fibrosing alveolitis in polymyositisa review of histologically confirmed cases
TRANSCRIPT

CASE REPORTS
Fibrosing Alveolitis in Polymyositis
A Review of Histologically Confirmed Cases
PHILIP E. DUNCAN, M.D.’
JOHN P. GRIFFIN, M.D.
ABRAHAM GARCIA, M.D.
STANLEY B. KAPLAN, M.D.
Memghis, Tennessee
From the Sections of Pulmonary Disease and Rheumatology, Medical Service, Veterans Ad- ministration Hospital, and Department of Medi- cine, University of Tennessee College of Medi- cine, Memphis, Tennessee. Requests for re- prints should be addressed to Dr. John P. Griffin, VA Hospital, 1030 Jefferson Avenue, Memphis, Tennessee 38104. Manuscript accepted De- cember 27, 1973.
* Present address: Fayetteville Diagnostic Center, 675 Lollar Lane, Fayetteville. Arkansas 72701.
Fibrosing alveolitis has been reported in polymyositis. Two such cases are presented and 12 previously reported cases in which there was histologic confirmation of these diagnoses are re- viewed. In half the cases, respiratory manifestations preceded
evidence of inflammatory muscle disease. In 12 of the 14 cases, respiratory signs and symptoms dominated the clinical picture and often initially masked the underlying rheumatologic disease. Patients with extensive, predominant pulmonary fibrosis fared poorly, and several did not respond to corticosteroid therapy; however, patients with actively inflamed lesions responded well. In cases of fibrosing alveolitis one should consider the possibility of an unrecognized connective tissue disorder, including polymy- ositis. Furthermore, pulmonary histology is useful in planning ther-
apy and in predicting prognosis.
The term fibrosing alveolitis (interstitial pneumonitis/fibrosis) has
been used to designate a spectrum of predominantly idiopathic pul-
monary diseases characterized by varying degrees of inflammation
and scarring in the interstitium and alveolar spaces of the lung [ 11.
In the broad sense, use of this term is appropriate to designate var-
ious expressions of pulmonary involvement in connective tissue
disorders. In contrast to the well documented occurrence of fibros-
ing alveolitis in systemic lupus erythematosus, scleroderma and
rheumatoid arthritis [ 2-41, its recognition in polymyositis has been
rarely reported. Only 12 histologically confirmed cases have ap-
peared in the literature over a 17 year period [ 5-151. We describe
two well studied patients with polymyositis and fibrosing alveolitis,
and discuss the implications of this association.
CASE REPORTS
Case 1. A 44 year old woman (T.M.) was admitted to the City of Mem-
phis Hospitals in June 1971 with progressive dyspnea of 3 weeks’ dura-
tion. One month prior to admission a pruritic, erythematous rash devel-
oped on her face and forearms followed by sore throat, myalgia, cough,
fever, and swelling and stiffness of her proximal interphalangeal joints.
She was treated for “bronchitis” with penicillin for 3 days; the rash and sore throat disappeared but the dry cough persisted and was followed by
the development of polyarthralgias, night sweats and progressive dysp-
nea. Her previous records included mild toxemia with pregn,ancy and the
documentation of AS hemoglobin by electrophoresis. Physical examination revealed an acutely ill, tachypneic woman with
October 1974 The American Journal of Medicine Volume 57 621
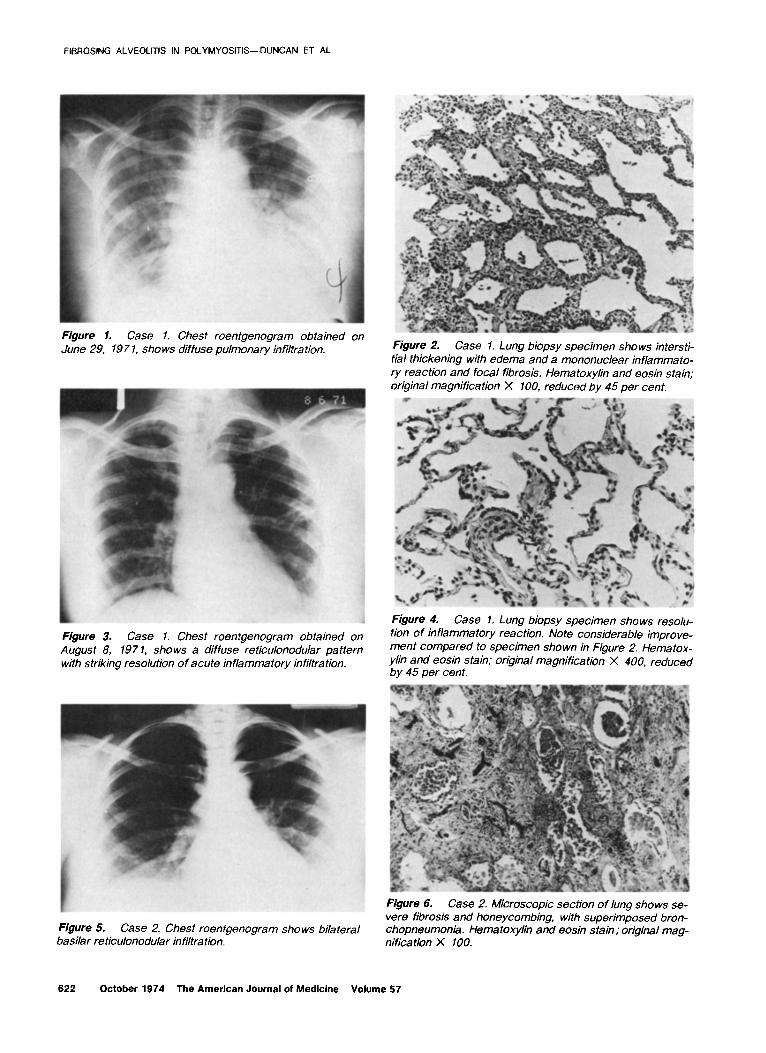
FIBROSING ALVEOLITIS IN POLYMYOSITIS-DUNCAN ET AL.
Figure 1. Case 1. Chest roentgenogram obtained on June 29, 197 1, shows diffuse pulmonary infiltration. Figure 2. Case 1. Lung biopsy specimen shows intersti-
tial thickening with edema and a mononuclear inflamma to- ry reaction and focal fibrosis. Hematoxylin and eosin stain; original magnification X 100, reduced by 45 per cent.
I
Figure 3. Case 1. Chest roentgenogram obtained on August 8, 197 1, shows a diffuse reticulonodular pattern with striking resolution of acute inflammatory infiltration.
Figure 4. Case 1. Lung biopsy specimen shows resolu- tion of inflammatory reaction. Note considerable improve- ment compared to specimen shown in Figure 2. Hematox- ylin and eosin stain; original magnification X 400, reduced by 45 per cent.
Figure 5. Case 2. Chest roentgenogram shows bilateral basilar reticulonodular infiltration.
Figure 6. Case 2. Microscopic section of lung shows se- vere fibrosis and honeycombing, with superimposed bron- chopneumonia. Hematoxylin and eosin stain; original mag- nification X 100.
622 October 1974 The American Journal of Medicine Volume 57

respirations 40/min, temperature 10l°F, pulse rate 1 lO/ min, restricted chest expansion, medium rales over the lower half of both lung fields, and striking weakness of the shoulder girdles. There were no skin lesions. The hemato- crit value was 35 per cent, leukocyte count 19,150/mm3 with 90 per cent neutrophils, and erythrocyte sedimenta- tion rate 54 mm/hour (Westergren). Urinalysis, serum urea nitrogen, electrolytes, serum protein electrophoresis and serum complement were within normal limits; Latex fixa- tion, antinuclear factor and lupus erythematosus cell tests were negative. Enzyme studies revealed creatine phos- phokinase (CPK) 580 mu/ml (normal 5 to 30), lactate de- hydrogenase (LDH) 1,200 mu/ml (normal 210 to 424), serum glutamic oxaloacetic transaminase (SGOT) 122 mu/ ml (normal 5 to 40) and aldolase 65 mu/ml (normal 1 to 8). The electrocardiogram showed sinus tachycardia. Chest roentgenogram (Figure 1) revealed diffuse and partially confluent infiltrates. Arterial blood gases showed arterial oxygen tension (Pa02) 38 mm Hg, arterial carbon dioxide tension (PaCOp) 28 mm Hg and pH 7.52. After the admin- istration of oxygen at 10 liters/min the Pa02 increased to 65 mm Hg. Sputum gram stain revealed mixed organisms. The initial impression was acute interstitial pneumonitis. The arthralgias and muscle weakness rapidly and com- pletely subsided during the first 3 days of hospitalization. The patient was treated initially with penicillin and later with cephalothin and kanamycin without any diminution in her temperature which was sustained at about 103’F, and the leukocyte count persisted at approximately 25,000/ mm3. Bacterial cultures of sputum and blood failed to re- veal pathogens, and studies for acid-fast bacilli and fungi were negative. On the 17th hospital day a trephine biopsy of the lung was performed in the right posterior axillary line. It revealed alveolar septal thickening and distortion, a marked inflammatory reaction with edema, areas of alveo- lar lining cell hyperplasia and focal fibrosis (Figure 2).
On the 18th hospital day, the administration of predni- sone 100 mg daily was begun with a dramatic resolution of fever and improvement in oxygenation and muscle strength. On the 30th hospital day, pulmonary function studies showed vital capacity (VC) 1.5 liters, total lung ca- pacity (TLC) 2.1 liters, forced expiratory volume at 1 sec- ond (FEV ,.,-,) 1.1 liters and maximum mid-expiratory flow rate (MMFR) 1.34 liters/set, consistent with a combined restrictive and obstructive ventilatory impairment. The sin- gle breath carbon monoxide diffusing capacity was mark- edly reduced at 10.7 ml/min/mm Hg (predicted 21.4 ml/ min/mm Hg), consistent with profound abnormality of re- spiratory gas exchange.
The dose of prednisone was gradualty reduced to 20 mg daily, and chest roentgenograms (Figure 3) taken 20 days after institution of therapy showed progressive clearing of the infiltrate. The striking clinical and roentgenologic im- provement as wetl as better oxygenation were associated with resolution of the obstructive ventilatory defect (FEV,,o 1.6 liters, MMFR 2.9 liters/set) without improvement in the restrictive impairment. Enzyme levels were persistently el- evated including a CPK of 290 mu/ml, SGOT 54 mu/ml and aldolase 41 mu/ml.
The patient was readmitted in October 1971 for persis- tent symptoms of fatigue and dyspnea on moderate exer-
FIBROSING ALVEOLITIS IN POLYMYOSITIS-DUNCAN ET AL.
tion. Repeat trephine lung biopsy (Figure 4) in the same
location showed a marked resolution in the acute inflam- matory reaction and focal areas of fibrosis.
During further reduction of the prednisone dose to 10 mg daily she experienced progressive generalized myal- gia, polyarthralgias and muscle weakness in her shoulders and hips. Fever, with temperatures to 104OF, with night sweats and associated dyspnea ensued, and she was readmitted to the hospital in January 1972. Physical exam- ination revealed generalized muscle tenderness, marked proximal muscle weakness, and effusions in the left elbow and knee. Fine rales were heard at the bases of both lungs. The blood leukocyte count was 16,000/mm3 with 84 per cent neutrophils and the erythrocyte sedimentation rate was 76 mm/hour (Westergren), but latex fixation, an- tinuclear factor and lupus erythematosus tests remained negative. PaOa had decreased to 55 mm Hg. Aspiration of the left knee revealed a turbid fluid with a white blood cell count of 31 ,800/mm3 and a glucose level of 80 mg/lOO ml. There was striking deterioration of enzymes with CPK 4,270 mu/ml, LDH 2,100 mu/ml and SGOT 570 mu/ml. Chest roentgenogram revealed no increase in\ infiltration. An electromyogram revealed changes consistent with polymyositis. A biopsy specimen of the deltoid muscle re- vealed the typical histologic appearance of polymyositis: endomysial, perimysial and perivascular acute and chronic inflammatory exudate with foci of degeneration and regen- eration within groups of fibers. Electron micrography con- firmed the findings by light microscopy. A profound weak- ness of the muscles of respiration was evident, with a concomitant deterioration in pulmonary function (VC 1.2 ti- ters, FEVlo 0.9 liter, MMFR 1.04 liters/set). Increasing the dose of prednisone to 60 mg daily resulted in resolution of fever and considerable gain in muscle strength, with some diminution in enzyme abnormalities. The patient continues to exhibit moderate proximal muscle weakness, fatigue and dyspnea on exertion.
In this case the diagnosis of a severe acute interstitial
pneumonitis by lung biopsy and its successful treat-
ment with corticosteroids overshadowed the underly-
ing rheumatologic disease. Only during reduction of
steroid dosage was the full blown picture of polymy-
ositis evident.
Case 2. A 39 year old woman (L.S.) experienced inter- mittent swelling, heat and soreness in both knees in July 1968. She was hospitalized in November of that year with a 1 month history of nonproductive cough, low grade fever and dyspnea on exertion without orthopnea. A chest roentgenogram (Figure 5) showed infiltrates at the base of both lungs. Work-up failed to reveal an etiology of the pul- monary problem. After treatment with antibiotics the fever subsided, but the cough and pulmonary infiltrates persist- ed. Muscle weakness in the shoulder girdles began in Jan- uary 1969 accompanied by gradual weight loss. By April of that year the patient was experiencing fever daily and Raynaud’s phenomenon had developed. By June she had lost 40 pounds in weight and required rehospitalization. Physical findings included respirations 26/min, tempera- ture 10i°F, fine rales at the bases of both lungs, and
October 1974 The American Journal of Medicine Volume 57 623

FIBROSING ALVEOLITIS IN POLYMYOSITIS-DUNCAN ET AL.
. . . . . . . . . . . . . . . . :I I I j I I I I
i I ill illll l I I I
. . . . . . . :+I i :I I+ l I+1
proximal muscle weakness and tenderness. The chest roentgenogram showed no interval change. Laboratory data included a normal hemogram, urinalysis and renal function studies; numerous lupus erythematosus cell prep- arations and antinuclear factor tests were negative, but latex fixation was positive at a titer of 1:2560. Enzyme studies revealed CPK 165 mu/ml, LDH 1,200 mu/ml and SGOT 177 mu/ml. An electromyogram showed changes consistent with polymyositis. A biopsy specimen of the deltoid muscle revealed the changes of polymyositis. Pul- monary function studies showed VC 1.84 liters, TLC 3.14 liters and FEVI.~ 1.31 liters; these findings were consid- ered indicative of restrictive ventilatory impairment. The resting PaOp was 78 mm Hg, PaCOp 28 mm Hg and pH 7.49. After exercise the Pa02 fell to 69 mm Hg; after the administration of 100 per cent oxygen for 7 minutes the ‘Pa02 rose to 525 mm Hg. These data were interpreted as consistent with a defect in diffusion.
After treatment with corticosteroids there was resolu- tion of fever and marked improvement in muscle strength; however, the pulmonary infiltration did not change.
In August 1969 the patient was readmitted with hemop- tysis, increasing dyspnea at rest, respirations 30/min and temperature IOl’F. Chest examination revealed medium rales over the lower half of both lung fields and an in- creased pulmonic component of the second heart sound. Chest roentgenograms revealed a marked increase in pul- monary infiltration consistent with a complicating bron- chopneumonia. The Pa02 had decreased to 58 mm Hg. The steroid dose was increased, and antibiotics were ad- ministered without significant improvement. On the 20th hospital day there was a spike in temperature to 105“F followed by progressive deterioration and death 3 days later. Blood cultures revealed Klebsiella pneumoniae, Pro- teus mirabilis and Listeria monocytogenes. Postmortem examination disclosed chronic polymyositis, cardiac fibro- sis and diffuse interstitial pulmonary fibrosis with superim- posed suppurative bronchopneumonia (Figure 6).
In this case a chronic pulmonary infiltration associ-
ated with symptoms of dyspnea and cough preceded
the development of polymyositis. After corticosteroid
treatment the manifestations of polymyositis dimin-
ished although the pulmonary abnormalities were un-
changed. At autopsy the lungs showed the typical
changes of fibrosing alveolitis.
COMMENTS
The thoracic manifestations in polymyositis are of
three general types: (1) secondary bronchopneumo-
nia, (2) involvement of the respiratory musculature, and (3) diffuse interstitial pneumonitis and fibrosis.
The first two complications are not specific for
polymyositis but may be observed in any chronic neuromuscular disorder. Interstitial pneumonitis and
fibrosis (fibrosing alveolitis) is a well documented fea-
ture of various connective tissue disorders including
systemic lupus erythematosus, scleroderma and rheumatoid arthritis [2-41; however, there are very
624 October 1974 The American Journal of Medicine Volume 57

few cases on record associated with polymyositis. In
a review of 133 cases of polymyositis, Pearson [ 161
noted pulmonary involvement in only two. Further-
more, Medsger et al. [ 171 found only 3 of 118 cases of polymyositis in which bilateral basilar interstitial in-
filtration might be considered consistent with the
roentgenologic changes of fibrosing alveolitis. Since
the first reported case of fibrosing alveolitis in
polymyositis by Mills [5] in 1956, this form of associ-
ated pulmonary involvement has been histologically
confirmed in only 12 cases in the English literature
[5- 151. Pertinent features of these cases and the
two reported herein are listed in Table I. Histologic study of muscle in every case showed
the typical changes of polymyositis. Electromyo-
graphic findings were consistent with polymyositis in
the seven patients in whom it was performed, and
muscle-related enzymes were elevated in the 12 pa-
tients in whom they were measured. The mean age
of patients in this series was 48 years (range 37 to
65 years). As is the usual experience in polymyositis
there was a predominance of females (11 of 14). In
seven patients the first symptom of their illness was
respiratory; in seven others the initial symptoms
were rheumatologic. Respiratory manifestations
were predominant in 12 of the 14 patients, and often
there was complicating respiratory failure. All pa-
tients had cough and dyspnea, usually with rales in
conjunction with the demonstrated pulmonary in- volvement. Chest roentgenograms usually showed
either a basilar or more generalized interstitial infiltra-
tion. Histologic study of the lung showed varying de-
grees of alveolar wall thickening and distortion, fibro-
sis and infiltration with lymphocytes and plasma cells.
Three patients had medial or intimal thickening of the
small pulmonary arteries and arterioles consistent
with changes of pulmonary hypertension, but vasculi-
tis was seen in only one specimen. The most impor-
tant clinicopathologic correlation was found between
the amount of fibrous tissue present and long-term
prognosis. Of the 10 patients with moderate to se-
vere pulmonary fibrosis, 8 died, 5 with respiratory
failure. Four of the eight who died were treated with
corticosteroids with no evidence of pulmonary im- provement, although two patients did show a diminu-
tion in rheumatologic manifestations. Of the five pa-
tients who were alive at the time of reporting, lung bi-
opsy in three showed a predominantly active inflam-
matory reaction with minimal fibrosis. This was in dis-
tinct contrast to the fatal cases. These patients re- sponded to corticosteroid therapy with resolution of
pulmonary infiltration as well as rheumatologic im-
provement. One patient with proved polymyositis, but with sclerodermatous features, failed to respond to
the administration of corticosteroids and at autopsy
FIBROSING ALVEOLITIS IN POLYMYOSITIS-DUNCAN ET AL.
showed predominantly pulmonary vasculitis [ 91. In
another patient, showing largely fibrosis, pulmonary
function improved with corticosteroid therapy, but
there was no change on the chest film [ 141. A final
patient had fibrosis and chronic inflammation [ 151.
After treatment there was improvement both in pul-
monary function and on the chest roentgenogram.
In addition to these histologically confirmed cases
of fibrosing alveolitis in polymyositis, the literature
does contain several reports in which no pulmonary
pathologic material was available. Some of these
cases do warrant mention. Dubowitz and Dubowitz
[ 181 described a 5 year old child with polymyositis
and a severe respiratory distress syndrome. A diffuse
pulmonary infiltration was visible on the chest roent-
genogram. Corticosteroid therapy produced a striking
clinical response with resolution of the abnormalities
noted on roentgenograms. Camp and a.ssociates
[ 191 described a 39 year old woman with derma-
tomyositis and progressive exertional dyspnea.
Roentgenograms of the chest revealed bilateral
widespread interstitial infiltrations. A dramatic clinical
response occurred with corticosteroid therapy. De-
spite the absence of pulmonary pathology. the re-
sponse to corticosteroids, including resolution of the
findings on chest roentgenograms, suggests a more
acute, inflammatory pulmonary lesion in t.hese two
patients. Hepper and colleagues [20] described a 56
year old woman with polymyositis and progressive
dyspnea for several months. Following steroid thera-
py her pulmonary symptoms subsided, and pulmo-
nary function testing and chest roentgenograms re-
vealed some improvement. In a less detailed report,
Brundin [21] described three patients with derma-
tomyositis, with dyspnea at work or rest and with pul-
monary crepitations and abnormalities on chest
roentgenograms. No treatment data were reported.
Four other cases recorded in the foreign literature
were not reviewed [ 22-251.
Rheumatoid factor, lupus erythematosus cell prep-
arations and serum antinuclear factor are reported in
Table I. In three of nine cases studied for rheumatoid
factor results were positive, but all tests for lupus er-
ythematosus cells and antinuclear factor were nega-
tive. The significance of these so-called “tests of au-
toimmunity” in this group of patients is uncertain.
Several studies have demonstrated the variable pres-
ence of these factors not only in diseases such as
systemic lupus erythematosus, scleroderma, rheu-
matoid arthritis and polymyositis [26], but also in fi-
brosing alveolitis without evidence of any coexisting
connective tissue disorder [27-291. However, any role of such factors in the causation of these diseas-
es is purely speculative at present. It has lbeen theo- rized that when fibrosing alveolitis is associated with
the collagen-vascular disorders, the pulmonary lesibn
October 1974 The American Journal of Medicine Volume 57 625

FIBROSING ALVEOLITIS IN POLYMYOSITIS-DUNCAN ET AL
might represent a manifestation of systemic immune
complex disease due to localization of materials such as antinuclear factor in the lungs. However, immu-
nofluorescence studies for deposited immunoglobulin
in the lung were negative in the one patient with fi-
brosing alveolitis in polymyositis so tested [ 121.
It appears that patients with fibrosing alveolitis as-
sociated with polymyositis are of two general types:
In one type there is a chronic progressive course,
and histologically pulmonary fibrosis is predominant.
These patients show a poor response to corticoste-
roids as exemplified by our patient (Case 2) who, al- though showing a rheumatologic response to this
treatment, shawed no pulmonary improvement. In
the other type there is generally a more acute
course, and histologically alveolar wall thickening,
distortion and acute inflammatory infiltration but little
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
fibrosis are evident. These patients usually have a
good response to corticosteroid therapy as shown by
our patient (Case 1) in whom the inflammatory reac-
tion resolved with only focal fibrosis remaining evi-
dent on post-treatment biopsy.
In the desperately ill patient, corticosteroid therapy
should be instituted empirically. Otherwise, a lung bi-
opsy is recommended in that the type of pathology
will provide a useful guideline for therapy. A reaction
showing active inflammation should be treated ag-
gressively with corticosteroids. However, the use of
corticosteroids in the presence of predominant, ex- tensive fibrosis would not be expected to improve
respiratory insufficiency, and if administered in large
doses and for extended periods might be expected to
greatly increase the probability of complicating bron-
chopneumonia.
REFERENCES
Scadding JG, Hinson KFW: Diffuse fibrosing alveolitis (dif- fuse interstitial fibrosis of the lungs). Correlation of histol- ogy at biopsy with prognosis. Thorax 22: 291, 1967.
Eisenberg H, Dubois EL, Sherwin RP, Balchum OJ: Diffuse interstitial lung disease in systemic lupus erythematosus. Ann Intern Med 79: 37. 1973.
&adding JG: The lungs in rheumatoid arthritis. Proc Roy Sot Med 62: 227, 1969.
Weaver AL, Divertie MB, Titus JL: The lung in scferoderma. Mayo Clin Proc 42: 754, 1967.
Mills ES, Mathews WH: Interstitial pneumonitis in derma- tomyositis. JAMA 160: 1467, 1956.
Goldfischer J, Rubin EH: Dermatomyositis with pulmonary lesions. Ann Intern Med 50: 194, 1959.
Pearson CM: Rheumatic manifestations of polymyositis and dermatomyositis. Arthritis Rheum 2: 127, 1959.
Hyun BH, Diggs CL, Toone EC: Dermatomyositis with cystic fibrosis (honeycombing) of lungs. Dis Chest 42: 449, 1962.
Pace WR, Decker JL, Martin CJ: Polymyositis: report of two cases with pulmonary function studies suggestive of pro- gressive systemic sclerosis. Am J Med Sci 245: 322, 1963.
Sandbank M, Grunebaum M, Katzenellenbogen. I. Derma- tomyositis associated with subacute pulmonary fibrosis. Arch Dermatol 94: 432, 1966.
Weaver AL, Brundage BH, Nelson RA, Bischoff MB: Pulmo- nary involvement in polymyositis: report of a case with response to corticosteroid therapy. Arthritis Rheum 11: 765, 1968.
Thompson PL. Mackay JR: Fibrosing alveolitis and polymy- ositis. Thorax 25: 504, 1970.
Thompson JE: Lung involvement in polymyositis. Med J Aust 1: 1332, 1971.
Olsen GN, Swenson EW: Polymyositis and interstitial lung disease. Am Rev Resp Dis 105: 611, 1972.
Webb DR, Currie GD: Pulmonary fibrosis masking polymy- ositis: remission with corticosteroid therapy. JAMA 222:
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
1146, 1972. Pearson CM: Patterns of polymyositis and their responses
to treatment. Ann Intern Med 59: 827, 1963. Medsger TA Jr, Robinson H, Masi AT: Factors affecting sur-
vivorship in polymyositis. A life-table study of 124 pa- tients. Arthritis Rheum 14: 249, 197 1.
Dubowitz LMS, Dubowitz V: Acute dermatomyositis pre- senting with pulmonary manifestations. Arch Dis Child 39: 394, 1964.
Camp AV, Lane DJ, Mowat AG: Dermatomyositis with pa- renchymal lung involvement. Br Med J 1: 155, 1972.
Hepper NGG, Ferguson RH, Howard FM Jr: Three types of pulmonary involvement in polymyositis. Med Clin North Am 13: 43, 1964.
Brundin A: Pulmonary fibrosis in scleroderma and derma- tomyositis. Stand J Resp Dis 51: 160, 1970.
Peralta MG, Carril RFF: Fibrosis pulmonar diffusary derma- tomiositis. Rev Clin Esp 84: 114, 1962.
Garcia EH, Toledo A: Manifestaciones pulmonares en un case de dermatomyositis en el nino. Medico 48: 1031, 1964.
Turiaf J, Basset F: Les fibroses pulmonaires insterstitielles diffuses des collagenoses. Poumon Coeur 21: 663, 1965.
Morawetz F: Die lungenmanifestationen des rheumatismus der sklerodermie und der mermatomyositis. Pneumonol- ogie 145: 244, 1971.
Caspary EA, Gubbay SS, Stern GM: Circulating antibodies in polymyositis and other muscle wasting disorders. Lan- cet 2: 941, 1964.
Mackay IR, Ritchie B: Diffuse fibrosing alveolitis (diffuse in- terstitial fibrosis of the lungs): two cases with autoim- mune features. Thorax 20: 200, 1965.
Turner-Warwick M, Doniach D: Auto-antibody studies in in- terstitial pulmonary fibrosis. Br Med J 1: 886, 1965.
Nagaya H, Sieker HO: Pathogenic mechanisms of intersti- tial pulmonary fibrosis in patients with serum antinuclear factor. Am J Med 52: 51. 1972.
626 October 1974 The American Journal of Medicine Volume 57