electrosynthesis and spectroelectrochemical characterization of poly(3,4-dimethoxy-thiophene),...
TRANSCRIPT

Electrosynthesis and spectroelectrochemical characterization ofpoly(3,4-dimethoxy-thiophene), poly(3,4-dipropyloxythiophene) and
poly(3,4-dioctyloxythiophene) films
Artur Szkurlat, Barbara Palys 1, Jozef Mieczkowski, Magdalena Skompska1 *
Department of Chemistry, Warsaw University, ul. Pasteura 1, 02 093 Warsaw, Poland
Received 4 June 2003; received in revised form 4 June 2003
Electrochimica Acta 48 (2003) 3665�/3676
www.elsevier.com/locate/electacta
Abstract
Poly(3,4-dialkoxythiophene) films with different length of alkyl chain (1,3 and 8 carbon atoms) were obtained on Pt and ITO
electrodes from the monomer solutions in acetonitrile by cyclic voltammetry (CV). The properties of the resulting films were studied
by electrochemical methods, UV�/Vis, FTIR and NMR spectra. The CVs were correlated with differential cyclic voltabsorptograms
(DCVA) recorded at the absorption maxima to explain the shape of the voltammograms of the polymers studied, dependent on the
alkyl-chain length in alkoxy group. The presence of the zones of different crystallinity in the polymer film was postulated. Significant
influence of the type of the solvent on asymmetry of the cyclic voltammograms for the polymer doping�/undoping has been
discussed in terms of the solvent interaction with radical cation (polaron) delocalized on the alkoxy side groups. The polaron
delocalization was proved by 1H-NMR spectra. Appearance of infrared activated vibrations (IRAVs) in the range 1500�/600 cm�1
and a characteristic electronic band at 3300 cm�1 at the polarization potential �/0.25 V versus Ag/Ag� and their gradual changes
upon further polymer oxidation were interpreted in terms of evolution of different charge carriers in lightly and heavily doped
polymer.
# 2003 Elsevier Ltd. All rights reserved.
Keywords: Poly(3,4-dialkoxythiophene); Electropolymerization; Redox behavior; NMR; UV�/Vis; IR spectra; Infrared activated vibration bands
1. Introduction
Polythiophene and its derivatives belong to attractive
group of polymers due to their high conductivity in the
oxidized form and electrochemical stability in doped
and undoped states. Substitution of the polythiophene
ring at b position with flexible alkyl, or alkyl containing
one or more ether groups leads to the conducting
polymers of unusual properties such as processability
or stereo- and ionoselectivity [1�/3]. Introduction of
electron-donating alkoxy substituent into the thiophene
ring results also in diminution of polymerization poten-
tial and significant increase of the polymer electroactiv-
ity in aqueous solution, in comparison with that of
poly(3-alkylthiophene) analogues [4,5]. The electroche-
mical properties of substituted thiophene depend on the
number of oxygen atoms in the side chain and their
position. If the oxygen is directly attached to the ring,
the conducting p-doping state of the polymer is sub-
stantially stabilized by stabilization of the positive
charge in the polymer backbone [4,6�/8].
Incorporation of the second alkoxy substituent into
the thiophene ring or cyclization between the 3 and 4
positions of the thiophene ring is a convenient way for
preparing the perfectly stereoregular, long conjugated
polymers by elimination of 2,4? couplings, [9�/11].
Poly(3,4-ethylenedioxythiophene) (PEDOT) is an excel-
lent example of highly conducting polymer (of conduc-
tivity ca. 500 S cm�1), very stable and optical
transparent in the oxidized state [12]. These properties
make the polymer attractive as an electrode material in
rechargeable polymer batteries, capacitors, electrochro-
mic devices and suitable as antistatic coatings [13,14].
* Corresponding author. Tel.: �/48-22-822-0211; fax: �/48-22-822-
5996.
E-mail address: [email protected] (M. Skompska1).1 ISE member.
0013-4686/03/$ - see front matter # 2003 Elsevier Ltd. All rights reserved.
doi:10.1016/S0013-4686(03)00504-8

However, a low oxidation potential of PEDOT is
responsible for instability of the polymer in the neutral
state at ambient conditions. Another disadvantage of
PEDOT is its insolubility in organic solvents. Someimprovement of the polymer properties may be achieved
by the change of substituents in the monomer molecule.
The conductive and magnetic properties of PEDOT and
poly(3,4-dimethoxythiophene) have been recently com-
pared by Zotti et al. [15]. The authors found rather small
influence of the dimethoxy- or ethylenedioxy-substituent
on the electrochemical properties of these two polymers.
Nevertheless, one may expect that the increase of thealkyl chain length in the alkoxy group will lead to the
polymer soluble in aprotic organic solvents. On the
other hand, this may also result in some difficulties in
electrosynthesis and shortening of conjugation length of
the resultant polymers due to the steric hindrances
[10,11,16]. However, the results discussed in the litera-
ture concern the properties of the polymers obtained in
different ways (by electrochemical or chemical synthesis)which may strongly influence on the polymer character-
istics. Therefore, it was reasonable to make a complex
comparative studies on electropolymerization and spec-
troelectrochemical properties of a series of poly(3,4-
dialkoxythiophenes) with different alkyl-chain length
obtained from the monomers synthesized by the same
procedure and electropolymerized in the same condi-
tions. In this work we compare the results of threepolymers of this group, poly(3,4-dimethoxythiophene)
(PDMT), poly(3,4-dipropyloxy-thiophene) (PDPT) and
poly(3,4-dioctyloxythiophene) (PDOT), electrodepos-
ited by cyclic voltammetry from acetonitrile solutions
of the monomers. We also discuss some advantages of
these polymers with respect to PEDOT and monosub-
stituted poly(3-alkylthiophenes). The electrochemical
and spectroscopic properties of poly(3,4-dialkoxythio-phenes) were investigated by cyclic voltammetry, in-situ
UV�/Vis absorption spectra, differential cyclic voltab-
sorptommetry (DCVA), NMR and ex-situ FTIR reflec-
tance spectroscopies.
2. Experimental
2.1. Synthesis of 3,4-dialkoxythiophenes
Synthesis of 3,4-dialkoxythiophenes has been de-
scribed in the literature [11,16�/18]. In our procedure,
presented in Scheme 1, the starting compound was
disodium 2,3-di(ethoxy-carbonyl)-3,4-thiophenediolate
(1) obtained according to Hinsberg [19]. A direct
alkylation of disodium salt leads to high synthesis yield
of about 30%.Characterization of the product: m.p.: 29.5�/30.5 8C.
Elemental Anal. C20H36O2S: Calc.: C, 70.58; H, 10.58.
Found: C, 70.39; H, 10.78%. IR (KBr, cm�1): 2925,
2850, 1580, 1510, 1470, 1380, 1210, 1155, 750; 1H-NMR
(CDCl3, d ): 6.15 (s, 2H), 3.96 (t, 4H), 1.81 (m, 4H),
1.51}/1.20 (m, 10H), 0.88 (t, 6H). 13C-NMR (CDCl3,
d ): 147.6, 96.8, 70.6, 31.8, 29.4, 29.3, 29.0, 26.9, 26.0,22.7, 14.1.
The same procedure was used for synthesis of two
other monomers of this group, 3,4-dimethoxythiophene
and 3,4-dipropyloxythiophene.
2.2. Spectroscopic and electrochemical measurements
Infrared (IR) spectra of byproducts and final product
of the synthesis were obtained using a Nicolet Magna IR500 spectrophotometer. NMR spectra were recorded on
a Varian Unity Plus spectrometer operating at 200 MHz
for 1H-NMR and at 125 MHz for 13C-NMR. Tetra-
methylsilane was used as an internal standard. Chemical
shifts are reported in ppm. TLC analyses were per-
formed on a Merck 60 silica gel glass plated and
visualised using iodine vapour. Column chromatogra-
phy was carried out at atmospheric pressure using silicagel (100�/200 mesh, Merck). Elemental analyses were
performed in a Microanalytical Laboratory of the
Institute of Organic Chemistry, Polish Academy of
Science, Warsaw. The determined elemental composi-
tion of the compounds synthesised confirmed the
expected molecular structure.
All electrochemical experiments were done in a
conventional, single compartment cell with a platinumgauze counter electrode and Ag/(0.1M AgNO3 in
CH3CN) double junction reference electrode, using an
AUTOLAB potentiostat (Ecochemie, The Netherlands).
Electrodeposition of poly(3,4-dimethoxythiophene)
(PDMT), poly(3,4-dipropyloxy-thiophene) (PDPT) and
poly(3,4-dioctyloxythiophene) (PDOT) was carried out
by cyclic voltammetry on a Pt disc with the surface area
of 0.02 cm2 or ITO (indium-tin oxide) electrodes fromdeaerated (with Ar) 0.05 M monomer solutions in
acetonitrile (AN) containing 0.1 M LiClO4 as the
supporting electrolyte. After deposition, the polymer
films were rinsed with acetonitrile and then studied by
cyclic voltammetry, chronopotentiometry or chronoam-
perometry in deaerated AN, propylene carbonate (PC)
or aqueous monomer-free solutions of 0.1 M LiClO4.
The absorption spectra of the polymer films weretaken using a double-beam UV/Vis spectrometer
(Lambda 12, Perkin�/Elmer). The electrochemical cell
containing the ITO/polymer working electrode, Pt gauze
counter electrode and a miniature Ag/Ag� reference
electrode in the supporting electrolyte was mounted in
the spectrophotometer sample compartment. The refer-
ence cell contained an identical uncoated ITO electrode
in the same electrolyte. Infrared reflectance spectra ofthe polymer layers were recorded with a FTIR 8400
Shimadzu spectrometer equipped with a specular reflec-
tance accessory (Spectra Tech) at 808 angle of incidence.
A. Szkurlat et al. / Electrochimica Acta 48 (2003) 3665�/36763666

3. Results and discussion
3.1. Electrodeposition of PDMT, PDPT and PDOT
Fig. 1a�/c show the cyclic voltammograms for electro-
deposition of PDMT, PDPT and PDOT on Pt electro-
des from 0.05 M monomer solutions in acetonitrilecontaining 0.1 LiClO4 as the supporting electrolyte. The
oxidation potentials of the monomers, determined from
oxidation peaks, decrease slightly with the length of the
alkyl chain, from 1.15 V for DMT to 1.12 V for DPT
and 1.09 V for DOT. In order to avoid degradation of
the polymers during deposition, the films were obtained
by cycling in the limited potential range, to 1.05 V for
PDMT, 1.03 V for PDPT and 1.0 V for PDOT at the
scan rate of 40 mV s�1.
As visible in Fig. 1, a character of the redox peaks,
developed gradually in the subsequent polymerisation
Scheme 1. Synthesis route of 3,4-dioctyloxythiophene.
Fig. 1. Cyclic voltammograms for electrodeposition of PDMT (a), PDPT (b) and PDOT (c) films on Pt electrodes from acetonitrile containing 0.05
M monomer�/0.1 M LiClO4, at the scan rate 40 mV s�1.
A. Szkurlat et al. / Electrochimica Acta 48 (2003) 3665�/3676 3667

scans, depends markedly on the side chain length of the
monomer. The longer chain, the sharper and more
symmetrical are the redox peaks. To compare the
behavior of the three polymers upon doping�/undoping
in the monomer-free solution, one should provide the
films of similar thickness. However, a gradual colour
change of the solutions in the vicinity of the working
electrode during polymerization suggest that any side
reaction, for example formation of soluble oligomers,
occurs. Therefore, the charge passed during polymeriza-
tion could not be used to control the film thickness.
Another reason of the charge loses may be a slow film
growth in comparison with the coupling process of
radical cations. In effect, the polymer would be formed
not only on the electrode but also in the solution. The
presence of oligomers in the solutions of 3,4-DMT and
3,4-DPT after three polymerisation cycles was con-
firmed by UV�/Vis spectra, presented in Fig. 2 (spectra
1 and 2, respectively). The absorption peaks are located
at 460 nm i.e. at the wavelength markedly lower than
that of the peak for PDOT dissolved in chloroform (536
nm, spectrum 4), as expected for short chain oligomers.
The two other polymers are not soluble in CHCl3 due to
insufficient length of alkyl chain. No oligomers were
detected in the solution after three polymerisation cycles
in 3,4-DOT (spectrum 3).
For comparison, according to the literature, the
absorption peak for poly(3,4-dibutyloxythiophene)
polymerized chemically (by means of FeCl3) and dis-
solved in chloroform is located at 480 nm [16]. Since the
absorption peaks of the monomers are located at the
same wavelength, 262 nm (see Table 1), one may
conclude that the electrochemical polymerisation of
poly(3,4-dialkoxythiophenes) leads to the polymers
with markedly longer conjugation than these obtained
by the chemical synthesis (according to the procedure
described by Daoust and Leclerc [16]).
Oligomerization in the solution is responsible for
relatively low polymerisation efficiency, h , determinedfrom EQCM experiments. The value of h for PDMT,
derived from the graph of Df versus Qdep, where Df is
the change of the resonant frequency of the quartz
resonator upon the passage of the deposition charge
Qdep, is 16%, being lower than that reported by Zotti et
al. (23%) [15].
Another factor, useful in estimation of the charge
loses during polymerisation is a charge yield, i.e. a ratioof the redox and deposition charges of the polymer, y�/
Qred/Qdep. These two charges are defined as Qred�/
zredFN and Qdep�/(2�/zred)FN(h)�1, where F is the
Faraday constant, zred is a number of electrons involved
in redox reaction of one monomer unit, N is a number
of monomer units deposited on the electrode and h is
polymerisation efficiency reflecting various loses of
electricity during deposition. According to the literature,zred for polythiophene derivatives usually ranges from
0.25 to 0.3 [20�/22]. This leads to the theoretical values
of y ranging from 11 to 13%, provided that there are no
charge loses during polymerization. The y values
obtained for the three polymer studied, listed in Table
1, are much lower than the theoretical ones. The
polymerisation efficiencies derived from these values
are 18�/21% for PDMT, 14�/16% for PDPT and 54�/63%for PDOT.
Because of the charge loses during polymerisation, the
amount of the film deposited on the electrode was
controlled not by the deposition charge but the value of
the surface coverage, G (mol cm�2). The values of G
were determined coulometrically, using the expression
G�/Qred/zredFS, where Qred is the charge passed during
reduction of the polymer in acetonitrile monomer-freesolution, S is the surface area of the working electrode
(0.02 cm2) and zred�/0.3.
3.2. Electrochemical and spectroscopic characterization
of PDMT, PDPT and PDOT films in acetonitrile solution
of LiClO4
The redox behaviors of the PDMT, PDPT and PDOT
films of G:/26�/10�8 mol cm�2 in acetonitrile solu-tion of 0.1 M LiClO4 are compared in Fig. 3. The cyclic
voltammogram of PDMT, with a broad redox peaks, is
similar to that presented by Zotti et al. [15] and
resembles the behaviour of PEDOT.
The increase of the number of carbon atoms in the
side groups gives rise to formation of a narrow oxida-
tion peak, preceded by a broad shoulder and followed
by a capacitive current. At the same time, electroactivityof the polymer shifts towards the more positive poten-
tials. Namely, the redox potentials of the polymers,
defined as Eredox�
/�/(Epa�/Epc)/2, increase from �/0.05 V
Fig. 2. Comparison of the spectra of the electrodeposition baths after
polymerisation of 3,4-DMT (1), 3,4-DPT (2) and 3,4-DOT (3) with the
spectrum of PDOT solution obtained by dissolution of the polymer
film in CHCl3 (4).
A. Szkurlat et al. / Electrochimica Acta 48 (2003) 3665�/36763668

for PDMT, to 0.26 V for PDPT and to 0.39 V for
PDOT. For comparison, the redox potential for
PEDOT is �/0.54 V. On the base of the monomer
spectra, the electronic effect of the substituents may be
excluded, as in the case of ethylenedioxy- and di-
methoxy-derivatives of polypyrrole and polythiophene
[15]. Therefore, the most probable reason of the anodic
shift of the redox potential is some distortion from the
planarity of the conjugated system due to the increasing
length of the side groups or/and decrease of the
polymerisation degree. Distortion from planarity leads
to diminution of donating effect of alkoxy group due to
less efficient overlapping between the p-orbital of
oxygen atom and the conjugated system [23]. According
to the literature, no disturbance from the planarity of
the main chain was found for 3,3?-bipentoxy-2,2?-bithienyl [24]. However, in the presence of two alkoxy
groups the steric hindrances may cause some distortion
in the backbone and this effect may be enhanced with
the increase of the alkyl chain length. Unfortunately, we
are not able to verify the second possibility, i.e. lowering
the polymerisation degree with the increase of the alkyl
chain length for the three polymers studied, due to
insolubility of PDMT and PDPT in organic solvents.
The next question to answer is the origin of the
shoulder and narrow redox peak in the cyclic voltam-
mograms of PDPT and PDOT, in contrast to broad
voltammograms for PDMT and PEDOT. A very useful
tool in explanation of the character of the redox
processes in the polymers is UV�/Vis spectroelectro-
chemistry. The spectra of PDMT and PDPT recorded at
various polarization potentials are presented in Fig. 4
(the spectroelectrochemical behavior of PDOT is similar
to that of PDPT). In the neutral state, under polariza-
tion of the films at �/0.6 V, the spectra reveal a very well
developed vibronic structure, characteristic of the rigid
polymers containing crystalline zones in amorphous
phase [25,26]. The distances between the vibronic peaks,
listed in Table 1, correspond to 0.16�/0.17 eV ascribed in
the literature to C�/C stretching mode in the thiophene
ring which confirms high regularity along the polymer
backbone [25,27,28]. The vibronic peaks of poly(3,4-
dialkoxythiophenes) are much sharper and better re-
solved than those obtained for poly(3-alkylthiophene)
and monosubstituted polyalkoxythiophenes [29]. In
spite of some distortion from planarity suggested above,
the alkoxy substituents hinder a free rotation of the
neutral polymer chains around the bonds between the
adjacent monomer units making undoped poly(3,4-
dialkoxythiophene) backbone more rigid than those of
the monosubstituted polythiophenes. Oxidation of the
polymer films results in the decrease of p0/p* transition
peaks intensity with simultaneous appearance of new
bands at l�/680 nm. A comparison of the maximum
intensity of the bands in this range shows that at high
doping levels PDPT is much more transparent than
PDMT.
It is also worth noting that evolution of the spectra at
the wavelength above 680 nm is different for PDMT
than for the two other polymers studied, PDPT and
PDOT. In the former case one can observe a develop-
ment of only one broad absorption peak at about 820
nm, as it has been reported for other electrosynthesised
polythiophene derivatives [20,30,31]. In contrast, two
separate peaks are formed for PDPT and PDOT*/one
at about 800 nm and the second at 980 nm. The first
band is less resolved due to overlapping with the region
of p0/p* transition peak. These two bands develop in
the spectra at relatively low oxidation potentials (for
PDPT in the range �/0.5}/0.16 V, corresponding to the
shoulder in the cyclic voltammogram) and overlap upon
Table 1
Maximum absorption (lmax) of the monomers and the polymers in the neutral state, oxidation potentials of the monomers (Ep(ox)), redox potentials
of the polymers in acetonitrile (Eredox� ) and the charge yields (y ) for the polymers studied
Polymer lmax/nm monomer Ep(ox)/V (monomer) /y�Qred=Qdep /Eredox�
//V a (polymer) lmax/nm polymer
PDMT 262 1.15 2.4% �/0.05 520, 558, 607
PDPT 262 1.12 1.8% 0.26 522, 560, 608
PDOT 264 1.09 7% 0.39 524, 565, 612
PEDOT 253[15] 1.09 13% �/0.54 610
a/Eredox
� ; is assumed as (Epa�/Epc)/2.
Fig. 3. Cyclic voltammograms of PEDOT, PDMT, PDPT and PDOT
films in the solution of 0.1 M LiClO4 in acetonitrile.
A. Szkurlat et al. / Electrochimica Acta 48 (2003) 3665�/3676 3669

gradual polymer oxidation. This gives a rather compli-
cated picture of spectroelectrochemical behavior of
poly(3,4-dialkoxythiophenes). Thus, in order to gain
more insight into the redox processes in the polymers
studied, we also made a series of experiments in which
the optical changes upon doping�/undoping were re-
corded simultaneously with the cyclic voltammograms.
The measurements were performed at several wave-
lengths, 560, 800, 980 nm for PDPT and at 560, 820 nm
for PDMT, corresponding to the maxima in the
absorbance spectra, and at 1100 nm. The obtained
absorbance�/time responses were differentiated, plotted
in the form of dA /dt versus E profiles (differential cyclic
voltabsorptograms, DCVA) and compared with the
cyclic voltammograms.
As visible in Fig. 5b, the shoulder and the main
oxidation peak in the voltammogram of PDPT have
their well developed counterparts in dA /dt �/E plots
obtained at 560 nm. The similar behavior was also
obtained for the spectra recorded at two other vibronic
peaks, at 522 and 608 nm. The shoulder in CV has also a
counterpart in a form of a small peak in the voltab-
sorptograms recorded at 800 and 980 nm. In contrast,
the maximum related to the main anodic peak nearly
disappeared in the voltabsorptograms recorded at these
two wavelengths. Some traces of this peak are visible
only in the curve obtained at 980 nm, probably due to
remarkable overlapping with the region of increased
absorbance above 1000 nm at E �/0.2 V. In turn, the
peak corresponding to the main current maximum is
very well developed in the voltabsorptogram obtained at
1100 nm. These observations and analysis of the spectra
presented in Fig. 4b allow us to conclude that the bands
at 800 and 980 nm correspond to the species created at
the beginning of oxidation, i.e. at relatively low doping
levels. According to the literature, in the polythiophene
Fig. 4. Evolution of UV�/Vis in-situ spectra of PDMT (a) and PDPT (b) upon gradual polymer oxidation in 0.1 M LiClO4 in AN.
Fig. 5. Cyclic voltammograms (dotted line) and voltabsorptograms (DCVAs) obtained for doping�/undoping of PDMT (a) and PDPT (b) at the
scan rate 10 mV s�1 in AN at the wavelengths: (a) 560, 820 and 1100 nm; (b) 560, 800, 980 and 1100 nm.
A. Szkurlat et al. / Electrochimica Acta 48 (2003) 3665�/36763670
derivatives the radical cations are created even at
extremely low doping level, below 0.1% [32]. The main
oxidation peak for PDPT located at 0.3 V corresponds
to the species created also directly from the neutralpolymer. At the more positive potentials, these species
are transformed into an another type of the charge
carriers, which are responsible for the increase of
absorbance at the wavelength above 1000 nm. This is
confirmed by appearance of isosbestic point at 912 nm.
Highly interesting voltabsorptograms were obtained
for PDMT. As visible in Fig. 5a, one can distinguish two
different redox couples in the dA /dt profiles recorded at820 and 1100 nm but only one in the cyclic voltammo-
gram. The oxidation potentials of these two waves are
located very close each other, at 0.03 and 0.18 V. Their
cathodic counterparts, respectively at �/0.23 and 0.18 V,
are better separated. These two peaks correspond
exactly to two reduction waves in the cyclic voltammo-
gram. This suggests that the anodic peak in the
voltammogram of PDMT is, in fact, a superpositionof two distinct peaks, as it has been postulated for
poly(3-methylthiopene) [33]. In the case of PDPT and
PDOT a distance between the two oxidation peaks is
much higher than for PDMT and additionally, the
second (more anodic) peak is sharper than the first one.
In consequence, one can distinguish two distinct redox
processes. The presence of overlapping redox peaks was
also observed for other poly(3-alkylthiophenes) andexplained as a result of oxidation-reduction of the zones
of different crystallinity and conjugation length [31,34].
In our opinion the redox behaviour of poly(3,4-dialkox-
ythiophenes) may be also elucidate by co-existence in the
polymer film of the regions of different ordering. The X-
ray diffraction data presented in the literature proved
the presence of the crystalline zones in poly(3-alkox-
ythiophene) films [29]. Moreover, it was found that thecrystallinity of the polymers increased with the increase
of the alkoxy side-chain length.
3.3. Stability of PDMT, PDPT and PDOT in the neutral
and oxidized states
A shift of the redox potential of poly(3,4-dialkox-
ythiophenes) with increasing side chain length is favour-
able because it allows for modification of the polymerproperties. For example, PEDOT, often used as a
reference polymer due to its unique properties, is very
stable in the doped state but not in the neutral form due
to very low oxidation potential. Therefore, in the neutral
state it should be handled only in oxygen-free condi-
tions. This limits the possibilities of practical applica-
tions of PEDOT. From this point of view, poly(3,4-
dialkoxythiophenes), whose redox potentials are morepositive, seem to be the promising materials. Thus, the
next step of the studies was directed to examination of
the stability of PDMT, PDPT and PDOT in the neutral
state. The films were deposited electrochemically on
ITO, then polarized for 180 s at �/0.8 V to transform the
polymer into the neutral state and finally, after inter-
ruption the external polarization, the relaxation of theopen circuit potential, Eocp, and absorbance at two
wavelengths, 560 and 820 nm, in non-deoxygenated
solution were monitored. These two parameters changed
very slowly and within 20 min the absorbance decreased
only slightly, from 0.75 to 0.73, and the open circuit
potential stabilized at the values within the region of the
neutral polymer.
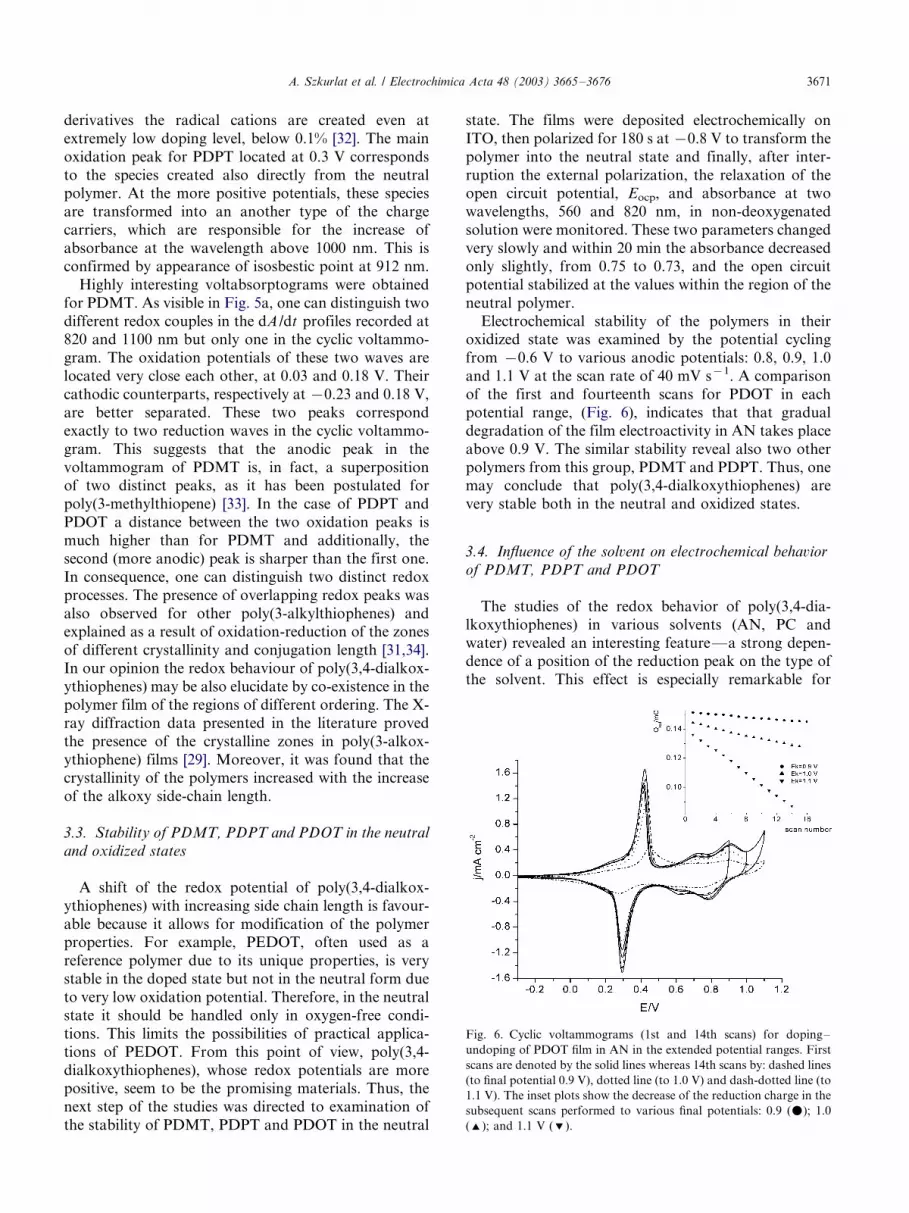
Electrochemical stability of the polymers in theiroxidized state was examined by the potential cycling
from �/0.6 V to various anodic potentials: 0.8, 0.9, 1.0
and 1.1 V at the scan rate of 40 mV s�1. A comparison
of the first and fourteenth scans for PDOT in each
potential range, (Fig. 6), indicates that that gradual
degradation of the film electroactivity in AN takes place
above 0.9 V. The similar stability reveal also two other
polymers from this group, PDMT and PDPT. Thus, onemay conclude that poly(3,4-dialkoxythiophenes) are
very stable both in the neutral and oxidized states.
3.4. Influence of the solvent on electrochemical behavior
of PDMT, PDPT and PDOT
The studies of the redox behavior of poly(3,4-dia-
lkoxythiophenes) in various solvents (AN, PC and
water) revealed an interesting feature*/a strong depen-dence of a position of the reduction peak on the type of
the solvent. This effect is especially remarkable for
Fig. 6. Cyclic voltammograms (1st and 14th scans) for doping�/
undoping of PDOT film in AN in the extended potential ranges. First
scans are denoted by the solid lines whereas 14th scans by: dashed lines
(to final potential 0.9 V), dotted line (to 1.0 V) and dash-dotted line (to
1.1 V). The inset plots show the decrease of the reduction charge in the
subsequent scans performed to various final potentials: 0.9 (m); 1.0
('); and 1.1 V (%).
A. Szkurlat et al. / Electrochimica Acta 48 (2003) 3665�/3676 3671

PDPT. In the case of PDMT, the cyclic voltammograms
are very broad and therefore, the solvent effect is rather
difficult to analyse. In turn, the long hydrophobic alkyl
chains in PDOT likely decrease the polymer swelling.
This is probably a reason of the lowering of redox
charge (from 140 to 93 mC) after the transfer of the film
from AN to the aqueous solution. In effect of dimin-
ished swelling, the resistance of the film in aqueous
solution increases and the redox peaks are shifted (Fig.
7b). In the case of shorter alkyl chain (propyl) the
hydrophilicity of the oxygen competes with hydropho-
bicity of alkyl and therefore, no change of the polymer
electroactivity was detected after the transfer of PDPT
from AN to aqueous solution. As visible in Fig. 7a, the
main oxidation peak for PDPT is located at about 0.3 V
irrespectively of the solvent, whereas the reduction peak
shifts from 0.27 V in aqueous solution to 0.2 V in AN
and to 0.02 V in PC, at the scan rate of 40 mV s�1.
Decrease of the scan rate to 1 mV s�1 only slightly
diminished the separation between oxidation and reduc-
tion peaks in PC. Thus, this fact excludes the possibility
that the strong asymmetry between the peaks in this
solvent is due to a slow diffusion of the counter ions to/
from the polymer film upon doping/undoping. There-
fore, the presence of alkoxy substituents seems to be
crucial for explanation of this asymmetry. Thus, let us
consider the phenomena occurring upon the polymer
oxidation and reduction. In general, the first stage of
oxidation is formation of the radical cations along the
polymer chain with simultaneous incorporation of the
counter anions into the polymer matrix to neutralize the
positive charge. In polyalkoxythiophenes the radical
cation created upon polymer oxidation may be deloca-
lized on the alkoxy group, according to Scheme 2, as has
been postulated in the literature [4,6,18,35].To specify the potential range in which delocalization
of the radical cation occurs, we registered the 1H-NMR
spectra of PDOT in the neutral state and after oxidation
for 5 min at two different potentials, 0.15 V (at the onset
of main oxidation peak) and 0.35 V (after oxidation
peak). No shift of the signal was found for polymer
oxidized at 0.15 V, whereas the polarization to 0.35 V
led to broadening and a profound shift of the proton
peak of �/OCH2�/ group from 4.11 to 4.4 ppm (see Fig.
8).
Fig. 7. Influence of the type of solvent on the redox behavior of PDPT (a) and PDOT (b) films in the solution of 0.1 M LiClO4. Scan rate 40 mV s�1.
Scheme 2. Formation of radical cation in poly(3-alkoxythiophene)
and its delocalization on the alkoxy group.
Fig. 8. 1H-NMR spectra of PDOT in the neutral state (A) and
oxidized for 300 s at and at 0.35 V (B).
A. Szkurlat et al. / Electrochimica Acta 48 (2003) 3665�/36763672

Based on the voltammograms carried out at very low
scan rate and 1H-NMR results presented above it is
reasonable to speculate that a remarkable shift of the
reduction peak in PC towards lower potentials is due tostrong interactions between the radical cations localized
on the alkoxy groups and the solvent molecules and/or
the anions accommodated by the oxidized polymer film.
It is highly plausible because PC has a very high dipole
moment (4.98 D) and the perchlorates are not solvated
in PC [36]. Since the perchlorates are also poorly
solvated in AN but a dipole moment of AN is much
lower (3.4 D) we suggest that the interactions of theradical cations with the solvent are of the main
importance. The dipole moment of the water molecule
is the lowest among the three solvent studied (1.76 D)
and probably therefore the reduction peak is the most
symmetrical with respect to the oxidation one. This
leads to the conclusion that poly(3,4-dialkoxythio-
phenes) are highly electroactive and stable both in the
presence of oxygen and the moisture. This is a greatadvantage, considering the possible practical application
of these polymers as, for example, chemical and
biosensors.
3.5. Infrared spectra
Fig. 9 presents the IR spectrum of pristine PDOT and
two monomer spectra: in a solid form (in KBr) and in
acetonitrile solution. The peak positions and proposed
assignments are listed in Table 2. A list of differences
between the polymer and monomer spectra starts from
the Ca�/H stretching band observed at 3105 cm�1 for
the monomer but missing in the spectrum of the
polymer. Two other bands, at 1205 and 870 cm�1,involving mainly the Ca�/H in-plane bending modes
(n15 and n17 in Refs. [37,38]) are strong in the monomer
spectrum but consequently not observed in the spectrum
of PDOT. In contrast, a new band at 1170 cm�1
attributable to the Ca�/Ca inter-ring stretching between
the 3,4-DOT units appears only in the polymer spec-
trum. The thiophene ring-stretching modes occur at
1565 and 1504 cm�1 in the monomer spectrum.
According to IR data for oligo- and polythiophenes,
these modes are expected to shift downwards upon
increasing conjugation length [39,41,42]. This is prob-
ably a reason of the shift of the two bands to 1437 cm�1
in the spectrum of PDOT.
Differences between the spectra of the solid monomer
and monomer in the solution, are visible mainly in the
range 1200�/600 cm�1 (see Fig. 9a and b) because it
comprises the conformation dependent bands. The C�/
O�/C motions usually mix with aromatic ring deforma-
tions involving C�/H or C-substituent bending modes
[43]. Frequencies of C�/O�/C vibrations are correlated
with the C�/O�/C angle. Therefore, in the monomer
solution, where the oxy-alkyl chain can rotate around
C�/O bond, the considered spectral region contains
broad overlapping bands. In contrast, relatively sharp
and well defined bands occur in the spectrum of a solid
3,4-DOT indicating that in the solid state a single
conformation is enforced. Moreover, in this wavenum-
ber range the polymer spectrum reveals numerous bands
forming quite different pattern than that in the mono-
mer spectrum. These differences likely result from the
changes in the thiophene ring deformation modes.
The bands due to the octyl chains are observed at the
similar positions in all spectra studied i.e. of the solid
monomer, monomer in the solution and the polymer
(see Table 2).
Fig. 9. Infrared spectra of 3,4-DOT in the acetonitrile solution (a) solid 3,4-DOT*/in KBr (b) and PDOT in KBr (c).
A. Szkurlat et al. / Electrochimica Acta 48 (2003) 3665�/3676 3673

The effect of electrochemical oxidation on the infra-
red spectra of PDOT is presented in Fig. 10. The spectra
were recorded ex-situ after conditioning the polymer
film for 1 min at the selected potentials. A bare platinum
electrode was used to record the background spectrum.
As it results from UV�/Vis data of Fig. 4, the polymer
conditioned at �/0.3 V is in the neutral form and
therefore, the corresponding infrared spectrum repro-
duces most features characteristic of undoped polythio-
phenes [44,47]. Polarization of PDOT film at 0.25 V,
located within the current shoulder (cf. Fig. 7b), leads to
development of a broad absorption with a maximum at
3300 cm�1, feature typical of conducting polymers due
to the lowest-energy electronic transition [48,49], (Fig.
10a). At the same time, some bands visible for the
neutral polymer within the range 1600�/600 cm�1 are
preserved, in particular the ring stretching mode at 1437
cm�1, although its relative intensity (with respect to the
band of CH2 bending at 1468 cm�1) is altered (Fig.
10b).New strong and very broad bands appear at 1326,
1180 and 1042 cm�1. Their intensity, shapes and
positions are similar to the p-doping induced bands,
so-called infrared activated vibration (IRAV) bands,
observed for several polythiophene derivatives. These
strong bands appear owing to the movement of the
electronic charge carriers along the polymer chains,
giving rise to large dipole changes in the vibrations [45�/
47,50�/52]. A new band appears also at 1491 cm�1 but
its origin is not obvious. It could be assigned to another
doping induced band, but its intensity and width is very
small in comparison to typical doping induced band.
Another possible explanation for the appearance of the
band at 1491 cm�1 is the conformation or orientation
change in the polymer film. The orientation or con-
formational changes could also explain the altered
relative intensity of ring stretching mode at 1437 cm�1.
When the potential value matches the main oxidation
peak in the cyclic voltammogram of PDOT (�/0.4 V),
the doping induced bands become broader and acquire
clear new components on their low wavenumber sides.
At higher doping levels, after the main oxidation peak at
0.5 V, these ‘new’ components become stronger than the
‘old’ ones and in effect, the bands at 1326, 1180 and
1042 cm�1 (observed at �/0.25 V) are replaced by the
bands at 1282, 1130 and 1001 cm�1 at 0.5 V. Such
strong changes in the doping induced bands cannot be
explained by simple increase of the charge carriers
concentration, but rather by a formation of an another
type of charge carrier. This behavior is similar to that
reported by Jones et al. [52] for polybenzo[c ]thiophene
but different from the spectral changes observed for
many polythiophene derivatives, where the increase of
the anodic polarization potential causes merely the
increase of the IRAV band intensities without clear
shift in their position, or development of new bands
[45�/51].
Polarization of PDOT above 0.25 V also gives rise to
the shift of the electronic absorption maximum from
3300 cm�1 towards lower wavenumbers, up to about
2000 cm�1 at �/0.7 V (Fig. 10a), with simultaneous
gradual increase of its intensity. In consequence, already
at �/0.4 V the electronic absorption covers the C�/H
stretching bands (around 2900 cm�1). Since the max-
Table 2
Infrared bands of 3,4-DOT and PDOT
Mon. sol. In acetonitrile Monomer solid Polymer (neutral) Assignment a
3105 3105 �/ Ca�/H stretching (thiophene ring)
2959 2953 2954 CH3 asymmetric stretching
2932 2920/2936 b 2918 CH2 asymmetric stretching
2875 2868 2873 CH3 symmetric stretching
2859 2855 2851 CH2 symmetric stretching
1565 1565 �/ Thiophene ring deformation
1500 1504/1495 b �/ Thiophene ring asymmetric stretching (Ca�/Cb)
1470 1470 1468 CH2 bending
1437 Thiophene ring deformation
1374 1396/1374 b 1374 CH2 wagging�/Cb�/Cb stretching
1269 Thiophene ring deformation
1205 1205 Ca�/H bending
1170 Ca�/Ca interring stretching
1154 1154/1126 b �/ C�/O�/C coupled with Ca�/H bending
1038, 961 1042, 1038, 990 1100, 1076, 1055, 1025, 1010, 990, 946 C(ring)�/O�/C bending modes
910 C�/S stretching
870 870 876 (weak) Ca�/H�/C�/S stretching
760 751/742 b �/ Ca�/H out-of-plane
723 723 723 CH2 out-of-plane deformations�/C�/S�/C deformation
a On the basis of Refs. [37�/42,44].b Crystal field splitting.
A. Szkurlat et al. / Electrochimica Acta 48 (2003) 3665�/36763674

imum of the electronic absorption and the dopinginduced bands for the second type of carrier (possibly
bipolarons) are located at the lower wavenumbers, these
species are characterized by lower ‘pinning’ and stronger
charge delocalization, as compared to the charge
carriers formed at the initial oxidation stage [53].
4. Conclusions
Our studies on electrosynthesis of poly(3,4-dialkox-
ythiophene) films have shown that electrochemically
stable, rigid and long conjugated polymer may be
obtained from acetonitrile solutions of the monomerin LiClO4 supporting electrolyte. Low polymerisation
efficiency results from formation of soluble oligomers in
the vicinity of the electrode.
By the increase of the alkyl-chain length in the alkyl
group one may modify the solubility of the resultant
polymer and its electroactivity in the aqueous solution.
The shift of the redox potential of poly(3,4-dialkox-
ythiophenes) to the more positive potentials with the
increase of the alkyl chain length of the alkoxy group
may result from some distortion of the polymer back-
bone from planarity or/and decreasing degree of poly-
merisation.
UV�/Vis in-situ spectra and DCVA profiles indicated
that the oxidation branch in the cyclic voltammograms
of poly(3,4-dialkoxythiophenes) consists of two over-
lapping oxidation peaks. The origin of the two peak may
be the same as in the case of regioregular poly(3-
alkylthiophenes) i.e. co-existence in the polymer film
of the zones of different crystallinity. The separation
between these two peaks is very small for PDMT and
Fig. 10. Infrared external reflectance spectra of PDOT film conditioned for 1 min at various potentials in the range 4000�/500 (a) and 1600�/600
cm�1 (b).
A. Szkurlat et al. / Electrochimica Acta 48 (2003) 3665�/3676 3675

increases with the increase of the alkyl chain length of
alkoxy group.
The shape of the cyclic voltammograms for the
polymer doping�/undoping depends markedly on thesolvent used. In our opinion a remarkable shift of the
reduction peak of PDPT in PC towards less positive
potentials in comparison to the peak obtained in AN
and in aqueous solution is due to strong interactions of
the solvent with the radical cations delocalised on the
alkoxy groups. Delocalization of the radical cations has
been proved by NMR spectra.
The changes in location and intensity of the infraredactivated vibrations (IRAVs) evidenced the changes in
the charge carrier identity during gradual polymer
oxidation.
Acknowledgements
This work was financially supported by grantsGR1686 and BST 761/16/2002.
References
[1] M. Lemaire, D. Delabouglise, R. Garreau, A. Guy, J. Roncali, J.
Chem. Soc. Chem. Commun. (1988) 658.
[2] J. Roncali, R. Garreau, D. Delabouglise, F. Garnier, M. Lemaire,
J. Chem. Soc. Chem. Commun. (1989) 679.
[3] R.D. McCullough, Adv. Mater. 10 (1998) 93.
[4] T. Yamamoto, A. Kashiwazaki, K. Kato, Macromol. Chem. 190
(1989) 1654.
[5] J. Roncali, L.H. Shi, R. Garreau, F. Garnier, M. Lemaire, Synth.
Met. 36 (1990) 267.
[6] S.-A. Chen, C.-C. Tsai, Macromolecules 26 (1993) 2234.
[7] S. Tanaka, M.A. Sato, K. Kaeriyama, Synth. Met. 25 (1988) 277.
[8] M. Feldheus, G. Kampf, H. Litterer, T. Mechlenburg, P. Wegner,
Synth. Met. 28 (1989) C487.
[9] L.B. Groenendaal, F. Jonas, D. Feitag, H. Pielartzik, J.R.
Reynolds, Adv. Mater. 12 (2000) 481.
[10] J. Roncali, Chem. Rev. 92 (1992) 711.
[11] M. Leclerc, G. Daoust, J. Chem. Soc. Chem. Commun. 3 (1990)
273.
[12] A.G. Bayer, European Patent, 339 340.
[13] M. Dietrich, J. Heinze, G. Heywang, F. Jonas, J. Ekectroanal.
Chem. 369 (1994) 87.
[14] F. Jonas, J.T. Morrison, Synth. Met. 85 (1997) 1397.
[15] G. Zotti, S. Zecchin, G. Schiavon, L.B. Groenendaal, Chem.
Mater. 12 (2000) 2996.
[16] G. Daoust, M. Leclerc, Macromolecules 24 (1991) 455.
[17] Q.T. Zhang, J.M. Tour, J. Am. Chem. Soc. 120 (1998) 5355.
[18] M. Fall, L. Assogba, J.-J. Aaron, M.M. Dieng, Synth. Met. 123
(2001) 365.
[19] O. Hinsberg, Ber. 43 (1910) 901.
[20] A.O. Patil, A.J. Heeger, F. Wudl, Chem. Rev. 88 (1988) 183.
[21] R.H.J. Schmitz, A.S. Hudson, K. Juttner, Synth. Met. 101 (1999)
102.
[22] C. Arbizzani, M. Catellani, M. Mastragostino, M.G. Cerroni, J.
Electroanal. Chem. 423 (1997) 23.
[23] E.E. Havinga, C.M.J. Mutsaers, L.W. Jenneskens, Chem. Mater.
8 (1996) 769.
[24] S.V. Meille, A. Farina, F. Bezziccheri, M.C. Gallazzi, Adv.
Mater. 6 (1994) 848.
[25] S.D.D. Rughooputh, S. Hotta, A.J. Heeger, F. Wudl, J. Pol. Sci.
B: Polym. Phys. 25 (1987) 1071.
[26] M. Sundberg, O. Inganas, S. Stafstrom, G. Gustafsson, B.
Sjogren, Solid State. Commun. 71 (1989) 435.
[27] R.D. McCullough, R.D. Lowe, M. Jayaraman, D.L. Anderson, J.
Org. Chem. 58 (1993) 904.
[28] R.D. McCullough, R.D. Lowe, J. Chem. Soc. Chem. Commun.
(1992) 70.
[29] X. Hu, L. Xu, Polymer 41 (2000) 9147.
[30] J. Lukkari, J. Kankare, C. Visy, Synth. Met. 48 (1992) 181.
[31] M. Skompska, A. Szkurlat, Electrochim. Acta 46 (2001) 4007.
[32] Y. Harima, T. Eguchi, K. Yamashita, K. Kojima, M. Shiotani,
Synth. Met. 105 (1999) 121.
[33] M. Skompska, Electrochim. Acta 44 (1998) 357.
[34] X. Jiang, Y. Harima, K. Yamashita, Y. Tada, J. Ohshita, A.
Kunai, Chem. Phys. Lett. 364 (2002) 616.
[35] T. Yamamoto, M. Omote, Y. Miyazaki, A. Kashiwazaki, B.-L.
Lee, T. Kanbara, K. Osakada, T. Inoue, K. Kubota, Macro-
molecules 30 (1997) 7158.
[36] H.A. Berman, T.R. Stengle, J. Phys. Chem. 79 (1975) 1001.
[37] M. Rico, J.M. Orza, J. Morcillo, Spectrochim. Acta 21 (1965) 689.
[38] P.A. Christensen, A. Hamnett, A.R. Hillman, J. Electroanal.
Chem. 242 (1988) 47.
[39] V. Hernandez, J. Casado, F.J. Ramirez, G. Zotti, S. Hotta, J.T.
Lopez-Navarrete, J. Chem. Phys. 104 (1996) 9271.
[40] E. Agosti, M. Rivola, V. Hernandez, M. Del Zoppo, G. Zerbi,
Synth. Met. 100 (1999) 101.
[41] V. Hernandez, Y. Kanemitsu, J.T. Lopez-Navarrete, J. Raman
Spectrosc. 29 (1998) 617.
[42] G. Louarn, M. Trznadel, J.P. Buisson, J. Laska, A. Pron, M.
Lapkowski, S. Lefrant, J. Phys. Chem. 100 (1996) 12532.
[43] L.J. Bellamy, The Infrared Spectra of Complex Molecules,
Chapman A. Hall, London, 1978.
[44] B.C. Uff, in: A.R. Katritzky, C.W. Rees (Eds.), Comprehensive
Heterocyclic Chemistry, vol. 2A, Pergamon Press, Oxford, 1886.
[45] C. Kvarnstrom, H. Neugebauer, A. Ivaska, N.S. Sariciftci, J.
Molec. Struct. 521 (2000) 271.
[46] A. Cravino, H. Neugebauer, S. Luzzati, M. Catellani, N.S.
Sariciftci, J. Phys. Chem. B 105 (2001) 46.
[47] M. Pohjakallio, G. Sundholm, P. Talonen, J. Electroanal. Chem.
406 (1996) 165.
[48] Y.H. Khim, S. Hotta, A.J. Heeger, Phys. Rev. B 36 (1987) 7486.
[49] H. Kuzmany, N.S. Sariciftci, H. Neugebauer, A. Neckel, Phys.
Rev. Lett. 60 (1988) 212.
[50] H. Neugebauer, C. Kvarnstrom, A. Cravino, T. Yohannes, N.S.
Sariciftci, Synth. Met. 116 (2001) 115.
[51] A. Cravino, H. Neugebauer, S. Luzzati, M. Catellani, A. Petr, L.
Dunsch, N.S. Sariciftci, J. Phys. Chem. B 106 (2002) 3583.
[52] C.L. Jones, S.J. Higgins, P.A. Christensen, J. Mater. Chem. 12
(2002) 758.
[53] Comprehensive review can be found in M. Del Zoppo, C.
Castiglioni, P. Zuliani, G. Zerbi, in: T.A. Skotheim, R.L. Else-
nbaumer, J.R. Reynolds (Eds.), Handbook of Conducting Poly-
mers, 2nd ed. (Chapter 28), New York, Marcel Dekker, 1988
(Chapter 28).
A. Szkurlat et al. / Electrochimica Acta 48 (2003) 3665�/36763676