exercise training boosts enos-dependent mitochondrial biogenesis in mouse heart: role in adaptation...
TRANSCRIPT

Exercise training boosts eNOS-dependent mitochondrial biogenesisin mouse heart: role in adaptation of glucose metabolism
Roberto Vettor,1* Alessandra Valerio,2* Maurizio Ragni,3 Elisabetta Trevellin,1 Marnie Granzotto,1
Massimiliano Olivieri,1 Laura Tedesco,2,3 Chiara Ruocco,3 Andrea Fossati,3 Roberto Fabris,1
Roberto Serra,1 Michele O. Carruba,3 and Enzo Nisoli31Internal Medicine Unit and Center for the Study and Integrated Treatment of Obesity, Department of Medicine, PaduaUniversity, Padua, Italy; 2Department of Molecular and Translational Medicine, Brescia University, Brescia, Italy; 3Centerfor Study and Research on Obesity, Department of Medical Biotechnology and Translational Medicine, Milan University,Milan, Italy
Submitted 11 November 2013; accepted in final form 28 December 2013
Vettor R, Valerio A, Ragni M, Trevellin E, Granzotto M, OlivieriM, Tedesco L, Ruocco C, Fossati A, Fabris R, Serra R, Carruba MO,Nisoli E. Exercise training boosts eNOS-dependent mitochondrial bio-genesis in mouse heart: role in adaptation of glucose metabolism. Am JPhysiol Endocrinol Metab 306: E519–E528, 2014. First published De-cember 31, 2013; doi:10.1152/ajpendo.00617.2013.—Endurance exer-cise training increases cardiac energy metabolism through poorlyunderstood mechanisms. Nitric oxide (NO) produced by endothelialNO synthase (eNOS) in cardiomyocytes contributes to cardiac adap-tation. Here we demonstrate that the NO donor diethylenetri-amine-NO (DETA-NO) activated mitochondrial biogenesis and func-tion, as assessed by upregulated peroxisome proliferator-activatedreceptor-� coactivator-1� (PGC-1�), nuclear respiratory factor 1, andmitochondrial transcription factor A (Tfam) expression, and by in-creased mitochondrial DNA content and citrate synthase activity inprimary mouse cardiomyocytes. DETA-NO also induced mitochon-drial biogenesis and function and enhanced both basal and insulin-stimulated glucose uptake in HL-1 cardiomyocytes. The DETA-NO-mediated effects were suppressed by either PGC-1� or Tfam small-interference RNA in HL-1 cardiomyocytes. Wild-type and eNOS�/�
mice were subjected to 6 wk graduated swim training. We found thateNOS expression, mitochondrial biogenesis, mitochondrial volumedensity and number, and both basal and insulin-stimulated glucoseuptake were increased in left ventricles of swim-trained wild-typemice. On the contrary, the genetic deletion of eNOS prevented allthese adaptive phenomena. Our findings demonstrate that exercisetraining promotes eNOS-dependent mitochondrial biogenesis in heart,which behaves as an essential step in cardiac glucose transport.
endothelial nitric oxide synthase; exercise training; heart; nitric oxide;mitochondrial biogenesis; glucose uptake
MODERATE ENDURANCE EXERCISE TRAINING fosters cardiovascularhealth and is prescribed for the prevention and the treatment ofheart diseases (13). Heart adaptation to chronic exercise im-plies increased glucose transport and mitochondrial respiratorycapacity, resulting in improved efficiency of cardiac musclecontraction, but our understanding of the underlying mecha-nisms is very limited and currently debated (1, 34). Metabolicremodeling of skeletal muscle to exercise training is the con-sequence of a coordinated genetic response that boosts perox-isome proliferator-activated receptor-� coactivator-1� (PGC-1�)-dependent mitochondrial biogenesis, increases the size and
number of mitochondria, and induces the GLUT4 (insulin-sensitive) isoform of the glucose transporters (15). Wu et al.has additionally shown that the transcriptional coactivatorPGC-1�, that is induced by exercise training in skeletal andcardiac muscle, differently contributes to the modulation ofendoplasmic reticulum homeostasis in these tissues (48). Thusthe distinctive molecular mechanisms governing cardiac adap-tation to exercise training, including the exercise-dependentincrease of myocardial substrate utilization and enhanced met-abolic capacity, are largely unresolved.
Nitric oxide (NO), tonically synthesized in cardiomyocytesby the constitutively expressed endothelial NO synthase(eNOS) (5, 27), mediates key aspects of the cardiac adaptiveresponses to exercise (13). We previously demonstrated thatNO increases PGC-1�, nuclear respiratory factor-1 (NRF-1),and mitochondrial transcription factor A (Tfam) expression,triggering mitochondrial biogenesis in a variety of cells (28,29), and found reduced PGC-1� levels in the heart of eNOSnull mutant (eNOS�/�) mice together with reduced mitochon-drial DNA (mtDNA) content and O2 consumption comparedwith wild-type (WT) animals (29).
Exercise training induces eNOS mRNA and protein expres-sion in mouse left ventricular tissue (18). Further studies haveobserved increased eNOS activation in hearts from trainedmice and suggested that exercise-stimulated GLUT4 translo-cation to the cell membrane and glucose uptake in the heartcould be mediated via a phosphatidylinositol 3-kinase-proteinkinase B (Akt)-NO-dependent mechanism (49). In spite of thisevidence, the contribution of the NO-mediated mitochondrialbiogenesis in heart adaptations to exercise and its possible rolein adaptive changes of glucose metabolism remain to beinvestigated. Here we show that the activation of a completeeNOS-dependent transcriptional program increasing cardiomy-ocyte mitochondrial mass and function is crucial for cardiacmetabolic adaptation to exercise.
MATERIALS AND METHODS
Mice and exercise protocol. Thirty-six adult (8-wk-old) male WT(F2 Hybrid B6.129S2 obtained from crossing C57BL/6J and 129S1/SvImJ mice) and male B6.129P2-Nos3tm1Unc/J (eNOS�/�) mice (38)(Jackson Laboratory) were treated according to the European Unionguidelines and with the approval of the Institutional Ethical Commit-tee. Body weight and food consumption were monitored throughoutthe experimental period. Swim training was chosen as an endurancetraining model since it is widely adopted for the study of physiologicalcardiac adaptation phenomena (1, 6, 13, 49). Both WT and eNOS�/�
* R. Vettor and A. Valerio contributed equally to this work.Address for reprint requests and other correspondence: E. Nisoli, Dept. of
Medical Biotechnology and Translational Medicine, Univ. of Milan, viaVanvitelli, 32-20129 Milan, Italy (e-mail: [email protected]).
Am J Physiol Endocrinol Metab 306: E519–E528, 2014.First published December 31, 2013; doi:10.1152/ajpendo.00617.2013.
0193-1849/14 Copyright © 2014 the American Physiological Societyhttp://www.ajpendo.org E519

mice (n � 18 mice/group) were assigned randomly to either swimtraining or to have no lifestyle modifications. Mice swam one time aday for 5 days/wk in a graduated protocol (6) starting at 10 min daily,with a 10-min increase each day until 90 min daily at the end of the2nd wk. Next, mice swam 30 days on the full training regimen (90min daily, 5 days/wk). Swimming sessions were supervised to preventdrowning.
Insulin tolerance test. Insulin tolerance test (ITT) was performed 2days after the last bout of exercise in 8-h-fasted animals (n � 6mice/group) by an intrperitoneal injection of insulin (0.5 U/kg bodywt diluted in 0.9% NaCl) (Actrapid HM; Novo Nordisk). Bloodsamples were obtained from the tail vein at 0, 5, 10, 15, 20, 30, 40, 60,and 90 min postinjection and used to determine plasma glucose levels.
Tissue glucose utilization index. At the end of the training periodand 2 days before the clamp studies, a catheter was inserted in theright femoral vein under general anesthesia with pentobarbital so-dium. Tissue glucose uptake studies were performed on mice underconscious and unstressed conditions after an 8-h fast. The nonmetabo-lizable glucose analog 2-deoxy-D-[1-3H]glucose ([3H]DG; 10 �Ci)(Amersham Biosciences) was injected as an intravenous bolus in thebasal condition or after hyperinsulinemic euglycemic clamp as previ-ously described with minor modifications (44). One hundred twentyminutes after tracer injection, animals were killed, and tissues werequickly collected in liquid nitrogen and kept at �80°C for subsequentanalysis. The glucose utilization index was derived from the amountof [3H]DG-6-phosphate ([3H]DGP) measured in the various tissues aspreviously described (14) thus using the accumulation of [3H]DGP asan index of the glucose metabolic rate.
Analytical procedures. Blood glucose was measured using the OneTouch Blood Glucose Monitoring System (LifeScan Italia; Johnson &Johnson Medical). Plasma insulin was measured by radioimmunoas-say (Linco Research). Plasma free fatty acids (FFA) were quantifiedusing the NEFA-HR(2) kit (Wako Chemicals).
Cell cultures and treatments. Adult mouse cardiomyocytes wereisolated and cultured as previously described (10). Hearts were ex-cised from heparinized and anesthetized mice, mounted on a steelcannula, and perfused for 3 min with a Ca2�-free buffer, previouslygassed with 95% O2-5% CO2. Enzymatic digestion was performed for10 min by adding liberase blendzyme 4 (Hoffmann-La Roche). Theleft ventricle was removed, cut into chunks, and further digested for10 min at 37°C. The dispersed myocytes were resuspended in bufferscontaining progressively increased concentration of Ca2�. Cardiomy-ocytes were plated in laminin-coated dishes with MEM containing2.5% FBS and antibiotics. After 1 h of culture, the medium waschanged to FBS-free MEM. Primary cardiomyocytes were exposed totest drugs the day after seeding. HL-1 cells (a gift from W. C.Claycomb) (9) were also used in the present study since they are
easily amenable to genetic manipulation and can be cultured forprolonged time while keeping a differentiated cardiomyocyte pheno-type and contracting in vitro (47). HL-1 cardiomyocytes were platedin fibronectin/gelatin-coated flasks and grown to 70–80% confluencein Claycomb medium (JRH Biosciences) supplemented with 100 �Mnorepinephrine, 2 mM L-glutamine, 100 U/ml penicillin, 100 �g/mlstreptomycin, and 10% FBS (JRH Biosciences) as described (10).Cells were exposed to diethylenetriamine-NO (DETA-NO), S-nitroso-N-acetylpenicillamine (SNAP) (both from Sigma-Aldrich), or vehicleas described in RESULTS. NO donor treatment left unchanged cardio-myocyte survival in culture (data not shown). For PGC-1� and Tfamknockdown experiments, HL-1 cells were transfected 24 h afterseeding with 50 nM PGC-1� or Tfam small-interference RNA(siRNA) SMARTpool (Dharmacon) or siGENOME nontargeting(NT) siRNA following the manufacturer’s instruction. At the end ofthe treatments, HL-1 cells were harvested for further analysis.
Cell glucose uptake. HL-1 cells were serum-starved for 2 h andincubated in the presence or absence of 300 nM insulin (NovoNordisk) for 30 min and then with 1.5 �Ci/ml [3H]DG (AmershamBiosciences) for 15 min. Cells were washed with ice-cold phosphate-buffered saline and lysed in 0.5 M NaOH. Radioactivity was mea-sured by liquid scintillation counting (Wallac, Turku, Finland). Eachcondition was assayed in three independent experiments in triplicate.
Cell GLUT4myc translocation. HL-1 cells were seeded on fi-bronectin-coated cover slips and transfected with 1.5 �g pCMV-GLUT4myc plasmid or empty vector (a gift from J. E. Pessin, AlbertEinstein College of Medicine, New York, NY) in Opti-MEM IReduced Serum medium with Lipofectamine-2000 (Invitrogen). Cellswere treated with vehicle or 100 �M DETA-NO for 72 h andsubsequently incubated with or without 300 nM insulin for 30 min.Cover slips were fixed in 4% paraformaldehyde, saturated with 5%horse serum-5% bovine serum albumin in PBS, incubated with Myc-Tag 9B11 mouse monoclonal antibody (Cell Signaling Technology)then with Alexa Fluor 546 anti-mouse IgG, counterstained withHoechst 33342, mounted with ProLong Gold antifade reagent (allfrom Invitrogen), and analyzed with the confocal laser-scanningmicroscope Leica DMI6000 CS SP8 (Leica Microsystem). Imageanalysis was performed with the NIH ImageJ software.
RNA analysis. Quantitative reverse transcription-PCR reactionswere performed as previously described (10, 42) and run with the iQSybrGreenI SuperMix (Bio-Rad Laboratories) on an iCycler iQ RealTime PCR detection system (Bio-Rad Laboratories, Hercules, CA).Calculations were performed by a comparative method (2���Ct) using18S rRNA as an internal control. Primers (Table 1) were designedusing Beacon Designer 2.6 software (Premier Biosoft International).
Immunoblot analysis. Western blotting was performed on proteinextracts as previously described (42). Primary antibodies were as
Table 1. Sequence of primers used for qPCR
Gene Symbol Forward Reverse
PGC-1� 5=-ACTATGAATCAAGCCACTACAGAC-3= 5=-TTCATCCCTCTTGAGCCTTTCG-3=Nrf-1 5=-ACAGATAGTCCTGTCTGGGGAAA-3= 5=-TGGTACATGCTCACAGGGATCT-3=Tfam 5=-AAGACCTCGTTCAGCATATAACATT-3= 5=-TTTTCCAAGCCTCATTTACAAGC-3=Cox IV 5=-GTGGTTAGCCATGACCTGAAAG-3= 5=-TTAGCATGGACCATTGGATACGG-3=Cyt c 5=-ATAGGGGCATGTCACCTCAAAC-3= 5=-GTGGTTAGCCATGACCTGAAAG-3=ATPase 5=-CGTGAGGGCAATGATTTATACCAT-3= 5=-TCCTGGTCTCTGAAGTATTCAGCAA-3=Mfn-1 5=-AGTCAGCGGTGAAAGCAAAGT-3= 5=-GGTCTTCCCTCTCTTCCATTGAAT-3=Mfn-2 5-ATATAGAGGAAGGTCTGGGCCG-3= 5=-CCGCATAGATACAGGAAGAAGGG-3=eNOS 5=-AGCGGCTGGTACATGAGTTC-3= 5=-GATGAGGTTGTCCTGGTGTCC-3=GLUT1 5=-GCTGGAATTACCGCGGCT-3= 5=-CACATACATGGGCACAAAGC-3=GLUT4 5=-ACATACCTGACAGGGCAAGG-3= 5=-CGCCCTTAGTTGGTCAGAAG-3=18S 5=-CTGCCCTATCAACTTTCGATGGTAG-3= 5=-CCGTTTCTCAGGCTCCCTCTC-3=Cyt b 5=-CTTCGCTTTCCACTTCATCTTACC-3= 5=-TTGGGTTGTTTGATCCTGTTTCG-3=36B4 5=-AGGATATGGGATTCGGTCTCTTC-3= 5=-TCATCCTGCTTAAGTGAACAAACT-3=
PGC-1�, peroxisome proliferator-activated receptor-� coactivator-1�; Nrf-1, nuclear respiratory factor 1; Tfam, mitochondrial transcription factor A; Cox IV,cytochrome c oxidase IV; Cyt c, cytochrome c; Mfn, mitofusin; eNOS, endothelial nitric oxide synthase; Cyt b, cytochrome b.
E520 eNOS, MITOCHONDRIA, AND CARDIOMETABOLIC ADAPTATION
AJP-Endocrinol Metab • doi:10.1152/ajpendo.00617.2013 • www.ajpendo.org

follows: anti-cytochrome c (Cyt c) (BD Pharmingen), anti-cyto-chrome c oxidase IV (COX IV) (Molecular Probes), anti-eNOS(Transduction Labs), anti-phospho-Akt (Ser473) and anti-Akt (CellSignaling Technology), anti-GLUT4, anti-PGC-1� (both Santa CruzBiotechnology), anti-Tfam (Aviva Systems Biology), anti-GAPDH(Histoline Laboratories), and anti--actin (Sigma-Aldrich). Myocar-dial membrane proteins were extracted using a membrane proteinextraction kit (Biovision). The membrane protein was subsequentlyused for Western blot analysis of GLUT4.
mtDNA. mtDNA copy number was measured by means of quantita-tive PCR from the Cyt b mtDNA gene compared with the large ribosomalprotein p0 (36B4) nuclear gene as previously described (10).
Electron microscopy. Electron microscopy studies were conductedon heart tissue processed as previously described (10). Ultrathinsections were doubly stained with uranyl acetate and lead citrate andexamined under a Philips CM10 transmission electron microscope(Philips, Eindhoven, the Netherlands). For morphometric analysis ofmitochondria, randomly selected areas of tissue derived from threeanimals per group were photographed at a 11,500 magnification andanalyzed with the NIH Image software.
Citrate synthase assay. Citrate synthase activity was measured ineither tissue or whole cell extracts and expressed as nanomole citrateproduced per minute per milligram protein as described (42).
Statistical analysis. Results were expressed as means � SE unlessotherwise specified. Data were analyzed by GraphPad Prism 5.0software using either unpaired Student’s t-test or one-way ANOVA ortwo-way ANOVA with Newman-Keuls’ post hoc test as appropriate.
RESULTS
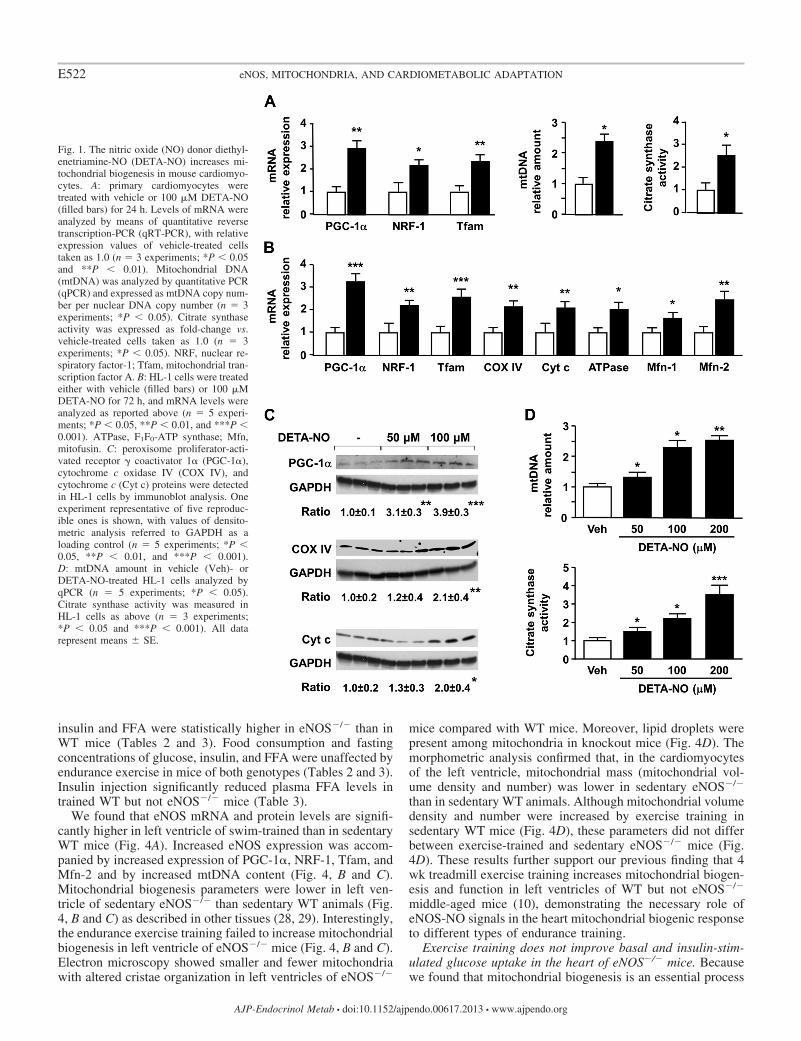
NO donors induce mitochondrial biogenesis in mousecardiomyocytes. NO is a crucial mediator of mitochondrialbiogenesis in various cell types of mammals (28, 29). Toinvestigate its contribution to mitochondrial biogenesis andfunction in cardiac cells, we exposed primary mouse cardio-myocytes to the slow-acting NO donor DETA-NO (half-timeof NO release, 20 h at 37°C) (17). DETA-NO (100 �M)treatment for 24 h induced PGC-1�, NRF-1, and Tfam mRNAexpression and increased mtDNA content and citrate synthaseactivity (i.e., indexes of mitochondrial mass and function) inprimary cardiomyocytes (Fig. 1A). This concentration ofDETA-NO is expected to result in �100 nM NO in vitro,which is within the wide range of NO concentrations that isknown to mediate discrete biological responses in vivo (43).Experiments in HL-1 cardiomyocytes (a murine cardiac-de-rived cell line that displays features similar to those of adultcardiomyocytes in terms of organized structure and ability tocontract in culture) (47) were preliminary conducted to assessdose and time dependency of the DETA-NO-mediated effecton gene expression (data not shown). Treatment with 100 �MDETA-NO for 72 h increased PGC-1�, NRF-1, Tfam, COXIV, Cyt c, F1F0-ATP synthase, and mitofusin (Mfn)-1 and -2mRNA levels in HL-1 cells (Fig. 1B). Moreover, treatmentwith DETA-NO dose-dependently increased PGC-1�, COXIV, and Cyt c protein levels (Fig. 1C) as well as mtDNAamount and citrate synthase activity (Fig. 1D). Similar resultswere obtained treating HL-1 cells for 72 h with a structurallyunrelated NO donor (SNAP, 1–100 �M) (2) (data not shown).
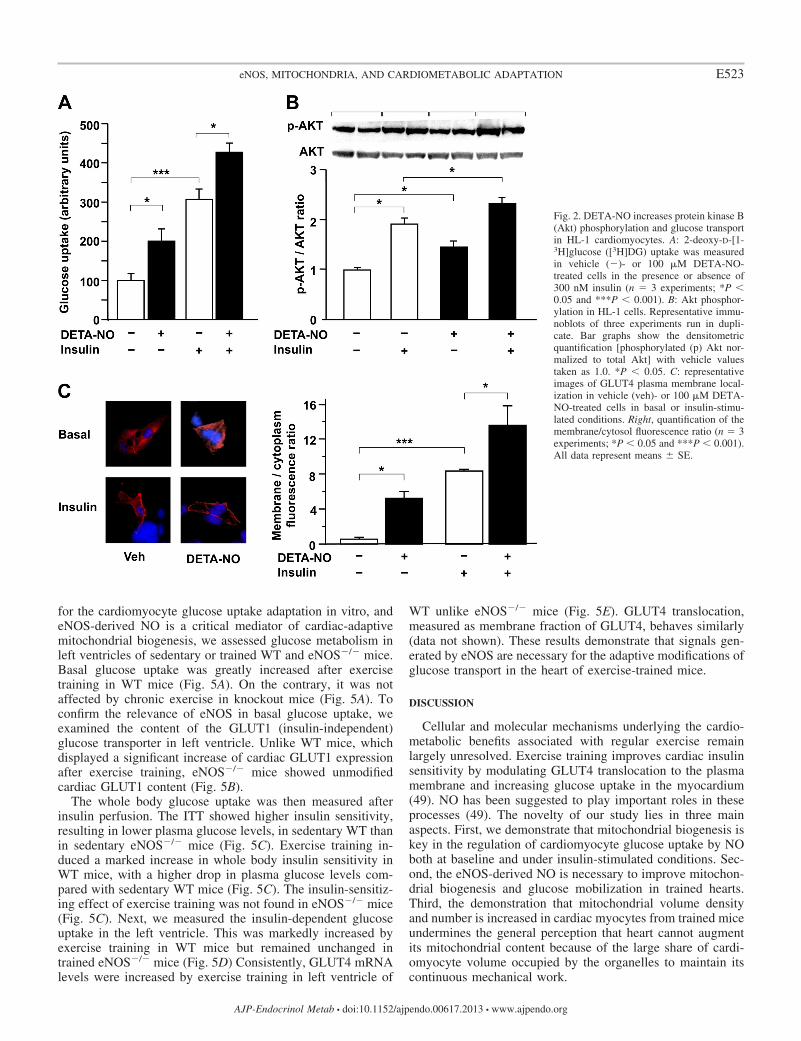
The NO donor DETA-NO induces cardiomyocyte glucoseuptake. We then investigated whether DETA-NO treatmentaffects glucose metabolism in HL-1 cardiomyocytes. Bothbasal and insulin-dependent [3H]DG uptake was increased byDETA-NO (100 �M for 72 h) (Fig. 2A). DETA-NO alsopromoted GLUT4 recruitment to the cell membrane in un-
stimulated HL-1 cells (Fig. 2C) and further enhanced theinsulin-mediated GLUT4 translocation (Fig. 2C). The serine/threonine protein kinase Akt/PKB is a crucial regulator ofinsulin-mediated GLUT4 trafficking (46). We observed thatDETA-NO significantly increased Akt phosphorylation overbasal values and enhanced Akt phosphorylation in insulin-stimulated HL-1 cells (Fig. 2B). These data demonstrate thatNO regulates multiple steps of the glucose uptake pathway incardiac cells.
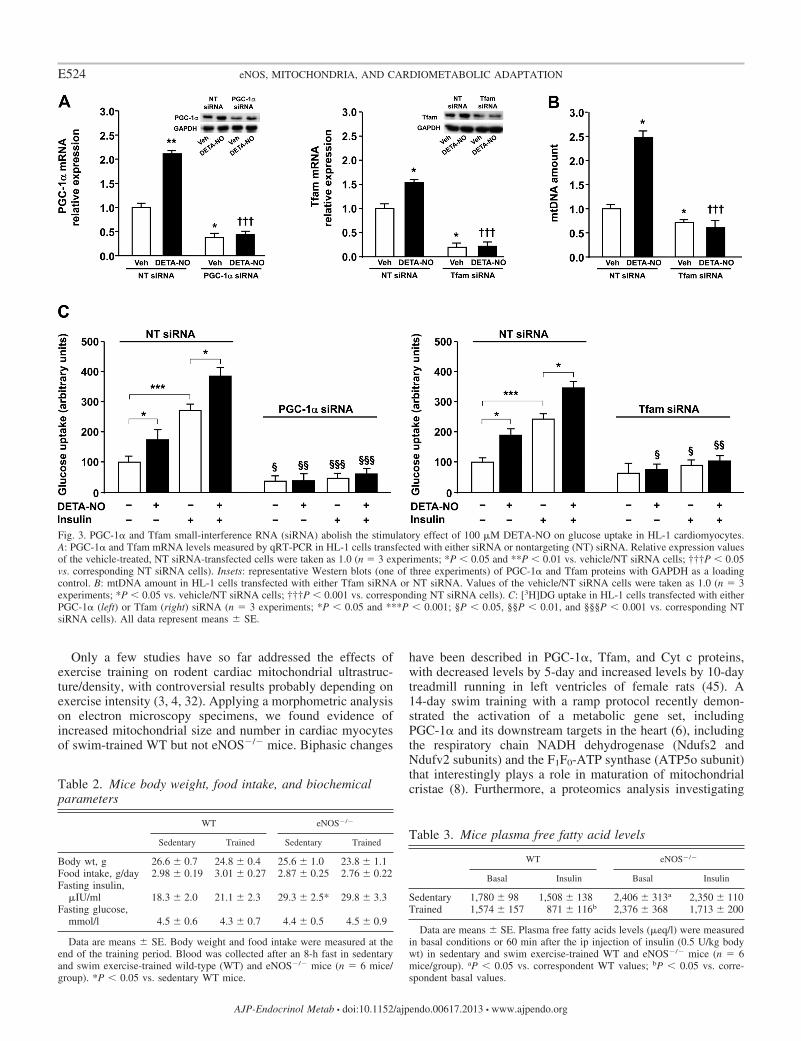
Mitochondrial biogenesis is necessary for DETA-NO-medi-ated glucose uptake in HL-1 cardiomyocytes. To assesswhether the NO-mediated induction of mitochondrial biogen-esis is relevant for cardiac glucose uptake, HL-1 cells weretransfected with siRNA against PGC-1�. The effectiveness ofPGC-1� knockdown by siRNA oligos was evaluated by mea-suring both mRNA and protein levels. Compared with cellstransfected with NT siRNA, PGC-1� knockdown cells dis-played an �60% reduction in PGC-1� mRNA (Fig. 3A).PGC-1� protein levels were concomitantly reduced by �55%(Fig. 3A). Furthermore, PGC-1� siRNA fully prevented theDETA-NO-mediated increase of PGC-1� expression (Fig. 3A).PGC-1� siRNA also prevented the insulin-mediated increase ofglucose uptake and fully blocked the DETA-NO-increased glu-cose uptake both in basal conditions and under insulin (Fig. 3C).
PGC-1� is known to affect GLUT4 expression (23). Toexclude that the inhibitory effects of PGC-1� knockdown werethe result of reduced GLUT4 expression, HL-1 cells weretransfected with Tfam siRNA. Tfam is critical for mitochon-drial biogenesis, being required for mtDNA replication (37).Furthermore, Shi et al. (39) have reported that GLUT4 trans-location, but not GLUT4 expression, is impaired in TfamsiRNA-transfected adipocytes. Tfam siRNA reduced TfammRNA and protein levels by �80 and 70%, respectively,compared with NT siRNA and prevented the DETA-NO-mediated increase of Tfam expression in HL-1 cells (Fig. 3A).Tfam knockdown also led to an �30% decrease in mtDNAcopy number (Fig. 3B) and abolished the DETA-NO-mediatedincrease of mtDNA amount (Fig. 3B). Moreover, a significantimpairment in citrate synthase activity was seen in Tfamknockdown cells (data not shown). Interestingly, Tfam deple-tion, although not significantly affecting basal glucose uptake,was able to reduce the insulin-stimulated glucose uptake andimpair the stimulatory effect of DETA-NO (Fig. 3C). Thesedata demonstrate that efficient mitochondrial biogenesis isessential for NO to induce glucose uptake in HL-1 cardiomy-ocytes.
Exercise training increases mitochondrial biogenesis inheart of WT but not eNOS�/� mice. To investigate the contri-bution of the NO-mediated mitochondrial biogenesis in car-diometabolic adaptations in vivo, we randomly assigned WTand eNOS�/� mice to sedentary conditions or to 6 wk ofgraduated swim training. Aged eNOS�/� mice (from 12.5 moonward) have reduced maximal work capacity in acute bouts oftreadmill exercise (30). The graduated swim training protocolwe used did not lead to exhaustion of the mice, and we did notobserve an obvious difference between the swimming perfor-mance of our 2-mo-old eNOS�/� mice compared with WTmice (data not shown). The body weight was comparablebetween WT and eNOS�/� mice and did not significantlychange after endurance exercise training (Table 2). As ex-pected (12, 27), the fasting plasma concentrations of both
E521eNOS, MITOCHONDRIA, AND CARDIOMETABOLIC ADAPTATION
AJP-Endocrinol Metab • doi:10.1152/ajpendo.00617.2013 • www.ajpendo.org
insulin and FFA were statistically higher in eNOS�/� than inWT mice (Tables 2 and 3). Food consumption and fastingconcentrations of glucose, insulin, and FFA were unaffected byendurance exercise in mice of both genotypes (Tables 2 and 3).Insulin injection significantly reduced plasma FFA levels intrained WT but not eNOS�/� mice (Table 3).
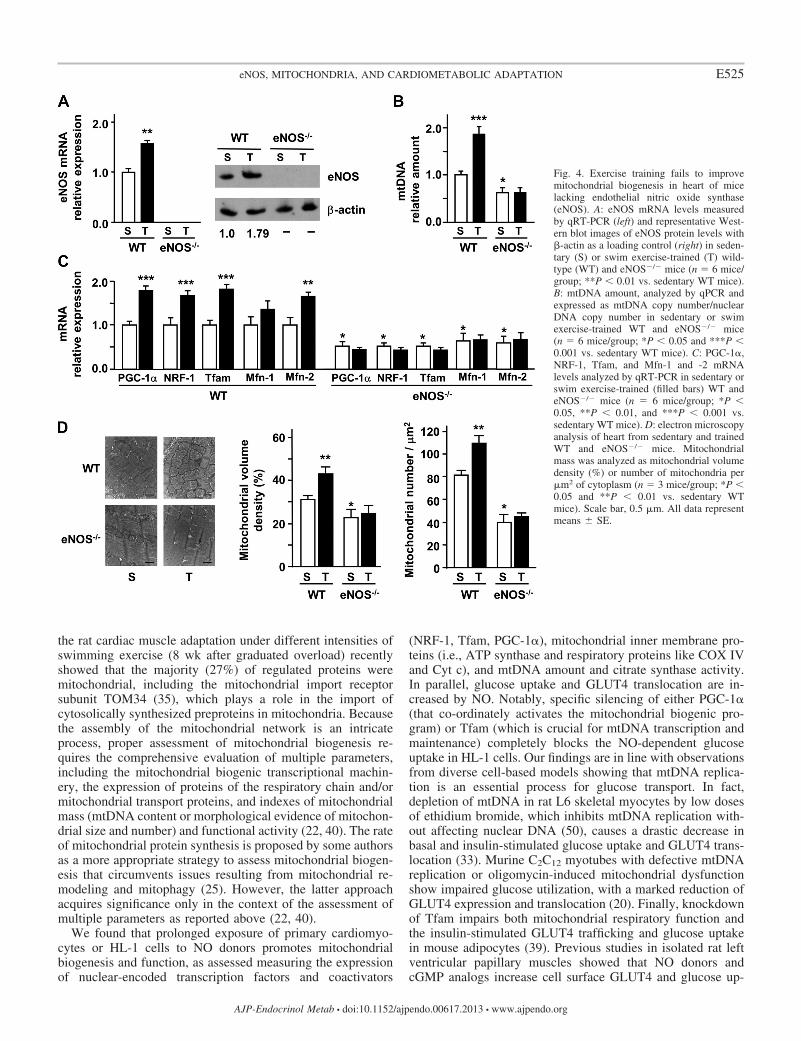
We found that eNOS mRNA and protein levels are signifi-cantly higher in left ventricle of swim-trained than in sedentaryWT mice (Fig. 4A). Increased eNOS expression was accom-panied by increased expression of PGC-1�, NRF-1, Tfam, andMfn-2 and by increased mtDNA content (Fig. 4, B and C).Mitochondrial biogenesis parameters were lower in left ven-tricle of sedentary eNOS�/� than sedentary WT animals (Fig.4, B and C) as described in other tissues (28, 29). Interestingly,the endurance exercise training failed to increase mitochondrialbiogenesis in left ventricle of eNOS�/� mice (Fig. 4, B and C).Electron microscopy showed smaller and fewer mitochondriawith altered cristae organization in left ventricles of eNOS�/�
mice compared with WT mice. Moreover, lipid droplets werepresent among mitochondria in knockout mice (Fig. 4D). Themorphometric analysis confirmed that, in the cardiomyocytesof the left ventricle, mitochondrial mass (mitochondrial vol-ume density and number) was lower in sedentary eNOS�/�
than in sedentary WT animals. Although mitochondrial volumedensity and number were increased by exercise training insedentary WT mice (Fig. 4D), these parameters did not differbetween exercise-trained and sedentary eNOS�/� mice (Fig.4D). These results further support our previous finding that 4wk treadmill exercise training increases mitochondrial biogen-esis and function in left ventricles of WT but not eNOS�/�
middle-aged mice (10), demonstrating the necessary role ofeNOS-NO signals in the heart mitochondrial biogenic responseto different types of endurance training.
Exercise training does not improve basal and insulin-stim-ulated glucose uptake in the heart of eNOS�/� mice. Becausewe found that mitochondrial biogenesis is an essential process
Fig. 1. The nitric oxide (NO) donor diethyl-enetriamine-NO (DETA-NO) increases mi-tochondrial biogenesis in mouse cardiomyo-cytes. A: primary cardiomyocytes weretreated with vehicle or 100 �M DETA-NO(filled bars) for 24 h. Levels of mRNA wereanalyzed by means of quantitative reversetranscription-PCR (qRT-PCR), with relativeexpression values of vehicle-treated cellstaken as 1.0 (n � 3 experiments; *P 0.05and **P 0.01). Mitochondrial DNA(mtDNA) was analyzed by quantitative PCR(qPCR) and expressed as mtDNA copy num-ber per nuclear DNA copy number (n � 3experiments; *P 0.05). Citrate synthaseactivity was expressed as fold-change vs.vehicle-treated cells taken as 1.0 (n � 3experiments; *P 0.05). NRF, nuclear re-spiratory factor-1; Tfam, mitochondrial tran-scription factor A. B: HL-1 cells were treatedeither with vehicle (filled bars) or 100 �MDETA-NO for 72 h, and mRNA levels wereanalyzed as reported above (n � 5 experi-ments; *P 0.05, **P 0.01, and ***P 0.001). ATPase, F1F0-ATP synthase; Mfn,mitofusin. C: peroxisome proliferator-acti-vated receptor � coactivator 1� (PGC-1�),cytochrome c oxidase IV (COX IV), andcytochrome c (Cyt c) proteins were detectedin HL-1 cells by immunoblot analysis. Oneexperiment representative of five reproduc-ible ones is shown, with values of densito-metric analysis referred to GAPDH as aloading control (n � 5 experiments; *P 0.05, **P 0.01, and ***P 0.001).D: mtDNA amount in vehicle (Veh)- orDETA-NO-treated HL-1 cells analyzed byqPCR (n � 5 experiments; *P 0.05).Citrate synthase activity was measured inHL-1 cells as above (n � 3 experiments;*P 0.05 and ***P 0.001). All datarepresent means � SE.
E522 eNOS, MITOCHONDRIA, AND CARDIOMETABOLIC ADAPTATION
AJP-Endocrinol Metab • doi:10.1152/ajpendo.00617.2013 • www.ajpendo.org
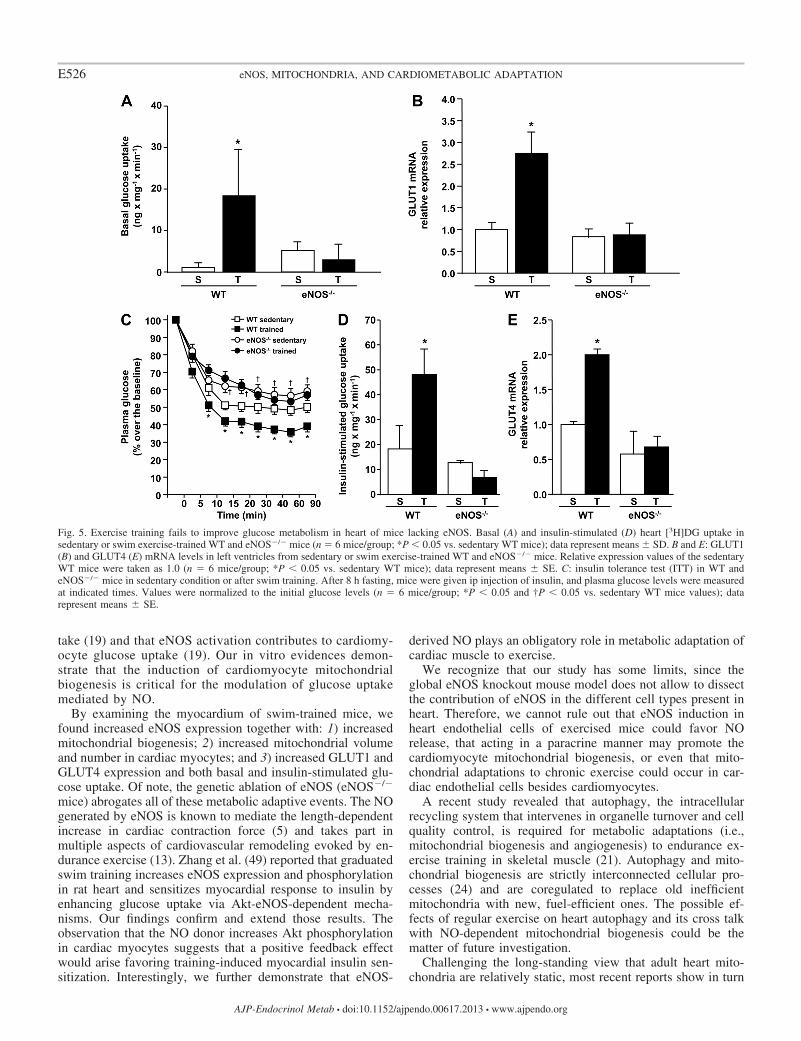
for the cardiomyocyte glucose uptake adaptation in vitro, andeNOS-derived NO is a critical mediator of cardiac-adaptivemitochondrial biogenesis, we assessed glucose metabolism inleft ventricles of sedentary or trained WT and eNOS�/� mice.Basal glucose uptake was greatly increased after exercisetraining in WT mice (Fig. 5A). On the contrary, it was notaffected by chronic exercise in knockout mice (Fig. 5A). Toconfirm the relevance of eNOS in basal glucose uptake, weexamined the content of the GLUT1 (insulin-independent)glucose transporter in left ventricle. Unlike WT mice, whichdisplayed a significant increase of cardiac GLUT1 expressionafter exercise training, eNOS�/� mice showed unmodifiedcardiac GLUT1 content (Fig. 5B).
The whole body glucose uptake was then measured afterinsulin perfusion. The ITT showed higher insulin sensitivity,resulting in lower plasma glucose levels, in sedentary WT thanin sedentary eNOS�/� mice (Fig. 5C). Exercise training in-duced a marked increase in whole body insulin sensitivity inWT mice, with a higher drop in plasma glucose levels com-pared with sedentary WT mice (Fig. 5C). The insulin-sensitiz-ing effect of exercise training was not found in eNOS�/� mice(Fig. 5C). Next, we measured the insulin-dependent glucoseuptake in the left ventricle. This was markedly increased byexercise training in WT mice but remained unchanged intrained eNOS�/� mice (Fig. 5D) Consistently, GLUT4 mRNAlevels were increased by exercise training in left ventricle of
WT unlike eNOS�/� mice (Fig. 5E). GLUT4 translocation,measured as membrane fraction of GLUT4, behaves similarly(data not shown). These results demonstrate that signals gen-erated by eNOS are necessary for the adaptive modifications ofglucose transport in the heart of exercise-trained mice.
DISCUSSION
Cellular and molecular mechanisms underlying the cardio-metabolic benefits associated with regular exercise remainlargely unresolved. Exercise training improves cardiac insulinsensitivity by modulating GLUT4 translocation to the plasmamembrane and increasing glucose uptake in the myocardium(49). NO has been suggested to play important roles in theseprocesses (49). The novelty of our study lies in three mainaspects. First, we demonstrate that mitochondrial biogenesis iskey in the regulation of cardiomyocyte glucose uptake by NOboth at baseline and under insulin-stimulated conditions. Sec-ond, the eNOS-derived NO is necessary to improve mitochon-drial biogenesis and glucose mobilization in trained hearts.Third, the demonstration that mitochondrial volume densityand number is increased in cardiac myocytes from trained miceundermines the general perception that heart cannot augmentits mitochondrial content because of the large share of cardi-omyocyte volume occupied by the organelles to maintain itscontinuous mechanical work.
Fig. 2. DETA-NO increases protein kinase B(Akt) phosphorylation and glucose transportin HL-1 cardiomyocytes. A: 2-deoxy-D-[1-3H]glucose ([3H]DG) uptake was measuredin vehicle (�)- or 100 �M DETA-NO-treated cells in the presence or absence of300 nM insulin (n � 3 experiments; *P 0.05 and ***P 0.001). B: Akt phosphor-ylation in HL-1 cells. Representative immu-noblots of three experiments run in dupli-cate. Bar graphs show the densitometricquantification [phosphorylated (p) Akt nor-malized to total Akt] with vehicle valuestaken as 1.0. *P 0.05. C: representativeimages of GLUT4 plasma membrane local-ization in vehicle (veh)- or 100 �M DETA-NO-treated cells in basal or insulin-stimu-lated conditions. Right, quantification of themembrane/cytosol fluorescence ratio (n � 3experiments; *P 0.05 and ***P 0.001).All data represent means � SE.
E523eNOS, MITOCHONDRIA, AND CARDIOMETABOLIC ADAPTATION
AJP-Endocrinol Metab • doi:10.1152/ajpendo.00617.2013 • www.ajpendo.org
Only a few studies have so far addressed the effects ofexercise training on rodent cardiac mitochondrial ultrastruc-ture/density, with controversial results probably depending onexercise intensity (3, 4, 32). Applying a morphometric analysison electron microscopy specimens, we found evidence ofincreased mitochondrial size and number in cardiac myocytesof swim-trained WT but not eNOS�/� mice. Biphasic changes
have been described in PGC-1�, Tfam, and Cyt c proteins,with decreased levels by 5-day and increased levels by 10-daytreadmill running in left ventricles of female rats (45). A14-day swim training with a ramp protocol recently demon-strated the activation of a metabolic gene set, includingPGC-1� and its downstream targets in the heart (6), includingthe respiratory chain NADH dehydrogenase (Ndufs2 andNdufv2 subunits) and the F1F0-ATP synthase (ATP5o subunit)that interestingly plays a role in maturation of mitochondrialcristae (8). Furthermore, a proteomics analysis investigating
Fig. 3. PGC-1� and Tfam small-interference RNA (siRNA) abolish the stimulatory effect of 100 �M DETA-NO on glucose uptake in HL-1 cardiomyocytes.A: PGC-1� and Tfam mRNA levels measured by qRT-PCR in HL-1 cells transfected with either siRNA or nontargeting (NT) siRNA. Relative expression valuesof the vehicle-treated, NT siRNA-transfected cells were taken as 1.0 (n � 3 experiments; *P 0.05 and **P 0.01 vs. vehicle/NT siRNA cells; †††P 0.05vs. corresponding NT siRNA cells). Insets: representative Western blots (one of three experiments) of PGC-1� and Tfam proteins with GAPDH as a loadingcontrol. B: mtDNA amount in HL-1 cells transfected with either Tfam siRNA or NT siRNA. Values of the vehicle/NT siRNA cells were taken as 1.0 (n � 3experiments; *P 0.05 vs. vehicle/NT siRNA cells; †††P 0.001 vs. corresponding NT siRNA cells). C: [3H]DG uptake in HL-1 cells transfected with eitherPGC-1� (left) or Tfam (right) siRNA (n � 3 experiments; *P 0.05 and ***P 0.001; §P 0.05, §§P 0.01, and §§§P 0.001 vs. corresponding NTsiRNA cells). All data represent means � SE.
Table 2. Mice body weight, food intake, and biochemicalparameters
WT eNOS�/�
Sedentary Trained Sedentary Trained
Body wt, g 26.6 � 0.7 24.8 � 0.4 25.6 � 1.0 23.8 � 1.1Food intake, g/day 2.98 � 0.19 3.01 � 0.27 2.87 � 0.25 2.76 � 0.22Fasting insulin,
�IU/ml 18.3 � 2.0 21.1 � 2.3 29.3 � 2.5* 29.8 � 3.3Fasting glucose,
mmol/l 4.5 � 0.6 4.3 � 0.7 4.4 � 0.5 4.5 � 0.9
Data are means � SE. Body weight and food intake were measured at theend of the training period. Blood was collected after an 8-h fast in sedentaryand swim exercise-trained wild-type (WT) and eNOS�/� mice (n � 6 mice/group). *P 0.05 vs. sedentary WT mice.
Table 3. Mice plasma free fatty acid levels
WT eNOS�/�
Basal Insulin Basal Insulin
Sedentary 1,780 � 98 1,508 � 138 2,406 � 313a 2,350 � 110Trained 1,574 � 157 871 � 116b 2,376 � 368 1,713 � 200
Data are means � SE. Plasma free fatty acids levels (�eq/l) were measuredin basal conditions or 60 min after the ip injection of insulin (0.5 U/kg bodywt) in sedentary and swim exercise-trained WT and eNOS�/� mice (n � 6mice/group). aP 0.05 vs. correspondent WT values; bP 0.05 vs. corre-spondent basal values.
E524 eNOS, MITOCHONDRIA, AND CARDIOMETABOLIC ADAPTATION
AJP-Endocrinol Metab • doi:10.1152/ajpendo.00617.2013 • www.ajpendo.org
the rat cardiac muscle adaptation under different intensities ofswimming exercise (8 wk after graduated overload) recentlyshowed that the majority (27%) of regulated proteins weremitochondrial, including the mitochondrial import receptorsubunit TOM34 (35), which plays a role in the import ofcytosolically synthesized preproteins in mitochondria. Becausethe assembly of the mitochondrial network is an intricateprocess, proper assessment of mitochondrial biogenesis re-quires the comprehensive evaluation of multiple parameters,including the mitochondrial biogenic transcriptional machin-ery, the expression of proteins of the respiratory chain and/ormitochondrial transport proteins, and indexes of mitochondrialmass (mtDNA content or morphological evidence of mitochon-drial size and number) and functional activity (22, 40). The rateof mitochondrial protein synthesis is proposed by some authorsas a more appropriate strategy to assess mitochondrial biogen-esis that circumvents issues resulting from mitochondrial re-modeling and mitophagy (25). However, the latter approachacquires significance only in the context of the assessment ofmultiple parameters as reported above (22, 40).
We found that prolonged exposure of primary cardiomyo-cytes or HL-1 cells to NO donors promotes mitochondrialbiogenesis and function, as assessed measuring the expressionof nuclear-encoded transcription factors and coactivators
(NRF-1, Tfam, PGC-1�), mitochondrial inner membrane pro-teins (i.e., ATP synthase and respiratory proteins like COX IVand Cyt c), and mtDNA amount and citrate synthase activity.In parallel, glucose uptake and GLUT4 translocation are in-creased by NO. Notably, specific silencing of either PGC-1�(that co-ordinately activates the mitochondrial biogenic pro-gram) or Tfam (which is crucial for mtDNA transcription andmaintenance) completely blocks the NO-dependent glucoseuptake in HL-1 cells. Our findings are in line with observationsfrom diverse cell-based models showing that mtDNA replica-tion is an essential process for glucose transport. In fact,depletion of mtDNA in rat L6 skeletal myocytes by low dosesof ethidium bromide, which inhibits mtDNA replication with-out affecting nuclear DNA (50), causes a drastic decrease inbasal and insulin-stimulated glucose uptake and GLUT4 trans-location (33). Murine C2C12 myotubes with defective mtDNAreplication or oligomycin-induced mitochondrial dysfunctionshow impaired glucose utilization, with a marked reduction ofGLUT4 expression and translocation (20). Finally, knockdownof Tfam impairs both mitochondrial respiratory function andthe insulin-stimulated GLUT4 trafficking and glucose uptakein mouse adipocytes (39). Previous studies in isolated rat leftventricular papillary muscles showed that NO donors andcGMP analogs increase cell surface GLUT4 and glucose up-
Fig. 4. Exercise training fails to improvemitochondrial biogenesis in heart of micelacking endothelial nitric oxide synthase(eNOS). A: eNOS mRNA levels measuredby qRT-PCR (left) and representative West-ern blot images of eNOS protein levels with-actin as a loading control (right) in seden-tary (S) or swim exercise-trained (T) wild-type (WT) and eNOS�/� mice (n � 6 mice/group; **P 0.01 vs. sedentary WT mice).B: mtDNA amount, analyzed by qPCR andexpressed as mtDNA copy number/nuclearDNA copy number in sedentary or swimexercise-trained WT and eNOS�/� mice(n � 6 mice/group; *P 0.05 and ***P 0.001 vs. sedentary WT mice). C: PGC-1�,NRF-1, Tfam, and Mfn-1 and -2 mRNAlevels analyzed by qRT-PCR in sedentary orswim exercise-trained (filled bars) WT andeNOS�/� mice (n � 6 mice/group; *P 0.05, **P 0.01, and ***P 0.001 vs.sedentary WT mice). D: electron microscopyanalysis of heart from sedentary and trainedWT and eNOS�/� mice. Mitochondrialmass was analyzed as mitochondrial volumedensity (%) or number of mitochondria per�m2 of cytoplasm (n � 3 mice/group; *P 0.05 and **P 0.01 vs. sedentary WTmice). Scale bar, 0.5 �m. All data representmeans � SE.
E525eNOS, MITOCHONDRIA, AND CARDIOMETABOLIC ADAPTATION
AJP-Endocrinol Metab • doi:10.1152/ajpendo.00617.2013 • www.ajpendo.org
take (19) and that eNOS activation contributes to cardiomy-ocyte glucose uptake (19). Our in vitro evidences demon-strate that the induction of cardiomyocyte mitochondrialbiogenesis is critical for the modulation of glucose uptakemediated by NO.
By examining the myocardium of swim-trained mice, wefound increased eNOS expression together with: 1) increasedmitochondrial biogenesis; 2) increased mitochondrial volumeand number in cardiac myocytes; and 3) increased GLUT1 andGLUT4 expression and both basal and insulin-stimulated glu-cose uptake. Of note, the genetic ablation of eNOS (eNOS�/�
mice) abrogates all of these metabolic adaptive events. The NOgenerated by eNOS is known to mediate the length-dependentincrease in cardiac contraction force (5) and takes part inmultiple aspects of cardiovascular remodeling evoked by en-durance exercise (13). Zhang et al. (49) reported that graduatedswim training increases eNOS expression and phosphorylationin rat heart and sensitizes myocardial response to insulin byenhancing glucose uptake via Akt-eNOS-dependent mecha-nisms. Our findings confirm and extend those results. Theobservation that the NO donor increases Akt phosphorylationin cardiac myocytes suggests that a positive feedback effectwould arise favoring training-induced myocardial insulin sen-sitization. Interestingly, we further demonstrate that eNOS-
derived NO plays an obligatory role in metabolic adaptation ofcardiac muscle to exercise.
We recognize that our study has some limits, since theglobal eNOS knockout mouse model does not allow to dissectthe contribution of eNOS in the different cell types present inheart. Therefore, we cannot rule out that eNOS induction inheart endothelial cells of exercised mice could favor NOrelease, that acting in a paracrine manner may promote thecardiomyocyte mitochondrial biogenesis, or even that mito-chondrial adaptations to chronic exercise could occur in car-diac endothelial cells besides cardiomyocytes.
A recent study revealed that autophagy, the intracellularrecycling system that intervenes in organelle turnover and cellquality control, is required for metabolic adaptations (i.e.,mitochondrial biogenesis and angiogenesis) to endurance ex-ercise training in skeletal muscle (21). Autophagy and mito-chondrial biogenesis are strictly interconnected cellular pro-cesses (24) and are coregulated to replace old inefficientmitochondria with new, fuel-efficient ones. The possible ef-fects of regular exercise on heart autophagy and its cross talkwith NO-dependent mitochondrial biogenesis could be thematter of future investigation.
Challenging the long-standing view that adult heart mito-chondria are relatively static, most recent reports show in turn
Fig. 5. Exercise training fails to improve glucose metabolism in heart of mice lacking eNOS. Basal (A) and insulin-stimulated (D) heart [3H]DG uptake insedentary or swim exercise-trained WT and eNOS�/� mice (n � 6 mice/group; *P 0.05 vs. sedentary WT mice); data represent means � SD. B and E: GLUT1(B) and GLUT4 (E) mRNA levels in left ventricles from sedentary or swim exercise-trained WT and eNOS�/� mice. Relative expression values of the sedentaryWT mice were taken as 1.0 (n � 6 mice/group; *P 0.05 vs. sedentary WT mice); data represent means � SE. C: insulin tolerance test (ITT) in WT andeNOS�/� mice in sedentary condition or after swim training. After 8 h fasting, mice were given ip injection of insulin, and plasma glucose levels were measuredat indicated times. Values were normalized to the initial glucose levels (n � 6 mice/group; *P 0.05 and †P 0.05 vs. sedentary WT mice values); datarepresent means � SE.
E526 eNOS, MITOCHONDRIA, AND CARDIOMETABOLIC ADAPTATION
AJP-Endocrinol Metab • doi:10.1152/ajpendo.00617.2013 • www.ajpendo.org

that they retain their ability to undergo fission and fusion,suggesting the relevance of mitochondrial dynamics in heartpathophysiology and cardioprotection (31). Accordingly, to-gether with evidence of ultrastructural changes in cardiomyo-cyte mitochondria, we found that exercise increases cardiacexpression of Mfn-2, an essential regulator of fusion andbiogenesis in the mitochondrial network, in an eNOS-depen-dent manner. Thus eNOS-derived NO might take part inmultiple PGC-1�-dependent mitochondrial adaptations to ex-ercise (i.e., mitochondrial biogenesis and fusion) in cardiacmyocytes, in analogy to what has been observed in the aorta oftrained mice (26).
Most recently, Kasahara et al. (16) reported that mitochon-drial fusion directs cardiomyocyte differentiation. Accord-ingly, mitochondrial activity is required for the differentiationand maturation in cardiomyocytes of a cardiac stem cell pop-ulation, and this phenomenon was increased by NO (36).Ongoing studies suggest that eNOS-mediated mechanisms areimplicated in swimming exercise-evoked activation of endog-enous cardiac stem/progenitor cells (13). The role of eNOS-mediated mitochondrial biogenesis in these phenomena takingpart in the beneficial adaptation of heart to exercise remains tobe investigated.
It is worth to note at this point that, different from physio-logical cardiac hypertrophy in response to exercise training,pathological cardiac hypertrophy resulting from transaorticconstriction is associated with reduced PGC-1� expression (6).This suggests that increasing the cardiomyocyte energetics viaexercise-induced PGC-1� cascade could be investigated as atherapeutic approach to improve the functional resilience tocardiac dysfunction. Interestingly, exercise training in mice canreverse the contractile abnormalities associated with diabeticcardiomyopathy (41), a condition characterized by mitochon-drial dysfunction in cardiomyocytes (11) and derangements infuel utilization because of GLUT4 depletion (7).
In summary, our results provide novel insights into themechanisms governing heart glucose homeostasis after exer-cise training, further supporting the health effects of the NO-dependent mitochondrial biogenesis (10, 42). The effects ofexercise training on metabolic cardiac adaptation mediated bythe eNOS signaling system are worth research also in patho-logical conditions and in clinical settings.
GRANTS
This work was supported by the Ministero dell’Istruzione, dell’Universita edella Ricerca (Italy), Grants 2009E48P9M_001 (E. Nisoli), 2009E48P9M_003(A. Valerio), and 2007BRR57M_004 (R. Vettor) and the CARIPARO Foun-dation (R. Vettor). L. Tedesco received a research contract cofinanced byNicOx Research Institute and Regione Lombardia.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.V. and E.N. contributed to the conception and design of the research;R.V., M.R., E.T., and E.N. analyzed the data; R.V., A.V., and E.N. interpretedthe results of the experiments; R.V., A.V., E.T., and E.N. drafted the manu-script; R.V., A.V., M.O.C., and E.N. edited and revised the manuscript; R.V.,A.V., M.O.C., and E.N. approved the final version of the manuscript; A.V. andE.T. prepared the figures; M.R., E.T., M.G., M.O., L.T., C.R., A.F., R.F., andR.S. performed the experiments.
REFERENCES
1. Abel ED, Doenst T. Mitochondrial adaptations to physiological vs.pathological cardiac hypertrophy. Cardiovasc Res 90: 234–242, 2011.
2. Al-Sa’doni HH, Khan IY, Poston L, Fisher I, Ferro A. A novel familyof S-nitrosothiols: chemical synthesis and biological actions. Nitric Oxide4: 550–560, 2000.
3. Arcos JC, Sohal RS, Sun SC, Argus MF, Burch GE. Changes inultrastructure and respiratory control in mitochondria of rat heart hyper-trophied by exercise. Exp Mol Pathol 8: 49–65, 1968.
4. Ascensão A, Ferreira R, Magalhães J. Exercise-induced cardioprotec-tion–biochemical, morphological and functional evidence in whole tissueand isolated mitochondria. Int J Cardiol 117: 16–30, 2007.
5. Balligand JL, Feron O, Dessy C. eNOS activation by physical forces:from short-term regulation of contraction to chronic remodeling of car-diovascular tissues. Physiol Rev 89: 481–534, 2009.
6. Boström P, Mann N, Wu J, Quintero PA, Plovie ER, Panáková D,Gupta RK, Xiao C, MacRae CA, Rosenzweig A, Spiegelman BM.C/EBP controls exercise-induced cardiac growth and protects againstpathological cardiac remodeling. Cell 143: 1072–1083, 2010.
7. Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. RevEndocr Metab Disord 11: 31–39, 2010.
8. Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR,Abramov AY, Tinker A, Duchen MR. Regulation of mitochondrialstructure and function by the F1F0-ATPase inhibitor protein, IF1. CellMetab 8: 13–25, 2008.
9. Claycomb WC, Lanson NA, Stallworth BS, Egeland DB, DelcarpioJB, Bahinski A, Izzo NJ. HL-1 cells: a cardiac muscle cell line thatcontracts and retains phenotypic characteristics of the adult cardiomyo-cyte. Proc Natl Acad Sci USA 95: 2979–2984, 1998.
10. D’Antona G, Ragni M, Cardile A, Tedesco L, Dossena M, Bruttini F,Caliaro F, Corsetti G, Bottinelli R, Carruba MO, Valerio A, Nisoli E.Branched-chain amino acid supplementation promotes survival and sup-ports cardiac and skeletal muscle mitochondrial biogenesis in middle-agedmice. Cell Metab 12: 362–372, 2010.
11. Dabkowski ER, Baseler WA, Williamson CL, Powell M, RazunguzwaTT, Frisbee JC, Hollander JM. Mitochondrial dysfunction in the type 2diabetic heart is associated with alterations in spatially distinct mitochon-drial proteomes. Am J Physiol Heart Circ Physiol 299: H529–H540, 2010.
12. Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M,Vollenweider P, Pedrazzini T, Nicod P, Thorens B, Scherrer U. Insulinresistance, hyperlipidemia, and hypertension in mice lacking endothelialnitric oxide synthase. Circulation 104: 342–345, 2001.
13. Ellison GM, Waring CD, Vicinanza C, Torella D. Physiological cardiacremodelling in response to endurance exercise training: cellular andmolecular mechanisms. Heart 98: 5–10, 2012.
14. Fabris R, Nisoli E, Lombardi AM, Tonello C, Serra R, Granzotto M,Cusin I, Rohner-Jeanrenaud F, Federspil G, Carruba MO, Vettor R.Preferential channeling of energy fuels toward fat rather than muscleduring high free fatty acid availability in rats. Diabetes 50: 601–608, 2001.
15. Holloszy JO. Regulation by exercise of skeletal muscle content of mito-chondria and GLUT4. J Physiol Pharmacol 59, Suppl 7: 5–18, 2008.
16. Kasahara A, Cipolat S, Chen Y, Dorn GW, Scorrano L. Mitochondrialfusion directs cardiomyocyte differentiation via calcineurin and notchsignaling. Science 342: 734–737, 2013.
17. Keefer LK. Progress toward clinical application of the nitric oxide-releasing diazeniumdiolates. Annu Rev Pharmacol Toxicol 43: 585–607,2003.
18. Kojda G, Cheng YC, Burchfield J, Harrison DG. Dysfunctional regu-lation of endothelial nitric oxide synthase (eNOS) expression in responseto exercise in mice lacking one eNOS gene. Circulation 103: 2839–2844,2001.
19. Li J, Hu X, Selvakumar P, Russell RR, Cushman SW, Holman GD,Young LH. Role of the nitric oxide pathway in AMPK-mediated glucoseuptake and GLUT4 translocation in heart muscle. Am J Physiol Endocri-nol Metab 287: E834–E841, 2004.
20. Lim JH, Lee JI, Suh YH, Kim W, Song JH, Jung MH. Mitochondrialdysfunction induces aberrant insulin signalling and glucose utilisation inmurine C2C12 myotube cells. Diabetologia 49: 1924–1936, 2006.
21. Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS,Hoehn KL, Yan Z. Autophagy is required for exercise training-inducedskeletal muscle adaptation and improvement of physical performance.FASEB J 27: 4184–4193, 2013.
E527eNOS, MITOCHONDRIA, AND CARDIOMETABOLIC ADAPTATION
AJP-Endocrinol Metab • doi:10.1152/ajpendo.00617.2013 • www.ajpendo.org

22. Medeiros DM. Assessing mitochondria biogenesis. Methods 46: 288–294, 2008.
23. Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Leh-man JJ, Kelly DP, Spiegelman BM. Restoration of insulin-sensitiveglucose transporter (GLUT4) gene expression in muscle cells by thetranscriptional coactivator PGC-1. Proc Natl Acad Sci USA 98: 3820–3825, 2001.
24. Michel S, Wanet A, De Pauw A, Rommelaere G, Arnould T, RenardP. Crosstalk between mitochondrial (dys)function and mitochondrialabundance. J Cell Physiol 227: 2297–2310, 2012.
25. Miller BF, Hamilton KL. A perspective on the determination of mito-chondrial biogenesis. Am J Physiol Endocrinol Metab 302: E496–E499,2012.
26. Miller MW, Knaub LA, Olivera-Fragoso LF, Keller AC, Balasubra-maniam V, Watson PA, Reusch JE. Nitric oxide regulates vascularadaptive mitochondrial dynamics. Am J Physiol Heart Circ Physiol 304:H1624–H1633, 2013.
27. Nisoli E, Clementi E, Carruba MO, Moncada S. Defective mitochon-drial biogenesis: a hallmark of the high cardiovascular risk in the meta-bolic syndrome? Circ Res 100: 795–806, 2007.
28. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C,Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO.Mitochondrial biogenesis in mammals: the role of endogenous nitricoxide. Science 299: 896–899, 2003.
29. Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M,Pisconti A, Brunelli S, Cardile A, Francolini M, Cantoni O, CarrubaMO, Moncada S, Clementi E. Mitochondrial biogenesis by NO yieldsfunctionally active mitochondria in mammals. Proc Natl Acad Sci USA101: 16507–16512, 2004.
30. Ojaimi C, Li W, Kinugawa S, Post H, Csiszar A, Pacher P, Kaley G,Hintze TH. Transcriptional basis for exercise limitation in male eNOS-knockout mice with age: heart failure and the fetal phenotype. Am JPhysiol Heart Circ Physiol 289: H1399–H1407, 2005.
31. Ong SB, Hall AR, Hausenloy DJ. Mitochondrial dynamics in cardiovas-cular health and disease. Antioxid Redox Signal 19: 400–414, 2013.
32. Oscai LB, Molé PA, Brei B, Holloszy JO. Cardiac growth and respira-tory enzyme levels in male rats subjected to a running program. Am JPhysiol 220: 1238–1241, 1971.
33. Park SY, Choi GH, Choi HI, Ryu J, Jung CY, Lee W. Depletion ofmitochondrial DNA causes impaired glucose utilization and insulin resis-tance in L6 GLUT4myc myocytes. J Biol Chem 280: 9855–9864, 2005.
34. Rimbaud S, Garnier A, Ventura-Clapier R. Mitochondrial biogenesisin cardiac pathophysiology. Pharmacol Rep 61: 131–138, 2009.
35. Rocha LA, Petriz BA, Borges DH, Oliveira RJ, de Andrade RV,Domont GB, Pereira RW, Franco OL. High molecular mass proteomicsanalyses of left ventricle from rats subjected to differential swimmingtraining (Abstract). BMC Physiol 12: 11, 2012.
36. San Martin N, Cervera AM, Cordova C, Covarello D, McCreath KJ,Galvez BG. Mitochondria determine the differentiation potential of car-diac mesoangioblasts. Stem Cells 29: 1064–1074, 2011.
37. Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mito-chondrial biogenesis. Trends Endocrinol Metab 23: 459–466, 2012.
38. Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE,Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in micelacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA 93:13176–13181, 1996.
39. Shi X, Burkart A, Nicoloro SM, Czech MP, Straubhaar J, Corvera S.Paradoxical effect of mitochondrial respiratory chain impairment on in-sulin signaling and glucose transport in adipose cells. J Biol Chem 283:30658–30667, 2008.
40. Short KR. Measuring mitochondrial protein synthesis to assess biogene-sis. Am J Physiol Endocrinol Metab 302: E1153–E1154, 2012.
41. Stølen TO, Høydal MA, Kemi OJ, Catalucci D, Ceci M, Aasum E,Larsen T, Rolim N, Condorelli G, Smith GL, Wisløff U. Intervaltraining normalizes cardiomyocyte function, diastolic Ca2� control, andSR Ca2� release synchronicity in a mouse model of diabetic cardiomy-opathy. Circ Res 105: 527–536, 2009.
42. Tedesco L, Valerio A, Cervino C, Cardile A, Pagano C, Vettor R,Pasquali R, Carruba MO, Marsicano G, Lutz B, Pagotto U, Nisoli E.Cannabinoid type 1 receptor blockade promotes mitochondrial biogenesisthrough endothelial nitric oxide synthase expression in white adipocytes.Diabetes 57: 2028–2036, 2008.
43. Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, SwitzerCH, Donzelli S, Hussain P, Vecoli C, Paolocci N, Ambs S, Colton CA,Harris CC, Roberts DD, Wink DA. The chemical biology of nitricoxide: implications in cellular signaling. Free Radic Biol Med 45: 18–31,2008.
44. Vettor R, Zarjevski N, Cusin I, Rohner-Jeanrenaud F, Jeanrenaud B.Induction and reversibility of an obesity syndrome by intracerebroventric-ular neuropeptide Y administration to normal rats. Diabetologia 37:1202–1208, 1994.
45. Watson PA, Reusch JE, McCune SA, Leinwand LA, Luckey SW,Konhilas JP, Brown DA, Chicco AJ, Sparagna GC, Long CS, MooreRL. Restoration of CREB function is linked to completion and stabiliza-tion of adaptive cardiac hypertrophy in response to exercise. Am J PhysiolHeart Circ Physiol 293: H246–H259, 2007.
46. Watson RT, Pessin JE. Bridging the GAP between insulin signaling andGLUT4 translocation. Trends Biochem Sci 31: 215–222, 2006.
47. White SM, Constantin PE, Claycomb WC. Cardiac physiology at thecellular level: use of cultured HL-1 cardiomyocytes for studies of cardiacmuscle cell structure and function. Am J Physiol Heart Circ Physiol 286:H823–H829, 2004.
48. Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, Boström P,Tyra HM, Crawford RW, Campbell KP, Rutkowski DT, KaufmanRJ, Spiegelman BM. The unfolded protein response mediates adaptationto exercise in skeletal muscle through a PGC-1�/ATF6� complex. CellMetab 13: 160–169, 2011.
49. Zhang QJ, Li QX, Zhang HF, Zhang KR, Guo WY, Wang HC, ZhouZ, Cheng HP, Ren J, Gao F. Swim training sensitizes myocardialresponse to insulin: role of Akt-dependent eNOS activation. CardiovascRes 75: 369–380, 2007.
50. Zylber E, Vesco C, Penman S. Selective inhibition of the synthesis ofmitochondria-associated RNA by ethidium bromide. J Mol Biol 44:195–204, 1969.
E528 eNOS, MITOCHONDRIA, AND CARDIOMETABOLIC ADAPTATION
AJP-Endocrinol Metab • doi:10.1152/ajpendo.00617.2013 • www.ajpendo.org