imiquimod-induced tlr7 signaling enhances repair of dna damage induced by ultraviolet light in bone...
TRANSCRIPT

The Journal of Immunology
Imiquimod-Induced TLR7 Signaling Enhances Repair ofDNA Damage Induced by Ultraviolet Light in BoneMarrow-Derived Cells
Rita Fishelevich,* Yuming Zhao,* Papapit Tuchinda,* Hannah Liu,* Ayako Nakazono,*
Antonella Tammaro,* Tzu-Ching Meng,† Jim Lee,† and Anthony A. Gaspari*,‡
Imiquimod is a TLR7/8 agonist that has anticancer therapeutic efficacy in the treatment of precancerous skin lesions and certain
nonmelanoma skin cancers. To test our hypothesis that imiquimod enhances DNA repair as a mechanism for its anticancer activity,
the nucleotide excision repair genes were studied in bone marrow-derived cells. Imiquimod enhanced the expression of xeroderma
pigmentosum (XP) A and other DNA repair genes (quantitative real-time PCR analysis) and resulted in an increased nuclear lo-
calization of the DNA repair enzyme XPA. This was dependent on MyD88, as bone marrow-derived cells fromMyD882/2 mice did
not increase XPA gene expression and did not enhance the survival of MyD882/2-derived bone marrow-derived cells after UV B
exposure as was observed in bone marrow-derived cells from MyD88+/+ mice. Imiquimod also enhanced DNA repair of UV light
(UVL)-irradiated gene expression constructs and accelerated the resolution of cyclobutane pyrimidine dimers after UVL expo-
sures in P388 and XS52. Lastly, topical treatment of mouse skin with 5% imiquimod cream prior to UVL irradiation resulted in
a decrease in the number of cyclobutane pyridimine dimer-positive APC that were found in local lymph nodes 24 h after UVL
irradiation in both wild-type and IL-12 gene-targeted mice. In total, these data support the idea that TLR7 agonists such as
imiquimod enhance DNA repair in bone marrow-derived cells. This property is likely to be an important mechanism for its
anticancer effects because it protects cutaneous APC from the deleterious effects of UVL. The Journal of Immunology, 2011, 187:
1664–1673.
Ultraviolet light (UVL) is a well-characterized carcinogenthat causes genetic lesions in epidermal keratinocytes,which over time accumulate and play a critical role in
the development of skin cancers (1, 2). Equally important in thedevelopment of skin cancer is the immunosuppressive cutane-ous microenvironment that develops as a result of chronic UVLexposures. Immunosuppressive cytokines such as IL-10 are in-duced after UVL exposure (3, 4). Additionally, there is impairedAg presentation by skin-derived dendritic cells such as Langer-hans cells (LC), and immune suppressive macrophages migrateinto skin after UVexposures (5, 6). As a result of UVL impairmentof cutaneous APC, there is impairment in the development ofTh1 immunity and loss of sensitization to potent topically appliedhaptens (7, 8). Instead of sensitization, there is hapten-specificimmune tolerance that can be adoptively transferred to naivehosts by regulatory T cells (9). This loss of contact hypersensi-tivity is thought to be a sentinel event that correlates well with theloss of immunologic reactivity against tumor Ags that develop
in parallel as a result of chronic UVL exposures. Thus, instead ofpreventing the development and progression of UVL-induced skintumors, there is immune tolerance and a permissive environmentfor skin cancer development.The fundamental lesion for both skin cancer development as
well as photoimmunosuppression is DNA damage. Within 24 h ofUVL exposures, dendriticAPCwith cyclobutane pyrimidine dimers(CBPD) have been found in the paracortical area of draininglymphnodes ofUVL-irradiatedmice (10). SuchAPC that bearDNAdamage are in part responsible for the loss of protective cutaneousimmunity against skin cancer development (11). A biomarker forthe loss of cutaneous immunity is the loss of contact sensitization.Local exposure of the skin to UVL results in both local and sys-temic effects. In model systems to protect mice from cutaneousimmunosuppression after UVL exposures, a variety of treatmentshave been applied in an attempt to enhance DNA repair by APC,potentially restoring immune competence after UVL exposures.DNA repair-promoting agents include bacterial DNA repairenzymes (T4 endonuclease encapsulated into liposomes) (12),green tea polyphenols (13), and cytokines such as IL-12 and IL-23, platelet-activating factor, and serotonin receptor antagonists(14–16).In recent studies, the effect of topically applied imiquimod was
studied in a mouse model of UVL-induced loss of hapten sensi-tization. This treatment prevented the loss of contact hypersen-sitivity as well as the development of hapten-specific tolerance(17). Even though imiquimod preserved contact hypersensitivityin vivo, it did not protect from UVL-induced depletion of LC fromthe epidermis of UVL-irradiated skin (17). In vitro, imiquimodactivation of TLR7 prior to UVL exposure induced resistant APCmaturation, preserving Th1 lymphocyte-derived cytokines. This isin marked contrast to UVL-irradiated APC that were not activated
*Department of Dermatology, School of Medicine, University of Maryland Balti-more, Baltimore, MD 21201; †Graceway Pharmaceutical Company, Exton, PA 19341;and ‡Department of Microbiology and Immunology, School of Medicine, Universityof Maryland Baltimore, Baltimore, MD 21201
Received for publication March 16, 2011. Accepted for publication June 15, 2011.
This work was supported by an unrestricted grant from Graceway Pharmaceuticals,Exton, PA (to A.A.G.).
Address correspondence and reprint requests to Dr. Anthony A. Gaspari, Departmentof Dermatology, University of Maryland Baltimore, 419 West Redwood Street, Suite240, Baltimore, MD 21201. E-mail address: [email protected]
Abbreviations used in this article: CBPD, cyclobutane pyrimidine dimers; LC, Lang-erhans cells; qPCR, quantitative real-time PCR; UVB, UV B; UVL, UV light; XP,xeroderma pigmentosum.
Copyright� 2011 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/11/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1100755

with imiquimod. These data lead us to hypothesize that imiqui-mod, via TLR7 activation, renders cutaneous APC resistant toUVL-induced immunosuppression because it enhances DNA re-pair. In this study, we describe the effects of imiquimod on APCDNA repair of UVL damage. We found that imiquimod inducesDNA repair gene expression that is dependent on MyD88 as wellas an intact NF-kB signaling pathway, which are early and latecomponents of TLR signaling pathway. As well, imiquimod-activated APC were resistant to UVL, and ionizing radiation in-duced loss of viability. Additionally, imiquimod-activated APCexhibited enhanced functional DNA repair compared with restingAPC. Lastly, topical application of imiquimod prior to UVL ex-posure resulted in a marked reduction of CBPD-positive APCand a reduction in suppressive cytokine gene expression in locallymph nodes of such treated mice when compared with vehicleor untreated, irradiated mice. These data lead us to concludeimiquimod-mediated, TLR7 signaling enhances repair of UVLdamage to genomic DNA and may play a therapeutic role in thepreservation of an intact cutaneous immune response after expo-sure to UVL.
Materials and MethodsExperimental mice
C57BL/6 female mice, 8 wk old, were purchased from The JacksonLaboratory (Bar Harbor, ME). These animals were chosen because theyhave been demonstrated to exhibit a phenotype of susceptibility to UV-induced suppression of contact hypersensitivity (18, 19). IL-12p40 gene-targeted female mice (B6.129S1IL2btm1Jm/J, stock number 002693) in theC57BL/6 background were purchased from The Jackson Laboratory (20).Animals were kept in standard housing conditions, fed mouse chow adlibitum, and exposed to a 12-h light/dark cycle. To study the appearance ofAPC in local lymph nodes, experimental mice (either wild-type or IL-12gene-targeted mice) were unirradiated or irradiated with a single dose ofUV B (UVB) at a dose of 150 mJ/cm2. Some of the UVB-irradiated micereceived topical applications of 5% imiquimod cream or its vehicle to thedorsal aspect of the pinna of the ear (once daily for 2 d); 24 h later, theanimals were then exposed to UVB. For the UVB exposure, the animalswere anesthetized with a single dose of avertin (i.p. injection), then theanimals’ ears were taped to a restraining chamber and the dorsum exposedto the indicated dose of UVB. Twenty-four hours after this, the animalswere sacrificed, the cervical lymph nodes were dissected, and DNA ex-tracted using standard methods. All experimental procedures were reviewedand approved by the University of Maryland Institutional Animal Careand Use Committee.
Cell lines
A20 was purchased from American Type Culture Collection. P388 cell linewas a generous gift from Dr. David Scott (University of Maryland Balti-more). XS52 was a generous gift from Dr. Akira Takashima (University ofToledo) andwas cultured using standard conditions.Wild-typemacrophagesand MyD88 knockout cell lines were a generous gift from Dr. Doug Gol-lenbach (University of Massachusetts). Xeroderma pigmentosum (XP) Alymphoblasts were purchased from Coreill Institute (Camden, NJ). Controllymphoblasts were produced by our laboratory using EBV transformation ofPBMCs from a healthy volunteer who had normal DNA repair (21).
Cytotoxicity assay
A thiazolyl blue tetrazolium bromide (MTTassay) was used to measure cellviability 24 h after UVL or ionizing radiation exposures (22). Either ad-herent cells (P388) or nonadherent lymphoblasts (EBV-immortalized cells)were used at a cell density of ∼5000 cells/well of a 96-well plate. For ad-herent P388 cells, the cells remained adherent to the microtiter wells; forthe nonadherent lymphoblasts (XPA or control lymphoblasts), the micro-titer plates were centrifuged (1200 rpm for 10 min) prior to any washes ormedium changes. Using standard methods (22), MTT solution (20 ml 5mg/ml MTT stock solution) was added to cell cultures, then incubated for4 h at 37˚C. The culture medium was then removed, and 150 ml MTTsolvent (4 mM HCl, 0.1% Nonidet P-40 in isopropanol) was added, and theplates were covered with tin foil and placed on an orbital shaker for 15min. The OD was read at 590 nm with a reference filter of 620 nm.Background OD for MTT was assayed in wells that contained no cells.
UVL source
Groups of mice were irradiated with a panel of 48” Q-Sun light bank (Q-Panel Laboratory Products, Cleveland, OH) (equipped with a UVCWG320filter) at a distance of 12 inches from the light source to their shaved ab-dominal skin. The spectral emission profile of this light source closelymimics that of natural sunlight, emitting predominantly UVA. A UVB ra-diometer (National Biologic Corporation, Twinsburg, OH) was used todetermine UVB output and calculate the time necessary to deliver the de-sired doses of UVB. Plasmid DNA, cell monolayers, or experimental micereceived UVL at the doses indicated in each of the individual experiments.
Ionizing radiation
A [137Cs] radiation source was used to deliver ionizing radiation to eithercontrol or XPA lymphoblasts. Exposure time of the irradiated cells wasadjusted to deliver the desired dose of radiation (see Results) using cali-bration curves.
Transient transfection by electroporation and luciferase assays
For the gene reactivation assay, control (medium alone) or imiquimod-treated cells (at the indicated doses for 24 h prior to electroporation) weretransfected with the following constructs: pGL3, an empty vector thatcontains no luciferase gene as a negative control; and pGL4.1[luc2], whichencodes for firefly luciferase as a positive control; these negative and positivecontrols were cotransfected with pGL4.70[hRluc] (Renilla luciferase),which serves as a control for transfection efficiency. In parallel, control orimiquimod-treated cells were transfected with UVL-irradiated pGL4.10[luc2] constructs, with UVL radiation doses ranging from 1000–5000 mJ/cm2. The UVL-irradiated constructs were produced by placing a 5-mlvolume of the plasmid DNA on a glass slide and exposing to the indicateddoses of UVL at a 12-inch distance from the UVL source. After UVL ir-radiation, these constructs were cotransfected with unirradiated pGL4.70[hRluc], which served as a control for transfection efficiency.
The luminometry data represent a normalized ratio of firefly luciferase toRenilla luciferase. These normalized data were then normalized to thenegative (pGL3/pGL4.7) and positive control values (pGL4.1/pGL4.7). Thenormalized expression of the UVL-irradiated (pGL4.1UVL/pGL4.7) wasthen compared with its respective positive and negative controls. Data arereported as percent of control because imiquimod treatment of host cellshad significant effects on transfection efficiency (expression of positivecontrol construct, data not shown). Thus, the formula for calculating genereactivation was as follows: [(pGL4.1UVL/pGL4.7) 2 (pGL3/pGL4.7)/(pGL4.1/pGL4.7) 2 (pGL3/pGL4.7)] 3 100 = percent gene reactivationcompared with control. We used methods as previously described for theluminometry assays (23).
Cell lines (P388, XS52, macrophage cell line) used in the gene reac-tivation assay were transfected at ∼70% confluence using the Amaxanucleofector kit (Amaxa) according to the manufacturer’s protocol.Briefly, cells were transfected with 2 mg supercoiled plasmid DNA perwell in transfection of six-well plates. A positive control plasmid pGL4.10[luc2] encodes a synthetic firefly luciferase gene. An internal controlplasmid, pGL4.70 [hRluc], which expresses the Renilla luciferase gene,was also used for correcting variations in the transfection efficiency. After15 min, 1 ml growth medium was added and incubated for 48 h. Lysateswere harvested, and extracts were prepared using freeze-thaw lysis.Lysates were monitored for luciferase activities using a commerciallyavailable Dual Luciferase (firefly and Renilla) Reagent kit (Promega) ina Turner 20/20n luminometer (Turner Biosystems, Sunnyvale, CA). Fireflyluciferase activity was normalized to the Renilla luciferase activity. Briefly,trypsinized 1 million cells were electroporated with 3 mg DNA and in-cubated for 48 h prior to harvesting the cellular lysates. All gene reporterassays were repeated at least three times.
Chemicals
Imiquimod (1-[2-methylpropyl]-1H-imidazo[4,5-c]quinolin-4-amine, R-837)5% cream and its vehicle cream were used for topical applications andwere provided by Graceway Pharmaceuticals (Exton, PA). For thein vitro studies, imiquimod powder was used (also provided by GracewayPharmaceuticals). It was prepared as a stock solution in sterile water at1 mg/ml and diluted to the appropriate working concentration in completemedium. BAY 11-7085 (Calbiochem) was dissolved in DMSO and addedto the culture medium of APC 30 min prior to exposure to imiquimodtreatment at the indicated concentrations to block NF-kB (24).
Western blot
Western blotting was performed as previously described (24, 25). Equalquantities of total protein from different samples were separated on a 10%
The Journal of Immunology 1665
Bis-Tris NuPage denaturing gel (Invitrogen Life Technologies, Carlsbad,CA). Gels were transferred to nitrocellulose membranes, probed with ap-propriate first Abs, and detected using a Western Breeze Chemilumines-cent detection kit (Invitrogen Life Technologies). The bands were detectedusing a Chemi-Doc densitometer and Quantity One software (Bio-Rad,Hercules, CA). b-actin signals from each sample were used for normal-izing the intensity of specific bands of interest. A mouse anti-XPA Ab(clone B1; Santa Cruz Biotechnology, Santa Cruz, CA) was used toWestern blot XPA in control or imiquimod-treated cells. This clone wasraised against human aa 1–123 derived from full-length human XPA. ThismAb reacts with a polypeptide of ∼38–40 kDa.
Cell fractionation
Nuclei from cultured cells were isolated using the Active Motif NuclearExtraction Kit (Active Motif North America, Carlsbad, CA). Nuclearextracts were prepared using the manufacturer’s instructions.
RNA extraction, cDNA synthesis, and quantitative real-timePCR
Standard methods were used to extract total cellular RNA and cDNAsynthesis. Detection of XPA, TNF-a, and TLR7 mRNA was performedusing quantitative real-time PCR (qPCR) according to previously pub-lished methods (26). The annealing temperature was 55˚C, followed by 35cycles of amplification using a PCR Sprint (Hybaid). Primers to amplifymouse DNA repair genes were as follows: XPA (reference number 592)and XPC (catalog number 04375e), XPG (catalog number 04636a), XPFRpa1 (catalog number 05392a), Rpa3 (catalog number 03481a), Xab2(catalog number 05491a), Rad23b (catalog number 04360a), and Ddb1(catalog number 04381a). Primers to amplify other genes included: TLR7(reference number 1136), TNF-a (reference number 879), Foxp3, IL-4, IL-10 (catalog #074), TGF-b (catalog number 035), and 18S (referencenumber X03205). All of these primers were purchased from SuperArrayBiosciences (Qiagen, Valencia, CA). Real-time PCR (LightCycler; Roche,Indianapolis, IN) was used to confirm differences in the levels of expres-
sion of selected mRNA. The primers, PCR protocol, and product quanti-fication for 18S rRNA were exactly as reported previously (SYBR Greendetection) (26). Amplification (40–50 cycles) of a single PCR product wasconfirmed by gel electrophoresis and melting curve analyses. The cDNAswere assayed in duplicate, and mean values are depicted in the graphics forthe quantitative PCR.
Statistical analyses
GraphPad Instat and Prism software programs (GraphPad, San Diego, CA)were used to compare quantitative data from experimental groups forstatistical significance. For comparisons between multiple groups, ANOVAwas used; p , 0.05 was considered to be statistically significant.
ResultsImiquimod induces DNA repair gene expression in a variety ofbone marrow-derived cells
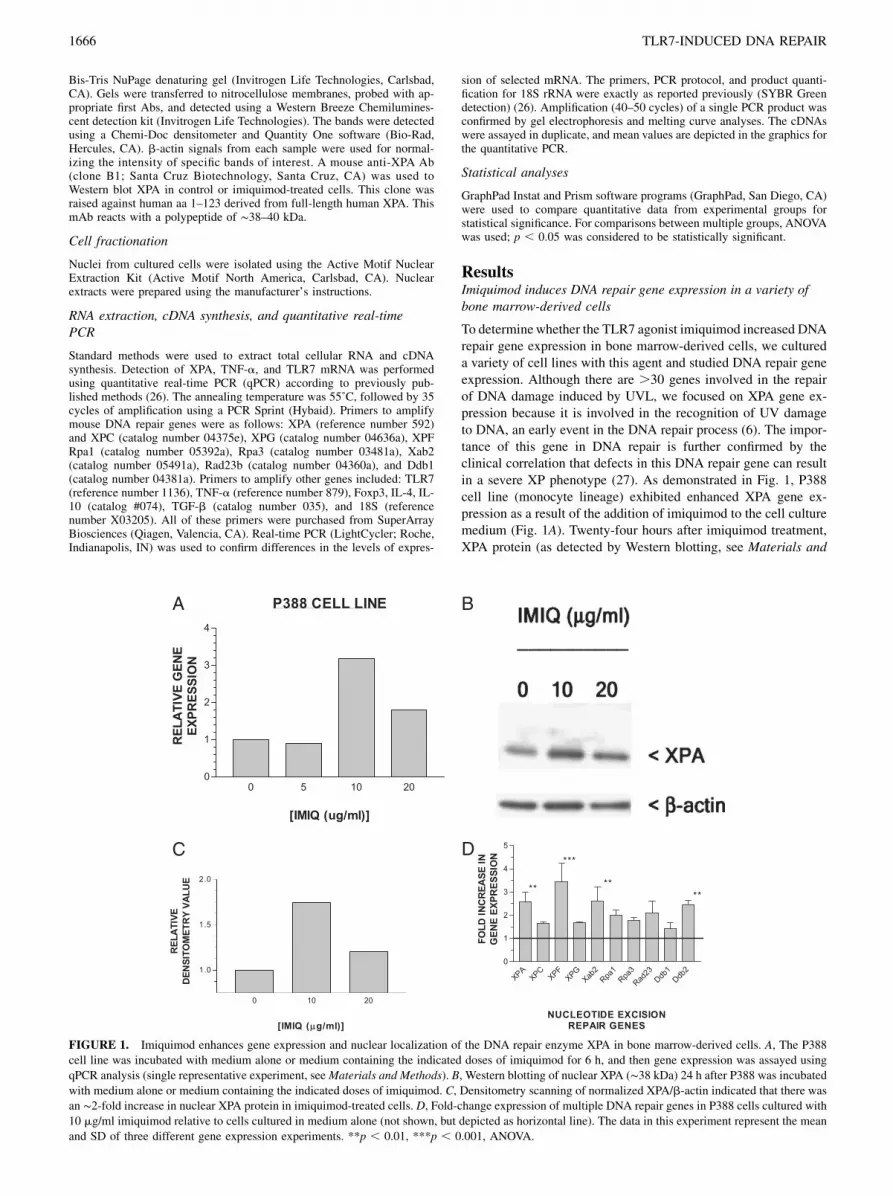
To determinewhether the TLR7 agonist imiquimod increased DNArepair gene expression in bone marrow-derived cells, we cultureda variety of cell lines with this agent and studied DNA repair geneexpression. Although there are .30 genes involved in the repairof DNA damage induced by UVL, we focused on XPA gene ex-pression because it is involved in the recognition of UV damageto DNA, an early event in the DNA repair process (6). The impor-tance of this gene in DNA repair is further confirmed by theclinical correlation that defects in this DNA repair gene can resultin a severe XP phenotype (27). As demonstrated in Fig. 1, P388cell line (monocyte lineage) exhibited enhanced XPA gene ex-pression as a result of the addition of imiquimod to the cell culturemedium (Fig. 1A). Twenty-four hours after imiquimod treatment,XPA protein (as detected by Western blotting, see Materials and
FIGURE 1. Imiquimod enhances gene expression and nuclear localization of the DNA repair enzyme XPA in bone marrow-derived cells. A, The P388
cell line was incubated with medium alone or medium containing the indicated doses of imiquimod for 6 h, and then gene expression was assayed using
qPCR analysis (single representative experiment, seeMaterials and Methods). B, Western blotting of nuclear XPA (∼38 kDa) 24 h after P388 was incubatedwith medium alone or medium containing the indicated doses of imiquimod. C, Densitometry scanning of normalized XPA/b-actin indicated that there was
an ∼2-fold increase in nuclear XPA protein in imiquimod-treated cells. D, Fold-change expression of multiple DNA repair genes in P388 cells cultured with
10 mg/ml imiquimod relative to cells cultured in medium alone (not shown, but depicted as horizontal line). The data in this experiment represent the mean
and SD of three different gene expression experiments. **p , 0.01, ***p , 0.001, ANOVA.
1666 TLR7-INDUCED DNA REPAIR

Methods) is increased in the nucleus of imiquimod-stimulatedP388 compared with control (unstimulated) P388 (Fig. 1B); den-sitometry scanning of the Western blot autoradiogram was thennormalized (XPA/b-actin ratio) and is presented as a histogram inFig. 1C, with an ∼2-fold increase in nuclear XPA protein at 24 hafter 10 mg/ml imiquimod exposure and a smaller increase in-duced by imiquimod at 20 mg/ml. Because DNA repair of UVLdamage is a complex process that involves a family of DNAdamage recognition and repair, we screened the P388 cell line forchanges in other repair genes at the 10 mg/ml dose of imiquimodand found that the transcription of the entire family of genes wasmodestly increased (range of increase between 1.5- and 3.5-foldabove control cells (Fig. 1D). We found that the optimal dose forthe increasing XPA gene expression, as well as the amplitude ofthe increase, varied among these three cell lines. The optimal doseof imiquimod for increasing XPA gene expression varied between10 and 30 mg/ml, which is slightly higher than the optimal dose(5–10 mg/ml) for triggering cytokine secretion by TLR7-expres-sing mouse APC (28).
Imiquimod-enhanced DNA repair gene expression and survivalafter UV exposure is dependent on MyD88
All of the TLR receptors, including TLR7 and -8, use MyD88 as anadaptor protein to initiate early events that eventually result inNF-kB signaling in the nucleus, which reprograms cells to expressinflammatory cytokine genes (29–31). Bone marrow-derived cellsfrom MyD88 gene-targeted mice exhibit impaired inflammatoryresponses after exposure to TLR ligands (3). Thus, if imiquimod in-duction of the DNA repair gene XPA is indeed dependent on TLR7signaling, then cells deficient in MyD88 would exhibit impairedability to upregulate XPA gene expression after stimulation by im-iquimod. Macrophage cell lines from wild-type mice (MyD88 +/+)and MyD88 knockout mice (MyD882/2) were exposed to mediumalone or medium containing imiquimod for 4 h and XPA gene ex-pression was assayed. Whereas macrophages from wild-type mice(MyD88+/+) responded by increasing XPA gene expression at both5 and 10 mg/ml, the macrophages from MyD882/2 knockout miceexhibited a blunted response to 5 mg/ml and an absent responseto the 10 mg/ml dose, which induced optimal gene expression inmacrophages from MyD88+/+ mice (Fig. 2A).After exposure to UVL, an irradiated cell will sense DNA
damage and can go on to repair the DNA damage and survive.Alternatively, if the DNA damage is too severe, an irradiated cellwill then undergo apoptosis (32, 33). We hypothesized that if DNArepair induced by TLR7 was functionally significant, there wouldbe enhanced survival after exposures to increasing doses of UVL.As shown in Fig. 2B, treatment of MyD88+/+ macrophages en-hanced the survival of these cells (MTT assay) compared withcontrol (medium alone) cells. In contrast, MyD882/2 macro-phages did not respond to imiquimod with enhanced survival toUVL (MTT assay) (Fig. 2C). Other cell lines such as P388 alsoexhibited imiquimod-induced enhanced survival after UVB ex-posure (data not shown).A late event in the TLR signaling pathway is translocation of
the transcription factor NF-kB from the cytoplasm to the nucleusof activated cells (28–30). Under steady-state conditions, NF-kBproteins are sequestered in the cytoplasm by members of the IkBfamily. Upon cellular stimulation by TLR agonists, activation ofthe IkB kinase phosphorylates IkB, which dissociates the NF-kB–IkB complexes. Free NF-kB heterodimers then translocate intothe nucleus, where they bind to enhancer elements of target genes(24). The chemical compound BAY 11-7085 inhibits the NF-kBsignaling pathway by preventing the degradation of IkB (24) andhence prevents the translocation of NF-kB from the cytoplasm to
the nucleus, thus blocking the activation of its target genes. If theinduction of XPA gene expression is dependent on classical TLRsignaling, we hypothesized that Bayer compound would block theupregulation of XPA gene expression after TLR7 stimulation byimiquimod. Consistent with our hypothesis, BAY 11-7085 blockedthe increase in XPA gene expression in the P388 cell line (Fig.3A). Similarly, BAY 11-7085 blocked the imiquimod-induced
FIGURE 2. MyD88 expression is necessary for the enhanced XPA gene
expression and survival after exposure to UVL in bone marrow-derived
cells. A, Macrophage cell lines derived from MyD88+/+ (wild-type) mice or
MyD882/2 (gene-targeted) mice were incubated with medium alone or
medium containing the indicated doses of imiquimod, and XPA gene
expression was assayed 6 h later. B, Bone marrow-derived cells from
MyD88+/+ (wild-type) mice were incubated with the indicated doses of
imiquimod for 24 h and then exposed to increasing doses of UVL, and
survival was measured after another 24 h using an MTT assay (see
Materials and Methods). C, Bone marrow-derived cells from MyD882/2
(gene-targeted) mice were incubated with the indicated doses of imiqui-
mod for 24 h, and then exposed to increasing doses of UVL, and survival
was measured after another 24 h using an MTT assay (see Materials and
Methods). Data depicted represent the mean 6 SD of six replicates from
a single representative experiment that was repeated three times. *p ,0.05, **p , 0.01, ***p , 0.001, ANOVA.
The Journal of Immunology 1667
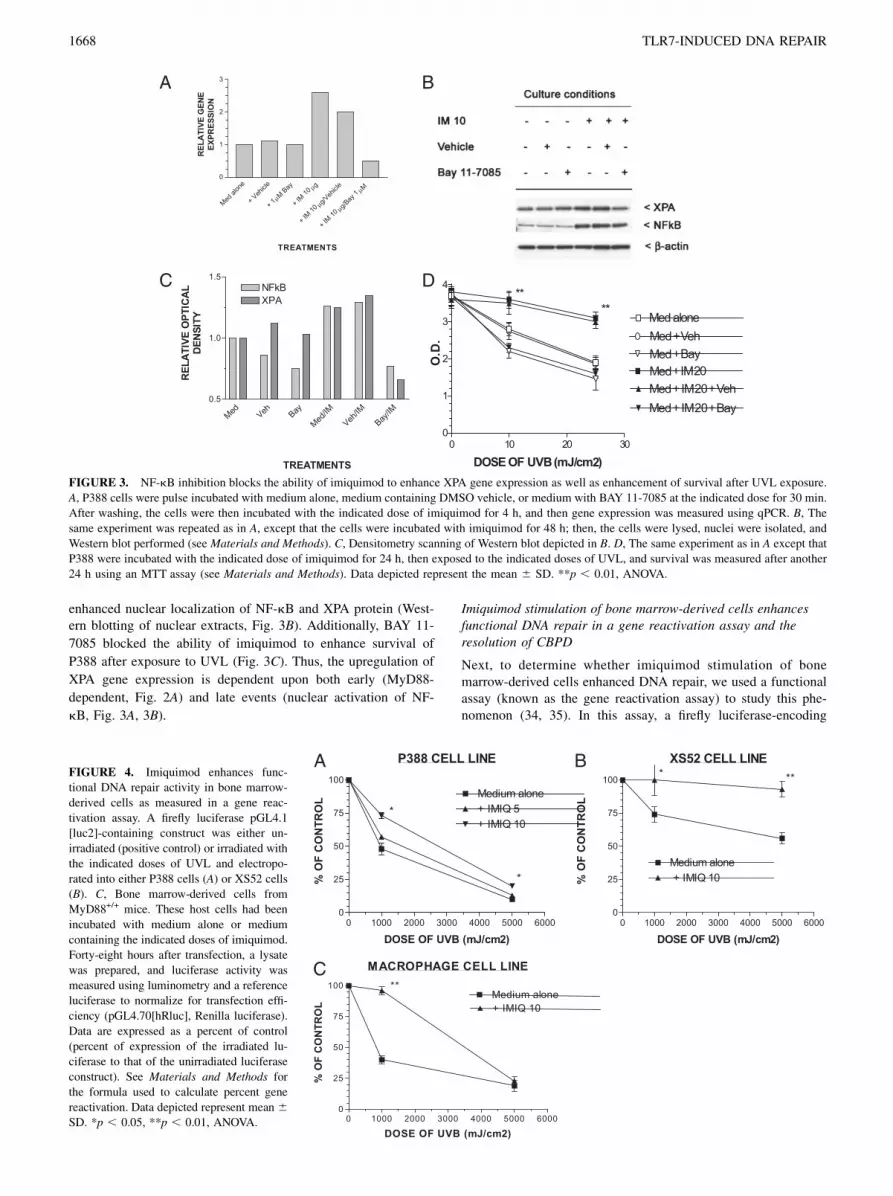
enhanced nuclear localization of NF-kB and XPA protein (West-ern blotting of nuclear extracts, Fig. 3B). Additionally, BAY 11-
7085 blocked the ability of imiquimod to enhance survival of
P388 after exposure to UVL (Fig. 3C). Thus, the upregulation of
XPA gene expression is dependent upon both early (MyD88-
dependent, Fig. 2A) and late events (nuclear activation of NF-
kB, Fig. 3A, 3B).
Imiquimod stimulation of bone marrow-derived cells enhancesfunctional DNA repair in a gene reactivation assay and theresolution of CBPD
Next, to determine whether imiquimod stimulation of bonemarrow-derived cells enhanced DNA repair, we used a functionalassay (known as the gene reactivation assay) to study this phe-nomenon (34, 35). In this assay, a firefly luciferase-encoding
FIGURE 4. Imiquimod enhances func-
tional DNA repair activity in bone marrow-
derived cells as measured in a gene reac-
tivation assay. A firefly luciferase pGL4.1
[luc2]-containing construct was either un-
irradiated (positive control) or irradiated with
the indicated doses of UVL and electropo-
rated into either P388 cells (A) or XS52 cells
(B). C, Bone marrow-derived cells from
MyD88+/+ mice. These host cells had been
incubated with medium alone or medium
containing the indicated doses of imiquimod.
Forty-eight hours after transfection, a lysate
was prepared, and luciferase activity was
measured using luminometry and a reference
luciferase to normalize for transfection effi-
ciency (pGL4.70[hRluc], Renilla luciferase).
Data are expressed as a percent of control
(percent of expression of the irradiated lu-
ciferase to that of the unirradiated luciferase
construct). See Materials and Methods for
the formula used to calculate percent gene
reactivation. Data depicted represent mean 6SD. *p , 0.05, **p , 0.01, ANOVA.
FIGURE 3. NF-kB inhibition blocks the ability of imiquimod to enhance XPA gene expression as well as enhancement of survival after UVL exposure.
A, P388 cells were pulse incubated with medium alone, medium containing DMSO vehicle, or medium with BAY 11-7085 at the indicated dose for 30 min.
After washing, the cells were then incubated with the indicated dose of imiquimod for 4 h, and then gene expression was measured using qPCR. B, The
same experiment was repeated as in A, except that the cells were incubated with imiquimod for 48 h; then, the cells were lysed, nuclei were isolated, and
Western blot performed (see Materials and Methods). C, Densitometry scanning of Western blot depicted in B. D, The same experiment as in A except that
P388 were incubated with the indicated dose of imiquimod for 24 h, then exposed to the indicated doses of UVL, and survival was measured after another
24 h using an MTT assay (see Materials and Methods). Data depicted represent the mean 6 SD. **p , 0.01, ANOVA.
1668 TLR7-INDUCED DNA REPAIR
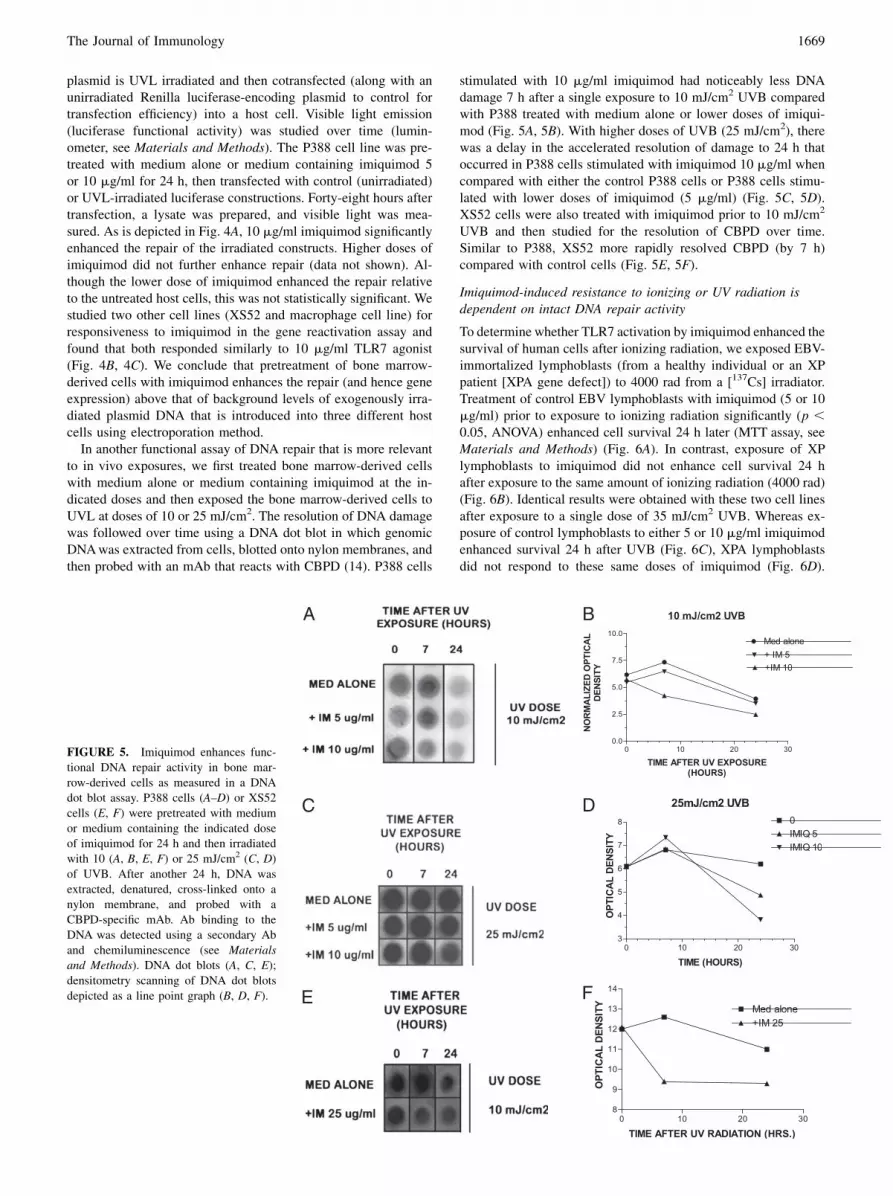
plasmid is UVL irradiated and then cotransfected (along with anunirradiated Renilla luciferase-encoding plasmid to control fortransfection efficiency) into a host cell. Visible light emission(luciferase functional activity) was studied over time (lumin-ometer, see Materials and Methods). The P388 cell line was pre-treated with medium alone or medium containing imiquimod 5or 10 mg/ml for 24 h, then transfected with control (unirradiated)or UVL-irradiated luciferase constructions. Forty-eight hours aftertransfection, a lysate was prepared, and visible light was mea-sured. As is depicted in Fig. 4A, 10 mg/ml imiquimod significantlyenhanced the repair of the irradiated constructs. Higher doses ofimiquimod did not further enhance repair (data not shown). Al-though the lower dose of imiquimod enhanced the repair relativeto the untreated host cells, this was not statistically significant. Westudied two other cell lines (XS52 and macrophage cell line) forresponsiveness to imiquimod in the gene reactivation assay andfound that both responded similarly to 10 mg/ml TLR7 agonist(Fig. 4B, 4C). We conclude that pretreatment of bone marrow-derived cells with imiquimod enhances the repair (and hence geneexpression) above that of background levels of exogenously irra-diated plasmid DNA that is introduced into three different hostcells using electroporation method.In another functional assay of DNA repair that is more relevant
to in vivo exposures, we first treated bone marrow-derived cellswith medium alone or medium containing imiquimod at the in-dicated doses and then exposed the bone marrow-derived cells toUVL at doses of 10 or 25 mJ/cm2. The resolution of DNA damagewas followed over time using a DNA dot blot in which genomicDNAwas extracted from cells, blotted onto nylon membranes, andthen probed with an mAb that reacts with CBPD (14). P388 cells
stimulated with 10 mg/ml imiquimod had noticeably less DNAdamage 7 h after a single exposure to 10 mJ/cm2 UVB comparedwith P388 treated with medium alone or lower doses of imiqui-mod (Fig. 5A, 5B). With higher doses of UVB (25 mJ/cm2), therewas a delay in the accelerated resolution of damage to 24 h thatoccurred in P388 cells stimulated with imiquimod 10 mg/ml whencompared with either the control P388 cells or P388 cells stimu-lated with lower doses of imiquimod (5 mg/ml) (Fig. 5C, 5D).XS52 cells were also treated with imiquimod prior to 10 mJ/cm2
UVB and then studied for the resolution of CBPD over time.Similar to P388, XS52 more rapidly resolved CBPD (by 7 h)compared with control cells (Fig. 5E, 5F).
Imiquimod-induced resistance to ionizing or UV radiation isdependent on intact DNA repair activity
To determine whether TLR7 activation by imiquimod enhanced thesurvival of human cells after ionizing radiation, we exposed EBV-immortalized lymphoblasts (from a healthy individual or an XPpatient [XPA gene defect]) to 4000 rad from a [137Cs] irradiator.Treatment of control EBV lymphoblasts with imiquimod (5 or 10mg/ml) prior to exposure to ionizing radiation significantly (p ,0.05, ANOVA) enhanced cell survival 24 h later (MTT assay, seeMaterials and Methods) (Fig. 6A). In contrast, exposure of XPlymphoblasts to imiquimod did not enhance cell survival 24 hafter exposure to the same amount of ionizing radiation (4000 rad)(Fig. 6B). Identical results were obtained with these two cell linesafter exposure to a single dose of 35 mJ/cm2 UVB. Whereas ex-posure of control lymphoblasts to either 5 or 10 mg/ml imiquimodenhanced survival 24 h after UVB (Fig. 6C), XPA lymphoblastsdid not respond to these same doses of imiquimod (Fig. 6D).
FIGURE 5. Imiquimod enhances func-
tional DNA repair activity in bone mar-
row-derived cells as measured in a DNA
dot blot assay. P388 cells (A–D) or XS52
cells (E, F) were pretreated with medium
or medium containing the indicated dose
of imiquimod for 24 h and then irradiated
with 10 (A, B, E, F) or 25 mJ/cm2 (C, D)
of UVB. After another 24 h, DNA was
extracted, denatured, cross-linked onto a
nylon membrane, and probed with a
CBPD-specific mAb. Ab binding to the
DNA was detected using a secondary Ab
and chemiluminescence (see Materials
and Methods). DNA dot blots (A, C, E);
densitometry scanning of DNA dot blots
depicted as a line point graph (B, D, F).
The Journal of Immunology 1669
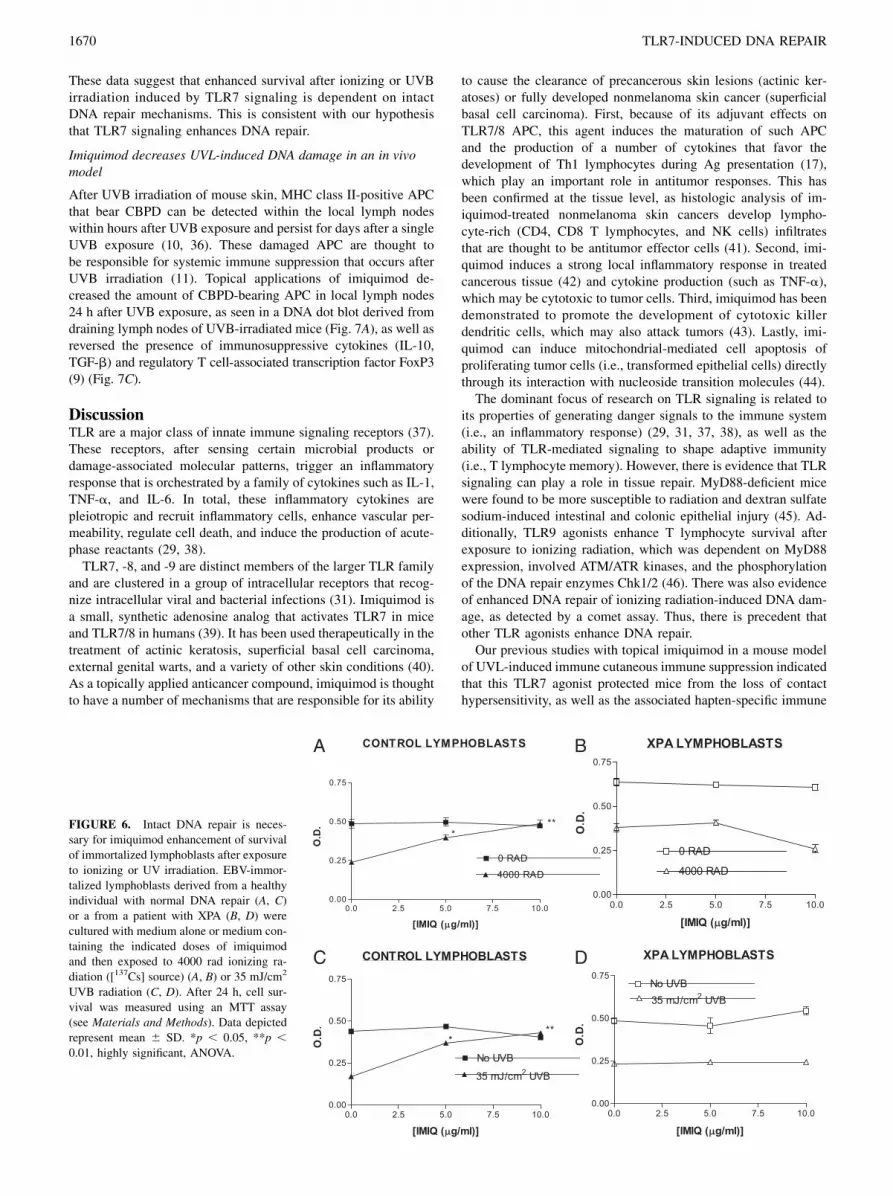
These data suggest that enhanced survival after ionizing or UVBirradiation induced by TLR7 signaling is dependent on intactDNA repair mechanisms. This is consistent with our hypothesisthat TLR7 signaling enhances DNA repair.
Imiquimod decreases UVL-induced DNA damage in an in vivomodel
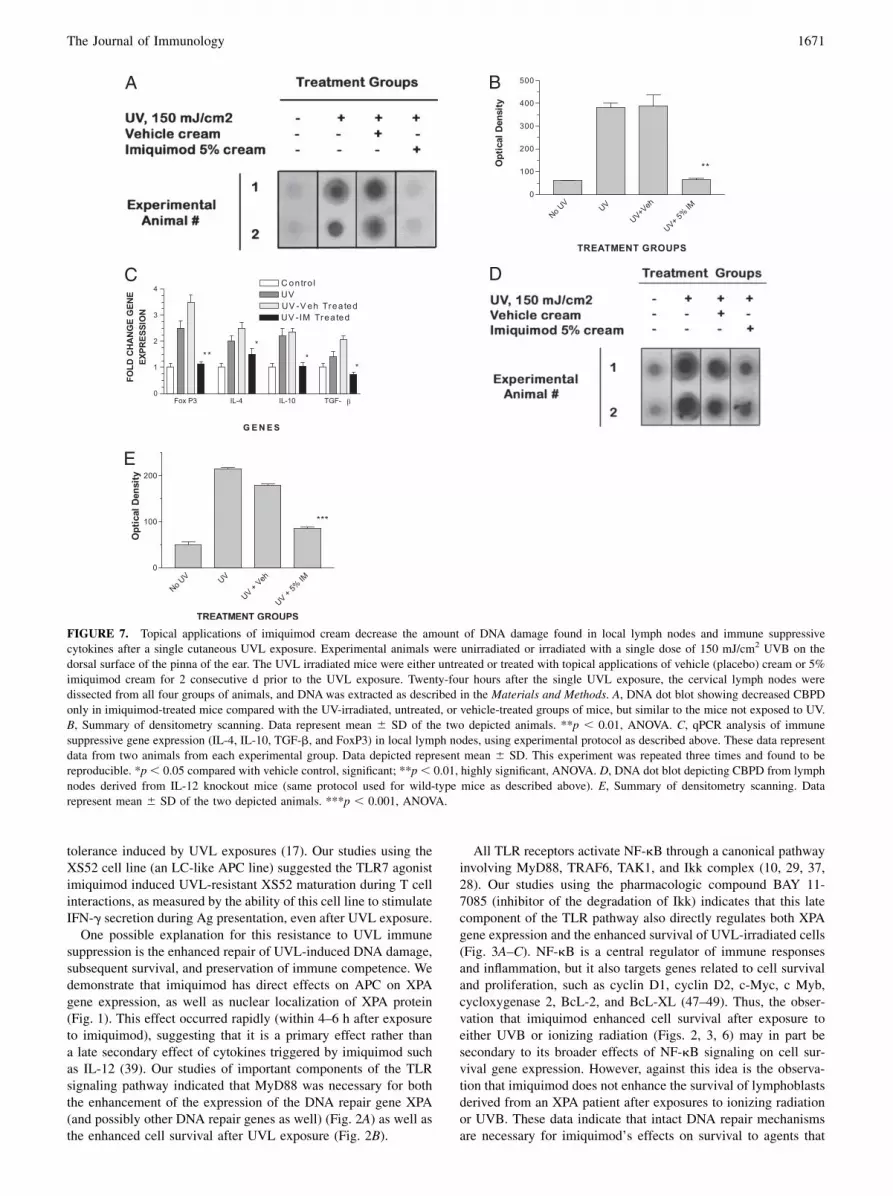
After UVB irradiation of mouse skin, MHC class II-positive APCthat bear CBPD can be detected within the local lymph nodeswithin hours after UVB exposure and persist for days after a singleUVB exposure (10, 36). These damaged APC are thought tobe responsible for systemic immune suppression that occurs afterUVB irradiation (11). Topical applications of imiquimod de-creased the amount of CBPD-bearing APC in local lymph nodes24 h after UVB exposure, as seen in a DNA dot blot derived fromdraining lymph nodes of UVB-irradiated mice (Fig. 7A), as well asreversed the presence of immunosuppressive cytokines (IL-10,TGF-b) and regulatory T cell-associated transcription factor FoxP3(9) (Fig. 7C).
DiscussionTLR are a major class of innate immune signaling receptors (37).These receptors, after sensing certain microbial products ordamage-associated molecular patterns, trigger an inflammatoryresponse that is orchestrated by a family of cytokines such as IL-1,TNF-a, and IL-6. In total, these inflammatory cytokines arepleiotropic and recruit inflammatory cells, enhance vascular per-meability, regulate cell death, and induce the production of acute-phase reactants (29, 38).TLR7, -8, and -9 are distinct members of the larger TLR family
and are clustered in a group of intracellular receptors that recog-nize intracellular viral and bacterial infections (31). Imiquimod isa small, synthetic adenosine analog that activates TLR7 in miceand TLR7/8 in humans (39). It has been used therapeutically in thetreatment of actinic keratosis, superficial basal cell carcinoma,external genital warts, and a variety of other skin conditions (40).As a topically applied anticancer compound, imiquimod is thoughtto have a number of mechanisms that are responsible for its ability
to cause the clearance of precancerous skin lesions (actinic ker-atoses) or fully developed nonmelanoma skin cancer (superficialbasal cell carcinoma). First, because of its adjuvant effects onTLR7/8 APC, this agent induces the maturation of such APCand the production of a number of cytokines that favor thedevelopment of Th1 lymphocytes during Ag presentation (17),which play an important role in antitumor responses. This hasbeen confirmed at the tissue level, as histologic analysis of im-iquimod-treated nonmelanoma skin cancers develop lympho-cyte-rich (CD4, CD8 T lymphocytes, and NK cells) infiltratesthat are thought to be antitumor effector cells (41). Second, imi-quimod induces a strong local inflammatory response in treatedcancerous tissue (42) and cytokine production (such as TNF-a),which may be cytotoxic to tumor cells. Third, imiquimod has beendemonstrated to promote the development of cytotoxic killerdendritic cells, which may also attack tumors (43). Lastly, imi-quimod can induce mitochondrial-mediated cell apoptosis ofproliferating tumor cells (i.e., transformed epithelial cells) directlythrough its interaction with nucleoside transition molecules (44).The dominant focus of research on TLR signaling is related to
its properties of generating danger signals to the immune system(i.e., an inflammatory response) (29, 31, 37, 38), as well as theability of TLR-mediated signaling to shape adaptive immunity(i.e., T lymphocyte memory). However, there is evidence that TLRsignaling can play a role in tissue repair. MyD88-deficient micewere found to be more susceptible to radiation and dextran sulfatesodium-induced intestinal and colonic epithelial injury (45). Ad-ditionally, TLR9 agonists enhance T lymphocyte survival afterexposure to ionizing radiation, which was dependent on MyD88expression, involved ATM/ATR kinases, and the phosphorylationof the DNA repair enzymes Chk1/2 (46). There was also evidenceof enhanced DNA repair of ionizing radiation-induced DNA dam-age, as detected by a comet assay. Thus, there is precedent thatother TLR agonists enhance DNA repair.Our previous studies with topical imiquimod in a mouse model
of UVL-induced immune cutaneous immune suppression indicatedthat this TLR7 agonist protected mice from the loss of contacthypersensitivity, as well as the associated hapten-specific immune
FIGURE 6. Intact DNA repair is neces-
sary for imiquimod enhancement of survival
of immortalized lymphoblasts after exposure
to ionizing or UV irradiation. EBV-immor-
talized lymphoblasts derived from a healthy
individual with normal DNA repair (A, C)
or a from a patient with XPA (B, D) were
cultured with medium alone or medium con-
taining the indicated doses of imiquimod
and then exposed to 4000 rad ionizing ra-
diation ([137Cs] source) (A, B) or 35 mJ/cm2
UVB radiation (C, D). After 24 h, cell sur-
vival was measured using an MTT assay
(see Materials and Methods). Data depicted
represent mean 6 SD. *p , 0.05, **p ,0.01, highly significant, ANOVA.
1670 TLR7-INDUCED DNA REPAIR
tolerance induced by UVL exposures (17). Our studies using theXS52 cell line (an LC-like APC line) suggested the TLR7 agonistimiquimod induced UVL-resistant XS52 maturation during T cellinteractions, as measured by the ability of this cell line to stimulateIFN-g secretion during Ag presentation, even after UVL exposure.One possible explanation for this resistance to UVL immune
suppression is the enhanced repair of UVL-induced DNA damage,subsequent survival, and preservation of immune competence. Wedemonstrate that imiquimod has direct effects on APC on XPAgene expression, as well as nuclear localization of XPA protein(Fig. 1). This effect occurred rapidly (within 4–6 h after exposureto imiquimod), suggesting that it is a primary effect rather thana late secondary effect of cytokines triggered by imiquimod suchas IL-12 (39). Our studies of important components of the TLRsignaling pathway indicated that MyD88 was necessary for boththe enhancement of the expression of the DNA repair gene XPA(and possibly other DNA repair genes as well) (Fig. 2A) as well asthe enhanced cell survival after UVL exposure (Fig. 2B).
All TLR receptors activate NF-kB through a canonical pathwayinvolving MyD88, TRAF6, TAK1, and Ikk complex (10, 29, 37,28). Our studies using the pharmacologic compound BAY 11-7085 (inhibitor of the degradation of Ikk) indicates that this latecomponent of the TLR pathway also directly regulates both XPAgene expression and the enhanced survival of UVL-irradiated cells(Fig. 3A–C). NF-kB is a central regulator of immune responsesand inflammation, but it also targets genes related to cell survivaland proliferation, such as cyclin D1, cyclin D2, c-Myc, c Myb,cycloxygenase 2, BcL-2, and BcL-XL (47–49). Thus, the obser-vation that imiquimod enhanced cell survival after exposure toeither UVB or ionizing radiation (Figs. 2, 3, 6) may in part besecondary to its broader effects of NF-kB signaling on cell sur-vival gene expression. However, against this idea is the observa-tion that imiquimod does not enhance the survival of lymphoblastsderived from an XPA patient after exposures to ionizing radiationor UVB. These data indicate that intact DNA repair mechanismsare necessary for imiquimod’s effects on survival to agents that
FIGURE 7. Topical applications of imiquimod cream decrease the amount of DNA damage found in local lymph nodes and immune suppressive
cytokines after a single cutaneous UVL exposure. Experimental animals were unirradiated or irradiated with a single dose of 150 mJ/cm2 UVB on the
dorsal surface of the pinna of the ear. The UVL irradiated mice were either untreated or treated with topical applications of vehicle (placebo) cream or 5%
imiquimod cream for 2 consecutive d prior to the UVL exposure. Twenty-four hours after the single UVL exposure, the cervical lymph nodes were
dissected from all four groups of animals, and DNAwas extracted as described in the Materials and Methods. A, DNA dot blot showing decreased CBPD
only in imiquimod-treated mice compared with the UV-irradiated, untreated, or vehicle-treated groups of mice, but similar to the mice not exposed to UV.
B, Summary of densitometry scanning. Data represent mean 6 SD of the two depicted animals. **p , 0.01, ANOVA. C, qPCR analysis of immune
suppressive gene expression (IL-4, IL-10, TGF-b, and FoxP3) in local lymph nodes, using experimental protocol as described above. These data represent
data from two animals from each experimental group. Data depicted represent mean 6 SD. This experiment was repeated three times and found to be
reproducible. *p, 0.05 compared with vehicle control, significant; **p, 0.01, highly significant, ANOVA. D, DNA dot blot depicting CBPD from lymph
nodes derived from IL-12 knockout mice (same protocol used for wild-type mice as described above). E, Summary of densitometry scanning. Data
represent mean 6 SD of the two depicted animals. ***p , 0.001, ANOVA.
The Journal of Immunology 1671

damage genomic DNA (in this case, ionizing radiation or UVB)(Fig. 6).Our data indicate that TLR7 signaling can enhance the XPA gene
expression and nuclear localization. XPA is involved in recognitionof UVB damage and is thought to be an important early componentof nucleotide excision repair of UV-induced CBPD (27). Althoughnucleotide excision repair genes are constitutively expressed, thereis evidence that they are inducible (49, 50). Cytokines such as IL-12 and IL-18, natural products such as green tea polyphenols, andDNA oligonucleotides all enhance nucleotide excision repair ofUV-damaged DNA (12–15, 51–54). Imiquimod also induces thenucleotide excision repair gene XPA (and presumably other nu-cleotide excision repair genes) as well as promoting functionalDNA repair (gene reactivation and DNA dot blot assays, Figs. 4,5).Our studies of the local (cervical) lymph nodes after UVB ir-
radiation of the dorsal surface of the pinna of the ear demonstratethat imiquimod treatment prevents the appearance of APC-bearingCBPD (Fig. 7A, 7B) and prevents the expression of immunosup-pressive genes (IL-4, IL-10, TGF-b, and Foxp3) (Fig. 7C) in theskin- associated lymphoid tissue, preventing systemic immunesuppression and the loss of contact sensitization, as we had pre-viously demonstrated (16). Although imiquimod induced less ef-ficient resolution of UV-induced CBPD in the local lymph nodesof IL-12 knockout mice (Figs. 7D, 7E) when compared with wild-type mice (Fig. 7A, 7B), there remained significant DNA repair inthese gene-targeted mice. These data indicate that there is TLR7-inducible DNA repair even in the absence of IL-12, suggestingthat TLR7 signaling has direct effects on DNA repair.In summary, the TLR7 agonist imiquimod enhances DNA ex-
cision repair of UVB-induced CBPD. Thus, in addition to trig-gering inflammation and acting as an adjuvant, this topicallyapplied anticancer agent also serves to protect the immune systemagainst the deleterious effects of UVB by promoting the repairand subsequent survival of UVB damage by APC. These data alsoimply that imiquimod and its related analogs can be used as atherapy to prevent skin cancer by accelerating the repair of UV-induced DNA damage in cutaneous APC, thus preserving im-mune competence.
DisclosuresT.-C.M. and J.L. are employees of Graceway Pharmaceuticals. The other
authors have no financial conflicts of interest.
References1. Beissert, S., and T. Schwarz. 2009. Ultraviolet-induced immunosuppression:
implications for photocarcinogenesis. Cancer Treat. Res. 146: 109–121.2. Murphy, G. M. 2009. Ultraviolet radiation and immunosuppression. Br. J.
Dermatol. 161(Suppl 3): 90–95.3. Ghoreishi, M., and J. P. Dutz. 2006. Tolerance induction by transcutaneous
immunization through ultraviolet-irradiated skin is transferable through CD4+CD25+ T regulatory cells and is dependent on host-derived IL-10. J. Immunol.176: 2635–2644.
4. Rivas, J. M., and S. E. Ullrich. 1992. Systemic suppression of delayed-typehypersensitivity by supernatants from UV-irradiated keratinocytes. An essen-tial role for keratinocyte-derived IL-10. J. Immunol. 149: 3865–3871.
5. Schwarz, A., M. Noordegraaf, A. Maeda, K. Torii, B. E. Clausen, andT. Schwarz. 2010. Langerhans cells are required for UVR-induced immuno-suppression. J. Invest. Dermatol. 130: 1419–1427.
6. Toichi, E., T. S. McCormick, and K. D. Cooper. 2002. Cell surface and cytokinephenotypes of skin immunocompetent cells involved in ultraviolet-induced im-munosuppression. Methods 28: 104–110.
7. Simon, J. C., T. Mosmann, D. Edelbaum, E. Schopf, P. R. Bergstresser, andP. D. Cruz Jr. 1994. In vivo evidence that ultraviolet B-induced suppression ofallergic contact sensitivity is associated with functional inactivation of Th1 cells.Photodermatol. Photoimmunol. Photomed. 10: 206–211.
8. Saijo, S., E. Kodari, M. L. Kripke, and F. M. Strickland. 1996. UVB irradiationdecreases the magnitude of the Th1 response to hapten but does not increase theTh2 response. Photodermatol. Photoimmunol. Photomed. 12: 145–153.
9. Maeda, A., S. Beissert, T. Schwarz, and A. Schwarz. 2008. Phenotypic andfunctional characterization of ultraviolet radiation-induced regulatory T cells. J.Immunol. 180: 3065–3071.
10. Sontag, Y., C. L. Guikers, A. A. Vink, F. R. de Gruijl, H. van Loveren,J. Garssen, L. Roza, M. L. Kripke, J. C. van der Leun, and W. A. van Vloten.1995. Cells with UV-specific DNA damage are present in murine lymph nodesafter in vivo UV irradiation. J. Invest. Dermatol. 104: 734–738.
11. Fisher, M. S., and M. L. Kripke. 1977. Systemic alteration induced in mice byultraviolet light irradiation and its relationship to ultraviolet carcinogenesis.Proc. Natl. Acad. Sci. USA 74: 1688–1692.
12. Yarosh, D., J. Klein, A. O’Connor, J. Hawk, E. Rafal, P. Wolf; XerodermaPigmentosum Study Group. 2001. Effect of topically applied T4 endonuclease Vin liposomes on skin cancer in xeroderma pigmentosum: a randomised study.Lancet 357: 926–929.
13. Schwarz, A., A. Maeda, D. Gan, T. Mammone, M. S. Matsui, and T. Schwarz.2008. Green tea phenol extracts reduce UVB-induced DNA damage in humancells via interleukin-12. Photochem. Photobiol. 84: 350–355.
14. Schwarz, A., S. Stander, M. Berneburg, M. Bohm, D. Kulms, H. van Steeg,K. Grosse-Heitmeyer, J. Krutmann, and T. Schwarz. 2002. Interleukin-12 sup-presses ultraviolet radiation-induced apoptosis by inducing DNA repair. Nat.Cell Biol. 4: 26–31.
15. Majewski, S., C. Jantschitsch, A. Maeda, T. Schwarz, and A. Schwarz. 2010. IL-23 antagonizes UVR-induced immunosuppression through two mechanisms:reduction of UVR-induced DNA damage and inhibition of UVR-induced regu-latory T cells. J. Invest. Dermatol. 130: 554–562.
16. Sreevidya, C. S., A. Fukunaga, N. M. Khaskhely, T. Masaki, R. Ono,C. Nishigori, and S. E. Ullrich. 2010. Agents that reverse UV-Induced immunesuppression and photocarcinogenesis affect DNA repair. J. Invest. Dermatol.130: 1428–1437.
17. Thatcher, T. H., I. Luzina, R. Fishelevich, and A. A. Gaspari. 2006. Topicalimiquimod treatment prevents ultraviolet-light induced loss of contact hyper-sensitivity and immune tolerance. J. Investig. Dermatol. 126: 821–831.
18. Toews, G. B., P. R. Bergstresser, and J. W. Streilein. 1980. Epidermal Langer-hans cell density determines whether contact hypersensitivity or un-responsiveness follows skin painting with DNFB. J. Immunol. 124: 445–453.
19. Elmets, C. A., P. R. Bergstresser, R. E. Tigelaar, P. J. Wood, and J. W. Streilein.1983. Analysis of the mechanism of unresponsiveness produced by haptenspainted on skin exposed to low dose ultraviolet radiation. J. Exp. Med. 158: 781–794.
20. Magram, J., S. E. Connaughton, R. R. Warrier, D. M. Carvajal, C. Y. Wu,J. Ferrante, C. Stewart, U. Sarmiento, D. A. Faherty, and M. K. Gately. 1996. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokineresponses. Immunity 4: 471–481.
21. Elliott, J., M. B. Coulter-Mackie, J. H. Jung, D. I. Rodenhiser, and S. M. Singh.1991. A method for transforming lymphocytes from very small blood volumessuitable for paediatric samples. Hum. Genet. 86: 615–616.
22. Heo, D. S., J. -G. Park, K. Hata, R. Day, R. B. Herberman, and T. L. Whiteside.1990. Evaluation of tetrazolium-based semiautomatic colorimetric assay formeasurement of human antitumor cytotoxicity. Cancer Res. 50: 3681–3690.
23. Sikder, H., Y. Zhao, A. Balato, A. Chapoval, R. Fishelevich, P. Gade, I. S. Singh,D. V. Kalvakolanu, P. F. Johnson, and A. A. Gaspari. 2009. A central role fortranscription factor C/EBP-b in regulating CD1d gene expression in humankeratinocytes. J. Immunol. 183: 1657–1666.
24. Ade, N., D. Antonios, S. Kerdine-Romer, F. Boisleve, F. Rousset, andM. Pallardy. 2007. NF-kappaB plays a major role in the maturation of humandendritic cells induced by NiSO(4) but not by DNCB. Toxicol. Sci. 99: 488–501.
25. Zhao, Y., R. Fishelevich, J. P. Petrali, L. Zheng, M. A. Anatolievna, A. Deng,R. L. Eckert, and A. A. Gaspari. 2008. Activation of keratinocyte protein kinaseC zeta in psoriasis plaques. J. Invest. Dermatol. 128: 2190–2197.
26. Fishelevich, R., A. Malanina, I. Luzina, S. Atamas, M. J. Smyth, S. A. Porcelli,and A. A. Gaspari. 2006. Ceramide-dependent regulation of human epidermalkeratinocyte CD1d expression during terminal differentiation. J. Immunol. 176:2590–2599.
27. Moriwaki, S. -I., and K. H. Kraemer. 2001. Xeroderma pigmentosum—bridginga gap between clinic and laboratory. Photodermatol. Photoimmunol. Photomed.17: 47–54.
28. Gorden, K. B., K. S. Gorski, S. J. Gibson, R. M. Kedl, W. C. Kieper, X. Qiu,M. A. Tomai, S. S. Alkan, and J. P. Vasilakos. 2005. Synthetic TLR agonistsreveal functional differences between human TLR7 and TLR8. J. Immunol. 174:1259–1268.
29. Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innateimmunity. Cell 124: 783–801.
30. Kawai, T., and S. Akira. 2010. The role of pattern-recognition receptors in innateimmunity: update on Toll-like receptors. Nat. Immunol. 11: 373–384.
31. Blasius, A. L., and B. Beutler. 2010. Intracellular toll-like receptors. Immunity32: 305–315.
32. Ziegler, A., A. S. Jonason, D. J. Leffell, J. A. Simon, H. W. Sharma,J. Kimmelman, L. Remington, T. Jacks, and D. E. Brash. 1994. Sunburn and p53in the onset of skin cancer. Nature 372: 773–776.
33. Brash, D. E., A. Ziegler, A. S. Jonason, J. A. Simon, S. Kunala, and D. J. Leffell.1996. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumorpromotion. J. Investig. Dermatol. Symp. Proc. 1: 136–142.
34. Carreau, M., E. Eveno, X. Quilliet, O. Chevalier-Lagente, A. Benoit,B. Tanganelli, M. Stefanini, W. Vermeulen, J. H. Hoeijmakers, A. Sarasin, et al.1995. Development of a new easy complementation assay for DNA repair de-ficient human syndromes using cloned repair genes. Carcinogenesis 16: 1003–1009.
1672 TLR7-INDUCED DNA REPAIR

35. Khan, S. G., H. L. Levy, R. Legerski, E. Quackenbush, J. T. Reardon, S. Emmert,A. Sancar, L. Li, T. D. Schneider, J. E. Cleaver, and K. H. Kraemer. 1998.Xeroderma pigmentosum group C splice mutation associated with autism andhypoglycinemia. J. Invest. Dermatol. 111: 791–796.
36. Kripke, M. L., P. A. Cox, L. G. Alas, and D. B. Yarosh. 1992. Pyrimidine dimersin DNA initiate systemic immunosuppression in UV-irradiated mice. Proc. Natl.Acad. Sci. USA 89: 7516–7520.
37. Takeuchi, O., and S. Akira. 2010. Pattern recognition receptors and in-flammation. Cell 140: 805–820.
38. Beutler, B., Z. Jiang, P. Georgel, K. Crozat, B. Croker, S. Rutschmann, X. Du,and K. Hoebe. 2006. Genetic analysis of host resistance: Toll-like receptorsignaling and immunity at large. Annu. Rev. Immunol. 24: 353–389.
39. Larange, A., D. Antonios, M. Pallardy, and S. Kerdine-Romer. 2009. TLR7 andTLR8 agonists trigger different signaling pathways for human dendritic cellmaturation. J. Leukoc. Biol. 85: 673–683.
40. A Gaspari, A., S. K. Tyring, and T. Rosen. 2009. Beyond a decade of 5% imi-quimod topical therapy. J. Drugs Dermatol. 8: 467–474.
41. Sullivan, T. P., T. Dearaujo, V. Vincek, and B. Berman. 2003. Evaluation ofsuperficial basal cell carcinomas after treatment with imiquimod 5% cream orvehicle for apoptosis and lymphocyte phenotyping. Dermatol. Surg. 29: 1181–1186.
42. Hurwitz, D. J., L. Pincus, and T. S. Kupper. 2003. Imiquimod: a topically appliedlink between innate and acquired immunity. Arch. Dermatol. 139: 1347–1350.
43. Stary, G., C. Bangert, M. Tauber, R. Strohal, T. Kopp, and G. Stingl. 2007.Tumoricidal activity of TLR7/8-activated inflammatory dendritic cells. J. Exp.Med. 204: 1441–1451.
44. Schon, M. P., and M. Schon. 2004. Immune modulation and apoptosis induc-tion: two sides of the antitumoral activity of imiquimod. Apoptosis 9: 291–298.
45. Rakoff-Nahoum, S., J. Paglino, F. Eslami-Varzaneh, S. Edberg, andR. Medzhitov. 2004. Recognition of commensal microflora by toll-like receptorsis required for intestinal homeostasis. Cell 118: 229–241.
46. Zheng, L., N. Asprodites, A. H. Keene, P. Rodriguez, K. D. Brown, andE. Davila. 2008. TLR9 engagement on CD4 T lymphocytes represses gamma-radiation-induced apoptosis through activation of checkpoint kinase responseelements. Blood 111: 2704–2713.
47. Li, X., S. Jiang, and R. I. Tapping. 2010. Toll-like receptor signaling in cellproliferation and survival. Cytokine 49: 1–9.
48. Clement, J. F., S. Meloche, and M. J. Servant. 2008. The IKK-related kinases:from innate immunity to oncogenesis. Cell Res. 18: 889–899.
49. Vallabhapurapu, S., and M. Karin. 2009. Regulation and function of NF-kappaBtranscription factors in the immune system. Annu. Rev. Immunol. 27: 693–733.
50. Schwarz, T., and A. Schwarz. 2009. DNA repair and cytokine responses. J.Investig. Dermatol. Symp. Proc. 14: 63–66.
51. Arad, S., N. Konnikov, D. A. Goukassian, and B. A. Gilchrest. 2006. T-oligosaugment UV-induced protective responses in human skin. FASEB J. 20: 1895–1897.
52. Goukassian, D. A., E. Helms, H. van Steeg, C. van Oostrom, J. Bhawan, andB. A. Gilchrest. 2004. Topical DNA oligonucleotide therapy reduces UV-induced mutations and photocarcinogenesis in hairless mice. Proc. Natl. Acad.Sci. USA 101: 3933–3938.
53. Katiyar, S. K., C. A. Elmets, and R. Agarwal. 1995. Protection againstultraviolet-B radiation-induced local and systemic suppression of contact hy-persensitivity and edema responses in C3H/HeN mice by green tea polyphenols.Photochem. Photobiol. 62: 855–861.
54. Meeran, S. M., S. K. Mantena, and S. K. Katiyar. 2006. (-)-Epigallocatechin-3-gallate prevents photocarcinogenesis in mice through interleukin-12-dependentDNA repair. Cancer Res. 66: 5512–5520.
The Journal of Immunology 1673