divergent adaptation of hepatitis c virus genotypes 1 and 3 to human leukocyte antigen-restricted...
TRANSCRIPT

VIRAL HEPATITIS
Divergent Adaptation of Hepatitis C Virus Genotypes 1 and 3to Human Leukocyte Antigen–Restricted Immune PressureAndri Rauch,1,2 Ian James,2* Katja Pfafferott,2* David Nolan,2* Paul Klenerman,3 Wendy Cheng,4 Lindsay Mollison,5
Geoff McCaughan,6 Nick Shackel,6 Gary P. Jeffrey,7 Ross Baker,8 Elizabeth Freitas,2 Isla Humphreys,3
Hansjakob Furrer,1 Huldrych F. Gunthard,9 Bernard Hirschel,10 Simon Mallal,2 Mina John,2 Michaela Lucas,2
Eleanor Barnes,3 and Silvana Gaudieri2,11
Many hepatitis C virus (HCV) infections worldwide are with the genotype 1 and 3 strains of the virus.Cellular immune responses are known to be important in the containment of HCV genotype 1infection, and many genotype 1 T cell targets (epitopes) that are presented by host human leukocyteantigens (HLAs) have been identified. In contrast, there is almost no information known about theequivalent responses to genotype 3. Immune escape mechanisms used by HCV include the evolutionof viral polymorphisms (adaptations) that abrogate this host–viral interaction. Evidence of HCVadaptation to HLA-restricted immune pressure on HCV can be observed at the population level asviral polymorphisms associated with specific HLA types. To evaluate the escape patterns of HCVgenotypes 1 and 3, we assessed the associations between viral polymorphisms and specific HLA typesfrom187 individualswithgenotype1aand136 individualswithgenotype3a infection.We identified51 HLA-associated viral polymorphisms (32 for genotype 1a and 19 for genotype 3a). Of theseputativeviral adaptation sites, six fellwithinpreviouslypublishedepitopes.Only twoHLA-associatedviral polymorphisms were common to both genotypes. In the remaining sites with HLA-associatedpolymorphisms, there was either complete conservation or no significant HLA association with viralpolymorphism in the alternative genotype. This study also highlights the diverse mechanisms bywhich viral evasion of immune responses may be achieved and the role of genotype variation in theseprocesses. Conclusion: There is little overlap in HLA-associated polymorphisms in the nonstructuralproteins of HCV for the two genotypes, implying differences in the cellular immune pressures actingon these viruses and different escape profiles. These findings have implications for future therapeuticstrategies to combat HCV infection, including vaccine design. (HEPATOLOGY 2009;50:1017-1029.)
Hepatitis C virus (HCV) is a genetically diverseRNA virus with six major genotype groups thatdiffer by approximately 30% at the nucleotide
(RNA) level.1 Among these, HCV genotypes 1 and 3 arethe most prevalent genotypes in Australia and northern
Europe. The extensive intergenotype genetic variation islikely to have important implications for the host’s cellu-lar immune responses to HCV, which are an importantcorrelate of infection outcome (reviewed by Klenermanand Hill2). These responses are stimulated by the presen-tation of internally processed viral peptides (epitopes) byhuman leukocyte antigen (HLA) class I molecules toCD8� cytotoxic T lymphocytes (CTLs). Viral polymor-phisms within or flanking these epitopes can result inescape from HLA-restricted CTL responses, providing aneffective mechanism for the virus to subvert host immunecontrol (reviewed by Bowen and Walker3). We and oth-ers4,5 have shown that viral polymorphisms in the HCVgenotype 1 genome are associated with specific HLA classI alleles, indicating adaptation of HCV to HLA-restrictedimmune pressure.
So far, the majority of studies examining cellular im-mune responses in individuals infected with HCV havefocused on genotype 1 and have used peptide librariesbased on a HCV genotype 1 sequence, even when exam-
Abbreviations: CTL, cytotoxic T lymphocyte, HCV, hepatitis C virus; HIV,human immunodeficiency virus; HLA, human leukocyte antigen; NS, nonstruc-tural; P, position; PCR, polymerase chain reaction.
From the 1University Clinic of Infectious Diseases, University Hospital Bern and Univer-sity of Bern, Bern, Switzerland; the 2Centre for Clinical Immunology and Biomedical Sta-tistics, Royal Perth Hospital and Murdoch University, Perth, Australia; the 3Oxford NIHRBiomedical Research Centre and Nuffield Department of Medicine, University of Oxford,Oxford, UK; the 4Department of Gastroenterology and Hepatology and the 8HaemophiliaCentre of Western Australia, Royal Perth Hospital, Perth, Australia; the 5Department ofGastroenterology and Hepatology, Fremantle Hospital, Fremantle, Australia; the 6AW Mor-row Gastroenterology and Liver Centre, Royal Prince Alfred Hospital, Centenary ResearchInstitute, University of Sydney, Sydney, Australia; the 7Department of Gastroenterology, SirCharles Gairdner Hospital, Nedlands, Australia; the 9Division of Infectious Diseases andHospital Epidemiology, University Hospital Zurich, University of Zurich, Zurich, Switzer-land; the 10Department of Infectious Diseases, Geneva University Hospital, Geneva, Swit-zerland; and the 11School of Anatomy and Human Biology and Centre for Forensic Science,University of Western Australia, Perth, Australia.
Received March 3, 2009; accepted May 16, 2009.
1017

ining individuals infected with genotype 3 (reviewed byWard et al.6 and Bowen and Walker7). Given the extent ofgenetic diversity distributed throughout their genomes, itwould be anticipated that the repertoire of HLA-re-stricted viral epitopes could be distinct for the differentHCV genotypes. Conversely, areas of the HCV genomethat are conserved across genotypes 1 and 3 can provide abasis for cross-genotype HLA-restricted CTL responses,as has recently been demonstrated for an HLA-A1–re-stricted epitope in the NS3 protein of HCV.8 Further-more, cellular immune responses are thought to play arole in response to interferon-based therapy, although thisremains controversial. Given the higher response rate tointerferon-� and ribavirin therapy in genotype 3 com-pared with genotype 1,9,10 understanding potential im-munological differences will be important. Betterunderstanding of the selection pressures on HCV is alsolikely to be relevant for new anti-HCV drugs11 and forHCV vaccine design.
We performed HLA typing and HCV sequencingfrom individuals with chronic HCV infection to comparethe viral adaptation profiles between genotypes 1a and 3a.
Subjects and MethodsSubjects
Individuals with chronic HCV genotype 1a (n � 187)or 3a (n � 136) infection were recruited from Australia(n � 145), Switzerland (n � 99), and the UK (n � 79).All HCV sequences were obtained from HCV treatment-naıve individuals. Written informed consent was ob-tained from participants, and local Institutional ReviewBoard approval was obtained by the contributing centers.
DNA and Viral RNA ExtractionDNA was obtained from whole blood using the
QIAamp DNA Blood Mini Kit following the manufac-turer’s guidelines. Viral RNA was extracted from plasmasamples using either the QIAamp Viral RNA Mini Kit(QIAGEN) or the COBAS AMPLICOR HCV Specimen
Preparation Kit version 2.0 (Roche) according to themanufacturer’s instructions.
HLA GenotypingSequenced-based four-digit HLA class I typing was
performed by direct DNA sequencing methods as de-scribed.4
Bulk Viral Sequencing and HCV GenotypingInitial reverse-transcription polymerase chain reaction
(PCR) using the SuperScript III One-Step RT-PCR Sys-tem with a Platinum Taq DNA Polymerase PCR kit (In-vitrogen) was performed. The first-round product wasthen used in several nested second-round PCRs contain-ing generic or genotype-specific primer pairs (primer se-quences available upon request) together with thePlatinum Taq DNA Polymerase High Fidelity Kit (In-vitrogen) to cover the area from NS2 to NS5B. ResultantPCR products were bulk (population)-sequenced usingthe BigDye Terminator version 3.1 cycle sequencing kit(Applied Biosystems) according to the manufacturer’srecommendations, and electropherograms were editedusing Assign software (Conexio Genomics). Mixtureswere identified where the secondary peak was �20% ofthe main peak. HCV subtypes were assigned by clinicaltests using commercial assays (INNO-LiPA HCV II, In-nogenetics Gent) and confirmed by way of phylogeneticanalysis (Supporting Fig. 3).
Due to the variability of the HCV genome, some sam-ples failed to produce a PCR product and as a result, someindividuals did not contribute sequences for all nonstruc-tural proteins. The number of individuals that contrib-uted sequences was as follows (genotype 1a/genotype 3a):NS2:170/129, NS3:181/136, NS4a:153/90, NS4b:178/108; NS5a:174/122, NS5b: 182/128. For the analysisthat adjusted for phylogenetic relatedness and for the as-sessment of sequence variation between and within geno-types, we only included sequences with �90% coveragefor the respective protein (see Statistical Methods). For
Supported by the National Health and Medical Research Council of Australia (Grants 334603 and 384702), the Haemophilia Foundation of Australia, the SwissNational Science Foundation (Grants 3347-069366 and 3247-116862 and Swiss HIV Cohort Study Grant 430), the National Institute for Health Research BiomedicalResearch Centre Programme, Wellcome Trust, James Martin School for the 21st century (Oxford), and the Medical Research Council (UK). A. R. was supported by a Grantfor Prospective Researchers from the Swiss National Science Foundation. M. L. was supported by a Hepatology/Immunology Research Fellowship from Royal Perth Hospitaland Murdoch University.
*These authors contributed equally to this work.Address reprint requests to: Dr. Silvana Gaudieri, Centre for Clinical Immunology and Biomedical Statistics, Royal Perth Hospital, Wellington St., Perth, Western
Australia 6000, Australia. E-mail: [email protected]; fax: (61)-8-9224 1981.Copyright © 2009 by the American Association for the Study of Liver Diseases.Published online in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/hep.23101Potential conflict of interest: Dr. McCaughan advises Roche and Schering-Plough. Dr. Gunthard advises and received grants from Gilead and Jansen Cilag. He also
received grants from Merck. Dr. Mallal is a consultant for, advises, and is on the speakers’ bureau of GlaxoSmithKline.Additional Supporting Information may be found in the online version of this article.
1018 RAUCH ET AL. HEPATOLOGY, October 2009

the NS5b protein, the analysis was restricted to residues2421-2870, because the number of individuals with se-quence coverage for the rest of NS5B was insufficient toperform the statistical analyses. The number of individu-als that contributed sequences for the analysis that ad-justed for phylogenetic relatedness was as follows(genotype 1a/genotype 3a): NS2: 149/108, NS3: 105/93,NS4a: 148/86, NS4b: 93/64; NS5a: 86/72, NS5b: 85/80.
Statistical Methods
HLA Association with Viral Polymorphism. Asso-ciations between HLA alleles and amino acid distributionat each residue of the HCV proteins were assessed viaFisher’s exact tests for classification as consensus versusnonconsensus amino acid using S-Plus 8.0 (InsightfulCorporation, Seattle, WA).
Assessment of Phylogenetic Relatedness. Polymor-phisms detected across viral genomes are likely to be theresult of natural selection (such as HLA-restricted im-mune pressure) and neutral evolution. The ability to dif-ferentiate between these two evolutionary processes couldbe compromised in cases where an HLA allele is overrep-resented in a subgroup of individuals that have viral se-quences sharing a recent common ancestor. In these cases,an association between an HLA allele and viral polymor-phism may reflect a founder effect rather than a true site ofviral adaptation to immune pressure. Others have previ-ously used phylogenetic methods to adjust for these po-tential confounding factors.5,12 In this study, weaddressed this issue by identifying clusters of possibly re-lated sequence and assessing the potential impact of suchrelatedness by performing analyses stratified by clusters.This approach is based on the notion that within rela-tively homogeneous clusters of possibly related sequences,the distributions of HLA types should be random. Clus-ters were determined using the robust partitioningaround medoids method of Kaufman and Rousseeuw13
based on the binary presence/absence of consensus at eachresidue. Optimal clustering was determined via the aver-age silhouette width, comparing the within to betweencluster dissimilarities. Numbers of clusters were deter-mined through plots of the silhouette values across num-bers of clusters, and individuals were assigned to theirnearest cluster. Genotype 3 sequences were alignedagainst genotype 1a sequences to maximize comparabilitybetween the genotypes.
Stratified Analysis by Way of Mantel-HaenszelTests. Associations between polymorphisms at eachamino acid residue and the HLA alleles in the populationadjusted for cluster strata were then assessed by Mantel-Haenszel tests. Because this procedure combines the asso-
ciations between viral polymorphisms and HLA alleleswithin the clusters of possibly related sequences, the riskof confounding through overrepresentation of HLA al-leles in phylogenetically related sequences was minimized.Because this analysis is based on calculating similaritiesbetween viral sequences that have sufficient overlap, weonly included sequences with �90% coverage for therespective protein (genotype 1a, n � 187; genotype 3a,n � 136). Analyses were performed by protein.
Only associations with P � 0.01 for both the Fisher’sexact test and the Mantel-Haenszel method were re-ported. In addition, because P values associated with rel-atively small frequencies can be biased and dominated bysmall numbers of misclassified cases, we restricted ourreport to associations for which there were at least fivenonconsensus amino acids and at least five carriers of theHLA allele.
False Discovery Rates and q Values. False discoveryrates (FDR) and associated q values14,15 need to take ac-count of both the discreteness of the test statistics and alsothe strong correlations between tests. We obtained thenull distributions by replicating the analysis to create theappropriate tables and marginal frequencies, fixing themargins but imputing random hypergeometric table val-ues subject to these fixed margins. Because of the replica-tion of similar tables with corresponding marginalfrequencies within each analysis, 50 imputed random ta-bles for each association were sufficient to provide an ac-curate estimate of the overall null P value distribution.FDR and q values were then obtained similarly to Storeyand Tibshirani.15
Inclusion of All HCV Sequences. Exclusion of HCVsequences with �90% coverage of a particular proteincould potentially bias the dataset toward individuals inwhom viral sequencing has been more successful (for ex-ample, those with more conserved sequences relative tothe primers or higher viral loads). We therefore also per-formed a subsequent analysis including all available HCVsequences. Because the cluster analysis requires higher se-quence coverage, we only report results based on P � 0.01and a q value cutoff of �0.2 for the entire dataset.
Data DepositionThe HCV sequences analyzed in this study have been
deposited in GenBank. For a listing of accession numbers,please see Supporting Information in the online version ofthis article.
ResultsCharacteristics of Study Population. This study
population is drawn from three cohorts of chronicallyHCV-infected individuals, comprising predominantly
HEPATOLOGY, Vol. 50, No. 4, 2009 RAUCH ET AL. 1019

males (73%) who had acquired HCV through injectiondrug use (65%), with a median age of 46 years (Table 1).There were 130 (40%) individuals coinfected with hu-man immunodeficiency virus (HIV) in the study, ofwhom the majority (91% in the Swiss HIV Cohort Studyand 75% in the Australian cohort) had a CD4� T cellcount above 200 cells/�L, which others have shown isgenerally sufficient to maintain HCV-specific CD8� Tcell activity.16,17 As shown in Supporting Fig. 1, the HLAclass I allele frequency distribution was typical of Cauca-sian populations and was similar across the study groups.Of the 45 HLA alleles with a frequency of 2% or more,only three alleles differed significantly between the geno-
types and four between the cohorts (Supporting Fig. 1).Hence, with only a few exceptions, the frequency of theHLA types of individuals infected with either genotypewas similar, providing the basis for comparison of HLA-restricted immune pressure on the two genotypes.
HLA-Associated Viral Polymorphisms Within theNonstructural Proteins of Genotypes 1a and 3a. Wefirst examined the viral adaptation pattern across the non-structural proteins for both genotypes. In this analysis, asso-ciations between HCV polymorphism and carriage ofspecific HLA class I alleles at each residue of the nonstruc-tural HCV proteins were investigated. Significant associa-tions with an odds ratio �1 are consistent with mutational
Table 1. Demographic and Clinical Characteristics of Study Participants
AustralianCohort(n � 145)
Swiss HIV CohortStudy (n � 99)
Oxford (UK) Cohort(n � 79)
HCV genotype 1a, n (%) 87 (60) 62 (63) 38 (48)HCV genotype 3a, n (%) 58 (40) 37 (37) 41 (52)Mode of transmission, n (%)*
Injection drug use 35 (49) 78 (79) 48 (61)Blood transfusion 21 (29) 1 (1) 8 (10)Other 16 (22) 20 (20) 23 (29)
Female sex, n (%) 28 (20) 37 (38) 23 (29)Median age (interquartile range), years 46 (39–52) 44 (40–48) 51 (45–56)HIV-infected (%) 31 (21) 99 (100) 0 (0)
*Unknown in 73 subjects from the Australian cohort.
Fig. 1. Comparison of P values between genotypes at HLA-associated viral polymorphism sites reveals limited overlap. The P values (listed in Table2) that define the significance of the HLA-associated polymorphism is shown as a black bar for genotype 1a and as a red bar for genotype 3a (�log10scale). The P value for the alternative genotype at the same site and for the same HLA type is indicated. A cross indicates where the sequences forthe alternative genotype at the selected site are completely conserved. A square indicates where the alternative genotype at the selected site hasa P value �0.9. In three sites (#4, #9, #31) there were fewer than three individuals carrying the respective HLA allele and the comparison wastherefore not performed. The two sites with significant associations for the same HLA type and genotype are indicated by an arrowhead; the P valuesof the corresponding genotype 3 associations (sites #38 and #51) are shown at the genotype 1 sites.
1020 RAUCH ET AL. HEPATOLOGY, October 2009

escape from HLA-restricted cellular immune pressure; viralpolymorphisms that abrogate HLA-restricted CTL re-sponses are overrepresented in the presence of the restrictingHLA allele. Associations with an odds ratio �1 indicateoverrepresentation of consensus in individuals that carry therespective HLA allele, indicating sites where the consensusviral sequence appears best adapted to T cell responses actingat these sites across the host population.4,5,18,19 In order tominimize confounding through founder effects,12 the anal-ysis was adjusted for viral phylogenetic relatedness (see Sta-tistical Methods).
We identified 51 HLA-associated viral polymor-phisms in total for both genotypes with P � 0.01 in theunadjusted and phylogeny-adjusted analysis (32 for ge-notype 1a and 19 for genotype 3a; Table 2A and 2B).Only two HLA-associated viral polymorphisms werecommon between the genotypes (HLA-A*0101 atNS3-1444 and HLA-B*1501 at NS5B-2467 [boxes inTable 2A and 2B]). For the remaining 47 sites where an
HLA-associated viral polymorphism was identified inone HCV genotype, 12 were completely conserved inthe alternative genotype, with no evidence for commonHLA-restricted polymorphism across genotypes at theremaining sites (Fig. 1). With respect to the differentHLA loci, we identified 14, 25, and 12 HLA-associatedviral polymorphisms for HLA-A, HLA-B, and HLA-C,respectively. However, in some instances (particularlyfor HLA-B and HLA-C), we have shown4 that associ-ations at the same site for HLA alleles of different locican, in part, be explained by the linkage disequilibriumthat is observed within the major histocompatiblitycomplex. Of those associations shown in Table 2A and2B, three are associated with strongly linked HLA-Band HLA-C alleles within common extended majorhistocompatiblity complex haplotypes (noted in Table2A).
Four HLA-associated polymorphisms for genotype1a were within previously published epitopes (marked
Table 2A. Significant HLA-Associated Viral Polymorphisms in HCV Genotype 1a
# Protein Residue HLACon-
sensusUnadjusted
P-valuePhylogeny-adjusted
P-value q-value OR
1 NS2 824 C1502 V 0.0012 0.0007 0.37 10.02 NS2 841 C0401 W 0.0014 0.0021 0.37 22.03 NS2 851 C1502 T 0.0016 0.0002 0.38 29.04 NS2 856 B3503 Q 0.0046 0.0031 0.59 23.05∧ NS2 957 B1302 R 0.0022 0.0008 0.42 10.06∧ NS2 957 C0602 R 0.0001 0.0002 0.04 8.87*∧ NS2 958 B3701 D 0.0000 0.0000 0.00 810.08∧ NS2 958 C0602 D 0.0000 0.0000 0.00 38.09 NS2 962 B3503 N 0.0042 0.0021 0.55 19.0
10 NS2 998 B1302 N 0.0090 0.0087 0.76 6.311∧ NS2 1006 B3701 R 0.0000 0.0004 0.01 44.012∧ NS2 1006 C0602 R 0.0042 0.0089 0.55 6.813 NS2 1017 B1501 G 0.0015 0.0098 0.37 7.314 NS3 1341 A1101 A 0.0054 0.0026 0.61 11.015 NS3 1366 C1502 A 0.0013 0.0009 0.37 36.016 NS3 1368 B5101 S 0.0031 0.0002 0.51 23.017* NS3 1398 B0801 K 0.0041 0.0061 0.55 8.018* NS3 1403 B0801 L 0.0091 0.0067 0.77 12.019* NS3 1444 A0101 F 0.0001 0.0005 0.04 0.120 NS3 1495 A0101 K 0.0013 0.0079 0.37 6.721 NS3 1503 C1203 A 0.0006 0.0000 0.18 31.022 NS3 1635 A1101 V 0.0070 0.0083 0.67 6.523 NS4A 1695 B2705 I 0.0041 0.0033 0.55 11.024 NS4B 1876 B4001 T 0.0003 0.0000 0.11 23.025 NS5A 2000 C0401 M 0.0056 0.0025 0.61 12.026 NS5A 2036 A2402 T 0.0026 0.0020 0.45 16.027 NS5A 2155 B3501 P 0.0003 0.0034 0.10 38.028 NS5A 2227 B4403 I 0.0005 0.0001 0.18 33.029 NS5A 2234 B5101 R 0.0035 0.0002 0.51 23.030$ NS5B 2467 B1501 Q 0.0001 0.0004 0.04 10.031 NS5B 2510 A3101 S 0.0000 0.0000 0.01 63.032 NS5B 2796 C0303 G 0.0018 0.0014 0.39 19.0
Adjusted for phylogenetic relatedness using sequences with �90% coverage for the respective protein. HLA associations common to genotypes are boxed.∧HLA alleles within an extended MHC haplotype.*Within known epitopes.$The HLA-B*1501 association at NS5B-2467 falls within a predicted epitope (SYFPEITHI and BIMAS).
HEPATOLOGY, Vol. 50, No. 4, 2009 RAUCH ET AL. 1021
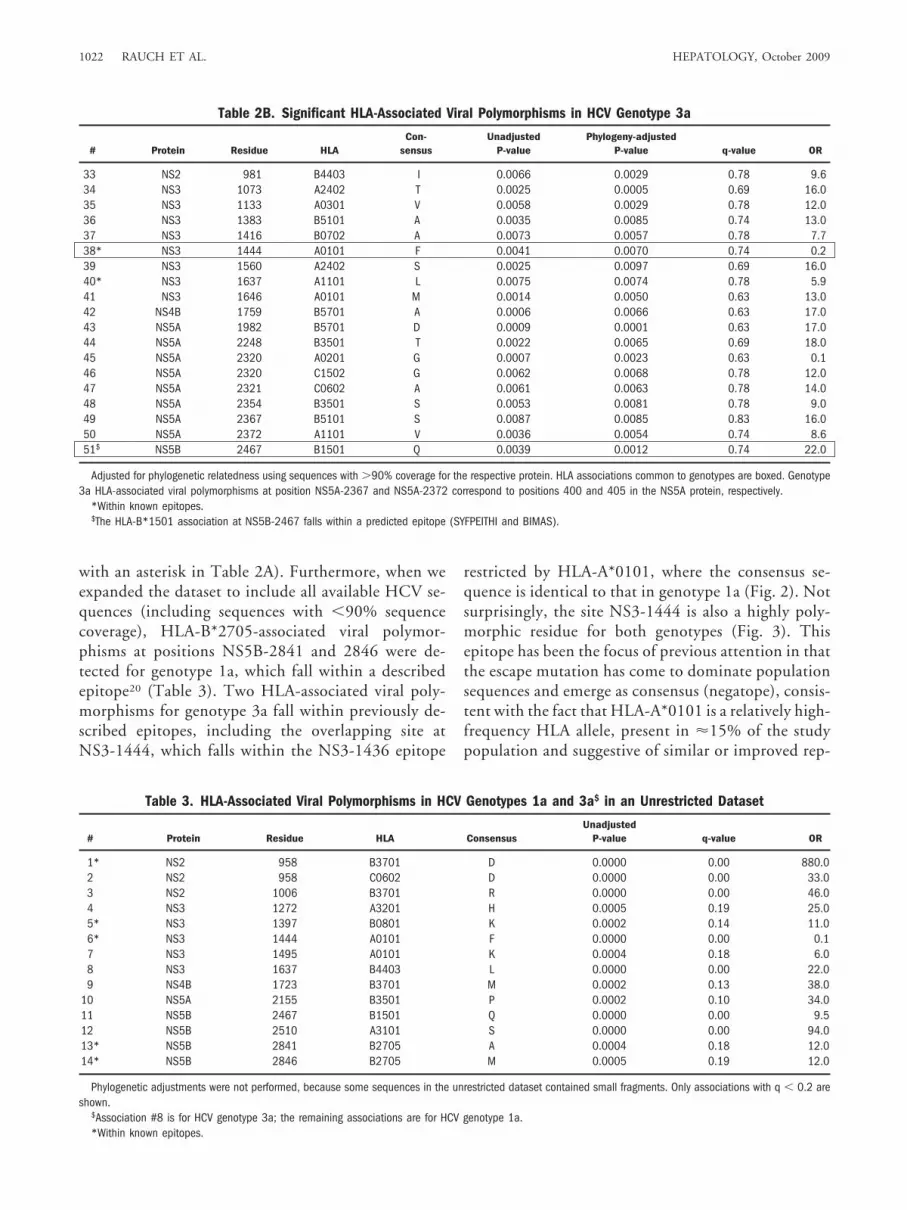
with an asterisk in Table 2A). Furthermore, when weexpanded the dataset to include all available HCV se-quences (including sequences with �90% sequencecoverage), HLA-B*2705-associated viral polymor-phisms at positions NS5B-2841 and 2846 were de-tected for genotype 1a, which fall within a describedepitope20 (Table 3). Two HLA-associated viral poly-morphisms for genotype 3a fall within previously de-scribed epitopes, including the overlapping site atNS3-1444, which falls within the NS3-1436 epitope
restricted by HLA-A*0101, where the consensus se-quence is identical to that in genotype 1a (Fig. 2). Notsurprisingly, the site NS3-1444 is also a highly poly-morphic residue for both genotypes (Fig. 3). Thisepitope has been the focus of previous attention in thatthe escape mutation has come to dominate populationsequences and emerge as consensus (negatope), consis-tent with the fact that HLA-A*0101 is a relatively high-frequency HLA allele, present in �15% of the studypopulation and suggestive of similar or improved rep-
Table 2B. Significant HLA-Associated Viral Polymorphisms in HCV Genotype 3a
# Protein Residue HLACon-
sensusUnadjusted
P-valuePhylogeny-adjusted
P-value q-value OR
33 NS2 981 B4403 I 0.0066 0.0029 0.78 9.634 NS3 1073 A2402 T 0.0025 0.0005 0.69 16.035 NS3 1133 A0301 V 0.0058 0.0029 0.78 12.036 NS3 1383 B5101 A 0.0035 0.0085 0.74 13.037 NS3 1416 B0702 A 0.0073 0.0057 0.78 7.738* NS3 1444 A0101 F 0.0041 0.0070 0.74 0.239 NS3 1560 A2402 S 0.0025 0.0097 0.69 16.040* NS3 1637 A1101 L 0.0075 0.0074 0.78 5.941 NS3 1646 A0101 M 0.0014 0.0050 0.63 13.042 NS4B 1759 B5701 A 0.0006 0.0066 0.63 17.043 NS5A 1982 B5701 D 0.0009 0.0001 0.63 17.044 NS5A 2248 B3501 T 0.0022 0.0065 0.69 18.045 NS5A 2320 A0201 G 0.0007 0.0023 0.63 0.146 NS5A 2320 C1502 G 0.0062 0.0068 0.78 12.047 NS5A 2321 C0602 A 0.0061 0.0063 0.78 14.048 NS5A 2354 B3501 S 0.0053 0.0081 0.78 9.049 NS5A 2367 B5101 S 0.0087 0.0085 0.83 16.050 NS5A 2372 A1101 V 0.0036 0.0054 0.74 8.651$ NS5B 2467 B1501 Q 0.0039 0.0012 0.74 22.0
Adjusted for phylogenetic relatedness using sequences with �90% coverage for the respective protein. HLA associations common to genotypes are boxed. Genotype3a HLA-associated viral polymorphisms at position NS5A-2367 and NS5A-2372 correspond to positions 400 and 405 in the NS5A protein, respectively.
*Within known epitopes.$The HLA-B*1501 association at NS5B-2467 falls within a predicted epitope (SYFPEITHI and BIMAS).
Table 3. HLA-Associated Viral Polymorphisms in HCV Genotypes 1a and 3a$ in an Unrestricted Dataset
# Protein Residue HLA ConsensusUnadjusted
P-value q-value OR
1* NS2 958 B3701 D 0.0000 0.00 880.02 NS2 958 C0602 D 0.0000 0.00 33.03 NS2 1006 B3701 R 0.0000 0.00 46.04 NS3 1272 A3201 H 0.0005 0.19 25.05* NS3 1397 B0801 K 0.0002 0.14 11.06* NS3 1444 A0101 F 0.0000 0.00 0.17 NS3 1495 A0101 K 0.0004 0.18 6.08 NS3 1637 B4403 L 0.0000 0.00 22.09 NS4B 1723 B3701 M 0.0002 0.13 38.0
10 NS5A 2155 B3501 P 0.0002 0.10 34.011 NS5B 2467 B1501 Q 0.0000 0.00 9.512 NS5B 2510 A3101 S 0.0000 0.00 94.013* NS5B 2841 B2705 A 0.0004 0.18 12.014* NS5B 2846 B2705 M 0.0005 0.19 12.0
Phylogenetic adjustments were not performed, because some sequences in the unrestricted dataset contained small fragments. Only associations with q � 0.2 areshown.
$Association #8 is for HCV genotype 3a; the remaining associations are for HCV genotype 1a.*Within known epitopes.
1022 RAUCH ET AL. HEPATOLOGY, October 2009
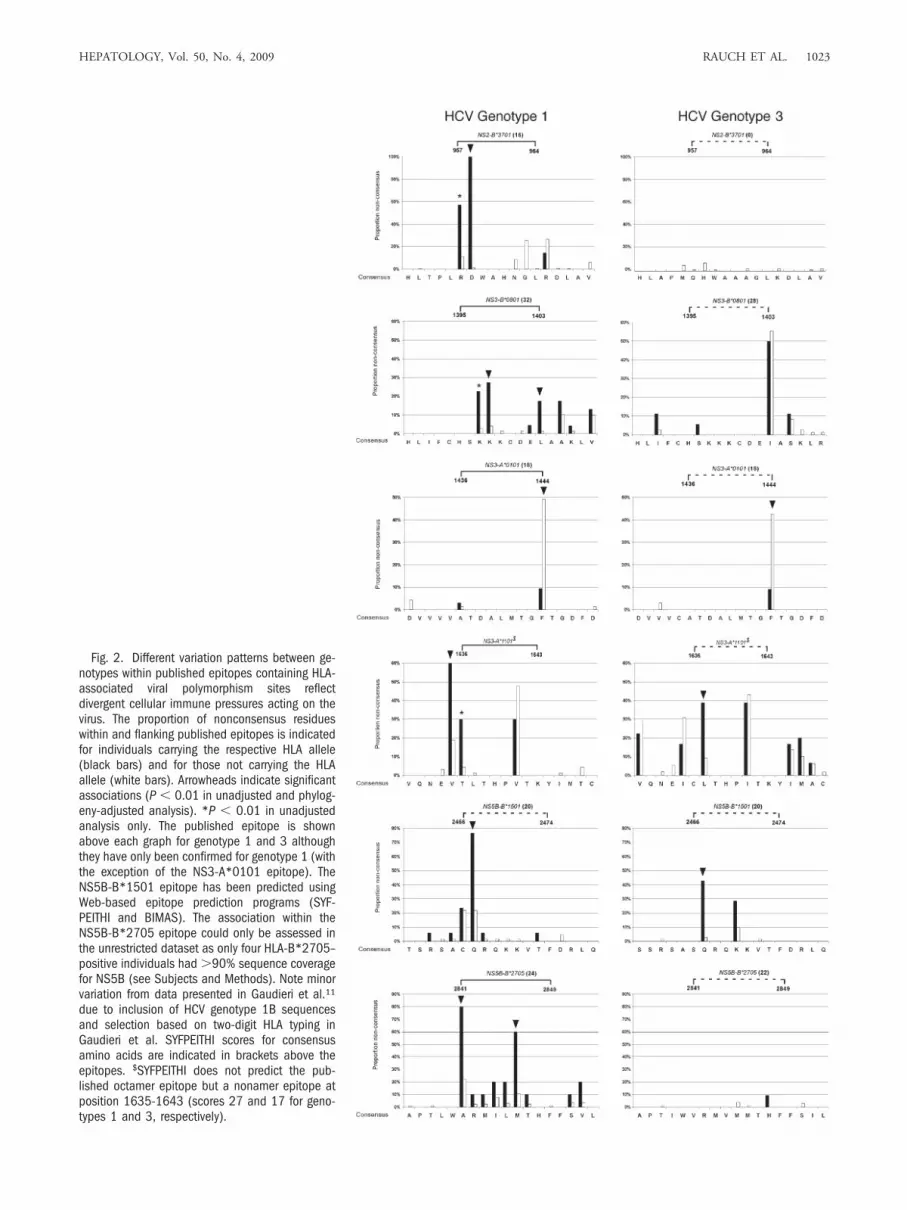
Fig. 2. Different variation patterns between ge-notypes within published epitopes containing HLA-associated viral polymorphism sites reflectdivergent cellular immune pressures acting on thevirus. The proportion of nonconsensus residueswithin and flanking published epitopes is indicatedfor individuals carrying the respective HLA allele(black bars) and for those not carrying the HLAallele (white bars). Arrowheads indicate significantassociations (P � 0.01 in unadjusted and phylog-eny-adjusted analysis). *P � 0.01 in unadjustedanalysis only. The published epitope is shownabove each graph for genotype 1 and 3 althoughthey have only been confirmed for genotype 1 (withthe exception of the NS3-A*0101 epitope). TheNS5B-B*1501 epitope has been predicted usingWeb-based epitope prediction programs (SYF-PEITHI and BIMAS). The association within theNS5B-B*2705 epitope could only be assessed inthe unrestricted dataset as only four HLA-B*2705–positive individuals had �90% sequence coveragefor NS5B (see Subjects and Methods). Note minorvariation from data presented in Gaudieri et al.11
due to inclusion of HCV genotype 1B sequencesand selection based on two-digit HLA typing inGaudieri et al. SYFPEITHI scores for consensusamino acids are indicated in brackets above theepitopes. $SYFPEITHI does not predict the pub-lished octamer epitope but a nonamer epitope atposition 1635-1643 (scores 27 and 17 for geno-types 1 and 3, respectively).
HEPATOLOGY, Vol. 50, No. 4, 2009 RAUCH ET AL. 1023

licative capacity of the adapted variant.8 The HLA-B*1501 association at NS5B-2467 falls within anepitope that has been predicted using Web-basedepitope prediction programs (SYFPEITHI and BI-MAS21,22).
Amino Acid Variation Within Published and Pre-dicted Viral Epitopes that Contain a Viral Adapta-tion Site Show Different Patterns of Viral Adaptationfor Genotype 1a and 3a. The proportion of noncon-sensus residues within and flanking published epitopes isindicated in Fig. 2 for individuals carrying the respective
HLA allele and for those not carrying the HLA allele.Carriage of the restricting HLA allele was associated withviral polymorphisms at several sites within immunogenicepitopes, consistent with viral escape from HLA-re-stricted immune pressure. At multiple sites, including ex-perimentally established viral escape sites (for example,NS3-1397-1398 for HLA-B*0801 and NS5B-2841 and2846 for HLA-B*270520,23-25), there was nearly completeconservation in the alternative genotype, even in individ-uals carrying the relevant HLA alleles (Fig. 2). Althoughin most epitopes only one site reached statistical signifi-
Fig. 3. (A) Polymorphism profile of the HCV NS3 protein for genotypes 1a and 3a shows differences in consensus amino acids and sites ofpolymorphism. Vertical bars indicate the proportion of sequences with nonconsensus residues for genotype 1a above the line and genotype 3a belowthe line. A red circle along the x-axis and red bars indicate residues with a different consensus amino acid for the genotypes. Similar plots for NS2,NS4, NS5A, and NS5B are provided in Supporting Fig. 2. (B) Correlation of polymorphism rates between genotypes for the HCV NS3 protein. Blackdots indicate residues with identical consensus amino acids for both genotypes; red dots indicate residues where the consensus amino acid differsbetween genotypes. Some residues are conserved for one genotype but display a high polymorphism rate for the alternative genotype (indicated bythe shaded areas). The two residues 1200 and 1403 indicate examples of residues with different consensus and with highly different polymorphismrates between the genotypes. Residue 1384 is highly polymorphic for both genotypes and has a different consensus. Residue 1444, which representsan escape site for an NS3-A*0101 epitope (Fig. 2) is polymorphic for both genotypes and shares the identical consensus amino acid. Similar plotsfor NS2, NS4, NS5a, and NS5b are provided in Supporting Fig. 2.
1024 RAUCH ET AL. HEPATOLOGY, October 2009

cance, the polymorphism rate in those individuals ex-pressing the relevant HLA type was clearly higher atadditional sites within the epitope (for example, position[P]1 and P2 for the NS2-957 HLA-B*3701; P2 and P3for the NS3-1395 HLA-B*0801 epitope; P1-P7 for theNS5B-2841 HLA-B*2705-epitope) (Fig. 2). Interest-ingly, the HLA-A*1101–associated viral polymorphismat NS3-1637 for genotype 3a falls within the describedNS3-1636 HLA-A*1101 epitope, but the HLA-A*1101-associated viral polymorphism for genotype 1a flanks thisepitope at 1635 (Fig. 2). Of note, the epitope predictionprogram SYFPEITHI21 predicts a nonamer epitope atpositions 1635-1643 for both genotypes, but not the pub-lished octamer epitope.
For the NS3-B*0801 and the NS5B-B*2705 epitopes,the Web-based epitope prediction program SYF-PEITHI21 predicted good epitope binding for both geno-types, mainly based on similar anchor residues.Nevertheless, genotype 3 sequences were completely con-served at sites with HLA-associated polymorphisms ingenotype 1 sequences. This may be due to poor immuno-genicity of the genotype 3 sequences despite good pre-dicted binding, divergent epitope processing pathways, orhigher fitness costs of potential escape mutations in geno-type 3 sequences. Furthermore, at least one substitutionwas present in every genotype 1–infected individual car-rying HLA-B*2705. Of the HLA-B*2705–positive indi-viduals infected with genotype 1, 60% carried more thanone substitution in the HLA-B*2705 epitope, whereasthere was almost complete conservation in genotype3–infected individuals.
From the seven putative viral adaptation sites identi-fied in our previous study within NS3,4 four sites (NS3-1397,1403,1444,1635) are replicated and associated withthe identical HLA allele using the more conservative cut-off in this study. Furthermore, in our recent study explor-ing the potential overlap between HLA-restrictedimmune pressure and anti-HCV drug resistance muta-tions,11 we tested for HLA associations at various drug-resistance sites. The HLA associations observed in thatstudy are reproduced in the analysis performed here (us-ing the Fisher’s exact test for the entire dataset) but are notincluded as they fall above the stringent cutoff set in thisstudy. Three sites shown in Tables 2 and 3 overlappedwith HLA-associated viral polymorphisms identified byTimm et al.5 (NS3-1398, NS3-1444, NS5B-2841). Inaddition, three sites identified in the report by Timm et al.were of borderline significance in our study (NS2-881,P � 0.04; NS3-1019, P � 0.07; NS5A-2153, P � 0.02).However, this comparison was hampered by the differentHLA typing resolution used.
Different Polymorphism Profiles for HCV Geno-types 1a and 3a Likely to Impact on Viral Presenta-tion to the Host’s Immune Response. In order to betterunderstand the limited overlap in the HLA-restricted im-mune pressure across the nonstructural proteins of geno-types 1 and 3, we investigated the genetic characteristicsof the two genotypes that could result in differences in theviral adaptation profiles observed in our analysis. We ini-tially examined the sequence variation between the geno-types, because this will have a significant effect on therepertoire of T cell epitopes that are presented by the hostHLA alleles. Viral polymorphism profiles for each of thenonstructural proteins were compared for the two HCVgenotypes, as shown in Fig. 3 and Supporting Fig. 2. Inthis analysis, we considered the rates of amino acid poly-morphism defined as the overall percentage of noncon-sensus amino acids at a particular site. This analysisrevealed substantial differences in the polymorphism ratesbetween the genotypes in that many polymorphic sites ingenotype 1 sequences were conserved in genotype 3 andvice versa (Fig. 3A,B).
We also noted that sites where the two HCV genotypesshared a common consensus amino acid were more thantwice as likely to be conserved at the population level(67% in genotype 1a; 74% in genotype 3a) comparedwith sites where the genotype consensus amino acids dif-fered (genotype 1a, 30%; genotype 3a, 36%) as shown inFig. 4A. Similarly, the majority of residues with highpolymorphism rates displayed different consensus aminoacids between the genotypes (Fig. 4B). Hence, the bulk ofintragenotype variation occurs at sites where the consen-sus sequence varies between the genotypes. These geno-type differences are likely to result in substantial variationin the escape potential of sites across the viral genome toHLA-restricted immune pressure.
Divergent Consensus Sequences, ProteasomalCleavage Sites, and Selective Pressures Between Ge-notypes 1a and 3a. Overall, the consensus amino acidsequence differed between HCV genotypes 1a and 3a at515 of 2,147 residues examined (24%; Supporting Table1). For a proportion of these sites (32%), the variantamino acid for one genotype was the consensus for theother. However, for many sites the mutational pathwayfor the genotypes was distinct. We also identified a largenumber of sites where different consensus sequence vari-ation was predicted to abrogate proteasomal cleavage (55for genotype 1a, 47 for genotype 3a; Supporting Table 1).Furthermore, when we tested for evidence of positive se-lection (using the Single-Likelihood Ancestor Countingalgorithm implemented in the program HyPhy26) actingat sites across the nonstructural proteins, only five out of34 sites were common to both genotypes. Overall, the
HEPATOLOGY, Vol. 50, No. 4, 2009 RAUCH ET AL. 1025

large number of different consensus amino acids and pre-dicted processing sites and the divergent selective pres-sures between the genotypes are likely to result indifferences in the repertoire of HLA-restricted viralepitopes across the genomes (see Supporting Material fora more detailed analysis).
Discussion
In this study, we have shown substantial differences inthe patterns of viral adaptation to HLA-restricted im-mune pressure between HCV genotypes 1 and 3. Fromthe 51 sites with HLA-associated viral polymorphisms,
Fig. 4. Residues with different consensus amino acids between genotypes are more polymorphic than residues with identical consensus. (A) Thepolymorphism rates of all residues from HCV-NS2 to HCV-NS5B are shown for residues where the consensus is identical and for residues where theconsensus differs between the genotypes. The residues are sorted by polymorphism rate, conserved residues are completely conserved in allindividuals. (B) The proportion of residues with different consensus between the genotypes is indicated on the y-axis for different polymorphism rateswithin a genotype on the x-axis. The majority of highly polymorphic residues display different consensus amino acids between the genotypes.
1026 RAUCH ET AL. HEPATOLOGY, October 2009

there were only two sites with the identical viral polymor-phism and HLA type between HCV genotypes 1a and 3a.In a large majority of sites with significant HLA associa-tions, the alternative genotype was either entirely con-served or there were no polymorphisms in individualscarrying the relevant HLA allele (Fig. 1). More directcomparisons of HCV amino acid variation within pub-lished epitopes (Fig. 2) identified similarly distinct poly-morphism profiles between HCV genotypes. Together,these findings suggested that the patterns of viral adapta-tion to the host’s immune pressure vary considerably be-tween the genotypes. This supported the hypothesis thatviral escape is driven by the host’s immune pressure andrestricted by viral (genotype) characteristics.
This study also served to highlight the diverse mecha-nisms by which viral evasion of CTL responses may beeffectively achieved, and the potential role of genotypesequence variation in these processes. First, the substantialdifferences in the consensus sequences for HCV geno-types 1a and 3a can abrogate HLA binding to criticalanchor residues within viral epitopes, as suggested by thecomplete abrogation of predicted HLA-B*3701 bindingto the NS2 957-964 epitope in genotype 3a shown in Fig.2. Second, genetic distance and physicochemical proper-ties may have an important influence on genotype-specificmutational pathways resulting in differences in the escapepotential of sites across the genome between the geno-types (Supporting Table 1). This is also highlighted inFig. 4, which shows that at sites of high intragenotypevariation the consensus amino acid of the two genotypestend to differ, implying divergent mutational potentialbetween the genotypes. Third, the divergent sequencecontext flanking immunogenic epitopes may have a sub-stantial impact on epitope processing, in keeping with thefinding that �20% of genotype-associated amino acidvariants are predicted to influence proteasomal cleavage oftarget peptides. Fourth, genotype-specific polymor-phisms outside the anchor positions may impair the rec-ognition of the HLA-epitope complex by CD8� T cells.
Dazert and colleagues25 have recently demonstratedthat CTL escape mutations within the HLA-B27 NS5B-2841 epitope occur in the majority of individuals carryingHLA-B*2705, but spare the binding anchors at positions2 and 9. The nearly complete conservation of the anchorpositions (P9 more so than P2) irrespective of carriage ofthe restricting allele for both genotypes (Fig. 2) reflects thesubstantial fitness cost of polymorphisms at these sites.25
However, more than one polymorphism was present inmost HLA-B*2705–positive genotype 1–infected indi-viduals, in line with the previous observation that clus-tered mutations outside the anchor position are requiredfor efficient escape.25 In contrast, a single mutation at
position 9 appears sufficient to abrogate T cell responsesin the A*0101–restricted NS3-1436 epitope8 (Fig. 2).
These analyses also indicated that the evolution ofadaptive HCV variants within the nonstructural proteinswas set against a background of overall negative selection,in keeping with the wide array of functional roles thatthese compact and tightly integrated proteins serve (Sup-porting Material).27 The fact that viral adaptation ishighly constrained may help to explain why persistentinfection is not always established following HCV infec-tion, and also suggests that viral adaptation to HLA-re-stricted CTL could frequently incur costs to viralreplicative capacity.
Given the similar function and structure of nonstruc-tural HCV proteins in genotypes 1 and 3, the large num-ber of amino acid differences in the genotype consensussequences might be surprising. However, we found that ina large proportion of these sites (32%) an intragenotypeamino acid polymorphism for one genotype was the con-sensus for the alternative genotype. This suggested thatfunctional constraints within the tightly integrated HCVprotein structure are likely to limit the possible alternativeamino acids that are compatible with the function of theprotein, and that mutational pathways would result in anaccumulation of alternative amino acids that were closestto the consensus sequence. In this respect, at some sitesthe consensus of one genotype might represent the escapevariant of the alternative genotype.
This study has some limitations. First, it is likely thatour analysis did not detect relevant HLA-associated viralpolymorphisms for rare HLA alleles due to low numbersof individuals carrying the respective allele. Second, theinclusion of a substantial proportion (40%) of individualscoinfected with HIV might influence the degree of HLA-associated viral polymorphisms as severe HIV-inducedimmunosuppression is associated with lower HCV-spe-cific T cell responses.16,17 However, the large majority(90%) of the coinfected individuals in this study had aCD4� T cell count above 200 cells/�L. We would there-fore expect that the HLA-restricted immune pressure inthe coinfected individuals would be similar to that formonoinfected individuals. In addition, HCV infectionprecedes HIV infection in the large majority of cases.28
Therefore, escape mutations that are established duringacute infection are likely to be under similar immunepressure in HCV-monoinfected and HIV/HCV-coin-fected individuals. Third, although we checked for phy-logenetic relatedness, such adjustments are alwaysestimations, and one cannot exclude the possibility that insome instances an association between an HLA allele andviral polymorphism may still retain an element of foundereffect confounding. Fourth, in many instances the HLA-
HEPATOLOGY, Vol. 50, No. 4, 2009 RAUCH ET AL. 1027

restricted polymorphisms represent putative escape mu-tations within potential epitopes that have not beenexperimentally confirmed. However, all these limitationswould affect both genotypes equally so the importantconclusion that the selection pressures differ markedlybetween genotypes should not be substantially affected.
In summary, we have identified major differences be-tween HCV genotypes 1a and 3a in the patterns of HLA-associated viral polymorphisms across the nonstructuralproteins indicating largely independent, nonoverlappingT cell responses. Differences in the polymorphism pro-files and consensus amino acids between the genotypescorrelated with the lack of overlap in viral adaptation sites.Accordingly, sequence content is highly relevant in deter-mining HLA-restricted immune pressure within the ge-notypes, and the epitope repertoire generated for the twogenotypes in the same HLA population would likely havelimited overlap. The two adaptation sites that overlapbetween the genotypes were found in areas of conserva-tion, and one falls within an HLA-A1–restricted epitopethat has recently been shown to induce CTL responses inindividuals infected with genotypes 1 and 3.8 Cross-gen-otype epitopes are likely to occur in predominantly con-served areas with similar consensus sequence. Theseresults may provide some explanation as to the relativelack of cross-protection between genotypes in HCV-in-fected individuals29,30 and may be relevant to the differ-ence in the response rates to immunomodulatory-basedtherapy with interferon-� and ribavirin.9,10,31
The different patterns of in vivo immune pressure onHCV as reflected by the limited overlap in viral adapta-tion between the genotypes also have implications forHCV vaccine design. T cell vaccine immunogens thatcontain escaped amino acids may be poorly immunogenicin some HLA contexts within a host population. In thisrespect, knowledge of viral escape patterns is importantfor generating immunogenic T cell vaccines. The limitedoverlap in putative escape patterns between genotypes ob-served in this study suggests that this information has tobe generated for the genotypes separately.
Acknowledgment: We thank the individuals and clin-ical staff at the different sites that have contributed to thisstudy and the Swiss HIV Cohort Study. We also thankDr. Larry Park and Kiloshni Naidoo for their contribu-tion. The members of the Swiss HIV Cohort Study are M.Battegay, E. Bernasconi, J. Boni, HC Bucher, P. Burgis-ser, A. Calmy, S. Cattacin, M. Cavassini, R. Dubs, M.Egger, L. Elzi, P. Erb, M. Fischer, M. Flepp, A. Fontana,P. Francioli (President of the Swiss HIV Cohort Study,Centre Hospitalier Universitaire Vaudois, CH-1011 Lau-sanne), H. Furrer (Chairman of the Clinical and Labora-tory Committee), C. Fux, M. Gorgievski, H. Gunthard
(Chairman of the Scientific Board), H. Hirsch, B. Hir-schel, I. Hosli, C. Kahlert, L. Kaiser, U. Karrer, C. Kind,T. Klimkait, B. Ledergerber, G. Martinetti, B. Martinez,N. Muller, D. Nadal, M. Opravil, F. Paccaud, G. Panta-leo, A. Rauch, S. Regenass, M. Rickenbach (Head of DataCenter), C. Rudin (Chairman of the Mother & ChildSubstudy), P. Schmid, D. Schultze, J. Schupbach, R.Speck, P. Taffe, P. Tarr, A. Telenti, A. Trkola, P. Ver-nazza, R. Weber, and S. Yerly.
References1. Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, et
al. Consensus proposals for a unified system of nomenclature of hepatitis Cvirus genotypes. HEPATOLOGY 2005;42:962-973.
2. Klenerman P, Hill A. T cells and viral persistence: lessons from diverseinfections. Nat Immunol 2005;6:873-879.
3. Bowen DG, Walker CM. Mutational escape from CD8� T cell immu-nity: HCV evolution, from chimpanzees to man. J Exp Med 2005;201:1709-1714.
4. Gaudieri S, Rauch A, Park LP, Freitas E, Herrmann S, Jeffrey G, et al.Evi-dence of viral adaptation to HLA class I-restricted immune pressure inchronic hepatitis C virus infection. J Virol 2006;80:11094-11104.
5. Timm J, Li B, Daniels MG, Bhattacharya T, Reyor LL, Allgaier R, et al.Human leukocyte antigen-associated sequence polymorphisms in hepatitisC virus reveal reproducible immune responses and constraints on viralevolution. HEPATOLOGY 2007;46:339-349.
6. Ward S, Lauer G, Isba R, Walker B, Klenerman P. Cellular immuneresponses against hepatitis C virus: the evidence base 2002. Clin Exp Im-munol 2002;128:195-203.
7. Bowen DG, Walker CM. Adaptive immune responses in acute and chronichepatitis C virus infection. Nature 2005;436:946-952.
8. Neumann-Haefelin C, Frick DN, Wang JJ, Pybus OG, Salloum S, NarulaGS, et al. Analysis of the evolutionary forces in an immunodominant CD8epitope in hepatitis C virus at a population level. J Virol 2008;82:3438-3451.
9. Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FLJr, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virusinfection. N Engl J Med 2002;347:975-982.
10. Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M,Reindollar R, et al. Peginterferon alfa-2b plus ribavirin compared withinterferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C:a randomised trial. Lancet 2001;358:958-965.
11. Gaudieri S, Rauch A, Pfafferot K, Barnes E, Cheng W, McCoughan G, etal. Hepatitis C virus drug resistance and immune-driven adaptations: rel-evance to new anti-viral therapy. HEPATOLOGY 2009;49:1069-1082.
12. Bhattacharya T, Daniels M, Heckerman D, Foley B, Frahm N, Kadie C, etal. Founder effects in the assessment of HIV polymorphisms and HLAallele associations. Science 2007;315:1583-1586.
13. Kaufman L, Rousseeuw PJ. Finding Groups in Data: An Introduction toCluster Analysis. New York: John Wiley & Sons, 1990.
14. Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the falsediscovery rate in behavior genetics research. Behav Brain Res 2001;125:279-284.
15. Storey JD, Tibshirani R. Statistical significance for genomewide studies.Proc Natl Acad Sci U S A 2003;100:9440-9445.
16. Kim AY, Lauer GM, Ouchi K, Addo MM, Lucas M, Wiesch JS, et al.Themagnitude and breadth of hepatitis C virus-specific CD8� T cells dependon absolute CD4� T-cell count in individuals coinfected with HIV-1.Blood 2005;105:1170-1178.
17. Kim AY, Schulze zur WJ, Kuntzen T, Timm J, Kaufmann DE, Duncan JE,et al. Impaired hepatitis C virus-specific T cell responses and recurrenthepatitis C virus in HIV coinfection. PLoS Med 2006;3:e492.
18. Leslie A, Kavanagh D, Honeyborne I, Pfafferott K, Edwards C, Pillay T, etal. Transmission and accumulation of CTL escape variants drive negative
1028 RAUCH ET AL. HEPATOLOGY, October 2009

associations between HIV polymorphisms and HLA. J Exp Med 2005;201:891-902.
19. Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S,et al. Dominant influence of HLA-B in mediating the potential co-evolu-tion of HIV and HLA. Nature 2004;432:769-775.
20. Neumann-Haefelin C, McKiernan S, Ward S, Viazov S, Spangenberg HC,Killinger T, et al. Dominant influence of an HLA-B27 restricted CD8� Tcell response in mediating HCV clearance and evolution. HEPATOLOGY
2006;43:563-572.21. Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S.
SYFPEITHI: database for MHC ligands and peptide motifs.Immunoge-netics 1999;50:213-219.
22. Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potentialHLA-A2 binding peptides based on independent binding of individualpeptide side-chains. J Immunol 1994;152:163-175.
23. Timm J, Lauer GM, Kavanagh DG, Sheridan I, Kim AY, Lucas M, etal.CD8 epitope escape and reversion in acute HCV infection. J Exp Med2004;200:1593-1604.
24. Salloum S, Oniangue-Ndza C, Neumann-Haefelin C, Hudson L, GiuglianoS, aus dem SM, et al. Escape from HLA-B*08-restricted CD8 T cells byhepatitis C virus is associated with fitness costs. J Virol 2008;82:11803-11812.
25. Dazert E, Neumann-Haefelin C, Bressanelli S, Fitzmaurice K, Kort J,Timm J, et al. Viral fitness cost and CD8� T cell cross-recognition limitescape from a protective HLA-B27 restricted response to hepatitis C virus.J Clin Invest 2009;199:376-386.
26. Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylog-enies. Bioinformatics 2005;21:676-679.
27. Campo DS, Dimitrova Z, Mitchell RJ, Lara J, Khudyakov Y. Coordinated evolu-tion of the hepatitis C virus. Proc Natl Acad Sci U S A 2008;105:9685-9690.
28. Villano SA, Vlahov D, Nelson KE, Lyles CM, Cohn S, Thomas DL.Incidence and risk factors for hepatitis C among injection drug users inBaltimore, Maryland. J Clin Microbiol 1997;35:3274-3277.
29. Farci P, Alter HJ, Govindarajan S, Wong DC, Engle R, Lesniewski RR, etal.Lack of protective immunity against reinfection with hepatitis C virus.Science 1992;258:135-140.
30. Accapezzato D, Fravolini F, Casciaro MA, Paroli M. Hepatitis C flare dueto superinfection by genotype 4 in an HCV genotype 1b chronic carrier.Eur J Gastroenterol Hepatol 2002;14:879-881.
31. Zinkernagel AS, von Wyl V, Ledergerber B, Rickenbach M, Furrer H,Battegay M, et al. Eligibility for and outcome of hepatitis C treatment ofHIV-coinfected individuals in clinical practice: the Swiss HIV cohortstudy. Antivir Ther 2006;11:131-142.
HEPATOLOGY, Vol. 50, No. 4, 2009 RAUCH ET AL. 1029