effects of busulfan dose escalation on engraftment of infant rhesus monkey hematopoietic stem cells...
TRANSCRIPT

Experimental Hematology 34 (2006) 369–381
Effects of busulfan dose escalation on engraftment of infant rhesusmonkey hematopoietic stem cells after gene marking by a lentiviral vector
Christoph A. Kahla, Alice F. Tarantalb,c,d, Chang I. Leeb,d, Daniel F. Jimenezb,d,Christopher Choia, Karen Peppera, Denise Petersena, Misty D. Fletcherb,d, Alyssa C. Leapleyb,d,
Jennifer Fisherb,d, Travis S. Burnsb,d, Man-Ni Ultschb,d, Frederick J. Doreye, and Donald B. Kohna,e
aDivision of Research Immunology/Bone Marrow Transplantation, The Saban Research Institute of
Childrens Hospital Los Angeles; bCalifornia National Primate Research Center; cDepartment of Pediatrics;dCenter for Fetal Monkey Gene Transfer for Heart, Lung, and Blood Diseases, University of California at Davis, Davis,
Calif., USA; eDepartment of Pediatrics, University of Southern California Keck School of Medicine, Los Angeles, Calif., USA
(Received 29 September 2005; revised 2 December 2005; accepted 6 December 2005)
Objective. Nonmyeloablative cytoreduction is used in clinical hematopoietic stem cell genetherapy trials to increase engraftment of gene-modified cells. We utilized an infant rhesusmonkey model to identify an optimal dosage of busulfan that results in efficient long-termgene marking with minimal toxicities.
Methods. Bone marrow (BM) was harvested, followed by a single 2-hour intravenous infusionof busulfan at escalating dosages of 0 to 160 mg/m2. CD34+ cells were immunoselected fromBM, transduced overnight with a simian immunodeficiency virus–based lentiviral vector car-rying a nonexpressed marker gene, and injected intravenously 48 hours post–busulfan admin-istration. Pharmacokinetics were assessed, as well as adverse effects and peripheral blood andBM gene marking.
Results. Increasing dosages of busulfan resulted in increased area-under-the-curve (AUC)with some variability at each dosage level, suggesting interindividual variation in clearance.Blood chemistries were normal and no adverse effects were observed as a result of busulfaninfusion. At 120 and 160 mg/m2, transient neutropenia and thrombocytopenia were noted butnot lymphopenia. Over the 6 months of study posttransplantation, a busulfan dosage-relatedincrease in gene marking was observed ranging from undetectable (no busulfan) up to 0.1%gene-containing cells in animals achieving the highest busulfan AUC. This corresponds toa more than 100-fold increase in gene marking over the busulfan dosage range studied.
Conclusions. These data indicate that increased gene marking of hematopoietic stem cellscan be achieved by escalating busulfan dosages from 40 to 160 mg/m2 without significanttoxicity in infant nonhuman primates. � 2006 International Society for ExperimentalHematology. Published by Elsevier Inc.
Genetic modification of hematopoietic stem cells (HSCs)has the potential to provide improved treatments for a vari-ety of genetic and infectious diseases. Gene transfer vectorsfor HSCs have been traditionally engineered from gamma-retroviruses because of their ability to integrate into chro-mosomal DNA and remain present as the stem cellsundergo proliferation. Recent studies using these vectors
Offprint requests to: Donald B. Kohn, M.D., Division of Research Im-
munology/Bone Marrow Transplantation, The Saban Research Institute
of Childrens Hospital Los Angeles, 4650 Sunset Boulevard, Mailstop
#62, Los Angeles, CA, 90027; E-mail: [email protected]
0301-472X/06 $–see front matter. Copyright � 2006 International Society for
doi: 10.1016/j.exphem.2005.12.005
in infants with X-linked severe combined immunodefi-ciency syndrome (X-SCID) have provided clinical proof-of-principle for the potential of genetic therapy, eventhough lymphoproliferative disease has been observed inseveral of these patients [1,2]. A new generation of vectorshas been developed derived from lentiviruses, such ashuman immunodeficiency virus (HIV)-type 1 and simianimmunodeficiency virus (SIV). In contrast to gamma-retroviruses, lentiviruses have the ability to integrate intonondividing cells such as quiescent HSCs [3,4]. This allowsa decreased time that target cells require stimulation in vi-tro, which better preserves the self-renewal and multiline-age potential of the HSCs [4].
Experimental Hematology. Published by Elsevier Inc.

370 C.A. Kahl et al./ Experimental Hematology 34 (2006) 369–381
One major challenge for successful HSC gene therapy isthe ability of the genetically modified cells to successfullyengraft in the host bone marrow (BM) compartment. In-fused gene-modified autologous HSCs must compete forengraftment with endogenous cells in the host. In some cir-cumstances, transduced cells expressing a particular trans-gene may have a selective advantage, leading to preferredengraftment of those cells. This occurred in the X-SCIDstudies, where hematopoietic cells were manipulated to ex-press the common g-chain of the lymphoid cytokine recep-tor, which is indispensable for the development of immunecompetent T lymphocytes. In most cases, however, theremay not be a selective advantage resulting from transgeneexpression. Therefore, it is crucial to provide an adequatehost environment that will be beneficial to engraftment ofthe gene-modified cells.
One way to aid engraftment is to ‘‘make space’’ in theBM by administering nonmyeloablative dosages of marrowcytoreduction, such as moderate dosages of total body irra-diation (TBI; 200 to 400 cGy) or chemotherapeutic agents,such as busulfan. Previously, oral administration of busul-fan was associated with relatively large inter- and intrapa-tient variability in bioavailability and unpredictablesystemic exposure. In recent years, an intravenous (IV) bu-sulfan formulation, Busulfex (ESP Pharma, Inc., Edison,NJ, USA), has become available and has been shown toprovide greater predictability in blood levels (area-under-the-curve [AUC]) achieved [5].
Prior studies in murine and nonhuman primate modelshave established that nonmyeloablative conditioning leadsto increased levels of engraftment of gene-marked HSCs[6–9]. In a landmark clinical gene therapy trial, Aiutiet al. combined low-dosage busulfan treatment (4 mg/kg)with retroviral gene therapy in two infants with adeno-sine-deaminase (ADA)-deficient SCID [10]. The studyshowed significant immune reconstitution in the two pa-tients. Although the high marking of T cells could be ex-plained by a selective advantage of lymphocytesexpressing the transgene, a significant frequency of gene-marked cells (up to 10%) was also found among myeloidcells, suggesting that the conditioning regimen had pro-vided enough ‘‘space’’ for HSCs to engraft. In contrast, inthe X-SCID gene therapy trials, patients did not receiveany myeloablative conditioning prior to transplant, and con-sequently significant gene marking was primarily restrictedto lymphoid cell populations that had likely been selectedfor transgene expression.
Despite its success, the study by Aiuti et al. chose thebusulfan dosage empirically, and it is unclear which dos-age(s) will provide optimal engraftment with minimal tox-icity. The objective of this study was to investigate therelationship between busulfan dosage and HSC engraftmentin a clinically relevant infant rhesus macaque bone marrowtransplant (BMT) model. The results of this study revealedthat submyeloablative busulfan dosages were well tolerated
and that busulfan dosages and gene marking were posi-tively correlated, with durable gene marking $ 0.1% of he-matopoietic cells. This corresponds to a more than 100-foldincrease in gene marking compared with animals withoutbusulfan conditioning. Together, this study providesa well-tolerated approach to achieving stable gene markingand potential efficacy in future gene therapy studies involv-ing HSCs in large-animal models. Insights gained from thisstudy will speed the translation of HSC gene therapies intothe clinical realm and aid in the rational design of new genetherapy protocols for humans.
Materials and methods
Vector constructionThe SIV vector packaging plasmid pSIV4D and the transfervector plasmid pSIVgaMES4SIN (both based on wild-typeSIVmac251) were kindly provided by Francois-Loıc Cosset (IN-SERM, Lyon, France), as previously described [11,12]. The plas-mid bgal-NeoB2-SK was generously provided by Cynthia Dunbar(National Institutes of Health, Bethesda, MD, USA), which con-tains the neomycin and b-galactosidase genes that have their startcodons (ATG) mutated to CTG to prevent expression [13]. Thenonexpressing neomycin (NeoNT) sequence was isolated frombgal-NeoB2-SK using the restriction enzymes Sma-1 and Xba-1and was subcloned into a shuttle plasmid. The fragment wasthen excised again with EcoRI and Bgl-II, blunted, and clonedinto pSIVgaMES4SIN in place of the enhanced green fluorescentprotein (eGFP) expression cassette, which had been removed bycutting with AgeI and XhoI and blunting. The resulting plasmidwas named pSIVGNeoNT or SIV-NoN (nonexpressed neomycin).
Vector supernatant productionLentiviral vector was produced by three-plasmid transient trans-fection of 293T cells (No. CRL-11268; American Type CultureCollection [ATCC], Manassas, VA, USA) under BSL-2 conditionsas previously described [14,15]. Production was scaled up to 500cm2 cell culture dishes coated with poly-L-lysine (Sigma Scien-tific, Inc., Brighton, MI, USA) and using 100 mg of transfer vector(pSIVGNeoNT), 100 mg of SIV packaging plasmid (pSIV4D),and 20 mg of envelope plasmid (pMD.G) expressing the vesicularstomatitis virus glycoproteins (VSV-G). Supernatants were col-lected approximately 72 hours after transfection and spun at3500 rpm to pellet cell debris, followed by filtration (0.45 mm)and preconcentration using Centricon Plus-70 centrifugal filterunits with a molecular weight cutoff of 30,000 kDa (Millipore,Billerica, MA, USA). Then, ultracentrifugation was performedat 25,000g for 90 minutes. Pellets were resuspended in approxi-mately 0.1 to 1% of starting volume of phosphate-buffered saline(PBS), aliquoted, and stored at #–80�C until use.
Vector titration293HEK cells (ATCC No. CRL-1573) were transduced with seri-ally diluted aliquots of concentrated vector supernatant for 16hours, using 8 mg/mL Polybrene (Sigma). Because the vector lacksa selectable marker or reporter, titers were measured using Taq-man real-time quantitative polymerase chain reaction (Q-PCR)as previously reported [16,17]. The primer and probe sequences
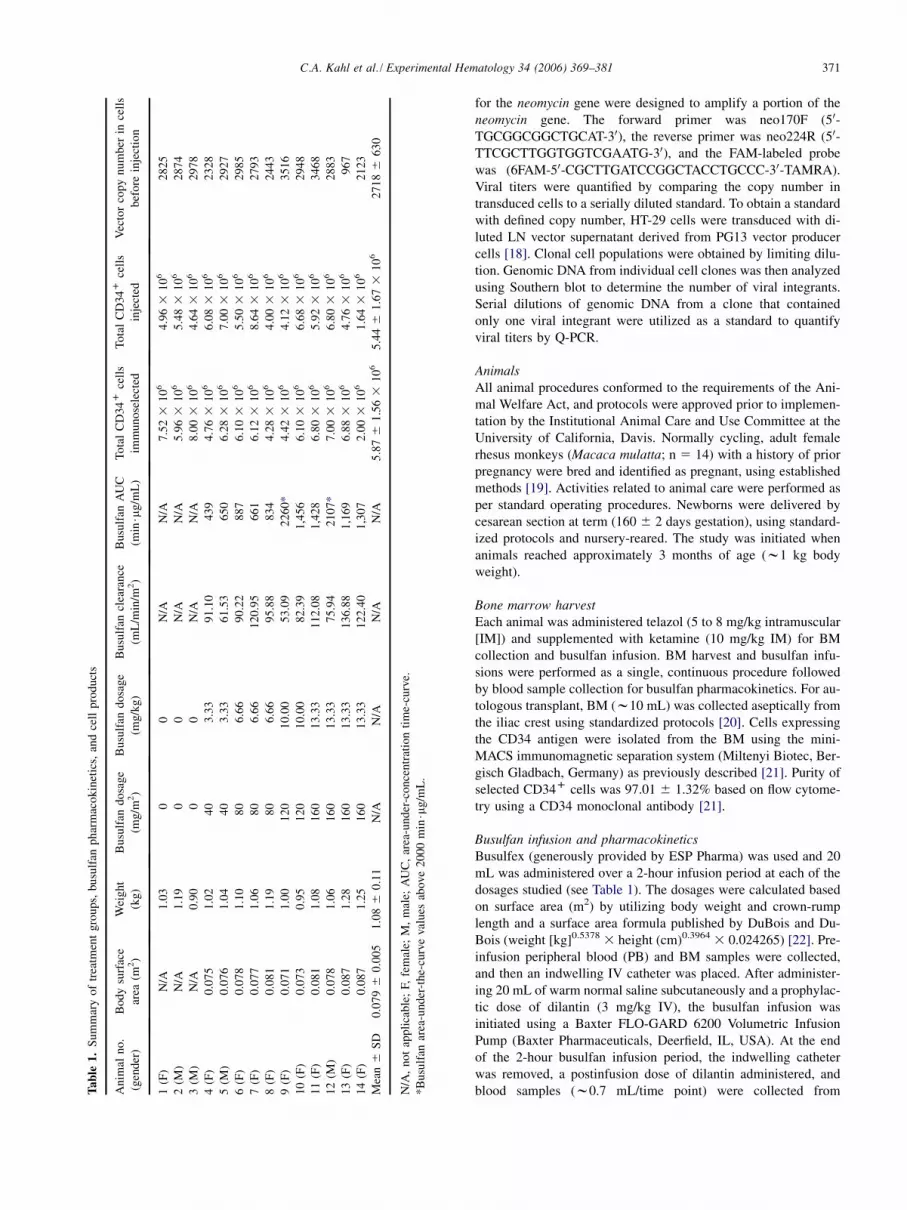
371C.A. Kahl et al. / Experimental Hematology 34 (2006) 369–381
Ta
ble
1.
Su
mm
ary
of
trea
tmen
tg
rou
ps,
busu
lfan
ph
arm
aco
kin
etic
s,an
dce
llp
rod
ucts
An
imal
no
.
(gen
der
)
Bod
ysu
rfac
e
area
(m2)
Wei
ght
(kg
)
Bu
sulf
and
osa
ge
(mg
/m2)
Bu
sulf
and
osa
ge
(mg
/kg)
Bus
ulf
ancl
eara
nce
(mL
/min
/m2)
Bus
ulf
anA
UC
(min
$mg
/mL
)
To
tal
CD
34D
cell
s
imm
un
ose
lect
ed
To
tal
CD
34D
cell
s
inje
cted
Vec
tor
copy
num
ber
ince
lls
bef
ore
inje
ctio
n
1(F
)N
/A1
.03
00
N/A
N/A
7.5
23
10
64
.96
31
06
28
25
2(M
)N
/A1
.19
00
N/A
N/A
5.9
63
10
65
.48
31
06
28
74
3(M
)N
/A0
.90
00
N/A
N/A
8.0
03
10
64
.64
31
06
29
78
4(F
)0
.07
51
.02
40
3.3
39
1.1
04
39
4.7
63
10
66
.08
31
06
23
28
5(M
)0
.07
61
.04
40
3.3
36
1.5
36
50
6.2
83
10
67
.00
31
06
29
27
6(F
)0
.07
81
.10
80
6.6
69
0.2
28
87
6.1
03
10
65
.50
31
06
29
85
7(F
)0
.07
71
.06
80
6.6
61
20
.95
66
16
.12
31
06
8.6
43
10
62
79
3
8(F
)0
.08
11
.19
80
6.6
69
5.8
88
34
4.2
83
10
64
.00
31
06
24
43
9(F
)0
.07
11
.00
12
01
0.0
05
3.0
92
26
0*4
.42
31
06
4.1
23
10
63
51
6
10
(F)
0.0
73
0.9
51
20
10
.00
82
.39
1,4
56
6.1
03
10
66
.68
31
06
29
48
11
(F)
0.0
81
1.0
81
60
13
.33
11
2.0
81
,42
86
.80
31
06
5.9
23
10
63
46
8
12
(M)
0.0
78
1.0
61
60
13
.33
75
.94
21
07*
7.0
03
10
66
.80
31
06
28
83
13
(F)
0.0
87
1.2
81
60
13
.33
13
6.8
81
,16
96
.88
31
06
4.7
63
10
69
67
14
(F)
0.0
87
1.2
51
60
13
.33
12
2.4
01
,30
72
.00
31
06
1.6
43
10
62
12
3
Mea
n6
SD
0.0
79
60
.00
51
.08
60
.11
N/A
N/A
N/A
N/A
5.8
76
1.5
63
10
65
.44
61
.67
31
06
27
18
66
30
N/A
,not
appli
cable
;F,
fem
ale;
M,
mal
e;A
UC
,ar
ea-u
nder
-con
centr
atio
nti
me-
curv
e.
*B
usu
lfan
area
-und
er-t
he-
curv
eva
lues
abov
e2
00
0m
in$m
g/m
L.
for the neomycin gene were designed to amplify a portion of theneomycin gene. The forward primer was neo170F (50-TGCGGCGGCTGCAT-30), the reverse primer was neo224R (50-TTCGCTTGGTGGTCGAATG-30), and the FAM-labeled probewas (6FAM-50-CGCTTGATCCGGCTACCTGCCC-30-TAMRA).Viral titers were quantified by comparing the copy number intransduced cells to a serially diluted standard. To obtain a standardwith defined copy number, HT-29 cells were transduced with di-luted LN vector supernatant derived from PG13 vector producercells [18]. Clonal cell populations were obtained by limiting dilu-tion. Genomic DNA from individual cell clones was then analyzedusing Southern blot to determine the number of viral integrants.Serial dilutions of genomic DNA from a clone that containedonly one viral integrant were utilized as a standard to quantifyviral titers by Q-PCR.
AnimalsAll animal procedures conformed to the requirements of the Ani-mal Welfare Act, and protocols were approved prior to implemen-tation by the Institutional Animal Care and Use Committee at theUniversity of California, Davis. Normally cycling, adult femalerhesus monkeys (Macaca mulatta; n 5 14) with a history of priorpregnancy were bred and identified as pregnant, using establishedmethods [19]. Activities related to animal care were performed asper standard operating procedures. Newborns were delivered bycesarean section at term (160 6 2 days gestation), using standard-ized protocols and nursery-reared. The study was initiated whenanimals reached approximately 3 months of age (w1 kg bodyweight).
Bone marrow harvestEach animal was administered telazol (5 to 8 mg/kg intramuscular[IM]) and supplemented with ketamine (10 mg/kg IM) for BMcollection and busulfan infusion. BM harvest and busulfan infu-sions were performed as a single, continuous procedure followedby blood sample collection for busulfan pharmacokinetics. For au-tologous transplant, BM (w10 mL) was collected aseptically fromthe iliac crest using standardized protocols [20]. Cells expressingthe CD34 antigen were isolated from the BM using the mini-MACS immunomagnetic separation system (Miltenyi Biotec, Ber-gisch Gladbach, Germany) as previously described [21]. Purity ofselected CD34D cells was 97.01 6 1.32% based on flow cytome-try using a CD34 monoclonal antibody [21].
Busulfan infusion and pharmacokineticsBusulfex (generously provided by ESP Pharma) was used and 20mL was administered over a 2-hour infusion period at each of thedosages studied (see Table 1). The dosages were calculated basedon surface area (m2) by utilizing body weight and crown-rumplength and a surface area formula published by DuBois and Du-Bois (weight [kg]0.5378 3 height (cm)0.3964 3 0.024265) [22]. Pre-infusion peripheral blood (PB) and BM samples were collected,and then an indwelling IV catheter was placed. After administer-ing 20 mL of warm normal saline subcutaneously and a prophylac-tic dose of dilantin (3 mg/kg IV), the busulfan infusion wasinitiated using a Baxter FLO-GARD 6200 Volumetric InfusionPump (Baxter Pharmaceuticals, Deerfield, IL, USA). At the endof the 2-hour busulfan infusion period, the indwelling catheterwas removed, a postinfusion dose of dilantin administered, andblood samples (w0.7 mL/time point) were collected from

372 C.A. Kahl et al./ Experimental Hematology 34 (2006) 369–381
a peripheral vessel at 30 minutes, then 1, 3, and 4 hours postinfu-sion for determination of busulfan pharmacokinetics. Plasma wascollected, frozen at #–80�C, and then shipped frozen for analysis(Clinical Pharmacokinetics Laboratory, Seattle Cancer CenterAlliance, Fred Hutchinson Cancer Research Center, Seattle, WA,USA) on the day of collection. Plasma busulfan concentrationswere determined as previously described [23] and the AUC wascalculated using noncompartmental methods within WinNonlin(Pharsight, Mountainview, CA, USA).
Blood and bone marrow sample collectionBlood samples were collected prior to busulfan infusion for com-plete blood counts (CBCs), chemistry panels, peripheral bloodmononuclear cells, immunophenotyping (CD3, 4, 8, 19, 20, 34;w2 mL), then at 4, 7, 11, 15, 21, and 30 days posttransplantationfor CBCs and chemistry panels (w1 mL). Blood and marrow (w3mL) were collected at monthly intervals beginning at 1 monthposttransplantation (see below).
Transduction and transplant of autologous CD34D cellsSelected CD34D cells were plated at 105 cells/cm2 in non-tissueculture-treated 25 cm2 flasks that had been coated with 4 mg/cm2 of the recombinant fibronectin fragment CH-296 (TakaraShuzo Co. LTD, Kyoto, Japan). Prestimulation was then per-formed overnight in X-Vivo 15 serum-free medium (CambrexBio Science Baltimore Inc., Baltimore, MD, USA) supplementedwith 2 mM L-glutamine, 100 units/mL penicillin, and 100 mg/mLstreptomycin (all from Gibco-BRL), and containing 100 ng/mLeach of the recombinant human cytokines: thrombopoietin(TPO), Flt3-Ligand (Flt3-L), and stem cell factor (SCF) (R&DSystems, Minneapolis, MN, USA). The next morning, cells wereresuspended in fresh cytokine-containing medium and transducedwith lentiviral vector supernatant at a concentration of 4.4 3 107
infectious units (IU)/mL and using 4 mg/mL protamine sulfate(American Pharmaceutical Partners, Inc., Schaumburg, IL,USA). After 2 hours the medium volume was doubled, and cellswere incubated at 37�C with 5% CO2 until the next day. Cellswere then washed and resuspended in PBS D 1% autologous se-rum (collected prior to busulfan infusion). Cell counts and viabil-ity were determined using trypan blue exclusion prior to eachtransplant. Small aliquots of cells were used for methylcellulosecolony assays and quantitative PCR analysis, while cell-free su-pernatant was utilized for aerobic and anaerobic culture (all neg-ative). Autologous cells were injected IV into each animal in anapproximate 2 mL volume, approximately 48 hours post–busulfaninfusion under ketamine.
Sample analysisFor evaluation of gene transfer, PB and BM samples were col-lected monthly as noted above. Mononuclear cells (MNCs) andgranulocytes from PB were isolated by density gradient centrifu-gation over Ficoll-Hypaque (1.077 g/cm3, Accurate Chemicals,Westbury, NY, USA) at 400g for 30 minutes at 18�C. Hematopoi-etic colony assays were performed on MNCs, as previously de-scribed [20]. Briefly, MNCs were resuspended in Roswell ParkMemorial Institute (RPMI) and washed. A total of 5 3 104
cells/plate (six plates, 3 3 105 MNCs) from PB and 2 3 104
cells/plate (four plates, 8 3 104 MNCs) from BM were platedin 1 mL MethoCult GFD H4435 containing human erythropoie-tin, granulocyte colony-stimulating factor (G-CSF), granulocyte-
macrophage colony-stimulating factor (CFU-GM), SCF, interleu-kin (IL)-3, and IL-6 (StemCell Technologies, Vancouver, BC,Canada) in fibroblast-free 35-mm culture plates. After a standard-ized 10-day incubation period, erythroid and myeloid progenitorcolonies were counted. Individual hematopoietic colonies werecollected and then lysed and genomic DNA isolated using theQIAamp DNA blood kit (Qiagen, Valencia, CA, USA), as recom-mended by the manufacturer. To detect proviral sequences, real-time Q-PCR using primers and probe for the neomycin gene wasperformed as described above. The 3-globin gene was utilized asan internal control and the primer and probe sequences havebeen previously described [24]. For each sample, parallel reactionswere run in triplicate in separate tubes for the detection of neomy-cin gene sequences in BM and PB fractions. DNA quantities werereported as the DNA genome equivalents per 25,000 cells by usinga conversion factor of 6.6 pg DNA per cell, as previously de-scribed [25].
Results
Study designThe study protocol is shown in Figure 1A. We hypothesizedthat by conditioning animals with escalating dosages of bu-sulfan we would find an optimal cytoreductive dosage thatprovides the highest level of engraftment of gene-markedtransplanted autologous CD34D cells, with minimal toxic-ity as evidenced by marrow suppression and other relatedside effects. The busulfan dosages administered rangedfrom 0 to 160 mg/m2 as studies have revealed that dosingbusulfan in relation to the body surface area, rather thanin relation to body mass, leads to better predictability ofsystemically delivered drug dosage, especially in infants[26–28]. Furthermore, because an expressed marker genesuch as eGFP can be immunogenic in nonhuman primateswithout immune suppression [8], we cloned a nonexpressedneomycin marker gene (NeoNT) into the SIV transfer vec-tor, which was termed the SIV-NoN vector (Fig. 1B).
Effects of busulfan conditioningThe busulfan concentrations in the plasma were sampled atmultiple time points 30 minutes to 4 hours postinfusion andwere used to calculate the AUC for each animal. The AUCis graphically shown for individual animals in Figure 2. Bu-sulfan AUC increased in the higher dosage groups, althoughthere was variability at each dosage level, similar to whathas been observed in human patients [26–28].
Both neutrophil and platelet counts were found to behighly negatively related to the AUC grouping (Spearmancorrelation 5 2.713, p 5 0.004 for neutrophil counts;Spearman correlation 5 2.536, p 5 0.048 for plateletcounts). Whereas there was no or little observable marrowsuppression in the lower busulfan dosage groups, absoluteneutrophil counts (ANCs, Fig. 3A) and platelet counts(Fig. 3B) significantly decreased in the two high-dosagegroups. The decreases were transient and values returnedto baseline by day 45. Animals #9 and #12 achieved

373C.A. Kahl et al. / Experimental Hematology 34 (2006) 369–381
Figure 1. Overall experimental plan and schematic of SIV-nonexpressing neomycin vector (SIV NoN vector). (A): Protocol for engraftment of gene-marked
CD34D cells with busulfan dosage escalation. BM (w10 mL/kg) was harvested from each monkey, followed by a single 20-mL IV infusion of busulfan over
2 hours. Monkeys were transplanted after preconditioning with busulfan at 0, 40, 80, 120, or 160 mg/m2. Peripheral blood busulfan levels were followed over
a period of 4 hours, and the AUC was determined. CD34D cells were isolated from the harvested bone marrow and cultured for 24 hours in serum-free
medium with 100 ng/mL of the cytokines SCF, Flt3-L, and TPO. Cells were then transduced overnight with 4 3 107 IU/mL of SIV-derived lentiviral vector
pseudotyped with the VSV-G and containing a nonexpressed neomycin gene on fibronectin-coated plates D 4 mg/mL protamine sulfate. The following morn-
ing, transduced cells were washed and reinfused IV approximately 48 hours after administration of busulfan. Animals were then monitored for myelosup-
pression, toxicity, and immune function. Gene marking in mononuclear cells, granulocytes, and CD34D and CD342 cells was assessed by determining the
number of integrated proviruses per cell with quantitative PCR. (B): SIV NoN vector. The gene encoding neomycin (neo) was cloned into a third-generation
self-inactivating SIV transfer vector [11]. The vector has a partially deleted gag-pol region (gag), and contains the central polypurine tract (cPPT) and the rev-
responsive element (RRE), as well as the early cytomegalovirus promoter (CMV). Also pictured is the deletion in the 30LTR (DU3) that is copied into the
50LTR during reverse transcription. The translation start codon (ATG) is mutated to a stop codon in order to abolish neo expression in transduced cells. Viral
vector supernatants were produced by cotransfecting 293T cells with SIV-NoN transfer vector, SIV4D packaging plasmid, and pMD.G plasmid expressing
the VSV-G envelope glycoproteins. Integrated provirus was detected by real-time quantitative PCR, using primers (arrowheads) and probe (rectangle) that
bind to the neomycin gene.
busulfan AUC greater than 2000 min$mg/mL and these twoanimals had the greatest evidence of marrow suppression,with ANCs reaching nadirs of less than 500/mm3 on daysD11 and D19, respectively (Figs. 3A and C). Plateletcounts in these animals also dropped significantly, but didnot decline below 100,000 cells/mL (Fig. 3B). The highestAUC of 2260 min$mg/mL was observed in animal #9.
By the day following BM harvest, the hemoglobin haddeclined below 10 gm/dL in most animals but returned tothe normative range within a week. Follow-up analysesalso included monthly CBCs, immunophenotyping, andblood chemistries. No lymphopenia was observed in anyof the animals, consistent with busulfan being myeloabla-tive but not immunosuppressive. Also, animals remainedhealthy, and no signs of clinical toxicities were detected
throughout the course of the study. There were no abnormalelevations of liver enzymes [alanine transaminase (ALT),alkaline phosphatase (ALP)] or bilirubin detected.
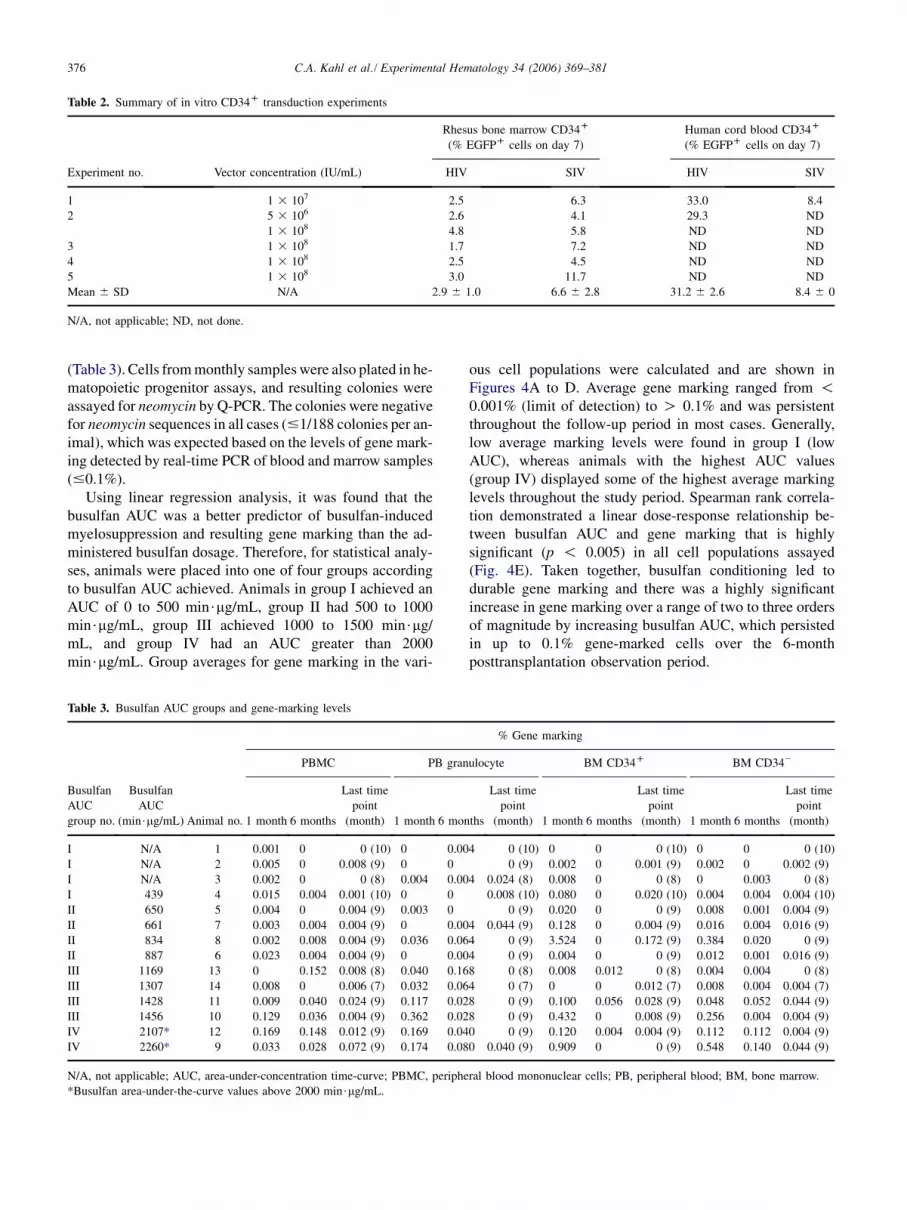
Quantitation of transduction efficiencyBecause we had previously determined that transduction byHIV-1-based lentiviral vectors of CD34D cells from theBM of rhesus macaques was quite low (1 to 3%; data notshown), we utilized an SIVmac251-based lentiviral vectorthat had been reported to yield effective gene transfer torhesus CD34D cells in short-term assays [29]. In prelimi-nary in vitro experiments, we compared transduction of rhe-sus BM CD34D cells by lentiviral vectors derived from theSIVmac251 vector and by a comparable HIV-1-derived vec-tor. Expression was assayed after 7 days of liquid culture

374 C.A. Kahl et al./ Experimental Hematology 34 (2006) 369–381
Figure 2. AUC positively correlates with busulfan dosage. Animals received IV infusions of busulfan at 0, 40, 80, 120, or 160 mg/m2. Peripheral blood
busulfan levels were then followed over a period of 4 hours and the AUC was determined. Plasma busulfan concentrations were determined as previously
described [23] and the AUC was calculated using noncompartmental methods within WinNonlin (Pharsight, Mountainview, CA, USA). The graph shows
busulfan AUC achieved as a function of the busulfan dosage. There was a positive correlation between AUC and busulfan dosage with R2 5 0.74. The ob-
served variability at each dosage level suggests variable clearance.
using an eGFP reporter transgene analyzed by flow cytom-etry. We found that transduction of rhesus CD34D cells bythe SIV-based vector was two- to fourfold more efficientthan by the HIV-1-derived vector. However, the percentagesof rhesus CD34D cells stably transduced by the SIV vectorwere consistently at or below 4 to 12%, even when usingvector concentrations up to 108 IU/mL (Table 2). Thesetransduction levels are manyfold lower than those weachieved in parallel transductions of human CD34D cellsusing HIV-1-type vector (30%, Table 2).
To estimate the efficiency of gene transfer in the BM sam-ples used for the transplants, CD34D cells before and aftertransduction were plated, and collected colonies were sub-jected to Q-PCR for detection of neomycin sequences. Bothcolony-forming unit erythroid (CFU-E) and CFU-GM werefound in anticipated quantities from all animals. Whereasnontransduced CD34D cells gave rise to colonies that wereuniformly negative for the neomycin gene, 100% of the col-onies from posttransduction cells were neomycin-positive inall cases (data not shown). This was striking, as normallyonly a fraction of the colonies would be expected to be trans-duced. In addition, DNAwas extracted from an aliquot of thetransduced cell product on the day of transplant, followed byquantitation of vector copy number by real-time Q-PCR. Ex-tremely high vector copy numbers were detected in cellsfrom all animals, with a mean of 2718 6 630 vector copiesper cell (Table 1).
We speculated that our results might reflect the presenceof nonintegrated, transient vector forms and plasmid carry-over from vector supernatant preparations that are detectedby the sensitive Q-PCR assay. To test this hypothesis, 5 3
106 rhesus BM CD34D cells were transduced in vitro underthe same conditions and with the identical vector superna-tant preparation as was used for the autologous transplants.Genomic DNA was extracted the day after transduction andsubjected to Q-PCR. Similar to our previous results, a high
viral copy number was detected (313 copies/cell). A South-ern blot analysis was performed on DNA from the trans-duced CD34D cells, using restriction enzymes that cuteither once or twice in the vector backbone and a probethat hybridizes to the neomycin gene. This study showedthe presence of vector plasmid DNA used for vector pro-duction associated with the CD34D cells, as well as bothlong terminal repeat (LTR) circles and linear forms, re-vealed with a restriction endonuclease that cut the vectoronce (BglII). These nonintegrated vector forms accountedfor at least 99% of the total vector present (by densitome-try) as revealed with a restriction endonuclease that cutthe vector twice, once in each LTR (StuI), and gave a singleband representing all vector forms (integrated provirus,LTR circles, linear form, and plasmid). In other related ex-periments, Southern blot analysis showed the persistence oftransient forms in lentivirally transduced rhesus CD34D
cells up to 4 weeks in culture.Together, these results suggest that the high vector copy
numbers in freshly transduced CD34D cells, as well as thehigh numbers of neomycin-positive progenitor colonies, arelikely due to the presence of carryover vector plasmid andtransient vector forms that are detected by the sensitiveQ-PCR method. These findings suggest that care must betaken when interpreting results from short-term transduc-tion assays using PCR methods.
Quantitation of gene markingSequential PB and BM samples were collected and ana-lyzed for 6 months posttransplantation, and gene markingwas evaluated in different cell populations using Q-PCR.Table 3 shows marking at 1 and 6 months posttransplanta-tion (4 and 9 months postnatal age) for each animal (#1through #14). Individual marking levels varied widely be-tween the animals but generally peaked in the first month.Control animals that did not receive busulfan displayed

375C.A. Kahl et al. / Experimental Hematology 34 (2006) 369–381
Figure 3. ANCs and platelet counts. (A): ANCs after busulfan infusion. Peripheral blood samples were obtained from animals at twice-weekly intervals in
the first month posttransplantation and then at monthly intervals. CBCs were performed and the ANC was determined. In animals that had received busulfan
dosages of 120 and 160 mg/m2, transient neutropenia was observed. (B): Platelet counts. Peripheral blood samples were collected from animals at twice-
weekly intervals in the first month posttransplantation and then at monthly intervals. Animals that received busulfan dosages of 120 and 160 mg/m2 showed
transient thrombocytopenia. (C): ANC nadirs of individual animals compared with busulfan AUC achieved. The nadirs of the ANC decreased as busulfan
AUC increased, with an R2 5 0.34.
either no gene marking or very low-level marking (!0.01%)of their PB and BM cells. For these animals, gene markingfell below the limit of detection (!0.001%) in most casesby month 6 posttransplantation. In contrast, most animalsin the two high-dosage groups (120 and 160 mg/m2) ex-
hibited up to 100- to 1000-fold higher levels of gene markedcells that remained present after 6 months posttransplanta-tion in most cases (Table 3). In addition, marking was oftenpresent at late time points up to 10 months posttransplanta-tion, suggesting that long-term gene marking was achieved
376 C.A. Kahl et al./ Experimental Hematology 34 (2006) 369–381
Table 2. Summary of in vitro CD34D transduction experiments
Rhesus bone marrow CD34D
(% EGFPD cells on day 7)
Human cord blood CD34D
(% EGFPD cells on day 7)
Experiment no. Vector concentration (IU/mL) HIV SIV HIV SIV
1 1 3 107 2.5 6.3 33.0 8.4
2 5 3 106 2.6 4.1 29.3 ND
1 3 108 4.8 5.8 ND ND
3 1 3 108 1.7 7.2 ND ND
4 1 3 108 2.5 4.5 ND ND
5 1 3 108 3.0 11.7 ND ND
Mean 6 SD N/A 2.9 6 1.0 6.6 6 2.8 31.2 6 2.6 8.4 6 0
N/A, not applicable; ND, not done.
(Table 3). Cells from monthly samples were also plated in he-matopoietic progenitor assays, and resulting colonies wereassayed for neomycin by Q-PCR. The colonies were negativefor neomycin sequences in all cases (#1/188 colonies per an-imal), which was expected based on the levels of gene mark-ing detected by real-time PCR of blood and marrow samples(#0.1%).
Using linear regression analysis, it was found that thebusulfan AUC was a better predictor of busulfan-inducedmyelosuppression and resulting gene marking than the ad-ministered busulfan dosage. Therefore, for statistical analy-ses, animals were placed into one of four groups accordingto busulfan AUC achieved. Animals in group I achieved anAUC of 0 to 500 min$mg/mL, group II had 500 to 1000min$mg/mL, group III achieved 1000 to 1500 min$mg/mL, and group IV had an AUC greater than 2000min$mg/mL. Group averages for gene marking in the vari-
ous cell populations were calculated and are shown inFigures 4A to D. Average gene marking ranged from !0.001% (limit of detection) to O 0.1% and was persistentthroughout the follow-up period in most cases. Generally,low average marking levels were found in group I (lowAUC), whereas animals with the highest AUC values(group IV) displayed some of the highest average markinglevels throughout the study period. Spearman rank correla-tion demonstrated a linear dose-response relationship be-tween busulfan AUC and gene marking that is highlysignificant (p ! 0.005) in all cell populations assayed(Fig. 4E). Taken together, busulfan conditioning led todurable gene marking and there was a highly significantincrease in gene marking over a range of two to three ordersof magnitude by increasing busulfan AUC, which persistedin up to 0.1% gene-marked cells over the 6-monthposttransplantation observation period.
Table 3. Busulfan AUC groups and gene-marking levels
% Gene marking
PBMC PB granulocyte BM CD34D BM CD342
Busulfan
AUC
group no.
Busulfan
AUC
(min$mg/mL) Animal no. 1 month 6 months
Last time
point
(month) 1 month 6 months
Last time
point
(month) 1 month 6 months
Last time
point
(month) 1 month 6 months
Last time
point
(month)
I N/A 1 0.001 0 0 (10) 0 0.004 0 (10) 0 0 0 (10) 0 0 0 (10)
I N/A 2 0.005 0 0.008 (9) 0 0 0 (9) 0.002 0 0.001 (9) 0.002 0 0.002 (9)
I N/A 3 0.002 0 0 (8) 0.004 0.004 0.024 (8) 0.008 0 0 (8) 0 0.003 0 (8)
I 439 4 0.015 0.004 0.001 (10) 0 0 0.008 (10) 0.080 0 0.020 (10) 0.004 0.004 0.004 (10)
II 650 5 0.004 0 0.004 (9) 0.003 0 0 (9) 0.020 0 0 (9) 0.008 0.001 0.004 (9)
II 661 7 0.003 0.004 0.004 (9) 0 0.004 0.044 (9) 0.128 0 0.004 (9) 0.016 0.004 0.016 (9)
II 834 8 0.002 0.008 0.004 (9) 0.036 0.064 0 (9) 3.524 0 0.172 (9) 0.384 0.020 0 (9)
II 887 6 0.023 0.004 0.004 (9) 0 0.004 0 (9) 0.004 0 0 (9) 0.012 0.001 0.016 (9)
III 1169 13 0 0.152 0.008 (8) 0.040 0.168 0 (8) 0.008 0.012 0 (8) 0.004 0.004 0 (8)
III 1307 14 0.008 0 0.006 (7) 0.032 0.064 0 (7) 0 0 0.012 (7) 0.008 0.004 0.004 (7)
III 1428 11 0.009 0.040 0.024 (9) 0.117 0.028 0 (9) 0.100 0.056 0.028 (9) 0.048 0.052 0.044 (9)
III 1456 10 0.129 0.036 0.004 (9) 0.362 0.028 0 (9) 0.432 0 0.008 (9) 0.256 0.004 0.004 (9)
IV 2107* 12 0.169 0.148 0.012 (9) 0.169 0.040 0 (9) 0.120 0.004 0.004 (9) 0.112 0.112 0.004 (9)
IV 2260* 9 0.033 0.028 0.072 (9) 0.174 0.080 0.040 (9) 0.909 0 0 (9) 0.548 0.140 0.044 (9)
N/A, not applicable; AUC, area-under-concentration time-curve; PBMC, peripheral blood mononuclear cells; PB, peripheral blood; BM, bone marrow.
*Busulfan area-under-the-curve values above 2000 min$mg/mL.
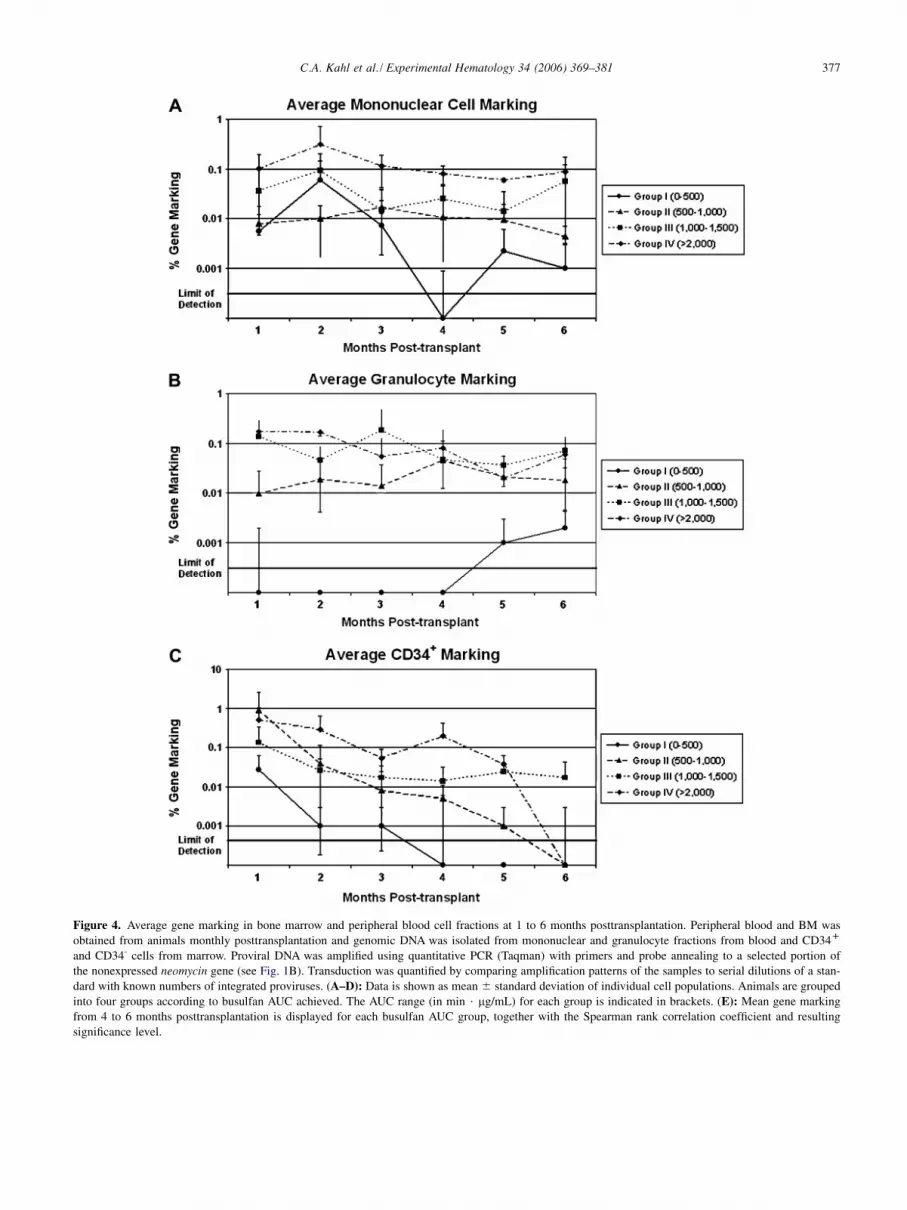
377C.A. Kahl et al. / Experimental Hematology 34 (2006) 369–381
Figure 4. Average gene marking in bone marrow and peripheral blood cell fractions at 1 to 6 months posttransplantation. Peripheral blood and BM was
obtained from animals monthly posttransplantation and genomic DNA was isolated from mononuclear and granulocyte fractions from blood and CD34D
and CD34- cells from marrow. Proviral DNA was amplified using quantitative PCR (Taqman) with primers and probe annealing to a selected portion of
the nonexpressed neomycin gene (see Fig. 1B). Transduction was quantified by comparing amplification patterns of the samples to serial dilutions of a stan-
dard with known numbers of integrated proviruses. (A–D): Data is shown as mean 6 standard deviation of individual cell populations. Animals are grouped
into four groups according to busulfan AUC achieved. The AUC range (in min $ mg/mL) for each group is indicated in brackets. (E): Mean gene marking
from 4 to 6 months posttransplantation is displayed for each busulfan AUC group, together with the Spearman rank correlation coefficient and resulting
significance level.
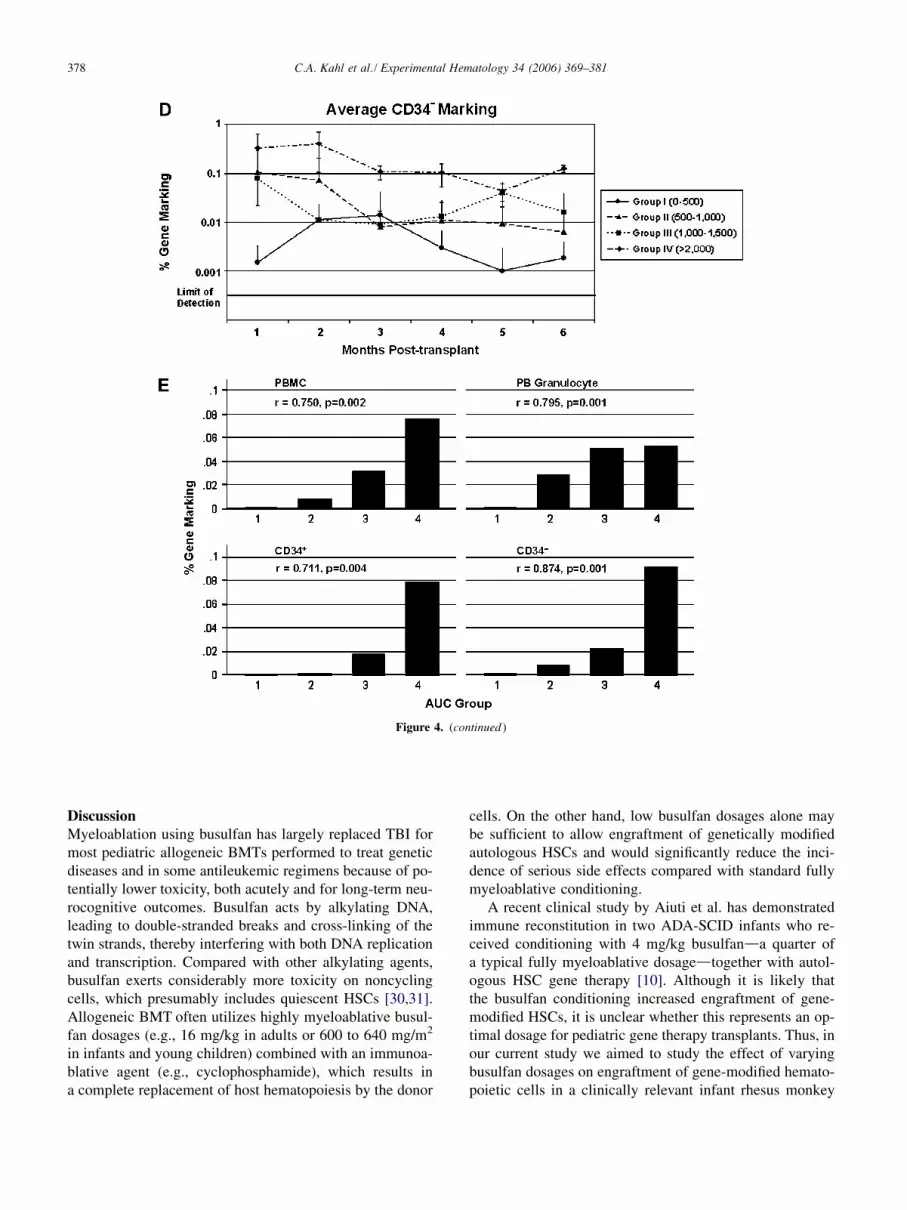
378 C.A. Kahl et al./ Experimental Hematology 34 (2006) 369–381
Figure 4. (continued )
DiscussionMyeloablation using busulfan has largely replaced TBI formost pediatric allogeneic BMTs performed to treat geneticdiseases and in some antileukemic regimens because of po-tentially lower toxicity, both acutely and for long-term neu-rocognitive outcomes. Busulfan acts by alkylating DNA,leading to double-stranded breaks and cross-linking of thetwin strands, thereby interfering with both DNA replicationand transcription. Compared with other alkylating agents,busulfan exerts considerably more toxicity on noncyclingcells, which presumably includes quiescent HSCs [30,31].Allogeneic BMT often utilizes highly myeloablative busul-fan dosages (e.g., 16 mg/kg in adults or 600 to 640 mg/m2
in infants and young children) combined with an immunoa-blative agent (e.g., cyclophosphamide), which results ina complete replacement of host hematopoiesis by the donor
cells. On the other hand, low busulfan dosages alone maybe sufficient to allow engraftment of genetically modifiedautologous HSCs and would significantly reduce the inci-dence of serious side effects compared with standard fullymyeloablative conditioning.
A recent clinical study by Aiuti et al. has demonstratedimmune reconstitution in two ADA-SCID infants who re-ceived conditioning with 4 mg/kg busulfanda quarter ofa typical fully myeloablative dosagedtogether with autol-ogous HSC gene therapy [10]. Although it is likely thatthe busulfan conditioning increased engraftment of gene-modified HSCs, it is unclear whether this represents an op-timal dosage for pediatric gene therapy transplants. Thus, inour current study we aimed to study the effect of varyingbusulfan dosages on engraftment of gene-modified hemato-poietic cells in a clinically relevant infant rhesus monkey

379C.A. Kahl et al. / Experimental Hematology 34 (2006) 369–381
model and to determine an optimal dosage that would pro-vide maximum efficacy with minimal toxicities from theconditioning regimen.
We found that nonmyeloablative dosages of busulfan ofup to 160 mg/m2 are well tolerated in infant rhesus mon-keys with no detectable toxicities, except for the expectedtransient myelosuppressive effects. Our key finding wasthat increasing dosages of busulfan resulted in a greaterAUC, which in turn was reflected in significantly highergene-marking levels. As may be predicted, animals withoutbusulfan preconditioning had little or no detectable markingthroughout the course of the study. Average gene-markinglevels in both the PB and BM compartments were over100- to 1000-fold higher in animals that achieved a busulfanAUC greater than 2,000 min$mg/mL compared with controlanimals, with durable gene marking of almost 0.1% onaverage beyond 6 months posttransplantation. A net busul-fan AUC in this range is what would typically be achievedfrom the first dose of the total 16 doses of busulfan given forfull myeloablation.
As we did not reach dose-limiting toxicity with our con-ditioning, it may be possible to further increase busulfandosage, potentially leading to higher AUC and increasedgene marking. We used a clinically approved IV formula-tion of busulfan (Busulfex) that is easier to administerand has a more predictable bioavailability than the oralform. However, due to the variability in disposition of thedrug, we found that AUC values varied within dosagegroups. In turn, we found that gene-marking levels corre-lated better with AUC than with actual busulfan dosage.To decrease the variability in AUC, it may be feasible tofirst give a test busulfan dosage to subjects and then adjustlater dosages based on the observed individual pharmacoki-netics, similar to approaches in clinical BMT [32].
Our highest treatment dosage of 160 mg/m2 correspondsto a dosage of over 13 mg/kg in the 1 kg infant monkeyswhen calculating the dosage based on body mass (Table 1).This far exceeds the 4 mg/kg dosage utilized by Aiuti et al.in their ADA-SCID clinical trial, although they observeda similar level and duration of neutropenia to what we ob-served in the monkeys that achieved AUC O 2000 min$mg/mL. It is possible that infant rhesus monkeys have a fasterbusulfan clearance than human children, and thus the netAUC achieved in the human ADA-SCID patients mayhave been higher per administered busulfan dosage thanin the rhesus infants [10].
A recent study by Huhn et al. also investigated nonmye-loablative conditioning in four rhesus macaques trans-planted with retrovirally marked CD34D cells [7]. Incontrast to our study, they used a moderate dosage of TBI(500 cGg) for conditioning. Marking levels assessed inthe PB mononuclear and granulocyte fractions were stableover 6 months posttransplantation and displayed a widerange from less than 1 in 100,000 marked cells up to 1 in10 marked cells. Another study by Rosenzweig et al.
used nonmyeloablative TBI (320 to 400 cGy) in four rhesusmacaques together with transplantation of autologousCD34D cells marked with a retrovirus expressing either hu-man truncated nerve growth factor receptor (htNGFR) orthe mCD24 surface antigen [8]. Whereas marking withthe htNGFR vector was minimal and did not persist, peakmarking levels in the whole leukocyte fraction with themCD24 vector ranged from 6% to over 60% at 6 to 8 weeksafter transplantation and persisted at modest levels (1 to5%) for the subsequent 6 months. Marking in BMCD34D cells dropped below 5% at the later time points.Similar to the Huhn et al. study, irradiated animals experi-enced a transient drop in neutrophil counts below 500/mL,but otherwise there were no significant toxicities found inthese studies. Both of these studies did not include controlgroups that did not receive cytoreduction, therefore thedose-related effects of TBI on engraftment of gene-markedcells could not be directly ascertained.
A major difference between the above studies and ourcurrent study is the source of CD34D cells. In the previousstudies of nonmyeloablative conditioning, prior to trans-plant of gene-marked cells in nonhuman primates,CD34D cells were obtained from the PB by priming withG-CSF and SCF, or with Flt3-L and G-CSF, whereas wecollected CD34D cells from nonmobilized BM. Also, inboth the Huhn and Rosenzweig studies, transplants wereconducted with older animals that weighed between 4 and6 kg and the total number of transplanted CD34D cellsper animal was up to 10-fold higher than in our study.Even on a mass-adjusted basis, animals received at leasttwice as many cells per kilogram as in the study describedherein. It has been shown previously that transplanted celldosage is a major contributing factor to engraftment andthat sufficient quantities of cells can outweigh the need tocreate a bone marrow ‘‘niche’’ [33]. On the other hand, itis possible that the different sources of CD34D cells (BMversus mobilized PB) have different contents of hematopoi-etic progenitor cells and HSCs. It is also possible that rhe-sus macaque BM-derived CD34D cells are less susceptibleto transduction than the mobilized PB-derived CD34D cellsused in most previous studies.
Prior studies of gene transfer using nonhuman primateCD34D cells have mainly used gamma-retroviral vectors,whereas we used an SIV-derived lentiviral vector. Rhesusmacaque cells express a potent restriction factor to lentivi-ral infection that has recently been identified. Although therestriction factor, termed TRIM5a, potently restricts infec-tion by HIV-1, it also has a modest inhibitory effect on in-fection by macaque-derived SIV but not on infection bymurine leukemia virus–derived gamma-retroviruses [34].As we demonstrated, the SIVmac251-based lentiviral vectorthat was used for our in vivo studies gave only modestlevels of gene transfer into rhesus CD34D cells assayedin vitro using short-term culture. Although this gene trans-fer capacity was better than what could be obtained using

380 C.A. Kahl et al./ Experimental Hematology 34 (2006) 369–381
HIV-1-based lentiviral vectors in rhesus CD34D cells, theyare relatively low compared with the highly efficient trans-duction of human CD34D cells by HIV-1-based lentiviralvectors [35]. Thus, relatively few engrafting HSCs mayhave been transduced, accounting for the low absolutelevels of gene marking seen in vivo.
A recent study by Hanawa et al. reported efficient trans-duction of rhesus macaque mobilized PB-derived CD34D
cells with an SIVmac1A11-derived lentiviral vector but notwith a corresponding HIV-1 vector [36]. At 6 months aftertransplantation, gene-marking levels in three fully mye-loablated rhesus macaques transplanted with transducedcells were in the 5 to 10% range in PB leukocytes and inthe 10% range in BM CD34D cells. Although the in vivogene-marking levels achieved in the Hanawa study [36]cannot be directly compared with our study, as they useda fully myeloablative TBI regimen (1000 cGy) and weused a nonmyeloablative conditioning regimen, it is possi-ble that the different SIV strains of origin of the vector orsome elements of their design may also have contributedto these differences in gene marking. We have recentlybeen provided with the SIVmac1A11-derived lentiviral vectorby Arthur Nienhuis (St. Jude Children’s Research Hospital)[36] and found significantly higher levels of gene transfer torhesus CD34D cells assayed in vitro compared with theSIVmac251-based vector used for the studies reported here(30 to 60% versus 4 to 12 %, respectively). Use of thismore efficacious vector in future studies may increase thelevels of gene marking achieved with busulfan given inthe well-tolerated dosage range identified here.
In conclusion, we performed a dosage-escalation studywith nonmyeloablative dosages of busulfan, demonstratinga dose-related increase in gene marking. Even at the highestdosage levels we used, busulfan conditioning was well tol-erated, except for the expected transient myelosuppression.The treatment schema led to measurable and durable en-graftment of gene-marked hematopoietic stem and progen-itor cells when busulfan AUC exceeded 2000 min $ mg/mL.Thus, nonmyeloablative conditioning represents a signifi-cant improvement toward safer autologous HSC gene ther-apy compared with the potential for serious side effectsassociated with full ablation with TBI or alkylating drugs.As nonmyeloablative conditioning is now being tested inseveral pediatric HSC gene therapy trials, this study shouldprovide important information to help in evaluating and op-timizing these treatment strategies for clinical gene therapy.This study also highlights the importance of preclinicalstudies using nonhuman primates.
AcknowledgmentsThese studies were supported by grants from the National Insti-tutes of Health, AI52798 (D.B.K.) and HL69748 (A.F.T.), andthe California National Primate Research Center (CNPRC) baseoperating grant (RR00169). We thank Francois-Loıc Cosset andCynthia Dunbar for generously sharing reagents and Sharon Roell
at ESP Pharma, Inc. for kindly providing Busulfex for these stud-ies. We also thank the animal care and clinical laboratory staff atthe CNPRC for expert technical assistance, and Jeannine McCune,Matthew Pawlikowski, and staff at the Busulfan PharmacokineticLaboratory at Fred Hutchinson Cancer Research Center, Seattle,WA, for the pharmacokinetic analyses.
References1. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associ-
ated clonal T cell proliferation in two patients after gene therapy for
SCID-X1. Science. 2003;302:415–419.
2. Cavazzano-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene
therapy of human severe combined immunodeficiency (SCID)-X1
disease. Science. 2000;288:669–672.
3. Naldini L, Bloemer U, Gallay P, et al. In vivo gene delivery and stable
transduction of nondividing cells by a lentiviral vector. Science. 1996;
272:263–267.
4. Miyoshi H, Smith KA, Mosier DE, Verma IM, Torbett BE. Transduc-
tion of human CD34D cells that mediate long-term engraftment of
NOD/SCID mice by HIV vectors. Science. 1999;283:682–686.
5. Schuler US, Renner UD, Kroschinsky F, et al. Intravenous busulphan
for conditioning before autologous or allogeneic human blood stem
cell transplantation. Br J Haematol. 2001;114:944–950.
6. Mardiney M, Malech HL. Enhanced engraftment of hematopoietic pro-
genitor cells in mice treated with granulocyte colony-stimulating factor
before low-dose irradiation: implications for gene therapy. Blood. 1996;
87:4049–4056.
7. Huhn RD, Tisdale JF, Agricola B, Metzger ME, Donahue RE, Dunbar
CE. Retroviral marking and transplantation of rhesus hematopoietic
cells by nonmyeloablative conditioning. Hum Gene Ther. 1999;10:
1783–1790.
8. Rosenzweig M, MacVittie TJ, Harper D, et al. Efficient and durable
gene marking of hematopoietic progenitor cells in nonhuman primates
after nonablative conditioning. Blood. 1999;94:2271–2286.
9. van Os R, Dawes D, Mislow JMK, Witsell A, Mauch PM. Host con-
ditioning with 5-fluorouracil and kit-ligand to provide for long-term
bone marrow engraftment. Blood. 1997;89:2376–2383.
10. Aiuti A, Slavin S, Aker M, et al. Correction of ADA-SCID by
stem cell gene therapy combined with nonmyeloablative conditioning.
Science. 2002;296:2410–2413.
11. Mangeot P-E, Negre D, Dubois B, et al. Development of minimal
lentivirus vectors derived from simian immunodeficiency virus (SIV-
mac251) and their use for gene transfer into human dendritic cells.
J Virol. 2000;74:8307–8315.
12. Negre D, Mangeot P-E, Duisit G, et al. Characterization of novel safe
lentiviral vectors derived from simian immunodeficiency virus (SIV-
mac251) that efficiently transduce mature human dendritic cells.
Gene Ther. 2000;7:1613–1623.
13. Hanazono Y, Brown KE, Handa A, et al. In vivo marking of rhesus
monkey lymphocytes by adeno-associated viral vectors: direct com-
parison with retroviral vectors. Blood. 1999;94:2263–2270.
14. Haas DL, Case SS, Crooks GM, Kohn DB. Critical factors influencing
stable transduction of human CD34D cells with HIV-1-derived lenti-
viral vectors. Mol Ther. 2000;2:71–80.
15. Soneoka Y, Cannon PM, Ramsdale EE, et al. A transient three-plasmid
expression system for the production of high titer retroviral vectors.
Nucleic Acids Res. 1995;23:628–633.
16. Price MA, Case SS, Carbonaro DA, et al. Expression from second-
generation feline immunodeficiency virus vectors is impaired in hu-
man hematopoietic cells. Mol Ther. 2002;6:645–652.
17. Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative
PCR. Genome Res. 1996;6:986–994.
18. Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer
and expression. Biotechniques. 1989;7:980–990.

381C.A. Kahl et al. / Experimental Hematology 34 (2006) 369–381
19. Tarantal AF, Hendrickx AG. Prenatal growth in the cynomolgus and
rhesus macaque (Macaca fascicularis and Macaca mulatta)da com-
parison by ultrasonography. Am J Primatol. 1988;15:309–323.
20. Tarantal AF, Goldstein O, Barley F, Cowan MJ. Transplantation of hu-
man peripheral blood stem cells into fetal rhesus monkeys (Macaca
mulatta). Transplantation. 2000;69:1818–1823.
21. Jimenez DF, Leapley AC, Lee CL, Ultsch M-N, Tarantal AF. Fetal
CD34D cells in the maternal circulation and long-term microchimerism
in rhesus monkeys (Macaca mulatta). Transplantation. 2005;79:142–146.
22. DuBois D, DuBois E. A formula to estimate the approximate surface
area if height and weight be known. Arch Intern Med. 1916;17:
863–871.
23. Slattery JT, Sanders JE, Buckner CD, et al. Graft-rejection and toxicity
following bone marrow transplantation in relation to busulfan pharma-
cokinetics. Bone Marrow Transplant. 1995;16:31–42.
24. Jimenez DF, Tarantal AF. Quantitative analysis of male fetal DNA in
maternal serum of gravid rhesus monkeys (Macaca mulatta). Pediatr
Res. 2003;53:18–23.
25. Lo YM, Tein MS, Lau TK, et al. Quantitative analysis of fetal DNA
in maternal plasma and serum: implications for noninvasive prenatal
diagnosis. Am J Hum Genet. 1998;62:768–775.
26. Hassan M, Ljungman P, Bolme P, et al. Busulfan bioavailability.
Blood. 1994;84:2144–2150.
27. Yeager AM, Wagner JE JR, Graham ML, Jones RJ, Santos GW, Gro-
chow LB. Optimization of busulfan dosage in children undergoing
bone marrow transplantation: a pharmacokinetic study of dose escala-
tion. Blood. 1992;80:2425–2428.
28. Vassal G, Deroussent A, Challine D, et al. Is 600 mg/m2 the appropri-
ate dosage of busulfan in children undergoing bone marrow transplan-
tation? Blood. 1992;79:2475–2479.
29. Sandrin V, Boson B, Salmon P, et al. Lentiviral vectors pseudotyped
with a modified RD114 envelope glycoprotein show increased stability
in sera and augmented transduction of primary lymphocytes and
CD34D cells derived from human and nonhuman primates. Blood.
2002;100:823–832.
30. Down JD, Boudewijn A, Dillingh JH, Fox BW, Ploemacher RE. Rela-
tionships between ablation of distinct haematopoietic cell subsets and
the development of donor bone marrow engraftment following recip-
ient pretreatment with different alkylating drugs. Br J Cancer. 1994;
70:611–616.
31. Down JD, Ploemacher RE. Transient and permanent engraftment po-
tential of murine hematopoietic stem cell subsets: differential effects
of host conditioning with gamma radiation and cytotoxic drugs. Exp
Hematol. 1993;21:913–921.
32. Slattery JT, Risler LJ. Therapeutic monitoring of busulfan in hemato-
poietic stem cell transplantation. Ther Drug Monit. 1998;20:543–549.
33. Rao SS, Peters SO, Crittenden RB, Stewart FM, Ramshaw HS, Ques-
enberry PJ. Stem cell transplantation in the normal nonmyeloablated
host: relationship between cell dose, schedule, and engraftment. Exp
Hematol. 1997;25:114–121.
34. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodros-
ki J. The cytoplasmic body component TRIM5alpha restricts HIV-1
infection in old world monkeys. Nature. 2004;427:848–853.
35. Case SS, Price MA, Jordan CT, et al. Stable transduction of quiescent
CD34DCD38- human hematopoietic cells by HIV-1-based lentiviral
vectors. Proc Natl Acad Sci U S A. 1999;96:2988–2993.
36. Hanawa H, Hematti P, Keyvanfar K, et al. Efficient gene transfer into
rhesus repopulating hematopoietic stem cells using a simian immuno-
deficiency virus-based lentiviral vector system. Blood. 2004;103:
4062–4069.