nied by weight gain and reductions in hematocritand total serum protein levels (Fig. 6b and Higginsand Mantzoros26). In contrast, no significant reduc-tion in hematocrit or total serum protein levels wasobserved in Zucker rats treated with equivalent highdoses of INT131. Increases in heart and lung weightswere substantially less with INT131 than with ro-siglitazone (Fig. 6c and Higgins andMantzoros26) aswell, suggesting an overall reduced propensity forfluid retention with INT131 treatment comparedwith rosiglitazone treatment.
Discussion
In this report, we describe the unique pharma-cological properties of a novel PPARγ ligand,INT131. A potent and selective ligand for PPARγ,INT131 is structurally distinct from the glitazoneclass of PPARγ agonists. We found that INT131 has a20-fold higher affinity for PPARγ compared withthat of rosiglitazone, but several in vitro biochemicaland cell-based assays reveal that INT131 can onlypartially activate PPARγ function. This was seen inan HTRF assay using DRIP205-derived peptide, in aHEK293 transfection assay using a Gal4-PPARγLBD-driven expression of a luciferase reportergene, and in preadipocyte differentiation assayswith the mouse 3T3L1 cell line. In all of thesesystems, INT131 potently—but only partially—activated receptor function compared with rosigli-tazone, and potently antagonized the effects ofrosiglitazone, consistent with a partial agonistprofile. This property of INT131 could be partlyexplained by the crystal structure of INT131 boundto PPARγ, which revealed that, in contrast torosiglitazone, INT131 does not form a direct H-bond with the Y473 residue of the AF2 activationhelix. Instead, only a water-mediated H-bond net-work was formed between the sulfonamide moietyof INT131 and the Y473 side chain. Due to thisweaker water-mediated H-bond, the AF2 activationhelix might be more mobile in solution with INT131bound than with rosiglitazone, and, therefore,under certain context/conditions, may yield thepartial agonist effect. The importance of the Y473residue in INT131 activity was further explored in a
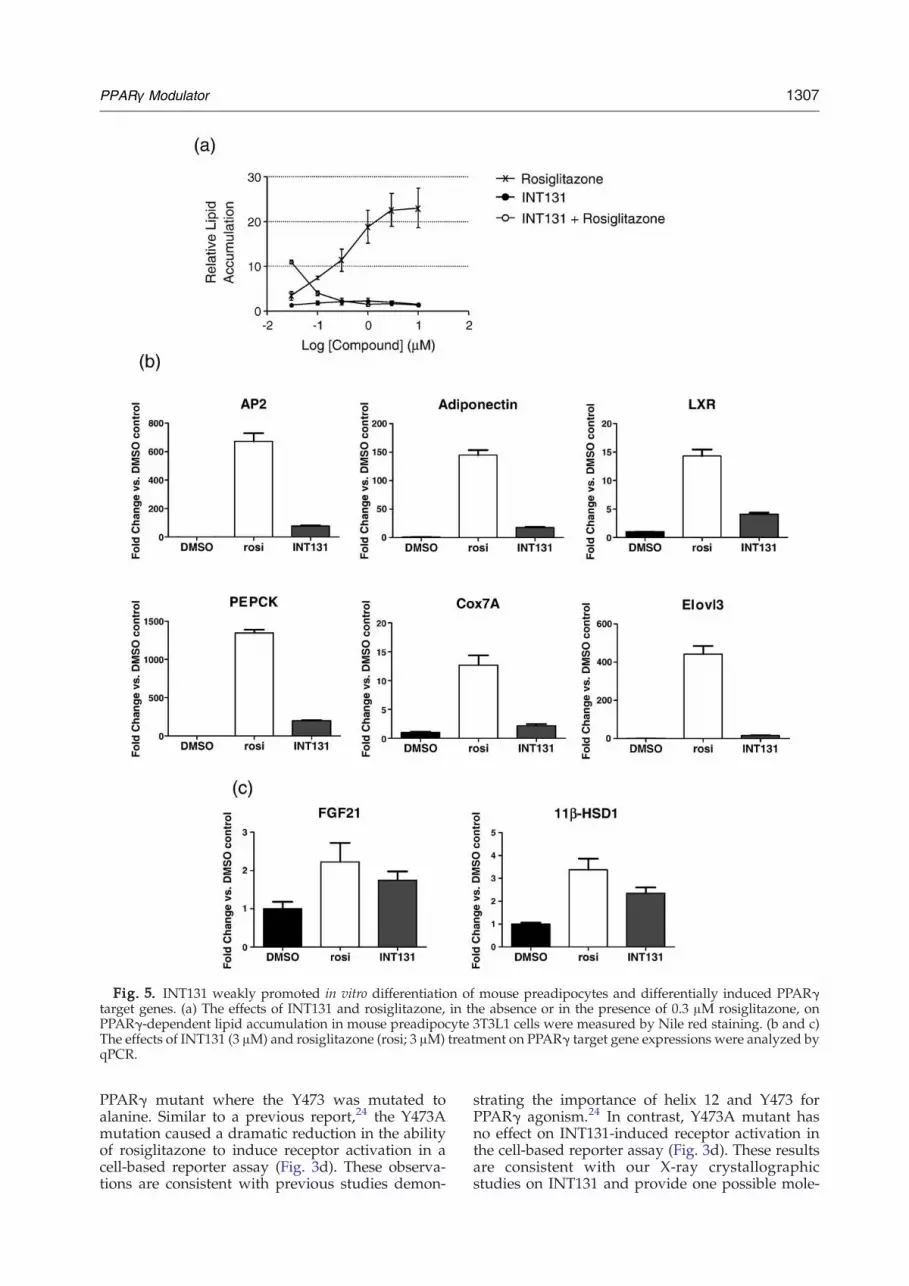
Fig. 5. INT131 weakly promoted in vitro differentiation of mouse preadipocytes and differentially induced PPARγtarget genes. (a) The effects of INT131 and rosiglitazone, in the absence or in the presence of 0.3 μM rosiglitazone, onPPARγ-dependent lipid accumulation in mouse preadipocyte 3T3L1 cells were measured by Nile red staining. (b and c)The effects of INT131 (3 μM) and rosiglitazone (rosi; 3 μM) treatment on PPARγ target gene expressions were analyzed byqPCR.
1307PPARγ Modulator
PPARγ mutant where the Y473 was mutated toalanine. Similar to a previous report,24 the Y473Amutation caused a dramatic reduction in the abilityof rosiglitazone to induce receptor activation in acell-based reporter assay (Fig. 3d). These observa-tions are consistent with previous studies demon-
strating the importance of helix 12 and Y473 forPPARγ agonism.24 In contrast, Y473A mutant hasno effect on INT131-induced receptor activation inthe cell-based reporter assay (Fig. 3d). These resultsare consistent with our X-ray crystallographicstudies on INT131 and provide one possible mole-
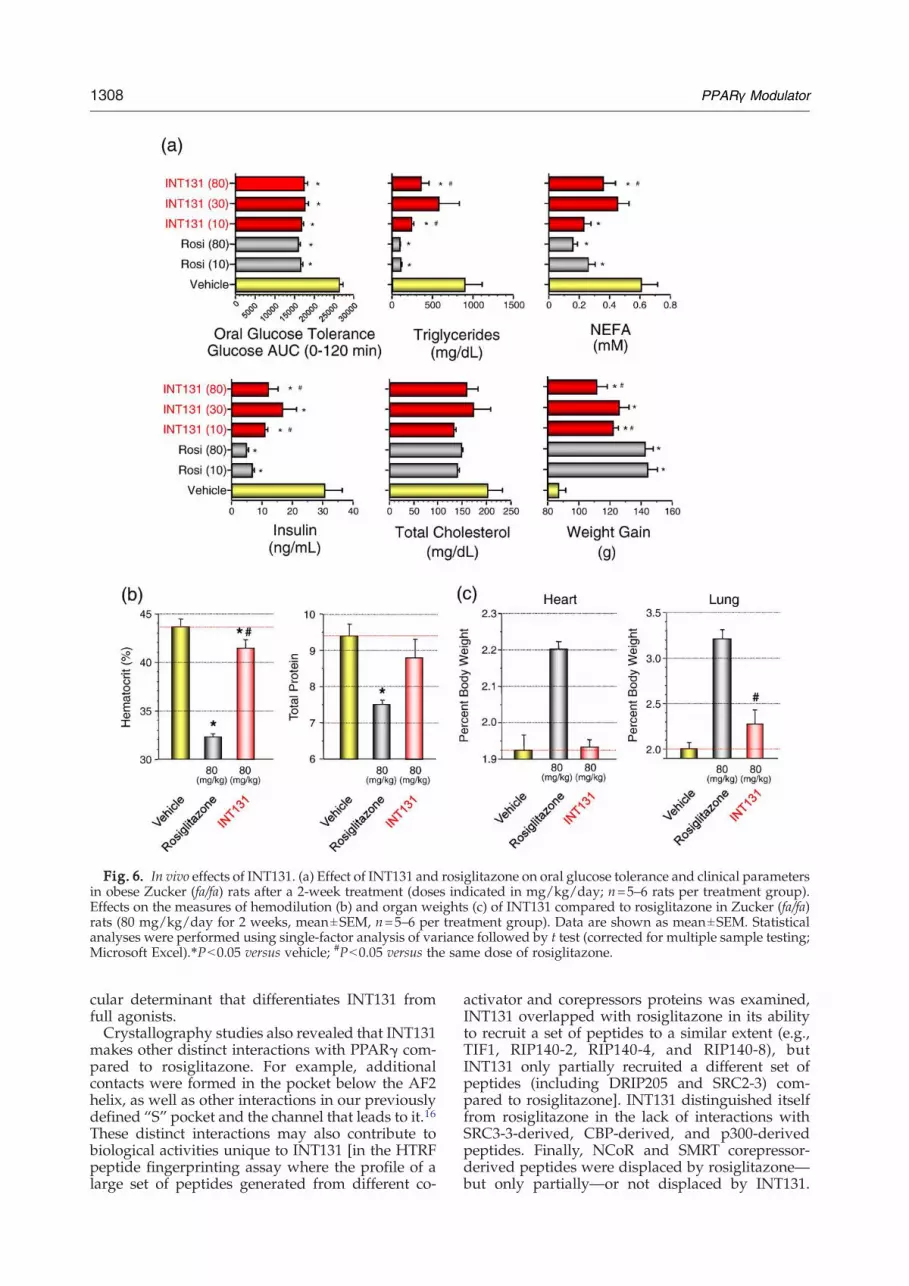
Fig. 6. In vivo effects of INT131. (a) Effect of INT131 and rosiglitazone on oral glucose tolerance and clinical parametersin obese Zucker (fa/fa) rats after a 2-week treatment (doses indicated in mg/kg/day; n=5–6 rats per treatment group).Effects on the measures of hemodilution (b) and organ weights (c) of INT131 compared to rosiglitazone in Zucker (fa/fa)rats (80 mg/kg/day for 2 weeks, mean±SEM, n=5–6 per treatment group). Data are shown as mean±SEM. Statisticalanalyses were performed using single-factor analysis of variance followed by t test (corrected for multiple sample testing;Microsoft Excel).⁎Pb0.05 versus vehicle; #Pb0.05 versus the same dose of rosiglitazone.
1308 PPARγ Modulator
cular determinant that differentiates INT131 fromfull agonists.Crystallography studies also revealed that INT131
makes other distinct interactions with PPARγ com-pared to rosiglitazone. For example, additionalcontacts were formed in the pocket below the AF2helix, as well as other interactions in our previouslydefined “S” pocket and the channel that leads to it.16These distinct interactions may also contribute tobiological activities unique to INT131 [in the HTRFpeptide fingerprinting assay where the profile of alarge set of peptides generated from different co-
activator and corepressors proteins was examined,INT131 overlapped with rosiglitazone in its abilityto recruit a set of peptides to a similar extent (e.g.,TIF1, RIP140-2, RIP140-4, and RIP140-8), butINT131 only partially recruited a different set ofpeptides (including DRIP205 and SRC2-3) com-pared to rosiglitazone]. INT131 distinguished itselffrom rosiglitazone in the lack of interactions withSRC3-3-derived, CBP-derived, and p300-derivedpeptides. Finally, NCoR and SMRT corepressor-derived peptides were displaced by rosiglitazone—but only partially—or not displaced by INT131.

1309PPARγ Modulator
This antagonist-like property of INT131 is quiteunique and distinct from those of full agonists suchas rosiglitazone, as well as those of the antagonistT070907 and the selective modulator T2384 that wehave described previously.25
These differences between INT131 and rosigli-tazone revealed by structural and biochemicalstudies may also explain the differences in theactivities induced by these different ligands in thecellular setting and suggest that the function ofINT131 may be context-dependent. Indeed, underthe in vitro differentiation conditions for the mouse3T3L1 cell line, INT131 activated very little adipo-genic programming, as seen by the minimalaccumulation of lipids in these cells, and partiallyinduced the expression of a group of PPARγ targetgenes, including aP2, adiponectin, PEPCK, LXR,Cox7A, and Elvol3 (Fig. 5b). However, INT131induced another group of PPARγ target genes, suchas 11β-HSD1 and FGF21, to a much higher extent—a level close to that of rosiglitazone (Fig. 5c). Not onlydid these differences provide additional support forthe context-dependent activation of receptor byINT131, but the reduced adipogenic potential ofINT131, coupled with the induction of 11β-HSD1and FGF21, could also directly contribute to thedifferences in in vivo pharmacology between INT131and rosiglitazone. Rosiglitazone and pioglitazonehave been clinically associatedwith an increased riskof peripheral edema and hemodilution.6 It has beenreported that these effects could contribute to theassociation of TZD treatment with an increased riskof congestive heart failure, and these drugs arecontraindicated in patients with New York HeartAssociation class III and class IV heart failure.11,12
Our own studies in the Zucker (fa/fa) rat modelreproduced the reported findings in numerousanimal models in which high doses of rosiglitazoneled to increases in heart weight, hemodilution(reduction in hematocrit and hemoglobin concen-tration), and plasma volume27 (Fig. 6 and Higginsand Mantzoros26). In contrast, our studies indicatedthat INT131 treatment had no effect on heart weightand showed reduced effects on body weight, lungweight, and hemodilution. These findings suggestthat INT131 may exhibit a more favorable hemody-namic and cardiovascular profile than the TZDs.Genetic evidence suggests that PPARγ-mediated
insulin sensitization may be decoupled from unde-sirable side effects associated with activation by fullagonists such as TZDs.18 Therefore, it appears thatthere may be an optimal PPARγ receptor activitylevel that would result in increased insulin sensitiv-ity with reduced adverse effects, as observed inPPARγ+/− heterozygous animals and humans car-rying the Pro12Ala allele of PPARγ. However, itmay be challenging to achieve this optimal receptoractivity level by pharmacological treatment, as toomuch activity could result in hyperactivation andundesirable side effects, while too little intrinsicactivity could result in lack of efficacy and insulinresistance.3,28 Whether the interesting pharma-cology displayed by INT131 is achieved by selective
modulation of PPARγ activity in different tissues, assuggested by the in vitro results, or as a result ofpartial activation of the receptor function remains tobe clarified. The data presented do support the ideathat, in the case of INT131, insulin sensitization canbe uncoupled, at least partly, from PPARγ-depen-dent side effects. These properties are currentlybeing examined in a clinical setting where INT131 isbeing evaluated as a therapeutic agent for the treat-ment of type 2 diabetes.
Materials and Methods
In vitro reagents and assay protocol
The design and method of generation of the plasmidsused and the discussions of the HTRF, ligand-binding, andtransient transfection assays have been reported.25
Adipocyte differentiation and gene expressionanalysis
3T3L1 preadipocytes were cultured and induced todifferentiate as follows. Cells were plated in DMEM with10% fetal bovine serum and 1% Pen-Strep at a density of3×105 cells/well on six-well plates. Two days postcon-fluence, differentiation was induced with either rosiglita-zone or INT131 at the concentrations indicated. Mediaand compounds were changed 48 h postinduction (andevery 2–3 days thereafter, for a total of 13 days). The lipidcontent of the cells was measured with Nile red staining(catalog no. N1142; Molecular Probes). RNA was pre-pared using the Qiagen RNeasy Midi Kit (catalog no.75142). Gene expression levels were measured usingquantitative PCR (qPCR). The primers used for qPCRwere from ABI: aP2 (Mm00445880_m1), PEPCK(Mm00440636_m1), LXR (Mm00443454_m1), Cox7a1(Mm00438296_m1), Elovl3 (Mm01194164_m1), adipo-nectin (Mm01343606_m1), FGF21 (Mm00840165_g1),and 11β-HSD1 (Mm00476182_m1). The primers used for18S were 5′-AGTCCCTGCCCTTTGTACACA-3′, 5′-GATCCGAGGGCCTCACTAAAC-3′, and 5′-6FAM-CGCCCGTCGCTACTACCGATTGG-BHQ1-3′. qPCRwas performed on Stratagene Mx3000P qPCR machineswith the Stratagene Brilliant II QRT-PCR Master Mix Kit,1-Step (catalog no. 600809), using 100 ng/well RNA andnormalized with mouse 18S.
Site-directed mutagenesis
Mutagenesis was performed on the hPPARγ LBD in thepM vector using the QuikChange XL site-directed muta-genesis kit from Stratagene (catalog no. 200517). Theprimers used to mutate the Tyr473 residue of the LBD toalanine were 5′-CTCCTGCAGGAGATCGCCAAGG-ACTTGTACTAG-3′ and 5′-CTAGTACAAGTCCTTGGC-GATCTCCTGCAGGAG-3′.
Cell culture and transient transactivation assay
HEK293 cells were cultured and maintained in DMEM,10% fetal bovine serum, and 1% Pen-Strep. Cells wereplated at a density of 5×106 cells per 100-mm plate in thesame medium and allowed to grow overnight. Cells were

1310 PPARγ Modulator
transfected with the pM-hPPARγWT or pM-hPPAR-γY473A expression vector, the pUAS(4X)-tk-luc reportervector, and pCMV-lacZ (as internal control) using Gene-PORTER II (catalog no. T202007; Genlantis). Following the24-h transfection, the cells were trypsinized and replatedonto 96-well plates, allowed to rest for 6 h, then exposed tocompounds for 18 h. Cell lysates were then produced andassayed for luciferase and β-galactosidase activities.
In vivo profiling in the Zucker (fa/fa) rat model
Male Zucker fatty (fa/fa) rats (Charles River Laboratories,Raleigh, NC) ages 7–8 weeks were allowed free access toirradiated Picolab 5053 diet and housed on a 12-h light/12-h dark cycle (0600–1800 h). Animal holding room tempera-ture and humidity were in conformance with the ”Guidefor the Care and Use of Laboratory Animals” (NationalResearch Council, National Academy Press, Washington,DC, 1996). Rats, while conscious, were prebled via a tailnick, and samples were analyzed for fed-state glucose,insulin, free fatty acids (NEFA), triglycerides, and totalcholesterol. Area under the glucose curve was determinedduring an oral glucose tolerance test (1 g/10 mL/kg, po)performed on rats fasted overnight. Blood glucose levelswere determined onwhole blood expressed from a tail nick(Accu-Check Blood Glucose Monitoring System; Roche).These eight clinical parameters, in addition to bodyweight,were used to randomize the rats into treatment groups.INT131 and rosiglitazone were both prepared in 1% Tween80/1% methylcellulose and administered by oral gavage(10 mL/kg), once daily, between 0900 and 1200 h at thedoses described. Bodyweight, food intake, and fluid intakewere recorded on a daily basis from the start of dosingthrough the day of sacrifice.
Clinical chemistry
Samples treated with ethylenediaminetetraacetic acidwere submitted for hematology, and serum samples weresubmitted for clinical chemistry (Quality Clinical Labs,Mountain View, CA). Specified organs were collected andweighed. The dry weights of heart and lung tissues weredetermined after desiccation of tissues overnight in aheated (50 °C) vacuum oven.
Protein preparation and crystallization
The human PPARγ LBD (residues 206–477) was PCR-cloned into a pET-30 vector (Novagen) with an N-terminalHis6 tag. The protein was expressed in Escherichia coli BL21(DE3) cells (Invitrogen) by growing in LB medium.Following two steps of purification by a Ni+ NTA-agarosecolumn (Qiagen) and an anion-exchange column of Mono-Q (Pharmacia), the protein was further purified by a gel-filtration column of Superdex 75 (Pharmacia). The purifiedprotein was concentrated to 1 mg/mL in 20 mM Tris–HCl(pH 7.9), 100 mM NaCl, 2 mM EDTA, and 5 mM DTTbefore mixing with a fivefold excess of INT131 and acoactivator peptide containing the LXXLL motif derivedfrom helical domain 1 (TSHKLVQLLTTT) of SRC-1. Theprotein mixture was further concentrated to 5–7 mg/mLfor crystallization. The cocrystal of PPARγ LBD withINT131 and the SRC-1 coactivator peptide were grown at20 °C in either hanging-drop or sitting-drop method with2.5 μL of the protein solution and 2.5 μL of the wellsolution containing 30% (wt/vol) polyethylene glycol4000, 0.1 M Tris–HCl (pH 8.5), and 0.2 M MgCl2. The
crystals were transferred into a well solution thatcontained an additional 20% (wt/vol) ethylene glycoland then flash frozen in liquid nitrogen.
Data collection, structure determination, andrefinement
An X-ray diffraction data set was collected on a RigakuRU-H3RHB generator/RAXIS-IV detector. The data wasintegrated using DENZO/SCALEPACK.29 Crystals grewin space group P212121 with cell dimensions of a=54.0 Å,b=68.1 Å, and c=87.0 Å. The cocrystal structure of PPARγLBD with INT131 was solved by molecular replacementusing previously published PPARγ coordinate 2PRG as asearch model. Manual model building was carried out inQuanta (Accelrys), and refinements were carried out withboth CNX30 and REFMAC5 in CCP4.31 The final R andRfree factors were 19% and 24%, respectively. The main-chain and side-chain atoms were visible for residues 206–258 and 275–477 of PPARγ, and for residues 1–12 of theSRC-1 peptide in the final electron density.
Accession numbers
Coordinates and structure factors have been depositedin the Protein Data Bank with accession number 3FUR.
Acknowledgements
We thank Tim Hoey, Xiao Hong Liu, Miki Rich,Linda Huang, and Haoda Xu for their technicalsupport and generous gifts of reagents. We wouldalso like to thank Pieter Timmermans, and JoyceChan for helpful discussions and critical reading ofthe manuscript.
References
1. Rosen, E. D. & Spiegelman, B. M. (2001). PPARgamma:a nuclear regulator of metabolism, differentiation,and cell growth. J. Biol. Chem. 276, 37731–37734.
2. Nuclear Receptors Nomenclature Committee. (1999).A unified nomenclature system for the nuclear recep-tor superfamily. Cell, 97, 161–163.
3. Evans, R. M., Barish, G. D. & Wang, Y. X. (2004).PPARs and the complex journey to obesity. Nat. Med.10, 355–361.
4. Semple, R. K., Chatterjee, V. K. & O'Rahilly, S. (2006).PPAR gamma and human metabolic disease. J. Clin.Invest. 116, 581–589.
5. Martens, F. M., Visseren, F. L., Lemay, J., de Koning,E. J. & Rabelink, T. J. (2002). Metabolic and additionalvascular effects of thiazolidinediones. Drugs, 62,1463–1480.
6. Nesto, R. W., Bell, D., Bonow, R. O., Fonseca, V.,Grundy, S. M., Horton, E. S. et al. (2004).Thiazolidinedione use, fluid retention, and conges-tive heart failure: a consensus statement from theAmerican Heart Association and American DiabetesAssociation. Diabetes Care, 27, 256–263.
7. Robinson, J. G. (2007). Update on PPAR agonists:the clinical significance of FIELD and PROACTIVE.Curr. Atheroscler. Rep. 9, 64–71.

1311PPARγ Modulator
8. Boden, G. & Zhang, M. (2006). Recent findingsconcerning thiazolidinediones in the treatment ofdiabetes. Expert. Opin. Invest. Drugs, 15, 243–250.
9. Patel, C., Wyne, K. L. & McGuire, D. K. (2005).Thiazolidinediones, peripheral oedema and conges-tive heart failure: what is the evidence? Diabetes Vasc.Dis. Res. 2, 61–66.
10. Shearer, B. G. & Billin, A. N. (2007). The nextgeneration of PPAR drugs: do we have the tools tofind them? Biochim. Biophys. Acta, 1771, 1082–1093.
11. Nissen, S. E. &Wolski, K. (2007). Effect of rosiglitazoneon the risk of myocardial infarction and death fromcardiovascular causes. N. Engl. J. Med. 356, 2457–2471.
12. Tang, W. H. & Maroo, A. (2007). PPARgammaagonists: safety issues in heart failure. Diabetes Obes.Metab. 9, 447–454.
13. Picard, F. & Auwerx, J. (2002). PPAR(gamma) andglucose homeostasis. Annu. Rev. Nutr. 22, 167–197.
14. Knouff, C. & Auwerx, J. (2004). Peroxisomeproliferator-activated receptor-gamma calls for acti-vation in moderation: lessons from genetics andpharmacology. Endocr. Rev. 25, 899–918.
15. Florez, J. C. (2004). Phenotypic consequences of theperoxisome proliferator-activated receptor-gammaPro12Ala polymorphism: the weight of the evidencein genetic association studies. J. Clin. Endocrinol.Metab. 89, 4234–4237.
16. Li, Y., Wang, Z., Furukawa, N., Escaron, P.,Weiszmann, J., Lee, G. et al. (2008). T2384, a novelantidiabetic agent with unique PPARγ bindingproperties. J. Biol. Chem. 283, 9168–9176.
17. Yamauchi, T., Kamon, J., Waki, H., Murakami, K.,Motojima, K., Komeda, K. et al. (2001). The mecha-nisms by which both heterozygous peroxisome pro-liferator-activated receptor gamma (PPARgamma)deficiency and PPARgamma agonist improve insulinresistance. J. Biol. Chem. 276, 41245–41254.
18. Argmann, C. A., Cock, T. A. & Auwerx, J. (2005).Peroxisome proliferator-activated receptor gamma:the more the merrier? Eur. J. Clin. Invest. 35, 82–92;discussion 80.
19. McGee, L. R., Rubenstein, S. M., Houze, J. B., Ye,G., Li, Y., Learned, R. M. et al. (2006). Proceedingsof the 231st American Chemical Society NationalMeeting, Atlanta, GA, USA.
20. Xu, H. E., Lambert, M. H., Montana, V. G., Parks, D. J.,Blanchard, S. G., Brown, P. J. et al. (1999). Molecularrecognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell, 3, 397–403.
21. Cronet, P., Petersen, J. F., Folmer, R., Blomberg, N.,Sjoblom, K., Karlsson, U. et al. (2001). Structure of thePPARalpha and -gamma ligand binding domain in
complex with AZ 242; ligand selectivity and agonistactivation in the PPAR family. Structure, 9, 699–706.
22. Oberfield, J. L., Collins, J. L., Holmes, C. P., Goreham,D. M., Cooper, J. P., Cobb, J. E. et al. (1999). Aperoxisome proliferator-activated receptor gammaligand inhibits adipocyte differentiation. Proc. NatlAcad. Sci. USA, 96, 6102–6106.
23. Nolte, R. T., Wisely, G. B., Westin, S., Cobb, J. E.,Lambert, M. H., Kurokawa, R. et al. (1998). Ligandbinding and co-activator assembly of the peroxisomeproliferator-activated receptor-gamma. Nature, 395,137–143.
24. Einstein, M., Akiyama, T. E., Castriota, G. A., Wang,C. F., McKeever, B., Mosley, R. T. et al. (2008). Thedifferential interactions of peroxisome proliferator-activated receptor gamma ligands with Tyr473 is aphysical basis for their unique biological activities.Mol. Pharmacol. 73, 62–74.
25. Lee, G., Elwood, F., McNally, J., Weiszmann, J.,Lindstrom, M., Amaral, K. et al. (2002). T0070907, aselective ligand for peroxisome proliferator-activatedreceptor gamma, functions as an antagonist ofbiochemical and cellular activities. J. Biol. Chem. 277,19649–19657.
26. Higgins, L. S. & Mantzoros, C. S. (2008). Thedevelopment of INT131 as a selective PPARgammamodulator: approach to a safer insulin sensitizer.PPAR Res. 2008, 936906.
27. Sotiropoulos, K. B., Clermont, A., Yasuda, Y., Rask-Madsen, C., Mastumoto, M., Takahashi, J. et al.(2006). Adipose-specific effect of rosiglitazone onvascular permeability and protein kinase C activa-tion: novel mechanism for PPARgamma agonist'seffects on edema and weight gain. FASEB J. 20,1203–1205.
28. Barroso, I., Gurnell, M., Crowley, V. E., Agostini, M.,Schwabe, J. W., Soos, M. A. et al. (1999). Dominantnegative mutations in human PPARgamma associatedwith severe insulin resistance, diabetes mellitus andhypertension. Nature, 402, 880–883.
29. Otwinowski, Z. A. M. W. (1997). Processing of X-raydiffraction data collected in oscillation mode. MethodsEnzymol. 276, 307–326.
30. Brunger, A. T., Adams, P. D., Clore, G. M., DeLano,W. L., Gros, P., Grosse-Kunstleve, R. W. et al. (1998).Crystallography and NMR system: a new softwaresuite for macromolecular structure determination.Acta Crystallogr. Sect. D, 54, 905–921.
31. Leslie, A. G. W. (1992). Recent changes to theMOSFLM package for processing film and imageplate data. Jt. CCP4+ESF-EAMCB Newsl. ProteinCrystallogr. 26.