experimental extracorporeal membrane oxygenation reduces central venous pressure: an adjunct to...
TRANSCRIPT

R E S EARCH ART I C L E
CARD IOVASCULAR D I S EASE
A Factor XIIa Inhibitory Antibody ProvidesThromboprotection in Extracorporeal CirculationWithout Increasing Bleeding RiskMagnus Larsson,1,2* Veronika Rayzman,3* Marc W. Nolte,4* Katrin F. Nickel,1,5,6*Jenny Björkqvist,1,5 Anne Jämsä,1,5 Matthew P. Hardy,3 Marion Fries,4 Stefan Schmidbauer,4
Patricia Hedenqvist,7 Michael Broomé,2,8 Ingo Pragst,4 Gerhard Dickneite,4 Michael J. Wilson,3
Andrew D. Nash,3 Con Panousis,3* Thomas Renné1,5,6*†
Currently used anticoagulants prevent thrombosis but increase bleeding. We show an anticoagulation therapywithout bleeding risk based on a plasma protease factor XII function-neutralizing antibody. We screened forantibodies against activated factor XII (FXIIa) using phage display and demonstrated that recombinant fullyhuman antibody 3F7 binds into the FXIIa enzymatic pocket. 3F7 interfered with FXIIa-mediated coagulation,abolished thrombus formation under flow, and blocked experimental thrombosis in mice and rabbits. Weadapted an extracorporeal membrane oxygenation (ECMO) cardiopulmonary bypass system used for infanttherapy to analyze clinical applicability of 3F7 in rabbits. 3F7 provided thromboprotection as efficiently as heparin,and both drugs prevented fibrin deposition and thrombosis within the extracorporeal circuit. Unlike heparin, 3F7treatment did not impair the hemostatic capacity and did not increase bleeding from wounds. These data establishthat targeting of FXIIa is a safe mode of thromboprotection in bypass systems, and provide a clinically relevantanticoagulation strategy that is not complicated by excess bleeding.
INTRODUCTION
Blood coagulation is not only essential for terminating bleeding frominjury sites (hemostasis) but also contributes to thrombosis-causingvascular occlusive diseases such as pulmonary embolism, myocardialinfarction, and stroke (1). Currently available anticoagulants used forprevention or treatment of thromboembolic events [heparins, vitaminK antagonists (for example, warfarin), and inhibitors of thrombin orfactor Xa] all target enzymes of the coagulation cascade that are crit-ical for formation of fibrin, a protein necessary for controlling injury-related blood loss. As a result, currently used anticoagulants increasethe risk of bleeding and are associated with an increase in potentiallylife-threatening hemorrhage, partially offsetting the benefits of reducedthrombosis (2).
Fibrin formation is initiated by two distinct pathways that are trig-gered by either tissue factor (TF) or the plasma protein factor XII[FXII or Hageman factor, the zymogen form of active FXII (FXIIa)].In the latter pathway (called the “intrinsic pathway of coagulation”),fibrin production is triggered by contact of FXII with polyanionic sur-faces such as glass, polyphosphate, or ellagic acid (referred to as contactactivation), resulting in formation of FXIIa, an active serine protease.FXIIa then initiates two physiological pathways: (i) the intrinsic coagu-lation pathway [by cleaving the FXIIa substrate factor XI (FXI) toform FXIa, another serine protease] and (ii) the kallikrein-kinin sys-
1Department of Molecular Medicine and Surgery, Karolinska Institutet and UniversityHospital, SE-171 76 Stockholm, Sweden. 2ECMO Department, Karolinska UniversityHospital, SE-171 76 Stockholm, Sweden. 3CSL Limited, Bio21 Institute, 30 FlemingtonRoad, Parkville, Victoria 3010, Australia. 4CSL Behring GmbH, Emil-von-Behring-Straße 76,35041 Marburg, Germany. 5Center of Molecular Medicine, Karolinska University Hospital,SE-171 76 Stockholm, Sweden. 6Institute of Clinical Chemistry, University Hospital Hamburg-Eppendorf, D-20246 Hamburg, Germany. 7Department of Clinical Sciences, SwedishUniversity of Agricultural Sciences, SE-750 07 Uppsala, Sweden. 8Department of Physiologyand Pharmacology, Karolinska Institutet, SE-171 76 Stockholm, Sweden.*These authors contributed equally to this work.†Corresponding author. E-mail: [email protected]
www.Scienc
tem, which produces the proinflammatory mediator bradykinin,which causes blood vessels to dilate (thus lowering blood pressure)(3). Further proteolytic cleavage of FXIIa at arginines 334, 343, and353 forms bFXIIa, which is composed of the serine protease domainattached by a disulfide bond to a short fragment of the FXII heavychain. FXII activation in vitro by contact with kaolin (a silicate) iscommonly used to trigger the activated partial thromboplastin time(aPTT) clotting assay, a standard laboratory measurement of plasmacoagulation.
Despite its importance for fibrin formation in vitro, FXII had beenconsidered to have no function for coagulation in vivo. This premise isbased on the fact that FXII-deficient patients have a normal hemosta-tic capacity and do not suffer from spontaneous or injury-related in-creased bleeding (4). Normal hemostasis in FXII-deficient individualshas led to the concept that fibrin formation in vivo is initiated largely,if not exclusively, by TF (5). We have generated FXII-deficient (FXII−/−)mice and found that thrombus formation is largely defective in theseanimals (6). FXII−/− mice are protected from experimental ischemicstroke (7) and pulmonary embolism (8). Despite the thromboprotec-tive effects, FXII−/− mice, like their human counterparts, do not bleedexcessively. In summary, the FXIIa-driven fibrin formation is essentialfor pathological thrombus formation and propagation but has nofunction for fibrin formation during “normal” hemostasis at a siteof injury. In contrast, deficiencies of other components of the coagu-lation cascade, such as factors VIII and IX, cause severe bleeding di-athesis (hemophilia A and B, respectively). This selective property ofFXII in mediating pathological thrombus formation, while being dis-pensable for hemostatic mechanisms, raises the possibility that inhi-bition of FXIIa activity offers a safe strategy for the prevention ofpathological thrombosis.
Extracorporeal membrane oxygenation (ECMO) is a life-supportingtreatment that uses a heart-lung machine to provide gas exchange andsystemic perfusion in patients with severe lung or heart failure. ECMO
eTranslationalMedicine.org 5 February 2014 Vol 6 Issue 222 222ra17 1

R E S EARCH ART I C L E
treatment produces a highly procoagulant condition by exposingblood to bio-incompatible surfaces and nonphysiological shear stress,turbulence, and osmotic forces (9). To prevent thrombotic occlusionsof the oxygenator and tubing in the extracorporeal circuit, anti-coagulation is required. Currently, unfractionated heparin is the stan-dard anticoagulant used in patients (10), and prostacyclin (11), aprotinin(12), contact activation inhibitors (13), a1-antitrypsin Pittsburgh (14),factor Xa inhibitors (15, 16), and nitric oxide donors (17–19) havebeen established for anticoagulation in experimental ECMO models.However, despite intensive monitoring as well as surgical and pharma-cological hemostatic therapies, life-threatening bleeding remains themajor threat to ECMO patients (20). Thus, new strategies for safe anti-coagulation in ECMO are urgently needed. We reasoned that agentstargeting FXIIa should provide thromboprotection without affectinghemostasis. Therefore, using phage display, we developed a recombi-nant fully human FXIIa activity neutralizing antibody (3F7) and showthat 3F7 provides safe anticoagulation in bypass systems.
RESULTS
3F7 blocks FXIIa active site and enzymatic activityTo generate a fully human recombinant antibody that specificallybinds to the catalytic site of human FXIIa and inhibits its proteolyticactivity, we screened the Dyax human Fab (fragment antigen binding)–based phage antibody library against plasma-derived human bFXIIausing a standard panning protocol. bFXIIa-binding phages were elutedwith the FXIIa inhibitor rHA-Infestin-4, which binds specifically to theFXIIa catalytic site and inhibits protease activity (21). FXIIa-specificphage clones were sequenced and analyzed for binding to immobilizedFXIIa and bFXIIa using direct binding assays and competitive enzyme-linked immunosorbent assay (ELISA). The entire light chain and thevariable domain of the heavy chain from 14 Fab clones that bound tobFXIIa were reformatted as intact human immunoglobulin G4 (IgG4)antibodies. The recombinant antibodies were expressed in 293T cellsand tested for interference with FXIIa proteolytic activity using inhi-bition of a chromogenic FXIIa substrate conversion (S-2302, Fig. 1A).All antibodies interfered with FXIIa proteolytic activity in a dose-dependent manner; however, only the 3F7 antibody completelyinhibited the protease activity at an IC50 (half maximal inhibitory con-centration) of 13 nM. Of all the bFXIIa-specific antibodies identified,3F7 had the longest CDR3 loop in its heavy chain (20 residues), whichmay promote access and blocking of the FXIIa catalytic cleft. To in-vestigate the specificity of 3F7 for targeting FXIIa, we tested the anti-body for inhibition of various human plasma proteases and found 3F7to be highly specific for activated FXII variants. We also tested 3F7 forits binding to bFXIIa across a number of species. The antibody bounddirectly to rabbit, mouse, and human activated FXII, but not to the ratprotein (Fig. 1B).
To identify key residues within the FXIIa light chain that areinvolved in the 3F7 epitope, we aligned the murine FXII catalytic do-main (recognized by 3F7) with that of rat FXII (not recognized by3F7). The sequences differed in 18 key positions (fig. S1A). We clonedwild-type murine FXII light chain and variants, where single or com-binations of these 18–amino acid residues were exchanged for their ratortholog. Constructs were expressed as C-terminally His-tagged pro-teins in transiently transfected FreeStyle 293 cells, and secreted solubleFXIIa mutants were tested for their ability to bind 3F7. Western
www.Scienc
blotting revealed that 3F7 was unable to bind to the N397K andI437A mutants, indicating that these residues are crucial for the3F7/FXIIa interaction (Fig. 1C). To confirm the critical role of N397and I437 in the murine FXIIa for 3F7 binding, we used a rescue ap-proach and mutated the two positions in rat FXII into the orthologousmurine residues. Exchange of K397N and K437I in rat FXII was suf-ficient to confer 3F7 binding to the variant (Fig. 1, B and D). Further-more, substitution of positions 397 and 437 in human bFXIIa with thecorresponding residues of the rat protein blunted 3F7 binding to theHu-bFXIIa (D397K/V437K) mutant (Fig. 1E and fig. S1B). Competi-tion ELISA revealed binding of immobilized 3F7 to plasmatic and re-combinant human bFXIIa, recombinant rabbit bFXIIa, and murineFXIIa to be in the low nanomolar range and comparable (Fig. 1F;12.4 to 4.8 nM). Surface plasmon resonance confirmed 3F7 high-affinitybinding to rabbit and human bFXIIa (fig. S1C) with KD (dissociationconstant) = 4.0 ± 0.1 nM and 6.2 ± 0.2 nM, respectively. Consistently,3F7 preferentially bound to contact-activated FXII compared to zymogenin human plasma (fig. S1D), reflecting a higher affinity of the antibodyfor FXIIa forms.
3F7 inhibits FXIIa-driven coagulation ex vivoWe performed plasma-clotting tests using rabbit and human blood toanalyze the effect of 3F7 on coagulation in vitro. The antibody dose-dependently interfered with FXIIa clotting activity in plasma of bothspecies, and the antibody (~50 mg/ml, 330 nM) was sufficient to re-duce protease activity to <5% in rabbit plasma, indicating that aboutan equimolar concentration of 3F7 to plasma FXII (~370 nM) is re-quired for efficient protease inhibition (Fig. 2A). 3F7 prolonged theaPTT up to maximal values (240 s) in both species and was more po-tent in plasma of rabbits as compared to humans (15 versus 240 mg/ml).The antibody (up to 500 mg/ml) did not affect the prothrombin time(PT; a measure of TF-initiated coagulation) in rabbit and human plasma,supporting the specificity of 3F7 for interference with FXIIa-mediatedclotting (Fig. 2, B and C). 3F7 (100 mg/ml) also inhibited dextransulfate–, ellagic acid–, long-chain (>150 phosphate units) and platelet-size (75 U) polyphosphate–driven FXII activation in human plasma(fig. S2A). In contrast, the antibody did not interfere with thrombin-mediated fibrin formation as assessed by thrombin time assays (fig. S2B).
To analyze the anticoagulant mechanisms of 3F7, we performedreal-time thrombin formation assays in human and rabbit plasma.3F7 dose-dependently reduced total (endogenous thrombin potential)and maximum (peak) thrombin formation and prolonged the lag timein plasma stimulated by the nonphysiological FXII activators kaolin(fig. S2, D and E) and ellagic acid (Fig. 2D). The antibody also inter-fered with thrombin formation initiated by the physiological contactactivators [platelet and long-chain polyphosphate] in a dose-dependentmanner (Fig. 2, E to G). Concentrations of 3F7 equimolar to plasmaFXII largely abolished contact-initiated thrombin formation, and evenat 30 times higher antibody concentrations, no measurable effect onTF-triggered thrombin formation was observed in human or rabbitplasma (Fig. 2, H and I). In contrast, heparin interfered with thrombingeneration in response to TF (fig. S2F).
The FXII contact activator dextran sulfate initiates the kallikrein-kinin system but does not trigger coagulation. 3F7 blunted complexformation of FXIIa with its endogenous plasma inhibitor C1 esteraseinhibitor, as well as high–molecular weight kininogen (HK) cleavagetriggered by dextran sulfate and various other contact activators(Fig. 2, J to L, and fig. S2, G to I). In contrast, heparin did not
eTranslationalMedicine.org 5 February 2014 Vol 6 Issue 222 222ra17 2

R E S EARCH ART I C L E
interfere with dextran sulfate–triggered FXII activation or HK cleavage(fig. S2, G to I).
Collagen is exposed in the subendothelial matrix at sites of vascularinjury. Therefore, we analyzed 3F7 for interference with thrombus for-mation on collagen-coated surfaces under flow (Fig. 3). Citrate anti-coagulated blood was recalcified before perfusion at an arterial andvenous shear rate of 1000 and 100 s−1, respectively. In untreated blood,platelets adhered to collagen fibers and aggregated, and fibrin formedwithin 4 min of the start of perfusion (46 ± 4% and 32 ± 2% surfacecovered). Consistent with earlier findings showing defective clot for-mation in blood of FXII-deficient mice (22), 3F7 dose-dependentlyreduced thrombus formation, and the antibody (2500 and 500 mg/ml)almost completely (<5% surface covered) abolished thrombus forma-tion at arterial (Fig. 3, A and C) and venous (Fig. 3, B and D) shearrates, respectively. Immunofluorescence microscopy confirmed the
www.Scienc
antithrombotic activity of 3F7 and revealed that the antibody reducedfibrin, platelet, and leukocyte accumulation under both arterial andvenous flow (Fig. 3, E and F, and fig. S3, E and F). 3F7 (≤500 mg/ml)did not affect adenosine diphosphate–, collagen-, or lipopolysaccharide-driven platelet or leukocyte activation in flow cytometry, suggestingthat reduced platelet and leukocyte accumulation is an indirect effectof anti-FXIIa, for example, mediated by impaired fibrin formation.There were no neutrophil extracellular traps (NETs) detected by anti-citrullinated histone H3 antibody, and nucleotide-specific DAPI andSytox Green staining in thrombi formed within 4 min under high orlow shear conditions (fig. S3, A to D).
3F7 inhibits experimental thrombosis in mice and rabbitsTo analyze 3F7 action in vivo, we induced thrombosis in the carotidartery of mice by topical application of 10% FeCl3, which triggers
Fig. 1. Generation, characterization, epitope mapping, and speciesspecificity of 3F7. (A) Inhibition of FXIIa protease activity with fully human
membranes were reprobed with anti-6×His antibodies to confirm equalprotein loading per lane (right panel). (D) Rescue of 3F7 binding to rat FXII.
antibodies. The Dyax human Fab-based phage antibody library wasscreened for human bFXIIa cross-reacting Fabs. Unique bFXIIa-specificphage clones were reformatted as intact human IgG4 antibodies and re-combinantly expressed in 293T cells. Purified antibodies were tested fortheir dose-dependent interference of FXIIa enzymatic activity. The FXIIa in-hibitor rHA-Infestin-4 (rHA-Inf4) (21) served as internal control. RemainingFXIIa activity was measured using hydrolysis of the chromogenic FXIIa sub-strate S-2302 at an absorbance l = 405 nm. Data are means ± SD (n = 3). (B)3F7 binding to bFXIIa from various species. Immunoplates were coated withrecombinant 8×His-taggedmurine FXIIa or rabbit, human, or rat bFXIIa (1 mg/mleach) overnight at 4°C and then probed with fourfold serially diluted 3F7starting at 20 mg/ml. Bound antibodies were detected by horseradish per-oxidase (HRP)–coupled detection antibodies and HRP-substrate reaction.Uncoated wells are given for comparison (Blank). Data are means ± SD(n = 3). (C) 3F7 epitope mapping. Single amino acids were mutated in murineFXII, and the variants were expressed in FreeStyle 293 cells, affinity-purified,and analyzed for binding to 3F7 by Western blotting (left panel). Stripped
Recombinant wild-type murine FXII (Mu-FXII-8His), rat FXII (Rat-FXII-8His),and a rat FXII variant in which residues 397 and 437 were mutated to theirmurine orthologous (Rat-FXII-8His K397N/K437I) were probed with 3F7 inWestern blot analysis (upper panel). Anti-6×His antibodies confirmed equalloading per lane (lower panel). A representative film of n = 3 is shown. (E)Loss of 3F7 binding in human bFXIIa. ELISA wells were coated with 1 mg/mleach of plasma-derived or recombinant wild-type bFXIIa or a mutated ver-sion of Hu-bFXIIa (D397K/V437K) in which amino acids at positions 397 and437 were changed to the homologous rat residues. Binding of a serial 3F7dilution series to immobilized proteins was performed as in (B). (F) Deter-mination of 3F7 binding affinity by competition ELISA. Recombinant 8×His-tagged murine FXIIa and rabbit, human, and rat bFXIIa were coatedovernight at 4°C with 1 mg/ml each. Wells were blocked with bovine serumalbumin, and a serial dilution of recombinant human-, murine-, and rabbit-activated FXII proteins starting from 100 nM was incubated together with asingle concentration of 3F7 determined from the titration ELISA (B) to givean absorbance of 1.5. Bound 3F7 was quantified as indicated above.
eTranslationalMedicine.org 5 February 2014 Vol 6 Issue 222 222ra17 3

R E S EARCH ART I C L E
formation of free radicals, thus injuring the vascular endothelium.All (10 of 10) FXII−/− mice were protected from vessel-occlusivethrombus formation. Intravenous injection of 3F7 dose-dependentlyreduced occlusion rates and prolonged the time to occlusion (Fig. 4A).At antibody doses ≥5 mg/kg, mice were completely protected fromFeCl3-induced thrombosis. In comparison, an isotype control anti-
www.Scienc
body (30 mg/kg) or vehicle was inactive. We collected blood of treatedanimals at the end of the observation period (at 60 min) and analyzedcoagulation activity. Injection of 3F7 at >5 mg/kg almost completelyabolished FXIIa clotting activity (<5% of FXIIa in saline/controlantibody–infused animals; Fig. 4B). 3F7 dose-dependently prolonged theaPTT up to maximal levels (>125 s) without affecting the PT (9.2 to
Fig. 2. Interference of 3F7 with FXIIa-driven procoagulant activity inplasma. (A to C) 3F7 inhibits FXIIa-driven clotting activity. Rabbit (red
(5 pM). Molar antibody concentrations are relative to plasma FXII (0.375 mM).A representative thrombin generation curve of a series of n = 5 is shown.
curves) and human (blue curves) plasma was incubated for 15 min at roomtemperature with a serial 3F7 antibody dilution starting at 0.5 mg/ml, andsubsequently, (A) FXIIa activity, (B) aPTT, and (C) PT were measured on anautomated blood coagulation analyzer (BCS). Data are means ± SEM (n = 3to 5). (D to I) 3F7 blocks contact-initiated thrombin formation. Real-timethrombin generation [in the absence or presence of increasing concentra-tions of 3F7] in platelet-poor human plasma stimulated with (D) ellagic acid(EA; 100 ng/ml), (E) platelet-size polyphosphate (polyP; 1 mg/ml), (F) long-chain polyphosphate (LC polyP; 0.5 mg/ml), or (H) TF (5 pM) or in rabbitplasma stimulated with (G) long-chain polyphosphate (0.5 mg/ml) or (I) TF
(J to L) 3F7 interferes with activation of the kallikrein-kinin system. Hu-man plasma was incubated with dextran sulfate (DXS; 10 mg/ml), ellagicacid (EA; 1.5 mg/ml), long-chain polyphosphate (LC polyP) and poly-phosphate (polyP) (10 mg/ml each), or buffer (w/o) in the absence or pres-ence of 3F7 (100 mg/ml) for 30 min at room temperature. Plasma sampleswere separated by reducing SDS–polyacrylamide gel electrophoresis(SDS-PAGE) analyzed by Western blot for (J) HK cleavage and (K) forma-tion of C1 inhibitor–FXIIa complexes. (L) Rabbit plasma was incubated withEA (1.5 mg/ml) or buffer (w/o), and HK cleavage dependent on 3F7 addition(100 mg/ml) was analyzed. w/o, buffer-treated samples.
eTranslationalMedicine.org 5 February 2014 Vol 6 Issue 222 222ra17 4

R E S EARCH ART I C L E
13.0 s) at the highest concentration tested (30 mg/kg; Fig. 4, C andD). Consistent with our initial phenotyping of FXII−/− mice (23),inherited deficiency in the protease neither prolonged the bleedingtime (190 ± 45 s versus 170 ± 30 s) nor increased blood loss (11 ±5 ml versus 13 ± 3 ml) compared to wild-type controls in a tail-bleedingassay. Pharmacological inhibition of FXIIa also did not impair the he-mostatic capacity, because bleeding times (180 ± 60 s and 200 ± 50 s)and blood loss (7 ± 2 ml and 10 ± 3 ml) of 3F7-treated wild-type mice(5 and 25 mg/kg), respectively, were not increased compared to saline-treated or FXII−/− mice (Fig. 4E).
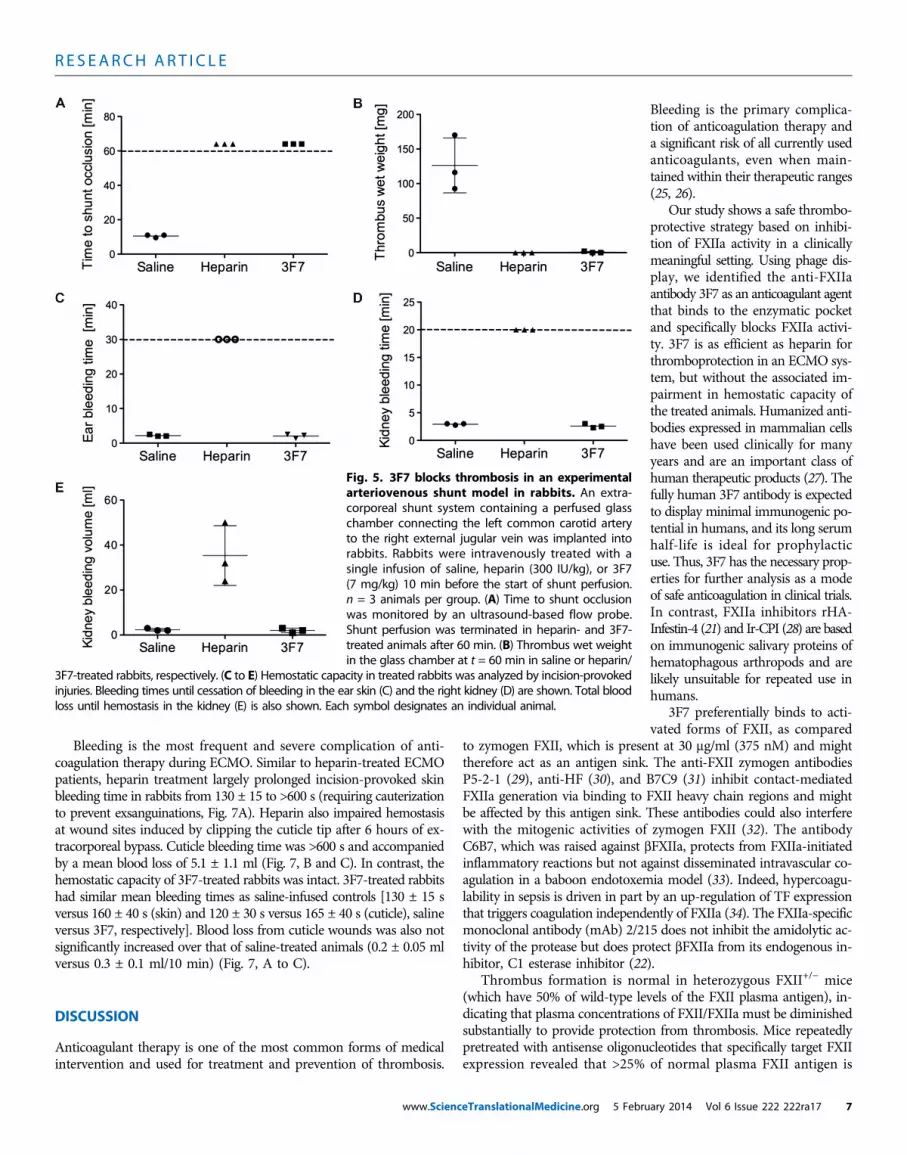
Larger animals are more predictive for anticoagulation-associatedbleeding in humans. Thus, we analyzed 3F7 for its anticoagulant effectsin rabbits using an arteriovenous shunt model. The shunt connects theleft carotid artery to the right external jugular vein and contains a mi-croglass chamber to assess thrombus formation. In saline-treatedcontrols, the chamber was occluded within 9 to 11 min with thrombiof a mean weight of 126 ± 23 mg (Fig. 5, A and B). In contrast, inrabbits treated with heparin (300 IU/kg) or 3F7 (7 mg/kg), shunts didnot occlude within the 60-min perfusion time, and only a minorthrombus of 2-mg weight was detectable in a single 3F7-treated
www.Scienc
animal. Although heparin and 3F7 provided similar thromboprotec-tion, the drugs had different effects on hemostasis (Fig. 5, C to E).Heparin largely prolonged bleeding times and increased blood lossfrom skin incisions and standardized kidney wounds. In contrast,bleeding from skin and kidney wounds was not increased in 3F7-treatedrabbits relative to saline-treated controls.
3F7 prevents occlusive clot formation in ECMOwithout increasing bleedingTo investigate the potential for clinical application of an FXIIa-inhibiting antibody, we adapted, for rabbits, an ECMO system usedfor providing pulmonary and circulatory support to infants. We recordedthe blood pressure gradient between inlet and outlet of the oxygenator(Medos hilite LT Infant 800) as a measure of occlusive thrombus for-mation. In animals without anticoagulation, the pressure gradientrapidly increased to >500 mmHg, the roller pump failed to maintaincirculation, and within <3 min, the extracorporeal circulation wascompletely occluded (Fig. 6A). In two other rabbits, we were unableto draw blood into the ECMO system because of thrombotic occlusionof the catheter at the cannulation site. In contrast, unfractionated
Fig. 3. Dose-dependent inhibition of thrombus formation under ar-terial and venous flow. 3F7 inhibits thrombus formation under high and
(C and D) Columns give the percentage of surface area covered by thrombi.Means ± SD (n = 5). **P < 0.01 versus buffer control, one-way analysis
low shear. (A to F) Citrated rabbit blood readjusted to physiological Ca2+
and Mg2+ concentrations (4.2 and 1.0 mM, respectively) was perfused for4 min over a surface coated with Horm’s type I collagen at an arterial (A,C, and E) (1000 s−1) or a venous (B, D, and F) (100 s−1) shear rate. (A and B)Representative phase-contrast images of thrombi formed during perfu-sion in the presence of indicated 3F7 concentrations. Scale bars, 20 mm.
of variance (ANOVA). (E and F) Bright-field image and immunofluores-cence microscopy of thrombi formed at t = 4 min under flow. Stainingfor fibrin (59D8, green), platelets (anti-CD41, red), DNA [4′,6-diamidino-2-phenylindole (DAPI), blue], and merged images (Merge) is shown.Scale bar, 20 mm. Representative images of n = 6 experiments areshown.
eTranslationalMedicine.org 5 February 2014 Vol 6 Issue 222 222ra17 5

R E S EARCH ART I C L E
www.ScienceTranslationalMedicine.org 5 Febru
heparin administered in identical dosesas those used in patients (50 IU/kg)inhibited occlusion of the cardiopul-monary bypass system. The blood pres-sure gradient over the oxygenatorremained low (<15 mmHg) through-out the 6-hour ECMO procedure.A single intravenous dose of 3F7(7 mg/kg) administered 5 min be-fore the start of ECMO providedsimilar thromboprotection to thatobserved with heparin. The pressuregradient over the oxygenator was<15 mmHg throughout the ECMO(Fig. 6A). Arterial oxygen saturationreached 100% in both heparin- and3F7-treated rabbits and was stablethroughout the 6-hour ECMO peri-od, indicating functional oxygena-tors (table S1).
Thrombosis in the oxygenatorwas evaluated by SEM after 6 hoursof ECMO. Large clots composed offibrin and blood cells were depositedat oxygenator capillaries of saline-treated rabbits (Fig. 6, B andC), whereasfibrin depositions were largely reducedin oxygenators of heparin- and 3F7-treated rabbits (Fig. 6, D to G). Wequantified the fibrin deposited in SEMimages and found that clots werelargely reduced in both heparin- and3F7-treated animals compared to salinecontrols (8 ± 6% and 4 ± 3% versus100 ± 19%; Fig. 6H). Oxygenator-extracted material was analyzed forfibrin deposition by Western blottingusing the fibrin-specific antibody 59D8(7). Consistent with the SEM images,the fibrin signal was high in the saline-treated group and largely reduced inthe heparin- and 3F7-treated animals(Fig. 6I). FXIIa was not detectablesystemically in the plasma of saline-,heparin-, or 3F7-treated animals, in-dicating that minor amounts of FXIIare locally activated (fig. S2C). FXIIahas been associated with complementactivation (24). There was an increasein plasma levels of the complementactivation biomarker C3a at 5 hoursof ECMO treatment in both theheparin- and 3F7-treated groups. 3F7therapy reduced complement acti-vation as compared to heparin, al-beit antibody-mediated inhibitionof C3a did not reach statistical sig-nificance (table S1).
Fig. 4. Defective thrombosis in 3F7-treated mice. (A) 3F7provides thromboprotection. Thrombus formation in the leftcarotid artery was initiated by topical application of 10% FeCl3for 3 min in FXII−/− mice and wild-type animals that were pre-treated with 3F7 (0.5 to 30 mg/kg), an isotype control antibody(30 mg/kg), or vehicle (saline). Time to vascular occlusion was
monitored using a flow probe. Twenty-five animals were used for the saline control, and n = 5 to 10 mice for the 3F7- and isotype control antibody–treated groups. (B to D)Effects of 3F7 treatment on plasma clotting. Blood of 3F7-treated and control mice was intravenously collected60 min after FeCl3 challenge and analyzed for (B) FXIIa, (C) aPTT, and (D) PT using an automated BCS. Means ±SD (n = 3 to 10 for antibody-treated groups and 18 to 23 for saline-infused animals). (E) 3F7 does not impair thehemostatic capacity. Tail bleeding times for saline- and 3F7-treated (5 or 25 mg/kg) wild-type and FXII−/− de-ficient mice were measured. Means ± SEM (n = 10 per group). P > 0.05, Kruskal-Wallis test for both total bloodloss and bleeding time.ary 2014 Vol 6 Issue 222 222ra17 6
R E S EARCH ART I C L E
Bleeding is the most frequent and severe complication of anti-coagulation therapy during ECMO. Similar to heparin-treated ECMOpatients, heparin treatment largely prolonged incision-provoked skinbleeding time in rabbits from 130 ± 15 to >600 s (requiring cauterizationto prevent exsanguinations, Fig. 7A). Heparin also impaired hemostasisat wound sites induced by clipping the cuticle tip after 6 hours of ex-tracorporeal bypass. Cuticle bleeding time was >600 s and accompaniedby a mean blood loss of 5.1 ± 1.1 ml (Fig. 7, B and C). In contrast, thehemostatic capacity of 3F7-treated rabbits was intact. 3F7-treated rabbitshad similar mean bleeding times as saline-infused controls [130 ± 15 sversus 160 ± 40 s (skin) and 120 ± 30 s versus 165 ± 40 s (cuticle), salineversus 3F7, respectively]. Blood loss from cuticle wounds was also notsignificantly increased over that of saline-treated animals (0.2 ± 0.05 mlversus 0.3 ± 0.1 ml/10 min) (Fig. 7, A to C).
DISCUSSION
Anticoagulant therapy is one of the most common forms of medicalintervention and used for treatment and prevention of thrombosis.
www.ScienceTranslationalMedicine.org 5 Febru
Bleeding is the primary complica-tion of anticoagulation therapy anda significant risk of all currently usedanticoagulants, even when main-tained within their therapeutic ranges(25, 26).
Our study shows a safe thrombo-protective strategy based on inhibi-tion of FXIIa activity in a clinicallymeaningful setting. Using phage dis-play, we identified the anti-FXIIaantibody 3F7 as an anticoagulant agentthat binds to the enzymatic pocketand specifically blocks FXIIa activi-ty. 3F7 is as efficient as heparin forthromboprotection in an ECMO sys-tem, but without the associated im-pairment in hemostatic capacity ofthe treated animals. Humanized anti-bodies expressed in mammalian cellshave been used clinically for manyyears and are an important class ofhuman therapeutic products (27). Thefully human 3F7 antibody is expectedto display minimal immunogenic po-tential in humans, and its long serumhalf-life is ideal for prophylacticuse. Thus, 3F7 has the necessary prop-erties for further analysis as a modeof safe anticoagulation in clinical trials.In contrast, FXIIa inhibitors rHA-Infestin-4 (21) and Ir-CPI (28) are basedon immunogenic salivary proteins ofhematophagous arthropods and arelikely unsuitable for repeated use inhumans.
3F7 preferentially binds to acti-vated forms of FXII, as compared
to zymogen FXII, which is present at 30 mg/ml (375 nM) and mighttherefore act as an antigen sink. The anti-FXII zymogen antibodiesP5-2-1 (29), anti-HF (30), and B7C9 (31) inhibit contact-mediatedFXIIa generation via binding to FXII heavy chain regions and mightbe affected by this antigen sink. These antibodies could also interferewith the mitogenic activities of zymogen FXII (32). The antibodyC6B7, which was raised against bFXIIa, protects from FXIIa-initiatedinflammatory reactions but not against disseminated intravascular co-agulation in a baboon endotoxemia model (33). Indeed, hypercoagu-lability in sepsis is driven in part by an up-regulation of TF expressionthat triggers coagulation independently of FXIIa (34). The FXIIa-specificmonoclonal antibody (mAb) 2/215 does not inhibit the amidolytic ac-tivity of the protease but does protect bFXIIa from its endogenous in-hibitor, C1 esterase inhibitor (22).
Thrombus formation is normal in heterozygous FXII+/− mice(which have 50% of wild-type levels of the FXII plasma antigen), in-dicating that plasma concentrations of FXII/FXIIa must be diminishedsubstantially to provide protection from thrombosis. Mice repeatedlypretreated with antisense oligonucleotides that specifically target FXIIexpression revealed that >25% of normal plasma FXII antigen is
Fig. 5. 3F7 blocks thrombosis in an experimentalarteriovenous shunt model in rabbits. An extra-corporeal shunt system containing a perfused glasschamber connecting the left common carotid arteryto the right external jugular vein was implanted intorabbits. Rabbits were intravenously treated with asingle infusion of saline, heparin (300 IU/kg), or 3F7(7 mg/kg) 10 min before the start of shunt perfusion.n = 3 animals per group. (A) Time to shunt occlusionwas monitored by an ultrasound-based flow probe.Shunt perfusion was terminated in heparin- and 3F7-treated animals after 60 min. (B) Thrombus wet weightin the glass chamber at t = 60 min in saline or heparin/
3F7-treated rabbits, respectively. (C to E) Hemostatic capacity in treated rabbits was analyzed by incision-provokedinjuries. Bleeding times until cessation of bleeding in the ear skin (C) and the right kidney (D) are shown. Total bloodloss until hemostasis in the kidney (E) is also shown. Each symbol designates an individual animal.
ary 2014 Vol 6 Issue 222 222ra17 7

R E S EARCH ART I C L E
necessary for occlusive thrombus formation in the FeCl3-injured ca-rotid artery (35). Our studies indicate that >95% of FXII/FXIIa mustbe neutralized to abolish thrombus formation in the same model sys-tem (Fig. 4).
Few clinical studies have analyzed thromboembolic disease in in-dividuals with inherited severe FXII deficiency (<25%). In contrast,FXII deficiency has long been suspected of having prothrombotic,rather than antithrombotic, properties on the basis of case reports ofvenous thrombosis (36) and myocardial infarction (37) in FXII-deficientindividuals. However, this hypothesis has been challenged by Girolamiand co-workers, who demonstrated that in most cases of thrombosisassociated with FXII deficiency, other congenital or acquired prothrom-botic risk factors are also present (38). Indeed, large clinical studies
www.Scienc
failed to identify a correlation between FXII deficiency and increasedrisk of bleeding or thrombosis (39, 40). In patients, severe FXI defi-ciency is associated with a reduced risk of ischemic stroke (41) anddeep vein thrombosis (42), supporting the notion that targeting FXIIa-driven fibrin production is a safe anticoagulation strategy in humans.
FXIIa operates via the intrinsic pathway of coagulation in a processthat involves the sequential activation of FXI and factor IX to formfibrin. The fact that a similar degree of thromboprotection is seen inFXII−/−, FXI−/−, and mice with combined deficiency in FXII and FXI(FXII−/−/FXI−/−) in a pulmonary embolisms model indicates that FXIbecomes predominantly activated by FXIIa during pathological throm-bus formation (8). Antibodies raised against the FXI zymogen that in-terfere with activation of FXI by FXIIa (14E11) or FXIa-mediated
Fig. 6. Inhibition of thrombosis in ECMO systems by 3F7 and heparin.An ECMO bypass system was established for rabbits as described in
3F7-treated animals. Scale bars, 100 mm (left panel) and 30 mm (right panel).Representative images of n = 25 are shown. (H) Fibrin depositions in oxy-
Materials and Methods. Animals were pretreated with a single bolus of ve-hicle (saline), heparin (50 IU/kg), or 3F7 (7 mg/kg) 5 min before the start ofECMO. The blood pressure gradient in the oxygenator was continuouslymonitored at the inflow and outlet by a pressure sensor. (A) Changes inblood pressure in the oxygenator over time for rabbits treated with heparin(n = 5), 3F7 (n = 4), and saline (n = 3). Means ± SD. (B to G) Scanningelectron microscopy (SEM) images of the gas-exchanging capillaries inthe oxygenators at time of occlusion in (B and C) saline-treated rabbitsand, after 6 hours of ECMO, for (D and E) heparin-treated and (F and G)
genators of heparin- and 3F7-treated rabbits relative to saline-infusedcontrols (set to 100%) were quantified from high-power field images suchas those in (B), (D), and (F). Means ± SD of 10 randomly taken SEM images.P values were determined using unpaired Student’s t test. n.s., nonsignif-icant. (I) Accumulation of fibrin in the oxygenator of saline-, heparin-, and3F7-treated rabbits. Fibrin formation was analyzed at the end of ECMO(6 hours for heparin- and 3F7-treated animals and 3 min for saline-treatedcontrols) by immunoblotting using the fibrin-specific antibody 59D8. Con-trol is thrombin-digested clotted rabbit plasma.
eTranslationalMedicine.org 5 February 2014 Vol 6 Issue 222 222ra17 8

R E S EARCH ART I C L E
factor IX activation (O1A6) or that deplete FXI from the circulation(aXIMab) (similarly to FXI antisense oligonucleotides) (43) interferewith thrombosis in a FeCl3-induced vessel injury model in miceand a collagen-coated graft occlusion model in baboon, respectively(44–46). Both experimental models address collagen-dependentthrombus formation. Collagen activates FXII either directly or in-directly through its effect on glycoprotein VI and protein kinaseC–stimulated platelets (4). Consistent with mouse and baboon models,treatment of rabbits with antibody XI-5108, which interferes withfactor XIa–mediated activation of factor X, reduces thrombus forma-tion in the jugular veins (47) and on blood vessel neointima exposedfrom repeated balloon injury (48). In addition to its role in thrombo-sis, FXIIa initiates the inflammatory kallikrein-kinin system. Thepotential ability of 3F7 or peptide-based FXIIa inhibitors (49) to mod-ulate both thrombotic (through FXI activation) and inflammatory ac-tivities (through bradykinin generation) is a distinct advantage overthe aforementioned FXI inhibitors. In support of this notion is the findingthat bradykinin plasma concentrations are largely elevated in patientson ECMO (50); thus, targeting FXIIa may provide an additional anti-inflammatory benefit to these patients.
Over the past 25 years, more than 50,000 patients have been treatedwith ECMO, and the modified heart-lung machine has salvaged manylives by providing gas exchange and systemic perfusion. However, theECMO circuit exposes blood to nonphysiological surfaces that directlyinduce FXII contact activation and thus coagulation. Currently,high-dose heparin is the standard anticoagulant in ECMO. Heparinincreases bleeding, which is the principal cause of morbidity and mor-tality in ECMO therapy. Bleeding from surgical sites occurs in asmany as 6.3 to 33% of ECMO patients, and furthermore, 3.9 to 7%of all patients suffer from intracranial hemorrhage, the most potentiallydevastating bleeding complication (20). Bleeding is associated with pooroutcome in ECMO (20), stressing the need for safer anticoagulants.
www.Scienc
Although coating of the gas-exchanging capillaries in ECMO withheparin (51), phosphorylcholine (52), or fibronectin (53) has improvedbiocompatibility, there is continuous FXIIa generation on bypass cir-cuit surfaces (54). Nonphysiological shear and mechanical forces acti-vate platelets in the bypass circulation. Activated platelets release theinorganic polymer polyphosphate, which provides an alternative sourceof FXIIa generation with implications for fibrin production within thegrowing thrombus and mechanical thrombus stability (8). Nitric oxideinhibits platelets and provides thromboprotection in experimentalECMO systems (17–19). In contrast, 3F7 interferes with both artificialsurface– and procoagulant platelet–produced FXIIa activities. Together,these observations suggest that the ECMO system is the ideal clin-ical setting for therapeutic use of an FXIIa-inhibiting agent for safeanticoagulation.
However, our study has some limitations. 3F7 provides safe thrombo-protection in an extracorporeal bypass system wherein blood isexposed to foreign surfaces, and FXII contact activation triggers de-velopment of thrombosis. Initiation of thrombosis in myocardial in-farction, ischemic stroke, or pulmonary embolism is more complexand involves multiple procoagulant mechanisms such as exposure ofTF on ruptured plaques, platelet and leukocyte activation, and dis-turbed blood flow. Although previous studies have shown FXII−/−
mice to be protected from experimental thromboembolic diseases(7, 8), it remains to be seen whether the thromboprotection affordedby 3F7 in ECMO translates to arterial or venous occlusive disease inhumans. Furthermore, targeting FXIIa provides thromboprotectionby reducing clot firmness (8). As revealed by intravital microscopy,thrombi in injured mesenteric vessels of FXII null mice are unstableand easily embolize, becoming lodged in other organs (6). Indeed,there are reports of pulmonary emboli in humans with congenitalFXII deficiency (55, 56). The fibrin degradation product D-dimerserves as a biomarker for pulmonary embolism and is elevated in
Fig. 7. Differential effects of 3F7 and heparin on hemostasis. Rabbitswere intravenously injected with a single bolus of heparin (50 IU/kg) or 3F7
ear. Injury was set using an automated incision device (Surgicutt Junior). (Band C) Cuticle bleeding time (B) and total blood loss (C) from the injured
(7 mg/kg), subsequently treated with ECMO, and analyzed for their hemo-static capacity via incision-triggered bleeding times and blood loss frominjury site at the end of the 6-hour ECMO procedure. Saline-infused rabbitsbefore ECMO treatment served as controls. (A) Skin bleeding time at the
cuticle within 10 min. Two bleeding tests were performed for each peranimal. Wounds of heparin-treated animals were cauterized after 600 s.Means ± SD of n = 8. P values were determined using Student’s t test. n.s.,nonsignificant.
eTranslationalMedicine.org 5 February 2014 Vol 6 Issue 222 222ra17 9

R E S EARCH ART I C L E
3F7-treated rabbits, raising the possibility that 3F7 therapy might beassociated with increased risk of embolization. 3F7 therapy is notassociated with excess fibrin formation, suggesting that elevated plasmaconcentrations of D-dimer are rather a consequence of an increased fi-brinolytic process. Indeed, the clot lysis time in plasma increases and themaximum rate of lysis decreases in an FXII concentration–dependentmanner, reflecting the multifaceted role of FXIIa or polyphosphate-FXIInot only in thrombosis but also in fibrinolysis (57, 58). Future studiesare required to dissect the precise origin of these activation markers andtheir in vivo implications. 3F7 antibody is fully humanized; however,affinity maturation for human FXIIa might be required to further in-crease its potency before application in a clinical setting.
MATERIALS AND METHODS
Study designWe tested the hypothesis that targeting FXIIa activity using a recom-binant anti-FXIIa antibody provides thromboprotection in a rabbitECMO model. In contrast to heparin-mediated anticoagulation, inhi-bition of FXIIa should not impair the hemostatic capacity in treatedanimals. We chose New Zealand White rabbits as a model system be-cause the rabbit is an established model in experimental ECMO (19)and the ECMO settings in these animals are similar to those for in-fants undergoing ECMO therapy, thus allowing for clinically meaning-ful results. Our study is a controlled laboratory experiment withoutrandomization or blinding. The control group treated with saline beforeECMO had to be closed after three animals because of ethical reasons.In all saline-treated control rabbits, immediate clotting was observed inthe oxygenators, and within <3 min, pumps failed and animals died.We excluded one animal because of severe bleeding during the cannu-lation procedure and another animal that was found to be pregnant. Nooutliers were excluded. Endpoints defined were excessive pressure gra-dient in the oxygenators and bleeding after ECMO treatment from stan-dardized injuries in the ear and cuticle.
Generation and selection of phage antibody binding tohuman bFXIIaA human Fab-based phage display library (Dyax) was screened withbiotinylated bFXIIa (Enzyme Research Laboratories) immobilized onM280 Streptavidin Dynabeads (Invitrogen, Life Technologies). Beadswith bFXIIa-phage complexes were collected using a Dynal magneticparticle separator (Invitrogen), extensively washed, and propagatedovernight in Escherichia coli TG1 after rescue with M13KO7 helperphage. The panning assay was repeated three times, and the third roundelution was performed in the presence of 10 mM rHA-Infestin-4 (CSLBehring, 100 times molar excess over bFXIIa coated on beads, 100 nM)(21). Phage clones were sequenced, and unique clones were further ana-lyzed for binding to immobilized bFXIIa by an ELISA. Candidates withthe strongest binding were reformatted into full-length human IgG4antibodies and tested using an in vitro FXIIa amidolytic activity assay.
Sequencing of output phage clonesThe polymerase chain reaction (PCR) amplification and sequencing ofthe Fab clones was carried out using standard PCR conditions with prim-ers pLacPCRfwd (5′-GTGAGTTAGCTCACTCATTAG-3′) and wtGIIIrev(5′-TTTTCATCGGCATTTTCGGTC-3′). PCR products were purifiedby ExoSAP-It (Affymetrix), according to the manufacturer’s instructions,
www.Science
and used as templates in subsequent sequencing reactions with BigDyeterminator (Invitrogen, Life Technologies). For the determination of se-quence diversity, amplified VH regions from round 3 output clones weresequenced using KpaCLfwd (5′-CCATCTGATGAGCAGTTGAAATCT-3′)and LdaCLfwd (5′-GTTCCCGCCCTCCTCTGAGGAGCT-3′) prim-ers in 1:1 molar ratio. The sequences were analyzed with Lasergenepackage software (DNASTAR Inc.). CDRH3 sequences were batch-translated with the Web-based EMBOSS Transeq tool (http://www.ebi.ac.uk/Tools/emboss/transeq/index.html), and diversity was ana-lyzed with a Web-based ExPASy Decrease Redundancy tool (http://ca.expasy.org/tools/redundancy/). To determine the full amino acid se-quences for Fab clones, double-stranded phagemid minipreps wereisolated from 5-ml overnight cultures. The Fab cassette DNA was se-quenced using 3254 (5′-GGTTCTGGCAAATATTCTG-3′) and SeqCLl (5′-GTTGCACCGACCGAATGTA-3′) primers, and sequences wereanalyzed with Lasergene package software.
IgG production and purification of phage-derived antibodiesVectors for recombinant antibody expression were constructed bycloning the entire light chain (variable and constant domains) andthe variable domain of the heavy chain from the selected phage-derivedFab constructs into the pRhG4 vector as described (59). Serum-freesuspension–adapted 293T cells (GeneChoice Inc.) cultured in FreeStyleExpression Medium (Invitrogen) were transiently transfected with293fectin transfection reagent (Invitrogen). mAbs were purified fromcell supernatants with protein A affinity chromatography (HiTrapMabSelect SuRe on an AKTA express, GE Healthcare). Antibodieswere eluted with 0.1 M sodium acetate (pH 3.0) and immediately ap-plied to desalting column chromatography (HiPrep 26/10, GE Healthcare).Protein fractions were pooled and concentrated with an Amicon UltraCel50K centrifugal device (Millipore) before sterile filtration with 0.22-mmfilters. The antibody concentration was determined chromatograph-ically by comparison to control antibody standards. No impuritieswere detectable by Coomassie Blue–stained SDS-PAGE loaded with10 mg of antibody per lane.
Phage ELISAThe reactivity of the selected Fab phage was investigated by ELISA,with FXIIa or bFXIIa coated overnight at 4°C on Nunc immunoplates(100 ml per well) at 1 mg/ml in phosphate-buffered saline (PBS). Neg-ative control wells coated with PBS alone were also included. Wellswere blocked for 2 hours at 37°C with 200 ml of 5% skim milk/PBSand washed three times in PBS + 0.05% Tween 20 (PBST). Fifty mi-croliters of 1% skim milk/PBST and 50 ml of phage culture supernatantwere added to each well, and plates were incubated with shaking atroom temperature for 2 hours. Plates were then manually washed fivetimes with PBST, and 100 ml of anti-M13 mAb diluted 1:5000 in 1%milk/PBST was added to each well, followed by 30-min incubation atroom temperature with shaking. Plates were then washed as before,100 ml of TMB substrate was added to each well, and the plates werethen incubated for 10 min at room temperature with shaking. Thereaction was stopped by the addition of 50 ml of 2 M phosphoric acid,and the absorbance was read at l = 450 nm with a microplate reader(Wallac Victor, PerkinElmer). Clones found to bind FXIIa in a single-well Fab-phage ELISA were further tested for reactivity to FXIIa in acompetition ELISA. Briefly, the phage titers from culture supernatantswere first determined using a titration ELISA, where phage super-natants were serially diluted fourfold in 1% skim milk/PBST, and
TranslationalMedicine.org 5 February 2014 Vol 6 Issue 222 222ra17 10

R E S EARCH ART I C L E
100 ml of each dilution was added to the blocked plate. The remainderof the ELISA protocol was followed as described above. The data wereplotted using KaleidaGraph software (Synergy Software) with Sigmoidalcurve fit, and the EC50 (half maximal effective concentration) valuewas recorded. For competition ELISA, Nunc 96-well immunoplateswere coated and blocked as above using phage concentrations fixedat a level determined from the titration ELISA. The competitor protein(rHA-Infestin-4) was serially diluted starting at 100 nM.
Epitope mapping of 3F7A synthetic and codon-optimized complementary DNA (cDNA) en-coding the entire wild-type murine FXII protein [National Center forBiotechnology Information (NCBI) sequence NP_067464], includinga C-terminal 8×His-tag (Mu-FXII-8His), was obtained from GeneARTAG. This cDNA was directionally cloned into the pcDNA3.1 expressionvector (Invitrogen) with a Kozak consensus sequence (GCCACC) up-stream of the initiating methionine and a double stop codon (TGA) atthe 3′ end of the open reading frame. The resulting plasmid sequencewas confirmed by automated sequencing. This expression plasmid wasthen used as a template to make the following point mutations withthe QuikChange Site-Directed Mutagenesis Kit (Stratagene): N375D,A384D, N397K, W419R, R426H, I437A, Q449R, delE450, S451G,K452R, T453K, G471S, N515S, T537A, and A588D. With the excep-tion of I438A (generated because of nonexpression of an I437K mutant),these residue changes represent mutations of the mouse residue to its ratortholog (GenBank accession no. 001014006). In the case of delE450,this involved a deletion of Glu450. Three mutations (E551D, T554V,and A555T) were introduced into the E551D_A555T Mu-FXII-8Hisvariant. All constructs encoded a C-terminal 8×His-tag, were clonedinto the mammalian expression vector pcDNA3.1 (Invitrogen), andhad their sequence confirmed by DNA sequencing. A synthetic andcodon-optimized cDNA encoding the entire wild-type rat (NCBIsequence NP_001014028) and human (NCBI sequence NP_000496)FXII protein with a C-terminal 8×His-tag (Rat-FXII-8His) was alsoobtained from GeneART AG and cloned into pcDNA3.1. A doubleamino acid mutant of Rat-FXII-8His was generated using standardPCR techniques, whereby K397 and K437 were both mutated to theirmurine ortholog (K397I/K437I). Vice versa, we exchanged residues D397and V437 of human bFXIIa to the corresponding residues of the ratsequence by PCR with the QuikChange Site-Directed Mutagenesis Kitto generate the Hu-bFXIIa D397K/V437K mutant. The construct wasthen cloned into pcDNA3.1 and sequenced as above. Plasma-derivedhuman bFXIIa was bought from Enzyme Research Laboratories.
For transient mammalian expression, FreeStyle 293 suspensioncells (Invitrogen) were grown to a density of 1.1 × 106 cells/ml in 5 mlof FreeStyle Expression Medium (Invitrogen). Seven microliters of293fectin transfection reagent (Invitrogen) was preincubated for 5 minwith 167 ml of Opti-MEM I medium (Invitrogen) and then added to5 mg of plasmid DNA encoding wild-type/mutant Mu-FXII-8His orRat-FXII-8His, and the mixture was incubated for a further 20 min.The DNA-293fectin complex was added to the cells, which werecultured for 6 days at 37°C, 8% CO2, in a shaking incubator at 250 rpm.Culture supernatants were harvested by centrifugation at 2000 rpm for5 min and stored at 4°C for analysis.
For Western blot analysis, cell supernatants containing recombi-nant wild-type/mutant Mu-FXII-8His or Rat-FXII-8His were addedto equal volumes of 2× nonreducing SDS-PAGE sample buffer, andthe mixture was incubated at 80°C for 10 min, then loaded onto pre-
www.Science
cast 4 to 12% bis-tris gels (Invitrogen), and electrophoresed for 1 hourat 200 V. Proteins were electrotransferred onto nitrocellulose membranesand blocked for 1 hour in 5%milk powder in tris-buffered saline (pH 7.4)with 0.05% Tween 20 (TTBS). The membranes were incubated for1 hour with either anti-HuFXII 3F7 mAb or an anti-His mAb 3H3 (bothat 1 mg/ml in TTBS with 5% milk powder), washed thoroughly withTTBS, and then incubated for a further hour with anti-human IgG-FITC(fluorescein isothiocyanate) or anti-mouse IgG-FITC, respectively (Millipore;both at 0.25 mg/ml in TTBS with 5% milk powder). After furtherwashing of the membranes in TTBS, IgG-FITC–bound proteins werevisualized with a Typhoon variable mode analyzer (GE Healthcare).
ECMO in rabbitsNew Zealand White rabbits (3500 to 4100 g) were used for the studythat was approved by the Ethics Committee for Experiments in Ani-mals, Stockholm. Anesthesia was performed with sufentanil (6.9 mg/kgper hour) (Sufenta, Jansen-Cilag) and midazolam (1.35 mg/kg perhour) (Actavis AB) as described (60). Animals were ventilated viaan endotracheal tube with 30% oxygen using a volume-cycled venti-lator. Heart rate, blood pressure, esophageal temperature, end-tidalcarbon dioxide, and saturation were recorded continuously, and bloodgases were checked every hour. A midline laparotomy was performed,and the vena cava caudalis and abdominal aorta were cannulated witha 12-French Fem-Flex II venous cannula and an 8-French Fem-Flex IIarterial cannula (Edwards Lifesciences) using the Seldinger technique.The tip of the venous cannula was controlled to be located in the rightheart atrium. This was confirmed during autopsy after termination.Two rabbits were used in each experiment. The first rabbit was intra-venously injected with either a single bolus of 3F7 (7 mg/kg), heparin(Heparin 5000 IE/ml, LEO Pharma A/S, 50 IU/kg), or 0.9% saline(control), and 120 ml of blood of this respective donor animal wasinfused into the ECMO circuit before the animal was sacrificed. Sub-sequently, the second animal was similarly treated with 3F7, heparin,or saline; connected to the ECMO circuit as described above; and per-fused with a constant flow rate of 50 ml/kg per minute. The ECMOcircuit consisted of a nonheparinized Medos hilite LT Infant 800 oxygen-ator (55-ml volume), a Stöckert roller pump, a heat exchanger (HirtzHICO-Aquatherm), and 200-cm (60-ml volume) nonheparinized stan-dard tubing. Oxygenator pressure gradients were continuously measuredwith DPT 6000 (Codan Triplus AB) as a marker of oxygenator clotting.Blood was collected from the oxygenator for biomarker analysis 15 minand 5 hours after commencement of ECMO (table S1).
Data analysisStatistical analyses were performed with GraphPad Prism 5.0 software.Bleeding times and total blood loss in mice studies were statisticallyanalyzed using the Kruskal-Wallis test. Flow chamber data were testedby one-way ANOVA, followed by Dunnett’s post hoc analysis. Coagu-lation markers were analyzed using unpaired Student’s t test, and P values<0.05 were considered statistically significant. Mean values ± SD arepresented unless otherwise indicated.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/6/222/222ra17/DC1Table S1. Clinical chemistry biomarkers during ECMO.Fig. S1. 3F7 binding characteristics.Fig. S2. Interference of 3F7 with FXIIa-driven contact system activation in plasma.
TranslationalMedicine.org 5 February 2014 Vol 6 Issue 222 222ra17 11

R E S EARCH ART I C L E
Fig. S3. NETs and leukocytes in thrombi generated under flow.Materials and MethodsReferences (61–65)
REFERENCES AND NOTES
1. N. Mackman, Triggers, targets and treatments for thrombosis. Nature 451, 914–918 (2008).2. J. W. Eikelboom, J. I. Weitz, New anticoagulants. Circulation 121, 1523–1532 (2010).3. L. M. Leeb-Lundberg, F. Marceau, W. Müller-Esterl, D. J. Pettibone, B. L. Zuraw, International
union of pharmacology. XLV. Classification of the kinin receptor family: From molecularmechanisms to pathophysiological consequences. Pharmacol. Rev. 57, 27–77 (2005).
4. T. Renné, A. H. Schmaier, K. F. Nickel, M. Blombäck, C. Maas, In vivo roles of factor XII. Blood120, 4296–4303 (2012).
5. N. Mackman, Role of tissue factor in hemostasis, thrombosis, and vascular development.Arterioscler. Thromb. Vasc. Biol. 24, 1015–1022 (2004).
6. T. Renné, M. Pozgajová, S. Grüner, K. Schuh, H. U. Pauer, P. Burfeind, D. Gailani, B. Nieswandt,Defective thrombus formation in mice lacking coagulation factor XII. J. Exp. Med. 202, 271–281(2005).
7. C. Kleinschnitz, G. Stoll, M. Bendszus, K. Schuh, H. U. Pauer, P. Burfeind, C. Renné, D. Gailani,B. Nieswandt, T. Renné, Targeting coagulation factor XII provides protection from patho-logical thrombosis in cerebral ischemia without interfering with hemostasis. J. Exp. Med.203, 513–518 (2006).
8. F. Müller, N. J. Mutch, W. A. Schenk, S. A. Smith, L. Esterl, H. M. Spronk, S. Schmidbauer,W. A. Gahl, J. H. Morrissey, T. Renné, Platelet polyphosphates are proinflammatory andprocoagulant mediators in vivo. Cell 139, 1143–1156 (2009).
9. R. M. Sniecinski, W. L. Chandler, Activation of the hemostatic system during cardiopulmonarybypass. Anesth. Analg. 113, 1319–1333 (2011).
10. J. Sanchez, G. Elgue, J. Riesenfeld, P. Olsson, Studies of adsorption, activation, and inhibi-tion of factor XII on immobilized heparin. Thromb. Res. 89, 41–50 (1998).
11. V. P. Addonizio, C. A. Fisher, J. C. Bowen, G. C. Palatianos, R. W. Colman, L. H. Edmunds Jr.,Prostacyclin in lieu of anticoagulation with heparin for extracorporeal circulation. Trans. Am.Soc. Artif. Intern. Organs 27, 304–307 (1981).
12. G. Fuhrer, M. J. Gallimore, W. Heller, H. E. Hoffmeister, Aprotinin in cardiopulmonary bypass—Effects on the Hageman factor (FXII)–kallikrein system and blood loss. Blood Coagul. Fibrinolysis3, 99–104 (1992).
13. Y. T. Wachtfogel, C. E. Hack, J. H. Nuijens, C. Kettner, T. M. Reilly, R. M. Knabb, R. Bischoff,H. Tschesche, H. Wenzel, U. Kucich, Selective kallikrein inhibitors alter human neutrophilelastase release during extracorporeal circulation. Am. J. Physiol. 268, H1352–H1357(1995).
14. Y. T. Wachtfogel, R. Bischoff, R. Bauer, C. E. Hack, J. H. Nuijens, U. Kucich, S. Niewiarowski,L. H. Edmunds Jr., R. W. Colman, Alpha 1-antitrypsin Pittsburgh (Met358®Arg) inhibits thecontact pathway of intrinsic coagulation and alters the release of human neutrophilelastase during simulated extracorporeal circulation. Thromb. Haemost. 72, 843–847(1994).
15. N. Gikakis, M. M. Khan, Y. Hiramatsu, J. H. Gorman III, C. E. Hack, L. Sun, A. K. Rao, S. Niewiarowski,R. W. Colman, L. H. Edmunds Jr., Effect of factor Xa inhibitors on thrombin formation andcomplement and neutrophil activation during in vitro extracorporeal circulation. Circulation94, 341–346 (1996).
16. G. Annich, T. White, D. Damm, Y. Zhao, F. Mahdi, J. Meinhardt, S. Rebello, B. Lucchesi,R. H. Bartlett, A. H. Schmaier, Recombinant Kunitz protease inhibitory domain of the amyloidbeta-protein precursor as an anticoagulant in venovenous extracorporeal circulation in rabbits.Thromb. Haemost. 82, 1474–1481 (1999).
17. G. M. Annich, J. P. Meinhardt, K. A. Mowery, B. A. Ashton, S. I. Merz, R. B. Hirschl, M. E. Meyerhoff,R. H. Bartlett, Reduced platelet activation and thrombosis in extracorporeal circuits coated withnitric oxide release polymers. Crit. Care Med. 28, 915–920 (2000).
18. E. D. Rauch, A. H. Stammers, B. L. Mejak, S. N. Vang, T. W. Viessman, The effects of nitricoxide on coagulation during simulated extracorporeal membrane oxygenation. J. ExtraCorpor. Tech. 32, 214–219 (2000).
19. A. M. Skrzypchak, N. G. Lafayette, R. H. Bartlett, Z. Zhou, M. C. Frost, M. E. Meyerhoff, M. M. Reynolds,G. M. Annich, Effect of varying nitric oxide release to prevent platelet consumption and pre-serve platelet function in an in vivo model of extracorporeal circulation. Perfusion 22, 193–200(2007).
20. S. A. Conrad, P. T. Rycus, H. Dalton, Extracorporeal Life Support Registry Report 2004. ASAIO J.51, 4–10 (2005).
21. I. Hagedorn, S. Schmidbauer, I. Pleines, C. Kleinschnitz, U. Kronthaler, G. Stoll, G. Dickneite,B. Nieswandt, Factor XIIa inhibitor recombinant human albumin Infestin-4 abolishes occlu-sive arterial thrombus formation without affecting bleeding. Circulation 121, 1510–1517(2010).
www.Science
22. M. P. Esnouf, A. I. Burgess, A. W. Dodds, A. F. Sarphie, G. J. Miller, A monoclonal antibodyraised against human b-factor XIIa which also recognizes a-factor XIIa but not factor XII orcomplexes of factor XIIa with C1-esterase inhibitor. Thromb. Haemost. 83, 874–881 (2000).
23. H. U. Pauer, T. Renné, B. Hemmerlein, T. Legler, S. Fritzlar, I. Adham, W. Müller-Esterl, G. Emons,U. Sancken, W. Engel, P. Burfeind, Targeted deletion of murine coagulation factor XII gene—Amodel for contact phase activation in vivo. Thromb. Haemost. 92, 503–508 (2004).
24. B. Ghebrehiwet, M. Silverberg, A. P. Kaplan, Activation of the classical pathway ofcomplement by Hageman factor fragment. J. Exp. Med. 153, 665–676 (1981).
25. A. Majeed, S. Schulman, Bleeding and antidotes in new oral anticoagulants. Best Pract. Res.Clin. Haematol. 26, 191–202 (2013).
26. S. Schulman, R. J. Beyth, C. Kearon, M. N. Levine; American College of Chest Physicians,Hemorrhagic complications of anticoagulant and thrombolytic treatment: American Col-lege of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest133, 257S–298S (2008).
27. R. D. Mayforth, J. Quintáns, Designer and catalytic antibodies. N. Engl. J. Med. 323, 173–178(1990).
28. Y. Decrem, G. Rath, V. Blasioli, P. Cauchie, S. Robert, J. Beaufays, J. M. Frère, O. Feron,J. M. Dogné, C. Dessy, L. Vanhamme, E. Godfroid, Ir-CPI, a coagulation contact phaseinhibitor from the tick Ixodes ricinus, inhibits thrombus formation without impairinghemostasis. J. Exp. Med. 206, 2381–2395 (2009).
29. H. Saito, T. Ishihara, H. Suzuki, T. Watanabe, Production and characterization of a murinemonoclonal antibody against a heavy chain of Hageman factor (factor XII). Blood 65,1263–1268 (1985).
30. E. J. Small, J. A. Katzmann, R. P. Tracy, O. D. Ratnoff, G. H. Goldsmith Jr., B. Everson, Amonoclonal antibody that inhibits activation of human Hageman factor (factor XII). Blood65, 202–210 (1985).
31. R. A. Pixley, L. G. Stumpo, K. Birkmeyer, L. Silver, R. W. Colman, A monoclonal antibodyrecognizing an icosapeptide sequence in the heavy chain of human factor XII inhibitssurface-catalyzed activation. J. Biol. Chem. 262, 10140–10145 (1987).
32. G. A. LaRusch, F. Mahdi, Z. Shariat-Madar, G. Adams, R. G. Sitrin, W. M. Zhang, K. R. McCrae,A. H. Schmaier, Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFRto initiate angiogenesis. Blood 115, 5111–5120 (2010).
33. R. A. Pixley, R. De La Cadena, J. D. Page, N. Kauffman, E. G. Wyshock, A. Chang, F. B. Taylor Jr.,R. W. Colman, The contact system contributes to hypotension but not disseminated intra-vascular coagulation in lethal bacteremia. In vivo use of a monoclonal anti-factor XII antibodyto block contact activation in baboons. J. Clin. Invest. 91, 61–68 (1993).
34. R. Pawlinski, J. G. Wang, A. P. Owens III, J. Williams, S. Antoniak, M. Tencati, T. Luther, J. W. Rowley,E. N. Low, A. S. Weyrich, N. Mackman, Hematopoietic and nonhematopoietic cell tissue factoractivates the coagulation cascade in endotoxemic mice. Blood 116, 806–814 (2010).
35. A. S. Revenko, D. Gao, J. R. Crosby, G. Bhattacharjee, C. Zhao, C. May, D. Gailani, B. P. Monia,A. R. MacLeod, Selective depletion of plasma prekallikrein or coagulation factor XII inhibitsthrombosis in mice without increased risk of bleeding. Blood 118, 5302–5311 (2011).
36. C. Kuhli, I. Scharrer, F. Koch, C. Ohrloff, L. O. Hattenbach, Factor XII deficiency: A thrombophilicrisk factor for retinal vein occlusion. Am. J. Ophthalmol. 137, 459–464 (2004).
37. L. T. Goodnough, H. Saito, O. D. Ratnoff, Thrombosis or myocardial infarction in congenitalclotting factor abnormalities and chronic thrombocytopenias: A report of 21 patients anda review of 50 previously reported cases. Medicine 62, 248–255 (1983).
38. A. Girolami, M. L. Randi, S. Gavasso, A. M. Lombardi, F. Spiezia, The occasional venousthromboses seen in patients with severe (homozygous) FXII deficiency are probablydue to associated risk factors: A study of prevalence in 21 patients and review of theliterature. J. Thromb. Thrombolysis 17, 139–143 (2004).
39. S. Zeerleder, M. Schloesser, M. Redondo, W. A. Wuillemin, W. Engel, M. Furlan, B. Lämmle,Reevaluation of the incidence of thromboembolic complications in congenital factor XIIdeficiency—A study on 73 subjects from 14 Swiss families. Thromb. Haemost. 82, 1240–1246(1999).
40. T. Koster, F. R. Rosendaal, E. Briët, J. P. Vandenbroucke, John Hageman’s factor and deep-veinthrombosis: Leiden Thrombophilia Study. Br. J. Haematol. 87, 422–424 (1994).
41. O. Salomon, D. M. Steinberg, N. Koren-Morag, D. Tanne, U. Seligsohn, Reduced incidenceof ischemic stroke in patients with severe factor XI deficiency. Blood 111, 4113–4117(2008).
42. O. Salomon, D. M. Steinberg, M. Zucker, D. Varon, A. Zivelin, U. Seligsohn, Patients withsevere factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb.Haemost. 105, 269–273 (2011).
43. J. R. Crosby, U. Marzec, A. S. Revenko, C. Zhao, D. Gao, A. Matafonov, D. Gailani, A. R. MacLeod,E. I. Tucker, A. Gruber, S. R. Hanson, B. P. Monia, Antithrombotic effect of antisense factor XIoligonucleotide treatment in primates. Arterioscler. Thromb. Vasc. Biol. 33, 1670–1678 (2013).
44. A. Gruber, S. R. Hanson, Factor XI–dependence of surface- and tissue factor–initiatedthrombus propagation in primates. Blood 102, 953–955 (2003).
45. E. I. Tucker, U. M. Marzec, T. C. White, S. Hurst, S. Rugonyi, O. J. McCarty, D. Gailani, A. Gruber,S. R. Hanson, Prevention of vascular graft occlusion and thrombus-associated thrombin gen-eration by inhibition of factor XI. Blood 113, 936–944 (2009).
TranslationalMedicine.org 5 February 2014 Vol 6 Issue 222 222ra17 12

R E S EARCH ART I C L E
46. Q. Cheng, E. I. Tucker, M. S. Pine, I. Sisler, A. Matafonov, M. F. Sun, T. C. White-Adams,S. A. Smith, S. R. Hanson, O. J. McCarty, T. Renné, A. Gruber, D. Gailani, A role for factorXIIa–mediated factor XI activation in thrombus formation in vivo. Blood 116, 3981–3989(2010).
47. M. Takahashi, A. Yamashita, S. Moriguchi-Goto, C. Sugita, T. Matsumoto, S. Matsuda, Y. Sato,T. Kitazawa, K. Hattori, M. Shima, Y. Asada, Inhibition of factor XI reduces thrombus forma-tion in rabbit jugular vein under endothelial denudation and/or blood stasis. Thromb. Res.125, 464–470 (2010).
48. A. Yamashita, K. Nishihira, T. Kitazawa, K. Yoshihashi, T. Soeda, K. Esaki, T. Imamura, K. Hattori,Y. Asada, Factor XI contributes to thrombus propagation on injured neointima of the rabbitiliac artery. J. Thromb. Haemost. 4, 1496–1501 (2006).
49. V. Baeriswyl, S. Calzavarini, C. Gerschheimer, P. Diderich, A. Angelillo-Scherrer, C. Heinis,Development of a selective peptide macrocycle inhibitor of coagulation factor XII towardthe generation of a safe antithrombotic therapy. J. Med. Chem. 56, 3742–3746 (2013).
50. R. de Vroege, R. Huybregts, W. van Oeveren, J. van Klarenbosch, G. Linley, J. Mutlu, E. Jansen,E. Hack, L. Eijsman, C. Wildevuur, The impact of heparin-coated circuits on hemodynamicsduring and after cardiopulmonary bypass. Artif. Organs 29, 490–497 (2005).
51. H. P. Wendel, A. M. Scheule, F. S. Eckstein, G. Ziemer, Haemocompatibility of paediatricmembrane oxygenators with heparin-coated surfaces. Perfusion 14, 21–28 (1999).
52. M. Pieri, O. G. Turla, M. G. Calabrò, L. Ruggeri, N. Agracheva, A. Zangrillo, F. Pappalardo, Anew phosphorylcholine-coated polymethylpentene oxygenator for extracorporeal membraneoxygenation: A preliminary experience. Perfusion 28, 132–137 (2013).
53. C. G. Cornelissen, M. Dietrich, K. Gromann, J. Frese, S. Krueger, J. S. Sachweh, S. Jockenhoevel,Fibronectin coating of oxygenator membranes enhances endothelial cell attachment. Biomed.Eng. Online 12, 7 (2013).
54. L. Irvine, S. Sundaram, J. M. Courtney, D. P. Taggart, D. J. Wheatley, G. D. Lowe, Monitoringof factor XII activity and granulocyte elastase release during cardiopulmonary bypass.ASAIO Trans. 37, 569–571 (1991).
55. A. K. Mangal, S. C. Naiman, Hageman factor deficiency and oral contraceptives. Lancet 1,774 (1980).
56. O. D. Ratnoff, The demise of John Hageman. N. Engl. J. Med. 279, 760–761 (1968).57. N. J. Mutch, R. Engel, S. Uitte de Willige, H. Philippou, R. A. Ariëns, Polyphosphate modifies
the fibrin network and down-regulates fibrinolysis by attenuating binding of tPA and plas-minogen to fibrin. Blood 115, 3980–3988 (2010).
58. J. Konings, J. W. Govers-Riemslag, H. Philippou, N. J. Mutch, J. I. Borissoff, P. Allan, S. Mohan,G. Tans, H. Ten Cate, R. A. Ariëns, Factor XIIa regulates the structure of the fibrin clot in-dependently of thrombin generation through direct interaction with fibrin. Blood 118,3942–3951 (2011).
59. T. Jostock, M. Vanhove, E. Brepoels, R. Van Gool, M. Daukandt, A. Wehnert, R. Van Hegelsom,D. Dransfield, D. Sexton, M. Devlin, A. Ley, H. Hoogenboom, J. Müllberg, Rapid generation offunctional human IgG antibodies derived from Fab-on-phage display libraries. J. Immunol.Methods 289, 65–80 (2004).
60. P. Hedenqvist, A. Edner, Å. Fahlman, M. Jensen-Waern, Continuous intravenous anaesthesiawith sufentanil and midazolam in medetomidine premedicated New Zealand White rabbits.BMC Vet. Res. 9, 21 (2013).
www.Science
61. C. Oschatz, C. Maas, B. Lecher, T. Jansen, J. Björkqvist, T. Tradler, R. Sedlmeier, P. Burfeind,S. Cichon, S. Hammerschmidt, W. Müller-Esterl, W. A. Wuillemin, G. Nilsson, T. Renné,Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo.Immunity 34, 258–268 (2011).
62. T. Renné, J. Dedio, J. C. Meijers, D. Chung, W. Müller-Esterl, Mapping of the discontinuousH-kininogen binding site of plasma prekallikrein. Evidence for a critical role of apple domain-2.J. Biol. Chem. 274, 25777–25784 (1999).
63. T. Renné, J. Dedio, G. David, W. Müller-Esterl, High molecular weight kininogen utilizesheparan sulfate proteoglycans for accumulation on endothelial cells. J. Biol. Chem. 275,33688–33696 (2000).
64. Y. Wang, M. Li, S. Stadler, S. Correll, P. Li, D. Wang, R. Hayama, L. Leonelli, H. Han, S. A. Grigoryev,C. D. Allis, S. A. Coonrod, Histone hypercitrullination mediates chromatin decondensation andneutrophil extracellular trap formation. J. Cell Biol. 184, 205–213 (2009).
65. E. R. Weibel, Stereological Methods: Practical Methods for Biological Morphometry (AcademicPress, London, 1979), vol. 1.
Acknowledgments: We are grateful to P. Janson and L. Labberton for support with animalexperiments and to K. Hultenby for skillful SEM analyses. We thank J. Hultman, B. Frenckner,and K. Palmér for continuous support establishing the ECMO system and S. Schenk, F. Kaspereit,E. Raquet, K. Theinert, K. Nowak, and E.-M. Norberg for their excellent technical assistance. Advicefrom T. Fuchs for NET analysis, J. Grundström for flow cytometry, and E. Hagel for statisticalanalysis is appreciated. Funding: This work was supported in part by grants from The CrownPrincess Lovisas Foundation for Sick Children to M.L., Vetenskapsrådet grant (2011-6373) to P.H.,and Hjärt Lungfonden (20110500), Stockholms läns landsting (ALF, 2110471), Cancerfonden(100615), Vetenskapsrådet (K2013-65X-21462-04-5), German Research Society (SFB 841 TP B8),and European Research Council grant (ERC-StG-2012-311575_F-12) to T.R. Author contribu-tions: M.L., P.H., and M.B. performed all ECMO studies; V.R., K.F.N., J.B., A.J., M.P.H., and S.S. con-ducted all ex vivo coagulation and biochemical analyses; M.W.N., M.F., and I.P. performedthrombosis studies in mice and rabbits; G.D., M.J.W., and A.D.N. provided critical tools and advise;C.P. and T.R. provided grant support, designed the experiments, and wrote the manuscript.Competing interests: V.R., C.P., M.W.N., M.F., M.P.H., S.S., I.P., G.D., M.J.W., and A.D.N. are employ-ees of CSL. T.R. is named as inventor on a patent application covering the use of FXII as an an-tithrombotic target. The other authors declare no competing financial interests. Data andmaterials availability: Address 3F7 antibody–related enquiries to C.P. at [email protected].
Submitted 14 June 2013Accepted 31 December 2013Published 5 February 201410.1126/scitranslmed.3006804
Citation: M. Larsson, V. Rayzman, M. W. Nolte, K. F. Nickel, J. Björkqvist, A. Jämsä, M. P. Hardy,M. Fries, S. Schmidbauer, P. Hedenqvist, M. Broomé, I. Pragst, G. Dickneite, M. J. Wilson,A. D. Nash, C. Panousis, T. Renné, A factor XIIa inhibitory antibody provides thromboprotectionin extracorporeal circulation without increasing bleeding risk. Sci. Transl. Med. 6, 222ra17(2014).
TranslationalMedicine.org 5 February 2014 Vol 6 Issue 222 222ra17 13