expression during erythroid differentiation and its role in
TRANSCRIPT

-sS-<,3
Regulation of Human S-Aminolevulinate Synthase 2
Expression During Erythroid Differentiation and Its Role in
X-Linked Sideroblastic Anaemia
A thesis submitted to the University of Adelaide for the degree of
Doctor of Philosophy
by
Tania Dell'Oso B.Sc. Hons (University of Adelaide)
School of Molecular and Biomedical Science
University of Adelaide
Adelaide, South Australia, Australia Marchr 2003

TABLE OF CONTENTS
THESTS SUMMARY .................. vr
DECLARATTON "" vlIrACKNOWLEDGEMENTS ........IX
ABBREVIATIONS.. ..................... xI
CHAPTER 1: GENERAL INTRODUCTI
1 . 3. I EnyTHROPOIETIN AND ERYTHROPOIETIN R¡CPPTOR SICN¡.TTNqC
1.4 TRANS CRIPTION FACTORS REGULATING ERYTIIROPOIESIS 7
1.4.1 SrBv C¡n L¡urpvn (SCL) ..... .7
I .4.2 LIM-oNLY PRoTEIN 2 (LM02)
r.4.3 GATA-2 9
1.4.4 c-Mve 9
1.4.s GATA-I .. 10
146FOG 11
1.4.7 EKLF 11
1.4.8 OrnBR KRUppEL-Lx¡ Fevlny MEvrssRs RpcurArn¡c ERvruRoPoIESIS......................13
1.4.9 NF-E2 t3
15
I.4,II Erppcr op e TRANSCRIPTIONAT CORCTIVATOR ON ERYTTTRON TNENSCRIPTION
F¡.croRs.. 15
1.5 GLOBIN GENE EXPRESSION DURING ERYTHROPOIESIS .............18
1.5.1 TsB GI-osnT GsNr CI-usrsR.....
1
8
t91.5.2 RpcULATIoN or B-Gronnl GnNB Expn¡sstoN ..........
1.7.1 PnopeRrIES on ALAS ..... 24
I,7 .2MqCIIANISM OP ALAS .. 26
1.7.3 ALAS GpNB SrRucruRE.... 27
1.8 REGULATTON OF 4L4S1............... ......28
I

1 .9. 1 TnnNSCRIPTIONAT RECUI¡.IION OF AL4S2..... 30
1 .9,2 R¡cuLATIoN o¡ ALAS2 TRRNsrerIoN..... ^^JJ
1.9.3 RgCULATION OP ALAS2 EY HRTN4 35
1.9.4 HvpoxIC REGULATIoN or ALAS2 ExPRESSIoN... .36
1.9.5 Rors or ALAS2 ExpRBsStoN nq ERyruRoIo Cplr Dm'ppRBNtIATIoN .........36
1.9.6 MoIEL FOR REGULATION OT ALAS2 37
1.10 STDEROBLASTTC 4N48MI4............. ..................37
1.10.1 CH¡.RIcIrRISTICS o¡'SII¡RoSLASTIC ANesun... ...'.................37
1.I0.2Itr¡prtceuoN or ALAS2 N XLSA...
I .10.3 A GENB INvorv¡p nt A. DIprpr¡Nr Fonv oF XLSA..
..41
..43
I .10.4 TnperupNr op SIo¡RoBLASTIC ANe¡vtl¡. 44
CHAPTER MATERIALS AND ME 47
2.1 MATERIALS ........47
2.1.1 Dnucs, CHSUIC,ILS AND Rp,tcsNrs 47
2. 1.2 ReoTocHEMICALS ........... 47
2.7.3 BN2yMES .................
2.1.4 BuppERS..................
2.1.10 Trssun CurruRE Cprr Lnqps eNt Mplni) Cell Lines
ii) Solutions.
iii) Media
47
48
2. 1.5 CroNrNG VECToRS........ 48
2.1.6 CroNno DNA SEqurNcEs. 48
2. 1 .7 SysrHETrC Oucor.rucLEoTIDES 49
i) Oligonucleotides for sequencing plasmid constructs 49
ii) Oligonucleotides used in the mutagenesis of human ALAS2lntron 8............... .................49
iii) Sequences of the sense strand of synthetic oligonucleotides used in gel shift assays ........49
iv) Oligonucleotides for the generation of XLSA point mutations..."...'. .'.'..'.......50
2.1.8 BacTERIAL SrnaNs ..... 50
2.1.9 B¡cTERIAL Gnowru Mnon...... 51
51
..52
52
52
53
il
2.1.1I Mrscprr¡.NBous

2.2 RECOMBINANT DNA METHODS ...............
2.2.1 GBNBn¡.r DNA MprHots
2.2.2 Pr¡sl¿rp DNA PRppeR¡.tloN ..........,
2.2.3 R¡sTRICTIoN ENzvtr¿s Dtc¡srtoNs op DNA
2.2.4 Pp'np IRATIoN or CI-oNnqc Vpcron s.....
2.5.1 Cnrr MerxrpNaNc¡
2.5.2 Iu Vtrno DmpSRBNTIATIoN op J2E CErrs
i) Erythropoietin induced differentiation of the J2E cellline...........
54
54
54
55
55
56
2,2.5 PISpARATION Or DNA R¡STzuCTION FRECIUBNTS 55
2.2.6LtcrtroN oF DNA
2.2.7 TnINSFoRMATIoN op E. cottwnn RScoNIBINANT Presups....
i) Preparation of Competent E. coli
ii) Transformation of Competent Bacteria....
2.2.8 DNA SEquENcr ANRrYsIs.
2.2.9 PISpARATION OP R¡.OIOTABELLED DNA PROS¡S
i) Oligo-Labelling DNA ...
ii) 5'End-Labelling of Synthetic DNA oligonucleotides
2.2.10 PRepenlroN oF R¡.oIoreeBLrso DNA MeRrc¡ns
2.2.1 | OlrcoNucLEorIDE SIrs-DlREcrso Mur,q'cENESIS
i) Mutagenesis Reaction .............
ii) Selection of Clones Containing the Mutation...............
2.3 REPORTER AND EXPRESSION CONSTRUCTS UTILISED IN THE
INVESTIGATION OF ALAS2 TRANSCRIPTIONAL RE,GULATION.
2.3 .l ALAS2 Pnovorsn/RnponrsR GpNB Preslr,tlos
2.3 .2 ALAS2 INrn oN/R¡ponrpn GsNs CoNsrRucrs .................
2.3.3 SIIr-DIRECTED MurecnNpSIS oF rn¡ Hulu,q.N ALAS2 INTRoN 8 SrquENcE.................
2.3.4Wruo Tvpp eNp MureNr EIA eNt p300 ExpnESSIoN CoNsrnucrs................
2.4 ALAS2 TARGETING CONSTRUCTS.
2.4.1 SyNrHESrs oF rHE ALAS2 ExoN 8 C1159G MurRNr Tlncprr¡{c CoNsrRucr............
2.4.2 Sv¡¡rHESrs oF rHE ALAS2 ExoN 9 Cl228T MureNr TRRcprrNc CoNsrRucr.............
2.4.3 CoNsTRUCTION op ALAS2 TeRcErNc VgcroR CONT¡.INING THE PGKNEOR/HSVTT
56
56
56
57
57
57
58
58
59
59
59
59
60
60
61
61
62
62
62
63
64
64
64
Snr-pcrroN Clss¡rrp. 63
2.5 METHODS FOR THE äT VITRO DIFFERENTIATION OF AN ERYTHROID
CELL LINE........ .........63
ii) Staining of cells with benzidine, a stain for haemoglobin production."..'.
ilI

2.6 METHODS FOR EXPRESSION OF REPORTER CONSTRUCTS IN TISSUE
CULTURE CELL LINES..... .......64
2.6. 1 TnINSIENT TReNsrpcuoN oF ruE J2E C¡rr Ln'rs 64
652.6.2LuctpERASE RepoRrEn GSNB Assev ru J2E Cprrs .
2.7 METIIODS FOR GENE TARGETING EMBRYONIC STEM CELLS....
2.7 .l Cnrr MRnqrpNeNCE ..................
1)El4TG2a Embryonic Stem Cells
iÐ W9.5 Embryonic Stem Cells
2J.2lvxtorATroN or SroR FleRosresr CsrLs
2.9.3 Erp.crRopHoRErIc MosIrtrv SUI¡T Assav .......
65
65
65
66
..66
66
66
67
68
68
69
69
69
69
70
70
70
7I
7l
..71
OF'THE HUMAN ALAS2 GENE
2.7.3 SrasIp TR¿,NSPECTION Or ES CELLS WTTTT TRRCETING V¡CTOR DNA VN
ErpcrRopoRATIoN.
1)EI4TG2a ES Cells
iÐ W9.5 ES Cells
2.7.4 Prcrnqc op S¡rpcrIoN RpslsraNr CoroNrcs
2.7.5 Fne¡zING oF T¡.Rcprpo CroNEs
2.7 .6 Htst ocHEMrcAL S ren tlNc non B - GITACToSIDASI Actlvtrv
2.1.7 K¡txvoryplNc oF ALAS2 Tlncprno ES Cen LrN¡s
2.8 SOUTHERN BLOT ANALYSIS AND TIYBRIDISATION CONDITIONS ..............
2.8.1 IsolerroN oF GsNoivtIc DNA FRoM ES csns .......
2.8.2 SourHpRN Bror ANervsIs
2.8.3 SrnIppING oF rup FII-rpn ...
2.9 METHODS FOR ELECTROPHORETIC MOBILITY SHIFT ASSAYS
2.9.1 PnppARATIoN op NucrEeR PRorpnq ExrRRcrs ..
2.9 .2 P yppARATION Or REPIOTAB ELLED AWNN,q.TEO OUCONUCLEOTIDE PROE¡ S
CHAPTER 3: TRANSCRIPTIONAL RE
IN RESPONSE TO ERYTHROPOIETIN INDUCED RF],NTIATION OF'
ERYTHROID CELLS 72
3.1 INTRODUCTION ..................72
3.2 R8SULTS............ .....................75
3.2.1Epo INoucpo DrpsRENrIArIoN op J2E ERvrnRoIo CPns....... ...................75
3.2.2 Trlr' Mnrrr\4ar LpNcrn oF THE Hurr¡RN ALAS2 Pnovor¡R Rrqunno FoR A RESPoNSE
.75ro Epo ts293 B¡.sB PnIRs ........
IV

3.2.3 LocILISATION or Epo RpspoNsIw ENUENCER ET¡Ir¿NNTS WITHIN THE HUVEU ALAS2
GBNp 77
3 .2.4 DnnRMrNATroN or TR¡NscRrprroN FecroR BnqorNc Strss Wtrsnq INrn oN 8 tulr eRp
CzurrcRr- ro Epo ENH,rNcEo TRANSCRIpTIoN oF rHE Hulr¡RN ALAS2 PRolr¿ornR .................79
3.2.5 BTNIING oF GATA-I To THE ENHRNcpR n,I HUN4EN ALAS2 INTRON 8 IS INONPENDENT
o¡' Epo Srltr,turerIoN..... .. 8l
3.2.6 TuF. Erpecr oF Epo oN THE BnqrrNc op CACCC-Assocnrpr PRorBnqs to Str¡s nl
Hurr¿eN ALAS2 INTRoN 8 .............. 82
3.2.7 INvEsTIGATING THE ROLE oT CBP/p3OO COICTryETOR IN THE TRANSCRIPTIONAL
REGULATToN op ALAS2
3.3 DISCUSSION.....
CHAPTER 4: GENERATION OF A MURINE MODEL FOR X.LINKED
86
90
SIDEROBLASTIC AEMIA
4.2.1 TencErrNG oF rHE ALAS2 Locus tN E14TG2a ES Cprrs....... 107
4.2.2 INrnoDUCrroN oF rHE XLSA-AssocrArED MurRrIoN (Cl228T) Nro ExoN 9 or rup
ALAS2 Locus oF rHE HPRT-Postrlvs R3 ES CErr LrNs........ 110
4,2.3 TencErrNc oF rHE ALAS2 Locus w W9.5 ES CBns .............113
4.2.4 SrnerEcy FoR rHE Corr¿rr¿ERCTAL GBNpR¿.rtoN oF AN ALAS2 TRRcBreo ES CErr LrNn
.tt7
4.2.5 CoNsrRUCTIoNornALAS2Cll59G T¡.Rcsrr¡qcV¡croR.... ...................118
4.2.6 Scn¡ENTNG SrRerpcy ro Ippxrmv ALAS2 TeRcpr¡r V/9.5 ES Cprrs Pnopucno Bv
Ozc¡Ns. 119
CHAPTER 5: F'INAL SUMMARY
103
127
REFERENCE LIST 133
V

THESIS SUMMARY
Haem is required for many cellular processes including haemoglobin synthesis in erythroid
cells, where the majority of the haem in the body is produced and utilised. Its synthesis
requires tight regulation to prevent toxic levels of free haem from arising. 5-Aminolevulinate
s¡mthase 2 (ALAS2) is the first and rate limitingenzpe in the biosynthesis of haem in
erythroid cells. Thus, the regulation of ALAS2 expression is critical for maintaining and
controlling haem production during the process of erythroid cell differentiation. A major aim
of this study was to identiff the regulatory elements within the ALAS2 gene that are involved
in controlling transcription of ALAS2 during erythropoietin (Epo) stimulated erythroid
differentiation. In order to investigate the regulation of ALAS2 transcription in the context of
red blood cell maturation, an erythroid cell line, J2E,that terminally differentiates in response
to Epo treatment was employed in this study.
Human ALAS2 promoter deletion studies demonstrated that the first 293bp of the proximal
promoter was sufficient to enhance transcription in response to Epo induced differentiation of
the J2E cells. Introns 1 and 8 exhibited Epo responsive enhancer activity with intron 8
proving to be the stronger transcriptional activator in response to Epo. Transcription factor
binding sites located in the 3' end of intron 8 that are critical to intron 8 Epo responsive
enhancer activity were also identified. Preliminary studies on the effect of the coactivators
CREB binding protein (CBP) and p300 on ALAS2 expression in response to Epo stimulation
were conducted and suggested a potential involvement of these factors in regulating ALAS2
transcription.
Defective haem synthesis, as a result of point mutations in the human ALAS2 gene, has been
implicated in a blood disorder called X-linked sideroblastic anaemia (XLSA). XLSA is
characterised by the presence of iron loaded mitochondria surrounding the nucleus in
erythroblasts of the bone marrow. Anaemia, associated with a cycle of ineffective
erythropoiesis that is linked to increased intestinal iron absorption and the secondary effect of
iron overload is exhibited by XLSA patients. Point mutations in the ALAS2 gene of XLSA
probands have been identified and two associated mutations, C1215G in exon 8 and Cl283T
in exon 9 were selected as a basis for a murine model for XLSA. Thus, the aim of this project
was to develop an animal model for XLSA to investigate the role of ALAS2 in this blood
disorder and the associated defects in iron metabolism. A gene targeting approach using
VI

embryonic stem (ES) cells was employed and several strategies trialed. A potential ALAS2
targeted ES cell line containing the Cl159G point mutation in exon 8 (equivalent to the
human mutation) was generated, with further characterisation required.
VII


ACKNOWLEDGEMENTS
I am grateful to Professor Rathjen for permission to undertake my PhD in the School of
Molecular and Biomedical Science. Sincere thanks to Dr. Brian May for providing me with
the opportunity to conduct research in his laboratory and for his supervision throughout my
PhD, particularly in the reading of this thesis.
To the past and present members of the lab - Thank You!! Particularly to Satish for his view
on life and many interesting chats, especially about matchmaking Indian style! Thanks Chris,
for still solving my little computer problems three years on, believe it or not I am catching on.
Prem, thanks for letting me think that I was the boss of the lab, now it is finally all yours,
ALAS2-free! Thank you Sophia for your smiling face and for always praising my cake
cooking, it made me feel like quite the chef. To Ingrid, I really enjoyed our time together in
(and outside) the lab, your sense of humour and attitude to life was a breath of fresh air -Good luck with the 'bump', how exciting! ! Josef, from a box of chicken crimpy biscuits to a
lunch box full of rabbit food, you are a changed man and I just wish I could say that it was my
years of subliminal healtþ lifestyle messages! It has been fun, lots of fun - nobody 'crumbs'
quite like you. It's your turn now, a little sorry I won't be here to see it but it's a sure bet that I
will hear all about it, Good Luck! Now there's one more, what's his name again. .that's
right, Tim! Forget you, I don't think so, but now I am finally out of your hair! I can never
thank you enough for your mentoring throughout these years. You were a wonderful teacher,
you only made me cry a few times and I think you even found my clumsiness endearing?!
Aside from the science stuft I just think you are an all round terrific guy and I was lucky
enough to have hooked up with you!
I would like to thank the Department for making it such a great and fun place to work in.
Always a friendly face in the corridor just waiting for a chat and I was certainly happy to
oblige. To Brian Denton, a big thank you for making every maintenance problem that I ever
had seem so easy to fix up and for the general chit chat. "Is it too late to order radiolabel" -thanks Jan, service was impeccable and always came with a smile!! Thanks to all TC support
staff, the old and the new.
To Emma and Francine, we have come along way since third year prac class and milkshakes
at the Equinox. I went to that class not wanting to make pals and I met you guys, two of my
greatest friends. Looking forward to us in NYC!!! Karen, your infectious giggle is a cure for
IX

bad times and an enhancer of happiness, thank you for that wonderful tonic. Kathryn, I knew
you'd be scanning this page for your name and here it is, you are quite a character and I don't
think I'll ever meet another quite like you! Look out for my emails, I have turned over a new
leaf, at least one ayear fiust kidding!). To Michael, you were there at the start and so happy
that I can say there at the end - thank you. With your philosophy on life you cannot go wrong
and I wish you only the very best in life. To Jan, you have been a strength and then some.
Your wisdom (George may want to have a chuckle about this!) helped me out of quite a few
pickles but we can keep that all to ourselves - you are quite a woman.
To my family- Yes, I have finally finished and thank you for your patience! I know how
fortunate I am to have you all, it's a good drawcard! Damian, I have found that hour for
dinner and Josephine we can do whatever you like now. You guys have been terrific through
everything, Hotel Passalacqua-Colangelo rocks! To my Mum and Dad, thank you for your
love and supporl - I know you rwere always close. Never ending story has ended John and
although it's not the 'real science' I hope you find it worthy! To Melinda, what a ride, but we
got there in the end! Sisters by nature, friends by choice - so true! Thank you for your support.
Ryan, your gorgeous face and cheeky personality always puts a smile on my face and laughter
in my heart!
To Andrew, how can I begin to thank you for all that you have done? Your support,
encouragement, love, good humour, patience, understanding and many extraordinary salads
got me where I am today. Lets have some fun!! Here's to our future and all that it holds.
Dedicated in loving memory of my Mum, Doralice Dell'Oso
X

ABBREVIATIONS
Abbreviations used throughout this thesis are in accordance with those described in The
Journal of Biological Chemistry Q99\. Additional abbreviations are listed below.
6-TG:
AGM:
ALA:
ALAS:
ALAS1:
ALAS2:
BFU-E:
bHLH:
BKLF:
bp:
CBP:
CFU-E:
Ci:
DMEM:
DMSO:
E:
EB:
eIF-2cr:
EKLF:
EMSA:
Epo:
EpoR:
ERC1:
ES Cell:
Fe-S:
FIAU:
FKLF:
FOG:
GM-CSF
hABCT:
6-thioguanine
aorta- gonad-mes enephro s
5-aminolevulinate
5 - aminolevulinate syrthase
housekeeping 5-aminolevulinate synthase
erythroid 5 -aminolevulinate synthase
erythroid burst forming unit
basis helix-loop-helix
Basic Kruppel-like factor
base pairs
CREB binding protein
erythroid colony forming unit
curie
Dulbecco's Modified eaglJs Medium
dimethyl sulphoxide
embryonic day
embryoid body
cr subunit of eukaryote initiation factor 2
Erythroid Kruppel-like factor
electrophoretic mobility shift assay
erythropoietin
erythropoietin receptor
EKLF coactivator remodelling complex- 1
embryonic stem cell
iron-sulphur
fialuridine
Foetal Kruppel-like factor
friend of GATA
granulocyte macrophage-colony stimulating factor
human ATP-binding cassette transporterT
XI

HAT:
HATs:
HIF:
HPTR:
HRI:
HRM:
HSV:
IL3:
IRE:
IRP:
Kb:
LMO2:
MEL:
MtF:
Neot:
PBG:
PBGD:
PE:
PLP:
lpm:
sau'.
SCL:
TBP:
TfR:
tk:
UTR:
UV:
XLSA/A:
XLSA:
histone acetyltransferase
hypoxanthine, aminopterin and thymidine
hypoxia inducible factor
hy,poxanthine- guanine phosphoribosyltransferase
erythroid-specific elF -2 a kinase
cysteine-proline motifs of the haem binding domain
hlpersensitivi
haematopoitic stem cell
herpes simplex virus
interleukin 3
iron responsive element L- CR Iiron regulatory protein
kilobase
LIM-only protein 2 .. )
murine erythroleukemia l¡A( '
mito chondri al ferritin
neomycin resistance
porphobilinogen l' ,
porphobilinogen deaminase
polychromatic erythroblast
pyridoxal phosphate
revolutions per minute
sauternes
stem cell leukemia
TATA binding protein
transferrin receptor
thymidine kinase
untranslated region
ultraviolet
X- I inked si derob I ast i c anaemi al spino c ereb ellar ataxia
X-linked sideroblastic anaemia
site:
XII

CHAPTER 1
GENERAL INTRODUCTION

CHAPTER 1: GENERAL INTRODUCTION
1.1 INTRODUCTION
Haem is a tetrapyrole ring compound that complexes with iron. The central ferrous iron atom
G"*) of haem can be reversibly oxidised to the ferric state (Fe***) by the transfer of a single
electron (Bottomley and Muller-Eberhard, 1988). This characteristic of haem facilitates its
ability to function as an active cofactor in both the electron transport chain and in reactions
with oxygen containing compounds. For example, in the ferrous state, haem has a high
affinity for oxygen allowing it to function as a carrier in the transport of oxygen in the form of
haemoglobin. Haem is assembled into a variety of proteins for diverse cellular functions
including oxygen transport and storage (haemoglobin and myoglobin), respiration (respiratory
cytochromes) and xenobiotic metabolism @a50s) (May et al., 1995).
All nucleated animal cells s¡mthesise haem as its absorption from serum is minimal. The
highest requirement for haem is in erythroid cells for the formation of haemoglobin,
synthesisingS0% of the body's total haem (Worwood, 1977). Therefore, intracellular iron
levels, de novo protoporphyrin IX production and globin sSmthesis need to be coordinately
controlled. This regulation is mediated at two levels, transcription at the onset of erythroid
differentiation and translation. Thus, common regulatory mechanisms are necessary to
coordinate haem and globin polypeptide expression. 5-Aminolevulinate synthase (ALAS) is
the first enzyme of the haem bios¡mthetic pathway. It has the lowest enzymatic activity in
both erythroid and non-erythroid tissues when compared to the activity of the other enzymes
in the pathway (May et al., 1995). It is accepted that ALAS is the rate limiting and key
regulatory enzyrne in haem synthesis. Two ALAS isoz¡rmes exist, ALASl which is
ubiquitously expressed and ALAS2, whose expression is restricted to erythroid tissue.
One of the major aims of this thesis is to examine the regulation of ALAS2 transcriptional
activity in the context of erythroid differentiation. Since defects in haem s¡mthesis can result
in a disease state, the role of ALAS2 in the blood disorder X-linked sideroblastic anaemia will
also be investigated. To appreciate the role of ALAS2 in haem synthesis and erythrocyte
differentiation, an overview of erythropoiesis and the current understanding of haem and
globin synthesis will be discussed.
1

1.2 HAEMATOPOIESIS
Haematopoeisis is the process by which a pluripotent haematopoietic stem cell (HSC)
differentiates in response to surrounding growth factors and transcriptional mediators,
resulting in commitment to a specific blood cell lineage. These multipotential haematopoietic
progenitors can become committed precursors of the lymphoid lineage to generate T and B
cells or of the myeloid lineage to produce a number of different blood cell types including
monocytes/macrophages, neutrophils, eosinphils, mast cells, megakaryocytes and
erythrocytes (Orkin, 2000) (Figure I .1). In vertebrae, embryonic hematopoiesis involves
primitive and definitive stages which are located in different regions of the embryo and body
(Palis and Segel, 7998; Migliaccio and Migliaccio, 1998; Dame and Juul, 2000). Primitive
haematopoiesis involves the production of large, nucleated erythroblasts that slmthesise
embryonic forms of globin. These cells arise from the extraembryonic mesoderm in the blood
islands of the yolk sac at embryonic day (E)
human gestation (Orkin, I996;Palis et al., 7
production has shifted to the foetal liver and
1.5 in the mouse embryo or day 15 to 18 of
E9.5 in the mouse, major blood cell
a multilineage stage referred to as
definitive haematopoiesis. Therefore, cells belonging to the lymphoid and myeloid lineages
can arise including enucleated erythrocytes synthesising adult globin chains (Dzierzak and
Medvinsky,lgg5). This shift to the foetal liver occurs at approximately five weeks gestation
in humans (Karlsson and Nienhuis, 1985). Late in murine foetal life, haematopoiesis moves
for the final time to the bone marrow where it remains throughout adulthood, becoming the
major haematopoietic site (Medvinsky andDzierzak,1996;Palis et al., 1999).In the human
embryo the shift of haematopoiesis to the bone marrow occurs in the third trimester (Karlsson
and Neinhuis, 1985). As mentioned earlier, transcriptional activators are involved in
controlling lineage commitment during haematopoiesis and this will be discussed at length in
relation to erythroid development in Section 1.4.
1.2.1 The Origin of Definitive Haematopoietic Stem Cells
In mammalian primitive haematopoiesis the nucleated red blood cells arise in the blood
islands of the extraembryonic yolk sac. A definitive, multilineage blood system that seeds the
foetal liver and bone marrow arises that is sustained by pluripotent HSCs with a capacity for
self-renewal and long term repopulation of haematopoietic tissues (Marshall and Thrasher,
2001). Traditional views on the origin of the definitive HSC have recently been challenged. It
was originally supposed that the definitive HSC originated in the blood islands of the yolk sac
2

oL'4)
Figure 1.1 Ilaematopoietic blood
The haernatopoietic stem cell (HSC) is a multipotent progenitor that can differentiate into any
one of the eight blood cell lineages. The initial effect of sunounding signalling molecules
influences the commitment of the HSC to formation of either myeloid or lymphoid
progenitors. Upon further lineage-specific signalling the myeloid and lymphoid progenitors
will become committed to a particular blood cell type. The lymphoid progenitor will
differentiate into either a T or B cell, whereas the myeloid progenitor will differentiate into a
precursor cell belonging to one of six potential blood cell lineages.

HaematopoieticStem Gell
B Gell
T Gell
Macrophage
Megakaryocyte
Erythrocyte
Neutrophil
Eosinophil
Lymphoid Progenitor
Myeloid Progenitor
Mast Gell

since this was where blood cells were first detectable in the embryo (Moore and Metcalf,
1910).It was proposed that the HSCs would then migrate to the foetal liver to propagate the
definitive haematopoietic system. However, grafting experiments with avian embryos
identified an alternate intra-embryonic HSC source in the dorsal aorta wall (Dietterlen-Lievre,
1975; Cormier et al., 1986). Thus, recent studies have focussed on the identification of an
intra-embryonic origin for definitive HSCs in the mouse embryo.
The equivalent region to the dorsal chick aortic wall was identified in the mouse embryo as
the source of definitive HSCs. This region is called the aorta-gonad-mesonephros (AGM) in
the mouse and is derived from para-aortic splanchnopleural mesoderm. It has been reported
that at E7.5, prior to the establishment of circulation between the embryo and yolk sac,
multipotential HSCs capable of lymphoid and myeloid differentiation are located in the AGM
region and not in the yolk sac of the murine embryo. Furthermore, at El0, spleen colony
forming unit (a blood cell) activity is greater in the AGM than the yolk sac or foetal liver
(Medvinsky et ql., 1993,1996;Dzierzak and Medvinsky, 1995). Experiments culturing AGM
tissue in vitro have found this region to be a rich source of HSCs that can arise autonomously
and independently from the yolk sac and foetal liver. These cells are also able to reconstitute
irradiated mice before the initiation of haematopoiesis in the liver (Muller et al., 1994;'
Medvinsky andDzierzak,1996 Sanchez et al., 1996). Together, these findings indicate that
HSCs detected in the AGM do not originate from the yolk sac but arise from within this intra-
embryonic tissue. In addition, the evidence suggests that the HSCs originating in the AGM is
the population of cells from which the foetal liver and ultimately the bone malrow are seeded
to generate the definitive blood cell system. Studies continue to nominate the embryonic yolk
sac as the source of HSCs supporting both primitive and definitive haematopoiesis (Palis e/
at., 1999).In the human embryo, pluripotent HSCs arise virtually exclusively from intra-
embryonic tissue between 25 to 35 days gestation (Huyhn et al., 1995).
1.3 ERYTHROPOIESIS
Erythropoiesis is a multi-stage process that involves the differentiation of pluripotent HSCs
into mature, circulating erythrocytes. As the committed precursor red blood cells progress
through a series of morphologically distinct stages, their function becomes increasingly
specialised to result in the primary role of oxygen delivery around the body. As described
earlier (Section L2),the bone marrow is the major site of erythropoiesis in the adult. Within
J

the bone matrow, stromal macrophages provide discreet domains known as 'erythroblastic
islands' where the process of erythroid differentiation occurs (Bessis et al., 1983).
Erythropoiesis is triggered by the release of the hormone erythropoietin (Epo) from the
kidney in response to low oxygen tension levels in the body (Jelkmann,1992; Porter and
Goldberg, 1993). Epo binds to the Epo receptor (EpoR) expressed on the cell surface of
immature erythroid cells to initiate a signalling cascade (Section 1.3.1) that ultimately
activates the genes required for erythropoiesis. The commitment of haematopoietic
progenitors to the erythroid lineage is associated with a vast increase in EpoR expression on
the cell surface (Heberlein et al., 1992).
Erythroid burst forming units (BFU-E) are the first progenitor cells to become committed to
the erythroid lineage and also the first to become Epo-responsive (Tilbrook and Klinken,
1999) (Figure 1.2). Differentiation to the next stage is also mediated by interleukin 3 and
granulocyte macrophage-colony stimulating factor (GM-CSF) to produce the late progenitor,
colony forming unit-erythroid (CFU-E) (Metcalf et al., 1986;Lopez et al., 1987). As will be
discussed in Section 1 .3.1, mice null for the EpoR will not progress past the CFU-E stage,
indicating that Epo is critical for later steps of erythropoiesis but is not essential for
progression prior to this point (Wu er al., 1995). Proerythroblast cells represent the next stage
of erythroid differentiation. These cells undergo a series of four mitotic divisions during
which the proerythroblast progresses through morphologically defined steps, basophilic
erythroblast, polychromatophilic erythroblast and orthochromatic erythrobl ast. The
differentiating red blood cells are responsive to Epo stimulation until the polychromatophilic
erythroblast stage (Tilbrook and Klinken,7999). V/ithin the next 72hours, the nucleus is
ejected, phagocytosed by perisinal bone marrow macrophages and the enucleated erythroid
cell becomes a reticulocfle. The reticulocyte enters the circulation via a process known as
diapedesis. Towards the end of erythroid differentiation, erythroblast cells condense, protein
s¡mthesis decreases, organelles begin to degenerate and the diameter of the erythroblast
reduces. In addition, as erythropoiesis progresses, the cells are less dependent on Epo
stimulation, supported by a reduction in cell surface EpoR expression (Landschulz et al.,
1989). The final stages of erythropoiesis occur in the circulation over 48 hours and involve
synthesis of the remaining 20o/o of haemoglobin content and ejection of residual organelles.
Haemoglobin comprises 90% of total protein in mature erythrocytes. As a result of the four
mitotic divisions, sixteen circulating erythrocytes are generated from each proerythroblast
4

Figure 1.2 Pathway for the process of erythopoiesis
In the adult, under hypoxic conditions, Epo binds to the EpoR expressed on the surface of
precursor erythroid cells to initiate red blood cell differentiation. The burst forming unit-
erythroid (BFU-E) is the first cell to become responsive to Epo signalling. The maturing
erythroid cells retain their ability to respond to Epo until the polychromatic erythoblast (PE).
Progression from the BFU-Es to the colony forming unit-erythroid (CFU-E) stage also
requires interleukin 3 (IL-3) and granulocyte macrophage-colony stimulating factor (GM-
CSF). The EpoR is critical for the late stages in differentiation as mice null for the receptor
cannot progress past the CFU-E stage of erythropoietic development. Adult erythropoiesis
occurs in the bone marrow followed by release of the mature enucleated red blood into the
circulation.
, lr',
\\1"
¡

Adult Bone Marrow Girculation
+>Stem Gell BFU-E CFU.E PE Erythrocyte
Obligatory role for Epo

(Bessis et a1.,1983; Lewis, 1990). It is estimated that 2x101r erythrocytes enter the circulation
each day, surviving for 120 days (Bessis et a1.,1983; Eckardt and Bauer, 1989).
1.3.1 Erythropoietin and Erythropoietin Receptor Signalling
Epo is a 166 amino acid, 3OkDa hormone glycoprotein that initiates erythropoiesis,
influencing the process that maintains the balance between red blood cell production and loss
to result in efficient oxygen delivery to the body (Kendall, 2001). Epo contains two internal
di-sulphide linkages between cysteine residues that are required for its function (Sfkowski,
1980), Epo is predominantly produced by the kidney (Jacobson et al., 1957; Erslev et al.,
1985) wilh 10o/o occurring in the liver (Jacobson et al., 1959). Production in the liver is
primarily during human foetal development(Zarfani et al., l9l4) and the switch to the kidney
occurs at approximately 30 weeks gestation via an unknown mechanism (Dame et al., 1998).
The Epo response is induced by low venous oxygen tension (Kurtz et al., 1988). Residing in
the 3'region of the Epo gene is an enhancer element that stimulates Epo mRNA levels in
response to hypoxia (Imagawa et al., l99l). Hypoxia inducible factor (HIF) has been
identified as the protein that interacts with this enhancer element and its binding to the Epo
gene is induced by hypoxia (Wang and Semenza,1993).In addition to the 3' enhancer there is
also a promoter element that increases transcription of the Epo gene in response to hypoxia
but this region does not contain a HIF binding site (Blanchard et al., 1992). Recently, a
protein termed hlpoxia-associated factor was shown to bind this element within the Epo gene
promoter (Gupta et a1.,2000). Interestingly, activation of the MAP kinase p38ct appears to
affect Epo gene expression as murine embryos null for p38cr die between El 1.5 and 812.5
and suffer severe anaemia due to failed definitive erythropoiesis and this is caused by
decreased Epo synthesis (Tamura et a1.,2000). Aside from developmental signals, Epo
expression is also stimulated by physiological and pharmacological agents such as carbon
monoxide and the iron chelator desferrioxamine (Ebert and Bunn, 1999).
The EpoR is encoded by a single copy 5 kb gene consisting of 8 exons (D'Andrea et al.,
1989). The gene encodes a 501 amino acid protein, between 72 to 78kDa which includes
modifications of the protein by glycosylation and phosphorylation (Tilbrook and Klinken,
1999). The EpoR is the founding member of the type I superfamily of single transmembrane
cytokines (D'Andrea et a1.,1989; Jones et al., 1990). It shares several features with other
5

cytokine receptor family members including a single transmembrane region and in the
extracellular domain, four conserved cysteines, a group of aromatic residues and a
tryptophan-serine-x-tryptophan-serine (V/SXWS) motif (Tilbrook and Klinken, 1999). The
intracellular domain consists of two functional regions. The first is the membrane-proximal
region containing two domains called box-1 and box-2 which are conseryed in several other
cytokine receptors. It is the region required for Epo stimulation, mitogenesis, differentiation
and initiation of signalling cascades (Tilbrook and Klinken, 1999). The second domain is the
membrane-distal region and is not essential for Epo signalling but has been demonstrated to
be a regulatoryregion (D'Andrea et a1.,1991). The 8 tyrosine residues present in the EpoR
are phosphorylated after Epo stimulation and function as docking sites for various signalling
molecules (Tilbrook and Klinken, 1999).
EpoR null mice exhibit premature termination of erythropoiesis at the CFU-E stáge,
signiffing the requirement of the EpoR in the later stages of red blood cell development (Wu
et al., 1995). Approximately 1200 EpoR molecules are expressed on the surface of an
immature erythroid progenitor (D'Andrea and Zon,1990) with the number dependent on the
stage of erythroid differentiation. In a state of hypoxia, Epo is produced by the kidney and
binds to the EpoR. This action leads to a dimerisation of the EpoR (Watowich,1999) and
signalling is initiated via two pathways, the janus kinase /signal transducer and activator of
transcription (JAK/STAT) and ras/MAP kinase pathways (Tilbrook and Klinken,1999).
JAK2 is the primary kinase responsible for phosphorylation of the tyrosine residues located
on the intracellular domain of the EpoR (Witthuhn et al., 1993). JAK2 binds to the EpoR
prior to Epo stimulation and is activated to phosphorylate the receptor once Epo binds. This
occurs within 30 seconds of exposure to Epo (Komatsu et al., 1992). It has been reported that
Lyn tyrosine kinase is required for differentiation but not viability of differentiating erythroid
cells (Tilbrook et al., 7997). Lyn, like JAK2, pre-associates with the EpoR and is only
activated once Epo binds to the EpoR and stimulates its activity (Tilbrook et al., 1997; Chin et
a1.,1998). Lyn is able to bind JAK2 and can affect STAT5 phosphorylation (Chin et a1.,1998).
In summary, Epo binds to the EpoR to cause receptor dimerisation. Pre-bound JAK2 is
activated and phosphorylates the EpoR. STAT5 binds to specific phosphorylated tyrosine
residues on the EpoR and is phosphorylated by JAK2 (Damen et al., 1995:. Barber et al.,
2001) and may also be activated by Lyn (Chin et al., 1998). This induces dimerisation of the
STAT5 molecules and localisation to the nucleus where they can directly stimulate
transcription of the genes required for erythroid differentiation.
6

The ras/MAP kinase pathway is more complex with many signalling effectors involved. Grb2
can bind either directly or indirectly via the Shc adaptor molecule to phosphorylated tyrosine
residues on the EpoR. It can also act as a linker molecule to SHP-2, cbl or SOS in order to
activate ras. Concurrently, ras GTPase activating protein is phosphorylated to prevent it from
deactivating ras. Raf is localised to the plasma membrane by ras-GTP where it is
phosphorylated and can activate MEK. MEK proceeds to activate MAP kinases. Other
signalling molecules activated by Epo stimulation of the EpoR include phospholipase Cy,
SHIP, JNK, vav, fes/fos and PI3 kinase (Tilbrook and Klinken,1999).
Experiments utilising mutant EpoR proteins that could not activate some of the signalling
molecules demonstrated that normal erythropoiesis was possible, suggesting that the
signalling pathways may be redundant (Constantinescu e/ al., 1999). There is also evidence
that the EpoR does not function in isolation and that cross-talk between different receptors is
required to initiate the desired response (Pircher et aL.,2001). For example, the EpoR
intracellular domain contains a c-kit receptor binding site. Binding of the ligand, stem cell
factor, to the c-kit receptor activates the receptor such that it is then able to tyrosine
phosphorylate the EpoR. Thus, an interaction between the EpoR and c-kit is required for
normal erythropoiesis (Damen et al., 1995; Chin et aL.,1998).
1.4 TRANSCRIPTION FACTORS REGULATING ERYTHROPOIESIS
The following section will discuss the contribution of various transcriptional mediators to the
control of red blood cell differentiation. The regulation of erythropoiesis requires the activity
of transcription factors that are both erythroid-specific and those that function in a broader
manner with respect to the haematopoietic process. Therefore, some of the factors described
are involved in regulating events that are centred on erlhroid differentiation as well as
mediating such processes as lineage commitment. A number of the nuclear DNA binding
proteins involved in this process and the consensus sequences they bind are summarised in
Table 1.1.
1.4.1 Stem Cell Leukemia (SCL)
SCL was originally identified in a chromosomal translocation in T-cell acute lymphoblastic
leukemia. It is a basic helix-loop-helix (bHLH) transcription factor that is encoded on
1

nl rtl
\'o"r"ln"Þn'{J Y J'
chromosome I (Hershfield et ø1., 1984; Begley et al., 1991). SCL binds to E-box DNA
elements as a heterodimer with El2lE4l, the alternately spliced products of the ElA gene
(Hsu e/ al., 1994; Shivdasani and Orkin, 1996).It also forms part of a complex with
transcription factors EI2|E47, GATA-1, Ldb-1 and LM02 (Wadman et al., 1997). SCL has
been detected in early haematopoietic progenitor cells, megakaryocles and mast cells. It is
also expressed in non-erythroid tissues such as endothelial cells and neurons (Green et al.,
1992; Drake et al., 19911' Shivdasani,l99T). During erythropoiesis, SCL expression increases,
resulting in enhanced proliferation and differentiation. Concomitantly, SCL represses
differentiation in myeloid progenitors and is undetectable in most mature myeloid and
lymphoid cells (Begley and Green, 1999).Interestingly, DNA binding independent roles for
SCL in primitive erythropoiesis have been identified (Porcher et al., 1999) but a contribution
to the maturation of definitive precursor erythroid cells requires DNA binding (Porcher et al.,
1999).In mice null for SCL, death occurs in utero at E8.5 with no evidence of blood
formation. In SCL null ES cells, no haematopoietic cell lineage is observed. This suggests a
critical role for SCL in primitive haematopoiesis and also in ensuring commitment to the
erythroid lineage and the process of erythropoiesis (Shivdasani et al., 1995; Porcher et al.,
1996). - t
Qç,a?/'-rl*- r iô\-J_¡;i ì'
l.4.2LlM-only protein 2 (LM02)
LM02 consists of two cysteine-rich LIM domains that are homologous to the DNA binding
domains of GATA transcription factors. It is involved in the translocation of childhood T cell
leukemia (Rabbitts, 1998). The highest level of LM02 expression is in haematopoietic tissues
(Foroni et al., 1992). It does not bind DNA directly but acts as a bridge between DNA binding
transcription factors such as SCL and GATA-1. Approximately 50o/o of erythroid Lilt402
protein associates with SCL (Shivdasani and Orkin,1996; Wadman et al., 1997). LM02 null
mice die due to severe anaemia at E9 with an absence of haematopoiesis in the yolk sac.
Thus, it is likely IhatLM)2 plays an essential role in the early stages of haematopoiesis
(Warren et al., 1994; Yamada et al., 1998). LM02 forms part of an erythroid transcription-
activation complex consisting of SCL, E2A, GATA-1 and Lbdl that recognises E box motifs
located approximately 10 bp upstream of a GATA site. This suggests that LM02 also
participates in a lineage-specific mechanism to regulate erythropoiesis (Wadman et al., l99l).
In further support of LM02 participation in erythropoiesis regulation, LI|l402 promoted
erythroid differentiation when ectopically expressed inXenopus (Mead et aL.,2001).
8

Table 1,.1. Regulatory proteins involved in haematopoiesis
A summary of a number of the transcriptional activators involved in blood cell development.
Indicated are the binding motif and DNA recognition sequence for each transcription factor.

Transcription Factor
SCL
LM02
GATA-2
c-Myb
GATA-1
FOG
EKLF
BKLF
NF-E2
Motifs
bHLH
LIM domain
zinc finger
helix-tu rn-helix/leucine zipper
zinc finger
zinc finger
zinc finger
zinc finger
leucine zipper
DNA Binding Sequence
CAGGTG
unknown
(r/A)GArA(A/G)
TAACGG
(T/A)GATA(A/G)
unknown
CCNCNCCCN
cAcc
(r/c)rGCrGA(c/G)TCA(r/C)
PU.1 winged helix-tu rn-helix unknown

1.4.3 GATA-2
Like other members of the GATA family of transcription factors, GATA-2 contains two
homologous zinc-finger domains and binds to the GATA consensus sequence
(T/AGATAA/G). It is expressed in both haematopoietic and endothelial cells (Perry and
Soreq, 2002). Forced expression of GATA-2 in erythroid precursors leads to increased
proliferation of the erythroid cells but inhibits differentiation (Shivdasani and Orkin, 1996).
GATA-2 is expressed prior to GATA-I such that its expression decreases simultaneously to
the increased expression of GATA-I, allowing erythroid differentiation to occur. GATA-2-1-
targeted mice die at around E10 to 11, during yolk sac haematopoiesis. Multipotent
progenitors derived from GAT A-2r- ES cells proliferate poorly and apoptose (Shivdasani and
Orkin, 1996; Shivdasani,lggl). These findings suggest that GATA-2 is primarily required for
the expansion and survival of early haematopoietic cells and not erythroid differentiation.
1.4.4 c-Myb
c-Myb is a proto-oncogene that is vastly expressed in immature haematopoietic cells of the
erythroid, myeloid and lymphoid lineages. As differentiation of these cells proceeds, c-Myb
expression is reduced. In addition, forced expression of c-Myb inhibits erythroid
differentiation (Shivdasani and Orkin, 1996). c-Myb is required for expansion of
haematopoietic cells and in order to achieve terminal differentiation its expression needs to be
down regulated (Mucenski et ql., I99l; Shivdasani and Orkin, 1996). Mice null for the c-Myb
gene exhibit normal primitive haematopoiesis but impaired definitive blood development with
death occurring in utero at E15. Defective circulating erythrocytes were observed together
with normal appearance of megakaryocfes, granulocytes and monocytes. Therefore, c-Myb
appears to be essential in the expansion of definitive haematopoietic cells but does not play a
role in primitive blood development (Perry and Soreq, 2002).
The following sections discuss the erythroid-specific transcriptional activators that mediate
haematopoiesis, specifically regulating the genes required for the process of erythroid
development.
9

1.4.5 GATA-I
The gene for GATA-1 is located on the X chromosome at position Xplt.23 (Zon et al.,
1990). It is expressed in erythrocytes, megakaryocytes, eosinophils, mast cells (Tsai et al.,
1989; Evans and Felsenfeld, 1989) and Sertoli cells in the testis (Ito et al., 1993). GATA-1
binding sites ((T/A)GATA(A/G)) can be found in almost all promoters and enhancers of
erythroid-specific genes including the globins (Tsai e/ al., 1989; Evans and Felsenfeld, 1989).
Gene targeting studies have demonstrated that GATA-I is critical for normal erythropoiesis
(Pevny et al., 1991). GATA-1 null mice die at El 1.5 and do not exhibit any erythropoiesis
due to arrested maturation and apoptosis at the proerythroblast stage (Fujiwara et al., 1996).
This work demonstrates that GATA-1 is essential for erythroid development via the
promotion of cell survival and differentiation. In addition to ablated erythropoiesis, GATA-l-/-
murine embryos demonstrate a blockage in megakaryocyte development at the mid-
maturation stage. However, ES cells lacking a functional GATA-I gene are able to
differentiate into other haematopoietic lineages. It has been proposed that GATA-I is able to
modulate the commitment of a progenitor cell to a particular lineage since forced GATA-1
expression in an early myeloid cell line results in megakaryocyte differentiation (Visvader er
al., 1992; Shivdasani et al., 1997).
GATA-1 is expressed in haematopoietic cells and the testis as two distinct transcripts. They
are regulated by different promoters and first exons but the coding exons are common to both
transcripts (Shimizu et aL.,2001). GATA-1 expression is regulated by a 5' enhancer in
primitive erythroid cells with additional elements in the first intron required for GATA-I
regulation in definitive erythroid cells. Together they form the GATA-I locus haematopoietic
regulatory domain (Shimizu et a|.,200I). The GATA-1 transcription factor contains two zinc
fingers that are localised to the C and N-termini. They are both required for DNA recognition,
binding and physical interaction with other transcription factors. They are of the Cys-X2-Cys-
XrzCys-Xz-Cys configuration. The N-terminal zinc finger is critical for interaction with
Friend of GATA (FOG), Erythroid Kruppel-like factor (EKLF), L}i402 and CREB binding
protein and provides specificity and stability of binding to the GATA recognition sequence on
DNA. Overall, the N-terminal zinc finger is essential for definitive but not primitive
erythropoiesis (Trainor et al., 1996; Crispino et al., 1999). The C-terminal zinc finger is
paramount to GATA-1 function and is responsible for high-affinity DNA binding (Martin and
Orkin, 1990).Therefore, different functional domains of GATA-1 are required for activation
of specific genes in primitive and definitive erythropoiesis as well as particular transcription
factor binding elements (Shimizu et aL.,2001).
10

1.4.6 FOG
The FOG protein contains 9 zinc fingers and at least one of these interacts with the N-terminal
zinc finger of GATA-I. FOG is co-expressed with GATA-I in the foetal liver, embryonic
erythroblasts, mast cells, megakaryocytes and adult spleen (Tsang et al., l99l). Thus, FOG
cooperates with GATA-1 to promote both erythroid and megakaryocyte differentiation.
Experiments utilising GATA-I mutants that could not interact with FOG found that terminal
erythroid differentiation was blocked. This occurred due to deregulation of GATA-I
expression and its target genes such as o and B-globin, suggesting FOG is required for
regulation of GATA-I and enables it to interact normally with its target genes (Crispino et al.,
tgee).
FOG null mice exhibit inhibited erythropoiesis and a complete lack of differentiation of the
megakaryocyte lineage (Tsang et al., 1998). This differs from mice deficient in GATA-I
where the megakaryocyte lineage is blocked midway through the differentiation process
(Fujiwara et al., 1996), indicating that FOG behaves independently of GATA-I in the early
stàges of megakaryocyte development.
1.4.7 EKLF
The EKLF gene is located on chromosome 19 and it encodes a zinc finger protein that
belongs to the Kruppel family of transcription factors. Human EKLF is362 amino acids in
length with three C2Þ2-type zinc frngers at the C-terminus. It has 690/o hotal identity aú 93Yo
identity with zinc fingers of the mouse EKLF protein. Each finger contains three key residues
that form sequence-specific contacts with DNA residues (Perkins, 1999).
EKLF binds the CACCC consensus sequence (5'-NCNCNCCCN-3') in the regulatory
domains of its target erythroid-specific genes, including adult B-globin. EKLF preferentially
binds CACCC elements in human and murine adult type B-globin promoters over murine
foetal Bhl-globin, human y-globin or EpoR gene promoters. It plays an essential role in the
regulation of human B-globin expression (Miller and Bieker,1993) with its expression
restricted to erythroid cells, megakaryocytes and mast cells (Turner and Crossley, 1999).
11

EKLF null mice die prior to El6 due to severe anaemia and B-globin deficiency. Embryonic
erythropoiesis and embryonic e and (-globin gene levels are normal (Perkins et al., 1995).
Therefore, EKLF is important to the activation of the adult B-globin gene in the later stages of
erythropoiesis. Naturally occurring mutations in CACCC boxes of the adult type B-globin
result in reduced B-globin expression coinciding with elevated y-globin mRNA levels and the
blood disorder B-thalassemia which is a result of poor EKLF binding (Huisman, 1997;
Perkins, 1999).In addition, overexpression of EKLF results in an earlier switch from foetal to
adult globin (Tewari et al., 1998). Thus, a role exists for EKLF in silencing of the y-globin
gene or in the switch from foetal to adult globin production.
EKLF activation of the B-globin gene ls y enhanced in the presence of hypersensitivity
(HS) site 2 of the locus control (LCR) et al., 1995). CACCC binding sites for
EKLF have been shown to physically th EKLF in the HS2 and HS3 sites of the B-
globin LCR (Lee et a\.,2000). EKLF activity also facilitates hypersensitivity formation at
both the HS3 site and the B-globin promoter, as evidenced in EKLF null mice (Wijerde et al.,
1996). A role for EKLF in the opening of chromatin surrounding the B-globin locus has been
put forward. Armstrong et al., (1998) purified an EKlF-dependent complex that could
activate transcription from a B-globin locus in a closed chromatin conformation. The purified
complex was called EKLF coactivator remodelling complex-1 (ERCI) (Armstrong et al.,
1998). This finding led to suggestions that the role of EKLF is to recruit this chromatin
modiffing complex to the B-globin locus, facilitating transcriptional activity of this domain.
An association between CACCC boxes and GATA motifs has been reported in many
erythroid promoters (de Boer et a1.,1988; Mignotte et a1.,1989a; Tsai et al., 1997;Zonet al.,
l99l; Rahuel et al., 1992; Max-Audit et al., 1993). There is also evidence to suggest that
transcription factors binding at these sites interact physically and functionally to cooperatively
play a critical role in transcriptional activation of erythroid-specific genes (Merika and Orkin,
1995; Gregory et al., 1996).
In addition to the participation of EKLF in B-globin regulation it may also play a role in cell
cycle control. For example, the re-introduction of EKLF into an EKlF-deficient erythroid cell
line containing the human B-globin locus results in increased differentiation and
haemoglobinisation but decreased cellular proliferation (Coghill et al., 2007).
I2

1.4.8 Other Kruppel-Like Family Members Regulating Erythropoiesis
Basic Ikuppel-like factor (BKLF) is expressed in erythroid cells, fibroblasts and the brain.
Like other members of the Kruppel family of transcription factors it binds to the CACCC
boxes of its target genes via its three highly conserved C-terminus Kruppel-like zinc fingers.
Expression of BKLF in erythroid cells is dependent on EKLF as BKLF levels are reduced in
erythroid cells but not in the brain of EKLF deficient mice (Crossley et al., 1996). BKLF
interacts with the corepressor CtBP to repress EKLF promoter activation ín vitro (Crossley e/
al., 1996; Tumer and Crossley, 1998).
(Foetd{@rppel-like factor (FKLF) in the human activates embryonic e-globin expression and
to a lesser extent foetal y-globin levels. Activation occurs via interaction of FKLF with
CACCC box elements of these particular globin genes but it cannot mediate expression of
other erythroid genes containing CACCC sites (Asano et al., 1999). The murine FKLF-2
protein can enhance y-globin expression 1O0-fold and also activate the promoters of e and B-
globin, GATA-I and enzymes of the haem biosynthetic pathway (Asano et a1.,2000).
1.4.9 NF-82
NF-E2 is a heterodimeric basic-leucine zipper transcription factor that consists of p45 NF-E2
and a member of the small Maf family of proteins (Blank and Andrews,1997; Motohashi er
al., 1997). p45 NF-E2 is mostly expressed in erythroid cells and megakaryocytes (Andrews et
al., 1993a), with Maf proteins expressed more widely. MafK, also known as pl8 NF-E2, and
MafG are the predominant partner factors to p45 NF-E2 in both erythroid cells and
megakaryocytes (Andrews et al., 1993b; Lecine et al.,1998; Shavit et a1.,1998). The
consensus NF-E2 binding site (5' (T/C)GCTGA(C/G)TCA(T aJ et al., 1993a)
has been identified and referred to as Maf recognition sites for
NF-E2 are present in the regulatory regions of erythroid and genes, ln
particular the HS2 site of the B-globin LCR where it is essential for enhancer function (Moi
and Kan, 1990; Ney e/ al., 1990; Talbot and Grosveld, 1991; Andrew et al., 1993a; Forsberg
et a\.,2000). NF-E2 binding sites have also been located in the promoters for the human
porphobilinogen deaminase (PBGD) (Mignotte et ql., 1989b) and ferrochelatase (Tugores e/
al., 1994) genes.
(MARES).
t3

In vitro studies have provided evidence to support the role of NF-E2 in enhancer-dependent
globin transcription. For example, the murine erythroleukemia (MEL) cell line, CB3, when
transformed by Friend murine leukemia virus is unable to express the p45 NF-82 protein (Lu
et al., 1994).In this cell line, c¡c and B-globin expression is reduced but can be partially
rescued by the introduction of a retroviral vector containing the p45 NF-E2 cDNA. In
addition, a dominant negative pl8 NF-E2 mutant that cannot bind DNA but can form
heterodimers with p45 NF-E2 was stably transfected into MEL cells. The resultant effect was
a reduction in functional NF-E2 protein and globin expression levels. Upon introduction of a
tethered p45-pl8 construct into these cells expression of the globin genes returned to wild
type levels (Kotkow and Orkin, 1995).
It has been postulated that NF-E2 is involved in the formation of HS sites in the B-globin
LCR. Using an in vitro Drosophila chromatin assembly system (Armstrong and Emerson,
1996) it was demonstrated that purified recombinant NF-E2 could form a HS2 site, suggesting
that it may be able to destabilise local nucleosome structure in an ATP-dependent process.
Other ATP-dependent chromatin remodelling factors have been identified in vitro including
the human (Kwon et al., 1994) and yeast (Cote et al., 1994) SWVSNF arrd Drosophilø NURF
(Tsukiyama and Wu, 1995) but NF-E2 interaction with them has not been investigated.
Interestingly, disruption of chromatin structure by NF-E2 factlitated GATA-1 binding to
inverted GATA motifs located approximately 60 bp downstream of the NF-E2 site
(Armstrong and Emerson, 1996). Furthermore, GATA-I and NF-E2 are both required for the
formation of HS4 in the human B-globin LCR (Stamatoyannopoulos e/ al., 1995). GATA-I
and NF-E2 sites localised to similar positions have also been found in the human B-globin
HSI and HS3, murine B-globin HS2, HS3 and HS4 and chicken B-globin enhancer
(Stamatoyannopoulos et al., 1995). This suggests a conserved mechanism may be utilised to
generate HS in regions pertaining to transcriptional control.
In vivo studies on each protein dimer have been conducted to further elucidate the role of NF-
E2 in globin regulation. Mice lacking either p45 or pl8 exhibited normal erythropoiesis,
comparable to red blood cell differentiation in wild tlpe mice (Shivdasani and Orkin, 1995;
Shivdasani et al., 1995; Chan et al., 7996; Kotkow and Orkin, 1996; Farmer et al., 1997).ln
mice lackingp45, platelet formation was not detectable (Shivdasani et al., 1995). Therefore,
results obtained from in vitro studies, suggesting that NF-E2 is essential for globin gene
l4

expression, were not supported by in vivo findings which implied that NF-E2 is not critical for
erythropoiesis or that functional redundancy exists among NF-E2 family members.
\,,. t i-'U
.,)
1.4.10 PU.l
PU.1 is a haematopoietic Ets factor that promotes the differentiation of the lymphoid and
myeloid lineages (Scott et al., 1994).It has been shown to inhibit erythroid differentiation and
needs to be suppressed to allow restoration of terminal differentiation in MEL cells (Ben-
David and Bernstein, 1991; Rekhtman et al., 1999). PU.1 interacts directlywith GATA-1,
using its DNA binding and transactivation domain to repress the function of GATA-I as a
transcriptional activator. PU.l does not affect the binding of other factors to GATA-I nor its
DNA binding capabilities (Rekhtman et al., 1999). Thus, it is most likely that PU.1 binds to
DNA bound GATA-I complexes and represses its function at this level. Further evidence for
the inhibitory effect of PU.1 on GATA-1 transactivating activity was observed when PU.1
was ectopically expressed in the Xenopus. Erythropoiesis was retarded but this inhibition was
relieved by exogenous GATA-I expressioninthe Xenopus embryo, suggesting lineage
commitment may be regulated by their respective levels (Rekhtman et al., 1999).
l.4.ll Effect of a Transcriptional Coactivator on Erythroid Transcription Factors
CREB binding protein (CBP) and p300 are closely related transcriptional coactivators that are
ubiquitously expressed and interact with a multitude of transcription factors via specific
domains. CBP/p300 are capable of a wide range of activities that can be generally applied to
any given gene. Firstly, CBP/p300 may provide a bridge between transcription factors and
components of the basal transcription machinery, thereby forming or maintaining the
transcription pre-initiation complex (Blobel, 2002). Certainly CBP has been shown to directly
or indirectly bind the general transcription factor TFIIB (Kwok et al., 1994), TATA binding
protein (TBP) and RNA polyrnerase II (Blobel, 2002). CBP/p300 can function as an eîzpe,
demonstrated by their ability to acetylate all four core histones (Bannister and Kouzarides,
1996; Ogryzko et al., 7996). Moreover, CBP/p300 associates with proteins harbouring
intrinsic acetyltransferase activity such as PCAF (Yang et al., 1996) and GCN5 (Xu et al.,
1998). This leads to formation of a large acetyltransferase complex with broad specificity.
Therefore, recruitment of acetyltransferases by transcriptional activators results in a localised
increase in histone acetylation, which may assist in opening up chromatin.
15

It is well documented that acetylation of transcription factors alters their activity, although the
mechanism by which this occurs is unclear. The possibilities include affecting DNA binding,
transcriptional activity, interaction with other proteins or nuclear transport (Blobel, 2002).
This section will focus on the effect of CBP/p300 on erythroid-specific transcription factors
during red blood cell differentiation.
In vitro and in vivo studies have demonstrated that CBP can bind GATA-I and increase its
transcriptional activity (Blobel et al., 7998). CBP acetylases GATA-I in vitro at conserved
lysine residues near the zinc frngers (Boyes et al., 1998; Hung et al., 1999). Transfection-
based assays have also shown that CBP can acetylate GATA-1 in vivo (Hung et al., 1999).
GATA-I that cannot be acetylated due to mutated acetylation sites was unable to restore
erythroid differentiation in a GATA-I-deficient cell line, indicating that acetylation of
GATA-I is essential for its function in erythroid differentiation (Hung et al., 1999). However,
how CBP/p300 acetylation of GATA-I results in a stimulation of its activityhas notbeen
resolved. For example, acetylation of chicken GATA-1 by p300 leads to an increase in DNA
binding affinity (Boyes et al., 1998) whereas acetylation of the murine GATA-I protein by
CBP did not alter DNA binding (Hung et al., 1999). Alternately, acetylation of GATA-I may
stimulate interaction with accessory proteins to aid in transcriptional activation and chromatin
remodelling of the target gene. Another possibility is that acetylation disrupts the interaction
of GATA-I with a repressor molecule (Blobel, 2002). Recently, restoration of GATA-I
activity in a GATA-I deficient cell line led to increased acetylation of histones H3 and H4 at
the B-globin promoter and LCR (Letting et aL.,2003). Time course experiments demonstrated
a direct GATA-I effect as histone acetylation occurred rapidly after GATA-1 activation,
coinciding with increased globin gene expression. In addition, a correlation was observed
between occupancy of the p-globin locus by GATA-I and CBP and increased histone
acetylation, suggesting that GATA-I and CBP are required for the formation of an erythroid-
specific acetylation pattern on the B-globin locus and facilitating high levels of gene
expression (Letting et a1.,2003).
In vitro binding studies have demonstrated a direct physical interaction between the N-
terminal domain of p45 NF-E2 and CBP (Cheng et al., 7991). Further support came from the
finding that E1A (an inhibitor of CBP/p300 activity) can inhibit the function of HS2 of the B-
globin LCR, whose activity is predominantly conferred byNF-E2 binding elements (Forsberg
et al., 1999).Interaction of CBP with p45 NF-E2 and its partner MafG in vivo was found to
t6

lead to acetylation of MafG and not p45 NF-E2 (Hung et a1.,200I). Acetylation of MafG
stimulates the binding of NF-E2 in vitro and mutation of the MafG acetylation site reduces
NF-E2 binding and the transcriptional activity it mediates (Hung et a1.,2001). Thus,
interaction of CBP with NF-82 may modulate NF-E2 activity and regulate chromatin
strucfure. Recently, studies have questioned the role of NF-E2 in establishing histone
acetylation at the B-globin LCR. Acetylation patterns at the LCR and B-globin gene promoter
in CB3 erythroleukemia cells were analysed (Johnson et aL.,2001). CB3 cells do not contain
NF-E2 and have low p-globin mRNA levels but upon re-introduction of NF-E2, p-globin
expression is restored (Lu et al., 1994; Kotkow and Orkin, 1995). A high level of H3 and H4
acetylation at HS2 and HS3 of B-globin was observed in these cells, indicating that NF-E2 is
dispensable for histone acetylation at the LCR. Histone acetylation at the B-globin promoter
was reduced and was raised to normal levels upon p45 NF-E2 expression (Johnson et al.,
2001). Interestingly, the promoter does not contain any NF-E2 sites. Therefore, the LCR
bound NF-E2 may interact with the B-globin promoter indirectly through promoter bound
transcription factors via an LCR looping mechanism (Sawado et al., 2001). A role for NF-E2
in establishing histone hyperacetylation at the LCR cannot be ruled out as it is likely the
globin locus is akeady in an open acetylated domain since CB3 cells are fully committed to
the erythroid lineage prior to retroviral infection used to derive this cell line (Johnson et al.,
2001).
CBP/p300 is able to bind EKLF and stimulate its activity (Zhang and Bieker, 1998).
Acetylation by CBP/p300 occurs in the zinc frnger and activation domain of the EKLF protein
(Zhang et al., 200I). Acetylation of EKLF has been shown to enhance its transcriptional
activity on chromatinised DNA templates in vitro (Zhang et al., 2001). A link between EKLF
acetylation and SWVSNF recruitment has been determined as in vitro acetylation of EKLF
has been found to stimulate its interaction with BRGI, a subunit of the SV/VSNF complex
(Zhang et al., 2001). A mechanism has been proposed for the effect of EKLF acetylation on
B-globin transcription where binding of CBP/p300 to EKLF results in acetylation of histones
and EKLF itself. Acetylation stimulates the recruitment of SWVSNF and chromatin
surrounding the B-globin locus is remodelled allowing transcriptional activation to proceed. It
is noteworthy that there is no direct evidence for an interaction between EKLF and the
components of the SWI/SNF complex (Blobel, 2002).
t7

1.5 GLOBIN GENE EXPRESSION DURING ERYTHROPOIESIS
1.5.1The Globin Gene Cluster
The globin polypeptides are encoded by two clusters of genes, cr and B, with each cluster
containing a small number of genes. Globin gene expression is activated at a specific stage of
development and then repressed once that developmental period ends. This process is termed
'developmental switching' and involves altemate expression of embryonic, foetal and adult
globins. As a result, the constituents of the haemoglobin tetramers that are formed within
erythrocytes are dependent upon the stage of development. Furthermore, this coincides with
the shifts observed in erythropoiesis location during vertebrate embryogenesis (Orkin 1995).
In mammals and avian, ø and p-globin gene clusters reside on different chromosomes, with a
single chromosome encoding both clusters in fish and frogs (Orkin 1995). The human cr-
globin cluster of genes is located on chromosome 16 and encompasses approximately 30 kb.
It encodes three functional genes, one embryonic (() gene located upstream of two duplicated
adult cr-globin genes. The human B-globin cluster is 60 kb in length and resides on
chromosome 11. It has five functional genes, embryonic (e) globin, two duplicated foetal (y)
globins, adult minor ô globin and adult major B-globin and their order of arrangement in the
locus follows their sequential pattern of expression (Figure 1.3) (Engel and Tanimoto, 2000).
As mentioned in Section 1.2, the embryonic yolk sac is the first site of erythropoiesis and
haemoglobin production. The first haemoglobin formed is a tetramer consisting of either two
(-globins or two cr-globins and two e-globin polypeptides, designated Çzez and uzez,
respectively. As development progresses, (-globin gradually decreases while o-globin
increases. The shift of erythropoiesis from the yolk sac to the foetal liver correlates to a
decrease in e-globin levels and activation of foetal y-globin. At this point, the major type of
haemoglobin synthesised is foetal haemoglobin(uzyù. The final migration of erythropoiesis
to the bone marrow coincides with the last globin switch from foetal globin to adult ô and B-
globin. Thus, 98Yo of total haemoglobin synthesised is haemoglobin A (q¿Þz) and 1% is
haemoglobin A2 (cxzôz). However, the remaining lo/o of haemoglobin synthesised in the bone
marrow expresses the y-globin gene (Stamatoyannopoulos and Nienhuis, 1994; Donze et al.,
1 ees).
18

1.5.2 Regulation of B-Globin Gene Expression
The B-globin genes are expressed in a stage-specific manner such that their production
coincides with the developmental process of the embryo. The regulation of B-globin
expression requires strict control as an oxcess ofglobin production can be cytotoxic
(V/eatherall,1994). A complex program of transcriptional regulation is necessary to ensure
the correct temporal expression of the globin genes. This process is mediated by both
ubiquitous and erythroid-specific transcription factors that bind DNA regulatory sequences
located proximal and distal to the globin gene coding regions. The mechanism underlying the
ordered expression of globin genes is not fully understood.
Initial studies were performed to identiflz cis-elements responsible for erythroid-specific
expression of the B-globin genes in erythroid progenitor cells. Analysis of the human
B-globin sequence in transgenic mice found that Bglobin expression was erythroid-specific,
low and varied between transgenic lines. The variation among different lines was attributable
to the integration site of the transgene. Overall, these transgenic mice studies implied that a
cis-regulating element was absent from the B-globin transgene (Magram et al., 1985; Townes
et al., 1985). DNase I h1-persensitivity studies identified four erythroid-specific sites (5'HSl
to HS4) spanning 15 kb and residing approximately 1 I kb to 1 8 kb upstream of the e-globin
gene (Figure 1.3). A fifth site (HS5) was later identified (Tuan et al., 1985; Fonester et al.,
1986).Jhis region of DNase I hypersensitivity was referred to as the locus control region
(LCR) foÌ the p-globin locus. The B-globin LCR and the transcription factor binding sites
it are conserved in every organism analysed to date (Hardison et al., 1997). An
additional two HS sites (HS6 and HS7) have been located further upstream from the B-globin
LCR and have been shown to not play an important role in globin gene expression and
switching (Bulger et al., 1999; Huang et a1.,2000). One HS site has been identified at the 3'
end of the B-globin gene (Bulger and Groudine, 1999; Engel and Tanimoto, 2000) (Figure
1.3).
Grosveld et al. (1987) generated mice that had incorporated a transgene containing four of the
erythroid-specific B-globin HS sites upstream of the B-globin locus. Resultant B-globin
expression was high, erythroid-specific and independent of the integration site of the
transgene. This suggested that the LCR can enhance B-globin expression irrespective of its
location in chromatin and possibly modulate chromatin structure to ensure it is permissive for
t9

Figure 1.3 The human p-globin gene locus
The five globin genes of the B-globin locus are represented as yellow boxes. They are
sequentially expressed in the same order as their arrangement in the locus. The red boxes refer
to the five HS sites that constitute the LCR located in the 5' region of the locus. Two
additional HS sites have been identified further 5' of the LCR and are denoted by blue boxes.
Lastly, there is a HS site located at the 3' end of the B-globin locus and it is represented in
green. The primary slte of erythropoiesis shifts from the embryonic yolk sac to the foetal liver
and finally to the bone marrow in the adult. The shift in erythropoiesis coincides with the
switch between expression of different globin chains.
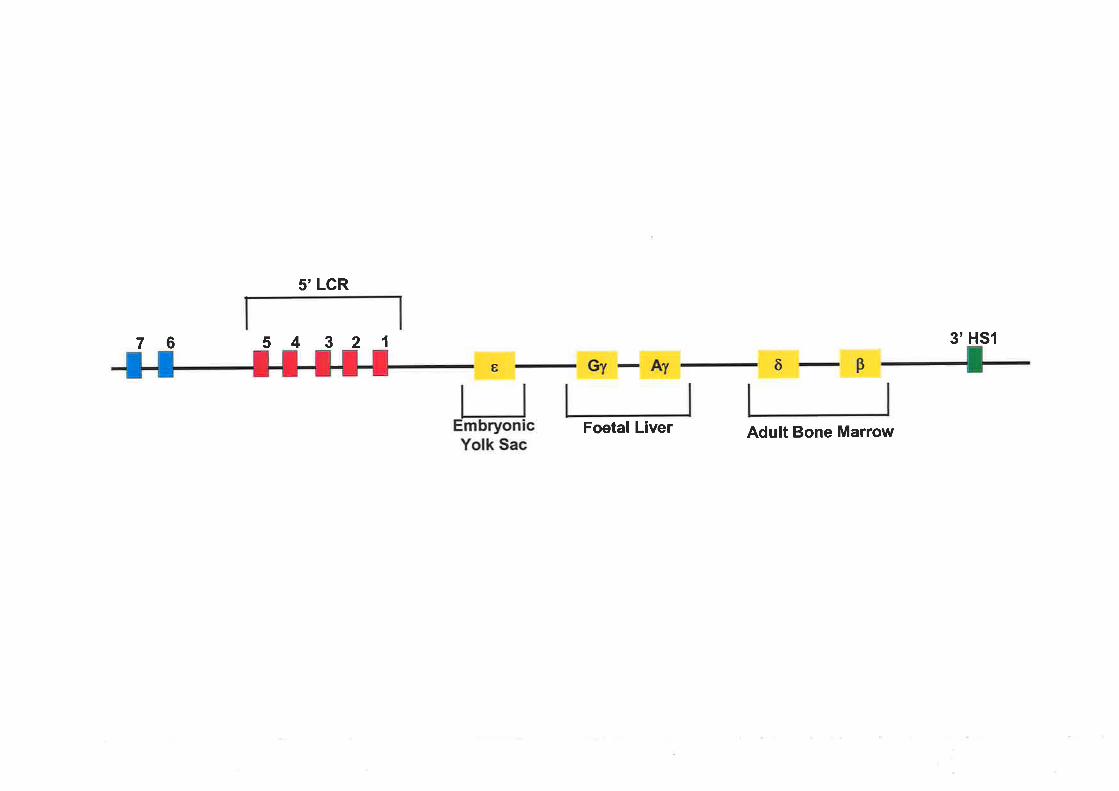
76
5'LCR
5 4 321 3'HS1
Foetal Liver Adult Bone Marrow

globin transcription. Supporting the notion that the original property of the LCR is to open
chromatin and facilitate gene transcription is the finding of a natural mutation in which the
LCR is deleted (Hispanic deletion). Associated with this mutation is a lack of globin gene
expression and the surrounding chromatin is found to be in an inactive state (Forrester et al.,
leeo).
Reik e/ al. (1998) deleted endogenous human HS2 to HS5 sites from the B-globin locus and
found that globin expression was lost in erythroid cells but no alteration in chromatin nuclease
sensitivity was observed. Furthermore, an in vivo study resulted in a decrease in
transcriptional activity upon deletion of the LCR but again, general DNase I sensitivity
throughout the murine B-globin locus was not affected (Bender et a1.,2000). These studies
suggested that the LCR is not required for unfolding of higþer order chromatin structure to
facilitate transcription of the desired genes in the locus. However, DNase I sensitivity is not
always linked to actively transcribing genes. For example, the LCR may not be involved in
general nuclease sensitivity but may function beyond this by regulating modifications to the
histone tails such as acetylation, methylation and phosphorylation (Levings and Bungert,
2002).
Thus, the role of the B-globin LCR in chromatin remodelling is currently unresolved.
However, a dual role for the LCR in B-globin regulation has been proposed. The first activity
of the LCR may be to establish and maintain the chromatin surrounding the globin locus in an
open configuration to facilitate transcription of the globin genes as required during erythroid
differentiation. Secondly, the LCR is proposed to strongly enhance the transcriptional activity
of the individual globin genes in an erythroid-specific manner. Evidence has been put forward
to support a'binary model' for the enhancing role of the LCR in B-globin gene regulation
(Walters et al., 1995; Martin et al., 1996; Milot et al., 1996; Sutherland et al., 1997). This
model suggests that the LCR acts to increase the probability that a promoter will be
transcriptionally active, increasing the proportion of expressing cells as opposed to enhancing
the level of expression in each cell. Therefore, the globin promoter is either 'on' such that the
LCR maybe maintaining the surrounding chromatin in an open configuration and permissive
for transcription or alternatively, the promoter is 'off due to absence of an interaction with
the LCR. This switch between inactive and active states may occur within open chromatin. [n
support of this, ectopic expression of the lacZ gene under the control of HS2 or HS3 of the
B-globin LCR has been shown to oscillate between 'on and ofP states in which changes in
nuclease sensitivity were not observed (Feng et al., 1999).
20

Erythroid-specific transcriptional activators are involved in regulating the B-globin locus.
Transcription factor binding sites located within the p-globin LCR and promoters recruit
proteins and protein complexes in a stage-specific manner to activate transcription of a
particular globin gene. The erythroid transcriptional mediators with pivotal roles in regulating
the B-globin locus include GATA-1, NF-E2 and EKLF. Protein-protein interactions of the
erythroid-specific transcription factors between themselves (Merika and Orkin, 1995) or with
coactivator complexes and acetyltransferases (Crossley et al., 1995; Blobel, 2000) may also
play arole in the function of the B-globin LCR. For an overview of the role of erythroid-
specific transcription factors in globin expression refer to Section 1.4.
Several regulatory mechanisms have been postulated to influence the expression of the B-
globin locus and the involvement of the LCR. Fluorescence in situ hybridisation studies have
indicated that the nucleus is divided into two compartments. Firstly, the 'permissive'
compartment is rich in euchromatin and factors to mediate chromatin opening. The second
'non-permissive' compartment consists of heterochromatin and factors to silence gene
expression (Francastel et a\.,2000). The LCR has been proposed to have a role in positioning
the B-globin locus in nuclear 'permissive domains' and this notion has been experimentally
supported (Francastel et al., 1999). However, Schubler et al. (2000) reported that the function
of the p-globin LCR in nuclear positioning is redundant. Alternatively, nuclear
compartmentalisation may be directed by transcriptional activators that bind regulatory
elements and disrupt interactions between the gene locus and heterochromatin, allowing the
gene to move into a permissive compartment of the nucleus (Francastel et al., 2000, 2001).
Boundary or insulator elements have been implicated in the regulation of the B-globin genes.
They are located at various positions in the B-globin loci and function to maintain a domain
within the locus in a steady state of open or closed chromatin, blocking the influence of
surrounding chromatin structure (Bell and Felsenfeld, 1999; Gribnau et a1.,2000). The 5' HS4
site in the chicken B-globin locus has been shown to contain insulator activity, suggesting that
boundary elements may facilitate gene expression from the B-globin locus (Pikaart et al.,
1 ee8).
Recently, the possibility of intergenic transcription regulating expression of the B-globin
locus has been investigated. The initiation of long transcripts was first observed in the HS2
2t

site of the B-globin LCR (Tuan et al., 1992). Following on from this finding was the
discovery of intergenic transcripts throughout the entire endogenous B-globin locus (Ashe e/
al., 1997). The pattern of intergenic transcripts in the B-globin locus was found to correlate
with DNase I sensitivity during development (Gribnau et a1.,2000), suggesting that intergenic
transcripts may assist in opening up the chromatin by disrupting its structure. However, the
function of intergenic transcription remains unclear as a recent study by Plant et al. (2001)
found no evidence for stage-specific generation of intergenic transcripts.
Two models have been proposed for the regulation of the B-globin locus by the LCR. The
first of these proposes a direct interaction between the proteins bound at the LCR and the
globin promoters. A holocomplex is proposed to form between the HS sites of the B-globin
LCR. The core elements of the HS sites of the B-globin LCR form an active site that binds
transcription factors and the flanking sequence of the HS sites provide the holocomplex with
its proper conformation (Grosveld et al., 1993; Bungert et al., 1995,1999; Harju et a1.,2002).
Currently, there is no direct evidence for the formation of a holocomplex (Tang et al., 2002).
This holocomplex is then able to loop through the nucleoplasm to directly activate or recruit
the basal transcriptional apparatus assembled at the globin gene promoters, looping out the
intervening DNA. This direct interaction between the LCR and globin promoters to activate
transcription is referred to as the 'loopilrg 'ìod"t. The looping model suggests that the globin
gene promoters are in competition for actiíation by the LCR since the LCR holocomplex
would provide only one activation centre for each locus.
The 'linking' model provides an alternative strategy by which the LCR could regulate
B-globin gene expression. In the context of this model, communication between the LCR and
a given globin gene promoter is facilitated by the formation of a chain of protein complexes
from the promoter to the enhancer. This occurs via a sequential, stage-specific binding of
transcription factors and chromatin facilitators along the chromatin fibre between the LCR
and a globin gene promoter to dehne a transcriptionally active domain (Bulger and Groudine,
t999; Dorsett, 1999). The transcription factors bound to a globin gene promoter and a HS site
of a transcriptionally primed locus are tethered to one another by a chain of non-DNA binding
facilitating factors. In vefiebrates these complexes are homologous to the Drosophila Chip
protein complexes and they act as guides for transcription initiation within the B-globin locus,
forming a bridge between factors binding the LCR and promoter-bound transcriptional
activators. These Chip-like factors may facilitate activation of one globin gene at a time while
simultaneouslyblocking transcription outside of this domain. The Chip-like proteins may
22

interact with transcription factors bound at the promoter at a specific developmental time
point, targeting that promoter for transcriptional activation via interaction with the LCR
(Harju et a1.,2002).
It is currently unresolved as to which model accurately resembles the mode in which the LCR
regulates B-globin gene expression in yiyo since the available data can be explained by either
model (Engel and Tanimoto, 2000). In addition, concepts such as intergenic transcription,
boundary elements and nuclear compartmentalisation also need to be accounted for in a
model for LCR regulation of the B-globin locus.
1.6 IIAEM BIOSYNTHESIS
The biosynthesis of haem requires eight steps (Figure 1.4). The hrst and last three steps occur
in the mitochondria with the remaining four steps taking place in the cytosol of the cell
(Bottomley and Muller-Eberhard, 1988). All enzymes are nuclear encoded and s¡mthesised in
the cytoplasm (May et al., 1995). The first reaction in the synthesis of haem is catalysed by
the enz¡rme 5-aminolevulinate synthase (ALAS) which is located on the matrix side of the
inner mitochondrial membrane (Rohde et a1.,1990). ALAS catalyses the condensation
reaction between glycine and succinyl CoA to produce 5-aminolevulinate (ALA). ALA
moves from the mitochondria into the cytoplasm where two molecules of ALA are condensed
by ALA dehydratase to yield porphobilinogen (PBG). Four molecules of PBG are linked
through the activity of PBGD to form the unstable intermediate hydroxymethylbilane.
Uroporphyrinogen III synthase converts hydroxyrnethylbilane to uroporphyrinogen III.
Uroporphyrinogen III decarboxylase catalyses the decarboxylation reaction of four acetic acid
side chains of uroporphyrinogen III to methyl groups to form coproporphyrinogen III. This is
the final reaction of the haem biosynthetic pathway that occurs in the cytoplasm of the cell.
Coproporphyrinogen III is imported into the mitochondria where coproporphyrinogen III
oxidase, residing in the intermembrane space, acts to generate protoporphyrinogen III.
Protoporphyrinogen III oxidase catalyses the next reaction to generate protoporphyrin IX.
Ferrochelatase, which spans the inner mitochondrial membrane, catalyses the insertion of
ferrous iron into protoporphynn IX and upon the release of two protons the end product of
haem is produced (Bottomley and Muller-Eberhard, 1988). The haem is then either utilised in
the mitochondria or exported to the cytosol via a mechanism that is poorly understood.
23

Figure 1.4 Haem Biosynthetic Pathway
Illustrated are the intermediate products of the eight steps of the haem biosynthetic pathway.
The specific compartmentilisation to either the mitochondrial matrix or the cytosol of the
individual steps and associated enzymes is shown. ALAS, 5-aminolevulinate synthase;ALA,
5-aminolevulinate; PBG, porphobilinogen; PBGD, porphobilinogen deaminase; URO'gen
III synthase, uropo{phyrinogen III synthase; URO'GEN III, uroporphyrinogen III;
URO'gen III decarboxylase, uroporphyrinogen III decarboxylase; COPRO'GEN III,
coproporphyrinogen; COPRO'gen III oxidase, coproporphyrinogen III oxidase;
PROTO'GEN III, protoporphyrinogen III; PROTO'gen oxidase, protoporphyrinogen
oxidase; A, acetate; M, methyl; P, propionate; V, vinyl. Adapted from May et al. (1995).
ALAoilc-cH2-NH2
-o
2
-ooc-cH2-cH2
CYTOPLASM
PBGD
UROS
P
P
UROD
PM
M
HYDROXYMETHYLBILANE
SUGGINYL GOA
GLYCINE
HEME
PROTOPORPHYRIN IX
Fo
PROTO'GEN
NH2-CH2-COO-
ALAS
M
Ferrochelatase Fe'*
M
M M
PPOX
CPX
M
+-oOc'cH2-cH2'f=O
SCoA
PBG
UROGEN
COPROGEN

All cells require haem for their function but strict regulation of haem production is imperative
as free haem is toxic to the cell. The toxicity of free haem is thought to be due to iron
catalysed free radical production that can cause lipid peroxidation, protein cross-linking and
DNA damage (Balla et al., I99l; Muller-Eberhard and Fraig, 1993;ili4ay et al., 1995).The
first enzyne of the haem biosynthetic pathway, ALAS, is the major control point for haem
production as its enzyme activity is lowest in comparison to the other enzymes in the pathway
(May et aL.,1995).
1.7 ALAS
ALAS is the first and rate-limitingeîzpe of the haem biosynthetic pathway (Bottomley and
Muller-Eberhard, 1988; Ponka et al., 1988; lll4ay et al., 1995). ALAS activity was first
identified in numerous bacterial and avian preparations. The ALAS enzyffìe was then purified
from several species including Rhodobacter sphaeroides (Warnick and Bumham, l97l;
Jordan and Laghai-Newton, 1986), rat liver (Ohashi and Kikuchi,7919; Srivastava et al.,
1982), chicken liver (Borthwick et al., 1983) and yeast (Volland and Felix, 1984). The
following section will focus on the properties of ALAS and its regulation since it is the
control point for haem biosynthesis.
1.7.1 Properties of ALAS
ALAS is a nuclear encoded enzyme (Bottomley et al., 7995) that is synthesised in the
cytoplasm in its precursor form. Precursor ALAS is targeted to the mitochondria via an
amphipathic N-terminal signal sequence which is proteolytically cleaved during import into
the mitochondria to generate the mature ALAS protein (May et al., 1995). The ALAS
substrate succinyl CoA is only found in the mitochondria, which prevents the function of
ALAS in the cytoplasm.
Two isoz¡rmes of ALAS have been identified and cloned. They are encoded by separate genes
that localise to different chromosomes. The first isozyme cloned was ALASI and its gene
localises to chromosome 3 in humans (Sutherland et al., 1988; Bishop et al., 1990). ALASI is
a housekeeping enzyme that is ubiquitously expressed in all cell types (Snvastava et al.,
1 e88).
24

The second isozyme is referred to as ALAS2 and is the erythroid-specific form of the enzyme.
The first reported cloning of ALAS2 was in the chicken (Riddle et al., 1989), followed by the
mouse (Schoenhaut and Curtis, 1989) and human (Cox et al., l99l). The human ALAS2 gene
is encoded on the X chromosome, banding at position Xpl1.2 (Bishop et al., 1990; Cox et al.,
1990). The mouse ALAS2 gene is also encoded on the X chromosome (Chapman et al.,
1994). ALAS2 expression is restricted to cells descending from the erythroid lineage in order
to support the high level of haem production that is required during erythropoiesis (Worwood,
te77).
ALASI and2 have limited N-terminal amino acid identity (Cox et al., I99l). The N-terminal
signal sequence that targets each ALAS isozyme to the mitochondria differs, in addition to the
N-terminal quarter of the mature proteins. The signal sequence of human ALAS2 is predicted
tobe 49 amino acids in length and cleavage is proposed to be between a serine and glutamine
residue (see Section 7.1 .3 for further discussion). The ALAS I enzyme has a signal sequence
that is seven amino acids longer with cleavage likely to occur between two glutamine residues
(Cox et al., I99l). Thus, ALASl is 64.4 kD in size, slightly larger than the ALAS2 enzyme at
59.5 kD. The C-terminal, encompassing approximately 75%o of the mature ALAS protein, is
highly conserved between the two isozymes in humans and mice (Cox et al., 1991).
Both isozymes, ALASI and2 are thought to function as homodimers. Electron microscopy
has demonstrated that the ALASI eîzyme is only catalytically active as a homodimer with its
subunits arranged in opposite polarities (Pirola et al., 1984). The development of systems for
overexpression and purification of mouse ALAS2 in E.coli (Ferreira and Dailey, 1993) has
facilitated study of the structural and functional requirements of the ALAS enzyme. For
example, Tan and Ferreira (1996) performed in vivo complementation experiments in which
two differing ALAS2 mutant subunits, both inactive as homodimers, were shown to form an
active heterodimer when brought together. Therefore, the above study suggests that ALAS2 is
functional as a dimer and that the active site is located at the interface between the two
subunits. Currently, a 3D structure of either isozyme has not been determined (Tan and
Ferreira, 1996).
25

1.7.2 Mechanism of ALAS
ALAS catalyses the condensation reaction between succinyl CoA and glycine to generate
ALA and the by-products, CO2 and CoASH in non-plant eukaryotes and some prokaryotes
(May et al., 1995). Pyndoxal phosphate (PLP), a derivative of vitamin 86, is the essential
cofactor for the catallic activity of ALAS (Tan and Ferreira, 1996). Steady-state kinetic
studies showed that glycine binds the ALAS-PLP complex prior to succinyl CoA and the
reaction of glycine with ALAS involves a 3-step kinetic process (Zhang and Ferreira,2002).
ALAS has high substrate specificity for glycine despite a relatively low binding affinity.
CoASH is released fìrst, followed by the release of COz and finally ALA (Figure 1.5).
Carbonyl and carboxylate groups of ALAS are able to induce a conformational change in
ALAS and this may modulate the release of products from the reaction (Zhartg and Ferreira,
2002).
In murine ALAS2, the lysine residue 313 forms a Schiff base linkage with PLP (Ferreira et
al., 1993). This was established by tryptic digestion, purification and sequencing of labelled
ALAS2 peptides in which PLP labelled with tritium was introduced. A lysine residue in other
PlP-dependent enzymes has been identified as the cofactor binding site (John, 1995). This
lysine residue, located in exon 9 of the ALAS2 C-terminal, is conserved in all known ALAS
sequences as determined from sequence alignment analysis (Cox et al., 1990) (Figure 1.6). As
mentioned earlier, glycine binds the ALAS-PLP complex prior to succinyl CoA where upon
an extemal aldimine is formed between glycine and PLP (Ferreira et al., 1995). The binding
of succinyl CoA facilitates a conformational change which accelerates conversion of the
external aldimine into the initial quinoid intermediate (Hunter and Ferreira, 1999). Site-
directed mutagenesis to change lysine 313 in ALAS2 to an alanine, histidine or glycine
demonstrated that lysine 313 is an essential catalytic residue (Ferreira et al., 7995). The study
reported that PLP could still bind the mutant ALAS2 enzpe but the complex was
catalytically inactive.
A conserved glycine-rich sequence has been identified in the ALAS gene of bacteria to
humans (Ferreira and Gong, 1995) (Figure 1.6). Similar glycine-rich sequences have been
shown to form part of the cofactor binding site in other PlP-dependent enzymes (V/eber er
aL.,I978; Marceau et aL.,1990). Therefore, the conserved sequence of glycines is proposed to
be part of the PLP binding site in ALAS2. Support of this notion arose from experiments in
which the eleven amino acids that span the glycine loop of murine ALAS2 underwent partial
random mutagenesis (Gong and Ferreira,1995). Randomly mutated ALAS2 expression
26

Figure 1.5 Mechanism of ALAS
Together with its cofactor pyridoxal phosphate (PLP), ALAS catalyses the condensation
reaction between succinyl CoA and glycine to yield 5-aminolevulinate. Initially, ALAS and
PLP form a complex via the generation of a Schiff base linkage between lysine 313 of ALAS
and PLP. Glycine is the first of the substrates to bind the ALAS-PLP complex followed by the
binding of succinyl CoA. The by-products of the reaction, COz and coenzyme A, are released
prior to the end-product, 5-aminolevulinate or aminolevulinic acid.

H*CO; o;
o
NH*
CO; HO
HO
OP
HR+
OP++NH
HO HO
H,Me
+NH
Me Me NH
I
glycine PLP
cocH,cH,co,-
CoAS-COCH,CH,CO;(Succinyl CoA)
I,CO,-.f H
H cocH,cHrco,- N
H
PHOPHO
+H + -co"
P
+NH
+NH
+NHaminolevulinic acid
Me Me Me

Figure 1.6 Amino Acid Sequence Comparison of Eukaryotic ALAS Proteins
Comparison of the amino acid sequence of several ALAS eukaryotic proteins derived from
CDNA clones. The ALAS sequences include 7: rat ALASI; 2: human ALAS2; 3: human
ALAS1; 4: chicken ALAS1; 5: chicken ALAS2; 6: mouse ALAS2; 7: Saccharomyces
cerevisae heml. The conserved lysine residue involved in formation of the Schiff base linkage
between PLP and ALAS is shaded and highlighted with a star. The glycine loop motif is
shaded. The human T388S (Ml) and R41lC (M2) XLSA associated mutations which reside
in exon 8 and 9, respectively, are boxed and labelled accordingly. The two predicted cleavage
sites in the human precursor ALAS2 isozyme are indicated and would generate either a 49 or
78 amino acid sequence.

Cleavage SiteI to 20 30 40 50 aol '10 80 90
1. METVVRRCPFLSRVPQAFLOKAGK**SLLFYAONCPKMME2 . MVTAAMLLQCCPVLARGPTSLLGKVVKTHQFLFGIGRCPI * * *3. MESVVRRCPFLSRVPQAELQKAGK**SLLFYAQNCPKMME4 . MEAVVRRCPFLARVSQAELQKAGP * * SLLFYAQHCPKMME5. AAFLR*CPLLARHP**PL IìAEAT******GARCPFMGE6 . VAAAMLLRSCPVLSNGPTGLLGKVAKTYQFLFS I GRCPI * * *.1 . MQRSIF
91
21r 280
110
290
AQTPDGTQLPPGHPSPSTSQS * SGSKC PFLAAQLS* * * * ** * * * **GGDSPSWAKG* ** *HCPFMLS* * SPDGTQLPSGHPLPATSQG*TASKCP* * NTDGSOPPAGHPPAAAVQS * SATKCP
r00
* * * ** * * ** **GGASPS!ùAKS* ** *HCP
Site130 140 150 160 170 180
EVAQSPVLPS * *LVNAKRDGEG* PS PLLKVQKAAPEVQEDVKAFKTD* * * * * * * * *LPS *SLVSVS * * * * * * * * *LRK
VECKASLELQEDVOEMNAVRKEVAETSAGPS * *VVSVKTDGGD* PS GLLKFCKASLELQEDVKEMQVDRKGKEFAK* I PTNSVVRNTEAEGEEQSGLLK
******HRAAPELQEDVERPQIPAVEVLEEL**********LRDGGA***ALNR] VQRAAPQVQEDVKTEKTD* * * * * * * * * LLS * TMDSTT * * * * * * * * * RSH
340 350 360
510 520 530 540YAAGFI FTTSLPPMLLAGALESVRT LKSNEGRAl,RRYAAGFT FTTSLPPMVLSGALESVRLLKGEEGQALRRYAAGFI FTTSLPPMLLAGALESVR] LKSAEGRVLRRHAAGFI FTTSLPPMLLAGALESVRTLKSAEGQVLRRLGPGFI FTTALPPQRGGGALAALOVVG SAEGAALRRYAAGFI ETTSLPPMMLSGALESVRLLKGEEGQALRREAPGEI FTTTLP PSVMAGATAAI RYQR* * CHI DLRT
CleavL20
300
1234561
*******t***************************
* * * ** * * ***** * ** ** ***t *TATG*ATAAAA
181 190 2oo 2Lo 220 230 240 250 260 2tO1. NF*QDIMRKQRPERVSHLLODNLPKVVSTFQYDHFFEKKIDEKKNDHT***YRVFKTVNRRAQIFPMADDYTDSL]TKKQVSV!{CSNDYL2 . PFSGPQEQEQI SGKVTHLIQNNMPGN* YVFSYDLFFRDKIMEKKQDHT* * * YRVFKTVNRWADAYPFAQHFSEASVASKDVSVWCSNDYL
3. NF*QDIMQKQRPERVSHLLODNLPKSVSTFQYDREFEKKIDEKKNDHT***YRVFKTVNRRAHIFPMADDYSDSL]TKKQVSVWCSNDYL4. KF*KDIMLKQRPESVSHLLQDNLPKSVSTFQYDQEFEKKIDEKKKDHT***YRVFKTVNRKAQIFPMADDYSDSLITKKEVSVWCSNDYL5 TV* RDCMDE* * * * * * * * * * * * * * * * * * DAFPYEEOFQAQLGALRRTHT * * * YRVFTAVGRRADAPPLGTRGTAPH* TS * * VELWCS SDYL
6. SFPFSQEPEQTEGAVPHLIQNNMTGS*QAFGYDQFFRDKIMGKKQDHT***YRVFKTVNRWANAYPFAQHFSEASMASKDVSVWCSNDYL'7. *********************NHSTQESGFDYEGLIDSELOKKRLDKS***YRYFNNINRLAKEFPLA****HRQREADKVTVÍ{CSNDYL
1234
561
GMSRHPRVCGAVGMSRHPQVLQAGMSRHPRVCGA
310 320 330SKFHVELEQELADLHGKDAALLFS SCEVANDSTLFTLAKMMPGCEI YS DSGNHASMI Q
SGTSKFHVELEQELAELHQKDSALLFS SCFVANDSTLFTLAKI LPGCEI YS DAGNHASMT Q
SGTSKFHVDLERELADLHGKDAALLFS SCFVANDSTLFTLAKMMPGCEI YS DSGNHASMT Q
SGTSKEHVDLEKELADLHGKDAALLES SCFVANDSTLFTLAKMLPGCE I YSDSGNHASMI QGMS RH PRVCGAVMDTLKGL S RH PAVLRAARAAL S PLHGALERALALLHRQPRAALES SC FAANDTALDTLARI LPGCQVYSDAGNHASMI Q
G] SRHPRVLQAI SGTSKEHVGLEQELAELHQKDSALLFS SC FVANDSTLFTLAKLLPGCE I YS DAGNHASMl Q
ALSKHPEVLDAMHKTI D PTLNLEAELATLHKKEGALVFS SCYVANDAVLSLLGQKMKDl,VI FS DELNHASM] V
361 370 380 390 400 410 420 430 440 4501. GIRNSRVPRY]FRHNDVNHLRELLQRSDPSVPKIVAEETVHSMDGAVCPLEELCDVAHEFGAITFVDEVHAVGLYGASGGGI********2. GIRNSGAAKFVFRHNDPDHLKKLLEKSNPKIPKIVAEETVHSMDGAICPLEELCDVSHQYGALTFVDEVHAVGLYGSRGAGI********3. G]RNSRVPKY]FRHNDVSHLRELLQRSDPSVPKIVAEETVHSMDGAVCPLEELCDVAHEFGAITFVDEVHAVGLYGARGGGI********4. GIRNSRVPKH]FRHNDVNHLRELLKKSDPSTPKIVAEETVHSMDGAVCPLEELCDVAHEHGAITFVDEVHAVGLYGARGGGI********5. GIRRRGVPKFIFRHNDPHHLEQLLGRSPPGVPKIVAFESLHSMDGSIAPLEELCDVAHAYGALTFVDEVHAVGLYGARGAGI********6. GIRNSGAAKFVFRHNDPGHLKKLLEKSDPKTPKIVAFETVHSMDGAICPLEELCDVAHQYGALTEVDEVHAVGLYGARGAGI********
4s1 460 4'70 qeo t( 490 soo
Ml M2541 550 560 570 580 590 600 610 620 630
I. QHQRNVKLMRQMLMDAGLPVIHCPSHITPVRVADAAKNTEICDELMTRHNIYVQAINYPTVPRGEELL*RIAPTPHHTPQMMNYELEKLL2. AHQRNVKHMRQLLMDRGLPVIPCPSHIIPIRVGNAALNSKLCDLLLSKHGIYVQAINYPTVPRGEELL*RLAPSPHHSPQMMEDFVEKLL3. QHQRNVKLMRQMLMDAGLPVVHCPSHIIPVRVADAAKNTEVCDELMSRHNIYVQAINYPTVPRGEELL*RIAPTPHHTPOMMNYFLENLL4. QHQRNVKLMRQMLMDAGLPVVHCPSHIIPIRVADAAKNTEICDKLMSQHSIYVQAINYPTVPRGEELL*RIAPTPHHTPQMMSYFLEKLL5. AHQRHAKHLRVLLRDRGLPAL**PSHIVPVRVÌ*DAEANTRLSRALLEEHGLYVQAINHPTVPRGQELLLRIAPTPHHSPPMl,ENLADKLS6. AHQRNVKHMRQLLMDRGFPVIPCPSHIIPIRVGNAALNSKICDLLLSKHSIYVQAINYPTVPRGEELL*RLAPSPHHSPQMMENFVEKLL7. SQQKHTMYVKKAFHELGIPVIPNPSHIVPVLIGNADLAKQASDILINKHQIYVQAINEPTVARGTERL*RITPTPGHT****NDLSDILI
631 640 650 660 670 680 6901 . LT!^IXRVGLELKPHSSAECN FCRXPLHFEVMSEREKAYFSGMSKM*VSAQA2 . LAWTAVGLPLODVSVAACNFCRRPVHFELMSEWERSYFGNMGPOYVTTYA3 . VTWKOVGLELKPHSSAECNECRRPLHFEVMSEREKSYESGLSKL*VSAQA4 . ATIIKDVGLELKPHSSAECNFCRRPLHFEVMSERERSYFSGMSKL*LSVSA5. ECWGAVGLPREDPPGPSCSSCHRPLHLSLLSPLERDQEG******VRGAAAG6 . LAWTGVGLPLQDVSVAACNFCHRPVHFELMS EWERSYFGNMGPQYVTTYA?. NAVDDVFNELQLPRVRDV.IESQGGLLGVGESGFVEESNLWTSSQLSLTNDDLNPNVRDPIVRQLEVSSGIKQ

constructs were transformed into a hemA- E.coli strain to select for functional ALAS
enzymes. The hemA- E.coli strain is unable to synthesise ALA such that the strain can only
grow if supplemented with ALA or transformed with an active ALAS expression plasmid.
From this study it was observed that the arginine residue at position 149 is critical for ALAS2
function and it is conserved in all functional ALAS2 mutants. Furthermore, glycine residues
at positions 142 and I43 arc also essential as they could only tolerate alanine replacement to
generate a functional ALAS2 enzyme. Site-directed mutagenesis performed on murine
ALAS2 to replace a conserved arginine residue at position 439 with either a lysine or leucine
residue demonstrated that arginine 439 plays an important role in substrate binding to ALAS2
(Tan et al., 1998). Currently, other residues in ALAS2 that may be involved in binding of the
PLP cofactor and processing of the substrates remain unclear.
1.7.3 ALAS Gene Structure
The ALASI and2 isozymes, encoded by separate genes, were isolated and characterised from
a number of different sources (Figure 1.7). The genomic organisation of the two genes are
well conserved, suggesting that they arose from gene duplication (Cox et al., l99l).
Nucleotide identity of 600/o exists between human ALAS1 and ALAS2 (Cox et al., l99l).The
following section will primarily discuss ALAS2 with reference to ALASl since the focus of
this study is ALAS2.
The human ALAS2 gene spans 22kb and consists of eleven exons (Conboy et al., 1992).
Exon and intron sizes range from 37 to 270 bases and 561 bases to 6 kb, respectively (Cox et
al., 1991). Exon I encodes the 5'-untranslated region (5'-UTR) that contains an iron
responsive element (IRE) associated with ALAS2 translational control (Cox et al., I99l).The
mitochondrial targeting sequence is located in exon 2 and this region is removed upon
ALAS2 import into the mitochondrial matrix. The length of the ALAS2 signal sequence is in
conflict, with protein alignment studies of various organisms suggesting it is 49 amino acids
in length (Cox et al., l99l; May et al., 1995; Goodfellow et a1.,2001) whereas others have
predicted a 78 amino acid targeting sequence (Schoenhaut and Curtis, 1986; Munakata et al.,
1993) (Figure 1.6). Recently, it has been shown that the first 49 amino acids of ALAS2 are
sufficient to target a heterologous enhanced green fluorescent protein to the mitochondria
(Cox et al., paper submitted). Exons 3 and 4 represent the N-terminal of the mature ALAS2
protein. This is absent in bacterial forms of the ALAS2 protein and the function of this
27

Figure 1,.7 Structure of the ALAS genes of Various Species
The two isotlpes of ALAS have been isolated and characterised in various orgaisma such as
the chicken rat, mouse and human. Exons are numbered and the start (ATG) and stop (TAA)
codons are noted. The proposed functions of the exons are indicated.
Exons
2Kb
ATGIT T ¡I T II TTI
IonchickenALAS 1
ratALAS 1
chickenALAS2
mouseALAS2
T-T--TI I TTI 2 345 67ut I
G
TT T8 910
1234s
678 910 11
6 1 I
t2 3 4 5
TAAI
9
TAA
t0 ll
ATGI
TAAI
I 23 4
34
t
50t 8 9 t0 ll
humanALAS2
Alternative Splicing
56
Regulatory ?
Signal sequence
TAA
ll1 tl2U
7 8 9 10
Catalytic Domain

domain is unknown (May et al., 1995). Exons 5 to 11 encompass the catalytic domain of
ALAS2 which is conserved from bacterial through to human ALAS2 genes (Cox et al., l99l).
Human ALAS2 mRNA is alternately spliced into five mRNA transcripts (Conboy et al.,
1992; Cox et al., paper submitted). One of the transcripts is the full length ALAS2 mRNA
and the remaining four lack either exon2, exon 4, both exons 2 and 4 or contain a 107 bp
insertion between exons I and2. ALAS2 transcripts without exon 4 equalled approximately
35Yoto 45o/o of total ALAS2 mRNA in different erythroid cell tlpes (Cox et al., paper
submitted). The altemate splicing of human ALAS2 is not phylogenetically conserved, being
absent in the mouse and dog. However, Schoenhaut and Curtis (1989) observed an altemate
splice variant of murine ALAS2 mRNA resulting in the loss of the first 45 nucleotides of
exon 3. This alternate murine ALAS2 transcript formed approximately l5o/o of total ALAS2
mRNA in mouse erythroid tissue (Schoenhaut and Curtis, 1989; Dzikaite et a1.,2000).In
humans, alternate ALAS2 mRNA transcripts are found at all stages of erythroid
differentiation, indicating that the transcripts are not developmental-specific. Currently, the
function of these alternate ALAS2 transcripts is unknown (Bottomley et al., 1995; Cox et al.,
paper submitted).
1.8 REGULATION OF ALAS1
The production of haem is coordinated with the cell's requirement for the molecule and is
regulated by the first and rate controlling enzyrne of the biosynthetic pathway, ALAS. The
two isoforms of ALAS, ALAS1 and ALAS2, supply haem for different processes such that
their regulation differs. For example, in the liver ALASI expression is induced by drugs (May
et al., 1995) whereas ALAS2 responds to signals for erythroid differentiation (Bottomley e/
al., 1995). ALASl was initially isolated from the liver but is ubiquitously expressed in all
tissue types to supply haem for mitochondrial respiratory clochromes and other cellular
haem containing proteins (May et al., 1995). The production of haem is vital for many
cellular proteins but an excess of free haem is toxic to the cell. Free haem can react with
hydrogen peroxide to result in reactive oxygen species which disrupt membrane lipids and
proteins (Muller-Eberhard and Fraig, 1993). Oxidation of porphyrinogens to porphyrins
enables them to absorb light which leads to the generation of free radicals that can cause
cutaneous damage (Darr and Fridovich,7994). Thus, the control of ALAS1 levels is primarily
regulated by the end-product of the pathway, haem.
28

ALASI regulation has been mostly studied in foetal and adult liver of the rat (May et al.,
1995) where activity can be induced by porphyrogenic drugs and repressed by haem (Granick,
1966; Srivastava et al., 1988, 1990). ALASl expression levels and activity are negatively
regulated by free haem at several stages including ALASI transcription, import of the
precursor ALASl into the mitochondrial matrix and ALASI mRNA stability (May et al.,
lees).
'When cellular levels of haem are high, haem negatively regulates ALASl transcription,
reducing the level of ALAS1 mRNA. This has been demonstrated in nuclear run-off studies
where drug-induced increases in ALAS I mRNA levels are inhibited by injection of either
exogenous haem (Srivastava et al., 1990) or the haem precursor ALA (Srivastava et al.,
1988). Haem has been shown to reduce ALASI mRNA stability with no alteration to the rate
of ALASI transcription in cultured chick embryo hepatocytes (Hamilton et al., l99l). More
recently, the half-life of ALASl mRNA was decreased in the presence of haem in an
immortalised rat hepatocyte cell line (Cable et a1.,2000), demonstrating that haem-regulated
ALASI mRNA stability is conserved in mammals and avian species. The mechanism of how
haem regulates ALASl mRNA stability has been investigated in our laboratoryusing the rat
hepatoma cell line FRL 4.1. The half-life of ALASI mRNA in FRL 4.1 cells was reduced
approximately 3-fold from 131 minutes to 42 minutes upon addition of exogenous haem and
de novo haem synthesis. In addition, expression of chimaeric genes containing ALASI and B-
globin sequences in FRL 4.1 cells identihed elements for both constitutively active instability
and for haem-regulated mRNA instability in the ALASl coding region (Sadlon et al.,
manuscript in preparation). Pulse-labelling studies in chicken embryonic livers found that
upon administration of haem, import of precursor ALASI into the mitochondria \¡/as
prevented (Srivastava et al., 1983). This is an extremely effective mechanism to control the
production of haem in the mitochondria, preventing the generation of excess free haem. In
summary, haem regulates ALASl expression which ultimately leads to haem autoregulating
its own synthesis in non-erythroid cell types.
1.9 REGULATION OF ALAS2
Upon the initiation of red blood cell differentiation several events, relating to specific gene
expression, are required to permit erythropoiesis to proceed. These include an increase in the
expression of ALAS2 (Elferink et al., 1988), other enzymes of the haem biosynthesis pathway
(Beaumont et al., 1984; Romeo et al., 7986; Raich et al., 1989) and globin (Karlsson and
29

Nienhuis, 1985). This occurs primarily at the transcriptional level. Transcription factors are
involved in the erythroid-specific transcription of the ALAS2 gene during erythropoiesis.
ALAS2 expression is also regulated post-transcriptionally via an IRE located in the 5'-UTR of
the ALAS2 precursor mRNA (Cox et al., l99l) which implies that ALAS2 expression is
coupled to iron availability. In addition, like globin, aspects of ALAS2 expression is regulated
by haem.
1.9.1 Transcriptional Regulation of ALAS2
Only a small number of erythroid-specific transcription factors are required to control the
expression of ALAS2 and other erythroid genes including, GATA-1, NF-E2 and the CACCC
box binding protein, EKLF. Isolation of the promoters for murine (Schoenhaut and Curtis,
1989), human (Cox et al., l99I) and chicken (Lim et al., 1994) ALAS2 demonstrated minor
similarities between their putative cis-acting elements. For example, the murine and chicken
ALAS2 promoter do not contain a canonical TATA box whereas the equivalent domain in the
human has a GATA-I binding motif at the TATA box location (Cox et al., 1991). GATA
motifs at the TATA position have also been localised to the promoters of chicken B-globin
(Fong and Emerson,1992), human glycophorin B (Rahuel et ql., 1992) and rat p¡rnrvate
kinase (Max-Audit et al., 7993), with GATA-I binding this site in preference to TFIID.
Interestingly, the assembly of a repressive chromosome is prevented when GATA-l binds to
the -30 GATA motif in the chicken B-globin promoter. This finding postulates a role for
GATA-1 in transcriptional activation of erythroid-specific genes through prevention of
nucleosome assembly on the promoter TATA box (Barton et al., 1993).
The 5'flanking region of the human ALAS2 promoter has been characterised and it was
determined that the first 300 bp upstream from the transcriptional start site is required to give
maximal activity in undifferentiated erythroid cells lines (Surinya et al., 1997). Two GATA-I
binding sites were localised to positions -1261-12l and-7021-97 inthe human ALAS2
promoter. GATA-I binding sites are found in regulatory regions of many erythroid genes
including the promoter of human PBGD (Mignotte et aL.,1989b), human EpoR (Zon et al.,
1991) and globins (deBoer et al., 1988; Talbot et al., 1990; Philipsen et al., 1990;Pruzina et
al., I99l; Gong et a|.,1991). Electrophoretic mobility shift assays (EMSA) determined that
each site could bind GATA-I in vitro (Surinya et al., 1997). The GATA-I binding sites in the
human ALAS2 promoter were shown to be functional via mutagenesis studies in both K562
30

'tt tr -.<-'', :
tf"t) ...-'
and MEL undifferentiated erythroid cells. In GATA-I expression was
able to increase human ALAS2 promoter activity through the GATA sites (Surinya et al.,
1997). The non-canonical TATA box¡r the human ALAS2 promoter was shown to bind/
GATA-I or TATA binding protein Çne¡ in vitro.In addition, conversion of this site to a
consensus TATA site resulted in reduced expression suggesting GATA-I is required for
ALAS2 promoter activity. However, conversion of this site to a consensus GATA site also
reduced expression from the ALAS2 promoter (Surinya et al., 1997). Thus, an interaction
between both factors may be required to obtain ALAS2 promoter activity.
A NF-E2 binding site was identified at position 491-39 in the human ALAS2 promoter (Cox
et al., I99l) with a mismatch at both ends of the 11 bp consensus sequence for NF-E2
(Andrews et al., 7993a). NF-E2 sites have also been found in the promoter of the human
PBGD gene (Mignotte et al., I989b) and enhancers of the cr (Strauss et al., 1992;Zhanget
aL.,1995) and B-globin genes (Talbot et a|.,1990;Talbot and Grosveld, 1991; Ney et al.,
1990). Mutagenesis and overexpression studies demonstrated that the NF-E2 site in the
human ALAS2 promoter is not functional in erythroid cell lines (Surinya et al., 1997). This
study supported an earlier finding in which this same site in the human ALAS2 promoter did
not bind NF-E2 in vitro (Andrews et al., 1993a).
Two overlapping CACCC boxes at position -591-48 on the non-coding strand of the human
ALAS2 promoter have been identified. The sequence of this CACCC box is similar to the
functional CACCC box site in the adult B-globin gene that binds EKLF (Miller and Bieker,
1993). Mutagenesis of this site and transient expression inK562 and MEL erythroid cell lines
showed it to be functional (Surinya et al., 7997). Binding of the ubiquitous Sp1 and BKLF
transcription factor to the CACCC box in the human ALAS2 promoter was detected using
erythroid cell nuclear extracts. EKLF binding was also detected using nuclear extracts
prepared from a non-erythroid cell line overexpressing EKLF. Competition binding
experiments demonstrated that EKLF binding to the human ALAS2 promoter CACCC
sequence was undetectable with erythroid extracts since levels of EKLF were too low in this
cell line (Surinya et al., 1997).
The murine ALAS2 promoter has been characterised, localising GATA-I, NF-E2 and EKLF
binding sites to the first 300 bp downstream of the transcription start site (Kramer et al.,
2000). For maximal transcriptional activation, an additional 418 bp (-300 to -718) of the
murine ALAS2 5' flanking region are required. This region does not confer erythroid-specific
31
¡

expression of ALAS2 since similar levels of expression were observed in non-erythroid cells
lines. Two GATA-I binding sites at position -1 18/-1 13 and -981-93 and a region located
within -5 1 8 to -3 1 5bp of mouse ALAS2 promoter are essential for transcriptional activation
during chemically induced differentiation of an erythroid cell line. Thus, these domains may
be important in conferring erythroid specificity to ALAS2 transcriptional activity in the
mouse (Kramer et a1.,2000).
Five DNase I hypersensitivity sites were identihed in the murine ALAS2 gene in non-induced
and dimethyl sulphoxide (DMSO) treated MEL erythroid cells (Schoenhaut and Curtis, 1989).
They were localised to the immediate promoter, two in the 5' end of intron 1, in intron 3 and
the 3' end of intron 8. DNase I hlpersensitivity sites are indicative of nucleosome-free DNA
that usually correspond to regions that interact with transactivating factors critical for
transcriptional regulation (Elgin, 1988; Gross and Garrad, 1988). As discussed previously, the
promoter of the human ALAS2 gene has been characterised to determine binding ability and
functionality of the various erythroid-specific transcription factor binding sites (Surinya et al.,
1997). The intronic regions identified as containing hypersensitivity sites have also been
investigated in the human ALAS2 gene (Surinya et al., 1998). Analysis of the sequence of
these domains identified the presence of seventeen putative consensus GATA sites and six
CACCC-like sequences in intron 1, two CACCC boxes and one GATA site in intron 3 and
finally two CACCC and four potential GATA sites in intron 8. Intron 1 and 8 sequence was
shown to be functional and positively enhance ALAS2 promoter activity in undifferentiated
erythroid cells, whereas the presence of intron 3 inhibited activity of the promoter (Surinya e/
al., 1998).
Human ALAS2 intron 8 was only functional in erythroid cell lines and could confer
erythroid-specific activity on the heterologous thymidine kinase promoter. Activity of intron 8
was localised to a 239bp region at the 3'end of the intron. This region encompassed two
GATA sites and CACCC boxes that were found to be conserved in human, mouse and canine
ALAS2 intron 8 sequences (Figure 1.8). Murine ALAS2 intron 8 contains additional 5'
sequence in comparison to the human and dog sequences. Upon mutagenesis of the GATA
and CACCC sites of the human ALAS2 intron 8, both CACCC boxes and the second GATA-
1 site withinthe23g bp active region were deemed to be functional whereas the first GATA
was found to be inhibitory of transcriptional activity (Surinya et al., 1998). In vitro binding of
GATA-I to both GATA sites was detected. Sp1 was able to bind each CACCC box but
binding of EKLF or BKLF was not observed. Furthermore, transactivation of the promoter
32

Figure 1.8 Comparison of the ALAS2 intron I sequence between the mouse, canine and
human
Alignment of human, dog and mouse ALAS2 intron 8 nucleotide sequence. Shaded sequence
represents regions of homology. Conserved GATA and CACCC motifs are boxed. In
addition, two GATA motifs that are not conseryed are boxed in black.

humao ALAS2 lnlron Idog ALAS2 ln{ron Imouse ALAS2 lntron I
5050
50
100
100100
150150
150
200200200
250250250
300300
300
350350
350
400400400
450
450450
500500
500
550
550550
600600
600
650650
650
750
750750
ð00800800
850
850850
900900
900
950
950950
51
51 GAAGCAAC TTGGGGAGGAAAGGGCTTACG TGGCTTACA TA TCC TG TATCA
101
101
101 C A AGC C C AC TG AGGGAA ACC AAGGC A TG A A C TC A A A C C AGG TA AG A A C C T
151't 5'l,I51 GG AGGC AGG AG C TG A T TC AG AGA TG T TGG T TA C TGG C T TA C TC C TC T TGG
TTTGf TCAGCC TGC TCGCTGA TAGAACACGG TAACA lGAGCCCACAG TGG
TA TTACCCCCAAAGG TC TGGGCCATCCCACA TCAATCAC TAA TTATAAAA
201201201
251251
251
301301
301
351351351
401401
401
45'l451
451
501501
501
ATGC TCTGCAGCCTAGTC TTACACATTTTCCCfCC TC TCAGATGAC TC TT
GC TTGCATCAAGTTGACATAAAGC TAGCCAGCAGTGAGCAAACAC TTGTA
TATAAAGGC TTCCCTTA TG TACAA TG TGGAGAAAGCACCCCCAGCAAACA
f TTTAAC TIGACATGAACCATf TTTA TTTTATTTATTTTTGA
Tc
CATT
c
TcG
551 C AGAGGAGGCffiC A - - - -
55lA-s51 T TTCCTCCTTffT CCCCTCTTTCCT
60l - cå'601 - Tå601 GTCATTAATATCTGTT T AT
651 cc65I AA651 AA
701701701
751751751
80180'l
80'l
ð51051851
901
90'l901
951
s51
951
ffi: ; J J#]låffiücËr A A -----tXAl
T
t+1000
1000
1000
10501050
1050
100'1
100't't 00'l
r05r C T1Os1-cA1051G-G
II:I ff¡ Jffiå:J Jlffi:ff:rror f;-
CAfGAC
100
100
100
11501150't'150

and intron 8 by endogenous EKLF was not any greater than the promoter alone (Surinya et
al., 1998). Therefore, it was concluded that EKLF may not function through the intron 8
CACCC sites in undifferentiated erythroid cells.
Thus far, regions within the ALAS2 gene have been investigated for their ability to confer
enhancer activity on transcription from the ALAS2 promoter in immature erythroid cells. One
of the primary aims of this thesis is to investigate the Epo responsiveness of the regulatory
domains that have been identified in the human ALAS2 gene during erythroid cell
differentiation, in particular the response of the enhancer region residing within intron 8.
1.9.2 Regulation of ALAS2 Translation
Following transcriptional regulation, ALAS2 expression is further controlled at the level of
mRNA translation. The high rate of erythroid haem production requires copious amounts of
iron, such that approximately 80% of plasma iron is utilised to s¡mthesise haem in erythroid
cells of the bone marrow (Finch, 1994),It is well established that iron is a major regulator of
haemoglobin formation in red blood cells and that translation of ALAS2 mRNA is dependent
upon iron since an IRE has been localised to the 52 bp 5'-UTR of the human ALAS2 gene
(Cox et al., l99I). As none of the other enzymes of the haem biosynthesis pathway contain an
IRE it demonstrates that iron-dependent translation of ALAS2 is an essential step in
protoporphyrin production in response to iron (Sadlon et a1.,1999). The IRE stem loop
structure is also conserved in the 5'-UTR of the mouse (Dierks, 1990) and chicken (Lim et al.,
1994) ALAS2 gene (Figure 1.9). In addition, the IRE identified in the ALAS2 genes has been
similarly documented in the 5'-UTR of the iron storage protein ferritin (Aziz and Munro,
1 9 87; Hent ze et al., 1987) (Figure I .9) and in the 3 '-UTR of the transferrin receptor (TfR)
(Casey et al., 1988).
Two cytoplasmic iron regulatory proteins, IRPI and IRP2, bind the IREs of iron regulated
genes (Henderson,I996a; Menotti et al., 1998). Despite the similarities of the two proteins
they are regulated by iron via different mechanisms. When cellular iron levels are elevated
IRP1 is inactivated by a conformational change that prevents it from binding RNA whereas
IRP2 is targeted for rapid degradation (Henderson, 7996a).In low iron, IRP binds the IRE in
the 5'-UTR of the ALAS2 mRNA preventing translation (Bhasker et al., I993;Melefors et al.,
1993; Smith and Cox, 19971, Vaisman et a\.,2000). The formation of the cap binding protein
JJ

Figure 1.9 Comparisonbetween the iron response elements (IRE) of ALAS2 in various
species
A conserved IRE, important in the translational control of ALAS2 has been identified in the
5'-UTR of the gene in several mammalian species including the chicken, mouse and human.
The stem loop structure of the ALAS2 IRE is highly conserved and is similar in sequence to
the IRE located in the 5'-UTR of human ferritin mRNA. The conserved sequence is shaded.
The first nucleotide of the mRNA is denoted as -r1.

Ferritin
l-h"r*l
A-UTæIlu-cl
U-AE
G-C
U
c
ALAS2
m0use human
U-Gc-G
U-AU-A
GCG-CGC
GG
GCc-G
G-CU
cU-GG-UA-UG-C
c
U
G
U
c-GfcEllu-cl
G-CE
U-AU-AG-C
cU-AU-G
Ac-GU-AG-C
UU
Uu-Ac-Gc-GA.U
c-G
tîflG-C
oU-AU-AG-C
U-AU.G
c-GU-AG-C
UU
Uc€c-G
c-G
chickenl-c clln cllc cl
U-Gc-G
rG6tlu-cl
G-CE
U-AU-AG-C
Gc-GG-CU.AG-CAAc-Gc-GU-AUCA GAUtt
+1 +44
G
UCAAGAUGI
cI
+'1
5
A
UI
+1
CTCAGGAUGI
+52
5'
+52
c-c 130ntCCACCC . .GAUG
A
c11 nt
GAGI
+1
5'I
+208
GUAGGUAGGUAG

complex on the IRP-bound ALAS2 mRNA occurs but recruitment of the small ribosomal
subunit is not facilitated, inhibiting translation. Alternatively, when cellular iron is high, the
ALAS2 IRE is free of IRP binding and translation can proceed as normal (Muckenthaler et
al., 1998). kon regulated translation of the ALAS2 gene ensures that in low iron
protorporphyrin does not accumulate to result in a potentially toxic pool. Secondly, it prevents
unnecessary production of globin since globin mRNA translation is regulated by the level of
haem (Chen and London, 1995).
In non-erythroid cells, TfR and ferritin mRNAs are post-transcriptionally regulated by iron
via their IREs to facilitate iron import and mobilisation. In low iron, IRP binds the 5'IRE in
ferritin mRNA which prevents its translation (Gray and Hentze, 1994). Concurrently, IRP
binds the IREs in the 3'-UTR of transferrin receptor mRNA enhancing its stability and
allowing translation of the TfR. (Theil, 1990). High iron levels inactivates the IRPs such that
translation of ferritin occurs and TfR mRNA is destabilised. In erythroid cells the mechanism
described above does not occur, instead uptake of iron into the haemoglobin producing cells
is mediated solely by the TfR pathway (Ponka, 1997). The existence of different regulatory
mechanisms for iron homeostasis in erythroid cells may be due to several reasons (Sadlon er
aI., 1999). Firstly, the TfR contains GATA-1 binding sites which may account for the
induction of the receptor during erythropoiesis (Chan and Gerhardt, 1992). Secondly, Epo
treatment increased IRP1 binding to the IRE in the TfR mRNA resulting in an increase in
mRNA stability and levels as well as enhanced TfR expression (Busfield et al., 1997; Weiss
et al., 1997). Finally, most of the iron imported into the erythroid cell may be directly targeted
to the mitochondria (Ponka, 1997) for haem production preventing a rise in cytoplasmic iron
levels in differentiating erythrocytes.
Recently, Epo induced differentiation of an erythroid cell line was shown to result in
increased ALAS2 activity, demonstrating that Epo directly affects ALAS2 expression at the
translational level (Zoller et a1.,2002).In addition, gel shift studies have shown that IRP2
binds to the IRE in the S'-UTR of ALAS2 mRNA with a higher affinity than IRPl.
Furthermore, Epo treatment reduced the binding of IRP2 to the ALAS2 IRE in a time-
dependent manner which ultimately resulted in increased expression of ALAS2 IRE-
controlled reporter gene constructs (Zoller et aL.,2002). Varyrng binding affinities of IRPl
and2 to different IREs hve been demonstrated (Butt et al., 1996; Henderson et al., 1996b).In
vivo IRPI repression of ALAS2 mRNA may not be predominant due to weak binding affrnity
of IRP1 to the ALAS2 IRE (Zoller et a1.,2002).Instead, the ALAS2 IRE maybe primarily
34

targeted by IRP2 which exhibits decreased binding affinity in the presence of Epo, facilitating
translation of ALAS2 mRNA as required. Currently, IRP control of ALAS2 translation has
been well documented but the precise mechanism of iron-dependent regulation in a
coordinated response to Epo and cellular iron levels in erythroid precursor cells has not been
clearly defined.
1.9.3 Regulation of ALAS2 by Haem
Neither ALAS2 gene transcription nor mRNA stability is regulated by haem but there is
evidence to link haem production to its utilisation in erythroid cells (Sadlon et al., 1999). For
example, normally there is no accumulation of haem in the erythroid cell as it is efficiently
utilised in haemoglobin production but in the case of overproduction or under-utilisation
feedback repression of haem synthesis seems to occur (Ponka, 1997). Moreover, there is
evidence that haem inhibits the import of precursor ALAS2 protein into the mitochondria.
Three cysteine-proline motifs were identihed in the N-terminal signal sequence of the ALASl
and ALAS2 proteins. This region resembles the seven cysteine-proline motifs (HRM) of the
haem binding domain found in the haem activated yeast transcription factor, HAPI, which is
involved in the transcriptional regulation of many nuclear encoded respiratory proteins
(Pfeifer et al., 1989).
In vitro studies in which the cysteine-proline motifs in the ALAS2 pre-sequence were either
mutated or transferred to heterologous proteins demonstrated that HRMs are able to mediate
inhibition of mouse ALAS2 precursor protein import into the mitochondria by the binding of
haem (Lathrop and Timko,1993; Goodfellow et a1.,2001). Furthermore, haem has been
shown to bind directly to HRM peptides, supporting the role of haem in preventing
mitochondrial import of ALAS2 (Zhang and Guarente, 1995).
In addition, the inhibition of ALAS2 translation by haem has also been reported, although the
mechanism is unknown (Smith and Cox, l99l). Lastly, high cellular haem levels block iron
uptake from transferrin by either inhibiting endocytosis or iron release from transferrin
(Ponka, 1997). This would inhibit the translation of ALAS2 due to reduced iron levels and
this may be a mechanism to prevent toxic amounts of free haem building up in the cell.
35

1.9.4 Hypoxic Regulation of ALAS2 Expression
Proteins involved in iron metabolism and erythropoiesis such as erythropoietin (Wang and
Semenza, 1993), transferrin (Rolfs et al., 1997), TfR (Lok and Ponka, 1999) and
ceruloplasmin (Mukhopadhyay et a\.,2000) are up-regulated in response to hypoxia. A
hypoxia response element is located in the 5'region of these genes facilitating transcriptional
activation by HIF-1. Since hypoxia induces the expression of several genes associated with
erythropoiesis the possibility of hypoxic regulation of ALAS2 was investigated. Exposure of
differentiated MEL cells to hypoxic conditions enhanced ALAS2 mRNA levels and increased
haem synthesis (Hofer et a1.,2003). Thus, hypoxic up-regulation of ALAS2 mRNA resulted
in the physiological effect of increased haem production during erythropoiesis. Furthermore, a
HIF-1 DNA-binding site was identified within the mouse ALAS2 promoter (Kramer et al.,
2000) and this region was shown to induce luciferase activity at the transcriptional level under
hl.poxic conditions. However, further investigation indicated that hypoxic induction of
ALAS2 transcription is independent of HIF-I (Hofer et a1.,2003).
1.9.5 Role of ALAS2 Expression in Erythroid Cell Differentiation
ALAS2 expression was demonstrated to be critical for haem production in DMSO-induced
differentiation of MEL erythroid cells using ALAS2 antisense RNA (Meguro et al., 1995). It
was observed that repression of ALAS2 synthesis inhibited mRNA levels for the following
erythroid genes PBGD, ferrochelatase, B-globin and p45 NF-E2. This confirmed that ALAS2
is necessary for haem synthesis which ultimately leads to globin production and the assembly
of haemoglobin. The generation of ALAS2 null ES cells and mice has demonstrated fuither
that ALAS2 is critical to the process of erythropoiesis. Differentiation of embryoid bodies
(EBs) from ALAS2/- ES cells resulted in only 3Yo of theEBs containing visible erythropoietic
cells in comparison to EBs derived from wild type ES cells in which 50% exhibited erythroid
cells. ln addition, levels of the remaining enzymes from the haem biospthetic pathway were
reduced and foetal B-globin mRNA was undetectable in erythroid differentiated ALAS2 null
ES cells (Harigae et al., 1998;Yin and Dailey, 1998). Mice null for ALAS2 die at El 1.5 with
arrested erythroid differentiation and an absence of haemoglobinised cells (Nakajima et al.,
1999).Interestingly, the ablation of ALAS2 expression resulted in an accumulation of
cytoplasmic iron and the presence of ring sideroblasts in adult mice chimaeric for ALAS2J-
mutant ES cells. In conclusion, studies targeting the ALAS2 locus have confirmed the
36

requirement for ALAS2 in erythropoiesis to synthesise haem and so facilitate the erythroid
differentiation process.
1.9.6 Model for Regulation of ALAS2
A model detailing the regulation of ALAS2 expression during erythropoiesis has been
detailed in Figure 1 .10. As previously described in detail (Section I .3), the gylcoprotein
hormone Epo is released from the kidney as a result of hlpoxia and binds to the EpoR
expressed on the cell surface of immature erythroblasts in the bone maffoìw. This leads to the
initialisation of signal transduction pathways to stimulate erythropoiesis, the process of red
blood cell maturation. Ultimately the required erlhroid and ubiquitous transcriptional
mediators are activated to enhance transcription of ALAS2 as well as other erythroid genes,
including globin. As the cell differentiates down the erythroid pathway the requirement for
haem increases. Concurrently, TfR levels increase due to the action of Epo, facilitating the
import of iron complexed to transferrin into the erythroid cell.
In order to synthesis haem, ALAS2 mRNA must be translated and this is initiated when the
intracellular iron concentration is sufficiently high such that the IRPs can be released from the
IRE motif in the 5'-UTR of the ALAS2 gene. Once the IRPs are removed translation initiation
factors can access ALAS2 mRNA and synthesise precursor ALAS2. Epo may play a role in
regulating ALAS2 translation by reducing the binding affinity of IRP2 for the ALAS2 IRE.
ALAS2 precursor protein is then translocated to the mitochondria where it catalyses the first
reaction in haem biosynthesis. Protoporphyrin is produced and iron is incorporated by
ferrochelatase to generate haem. Haem in the cytoplasm is then able to activate globin mRNA
translation. Haem and globin can then assemble to produce haemoglobin, resulting in an
abundantly haemoglobinised mature erlhroid cell. Lastly, free haem levels may be regulated
by a haem-mediated block on ALAS2 import (May et al., 1995).
1.10 SIDEROBLASTIC ANAEMIA
1.10.1 Characteristics of Sideroblastic Anaemia
The sideroblastic anaemias are aÍate, heterogenous group of blood disorders that are
characterised by the accumulation of iron deposits in the mitochondria of developing
-tt

Figure 1.10 A model for regulation of ALAS2 expression
Upon Epo activation of the EpoR on the cell surface of precursor erythroid cells in the bone
marrow a signal transduction pathway is initiated. This ultimately results in activation of
erythroid transactivating factors and transcription of the ALAS2 gene, the rate-limiting
enzyme in haem synthesis. ALAS2 mRNA is exported to the cytoplasm where iron, that has
been imported into the cell via the transferrin (Tf)transferrin receptor (TfR) mechanism,
initiates translation of ALAS2 by removing the iron regulatory protein (IRP) bound to the IRE
in the 5'-UTR of ALAS2 mRNA. The mitochondrial signal sequence of ALAS2 is removed
upon its import into the mitochondria. Once in the mitochondrial matrix, ALAS2 catalyses the
first reaction of the haem biosynthetic pathway to result in the end product of haem. Haem
initiates the translation of globin mRNA so that globin polypeptides can be synthesised and
assembled with haem to form the oxygen carrying molecule of the erythroid cell,
haemoglobin. The rise in haem levels also inhibits the mitochondrial import of ALAS2 and
may negatively affect ALAS2 translation, although this is not well understood. Such
regulation of ALAS2 by haem ensures the level of haem synthesised does not exceed the
cell's requirements, preventing a toxic accumulation of free haem in the erythroid cell.
r t0",!É'''
(ài
. ^.E-0

lron-Tf ? Tf
oALAS2 pre-mRNA
pre-ALAS2
to+
^/Signal transductionpathways initiated
frr
Nucleus
ALAS2
Transcription factors activated andALAS2 transcribed
+
III
I
ALAS2 Translation GlobinTranslation
cooH
Heme
ALAS2 > q¡¡
-r¡' + pp J> Heme
Heme
Heme
Heme

erythroblasts in the bone marrow (Koc and Harris, 1998). These iron-laden mitochondria
usually localise around the nucleus of the cell and so are termed ringed sideroblasts. The
ringed sideroblasts comprise between l5Yo to 50% of the erythroblasts of most sideroblastic
patients, with some individuals exhibiting solely ringed sideroblasts in the bone marrow
(Alcindor and Bridges,2002). Some of the cells survive and are released into the circulation
as small, hypochromic erythrocytes. The defective nature of these cells leads to the anaemia
observed in this disease. Interestingly, a dimorphic population of normal developing
erythroblasts and ring sideroblasts are observed concomitantly in the bone marrow of
sideroblastic anaemia patients. In addition, apoptosis of bone maffow cells is enhanced with
sideroblastic sufferers in comparison to normal controls (Fontenay-Roupie et al., 7999;
Matthes et a1.,2000).
It has been proposed that sideroblastic anaemia is a result of disturbed haem synthesis since
iron is still delivered to the erythroblast and mitochondria but is not incorporated into
protoporphyrin to form haem, instead accumulating in the mitochondria (May et al., 1982).
Abundant delivery of iron to the mitochondria occurs in normal cells for haem synthesis. A
gene has been isolated that encodes a mitochondrial form of ferritin, MtF (Arosio et al., 1978;
Boyd et al., 7985). Studies have localised MtF to the mitochondrial matrix (Levi et al., 2001)
where it can efficiently oxidise and store iron (Harrison and Arosio, 1996). Studies comparing
the rate of iron uptake by the cytosolic and mitochondrial ferritins found that exogenous iron
gains access to both compartments of the cell at the same time (Drysdale et a|.,2002).ln
sideroblastic anaemia, following the disruption of haem s¡mthesis, iron continues to enter the
erythroblast and is deposited in the mitochondria to form the characteristic ringed sideroblast.
It has recently been shown that the iron deposits accumulating in the mitochondria of the
sideroblast are in the form of MtF (Levi et al., 2007; Cazzola et al., 2002). Furthermore, it has
been observed that disrupted haem s¡mthesis in sideroblastic anaemia results in increased
levels of cytoplasmic and mitochondrial iron and ferritin (Cazzola et al., 2002) and since MtF
mRNA, unlike the cytoplasmic form, does not contain an IRE it must be regulated by an
alternate process. A role for MtF in iron homeostasis has not been defined but it is proposed
that it may sequester and detoxi$z free iron to protect against iron-mediated damage to the
iron-laden mitochondria in sideroblastic anaemia (Drysdale et aL.,2002).
The level of globin synthesis is also reduced in sideroblastic anaemia. This is a secondary
effect of the blood disorder since addition of haem to proerythroblast cells obtained from
sideroblastic patients has been shown in vitro to cause an increase in globin translation (White
38

et al., l97l; White and Ali, 1973). Synthesis of proteins, including globin, in erythroid cells is
dependent upon the level of haem (Chen and London, 1995). Translation of globin is initiated
by the binding of the cr subunit of the eukaryote initiation factor 2 (elF-2u) to the translational
pre-initiation complex. In red blood cells, low
erythroid- specific elF -2 u
activity. 'When
haem is at a normal in erythrocfles, HRI is inactive and globrn
translation can proceed. Therefore, the levels ofhaem and globin expression are coordinated.
The degrees of anaemia observed in sideroblastic cases varies from mild to severe. Anaemia
results due to the reduced capacity of abnormal blood cells to transport and deliver oxygen to
the body. In addition, the defective erythroblasts in the bone marrow are removed by
macrophages. The resultant anaemia and the accompanying hypoxia triggers the release of
Epo from the kidney which further stimulates erythropoiesis, perpetuating a cycle known as
ineffective erythropoiesis (Bottomley, 1998a). Linked to ineffective erythropoiesis is the
secondary effect of iron overload referred to as erythropoietic haemochromatosis (Finch,
1994; Bottomley, 1998a). The cycle of defective erythropoiesis in the bone maffo\M causes an
increase in the amount of iron absorbed by the gastrointestinal tract (Finch, 1994). Although
the components and mechanism by which iron is absorbed from the intestine is becoming
clearer (reviewed in Crichton et al., 2002), the link between ineffective erythropoiesis and
increased intestinal iron absorption has not been elucidated. Some of this iron absorbed from
the intestine is taken up by the developing erythrocles in the bone malrow but is not
incorporated into protoporphyrin since haem synthesis is perturbed as a result of defective
ALAS2 protein. Serum iron levels are increased which leads to saturation of the blood iron
transport protein, transferrin and increased serum ferritin. The iron accumulates and leads to
pathologies of rheumatoid arthritis, growth disturbances, diabetes, liver cirrhosis and heart
failure due to toxic iron levels (Cotter et al., 1999).
The erythropoietic haemochromatosis observed with sideroblastic anaemia is similar to the
clinical symptom of iron overload associated with other blood disorders such as thalassemia
major and congenital dyerythropoietic anaemia and is identical to hereditary
haemochromatosis (Bottomley, 1998a). Hereditary haemochromatosis is an autosomal
recessive disorder that results in multi-organ dysfunction caused by iron deposition. It is a
common disease, affecting one in every 400 (Powell et al., 1994) with a carrier frequency of
one in 200 individuals of Northern European descent (Crichton et a1.,2002). It was initially
described as a singular disease but it is now known to be a heterogeneous disorder that is due
haem levels orevent inactivation of the: I iì..t ?1
ordinarily phosphorylates and inhibits elF-2u
39

to mutations in a variety of genes (Roy and Andrews, 2001). One of the genes identified as
causative of haemochromatosis, Hfe, has been cloned (Feder et al., 1996; Jaswinska and
Powell, 1997) and there is a suggestion that erythropoietic haemochromatosis identified in
sideroblastic anaemia may be due to the presence of one or more mutant alleles of the Hfe
gene. A point mutation which results in a cysteine to tyrosine amino acid substitution at
position 282 (C282Y) of the Hfe protein prevents the formation of a intramolecular disulphide
bond that is critical for expression and proper function of Hfe (Feder et al., l99l;Lebron et
al., 1998;Levy et al., 1999). Other mutations in Hfe have been identified but their
contribution to the hereditary haemochromatosis is unknown.The C282Y and histidine to
asparagine mutation at residue 63 of the Hfe protein causes increased transferrin binding to
the transferrin receptor, increasing iron uptake (Feder et al., 7998). This leads to an increase
in circulating iron levels which eventually builds up in the tissues of the body. It is uncertain
if the mutant Hfe allele is a pre-requisite for iron overload in sideroblastic anaemia. A study
of several sideroblastic anaemia probands by Cotter et al. (1999) found a three-fold increase
in the frequency of the Hfe allele with the C282Y mutation. Although this particular finding
supports the coinheritance of the Hfe C282Y mutant allele with sideroblastic it has also been
demonstrated that the Hfe allele is not a pre-requisite for iron overload in sideroblastic
anaemia as there is not an increased prevalence of the mutation in the sideroblastic population
(Bottomley, 1998b). However, a Hfe mutant allele may increase the severity of the
erythropoietic haemochromatosis in individuals suffering from sideroblastic anaemia
(Yaouanq et al., l99l; Beris et al., 1999).
Sideroblastic anaemia is both an acquired and inherited disorder. Factors responsible for the
onset of acquired sideroblastic anaemia are poorly understood. It has been reported that
ethanol, copper deficiency, certain drugs (antituberculosis agents and chloroamphenicol) and
hypothermia may influence the pathogenesis of acquired sideroblastic anaemia cases
(Bottomley, 1998a).
There are several modes of inheritance for sideroblastic anaemia including X-linked,
autosomal recessive, and dominant, with X-linked being the most common and the focus of
the present study. The age at which the anaemia presents varies between individual kindreds
of X-linked sideroblastic anaemia (XLSA). For example, in the first case of XLSA described,
anaemia was observed in males in infancy and early childhood (Cooley, 1945; Cotter et al.,
1994) but the onset of anaemia has also been reported in adulthood, past the hfth decade
(Edgar et al., 1997). XLSA primarily effects males and if carrier females exhibit anaemia it is
40

usually a consequence of non-random X-inactivation (May et al., 1995; Bottomley et al.,
1998; Cazzola et al., 2000). In some kindreds only the females are affected and it is proposed
that the XLSA is so severe that males die in utero (May et al., 1995).
l.l0.2Implication of ALAS2 in XLSA
Abnormalities in haem synthesis are believed to be responsible for causing sideroblastic
anaemia. More specifically, several lines of evidence to support the proposition of ALAS2
involvement in the pathogenesis of XLSA have arisen. Firstly, the incorporation of
radiolabelled glycine but not ALA into haem was found to be reduced in vitro in reticulocytes
obtained from XLSA patients, implicating defects in ALAS2 as the cause (Volger and
Mingioli, 1965).
Secondly, a third of XLSA patients are responsive to pyridoxine, the precursor to the ALAS2
cofactor pyridoxal phosphate (Bottomley, 1998a). Administration of pyridoxine to responsive
patients increases ALAS2 enzymalic activity which is normally reduced in bone maffow
lysates to normal or supranormal levels (Aoki et al., 7979; Fitzsimons et al., 1988; Bottomley
et al., 1992). Decreased levels of ALAS2 mRNA have also been observed in response to
pyridoxine. In one case, ALAS2 mRNA levels in the bone marrow were found to be two-fold
less than levels observed in three normal controls. Interestingly, mRNA levels of the
housekeeping ALASI isotlpe were two to three-fold higher than average levels (Bottomley e/
al., 1992). Although levels of ALAS1 mRNA may increase to compensate for the low levels
of ALAS2 it is not sufficient to prevent the symptoms of XLSA.
Thirdly, a highly polyrnorphic CA repeat in intron 7 of ALAS2 was used to perform genetic
linkage analysis of a XLSA kindred. The study demonstrated that all affected males and two
obligate female carriers of XLSA had the same ALAS2 allele carrying a specific point
mutation (Cox et al., 1994). The mutation in the ALAS2 gene was not observed in unaffected
family members (Cox et al., 1994). Thus, this provided strong evidence for an association
between ALAS2 and XLSA.
Lastly, the ALAS2 gene has been mapped to the Xpll.2 region on the X chromosome
(Bishop et al., 1990; Cox et al., 1990) adding further merit to the notion that defective
ALAS2 activity is responsible for XLSA.
41

Once a potential role for ALAS2 in XLSA had been established, the sequence of the ALAS2
gene was investigated in several XLSA patients and kindreds. To date, over 20 different point
and missense mutations have been identified in the ALAS2 gene of individuals effected with
XLSA (Figure 1.11). All of the mutations reside in the catalytic domain except for one
(T372C in exon 3) and lead to amino acid substitutions in the ALAS2 protein. There are
several ways in which mutations in the ALAS2 gene can affect ALAS2 activity in the
mitochondria. For example, the mutations may alter the ALAS2 catalytic site, decrease the
affinity of ALAS2 for its substrates, reduce enzpe stability, modiff the mitochondrial
processing of ALAS2 or increase ALAS2 susceptibility to mitochondrial processes (Aoki e/
al., 7979; Cox et al., 1994; Furuyama et al., 1997; Furuyama and Sassa, 2000; Alcindor and
Bridges,2002).
In further support for the role of ALAS2 in perpetuating XLSA, a large scale genetic screen
for zebrafish mutants with developmental defects identihed the sauternes (sau) gene
(Brownlie et al., 1998). Upon analysis, this zebrafish gene was found to be a homologue of
ALAS2. The phenotype of the resultant sau mutants was similar to the biological effects
associated with sideroblastic anaemia in humans. For example, haem and embryonic B-globin
levels were reduced, developing erythrocytes were found to be hypochromic and microcytic
leading to an anaemic state and embryonic erythrocytes failed to mature morphologically.
Furthermore, the two mutations found in sau lead to single amino acid substitutions in a
region of the gene that is highly conserved across several species (Brownlie et al., 1998).
However, the characteristic ringed sideroblast associated with sideroblastic anaemia was not
observed with the sau mutants. Thus, it is inconclusive as to whether the congenital
sideroblastic anaemia associated with the sau gene in the zebrafish is analogous to the human
cases of XLSA potentially mediated by mutations in the ALAS2 gene.
On the basis of the evidence presented a strong proposal emerges that suggests ALAS2 is the
gene responsible for mediating XLSA. Thus, a model describing how defects in the ALAS2
gene may result in XLSA has been proposed (Figure l.l2). Mutant ALAS2 results in
decreased levels of ALA which eventually leads to lowered protoporphyrin levels. Iron
complexed to transferrin continues to be imported into the cytosol of the differentiating
erythrocyte (May et al., 1995) via the transferrin receptor expressed on the surface of the cell.
The iron is then transported to the mitochondria where it is ordinarily incorporated into
protoporphyrin to form haem. Since protoporphyrin levels are low in the case of XLSA, only
42

Figure 1.1L Location of point nmtations identified in the ALAS2 gene of XLSA patients
The table represents twenty-one of the identified XLSA associated point mutations in
ALAS2. The mutations are represented as black stars above the exon in which they are
located. The corresponding amino acid substitution and the reference for each mutation are
detailed.

XLSA Mutation #
1
2
3
4
5
6
7
I9
10
11
12
13
14
15
16
17
18
19
20
21
Nucleotide Chanqe
T372C
A523G
G5277
c547A-
G5617
G5664
46217
T647C
G8714
A947C
c12'l5G
c12837
Gl299A
Gl39sA
cl406A
c14067
G14074
T14794
cl622G
G17314
A1754G
Amino Acid Chanqe
L124P
T1754
Y159D
F165L
Rl70L
A'1727
D1gOV
Y199H
G291S
K299Q
T388S
R411C
G416D
R448Q
R452S
R452C
R452H
1476N
H524D
R56OH
S568G
Reference
Zhu and Bu, 2000
Zhu and Bu, 2000
Hurford et a|.,2002
Gotter et a|.,1994
Edgar et a1.,1998
May eú a1.,1994
Furuyama et a1.,1997
Cotter eú a/., 1999
Prades et al., 1995
Gotter et a1.,1992a
Cox ef a1.,1994
Cotter et al., 1992
Bottomley et a1.,1993
Bottomley et a|.,1993
Bottomley et al., 1995
Gotter eú al., 1999
Bottomley et al.,'1993
Gotter et a1.,1992b
Edgar and Wickramasinghe, 1998
Cazzola et a1.,2002
Harigae ef a/., 1999

Figure 1.12 The model proposed for the role of defective ALAS2 in XLSA
A model has been proposed outlining how mutations in the ALAS2 gene may result in XLSA.
Mutant ALAS2 results in decreased levels of aminolevulinate (ALA) and ultimately reduced
protoporphyrin (PP) levels which leads to lowered haem levels. Despite the cell's reduced
requirement for iron, iron complexed to transferrin is still imported into the developing
erythroid cell via the transferrin receptor (TfR). The pool of iron is transported into the
mitochondria and since it is not being used to form haem it accumulates in the mitochondria.
Ordinarily haem positively regulates translation of globin mRNA but since haem levels are
low globin production is hindered as a secondary effect of the blood disorder XLSA.
I1¿
Í,

lron-Tf Tf
ä(r)
--
ts
Plasma Membrane
Globin
\
GIobin mRNA
TfR TfR
MutantilÀË;>^'4++"J t Heme
oo oo o
Iooo o o o oo
o
a ooooo

a proportion of the iron is utilised to result in reduced levels of haem and an accumulation of
iron in the mitochondria to give the characteristic ringed sideroblast. Associated with low
levels of haem is a decrease in globin production as haem is involved in the regulation of
globin translation (see Section 1.10.1). Reduction in the levels of haem and globin results in
decreased haemoglobin formation which accounts for the microcytic and hlpochromic
developing erythrocytes observed in the bone marrow of XLSA probands. Since only a small
number of mature erythrocfles, which are mostly defective, are released into circulation
anaemia results. This prompts the release of Epo from the kidney to overcome the resultant
hlpoxia and a cycle of ineffective erythropoiesis is perpetuated. Lastly, via an unknown
mechanism ineffective erythropoiesis leads to the secondary effect of erythropoietic
haemochromatosis.
1.10.3 A Gene Involved in a Different Form of XLSA
A rare form of XLSA associated with spinocerebellar ataxia (XLSA/A) has been reported
(Pagon et a1.,1985). It is characterised bynon-progressive cerebellar ataxia, reduced deep
tendon reflex, incoordination, elevated levels of free erythrocyte portoporphyrin, non-
pyridoxine responsiveness, mild anemia and elevation of mitochondrial iron (Allikmets et al.,
1999; Bekri et a1.,2000). These are in addition to the typical synptoms observed with XLSA
such as the hypochromic, microcytic erythrocles and formation of ring sideroblasts (Lill and
Kispal,2001). Linkage analysis has mapped the XLSA/A condition to Xq13 (Raskind et al.,
1991) which is distinct from the region on the X chromosome associated with XLSA. A
human ortholog of the yeastATMI gene has been cloned and localised to the same region as
the XLSA/A disorder (Csere et al., 1998; l;4ao et al., 1998; Shimada et ql., 1998; Allikmets e/
al., 1999). The gene encodes a human ATP-binding cassette transporter, hABCT and it resides
in the inner mitochondrial membrane where it contributes to iron homeostasis (Shimada et al.,
1998; Allikmets et al., 1999). It has been proposed that the hABCT transporter is involved in
the formation and export of iron-sulphur (Fe-S) clusters generated in the mitochondria for
assembly of closolic Fe-S proteins (Kispal et al., 1999; Bekri et aL.,2000; Lill and Kispal,
2000). Several studies have reported the location of mutations in the hABCT gene of
individual XLSA/A kindreds (Allikmets et al., 1999; Bekri et a1.,2000; Maguire et a1.,2001).
The finding of XLSA/A associated mutations in the hABCT gene and its localisation to the
identical region of the X chromosome as the disorder has lead to the proposal that mutations
in the hABCT transporter are responsible for XLSA/4. The link between mutations in hABCT
43

and defective haem synthesis is unclear. Bekri et al. (2000) has proposed that mediation of
iron metabolism by IRP1 may be involved. For example, when iron is abundant the Fe-S
cluster may assemble on IRP1 and convert it to an aconitase-like protein that cannot bind to
the IRE found in the 5'-UTR of the ALAS2 mRNA, resulting in ALAS2 synthesis and haem
production for terminal differentiation of erythrocytes. However, failure of Fe-S cluster
formation due to mutations in the hABCT transporter would allow IRPI to bind the IRE in
ALAS2 and prevent its synthesis when iron is abundant. Unfortunately this hypothesis does
not account for the high free protoporphyrin levels that are characteristically observed in the
developing erythroblasts of XLSA/A patients (Bekri et a1.,2000). Most recently a study has
reported the involvement of the hABCT transporter in the biosynthesis of haem via an
interaction with the final enzyme of the haem biosynthetic pathway, ferrochelatase, which
catalyses the insertion of iron into protoporphyrin to form haem (Taketani et a1.,2002).
hABCT was shown to positively regulate the expression of ferrochelatase such that mutations
in the transporter may inhibit enzyme production and account for elevated levels of free
protoporphyrin. Thus, the molecular mechanism underlying the role of hABCT transporter
mutations in the erythropoietic aberrations associated with XLSA/A requires further
investigation.
1.10.4 Treatment of Sideroblastic Anaemia
,t II -U-'',
LiJ,,
As mentioned previously, a third of hereditary sideroblastic anaemia patiepts ur" r"r¡lnriu"
to pryridoxine, the precursor to the ALAS2 cofactor pyridoxal phosphateiBottomley, 1998a).
Therefore, oral pyridoxine ranging from 50 to 200mdday is administered as the initial
treatment regime with few side effects reported (Alicindor and Bridges,2002). The response
to pyridoxine treatment is particular to each case of sideroblastic anaemia. When the reaction
to pyridoxine administration is optimal reticuloclosis is observed, concentration of
haemoglobin in the blood increases to almost normal levels, serum iron levels are reduced and
low free erythrocyte protoporphyrin levels are returned to the expected range. However,
defects in the morphology of the erythrocytes is never completely abolished. Those
individuals who do not respond as well to pyridoxine treatment demonstrate an increase in
haemoglobin concentration that is less than the normal levels (Bottomley, 1998a).
Administration of pyridoxine must be continued as a relapse of sideroblastic anaemia
symptoms will occur within several months of ceasing vitamin supplementation.
,l
L,
¡!
44

In cases of severe anaemia blood transfusions are required for relief of the anaemia. Children
with sideroblastic anaemia also require such treatment to promote normal growth and
development (Bottomley, 1998a). The negative consequence of such an approach to treat
severe cases of anaemia in the context of sideroblastic anaemia is the vast increase in iron that
occurs. To combat the iron overload, iron chelation therapy can be applied. This involves a 12
hour, 5 days/week subcutaneous injection via a syringe infusion pump of the iron chelation
drug desferrioxamine (Bottomley, 1 998a).
For sideroblastic anaemia individuals with mild to severe forms of anaemia, an alternative
iron depletion program is employed if the assessed level of iron overload proves to be
detrimental. Phlebotomy, which involves bleeding the patient to remove excess iron, can be
administered (Bottomley, I 998a).
Lastly, allogenic bone marrow or stem cell transplantation for XLSA have also been reported
(Urban et al., 1992; Gonzalez et aL.,2000) and may potentially offer a cure for this blood
disorder, although a major disadvantage is the rejection of such a transplant.
1.11 AIMS
There are two major aims to this study of ALAS2 regulation and its role in disease. The first
of these is to further investigate the transcriptional regulation of ALAS2 expression in the
context of erythroid differentiation. The J2E cell line was selected for this study as it is an
immature erythroid cell that can be terminally differentiated with Epo, the natural stimulus of
erythropoiesis. Regions of the human ALAS2 gene were previously identified as regulators of
ALAS2 transcription in undifferentiated erythroid cell lines. These domains included the
promoter, intron 1 and intron 8. Thus, these same regions were transiently transfected into
J2E cells induced to differentiate with Epo and the effect on ALAS2 activity examined.
Erythroid-specific transcription factor binding sites within the conserved human ALAS2
intron 8 enhancer were also investigated to ascertain their functional contribution and binding
profile in a differentiating erythroid environment. In addition, preliminary studies to
determine the effect of the CBP/p300 coactivator on ALAS2 transcriptional regulation were
undertaken.
The second aim of this project was to develop a murine model for the blood disorder XLSA.
Mutations in the ALAS2 gene of XLSA patients, together with additional evidence discussed
45

above, implicated ALAS2 in the pathogenesis of this disease. Two mutations in ALAS2 that
had been associated with independent XLSA kindreds were selected as the basis for the
XLSA model. Gene targeting technology utilising various strategies was to be employed to
generate ES cell lines that had incorporated the XLSA associated mutations. These targeted
ES cell lines could then be injected into host blastocysts and the subsequent chimaeric mice
bred to obtain germline transmission of the ES cells with a manipulated ALAS2 gene. The
resulting XLSA murine models would then be analysed to assess the effect of the ALAS2
mutations on the erythropoietic function of the animal. This would include an examination for
the morphological characteristics associated with XLSA such as the dimorphic population of
microcytic, hlpochromic and normal erythrocytes in the bone marrow and circulation. The
severity of the anaemia and iron levels would also be assessed and compared to the effect of
the corresponding ALAS2 mutations in humans. The XLSA murine model would enable the
control of iron metabolism to be investigated since the regulation of iron absorption in the
intestine is altered in XLSA via an unknown mechanism. Ultimately, the XLSA murine
model may be useful for the development of more effective treatments for the disorder.
46

CHAPTER 2
MATERIALS AND METHODS

CHAPTER TWO: MATERIALS AND METHODS
2.1 MATERIALS
2.1.1 Drugs, Chemicals and Reagents
The following products were obtained from Sigma Chemical Co.: 4-(2-aminoethyl)-
benzenesulfonyl fluoride (AEBSF), Adenosine triphosphate (ATP), agarose (T¡pe 1),
ammonium persulphate (APS), ampicillin, aprotinin, benzamidine, benzidine, bestatin, B-
glycerophosphate, B-mercaptoethanol, bovine serum albumin (BSA), colcemid, dithiothreitol
(DTT), ethidium bromide, ethylenediaminetetra-acetic acid (EDTA), fialuridine, G418,
leupeptin, levamisole, N-2-hydroxyethylpiperazine-N-2-ethane sulphonic acid (Hepes),
pepstatin, phenylmethylsulfonyl fluoride (PMSF), salmon sperm DNA, sodium dodecyl
sulphate (SDS), spermidine, 6-thioguanine, Tris-base.
Sources of other reagents were as follows: caesium chloride: Roche Diagnostics;
deoxyribonucleoside triphosphates (dNTPs): GeneWorks; Erythropoietin (Epo): Janssen
Cilag; gentamicin: Pharmacia & Upjohn; penicillin/streptomycin: GIBCO; phenol: Wako
pure chemicals; polyethylene glycol6000 (PEGoooo): BDH chemicals; poly(dl-dC)
Pharmaciai N,N, N',N'-tetramethylethylethenediamine (TEMED): Tokyo Kasei.
All other chemicals and reagents were of analytical grade.
2,l.2Radiochemicals
¡cr-32r1-dCTP (3000ci/mmol) and ¡y-32P1-dATP (3000Ci/mmol) were purchased from
GeneWorks.
I
u''' n' '
2.1.3 Enzymes ,/,Restriction enzyrnes were purchased from Pharmacia, New England Biolabs or GeneWorks.
Other enzyrnes were obtained from the following sources: Calf intestinal phosphatase: New
U\,,.
47

England Biolabs Mannheim; Klenow: GeneWorks; lysozyme: Sigma; Pfu Turbo polymerase:
Stratagene; proteinase K: Boehringer Mannheim; ribonuclease A (RNase A): Sigma; Taq
polymerase: GeneWorks; T4 DNA ligase andT4 Polynucleotide Kinase: GeneWorks.
2.1.4 Buffers
Denhardt's solution: 0.1%(wlv) Ficoll, 0.1%(wlv) polyvinylpyrrolidine,0.1%(w/v) BSA.
SSPE: 150mM NaCl, 10mM NaHzPO¿, lmM EDTA.
TBE: 90mM Tris, 90mM boric acid, 2.5mM EDTA, pH 8.3.
TE: 10mM Tris-HCl, p}{7.5,0.lmM EDTA.
TES: 25mM Tris-HCl, pH 8.0, 10mM EDTA, 15olo sucrose.
3x Urea Loading Buffer: 4M urea, 50olo sucrose, 50mM EDTA, 0.1% bromophenol blue.
5x Annealing Buffer: 200mM Tris-HCL, pH7.5,100mM MgC12, 250mM NaCl.
10x Klenow Buffer: 0.5M Tris-HCL, 0.lM MgCl2, lmM DTT
10x Ligation Buffer: 500mM Tris-HCL, pH7.6,100mM MgC12, 10mM DTT, 50% (wlv)
PEG-6000.
10x Restriction enzyme digestion buffer: 330mM Tris-HCL, pH 7.8, 625mM KAc, 100mM
MgAc, 0.1M spermidine, 0.1M DTE.
2.1.5 Cloning Vectors
pBluescript KS- was purchased from Stratagene. pSP72 was purchased from GeneWorks.
pGl2-Basic vector was purchased from Promega. pmALAS2X/S-7 and pmALAS2/2.8E8-I10
were provided by H.C. Chandler, University of Adelaide, Australia.
2.1.6 Cloned DNA Sequences
The following cloned DNA sequences, used throughout this study, were generous gifts from
the following:
pTC-EA1: human ALAS2 genomic clone (Cox et al., 1991) was provided by Dr. K Surinya,
University of Adelaide, Australia
murine ALAS2 genomic clone: provided by H. Chandler, University of Adelaide, Australia
48

ploxPneo-I: obtained from Dr. A. Nagy, Samuel Lunenfield, Research Institute, Toronto,
Canada
pKS\eotk: provided by Dr. I. Atmosukarto, University of Adelaide, Australia
2.1.7 Synthetic Oligonucleotides
Synthetic primers were obtained from Geneworks, IDT or Sigma. The primer sequences are
listed below:
í) Olígonucleotides for sequencing pløsmid constructs
M I 3 Universal sequencing primer (1 Tmer) : 5'-dGTAAAACGACGGCCAGT-3'
T3 primer: 5'-dATTAACCCTCACTAAAG-3'
T7 primer: 5'-GATATCACTCAGCATAA-3'
íí) Oligonucleotides used in the mutøgenesis of humøn ALAS2 Intron IThe introduced mutations are underlined.
I8 CACCC site A: 5'-dTAAACCCCTCCTCAGCTGTAGCCCCAAGCTT-3'
I8 CACCC site B: 5'-dCAGCTAAAGGTTCAGCTGAGCTACTGCCT-3'
I8 GATA site A: 5'-dCCAGCTACTGCCAGCTGAGTCATTGCAT-3'
I8 GATA site B : 5'-dACTTGTAAGTCCAGCTGCAAAGCAGCAG-3'
íiì) Sequences of the sense strand of synthetíc olìgonucleotídes used ín gel shíft ussays
The binding motifs are underlined.
Consensus DNA Binding Sites:
B-globin GATA-cons (Wall et al., 1 988): 5'-dTTGGCTCCCTTATCATGTCCCTG-3'
B-globin CACCC (Miller & Bieker, 7993): 5'-dAGCTAGCCACACCCTGAAGCT-3'
49

Sp-l consensus (Promega): 5'-dATTCGATCGGGGCGGGGCGAGC-3'
Iluman ALAS2Intron 8:
18 CACCC site A-S: 5'-dCTAGTCCCCCACCCTAGCGAA-3'
I8 CACCC site B-S: 5'-dAAAGGTCCCCACCCAGCTACT-3'
18 GATA site A-S: 5'-dAGCTACTGCCTATCTAGTCATTGC-3'
I8 GATA site B-S : 5'-dTTGAAAGTCCTATCTC tuAAGCAGC-3'
iv) Olígonucleotides for the generøtìon of XLSA poínt mutations
The introduced point mutation is underlined.
Primers used to generate the exon 8 Cl159G mutation
mE8 C I 1 5 9G-A: 5'-dCATCATCTCTGGAAGTCTTGGTGAGTAGC-3'
mESC 1 I 59G-B: 5'-dGCTACTCACCAAGACTTCCAGAGATGATG-3'
Primers used to generate the exon 9 Cl228T mutation
nE9CI228T-A: 5'-dGGACATGGTGTGCTCCTACGC-3'
mE9Cl228T-B : 5'-dGCGTAGGAGCACACCATGTCC-3'
2.1.8 Bacterial Strains
The following E. coli Kl2 strains were used as hosts for recombinant plasmids in
recombinant DNA procedures :
(a) E. coliXL-l Blue: supE44 hsdRl7 recAl gyrL46 thi relAl lac-F' [proAB* laclqlacZ
M15 (et)l host for recombinant plasmids, purchased from Stratagene.
50

(b) ¿'. colíDlHsoc: supE441acU169 (p80lacZ M15) hadRlT recAl gyrA96 thi-l relAl host
for recombinant plasmids, obtained from the E. coli Genetic Stock Centre, Yale
University, New Haven.
Storage of stock cultures and plasmid transformed bacteria were prepared by dilution of an
overnight culture with an equal amount of 80% glycerol and maintained at -80'C. Single
colonies of bacteria were obtained by streaking the glycerol stock onto agar plates (Section
2|r9) containing the appropriate media and antibiotic selection. Colonies obtained were then
used to innoculate liquid growth medium. These cultures were grownat3T"C with continuous
shaking to allow adequate aeration.
2.1.9 Btcterial Growth Media
Growth media were prepared in double-distilled water and sterilised by autoclaving.
Antibiotics were added after the solution had cooled to 50"C.
Luria (L) broth: contained l% (wlv) Bacto-tryptone (Difco),0.5yo (w/v) yeast extract
l% (wlv) NaCl, adjusted to pH 7.0 withNaOH')
Ut Jt'Agar plates were prepared by adding 75% (wlv) Bacto-agar (Difoo) to the L broth.
2YT mediam: l.6Yo (w/v) Bacto:tryptone (Difco), 15& (w/v) yeast extract (Difco), 0.5%
NaCl, adjusted to pH 7.5 withNaOH.
Psi (y) broth: 2% (wlv) Bacto-tryptrone (Difco),0.5o/o (w/v) yeast extract (Difco), 0.5%
MgSOa, adjusted to pH 7.6 wilh 0.1M KOH.
Ampicillin (1OOug/ml) was added where appropriate to maintain selective pressure.
(Difccþ,
2.1.10 Tissue Culture Cell Lines and Media
51

i) Cell Lines
The sources of the following cell lines used throughout the course of this work are as
indicated:
(a) Mouse W9.5 embryonic stem cells, Dr Richard Harvey,'WEHI, Melbourne, Australia
(b) Mouse EI4TG2a embryonic stem cells, Dr. Austin Smith, CGR Edinburgh, UK
(c) StoR fibroblast cells, Dr. Richard Harvey, WEHI, Melboume, Australia
(d) murine erythroleukemia J2E-3 cells, Dr. S.P. Klinken, University of Westem Australia,
Australia
(e) monkey COS-I kidney cells, American Type Cell Culture (ATCC) Laboratory
(f) monkey CV-1 kidney fibroblast cells, Dr. O. Bernard, WEHI, Melbourne, Australia
iì) Solutions
ES Cell Lysis Buffer: 100mM Tris-HCL (pH 8.5), 50mM EDTA, 0.2% SDS, 200mM NaCl
and 100pg/ml Proteinase K.
Hypoxanthine amethopterin thymidine (HAT): 100FM hypoxanthine, 0.4pM amethopterin
and 16pM thymidine.
Phosphate buffered saline (PBS): 136mM NaCl, 2.6mM KCl, 1.5mM KHzPO+ and 8mM
Na2HPO4, pH 7 .4, was sterilized by autoclaving (20 psi for 25 minutes at 140"C).
Trypsin/EDTA solution: 0.1% trypsin (Difco) and lx EDTA Versene buffer solution (CSL),
was sterilized by filtration through a0.2¡tm filter (Whatman).
iii) Media
Dulbecco's Modified Eagle's Medium (DMEM) (GIBCO). Media contains 4500mg/ml D-
glucose and L-glutamine, 110mg/L Sodium pynrvate and pyridoxine hydrochloride.
Supplemented with 100 units/ml of penicillin and 100pg/ml of streptomycin (GIBCO). The
media was supplemented with 5%o foetal calf serum (CSL) that had been batch tested to
52

determine its ability to promote erythroid differentiation of the J2E cells. The batch testing
was performed by the laboratory of Dr. Klinken, University of Western Australia.
Incomplete ES mediaz 67 .4gDMEM, 1 8.5g NaHCO: and 6.25m1 gentamycin, p}J7 .4.
Volume was made up to five litres with water.
Complete ES media: Incomplete ES media was supplemented withl5o/o FCS (CSL). For
every 100m1of media 100pL of lM B-mercaptoethanol and lml each of L-glutamine and
non-purified LIF was added. Media contained 5x104 units/ml of Gentamicin. This media was
used to culture the E14TG2aES cells. The FCS was batch tested to ensure the solution did not
promote differentiation of the ES cells, maintaining their pluripotent state.
Knock-Out DMEM (KO-DMEM) (GIBCO): For every 100m1 of media 100u1 of lM B-
mercaptoethanol, 0.7m1of NaCO¡ (7.5% w/v), lml each of 200mM L-glutamine and 10mM
non-essential amino acids and 1x non-purified Leukemia Inhibitory Factor (LIF) was added.
This media was used to culture the W9.5 ES cells.
All media was filter sterilized prior to use.
LIF was prepared from transfected COS-1 cells according to the method of Smith (1991) with
the exception that transfection was via electroporation.
2.l.ll Miscellaneous
Film:AGFA Curix Blue HC-S Plus
Nytran 0.45pm: Schleicher and Schuell
3MM paper: Whatman Ltd.
53

2.2 RECOMBINANT DNA METHODS
2.2.1 General DNA Methods
The following methods were performed as describedin"Molecular Cloning: A Laboratory
Manual" Sambrook et al., (1989): Growth, maintenance and preseruation of bacteria;
quantitation of DNA; autoradiography; agarose and polyacrylamide gel electrophoresis;
precipitation of DNA;phenol/chloroform extractions; end-filling or end-labelling of DNA
fragments using the Klenow fragment of E.coli DNA polSrmerase I.
All manipulations involving viable organisms which contained recombinant DNA were
carried out in accordance with the regulations and approval of the Australian Academy of
Science Committee on Recombinant DNA and the University Council of the University of
Adelaide.
2.2.2 Plasmid DNA Preparation
Either the rapid alkaline hydrolysis procedure of Birnboim and Doly (1919) or the Ultra Clean
Mini Plasmid Prep Kit (MoBio Laboratories) was used for the isolation of plasmid DNA from
2ml overnight cultures for analytical restriction digests and for the preparation of plasmid
cDNAs for use as probes in Southem hybridization analysis.
Plasmid DNA used for the transfection of tissue culture cell lines was routinely grown up in
250m1 cultures of 2YT inoculated with 50¡rl from a 5ml 8 hour culture. Plasmid DNA was
extracted using the alkaline lysis procedure described above and further purified by the
cesium chloride/ethidium bromide density gradient procedure (as described in Sambrook e/
al., 1989) in a Beckman TL-100 benchtop ultracentrifuge and TLA-I00.2 rotor. Plasmid DNA
was then butanol extracted to remove ethidium bromide and precipitated twice with 0.1
volume of 3M sodium acetate, pH 5.2 and2.5 volumes of 100% ethanol. Plasmid DNA was
then washed with l}Yo elhanol, resuspended in TE buffer, quantified by spectrophotometry
and analysed by agarose gel electrophoresis to confirm concentration and supercoiling.
54

2.2.3 Restriction Enzyme Digestions of DNA
In analytical digests, 0.5-lpg of DNA was incubated with 2-5 units each of the appropriate
restriction enzyme(s) for a minimum of 2 hours in the buffer conditions specified by the
manufacturer. Reactions were terminated with the addition of 1/3 volume of urea load buffer
and electrophoresed on I or 2o/o mini-agarose gels depending on the size of the restricted
DNA fragment(s) in IxTBE buffer.
In preparative digests, 5-10 pg of DNA was restricted in a reaction volume of 50p1, and the
desired DNA fragments were isolated as detailed below.
2.2.4 Preparation of Cloning Vectors
Plasmids were linearised with the appropriate restriction enz¡rme(s) to prevent selÊligation of
the vector, 5'terminal phosphate groups were removed by incubation in 50mM Tris-HCL, pH
9.0, lmM MgCl2,O.lmM ZnCl2,with I unit of calf intestinal phosphatase (CIP), in a final
volume of 50¡rl for 15 minutes at37"C followed by 15 minutes at 56'C. Another 1 unit of CIP
was added and the incubations repeated. The vector DNA was electrophoresed on a l%o
agarose TBE gel and the gel stained with ethidium bromide. The linearised vector DNA was
visualized under UV light, excised and purified using the QIAEX II gel extraction kit
(QIAGEN) according to the manufacturer's instructions.
2.2.5 Pr eparation of DNA Restriction Fragments
DNA was incubated with the appropriate restriction enzyme(s) as described in Section 2,2.3,
and restriction fragments were isolated from a horizontal 0.8%-2.0% agarose 1x TBE gel,
depending on the size of the restriction fragment(s). Bands representing restriction fragments
were visualized under UV light following staining with ethidium bromide and the appropriate
fragment(s) excised from the gel. DNA fragments were isolated from agarose gels using the
QIAEX II gel extraction kit (QIAGEN) according to the manufacturer's instructions.
55

2.2.6Ligation of DNA
A 1Opl reaction contained 20ng of vector DNA, a 3 molar excess of the insert DNA, 50mM
Tris-HCL, p}J7.6,10mMMgCl2, lmMDTT,5o/o(wlv) PEGoooo, lmMATP, and l unitofT4
DNA ligase. The reactions were incubated for either 4 hours at room temperature or overnight
at l6oC. A control ligation with vector only was set up and included in the subsequent
transformation to determine the background levels of uncut or recircularised vector DNA.
2.2.7 Transformation of E. colí with Recombinant Plasmids
i) Prepøratìon of Competent E, coli
E. coli cells were made competent by the rubidium chloride method. A single colony of the E.
colihost strain was inoculated into 5ml pf Psi (y) broth and the culture incubated with
continuous shaking at3J"C overnight. 1.6 ml of the overnight culture was subcultured into
50ml of y-broth and incubated at37"C until the culture reached an absorbance at 600nm
(ODooo) of 0.4-0.6. 25ml of this culture was then used to inoculate 500m1 of y-broth and
again the culture was grown up to an ODooo of 0.4-0.6. The cells were chilled on ice for 5
minutes prior to centrifugation at 4000 rpm for 5 minutes at 4"C. The cells were then
resuspended in 0.4 volumes of ice cold Tfb 1 (30mM KAc, 100mM RbCl, 1OmM CaClz,
50mM MnCl, l5Yo glycerol, adjusted to pH 5.8, with 0.2M acetic acid), incubated on ice for
5 minutes and pelleted at 4000 rpm, at 4'C for 5 minutes. Cells were then resuspended in 0.04
volumes of ice cold Tfb2 (10mM MOPS free acid, 10mM RbCl, 75mM CaCl2,15% glycerol
adjusted to pH 6.5, with 0.1M KOH), and kept on ice for a further 15 minutes, before being
stored at -80'C in 500p1 aliquots.
ii) Transþrmøtìon of Competent Bøcteríø
100-150p1 of the cell suspension prepared using the rubidium chloride method was mixed
with 5pl of the DNA ligation reaction mix (Section2.2.6) and left on ice for 20 minutes. The
cells were then heat shocked at 42"C for 2 minutes, placed on ice for 2 minutes and lml of LB
56

medium added. The cells were then incubated at 37"C for 20 minutes, centrifuged for I
minute at 6000 rpm and 900p1 of supernatant removed. The transformed cells were gently
resuspended in the remaining medium and then plated onto L-agar containing 100¡.tglml of
ampicillin by spreading with a wire spreader. The agar plates were routinely incubated
ovemight at 37 " C overnight.
2.2.8 DNA Sequence Analysis
DNA sequencing was performed using the ABI PRISMTM Dye Terminator Cycle Sequencing
Ready Reaction Kit (Perkin Elmer). To a 20pl reaction, 0.5pg template DNA, 100ng of
primer and 8pl of Dye Terminator reaction mix was added. This reaction underwent the
following PCR conditions for 25 cycles: Denaturation at 96oC for 30 seconds, annealed at
50oC for l5 seconds and extension at 60oC.
At the completion of the PCR the reaction was precipitated with isopropanol and the
dehydrated pellet taken to the DNA Sequencing Facility at the Institute if Medical and
Veterinary Science where it was analysed on an ABI Prism 3700 DNA Analyser and the
sequence provided as an electropherogram.
2.2.9 Preparation of Radiolabelled DNA Probes
Ni) Olígo-Løbelling DNA
I'
Isolated oDNA inserts were labeled with ¡cr-3211-dCTP using the Megaprime DNA
Labelling kit (Amersham). Briefly, 50-100ng of DNA was denatured in the presence of
random nonamers in a total volume of 25¡tI at 100"C for 4 minutes. Reactions were set up
at room temperature as per the instruction manual, using 50pCi of ¡ct-32e1-dCTP and 2
units of Klenow fragment in a total volume of 50p1. The reactions were then incubated at
37"C for 10 minutes before being terminated by the addition of 2¡l of 0.5M EDTA, pH
8.0 (final concentration 20mM). The probes were then denatured by incubation at 100"C
for 5 minutes and quenched on ice before adding to the hybndization solution.
57

ä) 5'End-Labelling of Synthetíc DNA olígonucleotídes
The synthetic DNA oligonucleotides used as probes were ¡32P1 labelled at the 5' end using
¡y-32f1-aRfP and T4 polynucleotide kinase. The annealing reaction contained 2.5¡tgof
both sense and anti-sense oligomer and lx annealing buffer (Section 2.1.4) in a final
volume of 10p1. The reaction was incubated at 95"C for 2 minutes and then transferred to
a 65"C heating block removed to the bench and allowed to cool to room temperature for
30 minutes.
For the kinase reaction, 500ng of annealed oligomer, 1x polynucleotide kinase buffer, 3
units of polynucleotide kinase and 50pCi of ¡y-32t1-dATP in a final volume of 10pl was
incubated at37"C for 30 minutes. The reaction was then precipitated with lpg of polydl-
dC, 0.1 volume of 3M NaAc, pH5.2,2.5x I00Yo ethanol, incubated for 15 minutes at -
80oC and centrifuged for 20 minutes at 4"C. The probe was washed with 70o/o ethano|
dried and resuspended in lx TE. The radioactivity of the probe was determined by a
scintillation counter.
2.2.10 Preparation of Radiolabelled DNA Markers
The reaction to radiolabel lpg of marker DNA consisted of lx Klenow buffer (Section 2.1.4),
50pCi of ¡cr-32P1 dNTP radiolabel, 6pM dNTPs and I unit of Klenow enzyme. This was
performed in 10¡rl, using sterilised water to make up the reaction volume. The reaction was
incubated at room temperature for l5 minutes at which point the volume was increased to
200p1using 1x TE. A phenol/chloroform extraction was conducted and the reaction
centrifuged for 10 minutes at 13000 rpm. The aqueous layer containing the radiolabelled
markers was then removed for storage at -20"C. The labelled dNTP required for this
procedure was dependent upon the DNA marker chosen.
58

2.2.11 Oligonucleotide Site-Directed Mutagenesis
i) Mutøgenesis Reøction
Site-directed mutagenesis was performed on double-stranded plasmid DNA using the
QuikChangerM Sited-Directed Mutagenesis Kit (Stratagene) to introduce point
mutations. The 50pl polymerase chain reaction contained 50ng double-stranded
plasmid DNA, P/u DNA polymerase buffer (20mM Tris-HCL, pH8.8, 2mM MgSOa,
1OmM KCL, 1OmM (NH+)zSO+,0.lyo Triton-X-l00, 100pg/ml nuclease-free BSA),
L25ngeach of sense and anti-sense strand oligonucleotides with the desired point
mutation, dNTP mix to a final concentration of 1OmM and2.5 units of native Pfu
DNA polymerase. Fifteen cycles of amplification were performed using an automated
PTC-I00 programmable thermal cycler (MJ Research Inc) under the following
conditions: Cycle 1: denaturation at 95oC for 30 seconds; Cycle 2-15: denaþxation at
95'C for 30 seconds, annealing at 55oC for 1 minute, extension at 68"C for 2
minutes/kb of plasmid length. A final extension at 68'C for 5 minutes.
At the completion of the PCR, 10 units of DpnI was added to the reaction and
incubated at37"C ovemight to remove any parental DNA from the amplification
reaction.
ii) Selection of Clones Contøíníng the Mutøtion
Digested mutagenesis reactions were transformed into E.coli XLl-Blue competent
cells (Section2.2.7). Plasmid DNA was prepared as detailed in Section2.2.2 and
using the appropriate primers the DNA was sequenced (Section2.2.8) to determine ifthe point mutation had been introduced.
2.3 REPORTER AND EXPRESSION CONSTRUCTS UTILISED IN THE
INVESTIGATION OF ALAS2 TRANSCRIPTIONAL REGULATION
The following section details the constructs used during the course of this study. Reporter and
expression vectors that were constructed specifically for this project have been described in
59

detail and illustrated accordingly. Constructs that were supplied either by our laboratory or
obtained from external research groups are indicated and listed below.
2.3.1 ALAS2 Promoter/Reporter Gene Plasmids
The ALAS2 promoter deletion constructs used in Section 3.2.2were constructed in our
laboratoryby Dr. K Surinya. The series of 5' flanking ALAS2 deletion plasmids were
generated from subcloned fragments isolated from the human genomic clone pTC-EA1 (Cox
et al., 1991) and ligated into pGl2-basic (Promega). Constructs generated contained from I24
bp to 10.3 kb of 5'ALAS2 flanking sequence and28 bp of the 5'ALAS2 UTR cloned
upstream of a luciferase reporter gene inpGL2-basic. The constructs were labeled as follows:
pALAS2-10.3LUC, pALAS2-5 .ILUC, pALAS2-1.9LUC, pALAS2-0.293LUC and pALAS2-
0.l24LU C (Figure 3.3 a).
2.3.2 AL^S2 Intron/Reporter Gene Constructs
A series of luciferase reporter gene constructs containing human ALAS2 intron 8 fragments
were generated from the human genomic clone, pTC-EAl (Cox et al., l99l) and ligated into
the plasmid pALAS2-O.293LUC which was constructed from the parental vector pGl2-basic
as described in Section2.3.l. The pALAS2-0.293LUC plasmid contained 293bp of the
human ALAS2 promoter and28 bp of the ALAS2 5'-UTR cloned upstream of a luciferase
gene. To synthesise a plasmid construct containing 239bp (+159/+398) of human ALAS2
intron 8 sequence, a vector with239 bp of intron 8 sequence in the reverse orientation (pKS-
IntS(239)) (constructed in our laboratory) was digested with SalI and BamHI (in the
polylinker) to release a25Ibp fragment encompassing239 bp of intron 8 sequence extending
from the PstI site to the SalI site (Figure 2.1). The pALAS2-0.293LUC vector was digested
with BamHI and SalI and the BamHVSalI intron 8 fragment isolated above was directionally
cloned downstream of the human 293bp ALAS2 promoter and luciferase gene in the forward
orientation. This construct was termed pI8+LUC.
To construct a luciferase reporter gene construct containing 460bp (+1021+562) of human
ALAS2 intron 8 sequence, an EcoRI fragment encompassing this length of intron 8 sequence
was isolated as an EcoRV/SmaI fragment (both sites are in the polylinker) from a vector
containing 460 bp of human intron 8 sequence (pKS-Int8(4ó0) (constructed in our
laboratory). This EcoRV/SmaI fragment was blunt end cloned into pSP72 (GeneWorks)
60

Figure 2.1 Construction of a human ALAS2 reporter gene construct containing 239 bp
of intron I
Details for the cloning procedures are outlined in the text. The bold lines represent human
ALAS2 fragments within the cloning vectors.
et tr ea¡ru
pKS-lnt8(239)
Digested with Sall and BamHland ligated into similarly digested
pAlAS2-O.2931UC
BamHlHindlll
+28 bp
+lntron 8239 bp
-293 bp +1 pl8+LUCLUC

digested with SmaI. A BamHI/BglII fragment containing460 bp of human ALAS2 intron 8
was isolated and cloned into BamHI digested pALAS2-O.293LUC, downstream of the
ALAS2 promoter and luciferase gene (Figure 2.2). Clones containing intron 8 in the forward
orientation were determined by restriction digest analysis. This reporter gene construct
containing 460 bp of human ALAS2 intron 8 in the native orientation was referred to as
pI8(46O)+LUC.
The following ALAS2 intron reporter gene plasmids were constructed in our laboratoryby
Dr. K. Surinya. The human ALAS2 intron 1 reporter gene construct (pI1+LUC) contained all
of intron I (4.9 kb) cloned in the forward orientation directly downstream of the human
ALAS2 293bp promoter and upstream of the luciferase gene in the pGl2-basic vector. The
pI1+I8(460)+LUC vector was generated from pIl+LUC. A fragment containing460 bp of
human intron 8 (+102l+562) was cloned downstream of the luciferase gene in the pIl+LUC
vector (Figure 3.4a).
2.3.3 Site-directed Mutagenesis of the Human ALAS2 Intron 8 Sequence
Site-directed mutagenesis was performed on the plasmid pKS-Int8(239) containing239 bp of
human ALAS2 intron 8 sequence. Oligonucleotides were designed (Section 2.1.7(ii)) to
mutate the CACCC and GATA sites either singularly or in combination by introduction of a
PvuII site. To generate constructs containing multiple mutated sites, site-directed mutagenesis
was repeated when required. Sequencing was conducted on the plasmid that had undergone
mutagenesis to determine if the desired mutation had been introduced. The mutated intron 8
fragment for each mutation was then isolated by BamHVSalI and cloned into the similarly
digested pALAS2-O .293LUC (Figure 2.3).
2.3.4 \üild Type and Mutant EIA and p300 Expression Constructs
The following constructs were provided by Dr Kouzarides, University of Cambridge, UK:
pElAwt - ElA wild type expression vector
pElAl5-35 - mutant EIA expression vector that cannot bind CBP/p300
pElAblk - control empty vector for ElA
The following constructs were obtained from Dr Eckner, University of Zunch, Switzerland
61

Figure 2.2 Construction of a human ALAS2 reporter gene construct containing 460 bp
of intron 8
Details for the cloning procedures are outlined in the text. The bold lines represent human
ALAS2 fragments within the cloning vectors. The dotted lines represent the pSP72 cloning
vector.

RV
Smal/EcoRV
+EcoRl lntron 8
| ''iöõ ËP e"Î
SmalRI
pKS-lnt8(a60)
Digested with EcoRV and Smal andligated into Smal digested pSP72
BamHlI
+lntron I460 bp
BamHlEc
Digested with BamHl and Bglll andligated into pALAS2-O.2931UC
digested with BamHl
Hindlll----+
lntron 8460 bp
Bg lll/BamHlB
LUC-293 bp +1
+28 bppl8(a60)+LUC

Figure 2.3 Generation of human ALAS2 intron I mutant reporter gene constructs
Details for the cloning procedures are outlined in the text. The bold lines represent human
ALAS2 fragments within the cloning vectors. The black cross represents the mutated site/s

<-lntron 8239 bp
il tl Ba HIm
I
1111lntroduced a Pvull site by site-directed mutagenesis
lsolated a BamHl and Sa/l fragment from pKS-lnt8(239)
harbouring a single or multiple mutations and ligated intosimilarly d igested pAl-AS2-O.293LUC
BamHlindlll
pKS-lnt8(239)
plScA+LUC
il
plScB+LUC
plSgA+LUC
Bg
-293 bP +1
+28 bp
Bg
-293 bp +1
indlll
+28 bp
BamHl
BamHl
-293 bp +1
indlll
+28 bp
Pstl
indlll
gA gBcBLUC
gA gBcALUC
gBcA cBLUC
gAcA cBLUC
-293 bP +1+28 bp
BamHl
plSgB+LUC

gA gB
LUC-293 bP +1
indlll
+28 bp
BamHl
BamHl
BamHl
BamHl
P S
plScAB+LUC
Sall
plScAgA+LUC
S ll
plScAgB+LUC
-293 bP +1
indlll
+28 bp
indlll
+28 bp
fl
-293 bP +1
B indlll
gBcBLUC
gAcBLUC
gAcALUG
-293 bP +1+28 bp
plScBgB+LUC

p300CMV - p300 wild type expression vector
p300de130 - mutant p300 expression vector that cannot bind ElAp300blk - control empty vector for p300
The Renilla reniþrmis luciferase reporter vector (pRL-TK) (Promega) contains the herpes
simplex virus thymidine kinase promoter located upstream of the luciferase gene and was
used as an intemal control to normalise activity of the experimental reporter in transient
transfection assays.
2.4 ALAS? TARGETING CONSTRUCTS
2.4.1 Synthesis of the ALAS2 Exon 8 C1159G Mutant Targeting Construct
The mouse ALAS2 genomic clone used was isolated from a El4TG2a lambda library (H.
Chandler). QuikChange'" (Stratagene) PCR based site-directed mutagenesis to introduce the
Cl159 point mutation into ALAS2 exon 8 was performed on pmALAS2/2.8E8-I10 (generated
by H. Chandler in our laboratory) which contained a 2.8 kb fragment of mouse ALAS2
genomic DNA ranging from the SmaI site in exon 8 to the BamHI site in intron l0 (Figure
2.4). The primer pair used to insert the mutation was mE8Cl 159G-A and mESCl 159G-B (see
Section 2.I.7(iv)).Introduction of the Cl159G mutation into exon 8 of the pmALAS2l2.8E8-
I10 vector was confirmed by DNA sequence analysis and then a I.2kb XmaI (an
isoschizomer of SmaI that produces a 5' overhang instead of a blunt end) to StuI ALAS2
fragment containing the C1159G mutation was isolated and cloned directionally into the
unique XmaI and StuI sites found in the vector pmALAS2ñS-7 (generated by H. Chandler in
our laboratory). The vector pmALAS2)lS-7 contained approximately 7 kb of murine ALAS2
genomic DNA ranging from exon 7 (XhoI) to intron 10 (SacI). The targeting construct
generated was denoted pmALASzl Cl I 59G.
2.4.2 Synthesis of the ALAS2 Exon 9 Cl228T Mutant Targeting Construct
The QuikChange'" (Stratagene) PCR based site-directed mutagenesis method was utilised to
introduce the CI228T point mutation into exon 9 of the murine ALAS2 gene. The
62

Figure 2.4 Construction of the targeting vector for introduction of the exon 8 C1159G
point mutation into the murine ALAS2 gene
Details for the cloning procedures are outlined in the text. The solid lines represent murine
ALAS2 fragments and the dotted lines indicate the cloning vector. The boxed numbers
represent ALAS2 exons.
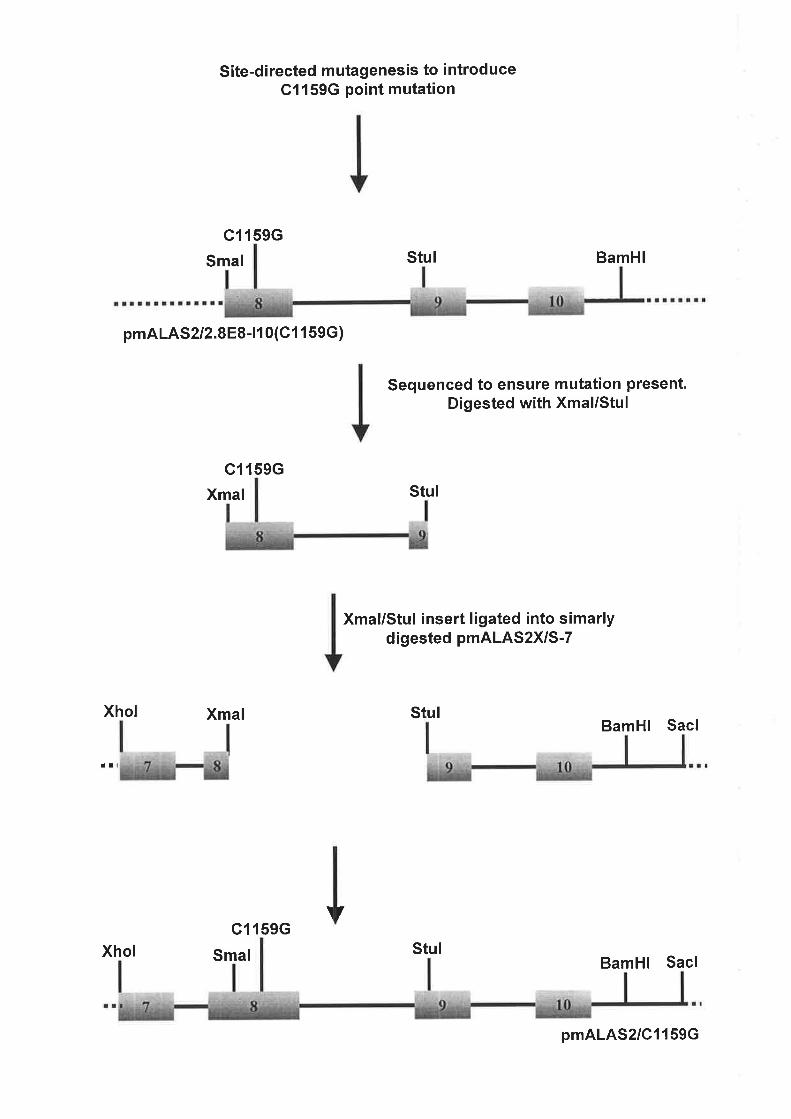
Site-directed mutagenesis to introduceC1159G point mutation
c1159G
Smal
pmA LAS2 12.888 -110(C 1 1 59G )
c1 l59G
Xmal
Stul BamHl
Sequenced to ensure mutation present.Digested with Xmal/Stul
Stul
Xmal/Stul insert ligated into simarlydigested pmALAS2X/S-7
StulBamHl Sacl
BamHl Sacl
Xhol Xmal
tl
\ /c1 159G
Xhol Smal Stul
pmALAS2/C1159G

mutagenesis was performed on pmALAS2/2.8E8-I10 using the primer pair mEx9Cl228T-A
and mEx9Cl228T-B (see Section2.l.7(iv)). DNA sequencing was performed to confirm the
introduction of the Cl228T mutation. Al.l kb StuI (exon 9) to BamHI (intron 10) fragment
containing the exon 9 mutation was isolated and directionally cloned into the similarly
digested pmALAS2XIS-7 construct to produce the targeting vector pmALAS2lCl22ST
(Figure 2.5).
2.4.3 Construction of ALAS2 Targeting Vector Containing the PGKneo'/HSVtk
Selection Cassette
A XhoI-NotI adaptor fragment was inserted into the XhoI site in ALAS2 exon 7 of
pmALAS2ñS-7 to enable release of a NotVSacI fragment for targeting. A 3.5 kb
HincII/Ecll36I fragment containing the PGKneo'/HSVtk selection cassette was isolated from
pKS\eotk and cloned into the SmaVPstI digested vector described above with the introduced
NotI site. Clones containing the PGKneo'/HSVtk fragment in the same orientation as the
direction of the ALAS2 gene were determined by restriction digest analysis. The vector was
referred to as pALAS2)lS-neotk (Figure 2.6).
2.5 METHODS FOR THE IN VITRO DIFFERENTIATION OF A}[ ERYTHROID
CELL LINE
2.5.1 Cell Maintenance
The mouse retroviral erythroid cell line, J2E was maintained in DMEM medium containing
5% FCS. Cells were routinely maintained in75cm2 flasks (Falcon) at37"C in an atmosphere
of 5o/o COz and subcultured every 24 hours to avoid reaching a density greater than
l06cells/ml. To subculture, a 50pl aliquo¡. of J2E cells were diluted into 50pl of trypan blue,
loaded onto a haemocytometer and counted under the microscope to obtain their
concentration. Cells were resuspended in fresh media at 10s cells/ml in 15ml of DMEM
supplemented with 5% FCS.
63

Figure 2.5 Construction of the targeting vector for introduction of the exon 9 Cl228T
point mutation into the murine ALAS2 gene
Details for the cloning procedures are outlined in the text. The solid lines represent murine
ALAS2 fragments and the dotted lines indicate the cloning vector. The boxed numbers
represent ALAS2 exons.
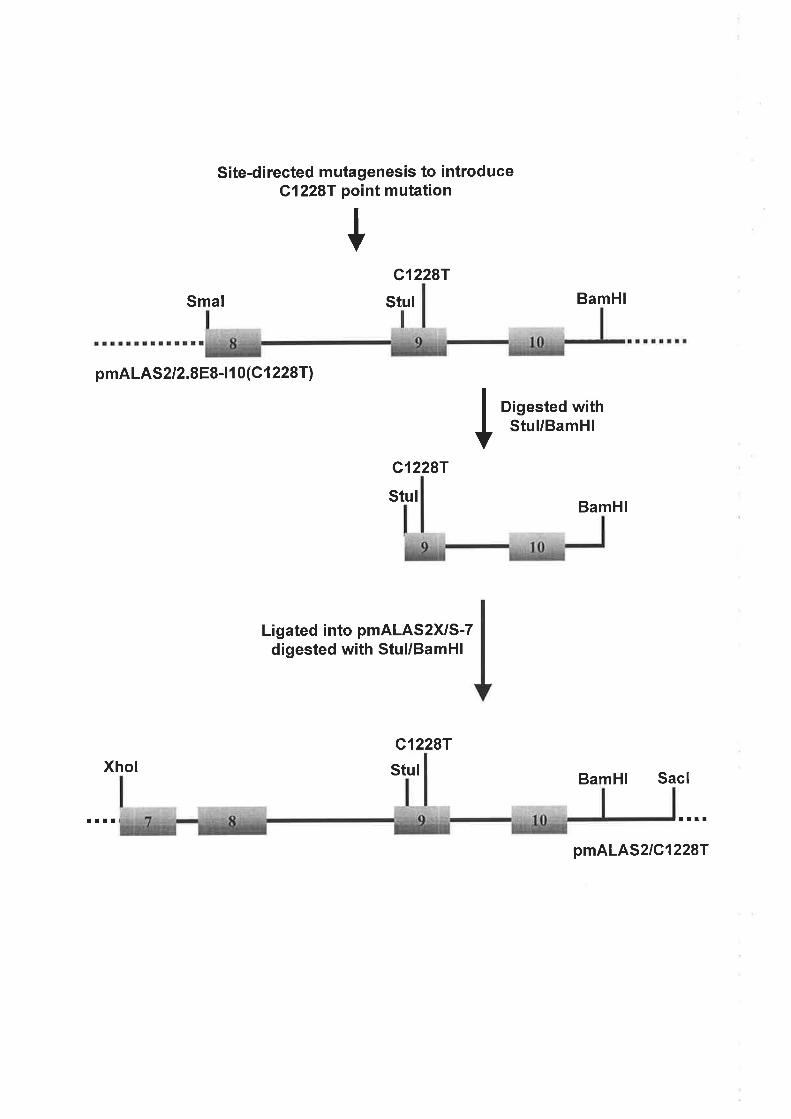
Site-directed mutagenesis to introduceC1228T point mutation
+c12287
StulSmal
p mA LA S 2 I 2.8E8 -11 0 (C 1 228T I
BamHl
Digested withStul/BamHlI
Xhol
c12287
Stul
Ligated into pmALAS2XS-7digested with Stul/BamHl
c12287
Stul
BamHl
BamHl Sacl
rtlrllrl
pmALAS2lC1228T

Figure 2.6 Generation of a targeting construct containing a PGKneo/HSYtk selection
cassette in the murine ALAS2 gene
Details for the cloning procedures are outlined in the text. The solid lines represent murine
ALAS2 fragments and the dotted lines indicate the cloning vector. Theboxed numbers are
representative of ALAS2 exons.
The XhoI-NotI adapter was purchased from Stratagene:
5'-TCGAGGCGGCCGC-3'
3'-CCGCCGGCG-5'
A: Adaptor

Xhol Smal Pstl
I
Digested with Xhol andligated in a Xhol-Notl
adaptor
Sacl
ttlttt!
pmALAS2X/S-7
Sacl
Sacl
pALAS2XS-neotk
¡ltlll
ttlltl
Notl Xhol
Notl Xhol
Smal Pstl
Digested with Smal/Pstl
Smal/Hincll Ecl136l/Pstl
Hincll Ecl136l
A 3.5 kb fragment digestedfrom pKS*Neotk with Hincll
and Ecl136l

2.5.2ln VitroDifferentiation of J2E Cells
í) Erythropoíetín índuced dífferentiation of the J2E cell líne
J2E cells were induced to differentiate with the natural hormone, erythropoietin (Epo)
(Janssen Cilag). The cells were seede d at adensity of 5 x 104 cells/ml in DMEM medium
supplemented with 5% FCS containing 5 units/ml of Epo. J2E cells were treated with Epo
over a 48 hour period and counted as described below.
íí) Støining of cells with benzídíne, a støin for haemoglobín production
A stock solution of benzidine was prepared by dissolving2}mg of benzidine into 10ml of
0.5o/o acetic acid and stored at -20"C. The benzidine stain working solution contained 500p1
of benzidine stock solution, 2¡i of 30% hydrogen peroxide and was made fresh prior to use.
To 50pl of benzidine stain solution, 50pl of cell suspension was added and incubated at room
temperature for approximately 1 minute and the number of blue stained cells counted using a
haemocytometer and Zeiss Axioplan Universal microscope.
2.6 METHODS FOR EXPRESSION OF REPORTER CONSTRUCTS IN TISSUE
CULTURE CELL LINES
2.6.1Transient Transfection of the J2E Cell Line
The J2E cells were gro\Mn to between 2xl0s and 106ce11s/ml and with greater than 85%
viability. The cells were resuspended in DMEM supplemented with 5% FCS at a
concentration of 5xl0acells/ml. 2mls of the cell suspension was aliquoted into the required
number of wells of 6-well trays to result in a total of lOscells/well.
In 200p1 of DMEM, 1.3pmol of DNA of the appropriate luciferase reporter constructs and
50ng of pRL-TK (internal control) was diluted. The volume of the transfection reagent
64

LIPOFECTAMINE 2000 used was 1 .2xthe number of micrograms of DNA transfected and
was diluted in 200¡rl of DMEM. The diluted DNA and LIPOFECTAMINE 2000 were mixed,
incubated for 20 minutes at room temperature and added to the J2E cells plated as described
above. To the appropriate wells, Epo was added to a final concentration of 5 units/ml.
Cells were routinely harvested at 48 hours following transfection and assayed for firefly
luciferase activity. As an internal control for transfection efficiency, reline luciferase activity
was also determined. Transfections were performed in triplicate.
2.6.2Lttciferase Reporter Gene Assay in J2E Cells
Transfected J2E cells were collected and centrifuged at 13000 rpm for 5 minutes tn a
Eppendorf 541 5D centrifuge" The cells were resuspended in 50¡rl of 1 x passive lysis buffer
(Promega), incubated for 15 minutes at room temperature, vortexed for 10 seconds and
centrifuged at 13000 rpm for 1 minute. 1Opl of cell extract was mixed with 25pl each of
Luciferase Assay Reagent II and Stop & Glo buffers and assayed for firefly and renilla
luciferase activity, respectively, using the luminometer (TD20120, Promega).
2.7 METHODS FOR GENE TARGETING EMBRYONIC STEM CELLS
2.7.1 Cell Maintenance
¡) E14TG2ø Embryonic Stem Cells
El4TG2aembryonic stem (ES) cells were maintained in complete ES media and grown at
3l"C in l0o/o COz. Cells were routinely passaged at the appropriate cell density to prevent
contact between established colonies. Cells were groìwn in gelatin (0.I%) coated plasticware
and harvested using trypsin. Briefly, ES cells were washed with PBS and incubated with
trypsin for one minute at37"C before being resuspended in media and pelleted at 1000rpm.
Cells were resuspended in fresh media and generally split 1 :10 and seeded into fresh gelatin
coated plates.
65

iÐ W9.5 Embryonic Stem Cells
W9.5 ES cells were maintained in complete KO-DMEM (as described in Section 2.1.10) and
grown at37"C in l\Yo COz. Cells were routinely passaged at the appropriate cell density to
prevent contact between established colonies. Briefly, ES cells were washed with PBS and
incubated with trypsin for 1 minute aI3J"C before being resuspended in media and pelleted.
Cells were resuspended in fresh media and generally split l:10 and seeded into plasticware
coated with gelatin and containing an irradiated StoR feeder fibroblast cell layer. The StoR
cells were plated approximately four hours prior to passaging of the V/9.5 ES cells.
2.7.2lrradiation of StoR Fibroblast Cells
StoR fibroblast cells were cultured in 175cm2 flasks in complete ES media. At 100%
confluency, cells were harvested by treatment with PBS and trypsin, centrifuged at 1200 rpm
for five minutes and resuspended in 50m1. The StoR cells were treated in a gamma irradiator
(LBL437C, Clsbiointernational) and then centrifuged at 1200 rpm for five minutes and
resuspended at 1xl07 cells/ml in equal amount of complete ES media and freeze mix (80%
FCS and 20% DMSO). Cells (2x107 cells) were aliquoted into freezing vials and stored in
liquid nitrogen.
2.7.3 Stable Transfection of ES cells with Targeting Vector DNA via Electroporation.
¡) E14TG2a ES Cells
Targeting vector DNA (150pg) was digested ovemight with XhoVSacI at 37"C and ethanol
precipitated. Following three washes in70%o ethanol the DNA was resuspended in sterile PBS
to a concentration of lpg/pl. ES cells were harvested by treatment with EGTA and trypsin,
centrifuged aI 1200 rpm for five minutes and resuspended in 1Oml of complete ES media. A
cell count was performed and cells were then pelleted by centrifugation at 1200 rpm for hve
minutes and resuspended in PBS at a concentration of 108 cells/650p1. To the cell suspension
of 650p1in a cuvette (Bio-Rad, 4mm disposable electroporation cuvette) the linearised
66

targeting vector DNA (150p1) was added and immediately electroporated by a single 800 volt
pulse at 3pF (Bio-Rad Genepulser).
ES cells transfected with the targeting vector were then mixed with 1Oml complete ES media
and 10 x I 50cm2 gelatin coated flasks (Falcon) were each seeded with 107 cells in complete
ES media. After three days each flask was passaged 1:15. Five days after transfection each
flask of cells was individually harvested as described above and three 1Ocm plates per flask
were seeded with 106 cells. The following day, 6-TG (20pM) selection was administered and
continued up to day seventeen post-transfection. The media was changed as required.
ES cells were also transfected with RSV-p-gal via electroporation to determine transfection
efhciency. Cells (5x106¡ *ere seeded into a 10cm dish and maintained in complete ES media
for two days prior to undergoing staining for B-galactosidase activity.
ií) ll/9.5 ES Cells
Targeting vector DNA (25ptg) for the first and second rounds of targeting in V/9.5 ES cells
was digested overnight with either NotVSacI or XhoVSacI, respectively, at 37"C and ethanol
precipitated. Following three washes in70o/o ethanol the DNA was resuspended in sterile PBS
to a concentration of O.5pg/pl. Prior to transfection, the media (KO-DMEM) on the ES cells
to be transfected was changed. Two hours later, ES cells were rinsed once in PBS followed by
a rinse in PBS + 0.5mM EGTA. For harvesting, cells were incubated with trypsin for two
minutes at 37"C and centrifuged at 1000 rpm for four minutes and resuspended in 1Oml of
KO-DMEM media. A cell count was performed and cells were then pelleted by centrifugation
at 1000 rpm for four minutes and resuspended in PBS at a concentration of 4x107 cells/650p1.
To the cell suspension of 650p1 in a pre-chilled cuvette (Bio-Rad, 4mm disposable
electroporation cuvette) the linearised targeting vector DNA (50p1) was added and incubated
at 4"C for five minutes. Cells were then electroporated by a single 220 volt pulse at 500pF
(Bio-Rad Genepulser).
ES cells transfected with the targeting vector were then mixed with 10ml KO-DMEM media
and 10 x 10cm gelatin (0.1%) coated plates (Falcon) containing 5x106 irradiated StoR feeder
cells were each seeded with approximately 4x106 cells in KO-DMEM media. After three days
each plate was passaged 1:8 as described above and a total of thirty 1Ocm plates (three for
67

each plate) were seeded. The following day, media containing G418 at 250pglml was
administered to the transfected cells and maintained up to day twelve post-transfection in the
first round of targeting. In the second round, media with 0.2pM of fialuridine was added five
days after transfection and continued up to day ten post-transfection. The media was changed
as required.
2.7.4Picking of Selection Resistant Colonies
Plates containing selection resistant colonies were rinsed in PBS. A few drops of tr5lpsin were
placed onto a single colony and left for approximately 20 seconds. The colony was then
aspirated from the dish using a automated pipette tip and transferred into the well of a 24 well
tray coated in gelatin and containing lml of complete ES media supplemented with the
selection reagent. The media was then pipetted up and down to disperse the colony. Half of
the cell suspension was transferred to the corresponding well in another tray 24 well tray. This
tray was grown for several days prior to being frozen down. ES cells in the original tray were
grown to confluency. Once confluent, genomic DNA was obtained from each of the clones as
described in Section 2.8.1 and screened for successful homologous recombination via
Southern blot analysis (Section 2.8.2).
For colonies obtained in targeting of the V/9.5 ES cells the picking procedure differed
slightly. The 24 well tray for freezing the picked clones was coated in gelatin and seeded with
1.5x10s irradiated StoR feeder cells. In addition, KO-DMEM media with the appropriate
selection reagent was used.
2.7.5 Freezing of Targeted Clones
Cells were washed in PBS and 100p1of trypsin was added and left for one minute at37"C.
after the addition of complete ES media (300p1) the cells were pipetted gently to disperse the
colonies. An equal volume (a00pl) of freezing mix(20o/o DMSO, 80% FCS) was added and
plates were stored at -80'C.
68

2.7.6 Histochemical Staining for p-Galactosidase Activity
B-galactosidase expression in transfected ES cells was detected by the following procedure.
Cells were rinsed three times in PBS and fixed for five minutes with}.2Yo glutaraldehyde.
After an additional three rinses in PBS the cells were incubated ovemight at 37'C in p-
galactosidase stain solution (0.45mM K3Fe(CN)6, 0.45mM IQFe(CN)6, 1mM MgClz)
containing X-gal (a0Opg/ml), Cells were viewed under a microscope.
2.7.7 Karyotyping of ALAS2 Targeted ES Cell Lines
Targeted ES cells to be introduced into mouse blastocysts were karyotyped according to the
method of Robertson (1937). Targeted ES cells (106) were seeded into a 60mm dish and
incubated overnight at37oC. The following day, cells were rinsed with PBS and fresh media
added to induce rapid growth. After three hours, 5ml of media containing 0.lpg/ml of
colcemid (Sigma) was added to the cells and incubated for one hour at 37oC to arrest cells in
the mitosis stage. Cells were then treated with trypsin and centrifuged in colcemid medium
for five minutes at 1100 rpm. The pelleted cells were resuspended in 9ml of 0.075M KCI and
incubated for 30 minutes at room temperature. A cold methanol:acetic acid (3:1) fixative
(1ml) was added to the cells and they were centrifuged as above. Cells were washed three
times in 1Oml of the fixative and at the end of the final wash, resuspended in 200p1 of the
fixative. The resuspended cells were dropped onto methanol treated slides from a height of
30cm to 50cm at a 45" angle. Slides were left to air dry at room temperature and then stained
with Giemsa for five minutes, rinsed in water and blotted d.y. A cover slip was mounted over
the cells and the karyotlpe analysed with a Zeiss Axioplan Universal microscope.
2.8 SOUTHERN BLOT ANALYSIS AND HYBRIDISATION CONDITIONS
2.8.1 Isolation of Genomic DNA from ES cells
Cells were washed in PBS and incubated overnight at37"C,I}Yo COz in ES cell lysis buffer
(Section 2.1.10(iÐ). The followingday,300pl of isopropanol was added and swirled to
precipitate out the DNA. The DNA was removed into 70Yo ethanol, centrifuged for 2 minutes
69

and supernatant removed. The DNA was resuspended overnight at37"C in 100p1 of TE and
stored at 4"C to be analysed by Southern hybridisation.
2.8.2 Southern Blot Analysis
Genomic DNA (50p1) was digested with 40 units of the appropriate enzpe at37"C
ovemight in a final volume of 150u1. The DNA was ethanol precipitated and resuspended in
14pl lxTE to which 7¡il3x urea loading buffer was added. The digested DNA was then
electrophoresed on 0.8Yo agarose gel in IxTBE at 40 volts. Following staining with ethidium
bromide, the gels were visualised under UV light and photographed. The gel was denatured
and neutralised prior to capillary transfer of the DNA to Nytran filters (Schleicher & Schuell),
performed as described in the manufacturer's protocol, except that it was carried out in 20x
SSPE. After transfer, the DNA was covalently cross-linked to the filter (120mJ, Stratagene
Inc., UV StratalinkerrM 1800). The filters were prehybridised for approximately 4 hours at
65"C in 250mM NaPOa(pH7.2), 7yo SDS, 10% PEG 6000, lmM EDTA and 0.1 pglml
sonicated salmon spenn DNA in roller bottles. Hybridisations were performed for 24 hours
under the same conditions, except for the addition of radiolabelled probe (Section 2.2.9 (1)).
Following hybridisation, the filter was washed twice in 2x SSPEl0.l% SDS at 65"C for 10
minutes and once in 0.5x SSPE/0.l% SDS at 65'C for 30 minutes. The filter was then dried,
sealed in a plastic bagand exposed to Storage Phosphor Screens (Molecular Dynamics) and
processed using a Molecular Dynamics Phosphorlmager.
2.8.3 Stripping of the Filter
The filter was stripped byboiling it in 1 litre of 0.01x SSPE, 0.1% SDS for approximately 15
minutes. The filter was then rinsed in 2x SSPE before prehybridisation.
2.9 METHODS FOR ELECTROPHORETIC MOBILITY SHIFT ASSAYS
70

2.9.lPreparation of Nuclear Protein Extracts
For preparation of nuclear extracts from Epo induced and non-induced J2E cells, a rapid
procedure described by Andrews and Faller (1991) was employed. This method was used to
generate nuclear extracts for the detection of GATA and CACCC binding proteins.
Nuclear extracts prepared from either COS-I cells transfected with an Spl expression clone
or CV-l cells expressing an EKLF clone were obtained from our laboratory stocks and
prepared as detailed in Surinya et al. (1997).
2.9.2 Preparation of Radiolabelled Annealed Oligonucleotide Probes
Oligonucleotide probes were radiolabelled with ¡y-32t1-aeTP using T4 polynucleotide kinase
as described in Section2.2.9 (i).
2.9.3 Electrophoretic Mobility Shift Assay
Binding reactions used in the detection of GATA binding proteins contained 10¡rg of nuclear
protein, 2pg poly(dl-dC) in 16 ¡l of 25mM Hepes,pH7.9,60mM KCL, 7 .5Yo glycerol,
0.1mM EDTA, 5mM MgC12, 0.75mM dithiothreitol and 2ml|l4 spermidine. The reaction was
incubated on ice for 10 minutes and2x70s cpm of radiolabelled probe was added to the
reaction and incubated on ice for an additional 30 minutes. In supershift assays,0.2trtg of the
GATA-I specific monoclonal antibody, (sc-265X Santa Cruz Biotechnology) was incubated
in the binding reaction prior to the addition of the probe. Retarded nuclear protein complexes
were resolved on a pre-electrophoresed 8olo non-denaturing polyacrylamide gel in 0.25xTBE
at 180 volts for 3 hours at 4"C.
For the detection of CACCC binding proteins the reaction contained 1Opg of nuclear extracts
from J2E cells and was performed as described by Crossley et al. (1996). For the supershift
assays 0.2ttg of monoclonal Spl antibody (sc-420X, Santa Cruz Biotechnology) or 2pl of
polyclonal EKLF antibody (provided by Dr M. Crossley) was used.
Following electrophoresis, the gels were transferred to'Whatman 3MM paper) dried and
exposed to X-ray film.
7l

CHAPTER 3
TRANSCRIPTIONAL REGULATION OF THE HUMAN ALAS2 GENE IN
RESPONSE TO ERYTHROPOIETIN INDUCED DIFFERENTIATION OF
ERYTHROID CELLS

CHAPTER 3: TRANSCRIPTIONAL REGULATION OF THE HUMAN ALAS2 GENE
IN RESPONSE TO ERYTHROPOIETIN INDUCED DIFFERENTIATION OF
ERYTHROID CELLS ilL
w
3.1 INTRODUCTION
Substantial expression of the ALAS2 gene is triggered by the differentiation of immature
erythroid precursors into circulating red blood cells by the hormone Epo. The resultant
increase in ALAS2 activity is critical for the production of haem for the formation of
haemoglobin, the crucial oxygen transport molecule in circulating erythroid cells (Hangae et
al., 1998;Yin and Dailey, 1998;Nakajima et a1.,1999). Consequently, the mechanisms
underlying the transcriptional regulation of ALAS2 expression during erythropoiesis, the
process of red blood cell differentiation, are of interest and the focus of this chapter.
The first study to identifu potential regulatory regions of ALAS2 involved DNase I
hypersensitivitymapping studies on the murine ALAS2 gene in undifferentiated MEL cells
(Schoenhaut and Curtis, 1989). Numerous studies have reported an association between
DNase I hypersensitivity sites and nucleosome-free DNA bound by transcription factors, the
latter being indicative of regulatory regions within the gene (Elgin, 1988; Gross and Garrard,
1988; Wei et aL.,2002). Five DNase I hypersensitivity sites located in the immediate
promoter, intron 1, intron 3 and the 3' end of intron 8 were identified by Schoenhaut and
Curtis (1989), indicating a number of potential regulatory regions in ALAS2. Sequence
analysis of the corresponding regions in the human ALAS2 gene identified in the immediate
promoter several potential binding sites for erythroid-specific regulatory proteins and non-
erythroid transcription factors (Figure 3.1a) (Surinya et al., 1997). Multiple putative
erythroid-specific binding sites have been identified includingTT consensus GATA sites and
6 CACCC-like elements in intron 1, two putative CACCC and one GATA site in intron 3 and
4 GATA consensus sites and 2 CACCC elements in the 3' end of intron 8 (Figure 3.lb)
(Surinya et a1.,1998). Within this 3'region of intron 8 two of each element (CACCC-A and
B, GATA-A and B) are conserved in the human, mouse and dog (Figure 1.8). The CACCC
boxes in human ALAS2 intron 8 in Figure 3.lb correspond to the consensus EKLF binding
site (Miller and Bieker, 1993) and also to the binding sites of other transcription factors like
Spl (Kadoîaga et al., 1987) and BKLF (Crossley et al., 1996). The GATA sites are
representative of consensus GATA-I binding elements (Evans et al., 1988; Tsai et al., 1989;
Ko and Engel, 1993; Merika and Orkin,,1993) (Table 1.1).
I¿
72

Figure 3.1 Location of transcription factor binding sites within the human ALAS2
promoter and intron 8
(a) Sequence of the first 156 bp of human ALAS2 promoter is represented with erythroid and
non-erythroid putative cis-acting elements denoted by coloured boxes. The legend is
outlined below. The transcriptional start site is represented as *1.
(b) The 3' end of human ALAS2 intron 8 is shown containing a cluster of erythroid-specific
transcription factor binding sites. The CACCC elements and GATA sites are represented
by coloured boxes as detailed in the legend below. The cores sequences have been
outlined in the diagram. The transcription factor binding sites were located at the
following positions in human ALAS2 intron 8: CACCC-A (+2681+275), CACCC-B
(+327t+334), GATA-A (+344t+349), GATA-B (+380/+385), GATA-C (+398/+403) and
GATA-D (+5871+592).
Legend
GATA NF-E2
CACCC TATA-like
Ets Thyroid response element
CCAAT
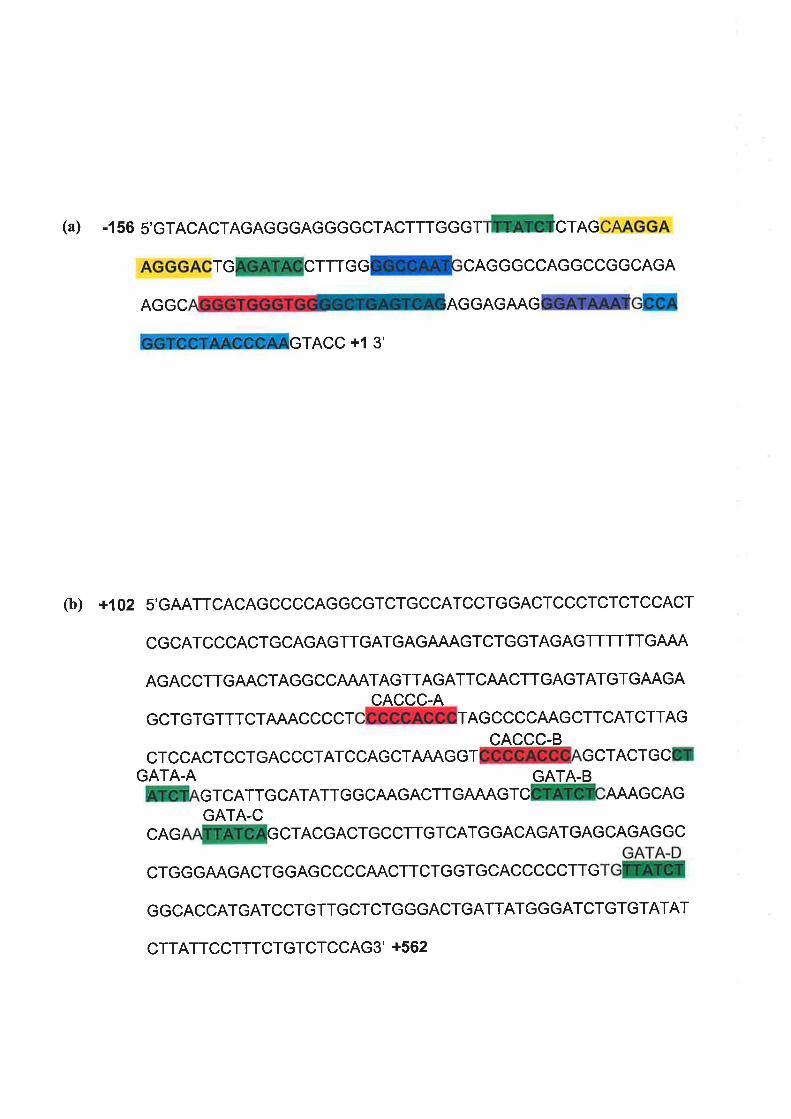
(a) .156 s'GTACACTAGAGGGAGGGGCTACTTTGGGT CTAG
TG CTTTGG CAGGGCCAGGCCGGCAGA
AGGC AGGAGAAG
GTACC +1 3'
(b) +102 5'GAATTCACAGCCCCAGGCGTCTGCCATCCTGGACTCCCTCTCTCCACT
CG CATC CCACTG CAGAGTTGATGAG MAGTCTG GTAGAGTTTTTTG AAA
AGACCTTGAACTAG G CCAAATAGTTAGATTCAACTTGAGTATGTGMGAcAccc-A
TTGCTGTGTTTCTAAACCCC AGCCCCAAGCTTCATCTTAG
CTCCACTC CTGAC CCTATCCAG CTAAAGGGATA-A
cAccc-BT GCTACTGC
GTCATTG CATATTG G CAAGACTTG AAAGGATA-C
CAG GCTACGACTGCCTTGTCATGGACAGATGAGCAGAGGC
GATA.BTC AAAGCAG
CTGG GAAGACTG GAGCCCCAACTTCTG GTGCACCCCCTTG
G G CAC CATGATCCTGTTG CTCTG G GACTGATTATG G GATCTGTGTATAT
CTTATTCCTTTCTGTCTCCAG3' +562

Transient transfection studies in undifferentiated erythroid cells were previously undertaken
to assess the ability of the human ALAS2 promoter, intron 1, intron 3 and intron 8 to function
as regulators of ALAS2 transciptional activity (Surinya et al., 7997, 1998). Briefly, it was
determined that the initial 293bp of the human ALAS2 promoter was the minimum length
required for maximal reporter activity in erythroid cells and the inclusion of either intron 1 or
intron 8 further increased activity levels, with intron 8 having the highest effect on ALAS2
promoter activity (Surinya et al., 1997,1998). Since the 3' end of intron 8 is conserved among
several species and was also found to be a strong enhancer of transcriptional activity when
compared to other regions of ALAS2, its function in gene regulation was studied further.
Transcription factor binding sites located within intron 8 were investigated for their
functionality and binding capabilities bymutational studies and EMSA. It was observed that
two of the GATA sites, designated A and B (see Figure 3.lb), bound GATA-1 invitrobut
only GATA-B proved to be functional in enhancing reporter activity in undifferentiated
erythroid cell types. Promoter activity inK562 and MEL cells was reduced in the absence of
either CACCC site, which in vitro were bound by the ubiquitous transcription factor Spl but
not the erythroid-specific factors EKLF or BKLF. This finding was supported by
transactivation studies inK562 cells in which EKLF was unable to transactivate intron 8
reporter constructs, suggesting the function of the CACCC sites in intron 8 is due to Spl and
not EKLF (Surinya et al., 1998).
The study by Surinya et al. (1998) established that intron 8 encompasses an erythroid-specific
enhancer element and identified sites within this element which play arole in the
transcriptional regulation of the human ALAS2 gene in erythroid cell lines. The studies , U., .' 't'' '
examining the transcriptional regulation of ALAS2 were conducted in undifferent iarcdf{62
and MEL erythroid cells (Surinya et al., 1997, 1998). These cell lines could only be
differentiated by chemical inducers such as DMSO but not by Epo, the natural stimulus of
erythropoiesis in the body. Whether these same elements and transactivating factors are
critical for the activation of the ALAS2 gene during erythroid maturation was not
investigated. Therefore, the aim of the current study was to investigate the transcriptional
regulation of ALAS2 during erythroôyte maturation using an erythroid cell line, J2E, which"
-!-"' '1'-^terminally differentiates in response to Epo (Klinken et al., 1988).
J2E cells are a conìmitted erythroid precursor cell line that was generated by infecting murine
foetal liver cells with the J2 retrovirus (Klinken et al., 1988). J2E cells are immortalised at the
73

proerythoblast stage of erythropoiesis (Klinken et al.,1993) but following exposure to Epo
undergo terminal erythroid maturation exhibited by increased cell proliferation, expression of
erythroid-specifi c genes, enhanced haemoglobin synthesis, morphological changes and
enhanced viability (Klinken et al., 7988; Busfield and Klinken,7992; Busfield et al., 1993;
Tilbrook et a|.,1996). A significant proportion of J2E cells enucleate in response to Epo,
which represents one of the final stages of the erythroid maturation process (Busfield and
Klinken, 1992).Increased activation of several erythroid-specific genes associated with the
haemoglobin producing stage of erythroid differentiation is observed in J2E cells stimulated
by Epo. Specifically, levels of adult cr and B-globin transcripts are elevated after treatment of
the J2E cells with Epo (Klinken et al., 1988; Busfield and Klinken, 1992; Spadaccini et al.,
1998) and also enzymes of the haem biosynthetic pathway, including ALAS2 (Busfield et al.,
1993; Klinken et al., 1993; Busfield et al., 1995a). A rise in haemoglobin levels, as a result of
increased haem and globin synthesis is observed upon Epo induced differentiation of J2E
cells and can be detected 24 hours post-Epo exposure (Busfield and Klinken,1992; Busfield
et al., 1995b).
Two erythroid-specihc transcription factors, GATA-1 and EKLF have been implicated in the
activation of these genes during Epo initiatedJ2E differentiation. For example, GATA-I
mRNA and protein levels rise significantly (2 to 3-fold) during maturation of the J2E cells,
with protein levels decreasing after 48 hours of Epo stimulation (Busfield et al., I995b). In
addition, studies using altemate erythroid cell lines found that an increase in GATA-I
activation upon Epo stimulation preceded the rise in globin levels (Whitelaw et al., 1990;
Chiba et al., 199I; Leonard et al., 1993; Dalyot et al., 1993). Interestingly, although EKLF
mRNA levels were not altered and protein levels were reduced upon Epo inducedJ2E
differentiation, expression of anti-sense EKLF inJ2E cells inhibited globin, ALAS2 and
ferrochelatase mRNA levels as well as haemoglobin production in response to Epo
(Spadaccini et aL.,199S). This suggests that EKLF is an important regulator of ALAS2 and
globin expression during Epo stimulated maturation of J2E erythroid cells. Thus, the Epo
responsive J2E cell line provides a model in which the molecular aspects associated with
erythroid di fferenti ation can b e investi gated.
The following chapter will focus on the effect that Epo induced differentiation of J2E
erythroid cells has on the ability of the human ALAS2 promoter, intron 1 and in particular
intron 8 to behave as regulatory regions in the transcriptional activation of ALAS2.
74

3.2 RESULTS
3.2.1 Epo Induced Differentiation of J2E Erythroid Cells
As a measure of erythroid differentiation, haemoglobin levels were monitored in Epo treated
J2E cells bybenzidine staining (Cooper et al., 1974) (Section 2.5.2). The number of
haemoglobin positive cells were counted and the level of differentiation expressed as a
percentage of positive cells over viable cells. Figure 3.2 represents induction levels typically
observed with Epo treated J2E cells and demonstrates that haemoglobin levels increase over a
period of Epo stimulation. After 48 hours of Epo stimulation,49yo+2 of the J2E cells stained
positive for haemoglobin. Since benzidine-positive cells are indicative of differentiation, it
can be said that at 48 hours approximately 50Yo of the J2E cells have undergone erythroid
differentiation. The level of differentiation observed is typical of J2E cells induced to
differentiate with Epo (Dr P. Tilbrook, University of 'Western Australia, personal
communication). Differentiation experiments were periodically conducted to ensure the J2E
cells maintained their ability to undergo Epo initiated erythroid differentiation.
3.2.2The Minimal Length of the Human ALAS2 Promoter Required for a Response to
Epo is 293 Base Pairs
Initial experiments were performed to ascertain if elements within the 5'flanking region of the
human ALAS2 gene were responsive to Epo induced differentiation. The ability of various
lengths of the human ALAS2 promoter to respond to Epo was investigated by transient
transfection analysis in differentiated J2E erythroid cells. The human ALAS2 promoter
deletion reporter gene constructs utilised in this study were constructed by Dr. K. Surinya in
our laboratory and are illustrated in Figure3.3a (see Section2.3.l).
The different lengths of the ALAS2 promoter expression plasmids were transiently
transfected (Section 2.6.1) into J2E erythroid cells. As a negative control the promoter-less
parental vector, pGl2-basic was also transfected (data not shown). To normalise transfection
efficiency, an alternative luciferase gene (from the sea pansy, Renilla reniformis) under the
control of the thymidine kinase promoter (pRL-TK) was co-transfected with each ALAS2
promoter reporter gene construct. 'Where
appropriate cells were treated with Epo to induce
75

*"1,l-f)Figure 3.2 Epo stimulated differentiation of J2E erythroid cells
J2E cells were seede d at aconcentration of 5x104 cells/ml in medium supplemented
wlth5%o FCS and treated with 5 units/mlof Epo (Eprex, J Cilag). Treated J2E cells
were incubated at 37"C, 5o/o COz and assayed at 24 and 48 hours to determine the percentage
of haemoglobin positive cells (Section 2.5.2). Haemoglobin synthesising cells is represented
as the number pf benzidine-positive cells/viable cells. The data on the graph represents the
average values of two independent differentiation experiments.
t\
P?t,r.À nf '
t'f_
^t,') f'f

% H
aem
og
lobi
n-po
sitiv
e G
ells
rtu(
rA(¡
lttoo
oooo
o
I o - tñ
o N .À ¡L ø

Figure 3.3 Deletion analysis of the human ALAS2 promoter in J2E erythroid cells
induced to differentiate with Epo
(a) ALAS2 reporter constructs consisted of fragments of the human ALAS2 promoter,
ranging from 10.3 kb to 124 bp which were fused to firefly luciferase reporter gene
(LUC). All constructs contained 28 bp of ALAS2 5'-UTR. The transcriptional start site is
indicated by +1 and is denoted by the affow.
(b) 1.3pmol of each human ALAS2 promoter deletion construct was transfected into J2E cells
and immediately treated with 5units/ml of Epo. After 48 hours, cells were harvested and
1Opl of whole cell extract was assayed for luciferase activity. Activity was normalised
against expression of a co-transfected Renilla luciferase reporter gene construct þRL-TK)
(50pg). Each transfection was performed in triplicate and the average of the values is
represented on the graph as relative luciferase activity (RLA). Each experiment was
repeated at least three times and a representative experiment graphed.
(c) Luciferase expression post-Epo stimulation for each promoter length is expressed as the
fold increase in activity.
,')
fl.

(a)
i"pALAS2-10.3LUC +28
-10.3 kb
+
pALAS2-5.7LUC
pALAS2-1.9LUC
pALAS2-0.293LUC
pALAS2-O.L24LUC
(b)
JÉ,
-5.7 kb
-1.9 kb
5.7 1.9 0.293 0.124
promoter length (kb)
r
+
0.70
0.60
0,50
0.40
0.30
0.20
0.10
0.00
-l24bP +28
10
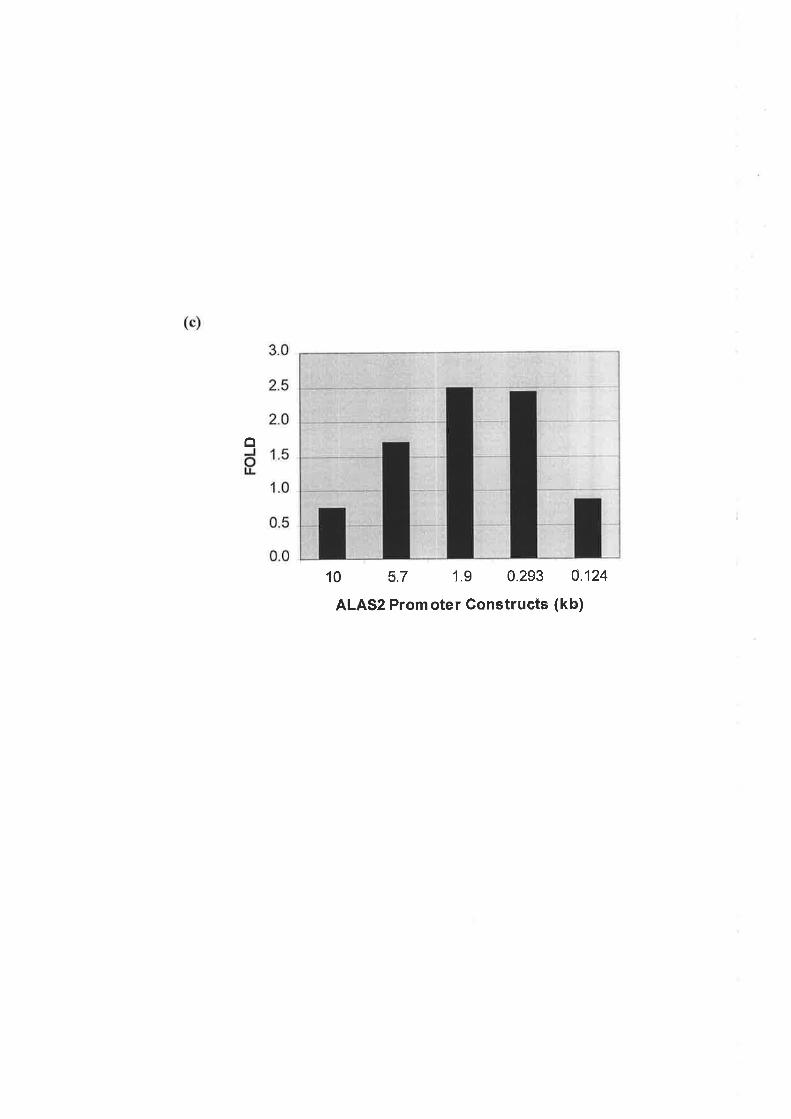
o
10 5.7 1.9 0.293 4124
ALAS2 Promoter Construets (kb)

erythroid differentiation, immediately following addition of the DNA and transfection
reagent.
In undifferentiated J2E erythroid cells, the proximal293 bp of the human ALAS2 promoter
generated the highest level of activity (Figure 3.3b). This was previously observed in non-
erythroid COS-I and undifferentiated K562 and MEL erythroid cells (Surinya et al., 1997).In
Epo treated J2E cells the maximal level of luciferase expression was also obtained with the
reporter construct containing 293bp of the human ALAS2 promoter (Figure 3.3b). In Epo
induced J2E cells the maximal increase (2.5 and2.4-fold) in luciferase activity was obtained
with 1.9 kb and 293 bp human ALAS2 promoter constructs, respectively (Figure 3.3c). This
demonstrated that the first 293 bp of the human ALAS2 promoter is sufficient to respond to
Epo signalling in J2E erythroid cells and this region contained all the Epo responsive
elements at least within the first 10 kb of ALAS2 5'flanking sequence. Thus, the initial293 ,/bp of human ALAS2 promoter will be referred to as the proximal ALAS2 promoter and
sequence 5'to this region as the distal promoter.
Interestingly, activity from the reporter construct containing 10.3 kb of ALAS2 distal
promoter sequence in both non-induced and Epo induced J2E cells was lower in comparison
to the shorter 5.7 kb, 1.9 kb and293 bp ALAS2 5' flanking deletions and in fact approximates
the activity obtained from the construct containing the initial l24bp of the human ALAS2
promoter sequence (Figure 3.3b). The construct containing 1.9 kb of ALAS2 distal promoter
was more active than the 10.3 kb of ALAS2 5' flanking sequence inJ2E cells induced to
differentiate with Epo but not in non-induced cells. Furthermore, a more critical observation
is the lack of response by the 10.3 kb ALAS2 distal promoter construct to Epo, suggesting
upstream elements may inhibit the promoter's response to Epo (Figure 3.3c). This trend was
observed in repeated experiments and with different plasmid preparations of the 10.3 kb
ALAS2 5'-UTR reporter construct. The inhibition generated may be due to the presence of
negative transcriptional control elements in the distal 5'-UTR of the human ALAS2 promoter
but the presence of such regions was not pursued during the course of this study.
l6

3.2.3Localisation of Epo Responsive Enhancer Elements within the Human ALAS2
Gene
The current study has shown that the maximum increase in activity by the 293 bp human
ALAS2 promoter in response to Epo induced differentiation of J2E cells was approximately
2.5-fold (Figure 3.3c). Since DNase I hypersensitivity sites have been identified in introns 1, 3
and 8 of the murine ALAS2 gene (Schoenhaut and Curtis 1989) and a previous study has
demonstrated that introns I and 8 of the human ALAS2 gene are able to enhance
transcriptional activity from the 293 bp ALAS2 promoter in immature erythroid cells (Surinya
et a1.,1998), it was of interest to determine if introns 1 and 8 could enhance transcriptional
activity further in response to Epo stimulated differentiation of J2E cells. Intron 3 was not
investigated as this domain actually inhibited transcriptional activity from the ALAS2
promoter (Surinya et a1.,1998). The human ALAS2 intron 8 sequence integrated into the
ALAS2 promoter luciferase reporter construct was a 460 bp fragment (+1021562) located at
the 3'end of the intron. This fragment contains two CACCC boxes (CACCC-A and B) and
four GATA sites (GATA-A, B, C and D sites) (Figure 3.1b).
To determine whether the previously identified intronic enhancer elements play a role in any
response of ALAS2 to Epo induced J2E differentiation, luciferase reporter constructs
containing either intron 1 (pI1+LUC), intron 8 (pI8(a60)+LUC) or both introns 1 and 8
(pI1+IS(460)+LUC) under the control of the human ALAS2 293bp promoter were transiently
transfected into J2E erythroid cells. Intron I containing constructs consisted of the entire
intron 1 sequence (4.9 kb) driven by 293 bp of the ALAS2 promoter, cloned in its native
orientation. Intron 8 sequence was incorporated downstream of the luciferase gene and the
ALAS2 promoter, also in its native 5' to 3' orientation (Figure 3.4a). Section 2.3.2 details the
construction of the ALAS2 intronic reporter gene constructs.
In untreated JzF. cells, intron 1 (0.310.08) did not increase activity beyond that observed
solely with the minimal ALAS2 promoter (0.3+0.06) (Figure 3.4b). However, the presence of
intron 8 (1.6+0.16) increased basal activity approximately 5-fold compared to the promoter
alone, suggesting a role for intron 8 as a transcriptional enhancer (Figure 3.4b).In the
presence of intron 1 and intron 8, basal luciferase activity driven by the ALAS2 promoter was
increased (0.3+0.06 compared to 1.1+0.7) but not to a level greater than activity generated by
intron 8 alone (Figure 3.4b). Thus, the presence of introns 1 and 8 did not result in a
slmergistic increase in reporter gene activity in undifferentiated J2E cells.
77

Figure 3.4 Localisation of an Epo-responsive element in the human ALAS2 gene
(a) ALAS2 promoter constructs containing various combinations of the intronic enhancer
regions were cloned into the luciferase reporter (LUC) vector to simulate the a:rangement of
the introns in the ALAS2 gene. The luciferase reporter constructs were under the control of
the minimal2g3 bp human ALAS2 promoter. Both intron I and intron 8 were cloned in their
5' to 3' native orientation (for details see Section 2.3.2). The arrow indicates the start site for
transcription.
(b) Each construct (1.3pmol) was transfected into J2E cells that were either treated with
5units/ml of Epo or left untreated. Cells were harvested after 48 hours and extracts assayed
for luciferase expression. Luciferase activity was nofinalised against the intemal Renilla
luciferase control (50pg) and expressed as relative luciferase activity (RLA). Each
transfection was conducted in triplicate and the average value graphed. The graph represents
one experiment that was repeated at least three times.

(a)
(b)
p293+LUC
pIl+LUC
pI8(460)+LUC
pI1+I8(460)+LUC
INTRON I
INTRON 1
p293 11
fú
30.0
25.0
20.0
15.0tr EPO-
r EPO+
10.0
5.0
0.0
18 11+18

As expected upon differentiation of the J2E cells with Epo, the ALAS2 promoter increased
luciferase activity approximately 3-fold relative to the levels observed in undifferentiated J2E
cells (1.1+0.2 compared to 0.3+0.06). The inclusion of intron 1, intron 8 or both introns
further enhanced luciferase activity generated by the ALAS2 promoter in Epo stimulated J2E
cells (Figure 3.4b). Intron 1 and intron 8 increased luciferase activity in Epo treated J2E cells
g-fold (2.5+0.7 compared to 0.3+0.08) and 13-fold (21+3 compared to 1.6+0.16) respectively,
compared to undifferentiated cells (Figure 3.4b). Transient transfection of pI1+I8(460)+LUC
increased luciferase activity 17-fold (19.1+6 compared to 1.1+0.7) in the presence of Epo
when compared to levels generated by this combination of introns in undifferentiated J2E
cells (Figure 3.4b). The inclusion of both intron 1 and intron 8 sequences resulted in a greater
Epo response than the construct containing solely intron 8. It is unclear if the activity of both
intronic regions increases the transcriptional response to Epo or is only a result of differences
between the basal activity levels of constructs containing either intron 8 or introns I and 8.
Interestingly, the level of luciferase activity generated by intron 8 was not enhanced upon the
inclusion of intron I in differentiated J2E cells.
In conclusion, Epo responsive elements were located in the promoter, intro\1..11$.,.,igtron 8 of
the ALAS2 gene. However, the 3'end of intron 8 was shown to exhibit the stronþest Epo
responsive enhancer activity in the regulation of ALAS2 transcription d.t.inffi'ài,iu,"¿
differentiation of the J2E cells. The two CACCC (CACCC-A and B) and two GATA (GATA-
A and B) transcription factor binding sites located within a239 bp conserved region at the 3'
end of intron 8 (+159/+393) were investigated further to determine their contribution to intron
8 in the regulation of ALAS2 transcription during erythroid differentiation. Intron 8 sequence
including the GATA-C and GATA-D sites was not analysed as these sites are not conserved
between species and mutagenesis studies showed they do not affect transcriptional activity of
the human ALAS2 promoter when transiently transfected into an erythroid cell line (Surinya
et aL.,1998). Furthermore, the 239bp region of intron 8 encompassing the two conserved
GATA and CACCC sites responds to Epo induced differentiation, enhancing transcriptional
activity to a level that is comparable to intron 8 sequence including the additional two GATA
sites (data not shown). Thus, the transcription factor binding sites within this region of intron
8 will be investigated for their contribution to intron 8 transcriptional enhancer activity in
response to Epo induced differentiation.
78

3.2.4 Determination of Transcription Factor Binding Sites \üithin Intron 8 that are
Critical to Epo Enhanced Transcription of the Human ALAS2 Promoter
Firstly, this study investigated the contribution of the ALAS2 intron 8 GATA sites and
CACCC boxes to basal expression, prior to analysis in Epo treated J2E erythroid cells. To
investigate the functionality of the 239bp intron 8 sequence these putative transcription factor
binding sites were mutated individually or in combination and the ability of these mutant
enhancer sequences to increase basal expression of the 293 bp human ALAS2 promoter was
tested in transiently transfected undifferentiated J2E cells. Activity values from the mutant
intron 8 constructs were expressed as a percentage of activity obtained from wild type intron
8. Section 2.2.3 detatls the construction of human ALAS2 intron 8 mutant reporter gene
constructs.
Transient transfection of either the pISoA+LUC or pIScB+LUC constructs resulted in
luciferase activities of 67Yo and 55o/o compared to wild type intron 8 containing constructs
(Figure 3.5). Interestingly, mutagenesis of the GATA-A site generated a slight increase in
activity of 600/o when compared to wild type, indicating that binding of transcription factors to
the GATA-A site may inhibit ALAS2 transcriptional activity. Luciferase activity was reduced
Io 25Yo of the wild type intron 8 construct when the GATA-B site was mutated (Figure 3.5).
The above findings suggest that GATA-B is a major contributor to intron 8 enhancer activity
in undifferentiated J2E cells, with CACCC-A and CACCC-B sites also required but in less
pivotal roles.
Mutagenesis of both CACCC sites (pISoAcB+LUC) decreased luciferase activity Io 43o/o of
wild type intron 8 constructs, which was slightly less than the reduction in activity upon
individual mutagenesis of these elements (Figure 3.5). The effect of mutating CACCC-A and
B concurrently did not have an additive or synergistic negative effect on luciferase activity.
Perhaps the remaining intact transcription factor binding sites within human ALAS2 intron 8
are able to compensate for the loss of CACCC-A and CACCC-B. Inactivation of both
CACCC-A and GATA-A (pISoAgA+LUC) reduced activity to 600/o of wild type intron 8
levels (Figure 3.5). This decrease was comparable to inactivation of CACCC-A alone,
indicating that GATA-A plays little role in the regulation of the ALAS2 intronic enhancer.
Inactivation of the CACCC-A and GATA-B sites (pISoAgB+LUC) reduced activity to 2Io/o of
wild type expression (Figure 3.5), equivalent to the decrease in activity observed upon
mutagenesis of GATA-B alone. Therefore, mutagenesis of CACCC-A in addition to GATA-B
did not reduce activity further, suggesting loss of CACCC-A is negligible and the presence of
79

Figure 3.5 Identifìcation of the transcription factor-binding sites critical to the function
of intron 8 as an enhancer of ALAS2 gene transcription
1.3pmol of the human ALAS2 intron wild-type and mutated reporter constructs (see Section
2.3.3 for construction details) were transfected into J2E cells which were either stimulated by
Epo to differentiate or remained untreated. Cells were haryested 48 hours post-transfection
and luciferase reporter activity assayed and normalised against expression of the internal
Renilla luciferase control (50pg). Wild-type intron 8 activity was assigned a value of 100 and
activities from the mutant intron 8 constructs \Mere expressed as a percentage of wild type
levels for both induced and non-induced J2E cells. X denotes mutation of the transcription
factor-binding elements into a PvuII restriction enzyme site. The 239bp intron 8 fragment
was cloned in its native 5' to 3' orientation. Each transfection was performed in triplicate and
the experiment repeated at least three times. Depicted is a representative experiment. The
transcription factor binding sites are located at the following positions in human ALAS2
intron 8 : CACCC - A (+268 I +27 5), C ACCC-B (+327 I +33 4), GATA-A (+3 44 I +3 49) and
GATA-B (+380/+38s).

plScA+LUG
pIScB+LUG
pl8
plSgB+LUG
plScAcB+LUG
A+LUC
plScAgB+LUG
JzE Gells
- Epo + Epo
100 100
67!4 3716
55r8 6517
160!47 151t34
25t3 911
43r5 22!2
60r7 66fl0
21t2 511
plScBgB+LUG
6r3 311

an intact GATA-B site is paramount to intron 8 enhancer activity in undifferentiated J2E
cells. Mutagenesis of both CACCC-B and GATA-B (pI8cBgB+LUC) virtually abolished
intron 8 activity, reducing luciferase activity levels to 60/o of wild type (Figure 3.5), which
was significantly lower than individual mutation of the above sites. This result indicates that
CACCC-B and GATA-B may be critical for human ALAS2 intron 8 enhancer function.
It can be concluded that three sites within human ALAS2 intron 8, CACCC-A, CACCC-B
and GATA-B, contribute positively to expression levels generated by intron 8 in '/undifferentiated J2E erythroid cells. Interestingly, the GATA-A site may behave as a negative
element in the regulation of ALAS2 transcription.
To determine if any of these sites within intron 8 are responsive to Epo induced maturation of
J2E cells, mutant intron 8 luciferase reporter constructs were transiently transfected into Epo
treated J2E erythroid cells. Luciferase expression'\À/as assayed and compared to wild type
intron 8 activity. Data obtained from the following experiments was expressed as a percentage
of wild tlpe intron 8 activity, where wild type expression was assigned a value of 100.
Mutagenesis of the GATA-B site virtually abolished activity from the intron 8 enhancer
region, resulting in only 9o/o of wild type activity in differentiated J2E errthroid cells (Figure
3.5). This suggests that GATA-B is critical for the Epo responsive enhancer activity of intron
8. This essential role for GATA-B was also seen with the double mutant constructs in which
either CACCC-A or CACCC-B were concurrentlymutated (Figure 3.5). Inactivation of either
CACCC site in combination with GATA-B did not significantly reduce activity further than
levels obtained with the mutagenesis of only the GATA-B site (Figure 3.5 - compare
pISoAgB+LUC (5%) and pI8cBgB+LUC (3%) with pI8gB+LUC (9%)), suggesting the IGATA-B site alone is critical for the response of intron 8 to Epo stimulation.
However, expression of the CACCC-A mutant construct in Epo induced J2E erythroid cells
resulted in activity that was 3lo/o of wlld type levels whereas inactivation of the CACCC-B
site reduced activity to 650/o (Figure 3.5), suggesting that in the presence of a functional
GATA-B site CACCC motifs may play a role in the complete response of the intron 8
enhancer to Epo induced differentiation. This is further supported by mutagenesis of the
CACCC-A and CACCC-B sites in tandem which resulted in reduced luciferase activity to
22o/o of wlld type intron 8 in differentiated J2E cells (Figure 3.5).
80

Similarly to the effect observed in untreated J2E-3 cells, mutagenesis of the GATA-A site
appeared to increase activityby 5l% when compared to wild type intron 8 (Figure 3.5),
suggesting that even under Epo signalling binding of transcription factors to this site may
contribute in a negative fashion to ALAS2 gene transcription. Indeed mutagenesis of
CACCC-A and GATA-A only reduced activity to 66%o of wild type in Epo treated J2E cells
in comparison to 3TYowith the single CACCC-A mutant construct, suggesting a possible
relief of GATA-A repression (Figure 3.5). Alternatively, activity observed from expression of
the pIScAgA+LUC mutant construct maybe due to the presence of intact CACCC-B and
GATA-B sites, implying that providing these two sites are present the Epo responsiveness and
activity generated by intron 8 can be partly maintained.
Thus, while both CACCC-A and B sites contribute to intron 8 enhancer activity in response to
Epo, GATA-B plays the major role in facilitating transcription of ALAS2 inJ2E erythroid
cells stimulated to differentiate by Epo signalling.
3.2.5 Binding of GATA-I to the Enhancer in Human ALAS2 Intron 8 is Independent of
Epo Stimulation
As described above, the GATA-B site in the conserved 239bp ALAS2 intron 8 enhancer
plays a critical role in the Epo induced increase in reporter gene expression. In addition, there
is some evidence that an intact GATA-A site modihes enhancer activity. In order to
determine if the binding of GATA-I or other protein complexes to the GATA-A and B sites
in intron 8 is altered by Epo induction, EMSAs (Section 2.9.3) were performed on double-
stranded oligonucleotides containing these sites with nuclear extracts prepared from Epo
stimulated J2E erythroid cells (Section 2..9.1). As a control probe, an oligonucleotide
encompassing a known GATA-1 binding site from the B-globin gene (Wall et al., 1988) was
included.
A major retarded complex was obtained with the GATA-A (Figure 3.6a,Lane 1 & 3) and
GATA-B oligonucleotides (Figure 3.6b, Lanes 1 & 3) using both control (undifferentiated)
and nuclear extracts prepared from Epo treated J2E cells. A less intensive band of slower
mobility was observed with the GATA-A oligonucleotides in all binding reactions but the
identity of the band is unknown. As a binding control, a binding reaction was conducted
omitting any nuclear extract and retarded bands were not observed (data not shown). To
81

Figure 3.6 Gel shift analysis of the GATA-A and GATA-B sites in human ALAS2 intron
I
(a) A radiolabelled double-stranded oligonucleotide containing the GATA-A sequence
(5'-AGCTACTGCCTATCTAGTCATTGC-3') was incubated with nuclear extracts
prepared from undifferentiated (Lanes 1 g, Ð and differentiated (Lanes 3 & 4) J2E
cells. For supershift assays, monoclonal GATA-I antibody (Ab) (0.2pg) was added to
the binding reaction 30 minutes prior to addition of the probe (Lanes 2 & 4). As
controls, binding reactions were performed either omitting the nuclear extract or
including a non-specific antibody (data not shown). The retarded complex
representing GATA-I is arrowed. The arrow followed by'?' indicates a less intense
band of unknown origin.
(b) A radiolabelled double-stranded oligonucleotide containing the GATA-B sequence
(5' -TTGAAAGTCCTATCTCAAAGCAGC-3') was incubated with nuclear extracts
prepared from undifferentiated (Lanes I 8.2) and differentiated (Lanes 3 e, Ð JzE
cells. For supershift assays, monoclonal GATA-1 antibody (0.2pg) was added to the
binding reaction 30 minutes prior to addition of the probe (Lanes 2 e, Ð. As controls,
binding reactions were performed either omitting the nuclear extract or including a
non-specific antibody (data not shown).The retarded complex representing GATA-1 is
arrowed.
(c) As a control for GATA-1 binding, a radiolabelled double-stranded oligonucleotide
containing the human B-globin GATA-I consensus sequence (5'-
TTGGCTCCCTTATCATGTCCCTG-3') was incubated with nuclear extracts
prepared from undifferentiated (Lanes I e. Ð and differentiated (Lanes 3 e, Ð J2E
cells. For supershift assays, monoclonal GATA-I antibody (0.2pg) was added to the
binding reaction 30 minutes prior to addition of the probe (Lanes 2 e. Ð. As controls,
binding reactions were performed either omitting the nuclear extract or including a
non-specific antibody (data not shown). The retarded complex representing GATA-1
is arrowed.
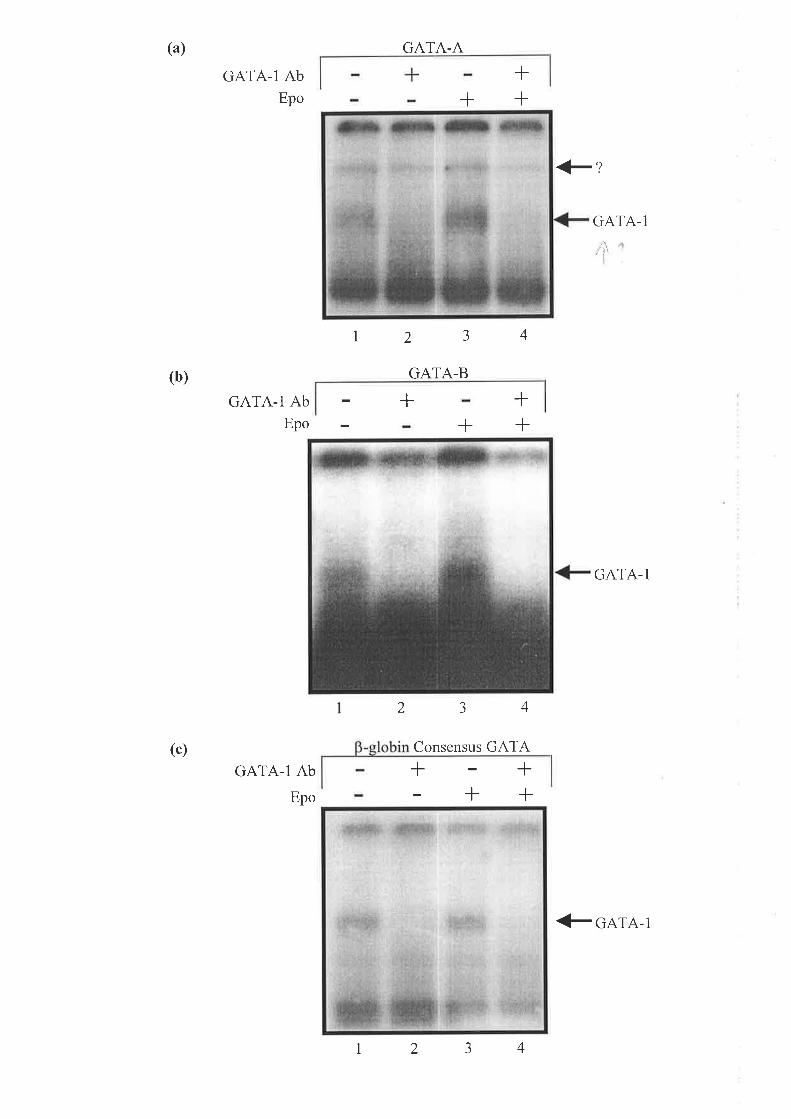
(a)
(b)
(c)
GATA-I AbEpo
GATA-I AbEpo
GATA-I Ab
Epo
GATA-A
GATA-B
+
#z
+++
GATA-1
I2 3 4
+++
,, ìl'l
2 J 4
Consensus GATA
GATA-I
<_ GATA-I
fl r-I 42 3

demonstrate that this major retarded complex was caused by GATA-I binding to the
supershift assay was attempted using a monoclonal GATA-I antibody, sc-265 (Santa
(Figure 3.6a, Lanes 2 &.4 and Figure 3.6b, Lanes 2 8.4). As a positive control, GATA-I
binding to the B-globin consensus GATA-1 site in both Epo induced and non-induced J2E
nuclear extracts was also investigated with the GATA-1 antibody (Figure 3.6c, Lanes 2 e, Ð.
The GATA-I antibody utilised in these experiments was purchased as a supershifting reagent
from Santa Cruz. Although the major retarded complex found with probes containing either
the ALAS2 GATA-A, GATA-B or B-globin consensus cATA-l binding siteËtolished
when the GATA-I antibody'l5in.to¿"¿ in the binding reaction, a shifted rlundfsíot I,ii 'n
observed. The explanation for the absence of a s
that binding of the GATA-1 antibody to GATA-l
or B-globin consensus GATA site alters the confo _ì_.
the protein-DNA interaction such that a supershifted complex is not observed. However, ç-¡'''.'l
given specific abolishment of the complex with GATA-I antibody it is likely to be
representative of GATA-1 binding and this was supported by a non-specific antibody control
in which a supershift of the retarded complexes was not observed (data not shown).
In conclusion, the results demonstrated that irrespective of the differentiation state of the J2E
erythroid cell, binding of the erythroid-specific protein, GATA-1, to sites in the human
ALAS2 intron 8 occurs prior to Epo stimulation and the amount of binding is apparer¡lly î ]"
unaltered upon Epo treatment. Interestingly, both GATA sites can bind GATA-I but only
ù
GATA-B is required for enhancement of transcriptional activity by intron 8 as shown by
mutational analysis (Figure 3.5).
3.2.6 The Effect of Epo on the Binding of CACCC-Associated Proteins to Sites in
Human ALAS2 Intron 8
Several transcription factors are known to bind to CACCC sequences in vitro, including the
erythroid-specific protein, EKLF (Miller and Bieker,1993), Sp1 (Kadonaga et al., 1987),
Spl-related proteins (Kingsley and'Winoto,1992), BKLF (Crossley et al., 1996), CAC C and
CAC D (Hartzog and Myers, 1993). The two CACCC sites in human ALAS2 intron 8,
CACCC-A and B have demonstrated approximately equal abilities in enhancing activity in
transiently transfected Epo treated and untreat ed J2E erythroid cells (Figure 3 .5). To
determine if binding of proteins to either CACCC-A or CACCC-B is affected by Epo induced
82

differentiation EMSAs were performed on double-stranded oligonucleotide probes using
nuclear extracts from Epo treated and untreated J2E cells. Included as control probes were the
CACCC site from the murine adult B-globin promoter which binds EKLF, BKLF and Sp1 iz
vitro (Crossley et al., 1996) and an Spl consensus sequence (Promega) (Section 2.I.7(111)).
Four retarded bands were obtained for both the CACCC-A and CACCC-B oligonucleotides
using Epo treated and untreatedJ2E nuclear extracts (Figure 3.7 and 3.8). Data from a number
of EMSAs indicated that neither the binding pattern nor intensity of protein binding was /
altered by Epo treatment. To determine if any of the retarded bands were specific for either
Spl or EKLF, supershift assays were conducted using a Sp1 monoclonal (sc-420, Santa Cruz)
and an EKLF polyclonal (supplied by Dr. M. Crossle¡ University of Sydney, Australia)
antibody. Neither the Spl nor EKLF antibodies supershifted any of the retarded complexes
formed on either the CACCC-A (Figure 3.7a,Lanes2 &,4 and Figure 3.7b, Lanes 2 & 4) or
CACCC-B (Figure 3.8a, Lanes 2 &,4 and Figure 3.8b, Lanes 2 e, Ð probes. In addition, the
retarded bands obtained with the consensus Spl (Figure 3.9, Lanes 2 &, 4) and B-globin
CACCC (Figure 3.10a, Lanes 2 &,4 and Figure 3.10b, Lanes 2 e. Ð control probes were not
shifted with either of the antibodies suggesting that none of the retarded bands corresponded
to Spl or EKLF using nuclear extracts prepared from Epo treated and untreatedJ2E cells. It
was expected that the consensus sequences for the B-globin CACCC box and Spl sites would
bind EKLF and Sp1 to provide a positive control for protein binding. In the following
experiments both antibodies are shown to be functional in EMSAs so the absence of a
supershift is not due to poor quality antibodies. Lack of detection of specific protein binding
may be due to a limitation of the J2E erythroid cell system in that Spl and EKLF levels may
be low, inhibiting their ability to bind to the consensus control probes or human ALAS2
intron 8 CACCC sites. Alternatively, the EMSA data may be indicative of a novel complex,
containing neither Spl nor EKLF, forming on the human ALAS2 intron 8 CACCC boxes in
undifferentiated and Epo stimulated J2E cells.
As an alternative method to determine if any of the retarded bands observed with the
CACCC-A and CACCC-B probes represented Spl and EKLF binding, nuclear extracts
isolated from COS-1 and CV-1 cells transiently transfected with Sp1 or EKLF expression
vectors, respectively, were utilised in EMSA experiments. The banding patterns obtained with
these nuclear extracts were then compared to shifts using nuclear extracts from Epo
stimulated and unstimulated J2E erythroid cells. In the first experiments, oligonucleotides for
the consensus B-globin CACCC box and Spl site were investigated using the nuclear extracts
83

Figure 3.7 Get shift analysis of the CACCC-A site in human ALAS2 intron 8
(a) A radiolabelled double-stranded oligonucleotide containing the CACCC-A sequence
(5'-CTAGTCCCCCACCCTAGCGAA-3') was incubated with nuclear extracts
prepared from either undifferentiated (Lanes I & 2) or differentiated (Lanes 3 & 4)
J2E cells. For supershift assays monoclonal Spl antibody (Ab) (0.zttÐ was added 30
minutes before the addition of the probe (Lanes 2 e, Ð. As a control, a binding
reaction omitting the nuclear extract was performed (data not shown). Retarded
complexes obtained from the mobility shift assay are arrowed.
(b) A radiolabelled double-stranded oligonucleotide containing the CACCC-A sequence
(5'-CTAGTCCCCCACCCTAGCGAA-3') was incubated with extracts prepared from
either undifferentiated (Lanes 1 & 2) or differentiated (Lanes 3 8L 4) J2E cells. For
supershift assays polyclonal EKLF antibody (2pl) was added before the addition of the
probe (Lanes 2 e, Ð. As a control, a binding reaction omitting the nuclear extract was
included (data not shown). Retarded complexes obtained from the mobility shift assay
are arrowed.

(a)
(b)
Spl AbEpo
EKLF AbEpo
CACCC-A
+
2a-)
CACCC-A
+
+++
<_+<-
<-4
+++
<-++
+1 42 a
J

Figure 3.8 Gel shift analysis of the CACCC-B site in human ALAS2 intron 8
(a) A radiolabelled double-stranded oligonucleotide containing the CACCC-B sequence
(5'-AAAGGTCCCCACCCAGCTACT-3') was incubated with extracts prepared from
either undifferentiated (Lanes I & 2) or differentiated (Lanes 3 e, Ð J2E cells. For
supershift assays monoclonal Spl antibody (Ab) (0.2pg) was added 30 minutes before
the addition of the probe (Lanes 2 8.4). As a control, a binding reaction omitting the
nuclear extract was performed (data not shown). Retarded complexes obtained from
the mobility shift assay have been arrowed.
(b) A radiolabelled double-stranded oligonucleotide containing the CACCC-B sequence
(5'-AAAGGTCCCCACCCAGCTACT-3 r) was incubated with extracts prepared from
either undifferentiated (Lanes 1 & 2) or differentiated (Lanes 3 e, Ð J2E cells. For
supershift assays polyclonal EKLF antibody (2pl) was added 30 minutes before the
addition of the probe (Lanes 2 & 4). As a control, a binding reaction omitting the
nuclear extract was performed (data not shown). Retarded complexes obtained from
the mobility shift assay have been arrowed.

(a)
(b)
Spl AbEpo
EKLF AbEpo
CACCC-B
+
2
CACCC-B
+
++ - _ií^ ¡ |
ßuu
+
<-
+++
<_
c)t
1 J 4
+++
<-<_
I 42aJ
<-

Figure 3.9 Binding of the Spl protein to the Spl consensus site
A radiolabelled double-stranded oligonucleotide containing a consensus Sp1 site (5'-
ATTCGAT ') was incubated with extracts prepared from either
undifferentiated (Lanes I & 2) or differentiated (Lanes 3 e, Ð J2E cells. For supershift assays
monoclonal Spl antibody (Ab) (0.2pg) was added 30 minutes before the addition of the probe
(Lanes 2 e, Ð. As a control, a binding reaction omitting the nuclear extract was included (data
not shown). Retarded complexes obtained from the mobility shift assay are arrowed.

++;
+Spl Ab
Epo
Consensus 1
2 J
+<-
<-4I

Figure 3.10 Gel shift analysis of the p-globin consensus CACCC site in undifferentiated
and differentiated J2E erythroid cells
(a) A radiolabelled double-stranded oligonucleotide containing the mammalian B-globin
CACCC site (5'-AGCTAGCç,{CACCCTGAAGCT-3') was incubated with extracts
prepared from either undifferentiated (Lanes 1 & 2) or differentiated (Lanes 3 e,4)
J2E cells. For supershift assays monoclonal Spl antibody (Ab) (0.2pg) was added 30
minutes before the addition of the probe (Lanes 2 e, Ð. As a control, abinding
reaction omitting the nuclear extract was included (data not shown). Retarded
complexes obtained from the mobility shift assay have been arrowed.
(b) A radiolabelled double-stranded oligonucleotide containing the mammalian B-globin
CACCC site (5' -AGCTAGCCACACCCTGAAGCT-3') was incubated with extracts
prepared from either undifferentiated (Lanes I & 2) or differentiated (Lanes 3 & 4)
J2E cells. For supershift assays polyclonal EKLF antibody (2pl) was added 30
minutes before the addition of the probe (Lanes 2 e, Ð. As a control, a binding
reaction omitting the nuclear extract was performed (data not shown). Retarded
complexes obtained from the mobility shift assay have been arrowed.
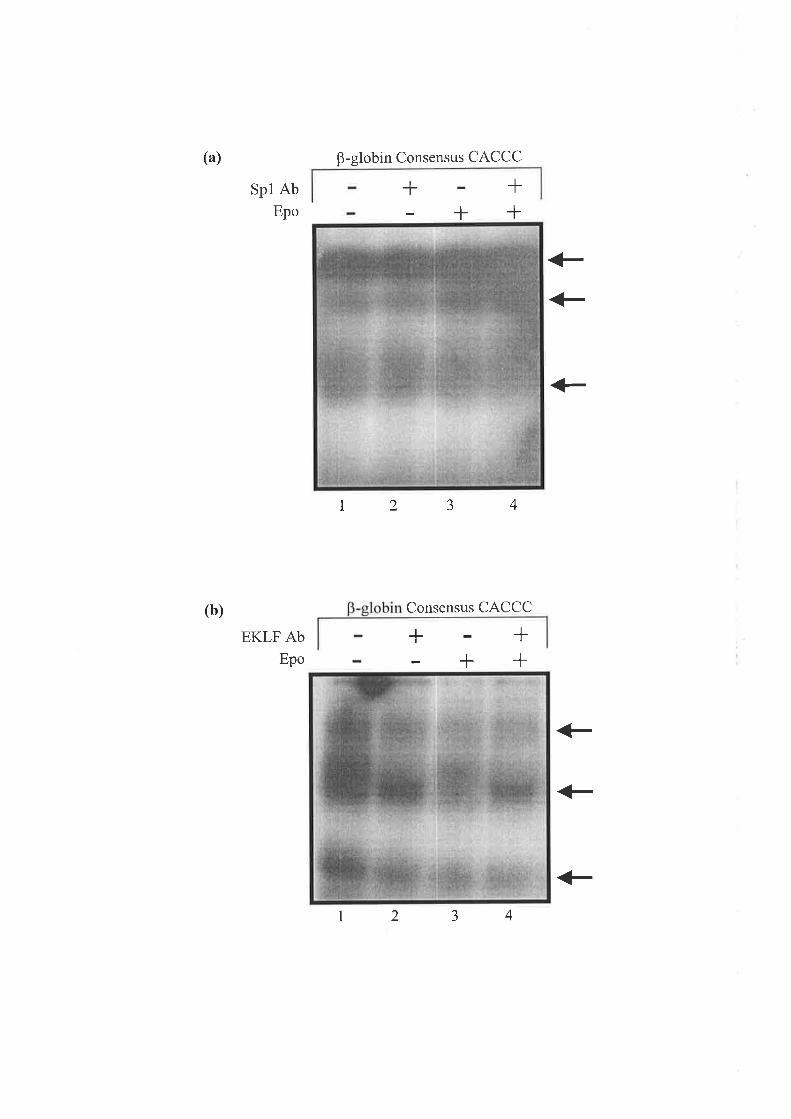
(a)
(b)
B-globin Consensus CACCC
Spl AbEpo
EKLF AbEpo
++
12
t2
<_<_
<-
+<_
+
aJ 4
Consensus CACCC
tlTl
++
34

described above. A faint band representing an Spl complex was detected and supershifted
with Spl antibody with the oligonucleotide containing the B-globin consensus CACCC site in
COS-I cells (Figure 11a, Lanes 5 & 6). A stronger Spl band was observed with the Spl
consensus oligonucleotide using COS-l nuclear extracts, although this band was not
completely shifted with Sp1 antibody (Figure 1lb, Lanes 5 &,6). A retarded complex that was
supershifted upon incubation with Spl antibody was detected with the B-globin CACCC
probe (Figure 3.Ila, Lanes 7 & 8) and the Sp1 probe (Figure 3.1lb, Lanes 7 e, Ð using COS-
l/Spl nuclear extracts. Only a partial supershift was observed with the consensus Spl probe.
Thus complexes forming on the consensus B-globin CACCC box and Spl consensus
oligonucleotides in COS-l/Spl nuclear extracts were shown to contain Spl as upon addition
of Sp1 antibody a supershift was detected. However, a coffesponding band was observed for
both the B-globin CACCC (Figure 3.lla, Lanes 1 & 3) and Spl consensus oligonucleotides
(Figure 3.1lb, Lanes I & 3) with untreated and treated J2E nuclear extracts but this complex
was not identified as Spl in supershift assays (Figure 3.lla, Lanes 2 &,4; Figure 3.11b, Lanes
2 & 4). Therefore, this finding suggests that the major complex formed on the B-globin
CACCC probe contains another protein/s that are enriched in J2E nuclear extracts. For the
consensus Sp1 oligonucleotide a similar observation was made, except that two major
complexes were obtained.
For the CACCC-B probe four major retarded bands were obtained using the COS-1/Sp1
nuclear extracts (Figure 3.l2a,Lane 7) and one of the bands was identified immunologically
as a Spl containing complex upon inclusion of Spl monoclonal antibody (Figure 3.l2a,Lane
8). A similar, very weak band was observed with control COS-I nuclear extracts (Figure
3.12a, Lane 5). The supershifted band was not observed in the assay (Figure 3.I2a, Lanes 6 &
8) and as discussed previously this maybe due to disruption of the Spl/DNA complex upon
binding of the Sp1 antibody. The inclusion of a binding reaction with non-specific antibody
did not supershift any of the bands (data not shown). A weak band that retarded to an identical
position was present in EMSAs using nuclear extracts prepared either Epo stimulated or
unstimulat ed J2E cells (Figur e 3 .I2a, Lanes 1 to 4) but did not appear to be supershifted with
Sp 1 antibody (Figure 3 .12a, Lanes 2 e, Ð. This result suggests that Sp 1 is able to bind to the
CACCC-B site in human ALAS2 intron 8 in vitro using COS-I cells overexpressing Spl but
an Spl containing complex was not detectable in either undifferentiated or differentiated J2E
cells. Therefore, in J2E cells alternate proteins may be binding weakly to the CACCC-B site
and this complex is comparable in size to the Sp1 complex forming on this same sequence in
COS-1 cells overexpressing Sp1.
1
)
84

Figure 3.11 Binding of Spl to the human p-globin consensus sequence and the consensus
Spl site in COS-I extracts overexpressing Spl
(a) A radiolabelled double-stranded oligonucleotide containing the mammalian B-globin
CACCC site (5'-AGCTAGCCACAççcTGAAGCT-3 r) was incubated with extracts
prepared from undifferentiated J2E cells (Lanes I 8.2), differentiated J2E cells (Lanes
3 &, 4), COS-I cells (Lanes 5 & 6) and COS-l/Spl cells (Lanes 7 e, Ð. For supershift
assays monoclonal Spl antibody (Ab) (0.2pg) was added 30 minutes prior to the
addition of the probe (Lanes 2, 4, 6 & 8). A solid arrow denotes the Spl band and the
supershifted Spl Band is represented by a dashed arrow. As a control, a binding
reaction omitting the nuclear extract was performed (data not shown).
(b) A radiolabelled double-stranded oligonucleotide containing a consensus Spl site (5'-
ATTCGATCGGGGCGGGGCGAGC-3') was incubated with extracts prepared from
undifferentiated J2E cells (Lanes 1 & 2), differentiated J2E cells (Lanes 3 & 4), COS-
I cells (Lanes 5 & 6) and COS-l/Spl (Lanes 7 &,8). For supershift assays monoclonal
Spl antibody (0.2pg) was added 30 minutes before the addition of the probe (Lanes 2,
4,6 8.8). A solid affow denotes the Spl band and a dashed alrow represents the
supershifted Spl band. The arrow followed by the '?' highlights the formation of an
unknown complex. As a control, a binding reaction omitting the nuclear extract was
included (data not shown).

(a)B-globin Consensus CACCC
J2E
++
34
COS-I COS-I
6 7
{- spl
ît''
ç0IÌ/"
#spt
1Extract
Spl Ab
Epo
(b)
1 2 5
Consensus 1
+
++ +
t/\-'
Extract
Spl Ab
Epo
J2E COS-1 COS-l/Sp1
+++
++
23456781
#z

Figure 3.12 Gel shift analysis of CACCC-A and CACCC-B sites in human ALAS2
intron 8 utilising COS-l/Spl nuclear extracts
(a) A radiolabelled double-stranded oligonucleotide containing the CACCC-B site (5'-
AAAGGTCCCCACCCAGCTACT-3') was incubated with extracts prepared from
undifferentiatedJ2E cells (Lanes I & 2), differentiatedJ2E cells (Lanes 3 & 4), COS-
I cells (Lanes 5 & 6) and COS-1/Sp1 cells (Lanes 7 8.8). For supershift assays
monoclonal Sp1 antibody (Ab) (0.2pg) was added 30 minutes before the addition of
the probe (Lanes 2, 4,6 & 8). An alrow denotes the Spl band. As controls, binding
reactions either omitting the nuclear extract or including a non-specific antibody were
performed (data not shown).
(b) A radiolabelled double-stranded oligonucleotide containing the CACCC-A site (5'-
CTAGTCCCCCACCCTAGCGAA-3') was incubated with extracts prepared from
undifferentiated I2E cells (Lanes 3 & 4), differentiatedJZB cells (Lanes 5 & 6), COS-
1 cells (Lanes 7 & 8) and COS-1/Sp1 cells (Lanes 9 & 10). As a control for Spl
binding, the consensus Spl oligonucleotide incubated with COS-l/Spl nuclear
extracts was included (Lanes I e,Ð. For supershift assays monoclonal Sp1 antibody
(0.2pg) was added 30 minutes before the addition of the probe (Lanes 2,4,6 & 8). As
a control, a binding reaction omitting the nuclear extract was included (data not
shown). A solid alrow denotes the Spl band and the Spl supershift is represented by a
dashed arrow.
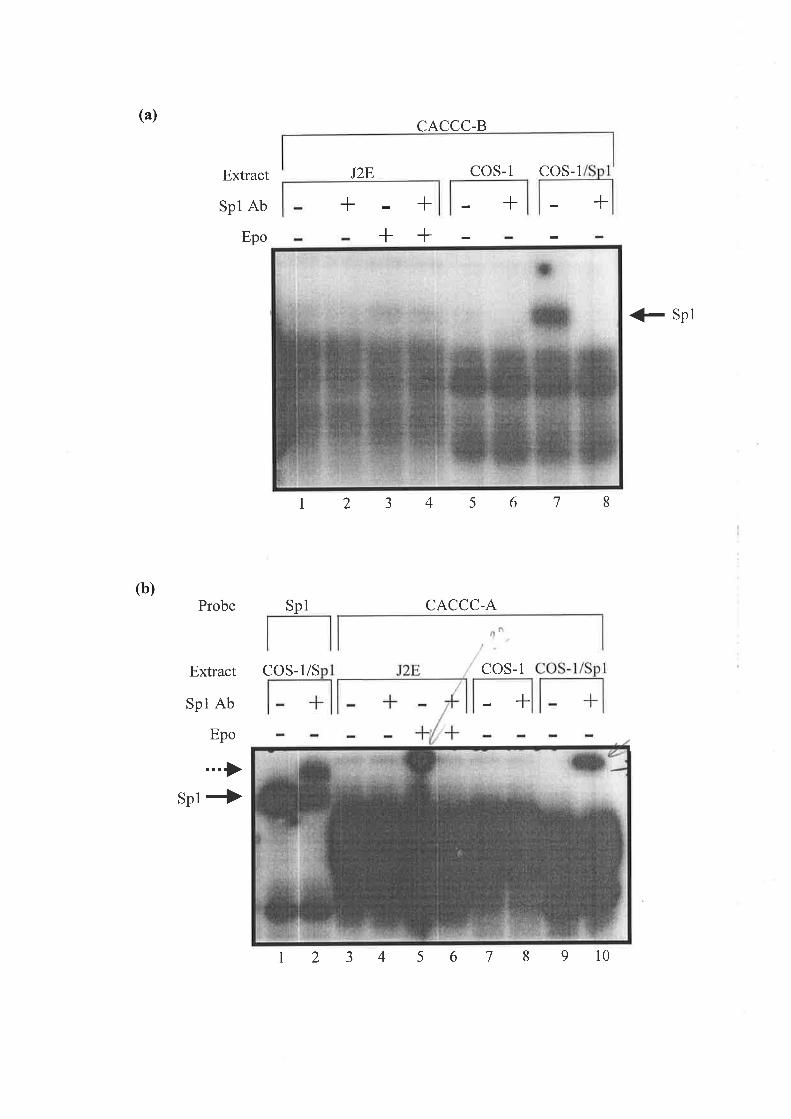
(a)
(b)
Extract
Spl Ab
Epo
'ìa
Probe Spl
CACCC-B
CACCC-A
COS-I COS- 1
+J2E
+++
+ +
{- spl
r2345678
Extract
Spl Ab
Epo
sp1*
COS-1/S COS-1
I 2 3 4 5 6 7 8 9 l0
r-J1r
I

A band representing Sp1 binding to the CACCC-A site of intron 8 was not observed with
COS-I nuclear extracts over-expressing Spl (Figure 3.I2b, Lanes 9 & 10) when compared to
the control reactions in which Spl was shown to bind the Spl consensus oligonucleotide
using COS-1/Spl nuclear extracts (Figure 3.12b, Lanes I e,Ð. Similarly, inbinding reactions
using nuclear extracts prepared from undifferentiated and Epo differentiated J2E cells a band
representing Sp1 binding was not observed (Figure 3.12b, Lanes 3 to 6). The majority of the
other bands formed when using the J2E, COS-I and COS-1/Sp1 nuclear extracts could not be
resolved for CACCC-A as after numerous attempts the quality of the EMSA could not be
improved. There appears to be a band at the top of Lanes 5 and 10 in Figure 3 .l2b but this
only represents an artefact of the assay and is not indicative of Spl binding to the CACCC-A
oligonucleotide. Thus, it is concluded that Spl does not bind to the CACCC-A site in vitro
irrespective of the differentiation state of the J2E nuclear extracts utilised.
The binding of EKLF to the CACCC-A and CACCC-B sites was next investigated. A major
retarded complex was observed with the control B-globin CACCC probe using nuclear
extracts from CV-1 cells overexpressing EKLF (CV-1/EKLF) (Figure 3.l3,Lane 7) but not
CV-l cells alone (Figure 3.13, Lane 5), suggesting this complex contained EKLF. In addition,
inclusion of a polyclonal EKLF antibody in the reaction markedly decreased the intensity of
this complex (Figure 3.l3,Lane 8). A supershift of the band representing EKLF was not
observed upon addition of EKLF antibody (Figure 3.l3,Lane 8), suggesting that binding of
the EKLF antibody to the protein-DNA complex may have disrupted its conformation. A non-
specific antibody control was performed and supershift of the retarded complexes was not
observed (data not shown). A similarly retarded complex was not present at detectable levels
in nuclear extracts prepared from non-induced (Figure 3.13, Lanes I e, Ð and Epo induced
(Figure 3.13, Lanes 3 8.4) J2E cells. This finding suggests that EKLF protein inJ2E cells is
at a level that is not detectable in EMSAs and this is irrespective of the Epo induced
differentiation state of the J2E cells. A retarded band with slow mobility was observed in all
the lanes running reactions incubated with EKLF antibody (Figure 3.13, Lanes 2, 4,6 &,8).
The identity of this band is unknown but may represent non-specific binding by the EKLF
polyclonal antibody.
Nuclear extracts prepared from CV-1/EKLF cells were tested with human ALAS2 intron 8
CACCC-A and B oligonucleotides in EMSA experiments. This was performed to determine
whether one of the retarded bands obtained with the J2E undifferentiated and differentiated
')J*1
85

Figure 3.13 Detection of EKLF binding to the human p-globin consensus CACCC site in
nuclear extracts from CV-l cells overexpressing EKLF
A radiolabelled double-stranded oligonucleotide containing the mammalian B-globin CACCC
site (5I-AGCTAGCCACA 3') was incubated with extracts prepared from
undifferentiatedJ2E cells (Lanes 1 & 2), differentiatedJ2E cells (Lanes 3 & 4), CV-l cells
(Lanes 5 & 6) and CV-l/ EKLF cells (LanesT & 8). For supershift assays polyclonal EKLF
antibody (Ab) (2pl) was added 30 minutes before the addition of the probe (Lanes 2,4,6 &,
8). An affow denotes the band representing EKLF binding and the unknown complex (?). As
controls, binding reactions either omitting nuclear extract or including a non-specific antibody
were performed (data not shown).

J2E
B-globin Consensus CACCC
CV-1
+
CV-1/EKLF
+Extract
EKLF Ab
Epo
+
23
+++
+?
+EKLF
1 45 6 78

nuclear extracts was representative of EKIF binding to the CACCC-A and B
oligonucleotides, since inclusion of EKLF antibody did not result in a supershift (Figure
3.14a, Lanes 2 & 4; Figure 3.14b, Lanes 2 &,4). A major retarded complex representing
EKLF was not observed with either the CACCC-A or CACCC-B sites using CV-I/EKLF
extracts when compared to control CV-l cells (Figure 3.14a, Lanes 5 &.7; Figure 3.14b,
Lanes 5 & 7). Furthermore, a supershift of any one of the retarded complexes was not
observed upon the addition of EKLF antibody. Therefore, from the binding data it appears
that EKLF does not bind either the CACCC-A or B sites in intron 8 invitro irrespective of the
differentiation state of the J2E nuclear extracts. Thus, it is likely that the retarded complexes
obtained represent the binding of other transactivating factors to the conserved CACCC sites
in intron 8 of the human ALAS2 gene.
In conclusion, GATA-A and B sites in human ALAS2 intron 8 bound GATA-I in vitro in
both Epo treated and untreated J2E cells. However, it is currently unclear as to the identity of
the proteins binding intron 8 CACCC-A and B sites but from the findings of the present
study, it is unlikely to be either EKIF or Spl. Interestingly the banding pattern of retarded
complexes obtained for the CACCC and GATA sites was consistently found to be unaltered ./.
in nuclear extracts from Epo stimulated J2E cells. This suggests that binding of transcriptional ' ',
activators to this region occurs independently of the differentiation state of the erythroid cells. ,
3.2.7 Investigating the role of CBP/p300 coactiv
ALAS2
The recruitment of coactivator complexes to the promoter and enhancer regions of specific
genes is linked to the activation of gene expression, resulting in histone acetylation and
chromatin remodeling (Wade and'Wolffe,7997; Grunstein, 1997; Kadonaga, 1998). Several
transcriptional coactivators, including CBP and its structural homologue p300 (Bannister and
Kouzarides,1996), have been shown to harbour intrinsic histone acetyltransferase (HAT)
activity which acts to alter chromatin structure via acetylation events and so regulate gene
expression (Ogryzko et al., 1996; Wang et al., 1998). Such activity from the coactivators
CBP/p300 has been proposed to foster a link between transcriptional activators and the basal
transcriptional machinery (Eckner,1996). The adenovirus ElA oncoprotein is able to bind
CBP/p300 and this event inhibits the ability of the coactivators to enhance gene expression
86

Figure 3.14 Gel shift analysis of CACCC-A and CACCC-B sites in human ALAS2
intron 8 utilising CV-I/EKLF nuclear extracts
(a) A radiolabelled double-stranded oligonucleotide containing the CACCC-A site (5'-
CCCTAGCÇ{,ry-3t ) was incubated with extracts prepared from
undifferentiatedJ2E cells (Lanes I & 2), differentiatedJ2E cells (Lanes 3 & 4), CV-1
cells (Lanes 5 & 6) and CV-I/EKLF cells (Lanes 7 e, Ð. For supershift assays
polyclonal EKLF antibody (Ab) (2p1) was added 30 minutes before the addition of the
probe (Lanes 2,4,6 & 8). As a control, a binding reaction omitting the nuclear extract
was performed (data not shown). Retarded complexes obtained from the mobility shift
assay have been arrowed.
(b) A radiolabelled double-stranded oligonucleotide containing the CACCC-B site (5'-
AAAGGTCCCCACCCAGCTACT-3') was incubated with extracts prepared from
undifferentiatedJ2E cells (Lanes I & 2), differentiated J2E cells (Lanes 3 & 4), CV-l
cells (Lanes 5 & 6) and CV-I/EKLF cells (Lanes i U. A¡.For supershift assays
polyclonal EKLF antibody (0.5p1) was added 30 minutes before the addition of the
probe (Lanes 2,4, 6 & 8). As a control, a binding reaction omitting the nuclear extract
was included (data not shown). Retarded complexes obtained from the mobility shift
assay have been arrowed.

(a) CACCC-A
CACCC-B
CV-1 CV-1/EKLF
+Extract
EKLF Ab
Epo
J2E
+
J2E
+++
+
++
<_
t2345678
(b)
Extract
EKLF Ab
Epo
r23456
CV-I CV-I/EKLF
+++
+++
++<-
+87

t -,.Ê. ;.,
J'r, L: a
a. '\'.' \
protein inJ2E cells was(Goodman and Smolik, 2000). Therefore, overexpression the EIA
used to determine the role of CBP/p300 in ALAS2 gene activation.
Initial experiments examined the effect of the EIA protein on the activity generated from the
ALAS2 293bp promoter and the enhancer element within intron 8. The ALAS2 promoter
(pALAS2-0.293LUC) and intron 8 (pI8+LUC) luciferase reporter constructs (1.3pmol of
each) were transiently transfected into J2E cells together with varying concentrations (50ng,
l25ng, and 250ng) of an ElA expression vector (pElAwt, a gift from Dr Kouzarides,
University of Cambridge). As a negative control, a vector lacking EIA (pElAblk, a gift from
Dr Kouzarides, Cambridge University) at 250ngwas also tested.
Activity from the 293 bp ALAS2 promoter (pALAS2-O .293LUC) was not significantly /affected by E1A at 50ng and l25ngbut moderate inhibition (approximately 40%) was
observed with the highest concentration of ElA expression vector (250ng) in both Epo treated
and untreatedJ2E cells (Figure 3.15a). In contrast, increasing concentrations of the E1A
expression vector resulted in a marked decrease in luciferase activity driven by the enhancer ,/element located within intron 8 (Figure 3.15b). This effect was most significantly observed in
Epo treated and in untreated J2E cells at l25ng and 250ng of ElA. Since ElA is an inhibitor
of CBP/p300, the data suggests that these coactivators may play a role in enhancing ALAS2
transcription via either direct interaction with intron 8 or factors that bind within this domain
but it is not required for the function of the promoter, at least not in transient transfection
assays. The control vector lacking EIA (pElAblk) had no effect on activity from either the
ALAS2 promoter or intron 8 constructs (data not shown). From this experiment we conclude
that the ALAS2 intron 8 enhancer is a target for ElA inhibitory action while the ALAS2
proximal promoter is, by comparison, only weakly affected.
To demonstrate that ElA is specifically interfering with the ability of CBP/p300 to mediate
intron 8 activity, a truncated E1A construct that is unable to bind CBP/p300 and therefore
inhibit their activity, was used. The CR1 domain (Offringa et al., 1990) of the EIA
oncoprotein is critical for repression of transcriptional activity mediated by CBP/p300 as it
contains the CBP/p300 binding site. To ascertain if EIA represses intron 8 enhancer activity
by specifically interacting with CBP/p300, a mutant of ElA with a CRI deletion (pElAl5-35,
a gift from Dr Kouzarides, University of Cambridge) which prevents it from interacting with
CBP/p300 was examined in transient transfection assays. A range of concentrations (50,125
and 250ng) of the mutant ElA expression construct (pElAl5-35) were transiently co-
87

Figure 3.15 The effect of ElA on the transcriptional regulation of human ALAS2
(a) The human ALAS2 293 bp proximal promoter vector (1.3pmol) (pALAS2-0 .293LUC)
was co-transfected into J2E cells with various concentrations of the EIA expression
construct (pElAwt). As a control,250ngof the ElA emptyvector (pElAblk) was co-
transfected (data not shown). Immediately following transfection, Epo (5units/ml) was
added to the appropriate cells to induce erythroid differentiation. The transfected cells
were incubated at 37"C for 48 hours. Cells were harvested and extracts assayed for
luciferase activity using the Dual-Luciferase@ Reporter Assay System (Promega).
Luciferase activity from the ALAS2 promoter reporter gene construct was normalised
against the internal Renilla luciferase control (50pg) and expressed as relative
luciferase activity (RLA). Displayed are the values calculated in the absence and
presence of Epo stimulated erythroid differentiation of the J2E cells. Transfections
were performed in triplicate and the experiment repeated at least three times. Graphed
is a representative experiment.
(b) The human ALAS2 intron 8 vector (1.3pmol) (pI8+LUC) was co-transfected with
various concentrations of the E1A expression construct (pElAwt) into the J2E cells.
As a control,250ngof the ElA empty vector (pElAblk) was co-transfected (data not
shown). Immediately following transfection, Epo (5units/ml) was added to the
appropriate cells to induce erythroid differentiation. The transfected cells were
incubated at37"C for 48 hours. Cells were harvested and extracts assayed for
luciferase activity. Luciferase activity was normalised against the internal Renilla
luciferase control (50pg) and expressed as relative luciferase activity (RLA).
Displayed are the values calculated in the absence and presence of Epo stimulated
erythroid differentiation of the J2E cells. Transfections were performed in triplicate
and the experiment repeated at least three times. Graphed is a representative
experiment.
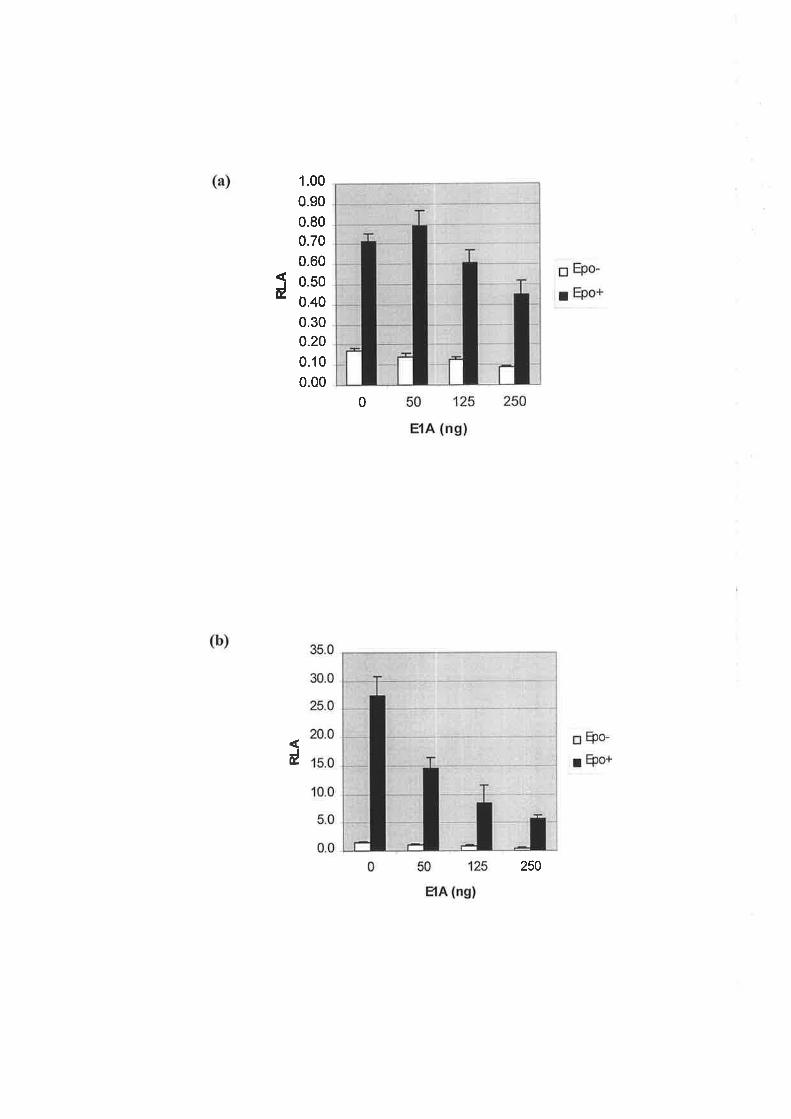
1.00
0.90
0.80
0.70
0.60
0.50
0.40
0.300.20
0.10
0.00
0t
0
d
250

transfected with the ALAS2 intron 8 reporter construct (pI8+LUC). The wild type EIA
expression construct (pElAwt) at I25ngwas co-transfected with the intron 8 reporter
construct also. As a control, the EIA parental empty vector (pElAblk) was included.
As previously noted, reporter gene activity driven by intron 8 was reduced considerably in the
presence of wild type EIA in undifferentiated and differentiated J2E cells (Figure 3.1ó). The
construct lacking ElA sequence (pElAblk) did not affect intron 8 enhancer activity (data not
shown). In contrast, expression of an E1A derivative that could not interact with CBP/p300
(pElAl5-35) did not alter the level of activity generated by ALAS2 intron 8 when compared
to intron 8 activity observed in the absence of pElAl5-35 (Figure 3.16). This was observed in
both Epo treated and untreated J2E cells. Hence, the domain of the E1A protein that interacts
with CBP/p300 is required for ElA repression of intron 8 enhancer activity. Therefore,
findings from this experiment lend further support to the notion that CBP/p300 interacts -/'positively with human ALAS2 intron 8 to increase transcriptional activity in undifferentiated
erythroid cells and in response to Epo induced differentiation.
Experiments conducted using wild type and mutant forms of the ElA expression vector
demonstrated a potential involvement of the CBP/p300 coactivator in stimulating activity
generated by the ALAS2 intron 8 enhancer. The subsequent experiment examined the effect
of overexpressing the coactivator p300 on the activity generated by ALAS2 intron 8. A p300
expression vector, a gift from Dr Eckner (University of Zunch), contains the cytomegalovirus
promoter to drive p300 expression (p300CMV). The intron 8 luciferase reporter construct was
co-transfected with a range of concentrations of the p300 expression vector (50, 125, 500ng).
As a control, a parental vector lacking p300 expression (p300blk, obtained from Dr. Eckner,
University of Zuricþ was included.
Overexpression of p300 did not significantly increase intron 8 enhancer activity in either î I'
undifferentiated or Epo stimul ated J2E cells (Figure 3.I7). Although luciferase activity was
slightly increased in the presence of the p300 expression construct this was equivalent to
levels observed with the parental p300 blank vector (p300blk), suggesting expression of p300
did not further enhance the ability of intron 8 to function as a regulator of gene transcription.
Concentrations of p300 expression vector between 50 to 500ng did not alter intron 8 enhancer
activity which may indicate that the maximal level of activation generated by the intron 8
enhancer is sufficiently met by endogenous levels of CBP/p300 and overexpression of p300 z
cannot increase activity further. Increasing the concentration of p300 expression vector to lpg
88

Figure 3.16 The effect of an ElA CBP/p300-binding mutant on human ALAS2 intron 8
enhancer activity
The human ALAS2 intron 8 vector (l.3pmol) (pI8+LUC) was co-transfected into the J2E
cells with 50 to 250ng of an ElA expression mutant (pElAl5-35). As a control, l25ng of the
E1A wild type expression construct was also co-transfected with the intron 8 reporter vector.
A ElA blank vector (pElAblk) (250ng) control was conducted (data not shown). Immediately
following transfection, the appropriate cells were treated with Epo (5units/ml). Cells were
harvested after 48 hours at37"C and extracts assayed for luciferase activity. Luciferase
activity from the ALAS2 promoter reporter gene construct was normalised against the internal
Renilla luciferase control (50pg) and expressed as relative luciferase activity (RLA). Graphed
is RLA in the presence and absence of Epo treatment. Transfections were performed in
triplicate and the experiment repeated at least three times. A representative experiment is
graphed.

12.0
10.0
8.0
6.0
4.0
2.0
0.0
pI8+LUC (1.3pmol) +
pElAl5-35 (ng)
pElAwt (ng)
EpG
EpotJÉ
+
50
+++
t25 250
t25

Figure 3.17 The effect of p300 overexpression on human ALAS2 intron 8 enhancer
activity
A range of concentrations (50,725 and 500ng) of the p300 expression vector (p300CMV)
were co-transfected with 1.3pmol of the intron 8 luciferase reporter construct þI8+LUC) into
the J2E cells. As a control, 500ng of the p300 blank vector O300blk) was also transfected.
The appropriate cells were treated with Epo (5units/ml) immediately following transfection.
After 48 hours at37"C, cells were harvested and extracts assayed for luciferase activity.
Luciferase activity was normalised against the internal Renilla luciferase control (50ng) and
expressed as relative luciferase activity (RLA). RLA obtained from the intron 8 reporter
construct has been graphed from Epo treated and untreatedJ2E cells. Each transfection was
conducted in triplicate and the experiment repeated at least three times. The graph represents
one of the experiments conducted.

45.4
40.0
35.0
30.0
d 2o.o
15.0
10.0
5.0
0.0
pI8+LUC (l.3ptrnol) +

caused a decrease in luciferase activity (data not shown) and this was presumably due to an
overabundance of transfected DNA being detrimental to transcriptional activity within the
J2E cells as the p300 empty vector (p300blk) at 7¡tgresulted in a similar reduction (data not
shown).
In order to further demonstrate that EIA inhibited ALAS2 intron 8 activity through binding to
CBP/p300 coactivators, a p300 mutant protein that cannot bind EIA þ300de130) was co-
expressed with E1A in undifferentiated and differentiated J2E erythroid cells in an attempt to
relieve EIA repression. The wild type EIA expression vector (125ng) was co-transfected with
1,3pmol of the ALAS2 intron 8 reporter construct and varying amounts of the p300 mutant
expression vector (125ng,500ng, lpg and 3pg). Although the p300 (p300CMV) vector at lpg
or above was observed to inhibit intron 8 activity (data not shown), most likely a result of
overloadingtheJ2E cells with DNA, quantities of p300de130 at 1 and 3pg were included in
this experiment in case higher concentrations were required to overcome EIA inhibition.
Expression vectors lacking either p300 (3pg) or E1A (l25ng) were included in the experiment
as negative controls. Wild type ElA reduced the level of activity generated by intron 8 in the /presence of Epo induced differentiation of J2E cells and only slightly in non-induced J2E
cells (Figure 3.18) which was previously observed (see Figure 3.15). The EIA empty vector
(pElAblk) did not inhibit intron 8 enhancer activity (data not shown). Expression of
p300del30 (500ng) did not alter the level of intron 8 activity obtained in Epo treated or '/
untreated J2E cells when compared to transfection of the intron 8 reporter construct alone
(Figure 3 . 1 8). Addition of the p3 00 E 1 A (p3 00de13 0) binding mutant at concentrations
ranging from 500ng to 3pg did not relieve the ElA inhibition of intron 8 activity in both J2E
cells induced to differentiate with Epo and undifferentiated cells (Figure 3.18). Ability to
overcome repression of ALAS2 intron 8 enhancer activity by EIA may require a higher
concentration than 3pg of the p300 deletion mutant expression vector but as observed with the
inclusion of the parental p300 blank vector (p300b1k) at3¡tg, high concentrations of
transfected DNA inhibit intron 8 activity in the J2E cells. As previously explained, this may
be due to transfecting a large quantity of the expression vector which appears to negatively
affect the function of the J2E cells. In addition, the inability of p300 to enhance intron 8
activity may be due to poor or no expression from the p300 expression vector and this needs
to be resolved prior to future work in this area.
Therefore, initial experiments taking advantage of the association between EIA and
CBP/p300 suggested that CBP/p300 interacts with the ALAS2 intron 8 enhancer element and
89

Figure 3.L8 Prevention of E1A inhibition of ALAS2 intron 8 activity by a p300 ElA-
binding mutant
The human ALAS2 intron 8 vector (1.3pmol) (pI8+LUC) was co-transfected into the J2E
cells with l}Sngof the E1A expression vector (pElAwt). A range of concentrations (125ng to
3pg) of the p300del30 expression vector (p300de130) were also transfected to counteract the
inhibitory effect of ElA. As controls, blank vectors for ElA (pElAblk) (data not shown) and
p300 (p300blk) at 125ng and 3pg, respectively were included. Following transfection, the
appropriate cells were treated with Epo to induce differentiation. After 48 hours at 37"C, cells
were harvested and extracts assayed for luciferase activity. Luciferase activity was normalised
against the internal Renilla luciferase control (50pg) and expressed as relative luciferase
activity (RLA). Graphed is RLA observed in the presence and absence of Epo induced
differentiation. Transfections were performed in triplicate and the experiment repeated at least
three times. An experiment representing the trend observed is graphed.

14.0
12.0
10.0
8.0
6.0
4.0
2.0
0.0
pI8+LUC (1.3pmol) +
pElAwt (125ng)
p300del30 (pg)
p300blk (pg)
fÉ
tr Epo-
¡ EPo+
+ +++ +
+ + +
++
+ +
0.5 0.125 0.5 I 3
aJ

not the ALAS2 promoter to increase transcriptional activity. This notion does not appear to be
supported by studies utilising p300 wild type and mutant expression clones. Thus, further
investigation is required to elucidate the role of CBP/p300 transcriptional coactivators in the
transcriptional regulation of ALAS2.
3.3 DISCUSSION
Erythropoiesis, the process of red blood cell maturation, is dependent upon the ordered up-
regulation of erythroid-specific genes. During the haemoglobin synthesising stage the genes
involved in the synthesis of haem and globin chains are expressed. Since ALAS2 is the
enzyme that controls the rate of haem production in erythroid cells (Bottomley and Muller-
Eberhard, 1988; Ponka et al., 1988) and expression levels increase during erythropoiesis
(Bottomley et al., 1995) the mechanism underlying the transcriptional regulation of ALAS2
with respect to erythroid differentiation is of interest. The critical importance of the up-
regulation of ALAS2 has also been demonstrated in ALAS2 gene targeting studies where
mice null for ALAS2 die during embryogenesis from severe anaemia as a result of decreased /haem synthesis disrupting erythropoiesis (Nakajima et al., 1999). The aim of this chapter was
to identifu and characterise elements within the human ALAS2 gene that elevate the level of
ALAS2 transcriptional activity in response to cellular differentiation induced by the hormone
Epo.
Although regions have been identified in human ALAS2 as important for ALAS2 regulation
in undifferentiated erythroid cells (Surinya et al., 1997,1998) little is known about how the
gene is activated during erythroid maturation. The process of erythropoiesis is triggered by
low oxygen tension and driven by the binding of Epo to the EpoR, which is expressed on the
surface of precursor erythroid cells in the bone marrow. This action results in the initiation of "/
a signalling cascade (Tilbrook and Klinken,1999) which ultimately facilitates the
transcription of ALAS2 as well as other erythroid genes. How EpoR signalling influences the
regulation of ALAS2 transcription is unclear and this chapter investigates this mechanism by
utilising the J2E erythroid cell line.
The J2E cell line terminally differentiates in response to Epo stimulation (Klinken et al.,
1988), exhibiting increased cell proliferation, morphological changes and enhanced
haemoglobin s¡mthesis (Klinken et al., 1988; Busfield and Klinken,1992;Busfield et al.,
1993; Tilbrook et al., 1996). Several enzymes of the haem bios¡mthetic pathway demonstrate
90

an increase in activity levels up to approximately 2-fold in response to Epo, which contributes
to the rise in haem levels observed in differentiated J2E cells (Busfield et al., 1993,1995a). In
the same study, ALAS2 mRNA levels were assayed and shown to be elevated by 3 to 4-fold
after Epo induced differentiation of J2E cells. Increases in globin transcripts and protein
levels were also detected in response to Epo induced differentiation of J2E cells (Klinken e/
al., 7993). The current study has shown that the J2E cells used were able to respond to Epo
with an increase in haemoglobin-positive J2E cells after 48 hours treatment. This
demonstrated that a proportion of the J2E cells underwent differentiation, providing a useful
model for investigating the molecular basis of ALAS2 regulation during erythroid
differentiation.
Initially regions within the human ALAS2 gene required for increased ALAS2 transcription
upon Epo induced differentiation of the J2E cells were identified. A number of ALAS2
domains were found to contribute to enhanced transcriptional activity in differentiated J2E
cells including the immediate promoter and introns I and 8. There was no evidence to support
the location of additional Epo responsive elements in at least the first 10 kb of ALAS2 5'
flanking sequence. These findings were consistent with DNase I hlpersensitivity studies /'conducted on the endogenous murine ALAS2 gene in undifferentiated MEL cells where
L*9 ":ì.... t
hlpersensitivity sites were shown to reside in the promoter, intron I and intron 8 and not
^upstream in the 5' flanking region (Schoenhaut and Curtis, 1989).
': ¡ ' *'t''î:"'''-'
dip#
The rise in activity generated by the 293 bp human ALAS2 proximal promoter upon Epo.- "f "stimulated differentiation of the J2E cells may be due to an increase in the levels of erythroid-
specific transcription factors, leading to a cellular environment that is conducive to increasing
transcriptional activity. An increase in erythroid-specific transcriptional activator levels could
regulate the expression of specific genes required during erythropoiesis, such as ALAS2 and
other enzSrmes involved in the haem s¡mthesis pathway. The levels of GATA-1 mRNA and
protein have been shown to increase in the erythroid J2E cell line upon Epo stimulation of
erythropoiesis (Busfield et a\.,1995b) and this maybe one of the erythroid-specific proteins
involved in regulating ALAS2 promoter activity. Surprisingly, EKLF mRNA levels are
unaffected by Epo stimulated differentiation of the J2E cell line (Spadaccini et al., 1998).
However, utilisation of EKLF antisense RNA to abrogate its activity resulted in reduced
haemoglobin production. This effect was due to a decrease in transcription of both the globin
genes and enzymes of the haem biosynthetic pathway, including ALAS2 (Spadaccini et al.,
1998). These results imply that EKLF is required for regulating globin and ALAS2 expression
qlr
9l

in order to form haemoglobin in the maturing erythroid cell. In addition, modifications such
as acetylation and phosphorylation that can activate transactivating factors may facilitate
increased ALAS2 transcription levels via activation of the ALAS2 promoter.
rJ\;i --,
1 l,¿
The binding of erythroid-specific factors to sites within the proximal293bp ALAS2 promoter
may contribute to the increase in ALAS2 expression observed during Epo-initiated
differentiation of J2E cells in several ways. For example, the basal transcription machinery
may be positioned at the immediate promoter and binding of erythroid-specific transcription
factors may activate the ALAS2 promoter in a tissue and stage-specific manner resulting in
enhanced transcription of the gene. Binding of transcription factors to the promoter may play
a role in initiating or maintaining the ALAS2 locus in an open chromatin configuration,
ensuring the DNA is accessible to binding by other transcription factors or chromatin
modifiers required for active transcription. Since DNase I hypersensitivity was detected in the
murine ALAS2 promoter in undifferentiated MEL cells (Schoenhaut and Curtis, 1989) it is
unlikely that unfolding of the ALAS2 locus is triggered by the commencement of erythroid
differentiation. Therefore, factors may initially bind to the ALAS2 promoter in immature
erythroid cells to initiate chromatin modiffing activities that result in an open chromatin
conformation in preparation for transcription of the ALAS2 gene during erythroid
differentiation. Upon differentiation more factors may be recruited to the promoter, perhaps
by transcriptional activators akeady bound, to both stabilise the surrounding chromatin
structure, recruit transcriptional components and activate the promoter. Levings and Bungert
(2002), in their model for regulation of the human B-globin locus, proposed that the 'first step
towards activation is the partial unfolding of the chromatin structure' into a DNase I sensitive
domain and that this is initiated by the diffusion of erythroid specific factors into domains of
the chromosome that are not permissive for transcription. This event was also reported to
potentially lead to hlperacetylation of the chromatin which is associated with an open
chromatin structure. A similar mechanism can be adopted to explain unfolding of chromatin
surrounding the ALAS2 locus, although what induces the initial change in chromatin structure
in immature erythroid cells is unknown.
An interesting trend of reduced luciferase reporter activity was observed with constructs
containing distal ALAS2 promoter sequence extending further than the initial 293bp.ln
particular, the 10.3 kb distal promoter construct generated approximately the lowest level of
luciferase activity in comparison to shorter lengths of the ALAS2 promoter in J2E cells and
levels were not raised in response to Epo. This suggests that elements inhibiting
92

transcriptional activity and Epo responsiveness may be located upstream in the 5' flanking ,/region of the human ALAS2 gene, beyond the initial 293bp that has been described as the
proximal promoter. The purpose of negative regulation of ALAS2 expression is uncertain but
it maybe involved in repressing ALAS2 activity at the end of erythroid maturation where
expression of ALAS2 in the mature erythroid cell is no longer required. Negative regulatory
elements have been identified in the upstream promoter region of the human adult B-globin
gene which cause a decrease in reporter gene expression when transiently transfected into
K562 erythroid cells (Ebb et al., 199S). Site-directed mutagenesis of binding sites for the BPI
andBP2 repressor proteins in the 5' flanking region of the p-globin promoter led to an
increase in promoter activity. Repression, mediated by binding of specific transcription
factors to the repressive sequences within the distal promoter, may be required to prevent B-
globin expression in later stages of development. Similarly, a 7.3 kb deletion of sequence
from the zebrafish GATA-1 proximal promoter resulted in expression of green fluorescent
protein in neural notochord tissue (Meng et al., 1999). This indicates that negative elements in
the GATA-1 promoter of zebraftsh may play arole in preventing expression of this erythroid-
specific transactivating factor in non-haematopoietic tissues where it is not functionally
required. The presence of these sites or other negative regulatory elements within the distal l0
kb promoter sequence of ALAS2 was not investigated during the course of this studybut may
be of interest in future research into the negative
ALAS2.
As previously described, the localisation of DNase I hypersensitivity sites in introns 1, 3 and
8 of the murine ALAS2 gene, identified these introns as potential regulatory domains in the
transcriptional activation and control of ALAS2 expression (Schoenhaut and Curtis, 1989).
Surinya et al. (1998) confirmed this notion, demonstrating that intron 1 and particularly intron
8 are able to enhance transcriptional activity from the ALAS2 promoter in undifferentiated .//'
erythroid cells. In addition, intronic transcriptional enhancer elements have been identified in
several genes including those for the erythroid alpha-fetoprotein (Scohy et a1.,2000) and
interleukin-4 (Hural et al., 2000).
In undifferentiated J2E cells intron 8 but not intron 1 was shown to increase basal
transcription beyond expression obtained solely with the minimal human ALAS2 promoter.
This finding was contrary to results previously reported in which both introns were found to
have a role in enhancing transcription in K562 and MEL erythroid cells, although intron 8
was also shown to contain a stronger enhancer element (Surinya et al., 7998). The use of
93

different erythroid cell lines in the current and earlier studies may account for the contrasting
results with K562 and MEL cells, presumably providing a cellular environment in which
intron 1 can also function as an enhancer of reporter gene activity. For example, mRNA and
protein levels of the erythroid-specific transcription factor GATA-1 are lower in J2E cells
than MEL cells (Busfield et al., I995b). Although GATA-I levels increase in differentiating
J2E cells, they always remain lower than the levels observed in MEL cells. Therefore,
differences in the GATA-I levels of J2E and MEL cells may account for the contrasting
results obtained for the ability of intron 1 to act as a transcriptional enhancer in î*undifferentiatedJ2B erythroid cells. ,: " i,'
'
In contrast to basal transcription, both intron 1 and intron 8 enhanced ALAS2 promoter
activity upon Epo stimulated differentiation of the J2E cells. Intron 1 and intron 8 do not
appear to function cooperatively in either undifferentiated or differentiated J2E cells since the
inclusion of intron 1 together with intron 8 did not increase the level of luciferase activity
obtained with intron 8 sequence alone. The combination of intron 1 and intron 8 appeared to
result in a greater response to Epo than intron 8 alone. However, the level of luciferase
activity generated by this combination was lower than intron 8 alone in undifferentiated J2E
cells and this may account for the larger response to Epo in the presence of both introns.
Overall, human ALAS2 intron 8 sequence had the strongest effect on transcriptional activity/
and it was decided to concentrate on how the intronic enhancer element within this domain
functioned in response to Epo induced differentiation.
A239 bp region in the 3'end of human ALAS2 intron 8 is responsive to Epo induced
differentiation of immature erythroid cells. In the human ALAS2 gene, intron 8 is located
approximately 16 kb downstream of the -|l transcriptional start site. This same region has
been shown by Surinya et al. (1998) to function as an enhancer of transcription in
undifferentiatedK1í2 and MEL cells, In the present study, one GATA site (GATA-B) and
two CACCC boxes (CACCC-A and B), conserved between species, i;:i.-""strated as
essential to enhancer function. Mutational studies were undertaken to identiff which of the
transcription factor binding sites were required to facilitate the increase in transcriptional
activity driven by intron 8 in response to Epo induced differentiation of the J2E cell line. Both
of the CACCC sites contributed to intron 8, increasing transcriptional activity in response to
Epo stimulation. Mutagenesis of the CACCC sites in tandem reduced the level of luciferase
activity further and this may indicate that both sites are required for complete intron 8
activity, with their loss preventing any interaction between proteins binding at these two sites.
94

A number of proteins have been shown to bind CACCC boxes in vitro including the
ubiquitous transcription factor Spl (Kadoîaga et al., 1987), BKLF (Crossley et al., 7996) and
the erythroid-specific protein EKLF (Miller and Bieker, 1993). Since EKLF plays a role in
erythroid gene expression, specifically B-globin, its ability to bind the CACCC sites in intron
8 of the human ALAS2 gene was assessed. EMSAs showed that the protein complexes
forming on either CACCC site did not change when nuclear extracts derived from J2E cells
treated with Epo were utilised, indicating no change in the amount or binding activity of the
ssays using nuclear extracts prepared from either
1 cells over-expressing EKLF demonstrated that
cleotide. It is interesting that CACCC-A and
ontribute to enhancing transcriptional activity yet do
sive CACCC box has been identified in the
,i,t promoter of the murine adult B-globin gene (Miller and Bieker,1993) and EKLF has beenH''\'
shown to bind to this site (Crossley et at., 1996).In addition, a role for EKLF in adult B-
globin gene transcription has been established (Kulozik et al. l99l; Feng et al., 1994; Beiker
and Southwood, 1995; Donze et al., 1995;Nuez et al., 1995; Perkins et al., 1995; Wijerde er
al., 1996). The role of EKLF in ALAS2 expression during erythroid terminal differentiation is
poorly understood. As discussed earlier, a study by Spadaccini et al. (1998) in which an
antisense EKLF construct was introduced into lhe J2E cell line to prevent its expression and
then treated with Epo, reported a reduction in ALAS2 and B-globin transcripts. This lead to a
decrease in haem and globin synthesis which ultimately resulted in reduced haemoglobin
levels in the cell, suggesting that EKLF is required for ALAS2 expression during
erythropoiesis. Perhaps binding of EKLF to the CACCC sites in intron 8 requires interaction
with other transactivating factors such as GATA-I. Physical and functional interactions
between EKLF and GATA-1 have been reported (Merika and Orkin, 1995; Gregory et ø1.,
1996) but this type of cooperation between proteins may go undetected in in vitro binding
assays. The finding in the current study that EKLF is unable to bind to the human ALAS2 ìi. nl(
intron 8 CACCC sites was consistent with Surinya et al. (1998) which could not detect EKLF ':_
binding to these CACCC sites using undifferentiated erythroid nuclear extracts.
Since it was determined that EKLF did not bind to either CACCC site in intron 8, the ability
of Spl to bind these sites was examined. Spl has been shown to bind CACCC sequences in
vitro (Kadonaga et al., 1987) and also shown to weaklybind human ALAS2 intron 8 CACCC
sites in K562 and MEL cells (Surinya et a1.,1998). It was shown via EMSAs that Spl could
95

not bind the intron 8 CACCC-A site in vitro in either undifferentiated or differentiated J2E
nuclear extracts. The inclusion of a Spl antibody in a EMSA with the intron 8 CACCC-B
oligonucleotide and nuclear extracts prepared from COS-1 cells overexpressing Sp1 identified
one of the retarded bands as Sp 1 . A similarly retarded band of weak intensity was also
detected in EMSAs using Epo treated and untreated J2E nuclear extracts but this
corresponding band was not shifted with Spl antibody. Therefore, it appears that the retarded
bands obtained for the human ALAS2 intron 8 CACCC-A and CACCC-B oligonucleotides
using J2E nuclear extracts represent protein complexes that do not contain Sp1. Thus,
altemate proteins may be binding to these sites and contributing to transcriptional regulation
of ALAS2. The intensity of protein binding to either the CACCC-A or B site is not markedlyT
altered by Epo stimulated differentiation of the J2E cells, suggesting that binding is
independent of erythroid maturation. An inability to detect Spl bindingmay also be due to
low Spl levels in the J2E cells, especially since this study showed CACCC-B could bind Spl
in COS-I cells overexpressing Spl and in addition Surinya et al. (1998) also showed that
CACCC-A and CACCC-B could bind Sp1 in vitro.
Thus the available data suggests that other transcription factors may be binding to the
CACCC sites and driving intron 8 to enhance transcription of ALAS2 when red blood cell
differentiation is stimulated by Epo. This may include other members of the Sp family of
transcription factors or alternatively, transcription factors within the Kruppel family of
transcriptional activators. Proteins that bind the CACCC sites of other erythroid genes
BKLF which has been shown to bind the CACCC site in the promoter of the human ankynn-
gene and increase transcriptional activity (Gallagher et a1.,2000). In addition, the recently
cloned members of the Kruppel family of transcription factors, FKLF and FKLF-2,have
shown to transactivate several erythroid genes including, PBGD, GATA-1, glycophorin B,
ferrochelatase, human embryonic and fetal B-like globin genes (Asano et al., 7999,2000).
Lastly, a novel transcriptional activator maybe binding the CACCC elements in intron 8 and
contribute to the transactivating ability of this region during erythroid differentiation.
The greatest decrease in Epo induced expression was caused by mutagenesis of the GATA-B
site in intron 8 of the human ALAS2 gene. This trend was repeated in both Epo treated and
untreated J2E cells and promotes GATA-B as the critical site for intron 8 enhancer activity in
response to intron 8. GATA-1 bound the GATA- B oligonucleotide whether nuclear extracts
were prepared from Epo stimulated or unstimulated J2E cells. This was not surprising as
Schoenhaut and Curtis (1989) reported the presence of a DNase I hypersensitivity site at the 3'
96

end of murine ALAS2 intron 8 prior to differentiation. This finding, together with the current
study, indicates that the ALAS2 gene is most likely in an open region of chromatin in
preparation for transcriptional activation before the immature erythroid cell is stimulated by
Epo to undergo differentiation. GATA-1 levels have been shown to increase during erythroid
maturation (Sposi et al., 1992; Leonard et al., 1993; Busfield et al., 1995b). However the
findings of this study show that binding of GATA-I to the GATA-B site in intron 8 of the
ALAS2 gene is unaffected by the differentiation state of the J2E cells. Since Epo does not
affect the binding of GATA-I to the GATA-B site the question arises as to how GATA-I in
response to Epo might cause an increase in ALAS2 transcriptional activity. Post-translational
modifications to the GATA-1 protein, such as acetylation and phosphorylation, may play a t fiJlrole in enhancing the transactivating ability of GATA-I. Firstly, it is well known that GATA- á1 interacts with and is acetylated by CBP/p300 in vivo and that this action leads to a direct
stimulation of GATA-I dependent transcription (Blobel et al., 7998; Boyes et q1.,1998).
Hung et al., (1999) reported that acetylation of GATA-1 by CBP/p300 does not affect the/
DNA binding capacity of GATA-1, although earlier findings suggested that DNA binding /increased upon CBP/p300 acetylation of GATA-I (Boyes et al., 1998). Secondly, the
phosphorylation of GATA-I has been reported in non-induced MEL cells with
phosphorylation of additional residues upon chemically stimulated differentiation (Crossley
and Orkin, 1994). Phosphorylation of GATA-1 appeared to have no affect on DNA binding
affinity or specificity or transactivation of transcription by GATA-I. However, the effect of
GATA-l phosphorylation on its DNA binding ability and function is unclear as Partington
and Patient (1999) reported a rise in the level of phosphorylated GATA-I after chemical ,/induction of K562 cells and this resulted in increased DNA binding affinity. Alternatively, the
ability of GATA-I to interact with itself or other transactivating factors may be enhanced by
phosphorylation of the protein, ultimately leading to increased transcriptional activity of the
target gene.
Surprisingly, the increase in reporter gene activity observed with mutagenesis of the GATA-A
site suggests that this binding site may function as a negative element, inhibiting transcription
of the ALAS2 gene. Although the GATA-A site within intron 8 was shown to be repressive in
mutational studies, EMSAs demonstrated that GATA-I still binds to this site using nuclear
extracts prepared from Epo treated and untreatedJ2E cells. These findings suggest thal in
vitro GATA-1 binds to the GATA-A site in intron 8 and acts to repress transcriptional
activity. Previous studies have shown that GATA-I can repress promoter activity by binding
to its target sequence within a regulatory region of a given gene. For example, located near
97

the HS5 site of the human B-globin LCR is a tandem GATA repeat that binds GATA-I and
represses HS2 enhancer activity in reporter gene assays (Ramchandran et al., 2000).
Preliminary studies have also been conducted proposing that GATA-1 can negatively regulate
expression of the non-erythroid anti-mullerian hormone gene in differentiating Sertoli cells of
the testes (Beau et a1..2000).
The present study has shown that GATA-B can bind GATA-I in vitro and that the function of
this site is crucial for enhancing transcriptional activity of the human ALAS2 promoter during
Epo stimulated differentiation. However, the intensity of binding and the pattern obtained for
GATA-B is unaltered by Epo stimulation of J2E cell differentiation, suggesting the rise in
transcriptional activity in response to Epo is not due to altered GATA-I binding. As discussed
above, modifications to the GATA-I protein bound at the GATA-B site may account for the
observed increase in transcriptional activity.
-, ,. , ,,1
Since GATA-1 is acetylated by the coactivators CBP/p300 and this results in enhanced 1*..
GATA-I activity, the effect of CBP/p300 on ALAS2 transcriptional regulation was ,,1
investigated. Using E1A as an inhibitor of CBP/p300 activity, experiments were conlucted to
ascertain the effect of this coactivator complex on regulation of ALAS2 transcription. Marked
inhibition by E1A was observed with the construct containing ALAS2 intron 8 and promoter
sequence but not with the promoter alone, implicating the enhancer element in intron 8 as a
major target for ElA. Furthermore, a mutant form of ElA, which is unable to bind CBP/p300,
supported this proposition. It has been reported that interfering with CBP/p300 function
through E1A blocks erythroid differentiation and this may explain the reduction in
transcriptional activity observed with the wild type ElA construct (Blobel et al., 1998).
However, a role for CBP/p300 in regulating ALAS2 expression was not supported by data
obtained from experiments in which reversal of E1A inhibition was attempted by the co-\
transfection of p300 deletion mutants unable to bind E1A or expression of exogenous p300 in )
order to transactivate ALAS2 intron 8. Since the studies conducted in regard to the role of /¡,¡ì i,._
CBP/p300 in the transcriptional regulation of ALAS2 by intron 8 were inconclusive, further t-,. "'
investigation is required to determine the precise effect of CBP/p300 on intron 8 enhancer , '''"'Y..o.
activity. í "t'*
To explore the role of CBP/p300 in ALAS2 transcriptional regulation further the initial step
would involve selection of an erythroid cell line that has low endogenous levels of CBP/p300
An erythroid cell line is required since the ALAS2 intron 8 enhancer is urnesponsive in non-
98

erythroid cells (Surinya et aL.,1998). In addition, low levels of endogenous CBP/p300 would
better facilitate experiments in which CBP/p300 wild tlpe and binding mutants were
exogenously expressed. For example, in the current study co-transfection of p300 at
concentrations of 500ng or less had no effect on intron 8 enhancer activity but it is unclear as
to whether this was a real effect or due to the maximal effect of CBP/p300 on intron 8 activity
being met by endogenous levels of the coactivator. An erythroid MEL cell line was employed
by Forsber g et al., (1999) to investigate the ability of CBP/p300 to facilitate B-globin HS2
mediated transactivation. Such a cell line could be selected for repeating experiments /'
involving E1A and CBP/p300 wild type and mutant proteins to determine if the effects
observed previously were cell line specific to the J2E cells. An additional consideration in the
selection of an appropriate erythroid cell line for future work investigating the effect of
CBP/p300 on ALAS2 expression is its differentiating capacity. A cell line that is responsive
to the natural stimulator of erythropoiesis as well as low endogenous levels of CBP/p300 ,ì,,'' "
would be ideal.
There are several mechanisms to account for CBP/p300 activity in erythroid differentiation.
The first of these describes CBP/p300 behaving as a bridge between transcription factors
binding at regulatory elements of erythroid-specific genes and the basal transcription
machinery bound at the immediate promoter. In this situation CBP/p300 is recruited by the
DNA bound transcription factors. CBP has been shown to interact with TFIIB (Kwok et al.
1994), TBP (Abraham et al., 1993; Swope et al., 1996; Dallas et al., 1997; Sang et al., 1997) 7'and RNA polymerase II (Kee et al., 1996;Nakajima et ql., 1997 ; Cho et al., 1998) as well as
the erythroid-specifc transcription factors GATA-1 (Blobel et al., 1998; Boyes et al., 1998;
Hung et al., 1999), NF-E2 (Hung et a1.,2001) and EKLF (Zhang and Bieker, 1998). Since the
ALAS2 promoter was insensitive to EIA treatment and it was indirectly shown that intron 8
interacts with CBP/p300 it indicates that this coactivator may provide a link between the
transcription factors binding within the intron 8 enhancer and the ALAS2 proximal promoter.
In addition, CBP/p300 may contribute to intron 8 enhancer activity by interacting with the
GATA-I transactivating factors bound at intron 8 during the differentiation process and acting
as a bridge to activate the basal transcription machinery positioned at the ALAS2 promoter.
This could be investigated by mutating the GATA sites in the intron 8 enhancer and 'f il I
*-t,E' r¡J' '
determining the effect of ElA and CBP/p300 expression constructs on the level of intron 8 , 'u
r€.rç ..¡,,
enhancer activity. -1i"tti'o.- ôt" i'l*'';i t'""-l
'- rQ rf 6'r
rr 't.i
r,
,.-..r
':99

The second function proposed for CBP/p300 is acetylation of histones which leads to an
alteration in chromatin structure (Bannister and Kouzarides, 1996; Ogryzko et al., 1996;
Goodman and Smolik, 2000). As it is well documented that CBP/p300 harbours intrinsic
HAT activity that modifies chromatin structure via acetylation it would be of interest to
determine if histone acetylation is required for ALAS2 intron 8 enhancer activity.
Experiments could be conducted using the histone deacetylase inhibitor, trichostatin A (TSA),
to determine if abrogation of histone deacetylase activity will increase activity generated by
the intron 8 enhancer and promote transcriptional activation of the ALAS2 gene. In order to
investigate the interaction between CBP/p300 and the intron 8 enhancer and the effect of
histone acetylation and altered chromatin structure, future experiments need to be conducted
within the context of a chromatin environment.
Although results from the current experiments involving the effect of CBP/p300 on ALAS2
intron 8 enhancer activity have been inconclusive a speculative model can be proposed
regarding the role of coactivators in the transcriptional regulation of ALAS2. Upon Epo
signalling the erythroid cells are induced to differentiate and the expression of ALAS2 is
required. CBP/p300, possibly together with additional coactivating factors, form a complex
with erythroid-specific transcription factors bound at the enhancer element within intron 8 i)
such as GATA-1. This complex then acts as a link between transcription factors and the basal
transcription machinerybound at the ALAS2 immediate promoter and the intron 8 enhancer
element, leading to establishment and maintenance of the transcription-pre-initiation complex
on the ALAS2 promoter. This may also facilitate the binding of additional transcription
factors to activate ALAS2 gene expression. Alternatively, CBP/p300 may acetylate histones
in chromatin surrounding the ALAS2 locus to generate a nucleosome-free locus in
preparation for transcription of the erythroid genes required for differentiation. This would
allow both the binding of erythroid transcription factors to the intron 8 enhancer element and
interaction with the promoter to stimulate ALAS2 transcription. However it appears that the
ALAS2 locus is in an open chromatin environment prior to Epo induced differentiation
(Schoenhaut and Curtis, 1989). Lastly, CBP/p300 may directly acetylate GATA-I, increasing
its activating capabilities. GATA-I would then bind the ALAS2 gene, perhaps in the enhancer
element in intron 8, to enhance transcription of the ALAS2 promoter. Future research to
determine the acetylation status of the ALAS2 proximal promoter and intron 8 enhancer in
erythroid cells differentiating in response to Epo will provide a strong model for the role of
CBP/p300 in the regulation of ALAS2 gene expression.
it
100

This study has demonstrated that intron 8 contains the most Epo responsive element of the
human ALAS2 gene in differentiatingJ2B erythroid cells. The mechanism by which intron 8
acts to enhance transcription of ALAS2 above basal levels in both immature and
differentiated erythroid cells is unclear. Currently there are two models on how enhancer
elements within a gene locus can act to enhance transcription in a temporal dependent manner
such that gene expression is elevated to coordinate with cellular events like differentiation.
Studies have primarily focused on how the LCR of the B-globin locus stimulates globin gene
transcription. The human B-globin locus contains five genes, e-globin, two y-globins, ô-globin
and B-globin, which are affanged in their order of expression during development
(Stamatoyannopoulos and Nienhuis, 1994). The LCR consists of five DNase I
hypersensitivity sites and is located upstream of the e-globin gene, spanning 6 to 22kb (Tuan
et a|.,1985; Forrester et al., 1986). The first of these models has been termed the 'looping /model' and is defined by direct interaction between transcriptional activators and chromatin
modif ing factors bound at the promoter and those attached to the LCR, such that the
intervening DNA is looped out (Engel and Tanimoto, 2000). The second model reported
relies on the binding of required transcription factors and chromatin modiffing proteins
throughout the locus to define it as region to be transcribed. These bound proteins are linked
to one another by protein-protein interactions to form a chain of complexes between two
regulatory regions and is referred to as the 'linking model'. An example of this is a chain of
chromatin modifuing factors forming from the LCR to the B-globin promoter linking the
transactivating factors that are bound at these domains (Bulger and Groudine,1999; Dorsett,
1999). Therefore, according to the models used to describe LCR activation of the B-globin
locus, intron 8 may function as a transcriptional enhancer of human ALAS2 by either directly
interacting with factors bound at the 293 bp ALAS2 promoter and looping out intervening
DNA (the looping model) or generating a chain of activating protein complexes along the
ALAS2 locus from intron 8 to the promoter (the linking model).
Recent evidence from studies on the role of enhancers in gene transcription have challenged
the traditional notion that enhancers act to increase the rate of transcription of a linked gene.
Studies employing single cell expression assays with either transient or transgenic mice
experiments have demonstrated that it is the number of cells expressing the reporter gene that
increases in the presence of an enhancer not the rate at which transcription is occurring
(V/alters et al., 1995,1996; Sutherland et al., I99l). Therefore, it has been proposed that
enhancers function to increase the probability of forming and maintaining a stable, active /chromatin domain without affecting the rate of transcription. Similarly, in vitro single cell
101

expression assays could be conducted in the future to determine if human ALAS2 intron 8
acts to increase the probability that an erythrocyte will express ALAS2 during erythropoiesis
or alternatively, enhance the overall rate of transcription.
ln conclusion, this study showed that a regulatory enhancer element within intron 8 enhances
ALAS2 transcription levels during Epo stimulated differentiation of the erythroid J2E cells. In
particular, a GATA site within intron 8, termed GATA-B, was critical for the function of
intron 8 as an enhancer in trànscription of the ALAS2 gene. Presently the effect of modifuing
coactivators such as CBP/p300 on ALAS2 transcription is unclear. Based on current evidence
two models, 'looping' and 'linking', have been proposed to explain the mode by which
enhancers interact with promoters to facilitate transcription. It is unknown which mechanism,
if either, intron 8 utilises to interact with the promoter region in its role as an enhancer of
ALAS2 gene transcription or whether intron 8 acts to either increase the rate or probability of
ALAS2 expression.
r02

CHAPTER 4
GENERATION OF A MURINE MODEL FOR X.LINKED SIDERBLASTIC
ANAEMIA

CHAPTER 4: GENERATION OF A MURINE MODEL FOR X-LINKED
SIDEROBLASTIC ANAEMIA
4.1 INTRODUCTION
Sideroblastic anaemias are a g:oup of rare, inherited and acquired disorders that exhibit varied
degrees of anaemia. They are characterised by the presence of iron-laden mitochondria
surrounding the nucleus of developing erythroblasts in the bone marrow. These defective cells
are termed ringed sideroblasts. A small proportion of these defective erythroblasts give rise to
mature, circulating red blood cells that are tlpically hypochromic and microcytic, reflecting a
deficit in normal haemoglobin complement (Bottomley, 1998a). The varying degrees of
anaemia and resultant hypoxia caused by sideroblastic anaemia triggers the release of Epo
from the kidney to stimulate differentiation of immature red blood cells in the bone marow.
Since many of the cells produced are defective in their maturation and are destroyed in the
bone marrow, the anaemia persists, stimulating further release of Epo to overcome the
resultant anaemia. This generates a cycle of abnormal red blood cell development known as
ineffective erythropoiesis (Sadlon et al., 1999). Linked to ineffective erythropoiesis, via an
unknown mechanism, is an increase in intestinal iron absorption. Once transferrin levels are
saturated iron begins to accumulate in the body and is referred to as iron overload or
erythropoietic haemochromatosis. If left untreated the build up of iron in the organs of the
body, particularly the heart and liver, can result in organ failure and death (Bottomley,
1 998a).
This study has focussed on the most common form of the inherited disorder, X-linked
sideroblastic anaemia (XLSA) (Bottomley, 1998a). XLSA predominantly affects males with
carrier females occasionally exhibiting anaemia, most likely as a result of skewed inactivation
of the wild type X chromosome (Bottomley et al., 1998; Cazzola et a1.,2000). As previously
discussed (see Section 1.10.2), mutations in the ALAS2 gene are associated with the majority
of XLSA cases. In this model, mutations in the ALAS2 gene decrease formation of 5-
aminolevulinate which leads to reduced levels of protoporphyrin IX in the developing
erythroblast. Therefore, if protoporphyrin levels are low the cell's requirement for iron is
reduced. Despite this, iron complexed to transferrin continues to be imported at normal levels
into the cell via the cell surface expressed transferrin receptor (May et al., 1995). Therefore,
unlike non-erythroid cells where there is tight control between iron uptake and utilisation, iron
import into erythroid cells can be uncoupled from utilisation. Iron that is not utilised, due to
103

defective haem synthesis, accumulates in the mitochondria to produce the characteristic
ringed sideroblast observed in the bone marrow of XLSA patients. In normal differentiating
erythroid cells haem regulates the translation of globin mRNA by inhibiting the haem-
regulated erythroid-specific eIF-2akinase (HRI) that inactivates the translation initiation
factor, eIF-2a (Chen et al., 1989; Crosby et al., 1994). Since cellular haem levels are low,
globin mRNA translation levels are also reduced and this further contributes to abnormal
erythroid development. As described above, a cycle of ineffective erythropoiesis, linked to
increased intestinal iron absorption, is generated and this eventually leads to erythropoietic
haemochromatosis.
The aim of this project was to generate a murine model for the human disorder XLSA. The
primary objective of developing such a model was to conclusively establish that mutations in
the ALAS2 gene are responsible for XLSA. The hallmark features of XLSA would be
examined using the murine model. This would include haematological parameters such as
haemoglobin levels and bone maffow smears to identiff red blood cell abnormalities in the
form of ringed sideroblasts. Iron levels in the serum and tissues, together with the degree of
intestinal iron absorption would be monitored. XLSA mice would also be assessed for
pyridoxine responsiveness and the model would facilitate investigation of how pyridoxine
alleviates the synptoms of XLSA, In addition, ALAS2 enzyme activity and mRNA levels
would be examined as well as expression of other erythroid-specific genes to determine the
consequence of reduced haem production on expression of these genes in XLSA mice.
úrterestingly, the age of onset of the clinical symptoms for XLSA varies from young children
to late adulthood. The presence of a mutant ALAS2 protein with decreased activity or
stability suggests that the XLSA phenotype (anaemia and red blood cell abnormalities) would
be present from birth, however patients can present with symptoms late in life (Cox et al.,
I994;Bottomley, 1998a). Thus, the XLSA murine model may enable investigation of the
phenomena of late-presenting XLSA patients. The XLSA murine model would facilitate
examination of the defects in iron metabolism observed with XLSA. For example, the link
between ineffective erythropoiesis and increased intestinal iron absorption within XLSA
could be investigated, particularly the molecular components involved in up-regulation of iron
absorption from the intestine in response to erythropoiesis. Lastly, a XLSA murine model
could prove to be beneficial for the development and improvement of treatments for XLSA.
Alternative methods to iron chelation therapy and phlebotomies for treating iron overload
could be developed and sampled using the XLSA animal model. Ultimately a gene therapy
r04

approach could be investigated to assist severe cases of XLSA that are non-responsive to
current treatments and the murine model could be utilised to test its validity.
As mentioned in Sectionl.l0.2, a heterogeneous group of point mutations in the ALAS2 gene
of XLSA patients have been identified (Figure 1.11). Two different point mutations in the
ALAS2 gene that have been linked to cases of XLSA were selected as the basis for the
development of a murine model for XLSA. Both of these mutations result in a single amino {t'c': ¡-'
acid substitution in the protein. The first is a cytosine to guanosine mutation at position I2l5
in exon 8 of the human ALAS2 gene, resulting in a threonine to serine amino acid substitution
at residue 388 of the protein (Cox et al., 1994) (Figure 4.1). This residue is highly conseryed
in all known ALAS2 proteins (Ferreira and Gong, 1995). The male proband first presented
with anaemia, haemoglobin levels at llgldl, and bone marrow ring sideroblasts at 55 years of
age. Treatment with pyridoxine overcame the anaemia and therapy ceased. At 69 years, the
patient again presented with severe anaemia (haemoglobin at 6.29/dl), ring sideroblasts (50%
of erythroid precursors) and iron overload (serum iron concentration 220% normal, transferrin
saturation 98%). Pyridoxine (300mg/day) was administered and haemoglobin levels returned
to normal with ringed sideroblasts rarely observed within 2 months of treatment. It was
determined that 4mglday of pyridoxine was required to maintain haemoglobin at normal
levels (Cox et al., 1994). ALAS activity in bone maffow lysates prior to pyridoxine treatment
ranged from 29Yo to 36Yo of normal enzyme activity and this was raised to 49o/o to 5lYo by the
addition of PLP to the lysate. During pyridoxine treatment, ALAS activity in the bone marrow
was within the normal range of enzyme activity (Cox et al., 1994). Genetic linkage studies
demonstrated a X-linked pattern of inheritance for this case study. A highly polymorphic CA
repeat motif in intron 7, in addition to other X-linked polymorphic markers, were used to
show transfer of a particular X chromosome through to an affected grandson and nephew and
obligate carrier daughter, but was not present in unaffected male family members (Cox et al.,
ree4).
The second mutation selected occurs in exon 9 of the human ALAS2 gene. It is a cytosine to
thymine point mutation at position 1283 and encodes an arginine to cysteine amino acid
substitution at residue 4Il, ahighly conserved site (Figure 4.1). The initial report of the
mutation in the ALAS2 gene of a XLSA patient with severe anaemia was by Cottet et ql.
(1992a) but the resultant phenotype was not characterised. More recently, Furuyama et al.
(1998) documented the same mutation in a 15 year old Japanese male proband. He presented
with severe anaemia (haemoglobin levels at4.8g/dl) and ring sideroblasts at22Yo of total
105

Figure 4.1 Location of the XLSA associated point mutations in exon 8 and exon 9 of the
human and mouse ALAS2 gene
Location of the two mutations chosen as a basis for the XLSA murine model. Represented at
the top are the human and mouse ALAS2 genes showing exon/intron structure. The exons are
represented as filled boxes. Initiation (ATG) and termination (TAA) codons are shown in
exons 2 and 11, respectively. The exon 8 mutation leads to a threonine to serine substitution at
position 310 and 388 in the mouse and human ALAS2 genes, respectively. The second
mutation occurs in exon 9 and results in a arginine to cysteine substitution at position 333 in
the mouse and 4ll in the human ALAS2 gene. Both mutations (boxed in blacþ reside in a
highly conserved region of the ALAS2 gene. Pyridoxal phosphate, the cofactor to ALAS2,
forms a Schiff base with an invariant lysine in exon 9 which is highlighted by the grey shaded
box. The position of exons 8 and 9 within the ALAS2 gene are also represented.
H:Human; M:Murine
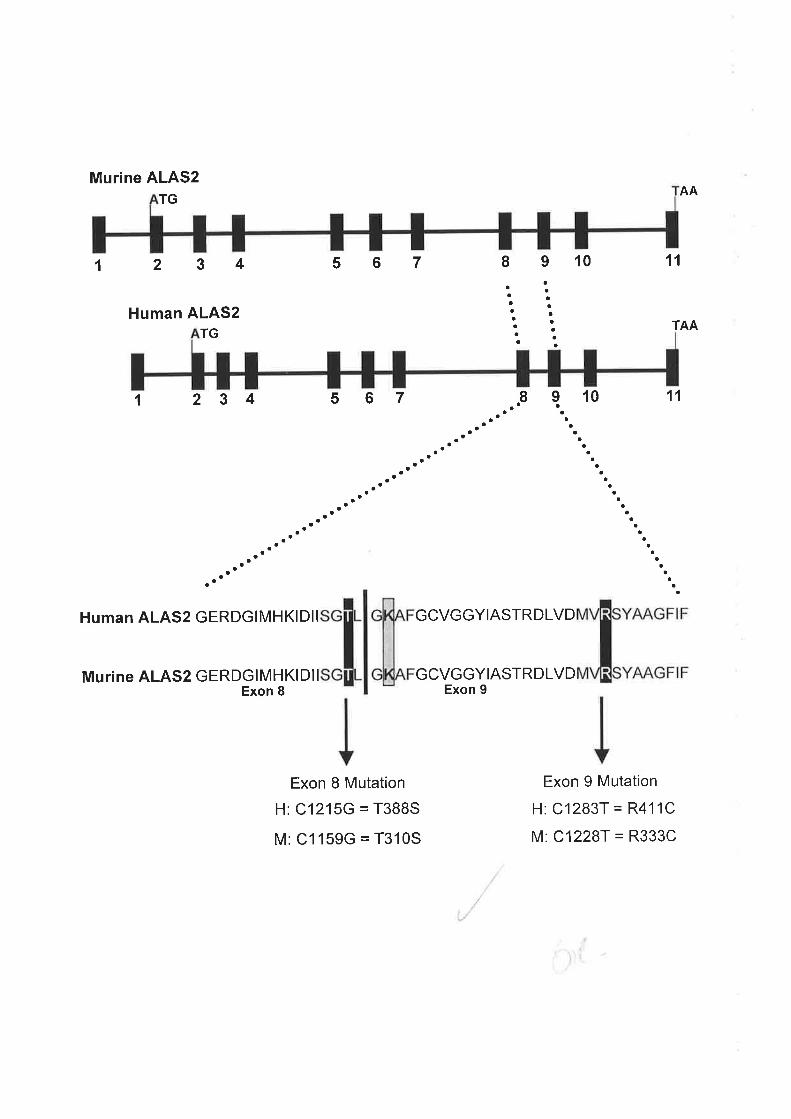
Murine ALAS2TG
1 234
Human ALAS2TG
1 2 3 4
Human ALAS2 GERDGIMHKIDI I
Murine ALAS2 GERDGIMHKI Dl I
Exon 8
567
s67
Exon 8 Mutation
H: C1215G = T388S
M: C1159G = T310S
8910
8 910
GCVGGYIASTRDLVD
GCVGGYIASTRDLVDExon 9
aa
aa
aa
aa
aa
aa
aa
aa
aa
aa
aa
aa
aa
aa
AA
'11
TAA
aaaaaaaaa
aaaaaaaa
aaaa
aaaaa
aaaaa
a
11
aaaaaaa
aaaaaa
aaaa
aaaa
Exon 9 Mutation
H: C1283T = R411C
M: C1228T = R333C

nucleated bone marrow cells. Assessment of serum iron demonstrated excess levels in the
blood. After 2 weeks of pyridoxine treatment (200mg/day) haemoglobin levels were raised to
T.sgldlwith a level of 10g/dl reached after five months. Pyridoxine treatment was continued
but the anaemia did not improve any further. "Ì
,, a.it"'I
These two mutations were selected as a basis for the XLSA models for several reasons.
Firstly, both mutations are associated with severe anaemia in humang which increases the)
likelihood that the effects of the mutations will be exhibited in the fnice. Secondly, both
mutations are partially responsive to pyridoxine in humans. Thus, if defects in the mice are
due to the ALAS2 mutations then it would be expected that the mice would be partially
pyridoxine responsive ;',, ') l
Thirdly, the mutations lie in a region of the ALAS2 gene that is highly conserved between ,f.'humans and mice. Therefore it is predicted that mutations that affect human ALAS2 should
also affect the mice protein. In support of this, our laboratory introduced the human mutations
into the murine ALAS2 protein and expressed them ínE. coli to determine if the mutations
also affect murine ALAS2. Measurement of protein levels and ALAS2 activity demonstrated
that the equivalent mutation (T310S) in the murine ALAS2 gene resulted in a 50olo reduction
in ALAS2 activity when compared to wild type murine ALAS2. Furthermore, there was no
difference in expression levels between the mutant and wild type ALAS2 protein. This /finding was in agreement with an earlier study in which a decrease in activity was observed
when the XLSA associated T388S mutation was introduced into the human ALAS2 gene and
expressed in E. coli (Cox et at., 1994)..
úrtroduction of the R333C mutation into the murine ALAS2 protein and expression in E. coli
produced u??% decrease in ALAS2 activity in comparison to wild type levels. In E. coli,
only very low expression levels of the murine R333C protein compared to the wild type
ALAS2 protein was obtained, indicating the mutation may destabilise the protein (Chandler,
1996). Similar experiments were performed by Furuyama et al. (1998) in which the
equivalent R411C mutation was introduced into the human ALAS2 protein and expressed in
E. coli. Activity of this ALAS2 mutant was 72Yo and25o/o of wild type levels in the absence
and presence of PLP, respectively. As both mutations decreased murine ALAS2 activity it
was feasible to continue with targeting of the ALAS2 gene.
106

Various gene targeting approaches were trialed in order to introduce the mutations into the
murine ALAS2 locus and generate an animal model for XLSA. The following chapter
documents the success of the methods employed.
4.2 RESULTS
4.2.lTargeting of the ALAS2 Locus in E14TG2a ES Cells
Initially, a two-step gene targeting approach was employed to introduce the XlSA-associated
mutations into the ALAS2 gene (Melton,1994) (Figure 4.2). This strategy involves a first
round of targeting to insert a selectable marker into the gene of interest within ES cells. To
achieve this a targeting construct is generated that contains two regions of homology to the
target gene separated by a selectable marker, which for the pu{poses of this study is the
hypoxanthine-guanine phosphoribosyltransferase (HPRT gene). The construct is then
introduced into a HPRT/- ES cell line. Cells that have incorporated the targeting construct
into their genome are selected for by incubating the cells in hypoxanthine, aminopterin and
thynidine (HATs) media. Cells lacking a functional HPRT gene due to an unsuccessful
recombination event are selected against in HAT media (Bronson and Smithies, 1994). HATs
resistant colonies are then screened by Southern blot analysis for cell lines that have
successfully undergone homolo gous recombination.
The ES cell lines that have correctly incorporated the HPRT cassette into the gene of interest
in the first round of targeting are then utilised in the second targeting event. This round of
targeting involves the introduction of a subtle alteration to the gene, such as a single
nucleotide change. Exchange of the HPRT gene at the target locus for the second round
targeting vector (alteration vector) by homologous recombination can be selected for by
treatment with 6-thioguanine (6-TG). Cells that have successfully introduced the specific
alteration into the gene will be determined by a Southern blot screen of genomic DNA.
The targeting strategy described above was initially used to introduce the exon 8 (Cl159G)
mutation into the ALAS2 locus of the murine genome. This work was initiated during my
honours degree in the laboratory of Dr Brian May. For the remainder of this chapter, the
ALAS2 exon 8 and exon 9 XlSA-associated point mutations will be referred to as Cl159G
andCl228T, the equivalent nucleotide changes in the murine ALAS2 gene' i'i li'
t07

Figure 4.2 Afwo step gene targeting strategy to introduce point mutations into a specifÏc
ES cell gene
In the first step, the HPRT gene is introduced into a gene of interest within the genome of a
HPRT- ES cell line. Cells that have undergone homologous recombination and incorporated
the HPRT cassette into their genome are selected for and used in the next round of targeting.
In the second step, the HPRT cassette within the gene of interest is exchanged for the
targeting vector containing a subtle alteration to the genomic sequence. Thus, an ES cell line
harbouring a mutation in a specific gene is generated and can be used to produce mice
carrying the particular mutation. The star is representative of the specific mutation introduced.

Target Gene in HPRT-ES Gell Genome
STEP 1
HPRT TargetingConstruct
lnactivated Gene
HPRT
HPRT
STEP 2
AlterationVector
Altered Gene in ES CellGenome

In order to generate a murine model for XLSA the feeder independent, male, HPRT null ES
ce|l line, El4TG2a(Thompson et aL.,1989) was utilised in the first round of gene targeting.
The first round (completed in our laboratory by Dr. T. Sadlon) involved insertion of a 6 kb
HPRT minigene cassette (Reid et a1.,1990) between exons 8 and 9 of the ALAS2 gene such
that a portion of each exon (covering both XLSA mutations) rwas removed (Figure 4.3). HATs
resistant ES cell colonies were screened by Southern blot analysis for correct insertion of the
HPRT cassette into the ALAS2 locus of theBT4TG2agenome. Two ES cell lines containing
the HPRT cassette in the ALAS2 locus were identified, R3 and R4, from 720 HATs resistant
colonies screened. Since the ES cell line employed in the study was generated from a male
embryo and the ALAS2 gene resides on the X chromosome, only a single copy of the ALAS2
gene was present in the cells.
The C1l59G point mutation was introduced into exon 8 of the ALAS2 gene to generate a
targeting construct (pmALAS2/C1159G) containing approximately 7 kb of mouse ALAS2
genomic DNA, ranging from exon 7 (XhoI) to intron 10 (SacI) (see Section2.4.l for
construction details). To generate an ES cell line harbouring the XlSA-associated point
mutation in exon 8 of the ALAS2 gene the next round of gene targeting was conducted in the
R4 HPRT-positive ES cell line produced from the first targeting event. This R4 targeted ES
cell clone was selected for the second round as upon blastocyst injection of the ES cell line a
greater percentage of chimaerism was observed in the resulting mice on the basis of coat
colour genetics when compared to the R3 clone. A high percentage of ES cell contribution to
the resulting chimaeras is thought to correlate with a high rate of germline transmission
(Stewart, 1993). Genomic DNA harvested from 28 6-TG resistant ES cell colonies was
screened by Southern hybridisation analysis to identiff cell lines that had undergone
homologous recombination, exchgnging the HPRT minigene for the ALAS2 targeting ''-ù
construct containing the exon 8 point mutation. Six ES cell lines containing the Cl159G
XlSA-associated point mutation in exon 8 of the ALAS2 gene were generated. As previously
mentioned, the work described above was conducted during my honours' degree. The
remainder of this chapter pertains to work conducted during the course of my PhD.
Two ES cell lines harbouring the exon 8 mutation in ALAS2, F2-Cl159G and H3-Cl159G'
were selected on the basis of their karyotype for injection into the inner cell mass of mouse
blastocysts. The karyotyping (Section 2.7.7) of the ALAS2 targeted ES cell lines was
performed to avoid cell lines which contain a high proportion of aneuploid cells which
108

Figure 4.3 The strategy employed for targeting of the ALAS2 locus in E14TG2a cells
A two step gene targeting approach to introduce a XLSA associated mutation into the ALAS2
gene of an ES cell line was described in Figure 4.2.lnthe first round of targeting an ES cell
line was generated that contained the HPRT selection cassette in the ALAS2 locus. The
cassette was inserted between exons 8 and 9, removing part of each exon and all of intron 8.
This ES cell line was used in the second round of targeting to produce ES cell lines that had
exchanged the HPRT minigene for an alteration vector carrying either point mutation
(C1159G or Cl228T) in the ALAS2 gene. Both mutations are indicated on the diagram of the
ALAS2 alteration vector and the mutated ALAS2 locus for the purpose of explaining the
targeting strategy but the constructs utilised in the experiments contained either of the XLSA
associated mutations.
E)l/'
tX
ti
t| ];/I.lu(
o93'''-ì\ '1"''
I;tlI
q
{
,),'ì \
I,.4-.-
rf".w
ir t
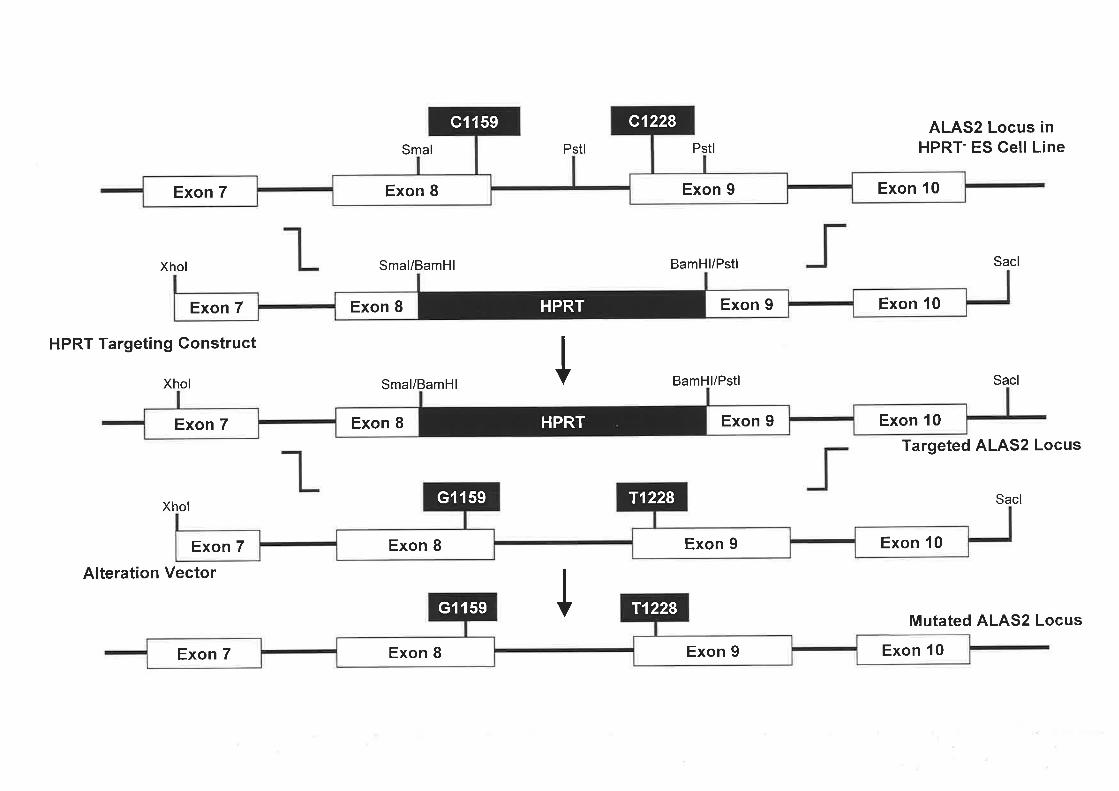
Exon 10Exon 9Exon 8Exon 7
c'1228c1159
Smal
Smal/BamHl
Pstl Pstl
BamHl/Pstl
ALAS2 Locus inHPRT-ES Gell Line
SaclXhol
Exon 10Exon 9Exon 8Exon 7 HPRT
HPRT Targeting Construct
Xhol Smal/BamHl I BamHl/Pstl Sacl
Exon 10Exon 9Exon 8Exon 7 HPRT
Xhol
Alteration Vector
Targeted ALAS2 Locus
Sacl
Mutated ALAS2 LocusIExon 10Exon 9Exon 8Exon 7
T1228G1 159
Exon 10Exon 9Exon 8Exon 7
T1228G1159

ultimately leads to the formation of weak chimaeras (Stewart .'1993). The karyotype of F2'
C1 159G and H3-Cl 159G was determined tobe 86Yo and 89Yo, respectively. Each line was
injected into F2CBA/C17b| niurine embryos at the blastocyst stage of development and
reimplanted into pseudo þiegnant Balb/c female mice to enable the targeted ES cells to
contribute to the growth of the embryo, generating chimaeric mice that are derived from both
the host embryo (F}CBNC57bI) and the manipulated E14TG2a ES cells (Melton, 1994). The
chimaeric mice are identifiable by their coat colour. Pups that were determined to be greater
than 5o/o chimaeric on the basis of their coat colour were mated with Balb/c mice to produce
progeny in which the injected ALAS2 targeted ES cells have contributed to the germline.
From blastocyst injection of the H3-C1159G ES cell line 52 chimaeric pups were born. Nine
male mice, exhibiting between 5 to 600/o chimaerism, rwere mated with female Balb/c mice.
Germline transmission of the mutant ALAS2 gene through the male chimaeric mice will
produce obligate female carriers of the mutation and males with a wild type loci. Germline
transmission was determined by the pattern of genetic inheritance of coat colour. Four of the
chimaeric male mice were deemed sterile as no litters were obtained after several mating
attempts with different female Balb/c mice (Figure 4.4a). No germline transmission by the
remaining five male chimaeric mice was observed after multiple litters indicating that H3-
Cl 159G cells contribute very poorly to the germline (Figure 4.4a).
Injection of the F2-CII59G targeted cell line into mouse blastocysts produced 34 pups with a
low level of chimaerism. Six of the male mice, exhibiting 5%o to 10% chimaerism, were
selected for mating with Balb/c female mice. Each chimaeric mouse produced at least three
litters but no germline transmission was obtained with the F2-Cl159G ES cell line (Figure
4.4b). Thus, germline transmission of the exon 8 C1159G mutant ALAS2 gene was ./.
unsuccessful using either the ALAS2 targeted H3-C1159G or F2-C1159G ES cell lines. /
As stated above, the second round of gene targeting utilised the ALAS2 targeted HPRT-
positive R4 ES cell line that had been generated from the parentalBl4TG2a ES cell line in
the first targeting step. This cell line was selected over the R3 ALAS2 targeted ES cell line
due to its ability to produce chimaeric pups of a higher percentage upon blastocyst injection,
which is believed to correlate to a greater level of germline transmission (Stewart, 1993).
Subsequently, mating of the chimaeric mice produced from the parental R3 and R4 ES cell
lines to assess germline transmission demonstrated that only the parental R3 cell line was able
to transmit through the germline to produce progeny with a manipulated ALAS2 gene (Dr. T.
109

Figure 4.4 Breeding of chimaeric mice to obtain germline transmission of the targeted
ALAS2 ES cell line
(a) The table lists the chimaeric mice obtained from the blastocyst injections of the H3-
Cl159G ES cell line containing the XLSA associated point mutation in exon 8 of the ALAS2
gene. The percentage of coat colour chimaerism is listed alongside each mouse that was
mated, where M followed by a number represents a mouse. The chimaeric mice were male
and bred for several rounds with female Balb/c mice. The number of litters and total pups
obtained from the matings of each chimaeric mouse are listed in the table.
(b) The table lists the chimaeric mice obtained from the blastocyst injections of the F2-
Cl159G ES cell line containing the XLSA associated point mutation in exon 8 of the ALAS2
gene. The percentage of coat colour chimaerism is listed alongside each mouse that was
mated, where M followed by a number represents a mouse. The chimaeric mice were male
and bred for several rounds with female Balb/c mice. The number of litters and total pups
obtained from the matings of each chimaeric mouse are listed in the table.
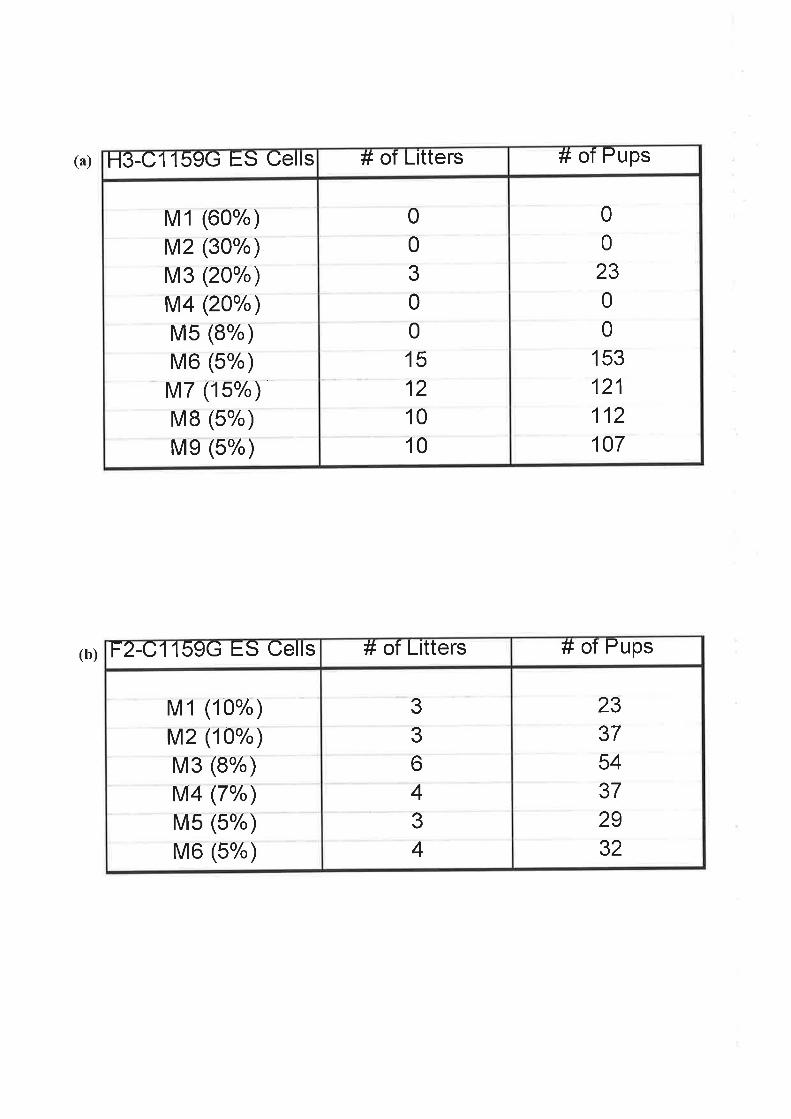
)(ù
(b)
H3-C1 159G ES Cells # of Litters # of Pups
M 1 (60%)M2 (30%)M3 (20%)1\A4 (20%)M5 (8%)M6 (5%)M7 (1 5%)M8 (5%)
Me (5%)
0
0
3
0
0
15
12
10
10
0
0
230
0
153
121
112107
F241159G ES Cells # of Litters # of Pups
M 1 (10%)r\A2 (10%)M3 (8%)l\A4 (7%)M5 (5%)M6 (5%)
3
3
643
4
23375437
29
32

Sadlon, personal communication). However, even germline transmission of the parental R3
ES cell line was arare event. This data was not available before coÍìmencement of the second
round of gene targeting involving the introduction of the exon 8 point mutation (Cl 159G) into
the ALAS2 locus. Thus, the inability of either the H3-C1159G or F2-C1159G cell line to
contribute to the germline may be related to the selection of the non-germline transmitting R4 ,/ES cells as the parental cell line in the second round of targeting i_ri
4.2.2lntroduction of the XlSA-Associated Mutation (C1228T) into Exon 9 of the
ALAS2 Locus of the HPRT-Positive R3 ES Cell Line
The exon 9 Cl228T point mutation was introduced into the murine ALAS2 locus using the
two-step strategy described in Section 4.2.1 (Figxe 4.3). A targeting vector to introduce the
exon 9 mutation into the ALAS2 locus was designed based on the restriction map of the
mouse ALAS2 gene and is detailed in Section 2.4.2. Briefly, the second round gene targeting
vector consisted of approximately 7 kb of mouse ALAS2 genomic DNA ranging from exon
7(XhoI) to intron 10 (SacI) containing the C7228T mutation in exon 9. The resultant exon 9 ,,,targeting vector was referred to as pmALAS2lCl228T.
The exon 9 targeting construct was then electroporated into the HPRT positive R3 ES cell line
generated from the first round of targeting. The R3 HPRT-positive ES cell line was selected
for this step as upon breeding of R3 chimaeras germline transmission was obtained. For
details of the transfection and selection procedure employed to introduce the exon 9 mutation
into the ALAS2 gene of the R3 genome see Section 2.7.The B-galactosidase expression
plasmid (RSV-Bgal) (150ug) was also transfected into the R3 ES cell line to determine
transfection efficiency. The transfected cells were stained for B-galactosidase expression
(Section 2.7.6) and transfection efficiency was determined to equal 0.5%. Six days after
transfection of the R3 cells with pmALAS2lCl229T, 6-TG (20pM) was added to the media to
select for cells that had undergone homologous recombination and exchanged the HPRT
cassette for the ALAS2 DNA fragment containing the Cl228T point mutation. Fifty 6-TG
resistant colonies were picked and each colony split into duplicate wells in separate 24-well
trays. One duplicate 24-well tray was frozen at -80oC and the second tray harvested for
genomic DNA (Section 2.5.1) once the cells had formed a confluent layer. However, of the
colonies originally picked only 32 grew.
110

To screen for ES cell clones that had exchanged the HPRT cassette for the ALAS2 fragment
harbouring the Cl228T point mutation, genomic DNA isolated from the picked 6-TG
resistant colonies was analysed by Southern blot hybridisation (Section 2.8.2). Genomic DNA
from each of the 32 colonies was digested with PstI. Digestion with PstI yielded fragments of
different lengths that were used to distinguish between 6-TG resistant colonies that had
undergone homologous recombination successfully and those that had lost the ability to
express HPRT due to chromosomal deletion or inactivation of the HPRT gene. The Southern
blot was initially hybridised with a radiolabelled 1.1 kb EcoRI (intron 6) to XhoI (exon 7)
mouse ALAS2 fragment which resides outside the ALAS2 sequence included in the targeting
vector. A7.2 kb band was expected to hybridise to the probe if homologous recombination
had occurred, while a larger fragment of 8.6 kb would hybridise if the HPRT cassette was still
present in the ALAS2 locus (Figure 4.5). As controls, genomic DNA obtained from wild type
E14TG2a cells, the parental R3 HPRT-positive ES cell line and a heterozygous female
ALAS2 knockout mouse were also digested with PstI and analysed by Southern blot
hybridisation. Digestion of wild tlpe DNA with PstI will produce the same size band as
correctly targeted ES cell lines. Alternatively, the R3 ES cell line would give the same band
as observed with 6-TG resistant colonies retaining the HPRT gene. DNA from the
heterozygous female ALAS2 knockout mouse will give bands representative of wild tlpe and
HPRT positive ALAS2 loci.
The 5'probe hybridised to a7.2 kb band in eight of the 32 6-TG resistant colonies (Figure 4.6,
Lanes 73,14,15,16,22,23,24 e.25). A similar sized band was also present in the lanes
containing the PstI digested wild ty,pe EI4TG2aDNA and heterozygous female ALAS2
knockout mouse DNA (Figure 4.6,Lanes 18 & 38, respectively). This indicated successful
homologous recombination at the ALAS2 locus, replacing the HPRT cassette in the ALAS2
gene with ALAS2 sequence containing the exon 9 Cl228T mutation. These eight clones were
found in two out of ten pools generated from the single electroporation. Therefore, at least
two independent targeting events were isolated. Since the cells were passaged twice prior to
6-TG selection it is unclear if the multiple positive clones found within a single pool
represented individual targeting events or originated from a single targeted cell. Fourteen of
theremainingclones (Figure 4.6,Lanes2,3,4,5,9, 10, 11,26,27,29,33,34,36 e'37)
contained a 8.6 kb band that hybridised to the probe. This band corresponded to the band
found with genomic DNA from the parental R3 HPRT:posit{€ ES cell line and the female
heterozygous ALAS2 knockout mouse (Figure 4.6'r.Lanes 19 & 38, respectively). This
indicated that atleast part of the HPRT gene was still preseát in the ALAS2 locus. Since
111

Figure 4.5 Restriction map of the Cl228T targeted ALAS2 locus in 6-TG resistant clones
1. Restriction map of the ALAS2 locus that has undergone homologous recombination and
introduced the Cl228T point mutation into exon 9 of the ALAS2 gene.
2. Restriction map of the ALAS2 locus containing the HPRT gene
The probes used are indicated in red and blue and are referred to as the 5' and 3'probes,
respectively. The PstI fragments are depicted in the same colour as the probe that they were
detected by. The boxed numbers are representative of ALAS2 exons.

1
ALAS2 TargetedLocus
Pstl
HPRT TargetedLocus
Pstl
5' 3'
c12287
Pstl Pstl
7.2kb
0.9kb
BamHl/Pstl
3'
BamHl Pstl
2
EcoRl Xhol
EcoRl Xhol
Smal
Smal/BamHl
BamHI Pstl
3.5kb
Pstl Pstl Pstl
8.6kb
11I rrzrr
119I5 rHPRTzr5.2kb

Figure 4.6 5t Southern blot analysis of 6-TG resistant ES cell colonies potentially
harbouringthe Cl228T point mutation in exon 9 of the ALAS gene
Genomic DNA was isolated (Section 2.8.1) from 6-TG resistant colonies and digested with
PstI. Digests were separated on a 0.8% agarose gel in TBE. The agarose gel was stained with
ethidium bromide and photographed. The gel was denatured, neutralised and transferred to a
nylon membrane by overnight capillary transfer. Filter was cross-linked, prehybridised and
hybridised with a radiolabelled with a 1.1 kb EcoRI/XhoI fragment probe. As controls, PstI
digested wild type E74TG2a, R3 HPRT positive and female heterozygous ALAS2 knockout
mouse genomic DNA were also included on the gel. Filters were washed twice at 65"C in2x
SSPE, 0.1% SDS for 15 minutes each. The filters were then washed once at 65'C in 0.2x
SSPE, 0.1% SDS for 30 minutes. Filters were exposed to a phosphoimager cassette and the
resultant image shown opposite. The7.2 kb fragment sizes expected for successful
homologous recombination at the 5' end is indicated by the numbered affow.
Lanes 1,20,21 and 39 : radiolabelled SppI molecular weight markers
Lanes 2-17,22-37 : 6-TG resistant colonies
Lane 18 : wild type E14TG2a genomic DNA
Lane 19: parental R3 genomic DNA
Lane 38 : female heterozygous ALAS2 knockout mouse genomic DNA

Lanesl2 345 67 8 910 1ll2 13 14 15 1617 18 1920 2t 2223 242526 2728 2930 31 3233 3435 3637 38 39
8.6 kb7.2kb
î

expression from the HPRT gene should have led to cell death in the presence of 6-TG, it
suggests that the HPRT gene may have been silenced in these clones. Interestingly, three of
the 6-TG resistant clones (Figure 4.6, Lanes 6,7 &,8) hybridised to a band at approximately 8
kb, slightly smaller than clones still harbouring the HPRT cassette. Although the reason for
this is unclear, it may be due to a partial deletion or random incorporation of the selection i,..:
cassette. No signal was detectable in three of the clones (Figure 4.6, Lanes 12,17 & 35) *- ('i i
despite the normal appearance of the DNA on the agarose gel prior to Southern transfer onto
the membrane. The remaining four clones (Figure 4.6,Lanes28,30,31 8.32) did not produce
a signal upon probing as the DNA was found to be degraded when visualised on the agarose
gel.
To confirm the 3' end of the construct had also been incorporated into the ALAS2 locus the
filter was probed with a radiolabelled 2.8 kb SmaVBamHI fragment ranging from the SmaI
site in exon 8 to the BamHI site in intron 10. Three bands of 7.2,3.5 and 0.9 kb were
expected to hybridise to this probe in a Southern blot, upon PstI digestion of genomic DNA
obtained from the 6-TG resistant colonies (Figure 4.5). Four of the clones that were positive
for homologous recombination at the 5' end were also found to contain correct insertion of the
targeting DNA at the 3' end of the ALAS2 locus. Figure 4.7 is a representative Southern blot
of the 3' screen for homologous recombinants. As expected the 3'probe hybridised to 7.2kb,
3.5 kb and 0.9 kb fragments in PstI digested wild type El4TG2a DNA (Figure 4.7,Lane 5). A
similar hybridisation pattern is seen for one ES cell clone (Figure 4.7,Lane 3), confirming
incorporation of the Cl228T point mutation into the ALAS2 locus. An additional band at
approximately 4.5 kb appears to be present in this representative Southern but is most likely
due to incomplete digestion of the DNA. This Southem also represents ES cell clones that
were 6-TG resistant but did not undergo homologous recombination at the 3' end (Figxe 4.7,
Lanes 1,2 &,4). Two of the clones (Figure 4.7 ,Lanes 1 &,2) have an additional band of 3 kb
suggesting they have randomly incorporated the targeting vector and thus, these clones were
not pursued. The fourth clone (Figure 4.7,Lane 4) did not hybridise with the probe which is
indicative of incomplete recombination at the ALAS2 locus resulting in a possible deletion at
the 3' end of the gene. Digestion of the parental R3 HPRT-positive cell line with PstI (Figure
4.7,Lane 6) resulted in the probe hybridising to a 5.2 kb band.
The four ES cell lines that had successfully undergone homologous recombination were also
screened for karyotype anomalies as before. The J9-C1228T taryeted ES cell line had the
highest percentage of cells (73%) with a complete set of forty chromosomes when compared
rt2

Figure 4.7 3' Southern blot analysis of 6-TG resistant colonies potentially harbouring
thLe Cl228T point mutation in exon 9 of the ALAS2 gene
Genomic DNA was isolated from 6-TG resistant colonies and digested with PstI. Digests
were separated on a0.8Yo agarose gel in TBE. The agarose gel was stained with ethidium
bromide and photographed. The gel was denatured, neutralised and transferred to a nylon
membrane by overnight capillary transfer. Filter was cross-linked, prehybridised and
hybridised with a radiolabelled with a 2.8 kb SmaI/ BamHI ALAS2 fragment probe. As
controls, PstI digested wild type E14TG2a andR3 HPRT+ genomic DNA were also included
on the gel. Filters were washed twice at 65'C in 2x SSPE , O.lyo SDS for 15 minutes each.
The filters were then washed once at 65"C in 0.2x SSPE ,0.1oÁ SDS for 30 minutes. Filters
were exposed to a phosphoimager cassette and a representative resultant image shown
opposite. The7.2,3.5 and 0.9 kb fragment sizes expected for successful homologous
recombination at the 3' end are indicated by the numbered alrorws. In addition, the 5.2 kb
band, representing the presence of the HPRT gene in the ALAS2 locus, is arrowed.
Lanes 1-4 : 6-TG resistant colonies
Lane 5 : wild type E14TG2a genomic DNA
Lane 6: parental R3 genomic DNA
LaneT: radiolabelled SppI molecular weight markers

.1
n",^U
67Lanes I 2 3 4 \,
7.zkb
s.2 kb
3.s kb
0.9 kb
sit
*
t'
xË
**.! :.
*.ï ,lÍiiaI

to the other correctly targeted cell lines. However, blastocyst injections of this ES cell line
was not pursued for reasons discussed in the following section.
4.2.3 Targeting of the ALAS2 Locus in \il9.5 ES Cells
The quality of the ES cell line selected for gene targeting is critical for maximising the
probability of achieving germline transmission. Several factors pertaining to the ES cells
influence the efficiency of germline transmission including, chromosomal abnormalities
(Suzuki et al., I99l) and the number of passages the cells have undergone (Nagy et al., 1990;
Fedorov et al., 1997). Taking these factors, as well as poor germline transmission of the
parental R3 ES cell line (discussed below), blastocyst injections using the cell line harbouring
the exon 9 C1228'l mutation (J9-CL228T) were not pursued. This decision was also prompted
by observation of a consistent decrease in germline transmission of targeted ES cells derived irtl
from the parental El4TG2a cell line in the gene targeting facility of the department during the
course of this study (Remizsewski, 2000; J. 'Wrin, personal communication). In the present
study, the parentalEl4TG2a ES cells utilised to generate the desired ALAS2 targeted cell
lines were already at a high starting passage number of 21. The passage number of the ES
cells was further increased by the two rounds of gene targeting required to introduce the
XlSA-associated point mutations into the ALAS2 locus. This resulted in ALAS2 targeted ES
cell lines with passage numbers ranging from 37 to 39. Thus, the high passage number of the
ALAS2 targeted ES cell lines was not a positive attribute when trying to maximise the chance
of obtaining germline transmission. In addition, further breeding of chimaeric mice produced
from the ALAS2 targeted R3 ES cell line generated in the first round and used as the parental
ES cell line for the second targeting step, indicated that this cell line was very inefficient at
producing chimaeric mice capable of germline transmission (Dr. T. Sadlon, personal
communication). Therefore, a high starting passage number, a l-oW rate of germline
transmission from the parental R3 ES cell line and the department's overall decline in . ,. .
t, -'
l
obtaining germline transmission with El4TG2a ES cells led to the utilisation of an altemative
targeting strategy to generate the murine model for XLSA.
Concurrently, the feeder cell dependent W9.5 ES cells were made available to the department
In initial tests, this ES cell line was associated with a higher rate of coat colour chimaerism
and germline transmission in comparison to the EL4TG2aES cell line (J. Wrin, personal
communication). In addition, the W9.5 ES cells were of a lower starting passage number
113

(passage number 9) which would increase the chance of obtaining germline competent ES
cells. Thus, targeting of the ALAS2 locus to introduce the XLSA associated point mutations
was performed in the W9.5 ES cells. Since W9.5 ES cells are HPRT-positive, the gene
targeting approach needed to be modified.
The first round of targeting in the W9.5 ES cells was conducted in collaboration with Dr. T.
Sadlon and involved replacing a region of the ALAS2 gene with a selectable marker
consisting of a fusion gene encoding for neomycin resistance (neo) and the viral herpes
simplex (HSV) th¡rmidine kinase (tk). Expression of this gene was driven by the PGK
promoter. This selection cassette was referred to as PGKneo'/HSVtk. The introduction of a
neo'cassette will enable selection of ES cell lines that have undergone homologous
recombination using G418, a neomycin derivative while HSVtk can be selected against by
treating cells with fialuridine (FIAU). Thus, a new targeting vector (pALASÐlS-neotþ was
constructed containing the 3.5 kb PGKneo'/HSVtk selection cassette inserted between the
SmaI site in exon 8 and the PstI site in exon 9 of the ALAS2 gene. This vector contained
approximately 2.9 kb of 5'homology to the mouse ALAS2 gene from the XhoI site in exon 7
to the SmaI site in exon 8 and approximately 3 kb of 3' homology from a PstI site in exon 9 to
a SacI site in intron 10. The region of the ALAS2 gene replaced by PGKneo'/HSVtk contains
sequence encoding both point mutations associated with XLSA in humans. Detailed
construction of the targeting vector can be found in Section 2.4.3.
The ALAS2 sequence harbouring the PGKneo/HSVtk selection cassette was isolated from
pALAS2)lS-neotk as a linear NotVSacI fragment for transfection via electroporation into the
W9.5 ES cells (Section 2.1.3(li)) (Figure 4.8). Four days after transfection the selection
reagent, G418 (250pglml), was added to the cells to select for colonies that had incorporated
the targeting construct into their locus. Of the G418 resistant colonies obtained, 720 colonies
were picked and genomic DNA harvested for screening by Southern blot analysis in order to
determine which cell line had correctly incorporated the PGKneo'/HSVtk cassette into the
desired position within the ALAS2 locus. Genomic DNA from each of the G418 resistant
colonies was digested with BglI and BgIII and a Southern blot performed using an external 5'
probe (a 1.1 kb Intron 6 (EcoRI) to Exon 7 (XhoI) ALAS2 fragment) to identifu clones in
which homologous recombination had occurred at the 5' end. Bands of 3.5 kb and 1 kb were
expected to hybridise to the probe if the PGKneo'/HSVtk cassette had been correctly
incorporated into the ALAS2 locus, while a 5.8 kb and 1 kb fragment would hybridise to the
probe if the ALAS2 locus was unaltered (Figure 4.9). Of the 720 colonies screened by
tr4

Figure 4.8 Targeting strategy employed in the generation of a W9.5 ES cell line with a
modified ALAS2locus
In the first round, the pALAS2)lS-neo/tk targeting vector was introduced into the parental
W9.5 ES cell line to generate ES cells that are with a PGKneo'/HSVtk insertion into the
ALAS2 locus. These cells are neo' and selected for with G418, a derivative of neomycin. This
ALAS2 targeted ES cell line then undergoes a second round of homologous recombination
where a second ALAS2 alteration vector containing either point mutation is introduced. Ifhomologous recombination is successful ES cells generated from this targeting round would
have exchanged the PGKneo'/HSVtk cassette for ALAS2 sequence harbouring the XLSA
associated point mutation and would be resistant to FIAU. The resultant ALAS2 targeted ES
cell lines will then be employed to generate XLSA mice with a specific point mutation in their
ALAS2 gene. Both mutations are indicated in the diagram of the alteration vector and the
mutated ALAS2 locus to assist in explaining the targeting strategy employed. However, the
actual targeting construct contained only one of the mutations such that ES cell lines
containing either of the XLSA associated mutations were generated.

Exon 10Exon 9Exon 8Exon 7
c1228c11591st TargetingRound Pstl Pstl
Ecl136l/Pstl
ALAS2 Locus inCell Line
SaclNotl
S
Smal/Hincll
Exon 10Exon 9Exon 8Exon 7 neotk
neo/tk Targeting Construct
Xhol
Alteration Vector
Xhol
Smal/Hincll t Ecll l/Pstl Sacl
Targeted ALAS2Locus
+
Sacl
Mutated ALAS2 LocusSacl
Exon 10Exon 9Exon 8Exon 7 neotk
Exon 10Exon 9Exon 8Exon 7
T1228G1159
2nd TargetingRound
Exon 10Exon 9Exon 8Exon 7
T1228G1159

Figure 4.9 Southern blot screen for identification of successfutly ALAS2 targeted W9.5
ES cells
1. Restriction map of the ALAS2 locus that has undergone homologous recombination and
introduced either of the XlSA-associated point mutations into the ALAS2 gene.
2. Restriction map of a ALAS2 locus that has incorporated the PGKneo/HSVtk selection
cassette.
The probes used are indicated in red and blue and are referred to as the 5' and 3'probes,
respectively. The BglI/BglII fragments detected by the 5'probe are depicted in red and the
PstI fragments detected by the 3'probe are illustrated in blue. The boxed numbers are
representative of ALAS2 exons. Both mutations are indicated in the diagram of the alteration
vector and the mutated ALAS2 locus to assist in explaining the targeting strategy employed.
However, the actual targeting construct contained only one of the mutations such that ES cell
lines containing either of the XLSA associated mutations were generated.
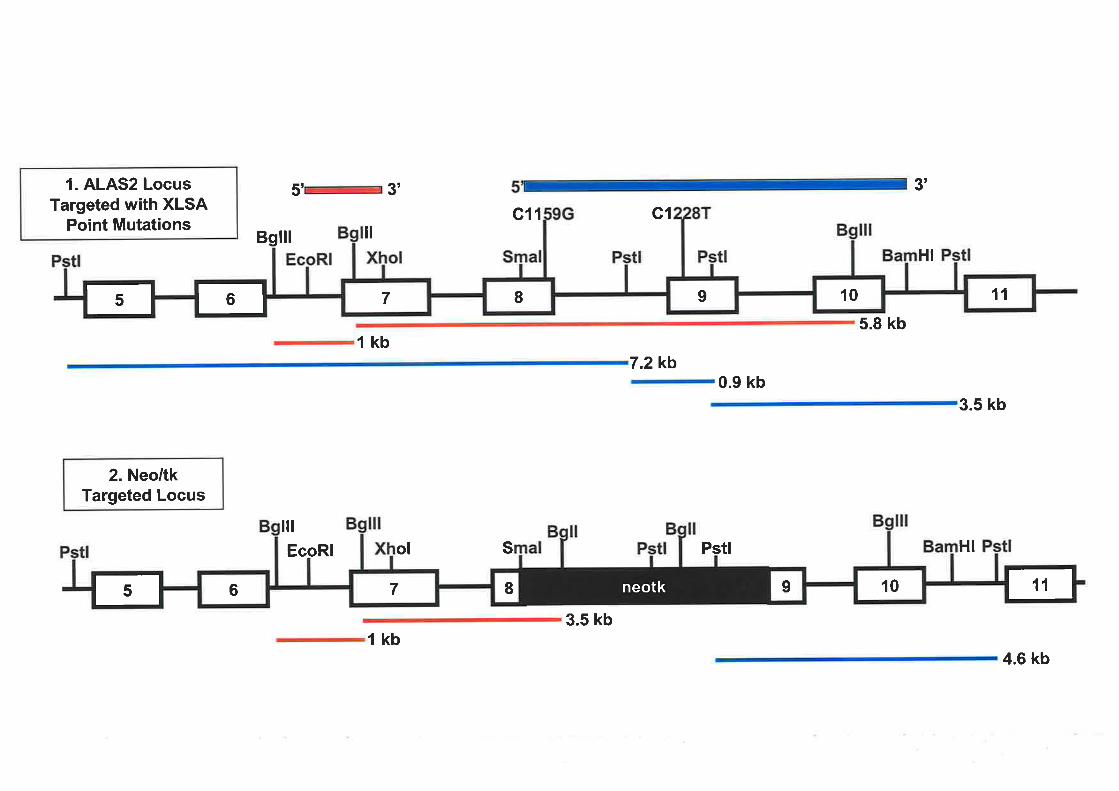
1. ALAS2 LocusTargeted with XLSA
Point Mutations
5' 3' 3'
c11 G1
7.zkb0.9 kb
Bglll ilHI
5.8 kb1kb
3.5 kb
2. Neo/tkTargeted Locus
11109I7rr
ilt
EcoRl Sol Pstl HI
3.5 kb1kb
10II76 rneotkr4.6 kb

Southern blot ten clones were identified as positive for homologous recombination at the 5'
end, having incorporated the PGKneo'/HSVtk cassette into the endogenous ALAS2 gene. A
representative Southern blot of the 5' screen in which one of the positive clones was identified
is presented in Figure 4.10 (Lane 14) with the remaining clones not having undergone r''
homologous recombination and so retaining an intact ALAS2 locus (Figure 10, Lanes 2-13 &.
rs-24).
A Southern blot was performed on the positive clones identified in the 5' screen to ensure
homologous recombination had been successful at the 3' end. Genomic DNA was digested
with PstI and a Southern blot conducted hybridising with a 2.8 kb SmaI (exon 8) to BamHI
(intron l0) fragment. If homologous recombination had occurred at the 3' end a single band of
4.6kb would be expected whereas three bands of 7.2 kb, 3.5 kb and 0.9 kb would be observed
in non-recombinants (Figure 4.9). Five of the positive ES cell clones detected in the 5' screen
were shown to have correctly undergone homologous recombination and four of the positives
are represented inFigure 4.11 (Lanes4,7,10 & 13). The remaining genomic DNA clones
presented on the Southern blot were known negative recombinants but were included as a
reference for identiffing those cell lines that had successfully incorporated the
PGKneo'/HSVtk cassette into the ALAS2 gene (Figure 4.1I, Lanes 3,5,6,8,9, 11, 12 &,14'
20). Wild type W9.5 genomic DNA was also included as a reference for homologous
recombination (Figure 4.Il,Lane 2). Therefore, ftve out of ten ALAS2 knockout ES cell
clones identified with the 5'probe were also shown to be correctly targeted at the 3' end.
A correctly targeted V/9.5 ES cell line was then selected for the second round of targeting in
which the XLSA associated point mutations would be introduced. The cell line chosen (Xl1-
W9.5neo/tk) was selected on the basis of its good ES colony morphology and high percentage
of cells (87%) with a normal karyotype. Initially, the exon 9 mutation was targeted into the
ALAS2 locus in W9.5 ES cells. The targeting vector (pmALAS2lCl228T) containing the
exon 9 CI228T point mutation was transfected into the W9.5 ES cells as described in Section
2.7.3(11). Five days after transfection, FIAU at 0.2pM was added to the cells to select against
cells expressing viral tk. Ninety FIAU resistant colonies were picked and out of these
colonies, 34 continued to grow in FIAU supplemented media and were subsequently treated
to obtain genomic DNA for Southern hybridisation analysis to determine if homologous
recombination had occurred. The isolated DNA was digested with BglI and BgIII and probed
with a radiolabelled 1.1 kb EcoRI (Intron 6) to Exon 7 (XhoI) ALAS2 fragment, located
outside of the targeting construct. If homologous recombination had occurred to recreate the
115

Figure 4.10 5' Southern blot analysis of G418 resistant colonies potentially containing
the PGKneo'/HSVtk cassette in the ALAS2 locus
Genomic DNA was isolated from G418 resistant colonies and digested with BglI and BgIIL
Digests rwere separated on a0.8o/o agarose gel in TBE. The ag¿ìrose gel was stained with
ethidium bromide and photographed. The gel was denatured, neutralised and transferred to a
nylon membrane by overnight capillary transfer. Filter was cross-linked, prehybridised and
hybridised with a radiolabelled with a 1.1 kb EcoRI/XhoI ALAS2 fragment probe. Filters
were washed twice at 65oC in 2x SSPE,0.lo/o SDS for 15 minutes each. The filters were then
washed once at 65"C in 0.2x SSPE,0.7o/o SDS for 30 minutes and exposed to a
phosphoimager cassette. A representative Southern is displayed. The 3.5 kb and 1 kb
fragment sizes expected for successful homologous recombination at the 5'end are indicated
by the numbered anows.
Lanes I &,25: radiolabelled SppI molecular weight markers
Lanes 2 to 24 : G4l8 resistant colonies

Lanes 1 234 5 6 78 910 1lr213r4rsl6 17 18 192021222324 25
<- 3.5 kb
G t.otu
tü*
tIË
rtI

Figure 4.113' Southern blot analysis of G418 ES cell resistant colonies potentially
harbouring the PGKneo/HSVtk selection cassette in the ALAS2 gene
Genomic DNA was isolated from G418 resistant colonies and digested with PstI. Digests
rwere separated on a0.8o/o agarose gel in TBE. The agarose gel was stained with ethidium
bromide and photographed. The gel was denatured, neutralised and transferred to a nylon
membrane by overnight capillary transfer. Filter was cross-linked, prehybridised and
hybridised with a radiolabelled with a2.8kb SmaI/ BamHI ALAS2 fragment probe. Filters
were washed twice at 65'C in 2x SSPE,0.lyo SDS for 15 minutes each. The filters were then
washed once at 65oC in 0.2x SSPE ,0Jyo SDS for 30 minutes and exposed to a
phosphoimager cassette. A representative Southern is displayed. The 4.6 kb fragment
expected for successful homologous recombination at the 3' end is indicated by the numbered
alïow
Lanes | &,2l: radiolabelled SppI molecular weight markers
Lane2: W9.5 genomic DNA
Lanes 3 to 20: G4l8 resistant colonies

Lanes 1 2345678 91011121314151617181920 2I
<- 4.6 kb
a<-

intact ALAS2 gene bands of 5 .8 kb and 1 kb would be expected. Bands of 3 .5 kb and 1 kb
would be detected if the selection cassette remained in the ALAS2 locus such that
homologous recombination had been unsuccessful. None of the FIAU resistant colonies
analysed had successfully undergone homologous recombination as illustrated by the
representative Southem blot in Figure 4.l2.In most lanes 3.5 kb and I kb bands were
detected indicating that the PGKneo/HSVtk selection cassette was still present in the ALAS2
locus (Figure 4.12, Lanes 2, 4, 5,7,9 - 15 &.78-21).In other lanes, no hybridisation to the
probe was observed suggesting FIAU resistance was due to loss or deletion of this region
(Figure 4.l2,Lanes 3, 6, 8, 16 & 17). Therefore, the second round of targeting in W9.5 ES
cells was unsuccessful.
A second attempt at targeting either of the XlSA-associated point mutations, C1159G or
Cl228T, into the ALAS2 locus of the W9.5 ES cells was undertaken as described above.
Forty five FIAU resistant colonies were picked (20 colonies from transfection of the C1159G
ALAS2 targeting construct and25 from the Cl228T mutation) and maintained in complete
ES media supplemented with FIAU (0.2pM) but after fifteen days in selection media only two
of the clones picked survived. Of the two surviving FIAU resistant W9.5 ES cell clones there
was one each of the XLSA associated point mutations. Since only two FIAU resistant
colonies were generated it was decided not to continue characterising these cells for correct
integration of the ALAS2 targeting vector. Normally, it is prudent to have at least two
independently generated targeted ES cell lines for blastocyst injection. This allows for
confirmation of results obtained and ensures that observations made of a genetically modified
mouse can be attributed solely to the alteration made and not an artefact of one particular cell
line. The reason for the loss of the majority of the FIAU resistant clones picked is unknown. It
may be that picking of the colonies after administration of FIAU was too soon, such that
FIAU sensitive colonies (still retaining the PGKneo/HSVtk cassette) were picked as resistant
colonies. However, for the reasons outlined below this approach was abandoned.
Although wild type W9.5 ES cells were tested by the department's targeting facility prior to
the commencement of this series of targeting experiments and demonstrated to produce a high
rate of germline transmission (J. Wrin, personal communication), attempts by several other-t
researchers within the department to obtain germline transmission using targeted derivatives I ,
of the W9.5 ES cell line failed to generate any germline transmission (J. Wrin, personal ';
communication). This was despite normal rates of chimaeric mouse production and a high
percentage of chimaerism observed. Further testing, including chimaera production and
tr6

Figure 4.12 5t Southern blot screen of FIAU resistant ES cell colonies potentially
harbouringthe Cl228T point mutation in exon 9 of the ALAS2 locus
Genomic DNA was isolated from FIAU resistant colonies and digested with BglI/BglII.
Digests were separated on a0.8%o agarose gel in TBE. The agarose gel was stained with
ethidium bromide and photographed. The gel was denatured, neutralised and transferred to a
nylon membrane by overnight capillary transfer. Filter was cross-linked, prehybridised and
hybridised with a radiolabelled with a 1.1 kb EcoRI/XhoI ALAS2 fragment probe. Filters
were washed twice at 65'C in 2x SSPE ,0.lyo SDS for 15 minutes each. The filters were then
washed once at 65'C in 0.2x SSPE,O.lyo SDS for 30 minutes and exposed to a
phosphoimager cassette. A representative Southern blot is displayed. Since successful
homologous recombinants were not obtained in the targeting round the numbered arrows
indicate the bands obtained for the clones retaining the PGKneo/HSVtk selection cassette.
Lanes I 8.22: radiolabelled SppI molecular weight markers
Lanes 2 to 2l: FIAU resistant ES cell colonies

Lanes I 2 3 4 5 6 7 8 910 1l12 13 14 1516 17 18 192021 22
<- 3.5 kb
< lkb

germline transmission, implicated a particular frozen stock of 'W9.5 ES cells as being
defective in germline transmission. The reason for this was never resolved and unfortunately
the original targeting experiments to produce the modified ALAS2 ES cell lines used this
stock. Since two rounds of targeting are necessary for the strategies adopted to generate ES
cells carrying a specific XLSA point mutation in the ALAS2 locus and other studies have
demonstrated a decrease in the ability of ES cells to transmit the germline as the passage
number of the cells increase (Nagy et al., 1993) we decided to employ an alternative approach
to develop a XLSA murine model.
4.2.4 Strategy for the Commercial Generation of an ALAS2 Targeted ES Cell Line
It was previously discussed that the quality of the ES cells used in gene targeting experiments
is paramount to their ability to contribute to the germline in the host blastocyst. During the
course of this research, germline transmission of targeted ES cells generated within the
department has been minimal (J. Wrin, personal communication). Thus, it was decided that an
ES cell line harbouring the exon 8 C1159G point mutation in the ALAS2 gene would be
commercially produced by Ozgene, an organisation specialising in gene targeting technology.
The XLSA associated mutation (Cl159G) that resides in intron 8 of the ALAS2 gene was
chosen as the initial mutation to be targeted by Ozgene since it has been well characterised
and it is likely that the XLSA mouse generated will survive into late adulthood as evidenced
with the human proband (Cox et al., 7994).
In consultation with Ozgene, a single round gene targeting approach was pursued (Figure
4.13).In this approach, the desired point mutation is introduced into the same targeting vector
as the selection cassette. The selection cassette is located in intronic sequence and most
importantly is flanked by loxP sites. This allows for the selection cassette to be deleted either
in ES cells by transient expression of cre recombinase or in mice by breeding with cre
expressing mice (Nagy, 2000). This approach was chosen over a two step strategy as it limits
the amount of in vitro manipulation of the ES cell, increasing the likelihood of successful
germline transmission.
The outcome of cre-initiated recombination is dependent on the orientation and location of the
/oxP sites within the DNA. When located in cis, as in this study, the result is excision of the
intervening sequence. For the pulposes of this study, the crelloxP recombinase system will
117

Figure 4.13 Targeting of the ALAS2 locus in W9.5 ES cells using the cre/loxP
recombination mechanism
Use of the crelloxP recombination system requires only one targeting step to generate an ES
cell line containing the C1159G XLSA associated point mutation in exon 8 of the ALAS2
gene. A targeting vector containing homologous murine ALAS2 sequence with the desired
point mutation and a loxPlneo'selection cassette in intron 7 is introduced into the ALAS2
locus of the'W9.5 ES cells. Successful homologous recombination will result in ES cells that
have incorporated the Cl 159G point mutation into exon 8 and the loxPlneo sequence into
intron 7 of the ALAS2 gene. The loxPlneo' cassette can be removed by either in vivo or in
vilro methods.

Exon 10Exon 9Exon IExon 7
c1159 ALAS2 Locus inW9.5 Gell Line
t_ rXhol
pCl 1 SgGloxneo Targeting Vector
Removal of loxPlneo by transientexpression of cre recombinase in
the ALAS2 targeted ES cells
I
Sacl
Mutated ALAS2 Locus inW9.5 ES Cells
Removal o'f loxPlneo by breedingwith cre expressing mice
Pstl
ln Vitro ln Vivo
Exon 10Exon 9Exon 8Exon 7
G1159
loxP/neo
Exon 10Exon 9Exon IExon 7
G1159
loxP/neo

enable removal of the selectable marker from the ALAS2 locus after introduction of the
XLSA associated point mutation into exon 8.
In the current study, the C1159G XLSA associated point mutation in exon 8 and a neo'gene
flanked by loxP sites will be introduced into the ALAS2 gene of 'W9.5 ES cells (Figure 4.13).
W9.5 ES cells that have incorporated the mutation will be selected on the basis of resistance
to neomycin. Once ES cells containing the Cl159G point mutation in exon 8 of the ALAS2
locus have been generated by Ozgene, injection into the blastocyst and breeding to obtain
germline transmission will be performed within the department. Subsequently, removal of the
selection cassette would be attempted by mating mice ubiquitously expressing cre
recombinase.
4.2.5 Construction of a ALAS2 C1159G Targeting Vector
As discussed in Section 4.2.4, the crelloxP recombinase system will be employed to generate
an ES cell line that has incorporated the Cl159G mutation into exon 8 of the ALAS2 locus.
The new targeting vector contained 7 kb of homologous murine ALAS2 DNA, the exon 8
mutation and the loxPlneo'selection cassette inserted into intron 7 (Figure 4.t4). To insert the
loxPlneo'cassette an EcoRV site was created in intron 7 sequence. Fragments of 2.7 kb and
288 bp encompassing ALAS2 sequence from the XhoI site in exon 7 to the XmaI site in exon
8 were amplified from the pmALAS2XIS-7 vector in order to introduce an EcoRV site into
intron 7. The 2.7 kb ALAS2 fragment was digested with XhoI and EcoRV and the 288 bp
amplified ALAS2 product was digested with EcoRV and XmaI. The digested PCR products
were purified and simultaneously ligated into pBluescript KS- (Stratgene) digested with XhoI
and XmaI to generate the vector pmALAS2ET-E8(EooRV). This vector contained 3 kb of
murine ALAS2 sequence from exon 7 (XhoI) to exon 8 (XmaI) with an EcoRV site
introduced into intron 7.
Site-directed mutagenesis was conducted to introduce the C1159G point mutation into exon 8
of a vector (pmALAS2 14 .4F,8-II0) containin g 4.4 kb of murine ALAS2 DNA ranging from
the XmaI site in exon 8 to the SacI site in intron 10. This region underwent automated
sequencing (Section 2.2.5) to ensure that only the exon 8 mutation had been introduced. The
4.4kb XmaI (exon 8) to SacI (intron 10) fragment containing the C1159G point mutation was
isolated and cloned into pmALAS2ET-E8(EcoRV) that had been digested with XmaI and
118

Figure 4.14 Generation of a targeting construct using the crelloxP recombinase system
to introduce the Cl159G point mutation into exon 8 of the ALAS2 locus in W9.5 ES cells
(a) Firstly, an EcoRV restriction site was introduced into intron 7. This was performed by
amplification of two ALAS2 fragments from the pmALAS2X/S-7 vector using primers
that included an EcoRV site. The red arrows depict primers A (mAETEcoRV-A) and C
(mAETEooRV-B) used to amplify Lhe2.7 kb ALAS2 fragment with an EcoRV site on the
3'end. The green affows represent primers B (mAESEooRV-A) and D (mAESEcoRV-B)
which amplified a 288 bp ALAS2 fragment with an EcoRV site at the 5' end. The ALAS2
fragments generated by PCR were digested with the appropnate enzymes and ligated into
pBluescriptKS- digested with XhoI and XmaI to produce the pmALAS2ET-E8(EcoRV)
vector. The Cl159G point mutation was introduced into exon 8 of the ALAS2 gene by
site directed mutagenesis of pmALAS2l2.8E8-I10 and a 4.4kb XmaI (exon 8) to SacI
(intron 10) fragment containing the mutation was isolated. This fragment was ligated into
pmALAS2ET-E8(EcoRV) that had been digested with XmaI/SacI. The construct produced
was named pmALAS2)lElSCl159G and consisted of approximately 7 kb of LAS2
homologous sequence, the Cl 159G point mutation and an EcoRV site introduced into
intron 7.
Primer A: mAETEooRV-A: 5' CCCCCTCGAGAAGTCTGATCCCAAGACAC 3'
Primer C: mAETEooRV-B: 5' CAAACATCAACCTGATATCCCACACCCATGCC 3'
Primer B: mAE8EcoRV-A: 5' ACCTGCACCCCGGGCTCCATACAGTCCTAC 3'
Primer D: mAE8EcoRV-B : 5' GGCATGGGTGTGGGATATCAGGTTGATGTTTG 3'
The EcoRV site is underlined.
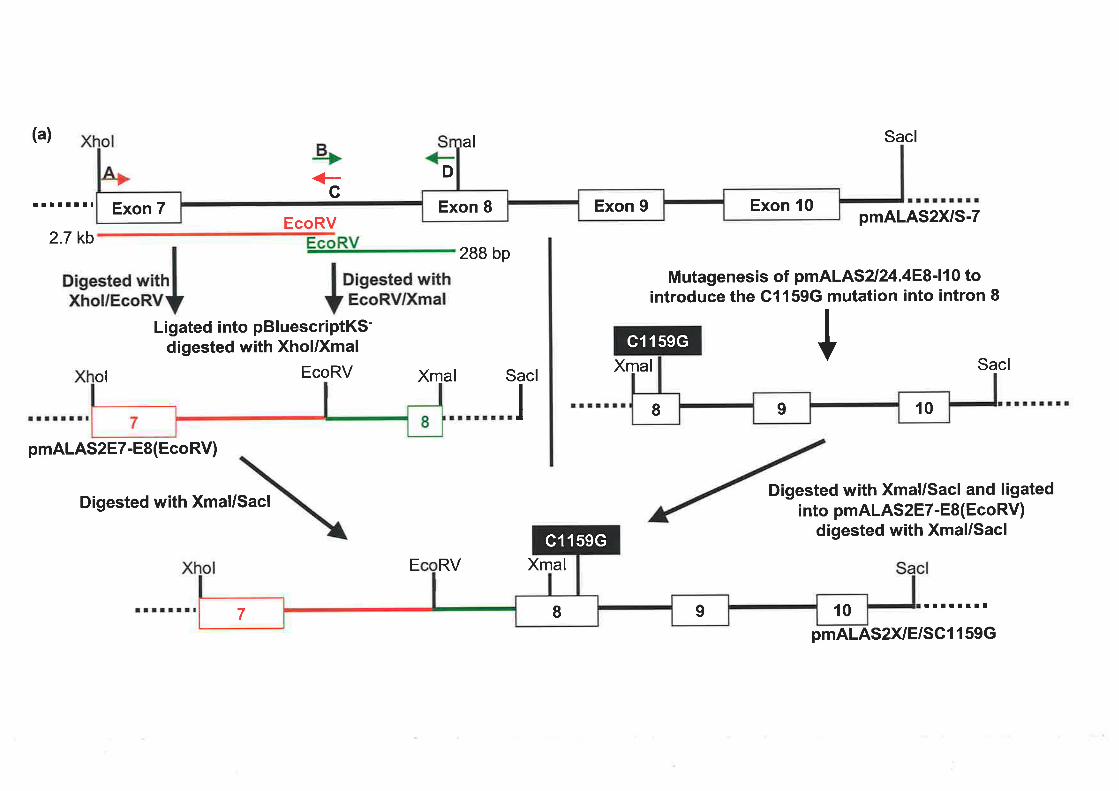
Exon 10Exon 9Exon 8Exon 7
(a)
!t¡tl¡¡t
2.7 kb
c
al
D
288 bp
Xmal Sacl
Sacl
pmALAS2XIS-7
Mutagenesis of pmALAS2l24.4E8'11 0 tointroduce the G1159G mutation into intron I
ISacl
+-EcoRV
Disested *¡thlXhol/EcoRVY
Ligated into pBluescriptKS-digested with Xhol/Xmal
EcoRV Xmalol
pmALAS2ET-E8(EcoRV)
Digested with Xmal/SaclDigested with Xmal/Sacl and ligated
into pmALAS2ET-E8(EcoRV)digested with Xmal/Sacl
E RV Xmal
rllttrr¡
pmALAS2X/E/SC1 159G
019I
c1159G
109I7
c1159G

(b) The next step introduced the loxPlneo'cassette into the EcoRV site in intron 7 of the
ALAS2 sequence in the pmALAS2)lElSC1159G vector. A2 kb Smal/Xhol loxPlneo
fragment was isolated from the vector ploxPneo-l and the XhoI end blunted. This fragment
was then ligated into pmALAS2)lElSCl159G digested with EcoRV to position the loxPlneo
sequence in intron 7. The resulting ALAS2 targeting construct was referred to as
pCl l59Gloxneo.
The solid black lines are representative of ALAS2 sequence. The dotted lines denote vector
backbone sequence.

Smal Xhol(b)
ploxPneo-l
Xhol/EcoRV EcoRV/Smal
Xmal
Digested with Smal/Xholand blunted Xhol end
Xmal
Digested with EcoRV
Sacl
¡tlllttl¡
pC1159Gloxneo
ol EcoRV Sacl
109I
c1159G
109I7
c1159G
pmALAS2X/E/SC1159G

SacL The vector contained approximately 7 kb of ALAS2 sequence with the Cl159G
mutation introduced into exon 8 and an EcoRV site inserted into intron 7
(pmALAS2)lElSC I I 59G) (Figure 4.14a).
The neo'cassette, driven by the PGK-I promoter, and flanked by loxP sites was isolated from
theploxPneo-l vector (gift from Andras Nagy) as a2 kb SmaVXhoI fragment (Figure 4.15b).
The XhoI end of this fragment was blunted and the fragment subsequently ligated into the
introduced EcoRV site in intron 7 of the pmALAS2)VE/SC1159G vector. The orientation of
lhe loxPlneo' sequence in the clones obtained was analysed by restriction enzyme digestion
and was found to be in the reverse orientation to the ALAS2 gene. The completed targeting
vector, containing the loxP/neo'' selection cassette intron 7 and the Cl159G mutation in exon
8 was referred to as pCl159Gloxneo. The pCl l59Gloxneo targeting vector was prepared
using the QIAGEN Endotoxin-Free Maxi Kit. Once prepped the construct was sequenced to
ensure the exon 8 mutation was present before sending the construct to Ozgene for targeting
into W9.5 ES cells. The vector (50pg) was digested with XhoI and SacI restriction enzymes,
gel purified twice and forwarded to Ozgene to begin the targeting process.
4.2.6 Screening Strategy to Identify ALAS2 Targeted W9.5 ES Cells Produced By
Ozgene
A 5' screen was devised to enable Ozgene to identiff targeted W9.5 ES cell lines that had
incorporated the ALAS2 targetingvector into the endogenous ALAS2 gene. Genomic DNA
prepared from the G418 resistant ES cell clones was digested with SphI and screened by
Southern blot analysis using a780 bp extemal probe spanning from an EcoRV site in intron 6
to a XhoI site in exon 7. This probe would hybridise to a 5.2 kb band in ES cell lines that had
correctly undergone homologous recombination and a 8.5 kb band in ES cell lines retaining a
wild type ALAS2 locus (Figure 4.15). Screening of 480 G418 resistant ES cell colonies by
Ozgene resulted in the identification of one ES cell line having correctly undergone
homologous recombination and is represented in the Southern blot depicted in Figure 4.16
(Lanes 2 e,3).
To confirm that homologous recombination had also occurred at the 3' end of the clone,
design of a 3' screen via Southern blot analysis was undertaken. Once a suitable restriction
digest and probe combination had been devised and tested on a sample of genomic W9.5
119

Figure 4.15 Restriction map for the 5r screen of Targeted ALAS2 G418 resistant ES cell
colonies
A 780 bp external ALAS2 EcoRV/XhoI fragment was used as a 5'probe for screening of
G418 resistant colonies that had successfully undergone homologous recombination and
incorporated the Cl159G point mutation and the loxPlneo' sequence into the ALAS2 locus of
the W9.5 ES cells. The SphI fragments expected to hybridise to the probe are depicted in red.
1. Restriction map of the wild type ALAS2 locus in parental W9.5 ES cells.
2. Restriction map of the ALAS2 gene that has been correctly targeted.

1. ALAS2 W¡ld TypeLocus in W9.5 ES Gells
c1159
Sphl EcoRV Xhol
2. Targeted ALAS2 Locusin W9.5 ES Gells
5' 3'
s', 3'
S
s ht
8.5 kb
c1159G
Sphl SphlSphl EcoRV Xhol
98 rEr
6 z I rr5.2 kb

Figure 4.16 Southern screen of G418 resistant colonies obtained from the transfection of
W9.5 ES cells with the ALAS2 targeting vector (pCl159Gtoxneo)
Genomic DNA was isolated from G418 resistant colonies and digested with SphI. The filter
was hybridised with a radiolabelled 780 bp EcoRV/XhoI ALAS2 fragment probe. A
representative Southern blot of the 5' screen for positive recombinants is depicted. The 5.2 kb
fragment size expected for successful homologous recombination at the 5' end is indicated by
the accordingly numbered arrow. The wild type ALAS2 results in a 8.5 kb band which is also
arrowed.
Lane 1:MarkerDNA
Lanes 2 and 3 : A single G418 resistant colony, repeated in each lane
Lane 4: Wild tlpe genomic DNA

Lanes I 2 3 4
+8.5 kb
f5.2 ¡6

DNA it was to be sent to Ozgene to enable confirmation of recombination at the 3'end. After , j",rl
testing of numerous combinations of different restriction enzymes and probes a suitable r '
screen using Southern blot analysis could not be determined. For the 3' screen a restriction
enzpe was required that cut within the loxPlneo'sequence inserted into intron 7 and cut
again 3'to this position, outside of the ALAS2 sequence included in the targeting construct.
Restriction enzymes were identified that cut at the positions specified but they were deemed
unsuitable for several reasons. Firstly, some of the enzymes were shown to be inefficient
cutters of genomic DNA as after several digestion attempts the DNA remained undigested.
The purchase of restriction enzymes that were quality tested to cut genomic DNA also proved
unsuccessful. Secondly, enzyÍres that were able to efficiently digest genomic DNA generated
fragment sizes for the ES cell lines harbouring either a wild type ALAS2 gene or loxPlneo'
modified ALAS2 locus that would not be distinguishable by Southern blot analysis.
Of the ALAS2 probes tested, one in particular proved to be binding non-specifically to
digested genomic DNA in test Southern hybridisations and was disregarded as a potential
probe in the screen for successful integration of the targeting vector at the 3' end. The
remaining probes tested were found to be specifically binding when tested on control genomic
Southern filters containing completely digested DNA but did not identiff the expected bands
with restriction enzymes deemed suitable for the 3' screen of the ALAS2 locus and the reason
for this is unclear.
Since difficulties in devising a suitable 3' screen using restriction eîzyme digestion and
Southern blot analysis of genomic DNA could not be resolved an alternate screening strategy
was undertaken. A PCR based method was adopted to determine if homologous
recombination at the ALAS2 locus in W9.5 ES cells had successfully occurred at the 3' end.
Two PCR screens were designed and initially optimised on parental W9.5 genomic DNA the
ALAS2 targeting vector (pC1l59Gloxneo) prior to Ozgene conducting the PCR screen. The
first screen was designed to ampliff a 1.8 kb fragment that spanned over the recombination
boundary between the targeting vector and endogenous ALAS2 attl;re 3' end and this was
performed on parental Wg.5 genomic DNA. The primers were designed to bind in exon 10
(mAExl0) of ALAS2 and to a position in intron 10 (mAsac7596) outside of the DNA
included in the targeting construct (Figure 4.17). Figure 4.18a illustrates the amplified 1.8 kb
band obtained in the test PCRs using parental W9.5 genomic DNA as a template. The
optimised PCR conditions are also detailed in Figure 4.I8a.
120

Figure 4.17 PCR screen of successful homologous recombination at the 3' end of
potential ALAS2 targeted W9.5 ES cells
A PCR screen was devised to confirm that homologous recombination had occurred at the 3'
end. The primer set A (mAloxneo3scr) and C (mAsac7596) would amplify up a 5 kb fragment
to establish that the loxPlneo sequence and Cl 159G mutation had been incorporated into
ALAS2 intron 7 and exon 8, respectively. To ensure that recombination had not altered the 3'
end where the targeting construct was introduced, an additional PCR amplification was
conducted. The reaction used primers B (mAExl0) and C (mAsac7596) to amplify a 1.8 kb
fragment spanning across this region. The lettered arrows represent the primers.
mAExlO : 5' GCGCTTGGCCCCCTCCCCCCACCACAGCCCTC 3'
mAsac7596 : 5' GGCTGTGGGAATAAGGGACTGGTGGAGTGGTGG 3'
mAloxneo3scr : 5' CGACCTGCAGCCAAGCTAGCTTGGCTGGACG 3'
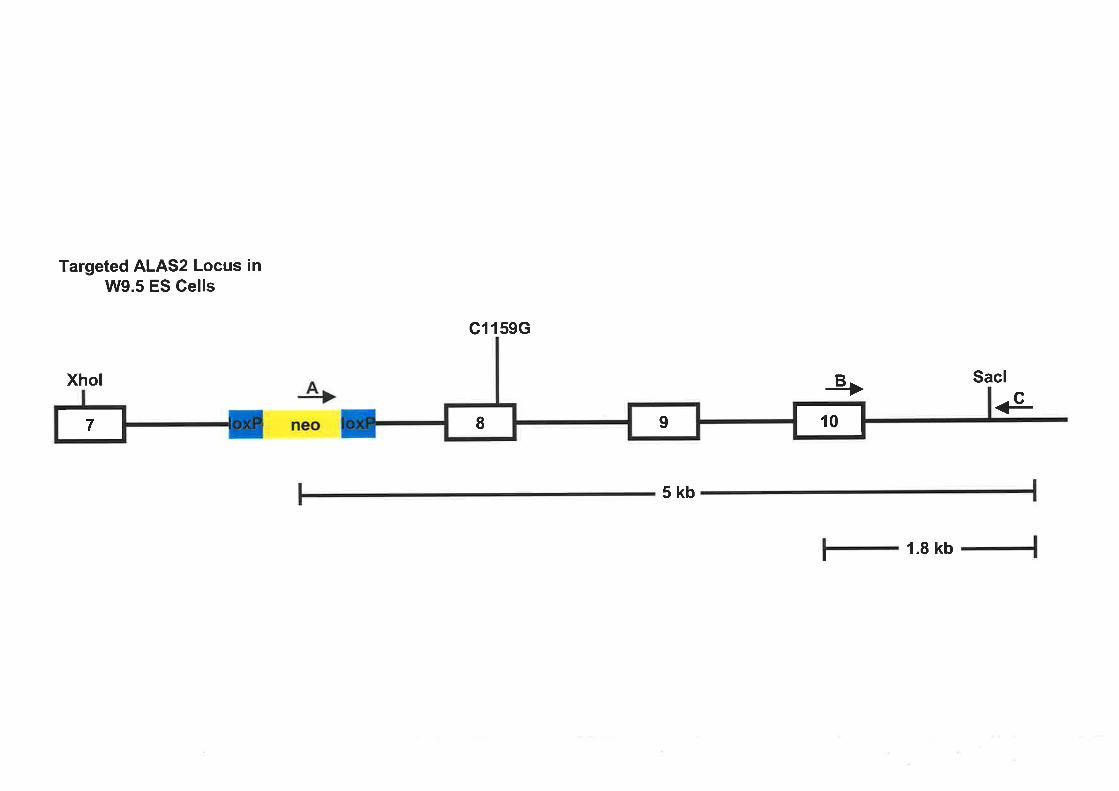
Targeted ALAS2 Locus inW9.5 ES Gells
Xhol
G1159G
+ Sacl4neo
é
5kb
1.8 kbI
z II r

Figure 4.18 Optimisation of a PCR based screen for assaying the success of homologous
recombination at the 3'end in ALAS2 targeted W9.5 ES cells
(a) The mAExl0 and mAsacT 596 pnmers at 100ng each were used to amplify a I .8 kb
ALAS2 fragment (labelled arrow) from parental W9.5 ES cell genomic DNA in a 50pl
reaction. Additional reagents included were:
-250pM dNTPs
-3.5mM MgCl2
-2 units of Taq polymerase
-Taq reaction buffer: 50mM KCl, l0mM Tris pH8.4
The reaction conditions \À/ere as follows: 1 minute at96oC,15 seconds at96"C,2 minutes at
68'C. This was performed for 45 cycles, followed by a final extension for 10 minutes at72"C.
The MgCl2was added after an initial step of 90 seconds at'75"C.
A control PCR amplification was performed in the absence of template DNA (data not
shown).
Lane 1 : Sppl marker (GeneWorks)
Lane 2 : PCR amplification

(a)
Lanes 1 2
+ 1.8kb

(b) The mAloxneo3scr and mAIl0sac primers at 100ng each were used to ampli$r a 4.8 kb
ALAS2 fragment (labelled arrow) from the ALAS2 targeting construct (pcl159c1oxneo)
(50ng) in a 50pl reaction. Additional reagents included were:
250pM dNTPs
4mM MgCl2
2 units of Taq/Pfu polymerase mix at 80:1
reaction buffer:25mM Tris pH9.1, 16mM (NH+)zSO+
Reaction condition were as follows: I minute at96"C,15 seconds aL96oC,45 seconds at
68"C, 5 minutes at72"C. This was performed for 45 cycles, followed by a final extension for
10 minutes at72"C. The MgCl2was added after an initial step of 90 seconds at'75"C. A
control PCR amplification was performed in the absence of template DNA (data not shown).
mAIl Osac : 5' GAACCCCTAGTGCCTATGTGACCATGGGCC 3'
Lane I : Sppl Markers (Geneworks)
Lane 2 : PCR amplification

(b)
Lanes 1 2
<- 4.8 kb

A second PCR screen was devised to ensure the loxPlneo'selection cassette was present in
the ALAS2 gene. Primers were designed to the loxPlneo'sequence (mAloxneo3scr) inserted
into intron 7 of the ALAS2 gene and the mAsac7596 pimer depicted in Figure 4.18 to result
in amplification of a 5 kb fragment. In order to optimise the screen using the mAloxneo3scr
primer the targeting vector (pC1l59Gloxneo) containing the loxPlneo'sequence was used as a
test template in conjunction with an alternate primer (mAIlOsac) partner that was able to bind
within this construct in intron 10 of the ALAS gene. this ùould result in amplification of a 4.8\.. .,'
kb fragment. Since a 4.8 kb product was amplified using the mAloxneo3scr primer (Figure
4.18b) partnered with a test primer it suggested that this primer would be functional in the
PCR screen when paired with the mAsac7596 primer and using ALAS2 targeted W9.5 DNA
as the template. The optimised PCR conditions are described in Figure 4.18b.
Upon consultation with Ozgene it was decided that I would perform the PCR screen on the
ALAS2 targeted W9.5 ES cell clone to confirm homologous recombination at the 3'end. The
PCR screen as optimised in Figure 4.18 was conducted on W9.5 genomic DNA that was
prepared from the ALAS2 targeted ES cell clone generated by Ozgene. 'I'he first PCR screen
conducted on the targeted ALAS2 W9.5 DNA was designed to amplifz a 1.8 kb band
spanning the recombination boundary @igure 4.I7)but no products were obtained (data not
shown). For the PCR using the internal primer (mAloxneo3scr) partnered with the primer
binding outside e!1tt" targeted region (mA2sac7596) many non-specific bands, smaller than
the expecfêd 5 kb frlernent were consistently observed (data not shown). Therefore, after'' \.
repeated atiêmpt5using the conditions optimised for the PCR screen successful homologous
recombination at the 3' end could not be confirmed by PCR analysis of the ALAS2 targeted
ES cell clone generated by Ozgene.
4.3 DISCUSSION
XLSA is an inherited blood disorder that leads to the production of defective red blood cells
that are morphologically small and poorly haemoglobinised. The differentiating erythroblasts
in the bone marrow of XLSA probands are characteristically represented by iron-laden
mitochondria surrounding their nucleus and are referred to as ringed sideroblasts (Bottomley,
1998a). The resultant hypoxia triggers the release of Epo from the kidney and a defective
cycle of erythropoiesis is stimulated. Linked to this cycle of ineffective erythropoiesis is an
increase in intestinal iron absorption which occurs via an unknown mechanism. If XLSA is
left untreated iron begins to accumulate in the body, particularly in the heart and liver, and
121

can lead to death. Point mutations in the ALAS2 gene of XLSA sufferers have been identified
and evidence suggests they are associated with the pathogenesis of the disease (see Section
1.10).
The aim of this project was to generate a mouso model for the blood disorder XLSA. Two
characterised XlSA-associated point mutations, Cl 159G and CI228T, were selected as a
basis for the XLSA model. A XLSA murine model would unequivocally establish mutations
in the ALAS2 gene as responsible for XLSA. Furthermore, a murine model for XLSA could
be used to investigate many aspects of the disease such as the late onset of the XLSA
phenotype in some probands and the abnormalities observed in iron regulation, including the
increase in intestinal iron absorption associated with ineffective erythropoiesis and iron
overload. The development of more effective treatments for XLSA, combating both the
anaemia and excess of iron that is observed with XLSA would also be benefited by an animal
model of this disease.
During my PhD, Brownlie et al. (1998) reported the establishment of azebraftsh embryo
model for a form of congenital sideroblastic anaemia. At both the molecular and
morphological level, zebrafish haematopoiesis resembles that of higher vertebrates (Catton,
l95l; Rowley et al., 1988; Detrich et al., 1995; Liao et al., 1998; Thompson et al., 1998). A
large scale genetic screen searching for zebrafish mutants with defects in normal development
identified the sauternes (sau) mutant. Zebrafish carrying the sau mutation are deficient in
haem and have decreased B-globin gene expression during embryogenesis. Subsequently, sau
was shown to encode a homologue of the ALAS2 eîzymq suggesting that mutations in saz
cause a similar phenotype to that observed in sideroblastic anaemia patients with a point
mutation in their ALAS2 gene. However, unlike human sideroblastic anaemia the
characteristic ringed sideroblasts were not evident in the analysis of adult red blood cells from
the zebraftsh sau mutants. Although the sau mutant zebrafish may represent the first animal
model for a type of congenital sideroblastic anaemia, the mutations found in the sau gene do
not correspond to any known human ALAS2 mutations associated with XLSA. Therefore, it
was worthwhile to pursue the development of a mouse model for XLSA based on the well
characterised Cl159G (Cox et al., 1994) and CI228T (Cotter et al,, I992a; Furuyama et al.
1998) XLSA associated point mutations.
Various gene targeting strategies were attempted to generate a XLSA murine model based on
either of the two ALAS2 mutations. The initial two-step strategy utilised the HPRT- ES cell
t22

line, E14TG2a, and introduction of the HPRT selection cassette into the ALAS2 locus. The
second round of targeting involved replacement of the HPRT sequence with ALAS2 sequence
containing the XLSA associated mutations to recreate a complete ALAS2 gene. ES cell lines,
derived from E14TG2a cells containing the HPRT cassette in the ALAS2 locus, were
produced that had incorporated either the Cl159G mutation into exon 8 or the Cl228T
mutation into exon 9. Two independently derived ES cell lines containing the exon 8 mutation
(C1159G) were injected into the inner cell mass of developing mouse blastocysts and the
resulting chimaeric mice were bred for several rounds but no germline transmission was
obtained. Although the exon 9 Cl228T mutation was incorporated into several independently
targeted ES cell clones it was decided to not pursue this targeting approach as a method for
developing a murine model of XLSA.
As discussed in Section 4.2.3, ES cells used in gene targeting need to be of exceptional
quality to increase the likelihood of obtaining cell lines that will contribute to the germline
following injection into murine blastocysts. This includes ES cells that have undergone a
limited number of passages (Nagy et al., 1990; F-edorov et al., 1991), cells that are able to
form morphologically sound colonies and cell lines devoid of chromosomal abnormalities
which can be common in ES cells (Suzuki et al., 1991). Although the ALAS2 exon 8 mutant
ES cell lines injected fulfilled two of these criteria, good morphology and high percentage of
cells with a normal karyotype, the second round of targeting produced cells of a very high
passage number. Our findings as well as those of other research groups within the department
indicated that the two round gene targeting approach was not very feasible as it produced ES
cells of a high passage number due to the high starting passage number of the stock E14TG2a
ES cells. To overcome this, anEl4TG2a or another ES cell line with a low starting passage
number was required. This observation, together with the department's procurement of an ES
cell line (W9.5) which was believed to have a comparably higher rate of germline "
transmission (J. Wrin, personal communication) lead to the decision to attempt targeting of
the ALAS2 locus in the W9.5 ES cells. The W9.5 ES cells were also available at a lower
starting passage number (9) than the E14TG2a ES cells (21)and thus, correctly targeted cells
for injection would be of a substantially lower passage number. A two-step gene targeting
approach, using the PGKneoYHSVtk selection cassette, was employed to manipulate the
ALAS2 gene in W9.5 ES cells. After several attempts at introducing either the Cl159G or
Cl228T mutation into the ALAS2 locus of the V/9.5 ES cells, suitable cell lines for injection
were not obtained. In addition, attempts at obtaining germline transmission from targeted
123

derivatives of W9.5 ES cells generated within other departmental research groups were
unsuccessful (J. Wrin, personal communication) (see Section4.2.3).
Up to this point, attempts to generate ALAS2 targeted ES cells had been conducted in two
types of ES cell lines, El4TG2a and \M9.5. At the time, these cell lines were available in the
department and being recommended for gene targeting experiments by the departmental
targeting facility. Since limited germline transmission \ryas obtained with modified derivatives
of E14TG2a ES cells and no contribution to the germline was observed with manipulated
W9.5 ES cell lines within the department's targeting facility, a commercial option to generate
ALAS2 targeted ES cells was undertaken. Ozgene was the commercial operation engaged to
target the exon 8 Cl159G mutation into the ALAS2 locus of W9.5 ES cells. The strategy for
introducing the point mutation into the ALAS2 gene took advantage of the Pl bacteriophage
crelloxP recombinase system. The main advantage of this approach is that only one step is
required to introduce the Cl159G mutation into exon 8 of the ALAS2 gene, unlike the
strategies employed with the El4TG2a and W9.5 ES cells. This would limit the amount of iz
vitro stltmng, producing ES cell lines with a low passage number and possibly a higher
potential for germline transmission. Ozgene generated one ES cell line that had potentially
incorporated the ALAS2 targeting construct, introducingaCll5gc point mutation into exon
8 and a loxPlneo' selection cassette into intron 7 of the ALAS2 gene. This was confirmed by
Southern hybridisation analysis of the 5' end of integration between the endogenous ALAS2
gene and the ALAS2 targeting vector.
However, the PCR screen devised to confirm integration at the 3'end was inconclusive. The
PCR conditions optimised on the test template DNA were not successful in amplif,iing the
desired fragments from the ALAS2 targeted W9.5 genomic DNA obtained from Ozgene.
Failure of the PCR screen to work effectively could indicate that homologous recombination
did not occur at the 3' end, although analysis of the 5' end by Southern hybridisation strongly
demonstrates that the ALAS2 alteration vector containing the loxPlneo'cassette had been
incorporated into the ALAS2 locus. An alternative explanation is that the conditions derived
for the PCR screen on the basis of the test templates (parental W9.5 genomic DNA and
pCl l59Gloxneo targeting vector DNA) were not suitable when applied to genomic DNA
from the ALAS2 targeted V/9.5 ES cell line. Therefore, prior to utilisation of this cell line to
generate a XLSA murine model it would be prudent to optimise the PCR screen using the
targeted ALAS2 W9.5 ES cell genomic DNA. Unfortunately, due to time constraints
characterisation of the 3' end of the ALAS2 targeted cell line could not be taken further.
ì ^,.,'.1 124

The targeting vector constructed to generate the ALAS2 targeted ES cell line contained a
loxPlneo' selection cassette inserted into intron 7, inlhe reverse orientation to the ALAS2
gene. It has been reported that mRNA levels are reduced to approximately 40Yo of wild type
alleles when the neo'gene is inserted in a non-coding region in the same orientation as the
gene of interest (Meyers et al., 19981, Nagy et al., 1998). This occurs due to the presence of a
splicing acceptor site between the PGK promoter and the neo'coding sequence which when
orientated in the same direction as the gene of interest generates an abnormal splicing event
(Nagy, 2000). Insertion of the neo'cassette in the reverse orientation, as in the ALAS2
targeted cell line, has also been reported to compromise the level of mRNA generated due to
creation of a splice acceptor site (Jacks et al., 1994; Carmeliet et al., 1996).
Although homologous recombination at the 3' end has not been confirmed it is highly likely,
due to a positive screen of the 5' region, that this study has produced one ALAS2 targeted ES
cell line containing the C1l59G point mutation in exon 8 and a loxPlneo'selection cassette in
intron 7 in the reverse orientation to the ALAS2 gene. As discussed above, the presence of the
neo' gene in the opposing direction may have a deleterious effect and may obscure the effect
of the introduced alteration on gene expression. The removal of a selection cassette flanked by
loxP sites from a targeted allele can be performed by either in vitro or in vivo expression of
cre recombinase. Common in vítro methods for removing foreign marker DNA from a
specific locus is to introduce the cre recombinase into the cell either transiently or stably via
electroporation or calcium precipitation (Nagy, 2000). This would enable removal of the
selection cassette from the targeted ES cell line prior to blastocyst injection and generation of
chimaeric mice. However, this would involve fuither invitro manipulation of the ES cells and
increase the passage number which may reduce the probability of the ES cell line contributing
to the germline (Kaartinen and Nagy, 2001).
Removal of the neo'gene from the ALAS2 locus via in vivo methods would require injection
of the current ES cell line generated by Ozgene into murine blastocysts to generate chimaeric
mice. Germline transmission of the male mice would produce female mice heterozygous for
the ALAS2 Cl159G point mutation. Since they are female heterozygotes, any effect of the
selection gene on expression of the modified ALAS2 gene will be masked by the presence of
a second wild type copy of the ALAS2 gene. These mice would then be bred with mice
ubiquitously expressing cre recombinase to enable the eventual removal of the marker DNA
125

from intron 7 of the ALAS2 gene. Therefore, in vivo removal of the neo' marker from the
ALAS2 gene would be the preferred strategy.
Post-targeting removal of the selection cassette by either in vitro or in vivo methods would
leave, in addition to the introduced Cl159G mutation, one of the /oxP sites inserted in intron
7 of the ALAS2 locus (Figure 4.19). Numerous gene targeted mice have been generated
indicating that the presence of a loxP site in intronic domains has no effect (Nagy, 2000).
Therefore, the loxP site remaining in intron 7 should not alter the effect of the Cl159G point
mutation on ALAS2 expression and hence generation of a murine model for XLSA.
In the future, a second commercial attempt should be made to target the ALAS2locus with
the Cl159G mutation as only one correctly targeted cell line was obtained from the initial
targeting experiment. As eluded to earlier, at least two independent targeted ES cell lines
should be generated prior to introduction into the mouse as a confirmation that the effects
observed in the mice generated are due to the mutation and not specific to the ES cell clone
chosen for blastocyst injection. This will also enable a cell line with the highest probability of
going germline to be selected for blastocyst injection and generation of the murine model for
XLSA.
In conclusion, several strategies were devised in an attempt to generate a mouse model for
XLSA. ES cell lines were produced that had been targeted at the ALAS2 locus incorporating
either the C1159G or the Cl228T mutation into exons 8 and 9, respectively. Majority of the
ALAS2 targeted ES cell lines were not suitable candidates for blastocyst injection and of
those injected, none transmitted to the germline. Future work in the area of XLSA should
focus on the continued development of a murine model to enable investigation of the
complexities associated with this disorder of the blood.
126

Figure 4.19 The introduction of a subtle change to a gene locus using the crelloxP
recombinase system
In order to incorporate a slight alteration into an endogenous gene using crelloxP recombinase
technology an antibiotic resistance gene, such as the neo'cassette, flanked by loxP sites needs
to be introduced along with the specific mutation. At a particular stage, cre recombinase is
used to excise out the loxPlneo'selection cassette from the targeted gene and remaining is the
introduced alteration as well as one of the /oxP sites. Therefore, the allele has been
specifically mutated and retains a loxP site in a non-coding region.

Locus of lnterest I II
xTarget Vector
Cre Excision
IMutated Allele
I\ I I
x
Targeted Locus
neo
neo

CHAPTER 5
FINAL SUMMARY

CHAPTER 5: FINAL SUMMARY
The regulation of ALAS2 expression during erythroid differentiation of precursor cells in the
adult bone maffow is of major interest to our laboratory since ALAS2 is the first and rate-
determining enzyme of haem s¡mthesis in red blood cells (May et al., 1995). Of the total
amount of haem produced in the body, 80% is utilised for the formation of haemoglobin in
red blood cells (V/orwood,1977). Although all cells require haem for specific cellular
functions, high levels of free haem are detrimental to the cell (Balla et al., l99l; Muller-
Eberhard and Fraig, 1993;May et al., 1995). Thus, strict regulation of haem synthesis is
crucial and in erythroid cells this is primarily controlled by expression and activity of ALAS2,
especially during erythropoiesis when production of haem is enhanced to form the oxygen
carrying molecule haemoglobin. In addition, aberration of haem synthesis can result in a
diseased state as observed with XLSA where a point mutation in the ALAS2 gene disrupts the
efficient production of haem, resulting in reduced haem levels and an anaemic phenotype
associated with iron overload. The work presented in this thesis will be discussed in the
context of the literature at the commencement of this project and will examine the
contribution of these findings to this area of research.
One of the major aims of this project was to investigate the regulation of ALAS2 during red
blood cell maturation, primarily focussing at the level of transcription. During erythropoiesis
ALAS2 expression is up-regulated to enable production of haem for formation of
haemoglobin in differentiating erythroid precursor cells. This regulation of ALAS2
expression occurs at the transcriptional, translational and post-translational level. It has been
well documented that cellular iron levels play a role in regulating translation of ALAS2
through the binding of IRPs to the IRE in the 5'-UTR of ALAS2 mRNA (Sadlon et al., 1999). ,
The action of Epo signalling has also been shown to affect the translation of ALAS2 possibly ,/mediated through weakened IRP2 binding to the IRE in the 5'-UTR of the mRNA (Zoller et
a1.,2002). At the post-translational level, haem itself has been shown to prevent the import of
ALAS2 precursor protein into the mitochondria by binding to a specific region of the protein
(Lathrop and Timko,1993;Zhang and Guarente,1995; Goodfellow et a1.,200I). ALAS2
expression during erythropoiesis is also regulated at the transcriptional level through the
effect of Epo (Zoller et a1.,2002) and hypoxia, although hypoxic up-regulation of ALAS2
mRNA levels is independent of HIF-I (Hofer et a1.,2003).
t27

DNase I hlpersensitivity site studies indicated the presence of potential transcriptional
regulatory regions within the immediate promoter, intron 1, intron 3 and intron 8 of the
murine ALAS2 gene (Schoenhaut and Curtis, 1989). Our laboratory extensively investigated
these regions to ascertain their contribution to the control of ALAS2 transcription. Surinya e/
al. (1997,199S) identified the promoter, intron 1 and the 3'end of intron 8 as control regions
within the human ALAS2 gene that activate and enhance the level of ALAS2 transcriptional
activity in an undifferentiated, erythroid cellular environment. Both erythroid-specific and
non-erythroid transcription factor binding sites were identified in these regions and specific
binding elements within the proximal ALAS2 promoter and intron 8 were shown to enhance
transcriptional activity of the ALAS2 gene in undifferentiated erythroid
1997,1998)
cells (Surin ya et al., '. "' : 't¿t.,r. I "" .,î "t.',:',.' , i/;'"-
h.:)
My doctoral research pursued the regulatory mechanism of ALAS2 transcription in the
context of erythroid differentiation. The J2E cell system was selected as a model for
erythropoiesis as upon Epo stimulation this cell line exhibits characteristics typical of
erythroid difTerentiation (Klinken et al., 1988; tsusfield and Klinken,1992; Busfield et al.,
1993; Tilbrook et al., 7996; Chappell et al., 1997). In Epo differentiatedJ2E erythroid cells,
the 293 bp ALAS2 proximal promoter generated the maximal increase in activity when ,'
compared to activity in undifferentiated J2E cells. This finding implied that the transcription
factor binding elements located within the ALAS2 293bp promoter, including CACCC boxes
and GATA-I binding sites, were adequate for facilitating the maximal level of transcriptional
activity in response to Epo induced differentiation. Furthermore, both introns I and 8 were
shown to enhance activity generated by the ALAS2 promoter in the presence of Epo treated i'
J2E cells. However, an enhancer element within intron 8 generated the strongest enhancement
of ALAS2 promoter activity in response to Epo stimulated differentiation. Investigation of the
contribution of individual transcription factor binding sites within the intron 8 Epo responsive
element to the promotion of ALAS2 transcription in response to Epo demonstrated that the
two CACCC box elements (CACCC-A and B) have minor roles in facilitating intron 8
enhancer activity, with the GATA-B site being the major element required. The GATA-A site
appeared to be inhibitory of intron 8 enhancer activity and like GATA-B, was shown to bind
GATA-1 in vitro and binding was unaffected by the use of nuclear extracts prepared from
Epo treated J2E cells. Interestingly, retarded complexes that formed with the human intron 8
CACCC-A and CACCC-B oligonucleotides in gel shift experiments using nuclear extracts
prepared from Epo stimulated or unstimulated J2E cells did not contain either the ubiquitous
r28

Sp1 or erythroid-specific EKLF transcription factors, which suggests a novel protein is
binding to these sites during erythroid differentiation.
It has been reported that the recruitment of coactivator complexes with intrinsic HAT activity
such as CBP/p300 to the promoter and enhancer regions of various genes is linked to
activation of gene expression, histone acetylation and chromatin remodelling (Wade and
V/olffe, t997; Grunstein, 1997; Kadonaga, 1998). Specifically, CBP/p300 has been shown to
acetylate and activate erythroid-specific transcription factors including GATA-1 (Blobel er
al., 1998; Boyes et a\.,1998; Hung et a\.,7999), NF-E2 (Hung et aL.,2001) and EKLF
(Zhang and Bieker, 7998; Zhang et al., 2001). Thus, preliminary experiments were conducted
in an attempt to evaluate the effect of CBP/p300 on the transcriptional regulatory elements of
the human ALAS2 gene. ElA, a known inhibitor of CBP/p300 activity (Goodman and
Smolik, 2000), markedly repressed the enhancer activity of intron 8 in transient transfection
of undifferentiated and Epo treated J2E cells. This was in contrast to the ALAS2 proximal
promoter where the effect of E1A on this region was negligible. An E1A mutant defective in
CBP/p300 binding was tested on human ALAS2 intron 8 reporter constructs and was shown
to have no effect on activity in either Epo treated or untreated J2E erythroid cells, suggesting
that EIA repression of intron 8 enhancer activity is mediated by CBP/p300. Howev.r, flité"'Idata supporting intron 8 as the element through which CBP/p300 regulates ALAS2 expression
"'-'
was contradictory to experimental results obtained with wild type p300 and a p300 mutant *,
, t -
expression construct that cannot bind ElA. Therefore, a definite role for CBP/p300 l,]"
coactivators in regulating ALAS2 transcription, particularly through the intron 8 Epo
responsive enhancer element during erythroid differentiation was not established in this study.
Prior to commencement of this study, transcriptional control elements within ALAS2 were
identified and characterised in undifferentiated erythroid cell types but little was known about
the identity and mechanism of the regulatory regions required for ALAS2 expression during
erythroid differentiation. The current study has provided insight into the major regulatory
elements within ALAS2 that are important for transcriptional activity during Epo induced
differentiation. In addition, specific transcription factor binding sites within the intron 8 Epo
responsive enhancer element and the transactivating factors they bind have been identified as
critical for this regulatory process. Coactivator proteins such as CBP/p300 may contribute to
the regulation of ALAS2 gene transcription but a dehnitive role was not established in this
study.
t29

Past studies have investigated the detailed regulation of ALAS2 expression in undifferentiated
erythroid cells while the current study has examined the effect of Epo, the physiological
stimulus of erflhropoiesis, on the regulatory elements controlling ALAS2 transcription.
However, these studies have not considered the effect of the chromatin environment on the
regulation of ALAS2 transcription as they have largely involved transient analysis of gene 'î"' i ''o
expression. Therefore, future studies investigating the transcriptional regulation of ALAS2 in
the context of chromatin would greatly benefit this area of research, advancing the current
knowledge of ALAS2 gene expression in differentiating erythroid cells.
Firstly, it would be of interest to determine whether the same Epo responsive elements
identified in the current study, particularly in intron 8, are also required to enhance ALAS2 _,,'
transcription when the gene is stably integrated into the chromosome of an erythroid cell 1..,,
l
induced to undergo erythroid differentiation. Experiments could also be conducted to , l' ,,
determine the in vivo profle of transactivating factors that bind at various sites within ALAS2
during erythroid differentiation. The current invitro binding studies showed that Epo
stimulation did not significantly alter the pattern or intensity of protein binding to the species
conserved transcription factor binding within intron 8, investigation of transcription factor
modifications such as acetylation and phosphorylation may provide an alternate mechanism
by which Epo induced differentiation mediates an increase in ALAS2 expression.
It would be of interest to determine the role, if any, that coactivator complexes play in ALAS2
gene transcription. If the involvement of a single or multiple coactivators was established,
investigation of the mechanism by which they regulate ALAS2 expression could be
undertaken. Possible roles for CBP/p300 in ALAS2 regulation include recruitment of
transcription factors to the ALAS2 gene, acetylation of ALAS2 bound transcription factors to
enhance their activity, provision of a 'bridge' to facilitate protein-protein interactions between
transactivating proteins binding at different regions of the ALAS2 gene and lastly,
modification of the chromatin structure surrounding the ALAS2 locus via histone acetylation.
In conclusion, the current knowledge regarding the transcriptional regulation of ALAS2
during erythropoiesis coupled with the findings from subsequent research conducted in the
context of a chromatin environment will provide a detailed and complete assessment of the
components and mechanisms important in regulating the expression of ALAS2 during
erythroid cell differentiation.
130

Defective haem synthesis has been linked to the blood disorder sideroblastic anaemia. Much
evidence has arisen to suggest that an inherited form of the disease, XLSA, may be associated
with the rate-limitingenzpe of haem synthesis in erythroblasts, ALAS2 (Bottomley, 1998a).
Our laboratory became involved in investigating the association between defective ALAS2
function and XLSA with particular interest in a XLSA case study involving a specific point
mutation in exon 8 of the ALAS2 gene (Cl2l5G). Although the available evidence implicated
defects in the ALAS2 gene as responsible for XLSA a direct relationship had not been
proven. Thus, the aim of this project was to generate a murine model for XLSA to definitively
establish that specific point mutations in the ALAS2 gene result in the blood disorder XLSA
and also to investigate various aspects of the disease such as perturbed iron homeostasis.
Two XlSA-associated mutations in the ALAS2 gene were selected as a basis for the model
(Cl159G in exon 8 and Cl228T in exon 9) and gene targeting technology employed to
introduce either mutation into the ALAS2 locus of the ES cell genome. Attempts were made
attargetingthe ALAS2 locus using the different targeting strategies made available through
the department's targeting facility and also a commercial gene targeting operation. However,
due to avanety of technical difficulties and time constraints the generation of a XLSA murine
model was not completed. Immediate future work should focus on completing the
characterisation of the ALAS2 targeted cell line generated through the commercial targeting
facility, Ozgene, to enable the continued development of the disease model.
During the course of this study, a zebrafish model for sideroblastic anaemia was reported
(Brownlie et al., 1998) which demonstrated that mutations in the sau Eene, a homologue of
the human ALAS2 gene, led to a similar phenotype associated with human XLSA, although
the characteristic ringed sideroblast was not observed. Although this claimed to be the first
report of an animal model for sideroblastic anaemia and documented a strong link between a
zebrafish homologue of human ALAS2 and a congenital form of sideroblastic anaemia, the
production of a XLSA murine model should still be pursued for several reasons. Firstly, this
study identified the disease phenotype as an inherited form of sideroblastic anaemia based on
the characteristics observed, however it did not specifically nominate an X-linked pattern of
inheritance.
Secondly, activity of the murine equivalent of the ALAS2 point mutations (Cl159G in exon 8
and Cl228T in exon 9) chosen for the model was reduced to levels comparable to the human
when expression studies were conducted in bacteria. However, equivalent mutations located
131

in the zebrafish sau geîe that resulted in congenital sideroblastic anaemia have not been
identified in the human ALAS2 gene for any known XLSA case studies so extrapolations of
the findings to the human form of the disease would be difficult.
Lastly, a zebraf,rsh model of congenital sideroblastic anaemia would limit the analysis of
perturbed iron metabolism associated with XLSA. Altematively, the murine model for XLSA
will permit investigation of the link between the cycle of ineffective erythropoiesis and the
increase in iron absorption from the gastrointestinal tract. Much is known about the process of
iron uptake into the intestinal mucosa cells from the intestine and recently new genes that are
crucial to the process of iron homeostasis have been identified (Ritter et al., 1999; Haile,
2000) and their functions characterised (Abboud and Haile, 2000; Donovan et a1.,2000;
McKie et aL.,2000, 2001). A murine model for XLSA may facilitate identification of genes
that when disrupted result in defective iron metabolism and this could provide insight into the
regulation of iron homeostasis. Such a model may even be used to investigate the molecular
basis of a broad range of diseases affecting iron metabolism.
In summary, this study has shown that transcription driven by the ALAS2 gene promoter
increases in response to Epo induced differentiation of erythroid cells. Furthermore, a region
at the 3' end of intron 8 and transcription factor-binding sites within this domain are critical
for enhancing the level of gene transcription during erlhroid differentiation. Contribution of
additional regulatory elements, such as coactivator complexes, to ALAS2 transcription is
unclear and their involvement needs to be investigated further. The major question that stands
to be addressed concerns the regulation of ALAS2 expression during erythropoiesis in the
context of a chromatin environment, such that the regulatory effect of chromatin structure on
ALAS2 transcription can be determined. Lastly, generation of a murine model for XLSA will
be pursued and primarily used to investigate the unknown aspects associated with XLSA such
as the link between ineffective erythropoiesis and the deregulation of iron absorption from the
intestine.
732

REFERENCE LIST

REFERENCE LIST
Abboud, S., and Haile, D. J. (2000). A novel mammalian iron-regulated protein involved in
intracellular iron metabolism. J Biol Chem 275,19906-12.
Abraham, S. E., Lobo, S., Yaciuk, P.,'Wang, H. G., and Moran, E. (1993)' p300, and p300-
associated proteins, are components of TATA-binding protein (TBP) complexes. Oncogene 8,
1639-47.
Alcindor, T., and Bridges, K. R. (2002). Sideroblastic anaemias. Br J Haematol 116,133-43.
Allikmets, R., Raskind,'W. H., Hutchinson, 4., Schueck, N. D., Dean, M., and Koeller, D. M.
(1999). Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked
sideroblastic anemia and ataxia (XLSA/A). Hum Mol Genet 8,143-9.
1'.
Andrews, N. C., Erdjument-Bromage, H., Davidson, M. 8., Tempst, P., and Orkin, S. H. \
(1993a). Erythroid transcription factor NF-E2 is a haematopoietic-specific basic-leucine
protein. Nature 3 62, 122-8.
Andrews, N. C., and Faller, D. V. (1991). A rapid micropreparation technique for extraction
of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res 19,
2499.
Andrews, N Kotkow, K. J., Ney, P. 4., Erdjument-Bromage, H., Tempst, P., and Orkin,
s. H. (1ee3b) ubiquitous subunit of erythroid transcription factor NF-E2 is a small basic-
related to the v-maf oncogene. Proc Natl Acad Sci U S A 90, 11488-92
Aoki, Y., Muranaka, S., Nakabayashi, K., and Ueda, Y. (1979). delta-Aminolevulinic acid
slmthetase in erythroblasts of patients with pyridoxine-responsive anemia. Hy,percatabolism
caused by the increased susceptibility to the controlling protease. J Clin Invest 64,1196-203.
Armstrong, J. 4., Bieker, J. J., and Emerson, B. M. (1998). A SWVSNF-related chromatin
remodeling complex, E-RC1, is required for tissue-specific transcriptional regulation by
EKLF in vitro. Cell95,93-104.
133

Armstrong, J. 4., and Emerson, B. M. (1996). NF-E2 disrupts chromatin structure at human
beta-globin locus control region hypersensitive site 2 in vitro. Mol Cell Biol 16,5634-44.
Arosio, P., Adelman, T. G., and Drysdale, J. W. (1978). On ferritin heterogeneity. Further
evidence for heteropol¡rmers. J Biol Chem 253,4451-8.
Asano, H., Li, X. S., and Stamatoyannopoulos, G. (2000). FKLF-2: a novel Kruppel-like
transcriptional factor that activates globin and other erythroid lineage genes. Blood 95,3518-
84.
Asano, H., Li, X. S., and Stamatoyannopoulos, G. (1999). FKLF, a novel Kruppel-like factor
that activates human embryonic and fetal beta-like globin genes. Mol Cell Biol 19,3571-9.
Ashe, H. L., Monks, J., Wijgerde, M., Fraser, P., and Proudfoot, N. J. (1997). Intergenic
transcription and transinduction of the human beta-globin locus. Genes Dev I I ,2494-509.
Aziz, N., and Munro, H.N. (1987). hon regulates ferritin mRNA translation through a
segment of its 5' untranslated region. Proc Natl Acad Sci U S A 84,8478-82.
Balla, G., Vercellotti, G. M., Muller-Eberhard, U., Eaton, J., and Jacob, H. S. (1991).
Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and
toxic oxygen species. Lab Invest 64,648-55.
Bannister, A. J., and Kouzarides, T. (1996). The CBP co-activator is a histone
acetyltransferase. Nature 3 84, 641-3.
Barber, D.L., Beattie,8.K., Mason, J.M., Nguyen, M.H., Yoakim, M., Neel,8.G.,
D'Andrea, A. D., and Frank, D. A. (2001). A common epitope is shared by activated signal
transducer and activator of transcription-5 (STAT5) and the phosphorylated erythropoietin
receptor: implications for the docking model of STAT activation. Blood 97,2230-7 .
Barton, M. C., Madani, N., and Emerson, B. M. (1993). The erythroid protein oGATA-1
functions with a stage-specific factor to activate transcription of chromatin-assembled beta-
globin genes. Genes Dev 7,1196-809.
t34

Beau, C., Rauch, M., Joulin, V., Jegou, 8., and Guerrier, D. (2000). GATA-1 is a potential
repressor of anti-Mullerian hormone expression during the establishment of puberty in the
mouse. Mol Reprod Dev 56,124-38.
Beaumont, C., Deybach, J. C., Grandchamp, 8., da Silva, V., de Vemeuil, H., and Nordmann,
Y. (1984). Effects of succinylacetone on dimethylsulfoxide-mediated induction of heme
pathway enzymes in mouse friend virus-transformed erythroleukemia cells. Exp Cell Res 154,
474-84.
Begley, C. G., and Green, A. R. (1999). The SCL gene: from case report to critical
hematopoietic regulator. Blood 9 3, 27 60-7 0.
Begley, C. G., Visvader, J., Green,4.R., Aplan, P.D., Metcalf, D., Kirsch,I. R., and Gough,
N. M. (1991). Molecular cloning and chromosomal localization of the murine homolog of the
human helix-loop-helix gene SCL. Proc Natl Acad Sci U S A 88, 869-73.
Bekri, S., Kispal, G., Lange, H., Fitzsimons, E., Tolmie, J., Lill, R., and Bishop, D. F. (2000).
Human ABCT transporter: gene structure and mutation causing X-linked sideroblastic anemia
with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood 96,3256-64.
Bell, A. C., and Felsenfeld, G. (1999). Stopped at the border: boundaries and insulators. Curr
Opin Genet Dev 9,191-8.
Ben-David, Y., and Bernstein, A. (1991). Friend virus-induced erythroleukemia and the
multistage nature of cancer. Ce||66,831-4.
Bender, M. 4., Bulger, M., Close, J., and Groudine, M. (2000). Beta-globin gene switching
and DNase I sensitivity of the endogenous beta-globin locus in mice do not require the locus
control region. Mol Cell 5, 387 -93.
Beris, P., Samii, K., Darbellây, R., Zoumbos, N., Tsoplou, P., Kourakli,4., Preud'homme, C.,
and Fenaux, P. (1999). Iron overload in patients with sideroblastic anaemia is not related to
the presence of the haemochromatosis Cys282Tyr and His63Asp mutations. Br J Haematol
104,97-9.
13s

Bessis, M., Lessin, L. S. and Beutler, E. (1983) in'. Hematology 3'd Ed.Wllliams, W. J.,
Beutler, E., Erslev, A. J. and Lichtman, M. A. (eds), McGraw-Hill, New York, pp:257-79.
Bhasker, C. R., Burgiel, G., Neupert,8., Emery-Goodman, 4., Kuhn, L.C., and May, B. K.
(1993). The putative iron-responsive element in the human erythroid 5-aminolevulinate
syrthase mRNA mediates translational control. J Biol Chem 268,12699-705.
Bieker, J. J., and Southwood, C.M. (1995). The erythroid Kruppel-like factor transactivation
domain is a critical component for cell-specific inducibility of a beta-globin promoter. Mol
Cell Biol 15,852-60.
Birnboim, H. C., and Doly, J. (1979). A rapid alkaline extraction procedure for screening
recombinant plasmid DNA. Nucleic Acids Res Z, l5l3-23
Bishop, D. F., Henderson, A. S., and Astrin, K. H. (1990). Human delta-aminolevulinate
synthase: assignment of the housekeeping gene to 3p2l and the erythroid-specific gene to the
X chromosome. Genomics 7,207-14.
Blanchard, K. L., Acquaviva, 4.M., Galson, D. L., and Bunn, H.F. (1992). Hypoxic
induction of the human erythropoietin gene: cooperation between the promoter and enhancer,
each of which contains steroid receptor response elements. Mol Cell Biol I2,5373-85.
Blank, V., and Andrews, N. C. (1997). The Maf transcription factors: regulators of
differentiation. Trends Biochem Sci 2 2, 437 -41 .
Blobel, G. A. (2002). CBP and p300: versatile coregulators with important roles in
hematopoietic gene expression. J Leukoc Biol 7l, 545-56,t-í-\.É\
\I
tiI
integratots'Blobel, G. A. (2000). CREB-binding protein and p300: molecular
hematopoietic transcription. Blood 9 5, 7 45 -5 5 .
Blobel, G.4., Nakajima, T., Eckner, R., Montminy, M., and Orkin, S. H. (1998). CREB-
binding protein cooperates with transcription factor GATA-I and is required for erythroid
differentiation. Proc Natl Acad Sci U S A 95, 2061-6.
of
t36

Borthwick, I. 4., Srivastava, G., Brooker, J. D., May, B. K., and Elliott, W. H. (1983).
Purification of 5-aminolaevulinate s¡mthase from liver mitochondria of chick embryo. Eur J
Biochem 129,615-20.
1
Bottomley, S (1ee8b). s iron overload disorders. Semin Hematol 35,17-86
Bottomtrey, S. S., Healy,H.M., Brandenburg, M. 4., and May, B. K. (1992).5-
Aminolevulinate synthase in sideroblastic anemias: mRNA and enzyme activity levels in
bone marrow cells. Am J Hematol 41,16-83.
Bottomley, S. S., May, B. K., Cox, T. C., Cotter, P. D., and Bishop, D. F. (1995). Molecular
defects of erythroid 5-aminolevulinate synthase in Xlinked sideroblastic anemia. J Bioenerg
, Biomembr 27,161-8.
Bottomley, S. S., and Muller-Eberhard, U. (1988). Pathophysiology of heme s¡mthesis. Semin
25,282-302
Bottomley, S. S. (1998a) Sideroblastic Anemias in: Wintrobe's Clínical Hematology, lTth Ed.
Lee, G. R., Bithell, T. C., Foerster, J., Athens, J. W. and Lukens, J. N. (eds), Williams and
Willeins, Baltimore, pp: 1022-45.
L
Bottomley, 3. S., Wir", P.D.,'Wasson, E.G,, and Carpenter, N. J. (1998). X-linked/t
')
sideroblastic anemia in ten female pro
chromosome American J
bands due to AIA¡!2 mutations
ournal of Genetics 63,'A352.
and skewed X
¿J4
Ælrv., .j1.
Boyd, D., Vecoli, C., Belcher, D.M., Jain, S. K., and Drysdale, J. W. (1985). Structural and
functional relqügnships of human ferritin H and L chains deduced from oDNA clones. J Biol
Chem 260,11155-61.
Boyes, J., Byheld, P., Nakatani, Y., and Ogryzko, V. (1998). Regulation of activity of the
transcription factor GATA-1 by acetylation. Nature 396,594-8.
Bronson, S. K., and Smithies, O. (1994). Altering mice by homologous recombination using
embryonic stem cells. J Biol Chem 269,27155-8.
t37

Brownlie,4., Donovan,4., Pratt, S. J., Paw,8.H., Oates,4.C., Brugnara, C., Witkowska,
H. 8., Sassa, S., and Zon,L.I. (1998). Positional cloning of the zebrafish sauternes gene: a
model for congenital sideroblastic anaemia. Nat Genet 20,244-50.
Bulger, M., and Groudine, M. (1999). Looping versus linking: toward a model for long-
distance gene activation. Genes Dev 13,2465-ll.
Bungert, J., Dave, U., Lim, K.C., Lieuw, K.H., Shavit, J.4., Liu, Q., and Engel, J. D.
(1995). Synergistic regulation of human beta-globin gene switching by locus control region
elements HS3 and HS4. Genes Dev 9,3083-96.
Bungert, J., Tanimoto, K., Patel, S., Liu, Q., Fear, M., and Engel, J. D. (1999). Hlpersensitive
site 2 specifies a unique function within the human beta-globin locus control region to
stimulate globin gene transcription. Mol Cell Bíol 19,3062-72.
Busfield, S. J., Chappell, D. S., Jennings, G., Trengove, N. J., Riches, K. J., Callus, B. 4.,
Tilbrook, P. 4., and Klinken, S. P. (1995a). Erythropoietin exerts transcriptional and
translational control over globin synthesis inJ2E cells. Cell Growth Differ 6,429-31.
Busfield, S. J., Farr, T. J., Singh, T., Sainsbury, A. J., and Klinken, S. P. (1993). Clonal
analysis of erythropoietin stimulated J2E cells reveals asynchrony during terminal
differentiation. Growth Factors 9, 307 -75.
Busfield, S. J., and Klinken, S. P. (1992). Erythropoietin-induced stimulation of
differentiation and proliferationinJ2E cells is not mimicked by chemical induction. Blood
80,4I2-g. t;
Busfield, S. J., Spadaccini, 4., Riches, K. J., Tilbrook, P. 4., and Klinken, S. P. (1995b). The
major erythroid DNA-binding protein GATA-1 is stimulated by erythropoietin but not by
chemical inducers of erythroid differentiation. Eur J Biochem 230, 415-80.
Busfield, S. J., Tilbrook, P. 4., Callus, B. 4., Spadaccini, 4., Kuhn, L., and Klinken, S. P.
(I 9 97 ). Complex re gul ation of transferrin receptors during erythropoietin-induced
138

differentiation of J2E erythroid cells--elevated transcription and mRNA stabilisation produce
only a modest rise in protein content. Eur J Biochem 249,17-84.
Butt, J., Kim, H. Y., Basilion, J. P., Cohen, S.,Iwai, K., Philpott, C. C., Altschul, S.,
Klausner, R. D., and Rouault, T. A. (1996). Differences in the RNA binding sites of iron
regulatory proteins and potential target diversity. Proc Natl Acad Sci U S A 93,4345-9.
Cable, E. E., Miller, T. G., and Isom, H. C. (2000). Regulation of heme metabolism in rat
hepatocytes and hepatocyte cell lines: delta-aminolevulinic acid s¡mthase and heme
oxygenase are regulated by different heme-dependent mechanisms. Arch Biochem Biophys
384,280-95.
Carmeliet, P., Ferreira, V., Breier, G., Pollefeyt, S., Kieckens, L., Gertsenstein, M., Fahrig,
M., Vandenhoeck,4., Harpal, K., Eberhardt, C., Declercq, C., Pawling, J., Moons, L., Collen,
D., Risau, W., and Nagy, A. (1996). Abnormal blood vessel development and lethality in
embryos lacking a single VEGF allele. Nature 380,435-9.
Casey, J.L.,Hentze, M. W., Koeller, D.M., Caughman, S.W.,Rouault, T. 4., Klausner, R.
D., and Harford, J. B. (1988). Iron-responsive elements: regulatory RNA sequences that
control mRNA levels and translation. Science 240,924-8.
Catton, W. T. (1951). Blood cell formation in teleost fishes. Blood 6,39-60.
Cazzola, M.,Invernizzi,R., Bergamaschi, G., Levi, S., Corsi,8., Travaglino, E., Rolandi, V.,
Biasiotto, G., Drysdale, J., and Arosio, P. (2003). Mitochondrial ferritin expression in
erythroid cells from patients with sideroblastic anemia. Blood l0l, 1996-2000.
Cazzola, M., May, 4., Bergamaschi, G., Cerani, P., Ferrillo, S., and Bishop, D. F. (2002).
Absent phenotypic expression of X-linked sideroblastic anemia in one of 2 brothers with a
novel ALAS2 mutation. Blood 100,4236-8. i'';
Cazzola, M., May, 4., Bergamaschi, G., Cerani, P., Rosti, V., and Bishop, D. F. (2000).
Familial-skewed X-chromosome inactivation as a predisposing factor for late-onset X-linked
sideroblastic anemia in carrier females. Blood 96,4363-5.
139

Chan, K., Lu, R., Chang, J. C., and Kan, Y. V/. (1996). NRF2, a member of the NFE2 family
of transcription factors, is not essential for murine erythropoiesis, growth, and development.
Proc Natl Acad Sci U S A 93,13943-8.
Chan, L.N., and Gerhardt, E. M. (1992). Transferrin receptor gene is hyperexpressed and
transcriptionally regulated in differentiating erythroid cells. J Biol Chem 267,8254-9.
Chandler, H. C., (1996). Honours' Thesis, University of Adelaide, South Australia, Australia.
Chapman, V. M., Keitz, B. T., and Bishop, D.F. (1994). Genetic linkage of the erythroid-
specific delta-aminolevulinate synthase gene (Alas2) to the distal region of the mouse X
chromosome. Mamm Genome 5,74I.
Chen, J. J., and London, I. M. (1995). Regulation of protein synthesis byheme-regulated eIF-
2 alphakinase. Trends Biochem Sci 20,105-8.
Chen, J. J., Yang, J.M., Petrysh¡m, R., Kosower, N., and London, L M. (1989). Disulfide
bond formation in the regulation of eIF-2 alpha kinase by heme. Joumal of Biological
, Chemistry 2 64, 9559-64.
Cheng, X., Reginato, M. J., Andrews, N. C., andLazar, M. A. (1997). The transcriptional
integrator CREB-binding protein mediates positive cross talk between nuclear hormone
receptors and the hematopoieticbZip protein p45A{F-E2. Mol Cell Biol 17,1407-16.
Chiba, T., Ikawa, Y., and Todokoro, K. (1991). GATA-I transactivates erythropoietin
NAR 9
gene, and erythropoietin receptor-mediated signals enhance GATA-l expression.
,3843-48.
Chin, H., Arai, 4.,'Wakao, H., Kamiyama, R., Miyasaka, N., and Miura, O. (1998). Lyn
physically associates with the erythropoietin receptor and may play a role in activation of the
Stat5 pathway. Blood 91,3734-45.
Cho, H., Orphanides, G., Sun, X., Yang, X. J., Ogryzko, V., Lees, E., Nakatani, Y., and
Reinberg, D. (1998). A human RNA polymerase II complex containing factors that modify
chromatin structure. Mol Cell Biol 18, 5355-63.
140

Cooley, T. B. (1945). A severe type of hereditary anemi
sequence of splenectom!. American Journal of Medical
Coghill, E., Eccleston, S., Fox, V., Cemrti, L., Brown, C., Cunningha-, J., Jane, S., and
Perkins, A. (2001). Erythroid Kruppel-like factor (EKLF) coordinates erythroid cell
proliferation and hemoglobinization in cell lines derived from EKLF null mice. Blood 97,
1 861-8.
Conboy, J. G., Cox, T. C., Bottomley, S. S., Bawden, M. J., and May, B.K. (1992). Human
erythroid 5-aminolevulinate synthase. Gene structure and species-specific differences in
alternative RNA splicing. J Biol Chem 267,18753-8.
Constantinescu, S. N., Ghaffari, S., and Lodish, H. F. (1999). The Erythropoietin Receptor:
Structure, Activation and lntracellular Signal Transduction. Trends Endocrinol Metab 10,18-
23.
a with elliptocytosis. Interesting
Science 2Q9,561.
¡-t',^+t'?Cooper, M. C., L"ty, J., Cantor, L.N., Marks, P. 4., and Rifkind, R. A. (1974). The effect of
erythropoietin on colonial growth of erythroid precursor cells in vitro. Proc Natl Acad Sci U S
A 71,1677-80.
Cormier, F., de Paz,P., and Dieterlen-Lievre, F. (1986). In vitro detection of cells with
monocytic potentiality in the wall of the chick embryo aorta. Dev Biol 118, 167-75.
Cote, J., Quinn, J., Workman , J . L., and Peterson, C. L. (1994). Stimulation of GAL4
derivative binding to nucleosomal DNA by the yeast SWVSNF complex. Science 265,53-60
Cotter, P. D., Baumann, M., Rucknagel, D. L., Fitzsimons, E. J., May, A. and Bishop, D. F
(1992a). Heterogeneity in X-linked sideroblastic anemia due to unique mutations in the
erythroid õ-aminolevulinic acid synthase gene. Am. J. Human Genetics 51, A4.
Cotter, P. D., Baumann, M., and Bishop, D.F. (1992b). Enzymatic defect in "X-linked"
sideroblastic anemia: molecular evidence for erythroid delta-aminolevulinate synthase
deficiency. Proc Natl Acad Sci U S A 89,4028-32.
t4l

Cotter, P. D., May, 4., Li,L, Al-Sabah, A. I., Fitzsimons, E. J., Cazzola, M., and Bishop, D.
F. (1999). Four new mutations in the erythroid-specific 5-aminolevulinate s¡mthase (ALAS2)
gene causing X-linked sideroblastic anemia: increased pyridoxine responsiveness after
removal of iron overload by phlebotomy and coinheritance of hereditary hemochromatosit. ñi\
Blood 93,1157-69. j
iCotter, P. D., Rucknagel, D. L., and Bishop, D.F. (1994). Xlinked sideroblastic anemia:
identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene
(ALAS2) in the original family described by Cooley. Blood 84,3975-24.
Cox, T. C., Bawden, M.J., Abraham, N.G., Bottomley, S.S., May, B. K., Baker, E., Chen, L'
2., and Sutherland, G.R. (1990). Erythroid 5-aminolevulinate synthase is located on the X
chromosome. Am J Hum Genet 46,107-ll.
Cox, T. C., Bawden, M. J., Martin,4., and May, B. K. (1991)' Human erythroid 5-
: promoter analysis and identification of an iron-responsive element
in the mRN Embo 10,189r-902.1
L-:\ Ito T
Cox, T. C., Bottomley, S. S., Wiley, J. S., Bawden, M. J., Matthews, C. S., and May, B. K.
(1994). X-linked pyridoxine-responsive sideroblastic anemia due to a Thr388-to-Ser
substitution in erythroid 5-aminolevulinate synthase. N Engl J Med 330,615-9.
Cox, T. C., Sadlon, T. J., Schwartz, Q. P., Matthews, C. S', Wise, P. D., Cox, L. L',
Bottomley, S. S. and May, B. K. Splice variants of human 5-aminolevulinate synthase-2: the
major variant contributes significantly to erythroid heme biosynthesis. Paper submitted
Crichton, R. R., Wilmet, S., Legssyer, R., and Ward, R. J. (2002). Molecular and cellular
mechanisms of iron homeostasis and toxicity in mammalian cells. J Inorg Biochem 91,9-18.
Crispino, J. D., Lodish, M. 8., MacKay, J. P., and Orkin, S. H. (1999). Use of altered
specificity mutants to probe a specific protein-protein interaction in differentiation: the
GATA-I:FOG complex. Mol Cell 3,219-28.
Crosby, J. S., Lee, K., London, I. M.,-.and Chen, J. J. (1994). Erythroid expression of the
heme-regulated eIF-2 alpha k-^i:.Y"lecular and Cell nio]oeV I 4, 3906-14.
r,i.l'.)ot t !^Q r - ''"" / '
ùtt 1' "r'142

Crossley, M., Merika, M., and Orkin, S. H. (1995). Self-association of the erythroid
transcription factor GATA-I mediated by its zinc finger domains. Mol Cell Biol 15, 2448-56.
Crossley, M., and Orkin, S. H. (1994). Phosphorylation of the erythroid transcription factor
GATA-I. J Biol Chem 269,16589-96.
Crossley, M., Whitelaw, E., Perkins,4., Williams, G., FujiwaÍa,Y., and Orkin, S. H. (1996)
Isolation and charactenzation of the cDNA encoding BKLF/TEF-2, amajor CACCC-box-
binding protein in erythroid cells and selected other cells. Mol Cell Biol 16,1695-105.
Csere, P., Lill, R., and Kispal, G. (1998). Identification of a human mitochondrial ABC
transporter, the functional orthologue of yeast Atmlp. FEBS Lett 441,266-70.
Dallas, P. 8., Yaciuk, P., and Moran, E. (1997). Charactenzation of monoclonal antibodies
raised against p300: both p300 and CBP are present in intracellular TBP complexes. J Virol
7l, r726-3t.
Dalyot, N., Fibach, E., Ronchi, A., Rachmilewltz, E.4., Ottolenghi, S., and Oppenheim, A.
(1993). Erythropoietin triggers a burst of GATA-I in normal human erythroid cells
differentiating in tissue culture. Nucleic Acids Res 21, 4031-1.
Dame, C., Fahnenstich, H., Freitag, P., Hofmann, D., Abdul-Nour, T., Bartmann, P., and
Fandrey, J. (1998). Erythropoietin mRNA expression in human fetal and neonatal tissue.
Blood 92,3218-25.
Dame, C., and Juul, S. E. (2000). The switch from fetal to adult erythropoiesis. Clin Perinatol
27,507-26.
Damen, J. E., Wakao, H., Miyajima,4., Krosl, J., Humphries, R. K., Cutler, R. L., and
Krystal, G. (1995). Tyrosine 343 inthe erythropoietin receptor positively regulates
erythropoietin-induced cell proliferation and Stat5 activation.
ï0" ,'44,5557-68.
D'Andrea,4.D., Lodish, H. F., and Wong, G. G. (1989). Expression cloning of the murine
erythropoietin receptor . Cell 5 7 , 271 -85 .
143

D'Andrea, A. D., Yoshimura,4., Youssoufian, H.,Zon, L. I., Koo, J.W., and Lodish, H.F.
(1991). The cytoplasmic region of the erythropoietin receptor contains nonoverlapping
positive and negative growth-regulatory domains. Mol Cell Biol I I,1980-7.
D'Andrea, A. D., andZon, L. I. (1990). Erythropoietin receptor. Subunit structure and
activation. J Clin Invest 86,687-7.
Darr, D., and Fridovich, I. (1994). Free radicals in cutaneous biology. J Invest Dermalol 102,
6ll-5.
deBoer, E., Antoniou, M., Mignotte, V., Wall, L., and Grosveld, F. (1988). The human beta-
globin promoter; nuclear protein factors and erythroid specific induction of transcription.
Embo J 7,4203-12.
Detrich, H.W., 3rd, Kieran, M.'W., Chan, F.Y., Barone, L.M., Yee, K., Rundstadler, J.4.,
Pratt, S., Ransom, D., and Zon,L.I. (1995). Intraembryonic hematopoietic cell migration
during vertebrate development. Proc Natl Acad Sci U S A 92,10713-1 .
Dierks, P. (1990) in: Biosynthesis of Heme and Chlorophylls.Dalley, H. A. (ed), McGraw-
Hill, New York, pp:201-33.
Dieterlen-Lievre, F. (1975). On the origin of haemopoietic stem cells in the avian embryo: an
experimental approach. J Embryol Exp Morphol 33,607-79.
Donovan,4., Brownlie,4., Zhou, Y., Shepard, J., Pratt, S. J., Moynihan, J., Paw, B. H.,
Drejer,4., Barut, B.,Zapata,A., Law, T.C., Brugnara, C., Lux, S. E., Pinkus, G. S., Pinkus,
J. L., Kingsley, P. D., Palis, J., Fleming, M. D., Andrews, N. C., andZon, L. I. (2000).
Positional cloning of zebrafish ferroportinl identifies a conserved vertebrate iron exporter.
Nature 403,776-81.
Donze, D., Townes, T. M., and Bieker, J. J. (1995). Role of erythroid Kruppel-like factor in
human gamma- to beta-globin gene switching. J Biol Chem 270,7955-9.
144

Dorsett, D. (1999). Distant liaisons: long-range enhancer-promoter interactions in Drosophila.
Curr Opin Genet Dev 9,505-14.
Drake, C. J., Brandt, S. J., Trusk, T. C., and Little, C.D. (1997). TALI/SCL is expressed in
endothelial progenitor cells/angioblasts and defines a dorsal-to-ventral gradient of
vasculogenesis. Dev Biol 1 92, 17 -30.
Drysdale, J., Arosio, P., Úrvernizzi,R., Cazzola, M., Volz, A', Corsi, 8., Biasiotto, G., and
Levi, S. (2002). Mitochondrial ferritin: a new player in iron metabolism. Blood Cells Mol Dis
29,376-83.
Dzieruak, E., and Medvinsky, A. (1995). Mouse embryonic hematopoiesis. Trends Genet 11,
359-66.
Dzlkaite,V., Kanopka,4., Brock, J.H., Kazlauskas,4., and Melefors, O. (2000). A novel
endoproteolytic processing activity in mitochondria of erythroid cells and the role in heme
s¡mthesis. Blood 96, 7 40-6.
Ebb, D., Tang, D. C., Drew, L., Chin, K., Berg, P. E., and Rodgers, G. P. (1998).
Identification of upstream regulatory elements that repress expression of adult betaJike
globin genes in a primitive erythroid environment. Blood Cells Mol Dís 24,356-69.
Ebert, B. L., and Bunn, H. F. (1999). Regulation of the erythropoietin gene. Blood 94,1864-
77.
Eckardt, K, U., and Bauer, C. (1989). Erythropoietin in health and disease. Eur J Clin Invest
lg,ll7-27.
Eckner, R. (1996). p300 and CBP as transcriptional regulators and targets of oncogenic
events. Biol Chem 377,685-8.
Edgar, A. J., Losowsky, M. S., Noble, J. S., and Wickramasinghe, S. N. (1997). Identihcation
of an arginine452 to histidine substitution in the erythroid 5-aminolaevulinate synthetase gene
in alargepedigree with X-linked hereditary sideroblastic anaemia. Eur J Haematol 58,l-4.
r45

Edgar,A. J., Vidyatilake, H.M., and Wickramasinghe, S. N. (1998). X-linked sideroblastic
anaemia due to a mutation in the erythroid 5-aminolaevulinate synthase gene leading to an
argininelT0 to leucine substitution. Eur J Haematol 61, 55-8.
Edgar, A. J., and.Wickramasinghe, S. N. (1998). Hereditary sideroblastic anaemia due to a
mutation in exon 10 of the erythroid 5-aminolaevulinate s¡mthase gene. Br J Haematol 100,
389-92.
Elferink, C. J., Sassa, S., and May, B. K. (1988). Regulation of 5-aminolevulinate synthase in
mouse erythroleukemic cells is different from that in liver. J Biol Chem 263,13012-6.
Elgin, S. C. (198S). The formation and function of DNase I hlpersensitive sites in the process
of gene activation. J Biol Chem 263,19259-62.
Engel, J. D., and Tanimoto, K. (2000). Looping,linking, and chromatin activity: new insights
into beta-globin locus regulation. CelI 100,499-502.
Epner, E., Reik, A., Cimbora, D., Telling,4., Bender, M. 4., Fiering, S., Enver, T', Martin,
D. I., Kennedy, M., Keller, G., and Groudine, M. (1998). The beta-globin LCR is not
necessary for an open chromatin structure or developmentally regulated transcription of the
native mouse beta-globin locus. Mol Cell 2,447-55.
Erslev, A. J., Caro, J., and Besarab, A. (1985). Why the kidney? Nephron 4I ,213-6.
Evans, T., and Felsenfeld, G. (1939). The erythroid-specific transcription factor Eryfl: a new
finger protein. Cell 58, 877 -85.
Farmer, S.C., Sun, C.'W., Winnier, G. E., Hogan,8.L., and Townes, T. M' (I997).ThebZIP
transcription factor LCR-FI is essential for mesoderm formation in mouse development.
Genes Dev 11,786-98.
Feder, J. N., Gnirke, A., Thomas,'W., Tsuchihashi,2., RuddY, D. 4., Basava, A', Dormishian,
F., Domingo, R., Jr., E|lis, M.C., Fullan,4., Hinton, L.M., Jones, N'L., Kimmel,8.E.,
Kronmal, G. S., Lauer, P., Lee, V.K., Loeb, D. 8., Mapa, F.4., McClelland, E., Meyer, N.
C., Mintier, G. 4., Moeller, N., Moore, T., Morikang, E., Wolff, R.K., and et al. (1996). A
146

novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat
Genet 13,399-408.
Feder, J. N., Penny, D.M., Irrinki, 4., Lee, V.K., Lebron, J. 4., Watson, N., Tsuchihashi, 2.,
Sigal, E., Bjorkmffi, P. J., and Schatzman, R. C. (1998). The hemochromatosis gene product
complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl
AcadSciUSA95, 1472-7.
Feder, J. N., Tsuchihashi, Z,Irrrnki,A., Lee, V.K., Mapa, F.4., Morikang, E., Prass, C.E.,
Starnes, S.M., Wolff, R.K., Parkkila, S., Sly, W. S., and Schatzman, R. C. (1997). The
hemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and
cell surface expression. J Biol Chem 272,14025-8.
Fedorov, L. M., Haegel-Kronenberger, H., and Hirchenhain, J. (7997). A comparison of the
germline potential of differently aged ES cell lines and their transfected descendants.
Transgenic Res ó, 223-31.
Feng, Vy'. C., Southwood, C. M., and Bieker, J. J. (1994). Analyses of beta-thalassemia mutant
DNA interactions with erythroid Kruppel-like factor (EKLF), an erlhroid cell-specific
transcription factor. J Biol Chem 269,1493-500.
Feng, Y. Q., Alami, R., and Bouhassira, E. E. (1999). Enhancer-dependent transcriptional
oscillations in mouse erythroleukemia cells. Mol Cell Biol 19,4901-17.
Ferreira, G. C., and Dailey, H.A. (1993). Expression of mammalian 5-aminolevulinate
synthase in Escherichia coli. Overproduction, purification, and charactenzation. J Biol Chem
268,584-90.
Ferreira, G. C., and Gong, J. (1995). 5-Aminolevulinate synthase and the first step of heme
biosynthesis. J Bioenerg Biomembr 27,151-9.
Ferreira, G. C., Neame, P. J., and Dailey, H. A. (1993). Heme bioslmthesis in mammalian
systems: evidence of a Schiff base linkage between the pyridoxal 5'-phosphate cofactor and a
lysine residue in 5-aminolevulinate sSmthase. Protein Sci 2,1959-65.
147

Ferreira, G. C., Vajapey, U.,Hafez, O., Hunter, G. 4., and Barber, M. J. (1995).
Aminolevulinate s¡mthase: lysine 313 is not essential for binding the pyridoxal phosphate
cofactor but is essential for catalysis. Protein Sci 4,1001-6.
Finch, C. (1994). Regulators of iron balance in humans. Blood 84,7691-702.
Fitzsimons,E.J., May, 4., Elder, G. H., and Jacobs, A. (1988). 5-Aminolaevulinic acid
synthase activity in developing human erythroblasts. Br J Haematol 69,281-5.
Fong, T. C., and Emerson, B. M. (1992). The erythroid-specific protein oGATA-1 mediates
distal enhancer activity through a specialized beta-globin TATA box. Genes Dev 6,521-32,
Fontenay-Roupie, M., Bouscary, D., Guesnu, M., Picard, F., Melle, J., Lacombe, C.,
Gisselbrecht, S., Mayeux, P., and Dreyfus, F. (1999). Ineffective erythropoiesis in
myelodysplastic syndromes: correlation with Fas expression but not with lack of
erythropoietin receptor signal transduction. Br J Haematol 106,464-73.
Foroni, L., Boehm, T., White, L., Forster,4., Sherrington, P., Liao, X.8., Brannan, C. I.,
Jenkins, N. 4., Copeland, N. G., and Rabbitts, T. H. (1992). The rhombotin gene family
encode related LlM-domain proteins whose differing expression suggests multiple roles in
mouse development. J Mol Biol 2 26, 7 4l -61.
Forrester, Vy'.C., Epner, E., Driscoll, M. C., Enver, T., Brice, M., Papayannopoulou, T., and
Groudine, M. (1990). A deletion of the human beta-globin locus activation region causes a
major alteration in chromatin structure and replication across the entire beta-globin locus.
Genes Dev 4,1631-49.
Forrester, W. C., Thompson, C., Elder, J. T., and Groudine, M. (1986). A developmentally
stable chromatin structure in the human beta-globin gene cluster. Proc Natl Acad Sci U S A
83, 1359-63.
Forsberg, E. C., Downs, K.M., and Bresnick, E. H. (2000). Direct interaction of NF-E2 with
hypersensitive site 2 of thebeta-globin locus control region in living cells. Blood 96,334-9.
148

Forsberg, E. C., Johnson, K,Zabolkina, T. N., Mosser, E.A., and Bresnick, E. H. (1999).
Requirement of an ElA-sensitive coactivator for long-range transactivation by the beta-globin
locus control region. J Biol Chem 274,26850-9.
Francastel, C., Magis, 'W.,
and Groudine, M. (2001). Nuclear relocation of a transactivator
subunit precedes target gene activation. Proc Natl Acad Sci U S A 98, I2I20-5.
Francastel, C., Schubeler, D., Martin, D. I., and Groudine, M. (2000). Nuclear
compartmentalization and gene activity. Nat Rev Mol Cell Biol 1, l3l-43.
Francastel, C., Walters, M. C., Groudine, M., and Martin, D. L (1999). A functional enhancer
suppresses silencing of a transgene and prevents its localization close to centrometric
heterochromatin. Cell 9 9, 259 -69 .
Fujiwara, Y., Browne, C. P., Cunniff, K., Goff, S.C., and Orkin, S. H. (1996). Arrested
development of embryonic red cell precursors in mouse embryos lacking transcription factor
GATA-I. Proc Natl Acad Sci U S A 93, 12355-8.
Furuyama, K., Fujita, H., Nagai, T., Yomogida, K., Munakata, H., Kondo, M., Kimura,4.,
Kuramoto, 4., Hayashi, N., and Yamamoto, M. (1997). Pyridoxine refractory X-linked
sideroblastic anemia caused by a point mutation in the erythroid S-aminolevulinate s¡mthase
gene. Blood 90,822-30.
Furuyama, K., and Sassa, S. (2000). Interaction between succinyl CoA synthetase and the
heme-biosynthetic enzyrne ALAS-E is disrupted in sideroblastic anemia. J Clin Invest 105,
757-64.
Furuyama, K.,lJno, R., Urabe,4., Hayashi, N., Fujita, H., Kondo, M., Sassa, S., and
Yamamoto, M. (1998). R41lC mutation of the ALAS2 gene encodes a pyridoxine-responsive
enzyme with low activity. Br J Haematol 103,839-4I.
Gallagher, P. G., Romana, M., Tse, W. T., Lux, S. E., and Forget, B. G. (2000). The human
ankyrin-l gene is selectively transcribed in erythroid cell lines despite the presence of a
housekeeping-like promoter. Blood 9 6, I 73 6-43 .
149

Gong, J., and Ferreira, G. C. (1995). Aminolevulinate synthase: functionally important
residues at a glycine loop, a putative pyridoxal phosphate cofactor-binding site. Biochemistry
34, t6l8-85.
Gong, Q. H., Stern, J., and Dean, A. (1991). Transcriptional role of a conserved GATA-1 site
in the human epsilon-globin gene promoter. Mol Cell Biol 11, 2558-66.
Gonzalez, M. I., Caballero, D., Vazquez,L., Canizo, C., Hernandez, R., Lopez, C.,lzaffa, 4.,
Arroyo, J. L., Gonzalez,M., Garcia, R., and San Miguel, J. F. (2000). Allogeneic peripheral
stem cell transplantation in a case of hereditary sideroblastic anaemia. Br J Haematol 109,
658-60.
Goodfellow, B. J., Dias, J. S., Ferreira, G. C., Henklein, P.,'Wray, V., and Macedo, A. L.
(2001). The solution structure and heme binding of the presequence of murine 5-
aminolevulinate synthase. FEBS Lett 5 0 5, 325 -3 I .
Goodman, R.H., and Smolik, S. (2000). CBP/p300 in cell growth, transformation, and
development. Genes Dev 1 4, 1553-77 .
Granick, S. (1966). The induction in vitro of the syrthesis of delta-aminolevulinic acid
s¡mthetase in chemical porphyria: a response to certain drugs, sex hormones, and foreign
chemicals. J Biol Chem 241,1359-75.
Gray, N. K., and Hentze, M. W. (I994).Iron regulatory protein prevents binding of the 43S
translation pre-initiation complex to ferritin and eALAS Embo'J 13,3882-91
Green, A. R., Lints, T., Visvader, J., Harvey, R., and Begley, C. G. (1992). SCL ts
coexpressed with GATA-I in hemopoietic cells but is also expressed in developing brain.
Oncogene 7,653-60.
Gregory, R.C., Taxman, D. J., Seshasayee, D., Kensinger, M. H., Bieker, J. J., and
Wojchowski, D. M. (1996). Functional interaction of GATA1 with erythroid Kruppel-like
factor and Spl at defined erythroid promoters. Blood 87,1793-807.
1s0

Gribnau, J., Diderich, K., Pruzina, S., Calzolari, R., and Fraser, P. (2000). lntergenic
transcription and developmental remodeling of chromatin subdomains in the human beta-
globin locus. Mol Cell 5,311-86.
Gross, D. S., and Garrard, W. T. (1988). Nuclease hypersensitive sites in chromatin. Annu
Rev Biochem 57,159-97.
Grosveld, F., Antoniou, M., Berry, M., De Boer, E., Dillon, N., Ellis, J., Fraser, P.,
Hanscombe, O., Hurst, J., Imam, 4., and et al. (1993). The regulation of human globin gene
switching. Philos Trans R Soc Lond B Biol Sci 339,183-91
Grosveld, F., van Assendelft, G. 8., Greaves, D. R., and Kollias, G. (1987). Position-
independent, high-level expression of the human beta-globin gene in transgenic mice. Cell 5l,
975-85.
Grunstein, M. (1997). Histone acetylation in chromatin structure and transcription. Nature
389,349-52.
Gupta, M., Mungai, P. T., and Goldwasser, E. (2000). A new transacting factor that modulates
hypoxia-induced expression of the erythropoietin gene. Blood 96,491-7.
Haile, D. J. (2000). Assignment of Slcl1a3 to mouse chromosome 1 band 1B and SLCl1A3
to human chromosome2q32 by in situ hybndizatíon. Cytogenet Cell Genet 88,328-9.
Hamilton, J.W., Bement, Vy'. J., Sinclair, P.R., Sinclair, J. F., Alcedo, J. 4., and Wetterhahn,
K. E. (1991). Heme regulates hepatic 5-aminolevulinate slmthase mRNA expression by
decreasing mRNA half-life and not by altering its rate of transcription. Arch Biochem
Biophys 289,381-92.
Hardison, R., Slightom, J. L., Gumucio, D.L., Goodman, M., Stojanovic, N., and Miller, W
(1991). Locus control regions of mammalian beta-globin gene clusters: combining
phylogenetic analyses and experimental results to gain functional insights. Gene 205,13-94.
Harigae, H., Furuyama, K., Kimura, A., Neriishi, K,, Tahara, N., Kondo, M., Hayashi, N.,
Yamamoto, M., Sassa, S., and Sasaki, T. (1999). A novel mutation of the erythroid-specific
151

delta-aminolaevulinate synthase gene in a patient with X-linked sideroblastic anaemia. Br J
Haematol 106,715-7.
Harigae, H., Suwabe, N.,'Weinstock, P. H., Nagai, M., Fujita, H., Yamamoto, M', and Sassa,
S. (1993). Deficient heme and globin synthesis in embryonic stem cells lacking the erythroid-
specific delta-aminolevulinate synthase gene. Bloo d 91 ,798-805.
Harju, S., McQueen, K. J., and Peterson, K. R. (2002). Chromatin structure and control of
beta-like globin gene switching. Exp Biol Med (Maywood) 227,683-700.
Harrison, P. M., and Arosio, P. (1996). The ferritins: molecular properties, iron storage
function and cellular regulation. Biochim Biophys Acta 1275,161-203.
]Hartzog, G. 4., and Myers, R. M. (1993). Discrimination among potential activators of the
beta-globin CACCC element by correlation of binding and transcriptional properties. Mol
Cell Biol 13,44-56.
Heberlein, C., Fischer, K.D., Stoffel, M., Nowock, J., Ford,4., Tessmer,lJ., and Stocking, C
(1992). The gene for erythropoietin receptor is expressed in multipotential hematopoietic and
embryonal stem cells: evidence for differentiation stage-specific regulation. Mol CellBiol 12,
r815-26.
Henderson, B. R. (1996a).Iron regulatory proteins 1 and 2. Bioessays 18,739-46
Henderson, B. R., Menotti, E., and Kuhn, L. C. (1996b). Iron regulatory proteins 1 and 2 bind
distinct sets of RNA Larget sequences. J Biol Chem 271,4900-8.
Hentze, M.Vy'., Rouault, T. 4., Caughman, S.W., Dancis, 4., Harford, J. 8., and Klausner, R.
D. (1987). A cis-acting element is necessary and sufficient for translational regulation of
human ferritin expression in response to iron. Proc Natl Acad Sci U S A 84,6730-4.
Hershfield, M. S., Kurtzberg, J., Harden, E., Moore, J. O., Whang-Peng, J', and Ha¡mes, B. F.
(1984). Conversion of a stem cell leukemia from a T-lynphoid to a myeloid phenotype
induced by the adenosine deaminase inhibitor 2'-deoxycoformycin. Proc Natl Acad Sci U S A
I I ,253-l .
t52

Hofer, T., Wenger, R.H., Kramer, M. F., Ferreira, G. C., and Gassmann, M. (2003). Hypoxic
up-regulation of erythroid 5-aminolevulinate synthase. Blood I 0 l, 3 48-50.
Hsu, H. L., Wadman, I., and Baer, R. (1994). Formation of in vivo complexes between the
TALI andB2|polypeptides of leukemic T cells. Proc Natl Acad Sci U S A 91, 3181-5.
Huang, Y., Liu, D.P., Wu, L., Li, T. C.,'Wu, M., Feng, D.X', and Liang, C. C. (2000). Proper
developmental control of human globin genes reproduced by transgenic mice containing a
160-kb BAC carrying the human beta-globin locus. Blood Cells Mol Dis 26,598-610.
Huisman, T.H. (1997). Levels of Hb A2 inheterozygotes and homozygotes for beta-
thalassemia mutations: influence of mutations in the CACCC and ATAAA motifs of the beta-
globin gene promoter. Acta Haematol 98,187-94.
Hung, H. L., Kim, A. Y., Hong, W., Rakowski, C., and Blobel, G. A. (2001). Stimulation of
NF-82 DNA binding by CREB-binding protein (CBP)-mediated acetylation. J Biol Chem.
276,10715-21.
jHung, H.L., La.u,J.,Kim, A. Y., Weiss, M.J., and Blobel, G.A.(1999). CREB-Binding
protein acetylates hematopoietic transcription factor GATA-I at functionally important sites
Mol Cell B.iol 19,3496-505.
Hunter, G. 4., and Ferreira, G. C. (1999). Pre-steady-state reaction of 5-aminolevulinate
s¡mthase. Evidence for a rate-determining product release. J Biol Chem 274,72222-8.
Hural, J. A., Kwan, M., Henkel, G., Hock, M. 8., and Brown, M.A. (2000). An intron
transcriptional enhancer element regulates IL-4 gene locus accessibility in mast cells. J
Immunol 165,3239-49.
Hurford, M.T., Marshall-Taylor, C., Vicki, S. L., Zhou,J.Z., Silverman, L.M., Rezuke, W.
N., Altman, 4., and Tsongalis, G. J. (2002). A novel mutation in exon 5 of the ALAS2 gene
results in X-linked sideroblastic anemia. Clin Chim Acta 321,49-53.
/\I
153

Huyhn,4., Dommergues, M'lzac,B., Croisille,L.,Kalz,4., Vainchenker,.W., and
Coulombel, L. (1995). Charactenzation of hematopoietic progenitors from human yolk sacs
and embryos. Blood 86,4414-85.
lmagawa, S., Goldberg, M. 4., Doweiko, J., and Bunn, H. F. (1991). Regulatory elements of
the erythropoietin gene. Blood 77,218-85.
Ito, E., Toki, T.,Ishihara, H., Ohtani, H., Gu, L., Yokoyama, M., Engel, J. D., and
Yamamoto, M. (1993). Erythroid transcription factor GATA-I is abundantly transcribed in
mouse testis. Nature 362,466-8.
Jacks, T., Shih, T. S., Schmitt, E. M., Bronson, R. T., Bemards,4., and'Weinberg, R. A.
(1994). Tumour predisposition in mice heterozygous for a targeted mutation in Nfl. Nat
Genet 7,353-61.
Jacobson, L. O., Goldwasser, E., Fried, W., and Plzak, L. (1959). Role of the kidney in
erythropoiesis. Nature I 79, 633.
Jacobson, L. O., Marks, E.K., Gaston, E. O., and Goldwasser, E. (1959). Studies on
erythropoiesis. Reticulocle response of transfusion induced pollhaemic mice to anemic
plasma from nephrectomised mice and to plasma from nephrectomised rats exposed to low
oxygen. Blood I 4, 635-43.
Jazwinska, E. C., and Powell, L.W. (1997). Hemochromatosis and "HLA-H": definite!
Hepatology 25,495-6.
Jelkmann, W. (1992). Erythropoietin: structure, control of production, and function. Physiol
R.ev 72,449-89.
John, R. A. (1995). Pyridoxal phosphate-dependent enzymes. Biochim Biophys Acta 1248,
81-96
Johnson, K.D., Christensen, H. M., Zhao,8., and Bresnick, E. H. (2001). Distinct
mechanisms control RNA polymerase II recruitment to a tissue-specific locus control region
and a downstream promoter. Mol Cell 8,465-77.
t54

Jones, S. S., D'Andrea, A. D., Haines,L.L., and Wong, G. G. (1990). Human erythropoietin
receptor: cloning, expression, and biologic characteization. Blood 76,31-5.
Jordan, P. M., and Laghai-Newton, A. (1986). Purification of 5-aminolevulinate synthase.
Methods Enz¡rmol I 23, 435-43.
Kaartinen, V., and Nugy, A. (2001). Removal of the floxed neo gene from a conditional
knockout allele by the adenoviral Cre recombinase in vivo. Genesis 31, 126-9.
Kadonaga, J. T. (1998). Eukaryotic transcription: an interlaced network of transcription
factors and chromatin-modif,zing machines. Cell 9 2, 307 -13 .
Kadonaga, J. T., Carner, K. R., Masiarz, F.R., and Tiian, R. (1987). Isolation of oDNA
encoding transcription factor Spl and functional analysis of the DNA binding domain. Cell
51, 1079-90.
Karlsson, S., and Nienhuis, A. V/. (1985). Developmental regulation of human globin genes.
Annu Rev Biochem 54,1071-108.
Kee, B. L., Arias, J., and Montminy, M. R. (1996). Adaptor-mediated recruitment of RNA
pol¡rmerase II to a signal-dependent activator. J Biol Chem 271,2373-5.
Kendall, R. G. (2001). Erythropoietin. Clin Lab Haematol 23,71-80
Kingsley, C., and Winoto, A. (1992). Cloning of GT box-binding proteins: a novel Spl
multigene family regulating T-cell receptor gene expression. Mol Cell Blol I 2, 4251-61.
Kispal, G., Csere, P., Prohl, C., and Lill, R. (1999). The mitochondrial proteins Atmlp and
Nfslp are essential forbiogenesis of cytosolic Fe/S proteins. Embo J 18,3981-9.
Klinken, P., Busfield, S., Keil,lJ., Farr, T., Colley, S., Callus,8., Chappell, D., and
Papadimitriou, J. (1993). Lineage Switching by Commitment Hemopoietic Precursors
Journal of Computer Assisted Microscopy 5, 81-84.
155

Klinken, S. P., Nicola, N. 4., and Johnson, G. R. (1988). In vitro-derived leukemic erythroid
cell lines induced by a raf- and myc-containing retrovirus differentiate in response to
erythropoietin. Proc Natl Acad Sci U S A 85, 8506-10.
Ko, L. J., and Engel, J. D. (1993). DNA-binding specificities of the GATA transcription
factor family. Mol Cell Biol 13, 40lt-22.
Koc, S., and Harris, J. V/. (1998). Sideroblastic anemias: variations on imprecision in
diagnostic criteria, proposal for an extended classification of sideroblastic anemias. Am J
Hematol 57,1-6.
Komatsu, N., Adamson, J. W., Yamamoto, K., Altschuler, D., Torti, M., Marzocchini, R., and
Lapetina, E. G. (1992). Erythropoietin rapidly induces tyrosine phosphorylation in the human
erythropoietin-dependent cell line, UT-7. Blood 80, 53-9.
Kotkow, K. J., and Orkin, S. H. (1996). Complexity of the erlhroid transcription factor NF-
E2 as revealed by gene targeting of the mouse p18 NF-E2 locus. Proc Natl Acad Sci U S A
93,3514-8 Àì
'.1,
Kotkow, K. J., and Orkin, S. H. (1995). Dependence of globin gene expression in mouse
erythroleukemia cells on the NF-E2 heterodimer. Mol Cell Biol 15, 4640-7.
Kramer, M.F., Gunaratne, P., and Ferreira, G. C. (2000). Transcriptional regulation of the
murine erythroid-specifi c 5-aminolevulinate synthase gene. Gene 2 4 7, 1 53 -66.
Kulozik, A. E., Bellan-Koch,4., Bail, S., Kohne, E., and Kleihauer, E. (1991). Thalassemia
intermedia: moderate reduction of beta globin gene transcriptional activity by a novel
mutation of the proximal CACCC promoter element. Blood 77,2054-8.
Kurtz,A., Eckardt, K. E., Tannahill, L., and Bauer, C. (1988). Regulation of erythropoietin
production. Contrib Nephrol 66, I -16.
Kwok, R. P., Lundblad, J. R., Chrivia, J. C., Richards, J. P., Bachinger, H. P., Brennan, R. G.,
Roberts, S. G., Green, M. R., and Goodman, R. H. (1994). Nuclear protein CBP is a
coactivator for the transcription factor CREB. Nature 370,223-6.
156

Kwon, H., Imbalzano, A. N., Khavari, P. 4., Kingston, R. E., and Green, M. R. (1994)
Nucleosome disruption and enhancement of activator binding by a human SWI/SNF
complex. Nature 370, 477 -81.
Landschulz, K.T., Noyes,4.N., Rogers, O., and Boyer, S. H. (1989). Erythropoietin
receptors on murine erythroid colony-forming units: natural history. Blood 73,7476-86'
Lathrop, J. T., and Timko, M. P. (1993). Regulation by heme of mitochondrial protein
transport through a conserved amino acid motif. Science 259,522-5.
Lebron, J.4., Bennett, M. J., Vaughn, D.E., Chirino, A' J., Snow, P.M., Mintier, G. 4.,
Feder, J. N., and Bjorkman, P. J. (1998). Crystal structure of the hemochromatosis protein
HFE and charactenzation of its interaction with transferrin receptor. Cell 93, lll-23.
Lecine, P., Blank, V., and Shivdasani, R. (1998). Characterizalion of the hematopoietic
transcription factor NF-E2 in primary murine megakaryocfles. J Biol Chem 273,7512-8
Lee, J. S., Ngo, H., Kim, D., and chung, J. H. (2000). Erythroid KruppelJike factor is
recruited to the CACCC box in the beta-globin promoter but not to the CACCC box in the
gamma-globin promoter: the role of the neighboring promoter elements. Proc Natl Acad Sci
u s A 97,2468-73.
Leonard, M., Brice, M., Engel, J. D., and Papayannopoulou, T. (1993). Dynamics of GATA
transcription factor expression during erythroid differentiation. Blood 82,1071-9.
Letting, D. L., Rakowski, C., Weiss, M.J., and Blobel, G.A. (2003). Formation of a Tissue-
Specific Histone Acetylation Pattern by the Hematopoietic Transcription Factor GATA-I.
Mol Cell Biol 2 3, 1334-40.
Levi, S., Corsi,8., Bosisio, M.,Invernizzi,R',Yolz,4., Sanford, D', Arosio, P.' and
Drysdale, J. (2001). A human mitochondrial ferritin encoded by an intronless gene. J Biol
Chem 276,24437-40.
157

Levings, P. P., and Bungert, J. (2002). The human beta-globin locus control region. Eur J
Biochem 269,1589-99.
L",.y, J. E., Montross, L. K., Cohen, D. E., Fleming, M.D., and Andrews, N. C. (1999). The
C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood
94,9-11.
Liao, E. C., Paw,8.H., Oates,4.C., Pratt, S. J., Postlethwait, J. H., and zon,L.I. (1998).
SCL/Ta|-1 transcription factor acts downstream of cloche to speciff hematopoietic and
vascular progenitors in zebrafish. Genes Dev 12,627-6.
Lill, R., and Kispal, G. (2000). Maturation of cellular Fe-S proteins: an essential function of
mitochondria. Trends Biochem Sci 25, 352-6.
Lill, R., and Kispal, G. (2001). Mitochondrial ABC transporters. Res Microbiol 152,331-40.
Lim, K. C.,Ishihara, H., Riddle, R.D., Yang, 2., Andrews, N., Yamamoto, M., and Engel, J'
D. (1994). Structure and regulation of the chicken erythroid delta-aminolevulinate syrthase
gene. Nucleic Acids Res 22,1226-33.
Lok, C. N., and Ponka, P. (1999). Identification of a hypoxia response element in the
transferrin receptor gene. J Biol Chem 274,24141-52.
Lopez,A.F., To, L. B., Yang, Y.C., Gamble, J. R., Shannon, M. F., Burns, G.F., Dyson, P.
G., Juttner, C. 4., Clark, S., and Vadas, M. A. (1987). Stimulation of proliferation,
differentiation, and function of human cells by primate interleukin 3. Proc Natl Acad Sci U S
A 84,2761-5.
Lu, S. J., Rowan, S., Bani, M. R., and Ben-David, Y. (1994). Retroviral integration within the
Fli-2 locus results in inactivation of the erythroid transcription factor NF-E2 in Friend
erythroleukemias: evidence that NF-E2 is essential for globin expression. Proc Natl Acad Sci
u s A 91,8398-402.
Magram, J., Chada, K., and Costantini, F. (1985). Developmental regulation of a cloned adult
beta-globin gene in transgenic mice. Nature 315,338-40.
158

Maguire,4., Hellier, K., Hammans, S., and May, A. (2001). X-linked cerebellar ataxia and
sideroblastic anaemia associated with a missense mutation in the ABCT gene predicting
V4llL. Br J Haematol I15,910-7.
Mao, M., Fu, G., Wu, J. S.,Zhang, Q. H., Zhou, J., Kan, L.X', Huang, Q. H., He, K. L., Gu,
B.W., Han,Z. G., Shen, Y., Gu, J., Yu, Y.P., Xu, S. H', Wang, Y'X., Chen, S. J., and Chen'
Z. (1998).Identification of genes expressed in human CD34(+) hematopoietic
stem/progenitor cells by expressed sequence tags and efficient full-length oDNA cloning.
Proc Natl Acad Sci U S A 95,8175-80.
Marceau, M., Lewis, S.D., Kojiro, C.L., Mountjoy, K., and Shafer, J' A. (1990). Disruption
of active site interactions with pyridoxal 5'-phosphate and substrates by conservative
replacements in the glycine-rich loop of Escherichia coli D-serine dehydratase. J Biol Chem
265,20421-9.
Marshall, C. J., and Thrasher, A. J. (2001). The embryonic origins of human haematopoiesis.
Br J Haematol 112,838-50.
Martin, D. I., Fiering, S., and Groudine, M. (1996). Regulation of beta-globin gene
expression: straightening out the locus. Curr Opin Genet Dev 6,488-95.
Martin, D. I., and Orkin, S. H. (1990). Transcriptional activation and DNA binding by the
erythroid factor GF-1/¡{F-EllErJf 1. Genes Dev 4,1886-98.
Matthes, T. W., Meyer, G., Samii, K., and Beris, P. (2000). Increased apoptosis in acquired
sideroblastic anaemia. Br J Haematol I I1,843-52.
Max-Audit, I., Eleouet, J. F., and Romeo, P. H. (1993). Transcriptional regulation of the
pynrvate kinase erythroid-specific promoter. J Biol Chem 268,5437-7.
May, 4., de Souza, P., Barnes, K., Kaaba, S., and Jacobs, A. (1982). Erythroblast iron
metabolism in sideroblastic marrows. Br J Haematol 52,611-21.
159

May,4., Al-Sabah,4., Fitzsimons, E. J., Houston, T., Woodcock, B., Cotter, P.D', Wong, L.
and Bishop, D. F. (1994). Erythroid ALA synthase gene mutations causing late-onset, X-
linked pyridoxine-responsive sideroblastic anemia. Br J Haematol 87(supp1.), 148
May, B. K., Dogra, S. C., Sadlon, T. J., Bhasker, C.R., Cox, T. C', and Bottomley, S. S.
(1995). Molecular regulation of heme biosynthesis in higher vertebrates. Prog Nucleic Acid
Res Mol Biol Sl, 1-51.
McKie, A.T., Barrow, D., Latunde-Dada, G. O., Rolfs,4., Sager, G., Mudaly, E., Mudaly,
M., Richardson, C., Barlow, D., Bomford,4., Peters, T. J., Raja, K.B., Shirali, S., Hediger,
M. 4., Farzaneh,F., and Simpson, R. J. (2001). An iron-regulated ferric reductase associated
with the absorption of dietary iron. Science 291 , l7 55-9. \I
McKie,4.T., Marciani, P., Rolfs,4., Brennan, K., Wehr, K'' Barrow, D., Miret, S.,
Bomford, 4., Peters, T. J.,Farzaneh, F., Hediger, M. 4., Hentze, M. W., and Simpson, R. J.
(2000). A novel duodenal iron-regulated transporter, IREGl, implicated in the basolateral
transfer of iron to the circulation. Mol Cell 5,299-309.
Mead, P.E., Deconinck,4.E., Huber, T.L., Orkin, S.H., andZon, L.I. (2001). Primitive
erythropoiesis in the Xenopus embryo: the synergistic role of LMO-2, SCL and GATA-
binding proteins. Development I 28, 2301 -8.
Medvinsky, 4., and Dzieruak, E. (1996). Definitive hematopoiesis is autonomously initiated
by the AGM region. Cell 86,891-906.
Medvinsky, A. L., Gan, O. I., Semenova, M. L., and Samoylina, N. L. (1996). Development
of day-8 colony-forming unit-spleen hematopoietic progenitors during early murin" ,fìembryogenesis: spatial and temporal mapping. Blood 87,557-66. I * t > --rl-i ',,ç,;ìi vMedvinsky, A.L., Samoylina, N.L., Muller,4.M., andDzietzak, E. A, (1993). An earlypre-
liver intraembryonic source of CFU-S in the developing mouse. Nature 364,64-7.
Meguro, K.,Igarashi, K., Yamamoto, M., Fujita, H., and Sassa, S. (1995)' The role of the
erythroid-specific delta-aminolevulinate synthase gene expression in erythroid heme
slmthesis. Blood 86, 940-8.
160

Melefors, O., Goossen, B., Johansson, H. E., Stripecke, R', Gray, N.K., and Hentze, M.W.
(1993). Translational control of 5-aminolevulinate synthase mRNA by iron-responsive
elements in erythroid cells. J Biol Chem 268,5974-8.
Melton, D. W. (1994). Gene targeting in the mouse. Bioessays 16,633-8.
Meng,4., Tang, H., Yuan, 8., Ong,8. 4., Long, Q., and Lin, S. (1999). Positive and negative
cis-acting elements are required for hematopoietic expression of zebrafish GATA-I. Blood
93, 500-8.
Menotti, E., Henderson, B. R., and Kuhn, L. C. (1993). Translational regulation of mRNAs
with distinct IRE sequences by iron regulatory proteins 7 and2. J Biol Chem 273, l82I-4.
Merika, M., and Orkin, S. H. (1993). DNA-binding specificity of GATA family transcription
factors. Mol Cell Biol 13, 3999-4010.
Merika, M., and Orkin, S. H. (1995). Functional synergy and physical interactions of the
erythroid transcription factor GATA-I with the Kruppel familyproteins Spl and EKLF. Mol
Cell Biol 15,2437-47.
Metcalf, D., Begley, C.G., Johnson, G.R., Nicola, N.A', Vadas, M.A', Lopez, A.F',
Williamson, D. J., Wong, G. G., Clark, S. C., and'Wang, E. A. (1986)' Biologic properties in
vitro of a recombinant human granulocyte-macrophage colony-stimulating factor. Blood 67,
37-45.
Meyers, E.N., Lewandoski, M., and Martin, G. R. (1998). An FgfS mutant allelic senes
generated by cre- and Flp-mediated recombination. Nat Genet 18,136-41.
Migliaccio, A. R., and Migliaccio, G. (1998). The making of an erythroid cell. Molecular
control of hematopoiesis. Biotherapy I 0, 251 -68.
Mignotte, V., Eleouet, J. F., Raich, N., and Romeo, P. H. (1989b). Cis- and trans-acting
elements involved in the regulation of the erythroid promoter of the human porphobilinogen
deaminase gene. Proc Natl Acad Sci U S A 8ó, 6548-52.
161

Mignotte, V., Wall, L., deBoer, E., Grosveld, F., and Romeo, P. H. (1989a). Two tissue-
specific factors bind the erythroid promoter of the human porphobilinogen deaminase gene.
Nucleic Acids Res 17,37-54.
Miller, I. J., and Bieker, J. J. (1993). A novel, erythroid cell-specific murine transcription
factor that binds to the CACCC element and is related to the Kruppel family of nuclear
proteins. Mol Cell Biol 13, 2716-86.
Milot, E., Strouboulis, J., Trimborn, T., Wijgerde, M., de Boer, E., Langeveld,4., Tan-Un,
K., Vergeer, W., Yannoutsos, N., Grosveld, F., and Fraser, P. (1996). Heterochromatin effects
on the frequency and duration of LCR-mediated gene transcription. Cell 87,105-14.
Moi, P., and Kan, Y. W. (1990). S¡mergistic enhancement of globin gene expression by
activator protein-l-like proteins. Proc Natl Acad Sci U S A 82, 9000-4.
Moore, M. 4., and Metcalf, D. (1970). Ontogeny of the haemopoietic system: yolk sac origin
of in vivo and in vitro colony forming cells in the developing mouse embryo. Br J Haematol
18,279-96.
Motohashi, H., Shavit, J. 4., Igarashi, K., Yamamoto, M., and Engel, J.D. (1997). The world
according to Maf. Nucleic Acids Res 25, 2953-59.
Mucenski, M.L., Mclain, K., Kier,4.8., Swerdlow, S.H., Schreiner, C.M., Miller, T.4.,
Pietryga, D.W., Scott, V/. J., Jr., and Potter, S. S. (1991). A functional c-myb gene is required
for normal murine fetal hepatic hematopoiesis. Cell 65,677-89.
Muckenthaler, M., Gray, N. K., and Hentze, M. W. (1998). IRP-I binding to ferritin mRNA
prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F.
Mol Cell 2,383-8.
Mukhopadhyay, C. K., Mazumder, 8., and Fox, P. L. (2000). Role of hypoxia-inducible
factor-l in transcriptional activation of ceruloplasmin by iron deficiency. J Biol Chem 275,
21048-54.
162

Muller,4.M., Medvinsky,4., Strouboulis, J., Grosveld, F., and Dzietzak, E. (1994).
Development of hematopoietic stem cell activity in the mouse embryo. Immunity 1,291-301.
Muller-Eberhard, U., and Fraig, M. (1993). Bioactivity of heme and its containment. Am J
Hematol 42,59-62.
Munakata, H., Yamagami, T., Nagai, T., Yamamoto, M., and Hayashi, N. (1993). Purification
and structure of rat erythroid-specific delta-aminolevulinate s¡mthase. J Biochem (Tokyo)
114, t03-7r.
Nugy, A. (2000). Cre recombinase: the universal reagent for genome tailoring. Genesis 2ó,
99-109
Nugy, A., Gocza,E.,Diaz, E. M., Prideaux, V. R., Ivanyi, E', Markkula, M., and Rossant, J.
(1990). Embryonic stem cells alone are able to support fetal development in the mouse.
Development I I 0, 81 5-21.
Nugy,4., Moens, C., Ivanyi, E., Pawling, J., Gertsenstein, M., Hadjantonakis, A. K., Pirity,
M., and Rossant, J. (199S). Dissecting the role of N-myc in development using a single
targeting vector to generate a series of alleles. Curr Biol 8,667-4.
Nugy,4., Rossant, J., Nagy, R., Abramow-Newerly, W., and Roder, J. C. (1993). Derivation
of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl
AcadSciUSA90,8424-8.
Nakajima, O., Takahashi, S., Harigae, H., Furuyama, K., Hayashi, N., Sassa, S', and
Yamamoto, M. (1999). Heme deficie4cy in erythroid lineage causes differentiation arrest and
cytoplasmic iron overload. Embo J 18\;6282-9.
Nakajima, T., Uchida, C., Anderson, S. F., Lee, C. G., Hurwitz, J., Parvin, J' D., and
Montminy,M. (1997). RNA helicase A mediates association of CBP with RNA polymerase
ll. Cell 90,1107-12.
163

Ney, P. 4., Sorrentino, B. P., McDonagh, K. T., and Nienhuis, A. W. (1990). Tandem AP-l-
binding sites within the human beta-globin dominant control region function as an inducible
enhancer in erythroid cells. Genes Dev 4,993-1006.
Nuez,8., Michalovich, D., Bygrave, A., Ploemacher, R., and Grosveld, F. (1995). Defective
haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature 375,316-8.
Offringa, R., Gebel, S., van Dam, H., Timmers, M., Smits, A.,Zwart, R., Stein, B., Bos, J' L.,
van der Eb,4., and Herrlich, P. (1990). A novel function of the transforming domain of Ela:
repression of AP-l activity. Ce||62,527-38.
Ogryzko, V. V., Schiltz, R. L., Russanova, V., Howard, B. H., and Nakatani, Y' (1996). The
transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87,953-9.
Ohashi,4., and Kikuchi, G. (1979). Purification and some properties of two forms of delta-
aminolevulinate synthase from rat liver cytosol. J Biochem (Tokyo) 85,239-47.
.<'
S. H. (1996). Development of the hematopoietic system. Curr Opin Genet Dev 6,597-
602.
Orkin, S. H. (2000). Diversification of haematopoietic stem cells to specific lineages. Nat Rev
Genet 1,57-64.
Orkin, S. H. (1995). Regulation of globin gene expression in erythroid cells. Eur J Biochem
231,271-81.
Pagon, R. 4., Bird, T. D., Detter, J. C., and Pierce, I. (1985). Hereditary sideroblastic anaemia
and ataxia: an X linked recessive disorder. J Med Genet 22,267-73.\)
Palis, J., Robertson, S., Kennedy, M., Wall, C., and Keller, G. (1999). Development of
erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse.
Development I 2 6, 507 3-84.
Palis, J., and Segel, G. B. (1998). Developmental biology of erythropoiesis. Blood Fiev 12,
106-14.
164

Partington, G. 4., and Patient, R.K. (1999). Phosphorylation of GATA-I increases its DNA-
binding affinity and is correlated with induction of humanK562 erythroleukaemia cells.
Nucleic Acids Ptes 27,1168-75.
Perkins, A. (1999). Erythroid Kruppel like factor: from fishing expedition to gourmet meal.
Int J Biochem Cell Biol -31, ll75-92.
Perkins, A. C., Sharpe, A. H., and Orkin, S. H. (1995). Lethal beta-thalassaemia ln mrce
lacking the erythroid CAccc-transcription factor EKLF. Nature 375,318-22.
Perry, C., and Soreq, H. (2002). Transcriptional regulation of erythropoiesis. Fine tuning of
combinatorial multi-domain elements. Eur J Biochem 269,3607-18.
Pevny, L., Simon, M.C., Robertson, E., Klein, W.H., Tsai, S. F', D'Agati, V., Orkin, S'H.,
and Costantini, F. (1991). Erythroid differentiation in chimaeric mice blocked by a targeted
mutation in the gene for transcription factor GATA-1. Nature 349,257-60.
Pfeifer, K., Kim, K. S., Kogan, S., and Guarente, L. (1989). Functional dissection and
sequence of yeast HAPI activator. Cell56,29l-301.
Philipsen, S., Talbot, D., Fraser, P., 4nd Grosv.eld, F. (1990). The beta-globin dominant
control region: hypersensitive site 2. Embo I g,\tSg-Al.
Pikaart, M. J., Recillas-TarEà,F., and Felsenfeld, G. (1998). Loss of transcriptional activity of
a transgene is accompanied by DNA methylation and histone deacetylation and is prevented
by insulators. Genes Dev 12,2852-62.
Pircher, T. J., Geiger, J. N., Zhang, D., Miller, C.P., Gaines, P., and Wojchowski, D. M'
(2001). Integrative signaling by minimal erythropoietin receptor forms and c-Kit. J Biol Chem
276,8995-9002.
Pirola,8. 4., Mayer, F., Borthwick, L A., Srivastava, G., May, B' K., and Elliott, W' H.
(1984). Electron microscopic studies on liver 5-aminolaevulinate synthase. Eur J Biochem
144,517-9.
165

Plant, K. E., Routledge, S. J., and Proudfoot, N. J. (2001). Intergenic transcription in the
humanbeta-globin gene cluster. Mol Cell Bíol21,6507-14.
Ponka, P. (1997). Tissue-specific regulation of iron metabolism and heme synthesis: distinct
control mechanisms in erythroid cells. Blood 89,1-25.
Ponka, P., Schulman, H. M., and Martinez-Medellin, J. (1988). Haem inhibits iron uptake
subsequent to endocytosis of transferrin in reticulocytes. Biochem J 25l, 105-9.
Porcher, C.,Liao, E. C., Fujiwara, Y.,Zon, L. I., and Orkin, S. H. (1999). Specification of
hematopoietic and vascular development by the bHLH transcription factor SCL without direct
DNA binding. Development I 2 6, 4603 -15.
Porcher, C., Swat, W., Rockwell, K., Fujiwara, Y., Alt, F.W., and Orkin, S. H. (1996). The T
cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic
lineages. Cell 86, 47 -57 .
Porter, D. L., and Goldberg, M. A. (1993). Regulation of erythropoietin production. Exp
Hematol 21,399-404.
Powell, L. W., Zazwinska, E. and Halliday, J. W. (1994) Primary iron overload in; Iron
Metabolism in Health and Disease. Brock, J. H., Halli day, J . W., Pippard, M. J. and Powell,
L. W. (eds), W. B. Saunders, Philadelphia, PA, pp:227-70.
Prades, E., Chambon, C., Dailey, T. 4., Dailey, H. 4., Briere, J., and Grandchamp, B. (1995).
A new mutation of the ALAS2 gene in alarge family with Xlinked sideroblastic anemia.
Hum Genet 95,424-8.
Pruzina, S., Hanscombe, O.,'Whyatt, D., Grosveld, F., and Philipsen, S. (1991).
Hypersensitive site 4 of the human beta globin locus control region. Nucleic Acids Res 19,
1473-9.
t66

Rabbitts, T. H. (1998). LMO T-cell translocation oncogenes typiry genes activated by
chromosomal translocations that alter transcription and developmental processes. Genes Dev
12,2651-7.
Rahuel, C., Vinit, M. 4., Lemarchandel, V., Cartron, J. P., and Romeo, P. H. (1992).
Erythroid-specific activity of the glycophorin B promoter requires GATA-1 mediated
displacement of a repressor. Embo J I I',4095-702.
Raich, N., Mignotte, V., Dubart,4., Beaupaitr, D., Leboulch, P., Romana, M', Chabret, C.,
Chamay, P., Papayannopoulou, T., Goossens, M., and et al. (1989). Regulated expression of
the overlapping ubiquitous and erythroid transcription units of the human porphobilinogen
deaminase (PBG-D) gene introduced into non-erythroid and erythroid cells. J BioIChem 264,
rot86-92
Ramchandran, R., Bengra, C., Whitney, B., Lanclos, K., and Tuan, D. (2000)' A (GATAXT)
motif located in the 5'boundary area of the human beta-globin locus control region exhibits
silencer activity in erythroid cells. Am J Hematol 65,14-24.
Raskind, W.H.,'Wijsman,8., Pagon, R.4., Cox, T. C., Bawden, M.J., May, B. K., and Bird,
T. D. (1991). X-linked sideroblastic aneniia and ataxia: linkage to phosphoglycerate kinase at
Xq13. Am J Hum Genet 48,335-47.
Reid, L. H., Gregg, R. G., Smithies, O., and Koller, B. H. (1990). Regulatory elements in the
introns of the human HPRT gene are necessary for its expression in embryonic stem cells.
Proc Natl Acad Sci U S A 87,4299-303.
Reik,4., Telling, A.,Zltnlk, G., Cimbora,D., Epner, E., and Groudine, M. (1998). The locus
control region is necessary for gene expression in the human beta-globin locus but not the
maintenance of an open chromatin structure in erythroid cells. Mol Cell Biol 18, 5992-6000.
Rekhtman, N., Radparvar, F., Evans, T., and Skoultchi, A. I. (1999). Direct interaction of
hematopoietic transcription factors PU.1 and GATA-I: functional antagonism in erythroid
cells. Genes Dev 13, 1398-411
Remizsewski, J., (2000). PhD Thesis, University of Adelaide, South Australia, Australia
r67

Riddle, R. D., Yamamoto, M., and Engel, J. D. (1989). Expression of delta-aminolevulinate
slmthase in avian cells: separate genes encode erythroid-specific and nonspecific isozymes.
Proc Natl Acad Sci U S A 86,792-6.
Ritter, M., Buechler, C., Langmann, T., and Schmitz, G. (1999). Genomic organization and
chromosomallocalization of the human CD163 (Ml30) gene: a member of the scavenger
receptor cysteine-rich superfamily. Biochem Biophys Res Commun 260,466-74.
Robertson, E. J. (1937) in Teratocarcinomas and Embryonic Stem Cells: a practical
approach. Robertson, E. J. (eds.), IRL Press, Oxford, UK.
Rohde, M., Srivastava,G.,Rylatt, D.B., Bundesen, P.,Zamattia,J.,Crane, D. I., and May, B.
K. (1990). Immunocytochemical studies on the localization of 5-aminolevulinate synthase in
rat liver. Arch Biochem Biophys 280,331-5.
Rolfs, 4., Kvietikova, L, Gassmann, M., and Wenger, R. H. (1997)' Oxygen-regulated
transferrin expression is mediated by hypoxia-inducible factor-1. J Biol Chem 272,20055-62
Romeo, P.H., Raich, N., Dubart, 4., Beaupain, D., Pryor, M., Kushner, J., Cohen-Solal, M.,
and Goossens, M. (1986). Molecular cloning and nucleotide sequence of a complete human
uroporphyrinogen decarboxylase cDNA. J Biol Chem 261,9825-31.
Rowley, A. F., Hunt, T. C., Page, M. and Mainwairing, G. (1988) in:. Vertebrate Blood Cells
Rowley, A. F. and Ratcliffe, N. A. (eds), Cambridge University Press, Cambridge, pp: 19-
t28.
Roy, C. N., and Andrews, N. C. (2001). Recent advances in disorders of iron metabolism:
mutations, mechanisms and modifiers. Hum Mol Genet 10,2781-6.
Sadlon, T. J., Dell'Oso, T., Surinya, K.H., and May, B. K. (1999). Regulation of erythroid 5-
aminolevulinate synthase expression during erythropoiesis. Int J Biochem Cell Biol 31,7153-
67.
168

r..l
Sadlon, T. J., Pyragius, T. and May, B. K. The coding region,óf theYo-arnìnolevulinate
s5mthase-l mRNA confers heme-regulated instability in hepatoma cells. Manuscript in
preparation.
Sambrook, J., Fritsch, E. F. and Maniatis T. (19S9). Molecular Cloning: a laboratory manual
2"d Ed. Cold Spring Harbour Laboratory Press, New York
Sanchez, M. J., Holmes,4., Miles, C., and Dzierzak, E. (1996). Charactenzation of the first
definitive hematopoietic stem cells in the AGM and liver of the mouse embryo. Immunity 5,
513-25.
S*g, N., Avantaggiati, M. L., and Giordano, A. (1997). Roles of p300, pocket proteins, and
hTBP in E1A-mediated transcriptional regulation and inhibition of p53 transactivation
activity. J Cell Biochem 66, 277-85.
Sawado, T., Igarashi, K., and Groudine, M. (2001). Activation of beta-major globin gene
transcription is associated with recruitment of NF-E2 to the beta-globin LCR and gene
promoter. Proc Natl Acad Sci U S A 98, 10226-31.
Schoenhaut, D. S., and Curtis, P. J. (1936). Nucleotide sequence of mouse 5-aminolevulinic
acid synthase oDNA and expression of its gene in hepatic and erythroid tissues. Gene 48,55-
63.
Schoenhaut, D. S., and Curtis, P. J. (1989). Structure of a mouse erythroid 5-aminolevulinate
synthase gene and mapping of erythroid-specific DNAse I hlpersensitive sites. Nucleic Acids
Res 17,7013-28.
Schubeler, D., Francastel, C., Cimbora, D.M., Reik,4., Martin, D.I., and Groudine, M.
(2000). Nuclear localization and histone acetylation: a pathway for chromatin opening and
transcriptional activation of the human beta-globin locus. Genes Dev 14,940-50.
Scohy, S., Gabant, P., Szpirer, C., and Szpirer, J. (2000). Identification of an enhancer and an
alternative promoter in the first intron of the alpha-fetoprotein gene. Nucleic Acids Res 28,
3743-51.
t69

Scott, 8.Vy'., Simon, M. C., Anastasi, J., and Singh, H. (1994). Requirement of transcription
factor PU.1 in the development of multiple hematopoietic lineages. Science 265,1573-7.
Shavit, J.4., Motohashi, H., Onodera, K., Akasaka,J.,Yamamoto, M., and Engel, J' D.
(1998). Impaired megakaryopoiesis and behavioral defects in mafG-null mutant mice. Genes
Dev 12,2164-74.
Shimada, Y., Okuno, S., Kawai,4., Shinomiya, H., Saito,4., Suzuki, M., Omori, Y.,
Nishino, N., Kanemoto, N., Fujiwara, T., Horie, M., and Takahashi, E. (1998). Cloning and
chromosomal mapping of a novel ABC transporter gene (hABC7), a candidate for X-linked
sideroblastic anemia with spinocerebellar ataxia. J Hum Genet 43,ll5-22.
Shimizu, R., Takahashi, S., Ohneda, K., Engel, J. D., and Yamamoto, M. (2001). In vivo
requirements for GATA-I functional domains during primitive and definitive erythropoiesis.
Embo J 20,5250-60.
Shivdasani, R. A. (1991). Stem cell transcription factors. Hematol Oncol Clin North Am 11,
tt99-206.
Shivdasani, R. 4., Fujiwara, Y., McDevitt, M. 4., and Orkin, S. H. (1997). A lineage-
selective knockout establishes the critical role of transcription factor GATA-1 in
megakaryocyte growth and platelet development. Embo J 16\,3965-13. \'\
I'
Shivdasani, R. 4., Mayer, 8.L., and Orkin, S. H.-(1995). Absence of blood formation in mice
lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature 373,432-4.
Shivdasani, R. 4., and Orkin, S. H. (1995). Erythropoiesis and globin gene expression in mice
lacking the transcription factor NF-E2. Proc Natl Acad Sci U S A 92,8690-4.
Shivdasani, R. 4., and Orkin, S. H. (1996). The transcriptional control of hematopoiesis.
Blood 87,4025-39.
Shivdasani, R.4., Rosenblatt, M.F., Zucker-Franklin, D., Jackson, C.W., Hunt, P., Saris, C.
J., and Orkin, S. H. (1995). Transcription factor NF-E2 is required for platelet formation
170

independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell 81,
695-704
Smith, A. G. (1991). Culture and Differentiation of Embryonic Stem Cells. J. Tiss. Culture
Methods 13,89-94.
Smith, S. J., and Cox, T. M. (1997), Translational control of erythroid delta-aminolevulinate
s¡mthase in immature human erythroid cells by heme. Cell Mol Biol (Noisy-le-grand) 43,
103-14.
Spadaccini,4., Tilbrook, P.4., Sarna, M.K., Crossley, M., Bieker, J.J., and Klinken, S. P
(1998). Transcription factor erythroid Kruppel-like factor (EKLF) is essential for the
erythropoietin-induced hemoglobin production but not for proliferation, viability, or
morphological maturation. ilournal of Biological Chemi stry i7g,23793-8.! '
/\,',,'-, ,
Sposi, N. M., Zon,L.I., Care,4., Valtieri, M., Testa, U., Gabbianelli, M., Mariani, G',
Bottero, L., Mather, C., Orkin, S.H., and et al. (1992). Cell cycle-dependent initiation and
lineage-dependent abrogation of GATA-I expression in pure differentiating hematopoietic
progenitors. Proc Natl Acad Sci U S A 89, 6353-1.
Srivastava, G., Borthwick, I. 4., Brooker, J. D., May, B. K., and Elliott, V/. H. (1982)'
Purification of rat liver mitochondrial delta-aminolaevulinate synthase. Biochem Biophys Res
Commun 109,305-12.
Srivastava, G., Borthwick, I. 4., Brooker, J. D., Wallace, J. C., May, 8.K., and Elliott, W. H.
(1983). Hemin inhibits transfer of pre-delta-aminolevulinate synthase into chick embryo liver
mitochondria. Biochem Biophys Res Commun I17,344-9.
Srivastava, G., Borthwick, I.4., Maguire, D. J., Elferink, C. J., Bawden, M. J., Mercer, J. F.,
and May, B. K. (1938). Regulation of 5-aminolevulinate synthase mRNA in different rat
tissues. J Biol Chem 263,5202-9.
Srivastava, G., Hansen, A. J., Bawden, M. J., and May, B. K. (1990). Hemin administration to
rats reduces levels of hepatic mRNAs for phenobarbitone-inducible enzyrnes. Mol Pharmacol
38, 486-93.
171

Stamatoyannopoulos, J. A., Goodwin, 4., Joyce, T., and Lowrey, C. H. (1995). NF-E2 and
GATA binding motifs are required for the formation of DNase I hl,persensitive site 4 of the
human beta-globin locus control region. Embo J 14,106-16.
Stamatoyannopoulos G. and Nienhuis, A. (1994) Hemoglobin Switchingin: The Molecular
Basis of Blood Diseases. Stamatoyannopoulos G., Nienhuis, 4., Majerus, P and Varmus, H
(eds.), W. B. Saunder, Philadelphia, PA, pp: 107-55.
Stewart, C. L. (1993). Production of chimeras between embryonic stem cells and embryos.
Methods Enzymol 2 2 5, 823-55.
Strauss, E. C., Andrews, N. C., Higgs, D. R., and Orkin, S. H. (1992). In vivo fooþrinting of
the human alpha-globin locus upstream regulatory element by guanine and adenine ligation-
mediated polymerase chain reaction. Mol Cell Biol 12,2135-42.
Surinya, K.H., Cox, T. C., and May, B. K. (199S). Identification and charactdzation of a
conserved erythroid-specific enhancer located in intron 8 of the human 5-aminolevulinate
synthase 2 gene. J Biol Chem 273,16198-809. It:/./
Surinya, K.H., Cox, T. C., and May, B. K. (1997). Transcriptional regulation of the human
erythroid 5-aminolevulinate synthase gene. Identification of promoter elements and role of
regulatoryproteins. J Biol Chem 272,26585-94.
Sutherland, G.R., Baker, E., Callen, D.F., Hyland, V. J., May,8.K., Bawden, M.J., Healy,
H. M., and Borthwick, I. A. (19S8). S-Aminolevulinate synthase is at 3p21 and thus not the
primary defect in X-linked sideroblastic anemia. Am J Hum Genet 43,331-5.
Sutherland, H. G., Martin, D. I., and Whitelaw, E. (1997). A globin enhancer acts by
increasing the proportion of erythrocytes expressing a linked transgene. Mol Cell Biol 17,
t607-14.
Suzuki, H., Takei, M., Yanagida, M., Nakahata, T., Kawakami, T., and Fukamachi, H. (1997).
Early and late events in Fc epsilon RI signal transduction in human cultured mast cells. J
Immunol159, 5881-8.
172

Swope, D. L., Mueller, C.L., and Chrivia, J. C. (1996). CREB-binding protein activates
transcription through multiple domains. J Biol Chem 271,28138-45.
Sytkowski, A. J. (1980). Denaturation and renaturation of human erythropoietin. Biochem
Biophys Res Commun 96, 143-9.
Taketani, S., Kakimoto, K., Ueta, H., Masaki, R., and Furukawa, T. (2002).Involvement of
ABCT in the Bios¡mthesis of Heme in Erythroid Cells: Interaction of ABCT with
ferrochelatase. Blood I 2, 12.
Talbot, D., and Grosveld, F. (1991). The 5'HS2 of the globin locus control region enhances
transcription through the interaction of a multimeric complex binding at two functionally
distinct NF-E2 binding site$. Embo J 10,l3gl-8. i'',i
ltTalbot, D., Philipsen, S., Fraser, P., and Grosveld, F. (1990).petailed analysis of the Síte 3
region of the human beta-globin dominant control region. Embo I i)Ztøg-ll.
Tamura, K., Sudo, T., Senftleben,IJ., Dadak, A. M., Johnson, R., and Karin, M. (2000)
Requirement for p3Salpha in erythropoietin expression: a role for stress kinases in
erythropoiesis. Cell I 02, 221-31.
Tan, D., and Ferreira, G. C. (1996). Active site of 5-aminolevulinate synthase resides at the
subunit interface. Evidence from in vivo heterodimer formation. Biochemistry 35,8934-4I.
Tan, D., Harrison, T., Hunter, G. 4., and Ferreira, G. C. (1998). Role of arginine 439 in
substrate binding of 5-aminolevulinate synthase. Biochemistry 37, 1478-84.
Tang, Y., Liu, D. P., andLiang, C. C. (2002). Further understanding of the beta-globin locus
regulation at the molecular level: looping or linking models? Genes Cells 7, 889-900'
Tewari, R., Gillemans, N., Wijgerde, M., Nuez, 8., von Lindern, M., Grosveld, F., and
Philipsen, S. (192!). Erythroid Kruppel-like factor (EKLF) is active in primitive and
definitive erirthroid cells and is required for the function of 5'HS3 of the beta-globin locus
control region. Embo J 1',7,2334-4L
173

Theil, E. C. (1990). Regulation of ferritin and transferrin receptor mRNAs. J Biol Chem 265,
4771-4.
Thompson, M.4., Ransom, D.G., Pratt, S. J., Maclennan, H., Kieran, M.W., Detrich, H.
'W., 3rd, Vail, B., Huber, T.L., Paw, B., Brownlie, A. J., Oates, A. C., Fntz, A., Gates, M.A''
Amores,4., Bahary, N., Talbot, W. S., Her, H., Beier, D.R., Postlethwait, J. H., andZon,L.
I. (199S). The cloche and spadetail genes differentially affect hematopoiesis and
vasculogenesis. Dev Biol I 97, 248-69.
Thompson, S., Clarke, A. R., Pow, A. M., Hooper, M. L., and Melton, D. W. (1989). Germ
line transmission and expression of a corrected HPRT gene produced by gene targeting in
embryonic stem cells. Cell 5 6, 313-2L
,-/' "l
Tilbrook, P. 4., Bittorf, T., Busfield, S. J., Chappell, D., and Klinken, S. P. (1996). Disrupted
signaling in a mutant J2E cell line that shows enhanced viability, but does not proliferate or
differentiate, with erythropoietin. J Biol Chem 271,3453-9.
P. 4., lngley, E., V/illiams, J. H., Hibbs, M. L., and Klinken, S' P. (1997).Lyn
is essential for erythropoietin-induced differentiation ofJ2E erythroid cells.
Embo J 1610-9
P. 4., and Klinken, S. P. (1999). The erythropoietin receptor. Int J Biochem Cell
Biol31, 1001-5
Townes, T.M., Lingrel, J. 8., Chen, H.Y., Brinster, R. L., and Palmiter, R. D. (19-85).
Erythroid-specific expression of human beta-globin genes in transgenic mice. Embo J 4,
t7t5-23. t.. ''
Trainor, C.D., Omichinski, J. G., Vandergon,T.L., Gronenborn,4.M., Clore, G. M., and
Felsenfeld, G. (1996). A palindromic regulatory site within vertebrate GATA-I promoters
requires both zinc fingers of the GATA-I DNA-binding domain for high-affinity interaction.
Mol Cell Biol 16,2238-47.
174

Tsai, S. F., Martin, D.l.,Zon, L. I., D'Andrea, A. D., Wong, G. G., and Orkin, S. H. (1989)'
Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through
expression in mammalian cells. Nature 339,446-51.
Tsai, S. F., Strauss, E., and Orkin, S. H. (1991). Functional analysis and in vivo footprinting
implicate the erythroid transcription factor GATA-I as a positive regulator of its own
promoter. Genes Dev 5, 919-31.
Tsang, A. P., Fujiwara, Y., Hom, D. 8., and Orkin, S. H. (1998). Failure of megakaryopoiesis
and arrested erythropoiesis in mice lacking the GATA-I transcriptional cofactor FOG. Genes
Dev 12,1176-88. p{-\
\,:,
Tsang, A. P., Visvader, J. E., Tumer, C. 4., Fujiwara, Y., Yu, C., Weiss, M. J., Crossley, M.,
and Orkin, S. H. (1997). FOG, a multitype zinc finger protein, acts as a cofactor for
transcription factor GATA-I in erythroid and megakaryocytic differentiation. Cell 90,109-19.
Tsukiyama, T., and'Wu, C. (1995). Purification and properties of an ATP-dependent
nucleosome remodeling factor. Cell 83, 1011-20.
Tuan, D., Kong, S., and Hu, K. (1992). Transcription of the hypersensitive site HS2 enhancer
in erythroid cells. Proc Natl Acad Sci U S A 89, ll2l9-23.
Tuan, D., Solomon,'W., Li, Q., and London, I. M. (1985). The "beta-like-globin" gene domain
in human erythroid cells. Proc Natl Acad Sci U S A 82, 6384-8.
Tugores, 4., Magness, S. T., and Brenner, D. A. (1994). A single promoter directs both
housekeeping and erythroid preferential expression of the human ferrochelatase gene. J Biol
Chem 269,30789-97.
Turner, J., and Crossley, M. (1999). Basic Kruppel-like factor functions within a network of
interacting haematopoietic transcription factors. Int J Biochem Cell Biol 31, 1169-74.
Turner, J., and Crossley, M. (1998). Cloning and characteizalionof mCtBP2, a co-repressor
that associates with basic Kruppel-like factor and other mammalian transcriptional regulators.
J_17,5129-40.
t75

Urban, C., Binder, 8., Hauer, C., and Larzer, G. (1992). Congenital sideroblastic anemia
successfully treated by allogeneic bone maffow transplantation. Bone Marrow Transplant 10,
373-5.
Vaisman, 8., Meyron-Holtz,E. G., Fibach, E., Krichevsky, A. M., and Konijn, A. M. (2000).
Ferritin expression in maturing normal human erythroid precursors. Br J Haematol I10,394-
401.
Visvader, J. E., Elefanty, A. G., Strasser,4., and Adams, J.M. (1992). GATA-l but not SCL
induces megakaryocytic differentiation in an early myeloid line. Embo J I l, 4557-64.
Voþr, W. R., and Mingioli, E. S. (1965). Heme s¡mthesis in pyridoxine -responsive anemia.
Ñew England Journal of Medicine 273,347. \
Volland, C., and Felix, F. (19S4).Isolation and properties of 5-aminolevulinate synthase from
the yeast Saccharomyces cerevisiae. Eur J Biochem 142,551-7.
'Wade, p. 4., and'Wolffe, A. P. (1997). Histone acetyltransferases in control. Curr Biol 7,
R82-4
wadman, I. 4., osada,H., Grulz, G. G., Agulnick, A. D.,'Westphal, H., Forster, 4., and
Rabbitts, T.H. (lgg7). The LIM-only protein Lmo2 is a bridging molecule assembling an
erythroid, DNA.bjnding complex which includes the TALI ,847, GATA-I and LdblA{LII
proteins. Embo I 1 6, 3145-51 .
Wall, L., deBoer, E., and Grosveld, F. (1938). The human b-globin geno 3' enhancer contains
multiple binding sites for an erythroid-specihc protein. Genes and Development 2,1089-
1 100.
Walters, M. C., Fiering, S., Eidemiller, J., Magis, W', Groudine, M., and Martin' D. I. (1995)'
Enhancers increase the probability but not the level of gene expression. Proc Natl Acad Sci U
s A 92,7125-9.
176

'Walters, M. C., Magis, W., Fiering, S., Eidemiller, J., Scalzo, D., Groudine, M., and Martin,
D. I. (1996). Transcriptional enhancers act in cis to suppress position-effect variegation.
Genes Dev 10,185-95.
Wang, G. L., and Semenza, G. L. (1993). General involvement of hlpoxia-inducible factor 1
in transcriptional response to hlpoxia. Proc Natl Acad Sci U S A 90, 4304-8.
Wang, L., Liu, L., and Berger, S. L. (1998). Critical residues for histone acetylation by Gcn5,
functioning in Ada and SAGA complexes, are also required for transcriptional function in
vivo. Genes Dev 12,640-53.
Warnick, G. R., and Burnham, B. F. (1911). Regulation of prophyrin biosynthesis.
Purification and characteization of -aminolevulinic acid synthase. J Biol Chem 246,6880-5
'Warren, A. J., Colledge,'W. H., Carlton, M. 8., Evans, M. J., Smith, A. J., and Rabbitts, T' H.
(1994). The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid
development. Cell 78, 45-57 .
Watowich, S. S. (1999). Activation of erythropoietin signaling by receptor dimerization. Int J
Biochem Cell Biol31, 1075-88.
Weatherall,D. (1994). Bone maffow transplantation for thalassemia and other inherited
disorders of hemoglobin. Blood 80,7379-87.
Weber, I. T., Johnson, L. N., Wilson, K. S., Yeates, D. G., Wild, D. L., and Jenkins, J. A.
(1978). Crystallographic studies on the activity of glycogen phosphorylase b. Natwe 274,
433-7.
Wei, X. C., Kishi, H., Jin, Z.X.,Zhao,W.P., Kondo, S., Matsuda, T., Saito, S., and
Muraguchi, A. (2002). Characterizationof chromatin structure and enhancer elements for
murine recombination activating gene-2. J Immunol 169,873-81.
Weiss, G., Houston, T., Kastner, S., Johrer, K., Grunewald, K., and Brock, J. H. (1997).
Regulation of cellular iron metabolism by erythropoietin: activation of iron-regulatory protein
and upregulation of transferrin receptor expression in erythroid cells. Blood 89,680-7 .
177

White, J. M., and Ali, M. A. (1973). Globin s¡mthesis in sideroblastic anaemia. II. The effect
of pyridoxine, -aminolaevulinic acid and haem, in vitro. Br J Haematol24,481-9.
White, J.M., Brain, M. C., and Ali, M. A. (1971). Globin synthesis in sideroblastic anaemia.
I. Alpha and beta peptide chain synthesis. Br J Haematol 20,263-75.
Whitelaw, E., Tsai, S. F., Hogben, P., and Orkin, S. H. (1990). Regulated expression of globin
chains and the erythroid transcription factor GATA-l during erythropoiesis in the developing
mouse. Mol Cell Biol 10,6596-606.
Wijgerde, M., Gribnau, J., Trimboffi, T., Nuez, 8., Philipsen, S., Grosveld, F., and Fraser, P.
(1996). The role of EKLF in human beta-globin gene competition. Genes Dev 10,2894-902,
Witthuhn,8.4., Quelle, F.W., Silvennoinen, O., Yi, T., Tang,8., Miura, O., and lhle, J. N,
(1993). JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and
activated following stimulation with erythropoietin. cell 7 4, 227 -3 6.
'Wu, H., Liu, X., Jaenisch, R., and Lodish, H. F. (1995). Generation of committed erythroid
BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor.
Cell 83, 59-61.
Xu, W., Edmondson, D. G., and Roth, S. Y. (1998). Mammalian GCN5 and P/CAF
acetyltransferases have homologous amino-terminal domains important for recognition of
nucleosomal substrates. Mol Cell Biol 18,5659-69.
Yamada, Y.,.Warren, A. J., Dobson, C., Forster,4., Pannell, R., and Rabbitts, T. H' (1998).
The T cell leukemia LIM protein Lmo2 is necessary for adult mouse hematopoiesis. Proc Natl
AcadSciUSA95,3890-5.
Yang, X. J., Ogryzko, V. V., Nishikawa, J., Howard, B. H., and Nakatani, Y. (199ó). A
p3QQ/CBP-associated factor that competes with the adenoviral oncoprotein ElA. Nature 382,
319-24.
778

Yaouanq, J., Grosbois, B., Jouanolle, A. M., Goasguen, J., and Leblay, R. (1997).
Haemochromatosis Cys282Tyr mutation in pyridoxine-responsive sideroblastic anaemia.
Lancet 349,7415-6.
Yin, X., and Dailey, H.A. (1998). Erythroid 5-aminolevulinate synthase is required for
erythroid differentiation in mouse embryonic stem cells. Blood Cells Mol Dís 24,41-53.
Zanjani,E.D., Peterson, E.N., Gordon, A. S., and Wasseñnan, L. R' (1974). Erythropoietin
production in the fetus: role of the kidney and maternal anemia. J Lab Clin Med 83,287-1.
Zhang, J., and Ferreira, G. C. (2002). Transient state kinetic investigation of 5-
aminolevulinate synthase reaction mechanism. J Biol Chem 277,44660-9.
Zhang,L., and Guarente, L. (1995). Heme binds to a short sequence that serves a regulatory
function in diverse proteins. Embo J 14,t373-20'(_
Zhang,Q., Rombel, I., Reddy, G.N., Gang, J. 8., and Shen, C. K. (1995). Functional roles of
in vivo footprinted DNA motifs within an alpha-globin enhancer. Erythroid lineage and
developmental stage specif,rcities. J Biol Chem 270,8501-5.
Zhang,W., and Bieker, J. J. (1993). Acetylation and modulation of erythroid KruppelJike
factor (EKLF) activity by interaction with histone acetyltransferases. Proc Natl Acad Sci U S
A 95,9855-60.
Zhang,W., Kadam, S., Emerson, B. M., and Bieker, J. J. (2001). Site-specific acetylation by
p300 or CREB binding protein regulates erythroid Kruppel-like factor transcriptional activity
via its interaction with the SV/I-SNF complex. Mol Cell Biol2l,2413-22.
Zhtt,P., and Bu, D. (2000). A novel mutation of the ALAS2 gene in a family with X-linked
sideroblastic anemia. Zhonghrta Xue Ye Xue Za Zhi 2 I , 47 8-81 (abstract)'
Zoller,H., Decristoforo, C., and Weiss, G. (2002). Erythroid 5-aminolevulinate synthase,
ferrochelatase and DMT1 expression in erythroid progenitors: differential pathways for
erythropoietin and iron-dependent regulation. Br J Haematol 118,619-26.
779

Zon,L.I., Tsai, S. F., Burgess, S., Matsudaira, P., Bruns, G.4., and Orkin, S. H. (1990).The
major human erythroid DNA-binding protein (GF-l): primary sequence andlocalization of
the gene to the X chromosome. Proc Natl Acad Sci U S A 82, 668'72.
Zon,L.I., Youssoufian, H., Mather, C., Lodish, H.F., and Orkin, S. H. (1991). Activation of
the erythropoietin receptor promoter by transcription factor GATA-I. Proc Natl Acad Sci U S
A 88, 10638-41.
180
