electrosynthesis and properties of poly(3,4-ethylenedioxythiophene) films functionalized with...
TRANSCRIPT

Electrochimica Acta 51 (2006) 2108–2119
Electrosynthesis and properties of poly(3,4-ethylenedioxythiophene)films functionalized with titanocene dichloride complex
Magdalena Skompskaa,∗, Mikhail A. Vorotyntsevb, Monika Refczynskaa, Jerome Gouxb,Eric Lesniewskac, Gilles Bonib, Claude Moiseb
a Department of Chemistry, Warsaw University, ul. Pasteura 1, Warsaw 02 093, Polandb LSEO-UMR 5188 CNRS-Universite de Bourgogne, Mirande, 9 Avenue A. Savary, BP 47 870, Dijon Cedex F-21078, France
c LPUB-UMR 5025 CNRS-Universite de Bourgogne, Mirande, 9 Avenue A. Savary, BP 47 870,Dijon Cedex F-21078, France
Received 21 October 2004; received in revised form 20 December 2004; accepted 1 January 2005Available online 31 August 2005
Abstract
e DCM) anda e reducedt lizedt pared witht merizationa regimesa ed by cyclicv BAPFi arison tot at of thec©
K tanocened
mtTopdMm
byallytive
ates,lec-es ofrob-
s thattheity
0d
Synthesis of a titanocene dichloride derivative functionalized with 3,4-etylenedioxythiophene group, Tc1EDOT (Cl2TiCpC5H4(CH2) (3,4-thylenedioxythiophene)) has been described. Redox behavior of the monomer in tetrahydrofuran (THF), dichloromethane (cetonitrile (AN) at different scan rates has been discussed in terms of different ability of these solvents to coordination with th
itanocene (Tc) complex and the solvation of Cl− anions. Electrooxidation of Tc1EDOT to get a conducting polymer film with immobiitanocene dichloride centers and electrochemical properties of its polymer matrix in background acetonitrile solution have been comhose of non-substituted PEDOT and PEDOT-methanol derivative (PEDOTMet), to elucidate the effect of substituents both on polynd redox potentials of the matrix. STM and AFM images of p(Tc1EDOT) films obtained with potentiodynamic and potentiostaticre compared to illustrate that the films deposited at constant potential are better ordered and more compact than those obtainoltammetry. A comparison of the cyclic voltammograms of p(Tc1EDOT) and poly(titanocene-propyl-pyrrole) (p(Tc3Py)) films in 0.1 T6
n THF has shown that the electroactivity of the polymer matrix of p(Tc1EDOT) is extended to more negative potentials in comphat of p(Tc3Py). This results in the anodic shift of redox potential of Tc centers immobilized in p(Tc1EDOT) film with respect to thenters fixed in p(Tc3Py).2005 Elsevier Ltd. All rights reserved.
eywords: Poly(3,4-ethylenedioxythiophene); Titanocene-EDOT derivative; EDOT-methanol derivative; Electrodeposition; STM; Immobilized tiichloride centers
Abbreviations: AN, acetonitrile; THF, tetrahydrofuran; DCM, dichloro-ethane; Cp, cyclopentadienyl, C5H5; Tc, titanocene = bis(cyclopen-
adienyl)titanium dichloride, Cp2TiCl2 or its radical; PPy, polypyrrole;c3Py, titanocene-propyl-pyrrole, Tc(CH2)3NC4H4; p(Tc3Py)polymerbtained from Tc3Py; EDOT, 3,4-ethylenedioxythiophene; PEDOT,oly(3,4-ethylenedioxythiophene); Tc1EDOT, titanocene-methyl-ethylene-ioxythiophene; p(Tc1EDOT), polymer obtained from Tc1EDOT; PEDOT-et, poly(3,4-ethylenedioxythiophene methanol); TBAPF6, tetrabuthylam-onium hexafluorophosphate∗ Corresponding author. Fax: +48 22 822 59 96.
E-mail address: [email protected] (M. Skompska).
1. Introduction
Functionalization of electronic conducting polymerscovalent bonding of the electrochemically or chemicactive groups (transition metal complexes, optically acside-chains, liquid crystalline groups, enzymes, cheletc.) is a very useful tool for preparation of modified etrodes used, for example, in electrocatalysis, novel typsensors, or electrochromic devices. One of important plems related to practical applications of these systems ithe functional group is electroactive in the range wherepolymer is in its neutral, insulating state. Low conductiv
013-4686/$ – see front matter © 2005 Elsevier Ltd. All rights reserved.oi:10.1016/j.electacta.2005.01.066

M. Skompska et al. / Electrochimica Acta 51 (2006) 2108–2119 2109
of the matrix may inhibit the redox reaction of electroactivecenters. In our previous papers we have reported the electro-chemical behavior of titanocene (bis-cyclopentadienyl tita-nium) dichloride (Tc) centers incorporated into the polypyr-role (PPy) matrix by covalent bonding, p(Tc3Py)[1–4]. Ourstudies revealed that the reduction of Tc centers in the poly-mer film and in the solution occurs according to the samechemical mechanism, consisting in the generation of a radicalanion, followed by its rapid dissociation (may be, incom-plete) and accommodation of solvent molecule as a ligandor formation of a dimeric complex[5–7]. Reversibility ofthe reaction depends on the binding strength of the ligand,namely, reaction is the slower, the stronger is coordination ofthe ligand. In the case of weakly bound ligands, tetrahydro-furan (THF) and dichloromethane (DCM), the redox transi-tion Ti(IV)/Ti(III) is reversible and a well developed pairof reduction-reoxidation peaks is observed at−1.16 and−1.08 V (in THF) and at−1.04 and−0.92 V (in DCM)(versus Ag/Ag+, 0.01 M, CH3CN), respectively. Incorpora-tion of Tc centers into the PPy matrix gave rise to a quasireversible redox reaction in THF with the peak separationof 300–400 mV (at the scan rate of 100 mV s−1) and notbalanced reduction and reoxidation charge due to an insulat-ing character of the matrix in this potential range[1,2]. Ourstudies revealed, however, that the remaining part of reducedcenters is reoxidized at the onset of the matrix oxidation andt amsi aveo
sis-t itys ty bya ten-t pene( triles oneo alsoni ,P con-d ofP hesiso ened OTv s oft icalp OTa T),i OTa OT-M d tod bothm tivityo nece We
also present the STM images of p(Tc1EDOT) films obtainedunder two different polymerization regimes.
2. Experimental
2.1. Synthesis of titanocene-ethylenedioxythiophenederivative, Tc1EDOT
Reactions were carried out under inert atmosphere (argon)using schlenk techniques. Solvents were dried and distilledunder argon. Paratoluenesulfonyl chloride (Acros) was usedas received. 3,4-Ethylenedioxythiophene methanol was pre-pared in the manner described by Chevrot and co-workers[10]. CpTiCl3 was prepared in the manner described by Car-doso et al.[11]. Nuclear magnetic resonance spectra wererecorded on a Bruker Advance DRX 300 instruments at300 MHz for 1H and 75 MHz for13C. The chemical shiftsare reported in ppm relative to SiMe4. Elemental analyseswere performed on a EA 1108 CHNS-O FISONS Instrumentsappartus.
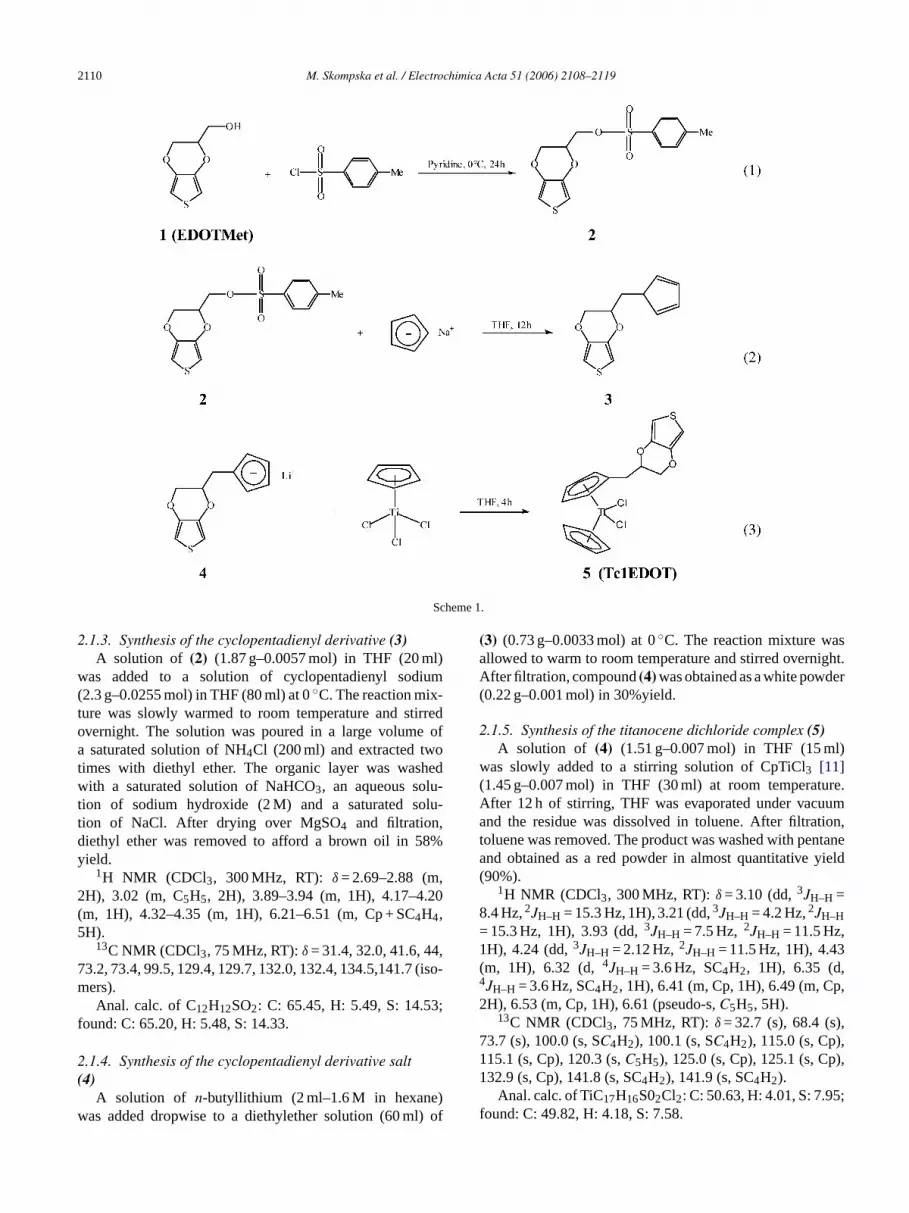
The further reaction path to Tc1EDOT is presented inScheme 1.
2.1.1. Synthesis of the 3,4-ethylenedioxythiophenemethanol, EDOTMet (1)
d byt
5( (s,2
,9
2was
a nps Ther ther( Cl3 olu-t rateds Cl.A nga tes
4 (d,J1P
,7
2;f
his phenomenon manifests itself in cyclic voltammogrn the form of sharp prepeaks at the foot of the oxidation wf the polymer matrix.
One possibility of overcoming the problem of a high reivity of the polymer matrix in the range of Tc electroactiveems to be given by the replacement of the pyrrole moienother monomer which is oxidized/reduced at a lower po
ial than PPy. One of candidates is 3,4-ethylenedioxythioEDOT), which can be easily polymerized from acetoniolution. Among other conducting polymers, PEDOT hasf the lowest redox potentials for p-doping and may be-doped in the range of negative potentials (below−2.0 V
n AN in TBAPF6 supporting electrolyte[9]). MoreoverEDOT is very stable in its doped state and reachesuctivity as high as 500 S cm−1 [8,9]. These propertiesEDOT motivated us to develop an approach to the syntf a new precursor molecule (Tc1EDOT) in which titanocichloride complex is linked to dioxyethylene group of EDia CH2 spacer. In this work, we describe the synthesihis new monomer, Tc1EDOT, as well as its electrochemolymerization. We compare polymerization of Tc1EDnd electrochemical behavior of the polymer, p(Tc1EDO
n the monomer-free AN solution with those of PEDnd poly(3,4-ethylenedioxythiophene methanol) (PEDet) to determine the role of the substituent attacheioxyethylene group on electrochemical properties ofonomer and polymer. Then, we discuss the electroacf the p(Tc1EDOT) film matrix and immobilized titanoceenters in THF in the presence of N(C4H9)4PF6 supportinglectrolyte and compare it with the behavior of p(Tc3Py).
3,4-Ethylenedioxythiophene methanol was preparehe procedure described by Chevrot and co-workers[10].
1H NMR (CDCl3, 300 MHz, RT):δ = 2.10 (s, 1H), 3.8m, 1H), 4.08 (dd, 1H), 4.18 (m, 1H), 4.25 (dd, 1H), 6.33H).
13C NMR (CDCl3, 75 MHz, RT): δ = 61.4, 65.8, 74.29.9, 141.5.
.1.2. Synthesis of the tosylate derivative (2)Paratoluenesulfonyl chloride (3.20 g–0.017 mol)
dded at 0◦C to the solution of(1) (1.46 g–0.0085 mol) iyridine (20 ml). The mixture was stirred 24 h at 0◦C. Theolution was poured in a large volume of water (50 ml).esulting solution was extracted three times with diethyl e100 ml). In order to eliminate pyridine, acidified water (H0%) was added slowly to the solution. After 1 h, the s
ion was extracted and washed repeatedly with a satuolution of NaHCO3, and with a saturated solution of Nafter drying over MgSO4, diethyl ether was removed usirotary evaporator. Compound(2) was obtained as a whi
olid in 80% yield.1H NMR (CDCl3, 300 MHz, RT):δ = 2.48 (s, CH3, 3H),
.06 (m, 1H), 4.19–4.27 (m, 3H), 4.39 (m, 1H), 6.29H–H = 3.7 Hz, SC4H2, 1H), 6.34 (d,JH–H = 3.6 Hz, SC4H2,H), 7.38 (d,JH–H = 8.4 Hz, Ph, 2H), 7.82 (d,JH–H = 8.32 Hz,h, 2H).
13C NMR (CDCl3, 75 MHz, RT): δ = 21.8, 65.0, 67.10.9, 100.3, 128.1, 130.1, 132.3, 140.4, 145.5.
Anal. calc. of C14H14S2O5: C: 51.53, H: 4.33, S: 19.6ound: C: 51.25, H: 4.32, S: 19.48.
2110 M. Skompska et al. / Electrochimica Acta 51 (2006) 2108–2119
Scheme 1.
2.1.3. Synthesis of the cyclopentadienyl derivative (3)A solution of (2) (1.87 g–0.0057 mol) in THF (20 ml)
was added to a solution of cyclopentadienyl sodium(2.3 g–0.0255 mol) in THF (80 ml) at 0◦C. The reaction mix-ture was slowly warmed to room temperature and stirredovernight. The solution was poured in a large volume ofa saturated solution of NH4Cl (200 ml) and extracted twotimes with diethyl ether. The organic layer was washedwith a saturated solution of NaHCO3, an aqueous solu-tion of sodium hydroxide (2 M) and a saturated solu-tion of NaCl. After drying over MgSO4 and filtration,diethyl ether was removed to afford a brown oil in 58%yield.
1H NMR (CDCl3, 300 MHz, RT): δ = 2.69–2.88 (m,2H), 3.02 (m, C5H5, 2H), 3.89–3.94 (m, 1H), 4.17–4.20(m, 1H), 4.32–4.35 (m, 1H), 6.21–6.51 (m, Cp + SC4H4,5H).
13C NMR (CDCl3, 75 MHz, RT):δ = 31.4, 32.0, 41.6, 44,73.2, 73.4, 99.5, 129.4, 129.7, 132.0, 132.4, 134.5,141.7 (iso-mers).
Anal. calc. of C12H12SO2: C: 65.45, H: 5.49, S: 14.53;found: C: 65.20, H: 5.48, S: 14.33.
2.1.4. Synthesis of the cyclopentadienyl derivative salt(4)
A solution of n-butyllithium (2 ml–1.6 M in hexane)w l) of
(3) (0.73 g–0.0033 mol) at 0◦C. The reaction mixture wasallowed to warm to room temperature and stirred overnight.After filtration, compound(4) was obtained as a white powder(0.22 g–0.001 mol) in 30%yield.
2.1.5. Synthesis of the titanocene dichloride complex (5)A solution of (4) (1.51 g–0.007 mol) in THF (15 ml)
was slowly added to a stirring solution of CpTiCl3 [11](1.45 g–0.007 mol) in THF (30 ml) at room temperature.After 12 h of stirring, THF was evaporated under vacuumand the residue was dissolved in toluene. After filtration,toluene was removed. The product was washed with pentaneand obtained as a red powder in almost quantitative yield(90%).
1H NMR (CDCl3, 300 MHz, RT):δ = 3.10 (dd,3JH–H =8.4 Hz,2JH–H = 15.3 Hz, 1H), 3.21 (dd,3JH–H = 4.2 Hz,2JH–H= 15.3 Hz, 1H), 3.93 (dd,3JH–H = 7.5 Hz, 2JH–H = 11.5 Hz,1H), 4.24 (dd,3JH–H = 2.12 Hz,2JH–H = 11.5 Hz, 1H), 4.43(m, 1H), 6.32 (d,4JH–H = 3.6 Hz, SC4H2, 1H), 6.35 (d,4JH–H = 3.6 Hz, SC4H2, 1H), 6.41 (m, Cp, 1H), 6.49 (m, Cp,2H), 6.53 (m, Cp, 1H), 6.61 (pseudo-s,C5H5, 5H).
13C NMR (CDCl3, 75 MHz, RT): δ = 32.7 (s), 68.4 (s),73.7 (s), 100.0 (s, SC4H2), 100.1 (s, SC4H2), 115.0 (s, Cp),115.1 (s, Cp), 120.3 (s,C5H5), 125.0 (s, Cp), 125.1 (s, Cp),132.9 (s, Cp), 141.8 (s, SC4H2), 141.9 (s, SC4H2).
Anal. calc. of TiC17H16S02Cl2: C: 50.63, H: 4.01, S: 7.95;f
as added dropwise to a diethylether solution (60 m ound: C: 49.82, H: 4.18, S: 7.58.
M. Skompska et al. / Electrochimica Acta 51 (2006) 2108–2119 2111
2.2. Electrochemical experiments
All electrochemical experiments were done in a con-ventional, one compartment cell with a platinum wirecounter electrode (CE) and Ag/(0.01 M AgNO3 + TBAPF6in CH3CN) double junction reference electrode (RE). Allpotentials below are referred to this reference electrode. Thepotential of this electrode is about 0.33 V versus SCE (aq)reference electrode[1].
The redox behavior of the monomer, titanocene-methyl-ethylenedioxythiophene (Tc1EDOT) of concentration0.001 M was studied at Pt disc electrode in different solvents:acetonitrile (AN), tetrahydrofuran (THF), dichloromethane(DCM) in the presence of 0.1 M tetrabuthylammoniumhexafluorophosphate (TBAPF6) as the supporting electro-lyte.
Electropolymerization of titanocene-methyl-ethylene-dioxythiophene was carried out on a Pt disc electrode (surfacearea: 0.005 or 0.02 cm2) from 0.001 M monomer solutionin acetonitrile containing 0.1 M TBAPF6, by means of anAUTOLAB potentiostat (Ecochemie, The Netherlands).
Electrodeposition of p(Tc1EDOT) was firstly studied bycyclic voltammetry to optimize the polymerization condi-tions and then, the films for further measurements in themonomer-free solutions were obtained potentiostatically at0.96 V at the polymerization charge 25 mC cm−2. Aftere ace-t r-t o themD or-o drya , thefi doxa0 ,f a-t thet
italI TMa TheA Sicr
3
3
lec-t nts( Fs ange0
The reduction peaks visible at about−1.15 V in THF andAN and at about−1.1 V in DCM at the scan rate of 0.1 V s−1,correspond to the reaction which consists of electron-transferstep followed by the complete or partial dissociation of anion-radical:
Cp2TiCl2 + e� Cp2TiCl2− (1)
Cp2TiCl2− � Cp2TiCl + Cl− (2)
as has been reported for Cp2TiCl2 [6,7]and titanocene centersimmobilized in polypyrrole matrix (Tc3Py)[1,2]. The chlo-ride ligand must be replaced by another moiety, either thesolvent molecule (S) or another Cp2TiCl molecule to formCp2TiCl(S) or dimeric species (Cp2TiCl)2, respectively.
The course of the reverse voltammetric scan stronglydepends on the solvent used in the experiment. A quasi-reversible voltammetric redox behavior, with anodic tocathodic peak separation of about 90 mV, linear depen-dences of anodic and cathodic peak currents on the squareroot of the scan rate and anodic-to-cathodic peak currentratio close to one (0.9 for 100 mV/s) is observed in THF(Fig. 1a).
More complicated picture of the redox process is obtainedin DCM (Fig. 1b). At low scan rates the curves resemble thequasi-reversible profiles, but with a greater peak separationthan that in THF (120 mV at the scan rate of 40 mV s−1).T oth ofp -t tureo CMiI nt oft nr
thew AN( oren eoxi-d thant
T entc lit-e atings gert reat HFt maya oft ofc to y ofC nTe is-
lectrosynthesis the polymer films were rinsed withonitrile (AN), then with the solvent in which the fuher measurements were performed, and placed intonomer-free solutions of 0.1 M TBAPF6 in AN, THF orCM. Prior to experiments all the solutions were thughly deaerated by vacuum pumping and filling withrgon. Before the measurements of Tc electroactivitylms were subject to three cycles in the range of rectivity of the polymer matrix, namely between−0.5 and.7 V in the presence of TBAPF6 or in limited range
rom −0.5 to 0 V in TEACl to avoid the film degradion at higher potentials in this solution (see further inext).
Nanoscope III and Nanoscope IIIA Quadrex (both Dignstruments/Veeco, USA) were used, respectively, for Snd AFM imaging the surfaces of the samples studied.FM image was done in oscillating contact mode withantilever (spring constant equal to 2.0 N m−1) in air at 33.5%elative humidity.
. Results and discussion
.1. Redox properties of Tc1EDOT in solution
Fig. 1presents cyclic voltammograms obtained on Pt erode in the solution of 0.001 M Tc1EDOT in various solveTHF, DCM and AN) in the presence of 0.1 M TBAP6upporting electrolyte, at several scan rates in the r.01–0.8 V s−1.
he increase of the scan rate results in the increase beak separation (240 mV at 600 mV s−1) and the cathodic
o-anodic peak ratio. However, the most interesting feaf the voltammograms performed at high scan rates in D
s appearance of an additional anodic peak at about−0.66 V.t is also worth noting that there is a gradual replacemehe peak at−0.92 V by the one at−0.66 V for higher scaates.
A drastic deviation from quasi-reversible behavior inhole scan range is observed for the redox process in
Fig. 1c). The potential of Tc reduction shifts towards megative values with the increase of the scan rate, the ration peak is located at a much more positive potential
hat in THF (−0.63 and−1.08 V, respectively).The differences in the redox behaviors of Cp2TiCl2 in
HF, DCM and AN can not be easily explained by differoordination properties of the solvents. According to therature, dichloromethane is one of the weakest coordinolvent, whereas the coordination ability of THF is stronhan that of AN[12,13]. Thus, if the solvent molecules accommodated by the reduced complex (to form Cp2TiClS)
he reoxidation reaction should be more hindered in Than in AN, but it is not the case. Another parameter thatffect the reversibility of the process is solvation ability
he solvents with respect to Cl− ions. It influences the rateleavage of Cl− from Cp2TiCl2− and equilibrium constanf reaction(2). It has been reported that the free energl− solvation (−�G◦
solv) is two times higher in AN than iHF (302.6 and 149.2 kJ mol−1, respectively)[14]. Thus, thequilibrium in AN is strongly shifted towards products of d

2112 M. Skompska et al. / Electrochimica Acta 51 (2006) 2108–2119
Fig. 1. Cyclic voltammograms of 0.001 M Tc1EDOT at Pt electrode in THF (a), DCM (b) and AN (c) +0.1 M TBAPF6 at different scan rates (indicated in thegraphs in V s−1).
sociation. Because of a low concentration of anion-radicalsCp2TiCl2− in the AN solution and their slow generation bythe association reaction the reoxidation in the course of theback scan involves the dissociated product, Cp2TiCl or itsdimer, giving rise a peak at a much less negative potential,compared to the case of the THF solution.
3.2. Electrochemical polymerization of Tc1EDOT on Ptelectrode. A comparison with polymerization of EDOTand hydroxymethylated EDOT
One of the reasons of our decision to attach the titanocenecomplex to EDOT moiety is a relatively low redox potentialof PEDOT. As visible inFig. 2, electroactivity of PEDOTextends over a broad potential range and the reduction peakof the polymer is located at about−0.8 V, being relativelyclose to the reduction wave of titanocene dichloride. There-fore, it seemed plausible that electroactivity of the polymermatrix in the p(Tc1EDOT) derivative would be also shiftedtowards more negative values with respect to that of p(Tc3Py)derivative. The higher conductivity of the polymer matrix inthe range of electroactivity of the titanocene centers wouldallow for more efficient reduction of the centers immobilizedin the polymer film and better reversibility of the process.
In order to check this supposition the p(Tc1EDOT) filmswere deposited on Pt substrates from acetonitrile solution ofthe monomer and then studied in monomer-free solutions of0.1 M TBAPF6. For better understanding of electrochemicalproperties of the monomer and polymer, the similar investiga-
Fig. 2. Comparison of the redox activity of PEDOT film (1) and Tc1EDOT(2) monomer in acetonitrile +0.1 M TBAPF6 at the scan rate 100 mV s−1.

M. Skompska et al. / Electrochimica Acta 51 (2006) 2108–2119 2113
Fig. 3. First voltammetric cycles for oxidation of EDOT (curve 1), Tc1EDOT (curve 2) and EDOTMet (curve 3) in AN solution in the potential range from−0.8 to 1.3 V (a) and from−0.8 to 1.8 V (b). Curve 0 corresponds to a baseline registered in the monomer-free solution of 0.1 M TBAPF6.
tions were performed with unsubstituted monomer (EDOT)and hydroxymethylated EDOT (EDOTMet)
The first voltammetric cycles for these three monomerspresented inFig. 3a shows that the oxidation potentials of theTc1EDOT and EDOTMet (identified with the peak potentialsof the monomers oxidation) are only slightly shifted (by about70–80 mV) in the positive direction with respect to the valueof unsubstituted EDOT.
This suggests that the electronic effect of both substituentsis rather weak. In spite of the same monomer concentra-tions, the monomer oxidation peak is the highest for EDOT,probably due to the highest value of diffusion coefficientof this unsubstituted molecule. Another possible origin ofthis greater oxidation rate of EDOT may be an acceleratingeffect of the subsequent chemical steps, dimerization of rad-ical cations and the deprotonation[15,16] which should bemuch faster for the unsubstituted species. As an indicationto such slower chemical steps for substituted monomers onemay point to the absence of the characteristic loop, the cross-ing of the oxidation and reduction branches near the positivepotential limit during the first cycle, which is observed forthe EDOT species.
The anodic extension of the polarization range to 1.8 Vreveals the presence of the second irreversible peak, at about1.6 V, corresponding to overoxidation of the monomeric oroligomeric species in solution or the polymer film alreadyd n ofa t int cur-r nglys theo micp in-i 8 Vf
Ta clicv
The shape of polymerization curves for EDOT derivativesin the range of the polymer matrix response is qualitativelydifferent from that of the parent compound. Formation ofthe film on the electrode gives rise to the redox peaks ofthe polymer matrix but the character of these peaks is dif-ferent for each of the three polymers studied. In the case ofp(Tc1EDOT), there is only one well developed and symmet-rical redox couple followed by a broad “capacitive” plateau,in contrast to double and non-symmetrical peaks for PEDOT.In the case of PEDOTMet, the redox peaks are broad and thedeposition of the polymer occurs rather slowly.
A more detailed picture of redox behaviors of the threepolymers obtained potentiostatically for the same deposi-tion charge density, 25 mC cm−2, is presented inFig. 5. Thevoltammograms were registered in the monomer-free solu-tion of 0.1 M TBAPF6 in AN. The ratio of the polymerreduction charge to polymerization charge may be used toestimate the polymerization yield. These values are 13% forPEDOT, 11% for p(Tc1EDOT) and 7% for PEDOTMet.
According to our previous discussion concerning thepTc3Py derivative[1,2] based on the treatment of Diaz etal. [17], the upper limit (ymax) of the ratio of the redox-to-deposition charge,y, for polypyrrole is within 9–11%,assuming that two electrons are involved in polymerizationof a monomer unit and aboutn = 0.2–0.25 of an electron isneeded to charge a unit inside the polymer:
y
w pro-c nt onv eac-t
wella ly. Inr posi-t thec au).
eposited on the electrode. This leads to the formatiothin overoxidized layer which is highly resistive so tha
he next scans after this extended first cycle the oxidationent is markedly lower and the oxidation peaks are strohifted to more positive potentials. Therefore, to avoidveroxidation process, the anodic limits for potentiodynaolymerization of the three polymers studied were dim
shed to 1.15 V for electrodeposition of p(Tc1EDOT), 1.1or p(EDOTMet) and 1.1 V for PEDOT.
Fig. 4 illustrates the formation of p(Tc1EDOT), PEDOnd PEDOTMet films on Pt electrodes by means of cyoltammetry.
= Qred
Qdep= ηymax, ymax = n
(2 + n)(3)
hereη < 1 characterizes the efficiency of the depositioness to produce redox-active units in the film dependearious losses (formation of soluble oligomers, side rions, deposition of redox-inactive material, etc.).
One should keep in mind that the latter parameter ass the redox charge are not defined quite unambiguouseality, they both depend essentially on the value of theive potential limit (0.6 V in our measurements) becauseurrent does not vanish in this range (“capacitive” plate

2114 M. Skompska et al. / Electrochimica Acta 51 (2006) 2108–2119
Fig. 4. Cyclic voltammograms for electrodeposition of p(Tc1EDOT) (a), PEDOT (b) and PEDOTMet (c) from acetonitrile solutions of 1 mM monomers inAN/0.1 M TBAPF6. The anodic potential limits were, respectively, 1.15, 1.1 and 1.18 V.
We have chosen this limit as allowing us to avoid the matrixdegradation (“overoxidation”).
Even for the same polymer this “safe potential limit”depends on experiment conditions, e.g. on the nature of
Fig. 5. Cyclic votammograms for PEDOT (curve 1), p(Tc1EDOT) (curve 2)and PEDOTMet (curve 3) films in 0.1 M TBAPF6/AN solution at the scanrate of 100 mV/s. The films were obtained by potentiostatic polymerizationat 0.91 V (1), 0.96 V (2) and 1.0 V (3) with the same deposition charge,25 mC/cm2.
the solvent and its purity, e.g. the water content in AN orpropylene carbonate influences dramatically the overoxida-tion process[16,18].
If this restriction is disregarded, one can integrate thecharging curve up to a much higher potential, in particular upto the potential range in which the current is vanishing (thisrange can be reached not for all polymers). Then, the valueof the electron charge per monomer unit in the film may bemuch higher (for the same polymer film), e.g. 0.6 for PEDOT[19] or even 1 for poly(4,4′-bimethoxybithiophene)[20].
Even for our restricted potential range, up to 0.6 V, thisvalue may be different for various polymers. For PEDOTwhose redox activity starts at very negative potentials onemay expect a bit higher value, e.g. 0.3[21] which wouldenhance the “theoretical” limit for the redox-to-depositioncharge ratio up toymax≈ 13%. This value corresponds per-fectly to the one found by us experimentally which testifiesin favor of the practical absence of losses during our PEDOTdeposition,η ≈ 1, Eq.(3). The ranges of electroactivity of twoother polymers are shifted strongly in the positive directionso that the standard value forn=0.25, should be reasonablefor our potential range. Then, the deposition efficiency,η,is also close to 1 for p(Tc1EDOT) but noticeably lower for

M. Skompska et al. / Electrochimica Acta 51 (2006) 2108–2119 2115
PEDOTMet, about 0.65. The latter may be explained by avery low deposition rate for this polymer (Fig. 4c) leading toa much higher loss of soluble oligomers to the bulk solution.
The potentials of the oxidation peaks of both functional-ized polymers are shifted by about 170 mV to more positivepotentials with respect to that of PEDOT. Since the size oftitanocene substituent is markedly larger than that of hydrox-ymethyl one, the anodic shift of oxidation potential of thePEDOT derivatives can not be simply explained by sterichindrances for planarization of polymer chains during oxida-tion due to the presence of side groups.
Hypothetically, the substitution may diminish the solubil-ity of oligomers (as one can see from a much lower solubilityof the substituted monomers in AN) thus resulting in theirearlier deposition, i.e. to the formation of the matrix withshorter chain length. In its turn, this factor should influencethe conjugation length and correspondingly the process of thematrix oxidation. This hypothesis might be checked if findinga solvent for the film dissolution to determine the molecularmass, not available actually for substituted polymers.
In the case of PEDOTMet (curve 3 inFig. 5), the redoxactivity is observed within a very broad range of potential. Itmay be related to a wide dispersion of the chain/conjugationlengths in this film.
In spite of the shift of the peak potential of p(Tc1EDOT)film in the positive direction compared to PEDOT, its redoxp Py)( cen-t edf ofa llert
i-n siont sulti es-e ins.
Fig. 6. Cyclic voltammograms for Pt electrodes modified with PEDOT(curve 1), (Tc1EDOT) (curve 2) and p(Tc3Py) (curve 3) films in 0.1 MTBAPF6/AN solution.
Therefore, it is recommended to decrease the polymeriza-tion potential by applying the potentiostatic polymerization,instead of potentiodynamic one. Potentiostatic polymeriza-tion allows also for an easy control of the charge passed duringelectrodeposition and in effect the thickness of the polymerfilm formed on the electrode.
A series of chronoamperometric curves obtained on Ptelectrode in 1 mM Tc1EDOT solution in AN +0.1 M TBAPF6for the steps from 0 V to different polymerization potentials,located in the range of increasing part of the cyclic voltam-mogram (Fig. 4a), are presented inFig. 7. The increase of thepolymerization potential results in the increase of the currentmaximum in the chronoamperograms and its shift to shortertimes. On the other hand, the resulting cyclic voltammogramsof the films obtained at different potentials but at the samepolymerization charge, 15 mC cm−2, are the same (Fig. 7b).
Since the film formation at the lower potentials is ratherslow, the polymer films for further studies were obtained at0.96 V.
F 1EDOT 0.98 V (4)f s of re
otential is less positive, by about 0.4 V, than that of p(Tc3Fig. 6). Thus, one may expect that the redox peaks of Tcers immobilized in p(Tc1EDOT) film will be still separatrom the redox activity of the polymer matrix by a windowrelatively low conductivity but its width should be sma
han that observed for p(Tc3Py).Cyclic voltammetry is a convenient method for prelim
ary studies of polymerization process. However, excuro the range of relatively high positive potentials may ren overoxidation of the polymer film, especially in the prnce of water traces, or cross-linking of the polymer cha
ig. 7. (a) Chronoamperometric curves for electrodeposition of p(Tcrom acetonitrile solution of 1 mM monomer. (b) Cyclic voltammogram
) by potential steps from 0 to: 0.92 V (1), 0.94 V (2), 0.96 V (3) andsultant polymer films in 0.1 M TBAPF6/AN at the scan rate of 100 mV s−1.

2116 M. Skompska et al. / Electrochimica Acta 51 (2006) 2108–2119
Fig. 8. AFM image 2.5�m× 2.5�m obtained in oscillating contact mode.Relative height: 10 nm. Mean roughness over 2�m× 2�m (512× 512points): 0.7 nm.
3.3. STM and AFM studies of morphology of the filmsobtained by potentiodynamic and potentiostaticpolymerization
Films for atomic-force microscopy (AFM) were depositedat Pt surface by the potentiodynamic procedure. The externalfilm surface (Fig. 8) is very flat, with the roughness below1 nm and the relative height within 10 nm at the 2�m scale.Along the surface one can see structural elements in the rangeof 100 nm.
The films for scanning tunneling microscopy (STM) wereobtained on indium–tin oxide (ITO)-glass coated substrate.Since ITO surface is not perfectly flat and its morphology mayinfluence the morphology of the film grown on it, we firstlyperformed the images of bare ITO substrate. The STM imageof its surface presented inFig. 9 shows a granular surface,similar to that presented in the literature[22,23], with a grainsize from several up to several tens of nanometers. The surfaceis relatively flat with variations up to 10 nm.
Deposition of the polymer film changes markedly theimage of the surface, as visible inFig. 10.
Moreover, morphology of the film depends on the poly-merization regime, by the cyclic voltammetry (images on theleft) or potentiostatically (images on the right). The formerfilm consists of the zones, each zone being rather uniformand composed of elements having a similar shape and size,whereas the orientation and the extension of structures in dif-ferent zones are different. The film obtained potentiostaticallyis well ordered and reveals a great compactness. As visible inthe image at the lower magnification (1�m× 1�m) it con-sists also of the zones but the orientation of structures in allzones is the same.
The observed difference between these two types of filmsmay be explained considering the peculiarities of the twopolymerization regimes. In potentiodynamic polymerizationin each scan the potential changes between−0.5 and 1.18 Vand in each scan the oxidation of monomer and likely thepropagation of polymer chains is interrupted for 20 or even30 s when the polymer already formed on the electrodesurface is transformed in the insulating state. In the sub-sequent scan the polymerization process is again initiatedin the range above 0.9 V. This probably results in diver-sity in orientation in the separate zones of the polymerfilm. In contrast, during the potentiostatic polymerization thefilm is growing without interruption, giving rise to a morec filmm
3fic
icals solu-ts t thep -i fort amec f the
ges of a
Fig. 9. STM imaompact polymer structure and a more homogeneousorphology.
.4. Electrochemical characterization of p(Tc1EDOT)lms in 0.1 M TBAFP6 in THF, DCM and AN: aomparison with behavior of p(Tc3Py)
The p(Tc1EDOT) and pTc3Py films for electrochemtudies in various solvents were obtained in acetonitrileion of the monomer (0.001 M) + 0.1 M TBAPF6 by potentio-tatic polymerization at 0.96 and 0.76 V, respectively, aolymerization charge of 25 mC cm−2. A good reproducibil
ty of the redox properties in the range of polymer matrixhe films with the same redox charge, obtained in the sonditions, allows us to assume that the films were o
bare ITO surface.

M. Skompska et al. / Electrochimica Acta 51 (2006) 2108–2119 2117
Fig. 10. The STM images of ITO covered by p(Tc1EDOT) films obtained by potentiodynamic (a, c) and potentiostatic polymerization at 0.96 V (b, d).
same thickness and having a similar structure, as confirmedby STM studies.
After deposition the films were cycled in 0.1 MTBAPF6/AN solution in the potential range from−0.6 to0.6 V to obtain the reproducible behavior and to checkwhether the electroactivity of all films was the same. Then,the films were rinsed with AN and with the appropriate sol-
Fig. 11. Cyclic voltammograms obtained for three identical p(Tc1EDOT)films obtained in AN solution (at the deposition charge density of25 mC cm−2) and transferred into the monomer free solutions of 0.1 MTBAPF6 in AN (1), THF (2) and DCM (3).
vent, placed in the solution of 0.1 M TBAPF6 in this solventand again cycled several times until obtaining a stable voltam-mogram.
As visible inFig. 11, the shape of voltammograms of thepolymer matrix is nearly independent of the type of the sol-vent, i.e. the electroactivity of the film in these media is verysimilar.
Electrochemical investigations of electroactivity of freetitanocene complexes in the solutions, presented in Sec-
Fig. 12. A comparison of first voltammetric cycles for p(Tc1EDOT) (curve1) and p(Tc3Py) (curve 2) films in 0.1 M TBAPF6/THF at the scan rate of100 mV s−1.

2118 M. Skompska et al. / Electrochimica Acta 51 (2006) 2108–2119
Fig. 13. An evolution of the consecutive cyclic voltammograms for p(Tc1EDOT) (a) and p(Tc3Py) (b) films in 0.1 M TBAPF6/THF.
tion 3.1, revealed that the most reversible behavior isobtained in THF (Fig. 1a). Therefore, this solvent was cho-sen for comparative studies of electrochemical behaviorof two polymers, p(Tc1EDOT) and poly(titanocene-propyl-pyrrole), p(Tc3Py). The properties of the latter have beendiscussed by us previously[1–3].
A comparison of electrochemical behaviors ofp(Tc1EDOT) and p(Tc3Py) films (obtained at deposi-tion charge density of 25 mC cm−2) in 0.1 M TBAPF6/THF,is presented inFig. 12.
General features of the graphs are the same: reduction peakof Tc is located at about−1.2 V for p(Tc3Py) and−1.15 Vfor p(Tc1EDOT), the reoxidation peaks respectively at about−0.95 and−0.8 V and pronounced prepeaks at the begin-ning of oxidation waves of the polymer matrices[1,2]. Asexpected, the prepeak for p(Tc1EDOT) is located at a lowerpotential than that for p(Tc3Py), due to the shift of the poly-mer matrix electroactivity to more negative potentials. Thisis probably also the reason of the shift of the reduction peakof Tc immobilized in the PEDOT matrix towards less nega-tive potentials. In contrast, a little surprising is a more anodicposition of reoxidation peak. This suggests that the chargetransfer between the Tc centers during reoxidation is moreimpeded than that in the case of p(Tc3Py). After polarizationof the polymer to the potential−1.6 V the polymer matri-ces are in their insulating state. Thus, the charge transferb cha-n s thed turnd lymec aceri pylc in am d inP
mso MT ceb
The redox activity of Tc centers immobilized in PEDOTmatrix is much less stable than that of the centers immobi-lized in PPy film. In the latter case, when the experimentswere performed in dry and oxygen-free conditions the elec-troactivity of the centers in the subsequent 10 scans changedonly slightly. In contrast, under the same conditions we werenot able to prevent a progressive decrease of the redox activityof centers in PEDOT matrix. This difference is rather difficultto explain. A further study is needed to establish the reasonof this different behavior.
4. Conclusions
We have performed the synthesis and electrochem-ical characterization of a new monomer (Tc1EDOT)with titanocene dichloride complex (Tc) linked to 3,4-ethylenedioxythiophene (EDOT) moiety by methyl chain.Electrochemical behavior of Tc centers attached to EDOTin solution is the same as that of unsubstituted titanocenedichloride or titanocene-propyl-pyrrole (Tc3Py) moiety. Inparticular, the reversibility of redox reaction of the new Tcderivative depends strongly on the properties of the solvent,namely on its solvation ability with respect to Cl− ion. Aquasi-reversible behavior was obtained in THF, whereas thevoltammograms obtained in AN are characteristic of an irre-v
andI en-t t thep pactm ymerm
OTm sul-t lt ofe f thec th as
etween Tc centers occurs exclusively by hopping meism. Therefore, the crucial parameter in this process iistance between neighboring redox centers which in itsepends on the character of the spacer between the pohain and Tc complex. In the case of p(Tc1EDOT) the sps more rigid and more voluminous than the flexible prohain between pyrrole and Tc centers. This may resultore difficult transport between the centers immobilizeEDOT matrix.A comparison of the consecutive cyclic voltammogra
f p(Tc1EDOT) and p(Tc3Py) films in THF solution of 0.1BAPF6, presented inFig. 13reveals an important differenetween the redox behaviors of the two polymer films.
r
ersible process.The Tc1EDOT species can be polymerized on Pt
TO electrodes from AN by potentiodynamic and potiostatic methods. The STM images have shown thaotentiostatic regime leads to the film of a more comorphology and better-ordered arrangement of the policrostructure.Attachment of Tc or methanol substituent to ED
onomer gives rise to a shift of the redox potential of reant polymer to more positive values. This is not a resulectronic effect of pendant group but rather decrease oonjugation length due to formation of the polymers wihorter chain/conjugation length.

M. Skompska et al. / Electrochimica Acta 51 (2006) 2108–2119 2119
The redox peaks of p(Tc1EDOT) matrix are shiftedtowards more negative potentials with respect to those ofp(Tc3Py), i.e. conductivity window of p(Tc1EDOT) filmextends in the range of more negative potentials. Thisresults in the shift of redox potential of titanocene cen-ters immobilized in the p(Tc1EDOT) matrix to less negativepotentials in comparison to that of the centers in p(Tc3Py)film.
Acknowledgements
This study was realized as a joint project within the frame-work of the bilateral Polonium program 5562.II; 07586QE.We are thankful to Evelyne Pousson for technical assistanceand the elemental analysis of the samples.
References
[1] M.A. Vorotyntsev, M. Casalta, E. Pousson, L. Roullier, G. Boni, C.Moise, Electrochim. Acta 46 (2001) 4017.
[2] M.A. Vorotyntsev, M. Skompska, E. Pousson, J. Goux, C. Moise, J.Electroanal. Chem. 552 (2003) 307.
[3] M. Skompska, M.A. Vorotyntsev, J. Goux, C. Moise, O. Heinz, Y.S.Cohen, M.D. Levi, Y. Gofer, G. Salitra, D. Aurbach, Electrochim.Acta 50 (2005) 1635.
C.
[5] E. Laviron, J. Besanson, F. Huq, J. Organometal. Chem. 159 (1978)279.
[6] Y. Mugnier, C. Moise, E. Laviron, J. Organometal. Chem. 204 (1981)61.
[7] R.J. Enemærke, J. Larsen, T. Skrydstrup, K. Daasbjerg,Organometallics 23 (2004) 1866.
[8] M. Dietrich, J. Heinze, G. Heywang, F. Jonas, J. Electroanal. Chem.369 (1994) 87.
[9] Q. Pei, G. Zuccarello, M. Ahlskog, O. Inganas, Polymer 35 (1994)1347.
[10] A. Lima, P. Schottland, P.S. Sadki, C. Chevrot, Synth. Met. 93 (1998)33.
[11] C. Cardoso, J.C.S. Moorhouse, Dalton Trans. (1980) 1156.[12] C. Reichardt, Solvents and solvent effects in organic chemistry, sec-
ond ed., Wiley-VCH, Weinheim, 1988.[13] Y. Marcus, Chem. Soc. Rev. (1993) 409.[14] J.C. Lauer, Electrochim. Acta 9 (1964) 1617.[15] M. Zhou, J. Heinze, Electrochim. Acta 44 (1999) 1733.[16] M. Zhou, J. Heinze, J. Phys. Chem. B 103 (1999) 8451.[17] A.F. Diaz, J.I. Castillo, J.A. Logan, W.Y. Lee, J. Electroanal. Chem.
129 (1981) 115.[18] M.D. Levi, E. Lankri, Y. Gofer, A. Aurbach, T. Otero, J. Elec-
trochem. Soc. 149 (2002) E204.[19] G. Zotti, G. Schiavon, S. Zecchin, L. Groenendaal, Chem. Mater. 11
(1999) 3624.[20] M. Dietrich, J. Heinze, Synth. Met. 41–43 (1991) 503.[21] H. Randriamahazaka, V. Noel, C. Chevrot, J. Electroanal. Chem. 472
(1999) 103.[22] L. Zhou, P.K.H. Ho, P.C. Zhang, S.F.Y. Li, G.Q. Xu, Appl. Phys. A
66 (1998) S643–S647.[23] J. Lukkari, M. Alanko, L. Heikkila, R. Laiho, J. Kankare, Chem.
[4] M.A. Vorotyntsev, M. Graczyk, A. Lisowska-Oleksiak, J. Goux,Moise, J. Solid State Electrochem. 8 (2004) 818.
Mater. 5 (1993) 289.